
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple magnesium alkoxides: synthesis, molecular structure, and catalytic behaviour in the ring-opening polymerization of lactide and macrolactones and in the copolymerization of maleic anhydride and propylene oxide†
Duleeka
Wannipurage
a,
Sara
D'Aniello
b,
Daniela
Pappalardo
 c,
Lakshani Wathsala
Kulathungage
a,
Cassandra L.
Ward
d,
Dennis P.
Anderson
d,
Stanislav
Groysman
*a and
Mina
Mazzeo
*b
c,
Lakshani Wathsala
Kulathungage
a,
Cassandra L.
Ward
d,
Dennis P.
Anderson
d,
Stanislav
Groysman
*a and
Mina
Mazzeo
*b
aDepartment of Chemistry, Wayne State University, 5101 Cass Ave., Detroit, MI 48202, USA. E-mail: groysman@wayne.edu
bDepartment of Chemistry and Biology “A. Zambelli” University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy. E-mail: mmazzeo@unisa.it
cDipartimento di Scienze e Tecnologie, Università del Sannio, via de Sanctis snc, 82100 Benevento, Italy
dLumigen Instrument Center, Wayne State University, 5101 Cass Avenue, Detroit, Michigan 48202, USA
First published on 9th May 2023
Abstract
The synthesis of two chiral bulky alkoxide pro-ligands, 1-adamantyl-tert-butylphenylmethanol HOCAdtBuPh and 1-adamantylmethylphenylmethanol HOCAdMePh, is reported and their coordination chemistry with magnesium(II) is described and compared with the coordination chemistry of the previously reported achiral bulky alkoxide pro-ligand HOCtBu2Ph. Treatment of n-butyl-sec-butylmagnesium with two equivalents of the racemic mixture of HOCAdtBuPh led selectively to the formation of the mononuclear bis(alkoxide) complex Mg(OCAdtBuPh)2(THF)2. 1H NMR spectroscopy and X-ray crystallography suggested the selective formation of the C2-symmetric homochiral diastereomer Mg(OCRAdtBuPh)2(THF)2/Mg(OCSAdtBuPh)2(THF)2. In contrast, the less sterically encumbered HOCAdMePh led to the formation of dinuclear products indicating only partial alkyl group substitution. The mononuclear Mg(OCAdtBuPh)2(THF)2 complex was tested as a catalyst in different reactions for the synthesis of polyesters. In the ROP of lactide, Mg(OCAdtBuPh)2(THF)2 demonstrated very high activity, higher than that shown by Mg(OCtBu2Ph)2(THF)2, although with moderate control degrees. Both Mg(OCAdtBuPh)2(THF)2 and Mg(OCtBu2Ph)2(THF)2 were found to be very effective in the polymerization of macrolactones such as ω-pentadecalactone (PDL) and ω-6-hexadecenlactone (HDL) also under mild reaction conditions that are generally prohibitive for these substrates. The same catalysts demonstrated efficient ring-opening copolymerization (ROCOP) of propylene oxide (PO) and maleic anhydride (MA) to produce poly(propylene maleate).
Introduction
Oil derived plastics are involved in almost every aspect of everyday life. However, their very broad utilization, combined with a lack of a forward-thinking strategy regarding their end life, has caused serious environmental pollution. An important challenge for the future is to improve the sustainability of plastics by designing new bio-based materials obtained by low environmental impact procedures.1,2 In this context, aliphatic polyesters represent the most promising materials. Depending on the structure of the repeating units, they show very different properties. Aliphatic polyesters having long methylene sequences between ester functionalities are highly hydrophobic materials with tensile properties similar to those of linear low-density poly(ethylene) (LLDPE), and may represent a biodegradable alternative to LLDPE.3–5 The useful synthetic routes for their preparation include the polycondensation of fatty acids6,7 and the ring-opening polymerization (ROP) of macrolactones promoted by metal-based catalysts,8,9 organic catalysts,10,11 or enzymes.12–15The chain-growth ROP of macrolactones offers the advantage of a good control over macromolecular parameters such as molecular weights and their dispersity, and end-group fidelity.8,11,16,17 Unfortunately, macrolactones are insufficiently reactive monomers because they typically do not exhibit ring strain. Therefore, they are rarely polymerized using traditional ROP catalysts and drastic reaction conditions are generally required.18,19 To date, a relatively few metal-based catalysts active in the ROP of macrolides have been reported, and most of them are based on early transition metals20 and main group metals (Zn, Al, Ca, and Mg).8,21,22
An alternative method for the synthesis of polyesters is the ring opening copolymerization of epoxides and anhydrides.23 The combination of two distinct monomers to form the repeating units of a polyester chain allows facile access to materials with properties and functionalities not easily achievable by the strict ROP of lactones.24–26 This synthetic methodology is particularly attractive given the large tolerance toward functional groups within the monomers offering a great opportunity for the synthesis of functionalized polymers.27 Recently, block co-polyesters have been achieved by a chemoselective switch catalysis between the ring opening copolymerization of epoxides and anhydrides and the ROP of lactones or macrolactones.28,29
Generally, the most investigated catalysts for the ROP of cyclic esters and for the ROCOP of epoxides and anhydrides are heteroleptic complexes of non-toxic metals such as magnesium30–32 and zinc,33–35 in which the metal center is coordinated to electronically and sterically tailored ancillary ligands and labile ligand/s that often behave as initiating groups; while this strategy offers the benefits of a very efficient control over polymerization behavior (such as tacticity),36–38 its disadvantages include somewhat less sustainable nature of the catalyst because of the required multistep synthesis of ancillary ligands. In contrast, recent studies have demonstrated that simple metal-alkoxides or metal-amides, which are commonly used as metal precursors in coordination chemistry, may represent a more sustainable route for polyester synthesis39–44 and/or their degradation by alcoholysis.45,46
In 2012, Chen and Cui reported a very simple binary catalyst MgnBu2/Ph2CHOH that showed very high activity in the ROP of lactide (LA), in the presence of a large excess amount of alcohol.47 In this system the choice of alcohol with bulky substituents proved to be crucial to promote immortal processes. Subsequently, Dove48 and Nifant'ev49 described the use of simple metal alkoxides such as magnesium 2,6-di-tert-butyl-4-methylphenoxide (Mg(BHT)2(THF)2) for the ‘immortal’ ring-opening polymerization of caprolactone (ε-CL) and pentadecalactone (PDL).
Our research groups have previously described the synthesis of a simple magnesium alkoxide Mg(OCtBu2Ph)2(THF)2 and its reactivity in the polymerization of lactides and the ring-opening copolymerization (ROCOP) of cyclic anhydrides with epoxides demonstrating high efficiency and control in the latter process.40 As the mononuclear complex Mg(OCtBu2Ph)2(THF)2 exhibited very high reactivity, we became interested in understanding whether a different steric encumbrance of the alkoxide ligand may affect the reactivity of the resulting Mg(OR)2 pre-catalyst in the ROP of lactones and lactide. Following these findings, we extended our investigations to additional monomers. Furthermore, we became interested in exploring whether a chiral alkoxide can lead to (1) a well-defined C2-symmetric structure of a pre-catalyst which could (2) lead to a stereoselective polymerization.
Herein we reported the synthesis of two new chiral bulky alkoxide ligands related to [OCtBu2Ph], [OCtBuAdPh] and [OCtBuMePh]. We demonstrated that while racemic [OCtBuAdPh] enabled the clean formation of the homochiral C2-symmetric complex Mg(OCtBuAdPh)2(THF)2, [OCtBuMePh] did not exhibit well-defined coordination chemistry. The new complex Mg(OCtBuAdPh)2(THF)2, along with the previously reported Mg(OCtBu2Ph)2(THF)2, was investigated as a catalyst in the polymerization of lactide, caprolactone and two macrolactones namely ω-pentadecalactone (PDL) and ω-6-hexadecenlactone (HDL). Both complexes, in combination with a primary alcohol, were also tested as catalysts for the copolymerization of maleic anhydride and propylene oxide to produce poly(propylene maleate). This polymer can be easily isomerized into poly(propylene fumarate), a 3D printable material to produce thin films and scaffolds that can be modified with bioactive groups by post-polymerization and post-printing functionalization for biomedical applications.27
Results and discussion
Design and synthesis of chiral alkoxide ligands [OCtBuAdPh] and [OCtBuMePh]
We have previously reported the synthesis of [OCtBu2Ph] via the reaction of PhLi with hexamethylacetone and the subsequent synthesis of its transition metal and magnesium complexes, all exhibiting the same mononuclear M(OCtBu2Ph)2(THF)2 structure.50–52 In an attempt to investigate the formation and coordination chemistry of asymmetric alkoxide ligands, we targeted two bulky asymmetric alkoxide ligands [OCAdtBuPh] and [OCAdMePh]. Both ligands feature a very bulky 1-adamantyl substituent and a planar phenyl group; the ligands differ by the third substituent: a relatively large tert-butyl group vs. smaller methyl. Given the (effectively) C2v-symmetric structures of M(OCtBu2Ph)2(THF)2 complexes, it is anticipated that the chiral ligands would form diastereomerically pure C2-symmetric complexes M(OCRR1R2R3)2(THF)2 and M(OCSR1R2R3)2(THF)2. Based on the quadrant model of the transmission of asymmetry, the resulting diastereomerically pure racemic C2-symmetric complexes should be capable of stereoselective polymerization if the catalysis takes place in the THF positions, and no exchange of the alkoxide ligands between different enantiomers occurs.The pro-ligands were synthesized via the intermediacy of the corresponding ketones (1-adamantyl tert-butyl ketone and 1-adamantyl methyl ketone), which can be obtained by the reaction of 1-adamantyl carboxylic acid with the corresponding lithium reagent (Scheme 1). The synthesis of the intermediate ketones and HOR2 was achieved by a modification of the previously reported procedures.53 Treatment of the ketones with phenyl lithium formed racemic HOCAdtBuPh (HOR2) and HOCAdtBuPh (HOR3) in 63% and 74% yields, respectively. We note that a different synthetic strategy toward HOR3 (via the treatment of methyl phenyl ketone via in situ obtained adamantyl lithium) was recently reported, using a flow microreactor system.54
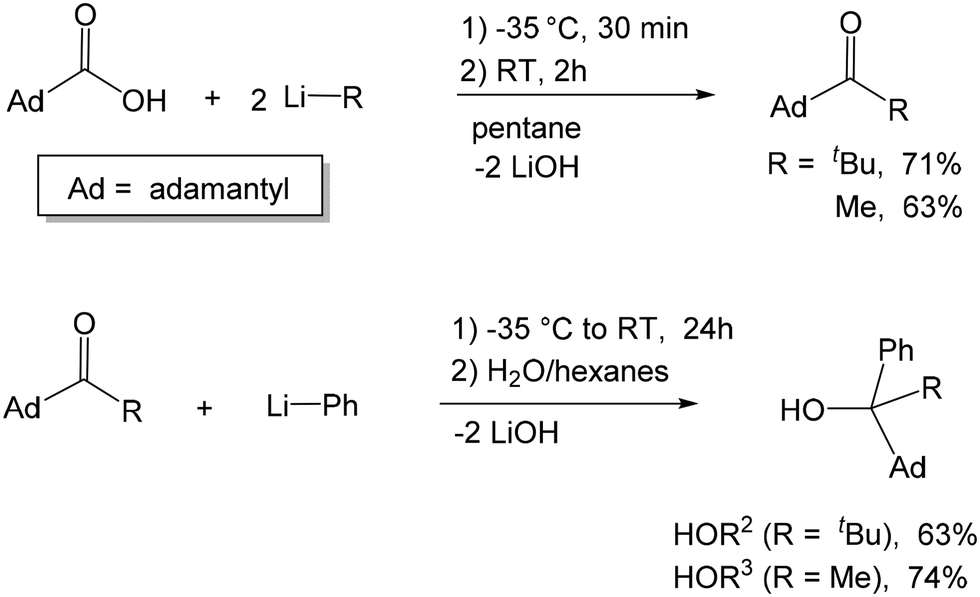 | ||
| Scheme 1 Syntheses of the racemic alkoxide pro-ligands HOCAdtBuPh and HOAdMePh. | ||
The pro-ligands were characterized by 1H and 13C NMR spectroscopy, IR and HRMS. The structure of HOR2 was also confirmed by X-ray structure determination. HOR2 crystallized as a racemic mixture in the polar space group Pna21.
