
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase evolution, speciation and solubility limit of aluminium doping in zinc oxide catalyst supports synthesized via co-precipitated hydrozincite precursors†
Benjamin
Mockenhaupt‡
 ab,
Jan Konrad
Wied‡
c,
Sebastian
Mangelsen
b,
Ulrich
Schürmann
d,
Lorenz
Kienle
d,
Jörn
Schmedt auf der Günne
*c and
Malte
Behrens
*ab
ab,
Jan Konrad
Wied‡
c,
Sebastian
Mangelsen
b,
Ulrich
Schürmann
d,
Lorenz
Kienle
d,
Jörn
Schmedt auf der Günne
*c and
Malte
Behrens
*ab
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, Universitätsstr. 7, 45141 Essen, Germany. E-mail: mbehrens@ac.uni-kiel.de
bInstitute of Inorganic Chemistry, Kiel University, Max-Eyth-Str. 2, 24118 Kiel, Germany
cDepartment of Chemistry and Biology, Inorganic Materials Chemistry, University of Siegen, Adolf-Reichwein-Straße 2, 57076 Siegen, Germany. E-mail: gunnej@chemie.uni-siegen.de
dDepartment of Materials Science, Kiel University, Kaiserstraße 2, 24143 Kiel, Germany
First published on 14th March 2023
Abstract
The preparation of Al-doped ZnO via thermal decomposition of crystalline precursors, with a particular emphasis on kinetic effects on the solubility limits, was studied. The promoting effect of Al3+ on the catalyst system is discussed for methanol synthesis where ZnO:Al is employed as a support material for copper nanoparticles. The synthesis of the Al-doped zinc oxides in this study was inspired by the industrial synthesis of the methanol synthesis catalyst via a co-precipitated crystalline precursor, here: hydrozincite Zn5(OH)6(CO3)2. To determine the aluminium speciation and the solubility limit of the aluminium cation on zinc positions, a series of zinc oxides with varying aluminium contents was synthesized by calcination of the precursors. Short precipitate ageing time, low ageing temperature and aluminium contents below 3 mol% metal were advantageous to suppress crystalline side-phases in the precursor, which caused an aluminium segregation and non-uniform aluminium distribution in the solid. Even if zinc oxide was the only crystalline phase, TEM revealed such segregation in samples calcined at 320 °C. Only at very low aluminium contents, the dopant was found preferably on the zinc sites of the zinc oxide structure based on the  signal dominating the 27Al NMR spectra. The solubility limit regarding this species was determined to be approximately xAl = 0.013 or 1.3% of all metal cations. Annealing experiments showed that aluminium was kinetically trapped on the
signal dominating the 27Al NMR spectra. The solubility limit regarding this species was determined to be approximately xAl = 0.013 or 1.3% of all metal cations. Annealing experiments showed that aluminium was kinetically trapped on the  site and segregated into ZnAl2O4 upon further heating. This shows that lower calcination temperatures such as applied in catalyst synthesis conserve a higher aluminium doping concentration on that specific site than is expected thermodynamically.
site and segregated into ZnAl2O4 upon further heating. This shows that lower calcination temperatures such as applied in catalyst synthesis conserve a higher aluminium doping concentration on that specific site than is expected thermodynamically.
Introduction
Zinc oxide is a typical semiconductor material with a wide direct band gap around 3.3 eV.1–4 Severalstudies addressed the band gap engineering to improve the optical or electronic properties.2,5 Incorporation of a trivalent cation like aluminium in the zinc oxide lattice introduces one extra electron, which results in a promoted n-type semiconductor.6Hence, the electronic conductivity of such a doped zinc oxide increases by the increased Fermi level and the facilitated excitation of the additional electron to the conducting band, which should be associated with a lowered band gap energy.7–10 Such improvement of the electronic properties of zinc oxide is not only of interest in the field of semiconductor applications, but also in the field of catalysis.
Typically, a copper/zinc oxide catalyst contains around 10% aluminium oxide for industrial methanol synthesis from COx/H2 mixtures (synthesis gas). It has been demonstrated that aluminium promotes the catalytic activity by improving and stabilizing the nano-structuring of the catalyst (structural promoter) as well as the strong metal–support interactions between zinc oxide and the copper nanoparticles (electronic promoter). Small amounts of this trivalent cation also increase catalyst lifetime and reduce copper sintering.11–13 The doping effect of bivalent (Mg2+) and trivalent cations (Al3+, Ga3+) in zinc oxide supports on the activity in methanol synthesis was further investigated,14 and it was found that a lower band gap energy of the doped zinc oxide is correlated with a higher catalytic activity after impregnating it with copper. Such a lowered band gap was discussed as a result of aluminium incorporation similar to semiconductor research. Contrarily, a bivalent cation does not lower the band gap nor improve the catalytic activity significantly.14 This finding is in line with studies, which found a correlation in conductivity increase by trivalent cationic-doped zinc oxides and a decrease for cationic dopants of lower valency.15,16 These results led to the hypothesis that beside the structural impact also electronic properties affect the catalysis in a positive way, which can be introduced by the incorporation of the Al3+ cation and could be related to the reducibility of doped zinc oxide under the hydrogenation conditions in methanol synthesis.14
In both fields, semiconductors and catalysis, the efficiency of the promoting effect depends on the maximum incorporation of aluminium ions in zinc oxide and on their lattice site substituting zinc ions (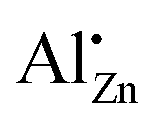 ) in the zinc oxide lattice.17 Contrarily, a segregated aluminium oxide side-phase in a doped zinc oxide sample could suppress the electronic promotion by its insulating properties.18,19 Thus, the determination of the maximum substitution limit of aluminium atoms in the zinc oxide lattice is of high importance. This can be a quite difficult task because of various side phases which can be formed. The solubility limit depends on the synthesis route and was estimated to be between 0.1 at% and 5.2 at% aluminium in zinc oxide.19–24 The determination of the substitution was performed with several techniques, like optical measurements,19 the reduction of dichromate,20 scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX) analysis,21 Raman-spectroscopy,22 resistivity measurements23 and 27Al solid state nuclear magnetic resonance (NMR) spectroscopy.24 With the help of 27Al NMR different aluminium environments can be resolved and assigned to the different coordination numbers of aluminium.24–28 In this regard, the 27Al NMR signal at δiso = 82.6 ppm could unambiguously be assigned to aluminium on a zinc position in the ZnO crystal structure
) in the zinc oxide lattice.17 Contrarily, a segregated aluminium oxide side-phase in a doped zinc oxide sample could suppress the electronic promotion by its insulating properties.18,19 Thus, the determination of the maximum substitution limit of aluminium atoms in the zinc oxide lattice is of high importance. This can be a quite difficult task because of various side phases which can be formed. The solubility limit depends on the synthesis route and was estimated to be between 0.1 at% and 5.2 at% aluminium in zinc oxide.19–24 The determination of the substitution was performed with several techniques, like optical measurements,19 the reduction of dichromate,20 scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX) analysis,21 Raman-spectroscopy,22 resistivity measurements23 and 27Al solid state nuclear magnetic resonance (NMR) spectroscopy.24 With the help of 27Al NMR different aluminium environments can be resolved and assigned to the different coordination numbers of aluminium.24–28 In this regard, the 27Al NMR signal at δiso = 82.6 ppm could unambiguously be assigned to aluminium on a zinc position in the ZnO crystal structure  ,27 and Knight shifted signal gave evidence of the targeted n-doping leading to an electronic conductivity.25
,27 and Knight shifted signal gave evidence of the targeted n-doping leading to an electronic conductivity.25
With that method, the solubility limit is determined in a closer range of 0.3 at% to <2 at% aluminium in ZnO depending on the synthesis procedure and the synthesis temperature.24,26–29 The inconsistencies with regard to the solubility limit are subject of this contribution. The hypothesis of a solubility limit is consistent with quantum-chemical investigations that suggested a solubility limitation by the formation of the spinel ZnAl2O4, which acts as “thermodynamic sink” and inhibits formation of highly doped ZnO variants.30 This is in agreement with earlier experimental work, where for a ZnO/Al2O3 mixture at temperatures lower than 1250 °C a solubility limit of ≤0.2% was suggested by X-ray diffraction.31 To the best of our knowledge, the solubility limit has not been investigated to that level of details in the context of catalyst synthesis with different occurring side-phases which impose a thermodynamic limit as suggested by theory.30 Clearly, such an investigation requires an analytical technique which can identify and quantify the different chemical environments of the Al atoms and a low-temperature synthesis approach to stabilize high Al substitution levels.
In the field of catalysis, the calcination temperatures are typically lower than the annealing temperatures of semiconductors. At lower temperatures such as 300–400 °C the  NMR signal was observed to have a high intensity.28,29 Furthermore it was found that the metallic character of ZnO:Al depends on the atmosphere, i.e. a reductive atmosphere may increase the metallicity of ZnO:Al as indicated from the formation of a Knight shifted 27Al NMR signal.27,32 For methanol synthesis, which inspired this work, the zinc oxide catalyst support is calcined in an oxidative atmosphere at a comparable temperature around 350 °C.33 It is the goal of this work to investigate the solubility limit of Al-doped zinc oxide for materials that represent the catalyst support formed by this method.
NMR signal was observed to have a high intensity.28,29 Furthermore it was found that the metallic character of ZnO:Al depends on the atmosphere, i.e. a reductive atmosphere may increase the metallicity of ZnO:Al as indicated from the formation of a Knight shifted 27Al NMR signal.27,32 For methanol synthesis, which inspired this work, the zinc oxide catalyst support is calcined in an oxidative atmosphere at a comparable temperature around 350 °C.33 It is the goal of this work to investigate the solubility limit of Al-doped zinc oxide for materials that represent the catalyst support formed by this method.
To determine the maximum amount of aluminium substituted zinc sites, a series of zinc oxides with varying aluminium content was synthesized using co-precipitation of crystalline hydroxycarbonate precursors, which is the established method for synthesizing Cu/ZnO:Al catalysts. In this case, the copper component was omitted to focus on the support.34 This has the advantage that the aluminium and zinc components are well distributed. In addition, no organic molecules are involved, which could interfere optical measurements. However, this procedure opens another question, namely that of the substitution chemistry in the hydroxy-carbonate precursor. The aluminium doping of a specific precursor phase, like the hydrozincite phase used here, should facilitate the formation of doped oxides upon thermal decomposition of the precursor and increase the inter-dispersion of both elements after calcination. Within our study, the question towards maximum substitution limit of Al3+ ions on the zinc sites in the hydrozincite precursor phase as well as in the zinc oxide structure will be addressed. We further aim at studying the aluminium dopant also under hydrogenation conditions that are relevant for methanol synthesis in forthcoming work. Finally, copper can be deposited on these supports with the aim to relate the catalytic properties to detailed knowledge of the aluminium species and amount. Here, we report on the as-prepared state of the catalyst support.
