
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal phosphine aldehyde complexes and their application in Cu-free Sonogashira and Suzuki–Miyaura cross-coupling reactions†‡
Saral
Baweja
 ,
Tom
Gabler
,
Peter
Lönnecke
and
Evamarie
Hey-Hawkins
*
,
Tom
Gabler
,
Peter
Lönnecke
and
Evamarie
Hey-Hawkins
*
Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Leipzig University, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Tel: (+49)341 97 36151
First published on 17th April 2023
Abstract
Transition metal coordination chemistry and catalysis are rife with phosphine ligands. One of the rather less studied members of the phosphine ligand family are phosphine aldehydes. We have synthesised 3-(diphenylphosphino)propanal (PCHO) with a slight modification of the known procedure and studied its complexation behaviour with palladium(II) and platinum(II). The catalytic activity of the palladium(II) phosphine aldehyde complexes was investigated in Cu-free Sonogashira and Suzuki–Miyaura cross-coupling reactions. Furthermore, the homogeneous nature of the catalytically active species was confirmed.
Introduction
In both extensive industrial processes and scientific research, homogeneous catalysis frequently employs phosphine ligands in a variety of significant reactions.1 Phosphines containing additional O-functionalities constitute a versatile class of ligands that has been extensively studied and utilised for applications in catalysis.2 One such class of ligands are phosphine aldehydes, in which an aldehyde moiety and a phosphanyl group are connected by a carbon backbone. When used as a ligand, phosphine aldehydes have three possible binding modes: The monodentate κ1-P coordination mode, the bidentate P,O-chelate or κ2-P,O coordination, in which complexation of the metal ion occurs via the phosphine and the oxygen atom through σ bonds, and the κ1-P,κ2-CO coordination, involving the π bond of the carbonyl group. Furthermore, chelate-assisted oxidative addition of the C–H bond of the aldehyde group can occur with formation of a metal–acyl hydride complex (κ1-P,κ1-Cacyl).3As phosphine aldehydes are generally very air-sensitive oils, their use in synthetic chemistry is limited.4 One of the more studied, commercially available and air-stable member of this class of ligands is 2-(diphenylphosphino)benzaldehyde which has been used to synthesise various complexes in different binding modes with a variety of transition metals in different oxidation states.5 Examples of other reported phosphine aldehydes are 2-(dicyclohexylphosphino)acetaldehyde4 and 3-(diphenylphosphino)propanal (PCHO).6 Established synthetic routes include the reaction of the acetal-protected Grignard reagent with diarylchlorophosphine,7 the reaction of lithium diphenylphosphanide with acetal-protected halo-substituted aldehydes,6 hydrophosphination of aldehydes containing olefinic bonds3 and from cyclic phosphonium salts.8
Phosphine aldehydes have been mainly employed as precursors for the synthesis of phosphine-imine ligands. Due to the presence of an aldehyde group which can easily react with various amines and hydrazines in condensation reactions, a plethora of bi-, tri-, and tetradentate ligands can be formed, especially for 2-(diphenylphosphino)benzaldehyde.9 These ligands have been used for the synthesis of a multitude of catalytically active complexes. The Morris lab reported the synthesis of Fe/P,N,N,P complexes via a template-directed condensation of an α-phosphinoacetaldehyde with suitable chiral amines and showed their high catalytic activity for asymmetric transfer hydrogenation of ketones and imines.10 We have previously reported the synthesis of heteroditopic ligands synthesised by condensation of pyridine aldehyde and different phosphine aldehydes with a dihydrazine moiety.11,12 These heteroditopic ligands were employed for the formation of various heterobimetallic complexes. We have shown that the Pd/Co heterobimetallic complex was catalytically active for a two-step one-pot sequential Cu-free Sonogashira cross-coupling reaction followed by semi transfer-hydrogenation to give Z-arylalkenes.11
However, metal phosphine aldehyde complexes have not been investigated in-depth especially in comparison to other phosphine ligand families.4 Only a few studies on the catalytic activity of Pd,13 Rh14 and Ru15 complexes exist in literature. Herein, we report the synthesis of palladium(II) and platinum(II) complexes with PCHO and the catalytic activity of well-defined palladium(II) phosphine aldehyde pre-catalysts for copper-free Sonogashira and Suzuki–Miyaura cross-coupling reactions.
Results and discussion
3-(Diphenylphosphino)propanal (PCHO) was synthesised following a modified procedure by Vaughn and Gladysz which utilises the reaction of lithium diphenylphosphanide with acetal-protected 3-bromopropanal.6 Due to the air-sensitivity of PCHO, the reaction as well as the subsequent work-up and purification was carried out under nitrogen atmosphere. PCHO was isolated as an off-white oil after performing column chromatography with dried degassed silica and degassed solvent under an inert atmosphere. The fractions were collected in Schlenk flasks under nitrogen flow. Due to the maintenance of an inert environment, the yield was improved to 57% in comparison to the previously reported 33% (Scheme 1). For further experiments, PCHO was stored as a 0.1 M solution in n-hexane at −20 °C.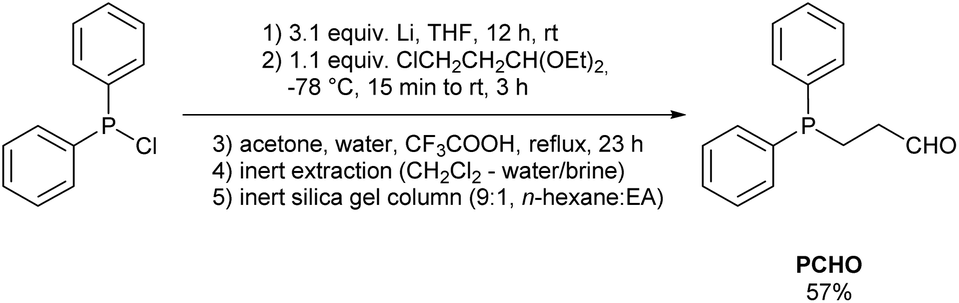 | ||
| Scheme 1 Modified synthesis of 3-(diphenylphosphino)propanal (PCHO). | ||
The reaction of [PdCl2(COD)] (COD = 1,5-cyclooctadiene) with one equivalent of PCHO resulted in the displacement of COD and formation of trans-[{PdCl(μ-Cl)(PCHO-κP)}2] (1) (Scheme 2). The 31P{1H} NMR spectrum contains a singlet at 31.4 ppm which is shifted downfield from −16.1 ppm for PCHO. The characteristic signal of the aldehyde proton in the 1H NMR spectrum at 9.54 ppm (in comparison to 9.67 ppm for PCHO) confirms the monodentate coordination via phosphorus.