Coordination chemistry of HOR2 and HOR3 with magnesium
The coordination chemistry of HOR2 and HOR3 was explored by treating n-butyl-sec-butylmagnesium (0.7 M solution in hexane) with two equivalents of the racemic mixture of HOR2 and HOR3 (Scheme 2). The previously reported synthesis of Mg(OR1)2(THF)2 (1) is also presented in Scheme 2. The reaction of Mg(n-Bu)(sec-Bu) with HOR2 led to the clean formation of Mg(OR2)2(THF)2 (2), which was isolated as colorless crystals from CH2Cl2 in 84% yield. 2 was characterized by NMR spectroscopy, X-ray crystallography, and elemental analysis. Elemental analysis confirms Mg(OR2)2(THF)2 formulation. Most significantly, the 1H NMR spectrum suggests the formation of a single diastereomer in solution.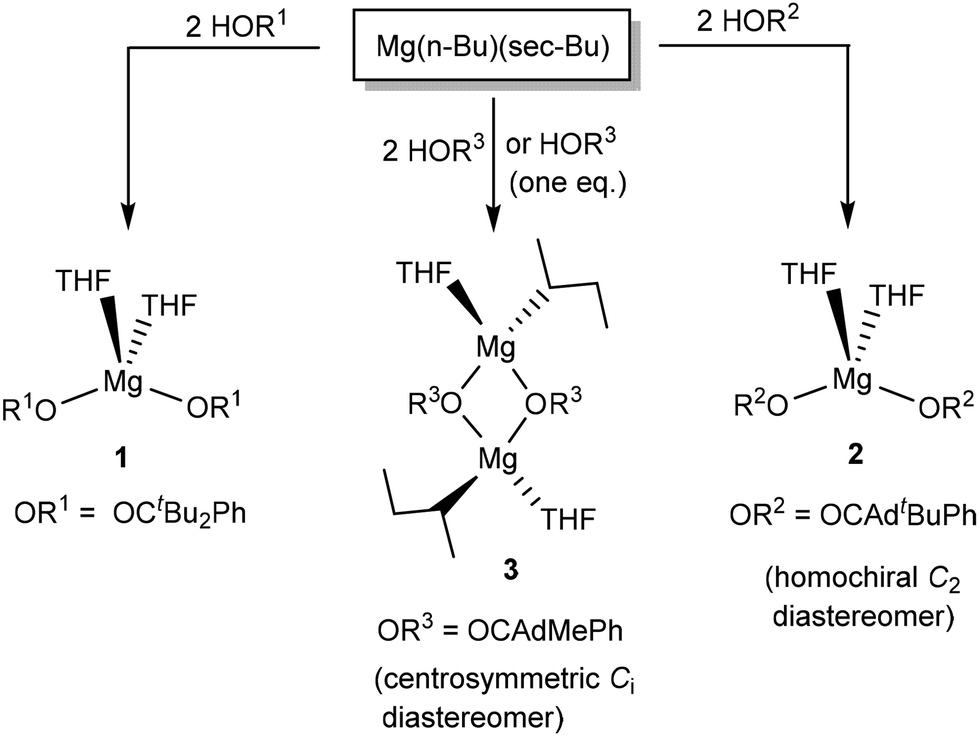 | ||
| Scheme 2 Reactivity of achiral alkoxides HOR1 and chiral (racemic) alkoxides HOR2 and HOR3 with n-butyl-sec-butylmagnesium. | ||
As a general rule, the combination of a racemic alkoxide mixture can lead to two different diastereomers in the resulting Mg(OR2)2(THF)2 product: a homochiral isomer of an approximate C2 symmetry and a meso isomer of an approximate Cs symmetry. Due to their different physical properties, different diastereomers should give rise to different NMR spectra. However, the 1H NMR spectrum of 2 suggests the presence of a single species in solution, exhibiting one singlet for the tBu groups (1.38 ppm), two signals for the THF ligands (3.84 and 1.28 ppm) and five aromatic signals for the alkoxide phenyl group. Five different aromatic signals for the phenyl group are generally consistent with its restricted rotation, as previously described for Mg(OR1)2(THF)2 (1, OR1 = OCtBu2Ph). This pattern is consistent with the presence of a single diastereomer in solution. The presence of a single species in solution indicates the chiral resolution of the ligands to create a single diastereomer.
The solid state structure of 2 is consistent with the solution structure, demonstrating the formation of a single homochiral diastereomer of C2 symmetry (Fig. 1). 2 crystallizes in the centrosymmetric group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; both enantiomers (RR and SS) are found in the unit cell. The structure of the RR enantiomer is presented in Fig. 1 and the selected bond distances and angles are provided in Fig. 1 caption.
; both enantiomers (RR and SS) are found in the unit cell. The structure of the RR enantiomer is presented in Fig. 1 and the selected bond distances and angles are provided in Fig. 1 caption.
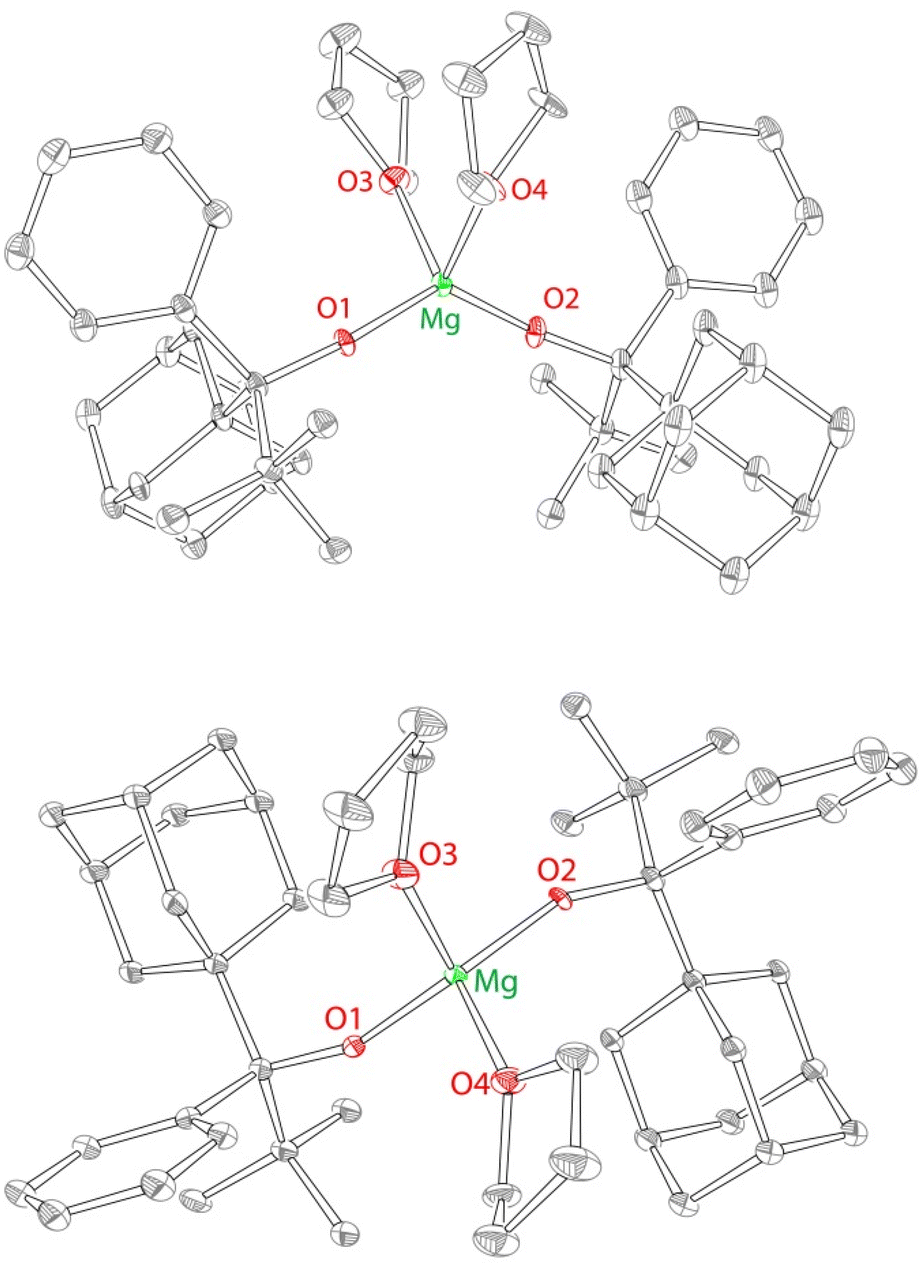 | ||
| Fig. 1 X-ray structure (50% probability ellipsoids) of the side view (left) and the top view (right) of 2. H atoms and the co-crystallized (disordered) CH2Cl2 solvent are omitted for clarity. Only one enantiomer (RR) of the structure is shown. Another enantiomer (SS) can be generated by the inversion operation (P). Selected bond distances (Å) and angles (°) for 2: Mg O1 1.842(4), Mg O2 1.831(4), O1 Mg O2 131.2(2), O3 Mg1 O4 90.5(1). | ||
Overall, the structure of 2 (Mg(OR2)2(THF)2) is in line with all previous structures of M(OR)2(THF)2 complexes,50–52 including a closely related magnesium complex Mg(OR1)2(THF)2 (1).40 Similarly to 1, complex 2 exhibits a distorted tetrahedral geometry, with a narrow THF–Mg–THF angle of 90.5(1)°, and a broader RO–Mg–OR/RO–Mg–C angle of 131.2(2)°. The examination of the structure of 2 clearly indicates that it is approximately C2-symmetric (see Fig. 1) although the C2 symmetry is not crystallographic. The C2 symmetry of 2 implies the exclusive formation of the homochiral diastereomer. We postulate that the C2-symmetric homochiral (RR and SS) diastereomer forms as a result of the steric gradient of the ligand, which pushes large adamantyl groups away from each other. One of the enantiomers (RR) is shown in Fig. 1; the presence of the other enantiomer is implied by the centrosymmetric nature of the space group (P).
In contrast to the reactivity of HOR2, the reaction of HOR3 (HOCAdMePh, two equivalents) with n-butyl-sec-butylmagnesium led to the formation of the product demonstrating broad NMR resonances. Recrystallization of the crude product produced colorless crystals of complex 3. 3 is a dimeric complex of Mg2(OR3)2(sec-Bu)2(THF)2 composition (Scheme 2), which was characterized by X-ray crystallography, elemental analysis, and NMR.