Experimental section
Sample labelling
The samples are labelled according to their nominal aluminium content relative to the total amount of metal atoms, i.e. the cation-based molar aluminium fraction xAl calculated as shown in eqn (1). | (1) |
Here, ni is the molar amount of element i (i = Zn, Al). Nominal values refer to the amount utilized during synthesis, which was found to match satisfactorily to the experimental values determined by ICP optical emission spectroscopy as discussed below in detail.
Precursor synthesis
The hydrozincite precursor was synthesized by co-precipitation with small amount of aluminium. The precipitation was carried out in a 1L automated stirred tank reactor (OptiMax1001, Mettler Toledo) using 1 M metal salt nitrate solutions at a temperature of 65 °C and at a constant pH of 6.5. Appropriate ratios of zinc and aluminium nitrate, with a purity ≥98% (ESI, Table S1†), were chosen to reach nominal compositions ranging from 0 to 10% Al (0 ≦ xAl ≦ 0.1). As precipitating agent, 1.6 M sodium carbonate solution was co-fed into the reactor. The dosing rate of metal salt solution was adjusted to 4.2 g min−1. After precipitation, approximately 300 mL of precipitate slurry was obtained and aged for 10 minutes without further pH control in the mother liquor. The precursor was washed with deionized water 10 times to reach a conductivity of the filtrate lower than 100 μS cm−1 and was subsequently dried at 80 °C for minimum 14 h. This synthetic procedure corresponds to the industrial preparation of the Cu/ZnO:Al catalyst for methanol synthesis35 with omittance of copper salts in the co-precipitation.For a time-resolved ageing experiment, a synthesis as described above was carried out with aluminium content of 2% (xAl = 0.02). After the finish of co-precipitation, the first sample (t = 0 min) was taken out of the reactor before precipitate ageing. At ageing times of 10 min, 30 min, 60 min and 120 min, additional samples were collected. The total amount of the slurry removed was around 30 ml. After 120 min, the remaining suspension was aged for additional 12 hours in a Teflon-lined steel autoclave (275 ml) under solvothermal conditions at 130 °C. The Teflon-lined steel autoclave was maximum filled by 2/3 of its total volume. The samples were centrifuged and three times washed with deionized water to get rid of excess ions, and afterwards dried at 80 °C for minimum 14 h.
A brief description of the hydrozincite crystal structure is provided as ESI and visualized in Fig. S1.†
Zinc oxide synthesis
The zinc oxide samples were synthesized by calcination of ground hydrozincite precursor obtained from the above-described co-precipitation synthesis. The calcination was performed at 320 °C with a heating ramp of 2 °C min−1 for 4 h in a muffle furnace of Narbertherm (LE 6/11/B150) in static air (without any volume flow). Such calcination temperature is known from the synthesis of typical industrial methanol catalysts.35 Further calcination experiments at higher temperatures were performed under similar conditions except for varying the target temperature.Characterization methods
X-ray diffraction analysis of the time resolved ageing samples and of the calcined samples was performed on a Bruker D8 advance with Cu-Kα radiation and a LYNXEYE XE-T detector. The diffractograms were recorded in Bragg–Brentano geometry at room temperature between 5° and 90°2θ. Phase analysis was performed by comparing the recorded pattern with structural data from ICSD and COD databases.
Pawley fits were carried out using TOPAS Academic version 6.0.36 Instrumental line broadening was described using the fundamental parameter approach37 as implemented in TOPAS and cross-checked against a measurement of LaB6 (NIST SRM 660c).
Brunauer–Emmet–Teller (BET) analysis
The specific surface areas were measured by nitrogen physisorption at 77 K in a Nova 3200e sorption station from Quantachrome. Before recording the isotherms, the samples were degassed under vacuum at 100 °C (hydrozincite) and 250 °C (zinc oxide) for 5 h. Afterwards the isothermal profiles between p/p0 = 0.0 and 1 referred to a reference cell were recorded. The multipoint BET surface area was determined by applying the Brunauer–Emmett–Teller equation to a specific range of each sample, determined by the micropore assistant of the NovaWin software only considering the volume increase.Scanning electron microscopy (SEM)
The SEM micrographs of the zinc oxides of the high temperature series and of the hydrozincites and their corresponding zinc oxides series of the time resolved ageing experiment were taken on an Apreo S LoVac electron microscope of Thermo Fisher Scientific. A suspension of around 1 mg sample in 2 ml ethanol was used for drop casting of 200 μL on single crystal silicon plates. Afterwards the sample holder was dried at 80 °C on a hot plate. The images of the hydrozincite precursors of the aluminium concentration series and their corresponding zinc oxides were taken on a Zeiss Gemini Ultra Plus with an Oxford EDX detector. A spatula tip of the sample was dispersed on a carbon covered stainless steel pin mount sample holder.Transmission electron microscopy (TEM)
TEM analyses were performed on a FEI Tecnai F30 G2 STwin (300 kV, FEG) equipped with an EDX detector (Si/Li, EDAX). The TEM samples were ground and dispersed in n-butanol (spatula tip sample in few drops of n-butanol) and prepared on Cu lacey TEM grids. The pure zaccagnaite reference sample was prepared in a dry state without n-butanol.Infrared resonance spectroscopy (IR)
Infrared spectroscopic characterization was performed on a Bruker Alpha FT-IR spectrometer with attenuate total reflection (ATR) unit. The spectra were recorded between 400 cm−1 and 4000 cm−1. To suppress water and CO2 signals from the atmosphere, the device was placed in a glovebox of MBraun with argon atmosphere.UV/Vis-spectroscopy
The UV/Vis spectra at ≤88 K were measured at a Varian Cary 5000 spectrometer. The sample was diluted with KBr and the pellet was adjusted in a solid sample holder. After flushing with nitrogen, the room temperature (25 °C) spectrum was recorded under vacuum. The 88 K (−185 °C) spectrum was recorded after cooling down with liquid nitrogen. When the sample was heated up to room temperature (27 °C) the spectrum was recorded to determine sample changes. The band gap determination was performed by differentiation of the raw data, to determine the changing point. At this point, where the first derivative shows a minimum, the wavelength was calculated to the electrical energy in eV. This method was used only to determine qualitatively the band gap energy change by UV/Vis spectroscopy in dependency of the aluminium incorporation.Raman-spectroscopy
Raman spectroscopy of the powders was performed in a powder sample holder positioned in 45° to the laser beam in a MacroRam spectrometer of Horiba Scientific. A 785 nm laser with an intensity of 90 mW was used. The Raman shifts were recorded in the range of 100 cm−1 to 3500 cm−1.Solid state NMR-spectroscopy
All 27Al solid-state NMR experiments were performed on the Bruker Avance Neo NMR spectrometer with a magnetic field of 14.1 T using a commercial Bruker MAS probe head with a non-Al-stabilized zirconia ceramic rotor with 3.2 mm outer diameter at a frequency of 156.375 MHz. The chemical shift of 27Al is reported relative to a 1.1 mol kg−1 solution of Al(NO3)3 in D2O on a deshielding scale.38,39 The referencing was done by using 1% TMS in CDCl3 as external secondary reference for the 1H resonance frequency using the Ξ scale as suggested by the IUPAC. All 27Al MAS NMR measurements were performed using 20 kHz spinning frequency and continuous wave 1H decoupling. For quantitative 27Al MAS NMR measurements the resonance frequency was selected according to the 27Al MAS NMR signal at δiso = 82.6 ppm and the nutation frequency was adjusted for the excitation of solely the central transition. The NMR data were processed with the topspin4.1.4 software. Typical recycle delays were of the order of 30 s (>3T1) and a few thousand scans were accumulated. The peak areas were determined by deconvolution into mixed Gaussian/Lorentzian profile functions with the program deconv2Dxy.40Results and discussion
Characterization of the Al doped hydrozincite precursors
 | ||
| Fig. 1 PXRD data of a co-precipitated zinc–aluminium precursor with an aluminium content of xAl = 0.02 at different ageing times. The ageing was performed between 0 min (immediately after co-precipitation) and 120 min. The remaining suspension was aged for additional 12 hours at 130 °C in a solvothermal reactor (120 min + solvothermal). The grey bars correspond to the zaccagnaite (ICSD#190041) and the black bars to hydrozincite (ICSD#16583) references. (a) is an overview, while (b) shows a more detailed zoom for better identification of the side-phase zaccagnaite, whose reflections are marked with stars. In (b) the patterns are sorted likewise as in (a). | ||
The morphology of the samples was investigated by SEM and the micrographs of the samples obtained at different ageing times are shown in Fig. 2. The morphology was similar up to an ageing time of 30 minutes. Aggregated platelets were intergrown to spheres and other larger aggregates, as they are shown in the ESI (Fig. S4 and S11†). After 60 minutes of ageing, the platelet size seemed to decrease and additional larger thin platelets are observed after 120 min of ageing. These larger platelets have grown even thicker and show well defined facets after the solvothermal treatment as shown in Fig. 2. In combination with the temporal evolution of the crystalline phases known from the PXRD results, the immediately formed aggregated spheres are assigned to the hydrozincite phase and the larger platelets formed between 60 and 120 min of ageing, which grow under solvothermal conditions to a crystal habitus well-known for hydrotalcite-like materials,44 are assigned to zaccagnaite.
 | ||
| Fig. 2 SEM images depicting the morphology of the precursor materials recovered at different ageing times after co-precipitation at 65 °C and a pH of 6.4 of a zinc–aluminium solution with an aluminium content of xAl = 0.02. | ||
The investigation of the ageing time series at 2% aluminium demonstrated that aluminium will thermodynamically favour an incorporation into the zaccagnaite phase, which evolved after 120 minutes ageing time and further grows by solvothermal treatment. This results in an aluminium segregation and in an inhomogeneous aluminium distribution in the solid. To suppress this untargeted phase and to receive a more homogeneously doped hydrozincite, the ageing time was set to 10 min for the aluminium concentration variation series. With that shortened ageing time, a kinetically controlled aluminium incorporation into the hydrozincite phase should be favoured.