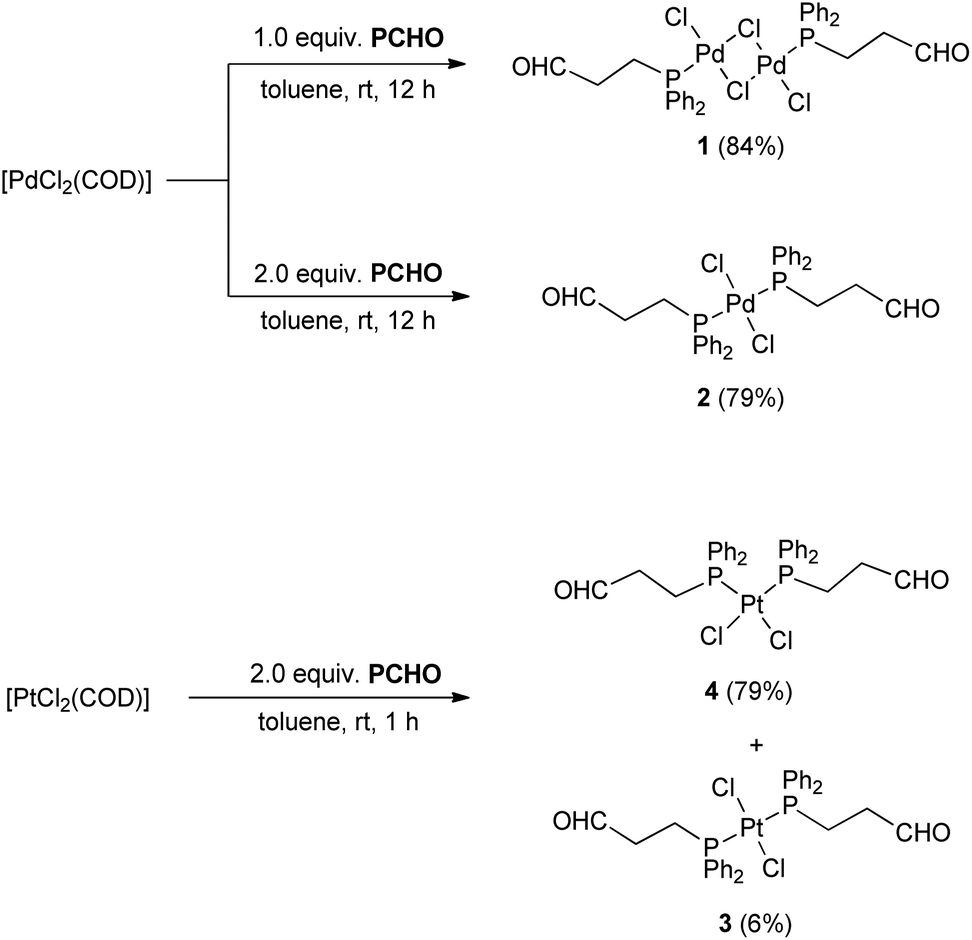 | ||
| Scheme 2 Synthesis of complexes 1, 2, 3 and 4. | ||
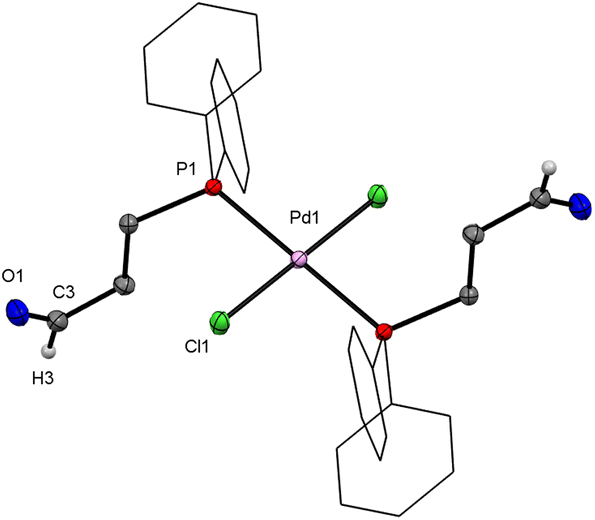
By vapour diffusion of Et2O into a toluene solution of 1, bright orange crystals were obtained that were suitable for single-crystal X-ray diffraction (XRD). The molecular structure shows a dinuclear Pd complex with two bridging chlorido ligands and a trans arrangement of the two monodentate phosphine ligands (Fig. 1). The Pd atoms have a square-planar geometry, and only a small deviation from the predicted value of 90° is observed between the angles of adjacent ligands. The Pd–Cl2′ bond trans to the phosphorus atom is slightly longer (11(1) pm) than the Pd–Cl2 bond cis to the phosphorus atom. This may be explained by the trans effect of the phosphorus ligands.
