
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantiopure cycloplatinated pentahelicenic N-heterocyclic carbenic complexes that display long-lived circularly polarized phosphorescence†
Debsouri
Kundu
a,
Natalia
del Rio
a,
Marie
Cordier
a,
Nicolas
Vanthuyne
 b,
Emma V.
Puttock
c,
Stefan C. J.
Meskers
d,
J. A. Gareth
Williams
*c,
Monika
Srebro-Hooper
*e and
Jeanne
Crassous
*a
b,
Emma V.
Puttock
c,
Stefan C. J.
Meskers
d,
J. A. Gareth
Williams
*c,
Monika
Srebro-Hooper
*e and
Jeanne
Crassous
*a
aUniversité de Rennes, CNRS, ISCR – UMR 6226, 35000 Rennes, France. E-mail: jeanne.crassous@univ-rennes1.fr
bAix Marseille University, CNRS Centrale Marseille, iSm2, 13284 Marseille, France
cDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
dMolecular Materials and Nanosystems and Institute for Complex Molecular Systems, Technische Universiteit Eindhoven, NL 5600, The Netherlands
eFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: srebro@chemia.uj.edu.pl
First published on 18th April 2023
Abstract
The preparation of the first enantiopure cycloplatinated complexes bearing a bidentate, helicenic N-heterocyclic carbene and a diketonate ancillary ligand is presented, along with their structural and spectroscopic characterization based on both experimental and computational studies. The systems exhibit long-lived circularly polarized phosphorescence in solution and in doped films at room temperature, and also in a frozen glass at 77 K, with dissymmetry factor glum values ≥10−3 in the former and around 10−2 in the latter.
Introduction
N-heterocyclic carbenes (NHCs) have become classical ligands for organometallic and coordination chemistry1 thanks to their strong σ-donor abilities that result in the formation of stable metal–carbon bonds. The introduction of chirality in NHC-based complexes has opened up a variety of applications for such molecules, for example in enantioselective catalysis2 and chiral materials science.3 Meanwhile, there has been intensive research into phosphorescent complexes of heavy metals,4,5 such as iridium(III) and platinum(II), including systems with NHC ligands, due to their importance as triplet-harvesting phosphors in organic light-emitting diodes (OLEDs).6,7The design of chiral emitters displaying intense circularly polarized luminescence (CPL) has attracted significant interest, thanks to the potential of circularly polarized (CP) light in a diverse range of applications from OLEDs and optical information processing to bio-imaging and chiral sensing.8 For example, CP-OLEDs offer an interesting approach to improve the performance of high-resolution displays by eliminating the need for anti-glare polarized filters, which can account for the loss of up to 50% of the emitted light.9 Although many types of chiral NHC-complexes with diverse stereogenic elements have been developed to date, especially for enantioselective catalysis, there are very few examples of enantiopure chiroptical materials based on NHC-Pt complexes, and they are mainly limited to those bearing monodentate NHCs.10
Our group has developed chiral versions of several classes of organometallic compounds,11 including complexes incorporating helical NHCs,12 with intriguing emission properties such as long-lived CP phosphorescence. Inspired by the work of Strassner and co-workers on phosphorescent Pt(II) complexes with (:C^C)-cyclometalating NHC ligands,7b,13 we tackled the synthesis of their helical analogues. As a result, we describe here the preparation and characterization of the first-of-their-kind enantiopure helicenic NHC-based Pt(II) complexes (see Scheme 1) displaying long-lived CP phosphorescence in solution (both at room and low temperatures) and in thin films (at room temperature). Enantiopure complexes of the form Pt(O^O)(:C^C) have been synthesized, where (:C^C) is an NHC ligand incorporating a [5]helicene unit, and (O^O) is a β-diketonate ligand, i.e. either acetylacetonate, acac (Pt1) or dibenzoylmethane anion, dbm (Pt2). The photophysical (absorption and emission) and chiroptical properties (optical rotation – OR, electronic circular dichroism – ECD, and CPL) of Pt1 and Pt2 have been studied both experimentally and computationally.
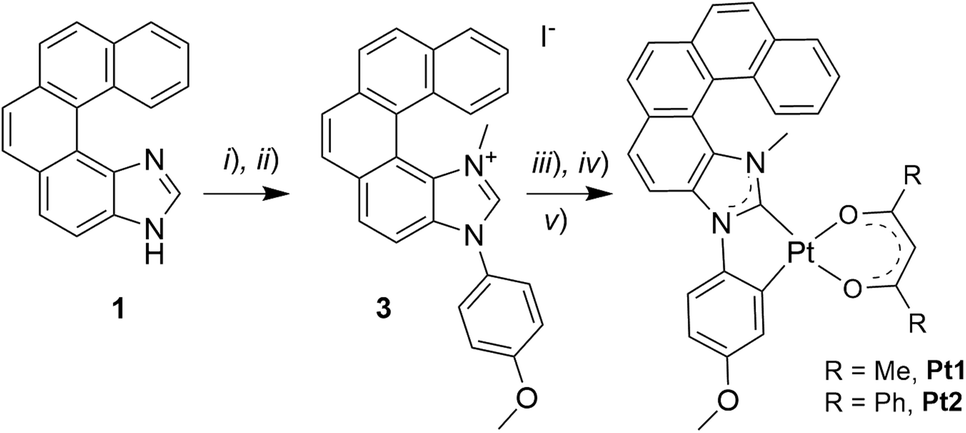 | ||
| Scheme 1 Synthesis of helicene-NHC-Pt(II) complexes Pt1 and Pt2: (i) 4-iodoanisole, CuI, L-proline, DMSO, 110 °C, 63 h; (ii) CH3I, CH3CN, reflux, 16 h (43%, over the 2 steps); (iii) Ag2O, 1,4-dioxane, rt, 16 h; (iv) cis-Pt(DMSO)2Cl2, toluene, reflux, 16 h; (v) 2,4-pentanedione (acacH) or dibenzoylmethane (dbmH), Na2CO3, toluene, reflux, overnight (35% for Pt1 and 58% for Pt2, over the 3 steps). | ||
Results and discussion
Synthesis and structural studies
Following the strategy previously developed in our group to prepare helical NHC ligands,12 the starting imidazole-fused pentahelicene 1 was subjected to N-arylation with 4-iodoanisole using CuI/L-proline as the catalytic system (Ullman-type coupling), as presented in Scheme 1,14 to yield 3N-(2-anisyl)-[5]helicene-imidazole (2). Methylation with MeI in acetonitrile led to the imidazolium iodide salt 3 with an overall yield of 43% over the two steps. Reaction of the salt with silver(I) oxide, followed by transmetalation with cis-Pt(DMSO)2Cl2,10d gave the cycloplatinated intermediate, which was treated without further purification with acacH or dbmH and Na2CO3 in refluxing toluene, to obtain (rac)-Pt1 or (rac)-Pt2, respectively, in moderate yields (35% for Pt1 and 58% for Pt2 for the three steps).15 The identity of the final complexes was confirmed by 1H and 13C NMR spectroscopy and mass spectrometry (see