
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization, and in vivo evaluation of the anticancer activity of a series of 5- and 6-(halomethyl)-2,2′-bipyridine rhenium tricarbonyl complexes†
Sara
Nasiri Sovari
a,
Isabelle
Kolly
a,
Kevin
Schindler
 a,
Ana
Djuric
b,
Tatjana
Srdic-Rajic
b,
Aurelien
Crochet
a,
Aleksandar
Pavic
*c and
Fabio
Zobi
*a
a,
Ana
Djuric
b,
Tatjana
Srdic-Rajic
b,
Aurelien
Crochet
a,
Aleksandar
Pavic
*c and
Fabio
Zobi
*a
aDepartment of Chemistry, University of Fribourg, Chemin du Musée 10, 1700 Fribourg, Switzerland. E-mail: fabio.zobi@unifr.ch; Fax: (+41) 26 300 97 37; Tel: (+41) 26 300 87 85
bDepartment of experimental oncology, Institute for Oncology and Radiology of Serbia, Pasterova 14, Beograd, Republic of Serbia
cInstitute of Molecular Genetics and Genetic Engineering, University of Belgrade, Vojvode Stepe 444a, 11000 Belgrade, Republic of Serbia. E-mail: sasapavic@imgge.bg.ac.rs; Fax: (+381) 11 397 58 08; Tel: (+381) 11 397 60 34
First published on 9th March 2023
Abstract
We report the synthesis, characterization, and in vivo evaluation of the anticancer activity of a series of 5- and 6-(halomethyl)-2,2′-bipyridine rhenium tricarbonyl complexes. The study was promoted in order to understand if the presence and position of a reactive halomethyl substituent on the diimine ligand system of fac-[Re(CO)3]+ species may be a key molecular feature for the design of active and non-toxic anticancer agents. Only compounds potentially able to undergo ligand-based alkylating reactions show significant antiproliferative activity against colorectal and pancreatic cell lines. Of the new species presented in this study, one compound (5-(chloromethyl)-2,2′-bipyridine derivative) shows significant inhibition of pancreatic tumour growth in vivo in zebrafish-Panc-1 xenografts. The complex is noticeably effective at 8 μM concentration, lower than its in vitro IC50 values, being also capable of inhibiting in vivo cancer cells dissemination.
1. Introduction
Cisplatin is well known for being the first metallodrug for treatment of neoplastic diseases,1 but after gaining clinical success, its use was limited due to the emergence of side effects (e.g., inherent nephrotoxicity and ototoxicity). The discovery of cisplatin, however, ignited a thriving field of research, with several medicinal inorganic chemists now exploring the potential anticancer efficacy of other transition metal species. Classic platinum(II) and new platinum(IV) complexes still dominate the literature2 with ruthenium, copper and gold being other prominent examples.3–5 Rhenium(I) tricarbonyl complexes are amongst the least explored in the field, but in the last decade have gained significant attention due to their unique and promising properties, which include high stability, low toxicity and rich spectroscopic and luminescent features. The surprising number of recent reviews6–15 on the anticancer potential of Re species corroborates the interest around such complexes.Apart from the constant fac-[Re(CO)3]+ core, little is known about what molecular features may be commonly required for an active compound. An examination of the published contributions in the field, generally indicates that the cytotoxicity of Re(I) tricarbonyl complexes positively correlates with lipophilic properties of the species,16–20 which is associated with an improved passive cellular uptake. Different Re(I) tricarbonyl molecules possessing neoplastic activities (often with in vitro anti-proliferative potency higher than cisplatin14) act by different mechanisms of action, including mitochondrial21–25 or enzymatic inhibition,26 DNA interaction27–30 or endoplasmic reticulum (ER) stress.31 Of the different complexes published to date, a few molecules are, in our view, of particular interest, because they have been studied in more details in vivo. These are the tricarbonyl rhenium isonitrile polypyridyl (TRIP) complex of Wilson et al., the diseleno-ether compound of Collery et al. (diSe-Re in Chart 1) and the N-heterocylic carbene complex (NHC-Re, Chart 1) of Falasca et al.
