
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium-(κ2-NSi) catalyzed dehydrogenation of formic acid: effect of auxiliary ligands on the catalytic performance†
Alejandra
Gomez-España
ab,
Jorge L.
Lopez-Morales
 a,
Belinda
Español-Sanchez
a,
Pilar
García-Orduña
a,
Fernando J.
Lahoz
a,
Manuel
Iglesias
a and
Francisco J.
Fernández-Alvarez
*a
a,
Belinda
Español-Sanchez
a,
Pilar
García-Orduña
a,
Fernando J.
Lahoz
a,
Manuel
Iglesias
a and
Francisco J.
Fernández-Alvarez
*a
aDepartamento de Química Inorgánica – Instituto de Síntesis Química y Catálisis Homogénea (ISQCH). Universidad de Zaragoza. Facultad de Ciencias 50009, Zaragoza, Spain. E-mail: paco@unizar.es
bUniversidad Pedagógica Nacional Francisco Morazán-UPNFM, 11101, Tegucigalpa, Honduras
First published on 26th April 2023
Abstract
The iridium(III) complexes [Ir(H)(Cl)(κ2-NSitBu2)(κ2-bipyMe2)] (2) and [Ir(H)(OTf)(κ2-NSitBu2)(κ2-bipyMe2)] (3) (NSitBu2 = {4-methylpyridine-2-yloxy}ditertbutylsilyl) have been synthesized and characterized including X-ray studies of 3. A comparative study of the catalytic activity of complexes 2, 3, [Ir(H)(OTf)(κ2-NSitBu2)(coe)] (4), and [Ir(H)(OTf)(κ2-NSitBu2)(PCy3)] (5) (0.1 mol%) as catalysts precursors for the solventless formic acid dehydrogenation (FADH) in the presence of Et3N (40 mol%) at 353 K has been performed. The highest activity (TOF5 min ≈ 3260 h−1) has been obtained with 3 at 373 K. However, at that temperature the FTIR spectra show traces of CO together with the desired products (H2 and CO2). Thus, the best performance was achieved at 353 K (TOF5 min ≈ 1210 h−1 and no observable CO). Kinetic studies at variable temperature show that the activation energy of the 3-catalyzed FADH process is 16.76 kcal mol−1. Kinetic isotopic effect (5 min) values of 1.6, 4.5, and 4.2 were obtained for the 3-catalyzed dehydrogenation of HCOOD, DCOOH, and DCOOD, respectively, at 353 K. The strong KIE found for DCOOH and DCOOD evidenced that the hydride transfer from the C–H bond of formic acid to the metal is the rate-determining step of the process.
Introduction
The potential of formic acid (FA) as an effective liquid organic hydrogen carrier (LHC) has been the subject of many scientific studies over the past decade.1 There are many advantages of using FA as LHC, for example, it has a high concentration of H2 (53 g L−1), and it can be easily prepared, stored, and transported.1 Among its disadvantages it stands out the fact that its dehydrogenation to produce H2 and CO2 competes with its dehydration to give CO and H2O (Scheme 1). Therefore, due to CO poisoning of the Pt electrode in fuel cells,2 the development of selective catalysts to produce H2 from FA is of great interest to apply FA in fuel cell-based technologies. | ||
| Scheme 1 Possible transformations for FA. | ||
To the best of our knowledge, the first studies on the potential of the catalytic formic acid dehydrogenation (FADH) to supply hydrogen to fuel cells were independently reported by Beller et al.3 and Laurenczy et al.4 in 2008. Since then, many examples of catalytic systems active for the FADH have been published.1,2 The most active catalytic systems for FADH so far reported are based on water soluble Ir–Cp* species with functionalized bipyridine or biimidazoline derivatives as ligands.5–9 Among them, the species [IrClCp*(2,2′-bi-2-imidazoline)]Cl (TOF ≈ 487![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1),6 the iridium-bipyridine catalysts described by Fujita and Himeda et al. (TOF ≈ 228000 h−1),5 and the DMSO soluble Ir–Cp* species reported by Albrecht el al. (TOF ≈ 300000 h−1)10 stand out.
500 h−1),6 the iridium-bipyridine catalysts described by Fujita and Himeda et al. (TOF ≈ 228000 h−1),5 and the DMSO soluble Ir–Cp* species reported by Albrecht el al. (TOF ≈ 300000 h−1)10 stand out.
On the other hand, one of the aims of our research group is to study the potential of iridium(III) complexes with pyridine-2-yloxy-silyl ligands (Ir-NSi) as homogenous catalysts.11–13 These species have been successfully used as CO211 or formamide12a hydrosilylation catalysts. Ir-NSi complexes are characterized by their short Ir–Si bond distances (in the range 2.25–2.28 Å),14 which has been explained by assuming a significant ionic contribution to the covalent bond,12b,15 and the strong trans effect exerted by the silicon atom. Theoretical calculations have shown that the presence of the silicon play a key role in the reactivity of iridium-formate intermediates in catalytic CO2 hydrosilylation11 and FADH processes.16 A challenge when using Ir-NSi complexes as FADH catalysts is that under the reaction conditions the active species can be reduced to inactive iridium nanoparticles.16 Therefore, with the purpose of preparing iridium(III)-NSi complexes stable under FADH conditions, we decided to use the monoanionic bidentate ligand (4-methylpyridine-2-yloxy)ditertbutylsilyl (NSitBu2), where the Ir–Si bond is sterically protected by the bulk of the two tert-butyl substituents on the silicon atom.