 | ||
| Fig. 1 Molecular structure of trans-[{PdCl(μ-Cl)(PCHO-κP)}2] (1). Hydrogen atoms are omitted for clarity, phenyl rings are drawn as wireframes; thermal ellipsoids are set at the 50% probability level. The molecule is located on an inversion centre. | ||
When the same reaction is performed with two equivalents of PCHO, a yellow compound is formed. The HR-ESI(+) mass spectrum showed the base peak at m/z 625.044 which corresponds to [PdCl(PCHO)2]+ and indicates the formation of the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex 2. In the 31P{1H} NMR spectrum a singlet is observed at 16.3 ppm which indicates the formation of one of the two possible isomers, cis and trans, only. As in 1, a singlet at 9.64 ppm for the aldehyde proton in the 1H HMR spectrum also suggest formation of a monodentate phosphine complex. Yellow crystals were obtained after slow vapour diffusion of Et2O into a toluene solution of 2. The molecular structure shows an almost perfect square-planar geometry of the Pd centre with a trans orientation of the ligands (Fig. 2). Both 1 and 2 are air-stable complexes and can be stored at room temperature.
:2 complex 2. In the 31P{1H} NMR spectrum a singlet is observed at 16.3 ppm which indicates the formation of one of the two possible isomers, cis and trans, only. As in 1, a singlet at 9.64 ppm for the aldehyde proton in the 1H HMR spectrum also suggest formation of a monodentate phosphine complex. Yellow crystals were obtained after slow vapour diffusion of Et2O into a toluene solution of 2. The molecular structure shows an almost perfect square-planar geometry of the Pd centre with a trans orientation of the ligands (Fig. 2). Both 1 and 2 are air-stable complexes and can be stored at room temperature.
 | ||
| Fig. 2 Molecular structure of trans-[PdCl2(PCHO-κP)2] (2). Hydrogen atoms are omitted for clarity, phenyl rings are drawn as wireframes; thermal ellipsoids are set at the 50% probability level. The molecule is located on an inversion centre. Complex 3 is isostructural (Fig. S26-a, ESI‡). | ||
In an attempt to prepare the corresponding platinum(II) complexes, a 1:1 reaction was conducted with [PtCl2(COD)] and PCHO. A colourless crystalline powder was isolated in a low yield of 42% and unreacted [PtCl2(COD)] was recovered. The 31P{1H} NMR spectrum showed a singlet at 7.5 ppm with 195Pt satellites (1JP,Pt 3646 Hz). These observations point towards the formation of a 1:2 complex, [PtCl2(PCHO-κP)2]. When the reaction was performed in a 1:2 ratio (metal precursor to ligand), two singlets (ratio 9:1) both showing 195Pt satellites were observed in the 31P{1H} NMR spectrum; the major one is centred at 7.5 ppm (1JP,Pt 3646 Hz) and the minor one at 12.4 ppm (1JP,Pt 2561 Hz). The major product can be assigned to the cis isomer (4), as the cis isomers of platinum complexes of the type [PtX2(PRR′R′′)2] (R, R′, R′′ = any organic substituent) generally exhibit larger coupling constants than their trans counterparts.13 Suitable single crystals for XRD were obtained from slow vapour diffusion of n-pentane into a toluene solution of [PtCl2(PCHO-κP)2]. The molecular structure confirmed the formation of the cis isomer (Fig. 3). The trans isomer was isolated from the ethereal washing solution of the reaction between PCHO and [PtCl2(COD)] (1:2 ratio). Single crystals suitable for X-ray diffraction were obtained by slow evaporation of diethyl ether from the ethereal washing solution. The molecular structure (Fig. S26-a, ESI‡) revealed that 3 is isostructural to the corresponding Pd complex 2.
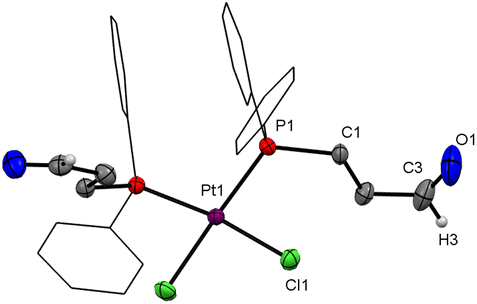 | ||
| Fig. 3 Molecular structure of cis-[PtCl2(PCHO-κP)2] (4). Hydrogen atoms are omitted for clarity, phenyl rings are drawn as wireframes; thermal ellipsoids are set at the 50% probability level. | ||
The calculated single point energies of the trans and cis isomers 3 and 4 at the PBE0 BJ3/ZORA-def2-TZVPP level of theory incorporating the conductor-like polarisable solvent model (CPCM, toluene as the solvent) indicated a slightly higher stability of the cis isomer (E = −13780284.4 kcal mol−1) over the trans configuration (E = −13780278.3 kcal mol−1), supporting the preferred formation of cis-[PtCl2(PCHO-κP)2] (4) (for details see ESI, chapter 5‡).