An increase in the BET surface area of the precursor after 120 minutes ageing time gives an additional indirect hint for a homogeneous aluminium distribution at short ageing times (Fig. S5†). The surface area of the precursor samples aged up to 60 minutes varied around 12 m2 g−1 and found to grow to 28 m2 g−1 after solvothermal treatment despite the newly grown phase clearly exhibiting larger particles. This might be explained by the effect of the aluminium ex-solution on the hydrozincite material leaving a more porous morphology. More evidence for an incorporation of aluminium into this phase is presented below for the aluminium concentration series.
Aluminium concentration variation
To avoid side-phase formation, the ageing time was reduced to 10 minutes, to kinetically trap the aluminium ions in the hydrozincite structure. The aluminium content was varied between 0% (xAl = 0) and 10% (xAl = 0.1) in different intervals and the recovered precursors were analysed regarding crystalline phase composition by PXRD (Fig. 3, selected range and Fig. S9† complete range). For all synthesized precursors, hydrozincite is determined as the main crystalline phase. An aluminium content higher than xAl = 0.02 resulted in small quantities of the above-described aluminium-rich side phase zaccagnaite despite the low ageing time (Fig. 3b).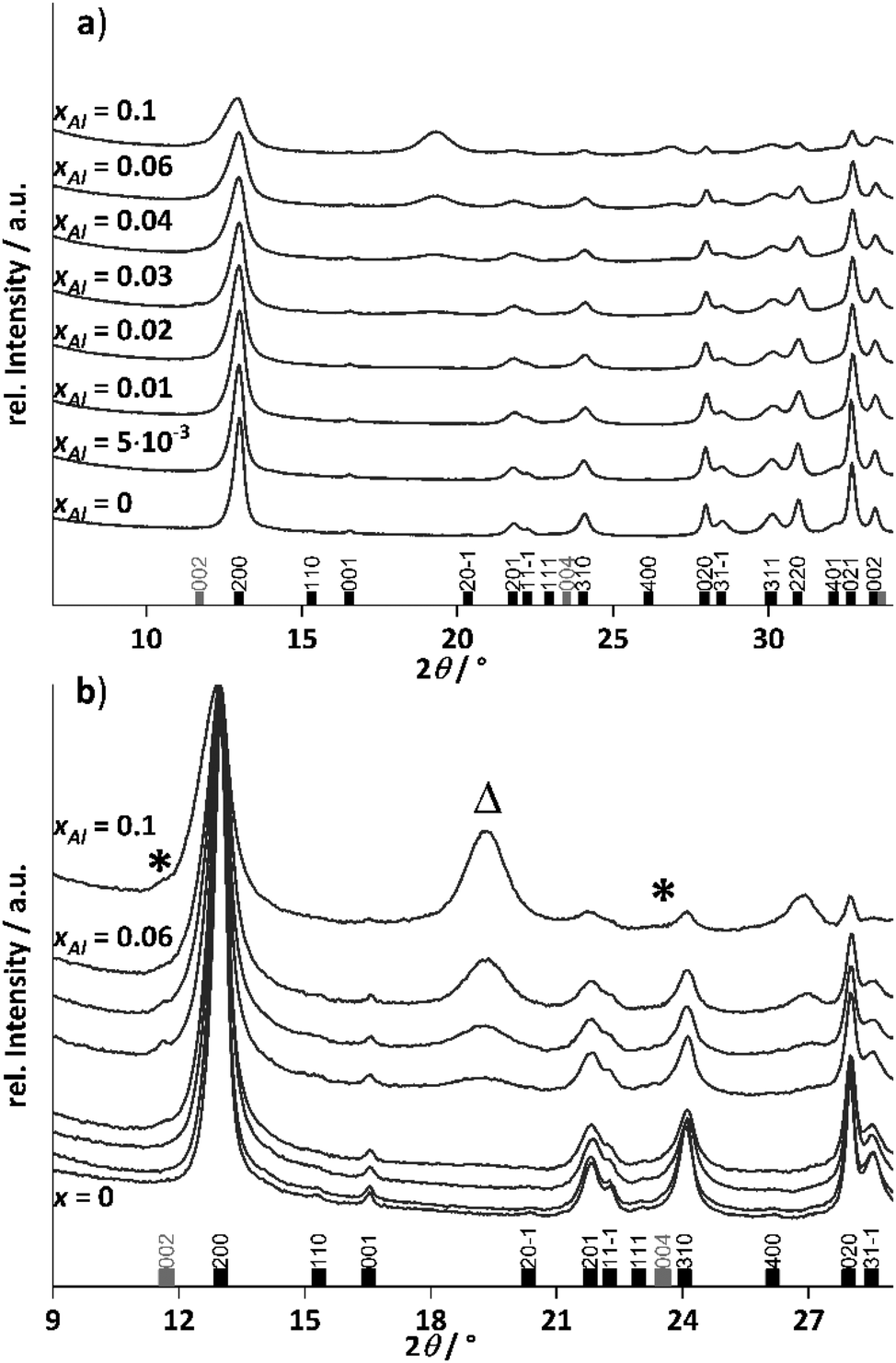 | ||
| Fig. 3 PXRD pattern of the co-precipitated aluminium-doped hydrozincite precursors with different cation-based molar aluminium fractions xAl. The reference pattern of the hydrozincite was taken from the ICSD (#16583, black bars) as well as the reference pattern of zaccagnaite (#190041, grey bars). (a) is an overview with indexation of the reflections, while (b) is a zoom at lower diffraction angles to better distinguish the zaccagnaite side-phase (marked with stars). The triangle marks an additional reflection discussed in the main text below. | ||
The weak reflections present as shoulders to the first hydrozincite peak indicate that the zaccagnaite structure is present in low concentration (marked with stars). Given that this reflection is hardly discernible and broadened, i.e. on the level of detection, it may account to ∼1–2 wt%. However, the relative intensity of the zaccagnaite reflections does not increase linearly with the aluminium content after their first appearance, which excludes a simple aluminium saturation of the hydrozincite phase with all excess aluminium being segregated into zaccagnaite.
The diffraction pattern of the hydrozincite shows noticeable changes with increasing incorporation of Al: there are slight shifts in the position of the reflections, which become noticeable in particular at higher diffraction angles. The cell volume of the samples was extracted via a Pawley fit and the results are summarized in Fig. 4.
 | ||
| Fig. 4 Cell volume and volume weighted average domain size estimated from the h00 reflections of the hydrozincite samples versus cation-based molar aluminium fraction xAl. | ||
The cell shrinks by ∼2 Å3 for the hydrozincite containing 10 at% Al compared to the unsubstituted compound, which can be expected from the lower ionic radius of Al3+ (54 pm) compared to Zn2+ (74 pm).45 For xAl = 0.1 the error becomes fairly large, which is expected due to the overall broadening of the reflections. This in turn hampers a precise determination of the lattice parameters. The line broadening was modelled assuming the effect to be caused by finite crystallite size, which can be justified by the information from electron microscopy. The crystallite size was found to be anisotropic, not unexpected for a layered material.
Furthermore, reflections of type h00, 0k0 and 00l are fairly sharp, where the first one describes the stacking of the layers while the latter two are related to the layer constitution. The cross plane reflections (e.g. 201, 301, 311) show stronger broadening, i.e. there is a loss of coherence among the layers, which is commonly observed for stacking faults in layered materials46,47 and was described earlier for synthetic hydrozincites.48 For an estimation of the crystallite size, the value for the h00 reflections is reported, which varies with a similar non-monotonous dependence on xAl as the cell volume does.
Aside of the zaccagnaite side phase, there are two prominent reflections located at 19.5 and 26.8°2θ appearing with increasing amount of Al3+ in the sample. They could not be assigned to any phase after extensive search in the COD and ICSD databases. Interestingly, the one located at 19.5°2θ has a d-spacing in excellent agreement with the a parameter and may be indexed as 300 reflection, which is forbidden in the space group C2/m that hydrozincite crystallizes in. For the second reflection, no such coincidence could be identified.
To further investigate the origin of these additional reflections, temperature resolved PXRD analysis of the sample with the highest aluminium content (xAl = 0.1) was performed (Fig. 5).
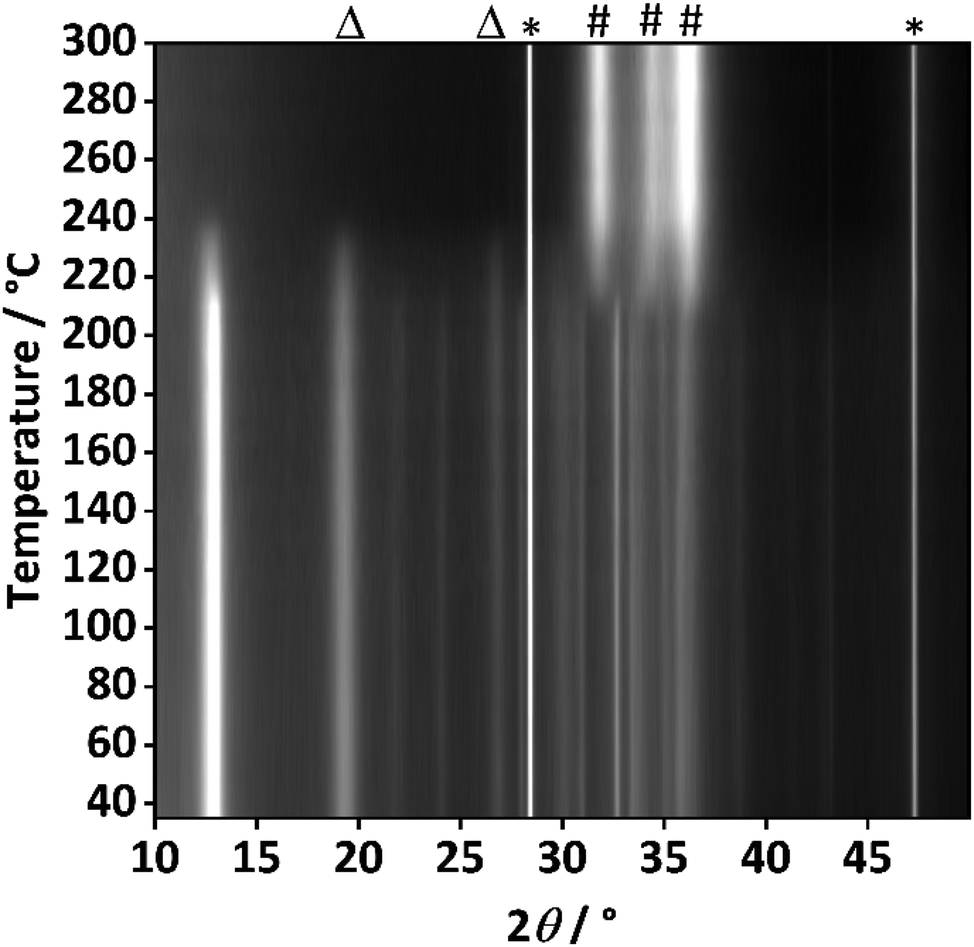 | ||
| Fig. 5 Temperature-resolved PXRD analysis of the thermal decomposition of the hydrozincite precursor containing 10% aluminium (xAl = 0.1). The PXRD analysis was performed with silicon as standard and in static air. The reflection positions are labelled as follows: silicon (*), ZnO (#) and additional unassigned reflections (Δ). | ||
At 215 °C the decomposition starts, which is evidenced by the most intense reflections of the hydrozincite phase (e.g. at 2θ = 12.9°) losing intensity. Complete decomposition is achieved at 250 °C. Simultaneously, the zinc oxide reflections evolved and further increase in intensity as the temperature was elevated, at 300 °C already well-defined reflections of ZnO are visible. This prompt crystallization is markedly different from the case of zaccagnaite (Fig. S8†), where even at 600 °C only very broad reflections of ZnO can be observed. This may be related to the presence of amorphous alumina, hampering the diffusion and crystallite growth of ZnO. Interestingly, the additional reflections at 2θ = 19.5° and 26.8° behaved like those assigned to the hydrozincite regarding the thermal decomposition, which further indicates that they do belong to a disordered hydrozincite phase and not to any side-phase like disordered aluminium hydroxides, although the presence of such phase cannot be ruled out completely based on the experimental evidence.