An additional difficulty when using homogeneous transition metal catalysts with silyl-based ligands in protic solvents is the breaking of the Ir–Si bond, which is favored in the presence of a base.17 Indeed, the hydrolysis of the Ir–Si bond in Ir-NSi species is favoured in basic-media, moreover the coordination of water to unsaturated Ir-NSi species has been recently reported.12b Thus, we decided to explore the potential of electronically and sterically saturated Ir–Si species as FADH catalyst in the absence and in presence of water. In this regard, it should be mentioned that examples of catalytic systems effective for catalytic FADH under neat conditions are scarce.16,18,19
Results and discussion
Synthesis of Ir-(NSitBu2) complexes with the 4,4′-dimethyl-2,2′-bipyridine (bipyMe2) ligand
Iridium-bipyridine derivatives have shown to be highly effective catalysts in FADH processes.5 Therefore, to prepare coordinative and electronically saturated Ir-NSitBu2 species we decided to use 4,4′-dimethyl-2,2′-bipyridine (bipyMe2) as auxiliary bidentate ligand. The reaction of the iridium(III) complex [Ir(H)(Cl)(κ2-NSitBu2)(coe)] (1)12a with one equivalent of bipyMe2 at 353 K for 48 h leads to an orange solid, which has been characterized as complex [Ir(H)(Cl)(κ2-NSitBu2)(κ2-bipyMe2)] (2) (Scheme 2). The reaction of light protected CH2Cl2 solutions of 2 with silver triflate affords a yellow solid of [Ir(H)(OTf)(κ2-NSitBu2)(κ2-bipyMe2)] (3) (Scheme 2). | ||
| Scheme 2 Preparation of complexes 2 and 3 and molecular structure of complex 3. | ||
The Ir-(κ2-NSitBu2) species 2 and 3 have been characterized by means of multinuclear NMR spectroscopy. The 1H NMR of 2 and 3 in CD2Cl2 show a singlet resonance corresponding to the Ir–H at δ −18.03 and −17.53 ppm, respectively. These values are low field shifted with respect to that observed for 1 (−20.68 ppm), 4 (−27.46 ppm) and 5 (−29.21 ppm). The 1H–29Si HMBC NMR spectra show the resonance due to the silicon atom at δ 46.8 (2) and 41.0 (3), which compare well with the value found for the unsaturated species 1 (δ 41.0),12a4 (δ 45.8),12a and 5 (δ 44.5).12b19F{1H} NMR spectra of 3 (at 298 K) in CD2Cl2 show a sharp resonance at δ −78.82. Considering that 19F{1H} NMR spectra of free TfO− in CD2Cl2 (298 K) show a singlet resonance at δ −79.0,20 the value found for 3 suggests a weak Ir–OTf bond.12b,20
In order to attain a deeper understanding about the metal coordination sphere, the solid-state structure of compound 3 has been determined by single crystal X-ray diffraction, using synchrotron radiation. Selected geometrical parameters are reported in Table 1. The metal atom exhibits a distorted octahedral geometry, with a κ2-Si,N and a κ2-N′,N′′ coordination of (NSitBu2) and bipyMe2, respectively, the hydride and an oxygen atom of the triflate fragment. The Ir–Si and Ir–N(1) bond lengths have been found to be slightly shorter than those reported in compound 1 (2.2853(6) and 2.0947(18) Å, respectively).12a Indeed, our previous studies have already revealed that the substitution of the chloride by a triflate ligand (which is a more electron-withdrawing group), reinforce the Ir–Si bond.12b On the contrary, the Ir–O(2) bond length in 3 (2.381(4) Å), with the oxygen trans to the silicon, is in the upper limit of Ir–O bond lengths reported in Ir–OTf fragments (in the 2.04–2.40 Å range),21 indicating a weak bonding, in good agreement with the aforementioned 19F{1H} NMR spectra of 3.
| Ir–Si | 2.2731(12) | O(2)–Ir–N(1) | 91.53(16) |
| Ir–O(2) | 2.381(4) | O(2)–Ir–N(2) | 86.98(16) |
| Ir–N(1) | 2.063(4) | O(2)–Ir–N(3) | 75.89(15) |
| Ir–N(2) | 2.044(4) | O(2)–Ir–H | 94(2) |
| Ir–N(3) | 2.148(4) | N(1)–Ir–N(2) | 175.12(15) |
| Ir–H | 1.53(7) | N(1)–Ir–N(3) | 96.75(16) |
| Si–Ir–O(2) | 171.94(13) | N(1)–Ir–H | 83(3) |
| Si–Ir–N(1) | 81.92(12) | N(2)–Ir–N(3) | 78.38(16) |
| Si–Ir–N(2) | 99.92(12) | N(2)–Ir–H | 102(3) |
| Si–Ir–N(3) | 109.42(11) | N(3)–Ir–H | 170(3) |
| Si–Ir–H | 81(2) |
Effect of the ancillary ligand in the performance of Ir-(NSitBu2) catalyzed FADH
A comparative study of the activity of complexes 2, 3, 4, and 5 (0.1 mol%) as catalyst precursors for the hydrogen generation from FA-Et3N (40 mol% of Et3N) at 353 K was performed using a man on the moon™ reactor (Fig. 1).22 It should be mentioned that while compounds 2 and 3 with bipyMe2 as auxiliary bidentate ligand are coordinatively saturated species, complexes 412a and 512b are unsaturated species with distorted trigonal bipyramidal and square pyramidal geometries in the solid state, respectively.