Since palladium catalysts are the backbone of various cross-coupling reactions, only 1 and 2 were tested in Sonogashira and Suzuki–Miyaura cross-coupling reactions. For the Sonogashira cross-coupling reaction, a condition scan was performed using bromobenzene and phenylacetylene as substrates (Table 1). The catalysis worked efficiently under a Cu-free regime. A variety of bases and solvents was screened with 1 and 2 as pre-catalysts. In all cases, 2 gave better yields than 1. Furthermore, the catalysis worked better in the presence of less polar solvents like toluene and 1,4-dioxane (Table 1, entries 2 and 4, GC-MS yields for diphenylacetylene of 94 and 99%). With DMF as solvent and 2 as a catalyst, an appreciable yield of 87% was observed (Table 1, entry 6), not observed for other polar solvents like MeCN and EtOH (Table 1, entries 5 and 7). Replacing DBU with cheaper and safer bases, such as TMG (1,1,3,3-tetramethylguanidine) in toluene, resulted in good yields (Table 1, entry 8). Using NEt3 or K2CO3 as bases resulted in low yields (Table 1, entries 10 and 11), and using [PdCl2(COD)] as a pre-catalyst resulted in only 4% yield employing optimised conditions (Table 1, entry 13). When the reaction was conducted with trans-[PdCl2(PPh2nBu)2] as pre-catalyst, similar yields as in the reaction catalysed by 2 were observed indicating no significant effect of the aldehyde functionality in the Sonogashira reaction under the tested reaction conditions (Table 1, entry 14 vs. entry 4). Furthermore, the reactions must be performed under a nitrogen atmosphere as no diphenylacetylene is formed in air (Table 1, entry 12 vs. entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Pre-catalyst | Yielda (%) |
| a Yields were determined by GC-MS with naphthalene as internal standard. b Reaction performed in air. | ||||
| 1 | DBU | Toluene | 1 | 69 |
| 2 | DBU | Toluene | 2 | 94 |
| 3 | DBU | 1,4-Dioxane | 1 | 78 |
| 4 | DBU | 1,4-Dioxane | 2 | 99 |
| 5 | DBU | CH3CN | 2 | 74 |
| 6 | DBU | DMF | 2 | 87 |
| 7 | DBU | EtOH | 2 | 47 |
| 8 | TMG | Toluene | 2 | 82 |
| 9 | TMG | 1,4-Dioxane | 2 | 73 |
| 10 | K2CO3 | Toluene | 2 | 9 |
| 11 | NEt3 | Toluene | 2 | 2 |
| 12b | DBU | 1,4-Dioxane | 2 | 2 |
| 13 | DBU | 1,4-Dioxane | [PdCl2(COD)] | 4 |
| 14 | DBU | 1,4-Dioxane | trans-[PdCl2(PPh2nBu)2] | 99 |
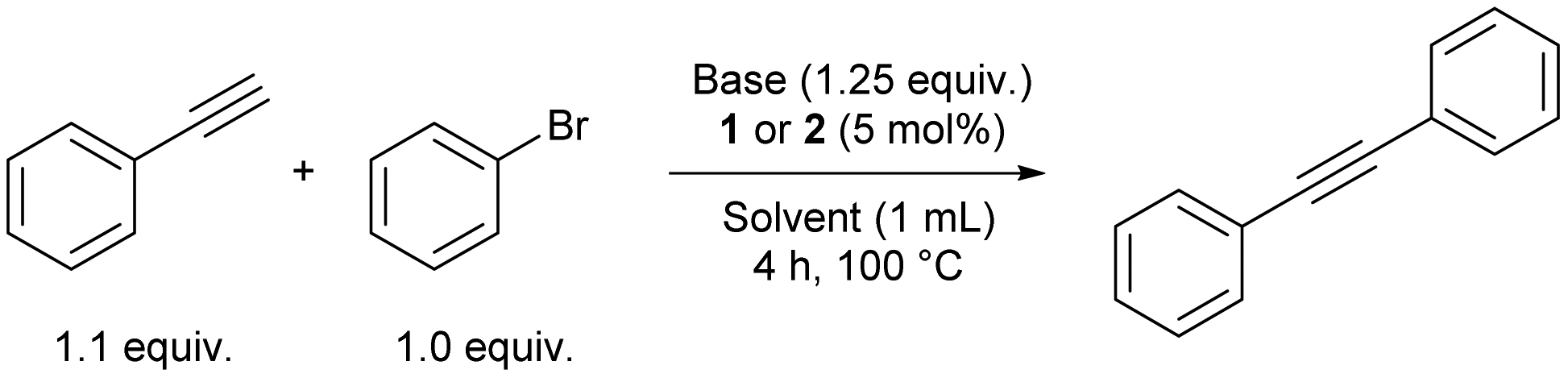
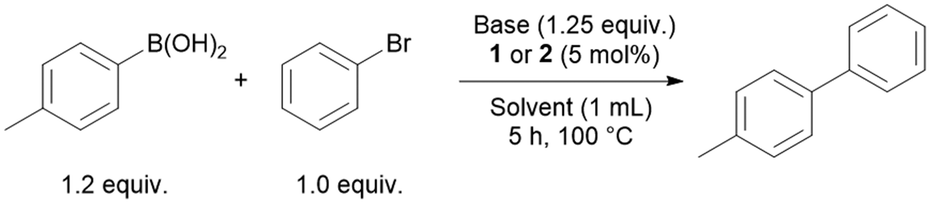
The palladium complexes were also tested in Suzuki–Miyaura cross-coupling reactions. Bromobenzene and p-tolylboronic acid were used as model substrates for the reaction to synthesise 4-methyl-1,1′-biphenyl (Table 2). While performing a solvent scan with TMG as base, 2 proved to be a better catalyst than 1 in almost all cases. The reaction worked best in toluene with a moderate yield of 57% (Table 2, entry 2). Different bases such as DBU, DABCO (1,4-diazabicyclo[2.2.2]octane) and NEt3 in toluene resulted in lower yields (Table 2, entries 4, 5 and 9). Using 1.5 equiv. of p-tolylboronic acid (instead of 1.2 equiv.) in toluene with TMG as a base and 2 as pre-catalyst gave yields up to 84% (Table 2, entry 10). For comparison, when the reaction was conducted under the same conditions replacing 2 with [PdCl2(COD)] as the pre-catalyst a yield of only 50% was obtained (Table 2, entry 12). When the reaction is conducted with [PdCl2(PPh2nBu)2] as the pre-catalyst a yield of 28% is observed in comparison to 84% with 2 as pre-catalyst indicating a positive effect of the aldehyde functionality in the catalytic reaction (Table 2, entry 13 vs. entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Pre-catalyst | Yielda (%) |
| a Yield was determined by GC-MS with naphthalene as internal standard. b 1.5 equiv. p-tolylboronic acid. c Reaction performed in air. | ||||
| 1 | TMG | Toluene | 1 | 47 |
| 2 | TMG | Toluene | 2 | 57 |
| 3 | DBU | Toluene | 1 | 8 |
| 4 | DBU | Toluene | 2 | 7 |
| 5 | DABCO | Toluene | 2 | 2 |
| 6 | DBU | DMF | 2 | 5 |
| 7 | TMG | DMF | 2 | 10 |
| 8 | TMG | MeCN | 2 | 22 |
| 9 | NEt3 | Toluene | 2 | 14 |
| 10b | TMG | Toluene | 2 | 84 |
| 11c | TMG | Toluene | 2 | 6 |
| 12b | TMG | Toluene | [PdCl2(COD)] | 50 |
