
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic reduction of CO2 to CO by a series of organometallic Re(I)-tpy complexes†
Shriya
Saha
 a,
Thomas
Doughty
b,
Dibyendu
Banerjee
c,
Sunil K.
Patel
a,
Dibyendu
Mallick
*c,
E. Siva Subramaniam
Iyer
a,
Souvik
Roy
*b and
Raja
Mitra
*a
a,
Thomas
Doughty
b,
Dibyendu
Banerjee
c,
Sunil K.
Patel
a,
Dibyendu
Mallick
*c,
E. Siva Subramaniam
Iyer
a,
Souvik
Roy
*b and
Raja
Mitra
*a
aSchool of Chemical and Materials Sciences, Indian Institute of Technology Goa, Farmagudi, Goa 403401, India. E-mail: rajamitra@iitgoa.ac.in
bSchool of Chemistry, University of Lincoln, Green Lane, Lincoln, Lincolnshire LN6 7DL, UK. E-mail: sroy@lincoln.ac.uk
cDepartment of Chemistry, Presidency University, Kolkata 700073, India. E-mail: dibyendu.chem@presiuniv.ac.in
First published on 3rd May 2023
Abstract
A series of organometallic Re(I)(L)(CO)3Br complexes with 4′-substituted terpyridine ligands (L) has been synthesised as electrocatalysts for CO2 reduction. The complexes’ spectroscopic characterisation and computationally optimised geometry demonstrate a facial geometry around Re(I) with three cis COs and the terpyridine ligand coordinating in a bidentate mode. The effect of substitution on the 4′-position of terpyridine (Re1–5) on CO2 electroreduction was investigated and compared with a known Lehn-type catalyst, Re(I)(bpy)(CO)3Br (Re7). All complexes catalyse CO evolution in homogeneous organic media at moderate overpotentials (0.75–0.95 V) with faradaic yields of 62–98%. The electrochemical catalytic activity was further evaluated in the presence of three Brønsted acids to demonstrate the influence of the pKa of the proton sources. The TDDFT and ultrafast transient absorption spectroscopy (TAS) studies showed combined charge transfer bands of ILCT and MLCT. Amongst the series, the Re-complex containing a ferrocenyl-substituted terpyridine ligand (Re5) shows an additional intra-ligand charge transfer band and was probed using UV-Vis spectroelectrochemistry.
Introduction
Carbon dioxide (CO2), one of the planet's most essential carbon sources, is also one of the key greenhouse gases contributing to global warming.1 Despite the existence of a natural carbon cycle that maintains the CO2 level in the atmosphere, the addition of an uncontrollable amount of anthropogenic CO2 is tilting the balance of CO2 in the Earth's atmosphere slowly but steadily in the wrong direction.2–5 The recent decline of CO2 emissions during the global pandemic in 2020![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 suggests a need to either find sustainable alternatives to fossil fuel (e.g. green H2) or upcycle the CO2 to fuel and feedstock to realise a carbon-neutral cycle. Without readily available alternative fuel sources, the dependence on fossil fuels cannot be eliminated drastically.6 Considering the current scenario, electroreduction presents a promising route to CO2 recycling for the production of value-added chemicals (CO, HCOO−) or carbon-neutral fuels (CH4, CH3OH).7–10
3 suggests a need to either find sustainable alternatives to fossil fuel (e.g. green H2) or upcycle the CO2 to fuel and feedstock to realise a carbon-neutral cycle. Without readily available alternative fuel sources, the dependence on fossil fuels cannot be eliminated drastically.6 Considering the current scenario, electroreduction presents a promising route to CO2 recycling for the production of value-added chemicals (CO, HCOO−) or carbon-neutral fuels (CH4, CH3OH).7–10
Nevertheless, the reduction of CO2 is associated with a high activation energy barrier due to the highly stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. One of the most prominent ways to overcome the high activation barrier is by an electrochemical reduction in the presence of a catalyst.11–16 In natural enzymatic systems, such as CO-dehydrogenase (CODH), CO2 activation is facilitated by hydrogen bonding interactions. This has inspired the molecular design of synthetic CO2R catalysts that involves tuning both the primary and secondary coordination spheres to modulate the electronic properties and harness intramolecular through-space interactions for improved activity. The tuneability of the ligand frameworks in transition metal complexes allows the introduction of tailored functional groups in the outer sphere that can promote selective CO2 reduction. Recently, a wide range of CO2R catalysts containing secondary sphere functionalities has been reported, such as cobalt–phosphine complexes with pendant amines,17 iron porphyrins with pendant phenols,18 phthalocyanines with trimethylammonium groups,19 and cobalt aminopyridine macrocycles.20
O bonds. One of the most prominent ways to overcome the high activation barrier is by an electrochemical reduction in the presence of a catalyst.11–16 In natural enzymatic systems, such as CO-dehydrogenase (CODH), CO2 activation is facilitated by hydrogen bonding interactions. This has inspired the molecular design of synthetic CO2R catalysts that involves tuning both the primary and secondary coordination spheres to modulate the electronic properties and harness intramolecular through-space interactions for improved activity. The tuneability of the ligand frameworks in transition metal complexes allows the introduction of tailored functional groups in the outer sphere that can promote selective CO2 reduction. Recently, a wide range of CO2R catalysts containing secondary sphere functionalities has been reported, such as cobalt–phosphine complexes with pendant amines,17 iron porphyrins with pendant phenols,18 phthalocyanines with trimethylammonium groups,19 and cobalt aminopyridine macrocycles.20
Since the discovery of the CO2R activity of fac-Re(I)(bpy)(CO)3X (X = Cl/Br) complexes by Lehn and coworkers, a range of Re(I)- and Mn(I)-carbonyl complexes with bidentate diamine N–N ligands have been synthesised and studied for electrochemical CO2-to-CO conversion.21–28 Among these molecular catalysts, 2,2′-bipyridine (bpy) and 1,10-phenanthroline (phen) remain the most widely used ligands.22,29–35 In particular, Re(bpy)(CO)3Cl has been demonstrated to display good selectivity towards CO evolution under electrochemical and light-driven conditions but with low catalytic rate constants and deactivation via dimerisation. To improve the performance of Re- and Mn-diimine systems, the direct electronic effect has been investigated by modifying the diimine ligand in the primary coordination sphere to introduce electron-donating or electron-withdrawing substituents.36–40 The outer-sphere interactions have also been exploited for Re- and Mn-diimine systems.39 Recent work on Mn and Re catalysts with bipyridine ligand scaffolds decorated with primary amines, hydroxyls, pendant imidazole and a thiourea tether shows a significant influence of the donor groups on CO2-to-CO activity.33,41–47
We envisioned that using substituted terpyridines (tpy) as the ligand offers a handle to tune the primary and secondary coordination structures in tandem. Terpyridine can coordinate to a [Re(CO)3X] unit in a robust bidentate fashion, leaving a pendant pyridine group near the active site.48 During catalysis, this pendant pyridine can facilitate CO2 activation in the protonated form, while the substituents on the terpyridine ligand can impact the electronics of the metal centre. A few Re(tpy)(CO)3X (X = Cl/Br) derivatives have been reported in the literature, but those have rarely been investigated for electrocatalytic CO2 reduction, despite their structural similarity to Lehn-type Re(bpy)(CO)3X catalysts.49–51 Structural characterisation of these complexes typically shows a facial arrangement of a bidentate tpy ligand and three COs.52,53 The uncoordinated pyridyl ring is forced to stay non-coplanar with the other two coordinated pyridine rings due to the sterically crowded Re(I) centre. Re(tpy)(CO)3X compounds show photochemical properties such as UV emitter, fluorescence, and NIR-emitter.54–58
Herein, we report the synthesis and electrocatalytic properties of a series of fac-Re(I)(L)(CO)3Br complexes containing 4′-substituted-2,2′:6′,2′′-terpyridine ligands (L). Five Re-complexes (Re1–5) containing 4′-substituents with varying electronic properties were synthesised, and their structures are shown in Fig. 1. The electrochemical CO2R activity of Re1–5 was compared with the well-known Lehn's catalyst, fac-Re(bpy)(CO)3Br (Re7). This study demonstrates the effect of a pendant pyridine ring and a range of 4′-substituents on the tpy ligands on CO2 reduction. A ferrocenyl-appended Re(I) compound (Re5) was also synthesised and studied using electrochemical techniques to understand the effect of the ferrocenyl moiety as multiple metal centres are known to alter the electrochemical and catalytic activities. This study explores the usefulness of having additional metal centres attached to terpyridine ligands at the 4′ position for the CO2R studies. The electrocatalytic investigations are complemented with computational studies, transient absorption spectroscopy (TAS) and spectroelectrochemistry to elucidate the mechanistic aspect of the electrochemical processes.
 | ||
| Fig. 1 Molecular structure of Re(I) complexes used as a catalyst in this study. Re7 is used as a standard for comparison. | ||
Results and discussion
Synthesis and characterisation
All ligands with a substitution (aryl or ferrocenyl) on the 4′ position of 2,2′:6′,2′′-terpyridine were synthesised using the modified Krönke pyridine synthesis that was reported by Hanan and co-workers (L2–5).59 Improved yields were obtained by refluxing the mixture for 12 h and using excess ammonia (50 instead of 2.5 equivalent). In most cases, the ligands precipitate out from the clear orange-coloured solution during the reaction due to the low solubility of the products (L2–4) in ethanol. Interestingly, the ligand L5 was obtained with a lower yield due to being more soluble in ethanol than other ligands. The yield of L5 was significantly improved by cooling the reaction mixture at 4 °C for 24 h. However, cooling had a minimal effect on the yield for other ligands (L2–4). The analytical data for precipitated ligands showed sufficient purity for further reaction.Rhenium(I) complexes were synthesised by refluxing Re(CO)5Br and ligands in dry toluene for 1–4 h following literature procedures (Scheme 1). All complexes, except Re5, precipitated as microcrystalline solids during the reaction or upon cooling to room temperature, with a yield of 56–98% (Re1–4). Compound Re5 was isolated in 74% yield that includes ∼0.5 molecules of toluene as a solvent of crystallisation (vide infra). The toluene was consistently present in different batches of synthesis. Complexes were characterised by infrared spectroscopy (IR), 1H NMR, mass spectroscopy, UV-Vis spectroscopy and elemental analysis (data can be found in ESI, Fig. S1–S9†).
