
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiano-stool ruthenium(II) complexes with maleimide and phosphine or phosphite ligands: synthesis and activity against normal and cancer cells†
Michał
Juszczak
 a,
Sujoy
Das
b,
Aneta
Kosińska
b,
Agnieszka J.
Rybarczyk-Pirek
c,
Kinga
Wzgarda-Raj
c,
Paulina
Tokarz
a,
Saranya
Vasudevan
d,
Arkadiusz
Chworos
d,
Katarzyna
Woźniak
*a and
Bogna
Rudolf
*b
a,
Sujoy
Das
b,
Aneta
Kosińska
b,
Agnieszka J.
Rybarczyk-Pirek
c,
Kinga
Wzgarda-Raj
c,
Paulina
Tokarz
a,
Saranya
Vasudevan
d,
Arkadiusz
Chworos
d,
Katarzyna
Woźniak
*a and
Bogna
Rudolf
*b
aUniversity of Lodz, Faculty of Biology and Environmental Protection, Department of Molecular Genetics, Pomorska 141/143, 90-236, Lodz, Poland. E-mail: katarzyna.wozniak@biol.uni.lodz.pl
bUniversity of Lodz, Faculty of Chemistry, Department of Organic Chemistry, Tamka 12, 91-403 Lodz, Poland. E-mail: bogna.rudolf@chemia.uni.lodz.pl
cUniversity of Lodz, Faculty of Chemistry, Department of Physical Chemistry, Pomorska 163/165, 90-236 Lodz, Poland
dCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Lodz, Poland
First published on 10th March 2023
Abstract
In these studies, we designed and investigated cyto- and genotoxic potential of five ruthenium cyclopentadienyl complexes bearing different phosphine and phosphite ligands. All of the complexes were characterized with spectroscopic analysis (NMR, FT-IR, ESI-MS, UV-vis, fluorescence and XRD (for two compounds)). For biological studies, we used three types of cells – normal peripheral blood mononuclear (PBM) cells, leukemic HL-60 cells and doxorubicin-resistance HL-60 cells (HL-60/DR). We compared the results obtained with those obtained for the complex with maleimide ligand CpRu(CO)2(η1-N-maleimidato) 1, which we had previously reported. We observed that the complexes CpRu(CO)(PPh3)(η1-N-maleimidato) 2a and CpRu(CO)(P(OEt)3)(η1-N-maleimidato) 3a were the most cytotoxic for HL-60 cells and non-cytotoxic for normal PBM cells. However, complex 1 was more cytotoxic for HL-60 cells than complexes 2a and 3a (IC50 = 6.39 μM vs. IC50 = 21.48 μM and IC50 = 12.25 μM, respectively). The complex CpRu(CO)(P(OPh)3)(η1-N-maleimidato) 3b is the most cytotoxic for HL-60/DR cells (IC50 = 104.35 μM). We found the genotoxic potential of complexes 2a and 3a only in HL-60 cells. These complexes also induced apoptosis in HL-60 cells. Docking studies showed that complexes 2a and CpRu(CO)(P(Fu)3)(η1-N-maleimidato) 2b have a small ability to degrade DNA, but they may cause a defect in DNA damage repair mechanisms leading to cell death. This hypothesis is corroborated with the results obtained in the plasmid relaxation assay in which ruthenium complexes bearing phosphine and phosphite ligands induce DNA breaks.
Introduction
In 1989, with discovery of NKP-1339 (sodium trans-[tetrachloridobis(indazole)Ru(III)]) and KP1019 (indazolium trans-[tetrachloridobis(indazole)Ru(III)]), a new class of metal-based anti-cancer drugs were unveiled to the world – ruthenium organic complexes,1,2 in the following years two other compounds were invented with promising anti-metastatic properties; NAMI-A (imidazoliumtrans-[tetrachlorido(dimethyl sulfoxide)(imidazole)Ru(III)]) and TLD1443 ([Ru(4,4′-dimethyl-2,2′-bipyridine)2(2-(2′,2′′:5′′,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]Cl2).3–5 These are the compounds which have proudly completed the Phase-I clinical trial and are knocking on the door for Phase-II.6Drug resistance of cancer cells is one of the major challenges of cancer therapy. Anti-cancer drug resistance can arise from a multitude of mechanisms like impaired drug uptake into the cancer cell, enhanced drug efflux, altered drug target, changed damage recognition/enhanced DNA repair, impaired induction of apoptosis as well as drug sequestration away from its target. Moreover, the entire composition of the tumor microenvironment (TME), containing multiple cell types such as fibroblasts or immune cells, has a strong impact on therapy success and failure.6
In recent years, several ruthenium complexes with anti-cancer activity against cisplatin-resistant cells have been synthesized.7–9 Some of them are also being studied to break down doxorubicin resistance.10 The anti-cancer activity of Doxorubicin (Dox), a member of the anthracycline family, is mainly exerted through the DNA intercalation and topoisomerase-II inhibition in fast-proliferating tumors. However, Dox causes cumulative and dose-dependent cardiotoxicity, which results in increased risks of mortality among cancer patients and thus limiting its wide clinical applications. Several mechanisms has been proposed for doxorubicin-induced cardiotoxicity and oxidative stress, free radical generation and apoptosis are the most widely reported. Apart from this, other mechanisms are also involved in Dox-induced cardiotoxicity such as impaired mitochondrial function, a perturbation in iron regulatory protein, disruption of Ca2+ homeostasis, autophagy, the release of nitric oxide and inflammatory mediators and altered gene and protein expression that involved apoptosis. Dox also causes downregulation of DNA methyltransferase 1 (DNMT1) enzyme activity which leads to a reduction in the DNA methylation process. This hypomethylation causes dysregulation in the mitochondrial genes like peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1-alpha (PGC-1α), nuclear respiratory factor 1 (NRF-1) and mitochondrial transcription factor A (TFAM) unit in the heart. Apart from DNA methylation, Dox treatment also alters the microRNAs levels and histone deacetylase (HDAC) activity.11
Piano-stool ruthenium(II) complexes are becoming notable in last few deacades for their structural flexibilities and bioactivities towards metastatic cells.12–14 They have been subdivided into types depending upon their structure–activity parameters: Ru(η6-arene) and Ru(η5-cyclopentadienyl) complexes.15,16 Within the first type, half-sandwich ruthenium complexes containing PTA (1,3,5-triaza-7-phosphaadamantane) ligands,17–19 and their functionalized derivatives (RAPTA complexes) are being widely acclaimed in anti-cancer experiments and advanced pre-clinical models.20,21 While, within the second type, most of the Ru(η5-cyclopentadienyl) half-sandwich complexes reported so far are found to be amply cytotoxic towards various human cancer cell lines like A2780CisR, MDA-MB-231, PC-3, and HT-29 etc.22,23 Their effectivity towards more than one cancer cell lines makes them potential broad-spectrum-anti-tumor agents in recent time.24,25
Some recent works have reported the use of phosphine or phosphite ligands at ruthenium complexes, and most of them concerned Ru(η6-p-cymeme) complexes.26–28 In 2011, Hanif et al. added phosphite moieties to sugar molecules attached to ruthenium centers in order to improve water solubility of the complexes.29 Recently, Klaimanee et al. developed three Ru(II) p-cymene complexes with different organophosphorus ligands possessing anti-cancer, antibacterial and anti-fungal activities.30
Most interestingly, Ribeiro et al. in 2019, found that the presence of triphenylphosphine significantly increases the antiproliferative potential of [RuCl(PPh3)(N–N)]Cl complexes, which is apparently due to the capability of triphenylphosphine to intercalate between DNA base pairs. Hence, triphenylphosphine acts as a vehiculizing agent for ruthenium complexes to specific tumor cells.31 Li et al. also reported the enhancement of anti-cancer activity with triphenylphosphine ligands in some copper complexes.32 On the other side, Küster's group reported various η6-areneruthenium(II) phosphite complexes for the treatment of Alveolar Echinococcosis with cytotoxicity effect on human fibroblasts, Vero cells, and rat hepatoma cells. Authors used triethyl phosphite, triphenyl phosphite and triisopropyl phosphite as ligands and observed that the complexes with triethyl phosphite and triisopropyl phosphite are the most cytotoxic towards cancer cells.33
Recently, we have found that cyclopentadienyl ruthenium complex bearing maleimidato ligand CpRu(CO)2(η1-N-maleimidato) (1) is highly cytotoxic and genotoxic, both for normal and cancer cells at the concentrations from 0.5 to 250 μM.34
Herein, we introduce five novel ruthenium cyclopentadienyl complexes bearing maleimide and different phosphine/phosphite ligands (2a, 2b, 3a, 3b and 3c) (Fig. 1), derivatized from complex 1 in UV-vis light induced CO ligand exchange process. Triphenylphosphine, tris(2-furyl)phosphine, triethyl phosphite, triphenyl phosphite and triisopropyl phosphite have been chosen for the study.
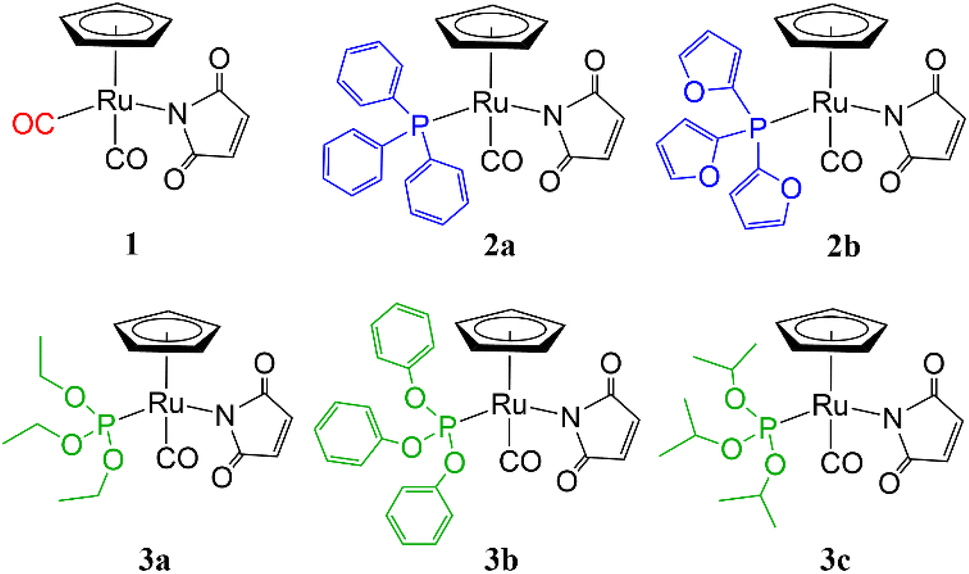 | ||
| Fig. 1 The structures of the ruthenium complexes 1, 2a–b and 3a–c. | ||
NMR (1H, 31P, 13C), FT-IR, ESI-MS, UV-vis, fluorescence and XRD studies were executed for the characterization and structural analysis of the complexes. The cytotoxic potential of these complexes was analyzed in peripheral blood mononuclear (PBM) cells as normal cells and leukemic HL-60 cells and also doxorubicin-resistant HL-60 cells (HL-60/DR cells). It has been observed that ruthenium complexes 2a and 3a are highly cytotoxic for cancer HL-60 cells and non-cytotoxic for normal PBM cells. Therefore, we examined the ability of these complexes to induce DNA damage and apoptosis in cancer cells. We also used the plasmid relaxation assay and docking studies to determine the potential of ruthenium complexes to directly damage DNA.
Results and discussion
Synthesis of complexes 1, 2a–b and 3a–c
Complex 1 CpRu(CO)2(η1-maleimidato) was obtained in previously described photochemical reaction of CpRu(CO)2I with maleimide in presence of diisopropylamine.12To obtain complexes 2a–b and 3a–c we performed the photochemical CO–phosphine/phosphite ligand exchange reaction of 1 (Scheme 1). Similar process was previously described for the iron analog where CpFe(CO)2(η1-maleimidato) was irradiated with triphenylphosphine by visible light to give CpFe(CO)(PPh)(η1-maleimidato).35 In present work, ruthenium complex 1 was irradiated by UV-light (360 nm) in the presence of chosen phosphines (triphenylphosphine, tris(2-furyl)phosphine), or phosphites (triethyl phosphite, triphenyl phosphite and triisopropyl phosphite) to give complexes 2a–b and 3a–c with good yields (Scheme 1). The crude products were purified by flash chromatography on silica gel. The pure products were characterised by spectroscopic methods (Fig. S2–S26†) and the crystals of 2a and 2b were analysed by X-ray (Fig. 2). All of the complexes were stable in the dark at room temperature. Because of some light sensitivity, the purification, characterisation, and crystallisation were performed in the absence of ambient light.
 | ||
| Scheme 1 Schematic diagram for the synthesis of complexes 2a–b and 3a–c. | ||
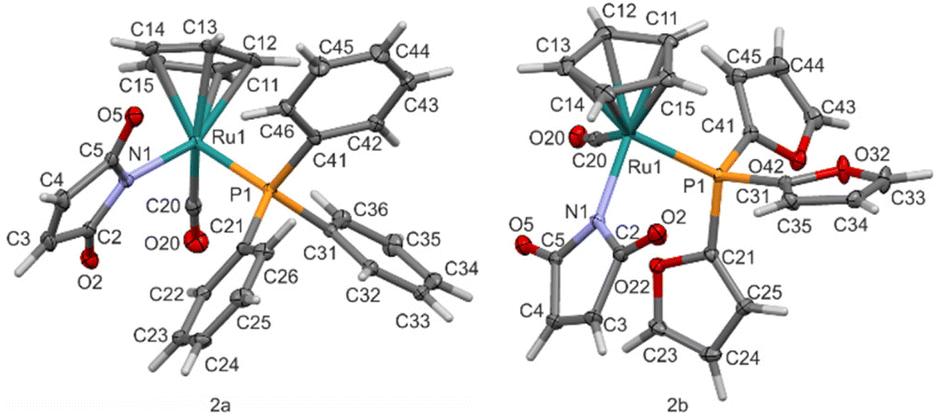 | ||
| Fig. 2 Molecular structure of 2a and 2b with atom labelling scheme. Displacement ellipsoid are drawn with 40% probability level. | ||
Prominent deshielding of the phosphorus atoms in phosphines and phosphites ligands has been observed upon reaction with complex 1. The 31P NMR analysis revealed that the peaks for the phosphorus atoms in all complexes have been shifted downfield compared to the starting phosphines or phosphites. It is evident that the extent of deshielding is much higher in the case of phosphines (2a, 2b) than that of phosphites (3a, 3b, 3c) as shown in Table 1.
| Phosphine/phosphites | 31P NMR (ppm) | Ru-complexes | 31P NMR (ppm) |
|---|---|---|---|
| P(Ph)3 | −5.26 | 2a | 56.94 |
| P(Fu)3 | −77 | 2b | 4.797 |
| P(OEt)3 | 139.1 | 3a | 148.307 |
| P(OPh)3 | 128 | 3b | 140.483 |
| P(OiPr)3 | 139.49 | 3c | 144.502 |
The absorbance and emission spectra of all complexes were recorded in chloroform. It was observed that complexes 2a–b and 3a–c misses characteristic emission peaks; only complex 1 has very weak emission at 358 nm upon excitation at 300 nm (Fig. S27†).
Crystallographic analysis
The results of crystal structure determination are presented in the Table 2. Compound 2a crystallizes in the non-centrosymmetric orthorhombic P212121 while 2b in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space groups. Molecular diagrams with atom labelling scheme are presented in Fig. 2. In the both cases ruthenium Ru1 atom is bonded to cyclopentadienyl ring (C11–C12–C13–C14–C15), carbonyl ligand (C20–O20), nitrogen atom N1 of maleimidato ligand and phosphorus P1 of phosphine ligand.
space groups. Molecular diagrams with atom labelling scheme are presented in Fig. 2. In the both cases ruthenium Ru1 atom is bonded to cyclopentadienyl ring (C11–C12–C13–C14–C15), carbonyl ligand (C20–O20), nitrogen atom N1 of maleimidato ligand and phosphorus P1 of phosphine ligand.
| 2a | 2b | |
|---|---|---|
| Formula/M [g mol−1] | C28H22NO3PRu/552.5 | C22H16NO6PRu/522.4 |
| Crystal system, space group | Orthorhombic, P212121 | Triclinic, P |
| a, b, c [Å] | 9.5419(1), 14.7114(2), 16.7725(2) | 8.1802(1), 8.7067(2), 14.9093(3) |
| α, β, γ [°] | 90, 90, 90 | 77.209(2), 83.852(2), 76.573(2) |
| V [Å3]/Z | 2354.44(5)/4 | 1005.49(4)/2 |
| F (000)/μ [mm−1] | 1120.0/1.559 | 524.0/1.725 |
| Crystal size [mm]/dx [mg m−3] | 0.263 × 0.131 × 0.074/1.559 | 0.107 × 0.077 × 0.063/1.725 |
| λ [Å], T [K] | MoKα/100 K | MoKα/100 K |
| 2θ range [°] | 4.858 to 61.448 | 4.912 to 61.824 |
| Data collected/unique | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 824/6306 (Rint = 0.0392) 824/6306 (Rint = 0.0392) |
22923/5196 (Rint = 0.0369) |
| R/wR2 [I > 2σ(I)] | R 1 = 0.0203/wR2 = 0.0438 | R 1 = 0.0233/wR2 = 0.0519 |
| R/wR2 (all data) | R 1 = 0.0214/wR2 = 0.0442 | R 1 = 0.0259/wR2 = 0.0530 |
| GooF on F2 | 1.015 | 1.071 |
| Δρmin/Δρmax [e Å−3] | 0.37/−0.33 | 0.44/−0.51 |
| Flack parameter | −0.011(10) | |
| CCDC number | 2209334 | 2209335 |
Mean bond length of ruthenium to carbon atoms of cyclopentadienyl rings equals 2.23 Å. In turn, bond lengths of ruthenium atom to carbonyl ligands and to the center of gravity of cyclopentadienyl ring (Cg1) are of a similar range, approximately 1.87 Å, while Ru–N1 and Ru1–P1 bonds are evidently longer respectively about 2.1 Å and 2.3 Å (see Table S1†). Valence angles around Ru1 and between N1 (maleimidato-), P1 (phosphine-) and C1 (carbonyl) atoms are very close to 90°. In turn, angles to the cyclopentadienyl ring (Cg1 in the Table S1†) are of significantly larger values – above 120°. Considering the three-dimensional arrangement of ligands around the metal atom, both compounds can be classified as “piano stool” complexes with cyclopentadienyl ring positioned on the top and other three ligands arranged on the opposite side almost perpendicularly each other (Fig. 2). This molecular conformation is similar to known structures for previously determined ruthenium complexes.34
Assuming cyclopentadienyl as a single ligand, the coordination sphere of Ru1 atom resembles strongly distorted tetrahedron in molecules of the both compounds. The presence of pseudo-tetrahedral arrangement of ligands around metal atom is the source of chirality in the molecule. A formal configuration may be then indicated in the same way as it is used for asymmetric tetrahedral carbon atoms. According the Cahn–Ingold–Prelog rules,36 the ruthenium ligands are listed in the following order: I – phosphine, II – maleimidato, III – carbonyl, IV – cyclopentadienyl. It is especially important for compound 2a which crystallizes a non-centrosymmetric space group as a conglomerate. It means that an analyzed crystal contains a single enantiomer. Thus, despite the non-stereospecific synthesis which results in both products with R and S configurations, the sample underwent spontaneous resolution upon crystallization and, as seen in Fig. 2, only R ruthenium stereoisomer is observed in the crystal.
The formation of conglomerates with spontaneous resolution is a relatively rare phenomenon which however yields enantiopure crystals. It was first demonstrated by Pasteur with sodium ammonium tartrate.37 Even though it is known that under favorable circumstances pure enantiomers can be obtained from a racemic mixture, factors responsible for spontaneous resolution are poorly understood and predictions are hardly possible.
The process of chiral self-sorting behavior has been recognized not only for organic compounds, but also for metal complexes and metalloorganic species. A spontaneous resolution of various cobalt, ruthenium, tungsten, molybdenum, cobalt, iron or zinc complexes of tetrahedral and octahedral coordination has been reported.38–43
Also seven-coordinate lanthanides complexes of praseodymium, samarium and erbium are chiral and crystallize as conglomerates in non-centrosymmetric R3 space group.44 The phenomenon of conglomerate crystallization was recognized in tetrahedral and octahedral metal–ligand cages.45,46 In addition, some approaches to asymmetric catalytic asymmetric synthesis with the use of transition-metal catalyst of ruthenium(II) and palladium, in particular chirality inducer, are known to be employed to achieve homochirality.47–49
As compound 2b crystalizes centrosymmetric P space group, due to crystallographic inversion symmetry both isomers (R and S) are present in the unit cell.
For further comparison of the overall molecular geometry between 2a and 2b the same isomers (R) have been taken to analysis (Table S1†–compare torsion angles). Small differences are observed in values of torsion angles around Ru1–P1 and Ru1–N1 bonds (approximately 20° and 4°) due to twist of phosphine aromatic rings along Ru–P bond. But in general, it does not change significantly the overall molecular conformation as seen in Fig. 3 presenting superposition of molecules along Ru–P1 and Ru–N1 bonds.
 | ||
| Fig. 3 Comparison of 2a (dark blue) and 2b (orange) molecular conformations. | ||
Both crystal structures are stabilized by C–H⋯O interactions of type hydrogen bods with oxygen of carbonyl groups of furane rings as hydrogen acceptors. Some geometric details of these interactions are presented in the Table S2.† The above interactions can be also described in terms of Hirshfeld surface analysis. Such a molecular surface (HS) represents the area where molecules in the crystal state come into mutual contacts. They are presented in Fig. S1† (right) and mapped with a colour scale of dnorm parameter for H⋯O contacts. As it can be seen red colors resulting from short C–H⋯O hydrogen bonds occur close to oxygen atoms. In the HS fingerprint plots (Fig. S1b† – left) H⋯O contacts are presented by two sharp, long distinct spikes in the bottom left area of picture. The greater number of oxygen atoms results in greater percentage of H⋯O contacts in 2b compared with 2a. Also, in this case peaks corresponding to C–H⋯O hydrogen bonds are elongated (∼1.3 Å) as compared to the other structure (>1.35 Å).
Cell viability
Using the method of reducing resazurin to resorufin by metabolically active cells, we examined the viability of cells after 2 and 24 h incubation with ruthenium complexes (Tables S3 and S4,† respectively). Then we determined IC50 doses for all ruthenium complexes against the three tested cell types after incubation for 24 h (Table 3). We have shown that the 3a complex, with an IC50 of 12.25 μM, is the most cytotoxic for HL-60 cells. This complex was much less cytotoxic to normal cells (IC50 > 250 μM).| Ruthenium complexes | HL-60 cells (μM) | HL-60/DR cells (μM) | PBM cells (μM) |
|---|---|---|---|
| 1 | 6.39 | 240.05 | 75.29 |
| 2a | 21.48 | >250 | >250 |
| 2b | 35.64 | 161.37 | 8.48 |
| 3a | 12.25 | >250 | >250 |
| 3b | 52.47 | 104.35 | 7.18 |
| 3c | 18.88 | 132.52 | 11.22 |
The complex 2a was also selected for further studies because it did not show cytotoxicity for normal PBM cells (IC50 > 250 μM). The IC50 for HL-60 cells for this complex was 21.48 μM. The remaining 2b, 3b and 3c ruthenium complexes were cytotoxic for both normal and cancer cells.
The HL-60/DR cell line that we obtained, resistant to doxorubicin, was characterized by about 100 times greater resistance to this drug compared to the original cell line (Fig. 4). In the case of HL-60/DR cells, all ruthenium complexes showed significantly lower cytotoxicity compared to HL-60 cells and PBM cells (Table 3). The 3b complex with an IC50 of 104.35 μM was the most cytotoxic for these cells. Our results indicate that ruthenium complexes do not overcome the doxorubicin resistance in HL-60 cells.
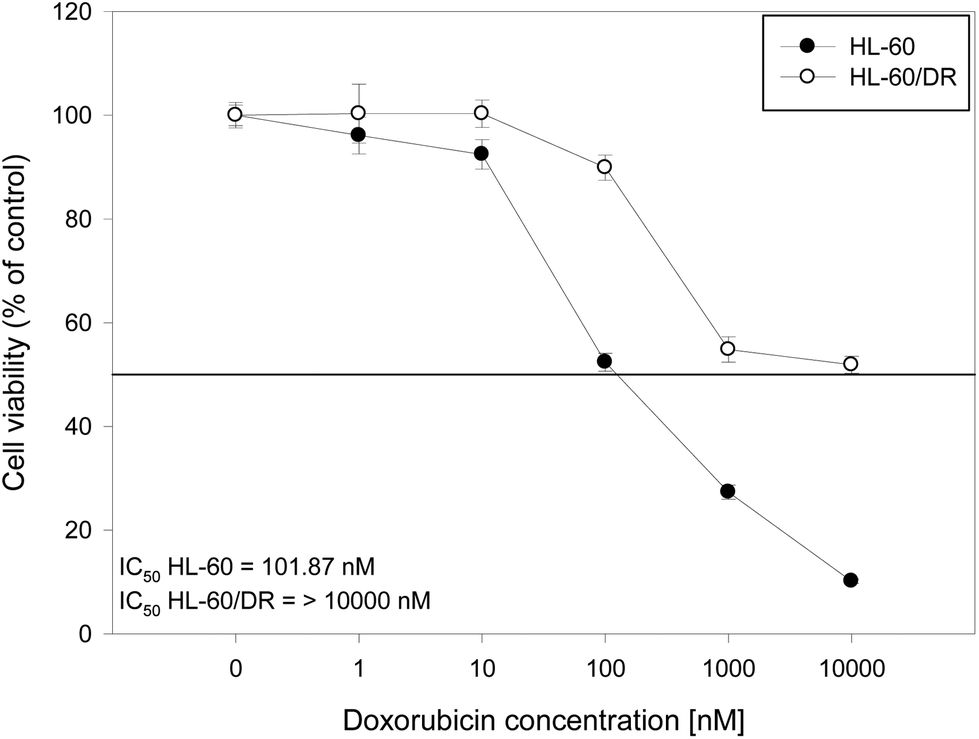 | ||
| Fig. 4 The comparison of HL-60 cells viability and doxorubicin-resistant HL-60/DR cells after incubation with the drug. The horizontal line represents viability at a level of 50%. | ||
The complex 1 which we studied previously34 was the most cytotoxic ruthenium complex among those currently under investigation for HL-60 cells. The IC50 for this complex was 6.39 μM.
In the case of a short 2 h incubation, an increase in the metabolic activity of cells was observed after incubation with all ruthenium complexes. This is especially noticeable in normal PBM cells (Table S3†). Given the high cytotoxicity to HL-60 cells and the lack of cytotoxicity to normal cells, two new ruthenium complexes 2a and 3a were selected for further studies.
DNA damage
Fig. 5a–c show the level of DNA damage analysed by the comet assay under alkaline conditions after incubation with 2a and 3a complexes. The comet assay in the alkaline version is a sensitive and simple method of determining the level of DNA damage, including single- and double-strand breaks and alkali-labile sites in living cells.50 The ruthenium complexes 2a and 3a in the concentration range from 5 to 50 μM were selected for the study of DNA damage. At all the concentrations used, the cells after 2 h incubation showed high viability over 90% (Table S3†). We observed a significant increase in the level of DNA damage in HL-60 cells incubated with ruthenium complexes 2a and 3a (Fig. 5a). The complex 1, which we studied previously,34 shows the highest genotoxic potential. | ||
| Fig. 5 DNA damage in (a) HL-60 cells (b) HL-60/DR cells and (c) PBM cells incubated for 2 h at 37 °C with ruthenium complexes 1, 2a and 3a analysed by the alkaline comet assay. The negative controls were cells incubated only with the cell culture medium for 2 h at 37 °C. The positive controls were cells incubated with H2O2 at 20 μM for 15 min on ice. The figure shows mean results ± SEM, n = 100; ** p < 0.01; *** p < 0.001. | ||
It induces DNA damage at all concentrations used, ranging from 5 to 50 μM (p < 0.001). At the highest concentration used, the complex 1 causes DNA damage at the level of 25% (p < 0.001). Two new ruthenium complexes 2a and 3a at this concentration induce DNA damage at the level of 20% (p < 0.001). In HL-60/DR cells resistant to doxorubicin, neither complex induces DNA damage (Fig. 5b). We obtained interesting results after incubation of PBM cells with the tested complexes (Fig. 5c). DNA damage was only observed in the case of the complex 1 at the level of 18% at the concentration of 50 μM. This result suggests a selective genotoxic activity of 2a and 3a complexes against HL-60 cancer cells. Fig. 6 shows the example images of comet's experiment.
 | ||
| Fig. 6 Representative photos of comet's effect obtained in the alkaline version of the comet assay after incubation of HL-60 cells and PBM cells with complex 2a at the concentrations of 5, 10, 25 and 50 μM for 2 h at 37 °C. The negative controls were cells incubated only with the cell culture medium for 2 h at 37 °C. The positive controls were cells incubated with H2O2 at 20 μM for 15 min on ice. | ||
We also investigated the possibility to DNA damage by ruthenium complexes in vitro. For this purpose, we used the plasmid relaxation assay. Results obtained from electrophoretic mobility shift analysis (EMSA) showed that pUC19 plasmid which we isolated from the DH5α E. coli cells is presented in supercoiled form (CCC). Overnight treatment at 37 °C with restrictase PstI led to linear form (L) of the plasmid. Incubation of CCC form with complexes 1, 2a–b, and 3a–c at concentration of 50 μM showed a possibility of DNA adducts or breaks, which affected topological changes of the plasmid (Fig. 7). After 2 h of incubation of the plasmid with ruthenium complexes, we see the appearance of the OC form in the case of complexes 2b and 3a–c (Fig. 7a). This result demonstrates the possibility of induction of DNA single-strand breaks by these ruthenium complexes in vitro. After longer incubation (24 h) the OC form of the plasmid appears in all complexes (Fig. 7b). In the case of the 3c complex, a linear form (L) of the plasmid is also visible, which proves the possibility of induction of DNA double-strand breaks by this complex.
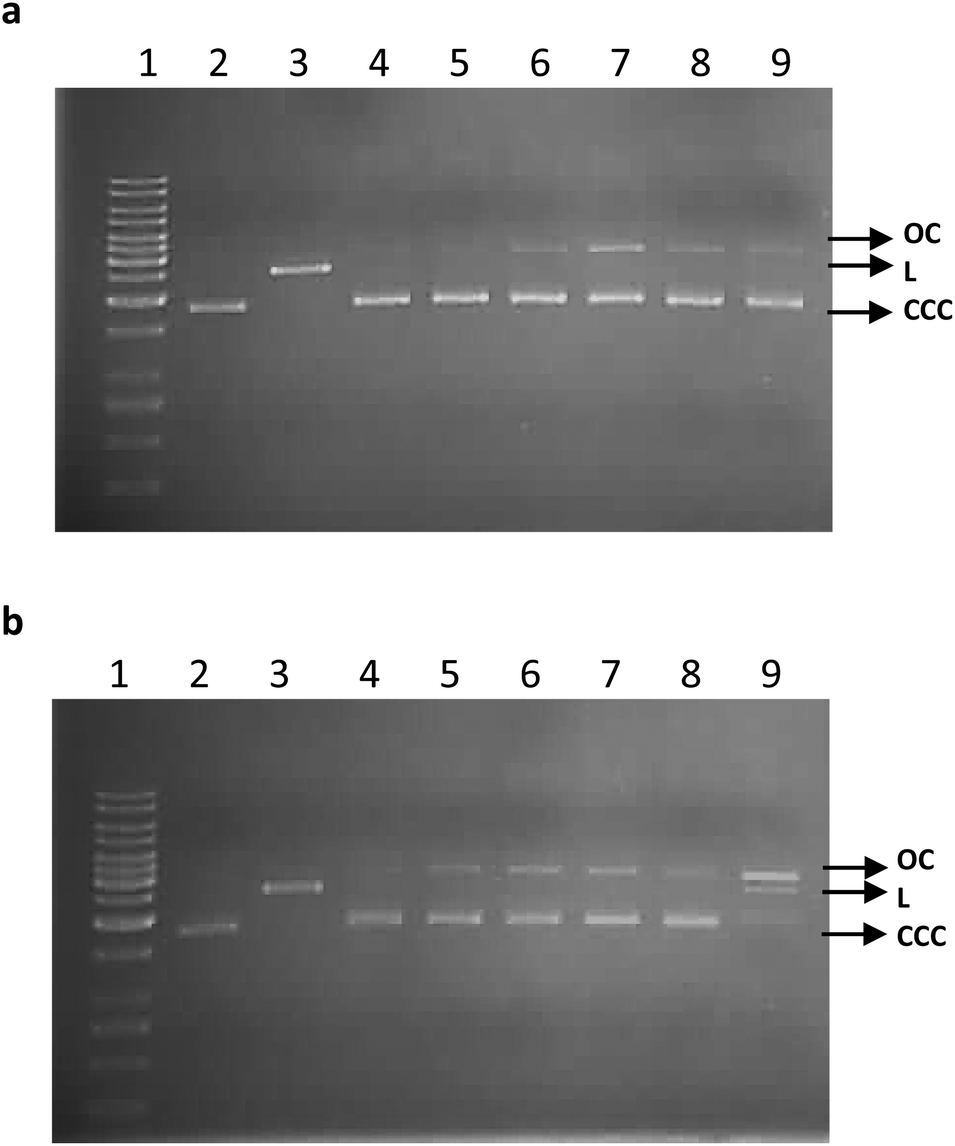 | ||
| Fig. 7 Plasmid relaxation assay. pUC19 plasmid was incubated for 2 h (a) and 24 h (b) (37 °C) with ruthenium complexes at 50 μM, and then was resolved on a 1% agarose gel, stained with ethidium bromide and visualized in UV light. Line 1 – DNA ladder; line 2 – pUC19 plasmid (the supercoiled form, CCC); line 3 – pUC19 plasmid incubated with restrictase PstI (the linear form, L); lines 4–9 – pUC19 plasmid incubated with complexes 1, 2a–b, and 3a–c, respectively. OC – the open circular form of pUC19 plasmid. | ||
Apoptosis
To quantify the number of cells in the different stages of apoptosis after incubation with ruthenium complexes, flow cytometry analysis of living cells double labelled with annexin V-FITC (annexin V) and propidium iodide (PI) was performed. In early apoptosis, the membrane phosphatidylserine (PS) translocates from the inner plasma membrane to the external leaflet. Annexin V is a specific fluorescence probe that can be used to detect PS on the plasma membrane surface. PI can pass through the plasma membrane of dead cells to stain the nucleus, which allows the further distinction of early apoptotic and late apoptotic/necrotic cells. The signals can be divided into four classes: viable cells, early apoptotic cells, late apoptotic cells and necrotic cells.We investigated the ability of 2a and 3a complexes to induce apoptosis in HL-60 and HL-60/DR cells. In HL-60 cells, both complexes induce apoptosis. The 2a complex showed a much greater potential for apoptosis. In the case of this complex, after 24 h at concentrations ranging from 10 to 50 μM, almost all cells are apoptotic (Fig. 8 and Fig. S29†). In the case of the 3a complex, apoptotic cells only dominate after incubation at the highest concentration of 50 μM. The ruthenium complex 2a shows greater potential for apoptosis compared not only to complex 3a but also to complex 1. No apoptosis was observed in HL-60/DR cells after incubation with the 1, 2a and 3a complexes (Fig. S30†). In these cells, we did not detect apoptosis even after incubation with 20 μM camptothecin for 24 h at 37 °C.
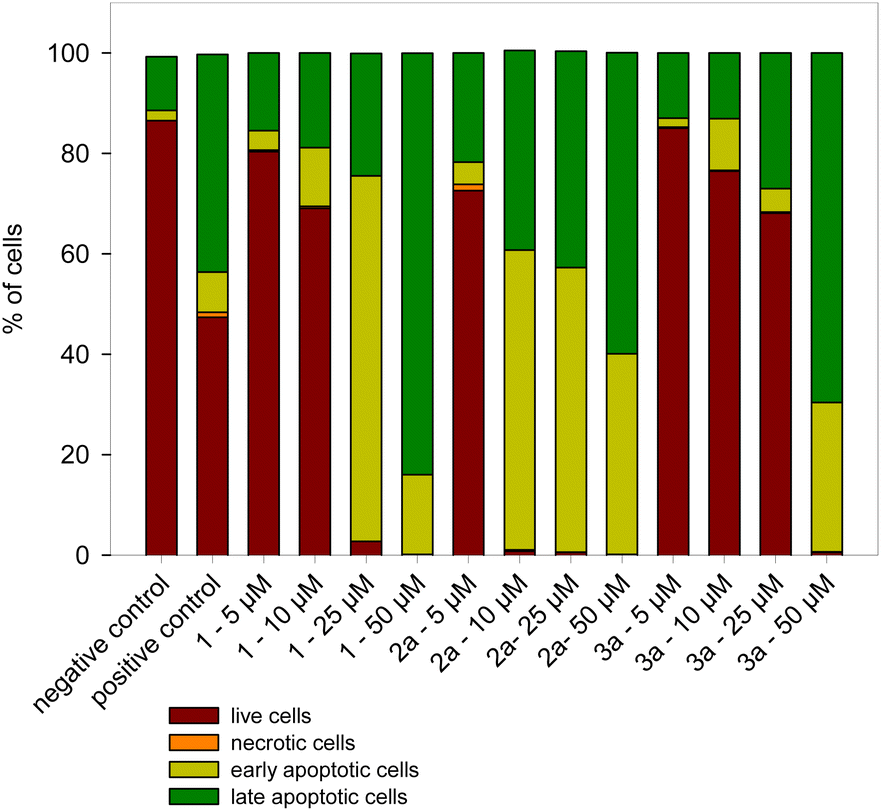 | ||
| Fig. 8 The effect of ruthenium complexes 1, 2a and 3a on the apoptosis of HL-60 cells. The positive control were cells incubated with 20 μM camptothecin for 24 h at 37 °C. Data represent means ± SD of three experiments. | ||
Ruthenium has the ability to form complexes at the different oxidative states (from II to IV) which exhibit different biological activities.51 Among them, the highest biological activity have complexes on the oxidative state(II), possessing the ability to interact with biomolecules such as DNA or proteins.52 The majority of novel synthesized ruthenium compounds, that can exhibit anti-cancer potential are on the oxidative state(II). One of the most common mechanisms of cytotoxicity is the induction of apoptosis. Novel synthesized fluorinated cyclometalated ruthenium(II) complexes led to a significant increase of apoptosis in A549 cells by induction of caspase 3/7 overexpression.7 Arene ruthenium(II) complex leads to a significant increase of apoptosis in MCF-7 cells, which is related to high genotoxicity displayed by severe DNA damage.53 Ruthenium(II)/allopurinol complex induced apoptosis in murine breast cancer cells by pro-caspase activation resulting in releasing of caspase 3 and 7 and Becklin-1 cleavage.54 Ruthenium(II) complex with lapachol induced reactive oxygen species (ROS)-mediated apoptosis in DU-145 cells.55 Moreover, complex increasing expression of cleaved caspases 3 and 9 and induced DNA damage. Ruthenium(II) methylimidazole complexes induce mitochondrial dysfunction in A549 cells, which results overexpression of caspase 9 and finally leads to apoptosis.56 These complexes also exhibit the potential to DNA damage. Polypyridyl ruthenium(II) complex disturbs the potential of the mitochondrial membrane in HepG2 cells.57 Moreover, the complex causes overproduction of ROS and leads to oxidative DNA damage. Cyclometalated ruthenium(II) β-carboline complex causes caspase 8 and caspase 9 activation which leads to the executioner caspase 3 and induction of apoptosis.58 Additionally, these metal complex increases the overproduction of ROS, which generate oxidative DNA damage. Recently, Li et al. presented very interesting results regarding the cycloruthenated complex RuZ.59 RuZ can self-assemble into nanoaggregates in the cell culture medium, resulting in a high intracellular concentration of RuZ in multi-drug resistance cancer cells. RuZ significantly increased the level of ROS and DNA damage, which caused apoptosis. RuZ inhibited the proliferation of 35 cancer cell lines, of which 7 cell lines were resistant to clinical drugs. Interestingly, the RuZ complex was also active in doxorubicin-resistant MDA-MB-231/Adr mouse tumor xenografts.
We observed that complex 2a exhibits the highest potential to induce caspase 3/7 activity at 5 and 10 μM (Fig. 9). This result corresponds with the measurement of apoptosis by flow cytometry, where complex 2a induced apoptosis in the most effective way (Fig. 8). Moreover, all complexes exhibit the potential to induce caspase 3/7 activity at 10 μM. Small absolute values of caspase 3/7 activity for 25 and 50 μM are the result of the high number of late apoptotic cells, which lost metabolic activity due to membrane perforation as determined by annexin V externalization (Fig. 8).
 | ||
| Fig. 9 Effect of ruthenium complexes 1, 2a and 3a on caspase 3/7 activity in HL-60 cells. The cells were incubated with complexes for 24 h at 37 °C. The positive control was sample of cells incubated with 20 μM camptothecin for 24 h at 37 °C. Data represent means ± SD of four experiments; * p < 0.05; *** p < 0.001. | ||
Evaluation of oxidative stress
We used an H2DCF-DA probe to determine the effect of ruthenium complexes 1, 2a, and 3a on the induction of ROS (Fig. 10). We performed a measurement of ROS generation kinetics, where HL-60 cells were incubated with complexes at the concentrations 5, 10, 25, and 50 μM for 15, 30, 45, 60, 90, and 120 min. Our results clearly showed that any of the tested complexes did not exhibit the potential to induction of ROS within 60 min. Statistically significant increase in ROS level was observed after 90 and 120 min only for complex 3a at 50 μM (Fig. 10c). Considering the short lifetime of ROS, this increase is more due to the secondary effect of the complex on cell metabolism than the ability of ROS generation by the complex itself. Our results indicate that both DNA damage and apoptosis in HL-60 cells are not caused by ROS formation induced by ruthenium complexes 1, 2a and 3a. | ||
| Fig. 10 ROS generation kinetics in HL-60 cells incubated with ruthenium complexes 1 (a), 2a (b) and 3a (c) at the concentrations 5, 10, 25, and 50 μM at 37 °C. The positive control were cells incubated with 5 mM H2O2 at 37 °C. Data represent means ± SD of six experiments; * p < 0.05; ** p < 0.01; *** p < 0.001. | ||
Docking studies
Interaction of newly developed ruthenium(II) complexes with DNA was analysed by computational methods using molecular docking and molecular dynamics, to ascertain whether there is a difference between fully paired DNA, as compared to genomic DNA, and a DNA mismatch, as compared to a damaged DNA fragment. Crystal structures for fully complementary and mismatched DNA were retrieved from the protein data bank, PDB ID: 1BNA (fully complemented) and 4E1U (for a mismatch).60,61 The calculated docking energy for selected ruthenium(II) complexes, compounds 1, 2a and 2b with fully complemented DNA (Fig. 11a and S31a, c†) show relatively low binding energy in the range −4.57 to −5.57 (Table 4). However, binding energy for the same complexes with mismatched DNA (Fig. 11b and S31b, d†) are stronger with the lowest difference for the compound 1 (ΔΔG = 3.98 kcal mol−1) and much greater for compounds 2a and 2b (ΔΔG = 9.60 kcal mol−1 and ΔΔG = 8.04 kcal mol−1, respectively). Clearly the mismatched DNA (A–A bulge) have higher docking score compared to the fully complementary DNA, which might suggest that the cytotoxic effect of compounds 2a and 2b are weakly related to the direct degradation of the genomic DNA but rather are interfering with the cellular repair mechanism and thus causing the damaged DNA accumulation and finally cell death. These results also corroborate with the EMSA analysis (gel study part of this work), where short exposure of plasmid DNA to these compounds does not cause the DNA breakage. This is a new hypothesis and has to be further investigated. These complexes were also analysed using Molecular Dynamics (data not shown), however the data is inconclusive probably due to the some discrepancy in the force field parameters for the ruthenium(II) complexes. To our knowledge such complexes were never analysed using MD with a DNA fragment.62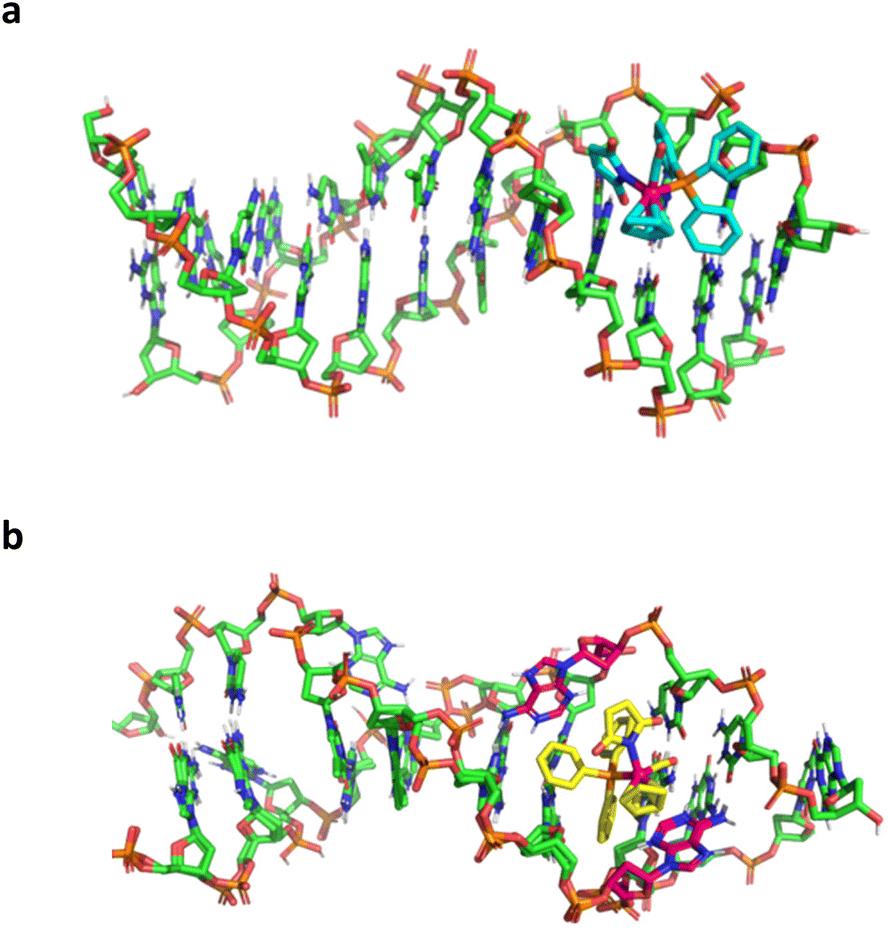 | ||
| Fig. 11 Ruthenium(II) complex 2a with fully complemented DNA (a) and mismatched DNA (b). | ||
| Ligand | Fully paired DNA (kcal mol−1) | Mismatched DNA (kcal mol−1) |
|---|---|---|
| 1 | −4.57 | −8.55 |
| 2a | −5.93 | −15.53 |
| 2b | −5.57 | −13.61 |
Experimental section
Materials and methods
Bis(cyclopentadienylrutheniumdicarbonyl) dimer, maleimide, chloroform-D and all the phosphines and phosphites were purchased from Sigma-Aldrich (Merck). Methanol, and toluene were purchased from POCH (Polish Chemical Reagents) and used as solvents without further purification. All syntheses were carried out under argon. Column chromatographic refinements of the crude products were performed on silica gel 60 (230–400 mesh) purchased from Merck. Infrared (IR) spectra were recorded in KBr on a Fourier Transform InfraRed (FTIR) NEXUS (Thermo Nicolet) spectrometer. Proton, Carbon and Phosphorus NMR spectra were recorded on Bruker Avance III (600 MHz) instrument. NMR data were collected in CDCl3 solution. The chemical shifts were stated in part per million (ppm). Coupling constants were calculated in Hertz (Hz). Electrospray ionization mass spectrometry (ESI-MS) spectra were recorded on the Varian 500-MS LC ion trap spectrometer. Absorbance and emission spectra were recorded on PerkinElmer Lambda 45 UV/vis spectrometer and PerkinElmer LS55 Fluorimeter, respectively. Photochemical syntheses were carried out using UV lamp TQ 150 Z3. Relevant guidelines and regulations were followed in all the performed methods. IMDM medium and fetal bovine seRpM (FBS) were obtained from Biowest (Cytogen, Zgierz, Poland). Dimethyl sulfoxide (DMSO), hydrogen peroxide (H2O2), low-melting-point (LMP), normal-melting-point (NMP) agarose, phosphate buffered saline (PBS), and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (USA). All other reagents were obtained at the highest commercially available grades. A stock solution of complexes (10 mM) was dissolved in DMSO. Complex 1 was synthesized following previously published method, by photochemical reaction of CpRu(CO)2I with maleimide in the presence of diisopropyl amine in toluene.34Synthesis of 2a–b and 3a–c
An argon-saturated solution of 1 (40 mg, 0.12 mmol) and phosphine or phosphite (30 mg, 0.11 mmol) in toluene (10 ml) was illuminated under UV lamp (λ = 350 nm) for 2 h. The progress of the reaction was continuously monitored with TLC. After completion of the reaction, solvent was evaporated in vacuum.:3) as eluent to afford a yellow solid. The product was recrystallized from chloroform/heptane.
Yield 32 mg (55%) 1H NMR (δ, ppm, CDCl3): 4.974 (5H, s, Cp), 6.219 (2H, s, maleimide), 7.331–7.335 (4H, d, J = 2.4 Hz), 7.342–7.349 (8H, d, J = 4.2 Hz), 7.366–7.383 (3H, m, J = 10.2 Hz). 13C NMR (δ, ppm): 205.39, 205.25, 183.93, 136.40, 134.41, 134.09, 133.81, 133.73, 130.20, 130.19, 128.35, 128.28, 85.21 (d). 31P NMR (δ, ppm): 56.939. IR (cm−1): 1958 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O); 1644 (C
O); 1644 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, imide); 1334, 694 (P–Ph). ESI-MS: m/z calcd for C28H22RuNO6P (M + H)+, 554.04; found, 554.27.
O, imide); 1334, 694 (P–Ph). ESI-MS: m/z calcd for C28H22RuNO6P (M + H)+, 554.04; found, 554.27.
:2) as eluent to afford a yellow solid. The product was recrystallized from chloroform/heptane.
Yield 15 mg (45%). 1H NMR (δ, ppm, CDCl3): 5.20 (5H, s, Cp), 6.435–6.446 (5H, m, maleimide, Fu), 6.797–6.803 (3H, dd, J = 3.6 Hz, Fu), 7.635–7.642 (3H, m, J = 4.2 Hz, Fu). 13C NMR (δ, ppm, CDCl3): 203.46, 203.31, 183.77, 148.01, 147.98, 147.20, 146.69, 136.65, 122.12, 121.99, 111.37, 111.32, 84.89, 84.87. 31P NMR (δ, ppm): 4.797. IR (cm−1): 1965 (CO); 1645 (CO, imide); 1330, 1007, 794 (P–Fu). ESI-MS: m/z calcd for C22H16RuNO6P (M + H)+, 523.98; found, 524.24.
:1) as eluent to afford a yellowish liquid. The product was recrystallized from chloroform/heptane to obtain bright yellow solid. Yield: 21 mg (79%). 1H NMR (δ, ppm, CDCl3): 1.209–1.233 (9H, t, J = 14.4 Hz, C2H5), 3.856–3.912 (6H, m, –CH2), 5.158–5.160 (5H, d, J = 1.2 Hz, Cp), 6.851 (2H, s, maleimide). 13C NMR (δ, ppm): 204.00, 203.80, 183.94, 136.91, 84.71, 84.70, 61.32, 61.29, 16.26, 16.22. 31P NMR (δ, ppm, CDCl3): 148.30. IR (cm−1): 1962 (CO); 1644 (CO, imide); 1333, 1023, 939 (P–OEt). ESI-MS: m/z calcd for C16H22RuNO6P (M + H)+, 458.03; found, 458.13.
:1) as eluent to afford a yellow solid. The product was recrystallized from chloroform/heptane.
Yield: 25 mg (51%). 1H NMR (δ, ppm, CDCl3): 4.905 (5H, s, Cp), 6.426 (s, 2H, maleimide), 7.141–7.184 (m, 9H, o-,p-phenyl), 7.302–7.328 (m, 6H, J = 15.6 Hz, m-phenyl). 13C NMR (δ, ppm): 85.080–85.099 (d), 121.339–121.369 (d), 125.137, 129.796, 136.671, 151.252–151.311 (d), 183.376, 202.128–202.324 (d). 31P NMR (δ, ppm): 140.48. IR (cm−1): 1972 (CO); 1652 (CO, imide); 1186, 921, 775 (P–OPh). ESI-MS: m/z calcd for C28H22RuNO6P (M + Na)+, 624.01; found, 624.31.
:1) as eluent to afford a yellow solid. The product was recrystallized from chloroform/heptane.
Yield 15 mg (50%). 1H NMR (δ, ppm, CDCl3): 6.587 (2H, s, maleimide), 5.127–5.129 (5H, d, Cp, J = 1.2 Hz), 4.455–4.511 (3H, m, iso-propyl–CH), 1.204–1.226 (18H, t, J = 13.2 Hz, isopropyl–CH3). 13C NMR (δ, ppm): 204.31, 204.11, 184.02, 136.99, 84.98, 84.96, 69.94, 69.90, 24.19, 24.15, 24.10, 24.09. 31P NMR (δ, ppm): 144.502. IR (cm−1): 1973 (CO); 1651 (CO, imide); 1008, 974 (P–OiPr). ESI-MS: m/z calcd for C19H28RuNO6P (M + Na)+, 522.06; found, 522.31.
X-ray structure determination
X-ray diffraction data for 2a and 2b compounds were measured on a four-circle Oxford Diffraction Supernova Dual diffractometer using a two-dimensional area CCD detector and a low-temperature device Oxford Cryosystem cooler. Integration of the intensities, corrections for Lorentz effects, polarization effects and analytical absorption were performed with CrysAlis PRO.63 The crystal structures was solved by direct methods and refined on F2 using a full-matrix least-squares procedure (SHELXL-2014).64 The positions of the hydrogen were introduced in the calculated positions with an idealized geometry and constrained using a rigid body model with isotropic displacement parameters equal to 1.2 of equivalent displacement parameters of their parent atoms. The molecular geometry was calculated by Platon65 and WinGX programs.66 The relevant crystallographic data are given in Table 2. Atomic coordinates, displacement parameters, a structure factors of the analysed crystal structures are deposited with Cambridge Crystallographic Data Centre CCDC https://www.ccdc.cam.ac.uk/conts/retrieving.html.Cell culture
HL-60 (human promyelocytic leukemia) cell line was obtained from the American Type Culture Collection (ATCC) and cultured in Iscove's Modified Dulbecco's Medium (IMDM) medium with 15% fetal bovine seRpM (FBS), streptomycin/penicillin solution (100 μg ml−1 and 100 U ml−1). HL-60 cells were cultured in flasks at 37 °C in 5% CO2 and sub-cultured every 2–3 days to maintain exponential growth.HL-60/DR (doxorubicin-resistant) cell line was derived from HL-60 cell line by long-term exposure to continuous stepwise increments of doxorubicin concentration. Procedure was similar to that previously described.67,68 HL-60 cells were incubated with 20 nM doxorubicin for three days, then cells were passage into new flask with 20 nM doxorubicin for another three days. After this time cells were collected and centrifuged in a density gradient of Lymphosep (Cytogen, Zgierz, Poland) at 1700 RPM for 35 min with the lowest values of acceleration and deceleration. The interface layer containing viable cells was transferred into 15 ml tubes. Then cells were washed two times with IMDM medium containing 50 nM doxorubicin and transferred into a new flask. Cells were harvested like normal HL-60 for one week. After that whole procedure was repeated with 50 nM doxorubicin. After this time, we observe a significant increase in the IC50 value (Fig. S28†). To maintain resistance to doxorubicin we added 50 nM doxorubicin every fourth passage.
Peripheral blood mononuclear (PBM) cells were isolated from a leucocyte-buffy coat collected from the blood of healthy, non-smoking donors from the Blood Bank in Lodz, Poland. The study protocol was approved by the Committee for Research on Human Subjects of the University of Lodz (17/KBBN-UŁ/III/2019). The first step of isolation of PBM cells was a mix of a fresh blood from buffy coats with PBS in a ratio of 1:1. In the next step, mixture was centrifuged in a density gradient of Lymphosep (Cytogen, Zgierz, Poland) at 2200 RPM for 20 min with the lowest values of acceleration and deceleration. Then the cells were washed three times by centrifugation with 1% PBS. After isolation cells were suspended in RPMI 1640 medium.
Cell viability
The resazurin reduction assay was performed similar to previously described.69 Resazurin salt powder was dissolved in sterile PBS. Cells were seeded on the 96-well plates in count of 1 × 103 in the case of HL-60 and HL-60/DR and of 5 × 103 for PBM cells per well. Ruthenium complexes were added to wells to obtain a final concentration of 0.5, 1, 2.5, 5, 10, 25, 50, 100 and 250 μM. In next step plates were incubated at 37 °C in 5% CO2 for 2 h and 24 h. After the desired time has elapsed 10 μl of resazurin salt was added to each well and plates again were incubated in 37 °C in 5% CO2 for 2 h. Next fluorescence was measured with microplate reader Synergy HT (Bio-Tek Instruments, USA) using an excitation wavelength of 530/25 and an emission wavelength of 590/35 nm.Comet assay
Ruthenium complexes 1, 2a, and 3a were added to the suspension of the cells to give final concentrations from the range 5–50 μM. PBM cells, HL-60 and HL-60/DR cells were incubated for 2 h at 37 °C in 5% CO2. The experiment included a positive control, i.e., a cell sample incubated with hydrogen peroxide (H2O2) at 20 μM or 200 μM in the case of HL-60/DR cells for 15 min on ice.The comet assay was performed under alkaline conditions according to the procedure of Tokarz et al.70 A freshly prepared cells suspension in 0.75% LMP agarose dissolved in PBS was layered onto microscope slides (Superior, Germany), which were pre-coated with 0.5% NMP agarose. Then, the cells were lysed for 1 h at 4 °C in a buffer containing 2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% Triton X-100, pH = 10. After cells lysis, the slides were placed in an electrophoresis unit. DNA was allowed to unwind for 20 min in the solution containing 300 mM NaOH and 1 mM EDTA, pH > 13.
Electrophoretic separation was performed in the solution containing 30 mM NaOH and 1 mM EDTA, pH > 13 at ambient temperature of 4 °C (the temperature of the running buffer did not exceed 12 °C) for 20 min at an electric field strength of 0.73 V cm−1 (28 mA). Then, the slides were washed in water, drained, stained with 2 μg ml−1 DAPI and covered with cover slips. In order to prevent additional DNA damage, the procedure described above was conducted under limited light or in the dark.
Comet analysis
The comets were observed at 200× magnification in an Eclipse fluorescence microscope (Nikon, Japan) attached to a COHU 4910 video camera (Cohu, Inc., San Diego, CA, USA) equipped with a UV-1 A filter block and connected to a personal computer-based image analysis system Lucia-Comet v. 6.0 (Laboratory Imaging, Praha, Czech Republic). Fifty images (comets) were randomly selected from each sample and the mean value of DNA in comet tail was taken as an index of DNA damage (expressed in percent).Plasmid relaxation assay
The pUC19 plasmid was isolated from the DH5α E. coli cells with AxyPrep Plasmid Miniprep Kit (Axygen) according to the manufacturer's instruction. The isolated plasmid quantity and quality were determined by A260/A280 ratio and gel electrophoresis, respectively. The native form of pUC19 exists mainly in the supercoiled form (CCC) which is characterized by a relatively high electrophoretic mobility. The plasmid was digested with the restrictase PstI (New English Biolabs) to induce linear (L) form. Topological differences between CCC and L forms of the plasmid account for their different electrophoretic mobility. The plasmid at 50 ng μl−1 was incubated for 2 h and 24 h with 1, 2a, 2b, 3a, 3b, and 3c at concentration of 50 μM. Then the samples were subjected to 1% agarose gel electrophoresis with ethidium bromide staining, visualization under UV light (302 nm), scanning by a CCD camera, and analysis with the GeneTools by Syngene (Cambridge, UK) software. During electrophoresis, we also separated 4 μl of 1 kb DNA ladder (GeneRuler 1 kb DNA Ladder, Thermo Scientific, Waltham, MA, USA).Apoptosis
The FITC Annexin V Apoptosis Detection Kit II (BD Biosciences, PA, USA) was used to evaluation of apoptosis using flow cytometry. HL-60 and HL-60/DR cells were seeded in each well of a 6-well plate at 2 × 105 cells per ml and incubated for 24 h with ruthenium complexes 1, 2a and 3a at concentrations from the range 5–50 μM. Cells were collected and washed three times with ice-cold PBS. Then cells were suspended in 100 μl of Binding Buffer and transferred to cytometry tubes. FITC annexin V (5 μl) and propidium iodide (5 μl) were added to cytometry tubes, gently vortexed and incubated at room temperature in the dark for 15 min. Then 400 μl of Binding Buffer was added and measured on the LSRII flow cytometer (Becton Dickinson, San Jose, CA, USA). The positive control were cells incubated with 20 μM camptothecin for 24 h at 37 °C.Caspase 3/7 activity
Caspase 3/7 activity was conducted using the Caspase-Glo®3/7 Assay kit (Promega, Madison, WI, USA) according to the manufacturer's protocol. HL-60 cells were seeded on a 96-well black plate in a count of 10000 per well. Then cells were incubated with complexes 1, 2a, and 3a for 24 h at 37 °C in 5% CO2 at the concentrations of 5, 10, 25, and 50 μM. Next 75 μL of Caspase-Glo®3/7 reagent was added to each well and then the plate was gently mixed by orbital shaking. After 30 min of incubation at room temperature, luminescence was measured with a microplate reader Synergy HT (Bio-Tek Instruments, USA). The positive control were cells incubated with 20 μM camptothecin for 24 h at 37 °C.
Evaluation of oxidative stress
In order to measure the production of reactive oxygen species (ROS), the fluorescence of 2′,7′-dichlorofluorescein diacetate (H2DCFDA) was measured. H2DCFDA is a cell-permeable non-fluorescent probe. 2′,7′-Dichlorofluorescein diacetate is de-esterified intracellularly and turns into highly fluorescent 2′,7′-dichlorofluorescein upon oxidation. HL-60 cells (final density 0.75 × 106 cells per ml) were stained with 20 μM H2DCFDA (Sigma-Aldrich, St Louis, MO, USA) for 30 min at 37 °C in darkness. Then, the cells were washed twice with HBSS and incubated with complexes 1, 2a and 3a at 37 °C in darkness at the concentrations of 5, 10, 25 and 50 μM. The intensity of fluorescence was measured after 30, 60, 90 and 120 min with λex = 495 nm and λem = 530 nm using a microplate reader Synergy HT (BioTek Instruments, USA). The data were analyzed according to the following formula: (TX − T0/T0) × 100, where TX is the DCF fluorescence measured at the indicated time and T0 is the DCF fluorescence measured at the beginning of the analysis.Docking studies
A molecular docking with AutoDock471,72 was used to analyse the interaction (shape and binding energy) of the ruthenium(II) complexes with maleimide and phosphines ligands (1, 2a and 2b) with normal and mismatched double stranded DNA fragment. To produce global poses of small molecules, a grid-based docking approach was designed using the Lamarckian genetic algorithm. All the needed parameters for ruthenium metal were added in the Autodock 4.2 parameters file.62 The optimum binding conformation was determined to be the one with the lowest binding energy and the highest number of docking poses and additional MD simulations were conducted on this conformation. The MD simulations of normal and mismatched DNA interacted ruthenium(II) complexes with maleimide and phosphine or phosphite ligands were studied using GROMACS 202173 for 100 ns.Statistical analysis
The values of the cell viability experiment were presented as mean ± SD from six repeats. The values of the comet assay were expressed as mean + standard error of the mean from three experiments; data from three experiments were pooled, and the statistical parameters were calculated. The statistical analysis was conducted using the Mann–Whitney test (samples with distributions departing from normality) and the Student's t-test (samples with the normal distribution). The differences were considered to be statistically significant when the p value was < 0.05.Conclusions
To summarize, our results suggest cyto- and genotoxic potential of ruthenium complexes 2a and 3a against HL-60 cancer cells but not against normal PBM cells. The plasmid relaxation assay demonstrated the possibility of induction of DNA single-strand breaks by ruthenium complexes 2a–b and 3a–c which increase with the incubation time. These findings corroborate with the docking studies performed for complexes 2a and 2b. Based on these results, we conclude that the cytotoxic effects of compounds 2a, 2b and/or 3a are weakly related to the direct degradation of genomic DNA or ROS generation, but may affect the DNA damage repair mechanisms leading to cell death. Our results also indicate that ruthenium complexes 1, 2a–b and 3a–c have small potential to overcome the resistance to doxorubicin in HL-60 cells. In the search for new anticancer drugs, their selective activity towards cancer cells and lack of effect on normal cells are of key importance. For this reason, these ruthenium complexes are expected to open new ways for anticancer research.Author contributions
Michał Juszczak: Investigation. Sujoy Das: Investigation. Aneta Kosinska: Investigation. Agnieszka Rybarczyk-Pirek: Crystallography, investigation. Kinga Wzgarda-Raj: Crystallography, investigation. Paulina Tokarz: Investigation. Saranya Vasudevan: Docking studies, investigation. Arkadiusz Chworos: Supervision, manuscript writing. Katarzyna Woźniak: Conceptualization, supervision, manuscript writing. Bogna Rudolf: Conceptualization; supervision, manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the University of Lodz, Poland, Faculty of Chemistry and Faculty of Biology and Environmental Protection. The research described in the article was carried out as part of the Interdisciplinary Research Grant of the University of Lodz (IDUB B2311013000182.07).References
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- D. Wernitznig, K. Kiakos, G. Del Favero, N. Harrer, H. Machat, A. Osswald, M. A. Jakupec, A. Wernitznig, W. Sommergruber and B. K. Keppler, Metallomics, 2019, 11, 1044–1048 CrossRef CAS PubMed.
- S. Y. Lee, C. Y. Kim and T. G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- J. P. C. Coverdale, T. Laroiya-McCarron and I. Romero-Canelón, Inorganics, 2019, 7(3), 31 CrossRef CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 2017, 1549–1560 CrossRef CAS.
- A. Valente, A. Podolski-Renić, I. Poetsch, N. Filipović, Ó. López, I. Turel and P. Heffeter, Drug Resist. Updates, 2021, 58, 100778 CrossRef CAS PubMed.
- Y. Wen, C. Ouyang, Q. Li, T. W. Rees, K. Qiu, L. Ji and H. Chao, Dalton Trans., 2020, 49, 7044–7052 RSC.
- L. Chen, J. Wang, X. Cai, S. Chen, J. Zhang, B. Li, W. Chen, X. Guo, H. Luo and J. Chen, Bioorg. Chem., 2022, 119, 105516 CrossRef CAS PubMed.
- J. Hildebrandt, N. Häfner, D. Kritsch, H. Görls, M. Dürst, I. B. Runnebaum and W. Weigand, Int. J. Mol. Sci., 2022, 23(9), 4976 CrossRef CAS PubMed.
- B. Ma, L. He, Y. You, J. Mo and T. Chen, Drug Delivery, 2018, 25, 293–306 CrossRef CAS PubMed.
- P. S. Rawat, A. Jaiswal, A. Khurana, J. S. Bhatti and U. Navik, Biomed. Pharmacother., 2021, 139, 111708 CrossRef CAS PubMed.
- B. Rudolf, A. Kubicka, M. Salmain, M. Palusiak, A. J. Rybarczyk-Pirek and S. Wojtulewski, J. Organomet. Chem., 2016, 801, 101–110 CrossRef CAS.
- M. Djukić, M. S. Jeremić, R. Jelić, O. Klisurić, V. Kojić, D. Jakimov, P. Djurdjević and Z. D. Matović, Inorg. Chim. Acta, 2018, 483, 359–370 CrossRef.
- L. Rafols, D. Josa, D. Aguilà, L. A. Barrios, O. Roubeau, J. Cirera, V. Soto-Cerrato, R. Pérez-Tomás, M. Martínez, A. Grabulosa and P. Gamez, Inorg. Chem., 2021, 60, 7974–7990 CrossRef CAS PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- J. Li, M. Tian, Z. Tian, S. Zhang, C. Yan, C. Shao and Z. Liu, Inorg. Chem., 2018, 57, 1705–1716 CrossRef CAS PubMed.
- G. Kovács, A. Rossin, L. Gonsalvi, A. Lledós and M. Peruzzini, Organometallics, 2010, 29, 5121–5131 CrossRef.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- N. Pagliaricci, R. Pettinari, F. Marchetti, C. Pettinari, L. Cappellacci, A. Tombesi, M. Cuccioloni, M. Hadiji and P. J. Dyson, Dalton Trans., 2022, 51, 13311–13321 RSC.
- M. Martínez-Alonso, N. Busto, F. A. Jalón, B. R. Manzano, J. M. Leal, A. M. Rodríguez, B. García and G. Espino, Inorg. Chem., 2014, 53, 11274–11288 CrossRef PubMed.
- M. Martínez-Alonso and G. Gasser, Coord. Chem. Rev., 2021, 434, 213736 CrossRef.
- P. R. Florindo, D. M. Pereira, P. M. Borralho, C. M. P. Rodrigues, M. F. M. Piedade and A. C. Fernandes, J. Med. Chem., 2015, 58, 4339–4347 CrossRef CAS PubMed.
- A. I. Tomaz, T. Jakusch, T. S. Morais, F. Marques, R. F. M. de Almeida, F. Mendes, É. A. Enyedy, I. Santos, J. C. Pessoa, T. Kiss and M. H. Garcia, J. Inorg. Biochem., 2012, 117, 261–269 CrossRef CAS PubMed.
- G. Albertin, S. Antoniutti, M. Bortoluzzi and G. Zanardo, J. Organomet. Chem., 2005, 690, 1726–1738 CrossRef CAS.
- K. M. Vyas, P. Mandal, R. Singh, S. M. Mobin and S. Mukhopadhyay, Inorg. Chem. Commun., 2020, 112, 107698 CrossRef CAS.
- M. Bispinghoff, Z. Benkő, H. Grützmacher, F. D. Calvo, M. Caporali and M. Peruzzini, Dalton Trans., 2019, 48, 3593–3600 RSC.
- M. Hanif, S. M. Meier, W. Kandioller, A. Bytzek, M. Hejl, C. G. Hartinger, A. A. Nazarov, V. B. Arion, M. A. Jakupec, P. J. Dyson and B. K. Keppler, J. Inorg. Biochem., 2011, 105, 224–231 CrossRef CAS PubMed.
- E. Klaimanee, T. Nhukeaw, S. Saithong, A. Ratanaphan, S. Phongpaichit, Y. Tantirungrotechai and N. Leesakul, Polyhedron, 2021, 204, 115244 CrossRef CAS.
- G. H. Ribeiro, L. Colina-Vegas, J. C. T. Clavijo, J. Ellena, M. R. Cominetti and A. A. Batista, J. Inorg. Biochem., 2019, 193, 70–83 CrossRef CAS PubMed.
- S. Li, J. Zhao, Y. Guo, Y. Mei, B. Yuan, N. Gan, J. Zhang, J. Hu and H. Hou, J. Inorg. Biochem., 2020, 210, 111102 CrossRef CAS PubMed.
- T. Küster, N. Lense, F. Barna, A. Hemphill, M. K. Kindermann, J. W. Heinicke and C. A. Vock, J. Med. Chem., 2012, 55, 4178–4188 CrossRef PubMed.
- M. Juszczak, M. Kluska, A. Kosińska, M. Palusiak, A. J. Rybarczyk-Pirek, K. Wzgarda-Raj, B. Rudolf and K. Woźniak, Appl. Organomet. Chem., 2022, e6595 CAS.
- B. Rudolf, J. Walendowska and J. Zakrzewski, J. Organomet. Chem., 2002, 648, 293–296 CrossRef CAS.
- R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl, 1966, 385–415 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 371–389 CrossRef CAS PubMed.
- I. Bernal, J. Cetrullo and W. G. Jackson, J. Coord. Chem., 1993, 28, 89–95 CrossRef CAS.
- J. W. Faller and P. P. Fontaine, Organometallics, 2005, 24, 4132–4138 CrossRef CAS.
- J. W. Faller and N. Sarantopoulos, Cryst. Growth Des., 2005, 5, 2356–2361 CrossRef CAS.
- J. W. Faller and P. P. Fontaine, J. Organomet. Chem., 2006, 691, 4667–4675 CrossRef CAS.
- K. A. Lazarou, K. González-Nieves, I. Chakraborty and R. G. Raptis, Angew. Chem., Int. Ed., 2019, 58, 7324–7328 CrossRef CAS PubMed.
- C. L. Yadav, G. Rajput, K. K. Bisht, M. G. B. Drew and N. Singh, Inorg. Chem., 2019, 58, 14449–14456 CrossRef CAS PubMed.
- A. Lennartson, M. Vestergren and M. Håkansson, Chem. – Eur. J., 2005, 11, 1757–1762 CrossRef CAS PubMed.
- P. Bonakdarzadeh, F. Pan, E. Kalenius, O. Jurček and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 14890–14893 CrossRef CAS PubMed.
- C. Xu, Q. Lin, C. Shan, X. Han, H. Wang, H. Wang, W. Zhang, Z. Chen, C. Guo, Y. Xie, X. Yu, B. Song, H. Song, L. Wojtas and X. Li, Angew. Chem., Int. Ed., 2022, 61, e202203099 CAS.
- J. W. Faller, A. R. Lavoie and J. Parr, Chem. Rev., 2003, 103, 3345–3368 CrossRef CAS PubMed.
- P. H. Dixneuf, T. Guyot, M. D. Ness and S. M. Roberts, Chem. Commun., 1997, 2083–2084 RSC.
- Z. Zhang, T. Roisnel, P. H. Dixneuf and J.-F. Soulé, Angew. Chem., Int. Ed., 2019, 58, 14110–14114 CrossRef CAS PubMed.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS PubMed.
- M. Abid, F. Shamsi and A. Azam, Mini-Rev. Med. Chem., 2016, 16, 772–786 CrossRef CAS PubMed.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692 CrossRef CAS.
- J. Qian, R. Liu, N. Liu, C. Yuan, Q. Wu, Y. Chen, W. Tan and W. Mei, Molecules, 2022, 27(10), 3046 CrossRef CAS PubMed.
- I. O. Travassos, F. Mello-Andrade, R. P. Caldeira, W. C. Pires, P. F. F. da Silva, R. S. Correa, T. Teixeira, A. Martins-Oliveira, A. A. Batista and E. P. de Silveira-Lacerda, J. Biol. Inorg. Chem., 2021, 26, 385–401 CrossRef CAS PubMed.
- R. A. De Grandis, K. M. Oliveira, A. P. M. Guedes, P. W. S. Dos Santos, A. F. Aissa, A. A. Batista and F. R. Pavan, Front. Oncol., 2021, 11, 682968 CrossRef PubMed.
- J. Chen, Y. Zhang, G. Li, F. Peng, X. Jie, J. She, G. Dongye, Z. Zou, S. Rong and L. Chen, J. Biol. Inorg. Chem., 2018, 23, 261–275 CrossRef CAS PubMed.
- Y. Li, Q. Wu, G. Yu, L. Li, X. Zhao, X. Huang and W. Mei, Eur. J. Med. Chem., 2019, 164, 282–291 CrossRef CAS PubMed.
- J. Chen, F. Peng, Y. Zhang, B. Li, J. She, X. Jie, Z. Zou, M. Chen and L. Chen, Eur. J. Med. Chem., 2017, 140, 104–117 CrossRef CAS PubMed.
- J. Li, L. Zeng, Z. Wang, H. Chen, S. Fang, J. Wang, C.-Y. Cai, E. Xing, X. Liao, Z.-W. Li, C. R. Ashby Jr., Z.-S. Chen, H. Chao and Y. Pan, Adv. Mater., 2022, 34, 2100245 CrossRef CAS PubMed.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179–2183 CrossRef CAS PubMed.
- H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed.
- D. Patel, M. Athar and P. C. Jha, ChemistrySelect, 2021, 6, 8189–8199 CrossRef CAS.
- CrysAlisPRO software system, 2020 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. J. Bodner, S. Tsai, R. C. Ting, S. J. Collins and R. C. Gallo, Leuk. Res., 1980, 4, 151–154 CrossRef CAS PubMed.
- K. Bhalla, A. Hindenburg, R. N. Taub and S. Grant, Cancer Res., 1985, 45, 3657–3662 CAS.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef PubMed.
- P. Tokarz, A. W. Piastowska-Ciesielska, K. Kaarniranta and J. Blasiak, Int. J. Mol. Sci., 2016, 17(6), 898 CrossRef PubMed.
- A. A. Adeniyi and P. A. Ajibade, Molecules, 2013, 18, 3760–3778 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- J. A. Lemkul, Living J. Comp. Mol. Sci., 2018, 1(1), 5068 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison table, 1H NMR, 13C NMR, and MS spectra, crystal data, cell viability and apoptosis. CCDC 22093342209335. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04083b |
| This journal is © The Royal Society of Chemistry 2023 |