| 13 | TMG | Toluene | trans-[PdCl2(PPh2nBu)2] | 28 |
For the Sonogashira and Suzuki–Miyaura cross-coupling reaction, the respective products diphenylacetylene and 4-methyl-1,1′-biphenyl were isolated by column chromatography with yields of 92% (for entry 4 in Table 1) and 79% (for entry 10 in Table 2), respectively (see ESI, chapter 3‡).
Last but not least, the nature of the catalyst was investigated. Initially, mercury poisoning studies were performed to decipher whether the catalyst is homogeneous or heterogeneous. The mercury test for both coupling reactions with 2 as a pre-catalyst showed a lowering of catalytic activity after mercury was added. This could have been due to the heterogeneous nature of the catalyst, but a loss of catalytic activity could also be attributed to a reaction of Hg with palladium(II) complexes.16 Therefore, a reaction of 2 with Hg was performed in deuterated toluene. Multiple signals were observed in the 31P{1H} NMR spectrum, implying that the Hg test is not reliable, as the pre-catalyst obviously already reacts with Hg to give multiple products. Crabtree's test17 was performed, but a false positive was observed, as no reaction occurred when 2 and DCT (dibenzo[a,e]cyclooctatetraene) were heated in toluene-d8. Finally, Maitlis’ test18 was performed, wherein the hot reaction mixture was filtered after half the completion time (2 hours for Sonogashira and 2.5 hours for Suzuki–Miyaura cross-coupling reaction) over a tightly packed Celite column. Fresh substrates, base, and solvent were then added to the filtrate and the mixture was heated at 100 °C for 4 hours for the Sonogashira reaction and 5 hours for the Suzuki–Miyaura cross-coupling reaction. Catalytic activity was retained and yields increased by 98% and 62% for Sonogashira and Suzuki–Miyaura cross-coupling reactions, respectively, in comparison to the half-time yields, indicating the homogeneous nature of the catalyst (see ESI, chapter 4‡).
Conclusions
In summary, we have reported the synthesis of palladium(II) and platinum(II) complexes with 3-(diphenylphosphino)propanal (PCHO). The synthetic route for the phosphine aldehyde was improved. Out of the various possible binding modes, it was seen that the complexation of the phosphine aldehyde with palladium(II) and platinum(II) has led to the formation of the κ1-P complex. These complexes exhibit an almost perfect square planar geometry. The air-stable palladium complexes were then studied for their catalytic activity for two cross-coupling reactions, namely Sonogashira and Suzuki–Miyaura cross-coupling. For the Sonogashira reaction, it was observed that the reaction works under a Cu-free regime. For all tested conditions, 2 outperformed 1. In comparison to 2, when the reaction was conducted with trans-[PdCl2(PPh2nBu)2] as pre-catalyst similar yields were obtained for the Sonogashira reaction, whereas a three-fold increase in yield is observed in case of the Suzuki cross-coupling reaction, showing a positive effect of the aldehyde functionality. Also, various tests were performed to understand whether the catalysis is homogeneous or heterogeneous. With a positive Maitlis’ test, it was concluded the (pre-)catalyst 2 is homogeneous. The synthesis and catalytic activity of these phosphine aldehyde complexes shed some light on the potential of these less-explored phosphine ligands.Experimental
General information
All reactions were conducted under nitrogen atmosphere in the absence of air and water using standard Schlenk techniques. Solvents (Et2O, dichloromethane, toluene, and n-hexane) were obtained from an MBraun Solvent Purification System (MB SPS-800). Dichloromethane and diethyl ether were stored on activated 4 Å molecular sieves, and n-hexane and toluene were stored over a potassium mirror. THF was distilled from sodium/benzophenone and stored over activated 4 Å molecular sieves, and MeCN was distilled from P2O5 and stored over 3 Å molecular sieves. PPh2Cl was obtained from Sigma Aldrich and distilled prior to use. 3-Chloro-1,1-diethoxypropane and [PdCl2(COD)] were obtained from Sigma Aldrich and used as received. Dichlorido(1,5-cyclooctadiene)platinum(II) was