 | ||
| Scheme 1 Synthesis of terpyridine ligands (L2–5) and fac-Re(I)(L)(CO)3Br complexes (Re1–5), where L = L1–5. *Re5 contains toluene (∼0.5 molecules, calculated from 1H NMR) as a solvent of crystallisation. | ||
The 1H NMR spectra in CDCl3 of the rhenium complexes show the deshielding of the ligand's pyridine protons, suggesting the coordination of terpyridine ligands to the Re(I) centre. The 1H NMR of the ligands shows a symmetric pattern due to the inherent plane of symmetry present on them, which is not observed in the proton NMR of the metal complexes, indicating that the metal has coordinated with the ligand in a κ2-NN coordination mode.60,61 Because of the κ2-NN coordination, one pyridine ring is available for further secondary interactions. The 13C peaks for CO ligands were not observed in the CDCl3 solvent for all Re complexes. Interestingly, the 13C peak corresponding to CO ligands was observed in DMSO-d6 solvent; hence, NMR was reported in both solvents. 1H NMR of Re5 showed three additional peaks at 7.24, 7.17 and 2.30 ppm in DMSO-d6, which upon analysis were found to be of toluene, confirmed by 2D NMR (see ESI†) and reported literature values.62 From 1H NMR, it was found out ∼0.5 molecule of toluene is present per molecule of the complex and it is present as a solvent of crystallisation.
IR comparison studies are carried out to probe the κ2-NN coordination further (Fig. 2). The presence of three carbonyl stretches confirms κ2-NN coordination and is consistent with the cis arrangement of the CO ligands, as reported in the recent literature.63 The starting material Re(CO)5Br shows two IR active νstr(CO) bands, which are assigned A1 (2017 cm−1) for the CO present in the axial position and a doubly degenerate E (1942 cm−1) for the other four CO in the equatorial position.64 With the coordination of the terpyridine unit to the Re(I) centre, three carbonyl frequencies are observed that are charcteristic of facial tricarbonyl complexes. The higher energy band, A1, does not shift significantly for Re1–5 (cf. 2017 cm−1 to 2013–2021 cm−1). The lower energy degenerate E band splits into two bands due to the presence of κ2-NN from the terpyridine derivatives (Fig. S10†).30 The shift in νstr(CO) frequency, particularly for the E band, for Re1–5 compared with the Re(CO)5Br can be attributed to the better back-donation from the Re(I) metal centre to the π* orbital of CO due to the increase in electron density on the metal centre. NMR and FTIR studies suggest that the complexes most probably stay in a distorted octahedral geometry, and all CO are cis in nature; hence all complexes are facial isomers.
 | ||
| Fig. 2 Representative partial ATR-IR (2100–1700 cm−1) comparison of the starting precursor, Re(CO)5Br, and the 4′-phenyl-2,2′:6′,2′′-terpyridine ligand-coordinated Re(I) complex, Re2. The complete range of ATR-IR spectra can be found in the ESI.† | ||
The UV-Vis spectra of ligands and metal complexes were recorded in organic solvents (DMF and acetonitrile, Table 1). The ligands and the Re(I) complexes showed similar absorption spectra in DMF and acetonitrile. The spectral features are typical of the rhenium complexes and the terpyridine ligands reported earlier. The higher energy transition band from 250 nm to 350 nm is assigned to π → π* transition in the terpyridine ligand. A bathochromic shift ∼30 nm is observed for Re complexes compared with the pure ligands that was ascribed to N^N ligand-centred π → π* transitions (307–388 nm in acetonitrile) (Table 1).53,65,66
| Ligands λmax /nm (103ε/L mol−1 cm−1) | Prediction from TDDFT calculation λmax/nm (oscillator strength) | Re(I) complexes λmax/nm (103ε/L mol−1 cm−1) | Prediction from TDDFT calculation λmax/nm (oscillator strength) | ||
|---|---|---|---|---|---|
| In DMF | In acetonitrile | In acetonitrile | In DMF | In acetonitrile | In acetonitrile |
| L1: | L1: | L1: | Re1: | Re1: | Re1: |
| 279 (37.8) | 235 (28.7) | 235 (0.412) | 309 (20.9) | 247 (21.5) | 249 (0.180) |
| 329 (4.36) | 277 (33.1) | 283 (0.323) | 375 (5.58) | 310 (15.5) | 256 (0.146) |
| 372 (2.80) | 303 (0.393) | ||||
| 431 (0.056) | |||||
| L2: | L2: | L2: | Re2: | Re2: | Re2: |
| 309 (25.3) | 252 (23.0) | 229 (0.274) | 384 (9.27) | 266 (47.6) | 281 (0.355) |
| 275 (22.5) | 264 (0.312) | 380 (7.08) | 309 (0.287) | ||
| 329 (1.8) | 438 (0.088) | ||||
| L3: | L3: | L3: | Re3: | Re3: | Re3: |
| 278 (25.3) | 253 (28.0) | 228 (0.33) | 307 (23.9) | 268 (28.6) | 287 (0.328) |
| 316 (10.2) | 278 (31.1) | 261 (0.21) | 382 (4.30) | 301 (29.3) | 309 (0.227) |
| 332 (1.5) | 274 (0.31) | 379 (4.91) | 436 (0.098) | ||
| 285 (0.53) | |||||
| L4: | L4: | L4: | Re4: | Re4: | Re4: |
| 275 (32.3) | 253 (28.1) | 231 (0.28) | 305 (32.6) | 269 (53.7) | 285 (0.437) |
| 312 (7.36) | 276 (30.8) | 262 (0.25) | 388 (5.84) | 297 (54.4) | 310 (0.211) |
| 330 (2.5) | 282 (0.58) | 378 (8.50) | 441 (0.095) | ||
| L5: | L5: | L5: | Re5: | Re5: | Re5: |
| 283 (17.9) | 250 (17.1) | 267 (0.174) | 311 (42.1) | 256 (23.4) | 282 (0.260) |
| 363 (1.29) | 280 (17.8) | 282 (0.318) | 370 (10.6) | 310 (21.8) | 309 (0.197) |
| 459 (0.56) | 365 (0.83) | 380 (0.015) | 516 (5.62) | 367 (5.02) | 438 (0.098) |
| 460 (0.26) | 506 (0.002) | 516 (2.35) | 516 (0.008) | ||
| 602 (0.013) | |||||
The Re5 complex shows a broad absorption band at λmax around 516 nm that extends up to 650 nm, which might have originated due to the presence of the ferrocenyl moiety. Compared with L5 (λmax = 460 nm) and free ferrocene (λmax = 440 nm), the bands for Re5 are substantially redshifted, which can be attributed to the extended conjugation of the ferrocene unit. The oscillator strength obtained from TDDFT studies agrees with the molar absorption coefficient (Table 1). TDDFT and ultrafast transient absorption spectroscopy studies were carried out to understand the absorption properties further assigned to the metal-to-ligand charge transfer (MLCT) and intra-ligand charge transfer (ILCT) processes within the metal complexes. Compounds Re1–5 showed no significant changes in the UV-Vis spectra even after 24 h, suggesting the stability of those compounds in acetonitrile (Fig. S11†).
The crystal obtained for Re5 is always associated with clusters or overlapping crystals unsuitable for single-crystal X-ray diffraction. A computational geometry-optimised structure for all was obtained using DFT calculation (vide infra) to understand the structural aspect of those Re(I) compounds. One of the representative DFT-optimised structures is shown in Fig. 3. It shows that the Re(I) is in an octahedral environment with terpyridine serving as a bidentate ligand, and all CO ligands are coordinated in a cis orientation.