ESI†). For instance, the imidazolium proton in compound 3 resonating at 10.70 ppm is not present in the 1H NMR spectra of Pt1 and Pt2, while a signal corresponding to the CH proton of the β-diketonate appears at respectively 5.53 and 6.80 ppm in the products.In addition, suitable crystals of racemic Pt2 for X-ray diffraction analysis were obtained by slow evaporation of a dichloromethane solution at room temperature (Fig. 1 and ESI†). The resulting (rac)-Pt2·CH2Cl2 crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) triclinic space group. The d8 Pt(II) ion adopts a slightly distorted square-planar geometry. The metric data of the Pt(II) coordination sphere (metal–ligand bond lengths and valence angles) are in the range typical for those reported for other (:C^C)Pt(acac) complexes,7b,13,15 with the NHC and (O^O) ligands situated within the same plane and the coordinated anisole phenyl and NHC rings being almost coplanar (twist angle of 1.58°). The Ccarbene–Pt bond distance of 1.940(3) Å is similar to that in other NHC-platinum compounds previously described.7b,13,15 The complex assembles into heterochiral dimers organized in a head-to-tail fashion, with π–π interactions between coordinated NHC and anisole phenyl rings of opposite helices (centroid–centroid distance of 3.625 Å); see Fig. 1b and c. The intramolecular Pt–Pt distances of 5.772(2) Å are far longer than in other NHC-Pt(II) derivatives reported in the literature (d(Pt–Pt) = 3.276–3.495 Å),7b,13,15 evidently showing that Pt–Pt interactions are inhibited by the steric hindrance imposed by the helicene unit. Regarding the helicene moiety, it exhibits a helicity angle (dihedral angle between terminal rings) of 49.3°, which is typical for [5]helicenes and consistent with those measured for similar helicene-NHC ligands.12 These observations indicate extended π-conjugation across the whole (anisole-NHC-helicene-Pt-β-diketonate) molecular system and efficient electronic interaction between the helicene-NHC ligand and the metal, as also supported by the calculated isosurfaces of the frontier molecular orbitals (vide infra, Fig. 2e). Note also that, according to the density functional theory (DFT) computations (see ESI† for a description of computational details used in this study and a full set of calculated results), Pt2 can be assumed to exist in solution as an equal mixture of practically isoenergetic rotamers of the phenyl groups in the dbm moiety (see Fig. S2.1 in ESI†), near-coplanarity of which ensures effective π-electron conjugation within this ligand.13b
triclinic space group. The d8 Pt(II) ion adopts a slightly distorted square-planar geometry. The metric data of the Pt(II) coordination sphere (metal–ligand bond lengths and valence angles) are in the range typical for those reported for other (:C^C)Pt(acac) complexes,7b,13,15 with the NHC and (O^O) ligands situated within the same plane and the coordinated anisole phenyl and NHC rings being almost coplanar (twist angle of 1.58°). The Ccarbene–Pt bond distance of 1.940(3) Å is similar to that in other NHC-platinum compounds previously described.7b,13,15 The complex assembles into heterochiral dimers organized in a head-to-tail fashion, with π–π interactions between coordinated NHC and anisole phenyl rings of opposite helices (centroid–centroid distance of 3.625 Å); see Fig. 1b and c. The intramolecular Pt–Pt distances of 5.772(2) Å are far longer than in other NHC-Pt(II) derivatives reported in the literature (d(Pt–Pt) = 3.276–3.495 Å),7b,13,15 evidently showing that Pt–Pt interactions are inhibited by the steric hindrance imposed by the helicene unit. Regarding the helicene moiety, it exhibits a helicity angle (dihedral angle between terminal rings) of 49.3°, which is typical for [5]helicenes and consistent with those measured for similar helicene-NHC ligands.12 These observations indicate extended π-conjugation across the whole (anisole-NHC-helicene-Pt-β-diketonate) molecular system and efficient electronic interaction between the helicene-NHC ligand and the metal, as also supported by the calculated isosurfaces of the frontier molecular orbitals (vide infra, Fig. 2e). Note also that, according to the density functional theory (DFT) computations (see ESI† for a description of computational details used in this study and a full set of calculated results), Pt2 can be assumed to exist in solution as an equal mixture of practically isoenergetic rotamers of the phenyl groups in the dbm moiety (see Fig. S2.1 in ESI†), near-coplanarity of which ensures effective π-electron conjugation within this ligand.13b
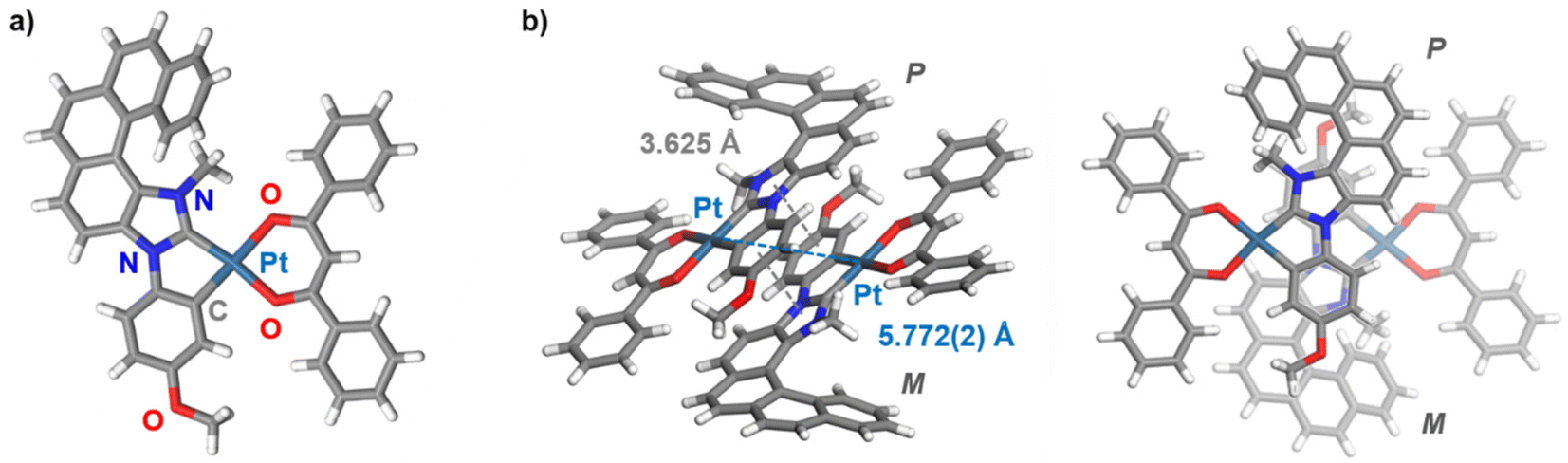 | ||
| Fig. 1 (a) X-ray crystallographic molecular structure of Pt2 in (rac)-Pt2·CH2Cl2 along with (b) its supramolecular (head-to-tail) organization into a heterochiral dimer (two views), with π–π interactions and Pt⋯Pt distances of 3.625 Å and 5.772(2) Å, respectively. | ||
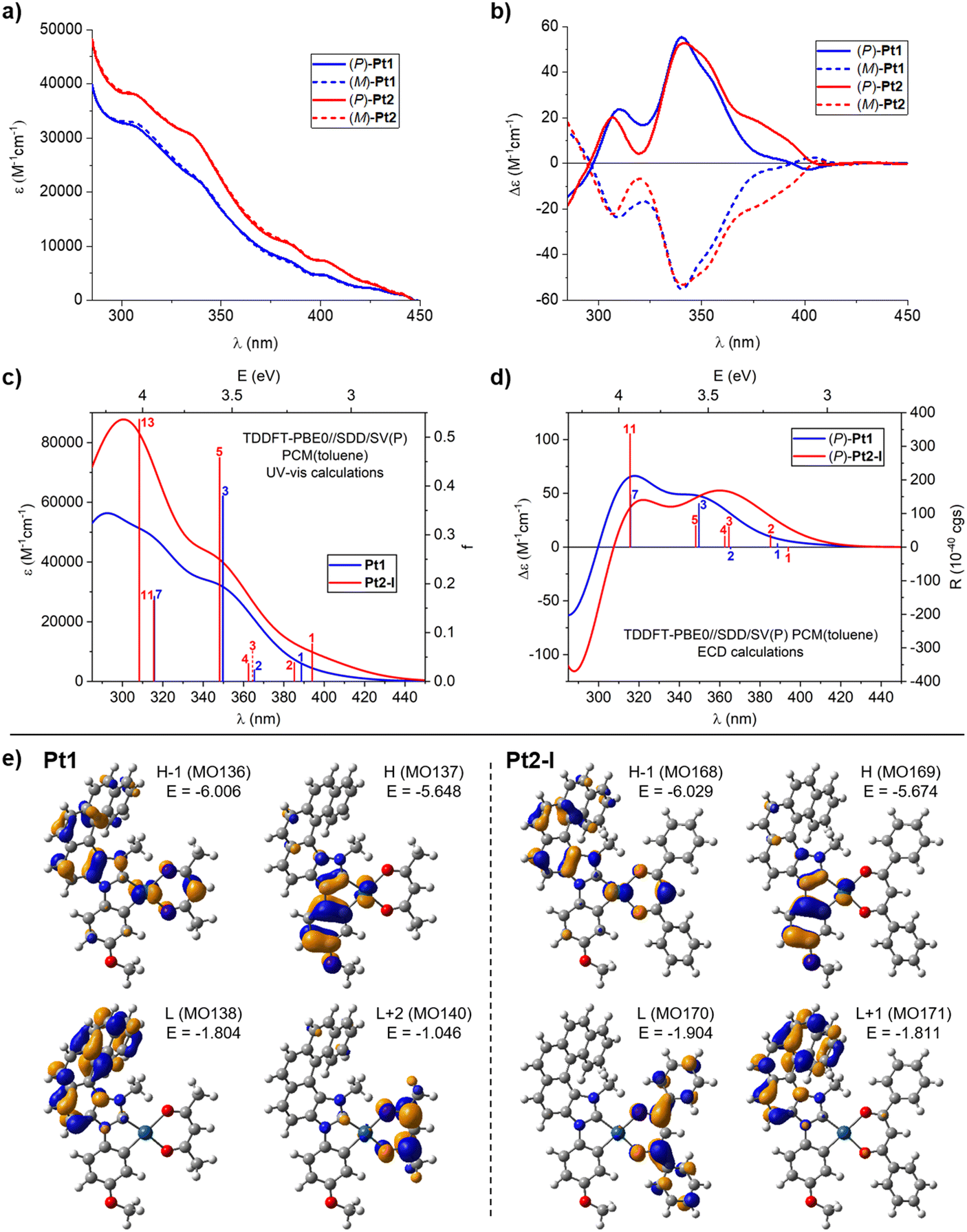 | ||
| Fig. 2 Experimental (a) UV-visible absorption and (b) ECD spectra of (P)-(+) and (M)-(−) enantiomers of Pt1 and Pt2 measured in toluene at room temperature (C = 1 × 10−5 M) along with the corresponding TDDFT-simulated spectra (panels (c) and (d), respectively; results for the lowest-energy rotamer (I) of Pt2 shown). (e) Isosurfaces (±0.04 au) of MOs involved in selected electronic transitions of Pt1 and Pt2. See ESI† for a full set of computed data including the electronic assignment of the spectra. | ||
Photophysical and chiroptical properties