synthesised from potassium tetrachloridoplatinate(II) according to the literature.19trans-[PdCl2(PPh2nBu)2] was synthesised according to literature20 (for details see ESI, chapter 6‡). All NMR spectra were recorded on a BRUKER Avance III HD 400 MHz NMR spectrometer at 25 °C (1H 400.13 MHz, 13C 100.63 MHz, and 31P 162.02 MHz, two-dimensional (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC). The chemical shifts δ are reported in parts per million (ppm). Tetramethylsilane (TMS) was used as the internal reference in 1H and 13C NMR spectra, all other nuclei spectra were referenced to TMS using the Ξ scale.21 The numbering scheme of all compounds is given in the ESI, chapter 1.‡ Electrospray ionisation mass spectrometry was carried out with an ESI-qTOF Impact II by Bruker Daltonics GmbH in positive ion mode. IR spectra were obtained with an FT-IR spectrometer Nicolet iS5 (ATR, Transmission, Thermo Fisher Scientific) scanning between 4000–400 cm−1 with a KBr beam splitter. Elemental analyses (C, H, and N) were performed with a Heraeus VARIO EL microanalyser. GC-MS data were measured with a GC-MS system GCMS-QP2010 PLUS by SHIMADZU. The GC was equipped with an Rtx®-5MS CB column (30 m × 0.25 mm × 0.25 μm) manufactured by RESTEK. The oven temperature was 50 °C and the injection temperature was 200 °C. The purge flow was 0.5 mL min−1, and the GC-MS was run with helium. The temperature programme was run starting at 50 °C with a rate of 5 °C min−1 up to 200 °C. The ion source and interface temperature were 200 °C. The MS was used in scan mode. Naphthalene was used as an internal standard.Single crystal X-ray diffractometry
The data were collected on a Gemini diffractometer (Rigaku Oxford Diffraction) using Mo-Kα radiation (λ = 71.073 pm) and ω-scan rotation. Data reduction was performed with CrysAlisPro22 including the program SCALE3 ABSPACK for empirical absorption correction. The structures were solved by dual-space methods with SHELXT-201823 and the refinement was performed with SHELXL-2018.24 Anisotropic refinement of all non-hydrogen atoms. Hydrogen atoms for 1 and 4 were calculated on idealised positions using the riding model, whereas for 2 and 3 a difference-density Fourier map was used to locate hydrogen atoms. Structure figures were generated with DIAMOND-4.25
Synthesis of ligand and complexes
:1). The column was eluted with n-hexane/ethyl acetate (9:1). The solvents were removed and the product was isolated as air- and temperature-sensitive off-white oil and stored as a 0.1 M solution in n-hexane at −20 °C. Yield: 3.04 g (57%). 1H NMR (400 MHz, CD3CN) δ = 9.67 (s, 1H, H3), 7.46–7.36 (m, 10H, H2′, H3′, H4′), 2.51–2.45 (m, 2H, H2), 2.33–2.29 (m, 2H, H1). 13C{1H} NMR (101 MHz, CD3CN) δ = 202.5 (d, 3JC,P = 12.4 Hz, C3), 139.2 (d, 1JC,P = 13.4 Hz, C1′), 133.5 (d, 2JC,P = 19.0 Hz, C2′), 129.9 (C4′), 129.6 (d, 3JC,P = 6.7 Hz, C3′), 40.6 (d, 2JC,P = 16.9 Hz, C2), 20.1 (d, 1JC,P = 11.6 Hz, C1). 31P{1H} NMR (162 MHz, CD3CN) δ = −16.1. HRMS (ESI(+), CH3CN): m/z calcd for [M + H]+: 243.0933; found: 243.0943.
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 1718 (s, νC
(cm−1) = 1718 (s, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (400 MHz, CD2Cl2) δ (ppm) = 9.54 (s, 2H, H3), 7.71–7.54 (m, 8H, H2′), 7.53–7.51 (m, 4H, H4′), 7.44–7.41 (m, 8H, H3′), 2.74–2.68 (m, 4H, H2), 2.58–2.51 (m, 4H, H1); 13C{1H} NMR (101 MHz, CD2Cl2) δ (ppm) = 198.3 (d, 3JC,P = 15.9 Hz, C3), 133.3 (d, 2JC,P = 10.0 Hz, C2′), 132.2 (s, C4′), 129.1 (d, 3JC,P = 11.6 Hz, C3′), 127.2 (d, 1JC,P = 58.3 Hz, C1′), 38.3 (s, C2), 20.1 (d, 1JC,P = 40.2 Hz, C1); 31P{1H} NMR (162 MHz, CD2Cl2) δ (ppm) = 31.4. HRMS (ESI(+), CH3CN): m/z calculated for [M − Cl]+: 802.886; found: 802.886.
O). 1H NMR (400 MHz, CDCl3) δ (ppm) = 9.64 (s, 2H, H3), 7.75–7.70 (m, 8H, H2′), 7.50–7.40 (m, 12H, H3′ and H4′), 2.72–2.70 (s, 8H, H1 and H2). 13C{1H} NMR (101 MHz, CDCl3) δ (ppm) = 199.8 (s, C3), 133.8 (m, C2′), 131.1 (s, C4′), 129.6 (m, C1′), 128.8 (m, C4′), 38.9 (s, C2), 18.6 (s, C1). 31P{1H} NMR (162 MHz, CDCl3) δ (ppm) = 16.3. HRMS (ESI(+), CH3CN): m/z calculated for [M − Cl]+: 625.054; found: 625.044.