 | ||
| Fig. 3 The DFT optimised structure of Re5 at the B3LYP/def2-TZVP level of theory. | ||
TDDFT studies
TDDFT calculations have been performed to investigate the photochemical properties of the complexes Re1–5. The major optical transitions above 250 nm with an oscillator strength f > 0.005 are tabulated in Fig. S12–S16.† The peak around 431 nm in Re1 originates due to the transition from HOMO−1, which is composed of 5dyz (Re) and 4py(Br) orbitals, to LUMO, i.e., the π*(tpy) orbital. Therefore, the band around 431 nm could be attributed to a metal-to-ligand charge transfer (MLCT) band. The MLCT band is slightly red-shifted (436–441 nm) for the complexes Re2, Re3 and Re4. Interestingly, for Re5, the MLCT band was observed at 438 nm, which predominantly arises due to the transitions from HOMO and HOMO−1 to LUMO, which is the π*(fctpy) orbital. The HOMO of Re5 is composed of the 3dxy (Fe) orbital, and the HOMO−1 is similar to the HOMO−1 of other rhenium complexes (Re1–4). Hence, the peak around 438 nm for Re5 can be characterised as a combination of intra-ligand charge transfer (ILCT) and MLCT transitions. The absence of the band around 438 nm for the L5 ligand further suggests the MLCT nature of this peak for the Re5 complex. A highly intense band is observed around the 303–309 nm region for all these complexes (Re1–5), which arises due to a combination of π(R-tpy) to π*(R-tpy) and πyz(Re–Br) to π*(R-tpy) transitions and hence can be assigned to a mixed ILCT and MLCT band. The most intense band is observed around 280 nm for all the complexes except Re1, which has the most intense band around 250 nm. The appearance of the 280 nm band for complexes Re2–5 is due to the transition from an orbital composed of 4py(Br) and π(R-tpy) to the π*(R-tpy) orbital. A similar transition is responsible for the 250 nm band in the Re1 complex. Two additional low-intensity bands observed around 516 nm and 602 nm regions for the Re5 complex arise due to d–d transitions from the ferrocene moiety of the fctpy ligand (L5). These bands are also found at 506 nm and 587 nm for the L5 ligand.Ultrafast transient absorption spectroscopy
The femtosecond transient absorption spectra resulting from the 320 nm (3.8 eV) pump and whitelight (350 nm to 650 nm) probe of Re1 and Re5 are shown in Fig. 4, while those of Re2 to Re4 are shown in Fig. S18–S20.† All measurements are performed in acetonitrile. The transients of Re1 to Re4 are dominated by excited state absorption (ESA) signals in the entire spectral range measured. The TA spectra of Re1 to Re4 reveal two distinct bands. One is a sharp band at <400 nm and a broad spectrum beyond 400 nm. The ESA at lower wavelengths exhibits a sharp peak at 375 nm for Re1 and around 365 nm for Re2, Re3, and Re4. This band is ascribed to the excitation to a higher energy state of the radical anion state of the terpyridine ligand resulting from charge transfer from the metal centre to the ligand. This assignment is reasonable as earlier reports on analogous molecules have shown similar features; e.g. the transient absorption of bipyridine (bpy) rhenium(I) analogue [Re(bpy)(CO)3Cl] also shows an ESA at 373 nm, which has been assigned to bpy˙−.65,67,68 A similar ESA in the sub-400 nm [Re(R-tpy-κ2N)(CO)3Cl] range is assigned to the ligand where the charge is concentrated over the bipyridine part of the ligand.65 The broad ESA in the mid-visible region (450 nm–600 nm) is assigned to the excitation of the MLCT state to higher states.69 A broad excited state absorption band from 450 nm to 600 nm in [Re(R-tpy-κ2N)(CO)3Cl] has been assigned to the lowest triplet MLCT state.67,68,70,71 | ||
| Fig. 4 2D contour plots of the transient absorption spectra of Re1 (a) and Re5 (b) measured in acetonitrile after 320 nm excitation and the corresponding globally fitted lifetime data in (c) and (d). | ||
The TA of Re5 has significantly different characteristics and warrants further investigation compared with the Re1 to Re4 complexes. Although the high-energy ESA is observed in the rest of the molecules studied in this work, this band is significantly red-shifted. This can be ascribed to the enhanced conjugation of the terpyridine ligand with ferrocene. Based on earlier work and the TDDFT calculations, the high energy band is assigned to a π → π* transition. The ferrocene is attached to the terpyridyl group directly through the cyclopentadienyl groups. Hence the red-shift of this band due to extended conjugation is trivial. However, unlike Re1 to Re4, where the ESAs of the MLCT states are observed, in Re5, this spectral region is dominated by two distinct negative signals (stimulated emission, SE). This is clearly due to the presence of the ferrocene moiety.72,73 It must be emphasised that the stimulated emission results only in the Re complex. In the presence of the ferrocene moiety in Re5, the MOs of the Re moiety, along with the 3d orbitals of the Fe, contribute to the MOs (Fig. S16†). This might lead to additional states in the complex and manifests as seen from the evolution of new negative bands centred at 590 nm in the transient spectra of the Re5 complex. Such a state is missing in the L5 compound (Fig. S21†). This new state might stabilise the 3CT state, which exhibits stimulated emission at a longer delay.
Interestingly, one of the negative bands develops only after 10 ps of the excitation by the pump pulse. At the pump wavelength of 320 nm, the direct excitation of ferrocene is inevitable. The initial negative signal centred at 525 nm closely matches the charge transfer band of lone ferrocene. It remains to be ascertained whether this band is a bleach signal or a stimulated emission from the ferrocene moiety. It is noteworthy that when the TA measurements are performed on ligand L5, no negative band is observed at these wavelengths. Had it been a ground-state bleach signal from the ferrocenyl group, such a state would have been observed in L5 as well (Fig. S21†). The computational calculations described in the earlier sections reveal the mixing of various orbitals of the two metal ions present in the molecule, which alter the MOs. Such mixing can result in the stabilisation of certain energy levels that are otherwise short-lived. In this experiment, the molecules are excited to energy states higher than the lowest energy transitions. Such a state can populate the CT state arising from ferrocene. This state is depopulated by the whitelight probe and appears as stimulated emission in the TA spectra of Re5, shown in Fig. 4. The emission band at 525 nm is also due to the presence of Re in the Re5 because it is not arising in the transient absorption of the ligand (L5) (Fig. S21†).
Unlike Re1 to Re4, the ILCT band is significantly redshifted in Re5 and can be attributed to enhanced conjugation resulting in a lowering of the energy gaps among the excited states. The ESA band in Re5 consequently has a peak at 415 nm. The most interesting feature is the appearance of a strong stimulated emission centred at 590 nm. This band evolves only beyond 10 ps post excitation. This shows that a new band is developed from the locally excited states and could not be formed from the direct excitation of any ground states. If this new band were the result of direct excitation, the signal would have evolved much earlier. The TDDFT investigations suggest a mixed nature of ILCT and MLCT transitions in Re5.
The wavelength-dependent kinetic traces were analysed to understand the excited states’ evolution, and global analyses were performed to obtain the decay-associated spectra. The global lifetime analysis spectra for Re1 and Re5 are given in Fig. 4(c and d), and the other compounds are in Fig. S18–S20.† The time constants at a few representative wavelengths are tabulated in Fig. S17–S20.† The evolution-associated decay spectra show that three main components characterised by distinct time constants can explain the excited state dynamics in these compounds. The TA features of Re1–4 are similar to the earlier reports on the structurally analogous [Re(C6H5-tpy-κ2N)(CO)3Cl].74
The various excited state processes are pictorially represented in a Jablonski diagram in Fig. 5. Upon 320 nm excitation, the molecules are excited to a 1MLCT state and then undergo intersystem crossing forming a hot triplet state within <200 fs. This evolution is missed in our experiments due to the instrument response. This nascent triplet state relaxes by internal vibrational relaxation, eventually forming a relaxed triplet 3MLCT state within a few picoseconds. This process takes place in ∼30 ± 1 ps for Re1, whereas for Re2–4, it is slower. The longer relaxation time for Re2–4 can be attributed to an enhanced conjugation upon substitution. The 3MLCT returns to the ground state by non-radiative pathways over the next few ns. These states appear as long-lived components in the global spectra (Fig. 4(c)).