Both of the racemic complexes, Pt1 and Pt2, were then resolved into their constituent enantiomers using HPLC over a chiral stationary phase, yielding (P)-(+) and (M)-(−) enantiomers with enantiomeric excess (ee) values between 98 and 99.5% (see ESI†), and their absorption and emission properties were examined.The UV-vis absorption spectra of Pt1 and Pt2 were recorded in toluene at room temperature (10−5 M), and they are presented in Fig. 2a; note that all ε and Δε values in this work are given in units of M−1 cm−1. Both complexes exhibit overall very similar spectral envelopes with intense bands at wavelengths shorter than 330 nm, attributable to π–π* transitions within (mainly) the extended π-helical unit (vide infra),11,16 and additional bands of lower intensity at longer wavelengths (ε = 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at λ > 380 nm). It can be seen that the Pt2 complex exhibits almost uniformly higher UV-vis intensity than Pt1.
000 at λ > 380 nm). It can be seen that the Pt2 complex exhibits almost uniformly higher UV-vis intensity than Pt1.
The ECD spectra of both complexes, Pt1 and Pt2, were also recorded in toluene at room temperature. As depicted in Fig. 2b, the (P) and (M) enantiomers of each system display the expected mirror-image ECD envelopes. For example, (P)-Pt1 demonstrates two strong positive bands at 309 (+23.9) and 341 (+55.1), accompanied by a positive shoulder at 356 nm (+35.6), and a weak negative band at 401 nm (−2.6). Similarly, (P)-Pt2 shows two strong positive bands at 307 (+20) and 341 (+52.8) with a positive shoulder at 353 nm (+44.4), additional positive bands at 376 (+19) and 390 nm (+11.1), and a very weak negative band at 408 nm (−0.7). As can be seen, both systems demonstrate very similar ECD both in shape and energetic position and magnitude of particular bands, except for the region between about 375 and 400 nm where Pt2 shows significantly increased intensity compared to Pt1. Finally, specific optical rotations (in deg cm3 g−1 dm−1) were measured in toluene for both complexes with values [α]23D = ±950 for Pt1 and ±749 for Pt2 (C = 1.0 mg mL−1).
Considering that specific solvent effects and vibronic contributions have not been taken into account in the calculations, the simulated UV-vis and ECD spectra (computed using time-dependent DFT (TDDFT) with PBE0//SDD/SV(P) and the continuum solvent model for toluene)17 agree satisfactorily with the experimental data (Fig. 2; see also the Computational details section and Fig. S2.2 in ESI†). In particular, energetic positions and signs of the bands are correctly reproduced by the theory along with an increased UV-vis intensity within the examined spectral range and enhanced ECD intensity of the low-energy tail of the spectra observed for Pt2vs.Pt1. The calculations also reproduce correctly the decrease in specific optical rotation values that accompanies the change from acac in Pt1 to dbm in Pt2 (with the better numerical agreement between experimental and theoretical results obtained for the BHLYP functional in line with the previously observed trends,18 see Table S2.1 in ESI†).
Analysis of molecular orbital (MO) pair contributions to computed excitations shows that for both complexes, the main UV-vis and (positive) ECD intensity observed experimentally between about 300 and 375 nm originates from excitations predominantly corresponding to π–π* transitions within NHC-helicene ligand mixed with anisole → NHC-helicene intraligand charge transfer (ILCT), β-diketonato → NHC-helicene ligand–ligand CT (LLCT) and platinum(II) → NHC-helicene metal–ligand CT (MLCT), vide infra. See, for example, excitations no. 3 and 7 for Pt1 and no. 5 and 11 for Pt2, calculated at respectively 349 and 316 nm (Fig. 2c and d, and ESI†), with the lower-energy excitation involving HOMO, HOMO−1, LUMO, LUMO+1 for Pt1 and HOMO, HOMO−1, LUMO+1, LUMO+2 for Pt2 (see Fig. 2e and ESI†). Note that the aforementioned Pt2 rotameric structures considered in the computations demonstrate practically identical UV-vis spectra and very similar ECD envelopes that differ mostly only in the intensity of the higher-energy bands but without a change in the dominant electronic character of the underlying excitations (see ESI†). The lowest-energy excitations calculated for Pt1 (no. 1 (389 nm) and 2 (365 nm)) also involve the aforementioned frontier MOs but they are mainly assigned to anisole-Pt → NHC-helicene ILCT and MLCT, and demonstrate rather modest oscillator (f) and rotatory strength (R) values. On the contrary, at wavelength longer than 350 nm, four excitations were computed for Pt2, all of enhanced f and/or R as compared to Pt1 in line with increased UV-vis and ECD intensity observed in this spectral region for the dbm-based Pt complex. These excitations (no. 1 (394 nm), 2 (385 nm), 3 (365 nm), 4 (362 nm)) also involve the frontier MOs and collectively show strong anisole-Pt → NHC-helicene ILCT and MLCT character but with admixture of anisole-NHC-helicene-Pt → dbm LLCT and MLCT due to the presence of the extended π-electron system within the phenyl-substituted β-diketonate ligand resulting in the dbm-centred LUMO13b (see Fig. 2 and ESI†). In particular, the lowest-energy excitation no. 1 for Pt2 corresponds to almost pure HOMO → LUMO anisole-NHC-Pt → dbm LLCT and MLCT and demonstrates negative rotatory strength. Finally, it should be highlighted that the aforementioned additional π-chromophoric dbm unit in Pt2 clearly contributes to an increased UV-vis intensity observed for this complex compared to Pt1 even at shorter wavelengths (around 300 nm); see for example excitation no. 13 computed at 308 nm (Fig. 2c and ESI†) and assigned to anisole-Pt-dbm → dbm π–π* transition mixed with ILCT, LLCT, and MLCT.