 | ||
| Fig. 1 Ir-(NSitBu2) catalyst precursors. | ||
The results of these experiments show that complex 3 (TOF5 min = 1210 h−1) is a more effective catalyst precursor than 2 (TOF5 min = 900 h−1) (Fig. 2), and both precursors are more active than [Ir(H)(OTf)(κ2-NSitBu2)(coe)] (4) (TOF5 min = 600 h−1) and/or [Ir(H)(OTf)(κ2-NSitBu2)(PCy3)] (5) (TOF5 min = 60 h−1) (Fig. 2). The systems based on 4 and 5 show the decomposition of the catalyst to give iridium nanoparticles, in a similar fashion to that previously observed for the related Ir-(κ2-NSiMe2)2 species.16 Conversely, the formation of iridium nanoparticles was not observed when using 2 or 3. These results show that the use of coordinatively saturated catalyst precursors prevents the reduction of iridium(III) and encouraged us to study the performance of 3 as catalyst for FADH.
 | ||
| Fig. 2 TON versus time (min) from the 2, 3, 4 or 5 catalyzed (0.1 mol%) solventless dehydrogenation of FA-Et3N adduct (Et3N 40 mol%) at 353 K. The gas evolution was measured using a man on the moon™ reactor22 (H2: CO2 = 1:1). | ||
Subsequently, we have studied the influence of the catalyst loading on the catalytic performance of 3. The activity of the 3-catalyzed FADH in presence of Et3N (40 mol%) depends on the catalyst loading, thus using a 0.05 mol%, instead of 0.1 mol%, a reduction of the activity was observed (TOF5 min = 820 h−1; Fig. S14†). Therefore, we decided to use a 0.1 mol% loading in the following studies.
Once the best catalyst precursor, 3, was chosen and the catalyst loading (0.1 mol%) optimized, we decided to study the effect of temperature on the performance. Thus, we have studied the catalytic FADH using 3 (0.1 mol%) as catalyst precursor in the presence of an initial concentration of Et3N of 40 mol% at different temperatures (Fig. 3). These studies revealed that, as expected, the temperature produces a positive effect on the activity of the reaction (Fig. 3) and allowed us to determinate the initial reaction rates (TOF5 min) at 323, 333, 343, 353, 363 and 373 K (Table S16†). The Arrhenius plot for these data23 yields an apparent activation energy of 16.76 ± 1.72 kcal mol−1, which is lower than that found for the related [Ir(CF3CO2)(κ2-NSiMe2)2] species (27.5 kcal mol−1)16 and agrees with the greater activity of 3 in comparison to that found for [Ir(CF3CO2)(κ2-NSiMe2)2], under the same reaction conditions (Table 2).
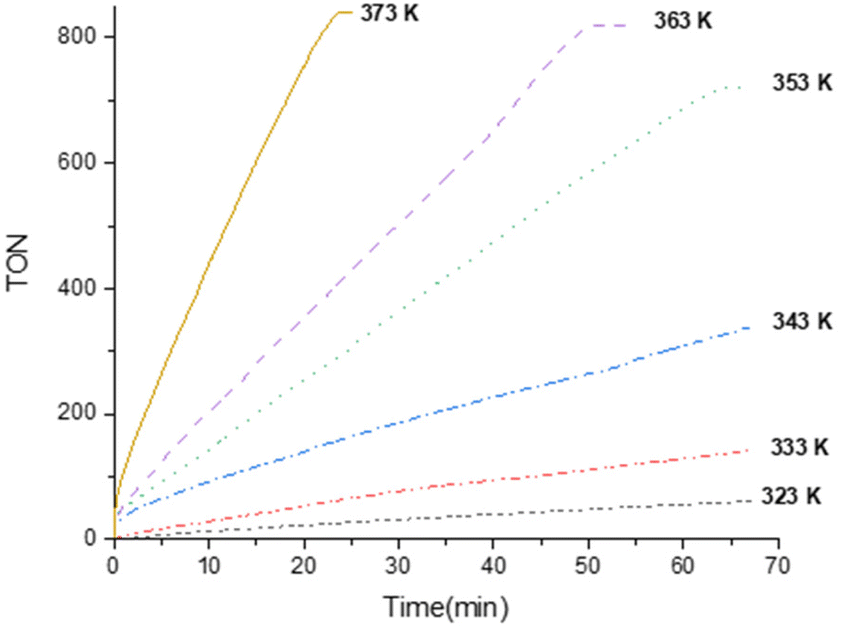 | ||
| Fig. 3 TON versus time (min) from the 3-catalyzed (0.1 mol%) solventless dehydrogenation of FA-Et3N adduct (Et3N 40 mol%) at different temperatures. The gas evolution was measured using a man on the moon™ reactor22 (H2:CO2 = 1:1). | ||
:2) adduct or sodium formate with 2, 4, 5 and other metal-based catalysts
| Catalyst | T (K) | TOF (h−1)/(time) | Ref. |
|---|---|---|---|
|
a sodium formate instead FA-Et3N (5:2) adduct,
b Maximum Turnover Frequency.
