
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopy, molecular structure, and electropolymerization of Ni(II) and Cu(II) complexes containing a thiophene-appending fluorinated Schiff base ligand†‡
Guillermo
Ahumada§
 *ab,
Paul
Hamon
b,
Thierry
Roisnel
b,
Vincent
Dorcet
b,
Mauricio
Fuentealba
c,
Loreto A.
Hernández
d,
David
Carrillo
a,
Jean-René
Hamon
*b and
Carolina
Manzur
*a
*ab,
Paul
Hamon
b,
Thierry
Roisnel
b,
Vincent
Dorcet
b,
Mauricio
Fuentealba
c,
Loreto A.
Hernández
d,
David
Carrillo
a,
Jean-René
Hamon
*b and
Carolina
Manzur
*a
aLaboratorio de Química Inorgánica, Instituto de Química, Facultad de Ciencias, Pontificia Universidad Católica de Valparaíso, Campus Curauma, Avenida Universidad 330, Valparaíso, Chile. E-mail: cecilia.manzur@pucv.cl
bUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: jean-rene.hamon@univ-rennes1.fr
cLaboratorio de Cristalografía, Instituto de Química, Facultad de Ciencias, Pontificia Universidad Católica de Valparaíso, Campus Curauma, Avenida Universidad 330, Valparaíso, Chile
dLaboratorio de Electroquímica, Instituto de Química y Bioquímica, Facultad de Ciencias, Universidad de Valparaíso, Avda. Gran Bretaña 1111, Playa Ancha, Valparaíso, Chile
First published on 3rd March 2023
Abstract
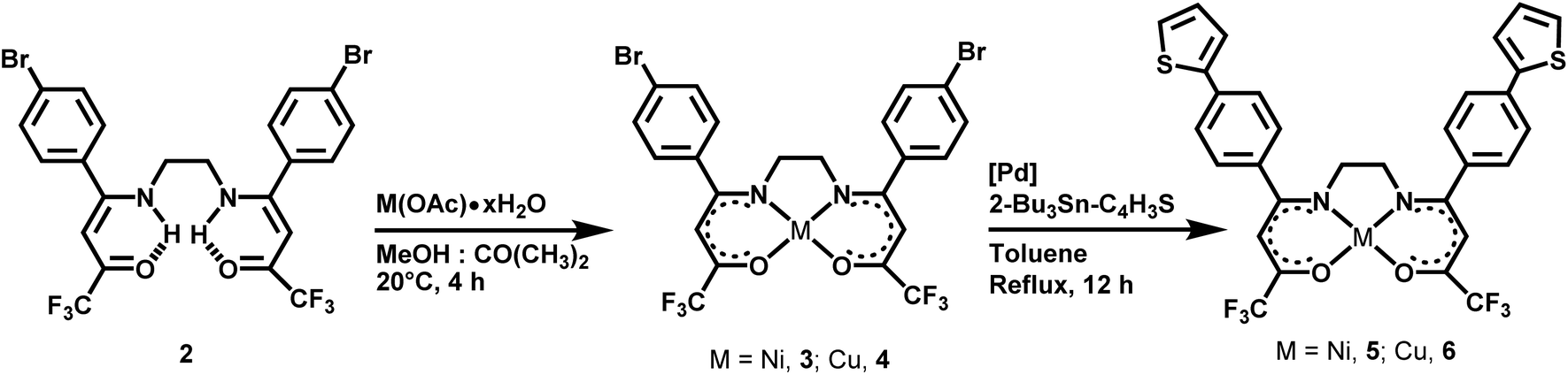
In this contribution, we describe the preparation, characterization, and electrochemical behavior of a series of four new mononuclear M(II) complexes featuring a symmetric substituted N2O2-tetradentate Schiff base ligand, bearing either trifluoromethyl and p-bromophenyl (M = Ni, 3; Cu, 4) or trifluoromethyl and the π-extended p-(2-thienyl)phenylene (M = Ni, 5; Cu, 6) substituents. Complexes 3 and 4 were readily synthesized by reacting the diprotic fluorinated Schiff base proligand 2 with the appropriate hydrated metal(II) acetates, whereas 5 and 6 were obtained upon Stille cross-coupling reaction of 3 and 4 with 2-(tributylstannyl)-thiophene, respectively. Compounds 3–6 were isolated as neutral, air, and thermally stable-coloured solids, with yields ranging from 60 to 80%. The four complexes, the diimine precursor 1 and its trifluoroacetylated derivative 2, were identified using analytical (EA, ESI-MS), spectroscopic (IR, 1H, 13C, and 19F NMR), and X-ray crystallographic methods. X-ray crystal structure determination of complexes 3–5 revealed that both four-coordinate Ni(II) and Cu(II) metal ions adopt a square planar geometry. The magnetic properties of powdered samples of the Cu(II) derivatives 4 and 6 have been investigated (2–300 K) and found consistent in both cases with a single isolated copper(II) ion (s = 1/2). DFT calculations were used to examine the optimal geometries of complexes 5 and 6, allowing for a consistent perspective of their structure and characteristics. The primary aspects of the UV-vis spectra were interpreted using TD-DFT computations. Finally, electrochemical data indicate that complexes 5 and 6 polymerize at high anodic potentials in acetonitrile (greater than 2.0 V vs. Ag/AgCl). Cyclic voltammetry, scanning electron microscopy, and energy-dispersive X-ray spectroscopy (SEM-EDS) analyses were used to characterize the obtained films poly-5 and poly-6.
Introduction
The incorporation of transition metal elements into conducting polymer chain provides an appealing platform,1 integrating the chemical, electrical, magnetic, optical, and redox properties of coordination compounds,2 with those of synthetic conducting polymers, such as significant strength, large Young's modulus, resistance to corrosion, low density and weight.3 These hybrid materials are attractive because of the feasibility of fine-tuning their electronic characteristics by modifying electronic interactions between the metal center and the polymer backbone's delocalized conjugated π-electrons.4,5 Furthermore, the interest in metal-containing polymers (MCP) lies in their promising applicability in electrical,6 optical,7,8 biological,9 and catalytic applications.10Conducting polymers can be easily obtained by the electrochemical oxidation of aromatic monomers on an electrode surface as a film.11,12 An attractive and well-known scaffold is thiophene,13 which can be electropolymerized at anodic potentials ranging between 1.2 to 1.6 V vs. a saturated calomel electrode (SCE, depending on the experimental conditions).14 Polythiophenes are especially attractive due to their lightweight, flexibility, structural versatility, and chemical, electrochemical and environmental stability,15–17 exceeding that of polypyrrole or polyaniline monomers.18 Also, these polymers are highly conductive (100 S cm−1) and have thermal and chemical stability.19 Moreover, electropolymerization is a proven method for assembling polymer structures, especially as thin insoluble films. Thiophene-functionalized monomers can integrate the functional and structural benefits of polymers with metals. As such, various structures and applications are feasible due to the broad spectrum of electropolymerizable monomeric thiophene-containing metal complexes.20–23
Complexes featuring N2O2-tetradentate Schiff bases ligands have gained interest in the field of coordination chemistry24–26 due to their impressive number of applications in different areas,27 such as in non-linear optics,28 biological chemistry,29 and catalysis.30,31 Besides, structural and electronic modifications over the Schiff base ligands bestow great versatility to the system in altering the architecture by modifying the substituents placed in the ligands.32,33 In addition, the electrochemical polymerization of Schiff base complexes containing 2-thienyl scaffolds has been leveraged in the fabrication of materials with applications in the fields of conducting metallopolymers,34–36 non-linear optics,37 transistors,38 spin-crossover,39 and catalysis.40,41
In recent years, our research team has been engaged in the preparation of metallopolymers using Schiff base complexes, specifically since they offer the ability to integrate catalytic properties10,42 to be used as glucose sensor43 or to modulate the non-linear optical responses in the preparation of main- and side-chain metal-containing polymers.44–46 In this context, we have previously disclosed the preparation of a series of cobalt(II), nickel(II), and copper(II) complexes of a fluorinated Schiff base ligand formally resulting from the condensation of 2-thenoyltrifluoroacetone (TTA) with 1,2-diaminoethane; cyclic voltammograms of these complexes displayed oxidation waves indeed, but none of them formed polymer films upon repeated cycling.47 Contrary to those, when the –CF3 groups were exchanged for –CH3 ones,48 the cyclic voltammetry exhibited a similar potentiodynamic behavior for the Ni(II) and Cu(II) complexes, where the oxidation produced a low conductivity oligomer film formed over fluorine tin oxide (FTO) electrodes. Those experiments demonstrate that electronics played an essential role in the outcome of the electropolymerization process and the stabilization of the thiophene radical formation. Pursuing along this line, we rationalize that extension of the conjugation between the electroactive 2-thienyl group and the metal core could facilitate the electrochemically-induced polymerization by the stabilization of the radical cation formed in the process of monomers containing the –CF3 groups and also could increase the conductivity of the final film. To test our hypothesis, we synthesized and fully characterized, including X-ray diffraction analysis, two metal complexes of Ni(II) and Cu(II) supported by a thiophene-appending fluorinated Schiff base ligand. The electrochemical properties of the complexes were interrogated by means of cyclic voltammetry, and to get a better insight into their electrochemical and electronic properties, DFT calculations were performed. The surface morphology and elemental composition of the obtained films were analysed by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS).