Both complexes are luminescent in deoxygenated toluene solution at room temperature. The emission spectrum recorded for Pt1 (Fig. 3a; see also Table 1 for numerical data) displays some vibrational structure, with a (0,0) band at about 530 nm, whilst the (0,1) component at about 560 nm is the most intense; these wavelengths correspond to a vibrational progression of 1300 cm−1 associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch of the aromatics, quite typical of many phosphorescent cyclometalated complexes.19 The quantum yield under these conditions is 5%. The spectrum of Pt2 covers a similar spectral region to that of Pt1 but lacks any clear vibrational structure, despite a higher quantum yield of 9%. Much of the previous literature on NHC-Pt(II) complexes reports quantum yields only in films, rather than in solution,7,13 so a direct comparison of the aforementioned quantum yield values for Pt1 and Pt2 with those obtained for related non-helicenic complexes is not readily possible. Compared to classical NHC-Pt systems, however, it appears that the helicene ligand imparts greater rigidity to the complex that helps to reduce some of the deactivation pathways. Overall, the obtained quantum yields are in the same range as those reported for other helicene-NHC-transition-metal complexes.11,12
C stretch of the aromatics, quite typical of many phosphorescent cyclometalated complexes.19 The quantum yield under these conditions is 5%. The spectrum of Pt2 covers a similar spectral region to that of Pt1 but lacks any clear vibrational structure, despite a higher quantum yield of 9%. Much of the previous literature on NHC-Pt(II) complexes reports quantum yields only in films, rather than in solution,7,13 so a direct comparison of the aforementioned quantum yield values for Pt1 and Pt2 with those obtained for related non-helicenic complexes is not readily possible. Compared to classical NHC-Pt systems, however, it appears that the helicene ligand imparts greater rigidity to the complex that helps to reduce some of the deactivation pathways. Overall, the obtained quantum yields are in the same range as those reported for other helicene-NHC-transition-metal complexes.11,12
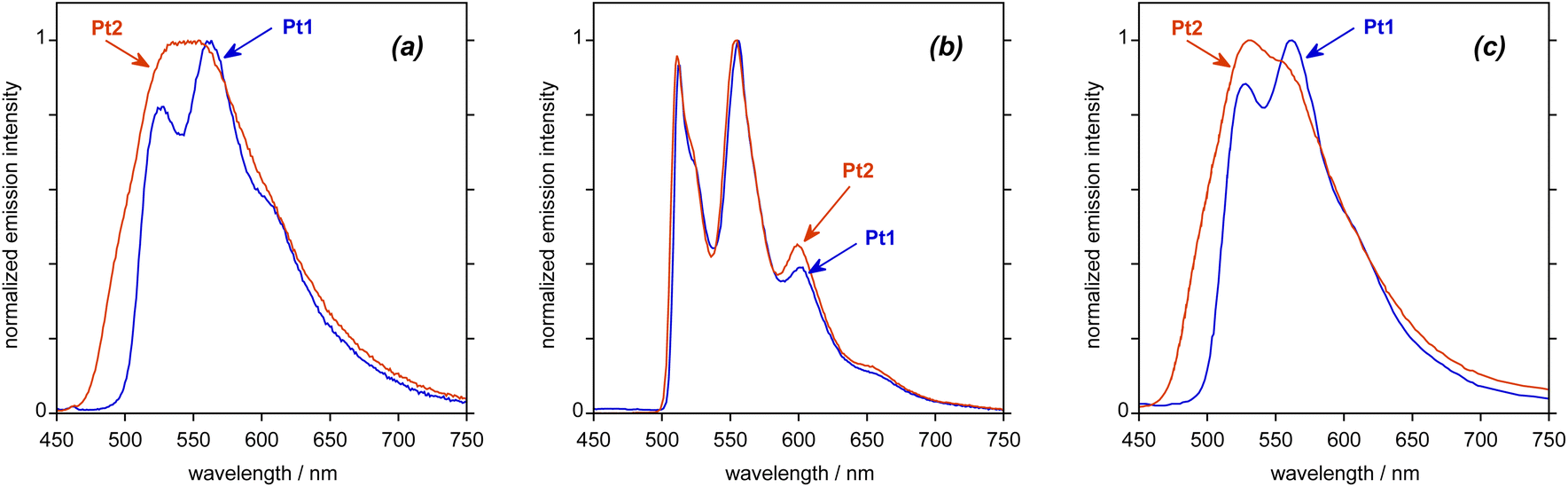 | ||
| Fig. 3 Experimental emission spectra of Pt1 and Pt2 upon excitation at 400 nm: (a) in deoxygenated toluene solution at 295 K, (b) in butyronitrile at 77 K, and (c) in PMMA film (2 wt%) at 295 K under an atmosphere of nitrogen gas. See also Table 1 for numerical data. | ||
| Complex | Absorption in toluene at 295 K λmax/nm (ε/103 M−1 cm−1) | Emission in toluene at 295 Ka |
Emission in PMMA film (2% w/w) at 295 Kb |
Emission in C3H7CN at 77 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max/nm | Φ lum | τ/μs | k r/s−1 | Σknr/s−1 | λ max/nm | Φ lum | τ/μs | k r/s−1 | Σknr/s−1 | λ max/nm | τ/μs | ||
| a Values refer to degassed solutions; the quantum yields Φlum were measured relative to [Ru(bpy)3]Cl2 in aqueous solution for which Φlum = 0.04. b Data were recorded for thin films under an atmosphere of nitrogen gas; spectra and quantum yields were measured using an integrating sphere. c The decays in films do not fit to a single-exponential decay owing to heterogeneity in the environment of the molecules in the film: the quoted values are amplitude-weighted average lifetimes obtained from fitting to the sum of two exponential decays, as shown in Fig. S1.15 in ESI.† | |||||||||||||
| Pt1 | 306 (32.9), 338 (22.4), 383 (7.24), 402 (4.56), 441 (1.1) | 526, 562, 605 | 0.05 | 290 | 170 | 3300 | 528, 562, 609 | 0.31 | 3200c | 97 | 216 | 512, 556, 601, 657 | 3700 |
| Pt2 | 308 (37.9), 336 (30.3), 385 (10.5), 403 (7.23), 441 (1.27) | 546 | 0.09 | 65 | 1400 | 14000 |
532, 555 | 0.23 | 1200c | 192 | 642 | 511, 554, 599, 654 | 6400 |
The luminescence decays mono-exponentially in both cases, with a lifetime of 290 μs for Pt1 and 65 μs for Pt2. The fact that the lifetime is significantly shorter for Pt2, despite its higher quantum yield, suggests that the emission is “more allowed” (exhibits a higher participation of Pt orbitals) in Pt2 than in Pt1. Further insight may be obtained by estimating the radiative kr and non-radiative ∑knr rate constants, assuming that the emitting state is formed with unit efficiency, through the relationships kr = Φ/τ and ∑knr = (1 − Φ)/τ. Such an analysis gives kr values of 1400 and 170 s−1 for Pt2 and Pt1, respectively. The order of magnitude difference suggests that the emissive excited state has significantly different orbital parentage in the two complexes. Conversely, the non-radiative decay is faster in Pt2 than in Pt1, the estimated ∑knr values being 14000 and 3300 s−1, respectively. Strassner and co-workers also observed a disappearance of a clear vibrational structure and substantial decrease in the lifetime of emission upon changing from acac to dbm in related Pt(II) complexes with a dibenzofuran NHC ligand, albeit in thin film rather than in fluid solution.13 In that case, however, the changes were accompanied by a drop in the quantum yield and a significant red-shift in the emission (not observed in Pt2), features that were attributed to the phenyl rings of the dbm extending the π-electron system of this ligand,13bvide infra.