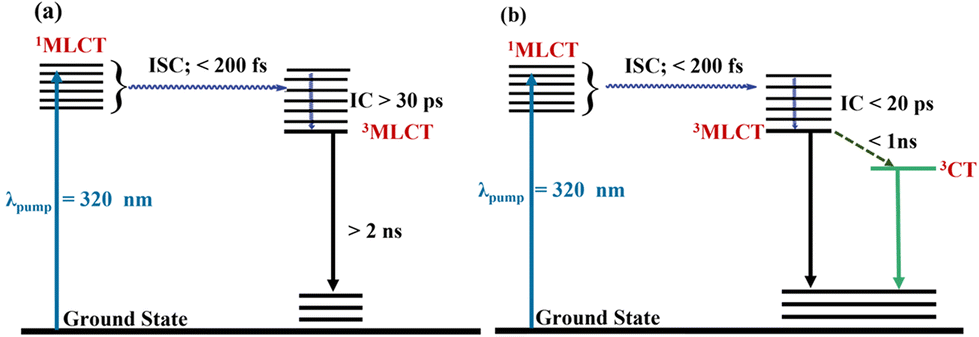 | ||
| Fig. 5 Proposed deactivation mechanism of excited state Re-complexes (Re1 to Re4) in (a) and Re5 in (b). | ||
In Re5, the TA measurements indicate that alternative pathways of de-excitation of the triplet state open up. We have observed an additional component appearing as a stimulated emission in Re5, as shown in Fig. 4(d). This additional state is formed possibly due to extensive long-range coupling between the orbitals of ferrocene and rhenium in the Re5 complex (Fig. S20†). This contention is bolstered by the theoretical investigations where the mixing of orbitals of the states involved in electronic transitions is observed. In addition to the fast internal conversion in <200 fs, a significantly fast ∼17 ± 1 ps process is observed in Re5. The shorter lifetime of the vibrationally relaxed state can be associated with the additional process leading to the formation of the mixed state. This new state is evolved in the presence of a ferrocenyl unit, which is long-lived and can alter the photo- and electrocatalytic activities of the rhenium complexes.75
Electrochemical properties
To assess the redox property of the complexes under noncatalytic conditions, cyclic voltammetry (CV) was performed in N2-saturated acetonitrile solutions containing a ∼0.6 mM Re-complex (0.5 mM for Re3 due to its lower solubility) and 0.1 M n-Bu4NBF4 supporting electrolyte at different scan rates (Fig. 6 and Fig. S22–S26†). All reported potentials are referenced against the ferrocene (Fc+/0) couple. The reduction potentials and diffusion coefficients (derived from scan rate-dependent CVs, Fig. S27†) for the complexes are summarised in Table 2. | ||
| Fig. 6 Cyclic voltammograms of Re1–5 and Re7 recorded in acetonitrile under N2 at scan rates (A) 10 V s−1 and (B) 0.1 V s−1 (supporting electrolyte 0.1 M n-Bu4BF4). Re7 is shown for comparison. | ||
| Complex | E (1)1/2 (V)a | E (2)p (V)b | D (cm2 s−1) |
|---|---|---|---|
| a Mid-point potential for the 1st quasi-reversible reduction. b Peak potential for the irreversible 2nd reduction. | |||
| Re1 | −1.75 | −2.13 | 9.87 × 10−6 |
| Re2 | −1.70 | −2.04 | 6.64 × 10−6 |
| Re3 | −1.70 | −2.04 | 5.61 × 10−6 |
| Re4 | −1.69 | −2.03 | 5.00 × 10−6 |
| Re5 | −1.77 | −2.13 | 2.67 × 10−6 |
| Re7 | −1.73 | −2.15 | 6.40 × 10−6 |
Under fast scan rate conditions (10 V s−1, Fig. 6A), all Re complexes display two reduction waves: the first reduction is quasi-reversible, and the second is irreversible. The reduction potentials for the two processes at 10 V s−1 are listed in Table 2 (E1/2 for the 1st reduction and peak potential (Ep) for the irreversible 2nd reduction). Re2–4 exhibit the 1st reduction feature at ∼−1.70 V and the 2nd irreversible reduction feature at ∼−2.04 V. Re1 and Re5 display the first reduction feature at −1.75 V and −1.77 V, respectively, and the second reduction feature at −2.13 V. The slight anodic shift of the reduction potentials for Re2–4 is rationalised by the electron-withdrawing nature of the phenyl substituent at the 4′-position of the tpy ligand. The more negative reduction potentials for Re5 can be attributed to the electron donation from the ferrocene moiety (vide infra). Based on previous reports on Re(bpy)(CO)3X-type complexes, the first reduction is attributed to a ligand (terpyridine)-centred process to form an anionic radical species, [ReI(tpy˙−)(CO)3Br]−, that can undergo slow Br− loss.30,76 The second event is assigned to a metal-centred reduction to Re0, leading to a rapid loss of Br− and the formation of a 5-coordinate species, [Re0(tpy˙−)(CO)3]−. This is consistent with the additional oxidation peak observed in the potential range −1.50 to −1.55 V during the return scan, which can be attributed to the oxidation of pentacoordinate species. Moreover, reversing the potential scan before the second reduction still resulted in the second reoxidation peak, which is in line with the quasi-reversibility of the first reduction process at scan rates >1 V s−1 due to the slow Br− loss from [ReI(tpy˙−)(CO)3Br]− (Fig. S28†).
When the voltammograms were recorded at slower scan rates (0.1 V s−1, Fig. 6B), a third irreversible reduction wave was observed for Re1–5 at a more cathodic potential (∼−2.15 V), suggesting the formation of new species on a longer time scale. This peak is more pronounced for Re2–4 than Re1 and Re5. We speculate that this reduction peak at <−2.15 V can be caused by Re–Re dimerisation, which has been reported previously in analogous bipyridine catalysts.77 This is further supported by the small oxidation wave observed at ∼−0.4 V during the return scan, which is assigned to the oxidation of a Re0–Re0 dimeric species.78,79 The additional reduction and oxidation features disappear at a faster scan rate (>2 V s−1; Fig. S28†), suggesting that the dimerisation step proceeds too slowly to be observed on the CV timescale. Interestingly, the first two reductions are well-separated for Re2 and Re3, whereas those two peaks coalesced to a broad wave for Re4 at 100 mV s−1. For Re5, oxidation of the ferrocene unit appended to the terpyridine framework is observed at ∼0.24 V, which indicates that the Fe-centre is significantly electron deficient compared with unmodified ferrocene, consistent with the more cathodic reduction potentials of Re5. Careful inspection of the oxidation wave suggests the presence of an overlapping irreversible peak which can be tentatively attributed to ReI/II oxidation.
The CVs of Re1–5 and Re7 at varying scan rates show that the reductive peak currents (ip) vary linearly versus the square root of the scan rate (Fig. S27†), consistent with diffusion-controlled homogeneous systems. The diffusion coefficients (D) for the Re1–5 are comparable to that determined for Re7 and those reported for analogous Re-complexes.41 The decrease in D values within the Re1–5 series is approximately in line with the steric bulk of the terpyridine ligand.
Electrocatalytic CO2 reduction
Following the studies under N2, cyclic voltammetry was performed in a CO2-saturated electrolyte to evaluate the catalytic activity of Re1–5 relative to Re7 for CO2 reduction. All complexes display current enhancement under CO2 at either the same or a more negative potential than their second reduction features, demonstrating electrocatalytic CO2 reduction. The catalytic response of Re2 under CO2-saturated conditions at different scan rates is shown in Fig. 7. Catalytic voltammograms for Re1, Re3–5 and Re7 are included in the ESI (Fig. S29 and S30†). As shown in Fig. 7, the first reduction process remains largely unaffected by the presence of CO2 and its quasi-reversibility is maintained at the scan rates investigated here (25–0.1 V s−1) (Fig. S30†). This is consistent with the hypothesis that the singly reduced Re species undergoes slow Br− loss to form the 5-coordinate species (too slow for the CV timescale), which is necessary for CO2 binding and activation at the Re centre. Interestingly, the 2nd oxidation peak at ∼−1.55 V during the return scan disappears at scan rates ≤2 V s−1, indicating that the doubly reduced species is consumed via a reaction with CO2 at slower scan rates (Fig. 7B). Higher scan rates (>2 V s−1) do not allow sufficient time for catalyst turnover, which provides a qualitative measure of the timescale of turnover frequency for Re2. Under faster scans (10 V s−1, Fig. 7C), Re2 exhibits a relatively modest current enhancement at the 2nd reduction feature. Similar responses are observed for the other Re catalysts under CO2 at different scan rates (Fig. S30†). | ||
| Fig. 7 Cyclic voltammograms of Re2 recorded in acetonitrile under N2 and CO2 at different scan rates: (A) 0.1 V s−1, (B) 2 V s−1 and (C) 10 V s−1 (supporting electrolyte 0.1 M n-Bu4BF4). | ||
The catalytic voltammograms under CO2 and N2 were analysed to compare the activity of the Re complexes. The ratio of the peak current in the presence of CO2 (icat) and the corresponding reductive peak in the absence of a substrate (ip) serves as a useful measure to assess and compare the activity of different Re complexes.80 These icat/ip ratios at a 0.1 V s−1 scan rate and the corresponding half-wave potentials for electrocatalytic CO2 reduction by the Re complexes are summarised in Table 3. The catalytic currents (icat) were determined from the plateau shape of the catalytic response in the CVs at strongly reducing potentials (<−2.15 V) (Fig. S31†). Re1–5 and Re7 display icat/ip ratios in the range of 7.1–9.6, which suggests that the rate of CO2 electroreduction is similar for these complexes, with Re4 exhibiting the largest enhancement and Re2 exhibiting the lowest. It should be noted that the icat/ip value only serves as a proxy for the catalytic rate, and there is a degree of error inherent in the values reported here due to the nature of overlapping catalytic waves at different potentials. During catalysis in the pure kinetic regime (CO2 concentration at the electrode surface is equal to the bulk concentration), the normalised peak catalytic current (icat/ip) can be used to derive the pseudo-first-order catalytic rate constant (kobs).80,81 However, the prerequisites for applying this method are the scan-rate independent catalytic current plateau and ‘S-shaped’ catalytic waves, which are not observed for the Re complexes investigated here. The catalytic currents for all complexes display a scan-rate dependence up to 25 V s−1, precluding the derivation of kobs from icat/ip. An alternative method to estimate kobs from CVs without ‘S-shaped’ catalytic waves is to apply ‘foot-of-the-wave analysis’ (FOWA), developed by Savéant and Costentin.81,82 However, FOWA requires an accessible ‘foot’ to the catalytic wave (i.e., the absence of redox processes distorting the foot of the wave). This is not applicable for the Re catalysts because the two reduction events overlap at the foot of the catalytic wave, and the onset occurs at a more cathodic potential than the 2nd reduction. Due to the lack of reliable methods for estimating kobs, the discussion on comparing the catalytic rate for Re complexes has been restricted to the difference in icat/ip.
| Catalyst | E cat (V vs. Fc+/0)a (at 0.1 V s−1) |
i
cat/ipa (at 0.1 V s−1) |
E
appb (V vs. Fc+/0) |
COb (μmol) | FYCOb |
TONCOb |
|---|---|---|---|---|---|---|
| a E cat (half-wave potential for the catalytic wave) and icat/ip values were determined from CVs recorded in acetonitrile. b E app (applied potential), amount of CO, Faradaic yields (FYCO) and catalyst turnover numbers (TONCO) were determined from 3.0 min CPE performed in CO2-saturated electrolyte using a glassy carbon plate working electrode. c CPE was not performed with Re7. | ||||||
| Re1 | −2.32 | 8.9 | −2.1 | 3.2 | 88% | 2.1 |
| −2.3 | 5.0 | 98% | 3.3 | |||
| Re2 | −2.20 | 7.1 | −2.1 | 4.1 | 87% | 2.7 |
| −2.3 | 3.5 | 78% | 2.3 | |||
| Re3 | −2.21 | 8.7 | −2.3 | 3.1 | 94% | 2.1 |
| Re4 | −2.14 | 9.6 | −2.1 | 0.9 | 62% | 0.6 |
| −2.3 | 1.8 | 70% | 1.2 | |||
| Re5 | −2.31 | 9.0 | −2.1 | 2.3 | 85% | 1.5 |
| −2.3 | 2.8 | 92% | 1.9 | |||
|
Re7c |
−2.42 | 8.1 | — | — | — | — |
Previous reports on Re(bpy)(CO)3X (X = Cl, Br) catalysts have suggested that CO2 reduction can proceed via two pathways: (1) a slow 1e− pathway that occurs after the 1st ligand-centred reduction and (2) a faster 2e− pathway that is activated after the 2nd metal-centred reduction and subsequent rapid loss of X−.77 This is consistent with the CV data for Re-complexes recorded at various scan rates, which show a minimal current increase at the 1st reduction step (ν = 0.1–25 V s−1). Significant current enhancement is only observed at further cathodic potentials than the 2nd reduction step and relatively slower scan rates (<1 V s−1) (Fig. 7B and S32†).