Results and discussion
Syntheses and characterization
To prepare the desired complexes with thiophene-appending symmetric tetradentate Schiff base ligands, we applied the synthetic methodology we developed in previous work, viz a cascade of three reactions starting from a carbonyl compound: (i) a Schiff condensation with a primary diamine, (ii) trifluoroacetylation of the formed diimine, and (iii) complexation of the ligand precursor with a metal(II) acetate salt.47 So, the known N,N′-bis[1-(p-bromophenyl)ethylidene]-1,2-diaminoethane 1 and the derivatized tetradentate Schiff base proligand 2 were prepared following modified procedures (Scheme 1).49 In addition to analytical and spectroscopic characterization data (see Experimental for details) that partly matches previously reported literature data,49 we report here the X-ray crystal structure of both 1 and 2 (see below). Coordination of 2 to nickel(II) and copper(II) metal ions afforded 3 and 4, on which the Stille cross-coupling reaction has been carried out to attach the terminal 2-thienyl moiety and obtain the desired final products 5 and 6, respectively (Scheme 2). To the best of our knowledge, this latter step represents a rare example of Stille coupling performed on a metal complex.50 | ||
| Scheme 1 Synthesis of the diimine 1 and of its derivatized fluorinated Schiff base proligand 2. | ||
 | ||
| Scheme 2 Synthesis of Schiff base complexes 3 and 4 and their respective thiophene-appending derivatives 5 and 6. | ||
N,N′-Bis[1-(p-bromophenyl)ethylidene]-1,2-diaminoethane 1 was obtained upon the reaction of p-bromoacetophenone with 1,2-ethylenediamine in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio in refluxing ethanol containing acetic acid as a catalyst for 18 h (Scheme 1). Diimine 1 was isolated as a white solid with an excellent yield of 85%. Despite a longer reaction time, this new procedure avoids using the highly toxic benzene solvent employed in the original report.49 The solid-state IR spectrum of 1 is characterized by two intense bands at 1625 and 1530 cm−1 due to the C
:1 stoichiometric ratio in refluxing ethanol containing acetic acid as a catalyst for 18 h (Scheme 1). Diimine 1 was isolated as a white solid with an excellent yield of 85%. Despite a longer reaction time, this new procedure avoids using the highly toxic benzene solvent employed in the original report.49 The solid-state IR spectrum of 1 is characterized by two intense bands at 1625 and 1530 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching vibrations. In the 1H NMR spectrum, the methyl and methylene protons gave rise to two sharp singlets at 2.25 and 3.88 ppm with a 3:2 integration ratio, respectively, while the imine carbon atom showed up at 163.09 ppm in the 13C NMR spectrum. Furthermore, the structure of 1 was authenticated by X-ray crystallographic analysis (see Experimental), and its molecular structure is depicted in Fig. S1 (ESI‡) with selected bond distances and angles given in the caption. Diimine 1 crystallizes in the monoclinic centrosymmetric space group P21/c, with half a molecule per asymmetric unit. The two halves of the molecule are related through an inversion center located in the middle of the H2C–CH2 bond. Compound 1 consists of two trisubstituted imine fragments connected by an ethylene linker, forming monomeric units. The p-bromophenylene and methylene groups adopt a trans configuration about the CN imine double bond (1.275(4) Å).
N and CC stretching vibrations. In the 1H NMR spectrum, the methyl and methylene protons gave rise to two sharp singlets at 2.25 and 3.88 ppm with a 3:2 integration ratio, respectively, while the imine carbon atom showed up at 163.09 ppm in the 13C NMR spectrum. Furthermore, the structure of 1 was authenticated by X-ray crystallographic analysis (see Experimental), and its molecular structure is depicted in Fig. S1 (ESI‡) with selected bond distances and angles given in the caption. Diimine 1 crystallizes in the monoclinic centrosymmetric space group P21/c, with half a molecule per asymmetric unit. The two halves of the molecule are related through an inversion center located in the middle of the H2C–CH2 bond. Compound 1 consists of two trisubstituted imine fragments connected by an ethylene linker, forming monomeric units. The p-bromophenylene and methylene groups adopt a trans configuration about the CN imine double bond (1.275(4) Å).
Treatment of diimine 1 with 1.5 equiv. of trifluoroacetic anhydride under dry and anaerobic conditions in THF, provided the desired known potentially tetradentate symmetrical Schiff base ligand precursor 2,49 as a white powder in 75% isolated yield (Scheme 1). Note that we confirm our previous observations47 in that an optimized refluxing time of 6 h, after the dropwise addition of trifluoroacetic anhydride at 0 °C, is necessary to reach the reported yield.49 The presence of the trifluoroacetyl group is evidenced by a sharp singlet at −77.37 ppm in the 19F NMR spectrum and two quartets assigned to the trifluoromethyl and carbonyl carbons in 13C NMR (see Experimental for details and Fig. S2, ESI‡). Compound 2 does exist as its enaminone tautomer in solution, as indicated by the intramolecular hydrogen-bonded amino N–H and vinylic C–H proton resonances of the enamine unit, showing up as singlets at 10.87 and 5.36 ppm, respectively (Fig. S3, ESI‡). Both solid-state IR spectroscopy (Fig. S4, ESI‡), with the ν(N–H) vibration observed at 3437 cm−1, and the X-ray diffraction study (see below) demonstrate that the enaminone tautomeric form is also present in the solid phase.
Nickel(II) and copper(II) complexes 3 and 4, respectively, were straightforwardly obtained by reacting the Schiff base proligand 2 with the appropriate hydrated metal(II) acetate salt in a 1:1 (v/v) acetone/methanol mixture at r.t. for 4 h (Scheme 2). During the work-up, an exhaustive washing of the formed solids with water is required in order to completely eliminate the formed acetic acid that is detrimental to purification and the follow-up Stille coupling reaction. Compounds 3 and 4 were isolated as light green and purple solids, respectively, in ∼80% yield. Reactions of 3 and 4 with a slight excess of 2-(tributylstannyl)-thiophene in the presence of 2 mol% PdCl2(PPh3)2 as the catalyst in refluxing toluene for 12 h afforded the coupling products 5 and 6 that were isolated as yellow-brown and purple powders in 60 and 65% yields, respectively (Scheme 2).
The four complexes 3–6 are air and thermally stable, showing good solubility in common polar organic solvents. They have been characterized on the basis of FT-IR, UV-Vis, and multinuclear NMR (for diamagnetic Ni(II) complexes 3 and 5) spectra, as well as by elemental analysis where all experimental results are in agreement with the calculated percentages (see Experimental for details). For the purpose of probing the molecular structure of paramagnetic Cu(II) complexes 4 and 6 in solution, positive mode ESI mass spectra were recorded and disclosed the sodium aggregate peaks [M + Na]+ at m/z = 695.852 and 704.006, respectively. Molecular structures of complexes 3–5 were further confirmed by single-crystal X-ray crystallography (see below).
The solid-state FT-IR spectra of 3–6 (Fig. S4, ESI‡) present similar absorption patterns, exhibiting three strong to medium intensity bands in the 1610–1490 cm−1 range attributed to ν(C![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) O), ν(CC) and/or ν(CN) stretching modes of the chelating Schiff base skeleton.47 The slight decrease in energy of those bands along with the disappearance of the ν(N–H) vibration seen at 3437 cm−1 in the Schiff base proligand 2 clearly suggest coordination of the dianionic Schiff base ligand to the Ni(II) and Cu(II) ions through the oxygen and nitrogen atoms.51 The strong bands observed in the 1190–1140 cm−1 region are due to the C–F stretching vibration mode of the CF3 groups.47 In both 5 and 6, the extra absorption band observed at 700 cm−1 is assigned to the out-of-plane bending vibration mode of the C–H groups of the 2-thienyl unit.52
O), ν(CC) and/or ν(CN) stretching modes of the chelating Schiff base skeleton.47 The slight decrease in energy of those bands along with the disappearance of the ν(N–H) vibration seen at 3437 cm−1 in the Schiff base proligand 2 clearly suggest coordination of the dianionic Schiff base ligand to the Ni(II) and Cu(II) ions through the oxygen and nitrogen atoms.51 The strong bands observed in the 1190–1140 cm−1 region are due to the C–F stretching vibration mode of the CF3 groups.47 In both 5 and 6, the extra absorption band observed at 700 cm−1 is assigned to the out-of-plane bending vibration mode of the C–H groups of the 2-thienyl unit.52
The 1H NMR spectrum of 3 (Fig. S5, ESI‡) consists of two singlets at δH 2.85 and 5.28 ppm and two doublets at δH 6.83 and 7.34 ppm, with an integral ratio of 2:1:2:2 attributed to the methylene, methine, and p-phenylene protons, respectively. That of 5 (Fig. S6, ESI‡) shows similar peak and integration patterns with two extra resonances, a double doublet at δH 7.10 ppm and a multiplet at δH 7.34 ppm (integral ratio 1:2), assigned to the protons of the appended 2-thienyl unit. The symmetrical nature of both 3 and 5 is confirmed by the 9 and 13 lines observed in their respective 13C{1H} NMR spectra (Fig. S7 and S8, ESI‡). In both cases, the carbonyl carbon that appears as a quartet (2JC–F = 33 Hz) is upfield shifted by about 10 ppm upon coordination to Ni(II).53 The –CF3 groups show up as singlets at δF −72.77 and −73.35 ppm in the 19F NMR spectra.