In a frozen glass in butyronitrile (C3H7CN) at 77 K, the vibronic structure becomes very well defined for both complexes, with λ(0,0) = 514 nm in both cases (Fig. 3b and Table 1). Under these conditions, the lifetimes for Pt1 and Pt2 become very long (3.7 and 6.4 ms, respectively). This behaviour is consistent with a largely ligand-centred state localized on an extended π-electron system, as observed in some of our previous studies on complexes of other metals like Ir(III) and Re(I) with helicene ligands.12a,c,e,20 Note that very similar vibronically structured phosphorescence signals were recorded in 2-methyltetrahydrofuran (2-MeTHF) at 77 K (see Fig. 4c).
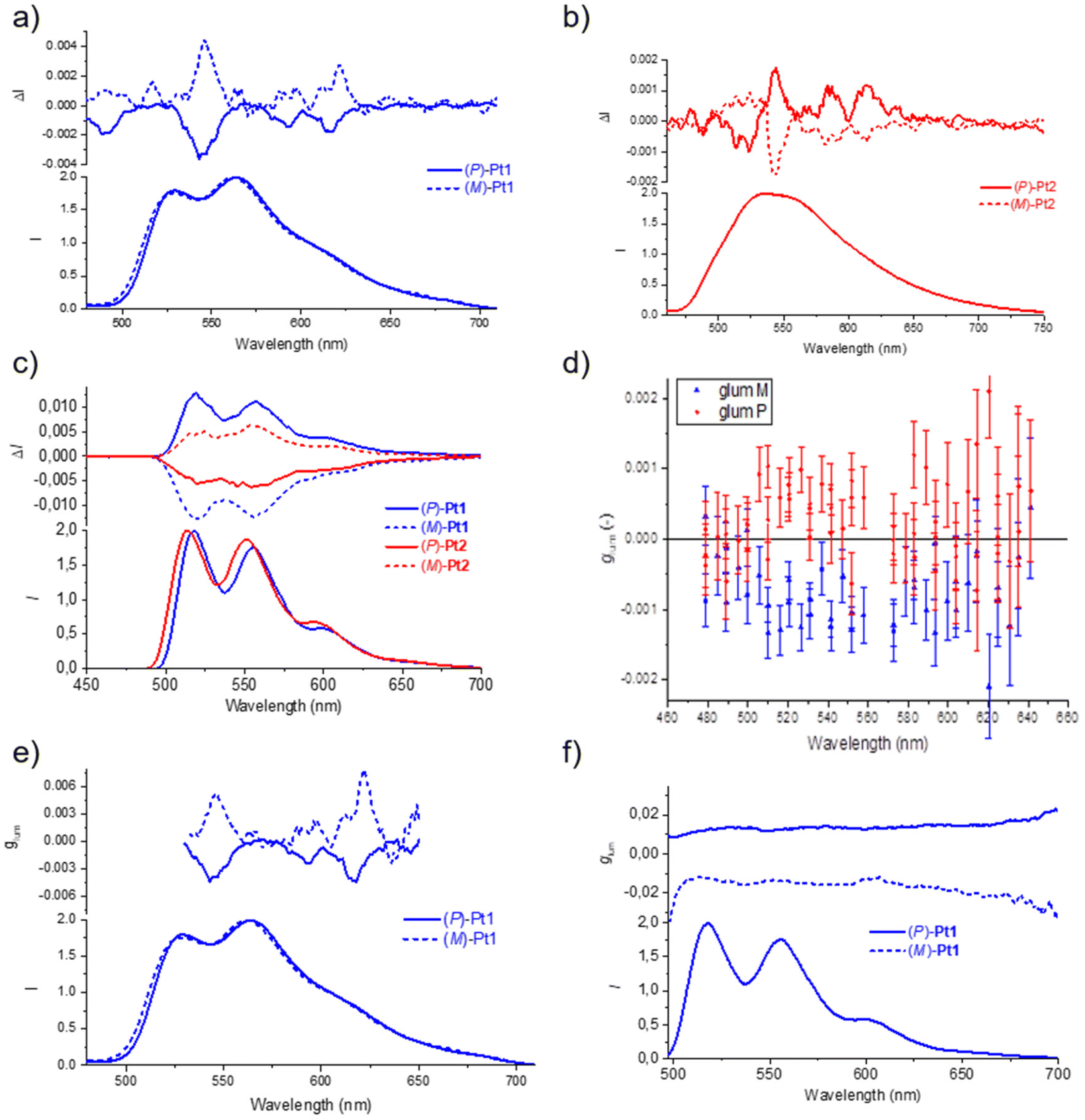 | ||
| Fig. 4 Experimental luminescence and CPL spectra of (P) and (M) enantiomers of (a) Pt1 and (b) Pt2 in degassed toluene at room temperature (298 K), and of (c) Pt1 and Pt2 in degassed 2-MeTHF at low temperature (77 K); C = 10−5 M, λex = 365 nm. (d) Plot of glum values with emission wavelength obtained for 2 wt% PMMA film of Pt1 enantiomers; λex = 313 nm. (e) and (f) Comparison of glum values for Pt1 enantiomers obtained at room temperature in toluene and at low temperature in 2-MeTHF, respectively. | ||
The CPL spectra of (P) and (M) enantiomers of the Pt1 and Pt2 complexes were initially recorded in degassed toluene at room temperature (Fig. 4a and b). Pleasingly, well-structured CPL responses were obtained for both complexes, with dissymmetry factors glum = 2(IL − IR)/(IL + IR) of +5 × 10−3 for (M)-Pt1 and −3.9 × 10−3 for (P)-Pt1 at 546 nm, and of +1.8 × 10−3 for (M)-Pt2 and −1.9 × 10−3 for (P)-Pt2 at 543 nm. Note that while the measured CPL spectra demonstrate overall similar shape, some bands have different signs for Pt1 and Pt2. The CPL responses were then also recorded in a glass of 2-MeTHF at 77 K (Fig. 4c).21 To our delight, the (P) and (M) enantiomers of both systems revealed mirror-image signals in the same region as the non-polarized fluorescence. In this case, the measured glum values were 1.3 × 10−2 for Pt1 and 6.2 × 10−3 for Pt2, the former being one of the highest values reported up to now for Pt-helicene derivatives.7a,11,16b Unlike for the signals recorded in toluene at room temperature, the corresponding enantiomers of Pt1 and Pt2 display opposite CPL signs at 77 K (for (P) enantiomer: positive for Pt1vs. negative for Pt2). Note also that, interestingly, the more resolved CPL spectra at room temperature offer the advantage of observing the vibronic structure, contrary to the non-polarized luminescence (Fig. 4a and b).16c Meanwhile, at 77 K, both CPL and non-polarized phosphorescence become well-resolved and the vibronic structure appears clearly in Fig. 4c.
Fig. 4e and f display the glum values of Pt1 enantiomers at room and low temperature, respectively, and the results are interesting to comment. Typically, if an optical transition derives from just two electronic states, ground and excited, then its glum value should be constant throughout the emission band. This is clearly not the case for the complexes examined here at room temperature, as visible in Fig. 4e for Pt1. There might be thus several electronic excited states involved in the signal under this condition. In addition, vibronic mixing may be involved in the excited states. Indeed, due to vibrational motion in the excited states, two different electronic states could get mixed. This very commonly leads to rapid variations in the glum value across the emission band.21,22 The electronic states that get mixed into the lowest excited state each bring their own electric and magnetic transition dipole moments.
To shed some light on the electronic origin of the emissive properties for Pt1 and Pt2, TDDFT calculations were then performed involving T1 excited-state geometry optimizations. Representative results can be found in Fig. 5, which presents the electron density differences between the ground state and the excited state computed for both complexes, with their negative (red)/positive (blue) values corresponding to outflow/inflow of electron density accompanying T1 → S0 phosphorescence transition, along with the corresponding dominant MO-pair contributions. See Fig. S2.8–S2.12 and Tables S2.7, S2.8 in ESI† for additional calculated data.