The potentially high degree of substitution of Zn2+ by Al3+ is likely to cause significant changes in the structure of hydrozincite. This raises the question of how the additional positive charge is compensated. Three possible scenarios, illustrated in Fig. 6, affecting the cation- or anion lattice shall be outlined: the surplus positive charge may be compensated by additional anions (OH− or CO32−) that may be introduced in the interlayer space and lead to a chemical formula (Zn5−5xAl5x(OH)6+5x(CO3)2) or (Zn5−5xAl5x(OH)6(CO3)2+2,5x). Also, deprotonation of hydroxyl groups could compensate the extra charge according to a chemical formula (Zn5−5xAl5x(OH)6−5x(O)5x(CO3)2). Alternatively, for any two Al3+ cations one Zn2+ cation might become a vacancy, which could affect in particular the Zn2+ in tetrahedral coordination since the layer made up of Zn–O octahedra already contains vacancies in the neighbourhood of the tetrahedrally coordinated Zn2+, which could be filled with Al3+: (Zn5−7,5xAl5x(OH)6(CO3)2). A loss of those tetrahedral sites would also disrupt the link between the layers facilitated by the carbonate anions, which would allow for an increased number of stacking faults. This in turn would be a viable explanation for the increasing line width and change of relative intensities in the diffraction patterns with increasing Al content. However, the full structure determination of the potentially modified hydrozincite phase is beyond the scope of the present work.
 | ||
| Fig. 6 Schematic depiction of potential defects in hydrozincite for balancing the surplus positive charge resulting from substitution of Zn2+ by Al3+: 1. Additional anions occupy the interlayer space. 2. Deprotonation of OH− to form O2− anions. 3. Vacancies on Zn2+ sites. The hydrogen atoms were placed at 0.9 Å distance and are not part of the crystal structure.43,48 | ||
SEM analysis of the concentration series shown in Fig. 7 revealed that the sample with an aluminium amount xAl = 0.01 contained aggregates of platelets, which formed spherical structures, as already observed in the ageing time series. Based on the assignment introduced above for the ageing time series, first additional larger platelets (highlighted by the arrows), which are encountered in the precursor with xAl = 0.03 aluminium content, are assigned to the zaccagnaite side-phase. Selected SEM-EDX spectra of these larger platelets are shown in the ESI in Fig. S12† and the results are listed in Table S2.† The median cationic ratio of Zn2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al3+ = 3 agreed well with the expected composition of zaccagnaite.44 TEM analysis of the precursors was complicated due to high beam sensitivity and is described as ESI (Fig. S7†).
:Al3+ = 3 agreed well with the expected composition of zaccagnaite.44 TEM analysis of the precursors was complicated due to high beam sensitivity and is described as ESI (Fig. S7†).
 | ||
| Fig. 7 Morphology evolution in dependency of the aluminium content of the hydrozincite precursors. The arrows show the regions containing larger platelets and xAl indicated the cation-based molar aluminium fraction. | ||
In order to characterize the aluminium environment in the hydrozincite precursors, a 27Al MAS NMR spectrum of the hydrozincite sample containing 3% aluminium (xAl = 0.03) was recorded (Fig. 8). The single peak at δobs = 14 ppm is indicative of aluminium occupying an octahedrally coordinated site, i.e. substituting a Zn atom in the hydrozincite structure.4027Al NMR peaks corresponding to a different coordination number are not observed. In comparison, the 27Al NMR spectrum of zaccagnaite Zn4Al2(OH)12(CO3)·3H2O exhibits two different signals at δobs = 15 ppm and δobs = 13 ppm. A 27Al 5QMAS NMR spectrum (Fig. S28†) does not resolve any further peaks and shows fairly broad signals, which is fully consistent with the published disordered crystal structure.42
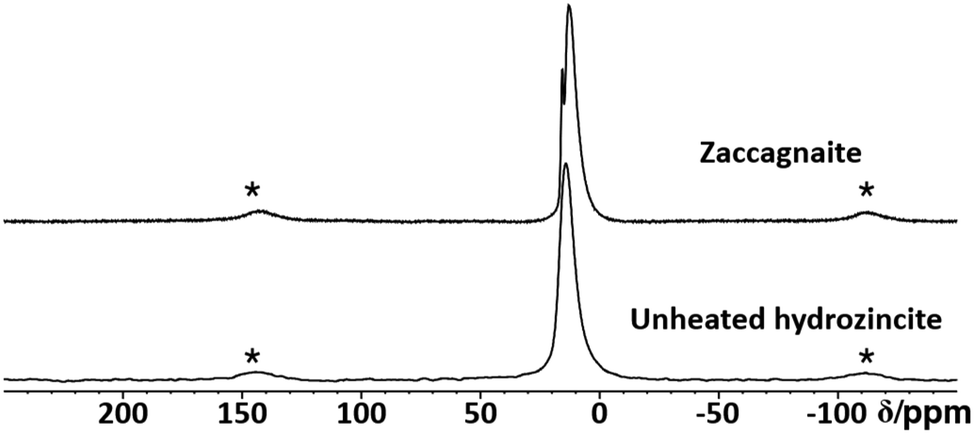 | ||
| Fig. 8 Stack plot of 27Al MAS NMR spectra of a coprecipitated hydrozincite precursor with xAl = 3% aluminium incorporation and zaccagnaite. Spinning side bands are labelled with a star (*). | ||
To further investigate the precursor samples, infrared and Raman spectroscopy have been applied. In the infrared spectra shown in Fig. 9, bands typical for hydrozincite were found.49 For a better comparison between hydrozincite and side-phases, a phase-pure zaccagnaite sample was synthesized as described in the ESI (Fig. S6†) and used as reference for the analysis of vibrational spectroscopy data. The infrared spectra (Fig. 9) show a rather gradual evolution with increasing aluminium content and a clear difference to the zaccagnaite reference pattern demonstrating that the observed bands can be assigned to the hydrozincite phase. The strongest changes are observed for the two antisymmetric carbonate stretching modes (ν3, 1502–1396 cm−1)50 when the aluminium content exceeds xAl = 0.03, i.e. in the same compositional range where the unassigned reflections in PXRD at 2θ = 19.5° and 2θ = 26.8° arise. At the same time, the band at 948 cm−1 disappears, which – in analogy to the hydrotalcite-like materials – is related to hydrogen bonds between hydroxyls and carbonate.51,52 Such hydrogen bonds are also present in hydrozincite48 and thus the vanishing of this band indicates breaking of these bonds. These gradual changes support the defective hydrozincite structure by aluminium incorporation in agreement with the observed additional reflections in PXRD (Fig. 3). An infrared band at 621 cm−1 appeared for xAl > 0.01 and increased with the aluminium content. For hydrotalcites, this band was assigned to hydroxyl groups between the sheets.53 These observations suggest that the anions of hydrozincite are affected by the charge compensating mechanism when zinc is substituted by aluminium through coordination changes of carbonate and hydroxyl. A similar gradual evolution with increasing aluminium content was also found by Raman spectroscopy (Fig. S17†). The Raman modes at 564 cm−1 (Zn-OH) and 494 cm−1 (Me-OH) were present in the samples with xAl = 0.06 and xAl = 0.1 and represent the side-phase54–56 zaccagnaite in agreement with the PXRD results.