X-ray crystallographic studies of compounds 2–5
Good X-ray quality crystals of compounds 2, 3, 4, and 5·CH2Cl2 were grown by slow evaporation of their respective saturated organic solution (see Experimental for details). Pictorial views of discrete Schiff base proligand 2, and mononuclear nickel(II) complexes 3 and 5·CH2Cl2 are displayed in Fig. 1 in similar orientations for the sake of comparison. The molecular structure of the copper(II) derivative 4 is presented in Fig. S9 (ESI‡). Selected bond distances and angles for the first M(II) coordination sphere (M = Ni, Cu) of compounds 3–5·CH2Cl2 are gathered in Table 1. Additionally, selected bond distances and angles for the free and complexed ligands in 2 and in 3–5·CH2Cl2 are listed in Tables S1 and S2 (ESI‡), respectively. | ||
| Fig. 1 Molecular structures of Schiff base proligand 2 (top), nickel(II) complexes 3 (middle), and 5·CH2Cl2 (bottom) with their respective labeling scheme. Hydrogen atoms (except those involved in hydrogen bonding interactions in 2), and solvent crystallization molecule for 5·CH2Cl2 are omitted for clarity. Thermal ellipsoids are drawn at 50% probability. | ||
| 3 | 4 | 5·CH2Cl2 | 6 | |
|---|---|---|---|---|
| a M = Ni, 3 and 5; Cu, 4 and 6. b No X-ray structure data. Symmetry transformations used to generate equivalent atoms: # −x, y, −z + 1/2. | ||||
| Bond distances | ||||
| M(1)–O(1) | 1.850(6) | 1.910(2) | 1.842(3) | — |
| 1.843 | 1.903 | |||
| M(1)–N(1) | 1.861(7) | 1.937(2) | 1.854(3) | — |
| 1.862 | 1.936 | |||
| Bond angles | ||||
| O(1)–M(1)–N(1)# | 177.50(3) | 178.68(10) | 175.22(14) | — |
| 172 | 158 | |||
| O(1)–M(1)–O(1)# | 82.10(4) | 84.89(12) | 83.30(19) | — |
| 84 | 92 | |||
| O(1)–M(1)–N(1) | 95.80(3) | 94.58(9) | 94.93(14) | — |
| 95 | 95 | |||
| N(1)–M(1)–N(1)# | 86.40(4) | 85.97(13) | 87.17(19) | — |
| 88 | 87 | |||
The Schiff base proligand 2 crystallizes in the monoclinic space group P21/c with one molecule per asymmetric unit. In contrast, all three complexes 3, 4, and 5·CH2Cl2 crystallize in the monoclinic crystal system in the centrosymmetric space group C2/c with half a molecule in the asymmetric unit. The symmetry-independent half molecule is related to its other half through a C2 axis passing through the metal atom and the middle of the CH2-CH2 bond. The single-crystal X-ray diffraction study confirms the double trifluoroacetylation of diamine 1 to form the potentially tetradentate Schiff base proligand 2. Complexes 3–5 consist of a metal(II) ion tetracoordinated by the N2O2 donor set of the dianionic Schiff base ligand, symmetrically substituted with trifluoromethyl and p-bromophenyl (3 and 4) or p-(2-thienyl)phenyl (5) groups, and forming monomeric units. Complexes 3 and 4 are isostructural, differing only by the nature of the M(II)-centered metal ion (M = Ni for 3 and Cu for 4). In any of the four compounds 2–5, the CF3 groups show rotational disorder. All the C–F bond distances are rather similar, varying between 1.307(8) and 1.339(10) Å (Tables S1 and S2, ESI‡). As previously reported, the orientational disorder of the 2-thienyl rings is observed in 5·CH2Cl2.47,48 In the four compounds, all close intermolecular contacts correspond to van der Waals interactions.
C–CC–NH⋯] fragment,57 with alternating double-, single-, double- and single bonds between the vicinal sp2-hybridized atoms (Table S1, ESI‡).58 Additionally, the crystal packing of 2 is stabilized by several intermolecular hydrogen bond interactions (Fig. S10 and Table S3, ESI‡). Each enaminone fragment, [O1C2C3C4N1H] and [O2C22C23C24N2H], is nearly flat, their respective mean plane presenting a root-mean-square deviation (RMSD) of 0.015 and 0.039 Å, respectively. They make dihedral angles of 52.46(12)° and 62.66(12)° with their corresponding attached phenyl ring plane. All the metrical parameters are in accordance with literature structural data reported for enaminone derivatives by ourselves,45,59,60 and others.55,56,61,62
Magnetic properties of Cu(II) derivatives 4 and 6
The magnetic properties of powdered samples of the two paramagnetic Cu(II) compounds 4 and 6 have been recorded as a function of the temperature and magnetic field. The temperature variations of χMT for both compounds are represented in Fig. 2. χMT's remain constant in the whole temperature range, and they are identical for 4 and 6 at ∼0.4 cm3 K mol−1. This value matches with a single isolated Cu(II) ion (s = 1/2) with a g value close to 2.06. Accordingly, the M vs. H curves (inset of Fig. 2) at 2 K follow Brillouin law with g = 2.04 for both compounds.64 | ||
| Fig. 2 Temperature variations of χMT for compounds 4 and 6 with the field variations at 2 K in inset (the best-fitted curves with Brillouin law are represented with a red line). | ||
Computational investigations
DFT calculations were performed for complexes 5 and 6 supported by a thiophene-appending symmetrical fluorinated Schiff base ligand, and the computational details are given in the Experimental section. The DFT-optimized geometry of compound 5 agrees with the experimental X-ray counterpart. Selected optimized metrical data associated with the nickel (for 5) and copper (for 6) coordination sphere are given in Table 1. Those of 5 are similar to those of their corresponding X-ray values and show even better homogeneity. The values computed for 6, for which no X-ray structure is available, fit well with those of the Ni(II) relative 5. Fully optimized geometries are provided as xmol files and depicted in Fig. S11 (ESI‡). The MO diagrams of 5 and 6 are shown in Fig. 3, and due to the spin-unrestricted nature of the calculations, each Kohn–Sham MO level in the diagram of 6 is split into two spin orbitals: one spin-up (left) and one spin-down (right). The computed spin density shape of 6 (Fig. S12, ESI‡) indicates that the first oxidation occurs on the metal coordination sphere of 6.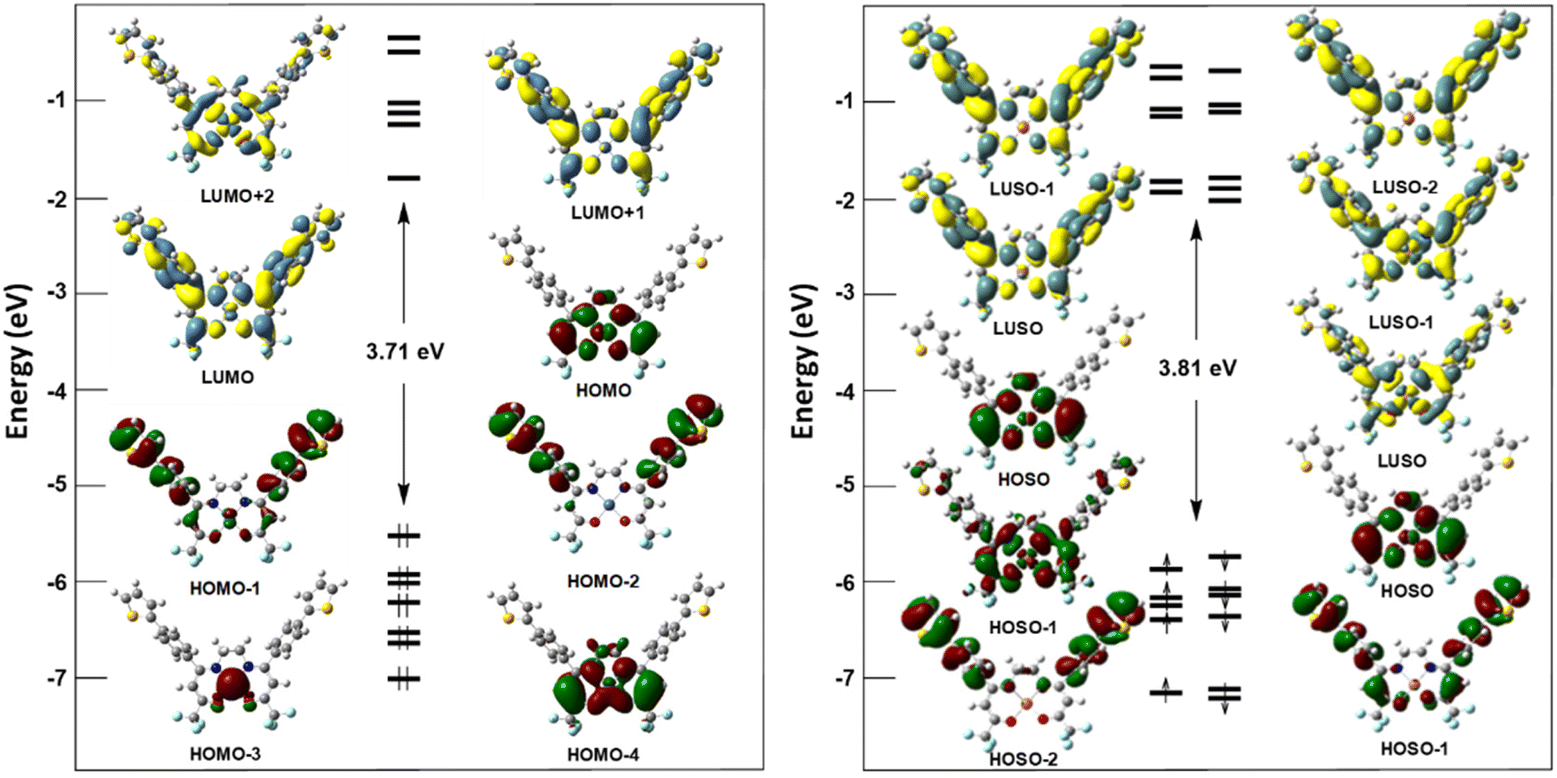 | ||
| Fig. 3 Kohn–Sham orbital diagrams of 5 (left) and 6 (right). HOSO: highest occupied spin–orbital; LUSO: lowest unoccupied spin–orbital. | ||
Time-dependent density functional theory (TD-DFT) calculations were subsequently performed at the B3LYP/6-31G(d,p) level on the optimized geometries to simulate the UV-vis spectra of complexes 5 and 6. The agreement with the experimental spectra is satisfying. The simulated spectra are shown in Fig. S13 (ESI‡), and their maximum absorption values and assignments are provided in Table 2 (see below).