Electrocatalytic CO2 reduction in the presence of proton sources
The influence of a proton source on electrocatalysis was evaluated using Re2 as the catalyst in the presence of water, trifluoroethanol (TFE), and acetic acid in a CO2-saturated acetonitrile electrolyte.35 Since an acidic environment can modulate the CO2 reduction activity on the electrode surface,83 we chose to evaluate three proton sources via cyclic voltammetry with increasing acidity: water (pKaqa 15.7) < trifluoroethanol (pKaqa 12.5) < acetic acid (pKaqa 4.7). In the presence of water, the hydration of CO2 forms H2CO3, which acts as a proton source with a pKa of 17.03 in acetonitrile. As discussed above, the quasi-reversibility of the 1st reduction is retained under CO2, but the second oxidation peak at −1.52 V in the return scan disappeared (Fig. 8A). Upon the incremental addition of water, a slight increase in current at −1.72 V was observed with a concomitant drop in current at −1.95 V and a gradual loss of reversibility of the 1st reduction. The variations in icat/ip at the two reduction steps are shown in the inset of Fig. 8A. It has been postulated that the 1e− pathway leads to a reductive disproportionation of CO2 to form CO and CO32−. We speculate that at higher proton concentrations, the pendant pyridine near the Re centre potentially facilitates CO32− release by forming a network of hydrogen-bonded water molecules.17 However, it should be noted that a pendant pyridine group has a pKa ∼ 5 (estimated based upon the pKa of pyridinium cations in aqueous solution84), and it is not expected to undergo direct protonation by H2CO3. Overall, the voltammograms suggest that adding water provides a minimal improvement in the catalytic activity. A similar effect of water was observed for other Re complexes (Fig. S33†). | ||
| Fig. 8 Catalytic CVs of Re2 (1 mM) in CO2-saturated acetonitrile containing three different proton sources: (A) CVs in the presence of increasing amount of water (0%, 0.1%, 0.2%, 0.4%, and 1%; v/v). The red trace presents 0% water and current traces with increasing water content is shown by the faded red traces. The inset figure shows the icat/ip ratio variation with water content at the two reduction steps. (B) CVs in the presnce of trifluoroethanol (0, 10, 20, 30, and 40 mM with the red trace presenting 0% water) The inset figure shows the shift of the peak potential of the second reduction step with incremental addition of TFE. (C) CVs under N2 and CO2 in the presence and absence of acetic acid (5 mM). All voltammograms were recorded at a 100 mV s−1 scan rate in 0.1 M n-Bu4BF4 supporting electrolyte. | ||
As shown in Fig. 8B, upon addition of TFE to a CO2-saturated solution of Re2, the voltammograms show no change in the reductive peak current at the 1st reduction (−1.73 V) and a significant decrease in the peak current at the 2nd reduction (−1.92 V). In the presence of TFE, the large catalytic wave at ∼−2.25 V displayed a decrease in current and shifted slightly to a more positive potential, forming a well-defined peak at −2.15 V. While increasing the concentration of TFE did not lead to any further increase in peak current at −2.15 V, the 2nd reduction feature (∼−1.92 V) underwent an incremental cathodic shift coupled with a gradual decrease of the peak current (Fig. 8B inset). This suggests that TFE is not directly involved in the rate-limiting step. At 50 mM TFE concentration, the 2nd reduction feature shifted to ∼−2.02 V and it was barely discernible (Fig. S34†). This can be tentatively described as the ‘EC’ step, where TFE reacts with the singly reduced species (‘C’ step), leading to its depletion at the electrode surface and a subsequent cathodic shift of the 2nd reduction. When the CVs were recorded at a faster scan rate (10 V s−1), the first two reduction peaks displayed minimal current enhancement in the presence of TFE, but a new reduction wave appeared at ∼−2.3 V with a concomitant loss of the reoxidation peak at −1.52 V (Fig. 9). Upon purging the electrolyte with N2, the peak current at −2.3 V was nearly halved, and the peak current at ∼−2.04 V (the 2nd reduction step) decreased to the pre-CO2 level (black and grey trace, Fig. 9). This suggests that the reduction wave at −2.3 V and the modest current enhancement at −2.04 V can be attributed to electrocatalytic CO2 reduction. Notably, there is a non-trivial contribution from proton reduction at −2.3 V as shown by the reduction wave under N2 (grey trace). Since this peak is only observed in the presence of TFE, we can speculate that it originates from the protonation of the doubly reduced species.
 | ||
| Fig. 9 CVs of Re2 (1 mM) in N2 or CO2-saturated acetonitrile containing varying amounts of TFE (0 or 20 mM). Voltammograms were recorded at a 10 V s−1 scan rate in 0.1 M n-Bu4BF4 supporting electrolyte. | ||
Interestingly, the reoxidation peak at −1.52 V reappeared when scanned under N2 (grey trace, Fig. 9), suggesting that the pentacoordinate species is regenerated. At a 100 mV s−1 scan rate (Fig. 8B), all three reduction steps display a current increase, which indicates that CO2 reduction can be observed for singly and doubly reduced species on slower CV timescales. For realising the catalysis in kinetic regimes and to achieve ‘S-shaped’ catalytic waves under CO2/TFE, the scan rate was increased up to 25 V s−1, but the plateau current kept increasing with the scan rate, which excluded the possibility of determining kobs (Fig. S35†) (higher scan rates were not measured due to instrument limitations).
Finally, acetic acid was evaluated as a proton source for CO2 due to its lower pKa, which allows protonation of the pendant pyridine. As shown in Fig. 8C, the CVs recorded in the presence of 5 mM AcOH under N2 and CO2 are nearly identical, suggesting that CO2 reduction is hindered under acidic conditions. Adding acetic acid renders the 1st reduction event irreversible, along with a ∼4-fold increase in the peak current (Ep = –1.70 V). However, no further increase in peak current was observed upon increasing the acetic acid concentration from 5 mM to 10 mM, suggesting that it is not a catalytic wave. We speculate that the protonation of the terpyridine ligand causes this proton-responsive behaviour, and the irreversible reduction is tentatively assigned to the multi-electron reduction of the protonated complex.
Controlled potential electrolysis
The catalytic performance of the compounds was investigated by 30 min controlled potential electrolysis (CPE) at −2.1 V and −2.3 V (vs. Fc+/0) in CO2-saturated acetonitrile containing 0.5% water (v/v) as the proton source. TFE was avoided as the proton source to minimise the competing proton reduction. The applied potentials correspond to overpotentials of 0.75 V and 0.95 V, respectively, for CO2 reduction to CO .73 After CPE, the headspace of the electrochemical cell was analysed by gas chromatography (GC), and the electrolyte was analysed by ion chromatography and 1H NMR spectroscopy for other liquid products (formic acid, methanol). CO was detected as the only CO2-reduction product from electrolysis. No H2 was detected for any Re catalysts. During electrolysis, the current density slowly decayed (Fig. S36†). This suggests slow catalyst decomposition, consistent with the change of colour of the electrolyte from bright yellow to pale brown. This is supported by the post-electrolysis characterisation of the electrolyte with UV-Vis (Fig. S37–S39†). To confirm that the molecular Re-complexes catalyse CO2-to-CO conversion in solution, a rinse test was performed with the post-electrolysis glassy carbon electrode under identical conditions for Re3 (Fig. S40†). The rinsed electrode showed negligible current with no CO2R activity, supporting the homogenous nature of the CO2R catalysis. No CO was detected in the control experiments performed under N2 or without a catalyst.
.73 After CPE, the headspace of the electrochemical cell was analysed by gas chromatography (GC), and the electrolyte was analysed by ion chromatography and 1H NMR spectroscopy for other liquid products (formic acid, methanol). CO was detected as the only CO2-reduction product from electrolysis. No H2 was detected for any Re catalysts. During electrolysis, the current density slowly decayed (Fig. S36†). This suggests slow catalyst decomposition, consistent with the change of colour of the electrolyte from bright yellow to pale brown. This is supported by the post-electrolysis characterisation of the electrolyte with UV-Vis (Fig. S37–S39†). To confirm that the molecular Re-complexes catalyse CO2-to-CO conversion in solution, a rinse test was performed with the post-electrolysis glassy carbon electrode under identical conditions for Re3 (Fig. S40†). The rinsed electrode showed negligible current with no CO2R activity, supporting the homogenous nature of the CO2R catalysis. No CO was detected in the control experiments performed under N2 or without a catalyst.