|
|||
| 4 | 353 | 600/(5 min) | This work |
| 5 | 353 | 60/(5 min) | This work |
| 2 | 353 | 900/(5 min) | This work |
| 3 | 353 | 1210/(5 min) | This work |
| 3 | 373 | 3260/(5 min) | This work |
| [Ir(CF3CO2)(κ2-NSiMe2)2] | 373 | 2900/(5 min) | 16 |
| [Ir(CF3CO2)(κ2-NSiMe2)2] | 353 | 600/(5 min) | 16 |
| [Mn(CO)2(tBuPNNOP)] | 353 | 8500/(3 min) | 24 |
| [RuCl2(PPh3)3] | 313 | 400/(2 h) | 3 |
| [Ir(COD)(NPtBu)][TfO]a |
363 | 3000b |
18 |
The activity of the catalytic system 3/Et3N (40 mol%) as FADH catalyst (373 K; TOF5 min = 3260 h−1) is higher than that reported for the iridium(III) complexes [Ir(CF3CO2)(κ2-NSiMe2)2] (373 K; TOF5 min = 2900 h−1)16 and [Ir(COD)(NPtBu)][TfO] (363 K; TOF5 min = 3000 h−1)18 but lower than that reported for the manganese compound [Mn(CO)2(tBuPNNOP)] (353 K; TOF5 min = 8500 h−1)24 (Table 2) and for the iridium(III) species reported by Fischmeister et al. (TOF ≈ 13300 h−1),19a Iglesias et al. (TOF ≈ 11600 h−1)19b and Gelman et al. (TOF ≈ 11800 h−1).19c
FTIR studies of the gases resulting from these 3-catalyzed reactions evidenced that the selectivity of the catalytic process depends on the temperature. Thus, while at 353 K, the formation of CO was below the detection limit of the FTIR (3 ppm) and, therefore, the selective formation of H2 and CO2 and residual Et3N was observed (Fig. 4), the FTIR spectra of the gases obtained from the reactions at 373 K shows the presence of traces of CO (Fig. S17†). It is known that the dehydration of neat FA to form H2O and CO is favored at high temperatures.25 Therefore, it is reasonable to assume that the traces of CO observed at 373 K may be formed by thermal, uncatalyzed decomposition pathways.26
 | ||
| Fig. 4 FTIR of the gas resulting from the solventless 3-catalyzed (0.1 mol%) FADH in presence of Et3N at 353 K. | ||
Kinetic isotope experiments on the 3-catalyzed FADH were performed at 353 K using 40 mol% of Et3N (Table 3 and Fig. 5). The results from these studies show that the initial TOF values (calculated after 5 min) dropped from 1210 to 770, 270 and 290 h−1 when HCOOH was replaced by HCOOD, DCOOH and DCOOD, respectively (Table 3). These TOF5 min values show a high kinetic isotopic effect (KIE) for DCOOH and DCOOD of 4.5 and 4.2, respectively (Table 3, entries 3 and 4), which evidenced that the hydride transfer from the CH of FA to the metal leading to CO2 is the rate-determining step of the process.26 Moreover, the KIE value (1.6) obtained when using HCOOD show a secondary KIE effect, analogous to that found for the related Ir-(κ2-NSiMe2)2 species (Table 3, entry 2).16
 | ||
| Fig. 5 TON versus time (min) from the 3-catalyzed (0.1 mol%) solventless dehydrogenation of HCOOH, HCOOD, DCOOH and DCOOD in presence of Et3N 40 mol% at 353 K. The gas evolution was measured using a man on the moon™ reactor.22 | ||
In all the studied 3-catalyzed (0.1 mol%) dehydrogenation of FA-Et3N (Et3N 40 mol%) reactions, once the gas evolution ends a blue residue remains in the reactor. 1H NMR spectra (DMSO-d6) of these blue residues show the presence of triethylammonium salts, a major component of the mixture, together with several unidentified Ir–H species. 1H–29Si HMBC spectra shows three different silicon resonances (Fig. S19†). Two of them correspond to compounds with tBu2Si groups not bonded to Ir (29Si{1H} NMR; δ −14.11 and −22.85 ppm), but the remaining resonance is due to a complex with an Ir–Si bond (29Si{1H} NMR; δ 45.11) and two inequivalent tBu substituents (Fig. 6).
 | ||
| Fig. 6 1H–29Si HMBC spectrum DMSO-d6 (400 MHz, 298 K) of the blue residue from the solventless 3-catalyzed (0.1 mol%) FADH in presence of Et3N (40 mol%) at 353 K. | ||
The addition of a second load of FA (50 μL) to this blue residue at 353 K evidenced that it catalyzed FADH. However, a decrease of the activity (TOF5 min ≈ 940 h−1) was observed (Fig. 7). This demonstrates that, although the use of 4,4′-dimethylbipyridine as ancillary ligand somehow protects the Ir–Si bond, the partial deactivation of the active species still occurs. It should be mentioned that the blue-residue is still active after 24 h (TOF5 min ≈ 270 h−1) (Fig. S15†).