| Compd. |
λ
max (exp.)/nm (logε) (CH2Cl2) |
λ (calcd)/nm (oscillator strength) [TD-DFT] | Major transition involved (from TD-DFT) |
|---|---|---|---|
| 5 | 306 (5.81) | 343 (8 × 10−2) | LLCT |
| 575 (1.95) | 483 (8 × 10−3) | MLCT (HOMO−3–LUMO−2) | |
| 577 (4 × 10−3) | MLCT (HOMO–LUMO) | ||
| 6 | 316 (5.46) | 374 (2 × 10−4) | LLCT |
| 559 (2.43) | 379 (1 × 10−3) | LLCT | |
| 537 (3 × 10−4) | LMCT (HOSO(β) → LUSO(β)) |
Electronic absorption spectra
The UV-vis spectra of complexes 5 and 6 were recorded in dichloromethane solutions at 20 °C. The experimental spectra are shown in Fig. 4, while the TD-DFT simulated ones are presented in Fig. S13 (ESI‡). Experimental absorption spectral and TD-DFT data are shown in Table 2. The bands found for both complexes at 306, and 316 nm are attributed to the π–π* transitions of the imine and aromatic groups, thus belonging to ligand-to-ligand charge transfer (LLCT) transitions.65 The low energy band of complexes 5 at 575 nm and 6 at 559 nm are assigned to d–d transition with metal to ligand charge transfer (MLCT) and ligand to metal charge transfer (LMCT) character, respectively. This difference comes from the fact that the d-type levels of the open-shell d9 Cu(II) derivative 6 lies at much lower energy than those of its close-shell d8 Ni(II) counterpart 5 (see Fig. 3). The presence of the bands in the visible region is typical for square planar [M(N2O2)] chromophores.66 | ||
| Fig. 4 UV spectra (top) and Vis spectra (bottom) of complexes 5 (dashed line) and 6 (solid line), measured in CH2Cl2 solutions at 20 °C. | ||
Electrochemical studies
Fig. 5a and 6a portray the electropolymerization of complexes 5 and 6, where an increase in current density is observed after 30 cycles, culminating in the formation of golden and golden-bronze-colored thin films on the Pt disk electrode surface, respectively. In addition, a greater increase was seen for complex 6, indicating that an electrogenerated film with higher conductivity has been produced. Both polymerizations did not produce thick films, presumably because, as noted previously, the HOMO and HOSO do not lie on the thienyl groups, hampering the polymer film formation.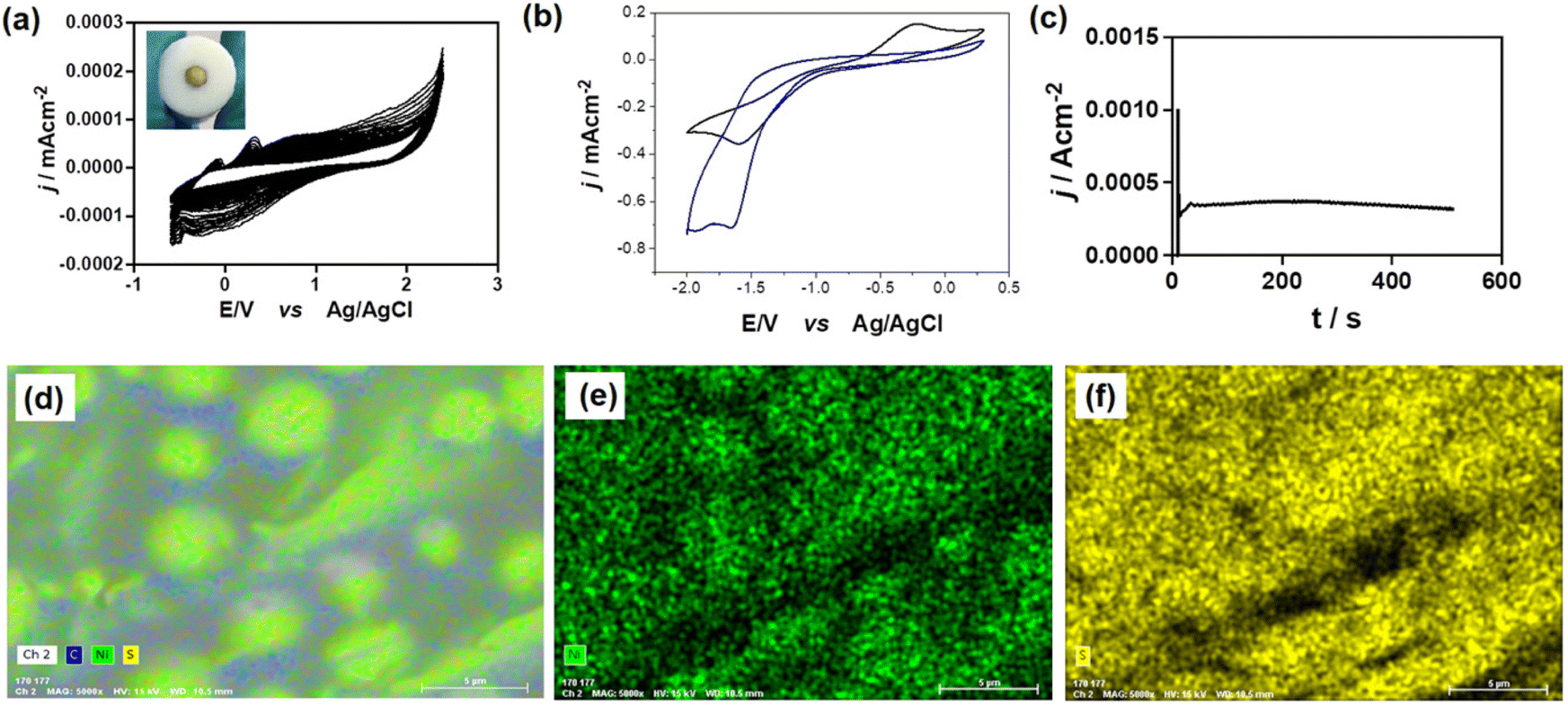 | ||
| Fig. 5 (a) Voltammetric profiles of Pt|poly-compound 5 prepared by CV vs. Ag/AgCl for 30 cycles. Interphase: 2 mM compound 5 + 0.02 M TBAPF6 in anhydrous CH3CN, v = 50 mV s−1. (b) Compound poly-5 (black line) and 3 (blue line) responses in 0.02 M TBAPF6 in CH3CN, v = 100 mV s−1. (c) j/t transient registered during electropolymerization of 5 on Pt. Interface: Pt|0.002 M 5 + 0.02 M TBAPF6 in CH3CN. (d)–(f) SEM-EDS analysis of poly-5 for: (C, Ni, S), Ni (Green dots), and S (yellow dots) elements. | ||
 | ||
| Fig. 6 (a) Voltammetric profiles of Pt|poly-compound 6 prepared by CV vs. Ag/AgCl for 30 cycles. Interfaces: 2 mM compound 6 + 0.02 M TBAPF6 in anhydrous CH3CN, v = 50 mV s−1. (b) Poly-6 (black line) and 4 (blue line) responses in 0.02 M TBAPF6 in CH3CN, v = 100 mV s−1. (c) j/t transient registered during electropolymerization of 6 on Pt. Interface: Pt|0.002 M 6 + 0.02 M TBAPF6 in CH3CN. (d)–(f) SEM-EDS analysis of poly-6 for: (C, Cu, S), S (yellow dots) and Cu (red dots) elements. | ||
Fig. 5b shows the voltammetric response of poly-5 in an acetonitrile solution containing TBAPF6 (0.01 M) as a supporting electrolyte versus the response of a 0.02 M solution of compound 5 without thiophene (compound 3), at a platinum electrode, as a way of comparing and evaluating whether nickel is still present in the polymer after the polymerization process.47
The electrochemical response of nickel(II) in poly-5 exhibits two waves or peaks. In fact, it is possible to observe in Fig. 5b (black line) one oxidation peak and one reduction peak at Pox = −1.1 V and Pred = −1.5 V, which could be attributed to the passage of Ni(I)/Ni(II) and Ni(II)/Ni(I), respectively. Furthermore, the response of the thiophene groups is observed at negative potentials (−0.2 V).68 If these results are compared with those of compound 3 (blue line), the oxidation and reduction peaks for the Ni(I)/Ni(II) and Ni(II)/Ni(I) processes are observed at Pox = −1.4 V and Pred = −1.7 V, respectively. Moreover, the signal attributed to the thienyl group observed in compound 5 does not appear in compound 3; due to the conductive polymer formation, this signal was correctly assigned to the thiophene response in the film.