 | ||
| Fig. 9 Vibrational infrared spectroscopy of aluminium doped hydrozincites with different cation-based molar aluminium fractions xAl in comparison with the aluminium rich zaccagnaite side phase Zn4Al2(OH)12(CO3)·3H2O. | ||
Summarizing the precursor part
It was established that short ageing time and low aluminium content can suppress the segregation of crystalline zaccagnaite. The samples without this aluminium side phase are interesting candidates for the synthesis of aluminium-doped zinc oxide due to their more uniform dopant distribution. The creation of structural defects is suggested for charge compensation upon substitution of zinc in crystalline hydrozincite.Characterization of the doped zinc oxide samples
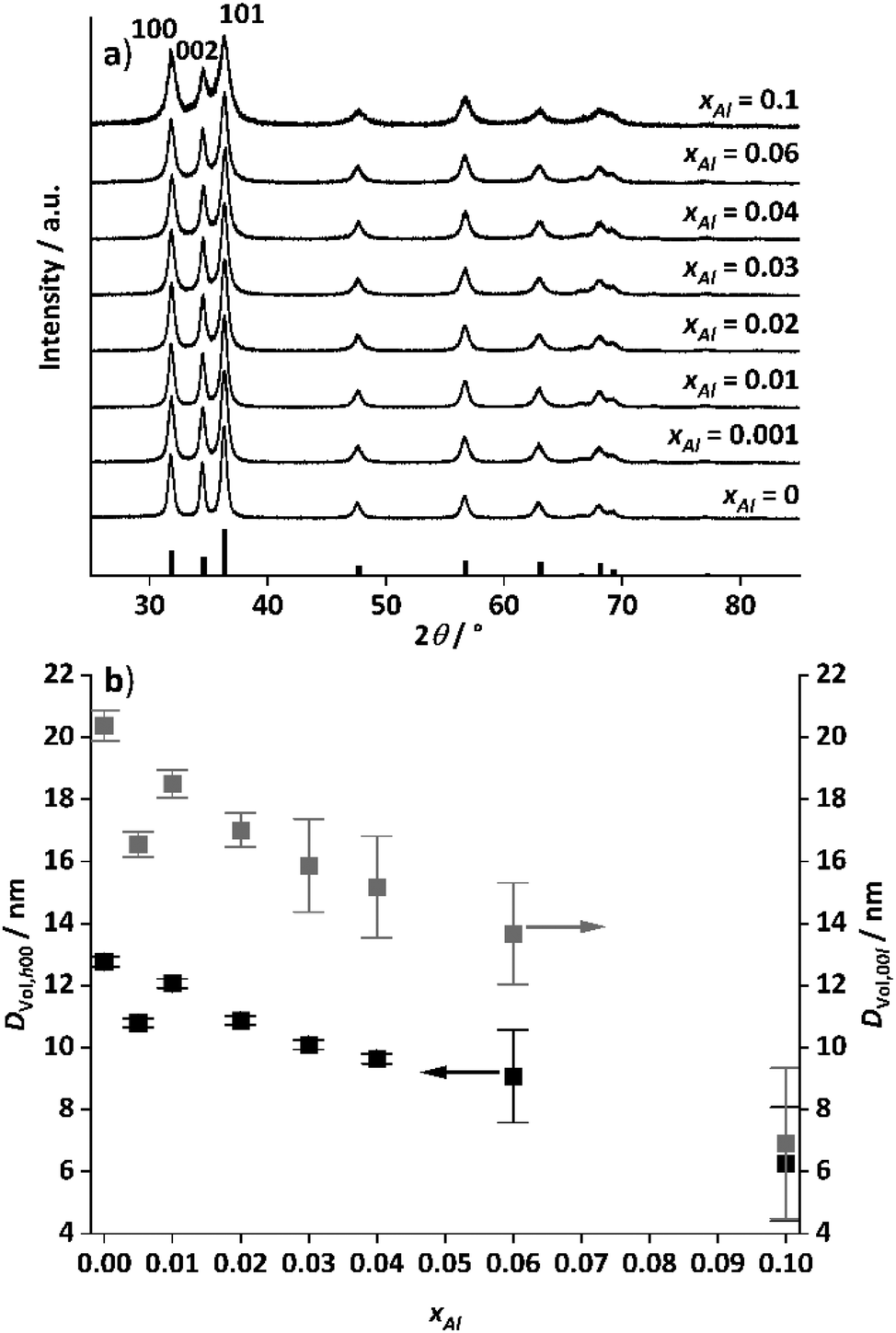 | ||
| Fig. 10 Powder XRD pattern of the aluminium doped zinc oxides (a) after calcination of the hydrozincite precursors at 320 °C with 2 °C min−1 heating ramp and 4 h holding time in static air. The reference pattern (black bars) was taken from COD, zinc oxide (#2107059). (b) Sizes of the coherently scattering domains for the h00 and 00l reflections extracted via Pawley fit from the patterns shown in (a) as function of the cation-based molar aluminium fraction xAl. | ||
Nitrogen physisorption measurements revealed a surface area enlargement after calcination as a result of the decomposition of the hydroxycarbonate by simultaneous pore formation due to water and carbon dioxide emission. There is a clear trend towards larger surface areas for increasing aluminium content with a local maximum at xAl = 0.005 ranging from 31 m2 g−1 (xAl = 0, Fig. S19†) to 106 m2 g−1 (xAl = 0.1, Fig. S19†). The main pore fraction are 10 nm mesopores for the samples up to xAl = 0.02. With an increase in the aluminium content, this fraction increased. For a higher amount than xAl = 0.03, the pore fraction of the 20–30 nm pores starts to increase with further increase in the total pore volume (Fig. S20†).
 | ||
| Fig. 11 TEM-analysis of an aluminium doped zinc oxide sample (with xAl = 0.03). Image (a) visualizes a typical aggregate as it was also found by SEM. The corresponding electron diffraction pattern is shown in figure (b). The image in (c) shows a large single crystal with some fractions of the typical aggregate structure on top. A HRTEM micrograph of a part of that single crystal is shown in (d) with the corresponding FFT pattern. | ||
The larger dimension of this platelet particle suggests that it originates from the zaccagnaite precursor side-phase. Indeed, EDX measurements were performed at several sample positions in the scanning TEM (STEM) mode and, hereby, a wide range of Al contents can be found that are all larger than the nominal 3% and span from 4.5% up to 38.6% (Fig. S22†). The highest aluminium concentrations were found where this thin ex-zaccagnaite platelet is not decorated by the nano-scaled ex-hydrozincite material indicated by the dark contrast in the high-angle annular dark field (HAADF)-STEM image. It is intriguing that it is at these aluminium-rich positions where the FFT has shown zinc oxide as only crystalline component. This suggests that the zinc and aluminium fractions of zaccagnaite have segregated upon decomposition into crystalline zinc oxide and amorphous alumina or nano-scale ZnAl2O4 spinel at a very small scale.
In summary, the electron microscopy investigation revealed the co-existence of two material systems in the sample with 3% aluminium. The major fraction of the material is ex-hydrozincite, i.e. crystalline zinc oxide with an aluminium content close to the nominal value. A minor, but increasing fraction, starting at xAl = 0.03 according to (precursor) PXRD, is ex-zaccagnaite and thus aluminium-richer and nano-structured in a complex manner containing crystalline zinc oxide and amorphous alumina segregated probably at the platelet surface.
 | ||
| Fig. 12 Stack plot of 27Al MAS NMR spectra of the unheated hydrozincite, zaccagnaite and after being annealed at 200 °C, 240 °C, 280 °C and 320 °C for 4 h in static air with a heating ramp of 2 °C min−1 with xAl = 0.005 aluminium incorporation. All spectra were recorded with a spinning frequency of 20 kHz. Spinning side bands are signed with a star (*). | ||
After the decomposition of the hydrozincite, there are two different signals observable: δobs = 82 ppm and δobs = 12 ppm. The sharp signal at δobs = 82 ppm has unambiguously been assigned to Al3+ on a zinc position inside the ZnO crystal structure,  .27 This small linewidth reflects the low quadrupolar coupling constant and an ordered environment of the
.27 This small linewidth reflects the low quadrupolar coupling constant and an ordered environment of the  defect. In contrast, zaccagnaite has a disordered crystal structure42 and even without Al substitution hydrozincite has been reported to have a strong tendency for stacking disorder.48 The second signal at δobs = 12 ppm can result from unreacted hydrozincite, but also from a disordered sixfold coordinated aluminium environment at the surface of ZnO particles.25 With increasing temperatures up to 320 °C, a growth of the
defect. In contrast, zaccagnaite has a disordered crystal structure42 and even without Al substitution hydrozincite has been reported to have a strong tendency for stacking disorder.48 The second signal at δobs = 12 ppm can result from unreacted hydrozincite, but also from a disordered sixfold coordinated aluminium environment at the surface of ZnO particles.25 With increasing temperatures up to 320 °C, a growth of the  signal is observed at the expense of the signal assigned to Al in unreacted hydrozincite (or in sixfold coordination). Here, the greatest build-up occurs between 200 and 240 °C, which is consistent with the formation of ZnO described by variable temperature PXRD (Fig. 5) and the decomposition of hydrozincite. The presence of a zaccagnaite side phase could not be observed by 27Al MAS NMR during the decomposition of the hydrozincite.
signal is observed at the expense of the signal assigned to Al in unreacted hydrozincite (or in sixfold coordination). Here, the greatest build-up occurs between 200 and 240 °C, which is consistent with the formation of ZnO described by variable temperature PXRD (Fig. 5) and the decomposition of hydrozincite. The presence of a zaccagnaite side phase could not be observed by 27Al MAS NMR during the decomposition of the hydrozincite.
In order to find out whether the in-literature-postulated solubility limit of Al3+ in ZnO25–30 is an explanation for the occurrence of extra peaks, a series of samples with different aluminium concentrations was investigated (Fig. 13).
 | ||
| Fig. 13 Stack plot of 27Al MAS NMR spectra of zinc oxide, annealed with 320 °C for 4 h, with different cation-based molar aluminium fractions xAl. All spectra were recorded with a spinning frequency of 20 kHz. Spinning side bands are signed with a star (*). | ||
With increasing aluminium concentration, two further signals are observed: δobs = 75 ppm and δobs = 47 ppm. Their chemical shift indicates that δobs = 75 ppm corresponds to a fourfold coordinated aluminium environment, while δobs = 47 ppm corresponds to a fivefold coordinated aluminium environment. Due to their broad line shape and their continuous growth with increasing aluminium concentrations δobs = 75 ppm, δobs = 47 ppm and δobs = 12 ppm, can be assigned to disordered aluminium environments not situated within the crystal structure of ZnO. This is consistent with the observation that with increasing aluminium concentration a steady particle growth is observed for xAl ≥ 0.02 (see previous section Fig. 2). This interpretation is consistent with the areas of high Al concentration in the TEM experiments (Fig. S22†), which is expected when the Al concentration is low within ZnO and the surplus of Al is found segregated from ZnO in form of side phases. TEM experiments provide information from local projections along the electron beam and are thus not expected to return the (lower) average bulk value.