The solid-state structure of 3 reveals incomplete substitution of the alkyl ligands in the Mg(n-Bu)(sec-Bu) precursor (Fig. 2). The reaction of Mg(n-Bu)(sec-Bu) with one equivalent of HOR3 similarly formed complex 3. We have previously shown that the protonolysis of the alkyl groups in Mg(n-Bu)(sec-Bu) with HOR1 takes place in two steps, with the more sterically accessible n-butyl group being replaced first.32
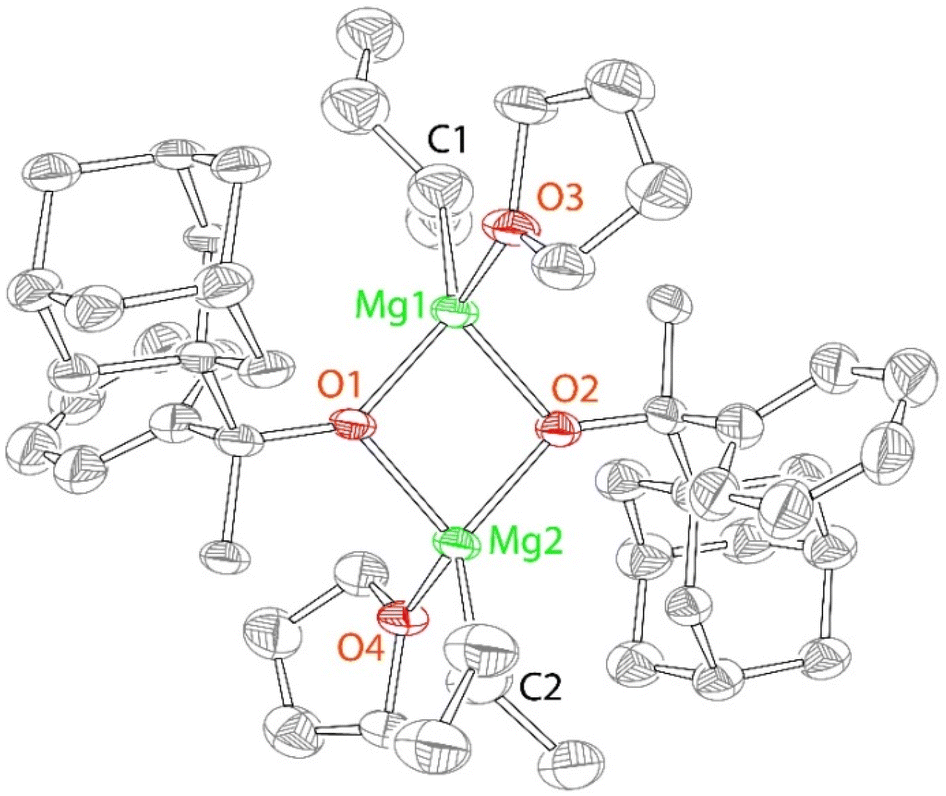 | ||
| Fig. 2 X-ray structure (50% probability ellipsoids) of 3. H atoms are omitted for clarity. Only one enantiomer (RR) of the structure is shown. Selected bond distances (Å) and angles (°) for 3: Mg1 O1 1.987(7), Mg1 O2 2.000(8), Mg1 O3 2.081(8), Mg1 C1 2.16(1), O1 Mg1 O2 84.4(3), O1 Mg2 O2 86.1(3), O3 Mg1 C1 128.0(5). | ||
Similarly, HOR3 replaces the n-butyl group first. However, the reaction of Mg(n-Bu)(sec-Bu) with one equivalent of HOR1 produced a mononuclear complex Mg(OR1)(sec-Bu)(THF)2, whereas 3 is a dimer, in which the alkoxides are bridging, and the sec-butyl and THF ligands are terminal. It is possible that it is due to the formation of the dimer that only one of the alkyl groups undergoes facile substitution in the present case. We also note that the reaction of mononuclear Mg(OR1)(sec-Bu)(THF)2 with one equivalent of HOR1 yielded complex 1, whereas no reaction between dinuclear 3 and HOR3 is observed at room or increased temperature (up to 80 °C) in toluene (Fig. S48†).
The close examination of the structure of 3 suggests that the presence of the less sterically demanding methyl group (that points towards sec-Bu and THF) is responsible for the dimeric structure. The reduced steric effect of the methyl-substituted [OR3] pro-ligand enables a relatively sharp angle (85 ± 1°) between the alkoxides at the same magnesium center. Finally, in a sharp contrast to the C2-symmetric structure of 2, the symmetry of 3 is Ci (non-crystallographic), implying the presence of both R and S enantiomers in the same structure. Crystalline and analytically pure 3 still exhibits a relatively broad and complicated 1H NMR spectrum, which is consistent with the presence of multiple species in solution.
It is possible that 3 undergoes monomer–dimer equilibrium in solution; such an equilibrium could further lead to the formation of other species (such as the homochiral dimer, or the mixture of Mg(OR3)2(THF)2 and Mg(sec-Bu)2). 1H NMR in toluene-d8 at varying temperatures (25–80 °C) has also shown broad and uninformative spectra (see Fig. S26†). We have also investigated the nature of complex 3 in solution by DOSY. The complex was prepared at concentrations of 5 and 10 mM, and DOSY experiments were performed on each. The resulting diffusion data were consistent between the samples (Fig. S49†). This suggests that the complex is intact in the toluene solution, without a significant population of dissociated components. However, the rapid dimer–monomer–dimer equilibrium in solution leading to the exchange of alkoxide/THF ligands cannot be ruled out by this experiment; it is also likely to result in a broad NMR spectrum. In light of the less well-defined structure of 3 (compared with 1 and 2) in solution, its reactivity in polymerization was not investigated.
Polymerization of rac-lactide
We have previously reported that complex 1 was a highly reactive catalyst for the ROP of racemic lactide (rac-LA), although the control degree over the polymerization process was modest. Herein, we explored the reactivity of complex 2 that features bulkier and chiral alkoxides and compared its behavior with complex 1. The representative ROP results are summarized in Table 1.| Runa | Cat. | rac-LA (eq.) | BnOH (eq.) | Time (min) | Solvent | Conv.b (%) | M n (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 10 μmol of Mg, 10 mL of the solvent, and T = 25 °C (reaction times not optimized). b Determined by 1H NMR. c Experimental Mn and Đ values were determined by GPC analysis in THF using polystyrene standards corrected with the factor of 0.58. d 10 μmol of Mg T = 150 °C, technical grade L-LA. | ||||||||
| 1 | 2 | 100 | — | 4 | DCM | >99 | 3.0 | 3.30 |
| 2 | 1 | 100 | — | 60 | DCM | 56 | 4.7 | 2.26 |
| 3 | 2 | 200 | — | 4 | DCM | >99 | 5.4 | 3.08 |
| 4 | 2 | 300 | — | 4 | DCM | >99 | 7.6 | 2.04 |
| 5 | 1 | 300 | — | 60 | DCM | 43 | 4.1 | 2.56 |
| 6 | 2 | 600 | — | 10 | DCM | >99 | 5.5 | 2.31 |
| 7 | 2 | 1000 | — | 15 | DCM | >99 | 9.1 | 2.10 |
| 8 | 2 | 5000 | — | 15 | DCM | >99 | 30.2 | 1.78 |
| 9 | 2 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
— | 15 | DCM | 97 | 72.6 | 1.83 |
| 10 | 2 | 100 | — | 30 | Tol. | >99 | 3.9 | 3.12 |
| 11 | 2 | 200 | — | 60 | Tol. | >99 | 6.5 | 2.62 |
| 12 | 2 | 300 | — | 60 | Tol. | >99 | 14.5 | 2.15 |
| 13 | 1 | 300 | — | 60 | Tol. | 20 | 17.1 | 1.96 |
| 14 | 2 | 200 | 1 | 2 | DCM | >99 | 18.6 | 1.79 |
| 15 | 2 | 200 | 1 | 0.5 | DCM | 70 | 8.1 | 1.59 |
| 16 | 2 | 200 | 1 | 0.5 | THF | 52 | 41.9 | 2.40 |
| 17 | 2 | 200 | 5 | 0.5 | THF | 87 | 5.3 | 1.23 |
| 18d | 2 | 10000 |
10 | 60 | — | 77 | 7.6 | 1.81 |
| 19d | 2 | 5000 | 50 | 60 | — | 48 | 3.6 | 1.56 |
Initially, the reactivity of complex 2 was explored under the same reaction conditions used for complex 1 in our previous work: in CH2Cl2 solution (10 mL), at 25 °C, and using 10 μmol of the catalyst and varying lactide:catalyst ratios. The obtained results revealed a very high activity for catalyst 2 that was able to convert quantitatively up to 10000 equivalents of the monomer within 15 minutes reaching the remarkable turnover frequency (TOF) of 39000 h−1 (see run 9 of Table 1), a value that is fully comparable to the most active magnesium complexes reported in the literature.47,55–57 The catalytic activity of complex 2 is significantly higher than that obtained for complex 1 (compare run 1 with 2 and run 4 with 5 of Table 1, respectively)40 and for Mg(BHT)2THF2,58 suggesting that the steric encumbrance around the magnesium center in the [Mg(OR)2] precatalyst plays an important role in the catalytic activity.
It is possible that the presence of bulky alkoxide groups around magnesium disfavors aggregation phenomena that can occur in the polymerization medium, above all when an alcohol is used as the cocatalyst, as observed by Miller58 and Nifant'ev59 who described the formation of dimeric species by the reaction of Mg(BHT)2THF2 with benzyl alcohol.
As already observed for complex 1, the activity decreased dramatically when the polymerizations were performed in toluene solution (runs 10–13, Table 1), while a little decrease was noted in the presence of a coordinating solvent namely THF (see runs 16 and 17, Table 1). By adding one or more equivalents of benzyl alcohol as the initiator, the performance of catalyst 2 improved in both solvents, DCM (see runs 14 and 15, Table 1) and THF (see runs 16 and 17, Table 1).
Subsequently, catalyst 2 was tested under more challenging industrial-like conditions: bulk conditions, 150 °C, unpurified monomer (technical grade) and in the presence of a large excess of alcohol as a chain transfer agent to improve the productivity of the catalyst (runs 18 and 19, Table 1). Also, in this case, the catalyst preserved its high activity showing a TOF of 7700 h−1.
All polymers produced were characterized by 1H NMR, GPC and MALDI-ToF-MS analyses.
The microstructures of the resulting PLA samples were analyzed by 1H NMR spectroscopy. For all samples, despite the chiral nature of complex 2, the Pm values were not higher than 0.56, suggesting the lack of stereochemical control (Fig. S27†). However, no epimerization phenomenon was detected in the samples obtained with L-LA.
The molecular masses of the PLA samples obtained in the absence of alcohol showed values significantly lower than those expected (although they increased with the number of equivalents of the reacted monomer), and relatively high dispersities (1.59 < Đ < 3.30). These features are indicative of a not well controlled process.
The MALDI-ToF spectra of the samples obtained in the exclusive presence of magnesium complex 2 (run 1, Table 1) revealed a main distribution of peaks, with a spacing of 72 g mol−1, corresponding to the cyclic species derived from the extensive intramolecular transesterification reactions (Fig. S28†).
A control over the properties of the resulting polymer can be improved significantly by the use of a coordinating solvent THF, and in the presence of 5 equivalents of alcohol as a chain transfer agent (see run 17, Table 1). These polymerization conditions led to a relatively narrow dispersity (Đ = 1.23). The molecular masses, evaluated by GPC and NMR, were consistent with the theoretical values calculated considering the amount of added alcohol. We postulate that the presence of five equivalents of alcohol as a chain transfer agent enables fast and reversible exchange reactions between the active species and the dormant hydroxyl-ended chains. They are much more rapid than the chain initiation and propagation steps thereby ensuring that the rapid growing/dormant interconversion goes on over the entire polymerization process. Consequently, better control over the molecular masses is achieved. The MALDI-ToF spectrum (Fig. S30†) described linear chains with BnO– and –H end groups, while the presence of a major and minor series with a separation of 72 Da indicated that transesterification reactions may still occur.
For the sample obtained from technical grade lactide, predominant –OH chain end groups were observed, as a consequence of the presence of a large number of protic impurities in the monomer (Fig. S31†).
To shed light on the mechanism of polymerization and the nature of the active species involved, alcoholysis experiments were performed with both complexes (2 and 1) and one equivalent of alcohol (BnOH or iPrOH) in C6D6 or CD2Cl2 solution.
The 1H NMR spectra of the reaction mixtures showed the disappearance of added alcohols (BnOH or iPrOH) and the production of HOR1 or HOR2 as free alcohols. At the same time, new metal species Mg(OBn)(OR2) were observed, suggesting the substitution of one OR ligand with an OBn or OiPr group at the Mg center (Fig. S32–S36†). Analogous results were described for the alcoholysis of Mg(BHT)2(THF)2.59
After the addition of 10 equivalents of lactide, the monomer was rapidly consumed while the ligand remained in the polymerization medium as a free ligand (Fig. S36 and S37†). Thus, when an exogeneous alcohol was added into the polymerization medium, new asymmetric magnesium alkoxides were produced, and the monomer insertion occurred in the new Mg-alkoxide bond formed in situ while the free ligand was not able to act as a chain transfer agent (Scheme 3).