Fig. 6b compares the electrochemical response of poly-6 to that of compound 4 under the same conditions as for poly-5. The black line (poly-6) shows two oxidation peaks and two reduction peaks at Pox = −1.6 V, Pox = −1.3 V, Pred = −1.8 V, and Pred = −1.9 V, respectively. In contrast, compound 4 exhibits only one anodic and cathodic peak, located at Pox = −1.3 V and Pred = −1.8 V, identical to the peak observed in the poly-6 film. According to Peters et al.,69 the presence of the two peaks in poly-6 film, which were not present in compound 4, may indicate the possibility of monomer occlusion on the electrode surface, resulting in signals with equal potentials. We reasoned that the oxidation and reduction peaks shifted to more negative potentials corresponding to the Cu(II)/Cu(I) oxidation and reduction processes,67 as illustrated in Fig. 6b.
For compounds 5 and 6, the formation of the film was also evaluated using chronoamperometry at the optimal potentials of 2.2 V and 2.1 V for extended working times (Fig. 5c and 6c), respectively. Both cases reaffirm the CV results, demonstrating that the polymerization process and formation of radical cations are more hampered in 5 than in 6, where an increase in current is observed as the potential is applied, which does not occur with 5, requiring a longer time to obtain films similar to those obtained with 6. The difference in current response can also be attributed to differences in the conductivity of the formed film, which is dependent on the structure of the monomer species and their ability to transport charge carriers. Indeed, different potentials were investigated, but only at the aforementioned potential was it possible to observe the formation of a thin polymer film when 5 was used as the monomer.
Finally, both electrodes modified with polymers generated from 5 and 6 were analysed by SEM-EDS to confirm the polymer's presence and the uniform distribution of Ni and Cu metals on the electrode surfaces. The images in Fig. 5d–f and 6d–f demonstrate the presence and homogeneous distribution of S on the electrode surface, indicating that both polymers contain thiophene rings. Additionally, as evidenced by the voltammetric responses obtained and discussed previously, we can confirm that Ni and Cu are evenly distributed on the surface of the generated polymer.
Conclusions
In summary, this contribution describes the synthesis of a series of four new monometallic nickel(II) and copper(II) complexes incorporating symmetrically-substituted N2O2-tetradentate Schiff base ligand bearing trifluoromethyl and p-bromophenyl or the π-extended π-(2-thienyl)phenyl substituents, and their complete characterization through various analytical, spectroscopic and X-ray diffraction techniques. Interestingly, the thiophene-appending ligand of compounds 5 and 6 has been constructed on the complexes through a Stille cross-coupling reaction. The crystal structures of the diimine precursor, the fluorinated Schiff base proligand, and two nickel(II) and one copper(II) complexes have been determined. The three complexes form discrete molecular entities. The four-coordinate central metal(II) ion sits in a quasi-perfect square planar environment with partial delocalization of bonding electron density throughout the six-membered heterometallacycle frameworks. Electrochemical polymerization of 5 and 6 was accomplished, yielding golden and golden-bronze-colored thin films, respectively. Compound 6 enables the formation of thicker coatings with increased conductivity in both the CV and chronoamperometry voltammetric profiles. Finally, the characterization of the polymeric films of 5 and 6 by SEM-EDS showed the even distribution of the Ni and Cu metals, in addition to also observing a homogeneous distribution of sulfur atoms on the surfaces, which allows asserting the successful modification of electrodes due to the polymerization processes of the thiophene termini. Our results establish a precedent for future work in the field of MCP of Schiff base complexes tethered to 2-thienyl groups. Future work should improve the redox compatibility of metal complexes and the electro-polymerizable fragments using, for instance, the 3,4-ethylene dioxythiophene (EDOT) scaffold.Experimental
Materials and physical measurements
Reactions were performed under dry dinitrogen or argon atmosphere using standard Schlenk techniques. Solvents were dried and distilled according to standard procedures.70 All starting materials were purchased from commercial sources and used as received. Checking of the purity of the organic compounds was carried out by TLC on silica gel-protected aluminum sheets (Type 60 F254, Merck), and the spots were detected by exposure to UV-lamp at λ = 254 nm. Solid-state FT-IR spectra were recorded with a PerkinElmer Model 1600 or a Bruker IFS28 FT-IR spectrophotometer with KBr disks in the 4000 to 450 cm−1 range. UV/Vis spectra were recorded using a Thermo Scientific, Helios Omega spectrophotometer. 1H, 13C, and 19F NMR spectra were recorded at 298 K with a Bruker 300 or Avance III 400 spectrometer. Chemical shifts are reported in parts per million (δ) relative to tetramethylsilane (TMS) for 1H and 13C NMR spectra and external CFCl3 for 19F spectra. Coupling constants (J) are reported in Hertz (Hz), and integrations are reported as the number of protons. High Resolution Electrospray Mass Spectra (HR-ESI-MS) were collected on a Bruker MAXI 4G (for 4) and a Thermo Fisher Scientific Q-Exactive (for 6) spectrometer at the Centre Régional de Mesures Physiques de l'Ouest (CRMPO) at the Université de Rennes 1, France. Elemental analyses were conducted on a Thermo-Finnigan Flash EA 1112 CHNS/O analyzer by the Microanalytical Service of the CRMPO. The temperature dependences of the magnetizations for powdered samples of 4 and 6 have been measured with a Quantum design MPMS-XL5 SQUID magnetometer operating between 2 and 300 K, with an applied magnetic field of 2 kOe below 20 K and of 10 kOe above 20 K. Diamagnetic corrections were applied by using Pascal's constants.71 Melting points were measured in evacuated capillaries on a Kofler Bristoline melting point apparatus, and the values obtained were not corrected.Synthesis of N,N′-bis[1-(p-bromophenyl)ethylidene]-1,2-diaminoethane (1)
A Schlenk tube was loaded with a magnetic stirring bar, 1.0 g (5.0 mmol) of p-bromoacetophenone, and 20 mL of ethanol. After 10 min of stirring, 0.16 mL (2.50 mmol) of ethylenediamine were added dropwise using a syringe. After 15 min of stirring, the transparent solution turned to a light-yellow color, and was refluxed for 18 h. The reaction mixture was cooled to r.t., the solvent was evaporated to dryness under vacuum, giving an oily product. It was washed several times with cold hexane (0 °C) before being re-dissolved in ethanol and kept at −30 °C overnight, producing (1) as a white solid. The white solid (Rf = 0.6, 1:1 hexane/dichloromethane) was filtered off, washed several times with cold hexane (0 °C), and dried under vacuum. Yield: 0.88 g (2.10 mmol, 85%). Single crystals suitable for X-ray diffraction analysis were grown by slow evaporation of a saturated ethanolic solution. m.p. 181–182 °C (lit. 192–194 °C).49 Elemental analysis (%) Calculated for C18H18Br2N2 (422.16 g mol−1): C, 51.21; H, 4.30; N, 6.64. Found: C, 51.45; H, 4.27; N, 6.33. FT-IR (KBr, cm−1): 3049(s) ν(C–H arom); 2911(m), 2888(m), 2826(w) ν(C–H aliph); 1625(vs) and 1583(s) ν(CN) and/or ν(CC). 1H NMR (300 MHz, CDCl3): 2.25 (s, 6 H, CH3), 3.88 (s, 4 H, CH2), 7.48 (d, JH,H = 8.3 Hz, 4 H, C6H4), 7.63 (d, JH,H = 8.3 Hz, 4 H, C6H4). 13C{1H} NMR (75.48 MHz, CDCl3): 14.70 (CH3), 48.27 (–CH2), 125.20 (C6H4), 131.40 (C6H4), 133.00 (C6H4), 137.00 (C6H4), 163.09 (CN).
Synthesis of fluorinated proligand 2
A Schlenk tube was placed in a water-ice bath and loaded with a magnetic stirring bar, 1.0 g (2.37 mmol) of 1 and 30 mL of dry THF. After 10 min of stirring, 0.99 mL (7.11 mmol) of trifluoroacetic anhydride was added dropwise by syringe (Caution: the reaction is strongly exothermic). The solution turned light yellow. After 15 min of stirring, the water ice bath was removed, and the solution was refluxed for 6 h. The reaction mixture was cooled to r.t., and the volatiles evaporated under vacuum. The yellow crystalline solid was dissolved in 10 mL of acetone and kept at −30 °C overnight, leading to the precipitation of a solid that was filtered off and discarded. The filtrate was evaporated, and the white solid residue was washed 3 times with 30 mL of cold hexane (0 °C). Yield: 1.09 g (1.78 mmol, 75%). Slow evaporation of a THF solution of 2 at r.t. produces suitable single crystals for X-ray diffraction study. m.p. 250–251 °C (lit. 180–182 °C).49 Elemental analysis (%) Calculated for C22H16Br2F6N2O2 (614.18 g mol−1): C, 43.02; H, 2.63; N, 4.56. Found: C, 42.75; H, 2.44; N, 4.45. FT-IR (KBr, cm−1): 3437 (w) ν(N–H); 3080(w) ν(C–H arom); 2959(w), 2942(w) ν(C–H aliph); 1611(vs), 1596(s) and 1582(s) ν(CO), ν(CN) and/or ν(CC); 1193, 1138 ν(C–F). 1H NMR (400 MHz, (CD3)2CO): 3.67 (s, 4 H, CH2), 5.36 (s, 2 H, CHC), 7.34 (t, JH,H = 8.6 Hz, 4 H, C6H4), 7.70 (dd, JH,H = 8.3 and 7.3 Hz, 4 H, C6H4), 10.87 (s, 2 H, N–H). 13C{1H} NMR (100 MHz, (CD3)2CO): 45.34 (–CH2), 89.77 (–CHC), 117.51 (q, 1JC,F = 286 Hz, –CF3), 129.56 (C–Br, Br–C6H4), 130.69 (Cipso, Br–C6H4), 132.07 (C–H, Br–C6H4), 132.65 (C–H, Br–C6H4), 169.60 (CHC), 175.40 (q, 2JC,F = 35 Hz, CO). 19F NMR (376 MHz, (CD3)2CO): −77.37 (s, CF3).
Synthesis of the Ni(II) Schiff base complex 3
A Schlenk tube was charged with a magnetic stirring bar, 1.0 g (1.63 mmol) of 2, 15 mL of a 1:1 (v/v) acetone/methanol mixture, and 0.40 g (1.63 mmol) of nickel(II) acetate tetrahydrate. The reaction mixture was stirred for 4 h at r.t. Then, the resulting green solution was evaporated under reduced pressure, and the solid residue was washed with 100 mL of water and filtered off using a glass frit. The collected material was dissolved in CH2Cl2 and dried over MgSO4. The suspension was filtered, the solvent was evaporated, and the product dried under vacuum, affording 0.86 g (1.28 mmol, 79% yield) of a pale green powder. Slow evaporation of an ethanolic solution of 3 at r.t. produces suitable single crystals for X-ray diffraction study. m.p. 279 °C. Elemental analysis (%) Calculated for C22H14Br2F6N2NiO2 (670.86 g mol−1): C, 39.39; H, 2.10; N, 4.18. Found: C, 40.84; H, 2.47; N, 4.02. FT-IR (KBr, cm−1): 2945 (w), 2922 (w) ν(C–H arom); 2875(w), 2851 (w) ν(C–H aliph); 1610(vs), 1525(s) and 1490(m) ν(CO), ν(CN) and/or ν(CC); 1187, 1140 ν(C–F). 1H NMR (300 MHz, (CD3)2CO): 3.01 (s, 4 H, CH2), 5.55 (s, 2 H, CHC), 7.29 (d, JH,H = 8.8 Hz, 4 H, C6H4), 7.67 (d, JH,H = 8.8 Hz, 4 H, C6H4). 13C{1H} NMR (75.48 MHz, (CD3)2CO): 55.17 (–CH2), 97.61 (–CHC), 118.99 (q, JC,F = 283 Hz, CF3), 123.74 (C–Br, Br–C6H4), 127.60 (Cipso, Br–C6H4), 132.02 (C–H, Br–C6H4), 134.79 (C–H, Br–C6H4), 160.68 (CHC), 168.03 (q, 2JC,F = 33 Hz, CO). 19F NMR (376 MHz, (CD3)2CO): −72.77 (s, CF3).
Synthesis of the Cu(II) Schiff base complex 4
Complex 4 was synthesized following an identical procedure to that described above for 3, using in this case 0.32 g (1.63 mmol) of copper(II) acetate monohydrate. Yield: 0.92 g (1.32 mmol, 81%) of 4 as a purple solid. Single crystals of 4 suitable for X-ray diffraction were grown as above for 3. m.p. 240–241 °C. Elemental analysis (%) Calculated for C22H14Br2CuF6N2O2·H2O (693.71 g mol−1): C, 38.05; H, 2.02; N, 4.03. Found: C, 38.38; H, 1.92; N, 3.86. ESI MS (m/z) calcd for C22H14N2O2F6Br2NaCu: 695.85144, found: 695.852 [M + Na]+. FT-IR (KBr, cm−1): 3490 (m) (H2O) 2945 (w), 2922 (w) ν(C–H arom); 2875(w), 2851 (w) ν(C–H aliph); 1610(vs), 1525(s) and 1490(m) ν(CO), ν(CN) and/or ν(CC); 1187, 1144 ν(C–F).
Synthesis of the Ni(II) thiophene-appending Schiff base complex 5
A Schlenk tube was charged with a magnetic stirring bar, 1.00 g (1.49 mmol) of 3, 20 mL of toluene and 20 mg (0.029 mmol) of PdCl2(PPh3)2. The solution was stirred 5 min before 1.09 mL (3.43 mmol) of 2-(tributylstannyl)-thiophene were added dropwise. The reaction mixture was refluxed for 12 h, becoming black. Upon cooling down to r.t., the volatils were removed under vacuum. The solid material was taken with diethyl ether, the suspension was filtered off and the filtrate dried over MgSO4. The organic phase was filtered and the solvent evaporated to dryness to afford 0.61 g (0.89 mmol, 60% yield) of a yellow-brown powder. Single crystal of 5 suitable for X-ray diffraction were grown by slow evaporation of a dichloromethane solution. m.p. 251 °C. Elemental analysis (%) Calculated for C30H20F6N2NiO2S2 (677.31 g mol−1): C, 53.20; H, 2.98; N, 4.14; S, 9.47. Found: C, 52.46; H, 3.60; N, 5.24; S, 8.32. FT-IR (KBr, cm−1): 3127 (w), 3109 (w) ν(C–H arom); 2926(w), 2852 (w) ν(C–H aliph); 1606(vs), 1518(s) and 1503(s) ν(CO), ν(CN) and/or ν(CC); 1184, 1140 ν(C–F); 700 δ(C–H C4H3S). 1H NMR (300 MHz, CDCl3): 2.97 (s, 4 H, CH2), 5.59 (s, 2 H, CHC), 7.10 (dd, JH,H = 5.1 and 3.7 Hz, 2 H, SC4H3), 7.16 (d, JH,H = 8.4 Hz, 4 H, C6H4), 7.34 (m, 4 H, SC4H3), 7.63 (d, JH,H = 8.4 Hz, 4 H, C6H4). 13C{1H} NMR (75.48 MHz, CDCl3): 55.45 (CH2), 97.91 (CHC), 117.20 (q, Jc,f = 281 Hz, CF3), 124.04 (C6H4), 125.91 (C4H3S), 126.09 (Cipso, C6H4), 126.80 (C4H3S), 128.32 (C4H3S), 134.93 (C6H4), 135.65 (Cipso, C6H4), 142.80 (Cipso, C4H3S), 160.85 (q, Jc,f = 33.5 Hz, CO), 168.75 (CHC). 19F NMR (376 MHz, (CD3)2CO): −73.35 (s, CF3).
Synthesis of Cu(II) thiophene-appending Schiff base complex 6
This purple complex 6 was prepared in a manner similar to that described above for 5, using in this case 1.00 g (1.48 mmol) of 4. Yield: 0.65 g (0.96 mmol, 65%). m.p. 310 °C. Elemental analysis (%) Calculated for C30H20CuF6N2O2S2 (682.16 g mol−1): C,52.13; H, 3.38; N,3.92; S, 8.97. Found: C, 51.44; H, 3.01; N, 3.49; S, 8.05. ESI MS (m/z) calcd for C30H20N2O2F6NaS263Cu: 704.00586, found: 704.0060 [M + Na]+. FT-IR (KBr, cm−1): 3127 (w), 3109 (w) ν(C–H arom); 2926(w), 2852 (w) ν(C–H aliph); 1606(vs), 1518(s) and 1503(s) ν(CO), ν(CN) and/or ν(CC); 1185, 1143 ν(C–F), 700 δ(C–H C4H3S).
X-ray crystal structure determination
A crystal of appropriate size and shape of each of the compounds 1, 2, 3, and 5·CH2Cl2 was coated with Paratone-N oil, mounted on a nylon loop, and transferred to the cold gas stream of a cooling device, while a crystal of 4 was mounted on top of glass fiber in a random orientation. Crystallographic measurements were carried out for 1, 2, and 3, at T = 150(2) K on an APEXII Bruker-AXS diffractometer equipped with a bidimensional CCD detector, for 4 at T = 296(2) K on a D8 VENTURE Bruker-AXS diffractometer and at T = 170 K for 5·CH2Cl2 on a Bruker D8 QUEST diffractometer, both equipped with a bidimensional CMOS Photon100 detector, using in the three cases graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Intensity data were corrected for absorption effects using multiscanned reflections. The structure of 1 was solved by direct methods using the SIR97 program,72 and then refined using full-matrix least-squares (L.S.) methods based on F2 (SHELXL-97),73 with the aid of WINGX program.74 The structures of 2, 3, and 4 were solved by dual-space algorithm using the SHELXT program,75 and refined with full-matrix L.S. methods based on F2 (SHELXL-2014).76 The structure of 5·CH2Cl2 was solved by direct methods using OLEX2,77 and refined by full-matrix l.s. methods based on F2.76 DFIX instruction restrains used on the bond lengths of one 2-thienyl moiety with a standard uncertainty of 0.01 Å. FLAT constrain instructions were applied in order to keep the planarity of one 2-thienyl ring with a standard uncertainty of 0.1 Å3. Finally, EADP constraints were used on the disordered ring in order to keep the same anisotropic displacement parameters. In all the compounds, non-hydrogen atoms were refined with anisotropic displacement parameters. H atoms were included in their calculated positions, assigned fixed isotropic thermal parameters, and constrained to ride on their parent atoms. A summary of the details about crystal data, collection parameters, and refinement are collected in Table 3, and additional crystallographic details are in the CIF files. ORTEP views were drawn using Olex2 software.77| 1 | 2 | 3 | 4 | 5·CH2Cl2 | |
|---|---|---|---|---|---|
| Empirical formula | C18H18Br2N2 | C22H16Br2F6N2O2 | C22H14Br2F6N2NiO2 | C22H14Br2CuF6N2O2 | C31H22Cl2F6N2NiO2S2 |
| Formula weight | 422.16 | 614.19 | 670.88 | 675.71 | 762.23 |
| Collection T (K) | 150(2) | 150(2) | 150(2) | 295(2) | 170(2) |
| Crystal size (mm) | 0.58 × 0.33 × 0.21 | 0.60 × 0.14 × 0.12 | 0.24 × 0.12 × 0.07 | 0.49 × 0.35 × 0.28 | 0.38 × 0.34 × 0.23 |
| Crystal color | Colorless | Colorless | Yellow | Orange | Light yellow |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | C2/c | C2/c | C2/c |
| a (Å) | 4.2470(2) | 12.9907(13) | 20.0985(14) | 19.941(3) | 18.4962(6) |
| b (Å) | 10.1878(5) | 18.1315(19) | 9.6051(6) | 9.7696(13) | 12.7149(6) |
| c (Å) | 19.0944(9) | 10.3924(12) | 11.9138(8) | 12.1230(16) | 14.2500(6) |
| β (°) | 92.379(2) | 109.105(4) | 100.023(2) | 100.103(5) | 105.310(2) |
| V (Å3) | 825.46(7) | 2313.0(4) | 2264.8(3) | 2325.1(6) | 3232.4(2) |
| Z | 2 | 4 | 4 | 4 | 4 |
| D calcd (g cm−3) | 1.699 | 1.764 | 1.968 | 1.930 | 1.566 |
| Abs. coeff. (mm−1) | 4.908 | 3.576 | 4.459 | 4.448 | 0.962 |
| F(000) | 420 | 1208 | 1312 | 1316 | 1544 |
| θ range (°) | 2.13 to 27.53 | 3.058 to 27.484 | 2.819 to 27.479 | 2.993 to 27.484 | 1.967 to 26.473 |
| Range h, k, l | −5/5, −12/13, −17/24 | −12/16, −12/23, −13/6 | −26/26, −12/12, −15/15 | −25/25, −12/13, −15/15 | −23/23, −15/15, −17/17 |
| No. refl. collected | 5459 | 8893 | 8649 | 11298 |
142898 |
| No. indepent refl. | 1908 | 5250 | 2580 | 2667 | 3317 |
| Comp. to θmax (%) | 99.9 | 99.2 | 97.8 | 99.9 | 99.6 |
| Max/min transmission | 0.357/0.142 | 0.651/0.452 | 0.732/0.536 | 0.315/0.111 | 0.7454/0.6192 |
| Data/restraints/parameters | 1908/0/101 | 5250/0/307 | 2580/0/160 | 2667/0/159 | 3317/1/213 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0324 | R 1 = 0.0424 | R 1 = 0.0754 | R 1 = 0.0339 | R 1 = 0.0740 |
| wR2 = 0.0617 | wR2 = 0.0764 | wR2 = 0.2461 | wR2 = 0.0848 | wR2 = 0.1875 | |
| R indices (all data) | R 1 = 0.044 | R 1 = 0.0776 | R 1 = 0.0855 | R 1 = 0.0468 | R 1 = 0.0961 |
| wR2 = 0.07349 | wR2 = 0.0861 | wR2 = 0.2542 | wR2 = 0.0920 | wR2 = 0.2184 | |
| Goodness-of-fit on (F2) | 1.086 | 1.026 | 1.157 | 1.090 | 1.073 |
| Largest diff. peak/hole (e A−3) | 0.474/−0.437 | 1.295/−1.115 | 1.904/−3.291 | 0.394/−0.671 | 0.969/−0.784 |
| CCDC number | 2180535 | 2180536 | 2180537 | 2180538 | 2180539 |
Computational details
DFT calculations were performed on compounds 5 and 6 using the Gaussian09 package,78 employing the hybrid functional Becke–3–Lee–Yang–Parr (B3LYP)79,80 functional and the double-ζ polarized basis set with six d-type Cartesian–Gaussian polarization functions 6-31G(d,p) basis set.81–84 The geometries were characterized as true minima on the potential energy surface using vibrational frequency calculations (no imaginary values). The UV-vis transitions were calculated by means of TD-DFT calculation on the optimized geometries at the same B3LYP/6-31G(d,p) level of theory.Electrochemical measurements
The electrochemical measurements were carried out on a CHI660 electrochemical workstation using a three-electrode system: platinum wire as the counter electrode, Ag/AgCl as the reference electrode, and Pt electrode with a geometric area of 0.03 cm2 as the working electrode. The experiments were conducted under a high purity argon atmosphere in a three-compartment, three-electrode anchor-type electrochemical cell setup. All electrochemical experiments started from the open circuit potential (OCP). Polymer films of complexes 5 and 6 were obtained electrochemically using cyclic voltammetry (CV) in anhydrous CH3CN containing 0.02 M of tetrabutylammonium hexafluorophosphate (TBAPF6), 2.0 mM of each compound during 30 cycles, with repeated scans ranges of −0.6 to +2.4 V (for compound 5) and 0.0 to +2.3 V (for compound 6) versus Ag/AgCl at a scan rate of 50 mV s−1. Subsequently, poly-5 and poly-6 modified electrode responses were studied employing a 0.02 M TBAPF6 solution in anhydrous CH3CN. Finally, a potentiostatic method was employed to synthesize polymer films on a Pt electrode disk (area = 0.03 cm2) using the same optimized conditions as for compounds 5 and 6 by CV. Images from the electrode surface were obtained on a fifty LEO VP1400 Scanning Electron Microscope Energy Dispersive Spectroscopy (SEM-EDS).Author contributions
Investigations (G.A., P.H., and L.A.H.), software (G.A., T.R., V.D., M.F. and L.A.H.), validation (D.C., C.M., and J.-R.H.), formal analysis (J.-R.H.), investigation (G.A., P.H., and L.A.H.), resources (C.M.), data curation (G.A. and J.-R.H), writing—original draft preparation (G.A., L.A.H., and J.-R.H.), writing—review, and editing (G.A., C.M., L.A.H. and J.-R.H.), supervision (J.-R.H. and C.M.), project administration (C.M.), conceptualization, methodology and funding acquisition (D.C. and C.M.). All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge Prof. J.-Y. Saillard (ISCR, Rennes) for pertinent advices concerning theoretical aspects of this work. We also thank Prof. O. Cador (ISCR, Rennes) and P. Jéhan (CRMPO, Rennes) for helpful assistance with SQUID and MS measurements. Prof. M. A. del Valle (PUC, Santiago de Chile) is thanked for her insightful insights and comments regarding the electrochemical analysis. This research has been performed as part of the Chilean–French International Research Project “IRP-CoopIC 2022-2026”. Financial support from the Fondo Nacional de Desarrollo Científico y Tecnológico [FONDECYT (Chile), grant no. 1140903 (C. M. and D. C.)], FONDEQUIP [EQM130154], the Vicerrectoría de Investigación y Estudios Avanzados, Pontificia Universidad Católica de Valparaíso, Chile (C. M. and D. C.), the CNRS and the Université de Rennes 1 is gratefully acknowledged. G. A. gratefully acknowledges CONICYT (Chile) for the graduate fellowship support no. 21120098. This work was performed on a volunteer basis in G. A. free time; this being said, G. A. also acknowledges the personal support from the Department of Chemistry, KTH Royal Institute of Technology at Stockholm, Sweden.References
- I. Manners, Synthetic Metal-Containing Polymers, Wiley, 2004 Search PubMed.
- G. A. Lawrance, Introduction to Coordination Chemistry, Wiley, 2013 Search PubMed.
- C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2011, 255, 2469–2502 CrossRef CAS.
- M. O. Wolf, Adv. Mater., 2001, 13, 545–553 CrossRef CAS.
- T. Hirao, Coord. Chem. Rev., 2002, 226, 81–91 CrossRef CAS.
- B. J. Holliday and T. M. Swager, Chem. Commun., 2005, 23–36 RSC.
- S. C. Yu, S. Hou and W. K. Chan, Macromolecules, 2000, 33, 3259–3273 CrossRef CAS.
- J.-K. Lee, D. Yoo and M. F. Rubner, Chem. Mater., 1997, 9, 1710–1712 CrossRef CAS.
- Y. Yan, J. Zhang, L. Ren and C. Tang, Chem. Soc. Rev., 2016, 45, 5232–5263 RSC.
- D. M. González, N. B. Cruz, L. A. Hernández, J. Oyarce, R. Benavente and C. Manzur, J. Polym. Sci., 2020, 58, 557–567 CrossRef.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- Y. Koizumi, N. Shida, M. Ohira, H. Nishiyama, I. Tomita and S. Inagi, Nat. Commun., 2016, 7, 10404 CrossRef CAS PubMed.
- G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581–1593 CrossRef CAS.
- J. Roncali, R. Garreau, A. Yassar, P. Marque, F. Garnier and M. Lemaire, J. Phys. Chem., 1987, 91, 6706–6714 CrossRef CAS.
- F. V. Oberhaus and D. Frense, Electrochim. Acta, 2022, 402, 139536 CrossRef CAS.
- J. L. Reddinger and J. R. Reynolds, Synth. Met., 1997, 84, 225–226 CrossRef CAS.
- J. L. Reddinger and J. R. Reynolds, Macromolecules, 1997, 30, 673–675 CrossRef CAS.
- J.-M. Moon, N. Thapliyal, K. K. Hussain, R. N. Goyal and Y.-B. Shim, Biosens. Bioelectron., 2018, 102, 540–552 CrossRef CAS PubMed.
- S. Jin and G. Xue, Macromolecules, 1997, 30, 5753–5757 CrossRef CAS.
- A. S. Abd-El-Aziz, S. Sezgin Dalgakiran and L. Bichler, Eur. Polym. J., 2012, 48, 1901–1913 CrossRef CAS.
- C. Friebe, M. D. Hager, A. Winter and U. S. Schubert, Adv. Mater., 2012, 24, 332–345 CrossRef CAS PubMed.
- W. Wang, V. M. Lynch, H. Guo, A. Datta and R. A. Jones, Dalton Trans., 2020, 49, 2264–2272 RSC.
- S. Napieraïa, M. Kubicki, V. Patroniak and M. Waïesa-Chorab, Electrochim. Acta, 2021, 369, 137656 CrossRef.
- I. Mondal and S. Chattopadhyay, J. Coord. Chem., 2019, 72, 3183–3209 CrossRef CAS.
- C. Freire, M. Nunes, C. Pereira, D. M. Fernandes, A. F. Peixoto and M. Rocha, Coord. Chem. Rev., 2019, 394, 104–134 CrossRef CAS.
- X. Liu, C. Manzur, N. Novoa, S. Celedon, D. Carrillo and J.-R. Hamon, Coord. Chem. Rev., 2018, 357, 144–172 CrossRef CAS.
- M. Abu-Dief and I. M. A. Mohamed, J. Basic Appl. Sci., 2015, 4, 119–133 Search PubMed.
- C. R. Nayar and R. Ravikumar, J. Coord. Chem., 2014, 67, 1–16 CrossRef CAS.
- A. de Fátima, C. de Paula Pereira, C. Raquel Said Dau Gonçalves Olímpio, B. Germano de Freitas Oliveira, L. Lopardi Franco and P. Henrique Corrêa da Silva, J. Adv. Res., 2018, 13, 113–126 CrossRef PubMed.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- E. Schulz, Chem. Rec., 2021, 21, 427–439 CrossRef CAS PubMed.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS.
- X. Liu and J.-R. Hamon, Coord. Chem. Rev., 2019, 389, 94–118 CrossRef CAS.
- M. L. Mejía, J. H. Rivers, S. F. Swingle, Z. Lu, X. Yang, M. Findlater, G. Reeske and B. J. Holliday, Main Group Chem., 2010, 9, 167–191 Search PubMed.
- M. L. Mejía, K. Agapiou, X. Yang and B. J. Holliday, J. Am. Chem. Soc., 2009, 131, 18196–18197 CrossRef PubMed.
- M. L. Mejía, G. Reeske and B. J. Holliday, Chem. Commun., 2010, 46, 5355–5357 RSC.
- A. Pietrangelo, B. C. Sih, B. N. Boden, Z. Wang, Q. Li, K. C. Chou, M. J. MacLachlan and M. O. Wolf, Adv. Mater., 2008, 20, 2280–2284 CrossRef CAS.
- A. K. Asatkar, S. P. Senanayak, A. Bedi, S. Panda, K. S. Narayan and S. S. Zade, Chem. Commun., 2014, 50, 7036–7039 RSC.
- B. Djukic and M. T. Lemaire, Inorg. Chem., 2009, 48, 10489–10491 CrossRef CAS PubMed.
- A. Voituriez, M. Mellah and E. Schulz, Synth. Met., 2006, 156, 166–175 CrossRef CAS.
- A. Zulauf, X. Hong, F. Brisset, E. Schulz and M. Mellah, New J. Chem., 2012, 36, 1399–1407 RSC.
- D. González, J. Cisterna, I. Brito, T. Roisnel, J.-R. Hamon and C. Manzur, Polyhedron, 2019, 162, 91–99 CrossRef.
- D. M. González, L. A. Hernández, J. Oyarce, A. Alfaro, N. Novoa, J. Cisterna, I. Brito, D. Carrillo and C. Manzur, Synth. Met., 2021, 271, 116633 CrossRef.
- S. Celedón, V. Dorcet, T. Roisnel, A. Singh, I. Ledoux-Rak, J.-R. Hamon, D. Carrillo and C. Manzur, Eur. J. Inorg. Chem., 2014, 4984–4993 CrossRef.
- S. Celedón, M. Fuentealba, T. Roisnel, I. Ledoux-Rak, J.-R. Hamon, D. Carrillo and C. Manzur, Eur. J. Inorg. Chem., 2016, 3012–3023 CrossRef.
- S. Celedón, T. Roisnel, I. Ledoux-Rak, J.-R. Hamon, D. Carrillo and C. Manzur, J. Inorg. Organomet. Polym. Mater., 2017, 27, 795–804 CrossRef.
- G. Ahumada, M. Fuentealba, T. Roisnel, S. Kahlal, D. Carrillo, R. Cordova, J.-Y. Saillard, J.-R. Hamon and C. Manzur, Polyhedron, 2018, 151, 279–286 CrossRef CAS.
- G. Ahumada, J. Oyarce, T. Roisnel, S. Kahlal, M. A. del Valle, D. Carrillo, J.-Y. Saillard, J.-R. Hamon and C. Manzur, New J. Chem., 2018, 42, 19294–19304 RSC.
- K. A. Khan, H. Faidallah and A. M. Asiri, J. Chem., 2013, 478635 Search PubMed.
- C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040–3053 CrossRef CAS.
- J. S. Danilova, S. M. Avdoshenko, M. P. Karushev, A. M. Timonov and E. Dmitrieva, J. Mol. Struct., 2021, 1241, 130668 CrossRef CAS.
- P. Molina, A. Arques and I. Cartagena, Thiophenes and their Benzo Derivatives: Structure, Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, New York, 2008, ch. 9, vol. 3, pp. 625–739 Search PubMed.
- S. Celedón, P. Hamon, V. Artigas, M. Fuentealba, S. Kahlal, I. Ledoux-Rak, D. Carrillo, J.-Y. Saillard, C. Manzur and J.-R. Hamon, Eur. J. Inorg. Chem., 2022, e202200478 Search PubMed.
- J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277–294 RSC.
- J. Mahrholdt, E. Kovalski, T. Ruffer, V. Vrček and H. Lang, Eur. J. Inorg. Chem., 2022, e202101059 CAS.
- Y.-P. Jiao, H.-Y. Shi, W.-Y. Zhou, A.-Q. Jia, H.-T. Shi and Q.-F. Zhang, J. Mol. Struct., 2022, 1259, 132695 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427–9477 CrossRef CAS PubMed.
- M. Fuentealba, A. Trujillo, J.-R. Hamon, D. Carrillo and C. Manzur, J. Mol. Struct., 2008, 881, 76–82 CrossRef CAS.
- N. Novoa, T. Roisnel, P. Hamon, S. Kahlal, C. Manzur, H. M. Ngo, I. Ledoux-Rak, J.-Y. Saillard, D. Carrillo and J.-R. Hamon, Dalton Trans., 2015, 44, 18019–18037 RSC.
- C.-H. Lin, H. Pan, V. N. Nesterov and M. G. Richmond, J. Organomet. Chem., 2013, 748, 56–62 CrossRef CAS.
- M. A. Gaona, F. Montilla, E. Álvarez and A. Galindo, Dalton Trans., 2015, 44, 6516–6525 RSC.
- τ 4 = 0 for a perfect square planar geometry; L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- O. Kahn, Molecular Magnetism, Dover Publications, 2021 Search PubMed.
- H. Kargar, M. Ashfaq, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, K. S. Munawar and M. N. Tahir, Inorg. Chim. Acta, 2022, 536, 120878 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Wiley- Interscience, Hoboken, NJ, 2006 Search PubMed.
- J. Oyarce, L. Hernández, G. Ahumada, J. P. Soto, M. A. del Valle, V. Dorcet, D. Carrillo, J.-R. Hamon and C. Manzur, Polyhedron, 2017, 123, 277–284 CrossRef CAS.
- K. Łe˛picka, P. Pieta, G. Francius, A. Walcarius and W. Kutner, Electrochim. Acta, 2019, 315, 75–83 CrossRef.
- C. E. Dahm, D. G. Peters and J. Simonet, J. Electroanal. Chem., 1996, 410, 163–171 CrossRef.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Elsevier Inc., Amsterdam, The Netherlands, 5th edn, 2003 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 839–854 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Jean-Yves Saillard, our esteemed colleague and friend, not only on the occasion of his 75th anniversary but also in recognition of his profound engagement in our fruitful long-term collaboration and for his invaluable contribution to inorganic and cluster chemistry. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S13 and Tables S1–S3. CCDC 2180535–2180539. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00224a |
| § Present address: Department of Chemistry, KTH Royal Institute of Technology, SE-10044, Stockholm, Sweden. E-mail: E-mail: gahumadat@gmail.com |
| This journal is © The Royal Society of Chemistry 2023 |