To obtain a better estimate for the maximum degree of Al substitution that can be achieved under these conditions, the amount of  signal at δobs = 82 ppm is determined as a function of the degree of substitution xAl (Fig. 14). What can clearly be seen is that the signal/mass ratio increases only up to values of 2%. By interpolation with two linear functions, the “saturation limit” can be determined to xAl = 0.013. This agrees with the reported rough estimates of <2 mol% aluminium content in zinc oxide from other NMR studies.25,27
signal at δobs = 82 ppm is determined as a function of the degree of substitution xAl (Fig. 14). What can clearly be seen is that the signal/mass ratio increases only up to values of 2%. By interpolation with two linear functions, the “saturation limit” can be determined to xAl = 0.013. This agrees with the reported rough estimates of <2 mol% aluminium content in zinc oxide from other NMR studies.25,27
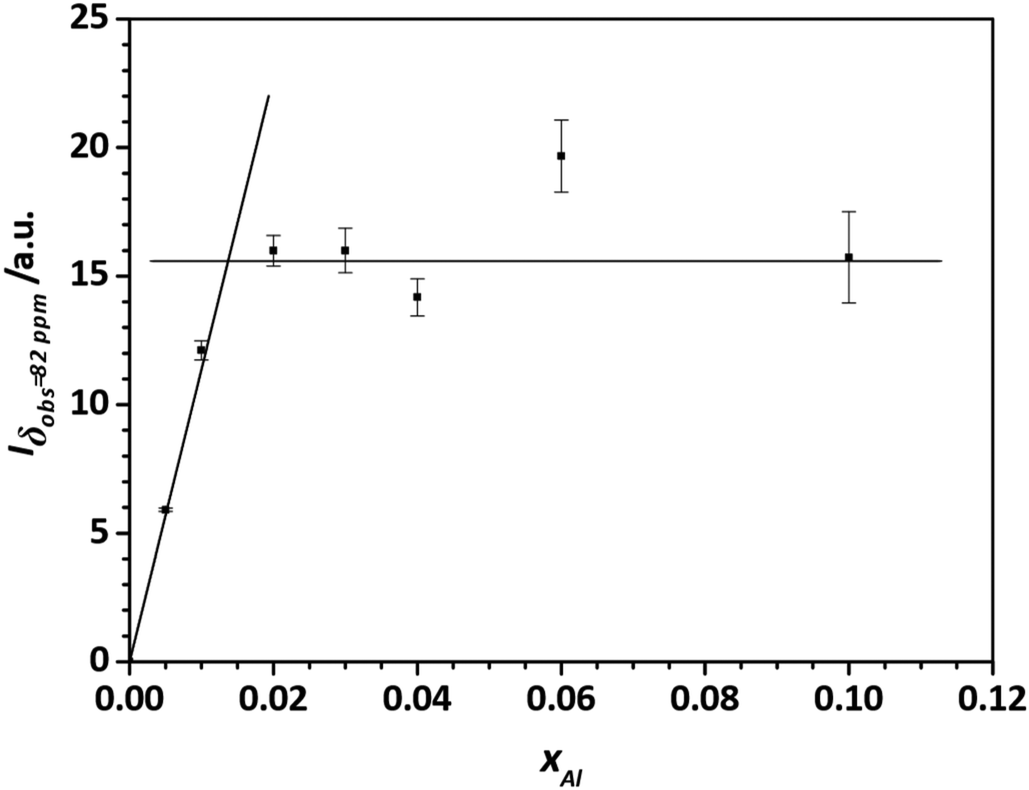 | ||
| Fig. 14 27Al-signal/mass ratio of the 27Al MAS NMR signal at δobs = 82 ppm of zinc oxide, annealed with 320 °C for 4 h, with different cation-based molar aluminium fractions xAl. The NMR spectra were excited with selective π/2 pulse and the peak area determined by deconvolution with Gaussian/Lorentzian profiles. The error bars are estimated, by taking an error of 1% of the four-fold coordinated aluminium 27Al MAS NMR signals. The lines to interpolate the substitution limit were obtained by fitting linear functions into the corresponding data points. | ||
Optical band gap of doped zinc oxides
Because of the expected influence of the aluminium doping on sites, UV/Vis spectroscopy was performed on the zinc oxide samples. The results of the band gap energies are shown in Fig. 15.
sites, UV/Vis spectroscopy was performed on the zinc oxide samples. The results of the band gap energies are shown in Fig. 15.
 | ||
| Fig. 15 Band gap energies of the aluminium doped zinc oxides with different cation-based molar aluminium fraction xAl. The measurements were performed at room temperature (25 °C, circles) at −185 °C (triangles) and at 27 °C after cryogenic temperature measurement (upside down triangle). The measurements were performed under vacuum in transmission mode on a pellet containing dried KBr as diluent. The lines are to guide the eye. | ||
The band gap determination was performed at room temperature and at −185 °C to exclude heat effects. Afterwards, the measurement was repeated upon temperature increase back to 27 °C to check for thermally induced changes. The similar band gap energies before (25 °C) and after (27 °C) cooling demonstrate good reversibility within an error of around 6 meV. A band gap near 3.30 eV was found for pure zinc oxide at room temperature, which is in alignment with reported values around 3.3 eV.1–4 No clear trend was observed at low doping level, while a slight increase in band gap energy was determined for aluminium contents above 1%. This might be caused to the segregation of aluminium in ex-zaccagnaite regions as it was suggested by the TEM results. Because of the wide band gap of aluminium oxide (ca. 5.6 eV), the presence of disordered alumina could shift the band gap energy of the material to higher values.59 Generally, the band gap can be affected by defects, free excitons, impurities and by the lattice site occupied by the dopant.17,60–65 To exclude free excitons, the band gap energies were recorded at −185 °C. Compared to the room temperature spectra, the absolute energies shifted to higher values. The band gap energy of the undoped ZnO was increased to 3.37 eV at −185 °C. However, the relative trend between the samples was mostly maintained with the exception that the lowest band gap was determined for the sample with xAl = 0.01 in the cryogenic measurements (3.36 eV), which is in the range of the highest occupancy of the  site. This finding is in agreement with the model of the band gap renormalisation, which predicts an optimal band gap for zinc oxide, if aluminium substitutes a zinc site.17
site. This finding is in agreement with the model of the band gap renormalisation, which predicts an optimal band gap for zinc oxide, if aluminium substitutes a zinc site.17
The behaviour of the zinc oxide sample with xAl = 0.005 aluminium with its wider band gap than xAl = 0 and xAl = 0.01, however, cannot be explained easily so far. This observation might be related to the Burnstein–Moss effect, which predicts a band gap widening if the lowest conduction band states are occupied by electrons introduced by the dopant and the next free lower level is at higher energies compared to the undoped sample.66,67 Altogether, the band gap trend is complex and likely caused by several effects such as the Burnstein–Moss effect, proper 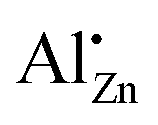 doping and the segregation of alumina when the solubility limit is exceeded.
doping and the segregation of alumina when the solubility limit is exceeded.
Doped zinc oxides at higher calcination temperature
Often, calcination temperatures higher than 320 °C are used to synthesize aluminium doped zinc oxides and to investigate their opto-electrochemical properties.22–24,27 A temperature series of aluminium doped hydrozincite with xAl = 0.03 was prepared to investigate the effect of higher calcination temperature and to learn about the thermodynamics of the doped system. Undoped zinc oxide was treated in the same way as a reference material. The xAl = 0.03 sample was chosen because this was the maximum amount aluminium which could be incorporated into the hydrozincite with only very little side-phase formation observable in PXRD. The calcination of the hydrozincite precursor was always performed by heating up in air from room temperature to the target dwell temperatures, which were held for four hours. PXRD shows that the reflections become sharper after calcination at higher temperatures (Fig. 16) indicating that the crystallinity of the sample increased with temperature as expected. At a temperature of 920 °C, reflections of the ZnAl2O4 spinel phase were observed, which increased in intensity with further temperature increase. Simultaneously, a change in morphology was observed by SEM. The images of the undoped sample (Fig. 17) demonstrate that the morphology at 420 °C is still similar to that described above for 320 °C calcination temperature. At 520 °C the platelets were thicker, and a few larger holes demonstrate a beginning sinter effect. Further increase in temperature resulted in an enhanced sinter effect and an enlargement of the particles, losing their original nanostructure. At 1100 °C the zinc oxide has crystallized into bulky, roundish particles with a clearly decreased porosity. | ||
| Fig. 16 PXRD pattern of the aluminium doped zinc oxide with a cation-based molar aluminium fraction xAl = 0.03 after calcination of the hydrozincite precursors at different temperatures with 2 °C min−1 heating ramp and 4 h holding time in static air. Reflections assigned to ZnAl2O4 spinel are marked with a star (*). The reference pattern of zinc oxide (black bars) was taken from COD (#2107059) and the reference pattern of zinc–aluminium spinel (grey bars and stars in pattern) was taken from ICSD database (#75628). In (a) the complete range is shown. In (b) a zoom of the pattern with spinel reflections is presented. | ||
 | ||
| Fig. 17 Morphology evolution depending on the calcination temperature of the undoped zinc oxide. Calcination was performed with 2 °C min−1 heating ramp and 4 h holding time in static air. | ||
In order to obtain insight into the effect of thermal annealing on the presence of the different aluminium species, 27Al MAS NMR was performed (Fig. 18). Up to 620 °C, the presence of the aforementioned four different aluminium species is observed. During this process, a decrease in the  signal is observed (Fig. S22†). In thermodynamic equilibrium, the Al substitution limit is expected to be determined by the formation of spinel ZnAl2O4 side phase,30 which under low-temperature conditions amounts to a concentration limit far below the observed values.31 The expected higher substitution levels of the low-temperature route can be explained by the higher chemical potential of Al in the amorphous surface layers formed, which are less favourable to Al then that of the spinel phase. The formation of the spinel phase as seen by 27Al NMR (Fig. 18) begins at annealing temperature of 720 °C upwards and drastically reduces the amount of Al in all phases but not in the spinel phase ZnAl2O4. This is shown by the presence of an additional signal at δobs = 15 ppm corresponding to ZnAl2O4.68