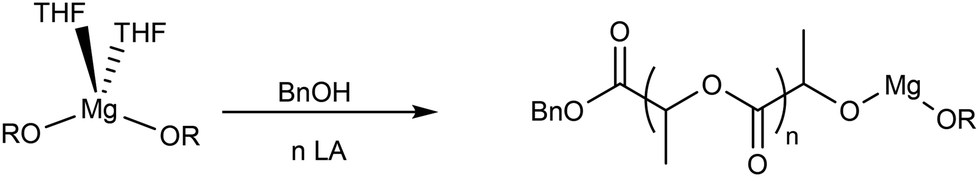 | ||
| Scheme 3 Mechanisms of polymerization in the presence of alcohol. | ||
Polymerization of lactones
Based on the high activities obtained in the ROP of rac-lactide, we decided to extend the application of these systems to ε-caprolactone (ε-CL) and to less reactive substrates such as macrolactones, namely ω-pentadecalactone (PDL) and ω-6-hexadecenlactone (6-HDL) (Scheme 4). Their polymers can be imagined as the sustainable alternative to linear low-density polyethylene. Moreover, HDL is an unsaturated macrolide that offers the chance of simple post-polymerization functionalization.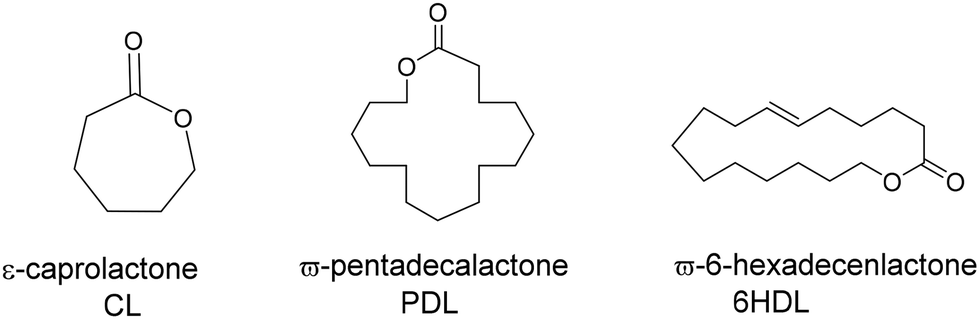 | ||
| Scheme 4 Structures of lactones investigated in this work. | ||
The polymerization of lactones was generally performed in toluene solution in the presence of benzyl alcohol (BnOH) as an initiator. Polymerization data are summarized in Table 2. Monomer conversions were evaluated during the polymerization using 1H NMR spectroscopy, by comparing the intensity of the signal related to methylene protons adjacent to the ester group of the monomer, and the signal of the same protons within the polymer.
| Runa | Cat. | Lactone (eq.) | T (°C) | Time (min) | Conv.b (%) | TOF (h−1) | M n (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|
|
a Reaction conditions: 10 μmol of Mg; 10 μmol of benzyl alcohol; [monomer]/[catalyst] = 200:1, 0.5 mL of toluene.
b Determined by 1H NMR.
c Experimental Mn and Đ values were determined by GPC analysis in THF using polystyrene standards, while for PDL in CHCl3 using polystyrene standards.
d Solvent DCM, 1 mL, reaction time 24 h.
|
||||||||
| 1 | 1 | ε-CL | 25 | 0.5 | 76 | 18240 |
23.3 | 1.73 |
| 2 | 1 | HDL | 110 | 10 | 54 | 648 | 29.7 | 2.51 |
| 3 | 1 | HDL | 110 | 30 | >99 | 400 | 66.0 | 3.19 |
| 4 | 2 | HDL | 110 | 10 | 56 | 672 | 31.0 | 2.26 |
| 5 | 1 | PDL | 110 | 10 | 48 | 600 | 26.4 | 2.13 |
| 6 | 2 | PDL | 110 | 10 | 74 | 920 | 37.2 | 2.18 |
| 7d | 1 | HDL | 25 | 1440 | 60 | 5 | 26.0 | 2.31 |
| 8d | 2 | HDL | 25 | 1440 | >99 | 8 | 49.8 | 2.09 |
In the ROP of ε-CL, the conversion of 160 equivalents of the monomer was achieved after 0.5 min at room temperature (run 1, Table 2) showing a catalytic activity analogous to that achieved in the ROP of rac-LA and higher than that reported for Mg(BHT)2(THF)2.60 In this case, a good control of the molecular masses was observed, and the experimental values were coherent with those expected.
Both magnesium complexes revealed high activity in the polymerization of HDL, allowing the conversion of approximately 100 equivalents of the monomer after 10 minutes (runs 2 and 4, Table 2) and showing remarkable turnover frequencies (TOF) of 648 and 672 h−1, respectively.
The quantitative conversion of HDL was achieved in 30 min (run 3, Table 2). Quite surprisingly, both complexes were able to promote the polymerization of HDL also at room temperature. These very mild reaction conditions are unusual for the ROP of macrolactones (runs 7 and 8, Table 2).20 As observed in other polymerizations, the activity of complex 2 was slightly higher than that of complex 1 (compare runs 5 and 6 and runs 7 and 8, Table 2).
The observed activities for complexes 1 and 2 were very high; a similar magnesium complex Mg(BHT)2(THF)2 was able to convert only 50 equivalents of PDL after 5 hours under analogous reaction conditions.48
The data suggest that the higher basicity of the OR ligands in comparison with phenoxy ligands could modulate more efficiently the Lewis acidity of the magnesium center with beneficial effects on the catalytic activity in the ROP of macrolactones.
Fig. 3 shows the 1H NMR spectrum of a typical poly(PDL) sample. In addition to the signals attributable to the methylene groups of the main chain, signals of low intensity are observed at 5.2 ppm and 3.5 ppm. These signals can be attributed to the methylene protons of the benzylic –OCH2Ph and alkyl CH2–CH2–OH end groups. In the 1H NMR spectrum of poly(HDL), in addition to the same main resonances observed for the poly(PDL), a signal was evident at 5.4 ppm for the protons of the double bond of the repeating unit (Fig. 4).
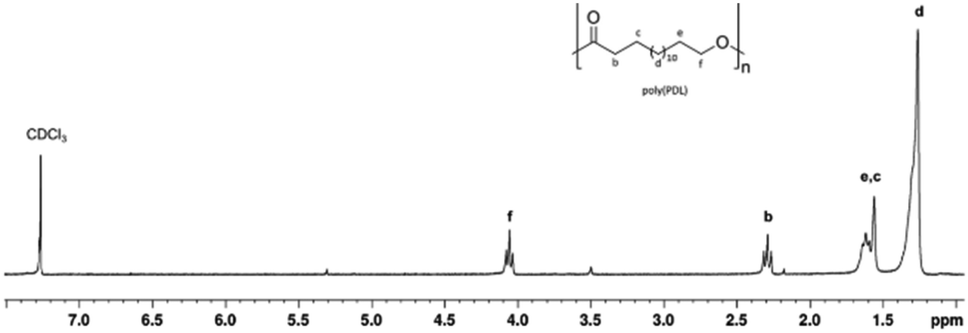 | ||
| Fig. 3 1H NMR (300 MHz, CDCl3, 298 K) spectrum of poly(ω-PDL). | ||
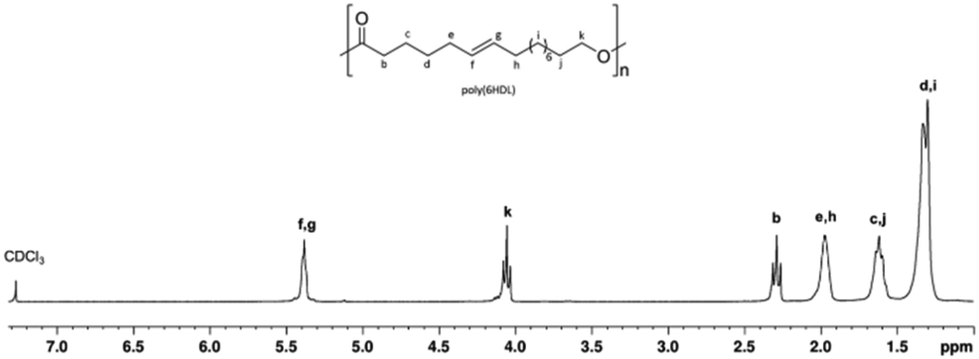 | ||
| Fig. 4 1H NMR (300 MHz, CDCl3, 298 K) spectrum of poly(HDL). | ||
The GPC analysis of these polymers showed molecular masses consistent with the theoretically expected values and monomodal distributions (Fig. S45 and S46†). The dispersity values were around 2, as expected for macrolactone ROP and can be understood in terms of relatively similar rates of propagation and transesterification.
The end-group analysis of a low molecular weight sample of poly(ω-6-HDL) (prepared with a low monomer/Mg ratio of 20) using MALDI-TOF mass spectrometry similarly showed mostly a distribution of OBn end-capped chains (Fig. 5). In the range of the analyzed masses (3000–8500 m/z), a second distribution was observed corresponding to the cyclic structures (Fig. S34†).
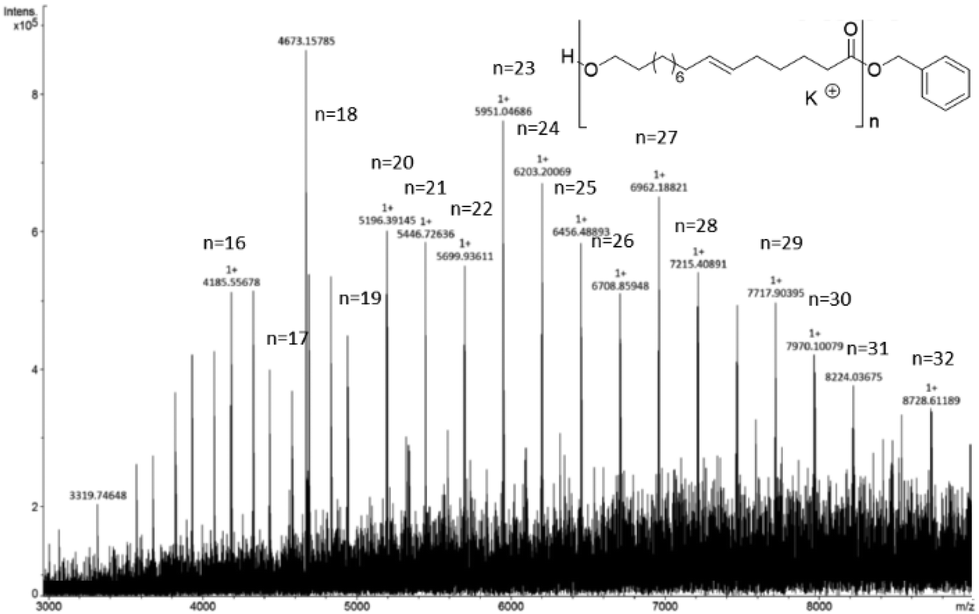 | ||
| Fig. 5 MALDI-TOF spectrum of poly(HDL) (for reaction conditions see run 5 of Table 2, [HDL]/[Mg] = 20). | ||
We note that in the ROP of macrolactones, linear chains are prevailingly produced. It is likely that the back-biting ring-closure reactions, responsible for the formation of cyclic polymers, are disfavored because of the long methylene sequences of the repeating units.48
Copolymerization of maleic anhydride and propylene oxide
Poly(propylene fumarate) (PPF) is a biodegradable and biocompatible polymer which has been largely investigated for the preparation of biological scaffolds since its unsaturated backbone can be used for photochemical cross-linking reactions in stereolithographic printing61,62 or suitable functionalizations.63,64 PPF was traditionally obtained by step-growth polycondensation, although this approach suffers from low yields, and a lack of control over molecular masses.In 2002, Hirabayashi and co-workers described a different strategy to obtain PPF by the ring-opening copolymerization of propylene oxide (PO) and maleic anhydride (MA) using magnesium diethoxide ([Mg(OEt)2]n) as the catalyst.65 A systematic exploration of several catalysts for MA/PO copolymerization was performed by Coates.66 Recently, Becker and co-workers described the synthesis of poly(propylene fumarate) by the ring-opening copolymerization PO/MA with 2,6-di-tert-butylphenoxide magnesium in combination with a functionalized primary alcohol as the initiator.27,63
Considering the structural analogy between the magnesium catalyst used by Becker and the complexes described in this work, we decided to explore their behavior in the copolymerization of maleic anhydride with racemic propylene oxide (Scheme 5).