O). 1H NMR (400 MHz, CDCl3) δ (ppm) = 9.52 (s, 2H, H3), 7.53–7.41 (m, 12H, C2′ and C4′), 7.32–7.25 (m, 8H, C3′), 2.93–2.87 (m, 4H, C2), 2.69–2.62 (m, 4H, C1). 13C{1H} NMR (101 MHz, CDCl3) δ (ppm) = 199.2 (m, C3), 133.3 (m, C2′), 131.5 (s, C4′), 128.7 (m, C1′ and C3′), 38.9 (s, C2), 23.2 (m, C1). 31P{1H} NMR (162 MHz, CDCl3) δ (ppm) = 7.5 (s + Pt satellites, 1JP,Pt = 3646 Hz).
O). 1H NMR (400 MHz, CD2Cl2) δ (ppm) = 9.54 (s, 2H, H3), 7.71–7.54 (m, 8H, H2′), 7.53–7.51 (m, 4H, H4′), 7.44–7.41 (m, 8H, H3′), 2.74–2.68 (m, 4H, H2), 2.58–2.51 (m, 4H, H1); 13C{1H} NMR (101 MHz, CD2Cl2) δ (ppm) = 198.3 (d, 3JC,P = 15.9 Hz, C3), 133.3 (d, 2JC,P = 10.0 Hz, C2′), 132.2 (s, C4′), 129.1 (d, 3JC,P = 11.6 Hz, C3′), 127.2 (d, 1JC,P = 58.3 Hz, C1′), 38.3 (s, C2), 20.1 (d, 1JC,P = 40.2 Hz, C1); 31P{1H} NMR (162 MHz, CD2Cl2) δ (ppm) = 31.4. HRMS (ESI(+), CH3CN): m/z calculated for [M − Cl]+: 802.886; found: 802.886.
O). 1H NMR (400 MHz, CDCl3) δ (ppm) = 9.64 (s, 2H, H3), 7.75–7.70 (m, 8H, H2′), 7.50–7.40 (m, 12H, H3′ and H4′), 2.72–2.70 (s, 8H, H1 and H2). 13C{1H} NMR (101 MHz, CDCl3) δ (ppm) = 199.8 (s, C3), 133.8 (m, C2′), 131.1 (s, C4′), 129.6 (m, C1′), 128.8 (m, C4′), 38.9 (s, C2), 18.6 (s, C1). 31P{1H} NMR (162 MHz, CDCl3) δ (ppm) = 16.3. HRMS (ESI(+), CH3CN): m/z calculated for [M − Cl]+: 625.054; found: 625.044.
O). 1H NMR (400 MHz, CDCl3) δ (ppm) = 9.52 (s, 2H, H3), 7.53–7.41 (m, 12H, C2′ and C4′), 7.32–7.25 (m, 8H, C3′), 2.93–2.87 (m, 4H, C2), 2.69–2.62 (m, 4H, C1). 13C{1H} NMR (101 MHz, CDCl3) δ (ppm) = 199.2 (m, C3), 133.3 (m, C2′), 131.5 (s, C4′), 128.7 (m, C1′ and C3′), 38.9 (s, C2), 23.2 (m, C1). 31P{1H} NMR (162 MHz, CDCl3) δ (ppm) = 7.5 (s + Pt satellites, 1JP,Pt = 3646 Hz).
General screening procedure for the Cu-free Sonogashira cross-coupling reaction
A mixture of bromobenzene (0.125 mmol, 1.0 equiv.), phenylacetylene (0.137 mmol, 1.1 equiv.), base (0.156 mmol, 1.25 equiv.), pre-catalyst (0.003 mmol Pd, 5 mol% Pd) and dry, degassed solvent (1.0 mL) was heated under nitrogen atmosphere for 4 h. The reaction process was monitored by GC-MS. The yields have been determined by GC-MS using naphthalene as internal standard (10 μL of the reaction solution have been diluted with 1.000 mL of a naphthalene stock solution in acetone).General screening procedure for the Suzuki–Miyaura cross-coupling reaction
A mixture of bromobenzene (0.125 mmol, 1.0 equiv.), p-tolylboronic acid (0.15 mmol, 1.5 equiv.), base (0.156 mmol, 1.25 equiv.), pre-catalyst (0.003 mmol, 5 mol%) and dry, degassed solvent (1.0 mL) were heated under nitrogen atmosphere for 5 h. The reaction process was monitored by GC-MS. The yields have been determined by GC-MS using naphthalene as internal standard (10 μL of the reaction solution have been diluted with 1.000 mL of a naphthalene stock solution in acetone).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Manuela Roßberg for the elemental analysis and Ramona Oehme for the measurement of the mass spectra. We also thank Aleksandr Kazimir for performing the DFT calculations. We appreciate the financial support from the Saxon Ministry of Science and the Fine Arts (doctoral grant for S. B.), the German-Israeli Foundation for Scientific Research and Development (GIF) and the Graduate School Building with Molecules and Nano-objects (BuildMoNa).References
- (a) J. Hagen, Industrial catalysis. A practical approach, Wiley-VCH, Weinheim, 2015 CrossRef; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (c) P. Kamer and P. W. N. M. van Leeuwen, Phosphorus(III) ligands in homogeneous catalysis. Design and synthesis, Wiley-Blackwell, Oxford, 2012 CrossRef; (d) M. Beller and H. U. Blaser, Organometallics as catalysts in the fine chemical industry, Springer, Heidelberg, 2012, vol. 42 CrossRef.
- (a) K. D. Hesp, R. McDonald and M. Stradiotto, Can. J. Chem., 2010, 88, 700–708 CrossRef CAS; (b) F. Kwong and A. Chan, Synlett, 2008, 1440–1448 CrossRef CAS; (c) J. Pietsch, P. Braunstein and Y. Chauvin, New J. Chem., 1998, 22, 467–472 RSC; (d) W.-H. Zhang, S. W. Chien and T. A. Hor, Coord. Chem. Rev., 2011, 255, 1991–2024 CrossRef CAS.
- F. Lorenzini, D. Moiseev, B. O. Patrick and B. R. James, Inorg. Chem., 2010, 49, 2111–2122 CrossRef CAS PubMed.
- K. Park, P. O. Lagaditis, A. J. Lough and R. H. Morris, Inorg. Chem., 2013, 52, 5448–5456 CrossRef CAS PubMed.
- M. A. Garralda, C. R. Chim., 2005, 8, 1413–1420 CrossRef CAS.
- G. D. Vaughn and J. A. Gladysz, J. Am. Chem. Soc., 1986, 108, 1473–1480 CrossRef CAS.
- G. P. Schiemenz and H. Kaack, Liebigs Ann. Chem., 1973, 1480–1493 CrossRef CAS.
- A. A. Mikhailine, P. O. Lagaditis, P. E. Sues, A. J. Lough and R. H. Morris, J. Organomet. Chem., 2010, 695, 1824–1830 CrossRef CAS.
- (a) C. Feng, S. Zhou, D. Wang, Y. Zhao, S. Liu, Z. Li and P. Braunstein, Organometallics, 2021, 40, 184–193 CrossRef CAS; (b) C. Li, K. Zhang, M. Zhang, W. Zhang and W. Zhao, Org. Lett., 2021, 23, 8766–8771 CrossRef CAS PubMed; (c) A. C. Matsheku, M. C. Maumela and B. C. Makhubela, Polyhedron, 2021, 205, 115280 CrossRef CAS; (d) N. L. Ngcobo, S. O. Akiri, A. O. Ogweno and S. O. Ojwach, Polyhedron, 2021, 203, 115243 CrossRef CAS; (e) W. Yang, I. Y. Chernyshov, R. K. A. van Schendel, M. Weber, C. Müller, G. A. Filonenko and E. A. Pidko, Nat. Commun., 2021, 12, 12 CrossRef CAS PubMed; (f) L. Zhang, L. Zhang, Q. Chen, L. Li, J. Jiang, H. Sun, C. Zhao, Y. Yang and C. Li, Org. Lett., 2022, 24, 415–419 CrossRef CAS PubMed.
- (a) A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed; (b) P. E. Sues, A. J. Lough and R. H. Morris, Organometallics, 2011, 30, 4418–4431 CrossRef CAS; (c) W. Zou, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1076–1080 CrossRef PubMed.
- R. Clauss, S. Baweja, D. Gelman and E. Hey-Hawkins, Dalton Trans., 2022, 51, 1344–1356 RSC.
- (a) D. J. Hutchinson, R. Clauss, M.-B. Sárosi and E. Hey-Hawkins, Dalton Trans., 2018, 47, 1053–1061 RSC; (b) R. Clauss and E. Hey-Hawkins, Dalton Trans., 2022, 51, 9632–9641 RSC; (c) R. Clauss, A. Kazimir, A. Straube and E. Hey-Hawkins, Inorg. Chem., 2021, 60, 8722–8733 CrossRef CAS PubMed.
- (a) Q. Dai, L. Liu, Y. Qian, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20645–20650 CrossRef CAS PubMed; (b) H. Luo, Y. Yu and S. Ma, Org. Chem. Front., 2016, 3, 1705–1710 RSC.
- (a) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed; (b) P. Usha Rani, P. Muralidhar Reddy, K. Shanker and V. Ravinder, Transition Met. Chem., 2008, 33, 153–160 CrossRef CAS.
- (a) P. Crochet, M. A. Fernández-Zumel, C. Beauquis and J. Gimeno, Inorg. Chim. Acta, 2003, 356, 114–120 CrossRef CAS; (b) P. M. Reddy, K. Shanker, R. Rohini, M. Sarangapani and V. Ravinder, Spectrochim. Acta, Part A, 2008, 70, 1231–1237 CrossRef PubMed.
- V. M. Chernyshev, A. V. Astakhov, I. E. Chikunov, R. V. Tyurin, D. B. Eremin, G. S. Ranny, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2019, 9, 2984–2995 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS.
- J. E. Hamlin, K. Hirai, A. Millan and P. M. Maitlis, J. Mol. Catal. A: Chem., 1980, 7, 534–544 Search PubMed.
- S. De, M. V. Babak, M. J. Hülsey, W. H. Ang and N. Yan, Chem. – Asian J., 2018, 13, 1053–1059 CrossRef CAS PubMed.
- (a) S. O. Grim and R. L. Keiter, Inorg. Chim. Acta, 1970, 4, 56–60 CrossRef CAS; (b) T. Bartik and T. Himmler, J. Organomet. Chem., 1985, 293, 343–351 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- CrysAlisPro data reduction software package, Rigaku Oxford Diffraction, Oxford, UK Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Brandenburg, DIAMOND 4, - Crystal and Molecular Structure Visualization, Crystal Impact GbR, Bonn, Germany Search PubMed.
Footnotes |
| † Dedicated to Professor Wolfgang Weigand on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2233898–2233900 and 2240480. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00507k |
| This journal is © The Royal Society of Chemistry 2023 |