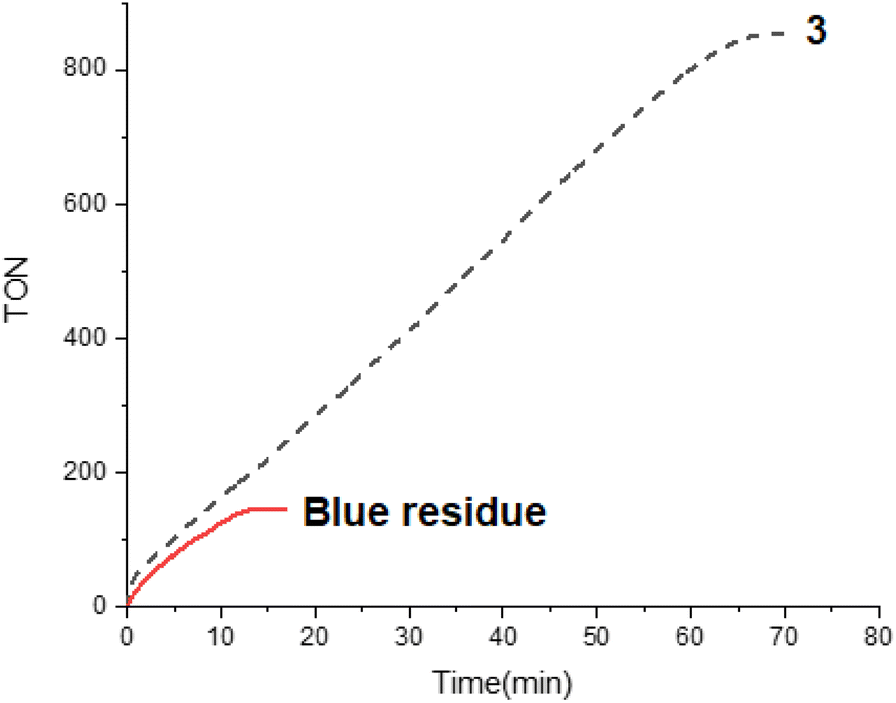 | ||
| Fig. 7 TON versus time (min) from the 3-catalyzed (0.1 mol%) of FA (250 μL) in presence of Et3N 40 mol% or the blue-residue catalyzed solventless dehydrogenation of HCOOH (50 μL) at 353 K. The gas evolution was measured using a man on the moon™ reactor.22 | ||
Therefore, the catalytic system based on 3, with bipyMe2 as auxiliary ligand, is more active (Table 2) and stable than [Ir(CF3CO2)(κ2-NSiMe2)2].16 This encouraged us to study its performance as FADH catalyst in an aqueous medium. These studies show that initially the system based on 3 (0.1 mol%), in presence of Et3N (40 mol%) at 353 K, is slightly more active in water solution (TOF5 min = 1500 h−1) than under solventless conditions. However, after 15 min of reaction catalyst deactivation is observed (Fig. 8).
 | ||
| Fig. 8 TON versus time (min) from the solventless 3-catalyzed (0.1 mol%) of FA (250 μL) in presence of Et3N 40 mol% (blue) at 353 K or 3-catalyzed (0.1 mol%) of FA (250 μL)/H2O (250 μL) in presence of Et3N 40 mol% at 353 K. The gas evolution was measured using a man on the moon™ reactor.22 | ||
Experimental
General information
Complexes 1,12a4,12a and 512b were prepared according to the reported method. HCOOH, HCOOD, DCOOH and DCOOD were purchased from commercial sources and used without further purifications. Et3N was purchased from commercial sources and distilled prior to use.
All reactions and manipulations were carried out under an argon atmosphere by using Schlenk-type techniques or in a Glovebox-MBraun UNIlab. Organic solvents were dried by standard procedures and distilled under argon prior to use or obtained oxygen- and water-free from a Solvent Purification System (Innovative Technologies). 1H, 13C, 29Si, 31P and 19F NMR spectra were obtained on a Bruker AV-300, AV-400 or AV-500 spectrometer using TMS as the internal reference in CD2Cl2, or DMSO-d6 as solvent. All chemical shifts (δ) are reported in ppm and coupling constants (J) are reported in Hz to apparent peak multiplets. 1H–1H-COSY, 13C-APT, 1H/13C HSQC, 1H/13C HMBC and 1H/29Si HMBC sequences were used for help in the assignments of the 1H, and 13C{1H} spectra. All catalytic experiments have been reproduced a minimum of three times.