signal is observed (Fig. S22†). In thermodynamic equilibrium, the Al substitution limit is expected to be determined by the formation of spinel ZnAl2O4 side phase,30 which under low-temperature conditions amounts to a concentration limit far below the observed values.31 The expected higher substitution levels of the low-temperature route can be explained by the higher chemical potential of Al in the amorphous surface layers formed, which are less favourable to Al then that of the spinel phase. The formation of the spinel phase as seen by 27Al NMR (Fig. 18) begins at annealing temperature of 720 °C upwards and drastically reduces the amount of Al in all phases but not in the spinel phase ZnAl2O4. This is shown by the presence of an additional signal at δobs = 15 ppm corresponding to ZnAl2O4.68
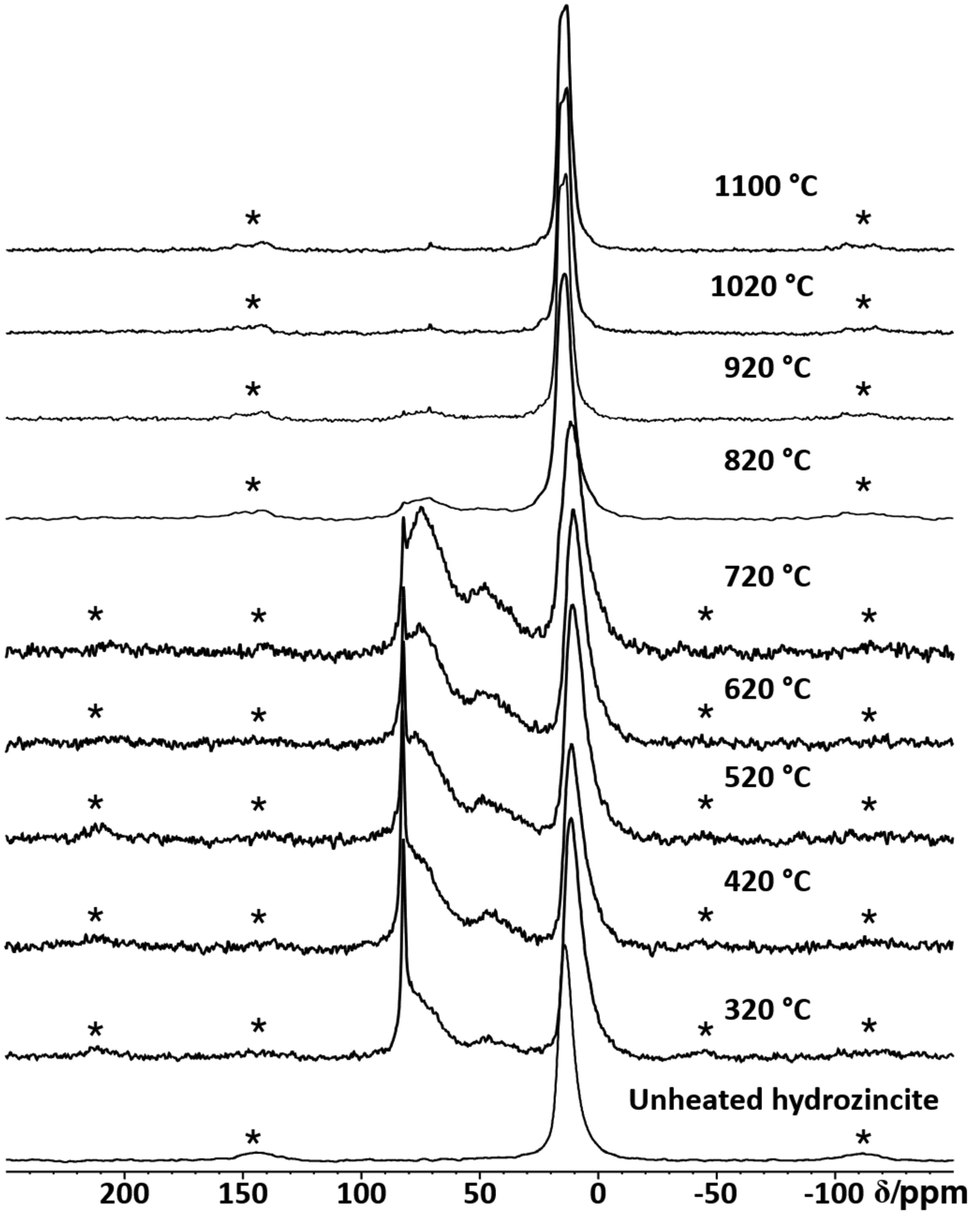 | ||
| Fig. 18 Stack plot of the 27Al MAS NMR spectra of aluminium doped zinc oxide with a cation-based molar aluminium fraction xAl = 0.03 after calcination at different temperatures. All spectra were recorded at a spinning frequency of 20 kHz. Spinning side bands are labelled with a star (*). | ||
At temperatures of 820 °C, the  signal in ZnO has dropped below the detection limit of xAl = 0.0005, which is consistent with the low equilibrium values determined in a previous study.24 We note that all (NMR visible) Al is consumed by the formed spinel ZnAl2O4 phase. At these high annealing temperatures, only two 27Al MAS NMR signals are observed: δobs = 15 ppm, which corresponds to the octahedral coordinated aluminium environment in the ZnAl2O4 crystal structure, and δobs = 75 ppm, which likely results from a cation inversion defect of the zinc and aluminium.68,69
signal in ZnO has dropped below the detection limit of xAl = 0.0005, which is consistent with the low equilibrium values determined in a previous study.24 We note that all (NMR visible) Al is consumed by the formed spinel ZnAl2O4 phase. At these high annealing temperatures, only two 27Al MAS NMR signals are observed: δobs = 15 ppm, which corresponds to the octahedral coordinated aluminium environment in the ZnAl2O4 crystal structure, and δobs = 75 ppm, which likely results from a cation inversion defect of the zinc and aluminium.68,69
What can be concluded is that the low-temperature approach achieves a much higher Al3+ substitution of the Zn2+ ions in ZnO than the equilibrium concentration would permit. Furthermore, this high substitution level can be maintained up to a temperature of about 720 °C when the activation energy for the formation of spinel crystallite is overcome and low thermodynamic substitution levels in ZnO are observed. It can be concluded that n-doping of ZnO by Al is thus stable under the conditions relevant for the catalytic process.
Together with the results from the composition series, where the 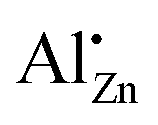 increased up to xAl = 0.01 after the calcination of an aluminium-doped hydrozincite, the observation made for the temperature series indicates that the substitution on a zinc site is rather a kinetic effect and can be facilitated by a proper formed precursor acting as a kinetic trap. A further increase in the aluminium content does not result in an increased number of substituted sites but increases the side-phase amount in the precursors phase which has a negative effect on the opto-electrochemical properties. A calcination temperature above 320 °C minimizes the number of substituted sites and is therefore disadvantageous. The results clearly demonstrate that the presence of aluminium-doped zinc oxide containing the
increased up to xAl = 0.01 after the calcination of an aluminium-doped hydrozincite, the observation made for the temperature series indicates that the substitution on a zinc site is rather a kinetic effect and can be facilitated by a proper formed precursor acting as a kinetic trap. A further increase in the aluminium content does not result in an increased number of substituted sites but increases the side-phase amount in the precursors phase which has a negative effect on the opto-electrochemical properties. A calcination temperature above 320 °C minimizes the number of substituted sites and is therefore disadvantageous. The results clearly demonstrate that the presence of aluminium-doped zinc oxide containing the  species has to be considered as component of a typical methanol catalyst support.11
species has to be considered as component of a typical methanol catalyst support.11
Conclusion
The aluminium speciation was found to be complex when co-precipitation of hydrozincite precursors, Zn5(OH)6(CO3)2, is used as a method inspired by catalyst synthesis to prepare aluminium-doped zinc oxide support materials. Thermodynamically favoured aluminium segregation into an aluminium-rich crystalline zaccagnaite side-phase, Zn4Al2(OH)12(CO3)·3H2O, was observed at aluminium contents higher than xAl = 0.02, for long ageing times and after solvothermal treatment. This side-phase was found to be responsible for a non-uniform aluminium distribution in the precursors and later in the calcined samples. Still, the results suggest that hydrozincite can take up aluminium in an octahedral coordination even for xAl > 0.02 leading to the formation of defects due to charge compensation in the hydroxycarbonate. Calcination at 320 °C leads to formation of zinc oxide as the only crystalline phase, but electron microscopy and 27Al NMR revealed non-uniform aluminium distribution and presence of diverse aluminium species for a doping level of xAl = 0.03 and larger. Such complexity is based on the ex-zaccagnaite regions, which were found to segregate into nano-crystalline zinc oxide with an aligned crystallographic orientation and into amorphous alumina. At lower aluminium contents, however, the dopant was found preferably on the zinc sites of the zinc oxide lattice based on the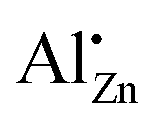 signal dominating the NMR spectra. The solubility limit regarding this species was determined to be approximately xAl = 0.013 or 1.3% of all metal cations. Annealing experiments showed that the substitution of zinc by aluminium is a kinetic driven process. The aluminium was kinetically trapped on the
signal dominating the NMR spectra. The solubility limit regarding this species was determined to be approximately xAl = 0.013 or 1.3% of all metal cations. Annealing experiments showed that the substitution of zinc by aluminium is a kinetic driven process. The aluminium was kinetically trapped on the  site up to a substitution limit which is much higher than the thermodynamic limit set by a segregation into zinc oxide and ZnAl2O4 spinel. This shows that lower calcination temperatures, such as applied in catalyst synthesis, favour the aluminium doping on that specific site. The investigation of these support materials under hydrogenation conditions and their application in catalytic reaction will be addressed in forthcoming work.
site up to a substitution limit which is much higher than the thermodynamic limit set by a segregation into zinc oxide and ZnAl2O4 spinel. This shows that lower calcination temperatures, such as applied in catalyst synthesis, favour the aluminium doping on that specific site. The investigation of these support materials under hydrogenation conditions and their application in catalytic reaction will be addressed in forthcoming work.
Author contributions
BM synthesized the materials and contributed to the characterization, data analysis and wrote the first draft of the manuscript. JKW conducted the NMR experiments, the data analysis and contributed to the preparation of the manuscript. SM conducted the PXRD refinements, contributed to experiments, design of experiments, data analysis and evaluation and preparation of the manuscript. US conducted the TEM measurements, contributed to design of experiments, data analysis and preparation of the manuscript. LK, JSG and MB contributed in conceptualization of the work, acquisition of the financial support, provision of study materials, reagents, materials, instrumentation, computing resources and other analysis tools, and preparation and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. B. gratefully acknowledges the financial support from the Mercator Research Centre Ruhr (MERCUR, Pe-2018-0034). Basic financial support from the federal state of Schleswig-Holstein is gratefully acknowledged. We also thank Dr K. Loza (University Duisburg-Essen) and Dr C. Szillus (Kiel University) for SEM analysis of the prepared materials and Maren Rasmussen, Benita Rönnau and Melina Ottenberg for help with the ICP and TG analysis at Kiel University.References
- V. Srikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447–5451 CrossRef CAS.