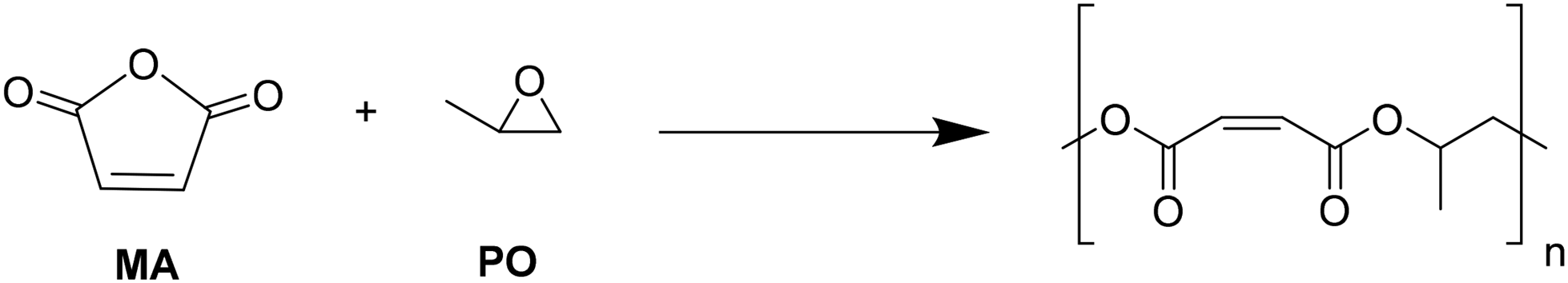 | ||
| Scheme 5 Copolymerization of propylene oxide and maleic anhydride. | ||
The polymerization reactions were initially performed at 80 °C and in the presence of a single equivalent of benzyl alcohol as the initiator (Table 3).
| Run | Catalyst | Cocat. | Solvent | T (°C) | Time (h) | Conv.b (%) | Ester (%) | M n (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 10 μmol of THE Mg complex; [MA]/[PO]/[Mg]/[Cocat.]/ = 200/1500/1/1 solvent = 1 mL. b Conv. (%) is the conversion of MA, and ester (%) is the percentage of the ester linkage in the polymer. c Experimental Mn and Đ values were determined by GPC analysis in THF using polystyrene standards. | |||||||||
| 1 | 1 | BnOH | Toluene | 80 | 24 | 80 | 78 | 3.1 | 1.89 |
| 2 | 1 | BnOH | Hexane | 80 | 24 | 17 | 81 | 4.1 | 2.04 |
| 3 | 1 | BnOH | — | 80 | 24 | >99 | 87 | 13.2 | 2.07 |
| 4 | 1 | PPNCl | — | 80 | 15 | >99 | >99 | 4.0 | 1.77 |
| 5 | 1 | PPNCl | — | 80 | 8 | 65 | >99 | 1.1 | 2.02 |
| 6 | 2 | PPNCl | — | 80 | 8 | 54 | >99 | 0.9 | 1.78 |
| 7 | 2 | PPNCl | — | 25 | 72 | 24 | >99 | 2.2 | 1.99 |
| 8 | — | PPNCl | — | 25 | 72 | <1 | — | — | — |
A strong solvent effect on activity was observed for catalyst 1; the best activity was achieved for the reactions performed in bulk, while in hexane it decreased significantly (runs 1–3, Table 3). A higher selectivity was achieved in the absence of the solvent while no difference was observed when a solvent was used.
The molecular masses were similar to those obtained with related Mg catalysts.27
A significant increase in the activity and selectivity was observed when the polymerization was performed in the presence of PPNCl (cf. runs 3 with 4 and 5, Table 3). A control experiment performed in the absence of the catalyst (with PPNCl only) showed an insignificant conversion of the monomers. A perfectly alternating structure was obtained, as evidenced by the absence of the resonances characteristic of polyether sequences at 3.5 ppm of the 1H NMR spectrum (Fig. 6) even when the copolymerization was run to full conversion with an excess of PO (run 4, Table 3). As a result, further polymerization experiments were conducted by adding the onium salt (PPNCl) as the cocatalyst.
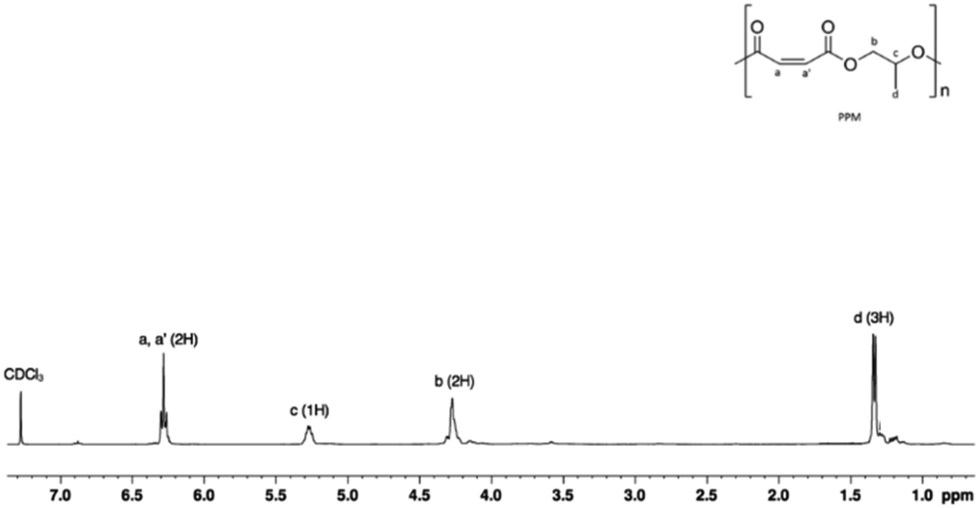 | ||
| Fig. 6 1H NMR (400 MHz, CDCl3, 298 K) spectrum of poly(propylene maleate). | ||
Both catalysts 1 and 2 showed the same reactivity and complete selectivity (runs 5 and 6, Table 3).
The regioregularity of the resultant PPMs was evaluated from the content of the head-to-tail (H–T) diads of PPM in the 1H and 13C NMR spectra (Fig. 7 and S39†). Both complexes were not regioselective. Consequently, atactic poly(propylene maleate)s were obtained in all cases as evidenced by the signals observed at 130 ppm in the 13C NMR spectrum (Fig. S39†).67
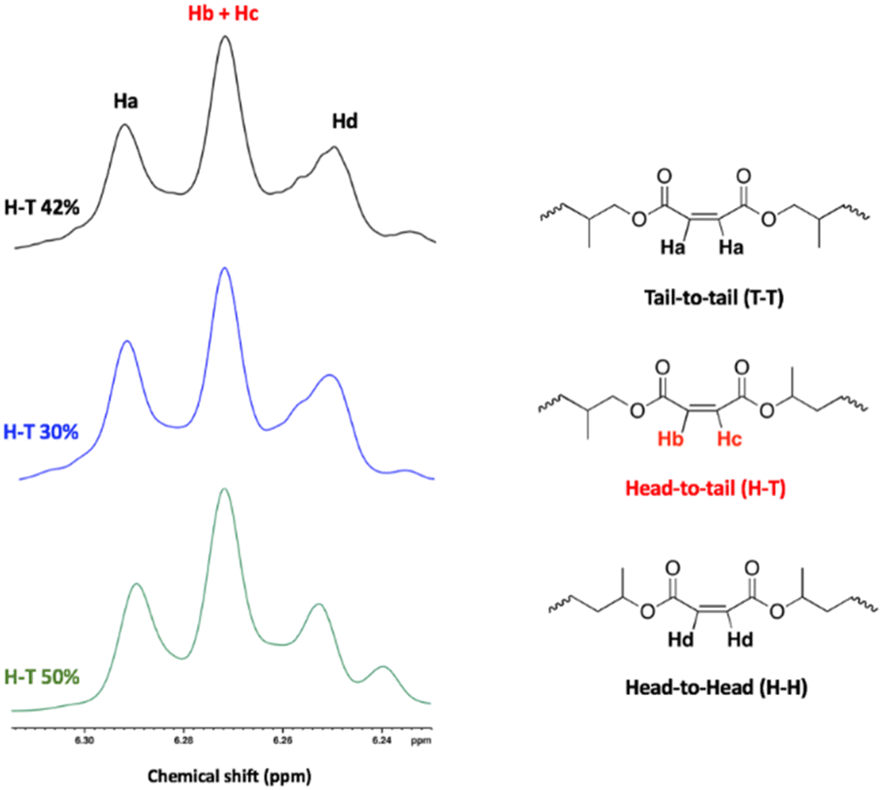 | ||
| Fig. 7 Analysis of the regiochemistry of PPMs using 1H NMR. Black curve: run 5, blue curve: run 6. Green curve: run 1. | ||
No significant differences were observed when PPNCl was used as the cocatalyst.
Subsequently, cis–trans isomerization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the polymer backbone of poly(propylene maleate) was performed (Scheme 6). Quantitative isomerization of the cis-maleate groups to form the related trans-fumarates was carried out by the addition of a catalytic amount of diethylamine, as described in the literature.66 A comparison of the proton spectra of PPM and PPF shown in Fig. 7 shows a shift in the alkene protons of the repeating unit, (from 6.28 to 6.86 ppm), while all other signals remain unchanged, confirming the isomerization of the chain. No change in either the molecular weight or the dispersity of the polymer was observed after the isomerization reaction.
C bonds in the polymer backbone of poly(propylene maleate) was performed (Scheme 6). Quantitative isomerization of the cis-maleate groups to form the related trans-fumarates was carried out by the addition of a catalytic amount of diethylamine, as described in the literature.66 A comparison of the proton spectra of PPM and PPF shown in Fig. 7 shows a shift in the alkene protons of the repeating unit, (from 6.28 to 6.86 ppm), while all other signals remain unchanged, confirming the isomerization of the chain. No change in either the molecular weight or the dispersity of the polymer was observed after the isomerization reaction.
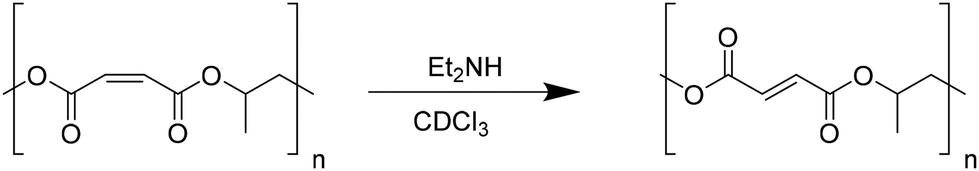 | ||
| Scheme 6 Isomerization of poly(propylene maleate) to poly(propylene fumarate). | ||
Finally, complexes 1 and 2 were tested in the chemoselective terpolymerization of maleic anhydride (MA) and propylene oxide (PO) with lactide (LA), in order to obtain a di-block polyester (Scheme 7).
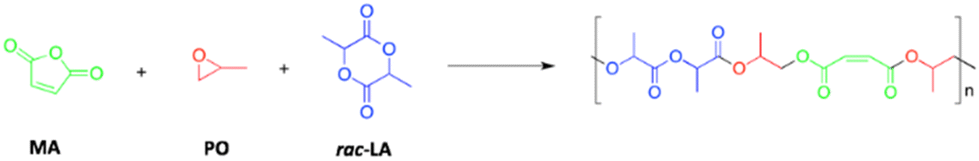 | ||
| Scheme 7 Terpolymerization of maleic anhydride (MA), propylene oxide (PO) and rac-lactide (rac-LA). | ||
The synthesis of poly(lactic acid)-block-poly(propylene fumarate) copolymers with well-defined composition was reported for the first time by Becker using copolymerization sequential procedures.68,69 Recently, block polyesters were obtained by chemoselective copolymerization from a multicomponent system formed by MA, PO, and LA with bipyridine bisphenolate aluminum.70
The polymerization tests were conducted at 80 °C and in the absence of a solvent. The reactions were carried out by mixing at the same time an excess of PO (1500 eq.), 200 equivalents of MA, 100 equivalents of rac-LA, and 1 equivalent of PPNCl as the co-catalyst. The polymerization was monitored by 1H NMR spectroscopy. After 16 hours, the anhydride conversion was quantitative for both catalysts while no conversion of the lactide was observed.
After 24 hours the rac-LA conversion was estimated to be around 50% for complex 1 and 62% for complex 2.
The 1H NMR spectra (Fig. 8) of the resulting polymers showed signals attributable to both blocks and were fully consistent with those previously reported.70
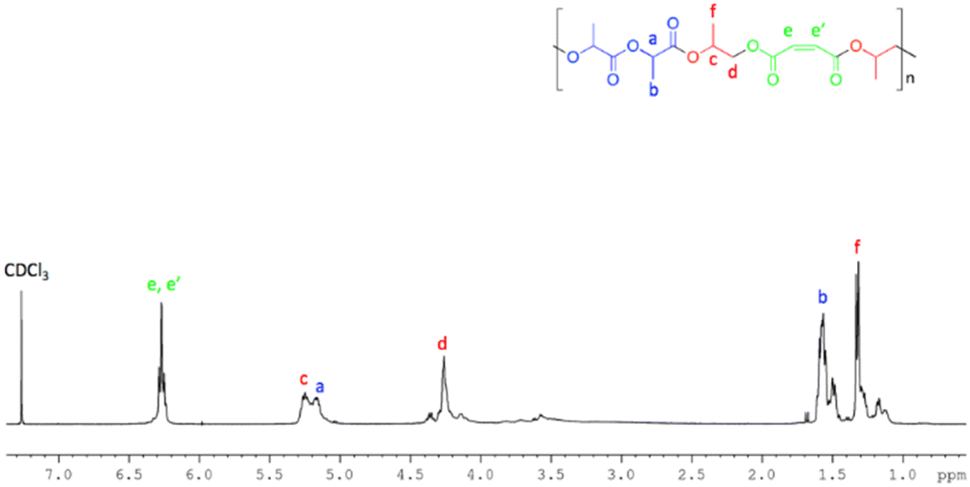 | ||
| Fig. 8 1H NMR (400 MHz, CDCl3, 298 K) of poly[(propylene maleate)-block-poly(lactic acid)] obtained by using 1. | ||
The DOSY spectrum (Fig. 9) indicated that the resonances of the PLA sequences and of PPM portion showed the same diffusion coefficient, indicating that they belong to the same polymer chains. This finding supported the formation of the di-block copolymer, namely poly(propylene maleate)-block-poly(lactic acid), by terpolymerization of PO, MA and rac-LA. Accordingly, the GPC analysis of the sample showed a monomodal distribution of the molecular masses with a Mn value of 3.5 kDa. This value agrees with the low molecular masses obtained in the ROCOP process that represents the first step of the whole terpolymerization, as already observed in other examples of switch catalysis between ROCOP and ROP.28,71–74
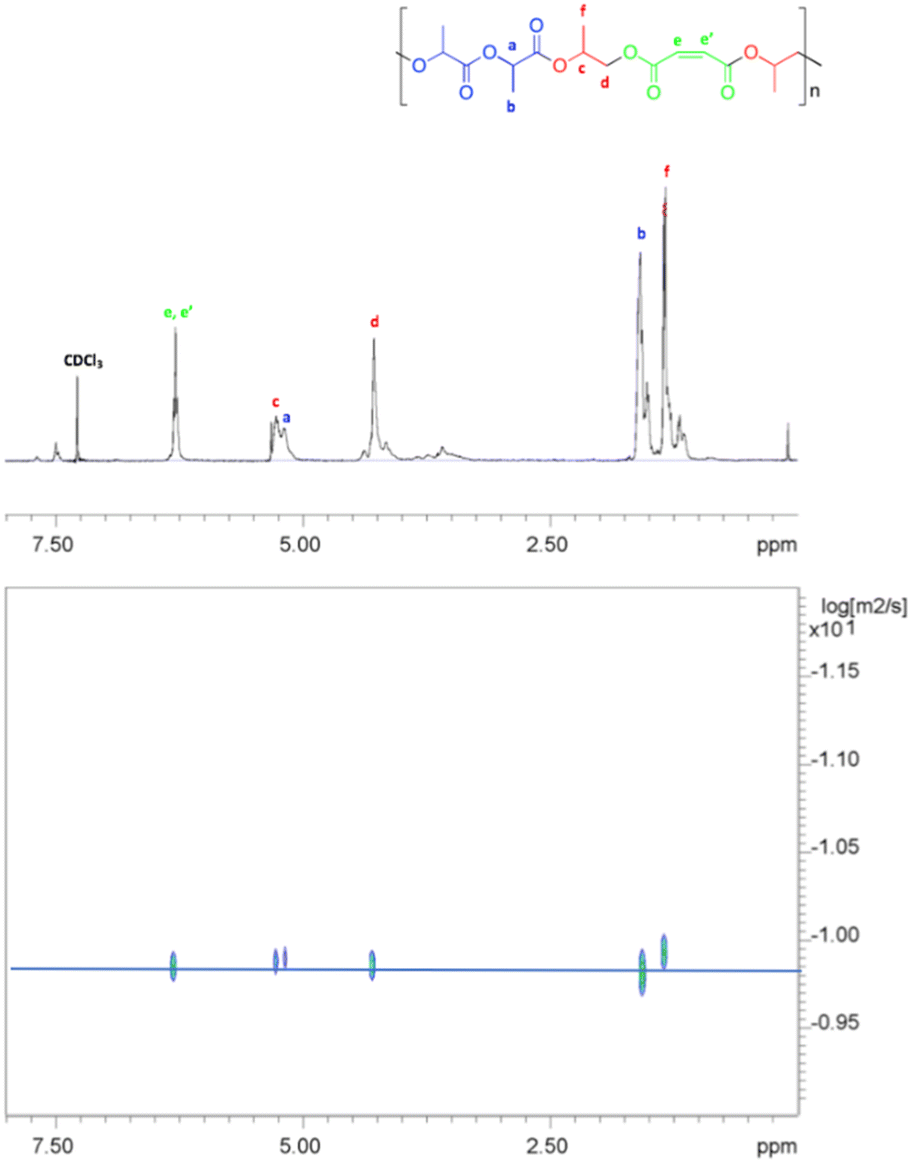 | ||
| Fig. 9 2D DOSY NMR (400 MHz, CDCl3, 298 K) of poly[(propylene maleate)-block-poly(lactic acid)]. | ||
Conclusions
In this work, we reported the synthesis of two new chiral bulky alkoxide ligands related to [OCtBu2Ph], [OCtBuAdPh] and [OCtBuMePh] and studied the coordination chemistry upon reaction with n-butyl-sec-butylmagnesium. We demonstrated that while racemic [OCtBuAdPh] enabled the clean formation of the homochiral C2-symmetric complex Mg(OCtBuAdPh)2(THF)2, [OCtBuMePh] did not exhibit well-defined coordination chemistry.The reactivity of the new precatalyst Mg(OCAdtBuPh)2(THF)2 (2), along with the reactivity of the previously reported Mg(OCtBu2Ph)2(THF)2 (1), was investigated in the homopolymerization of lactide and lactones and copolymerization of maleic anhydride and propylene oxide. Likely due to the bulkier nature of the alkoxides, catalyst 2 revealed somewhat higher activity compared with catalyst 1 in the ROP of lactide. When the polymerization reactions were performed in non-coordinating solvents, the molecular masses of PLAs were always significantly lower than theoretically expected values, because of extensive intramolecular transesterification phenomena. In contrast, with the use of THF as the solvent and benzyl alcohol as the chain transfer agent, a better control of the molecular masses was achieved.
Both complexes showed high activity in the ROP of macrolactones such as ω-pentadecalactone (PDL) and ω-6-hexadecenlactone (6-HDL). In this case, linear polymeric chains with molecular masses consistent with the expected values were obtained.
Importantly, these catalysts were also active at room temperature. These reaction conditions are uncommon in the polymerization of these (relatively unreactive) monomers. This finding further contributes to the overall sustainability of our simple magnesium-alkoxide catalysts.
Finally, these complexes exhibited efficient copolymerization of maleic anhydride and propylene oxide, producing polypropylene fumarate with a perfectly alternating structure when the polymerization was performed in the absence of a solvent or in the presence of PPNCl as the cocatalyst. A fully biocompatible diblock polyester poly(propylene maleate)-block-polylactide was obtained by combining the two synthetic routes in a one-pot procedure. In our future work, we will continue to investigate homo- and copolymerization using these efficient, non-toxic, and cost-effective catalysts.
Experimental details
Ligands and complexes: materials and methods
Reactions involving air-sensitive materials were performed under oxygen-free conditions in a MBraun N2-filled glovebox. n-Butyl-sec-butylmagnesium (0.7 M solution in hexane) was obtained from Sigma and used as received. All non-deuterated solvents (HPLC grade) were obtained from Sigma and dried using an MBraun solvent purification system. Deuterated solvents C6D6 and CDCl3 were obtained from Cambridge Isotope Laboratories and were dried over activated molecular sieves. All solvents were stored over 3 Å molecular sieves. The complexes were characterized by 1H and 13C NMR, X-ray crystallography, and elemental analysis. NMR spectra for the metal complexes were recorded at the Lumigen Instrument Centre (Wayne State University) on Agilent 400 and 600 MHz spectrometers in C6D6 at room temperature, and on Bruker AVANCE NEO 500 spectrometer (DOSY). Chemical shifts and coupling constants (J) were reported in parts per million (δ) and Hertz respectively. Elemental analysis was performed under ambient air-free conditions by Midwest Microlab LLC. HOR1 and Mg(OR1)2(THF)2 (1) were prepared as previously described.The number-average molecular weights (Mn) and molecular weight distributions of the polymers (dispersity, Đ) were evaluated by size exclusion chromatography (SEC), using an Agilent 1260 Infinity Series GPC (ResiPore 3 μm, 300 × 7.5 mm, 1.0 mL min−1, UV (250 nm) and refractive index (RI, PLGPC 220)) detector. All measurements were performed with THF as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. Monodisperse poly(styrene) polymers were used as calibration standards. MALDI-ToF-MS analysis was performed on a Waters Maldi Micro MX equipped with a 337 nm nitrogen laser. An acceleration voltage of 25 kV was applied. The polymer sample was dissolved in THF with Milli-Q water containing 0.1% formic acid at a concentration of 0.8 mg mL−1. The matrix used was 2,5-dihydroxybenzoic acid (DHBA) (Pierce) and was dissolved in THF at a concentration of 30 mg mL−1.
Polymerization and polymer characterization: materials and methods
rac-Lactide was obtained from Sigma and purified by recrystallization from toluene, followed by drying over P2O5 for 72 h. Toluene and hexane (Sigma) were distilled under nitrogen over sodium. Cyclohexene oxide (CHO) and propylene oxide (PO) were purchased from Sigma-Aldrich and freshly distilled over CaH2. Phthalic anhydride and maleic anhydride were purchased from Sigma and purified according to the published procedure. Tetrahydrofuran (THF) was refluxed over Na and benzophenone and distilled under nitrogen. Monomers (Sigma-Aldrich) were purified before use: ω-6-hexadecenlactone (6HDL), ω-pentadecalactone, and cyclohexene oxide were distilled under vacuum on CaH2 and stored over 4 Å molecular sieves. Phthalic anhydride (PA) was crystallized from dry toluene. CDCl3 and toluene-d8 were purchased from Eurisotop and used as received. Benzyl alcohol was purified by distillation over sodium. All other chemicals were commercially available and used as received. Mass spectra were acquired using a Bruker solariX XR Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7 T refrigerated actively-shielded superconducting magnet (Bruker Biospin, Wissembourg, France). The polymer samples were ionized in positive ion mode using the MALDI ion source. The mass range was set to m/z 200–5000. The laser power was 12% and 18 laser shots were used for each scan. Mass spectra were calibrated externally using a mixture of peptide clusters in MALDI ionization positive ion mode. A linear calibration was applied. The polymer samples were dissolved in THF at a concentration of 1 mg mL−1. The cationizing agent used was potassium trifluoroacetate (Fluka, >99%) dissolved in THF at a concentration of 5 mg mL−1. The matrix used was trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) (Fluka) and was dissolved in THF at a concentration of 40 mg mL−1. Solutions of the matrix, salt and polymer were mixed in a volume ratio of 4:1:4, respectively. The mixed solution was hand-spotted on a stainless steel MALDI target and allowed to dry. NMR spectra were recorded on a Bruker Advance 400 spectrometer at 25 °C, unless otherwise stated. Chemical shifts (δ) are expressed in parts per million and coupling constants (J) in Hertz. 1H NMR spectra are referenced using the residual solvent peak at δ = 7.27 for CDCl3. Moisture and air-sensitive materials were manipulated under nitrogen using Schlenk techniques or an MBraun Labmaster glovebox.
X-ray crystallographic details
The structures of HOCAdtBuPh (HOR2), Mg(OR2)2(THF)2 (2), and Mg2(OR)3(THF)2(sec-Bu)2 (3) were determined by X-ray crystallography (Table 4). A Bruker Kappa APEX-II CCD diffractometer was used for data collection. A graphic monochromator was employed for wavelength selection (MoKα radiation, λ = 0.71073 Å). The data were processed using the APEX-2/3 software. The structures were solved and refined using SHELXT75 and difference Fourier (ΔF) maps, as embedded in SHELXL-201876 running under Olex2.77 The carbon hydrogen atoms were placed in calculated positions using a standard riding model and refined isotropically; all other atoms were refined anisotropically. The hydrogen on the oxygen in structure HOR2 was located using the ΔF maps. The structure of 2 contained a co-crystallized disordered CH2Cl2 molecule; the disorder was modeled by two alternate conformations. The crystal structure of 2 is a two-component non-merohedral twin (180° rotation around the [1 0 1] reciprocal rotation vector). Refinement was performed using the HKLF-5 file with reflections from both domains, which lead to a batch scale factor (BASF) parameter of 0.423(2). A solvent mask in Olex2 was applied for the structure Mg2(OR)3(THF)2(sec-Bu)2 to remove a disordered ether (1.33 ethers/asymmetric unit) located along a solvent channel. A sec-Bu group was also disordered between two conformations.
| Complex | HOR2 | 2 | 3 |
|---|---|---|---|
| a R 1 = ∑||Fo − |Fc||/∑|Fo|. b wR2 = (∑(w(Fo2 − Fc2)2)/∑(w(Fo2)2))1/2. c GOF = (∑w(Fo2 − Fc2)2/(n − p))1/2 where n is the number of data and p is the number of parameters refined. | |||
| Formula | C21H30O | C50H74MgO4·CH2Cl2 | C52H80Mg2O4 |
| F w, g mol−1 | 298.45 | 848.32 | 914.34 |
| Temperature | 100 K | 100 K | 100(2) K |
| Cryst. syst. | Orthorhombic | Triclinic | Monoclinic |
| Space group | Pna21 |
P |
Pc |
| Color | Colorless | Colorless | Colorless |
| Z | 4 | 2 | 2 |
| a, Å | 9.3463(5) | 12.352(6) | 12.5905(10) |
| b, Å | 13.8584(7) | 13.519(6) | 10.4610(9) |
| c, Å | 12.5331(6) | 15.032(7) | 20.3664(17) |
| α, deg. | 90.00 | 67.339(13) | 90 |
| β, deg. | 90.00 | 83.200(15) | 90.778(2) |
| γ, deg. | 90.00 | 82.150(14) | 90 |
| V, A3 | 1623.35(14) | 2288.8(18) | 2682.2(4) |
| d calcd, g cm−3 | 1.221 | 1.231 | 1.132 |
| μ, mm−1 | 0.072 | 0.200 | 0.091 |
| 2θ, deg. | 52.74 | 51.112 | 51.016 |
| R 1 (all data) | 0.0728 | 0.1295 | 0.2148 |
| wR2b (all data) | 0.0976 | 0.2400 | 0.3109 |
| R 1 [(I > 2σ)] | 0.0604 | 0.0830 | 0.0959 |
| wR2 [(I > 2σ)] | 0.0933 | 0.2068 | 0.2430 |
| GOFc (F2) | 1.059 | 1.045 | 0.982 |
856 (m), 709 (s).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 molar ratio: A 1 mL solution of HOR3 (60 mg, 0.234 mmol, 2.0 equiv.) in diethyl ether and a 1 mL solution of Mg(n-butyl)(sec-butyl) (0.125 mmol, 1 equiv.) in hexane were prepared. The solution of HOR3 was then added dropwise to a stirring solution of Mg(n-butyl)(sec-butyl). Following the addition of the ligand, 0.5 ml of THF was added to the reaction mixture. The reaction mixture was stirred for 2 hours, upon which the volatiles were removed in vacuo. The resulting oily solid was extracted with diethyl ether, filtered and concentrated in vacuo to get a white solid. Recrystallization from diethyl ether overnight produced 3 in 58% yield. The nature of 3 was confirmed by NMR (broad peaks), elemental analysis and X-ray crystallography. Reaction at a 1:1 molar ratio: A 1 mL solution of HOR3 (60 mg, 0.234 mmol, 1.0 equiv.) in diethyl ether and a 1 mL solution of Mg(n-butyl)(sec-butyl) (0.238 mmol, 1.0 equiv.) in hexane were prepared. The solution of HOR3 was then added dropwise to a stirring solution of Mg(n-butyl)(sec-butyl). Following the addition of the ligand, 0.5 ml of THF was added to the reaction mixture. The reaction mixture was stirred for 2 hours, upon which the volatiles were removed in vacuo. The resulting oily solid was extracted with diethyl ether, filtered and concentrated in vacuo to get a white solid. Recrystallization from diethyl ether overnight produced 3 in 46% yield. 1H NMR (400 MHz, C7D8, room temperature) δ 7.70 (br s, 4H, OCAdMePh), 7.18 (br s, 4H, OCAdMePh), 7.08 (br s, 2H, OCAdMePh), 3.67 (s, 8H, THF), 2.02 (s, 6H, OCAdMePh), 1.19 (s, 8H, THF), 1.75–0.89 (Ad + sec-Bu resonances) ppm. 1H NMR (400 MHz, C7D8, 80 °C) δ 7.55 (br s, 4H, OCAdMePh), 7.15 (br s, 4H, OCAdMePh), ∼7.08 (br s, 2H, OCAdMePh), 3.67 (s, 8H, THF), 1.95 (s, 6H, OCAdMePh), 1.32 (s, 8H, THF), 1.69–0.86 (Ad + sec-Bu resonances) ppm. 13C{1H} (C6D6, 100 MHz) δ 149.94, 128.92, 127.25, 127.05, 126.09, 80.12, 69.31, 40.27, 37.38, 34.10, 33.06, 29.58, 26.61, 25.14, 20.88, 17.33, 14.71 ppm. Anal. calcd for: C52H80Mg2O4 C, 76.37; H, 9.86; found: C, 76.69; H, 9.41.
:2 molar ratio: A 1 mL solution of HOR3 (60 mg, 0.234 mmol, 2.0 equiv.) in diethyl ether and a 1 mL solution of Mg(n-butyl)(sec-butyl) (0.125 mmol, 1 equiv.) in hexane were prepared. The solution of HOR3 was then added dropwise to a stirring solution of Mg(n-butyl)(sec-butyl). Following the addition of the ligand, 0.5 ml of THF was added to the reaction mixture. The reaction mixture was stirred for 2 hours, upon which the volatiles were removed in vacuo. The resulting oily solid was extracted with diethyl ether, filtered and concentrated in vacuo to get a white solid. Recrystallization from diethyl ether overnight produced 3 in 58% yield. The nature of 3 was confirmed by NMR (broad peaks), elemental analysis and X-ray crystallography. Reaction at a 1:1 molar ratio: A 1 mL solution of HOR3 (60 mg, 0.234 mmol, 1.0 equiv.) in diethyl ether and a 1 mL solution of Mg(n-butyl)(sec-butyl) (0.238 mmol, 1.0 equiv.) in hexane were prepared. The solution of HOR3 was then added dropwise to a stirring solution of Mg(n-butyl)(sec-butyl). Following the addition of the ligand, 0.5 ml of THF was added to the reaction mixture. The reaction mixture was stirred for 2 hours, upon which the volatiles were removed in vacuo. The resulting oily solid was extracted with diethyl ether, filtered and concentrated in vacuo to get a white solid. Recrystallization from diethyl ether overnight produced 3 in 46% yield. 1H NMR (400 MHz, C7D8, room temperature) δ 7.70 (br s, 4H, OCAdMePh), 7.18 (br s, 4H, OCAdMePh), 7.08 (br s, 2H, OCAdMePh), 3.67 (s, 8H, THF), 2.02 (s, 6H, OCAdMePh), 1.19 (s, 8H, THF), 1.75–0.89 (Ad + sec-Bu resonances) ppm. 1H NMR (400 MHz, C7D8, 80 °C) δ 7.55 (br s, 4H, OCAdMePh), 7.15 (br s, 4H, OCAdMePh), ∼7.08 (br s, 2H, OCAdMePh), 3.67 (s, 8H, THF), 1.95 (s, 6H, OCAdMePh), 1.32 (s, 8H, THF), 1.69–0.86 (Ad + sec-Bu resonances) ppm. 13C{1H} (C6D6, 100 MHz) δ 149.94, 128.92, 127.25, 127.05, 126.09, 80.12, 69.31, 40.27, 37.38, 34.10, 33.06, 29.58, 26.61, 25.14, 20.88, 17.33, 14.71 ppm. Anal. calcd for: C52H80Mg2O4 C, 76.37; H, 9.86; found: C, 76.69; H, 9.41.
General procedure for the polymerization of lactide in solution
A dichloromethane/toluene solution of 10 μmol of the catalyst was mixed with a solution containing 100 equivalents (144 mg) of lactide in dichloromethane/toluene (the total volume of the reaction was 10 mL, [LA] = 0.1 M). The reaction mixture was stirred at room temperature for a given time after which it was stopped by adding 2–5 mL of methanol. PLA was precipitated in methanol and washed with an excess of methanol to remove all the impurities. For further purification, the polymer was dissolved using a minimal amount of DCM and then added to 20 mL of methanol to precipitate pure PLA. Excess methanol was decanted, and the polymer was dried for 1 hour under vacuum. The reaction with 200, 300, 600, 1000, 5000, and 10000 equivalents of lactide (0.2 M, 0.3 M, 0.6 M, 1 M, 5 M, and 10 M respectively) in dichloromethane and 200, 300, and 600, (0.2 M, 0.3 M, and 0.6 M) toluene solutions was carried out in a similar fashion. The resulting polymer was characterized by 1H NMR spectroscopy, to determine the degree of polymerization. The methine region was also analyzed by homonuclear decoupled 1H NMR, to determine the tacticity of the polymer.
General procedure for the polymerization of lactide in bulk
10 μmol of the catalyst was mixed with 10000 equivalents (14.4 g) of lactide and 10 equivalents of benzyl alcohol in a pressure vessel. The reaction mixture was heated at 150 °C for one hour.
General procedure for the co-polymerization of epoxides with cyclic anhydrides
Procedure for the terpolymerization of epoxides with cyclic anhydride and cyclic esters
Terpolymerization was performed in a MBraun MBG20 glovebox. A magnetically stirred vessel (10 mL) was charged with the anhydride and ester. Subsequently, the catalyst dissolved in neat epoxide was added, followed by the co-catalyst. The reaction mixture was stirred at the desired temperature. At desired times, small aliquots of the reaction mixture were sampled, dissolved in CDCl3 and analyzed by 1H NMR spectroscopy. At the end of the polymerization, the product was dissolved in CH2Cl2, coagulated in diethyl ether and dried in a vacuum oven. All analyses were performed on the crude samples.Author contributions
Mina Mazzeo and Stanislav Groysman: conceptualization, supervision and original draft preparation; Duleeka Wannipurage, Sara D'Aniello and Lakshani Wathsala Kulathungage: data curation, investigation and methodology; Daniela Pappalardo: writing; Dennis P. Anderson and Cassandra L. Ward: investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. G. is grateful to the National Science Foundation (NSF) for current support under grant number CHE-1855681. Pro-ligands and Mg complexes were characterized at the Lumigen Instrument Center. NIH support (NIH S10OD028488) for the purchase of Bruker AVANCE NEO 500 is acknowledged. M. M. thanks Dr Patrizia Iannece for Maldi ToF spectra, Dr Patrizia Oliva for NMR assistance, Dr Mariagrazia Napoli for GPC analysis.Notes and references
- L. Filiciotto and G. Rothenberg, ChemSusChem, 2021, 14, 56–72 CrossRef CAS PubMed.
- P. B. V. Scholten, J. Cai and R. T. Mathers, Macromol. Rapid Commun., 2021, 42, 2000745 CrossRef CAS PubMed.
- I. van der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS.
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef CAS PubMed.
- J. A. Wilson, Z. Ates, R. L. Pflughaupt, A. P. Dove and A. Heise, Prog. Polym. Sci., 2019, 91, 29–50 CrossRef CAS.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- C. Liu, F. Liu, J. Cai, W. Xie, T. E. Long, S. R. Turner, A. Lyons and R. A. Gross, Biomacromolecules, 2011, 12, 3291–3298 CrossRef CAS PubMed.
- M. Bouyahyi and R. Duchateau, Macromolecules, 2014, 47, 517–524 CrossRef CAS.
- T. Fuoco, A. Meduri, M. Lamberti, V. Venditto, C. Pellecchia and D. Pappalardo, Polym. Chem., 2015, 6, 1727–1740 RSC.
- V. Ladelta, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2017, 8, 511–515 RSC.
- V. Ladelta, J. D. Kim, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2018, 51, 2428–2436 CrossRef CAS.
- T. Witt, M. Haeussler and S. Mecking, Macromol. Rapid Commun., 2017, 38, 1600638 CrossRef PubMed.
- A. E. Polloni, V. Chiaradia, E. M. Figura, J. P. De Paoli, D. de Oliveira, J. Vladimir de Oliveira, P. H. Hermes de Araujo and C. Sayer, Appl. Biochem. Biotechnol., 2018, 184, 659–672 CrossRef CAS PubMed.
- M. L. Focarete, M. Gazzano, M. Scandola, A. Kumar and R. A. Gross, Macromolecules, 2002, 35, 8066–8071 CrossRef CAS.
- Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed.
- S. Torron, M. K. G. Johansson, E. Malmstroem, L. Fogelstroem, K. Hult and M. Martinelle, Handbook of Telechelic Polyesters, Polycarbonates, and Polyethers: (pp. 29-64). Pan Stanford Publishing Pte. 2017.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, ACS Macro Lett., 2016, 5, 346–350 CrossRef CAS PubMed.
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS PubMed.
- M. P. F. Pepels, I. Hermsen, G. J. Noordzij and R. Duchateau, Macromolecules, 2016, 49, 796–806 CrossRef CAS.
- D. Myers, T. Witt, A. Cyriac, M. Bown, S. Mecking and C. K. Williams, Polym. Chem., 2017, 8, 5780–5785 RSC.
- M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS.
- F. Van Der Sanden, E. Gubbels and R. Duchateau, Macromolecules, 2016, 49, 4441–4451 CrossRef.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- R. A. Dilla, C. M. M. Motta, S. R. Snyder, J. A. Wilson, C. Wesdemiotis and M. L. Becker, ACS Macro Lett., 2018, 7, 1254–1260 CrossRef CAS PubMed.
- J. Guo, X. Liu, A. L. Miller, II, B. E. Waletzki, M. J. Yaszemski and L. Lu, J. Biomed. Mater. Res., Part A, 2017, 105, 226–235 CrossRef CAS PubMed.
- J. A. Wilson, D. Luong, A. P. Kleinfehn, S. Sallam, C. Wesdemiotis and M. L. Becker, J. Am. Chem. Soc., 2018, 140, 277–284 CrossRef CAS PubMed.
- I. D'Auria, F. Santulli, F. Ciccone, A. Giannattasio, M. Mazzeo and D. Pappalardo, ChemCatChem, 2021, 13, 3303–3311 CrossRef.
- S. Kaler and M. D. Jones, Dalton Trans., 2022, 51, 1241–1256 RSC.
- C. Gallegos, V. Tabernero, M. E. G. Mosquera, T. Cuenca and J. Cano, Eur. J. Inorg. Chem., 2015, 5124–5132 CrossRef CAS.
- C. Gallegos, V. Tabernero, F. M. Garcia-Valle, M. E. G. Mosquera, T. Cuenca and J. Cano, Organometallics, 2013, 32, 6624–6627 CrossRef CAS.
- I. D'Auria, C. Tedesco, M. Mazzeo and C. Pellecchia, Dalton Trans., 2017, 46, 12217–12225 RSC.
- S. D’Aniello, S. Laviéville, F. Santulli, M. Simon, M. Sellitto, C. Tedesco, C. M. Thomas and M. Mazzeo, Catal. Sci. Technol., 2022, 12, 6142–6154 RSC.
- F. Santulli, M. Lamberti and M. Mazzeo, ChemSusChem, 2021, 14, 5470–5475 CrossRef CAS PubMed.
- I. D'Auria, M. Lamberti, M. Mazzeo, S. Milione, G. Roviello and C. Pellecchia, Chem. – Eur. J., 2012, 18, 2349–2360 CrossRef PubMed.
- T. Rosen, I. Goldberg, W. Navarra, V. Venditto and M. Kol, Angew. Chem., Int. Ed., 2018, 57, 7191–7195 CrossRef CAS PubMed.
- T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem. – Eur. J., 2016, 22, 11533–11536 CrossRef CAS PubMed.
- T. Rosen, J. Rajpurohit, S. Lipstman, V. Venditto and M. Kol, Chem. – Eur. J., 2020, 26, 17183–17189 CrossRef CAS PubMed.
- I. E. Nifant'ev, A. V. Shlyakhtin, A. N. Tavtorkin, P. V. Ivchenko, R. S. Borisov and A. V. Churakov, Catal. Commun., 2016, 87, 106–111 CrossRef.
- D. Wannipurage, T. S. Hollingsworth, F. Santulli, M. Cozzolino, M. Lamberti, S. Groysman and M. Mazzeo, Dalton Trans., 2020, 49, 2715–2723 RSC.
- H. Xie, C. Wu, D. Cui and Y. Wang, J. Organomet. Chem., 2018, 875, 5–10 CrossRef CAS.
- R. Petrus and P. Sobota, Coord. Chem. Rev., 2019, 396, 72–88 CrossRef CAS.
- Y. Gao, Z. Dai, J. Zhang, X. Ma, N. Tang and J. Wu, Inorg. Chem., 2014, 53, 716–726 CrossRef CAS PubMed.
- C. W. Lee, S. Kuno and Y. Kimura, Macromol. Res., 2013, 21, 385–391 CrossRef CAS.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347–18353 CrossRef CAS.
- F. Santulli, M. Lamberti, A. Annunziata, R. C. Lastra and M. Mazzeo, Catalysts, 2022, 12, 1193 CrossRef CAS.
- Y. Wang, W. Zhao, X. Liu, D. Cui and E. Y. X. Chen, Macromolecules, 2012, 45, 6957–6965 CrossRef CAS.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Polym. Chem., 2014, 5, 2691–2694 RSC.
- I. E. Nifant'ev, A. V. Shlyakhtin, A. N. Tavtorkin, P. V. Ivchenko, R. S. Borisov and A. V. Churakov, Catal. Commun., 2016, 87, 106–111 CrossRef.
- J. A. Bellow, D. Fang, N. Kovacevic, P. D. Martin, J. Shearer, G. A. Cisneros and S. Groysman, Chem. – Eur. J., 2013, 19, 12225–12228 CrossRef CAS PubMed.
- J. A. Bellow, P. D. Martin, R. L. Lord and S. Groysman, Inorg. Chem., 2013, 52, 12335–12337 CrossRef CAS PubMed.
- J. A. Bellow, M. Yousif, D. Fang, E. G. Kratz, G. A. Cisneros and S. Groysman, Inorg. Chem., 2015, 54, 5624–5633 CrossRef CAS PubMed.
- J. S. Lomas and V. Bru-Capdeville, J. Chem. Soc., Perkin Trans. 2, 1994, 459–466, 10.1039/P29940000459.
- A. Nagaki, H. Yamashita, Y. Tsuchihashi, K. Hirose, M. Takumi and J.-i. Yoshida, Chem. – Eur. J., 2019, 25, 13719–13727 CrossRef CAS PubMed.
- Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- M.-L. Shueh, Y.-S. Wang, B.-H. Huang, C.-Y. Kuo and C.-C. Lin, Macromolecules, 2004, 37, 5155–5162 CrossRef CAS.
- V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2011, 40, 523–534 RSC.
- H.-Y. Chen, L. Mialon, K. A. Abboud and S. A. Miller, Organometallics, 2012, 31, 5252–5261 CrossRef CAS.
- I. E. Nifant'ev, A. V. Shlyakhtin, V. V. Bagrov, M. E. Minyaev, A. V. Churakov, S. G. Karchevsky, K. P. Birin and P. V. Ivchenko, Dalton Trans., 2017, 46, 12132–12146 RSC.
- H.-J. Fang, P.-S. Lai, J.-Y. Chen, S. C. N. Hsu, W.-D. Peng, S.-W. Ou, Y.-C. Lai, Y.-J. Chen, H. Chung, Y. Chen, T.-C. Huang, B.-S. Wu and H.-Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2697–2704 CrossRef CAS.
- Y. Luo, C. K. Dolder, J. M. Walker, R. Mishra, D. Dean and M. L. Becker, Biomacromolecules, 2016, 17, 690–697 CrossRef CAS PubMed.
- Y. Luo, G. Le Fer, D. Dean and M. L. Becker, Biomacromolecules, 2019, 20, 1699–1708 CrossRef CAS PubMed.
- Y. Chen, J. A. Wilson, S. R. Petersen, D. Luong, S. Sallam, J. Mao, C. Wesdemiotis and M. L. Becker, Angew. Chem., Int. Ed., 2018, 57, 12759–12764 CrossRef CAS PubMed.
- Z. Cai, Y. Wan, M. L. Becker, Y.-Z. Long and D. Dean, Biomaterials, 2019, 208, 45–71 CrossRef CAS PubMed.
- S. Takenouchi, A. Takasu, Y. Inai and T. Hirabayashi, Polym. J., 2002, 34, 36–42 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- N. D. Harrold, Y. Li and M. H. Chisholm, Macromolecules, 2013, 46, 692–698 CrossRef CAS.
- S. R. Petersen, J. A. Wilson and M. L. Becker, Macromolecules, 2018, 51, 6202–6208 CrossRef CAS.
- S. R. Petersen, J. Yu, T. R. Yeazel, G. Bass, A. Alamdari and M. L. Becker, Biomacromolecules, 2022, 23, 2388–2395 CrossRef CAS PubMed.
- Y.-B. Wang, M.-Q. Wang, Y.-B. Shi, X.-L. Chen, D.-P. Song, Y.-S. Li and B. Wang, Macromol. Chem. Phys., 2022, 223, 2200079 CrossRef CAS.
- F. Isnard, M. Lamberti, C. Pellecchia and M. Mazzeo, ChemCatChem, 2017, 9, 2972–2979 CrossRef CAS.
- F. Isnard, F. Santulli, M. Cozzolino, M. Lamberti, C. Pellecchia and M. Mazzeo, Catal. Sci. Technol., 2019, 9, 3090–3098 RSC.
- F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213–1220 CrossRef CAS.
- I. D’Auria, S. D’Aniello, G. Viscusi, E. Lamberti, G. Gorrasi, M. Mazzeo and D. Pappalardo, Polymers, 2022, 14, 4911 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. A. Olah, A. H. Wu and O. Farooq, J. Org. Chem., 1989, 54, 1375–1378 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2201952–2201954. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00785e |
| This journal is © The Royal Society of Chemistry 2023 |