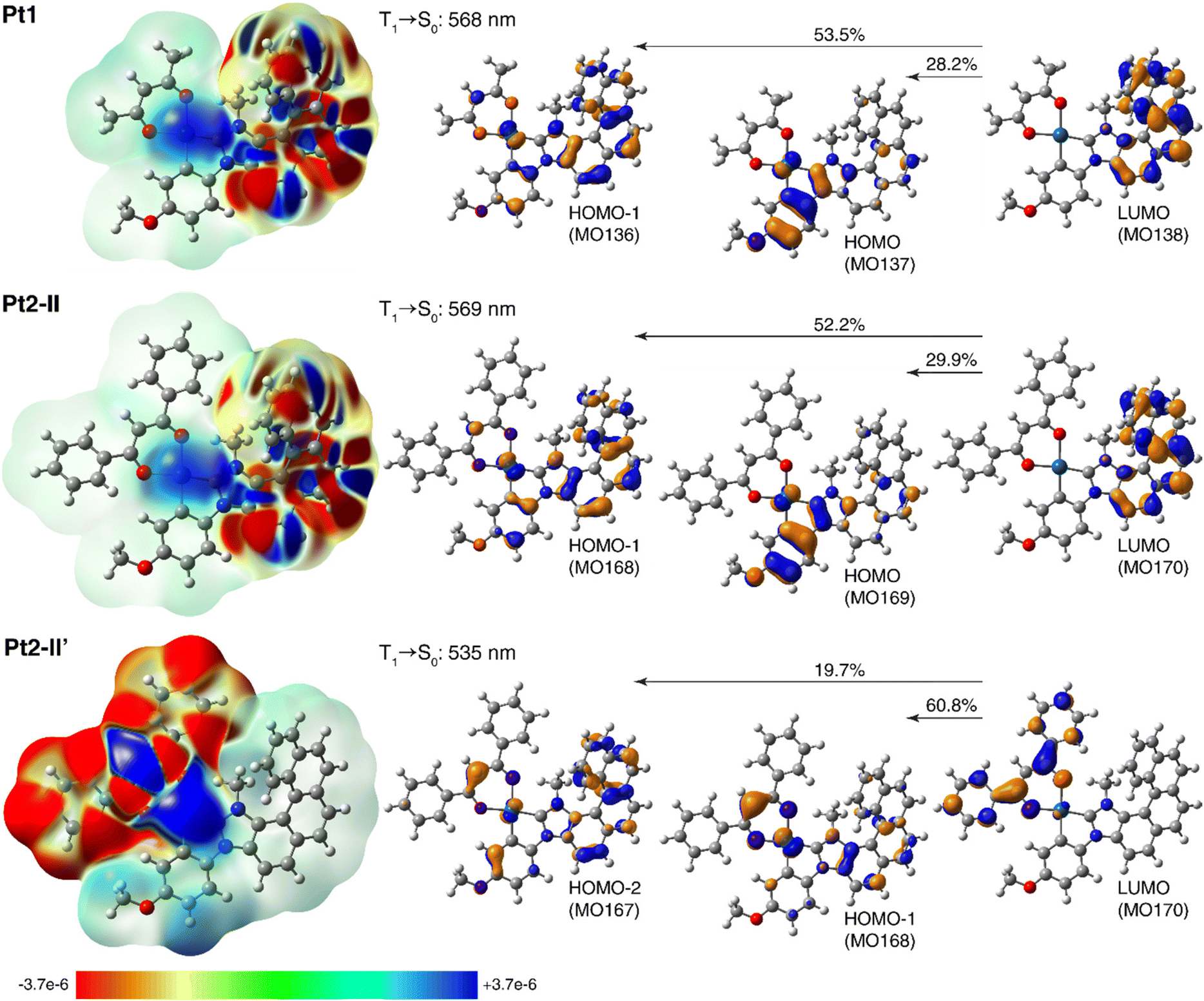 | ||
| Fig. 5 Left: Electron density differences between the S0 ground state and T1 excited state, Δρ = ρg − ρe, color-mapped on ρg (isosurfaces: ±0.0001 au) for Pt1 and Pt2 (in two conformations) based on the TDA-TDDFT-PBE0//SDD/SV(P) PCM(toluene) calculations. Electron density moves from the red region to the blue region when moving from the excited state to the ground state. Right: The corresponding computed T1 → S0 phosphorescence wavelengths along with dominant MO-pair contributions (isosurfaces: ±0.04 au) to emission transition. See ESI† for a full set of calculated data. | ||
As can be seen from Fig. 5 and data presented in the ESI,† the computations reproduce correctly the experimental energies of Pt1 and Pt2 emission, and in the case of Pt2 seem to confirm that the existence of (structurally and/or electronically) different excited states may indeed be responsible for its experimentally observed emission features. Namely, for this system several T1 excited-state structures were obtained including the expected (practically isoenergetic) rotamers (I–IV) of the phenyl groups in the dbm moiety corresponding to S0 ground-state geometries, but also structures with the carbon C(Ph)C(H)C(Ph) chain of the dbm core deviated out the plane of the metalacycle OPtO and the two phenyl rings in the dbm ligand arranged more closely to planarity with this chain (II′ and II′′ of slightly higher energy compared to I–IV); see Fig. S2.8 and Tables S2.7, S2.8.† The aforementioned rotameric T1 structures of Pt2, as illustrated for the conformer II in Fig. 5 (see also ESI†), show essentially the same emission wavelength as that calculated for Pt1 along with the same electronic origin that corresponds to predominantly NHC-helicene-centred 3ππ* state with some NHC-helicene → anisole-Pt 3MLCT and 3ILCT character (see alternating inflow (blue) and outflow (red) of electron density within the π-system of NHC-helicene moiety along with accumulation of electron density around the metal centre accompanying T1 → S0 transition visible in the electron difference densities for Pt1 and Pt2-II shown in the left part of Fig. 5; compare also with Fig. S2.12†). Note that going to the T1 equilibrium Pt2 structures of this type, there is some reorganization among the frontier unoccupied MOs (LUMO and LUMO+1) as compared to their corresponding S0 equilibrium structures, with a significant energetic stabilization of the NHC-helicene-centred π*-orbital, which accordingly becomes LUMO and results in a change of the assignment for T1 → S0 phosphorescence transition vs. S0 → S1 absorption (vide supra). The structural modifications observed in the T1Pt2-II′ and Pt2-II′′ geometries, on the other hand, seem to favour extended π-delocalization within the β-diketonate ligand leading to a decrease in its corresponding π*-orbital energy, such that in these structures the LUMO is localized on the (O^O) ligand, as observed in the ground-state structures of this system. Consequently, the corresponding T1 → S0 transition in such conformers appears blue-shifted as compared to that in rotamers I–IV, and is assigned as predominantly dbm-centred (phenyl → core) 3ILCT admixed with dbm → anisole-NHC-helicene-Pt 3MLCT and 3LLCT, as visible in Fig. 5 and ESI.† Note that similar assignment was proposed for the related dbm-NHC-dibenzofuran Pt(II) complex.13b Finally, it is worth noting that while solvation (toluene vs. THF used as a model for 2-MeTHF) effects simulated in the computations via polarizable continuum model do not appear to affect the calculated emission wavelength, they significantly change the relative percentage of MO-pair contributions to the emission transition (see ESI†), thus additionally influencing the electronic character of the emitting state depending on the solvent. All this indicates that, depending on measurement conditions such as temperature and environment/solvent, the emission signal observed for Pt1 and Pt2 may stem either from one or from several emitting states of similar or different orbital parentage. This conclusion accords well with the experimental findings. For example, the phosphorescence emission originated from Pt2-II′-type T1 structure (with visibly enhanced metal orbitals' involvement as compared to Pt1, see Fig. 5) is consistent with the higher radiative rate constant observed for Pt2vs.Pt1, and (cautiously speculating) it may be a reason for a change in the sign of the CPL signal observed experimentally for both systems (for (P) enantiomer: positive for Pt1vs. negative for Pt2). The co-existence of several emitting states of slightly different emission wavelengths and electronic origins accounts also for a complicated picture of the CPL responses observed experimentally for both complexes at room temperature. Of importance, might be also the aforementioned mixing of different (higher-energy) electronic states into the emitting excited-state.21,22 Further computational studies, including spin–orbit coupling effects and possibly higher-order correlated methods, are needed to provide a detailed rationalization of all the experimental results for Pt1 and Pt2,20c,21,23 which we plan to report for these and other related systems elsewhere in the future.