- K. Davis, R. Yarbrough, M. Froeschle, J. White and H. Rathnayake, RSC Adv., 2019, 9, 14638–14648 RSC.
- M. Izaki and T. Omi, Appl. Phys. Lett., 1996, 68, 2439–2440 CrossRef CAS.
- A. Arif, O. Belahssen, S. Gareh and S. Benramache, J. Semicond., 2015, 36, 13001 CrossRef CAS.
- A. B. Djurisić and Y. H. Leung, Small, 2006, 2, 944–961 CrossRef PubMed.
- K. Hagedorn, W. Li, Q. Liang, S. Dilger, M. Noebels, M. R. Wagner, J. S. Reparaz, A. Dollinger, J. Schmedt auf der Günne, T. Dekorsy, L. Schmidt-Mende and S. Polarz, Adv. Funct. Mater., 2016, 26, 3424–3437 CrossRef CAS.
- P. K. Samanta, Optik, 2020, 221, 165337 CrossRef CAS.
- E. R. Martins, Essentials of Semiconductor Device Physics, Wiley, 2022 Search PubMed.
- R. P. Huebener, Conductors, Semiconductors, Superconductors: An Introduction to Solid-State Physics, Springer International Publishing, 2019 Search PubMed.
- S. Rajendran, M. Naushad, L. C. Ponce and E. Lichtfouse, Green Photocatalysts for Energy and Environmental Process, Springer International Publishing, 2019 Search PubMed.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS PubMed.
- M. Behrens, Catal. Today, 2015, 246, 46–54 CrossRef CAS.
- T. Lunkenbein, F. Girgsdies, T. Kandemir, N. Thomas, M. Behrens, R. Schlögl and E. Frei, Angew. Chem., Int. Ed., 2016, 55, 12708–12712 CrossRef CAS PubMed.
- J. Schumann, M. Eichelbaum, T. Lunkenbein, N. Thomas, M. C. Álvarez Galván, R. Schlögl and M. Behrens, ACS Catal., 2015, 5, 3260–3270 CrossRef CAS.
- H. K. Park and J. Heo, Appl. Surf. Sci., 2014, 309, 133–137 CrossRef CAS.
- I. Y. Kim, S. W. Shin, M. G. Gang, S. H. Lee, K. V. Gurav, P. S. Patil, J. H. Yun, J. Y. Lee and J. H. Kim, Thin Solid Films, 2014, 570, 321–325 CrossRef CAS.
- A. Walsh, J. L. F. Da Silva and S.-H. Wei, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075211 CrossRef.
- K. E. Lee, M. Wang, E. J. Kim and S. H. Hahn, Curr. Appl. Phys., 2009, 9, 683–687 CrossRef.
- Y. Okuhara, T. Kato, H. Matsubara, N. Isu and M. Takata, Thin Solid Films, 2011, 519, 2280–2286 CrossRef CAS.
- V. J. Norman, Aust. J. Chem., 1969, 22, 325–329 CrossRef CAS.
- K. Shirouzu, T. Ohkusa, M. Hotta, N. Enomoto and J. Hojo, J. Ceram. Soc. Jpn., 2007, 115, 254–258 CrossRef CAS.
- M. H. Yoon, S. H. Lee, H. L. Park, H. K. Kim and M. S. Jang, J. Mater. Sci. Lett., 2002, 21, 1703–1704 CrossRef CAS.
- S. B. Majumder, M. Jain, P. S. Dobal and R. S. Katiyar, Mater. Sci. Eng., B, 2003, 103, 16–25 CrossRef.
- H. Serier, M. Gaudon and M. Ménétrier, Solid State Sci., 2009, 11, 1192–1197 CrossRef CAS.
- S. Ghosh, J. Sebastian, L. Olsson and D. Creaser, Chem. Eng. J., 2021, 416, 129120 CrossRef CAS.
- C. Schilling, M. Zähres, C. Mayer and M. Winterer, J. Nanopart. Res., 2014, 16, 2506 CrossRef.
- T. Tsubota, M. Ohtaki, K. Eguchi and H. Arai, J. Mater. Chem., 1997, 7, 85–90 RSC.
- Y. S. Avadhut, J. Weber, E. Hammarberg, C. Feldmann and J. Schmedt auf der Günne, Phys. Chem. Chem. Phys., 2012, 14, 11610–11625 RSC.
- R. Noriega, J. Rivnay, L. Goris, D. Kälblein, H. Klauk, K. Kern, L. M. Thompson, A. C. Palke, J. F. Stebbins, J. R. Jokisaari, G. Kusinski and A. Salleo, J. Appl. Phys., 2010, 107, 074312 CrossRef.
- A. Walsh and A. Zunger, Nat. Mater., 2017, 16, 964–967 CrossRef CAS PubMed.
- R. Hansson, P. C. Hayes and E. Jak, Metall. Mater. Trans. B, 2004, 35, 633–642 CrossRef.
- N. Roberts, R.-P. Wang, A. W. Sleight and W. W. Warren, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 5734–5741 CrossRef CAS.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS.
- S. J. Miao, R. N. d'Alnoncourt, T. Reinecke, I. Kasatkin, M. Behrens, R. Schlögl and M. Muhler, Eur. J. Inorg. Chem., 2009, 2009, 910–921 CrossRef.
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- R. W. Cheary, A. A. Coelho and J. P. Cline, J. Res. Natl. Inst. Stand. Technol., 2004, 109, 1–25 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- D. Jardon-Alvarez and J. Schmedt auf der Günne, Solid State Nucl. Magn. Reson., 2018, 94, 26–30 CrossRef CAS PubMed.
- S. Merlino and P. Orlandi, Am. Mineral., 2001, 86, 1293–1301 CrossRef CAS.
- R. P. Lozano, C. Rossi, Á. La Iglesia and E. Matesanz, Am. Mineral., 2012, 97, 513–523 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Layered Materials, Taylor & Francis, 2004 Search PubMed.
- E. J. Riedel and C. Janiak, Anorganische Chemie, Walter de Gruyter, Berlin, New York, 2007 Search PubMed.
- S. Bette, B. Hinrichsen, D. Pfister and R. E. Dinnebier, J. Appl. Crystallogr., 2020, 53, 76–87 CrossRef CAS PubMed.
- S. Mangelsen, B. R. Srinivasan, U. Schürmann, L. Kienle, C. Näther and W. Bensch, Dalton Trans., 2019, 48, 1184–1201 RSC.
- S. Ghose, Acta Crystallogr., 1964, 17, 1051–1057 CrossRef CAS.
- R. Sanna, G. De Giudici, A. M. Scorciapino, C. Floris and M. Casu, Am. Mineral., 2013, 98, 1219–1226 CrossRef CAS.
- D. Stoilova, V. Koleva and V. Vassileva, Spectrochim. Acta, Part A, 2002, 58, 2051–2059 CrossRef CAS PubMed.
- J. T. Kloprogge, D. Wharton, L. Hickey and R. L. Frost, Am. Mineral., 2002, 87, 623–629 CrossRef CAS.
- W. Kagunya, R. Baddour-Hadjean, F. Kooli and W. Jones, Chem. Phys., 1998, 236, 225–234 CrossRef CAS.
- J. T. Kloprogge and R. L. Frost, J. Solid State Chem., 1999, 146, 506–515 CrossRef CAS.
- J. T. Kloprogge, L. Hickey and R. L. Frost, J. Raman Spectrosc., 2004, 35, 967–974 CrossRef CAS.
- N. Buzgar and A. Apopei, The Raman study of certain carbonates, 2014, pp. 98–112 Search PubMed.
- S. J. Palmer, R. L. Frost and L. M. Grand, J. Raman Spectrosc., 2011, 42, 1168–1173 CrossRef CAS.
- J. P. Cline, M. Leoni, D. Black, A. Henins, J. Bonevich, P. Whitfield and P. Scardi, Powder Diffr., 2013, 28, S26–S32 CrossRef.
- J. Langford, A. Boultif, J. Auffrédic and D. Louër, J. Appl. Crystallogr., 1993, 26, 22–33 CrossRef CAS.
- M. Aguilar-Frutis, M. Garcia and C. Falcony, Appl. Phys. Lett., 1998, 72, 1700–1702 CrossRef CAS.
- Y. Ishikawa, K. Wada, D. D. Cannon, J. F. Liu, H. C. Luan and L. C. Kimerling, Appl. Phys. Lett., 2003, 82, 2044–2046 CrossRef CAS.
- A. P. Roth, J. B. Webb and D. F. Williams, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 25, 7836–7839 CrossRef CAS.
- W. Shan, W. Walukiewicz, J. W. Ager, K. M. Yu, H. B. Yuan, H. P. Xin, G. Cantwell and J. J. Song, Appl. Phys. Lett., 2005, 86, 191911 CrossRef.
- S. E. Aw, H. S. Tan and C. K. Ong, J. Phys.: Condens. Matter, 1991, 3, 8213–8223 CrossRef CAS.
- Y. Gu, I. L. Kuskovsky, M. Yin, S. O'Brien and G. F. Neumark, Appl. Phys. Lett., 2004, 85, 3833–3835 CrossRef CAS.
- B. K. Meyer, H. Alves, D. M. Hofmann, W. Kriegseis, D. Forster, F. Bertram, J. Christen, A. Hoffmann, M. Straßburg, M. Dworzak, U. Haboeck and A. V. Rodina, Phys. Status Solidi B, 2004, 241, 231–260 CrossRef CAS.
- E. Burstein, Phys. Rev., 1954, 93, 632–633 CrossRef CAS.
- D. G. Avery, D. W. Goodwin, W. D. Lawson and T. S. Moss, Proc. Phys. Soc., London, Sect. B, 1954, 67, 761–767 CrossRef.
- C. Yang, W. Zhu, S. Sen and R. H. Castro, J. Phys. Chem. C, 2019, 123, 8818–8826 CrossRef CAS.
- S. Sommer, E. D. Bøjesen, N. Lock, H. Kasai, J. Skibsted, E. Nishibori and B. B. Iversen, Dalton Trans., 2020, 49, 13449–13461 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00253e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |