
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew copper(I) complexes of bulky 5-substituted-2-iminopyrrolyl ligands as catalysts for azide–alkyne cycloaddition†
Tiago F. C.
Cruz
 a,
Patrícia S.
Lopes
a,
M. Amélia N. D. A.
Lemos
b,
Luís F.
Veiros
a and
Pedro T.
Gomes
*a
a,
Patrícia S.
Lopes
a,
M. Amélia N. D. A.
Lemos
b,
Luís F.
Veiros
a and
Pedro T.
Gomes
*a
aCentro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1000-049 Lisboa, Portugal. E-mail: pedro.t.gomes@tecnico.ulisboa.pt
bCERENA, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1000-049 Lisboa, Portugal
First published on 18th May 2023
Abstract
Five dinuclear copper(I) complexes of the type [Cu{κN,κN′-5-R-NC4H2-2-C(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(2,6-iPr2C6H3)}]2 (1a–e; R = 2,4,6-iPr3C6H2 (a), R = 2,6-Me2C6H3 (b), R = 3,5-(CF3)2C6H3 (c), R = 2,6-(OMe)2C6H2 (d), R = CPh3 (e)) were prepared by the reaction of the respective 5-R-2-iminopyrrolyl potassium salts KLa–e and [Cu(NCMe)4]BF4 in moderate yields. These new copper(I) complexes were characterized by NMR spectroscopy, elemental analysis and, in selected cases, by single crystal X-ray diffraction and their structural and electronic features further analyzed by DFT calculations and cyclic voltammetry, respectively. X-ray diffraction studies reveal dimeric Cu structures assembled by 2-iminopyrrolyl bridging ligands adopting a transoid conformation (complexes 1a and 1d), while complexes 1c and 1e displayed a cisoid conformation of those moieties, with respect to the Cu(I) centers. Additionally, VT-1H NMR and 1H–1H NOESY NMR experiments on complexes 1a–e exhibited complex fluxional processes in solution, assigned to a conformational inversion of the respective Cu2N4C4 metallacycles in all complexes but 1c, accompanied by a cisoid–transoid isomerization in the cases of complexes 1d,e. The Cu(I) complexes were also analyzed by cyclic voltammetry, where all complexes exhibit two oxidation processes, where the first oxidation is reversible, with the exception of 1b and 1c, which also show the highest oxidation potentials. The oxidation potentials follow clear trends related to the structural parameters of the complexes, in particular the Cu⋯Cu distance and the Cu2N4C4 macrocycles torsion angles. All new 5-substituted-2-iminopyrrolyl Cu(I) complexes 1a–e served as catalysts for azide–alkyne cycloaddition (CuAAC) reactions, being able to generate the respective 1,2,3-triazole products in yields as high as 82% and turnover frequencies (TOFs) as high as 859 h−1, after optimizing the conditions. The activity, as measured by the TOF, is in accordance with the oxidation potential of the corresponding complexes, the easier the oxidation, the higher the TOF value. Complex 1-H, where R = H, proved to be a poor catalyst for the same reactions, indicating that the 5-substitution in the ligand framework is crucial in stabilizing any potential catalytic species.
N(2,6-iPr2C6H3)}]2 (1a–e; R = 2,4,6-iPr3C6H2 (a), R = 2,6-Me2C6H3 (b), R = 3,5-(CF3)2C6H3 (c), R = 2,6-(OMe)2C6H2 (d), R = CPh3 (e)) were prepared by the reaction of the respective 5-R-2-iminopyrrolyl potassium salts KLa–e and [Cu(NCMe)4]BF4 in moderate yields. These new copper(I) complexes were characterized by NMR spectroscopy, elemental analysis and, in selected cases, by single crystal X-ray diffraction and their structural and electronic features further analyzed by DFT calculations and cyclic voltammetry, respectively. X-ray diffraction studies reveal dimeric Cu structures assembled by 2-iminopyrrolyl bridging ligands adopting a transoid conformation (complexes 1a and 1d), while complexes 1c and 1e displayed a cisoid conformation of those moieties, with respect to the Cu(I) centers. Additionally, VT-1H NMR and 1H–1H NOESY NMR experiments on complexes 1a–e exhibited complex fluxional processes in solution, assigned to a conformational inversion of the respective Cu2N4C4 metallacycles in all complexes but 1c, accompanied by a cisoid–transoid isomerization in the cases of complexes 1d,e. The Cu(I) complexes were also analyzed by cyclic voltammetry, where all complexes exhibit two oxidation processes, where the first oxidation is reversible, with the exception of 1b and 1c, which also show the highest oxidation potentials. The oxidation potentials follow clear trends related to the structural parameters of the complexes, in particular the Cu⋯Cu distance and the Cu2N4C4 macrocycles torsion angles. All new 5-substituted-2-iminopyrrolyl Cu(I) complexes 1a–e served as catalysts for azide–alkyne cycloaddition (CuAAC) reactions, being able to generate the respective 1,2,3-triazole products in yields as high as 82% and turnover frequencies (TOFs) as high as 859 h−1, after optimizing the conditions. The activity, as measured by the TOF, is in accordance with the oxidation potential of the corresponding complexes, the easier the oxidation, the higher the TOF value. Complex 1-H, where R = H, proved to be a poor catalyst for the same reactions, indicating that the 5-substitution in the ligand framework is crucial in stabilizing any potential catalytic species.
Introduction
The catalytic importance of copper arises from its ability to activate molecular oxygen in copper oxygenase systems.1 Crucially, copper(I) species play important roles as catalysts for many organic transformations, such as C–H activation,2 living radical polymerization3,4 and oxidation reactions.3,5 Of those catalytic systems, the copper-catalyzed azide–alkyne cycloaddition reaction (CuAAC) has become the most sought-after copper-based catalytic system.6 CuAAC reactions paved the way to unprecedented catalytic activity and selectivity towards the formation of 1,2,3-triazoles under mild conditions,7,8 having been applied in both homogeneous7 and heterogeneous9 platforms. The CuAAC reaction has had tremendous impact in the synthesis of heterocycles,10 natural products,11 porphyrin-based derivatives,12 dendrimers13 and polymers14 for example, with important implications in biological and material sciences. These types of efficient, atom-economical and thermodynamically favored reactions that lead specifically to one product are generally known as “click chemistry”. The chemical competence, versatility and implications of CuAAC reactions in click chemistry were greatly emphasized very recently by being the subject of the last Nobel Prize in Chemistry 2022.15The early copper sources used in CuAAC reactions consist either in the CuSO4/sodium ascorbate16 or in the CuI/diisopropylethylamine17 catalyst systems, which have led to near quantitative yields in the respective 1,2,3-triazole products using catalyst loadings as low as 1 mol%.7 Despite their efficiency, these catalyst systems display a maximum turnover frequency (TOF) of 27 h−1 and their multicomponent nature requires the use of excess amounts of the respective reducing agent, since the catalytically active CuAAC systems are usually Cu(I)-based. Furthermore, the separation of the catalyst from the reaction products remains a challenge. To overcome these hurdles, several works reported the addition of nitrogen-based ligand systems of variable denticity, which have also led to the acceleration of the respective CuAAC reactions.18
The catalyst systems mentioned above are prepared in situ, which means they are somewhat limited in terms of catalytic activity, and it is nearly impossible to understand the mechanism behind the respective reactions. To gain mechanistic insight into CuAAC reactions and improve their activity, the study of well-defined Cu(I) molecular systems is imperative.19 Some examples of well-defined copper(I) complexes capable of performing CuAAC reactions have been reported in the literature, in which the respective copper centers were supported by N-heterocyclic carbenes (Chart 1, A, maximum TOF = 950 h−1),20 phosphines/phosphonites/phosphinites (Chart 1, B, maximum TOF = 313 h−1),21 or nitrogen-based diimine ligands (Chart 1, C, maximum TOF = 1920 h−1).22 As a representative example of the development of nitrogen-based ligands toward CuAAC reactions, Tilley and co-workers, isolated a series of dinuclear copper(I) intermediates using the dinucleating ligand 2,7-bis(fluoro-di(2-pyridyl)-methyl)-1,8-naphthyridine (Chart 1, D),23 which supported the propensity of catalytic dinuclear species in CuAAC reactions, with cooperation between the two copper centers.24 When considering neutral dinuclear copper(I) complexes, several examples of their preparation and structural characterization are to be noted, the vast majority utilizing monoanionic ligand systems such as amidinates (X = CR) or guanidinates (X = N) (Chart 1, E).25
 | ||
| Chart 1 (A–C) Examples of CuAAC catalyst precursors and their maximum TOF for the formation of 1-benzyl-4-phenyl-1H-1,2,3-triazole values in italics; (D and E) nitrogen-based ligands used to stabilize dinuclear copper complexes; and (F–I) 2-iminopyrrolyl copper complexes reported in the literature. | ||
On the other hand, the copper chemistry with 2-iminopyrrolyl ligands, another monoanionic framework, is still quite limited. The literature concerning this ligand system is dominated by Cu(II) complexes of the type [Cu(2-iminopyrrolyl)2] (Chart 1, F).26 Also referring to Cu(II) complexes bearing these chelates, Schaper et al. reported several dinuclear complexes of the type [{Cu(2-iminopyrrolyl)}2(μ-O,κN)2] (Chart 1, G), which served as catalysts for the polymerization of rac-lactide.27 The first record of an isolable Cu(I) complex with 2-iminopyrrolyl ligands came at the hands of Emslie, Blackwell and co-workers when reporting the transoid dinuclear Cu(I) complex transoid-[Cu{κN,κN′-NC4H3-2-C(H)NiPr}]2 (Chart 1, H).28 A few years later, our group synthesized and characterized the Cu(I) complex [Cu{κN,κN′-NC4H3-2-C(H)N(2,6-iPr2C6H3)}]x, which, upon exposure to air, led to the formation of the bis chelated Cu(II) complex [Cu{κ2N,N′-NC4H3-2-C(H)N(2,6-iPr2C6H3)}2].29 More recently, Heinze and co-workers synthesized the dinuclear Cu(I) complex cisoid-[Cu{κN,κN′-NC4H3-2-C(H)N(4-tBuC6H4)}]2 (Chart 1, I) and studied its redox chemistry and electronic structure.30 Of all previously considered complexes, to the best of our knowledge, no studies including CuAAC reactions have been reported.
Over the last years, our group has synthesized numerous 5-substituted-2-iminopyrrolyl ligand precursors and their subsequent use as ligands for various coordination/organometallic molecular systems. In one aspect, some B(III) complexes have been prepared, characterized and studied as components of emissive layers in light emitting diodes.31 On the other hand, we have also reported several molecular systems with Mn,32 Fe33,34 and Co,33,35,36 and with Ni37 with applications in catalysis (as catalysts for hydrosilylation and hydroboration, and for oligo-/polymerization) and in magnetism (as single-ion magnets, i.e. SIMs), respectively. Considering the great stereochemical protection and electronic tuning of the 5-substituted-2-iminopyrrolyl framework, we present herein the first examples of Cu(I) complexes of this type. Aside from the structural characterization and electronic structure determination of the new Cu(I) complexes, we also present their catalytic competence towards CuAAC reactions.
Results and discussion
Synthesis and characterization of the copper(I) complexes
The reaction of the 5-substituted-2-iminopyrrolyl potassium salts KLa–e with [Cu(NCMe)4]BF4 in THF gave rise, after work-up, to the dinuclear copper complexes 1a–e (Scheme 1). These complexes were isolated as crystalline pale-yellow to yellow solids in good yields from concentrated toluene solutions cooled to −20 °C, except in the case of complex 1c, bearing 5-[3,5-(CF3)2C6H3] substituents, which was isolated from n-hexane at −20 °C. After crystallization from toluene, all complexes (except from 1c, having crystallized from n-hexane) are only partially soluble in that solvent, but well soluble in THF or halogenated solvents. Complexes 1a–e exhibited different structural isomers. According to a combination of NMR spectroscopy and single crystal X-ray diffraction studies (see below in the text and in the “X-ray diffraction structural studies” and “Dynamic behavior of the copper(I) complexes in solution” subsections), we have assigned the configuration of the two 2-iminopyrrolyl ligands to be transoid in complexes 1a,b and cisoid in complex 1c. Complexes 1d,e were isolated as mixtures of interconvertible cisoid and transoid conformers, as presented in Scheme 1. | ||
| Scheme 1 Synthesis of the Cu(I) complexes 1a–e. | ||
All complexes were characterized by 1H, 13C and 19F (for complex 1c) and multidimensional NMR spectroscopy, the spectra of which being presented in Fig. S7–S25 in the ESI.† The 1H NMR spectra of complexes 1a–c present the expected resonances for their respective 5-substituted-2-iminopyrrolyl moieties (see the stacked spectra in Fig. 1). The resonances corresponding to the iminic protons appear at 7.57–7.76 ppm for the three complexes. In complexes 1a and 1b the H3 and H4 pyrrolyl protons appear at 6.80–6.85 and 6.08–6.13 ppm, respectively, while the same protons in complex 1c appear at 6.98 and 6.70 ppm, respectively. This downfield shift of the pyrrolyl protons and, generally, of most of the aromatic moieties in complex 1c is attributed to the high degree of electron withdrawing character of the 5-[3,5-(CF3)2C6H3] substituent. The methine isopropyl protons of the N-(2,6-iPr2C6H3) substituent are inequivalent at room temperature, being characteristically split into two heptets. Generally, the 13C{1H} NMR spectra of complexes 1a–c follow the same trends as those observed for their respective 1H NMR spectra. Additionally, complex 1c presents a 19F{1H} trifluoromethyl resonance at −63.7 ppm. The 1H–1H NOESY spectra of complexes 1a,b present NOE through-space off-diagonal cross peaks between resonances of the respective 5-aryl and N-aryl rings, diagnostic of transoid configurations. Namely, the meta proton resonance of the 5-aryl ring in complex 1a (5-Ph-Hmeta), at δ 6.76, presents a spatial correlation with N-aryl methine and methyl resonances, at δ 1.12 and the methyl resonances of the 5-aryl substituents in complex 1b, at δ 2.35 and 1.62, present spatial correlations with N-aryl methine and methyl resonances, at δ 3.39 and 2.91 and at δ 1.21, respectively (see Fig. S10 and S14 in ESI†). On the other hand, the 1H–1H NOESY spectrum of complex 1c does not present any NOE through-space off-diagonal cross peaks between resonances of the respective 5-aryl and N-aryl rings, which is, conversely, diagnostic of a cisoid configuration.
 | ||
| Fig. 1 Stacking of the 1H NMR spectra (300 MHz, CDCl3, 298 K) of complexes 1a–c. | ||
On the other hand, compounds 1d,e display much more complex 1H NMR spectra (see the stacked spectra in Fig. 2). The 1H NMR spectra of complexes 1d,e systematically display two sets of resonances corresponding to two different 2-iminopyrrolyl moieties, with one major and another minor species being observed. Even considering this intriguing observation, while the two sets of resonances are well resolved in complex 1e, compound 1d only displays spectral resolution for one of the observed species, suggesting a complex fluxional behavior of the latter. The dynamic processes occurring in complexes 1d,e shall be further explored in a separate section on the article (see below in the “Dynamic behavior of the copper(I) complexes in solution” subsection). Again, the 13C APT NMR spectra of complexes 1d,e follow the same trends as those observed for their respective 1H NMR spectra.
 | ||
| Fig. 2 Stacking of the 1H NMR spectra (300 MHz, CDCl3, 298 K) of complexes 1d,e: (a) full view of the spectra, and (b) expansion of the pyrrolyl/aromatic, O(CH3)/CH(CH3)2 and CH(CH3)2 sections. | ||
The 1H–1H NOESY spectra of complexes 1d,e present NOE through-space off-diagonal cross peaks between resonances of the respective 5-aryl, trityl and N-aryl rings, diagnostic of transoid conformations. Namely, the methoxy resonance of the 5-aryl ring of the major species in complex 1d, at δ 3.67, presents a spatial correlation with the methyl resonance of the N-aryl ring, at δ 0.98, and some of the 5-trityl resonances of the major species in complex 1e, at δ 6.97, present a spatial correlation with the methyl resonances of the N-aryl ring, at δ 0.36, 0.74, 0.91 and 1.06 (see Fig. S21 and S25 in ESI†). On the other hand, the methoxy resonance of the 5-aryl ring of the minor species in complex 1d, at δ 3.49, does not present any NOE through-space off-diagonal cross peak with an N-aryl ring resonance, which is diagnostic of a cisoid conformation for the minor species. We therefore conclude that both species observed in solution for complexes 1d,e are the cisoid and the transoid conformers.
Considering that all complexes were easily characterized by NMR spectroscopy, i.e. no paramagnetism whatsoever was observed in the solution samples, it is clear that we are in the presence of Cu(I)–Cu(I) complexes. Even though complexes 1a–e are in the +1 oxidation state and present a potentially reactive “[Cu{κ2N,N′-5-R-NC4H2-2-C(H)N(2,6-iPr2C6H3)}]” synthon, attempts to react them with small molecules, such as N2, O2, CS2 or S8, or with weakly coordinating molecules, such as THF or acetonitrile, were unsuccessful, proving their chemical inertness toward these substrates. However, prolonged heating of these complexes in halogenated solvents led to the formation of very dark red solutions, soon followed by decomposition of the respective mixtures. In toluene solutions, however, the complexes are indefinitely stable under dinitrogen, even after prolonged heating at 100 °C.
X-ray diffraction structural studies
Complexes 1a and 1c–e were obtained as pale-yellow to yellow crystalline solids suitable for single crystal X-ray diffraction and were, therefore, characterized by this technique. Complexes 1d,e crystallized in the triclinic system, in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, while complexes 1a and 1c crystallized in the monoclinic system, in the P21/c and C2/c space groups, respectively. The structures of complexes 1a, 1c, 1d and 1e are presented in Fig. 3, a selection of bond lengths and angles, as well as their crystallographic data, being listed in Tables S1 and S2 (in the ESI†), respectively. Additionally, the molecular structure of complex 1c is generated by a −x, −y, −z symmetry operation of the “[Cu{κN-5-[3,5-(CF3)2C6H3]-NC4H2-2-C(H)N(2,6-iPr2C6H3)}]” fragment, with an inversion center at [0, 0, 0].
space group, while complexes 1a and 1c crystallized in the monoclinic system, in the P21/c and C2/c space groups, respectively. The structures of complexes 1a, 1c, 1d and 1e are presented in Fig. 3, a selection of bond lengths and angles, as well as their crystallographic data, being listed in Tables S1 and S2 (in the ESI†), respectively. Additionally, the molecular structure of complex 1c is generated by a −x, −y, −z symmetry operation of the “[Cu{κN-5-[3,5-(CF3)2C6H3]-NC4H2-2-C(H)N(2,6-iPr2C6H3)}]” fragment, with an inversion center at [0, 0, 0].
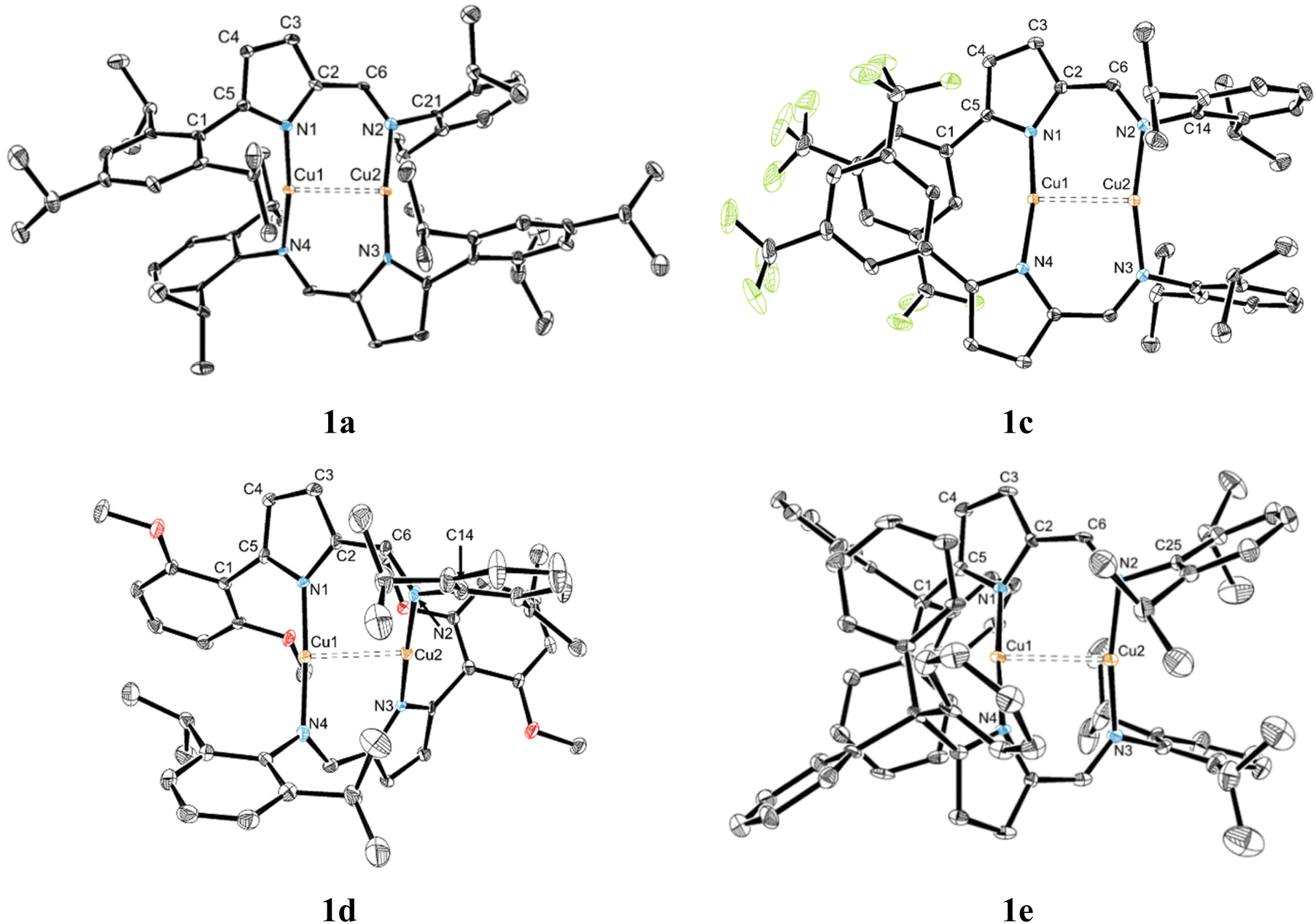 | ||
| Fig. 3 ORTEP-3 diagrams of complexes 1a, 1c, 1d and 1e, showing 30% probability ellipsoids. All hydrogen atoms were omitted for clarity. | ||
All complexes contain two copper atoms and two bridging 5-substituted-2-iminopyrrolyl ligands, with one dinuclear entity in the respective asymmetric units. The copper centers in all structures exhibit a nearly linear coordination geometry, as the N–Cu–N bond angles in all complexes range from 166.1(2) to 176.7(2)°. In all complexes, each copper center has two coordinated nitrogen atoms from two different 2-iminopyrrolyl ligands and a cuprophilic pseudo-contact to the other copper atom present in the molecule (see “DFT structural studies” subsection). In general, the Cu–N bond lengths for all complexes are in the range of 1.841(4)–1.917(5) Å. This observation is in accordance with the other crystallographically characterized dinuclear Cu(I) complexes of the same type.25,30 The Cu1⋯Cu2 distances of all complexes range between 2.4806(10)–2.5677(11) Å, rendering them longer than the other reported dinuclear Cu(I) complexes bearing 2-iminopyrrolyl ligands.28,30
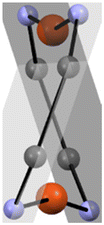
All complexes form Cu2N4C4 ten-membered macrocyclic cores (in which C corresponds to the two iminic and the two ipso C2 carbon atoms of each of the 2-iminopyrrolyl ligands) with twisted conformations, with varying degrees of torsion. The different torsions of the twisted Cu2N4C4 macrocycles were measured by considering the two planes containing both nitrogen atoms of the two different ligands, i.e. the N2C2 coordinating planes of the respective 2-iminopyrrolyl fragments, and are summarized in Table 1.
| Complex | Torsion (°) | |
|---|---|---|
| 1a | 49.50(2) |
|
| 1c | 36.30(2) | |
| 1d | 67.51(2) | |
| 1e | 64.50(2) |
The crystalline state structures of complexes 1a and 1d present a transoid conformation. Complex 1a displays the longest Cu1⋯Cu2 distance of all complexes but, by contrast, has the shortest Cu–N bond lengths, with the Cu1–N1/Cu2–N3 and Cu1–N2/Cu2–N4, i.e. the Cu–Npyrrolyl and Cu–Nimine lengths being equal to 1.841(5)–1.844(4) and 1.873(4)–1.876(5), respectively. Complex 1d, also with a transoid conformation, has the shortest Cu1⋯Cu2 distance of all complexes, whilst displaying slightly longer Cu–N bonds than complex 1a, in the range of 1.865(5)–1.891(5) Å. Complex 1a, owing to the high rigidity, electron donating capability and stereochemical repulsion imparted by the 5-(2,4,6-iPr3C6H2) substituents, is able to stabilize a transoid conformation with a Cu2N4C4 macrocycle torsion angle equal to 49.6°, much lower than that of complex 1d (the other transoid structure with a Cu2N4C4 macrocycle torsion angle of 67.5°), at the expense of the expansion of the Cu2N4C4 macrocycle, resulting in a Cu1⋯Cu2 distance of 2.5677(11) Å. The dihedral angles formed between either both aryl ring planes in complex 1a or the N-(2,6-iPr2C6H3) ring plane of 1d with the iminopyrrolyl plane are in the range of 61.86–72.89°. On the other hand, the dihedral angles of the 5-[2,6-(MeO)2C6H3] substituent ring planes with the iminopyrrolyl plane in 1d are in the range of 44.79–47.83°, justified by the observation of Cu1⋯O2 and Cu2⋯O4 contacts, with lengths in the range of 2.766–2.984 Å.
Conversely, complexes 1c and 1e exhibit a cisoid conformation in the crystalline state. In the case of complex 1c, all its Cu–N bond lengths are very similar, in the range of 1.8616(15)–1.8660(16) Å (Δ < 0.01 Å), indicating a considerable degree of electronic delocalization. On the other hand, the Cu–N bond length in complex 1e is slightly different, thus implying electronic differentiation of the two copper centers of those complexes. Complexes 1d and 1e present the most twisted configurations, with torsion angles in the 64.5–67.5° range. In addition, in complex 1c, a weak π-stacking of adjacent 5-[3,5-(CF3)2C6H3] rings is observed, the distances between the respective centroids being equal to 3.844 Å and the angle between those two rings equal to 5.93°. In the case of complex 1c, the stacking of adjacent 5-[3,5-(CF3)2C6H3] rings in a nearly staggered conformation is an important driving force for the stabilization of the observed cisoid isomer. The dihedral angles of the N-(2,6-iPr2C6H3) rings with the respective iminopyrrolyl planes in complexes 1c and 1e are in the range of 63.74–86.70°. By contrast, as expected from the stacking capability of the ortho-unencumbered 5-[3,5-(CF3)2C6H3] substituents in 1c, the corresponding dihedral angles are much smaller, being equal to 27.27°. It is also observed that the unit cells of all crystal structures derived from single-crystal X-ray diffraction display both conformations of the Cu2N4C4 macrocycle, generated by a symmetry operation along the Cu⋯Cu axis.
DFT structural studies
All new complexes were also the subject of computational studies by DFT calculations.38 The structures of the complexes were first optimized to their respective energy minima and, subsequently, appropriate single-point calculations were further performed (see “Computational details” of the Experimental section). This methodology was performed on a total of twelve structures, i.e. the cisoid and transoid isomers of complexes 1a–e and of 1-H, the latter corresponding to the 5-unsubstituted (R = H) analogue of complexes 1a–e, previously reported by us.29 The coordinates of all optimized structures are presented in the ESI.† Additionally, a summary of the comparisons between experimental data derived from X-ray diffraction and the calculated parameters for the corresponding isomers is presented in Table 2. It was also possible to derive the relative stability of the cisoid and transoid isomers from DFT calculations, and the corresponding Gibbs energy differences ( ). For complexes 1b and 1-H, for which no experimental structural data was available, the values of the computationally calculated most stable cisoid isomers are listed.
). For complexes 1b and 1-H, for which no experimental structural data was available, the values of the computationally calculated most stable cisoid isomers are listed.
| Complex | Cu1⋯Cu2 distancec (Å) | Cu–Npyrrolyl lengthc (Å) | Cu–Nimine lengthc (Å) | Torsionc (°) |
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc. | Exp.d | Calc. | Exp.d | Calc. | Exp. | Calc. | ||
| a Determined by single crystal X-ray diffraction. b Determined by DFT calculations (see “Computational details” in the Experimental section). c Transoid isomer for 1a and 1d, and cisoid isomer for 1c and 1e. d Average value. | |||||||||
| 1a | 2.568(1) | 2.630 | 1.843(5) | 1.876 | 1.875(5) | 1.890 | 50 | 47 | −3.8 |
| 1b | — | 2.564 | — | 1.880 | — | 1.882 | — | 48 | 5.6 |
| 1c | 2.497(1) | 2.523 | 1.862(2) | 1.883 | 1.867(2) | 1.881 | 36 | 48 | 6.0 |
| 1d | 2.481(2) | 2.476 | 1.865(5) | 1.889 | 1.891(6) | 1.907 | 68 | 72 | 7.7 |
| 1e | 2.535(2) | 2.580 | 1.917(5) | 1.938 | 1.861(5) | 1.873 | 65 | 60 | 0.7 |
| 1-H | — | 2.537 | — | 1.867 | — | 1.883 | — | 48 | 2.2 |
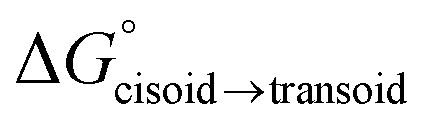
In another aspect, whilst observing the relative Gibbs energies of the cisoid or the transoid isomers of the copper complexes, it is clear that the former are more thermodynamically favored than the latter, except for complex 1a, for reasons discussed above in the “X-ray diffraction structural studies” subsection. For complexes 1a and 1c, the preferred isomers calculated by DFT studies seem to be in accordance with those observed by single-crystal X-ray diffraction and NMR spectroscopy. For complex 1d, the DFT calculations do not necessarily corroborate the experimental results. Finally, for complex 1e, DFT correctly predicted the transoid–cisoid equilibrium observed in solution by NMR spectroscopy. Since the present DFT calculations only account for isolated molecules and not their intermolecular interactions in solution or in solid state, the small energy differences obtained might have inappropriately reproduced the molecular systems at stake. Unfortunately, the size of the molecules studied in this work precluded a DFT mechanistic study of the cisoid/transoid isomerization equilibria.
A Natural Population Analysis (NPA) was performed on all complexes in their favored configurations (transoid for complexes 1a,b and cisoid for complexes 1c–e) in order to further understand the electronic nature and bonding of the Cu centers. For all complexes, the charges of the copper atoms bonded to pyrrolyl or iminic nitrogen atoms vary between 0.65–0.74 and 0.67–0.70, respectively. These results reinforce the fact that both copper centers are in the +1-oxidation state. Additionally, the Cu–Cu Wiberg indices for all complexes are in the range of 0.06–0.07, which clearly indicates that no copper–copper bonding is present. In view of these results and considering that the C2–C6 bond lengths are in the range of 1.393(9)–1.419(8) Å, somewhat shorter than expected for a C(sp2)–C(sp2) single bond,39 two observations regarding the bonding of the complexes may be noted: (1) the Cu(I) moieties in the complexes are neutral, and (2) the negative charge of the anionic 2-iminopyrrolyl ligands is delocalized across both the pyrrolyl backbone and the iminopyrrolyl synthon, i.e. the N1–C2–C6–N2 moieties. Whereas Heinze and co-workers, while mentioning complex cisoid-[Cu{κN,κN′-NC4H3-2-C(H)N(4-tBuC6H4)}]2 (see Chart 1, I) with similar C2-C6 bond lengths, interpreted these Cu(I)–Cu(I) dimeric entities as zwitterions,30 we believe that our present interpretation more broadly describes the bonding in these copper complexes.
Electrochemical cyclic voltammetry studies
The copper(I) complexes were also analyzed by cyclic voltammetry to gain insight regarding their electronic structure. The experiments were performed in dichloromethane utilizing [N(n-Bu)4][BF4] as a supporting electrolyte. As an example, the cyclic voltammogram of complex 1a, at a scanning rate of 200 mV s−1, is presented in Fig. 4. | ||
| Fig. 4 Cyclic voltammogram for the oxidation of complex 1a in a [N(n-Bu)4][BF4]/CH2Cl2 solution at 200 mV s−1 scan rate. Potentials in volts measured versus FcH/FcH+. The black arrow indicates the initial potential scanning direction. | ||
All complexes undergo a first oxidation process in a range of potentials from 0.37 V for complex 1d to 1.00 V for complex 1c, followed by a second oxidation process occurring from 1.02 V for complex 1d to 1.31 V for complex 1c (see Table S3 in the ESI†). The first oxidation is reversible in the analyzed range of scan rates, from 50 mV s−1 to 1 V s−1, for all complexes, except for 1b and 1c, which are the two complexes exhibiting the highest first oxidation potentials, showing that oxidation is more difficult to occur in the latter complexes (see Fig. S26†). The electrochemical behavior of complex 1-H is similar to that of complexes 1b and 1c showing an irreversible oxidation at 0.93 V.
The range of oxidation potentials is consistent with other studies on bimetallic copper(I) complexes.30 The complexes exhibiting a reversible first oxidation process (1a, 1d and 1e) exchange a single electron, whereas two electrons are involved in the second oxidation wave, as can be seen in Table S3 (ESI†) from the corresponding ip [ox (I)]/ip [ox (II)] ratio. For the remaining complexes 1b and 1c, the first oxidation involves two electrons and is irreversible, while the second oxidation involves only one electron (see the corresponding ip [ox (I)]/ip [ox (II)] ratio in Table S3 in the ESI†). Despite this, the two oxidation potentials are clearly correlated, as can be seen in Fig. S27,† for all complexes under study.
Complex 1d, which has the smallest Cu1⋯Cu2 interatomic distance, is the one that also shows the lowest first oxidation potential. Additionally, as the Cu1⋯Cu2 distance increases, the first oxidation potential also increases (see Fig. S28 in the ESI†). A notable exception is the case of complex 1c, which is much harder to oxidize than expected in this trend, owing to the high electron-withdrawing ability of the ligand. A trend is also observed with the measured torsion angle: the higher the torsion angle, the lower the oxidation potential, i.e., the easier the oxidation (see Fig. S29 in the ESI†).
Dynamic behavior of the copper(I) complexes in solution
While characterizing the new copper complexes in solution by NMR spectroscopy at room temperature, we noticed that complexes 1a–c showed a single entity, while complexes 1d,e showed a mixture of two species. To understand these observations, we further explored these results by performing variable-temperature NMR (VT-NMR) spectroscopy studies. Owing to the persistent decomposition of all complexes after prolonged heating in halogenated solvents, the VT-NMR experiments were performed in toluene-d8, in the temperature range of −60 to 90 °C. The complete VT-NMR spectra for all complexes are presented in Fig. S30–S34 in the ESI.†In all complexes except 1c, the isopropyl groups of the respective iminic aryl groups are inequivalent below room temperature and tend to coalesce at higher temperatures. The values of the Gibbs energies of activation (ΔG‡) for the dynamic processes operating in the copper complexes were estimated using the Eyring equation, considering the respective rate constants of the frozen process at the resonances coalescence temperature (see “Kinetic and thermodynamic data derived from VT-NMR studies in complexes 1a–e” subsection in the ESI†). The values of ΔG‡ for the fluxional processes associated with resonances coalescence for complexes 1a,b and 1e are in the range of 15.9–17.9 kcal mol−1 (see Table S4 in the ESI†). The fluxional processes are possibly associated with the rotation of the respective aryl rings, since all calculated barriers are within the expected values for the rotation of an aromatic ring.40 The presented calculations only considered the coalescence of the methinic resonances of the respective N-(2,6-iPr2C6H3) substituents. Even though the methyl resonances of the respective N-(2,6-iPr2C6H3) substituents also exhibited coalescence at higher temperatures, their complex overlapping nature prevented the gathering of accurate parameters. No parameters were determined for complex 1d, owing to the overlapping and broadening of the respective CH(CH3)2, CH(CH3)2 and O(CH3) 1H NMR resonances. In addition, it is observed that the methyl protons of the 5-(2,6-Me2C6H3) substituents in complex 1b are also inequivalent, implying a hindered rotation of those rings, with a rotation barrier of 15.5 kcal mol−1. By contrast with all the remaining compounds, the VT-NMR spectra of complex 1c revealed to be completely invariant with temperature, no dynamic processes whatsoever being observed. In summary, the rotational barriers recorded for all complexes except for complex 1c are likely associated to a conformational inversion of the Cu2N4C4 metallacycle (i.e. inversion of the torsion angle evidenced in Table 1). This fact is supported by (1) the same energy barriers recorded across different resonances of the complexes and (2) the observation of both conformers in the unit cells of the molecular structures studied by single crystal X-ray diffraction, both expected to be equivalent species in solution.
The VT-NMR spectra of complexes 1d,e are much more complex than those of complexes 1a–c, with additional processes occurring, aside from aryl ring rotations. Fig. 5 shows sections of the VT-1H NMR and of the room temperature 1H–1H NOESY NMR spectra of complex 1e, as a representative example.
 | ||
| Fig. 5 Sections of the (a) VT-1H NMR spectra of complex 1e (300 MHz, toluene-d8) highlighting the chemical equilibrium taking place, and of the (b) 1H–1H NOESY spectrum of complex 1e (300 MHz, CDCl3, room temperature) displaying chemical exchange off-diagonal cross peaks between the minor (cisoid-1e) and major (transoid-1e) species. The assignment of the NCH (orange circles), H3 + H4 (red circles), CH(CH3)2 (green circles) and CH(CH3)2 (blue circles) resonances of the major and minor isomers are distinguished by the respective darker and lighter colored circles, respectively. The dashed lines in (b) are a guide to the eye. | ||
From the spectra shown in Fig. 5(a), we can depict that the two conformers in complex 1e, transoid-1e and cisoid-1e, are components of a chemical equilibrium, as their relative concentrations vary with temperature. In the case of complex 1d, it is possible to almost exclusively observe transoid-1d or cisoid-1d at low (<−60 °C) or at high (>90 °C) temperatures, respectively. We have established above that the transoid conformer was the major species for both complexes 1d,e. The thermodynamic parameters for the equilibria observed in complexes 1d,e were estimated via van't Hoff analyses (see “Isomer equilibria observed in complexes 1d,e determined by van't Hoff analysis” subsection in the ESI†), utilizing different resonances whenever possible, and are presented in Table S5 and Fig. S35, S36 in the ESI.† Further evidence that chemical equilibria are taking place in complexes 1d,e is offered by the presence of chemical exchange off-diagonal cross peaks between the same resonances of cisoid and transoid conformers in the 1H–1H NOESY NMR spectra of those complexes. As an example, such cross peaks were clearly observed for several resonances across complex 1e spectrum, namely those corresponding to NCH, H3, H4, CH(CH3)2 and CH(CH3)2 protons, as demonstrated in Fig. 5(b).
From the thermodynamic data, it is observable that the chemical exchange process occurring in complexes 1d,e is nearly at equilibrium, as the respective average of the calculated ΔG° values are nearly 0 kcal mol−1 (−0.7 to −0.1 kcal mol−1), being endothermic (ΔH° = 1.2 to 2.4 kcal mol−1) processes with small entropy changes (ΔS° = 6.4 to 8.5 cal mol−1 K−1).
In summary, the observed equilibria in complexes 1d,e is a transoid–cisoid isomerization and is likely assisted by the coordinative stabilization of the ortho-methoxide groups present in complex 1d or a cogged-wheel rotation of the adjacent CPh3 groups in complex 1e. The equilibria in complexes 1d,e is also likely accompanied by the same conformational metallacycle inversion process proposed for complexes 1a,b.
The occurrence of this equilibrium process in the remaining complexes 1b,c cannot be ruled out. In these complexes, where a single species was observed, the conformational process may be operating much too quickly for it to be observed in the NMR timescale, owing to smaller activation energy barriers. The lack of solution dynamic behavior observed in complex 1c is very likely related with the rigidity imparted by the stacking of the 5-[3,5-(CF3)2C6H3], whose ortho protons remain inequivalent in the range of temperatures studied. This rigidity blocks the concerted process of change in the conformation of complex 1c.
Azide–alkyne cycloaddition catalyzed by the copper(I) complexes
Complexes 1a–e were tested as catalysts for azide–alkyne cycloaddition reactions. The results of the catalytic runs for complexes 1a–e using phenylacetylene as the alkyne and (azidomethyl)benzene as the azide are presented in Table 3, with catalytic activities measured in turnover frequencies (TOF). In a preliminary reaction, when 1 mol% of complex 1b was used as catalyst for the bulk reaction of phenylacetylene with (azidomethyl)benzene, the formation of the respective 1,2,3-triazole product, 1-benzyl-4-phenyl-1H-1,2,3-triazole, was observed in a near quantitative yield in half an hour (entry 1). By reducing the loading of complex 1b to 0.5 and 0.1 mol%, we observed yields of the respective 1,2,3-triazole product equal to 67 and 42%, respectively (entries 2 and 3). To optimize the reaction conditions, we performed subsequent catalytic runs in dichloromethane and varied the catalyst loading, using complex 1b as reference. It was observed that by carrying out the catalytic runs in dichloromethane with 1 mol% of complex 1b, the yield decreased to 70% (entry 4), but the reaction proceeded in a much more controlled fashion. Also, by reducing the loading of 1b, the catalysis yields were as low as 29% (entry 5). Taking these optimizations into account, we proceeded our studies in dichloromethane and using 0.5 mol% of catalyst loading.|
|
|||||
|---|---|---|---|---|---|
| Entry | Complex | mol% | Solvent | Yielda (%) | TOF (h−1) |
| Conditions: 0.9 mmol of phenylacetylene; 0.9 mmol of (azidomethyl)benzene; reaction time: 0.5 h; temperature: 25 °C; solvent volume = 5 mL.a Yields determined by weighing the isolated reaction products. | |||||
| 1 | 1b | 1 | Neat | >99 | >300 |
| 2 | 1b | 0.5 | Neat | 67 | 266 |
| 3 | 1b | 0.1 | Neat | 42 | 845 |
| 4 | 1b | 1 | CH2Cl2 | 70 | 208 |
| 5 | 1b | 0.1 | CH2Cl2 | 29 | 859 |
| 6 | 1b | 0.5 | CH2Cl2 | 60 | 358 |
| 7 | 1a | 0.5 | CH2Cl2 | 82 | 490 |
| 8 | 1c | 0.5 | CH2Cl2 | 47 | 281 |
| 9 | 1d | 0.5 | CH2Cl2 | 80 | 481 |
| 10 | 1e | 0.5 | CH2Cl2 | 69 | 412 |
| 11 | 1-H | 0.5 | CH2Cl2 | 12 | 73 |

Using the optimized conditions for complex 1b, we screened the catalytic reactions with the remaining complexes and observed that all of them promoted the formation of 1-benzyl-4-phenyl-1H-1,2,3-triazole, with isolated yields as high as 82% (entries 6–11). Additionally, the TOFs observed for the catalytic runs using complexes 1a–e were in the range of 281–490 h−1. The least active catalyst is complex 1c, leading to a yield of 47% (entry 8, TOF = 281 h−1), a fact justified by the high electron withdrawing properties of the 5-[3,5-(CF3)2C6H3] substituent, being therefore less efficient in stabilizing any possible catalytic intermediates. Moreover, given the similar catalytic results obtained for complex 1a (in the transoid form) and 1e (cisoid and transoid forms in equilibrium), the structural preference of the complexes apparently does not affect the catalytic performance.
For the sake of comparison, complex 1-H, the one containing 5-unsubstituted pyrrolyl rings (R = H), was also tested as a catalyst, only leading to the formation of the respective 1,2,3-triazole in 12% yield (entry 11). This latter fact is due to the very low stereochemical protection of the copper centers (and also to an irreversible oxidation process), whereby reaction with the substrates used in this catalyst system might promote the irreversible disproportionation of complex 1-H to form the inactive Cu(II) homoleptic species [Cu{κ2N,N′-NC4H3-2-C(H)N(2,6-iPr2C6H3)}2] and Cu(0).29
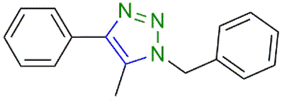
The substrate scope of the present catalyst system was also investigated by varying both the nature of the alkynes and of the azides present in the reactions, using complex 1a as the reference, since it recorded the highest yield of 1-benzyl-4-phenyl-1H-1,2,3-triazole. The catalytic results for the substrate scope of the present catalyst system are presented in Table 4 and the 1H NMR data of the products of catalysis are presented in Fig. S37–S42 in the ESI.†
|
|
|||
|---|---|---|---|
| Entry | Product | Yielda (%) | TOF (h−1) |
| Conditions: 0.5 mol% of 1a; 0.9 mmol of alkyne; 0.9 mmol of azide; reaction time: 0.5 h; temperature: 25 °C; solvent volume = 5 mL.a Yields determined by weighing the isolated reaction products.b No cycloaddition product was observed. | |||
| 1 |

|
82 | 490 |
| 2 |

|
91 | 545 |
| 3 |
|
75 | 450 |
| 4 |

|
25 | 154 |
| 5 |

|
28 | 161 |
| 6 |

|
92 | 554 |
| 7 |
|
||
| 8 |

|
||

Complex 1a was also able to catalyze the cycloaddition of phenylacetylene with different azides in respectable yields (entries 1–3 and 6, 75–92%), the latter being lower when aliphatic or polar group-substituted azides were used (entries 4 and 5, 25–28%). The catalyst system also promoted the cycloaddition of 2-ethynylaniline with (azidomethyl)benzene (entry 6), but was completely ineffective when internal alkynes, such as prop-1-yn-1-ylbenzene or but-2-yne, were used (entries 7 and 8).
The catalytic results obtained with the present system led a TOF value as high as 859 h−1 for the production of 1-benzyl-4-phenyl-1H-1,2,3-triazole, resulting from the cycloaddition of phenylacetylene with (azidomethyl)benzene, operating at concentrations of catalyst as low as 0.1 mol%. Considering the set of activities recorded for the present catalyst system, we can conclude it is less efficient than the mononuclear copper complexes bearing NHC (maximum TOF = 950 h−1)20 or diimine ligands (maximum TOF = 1920 h−1),22 slightly better than the catalysts bearing phosphine/phosphonite/phosphinite ligands (maximum TOF = 313 h−1)21 and considerably better than the in situ prepared ones (maximum TOF = 27 h−1).7
The increased activity of the complexes reported in this work indicates that the stereochemical hindrance imparted by the pyrrolyl substitution properly stabilized the respective Cu2N4C4 macrocycle, providing strong evidence that the mechanism always involves a dimeric entity and that cooperation between the two copper centers of the complexes is entirely possible. From the cyclic voltammetry studies, it is also interesting to conclude that, in general, the easier the complexes are to oxidize, i.e. the lower the oxidation potential is, the higher the catalytic activity (TOF) obtained, as depicted in Fig. S43 in the ESI.† Moreover, the most active catalysts (1a, 1d and 1e) are those displaying a reversible one-electron oxidation, pointing out to the possible formation of stabilized mixed-valence Cu(II)/Cu(I) intermediate dimers. Such species are proposed in catalytic dinuclear models for the CuAAC reactions, previously established in the literature as one of the possible pathways.24
Conclusions
Five new dinuclear Cu(I) complexes bearing 5-substituted-2-iminopyrrolyl ligands were synthesized and characterized, serving as catalyst precursors for azide–alkyne cycloaddition (CuAAC) reactions.The Cu(I) complexes 1a–e were synthesized by reacting the respective potassium salts KLa–e with [Cu(NCMe)4]BF4 in moderate yields. All complexes were isolated as pale-yellow to yellow microcrystalline solids and were structurally characterized by elemental analysis, 1H, 13C and 19F (for complex 1c) and multidimensional NMR spectroscopy, and selected cases by single crystal X-ray diffraction. The Cu(I) complexes are dinuclear entities, with two two-coordinate copper centers and two bridging 2-iminopyrrolyl ligands, forming Cu2N4C4 macrocycles. The X-ray structures of complexes 1a and 1d display dimers with the 2-iminopyrrolyl ligands adopting a transoid conformation, while those of complexes 1c and 1e show cisoid isomers. In solution, as attested by NMR spectroscopy, complexes 1a,b were identified as the transoid isomers, complex 1c as the cisoid isomer and complexes 1d,e as mixtures of interconvertible cisoid and transoid conformers.
VT-1H NMR and 1H–1H-NOESY NMR studies performed on the Cu(I) complexes indicated that complexes 1d,e exhibit a cisoid–transoid isomerization process. For all complexes but 1c, a conformational inversion of the respective Cu2N4C4 metallacycles was also observed. Complex 1c, however, owing to a very rigid conformation imparted by π-stacking of consecutive 5-[3,5-(CF3)2C6H2] substituents remained perfectly static in solution, on the NMR timescale. The electronic features and bonding of the Cu(I) complexes was further detailed by cyclic voltammetry and DFT calculations, indicating that the redox potential has a clear relationship with the structural features of the complexes, in particular with the Cu⋯Cu distance and the Cu2N4C4 macrocycle torsion angle and that all copper centers are formally neutral.
The new Cu(I) complexes 1a–e were tested as catalysts in azide–alkyne cycloaddition reactions. The CuAAC reactions catalyzed by 0.5 mol% of complex 1a gave rise to the respective 1,2,3-triazole products in yields in the range of 25–92%, with TOFs equal to 154–554 h−1. When extending the cycloaddition reaction of phenylacetylene with (azidomethyl)benzene using the remaining complexes 1a and 1c–e as catalysts, the yields in the respective addition product were as high as 82%, with TOFs as high as 859 h−1. Complex 1-H, the 5-unsubstituted analogue of complexes 1a–e (R = H), was a very poor catalyst in the CuAAC reaction, indicating that the stereochemical protection imparted by the 5-substituent is crucial in stabilizing any hypothetical catalytic intermediates. Interestingly, the most active catalyst precursors presently studied (1a, 1d and 1e) are those displaying a reversible one-electron oxidation, which are also characterized by presenting the lowest oxidation potentials.
Experimental
General procedures
All operations were performed under dry dinitrogen atmosphere using standard glovebox and Schlenk techniques unless otherwise noted.41 Dinitrogen gas for all operations (purity: <1 ppm O2 and H2O) was supplied by Air Liquide and purified by passage through 4 Å molecular sieves. Solvents were pre-dried with activated 4 Å molecular sieves and distilled by heating under dinitrogen over suitable drying agents (sodium/benzophenone for toluene and THF; CaH2 for n-hexane and dichloromethane). Solvents and solutions were transferred using a positive pressure of dinitrogen through stainless steel cannulae and mixtures were filtered in a similar way using modified cannulae that could be fitted with glass fiber filter disks. Elemental analyses were obtained from the elemental analysis service of Instituto Superior Técnico (IST) on a Fisons Instrument Mod EA-1108.The 5-substituted-2-iminopyrrolyl potassium salts KLa–c and KLe,35,37c complex 1-H29 and [Cu(NCMe)4]BF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 were prepared as described in the literature (or slightly adapted from those procedures). The syntheses and characterization data of the potassium salt KLd and its precursors are presented in the ESI.†
42 were prepared as described in the literature (or slightly adapted from those procedures). The syntheses and characterization data of the potassium salt KLd and its precursors are presented in the ESI.†
NMR spectroscopy measurements
NMR spectra were recorded on a Bruker “AVANCE III” 300 MHz spectrometer at 299.995 MHz (1H), 75.4296 MHz (13C) and 282.218 MHz (19F). The spectra were referenced internally using the residual protio-resonances (1H) and the solvent carbon (13C) resonances of the corresponding solvents to tetramethylsilane (δ = 0), and referenced externally using CFCl3 (δ = 0), for 19F. All solution samples, excluding those involving products of the catalytic reactions, were prepared inside a glovebox and transferred to J. Young NMR tubes. All chemical shifts are quoted in δ (ppm) and coupling constants (J) in Hz with multiplicities abbreviated as br (broad), s (singlet), d (doublet), t (triplet), q (quartet), h (heptet) and m (multiplet).X-ray diffraction
Crystallographic and experimental details of crystal structure determinations are listed in Tables S1 and S2 of the ESI.† The crystals were selected under an inert atmosphere, covered with polyfluoroether oil and mounted on a nylon loop. Crystallographic data were collected using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) on a Bruker AXS-KAPPA APEX II diffractometer equipped with an Oxford Cryosystem open-flow dinitrogen cryostat, at 150 K. Cell parameters were retrieved using Bruker SMART43 software and refined using Bruker SAINT44 on all observed reflections. Absorption corrections were applied using SADABS.45 Structure solution and refinement were performed using direct methods with the programs SIR201446 and SHELXL47 included in the package of programs WINGX-Version 2014.1.48 All complexes, except for complex 1c, contained highly disordered toluene solvate molecules in an intermediate electron density map and, therefore, the SQUEEZE routine, included in PLATON,49 was included, since no appropriate disorder model could be applied. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were inserted in idealized positions and allowed to refine riding on the parent carbon atom. Graphic presentations were prepared with ORTEP-3.50 Data was deposited in CCDC under the deposit numbers 2243570 for 1a, 2243571 for 1c, 2243572 for 1d and 2243573 for 1e.†
Computational details
All calculations were performed using the Gaussian 09 software package51 and the PBE0 functional. This functional uses a hybrid generalized gradient approximation (GGA), including 25% mixture of Hartree–Fock52 exchange with DFT38 exchange–correlation, given by Perdew, Burke and Ernzerhof functional (PBE).53 The geometry optimizations were accomplished without symmetry constraints using a standard 6-31G** basis set54 for all atoms except for copper, that used the SDD55 basis set with a f-polarization function56 for Cu (basis b1). Frequency calculations were performed to confirm the nature of the stationary points. The electronic energies obtained (Eb1) were converted to Gibbs energy at 298.15 K and 1 atm (Gb1) by using zero-point energy and thermal energy corrections based on structural and vibration frequency data calculated at the same level. A Natural Population Analysis (NPA)57 and the resulting charges and Wiberg indices58 were used to study the electronic structure and bonding of the optimized species.Single point energy calculations were performed on the geometries optimized at the PBE0/b1 level, using the same functional and a standard 6-311++G(d,p) basis set.59 Solvent effects (toluene) were considered in the single point calculations using the Polarizable Continuum Model (PCM) initially devised by Tomasi and co-workers60 with radii and non-electrostatic terms of the SMD solvation model, developed by Truhlar et al.61 The Gibbs energy values presented (Gb2) were corrected for dispersion by means of Grimme DFT-D3 method62 with Becke and Johnson short distance damping,63 and were derived from the electronic energy values obtained at the PBE0-D3/6-311++G(d,p)//PBE0/b1 level (Eb2) according to the following expression: Gb2 = Eb2 + Gb1 − Eb1.
Cyclic voltammetry measurements
Cyclic voltammetry experiments on ca. 3 mM solutions of complexes 1a–e in dichloromethane, using [N(n-Bu)4][BF4] (0.2 M) as a supporting electrolyte, were performed at a scanning rate of 200 mV s−1 (unless otherwise specified) with a three compartment electrochemical cell, under dinitrogen atmosphere, at room temperature, using a Pt disc working electrode and a Pt wire counter electrode, with a Ag pseudo-reference electrode connected to the main compartment by a Luggin capillary. The redox potentials were calculated using the reference potential of the ferrocene/ferrocenium couple measured under the same experimental conditions for each of the complexes.General procedure for the synthesis of complexes 1a–e
A solution of the appropriate 5-substituted-2-iminopyrrolyl potassium salt KLa–e in THF (one equivalent) was added dropwise to a solution of [Cu(NCMe)4]BF4 (one equivalent) in THF, cooled to −20 °C, giving rise to a light grey suspension. The suspension was stirred overnight at room temperature. The reaction mixture was evaporated to dryness and the residue dried under vacuum. The residue was extracted with the appropriate solvent, concentrated and stored at −20 °C, yielding pale-yellow to yellow microcrystalline solids.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N(2,6-iPr2C6H3)}]2 (1a).
The general procedure was followed, using 0.57 g (0.96 mmol) of the potassium salt KLa and 0.30 g (0.96 mmol) of [Cu(NCMe)4]BF4. The residue was washed with n-hexane, extracted with toluene, the extracts combined, concentrated and stored at −20 °C, yielding a pale-yellow microcrystalline solid suitable for X-ray diffraction. Yield: 0.19 g (39%).
N(2,6-iPr2C6H3)}]2 (1a).
The general procedure was followed, using 0.57 g (0.96 mmol) of the potassium salt KLa and 0.30 g (0.96 mmol) of [Cu(NCMe)4]BF4. The residue was washed with n-hexane, extracted with toluene, the extracts combined, concentrated and stored at −20 °C, yielding a pale-yellow microcrystalline solid suitable for X-ray diffraction. Yield: 0.19 g (39%).
Anal. calc. for C64H86Cu2N4·0.5C7H8, obtained (calculated): C 74.54 (74.75), H 8.14 (8.36), N 5.00 (5.17). 1H NMR (300 MHz, CDCl3, 298 K): δ 7.57 (s, 2H, NCH), 7.07 (s, 4H, 5-Ph-Hmeta + N-Ph-Hpara), 6.97 (d, 4H, N-Ph-Hmeta), 6.80 (d, 2H, H3, 3JHH = 3.6 Hz), 6.76 (br, 2H, 5-Ph-Hmeta), 6.08 (d, 2H, H4, 3JHH = 3.6 Hz), 3.59 (h, 2H, N-Ph-(CH(CH3)2), 3JHH = 6.9 Hz), 2.99 (h, 2H, 5-Ph-(CH(CH3)2ortho), 3JHH = 6.6 Hz), 2.91–2.71 (m, 4H, N-Ph-(CH(CH3)2) + 5-Ph-(CH(CH3)2para)), 2.35 (h, 2H, 5-Ph-(CH(CH3)2ortho), 3JHH = 6.6 Hz), 1.35–1.20 (m, 30H, N-Ph-(CH(CH3)2) + 5-Ph-(CH(CH3)2ortho) + 5-Ph-(CH(CH3)2para)), 1.12 (d, 12H, N-Ph-(CH(CH3)2), 3JHH = 6.0 Hz), 0.99 (d, 6H, 5-Ph-(CH(CH3)2ortho), 3JHH = 6.3 Hz), 0.35 (d, 6H, N-Ph-(CH(CH3)2), 3JHH = 6.6 Hz), 0.19 (d, 6H, 5-Ph-(CH(CH3)2ortho), 3JHH = 6.6 Hz). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 162.0 (NCH), 152.1 (C5), 148.6 (N-Ph-Cortho), 148.2 (5-Ph-Cortho), 148.0 (5-Ph-Cipso), 146.2 (N-Ph-Cipso), 142.2 (5-Ph-Cortho), 141.8 (N-Ph-Cortho), 133.9 (C2), 132.2 (5-Ph-Cpara), 128.2 (C3), 126.2 (N-Ph-Cpara), 124.7 (N-Ph-Cmeta), 123.5 (N-Ph-Cmeta), 121.0 (5-Ph-Cmeta), 120.7 (5-Ph-Cmeta), 116.2 (C4), 34.3 (5-Ph-CH(CH3)2para), 30.7 (5-Ph-CH(CH3)2ortho), 30.2 (5-Ph-CH(CH3)2ortho), 28.2 (N-Ph-CH(CH3)2), 27.9 (N-Ph-CH(CH3)2), 26.0 (N-Ph-CH(CH3)2), 24.9 (5-Ph-CH(CH3)2ortho), 24.5–23.9 (set of resonances corresponding to the remaining CH(CH3)2 carbons).
N(2,6-iPr2C6H3)}]2 (1b).
The general procedure was followed, using 0.29 g (0.75 mmol) of the potassium salt KLb and 0.24 g (0.75 mmol) of [Cu(NCMe)4]BF4. The residue was washed with n-hexane, extracted with toluene, the extracts combined, concentrated and stored at −20 °C, yielding a pale-yellow microcrystalline solid. Yield: 0.23 g (55%).
Anal. calc. for C50H58Cu2N4, obtained (calculated): C 71.43 (71.31), H 7.29 (6.94), N 6.56 (6.65). 1H NMR (300 MHz, CDCl3, 298 K): δ 7.58 (s, 2H, NCH), 7.16–7.05 (m, 6H, 5-Ph-Hmeta + 5-Ph-Hpara), 7.04–6.91 (m, 6H, N-Ph-Hmeta + N-Ph-Hpara), 6.85 (d, 2H, H3, 3JHH = 3.6 Hz), 6.13 (d, 2H, H4, 3JHH = 3.6 Hz), 3.39 (h, 2H, CH(CH3)2, 3JHH = 6.6 Hz), 2.91 (h, 2H, CH(CH3)2, 3JHH = 6.0 Hz), 2.35 (s, 6H, CH3), 1.62 (s, 6H, CH3), 1.22 (d, 6H, CH(CH3)2, 3JHH = 6.3 Hz), 1.19 (d, 6H, CH(CH3)2, 3JHH = 5.7 Hz), 1.06 (d, 6H, CH(CH3)2, 3JHH = 5.1 Hz), 0.32 (d, 6H, CH(CH3)2, 3JHH = 5.1 Hz). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 160.9 (NCH), 152.0 (C5), 145.8 (N-Ph-Cipso), 141.9 (N-Ph-Cortho), 141.8 (N-Ph-Cortho), 137.1 (5-Ph-Cortho), 137.0 (5-Ph-Cortho), 134.3 (C2), 127.3 (C3), 127.2 (N-Ph-Cmeta) 126.4 (5-Ph-Cmeta), 124.0 (5-Ph-Cpara), 123.6 (N-Ph-Cpara), 114.3 (C4), 28.2 (CH(CH3)2), 24.4 (CH(CH3)2), 24.1 (CH(CH3)2), 23.7 (CH(CH3)2), 22.1 (CH(CH3)2), 21.3 (CH3), 21.1 (CH3). 5-Ph-Cipso resonance absent.
N(2,6-iPr2C6H3)}]2 (1c).
The general procedure was followed, using 0.51 g (1 mmol) of the potassium salt KLc and 0.32 g (1 mmol) of [Cu(NCMe)4]BF4. The residue was extracted with n-hexane, the extracts combined, concentrated and stored at −20 °C, yielding a yellow-light green microcrystalline solid suitable for X-ray diffraction. Yield: 0.33 g (63%).
Anal. calc. for C50H46Cu2F12N4, obtained (calculated): C 56.97 (56.76), H 4.49 (4.38), N 5.23 (5.30). 1H NMR (300 MHz, CDCl3, 298 K): δ 8.03 (br, 4H, 5-Ph-Hortho), 7.76 (s, 2H, NCH), 7.45 (br, 2H, 5-Ph-Hpara), 7.15 (m, 4H, N-Ph-Hmeta), 7.03 (m, 2H, N-Ph-Hpara), 6.98 (d, 2H, H3, 3JHH = 3.6 Hz), 6.70 (d, 2H, H4, 3JHH = 3.9 Hz), 3.38 (h, 2H, CH(CH3)2, 3JHH = 6.9 Hz), 2.89 (h, 2H, CH(CH3)2, 3JHH = 6.6 Hz), 1.26 (d, 6H, CH(CH3)2, 3JHH = 7.2 Hz), 1.05 (d, 6H, CH(CH3)2, 3JHH = 7.2 Hz), 0.43 (d, 6H, CH(CH3)2, 3JHH = 6.9 Hz), 0.41 (d, 6H, CH(CH3)2, 3JHH = 6.9 Hz). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 161.69 (NCH), 148.7 (C5), 145.2 (N-Ph-Cipso), 141.4 (N-Ph-Cortho), 141.2 (N-Ph-Cortho), 138.5 (5-Ph-Cipso), 136.5 (C2), 131.3 (q, 5-Ph-Cmeta, 2JCF = 33 Hz), 128.9 (C3), 126.7 (N-Ph-Cmeta), 125.8 (br, 5-Ph-Cortho), 123.9 (N-Ph-Cpara), 123.8 (N-Ph-Cmeta), 123.2 (q, CF3, 1JCF = 271 Hz), 119.7 (h, 5-Ph-Cpara, 3JCF = 3.0 Hz), 110.1 (C4), 28.8 (CH(CH3)2), 28.4 (CH(CH3)2), 24.0 (CH(CH3)2), 23.3 (CH(CH3)2), 23.2 (CH(CH3)2), 21.6 (CH(CH3)2). 19F{1H} NMR (282 MHz, CDCl3, 298 K): δ −63.7 (CF3).
N(2,6-iPr2C6H3)}]2 (1d).
The general procedure was followed, using 0.36 g (0.84 mmol) of the potassium salt KLd and 0.27 g (0.84 mmol) of [Cu(NCMe)4]BF4. The residue was washed with n-hexane, extracted with toluene, the extracts combined, concentrated and stored at −20 °C, yielding a yellow microcrystalline solid. Yield: 0.15 g (40%).
Anal. calc. for C50H58Cu2N4O4·0.5C7H8, obtained (calculated): C 67.89 (67.48), H 6.87 (6.56), N 5.56 (5.88). At 298 K, the NMR spectra of complex 1d consists of two different conformers (transoid-1d and cisoid-1d), in a 4.0:1 ratio of transoid-1d:cisoid-1d, which varies with temperature, being reported as follows: 1H NMR (300 MHz, CDCl3, 298 K): δ 7.69 (s, 2H, NCH of cisoid-1d), 7.52 (s, 2H, NCH of transoid-1d), 7.46–6.99 (m, 6H of transoid-1d + 10H of cisoid-1d, complex set of overlapping resonances corresponding to H4 of cisoid-1d, 5-Ph-Hpara of cisoid-1d and N-Ph-Hmeta + N-Ph-Hpara of transoid-1d and cisoid-1d), 6.95 (t, 2H, 5-Ph-Hpara of transoid-1d, 3JHH = 8.4 Hz), 6.87 (d, 2H, H3 of cisoid-1d, 3JHH = 3.9 Hz), 6.84 (d, 2H, H3 of transoid-1d, 3JHH = 3.6 Hz), 6.56 (d, 4H, 5-Ph-Hmeta of cisoid-1d, 3JHH = 5.1 Hz), 6.54 (d, 2H, H4 of transoid-1d, 3JHH = 3.9 Hz), 6.32 (d, 4H, 5-Ph-Hmeta of transoid-1d, 3JHH = 8.4 Hz), 3.67 (s, 12H, OCH3 of transoid-1d), 3.49 (s, 12H, OCH3 of cisoid-1d), 3.48–2.93 (m, 4H of transoid-1d + 4H of cisoid-1d, complex set of overlapping resonances corresponding to CH(CH3)2 of transoid-1d and cisoid-1d), 1.40–0.13 (m, 24H of transoid-1d + 24H of cisoid-1d, complex set of overlapping resonances corresponding to CH(CH3)2 of transoid-1d and cisoid-1d). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 162.5 (NCH of cisoid-1d), 160.6 (NCH of transoid-1d), 158.5 (5-Ph-Cortho of transoid-1d), 157.5 (5-Ph-Cortho of transoid-1d), 147.6 (C2 of cisoid-1d), 147.1 (C2 of transoid-1d), 145.3 (C5 of transoid-1d), 144.5 (C5 of cisoid-1d), 142.2 (N-Ph-Cortho of transoid-1d), 142.0 (N-Ph-Cortho of cisoid-1d), 141.6 (overlapping of 5-Ph-Cipso of transoid-1d and cisoid-1d), 135.0 (N-Ph-Cipso of cisoid-1d), 133.61 (N-Ph-Cipso of transoid-1d), 129.18 (5-Ph-Cmeta of cisoid-1d), 129.0 (N-Ph-Cpara of transoid-1d), 128.4 (N-Ph-Cpara of cisoid-1d), 128.1 (5-Ph-Cpara of transoid-1d), 127.1 (C3 of transoid-1d), 126.1 (N-Ph-Cmeta of transoid-1d), 125.4 (5-Ph-Cpara of cisoid-1d), 124.0 (C3 or C4 of cisoid-1d), 123.6 (C3 or C4 of cisoid-1d), 116.7 (C4 of transoid-1d), 104.2 (N-Ph-Cmeta of cisoid-1d), 104.1 (5-Ph-Cmeta of transoid-1d), 55.3 (OCH3 of cisoid-1d), 55.2 (OCH3 of transoid-1d), 28.3 (CH(CH3)2 of cisoid-1d), 28.0 (CH(CH3)2 of transoid-1d), 25.22 (CH(CH3)2 of cisoid-1d), 24.94 (CH(CH3)2 of transoid-1d), 23.12 (CH(CH3)2 of cisoid-1d).
N(2,6-iPr2C6H3)}]2 (1e).
The general procedure was followed, using 0.46 g (0.87 mmol) of the potassium salt KLe and 0.27 g (0.87 mmol) of [Cu(NCMe)4]BF4. The residue was washed with n-hexane, extracted with toluene, the extracts combined, concentrated and stored at −20 °C, yielding a pale-yellow microcrystalline solid suitable for X-ray diffraction. Yield: 0.31 g (64%).
Anal. calc. for C72H70Cu2N4·C7H8, obtained (calculated): C 78.57 (78.38), H 6.83 (6.49), N 4.71 (4.63). At 298 K, the NMR spectra of complex 1e consists of two different conformers (transoid-1e and cisoid-1e), in a 2.5:1 ratio of transoid-1e:cisoid-1e, which varies with temperature, being reported as follows: 1H NMR (300 MHz, CDCl3, 298 K): δ 7.68 (s, 2H, NCH of cisoid-1e), 7.51 (s, 2H, NCH of transoid-1e), 7.49–6.80 (m, 36H of transoid-1e + 36H of cisoid-1e, overlapping set of resonances corresponding to the CPh3 + N-Ph protons of transoid-1e and cisoid-1e), 6.75 (d, 2H, H3 of transoid-1e, 3JHH = 3.3 Hz), 6.69 (d, 2H, H3 of cisoid-1e, 3JHH = 2.7 Hz), 6.18 (d, 2H, H4 of cisoid-1e, 3JHH = 2.7 Hz), 5.66 (d, 2H, H4 of transoid-1e, 3JHH = 3.3 Hz), 3.46–3.24 (m, 2H of transoid-1e + 2H of cisoid-1e, CH(CH3)2 of transoid-1e + CH(CH3)2 of cisoid-1e), 2.92 (h, 2H, CH(CH3)2 of transoid-1e, 3JHH = 6.9 Hz), 2.35 (m, 2H, CH(CH3)2 of cisoid-1e), 1.25 (d, 6H, CH(CH3)2 of cisoid-1e, 3JHH = 6.3 Hz), 1.12 (d, 6H, CH(CH3)2 of cisoid-1e, 3JHH = 6.3 Hz), 1.06 (d, 6H, CH(CH3)2 of transoid-1e, 3JHH = 6.9 Hz), 1.02 (d, 6H, CH(CH3)2 of cisoid-1e, 3JHH = 6.3 Hz), 0.91 (d, 6H, CH(CH3)2 of transoid-1e, 3JHH = 6.9 Hz), 0.74 (d, 6H, CH(CH3)2 of transoid-1e, 3JHH = 6.9 Hz), 0.65 (d, 6H, CH(CH3)2 of cisoid-1e, 3JHH = 6.3 Hz), 0.36 (d, 6H, CH(CH3)2 of transoid-1e, 3JHH = 6.9 Hz). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 163.9 (NCH of 1eB), 160.5 (NCH of transoid-1e), 160.0 (C5 of transoid-1e), 158.4 (C5 of cisoid-1e), 147.2 (CPh3 of transoid-1e), 145.9 (N-Ph-Cipso of transoid-1e), 142.0 (N-Ph-Cortho of transoid-1e), 141.9 (N-Ph-Cortho of cisoid-1e), 141.8 (N-Ph-Cortho of transoid-1e), 141.5 (N-Ph-Cortho of cisoid-1e), 135.5 (C2 of cisoid-1e), 135.1 (C2 of transoid-1e), 131.1–129.1 (set of resonances corresponding to CPh3 + N-Ph CH carbons of transoid-1e and cisoid-1e), 128.6 (C3 of transoid-1e), 128.5–125.9 (set of resonances corresponding to CPh3 + N-Ph CH carbons of transoid-1e and cisoid-1e), 125.8 (C3 of cisoid-1e), 125.5–123.1 (set of resonances corresponding to CPh3 + N-Ph CH carbons of transoid-1e and cisoid-1e), 119.6 (C4 of transoid-1e), 116.6 (C4 of cisoid-1e), 28.1 (CH(CH3)2 of transoid-1e), 28.0 (CH(CH3)2 of cisoid-1e), 27.9 (CH(CH3)2 of cisoid-1e), 27.8 (CH(CH3)2 of transoid-1e), 27.3 (CH(CH3)2 of cisoid-1e), 25.2 (CH(CH3)2 of cisoid-1e), 24.7 (CH(CH3)2 of cisoid-1e), 24.1 (CH(CH3)2 of transoid-1e), 24.0 (CH(CH3)2 of cisoid-1e), 23.4, (CH(CH3)2 of transoid-1e), 23.2 (CH(CH3)2 of transoid-1e), 22.4 (CH(CH3)2 of transoid-1e). CPh3 and N-Ph-Cipso of cisoid-1e absent.
General procedure for the alkyne–azide cycloadditions catalyzed by complexes 1a–e
In a dinitrogen-filled glove box, a glass ampoule was charged with the desired amount of copper complex, the azide (0.9 mmol) and the alkyne (0.9 mmol) and the appropriate solvent was added. The ampoule was closed with a glass stopper and stirred for the desired time at 25 °C. After the desired time, the mixture was exposed to air and all volatile materials were evaporated under reduced pressure. The resulting solids were dried under vacuum, to give the respective 1,2,3-triazoles. The yields were determined by weighing the respective solids. The 1H NMR spectra of the products of the catalytic reactions are presented in Fig. S37–S42 of the ESI,† corresponding to those reported in the literature.64Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Fundação para a Ciência e a Tecnologia (FCT) for financial support (project PTDC/QUI-QIN/31585/2017). Centro de Química Estrutural (CQE) and Institute of Molecular Sciences (IMS), and CERENA acknowledge the FCT for financial support (respectively: projects UIDB/00100/2020, UIDP/00100/2020 and LA/P/0056/2020; and UIDB/04028/2020 and UIDP/04028/2020.References
- (a) R. L. Peterson, S. Kim and K. D. Karlin, Copper Enzymes, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 149–177 Search PubMed; (b) S. E. N. Brazeau and L. H. Doerrer, Dalton Trans., 2019, 48, 4759–4768 RSC.
- X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- T. J. Zerk and P. V. Bernhardt, Coord. Chem. Rev., 2018, 375, 173–190 CrossRef CAS.
- C. Boyer, N. Alan, C. Kenward, J. Diep, N. Thuy-Khanh, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- For example: (a) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed; (b) J. M. Hoover and S. S. Stahl, Nat. Protoc., 2012, 7, 1161–1166 CrossRef CAS PubMed.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- B. Dervaux and F. E. D. Prez, Chem. Sci., 2012, 3, 959–966 RSC.
- R. M. Cherian, N. A. Harry, S. Saranya, K. R. Rohit and G. Anilkumar, Asian J. Org. Chem., 2019, 8, 197–233 CrossRef CAS.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystro, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed.
- For example: (a) W. H. Binder and R. Zirbs, “Click” Chemistry in Macromolecular Synthesis, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Hoboken, New Jersey, 2014, vol. 3, pp. 186–230 Search PubMed; (b) D. Döhler, P. MichaelOrcid and W. H. Binder, Acc. Chem. Res., 2017, 50, 2610–2620 CrossRef PubMed; (c) S. Neumann, M. Biewend, S. Rana and W. H. Binder, Macromol. Rapid. Commun., 2020, 41, 1900359 CrossRef CAS PubMed.
- The Nobel Prize in Chemistry 2022, NobelPrize.org, Nobel Prize Outreach AB, 2023, https://www.nobelprize.org/prizes/chemistry/2022/summary/, last accessed 22/05/2023.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 14397–14402 CrossRef CAS PubMed.
- (a) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (b) A. N. Semakin, D. P. Agababyan, S. Kim, S. Lee, A. Y. Sukhorukov, K. G. Fedina, J. Oh and S. L. Ioffe, Tetrahedron Lett., 2015, 56, 6335–6339 CrossRef CAS; (c) D. Wang, M. Zhao, X. Liu, Y. Chen, N. Li and B. Chen, Org. Biomol. Chem., 2012, 10, 229–231 RSC; (d) D. Wang, N. Li, M. Zhao, W. Shi, C. Ma and B. Chen, Green Chem., 2010, 12, 2120–2123 RSC.
- S. Díez-González, Catal. Sci. Technol., 2011, 1, 166–178 RSC.
- (a) S. Díez-González, E. D. Stevens and S. P. Nolan, Chem. Commun., 2008, 4747–4749 RSC; (b) S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawinc and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC; (c) J.-M. Collinson, J. D. E. T. Wilton-Ely and S. Díez-González, Chem. Commun., 2013, 49, 11358–11360 RSC; (d) S. Lal, H. S. Rzepa and S. Díez-González, ACS Catal., 2014, 4, 2274–2287 CrossRef CAS; (e) F. Sebest, J. J. Dunsford, M. Adams, J. Pivot, P. D. Newman and S. Díez-González, ChemCatChem, 2018, 10, 2041–2045 CrossRef CAS PubMed.
- (a) S. Lal, J. McNally, A. J. P. White and S. Díez-González, Organometallics, 2011, 30, 6225–6232 CrossRef CAS; (b) S. Lal and S. Díez-González, J. Org. Chem., 2011, 76, 2367–2373 CrossRef CAS PubMed; (c) J. M. Pérez, P. Crosbie, S. Lal and S. Díez-González, ChemCatChem, 2016, 8, 2222–2226 CrossRef PubMed; (d) A. Beltrán, I. Gata, C. Maya, J. Avó, J. C. Lima, C. A. T. Laia, R. Peloso, M. Outis and M. C. Nicasio, Inorg. Chem., 2020, 59, 10894–10906 CrossRef PubMed.
- (a) C. G. Bates, P. Saejueng, J. M. Murphy and D. Venkataraman, Org. Lett., 2002, 4, 4727–4729 CrossRef CAS PubMed; (b) L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Avilés and P. T. Gomes, Dalton Trans., 2012, 41, 5144–5154 RSC; (c) L. Li, P. S. Lopes, C. A. Figueira, C. S. B. Gomes, M. T. Duarte, V. Rosa, C. Fliedel, T. Avilés and P. T. Gomes, Eur. J. Inorg. Chem., 2013, 1404–1417 CrossRef CAS; (d) J. M. Barta and S. Díez-González, Molecules, 2013, 18, 8919–8928 CrossRef PubMed; (e) M. S. Viana, C. S. B. Gomes and V. Rosa, Catalysts, 2023, 13, 386–404 CrossRef CAS.
- (a) M. S. Ziegler, D. S. Levine, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2016, 138, 6484–6491 CrossRef CAS PubMed; (b) M. S. Ziegler, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 5378–5386 CrossRef CAS PubMed; (c) M. S. Ziegler, N. A. Torquato, D. S. Levine, A. Nicolay, H. Celik and T. D. Tilley, Organometallics, 2018, 37, 2807–2823 CrossRef CAS; (d) A. Nicolay, M. S. Ziegler, D. W. Small, R. Grunbauer, M. Scheer and T. D. Tilley, Chem. Sci., 2020, 11, 1607–1616 RSC; (e) A. N. Desnoyer, A. Nicolay, M. S. Ziegler, N. A. Torquato and T. D. Tilley, Angew. Chem., Int. Ed., 2020, 59, 12769–12773 CrossRef CAS PubMed; (f) A. N. Desnoyer, A. Nicolay, M. S. Ziegler, K. V. Lakshmi, T. R. Cundari and T. D. Tilley, J. Am. Chem. Soc., 2021, 143, 7135–7143 CrossRef CAS PubMed; (g) M. Piesch, A. Nicolay, M. Haimerl, M. Seidl, G. Balázs, T. D. Tilley and M. Scheer, Chem. – Eur. J., 2022, 28, e202201144 CrossRef CAS PubMed; (h) P. Ríos, M. S. See, R. C. Handford, S. J. Teat and T. D. Tilley, Chem. Sci., 2022, 13, 6619–6625 RSC.
- (a) M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS; (b) B. F. Straub, Chem. Commun., 2007, 3868–3870 RSC; (c) B. R. Buckley, S. E. Dann and H. Heaney, Chem. – Eur. J., 2010, 16, 6278–6284 CrossRef CAS PubMed; (d) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed; (e) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 CrossRef PubMed; (f) M. González-Lainez, M. Gallegos, J. Munarriz, R. Azpiroz, V. Passarelli, M. Victoria Jiménez and J. J. Pérez-Torrente, Organometallics, 2022, 41, 2154–2169 CrossRef PubMed.
- (a) S. K. Adas, J. A. Ocana and S. D. Bunge, Aust. J. Chem., 2014, 67, 1021–1029 CrossRef CAS; (b) T. J. J. Whitehorne, J. P. Coyle, A. Mahmood, W. H. Monillas, G. P. A. Yap and S. T. Barry, Eur. J. Inorg. Chem., 2011, 3240–3247 CrossRef CAS; (c) H. S. Lee and M. Niemeyer, Inorg. Chim. Acta, 2011, 374, 163–170 CrossRef CAS; (d) A. M. Willcocks, T. P. Robinson, C. Roche, T. Pugh, S. P. Richards, A. J. Kingsley, J. P. Lowe and A. L. Johnson, Inorg. Chem., 2012, 51, 246–257 CrossRef CAS PubMed; (e) J. P. Coyle, W. H. Monillas, G. P. A. Yap and S. T. Barry, Inorg. Chem., 2008, 47, 683–689 CrossRef CAS PubMed; (f) L. R. Falvello, E. P. Urriolabeitia, U. Mukhopadhyay and D. Ray, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 170–172 CrossRef; (g) Z.-M. Cai, G. Yu, Q.-Y. Lv, W.-Q. Jiang and S.-Z. Zhan, Z. Anorg. Allg. Chem., 2012, 638, 1519–1522 CrossRef CAS; (h) A. L. Johnson, A. M. Willcocks and S. P. Richards, Inorg. Chem., 2009, 48, 8613–8622 CrossRef CAS PubMed; (i) M. Fan, Q. Yang, H. Tong, S. Yuan, B. Jia, D. Guo, M. Zhou and D. Liu, RSC Adv., 2012, 2, 6599–6605 RSC; (j) A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler, Jr., X. Yang, G. A. Baker and J. R. Walensky, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS PubMed.
- (a) C. H. Wei, Inorg. Chem., 1972, 11, 2315–2321 CrossRef CAS; (b) C. M. Wansapura, C. Juyoung, J. L. Simpson, D. Szymanski, G. R. Eaton, S. S. Eaton and S. Fox, J. Coord. Chem., 2003, 56, 975–993 CrossRef CAS; (c) V. V. Grushin and W. J. Marshall, Adv. Synth. Catal., 2004, 346, 1457–1460 CrossRef CAS; (d) J. A. Castro, J. Romero, J. A. Garcia-Vazquez, A. Castiñeiras, M. L. Duran and A. Sousa, Z. Anorg. Allg. Chem., 1992, 615, 155–160 CrossRef CAS; (e) P. J. Figiel, A. Sibaouih, J. U. Ahmad, M. Nieger, M. T. Risnen, M. Leskel and T. Repo, Adv. Synth. Catal., 2009, 351, 2625–2632 CrossRef CAS.
- (a) P. Daneshmand, S. Fortun and F. Schaper, Organometallics, 2017, 36, 3860–3877 CrossRef CAS; (b) P. Daneshmand, J. L. Jiménez-Santiago, M. Aragon-Alberti and F. Schaper, Organometallics, 2018, 37, 1751–1759 CrossRef CAS.
- B. Vidjayacoumar, D. J. H. Emslie, J. M. Blackwell, S. B. Clendenning and J. F. Britten, Chem. Mater., 2010, 22, 4854–4866 CrossRef CAS.
- C. S. B. Gomes, M. Teresa Duarte and P. T. Gomes, J. Organomet. Chem., 2014, 760, 167–176 CrossRef CAS.
- O. Back, J. Leppin, C. Förster and K. Heinze, Inorg. Chem., 2016, 55, 9653–9662 CrossRef CAS PubMed.
- A. I. Rodrigues, C. A. Figueira, C. S. B. Gomes, D. Suresh, B. Ferreira, R. E. Di Paolo, D. de Sá Pereira, F. B. Dias, M. J. Calhorda, J. Morgado, A. L. Maçanita and P. T. Gomes, Dalton Trans., 2019, 48, 13337–13352 RSC.
- T. F. C. Cruz, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2022, 61, 1195–1206 CrossRef CAS PubMed.
- T. F. C. Cruz, C. A. Figueira, J. C. Waerenborgh, L. C. J. Pereira, Y. Li, R. Lescouëzec and P. T. Gomes, Polyhedron, 2018, 152, 179–187 CrossRef CAS.
- T. F. C. Cruz, L. C. J. Pereira, J. C. Waerenborgh, L. F. Veiros and P. T. Gomes, Catal. Sci. Technol., 2019, 9, 3347–3360 RSC.
- (a) P. S. Ferreira, A. C. Cerdeira, T. F. C. Cruz, N. A. G. Bandeira, D. Hunger, A. Allgaier, J. van Slageren, M. Almeida, L. C. J. Pereira and P. T. Gomes, Inorg. Chem. Front., 2022, 9, 4302–4319 RSC; (b) P. S. Ferreira, J. F. Malta, N. A. G. Bandeira, A. Allgaier, J. van Slageren, J. A. Paixão, M. Almeida, L. C. J. Pereira and P. T. Gomes, Chem. Commun., 2022, 58, 9682–9685 RSC.
- (a) T. F. C. Cruz, P. S. Lopes, L. C. J. Pereira, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2018, 57, 8146–8159 CrossRef CAS PubMed; (b) T. F. C. Cruz, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2018, 57, 14671–14685 CrossRef CAS PubMed.
- (a) C. A. Figueira, P. S. Lopes, C. S. B. Gomes, J. C. S. Gomes, F. Lemos and P. T. Gomes, Dalton Trans., 2018, 47, 15857–15872 RSC; (b) C. A. Figueira, P. S. Lopes, C. S. B. Gomes, J. C. S. Gomes, L. F. Veiros, F. Lemos and P. T. Gomes, Organometallics, 2019, 383, 614–625 CrossRef; (c) T. F. C. Cruz, C. A. Figueira, L. F. Veiros and P. T. Gomes, Organometallics, 2021, 40, 2594–2609 CrossRef CAS; (d) T. F. C. Cruz, P. S. Lopes and P. T. Gomes, Polyhedron, 2021, 207, 115357 CrossRef CAS.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- S. Kulpe and S. Dähne, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3616–3623 CrossRef.
- Dynamic Nuclear Magnetic Resonance Spectroscopy, ed. G. Binsch, L. M. Jackman and F. A. Cotton, Academic Press, New York, 1975 Search PubMed.
- For example: (a) P. T. Gomes, M. L. H. Green and A. M. Martins, J. Organomet. Chem., 1998, 551, 133–138 CrossRef CAS; (b) M. M. Marques, S. Fernandes, S. G. Correia, J. R. Ascenso, S. Caroço, P. T. Gomes, J. F. Mano, S. G. Pereira, T. Nunes, A. R. Dias, M. D. Rausch and J. C. W. Chien, Macromol. Chem. Phys., 2000, 201, 2464–2468 CrossRef CAS; (c) R. Garçon, C. Clerk, J.-P. Gesson, J. Bordado, T. Nunes, S. Caroço, P. T. Gomes, M. E. Minas da Piedade and A. P. Rauter, Carbohydr. Polym., 2001, 45, 123–127 CrossRef; (d) V. Rosa, P. J. Gonzalez, T. Avilés, P. T. Gomes, R. Welter, A. C. Rizzi, M. C. G. Passeggi and C. D. Brondino, Eur. J. Inorg. Chem., 2006, 4761–4769 CrossRef CAS; (e) C. S. B. Gomes, P. T. Gomes and M. T. Duarte, J. Organomet. Chem., 2014, 760, 101–107 CrossRef CAS; (f) D. Suresh, B. Ferreira, P. S. Lopes, P. Krishnamoorthy, C. S. B. Gomes, A. Charas, D. Vila-Viçosa, J. Morgado, M. J. Calhorda, A. L. Maçanita and P. T. Gomes, Dalton Trans., 2016, 45, 15603–15620 RSC.
- G. J. Kubas, B. Monzyk and A. L. Crumblis, Inorg. Synth., 1990, 28, 68–70 CAS.
- SMART Software for the CCD Detector System Version 5.625, Bruker AXS Inc., Madison, WI, USA, 2001 Search PubMed.
- SAINT Software for the CCD Detector System, Version 7.03, Bruker AXS Inc., Madison, WI, USA, 2004 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction, University of Göttingen, Göttingen, 1996 Search PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (b) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- (a) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS; (b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- (a) A. L. Spek, PLATON – A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1998 Search PubMed; (b) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- (a) M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustration, Report ORNL-6895, Oak Ridge National Laboratory, 1996; (b) L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, John Wiley & Sons, New York, 1986 Search PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS; (c) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- (a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (c) P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209–214 CrossRef CAS; (d) M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163–168 CrossRef CAS; (e) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- (a) U. Haeusermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger and R. M. Pitzer, Mol. Phys., 1993, 78, 1211–1224 CrossRef; (b) W. Kuechle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef CAS; (c) T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- (a) J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41–62 CrossRef; (b) J. E. Carpenter, PhD thesis, University of Wisconsin, Madison, WI, 1987 Search PubMed; (c) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS; (d) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS; (e) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 1736–1740 CrossRef; (f) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (g) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (h) F. Weinhold and J. E. Carpenter, The Structure of Small Molecules and Ions, Plenum Press, 1988, p. 227 Search PubMed.
- (a) K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS; (b) Wiberg indices are electronic parameters related with the electron density in between two atoms, which scale as bond strength indicators. They can be obtained from a Natural Population Analysis.
- (a) A. D. McClean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (c) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS; (d) P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS; (e) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef; (f) R. C. Binning Jr. and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206–1216 CrossRef; (g) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511–516 CrossRef CAS; (h) L. A. Curtiss, M. P. McGrath, J.-P. Blaudeau, N. E. Davis, R. C. Binning Jr. and L. Radom, J. Chem. Phys., 1995, 103, 6104–6113 CrossRef CAS; (i) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS; (j) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- (a) M. T. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef; (b) M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS; (c) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS; (d) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154101 CrossRef PubMed; (b) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 24101 CrossRef PubMed; (c) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed.
- (a) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed; (b) L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 2139–2141 RSC; (c) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2006, 12, 7558–7564 CrossRef PubMed; (d) B. Sreedhar, P. S. Reddy and V. R. Krishna, Tetrahedron Lett., 2007, 48, 5831–5843 CrossRef CAS; (e) L. Li, C. S. B. Gomes, P. T. Gomes, M. T. Duarte and Z. Fan, Dalton Trans., 2011, 40, 3365–3380 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2243570 (1a), 2243571 (1c), 2243572 (1d) and 2243573 (1e). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00617d |
| This journal is © The Royal Society of Chemistry 2023 |