Due to the long-lived phosphorescence of these helicene-NHC-based platinum(II) complexes, and hence the potential for triplet–triplet quenching, the solid-state luminescence properties of compounds Pt1 and Pt2 were better studied in amorphous poly(methyl methacrylate) (PMMA) films at room temperature with 2 wt% emitter concentration under inert conditions. The recorded emission spectra are shown in Fig. 3c. As can be seen, the spectrum of Pt1 is similar to the spectrum obtained in toluene solution, with three vibrational components resolved. The spectrum of Pt2 shows a more resolved spectral profile than the largely featureless spectrum observed in solution, no doubt reflecting the more rigid environment in the polymer host. The quantum yields are elevated compared to those recorded in solution, rising to 31 and 23% for Pt1 and Pt2, respectively, which can reasonably be attributed to a decrease in the non-radiative decay processes. The emission in the films under an inert atmosphere is very long-lived. Neither case shows the temporal decay following single exponential kinetics, no doubt reflecting heterogeneity in the environment of the complexes in the film. The decays fit well to two exponents (see Fig. S1.15†), from which amplitude-weighted average lifetimes of 3200 and 1200 μs are estimated for Pt1 and Pt2, respectively. These values are of a similar order of magnitude to those at 77 K, confirming that the incorporation of the compounds into the film leads to an environment that is sufficiently rigid to greatly attenuate non-radiative decay processes. On the other hand, even in the films, the emission is very efficiently quenched by oxygen. The decay kinetics in air-equilibrated vs. inert atmosphere conditions are compared in Fig. S1.15,† and average lifetimes of around 80 μs are obtained for the films in air.
Finally, the CPL responses of both complexes were also examined in PMMA films at room temperature with 2 wt% emitter concentration. The compounds were excited at 313 nm, and the samples were measured using an in-line geometry for excitation and emission collection, while the excitation light was depolarized using a bundle of optical fibers. The (P) and (M) enantiomers of Pt1 show signals of opposite sign, similar to those for CPL at low temperature (Fig. 4d), with λmax at 520 nm and glum of the order of 10−3. However, for Pt2, the glum values were so small that they could not be determined. Note that while for Pt2 the CPL signs remain unchanged (+ for (M) and − for (P)), for Pt1 they have been inverted when going from room to low temperature, but they are the same in PMMA as at low temperature, thus again highlighting the influence of the environment.
Conclusions
In summary, we have successfully synthesized two novel, chiral helicene-NHC-based platinum(II) complexes (Pt1 and Pt2, bearing acac and dbm ligands, respectively), and studied in detail their photophysical and chiroptical properties. They exhibit long-lived phosphorescence with moderate quantum yields. Overall, the helicenic backbone in the N-heterocyclic carbene ligand allowed us to introduce chirality within the structure, generating strong chiroptical responses (intense electronic circular dichroism and circularly polarized phosphorescence). Complex Pt1 was found to exhibit strong CPL response both in solution (glum of the order of 10−2 at low temperature and around 10−3 at room temperature) and in the solid state (glum of the order of 10−3 in thin films at room temperature). Theoretical analysis enabled the different behaviors of Pt1 and Pt2 to be accounted for in terms of structural and electronic origins. Compared to the platinum(II) complexes already reported in the literature, thanks to the presence of the helicenic moiety, we observed one of the strongest circularly polarized luminescence glum (0.013 at low temperature).7a In addition, the steric protection imparted by the helicene allowed to study, for the very first time, the photophysics of square-planar cyclometalated NHC-Pt complexes in solution. Finally, the CPL measurements in solution allowed us to clearly observe the vibronic structure. Our further efforts are directed towards developments of chiral supramolecular assemblies of helical systems displaying Pt–Pt interactions for amplifying the circularly polarized phosphorescence.24Author contributions
Conceptualization, formal analysis, validation, resources, writing – review & editing: J. C., M. S.-H., S. C. J. M., J. A. G. W.; investigation: D. K. (synthesis, characterization, computations), N. d. R. (synthesis, characterization), M. C. (X-ray structure analysis), N. V. (chiral HPLC separations), S. C. J. M. (solid-state CPL), E. V. P. & J. A. G. W. (photophysics in solution and films), M. S.-H. (computations); methodology: D. K., M. S.-H.; writing – original draft: D. K., N. d. R., J. C.; visualization: D. K., M. S.-H., S. C. J. M., E. V. P.; supervision: J. C., J. A. G. W., M. S.-H.; funding acquisition: J. C., E. V. P.; project administration: J. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l'Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS), and the European Commission Research Executive Agency (grant agreement number: 859752—HEL4CHIROLED—H2020-MSCA-ITN-2019) for financial support. The computational part of the study was supported by PL-Grid Infrastructure and the ACC Cyfronet AGH in Krakow, Poland. E. V. P. and J. A. G. W. acknowledge the EPSRC grant ref: EP/S012788/1.References
- (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed; (b) N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, RSC, Cambridge, UK, 2011 Search PubMed; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- Selected examples: (a) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (c) S. J. C. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, 2011 CrossRef; (d) F. Wang, L.-J. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804–853 CrossRef CAS; (e) D. Zhao, L. Candish, D. Paul and F. Glorius, ACS Catal., 2016, 6, 5978–5988 CrossRef CAS; (f) M. Karras, M. Dąbrowski, R. Pohl, J. Rybácek, J. Vacek, L. Bednárová, K. Grela, I. Starý, I. G. Stará and B. Schmidt, Chem. – Eur. J., 2018, 24, 10994–10998 CrossRef CAS PubMed; (g) J. Thongpaen, R. Manguin and O. Baslé, Angew. Chem., Int. Ed., 2020, 59, 10242–10251 CrossRef CAS PubMed; (h) A. M. Ruiz-Varilla, E. A. Baquero, B. Chaudret, E. de Jesus, C. Gonzalez-Arellano and J. C. Flores, Catal. Sci. Technol., 2020, 10, 2874–2881 RSC.
- (a) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed; (b) R. Tarrieu, I. H. Delgado, F. Zinna, V. Dorcet, S. Colombel-Rouen, C. Crevisy, O. Basle, J. Bosson and J. Lacour, Chem. Commun., 2021, 57, 3793–3796 RSC; (c) S. Dery, P. Bellotti, T. Ben-Tzvi, M. Freitag, T. Shahar, A. Cossaro, A. Verdini, L. Floreano, F. Glorius and E. Gross, Langmuir, 2021, 37, 10029–10035 CrossRef CAS PubMed.
- Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley VCH, 2008 Search PubMed.
- Selected examples of phosphorescent chiral Pt complexes in OLEDs: (a) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed; (b) Z.-P. Yan, X.-F. Luo, W.-Q. Liu, Z.-G. Wu, X. Liang, K. Liao, Y. Wang, Y.-X. Zheng, L. Zhou, J.-L. Zuo, Y. Pan and H. Zhang, Chem. – Eur. J., 2019, 25, 5672–5676 CrossRef CAS PubMed.
- Selected examples of cycloiridiated NHC complexes in OLEDs: (a) T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed; (b) C.-F. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Chiu, C.-C. Lin, G.-H. Lee, P.-T. Chou, C.-C. Chen, C.-H. Chang and C.-C. Wu, Angew. Chem., Int. Ed., 2008, 47, 4542–4545 CrossRef CAS PubMed; (c) T.-Y. Li, X. Liang, L. Zhou, C. Wu, S. Zhang, X. Liu, G.-Z. Lu, L.-S. Xue, Y.-X. Zheng and J.-L. Zu, Inorg. Chem., 2015, 54, 161–173 CrossRef CAS PubMed; (d) J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed; (e) P.-H. Lanoë, J. Chan, G. Gontard, F. Monti, N. Armaroli, A. Barbieri and H. Amouri, Eur. J. Inorg. Chem., 2016, 1631–1634 CrossRef; (f) M. Zhang, S.-W. Zhang, C. Wu, W. Li, Y. Wu, C. Yang, Z. Meng, W. Xu, M.-C. Tang, R. Xie, H. Meng and G. Wei, ACS Appl. Mater. Interfaces, 2022, 14, 1546–1556 CrossRef CAS PubMed.
- Selected examples of cycloplatinated NHC complexes in OLEDs: (a) X. Wang, S. Ma, B. Zhao and J. Deng, Adv. Funct. Mater., 2023, 2214364 CrossRef; (b) T. Strassner, Acc. Chem. Res., 2016, 49, 2680–2689 CrossRef CAS PubMed; (c) G. Li, S. Liu, Y. Sun, W. Lou, Y.-F. Yang and Y. She, J. Mater. Chem. C, 2022, 10, 210–218 RSC; (d) R. He, R. A. Domingues, S. Valandro and K. S. Schanze, Macromolecules, 2021, 54, 9888–9895 CrossRef CAS; (e) V. Sicilia, S. Fuertes, A. J. Chueca, L. Arnal, A. Martín, M. Perálvarez, C. Botta and U. Giovanella, J. Mater. Chem. C, 2019, 7, 4509–4516 RSC; (f) X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753–6756 CrossRef CAS PubMed; (g) T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- (a) M. Lindemann, G. Xu, T. Pusch, R. Michalzik, M. R. Hofmann, I. Žutić and N. C. Gerhardt, Nature, 2019, 568, 212–215 CrossRef CAS PubMed; (b) H. Wang, L. Liu and C. Lu, Procedia Comput. Sci., 2018, 131, 511–519 CrossRef; (c) J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef; (d) T. Novikova, A. Pierangelo, S. Manhas, A. Benali, P. Validire, B. Gayet and A. D. Martino, Appl. Phys. Lett., 2013, 102, 241103 CrossRef; (e) B. Kunnen, C. Macdonald, A. Doronin, S. Jacques, M. Eccles and I. Meglinski, J. Biophotonics, 2015, 8, 317–323 CrossRef PubMed; (f) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC; (g) T. Mori, Circularly Polarized Luminescence of Isolated Small Organic Molecules, Springer, Singapore, 2020 CrossRef.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- Chiral platinum complex bearing chiral monodentate NHCs: (a) D. Brissy, M. Skander, P. Retailleau and A. Marinetti, Organometallics, 2007, 26, 5782–5785 CrossRef CAS; (b) A. Meyer, M. A. Taige and T. Strassner, J. Organomet. Chem., 2009, 694, 1861–1868 CrossRef CAS; (c) S. K. U. Riederer, B. Bechlars, W. A. Herrmann and F. E. Kühn, Eur. J. Inorg. Chem., 2011, 249–254 CrossRef CAS; (d) P. Marshall, R. L. Jenkins, W. Clegg, R. W. Harrington, S. K. Callear, S. J. Coles, I. A. Fallis and A. Dervisi, Dalton Trans., 2012, 41, 12839–12846 RSC.
- (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC; (b) H. Isla and J. Crassous, C. R. Chim., 2016, 19, 39–49 CrossRef CAS; (c) J.-K. Ou-Yang and J. Crassous, Coord. Chem. Rev., 2018, 376, 533–547 CrossRef CAS; (d) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (e) E. S. Gauthier, R. Rodríguez and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 22840–22856 CrossRef CAS PubMed.
- (a) N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2017, 56, 8236–8239 CrossRef CAS PubMed; (b) A. Macé, N. Hellou, J. Hammoud, C. Martin, E. S. Gauthier, L. Favereau, T. Roisnel, E. Caytan, G. Nasser, N. Vanthuyne, J. A. G. Williams, F. Berrée, B. Carboni and J. Crassous, Helv. Chim. Acta, 2019, 102, e1900044 CrossRef; (c) E. S. Gauthier, L. Abella, N. Hellou, B. Darquié, E. Caytan, T. Roisnel, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 8394–8400 CrossRef CAS PubMed; (d) E. S. Gauthier, M. Cordier, V. Dorcet, N. Vanthuyne, L. Favereau, J. A. G. Williams and J. Crassous, Eur. J. Org. Chem., 2021, 4769–4776 CrossRef CAS; (e) E. S. Gauthier, N. Hellou, E. Caytan, S. Del Fré, V. Dorcet, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams and J. Crassous, Inorg. Chem. Front., 2021, 8, 3916–3925 RSC.
- (a) A. Tronnier, A. Poethig, S. Metz, G. Wagenblast, I. Muenster and T. Strassner, Inorg. Chem., 2014, 53, 6346–6356 CrossRef CAS PubMed; (b) A. Tronnier, U. Heinemeyer, S. Metz, G. Wagenblast, I. Muenster and T. Strassner, J. Mater. Chem. C, 2015, 3, 1680–1693 RSC.
- H. Zhang, Q. Cai and D. Ma, J. Org. Chem., 2005, 70, 5164–5173 CrossRef CAS PubMed.
- (a) Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Munster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed; (b) G. L. Petretto, M. Wang, A. Zucca and J. P. Rourke, Dalton Trans., 2010, 39, 7822–7825 RSC; (c) S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885–9895 CrossRef CAS PubMed.
- (a) L. Norel, M. Rudolph, N. Vanthuyne, J. A. G. Williams, C. Lescop, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Angew. Chem., Int. Ed., 2010, 49, 99–102 CrossRef CAS PubMed; (b) J. Crassous, I. G. Stará and I. Starý, Helicenes: Synthesis, Properties and Applications, John Wiley & Sons, Ltd, 2022 CrossRef; (c) D. Schnable, N. D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 10718–10722 CrossRef PubMed.
- (a) M. Srebro-Hooper, J. Crassous and J. Autschbach, in Helicenes: Synthesis, Properties and Applications, ed. J. Crassous, I. G. Stará and I. Starý, John Wiley & Sons, Ltd, 2022, vol. 12, pp. 395–421 Search PubMed; (b) M. Srebro-Hooper and J. Autschbach, Annu. Rev. Phys. Chem., 2017, 68, 399–420 CrossRef CAS PubMed; (c) J. Autschbach, Chirality, 2009, 21, E116–E152 CrossRef CAS PubMed.
- M. Srebro, N. Govind, W. A. de Jong and J. Autschbach, J. Phys. Chem. A, 2011, 115, 10930–10949 CrossRef CAS PubMed.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2009, 48, 11407–11414 CrossRef CAS PubMed.
- (a) N. Saleh, M. Srebro, T. Reynaldo, N. Vanthuyne, L. Toupet, V. Y. Chang, G. Muller, J. A. G. Williams, C. Roussel, J. Autschbach and J. Crassous, Chem. Commun., 2015, 51, 3754–3757 RSC; (b) N. Saleh, D. Kundu, N. Vanthuyne, J. Olesiak-Banska, A. Pniakowska, K. Matczyszyn, V. Y. Chang, G. Muller, J. A. G. Williams, M. Srebro-Hooper, J. Autschbach and J. Crassous, ChemPlusChem, 2020, 85, 2446–2454 CrossRef CAS PubMed; (c) H. D. Ludowieg, M. Srebro-Hooper, J. Crassous and J. Autschbach, ChemistryOpen, 2022, 11, e202200020 CrossRef CAS PubMed.
- K. Dhbaibi, P. Morgante, N. Vanthuyne, J. Autschbach, L. Favereau and J. Crassous, J. Phys. Chem. Lett., 2023, 14, 1073–1081 CrossRef CAS PubMed.
- H. P. J. M. Dekkers and L. E. Closs, J. Am. Chem. Soc., 1976, 98, 2210–2219 CrossRef CAS.
- F. Gendron, B. Moore II, O. Cador, F. Pointillart, J. Autschbach and B. Le Guennic, J. Chem. Theory Comput., 2019, 15, 4140–4155 CrossRef CAS PubMed.
- G. Park, H. Kim, H. Yang, K. R. Park, I. Song, J. H. Oh, C. Kim and Y. You, Chem. Sci., 2019, 10, 1294–1301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2207037. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00577a |
| This journal is © The Royal Society of Chemistry 2023 |