Synthesis of [Ir(H)(Cl)(κ2-NSitBu2)(κ2-bipyMe2)] (2)
Toluene (10 mL) was added to a mixture of [Ir(Cl)(H)(κ2-NSitBu2)(coe)] (1) (0.538 g, 0.913 mmol) and 4,4′-dimethylbypyridine (0.185 g, 1.004 mmol). The reaction mixture was warmed at 353 K and stirred for 48 hours to give a dark-red solution and an orange precipitate. After that, the reaction mixture was cooled to 273 K. The red solution was filtered through Celite and the residue was washed with hexane (2 × 5 mL) and CH2Cl2 (2 × 2 mL) to give an orange solid of 2. 425 mg (70%). Anal. calcd for C26H37ClIrN3OSi: C, 47.08; H, 5.62; N, 6.33, found C, 46.93; H, 5.68; N, 6.48. 1H NMR plus COSY (300 MHz, CD2Cl2, 298 K): δ 9.33 (d, 3JH–H = 6.3 Hz, 1H, H6–Py), 9.18 (d, 3JH–H = 6.0 Hz, 1H, H6–Bpy), 7.96 (s, 1H, H3–Bpy), 7.91 (d, 3JH–H = 5.6 Hz, 1H, H6–Bpy), 7.87 (s, 1H, H3–Bpy), 7.20 (d, 3JH–H = 5.6 Hz, 1H, H5–Bpy), 7.02 (d, 3JH–H = 6.0 Hz, H5–Bpy), 6.69 (s, 1H, H3–Py), 6.58 (d, 3JH–H = 6.5 Hz, 1H, H5–Py), 2.48 (s, 3H, Me–Bpy), 2.44 (s, 3H, Me–Bpy), 2.32 (s, 3H, Me–Py), 1.09 (s, 9H, tBu–Si), 0.44 (s, 9H, tBu–Si), −18.03 (s, 1H, Ir–H). 13C{1H} NMR APT plus HSQC, COSY and NOESY (75 MHz, CD2Cl2, 298K): δ 158.7 (C6–Bpy), 151.2 (Cipso), 150.7 (C6–Py), 150.7 (C6–Bpy), 149.2 (Cipso), 147.8 (Cipso), 147.7 (C6–Bpy), 128.0 (C5–Bpy), 127.4 (C5–Bpy), 124.3 (C3–Bpy), 124.0 (C3–Bpy), 117.9 (C5–Py), 111.0 (C3–Py), 29.9 (CMe3), 29.0 (CMe3), 25.6 (CMe3), 25.3 (CMe3), 21.9 (Me–Bpy), 21.7 (Me–Bpy), 21.20 (Me–Py). 1H–29Si HMBC (60 MHz, CD2Cl2, 298 K): δ 46.8. High-resolution mass spectrometry (ESI+): calcd m/z = 663.2024; found m/z = 628.2339 (M ± Cl).Synthesis of [Ir(H)(OTf)(κ2-NSitBu2)(κ2-bipyMe2)] (3)
CH2Cl2 (8 mL) was added to a mixture of complex 2 (0.370 g, 0.558 mmol) and silver triflate (0.157 g, 0.614 mmol). The resulting suspension was stirred overnight at room temperature. The orange solution was filtered through Celite and the solvent was removed in vacuo, washed with hexane (2 × 5 mL) and dried in vacuo to give a yellow powder of 3.320 mg (74%). Anal. calcd for C27H37F3IrN3O4SSi: C, 41.74; H, 4.80; N, 5.41; S, 4.13; found C, 41.23; H, 4.78; N, 5.18; S, 4.36. 1H NMR plus COSY (300 MHz, CD2Cl2, 298 K): δ 9.08 (d, 3JH–H = 5.9 Hz, 1H, H6–Bpy), 8.43 (d, 3JH–H = 6.2 Hz, 1H, H6–Py), 8.02 (s, 1H, H3–Bpy), 7.94 (s, 1H, H3–Bpy), 7.82 (m, 1H, H6–Bpy), 7.32 (m, 1H, H5–Bpy), 7.10 (m, 1H, H5–Bpy), 6.76 (m, 1H, H3–Py), 6.71 (m, 1H, H5–Py), 2.52 (s, 3H, Me–Bpy), 2.47 (s, 3H, Me–Bpy), 2.36 (s, 3H, Me–Py), 1.07 (s, 9H, tBu–Si), 0.47 (s, 9H, tBu–Si), −17.40 (br, 1H, Ir–H). 13C{1H} NMR APT plus 1H–13C HSQC: δ 169.3 (Cipso), 159.9 (C6–Bpy), δ 159.1 (Cipso), 158.0 (Cipso), 148.3 (C6–Py), 147.7 (C6–Bpy), 128.5 (C5–Bpy), 127.8 (C5–Bpy), 124.4 (C3–Bpy), 124.2 (C3–Bpy), 118.9 (C5–Py), 111.7 (C3–Py), 29.7 (CMe3), 28.8 (CMe3), 25.4 (CMe3), δ 24.3 (CMe3), 22.2 (Me–Bpy), 22.0 (Me–Bpy), 21.4 (Me–Py). 1H–29Si HMBC (60 MHz, CD2Cl2, 298 K): δ 41.0. 19F{1H} NMR (300 MHz, CD2Cl2, 298 K): δ −78.82 (s, Ir–OTf). High-resolution mass spectrometry (ESI+): calcd m/z = 777.1855; found m/z = 628.2339 (M ± OTf).Single crystal structure determination of 3
X-ray data of 3 were collected in the XALOC beamline at ALBA synchrotron (Spain).27 Data were collected at 100 K with 0.72940 Å wavelength, using the Dectris Pilatus 6 M detector placed at 122.1 mm from the crystal. The complete φ scans were performed in steps of 0.5°, with an exposure time of 0.1 s per frame. Intensity transmission was attenuated to 20%. Sample crystallized as weakly diffracting thin needles, which suffered radiation damage during data collection. Several crystals were measured and subsequently merged. However, this strategy led to worst agreement factors. Therefore, the best data set was selected to perform the structural refinement. Small redundancy in the measured diffraction of this anisotropic sample may affect the absorption correction process. Data were indexed, integrated, and scaled using APEX4 package.28 The crystal structure was solved and refined using SHELXS29 and SHELXL30 in OLEX2 program.31433 reflections measured, 11179 unique; Rint = 0.0781; number of data/restraint/parameters: 11179/0/438; R1 = 0.0595 [9705 reflections, I > 2σ(I)], wR(F2) = 0.1756 (all data); largest difference peak: 6.91 e Å−3. Highest residual density peaks are found close to the metal atom.
General procedure for the formic acid dehydrogenation under neat conditions
Catalytic reactions were carried out on a microreactor (man on the moon™ series X102 Kit)22 with a total volume of 16.2 mL. Under an argon atmosphere, the reactor was filled with 250 μL of formic acid and the corresponding amount of the catalyst precursor. The reactor was then closed and heated to the desired temperature in an oil bath, and when the temperature and pressure of the system are stabilized, Et3N (370 μL, 40 mol% to FA) is injected with a microsyringe.Procedure for the formic acid dehydrogenation in water
Catalytic reactions were carried out on a microreactor (man on the moon™ series X102 Kit)22 with a total volume of 16.2 mL. Under an argon atmosphere, the reactor was filled with 250 μL of formic acid, 250 μL of distilled water, and the corresponding amount of the catalyst precursor. The reactor was then closed and heated to the desired temperature in an oil bath, and when the temperature and pressure of the system are stabilized, Et3N (370 μL, 40 mol% to FA) is injected with a microsyringe.Conclusions
The iridium(III) complexes 2, 3, 4 and 5 (0.1 mol%) are effective catalyst precursors for the solventless FADH in presence of Et3N (40 mol%). The catalytic system based on 3 has proven to be the most active of them all. The best catalytic performance (activity/selectivity) was achieved using 3 at 353 K (TOF5 min ≈ 1210 h−1). The highest activity (TOF5 min ≈ 3260 h−1) has been obtained with 3 at 373 K. However, at that temperature the FTIR spectra of the resulting gas show that the desired products (H2 and CO2) are impurified by traces of CO.Kinetic studies at variable temperature show that the activation energy of the 3-catalyzed FADH process is 16.76 kcal mol−1. KIE values of 1.6, 4.5, and 4.2 were obtained for the 3-catalyzed dehydrogenation of HCOOD, DCOOH, and DCOOD, respectively, at 353 K. The strong KIE found for DCOOH, and DCOOD (>4.0) evidenced that the activation of the C–H bond of FA is the rate-determining step of the process.
In conclusion, the use of the ligand κ2-NSitBu2 with two tert-butyl substituents on the silicon atom and 4,4′-dimethylbipyridine as ancillary ligand enhances the catalytic performance of Ir-NSi species as FADH catalyst precursors under solventless conditions. However, their use in an aqueous medium is hampered due to the short life of the catalyst under these conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from projects PID2021-126212OB-I00 (AEI-Spain) and DGA/FSE project E42_20R (Gobierno de Aragón) is gratefully acknowledged. A. G.-E. thankfully acknowledges the Universidad de Zaragoza and Banco Santander for a predoctoral fellowship “Ayudas para iberoamericanos y ecuatoguineanos en Estudios de Doctorado. Universidad de Zaragoza – Santander Universidades (2022–2023)”. The single crystal X-ray diffraction data were measured at XALOC bemline at ALBA synchrotron with the collaboration of ALBA staff. We are indebted to Dr Carsten Lenczyk (Bruker) for his strong support and fruitful discussions about the XR data treatment.References
- For recent reviews see: (a) M. Iglesias and F. J. Fernández-Alvarez, Catalysts, 2021, 11, 1288, DOI:10.3390/catal11111288; (b) K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef; (c) C. Guan, Y. Pan, T. Zhang, M. J. Ajitha and K.-W. Huang, Chem. – Asian J., 2020, 15, 937–946 CrossRef CAS PubMed; (d) P. Stathi, M. Solakidou, M. Louloudi and Y. Deligiannakis, Energies, 2020, 13, 733 CrossRef CAS; (e) M. Iglesias and L. A. Oro, Eur. J. Inorg. Chem., 2018, 2125–2138 CrossRef CAS; (f) K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed; (g) N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Coord. Chem. Rev., 2018, 373, 317–332 CrossRef CAS; (h) P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed; (i) J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS; (j) D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC; (k) A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC.
- (a) G. Kim and S.-H. Jhi, ACS Nano, 2011, 5, 805–810 CrossRef CAS PubMed; (b) S. K. Das, A. Reis and K. J. Berry, J. Power Sources, 2009, 193, 691–698 CrossRef CAS; (c) J. J. Baschuk and X. Li, Int. J. Energy Res., 2001, 25, 695–713 CrossRef CAS.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS PubMed.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS.
- Z. Wang, S.-M. Lu, J. Li, J. Wang and C. Li, Chem. – Eur. J., 2015, 21, 12592–12595 CrossRef CAS PubMed.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- A. Matsunami, Y. Kayaki and T. Ikariya, Chem. – Eur. J., 2015, 21, 13513–13517 CrossRef CAS.
- A. Matsunami, S. Kuwata and Y. Kayaki, ACS Catal., 2017, 7, 4479–4484 CrossRef CAS.
- N. Lentz and M. Albrecht, ACS Catal., 2022, 12, 12627–12631 CrossRef CAS.
- (a) J. Guzmán, P. García-Orduña, V. Polo, F. J. Lahoz, L. A. Oro and F. J. Fernández-Alvarez, Catal. Sci. Technol., 2019, 9, 2858–2867 RSC; (b) J. Guzmán, P. García-Orduña, F. J. Lahoz and F. J. Fernández-Alvarez, RSC Adv., 2020, 10, 9582–9586 RSC; (c) J. Guzmán, A. Urriolabeitia, M. Padilla, P. García-Orduña, V. Polo and F. J. Fernández-Alvarez, Inorg. Chem., 2022, 61(50), 20216–20221 CrossRef PubMed.
- (a) J. Guzmán, A. M. Bernal, P. García-Orduña, F. J. Lahoz, L. A. Oro and F. J. Fernández-Alvarez, Dalton Trans., 2019, 48, 4255–4262 RSC; (b) A. Gómez-España, P. García-Orduña, J. Guzmán, I. Fernández and F. J. Fernández-Alvarez, Inorg. Chem., 2022, 61, 16282–16294 CrossRef PubMed.
- F. J. Fernández-Alvarez, R. Lalrempuia and L. A. Oro, Coord. Chem. Rev., 2017, 350, 49–60 CrossRef.
- J. Guzmán, A. M. Bernal, P. García-Orduña, F. J. Lahoz, V. Polo and F. J. Fernández-Alvarez, Dalton Trans., 2020, 49, 17665–17673 RSC.
- For related studies see: P. García-Orduña, I. Fernández, L. A. Oro and F. J. Fernández-Alvarez, Dalton Trans., 2021, 50, 5951–5959 RSC.
- J. Guzmán, A. Urriolabeitia, V. Polo, M. Fernández-Buenestado, M. Iglesias and F. J. Fernández-Alvarez, Dalton Trans., 2022, 51, 4386–4393 RSC.
- L. Turculet, PSiP Transition-Metal Pincer Complexes: Synthesis, Bond Activation, and Catalysis, Chapter 6, in Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. F. Wendt, John Willey and Sons, 1st edn, 2014 Search PubMed.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- (a) S. Wang, H. Huang, T. Roisnel, C. Bruneau and C. Fischmeister, ChemSusChem, 2019, 12, 179–184 CrossRef CAS PubMed; (b) A. Iturmendi, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Green Chem., 2018, 20, 4875–4879 RSC; (c) S. Cohen, V. Borin, I. Schapiro, S. Musa, S. De-Botton, N. V. Belkova and D. Gelman, ACS Catal., 2017, 7, 8139–8146 CrossRef CAS; (d) A. Luque-Gómez, S. García-Abellán, J. Munarriz, V. Polo, V. Passarelli and M. Iglesias, Inorg. Chem., 2021, 60, 15497–15508 CrossRef PubMed.
- E. Suárez, P. Plou, D. G. Gusev, M. Martín and E. Sola, Inorg. Chem., 2017, 56, 7190–7199 CrossRef PubMed.
- (a) Cambridge Structural Database. CSD version 5.43 update Nov 2022; ; (b) C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- https://www.manonthemoontech.com/ .
- For recent examples of catalytic FADH activation parameters obtained from Arrhenius plot using initial TOF values see: (a) M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. – Eur. J., 2017, 23, 17017–17021 CrossRef CAS PubMed; (b) Y. Manaka, W.-H. Wang, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2014, 4, 34–37 RSC; (c) H. Junge, M. Beller and H. Grützmacher, ChemSusChem, 2018, 11, 3092–3095 CrossRef PubMed; (d) W.-H. Wang, H. Wang, Y. Yang, X. Lai, Y. Li, J. Wang, Y. Himeda and M. Bao, ChemSusChem, 2020, 13, 5015–5022 CrossRef CAS PubMed.
- N. H. Anderson, J. Boncella and A. M. Tondreau, Chem. – Eur. J., 2019, 25, 10557–10560 CrossRef CAS PubMed.
- (a) J. Yu and P. E. Savage, Ind. Eng. Chem. Res., 1998, 37, 2–10 CrossRef CAS; (b) N. Akiya and P. E. Savage, AIChE J., 1998, 44, 405–415 CrossRef CAS.
- G. Menendez Rodriguez, F. Zaccaria, L. Tensi, C. Zuccaccia, P. Belanzoni and A. Macchioni, Chem. – Eur. J., 2021, 27, 2050–2064 CrossRef CAS PubMed.
- J. Juinhuix, F. Gil-Ortiz, G. Cuní, C. Colldelram, J. Nicolás, J. Lidón, E. Boter, C. Ruget, S. Ferrer and J. Benach, J. Synchrotron Radiat., 2014, 21, 679–686 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 871, 3–8 Search PubMed.
- APEX 4 v2021.10-0. Bruker AXS Search PubMed.
- O. V. Dolomanov, L. J. Bourhus, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2247182 for compound 3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00744h |
| This journal is © The Royal Society of Chemistry 2023 |