The electrolysis results are summarised in Table 3. All Re complexes, except Re4, produced CO with a high FYCO (>85%) after 30 min electrolysis. In contrast, the FYCO after electrolysis with Re4 was 62% and 70% at −2.1 V and −2.3 V, which could be attributed to the lower stability of the complex under reductive conditions. This is consistent with the fast decay of current density observed during electrolysis with Re4. Total catalytic turnover numbers for CO (TONCO) were determined from the amount of CO produced after 30 min electrolysis. The TONCO values were in the range of 1.2–3.3, with Re1 displaying the best activity at −2.3 V. Overall, the performance of the catalysts under electrolysis conditions does not vary significantly within the series. Interestingly, applying a more reductive potential during electrolysis (−2.1 V versus −2.3 V) had a minimal influence on FYCO and TONCO. Electrolysis at −2.1 V is expected to follow the 2e− pathway for Re2, Re3, and Re4 (Table 3), whereas it would proceed via the 1e− pathway for Re1 and Re5 as the second metal-centred reduction occurs at a more negative potential for the latter two complexes (∼−2.3 V, Table 3). This is consistent with the slightly improved CO yield, and the TONCO observed for electrolysis at −2.3 V for Re1 and Re5.
Spectroelectrochemistry of Re5
A distinct colour change was observed for Re5 before and after the electrolysis experiments, which prompted us to investigate Re5 using UV-Vis spectroelectrochemistry in acetonitrile. The UV-Vis spectra of Re5 at reducing potentials are shown in Fig. 10. At an open circuit voltage, the starting solution of Re5 displays a peak at 516 nm in the visible region. Upon reduction, a new absorbance appears at ∼580 nm at <−1.9 V (vs. Fc+/0) and shows complete conversion at about −2.3 V. The final UV-Vis spectrum also shows a broad absorbance in the 400–500 nm range. The CV of Re5 suggests that this is likely to be a 2e− reduced species. Interestingly, the UV-Vis spectra show very little change after the first reduction at −1.75 V (Fig. S36†). | ||
| Fig. 10 UV–Vis spectroelectrochemical data for Re5 (1 mM), showing the steady-state relative absorbance at different applied potentials under N2. The inset figure shows the cyclic voltammogram of Re5 at the reductive potential. | ||
TDDFT data suggest that the 516 nm absorbance majorly corresponds to a d–d transition within the ferrocenyl moiety in the ligand. We speculate that the extra electron density in the reduced Re5 species (at <−2.1 V) is partly delocalised on the ferrocenyl moiety through the coordinating terpyridine unit. Unfortunately, we were unable to isolate the reduced species for further characterisation. The spectroelectrochemical measurements show the evolution of a new peak at 590 nm in the ground state absorption spectrum under higher applied potentials. This wavelength is the same region where a delayed stimulated emission is observed in our measurements. It is difficult to predict if the bands at 590 nm in the TA and spectroelectrochemistry originate from the same state. Currently, detailed transient spectro-electrochemical measurements are carried out to ascertain 590 nm bands for Re5. If these bands correspond to the same state, then the same molecule could be used as an electrochemical and photochemical catalyst for fixing atmospheric CO2.
Conclusions
A series of Re(I)(L)(CO)3Br complexes containing 4′-substituted-2,2′:6′,2′′-terpyridine ligands (L2–5) were synthesised and characterised (Re2–5). Analytical data suggest that the tpy ligand coordinates to the Re centre in κ2-NN mode with all CO ligands in the cis position. TAS and TDDFT studies indicate that a highly intense band around 303–310 nm is a combination of π(tpy)–π*(tpy) and πyz(Re–Br)–π*(tpy), categorising them to ILCT and MLCT bands for all complexes. According to the TDDFT calculation, another MLCT band is observed around 430 nm for Re1–4 due to the 5dyz(Re)–π*(tpy) transition. Interestingly, for Re5, the second MLCT band also involves the 3dxy orbitals of the iron centre in the ferrocene substituent along with the 5dyz(Re)–π*(tpy) transition. Re5 also showed a d–d transition around 516 nm owing to a ferrocenyl moiety probed by in situ spectroelectrochemistry. An additional excited state was observed for the Re5 complex in transient absorption spectroscopy due to the presence of the ferrocenyl moiety. The electrochemical data demonstrate the influence of the substitution on the tpy ligand on the redox property of the complexes. The catalytic activity of all Re complexes towards CO2-to-CO electroreduction in acetonitrile is demonstrated using cyclic voltammetry and controlled potential electrolysis, which reveal comparable catalytic currents for tpy-substituted Re-complexes (Re1–5) and the bipyridine analogue (Re7). Within the series, the best activity towards CO2 reduction is observed for Re4 based on the icat/ip ratios determined from cyclic voltammetry, and the best TONCO and FYCO were obtained for Re1 using controlled potential electrolysis. The electrocatalysis was mostly unaffected by adding water (Brønsted acid), suggesting that the pendant pyridine group does not directly engage in CO2 reduction under weakly acidic conditions. However, the electrocatalytic activity of the complexes can be modulated by using proton sources with lower pKa (TFE and AcOH), as demonstrated by the voltametric data recorded here. In summary, this work expands the library of Lehn-type Re-catalysts for CO2 reduction and demonstrates the effect of integrating terpyridine ligands with an uncoordinated pyridine towards electrocatalytic CO2 conversion.Experimental section
Materials
Ethanol was received from LabChem; toluene and diethyl ether were received from SRL Chemicals. Ethanol and diethyl ether were used as received, and toluene was dried over sodium before use. For the cyclic voltammetric measurements, acetonitrile was dried over phosphorous pentoxide, and dried DMF was used as obtained from SRL Pvt. Ltd. Rhenium pentacarbonyl bromide, Re(CO)5Br, was purchased from Sigma Aldrich, aldehydes from Spectrochem Pvt. Ltd, potassium hydroxide from Avra Synthesis Pvt. Ltd, and aqueous ammonia from Qualigen Laboratories. All reagents were used without further purification. The ligand L1 was obtained from BLD Pharmatech (India) Pvt. Ltd through Carbanio. All metal complex synthesis reactions were carried out using the Schlenk technique. The filtration and other manipulations were done in the open air.Instrumentation
Melting points of the ligands were recorded using the DBK-programmable melting point (max = 320 °C) apparatus. 1H and 13C{1H} NMR spectra were acquired on a Bruker AVANCE III HD spectrometer using 500 MHz (1H NMR) or a Bruker ASCEND™ spectrometer using 400 MHz (1H NMR) in CDCl3 (δ = 7.26 ppm); DMSO-d6 (δ = 2.50 ppm) or 125 MHz or 101 MHz (13C{1H} NMR) in CDCl3 (δ = 77.1 ppm); and DMSO-d6 (δ = 39.5 ppm) at 298 K. Multiplicities of the signals are reported using the following abbreviations: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; dt, doublet of triplet; m, multiplet; br, broad. The UV-Vis absorption spectral measurements of ligands and complexes in DMF or acetonitrile solution were performed with a JASCO – V-770 UV-Visible/NIR double-beam spectrophotometer. In the ATR mode, the infrared (IR) spectra were recorded using a Shimadzu IRAffinity-1S spectrophotometer (40 scans, scanned in 4000–400 cm−1, resolution 4 cm−1). The reverse phase HPLC method was carried out on a C18 column (100 mm × 2.1 mm i.d., 3 μm), maintained at 40 °C, using aqueous acetonitrile gradients containing 1% ammounium formate as the mobile phase with a flow rate of 0.2 mL min−1. A photodiode array (PDA) detector set at 254 nm was used, unless stated otherwise. Elemental analysis was performed using an Elementar Vario Macro cube organic chemical analyser (BITS Goa). Elemental analysis for Re5 was done at Mikrolab Kolbe, Oberhausen, Germany. MS were recorded using a Bruker Daltonics – micrOTOF-Q II – ESI-Qq-TOF mass spectrometer (IISER Bhopal) or Shimadzu LCMS – 2020 with ESI ionisation mode.Synthesis and characterisation of ligands
In a Schlenk flask with a magnetic stirbar, aldehyde (1.0 equiv.) was dissolved in 5.0 mL of EtOH at room temperature. To this solution, 2-acetylpyridine (2.0 equiv.) was added and stirred to complete the dissolution. KOH pellets (2.0 equiv.) and aq. NH3 (60%, 50 equiv.) were added to the solution. The mixture was refluxed at 70 °C for 12 h. The clear orange-coloured solution changes to a turbid mixture during the reaction, indicating the desired product formation. After cooling the solution to room temperature, it is kept at 4 °C overnight, which helps to form more precipitate. The white or off-white solid was collected by filtration, washed with cold EtOH (3 × 10 mL), and air-dried for 24 h.:5 (v/v). ESI-MS (m/z): 310 (expected 310 (M + H+), C21H16N3+) and 332 (expected 332, (M + Na+), C21H15N3Na+).
:5 (v/v). ESI-MS (m/z): 324 (expected 324 (M + H+), C22H18N3+) and 346 (expected 346, (M + Na+), C22H17N3Na+).
:5 (v/v). ESI-MS (m/z): 388 (expected 388, (M + H+), C21H15BrN3+).
:5 (v/v). ESI-MS (m/z): 418 (expected 418, (M + H+), C25H20FeN3+) and 440 (expected 440, (M + Na+), C25H19FeN3Na+).
Synthesis and characterisation of Re(I) complexes
The Re(I) complexes were synthesised following modified literature procedures: Re1,89Re2,61Re390 (Cl variation), and Re491 (Cl variation). A Schlenk flask equipped with a magnetic stirbar was charged with ligand (1.0 equiv.) and dissolved in 3 mL of dry toluene under argon. To this solution, Re(CO)5Br (1.0 equiv.) was added, and the reaction mixture was refluxed at 100 °C, wherein the colour changes from colourless to a yellow to dark orange solution for Re1 to Re4. For Re5, the colour of the solution changed from brown to red. After the colour change was observed, it was stirred for more time and then cooled down to room temperature, leading to a precipitate, except for Re5. The solution was then decanted, and the precipitate was washed with toluene (2 × 5 mL), and then cold diethyl ether (2 × 5 mL). The compound was dried under vacuum at 340 mbar for 20 minutes.
:5 (v/v). ESI-MS (m/z): 768 (expected 767, (M + H+), C28H20BrFeN3O3Re+) and 688 (expected 688, (M − Br+), C28H19FeN3O3Re+). HRMS (m/z): calcd for [M + Na+] 789.9394, found [M + Na+] 789.9397 and calcd for [M − Br]+ 688.0352, found [M − Br]+ 688.0422. Anal calcd (in %) for C28H19BrFeN3O3Re: C, 43.81; H, 2.50; N, 5.48. Found: C, 43.71; H, 2.51; N, 5.43.
Computational details
All the structures were optimised using B3LYP functional93–95 using the ORCA package.96 The def2-TZVP basis set is used for the optimisation.97 The optimisations were carried out in an implicit solvent using the SMD solvent model,98 with parameters corresponding to acetonitrile used to mimic the experimental conditions. The natures of the stationary points were characterised by frequency calculations at the same level of theory using the Gaussian 16 package.99 Time-dependent density functional theory (TDDFT) calculations were carried out at the B3LYP/def2-TZVP level to calculate the absorption spectra of all the complexes using the ORCA package. The lowest 50 transitions were taken into account in the calculations of the absorption spectra.Transient absorption spectroscopy
The ultrafast transient absorption (TA) was performed on a transient absorption spectrometer (Ultrafast Systems, USA). The pump pulses of 320 nm were generated from an OPerA Solo OPA (Coherent, USA) pumped by 35 fs, 5 mJ per pulse, and centred at 800 nm operating at 1 kHz generated from an Astrella amplified laser system (Coherent, USA). A part of the amplified output is used to generate broadband (340 to 680 nm) probe pulses by focusing it on the CaF2 window. The transmission of the probe in the presence and absence of a pump was recorded on a CCD spectrograph using a synchronised mechanical chopper operating at 500 Hz. All samples were prepared in freshly distilled acetonitrile (HPLC, Spectrochem). The pump and probe polarisations are kept vertical with respect to each other. The OD of the samples were maintained at around 0.5 at the excitation wavelength in a 2 mm path length cuvette. The samples were stirred continuously using a magnetic stirrer throughout the experiment. The sample integrity was checked by recording its steady-state spectra before and after the experiment. The obtained transient absorption spectra were analysed using Surface Xplorer (V 4.0) and Origin 2019.Electrochemistry
Electrochemical analysis was performed using a CH760E potentiostat or a BioLogic VSP potentiostat. Cyclic voltammetry measurements were performed in a one-compartment three-electrode setup using a glassy carbon working electrode (3 mm diameter), a platinum wire as the auxiliary electrode, and a non-aqueous Ag|Ag+ pseudo-reference electrode, Ag|AgNO3 (10 mM) in 0.1 M n-Bu4NBF4 in acetonitrile (or DMF). The voltammograms were referenced by the addition of ferrocene as an internal standard after the final experiment, and all potentials given in this work are reported against the Fc+/0 couple. The potential of the Fc+/0 couple was found to be ∼0.07 V vs. the non-aqueous Ag|Ag+ pseudo-reference. The supporting electrolyte was n-Bu4NBF4 (0.1 M), which was purified by recrystallisation from 1:2 ethanol/water followed by drying at 80 °C in a vacuum oven.
The diffusion coefficients (D) were determined by the Randles−Sevcik equation shown below:

The observed rate constant (kobs) was calculated using the following equation:

Controlled potential electrolysis
Controlled potential electrolysis measurements were performed in a custom two-compartment cell separated by a glass frit. A glassy carbon plate (2 mm thick Sigradur G-plates from HTW GmbH, Germany) was used as the working electrode, Pt mesh as the counterelectrode, and non-aquoeus Ag/Ag+ as the reference. The effective geometric surface area of the electrode during electrolysis was 2 cm2. Typically, the volumes of electrolytes in the working and counter compartments were 6 mL and 2 mL, respectively, with a headspace volume of 23 mL. The concentration of the catalyst during electrolysis was 0.25 mM.The quantification of H2 and CO was conducted using an SRI gas chromatograph (multiple gas analyser #1) equipped with a thermal conductivity detector (TCD) and a flame ionisation detector (FID) with a built-in methaniser attachment. A silica gel column (6 ft) column was used to block CO2 and H2O, and a molecular sieve 13X (6 ft) main column was used to separate the H2, and CO. N2 was used as the carrier gas. The GC was calibrated regularly using a known standard for H2, CO and CH4 (2000 ppm H2/2000 ppm CO/2000 ppm CH4 in balance gas N2).
Spectroelectrochemistry
Spectroelectrochemical analysis was performed in a thin-layer quartz glass cuvette (path length 1 mm) using a Pt mesh working electrode, Ag|Ag+ pseudo-reference electrode, and a Pt wire counterelectrode. An AvaLight-DHC (deuterium and halogen light source) was used as the light source, and the spectra were recorded using an AvaSpec-2048 spectrometer. The electrode was held at a set potential for 2 min before recording the UV-Vis spectrum.Author contributions
RM conceived and supervised the project. SS synthesised and characterised all ligands and Re(I) complexes and observed the initial CO2 reduction. TD and SR performed electrocatalysis and spectroelectrochemical experiments. DB and DM performed the computational studies. SKP and ESSI carried out ultrafast transient absorption spectroscopy. The manuscript was written through contributions from all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SS, RM, SKP and ESSI thank Indian Institute of Technology Goa (IIT Goa) for the infrastructure and teaching assistant fellowship. RM thanks SERB for the initial funding through a start-up research grant (SERB SRG/2019/000604). TD and SR acknowledge support from the EPSRC Doctoral Training Program (EP/T518177/1) and the Royal Society (RGS\R2\202350). DM acknowledges the National Supercomputing Mission (DST/NSM/R&D_HPC_Applications/2021/08) for providing computing resources of ‘PARAM Shakti’ at the Indian Institute of Technology Kharagpur (IIT Kgp) and ‘PARAM Brahma’ at IISER Pune, which are implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India. SERB SRG/2019/000604; EPSRC EP/T518177/1.References
- T. R. Anderson, E. Hawkins and P. D. Jones, Endeavour, 2016, 40, 178–187 CrossRef PubMed.
- I. P. on C. C. W. G. 1 Science, I. P. on C. Change and I. P. on C. C. W. G. I, Climate Change 2007 - The Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the IPCC, Cambridge University Press, 2007 Search PubMed.
- Climate Change 2022, https://www.ipcc.ch/report/ar6/wg3/, (accessed 3 October 2022).
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS PubMed.
- J. L. Sarmiento, T. M. C. Hughes, R. J. Stouffer and S. Manabe, Nature, 1998, 393, 245–249 CrossRef CAS.
- G. Centi and S. Perathoner, Green Chem., 2022, 24, 7305–7331 RSC.
- J. K. Nganga, L. M. Wolf, K. Mullick, E. Reinheimer, C. Saucedo, M. E. Wilson, K. A. Grice, M. Z. Ertem and A. M. Angeles-Boza, Inorg. Chem., 2021, 60, 3572–3584 CrossRef CAS PubMed.
- Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905–3926 RSC.
- N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CrossRef CAS PubMed.
- H.-Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202200723 CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- D. Grammatico, A. J. Bagnall, L. Riccardi, M. Fontecave, B.-L. Su and L. Billon, Angew. Chem., Int. Ed., 2022, 61, e202206399 CrossRef CAS PubMed.
- K. Lei and B. Y. Xia, Chem. – Eur. J., 2022, 28, e202200141 CAS.
- J. P. Collin and J. P. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- W. Nie and C. C. L. McCrory, Dalton Trans., 2022, 51, 6993–7010 RSC.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- B. Siritanaratkul, C. Eagle and A. J. Cowan, Acc. Chem. Res., 2022, 55, 955–965 CrossRef CAS PubMed.
- M. R. Madsen, M. H. Rønne, M. Heuschen, D. Golo, M. S. G. Ahlquist, T. Skrydstrup, S. U. Pedersen and K. Daasbjerg, J. Am. Chem. Soc., 2021, 143, 20491–20500 CrossRef CAS PubMed.
- M. Boraghi, T. A. White, S. Pordel and H. Payne, ACS Appl. Energy Mater., 2021, 4, 13725–13734 CrossRef CAS.
- M. Guyot, M.-N. Lalloz, J. S. Aguirre-Araque, G. Rogez, C. Costentin and S. Chardon-Noblat, Inorg. Chem., 2022, 61, 16072–16080 CrossRef CAS PubMed.
- C. J. Stanton, C. W. Machan, J. E. Vandezande, T. Jin, G. F. Majetich, H. F. Schaefer, C. P. Kubiak, G. Li and J. Agarwal, Inorg. Chem., 2016, 55, 3136–3144 CrossRef CAS PubMed.
- J. Mukherjee and I. Siewert, Eur. J. Inorg. Chem., 2020, 2020, 4319–4333 CrossRef CAS.
- E. Quartapelle Procopio, A. Boni, L. Veronese, M. Marcaccio, P. Mercandelli, G. Valenti, M. Panigati and F. Paolucci, ChemElectroChem, 2021, 8, 2065–2069 CrossRef CAS.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, Chem. Sci., 2019, 10, 3080–3088 RSC.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- F. P. A. Johnson, M. W. George, F. Hartl and J. J. Turner, Organometallics, 1996, 15, 3374–3387 CrossRef CAS.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- L. Rotundo, C. Garino, E. Priola, D. Sassone, H. Rao, B. Ma, M. Robert, J. Fiedler, R. Gobetto and C. Nervi, Organometallics, 2019, 38, 1351–1360 CrossRef CAS.
- L. Rotundo, D. E. Polyansky, R. Gobetto, D. C. Grills, E. Fujita, C. Nervi and G. F. Manbeck, Inorg. Chem., 2020, 59, 12187–12199 CrossRef CAS PubMed.
- C. Riplinger and E. A. Carter, ACS Catal., 2015, 5, 900–908 CrossRef CAS.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- S. E. Tignor, H.-Y. Kuo, T. S. Lee, G. D. Scholes and A. B. Bocarsly, Organometallics, 2019, 38, 1292–1299 CrossRef CAS.
- C. Jiang, A. W. Nichols and C. W. Machan, Dalton Trans., 2019, 48, 9454–9468 RSC.
- S. Amanullah, P. Saha, A. Nayek, M. E. Ahmed and A. Dey, Chem. Soc. Rev., 2021, 50, 3755–3823 RSC.
- W. L. J. Loke, W. Guo, M. Sohail, A. A. Bengali and W. Y. Fan, Inorg. Chem., 2022, 61, 20699–20708 CrossRef CAS PubMed.
- K. Talukdar, S. Sinha Roy, E. Amatya, E. A. Sleeper, P. Le Magueres and J. W. Jurss, Inorg. Chem., 2020, 59, 6087–6099 CrossRef CAS PubMed.
- S. Sinha Roy, K. Talukdar and J. W. Jurss, ChemSusChem, 2021, 14, 662–670 CrossRef PubMed.
- A. N. Hellman, J. A. Intrator, J. C. Choate, D. A. Velazquez and S. C. Marinescu, Polyhedron, 2022, 223, 115933 CrossRef CAS.
- A. N. Hellman, R. Haiges and S. C. Marinescu, ChemElectroChem, 2021, 8, 1864–1872 CrossRef CAS.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451–12456 CrossRef CAS PubMed.
- J. O. Taylor, G. Neri, L. Banerji, A. J. Cowan and F. Hartl, Inorg. Chem., 2020, 59, 5564–5578 CrossRef CAS PubMed.
- C. C. Addison, R. Davis and N. Logan, J. Chem. Soc., Dalton Trans., 1974, 2070–2071 RSC.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- T. Yoshida, K. Tsutsumida, S. Teratani, K. Yasufuku and M. Kaneko, J. Chem. Soc., Chem. Commun., 1993, 631–633 RSC.
- S. Sinha, A. Sonea, C. A. Gibbs and J. J. Warren, Dalton Trans., 2020, 49, 7078–7083 RSC.
- E. W. Abel, N. J. Long, K. G. Orrell, A. G. Osborne, H. M. Pain and V. Šik, J. Chem. Soc., Chem. Commun., 1992, 303–304 RSC.
- K. Choroba, S. Kotowicz, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, K. Sulowska, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Dyes Pigm., 2021, 192, 109472 CrossRef CAS.
- A. P. Sadimenko, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic Press, 2007, vol. 94, pp. 107–172 Search PubMed.
- R. Fernández-Terán and L. Sévery, Inorg. Chem., 2021, 60, 1334–1343 CrossRef PubMed.
- R. J. Fernández-Terán and L. Sévery, Inorg. Chem., 2021, 60, 1325–1333 CrossRef PubMed.
- K. Choroba, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, S. Maćkowski, T. Pedzinski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2021, 50, 3943–3958 RSC.
- M. Małecka, A. Szlapa-Kula, A. M. Maroń, P. Ledwon, M. Siwy, E. Schab-Balcerzak, K. Sulowska, S. Maćkowski, K. Erfurt and B. Machura, Inorg. Chem., 2022, 61, 15070–15084 CrossRef PubMed.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251–1254 CAS.
- A. Gelling, K. G. Orrell, A. G. Osborne and V. Šik, J. Chem. Soc., Dalton Trans., 1998, 937–946 RSC.
- S. A. Moya, R. Pastene, H. Le Bozec, P. J. Baricelli, A. J. Pardey and J. Gimeno, Inorg. Chim. Acta, 2001, 312, 7–14 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- T. Auvray, B. Del Secco, A. Dubreuil, N. Zaccheroni and G. S. Hanan, Inorg. Chem., 2021, 60, 70–79 CrossRef CAS PubMed.
- M. Feng, F. Yang and J. Wang, Chin. J. Chem. Phys., 2016, 29, 81–86 CrossRef CAS.
- A. El Nahhas, A. Cannizzo, F. Van Mourik, A. M. Blanco-Rodríguez, S. Záliš, A. Vlček and M. Chergui, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef CAS PubMed.
- A. M. Maroń, A. Szlapa-Kula, M. Matussek, R. Kruszynski, M. Siwy, H. Janeczek, J. Grzelak, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2020, 49, 4441–4453 RSC.
- A. N. Tarnovsky, W. Gawelda, M. Johnson, C. Bressler and M. Chergui, J. Phys. Chem. B, 2006, 110, 26497–26505 CrossRef CAS PubMed.
- A. El Nahhas, A. Cannizzo, F. van Mourik, A. M. Blanco-Rodríguez, S. Záliš, A. Vlček Jr. and M. Chergui, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef CAS PubMed.
- J. Palion-Gazda, A. Szłapa-Kula, M. Penkala, K. Erfurt and B. Machura, Molecules, 2022, 27, 7147 CrossRef CAS PubMed.
- R. Fernández-Terán and L. Sévery, Inorg. Chem., 2021, 60, 1334–1343 CrossRef PubMed.
- A. Szlapa-Kula, M. Małecka, A. M. Maroń, H. Janeczek, M. Siwy, E. Schab-Balcerzak, M. Szalkowski, S. Maćkowski, T. Pedzinski, K. Erfurt and B. Machura, Inorg. Chem., 2021, 60, 18726–18738 CrossRef CAS PubMed.
- O. F. Mohammed and A. A. O. Sarhan, Chem. Phys., 2010, 372, 17–21 CrossRef CAS.
- A. J. King, Y. V. Zatsikha, T. Blessener, F. Dalbec, P. C. Goff, M. Kayser, D. A. Blank, Y. P. Kovtun and V. N. Nemykin, J. Organomet. Chem., 2019, 887, 86–97 CrossRef CAS.
- J. Palion-Gazda, A. Szłapa-Kula, M. Penkala, K. Erfurt and B. Machura, Molecules, 2022, 27, 7147 CrossRef CAS PubMed.
- K. Choroba, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, S. Maćkowski, T. Pedzinski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2021, 50, 3943–3958 RSC.
- T. R. O'Toole, L. D. Margerum, T. D. Westmoreland, W. J. Vining, R. W. Murray and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1416–1417 RSC.
- B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1414–1416 RSC.
- P. Christensen, A. Hamnett, A. V. G. Muir and J. A. Timney, J. Chem. Soc., Dalton Trans., 1992, 1455–1463 RSC.
- A. N. Hellman, R. Haiges and S. C. Marinescu, Dalton Trans., 2019, 48, 14251–14255 RSC.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- J. T. Muckerman, J. H. Skone, M. Ning and Y. Wasada-Tsutsui, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 882–891 CrossRef CAS PubMed.
- A. G. Al-Sehemi, A. Irfan, A. M. Asiri and Y. A. Ammar, Spectrochim. Acta, Part A, 2012, 91, 239–243 CrossRef CAS PubMed.
- S. Vaduvescu and P. G. Potvin, Eur. J. Inorg. Chem., 2004, 2004, 1763–1769 CrossRef.
- M. N. Patel, P. A. Parmar and D. S. Gandhi, J. Enzyme Inhib. Med. Chem., 2011, 26, 734–741 CrossRef CAS PubMed.
- A. Juneja, T. S. Macedo, D. R. M. Moreira, M. B. P. Soares, A. C. L. Leite, J. K. de A. L. Neves, V. R. A. Pereira, F. Avecilla and A. Azam, Eur. J. Med. Chem., 2014, 75, 203–210 CrossRef CAS PubMed.
- P. Bulsink, A. Al-Ghamdi, P. Joshi, I. Korobkov, T. Woo and D. Richeson, Dalton Trans., 2016, 45, 8885–8896 RSC.
- T. Klemens, A. Świtlicka, A. Szlapa-Kula, S. Krompiec, P. Lodowski, A. Chrobok, M. Godlewska, S. Kotowicz, M. Siwy, K. Bednarczyk, M. Libera, S. Maćkowski, T. Pędziński, E. Schab-Balcerzak and B. Machura, Appl. Organomet. Chem., 2018, 32, e4611 CrossRef.
- T. Klemens, A. Świtlicka-Olszewska, B. Machura, M. Grucela, H. Janeczek, E. Schab-Balcerzak, A. Szlapa, S. Kula, S. Krompiec, K. Smolarek, D. Kowalska, S. Mackowski, K. Erfurt and P. Lodowski, RSC Adv., 2016, 6, 56335–56352 RSC.
- E. W. Abel, V. S. Dimitrov, N. J. Long, K. G. Orrell, A. G. Osborne, H. M. Pain, V. Šik, M. B. Hursthouse and M. A. Mazid, J. Chem. Soc., Dalton Trans., 1993, 597–603 RSC.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00441d |
| This journal is © The Royal Society of Chemistry 2023 |