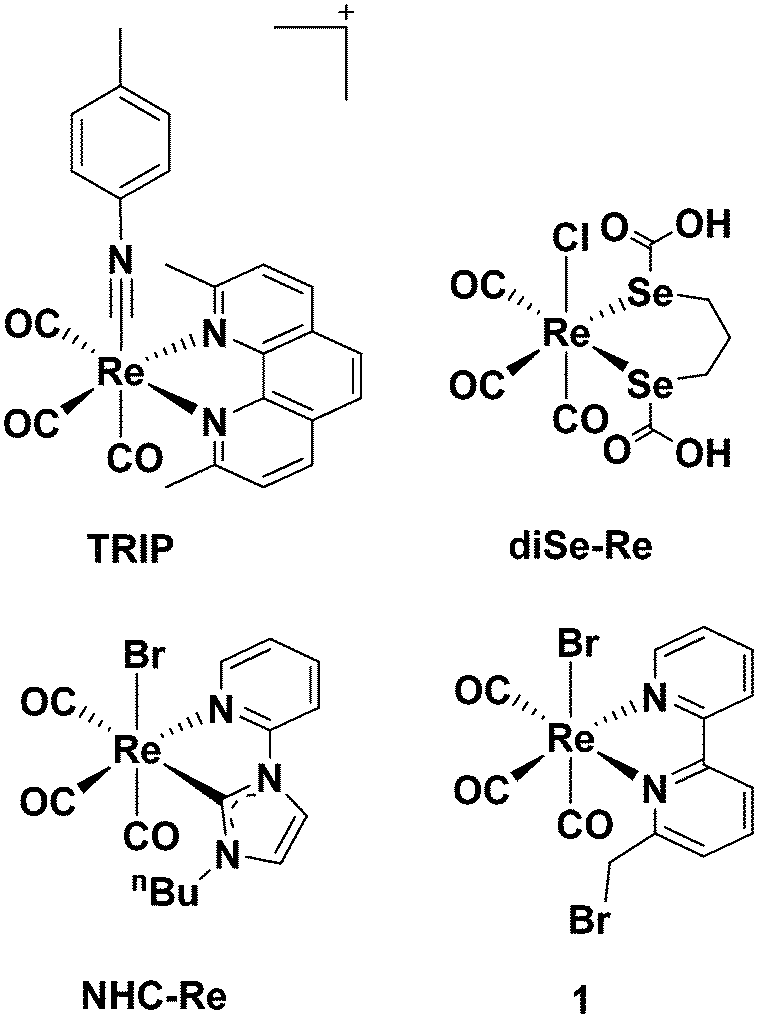 | ||
| Chart 1 Selected structures of anticancer Re complexes evaluated in vivo. | ||
TRIP exhibits potent in vitro anticancer activity in a variety of cell lines (IC50 value 1.4–1.9 μM) and acts by triggering the accumulation of misfolded proteins, which causes endoplasmic reticulum (ER) stress, unfolded protein response, and apoptotic cell death in addition to mitochondrial fission.31 The compound remains intact in vitro as demonstrated by X-ray fluorescence microscopy (XFM),32 and when administered in NSG mice bearing A2780 ovarian cancer xenografts (20 mg per kg twice weekly), it is able to inhibit tumor growth and prolong mouse survival by 150% compared to control.33 DiSe-Re exhibits activities against several solid tumor cell lines34 and a potent inhibitory effect on breast cancer MDA-MB231 cell division.35 Moreover, diSe-Re promotes in vivo (10 mg kg−1 d−1) a remarkable reduction of tumors volume in mice-bearing a MDA-MB231 Luc+ xenografts and pulmonary metastases without signs of clinical toxicity.35 The compound acts as an anti-oxidant agent (decreasing ROS production) and significantly decreases the levels of the transforming growth factor beta 1 (TGF-β1), vascular endothelial growth factor A (VEGF-A) and insulin-like growth factor 1 (IGF-1).36,37 NHC-Re shows low μM activity against pancreatic cancer cell lines where it induces cell cycle arrest at the G2/M phase by inhibiting the phosphorylation of Aurora-A kinase,38 and blocks growth of aggressive cancers in vivo (mice bearing HPAF-II human pancreatic cancer xenografts) by inhibiting FGFR- and SRC-mediated signaling.39
We have also been interested in the development of antibiotic40 and anticancer rhenium species.41 In a recent study, we have reported the anti-proliferative efficacy of a series of fac-[Re(I)(CO)3]+N-derivatized N-([2,2′-bipyridin]-6-ylmethyl)-species against colorectal carcinoma (CRC) and identified complex 1 (Chart 1) as a potent in vivo (zebrafish xenograft model of human CRC) anticancer, anti-angiogenic and antimetastatic compound.42In vivo, compound 1 (1–3 μM concentration) is more potent than clinical drugs, such as cisplatin and sunitinib malate, and shows no signs of clinical toxicity (cardio-, hepato-, and myelotoxicity) at high concentrations (i.e., 250 μM).
The identification of 1 amongst a series of related species, made us wonder if the presence and position of a reactive halomethyl substituent on the diimine ligand system of fac-[Re(CO)3]+ core may be a/the key molecular feature for the activity of 1 and for design of similar active and non-toxic anticancer agents. After all, 1 is of a relatively simple design, not dissimilar from other diimine rhenium compounds that, however, do not appear to be as promising as 1, and the presence of the reactive halomethyl substituent is the unique feature of 1 in the series of compounds investigated. To this end, we have prepared a small library of 5- and 6-(halomethyl)-2,2′-bipyridine rhenium tricarbonyl complexes, and evaluated in vivo their anticancer activity against pancreatic and CRC tumors. Of the new species presented in this study, only one compound (5-(chloromethyl)-2,2′-bipyridine derivative) shows significant inhibition of pancreatic tumour growth in zebrafish-Panc-1 xenografts. The complex is also capable of inhibiting in vivo cancer cells dissemination, but it does not surpass the genuine potential of 1. We describe our findings in the following sections.
2. Results and discussion
2.1. Chemistry
Ligands and rhenium complexes 1–10 were prepared according to the synthetic protocols illustrated in Scheme 1. The 2,2′-bipyridine derivatives were obtained via the well-established Pd-catalyzed Stille's C–C coupling reaction,43 starting from commercially available reagents. The 5- and 6-(difluoromethyl)-2,2′-bipyridines (L5 and L10 in Scheme 1) were obtained in a single step in moderate yields of 17% and 38% respectively. Similarly [2,2′-bipyridin]-5- and 6-ylmethanol (L3 and L8) were obtained in good yield from the corresponding methyl [2,2′-bipyridine]-#-carboxylate following its reduction with NaBH4. Ligands L3 and L8 were then converted to the 5- and 6-(halomethyl)-2,2′-bipyridines (L1, L2, L6 and L7) either by treatment with PBr3 (L1 and L6) or thionyl chloride (L2 and L7). Finally, the rhenium complexes were obtained in high yield by treatment of fac-[Re(CO)5Br] with the corresponding 2,2′-bipyridine ligand in hot toluene42 (Scheme 1).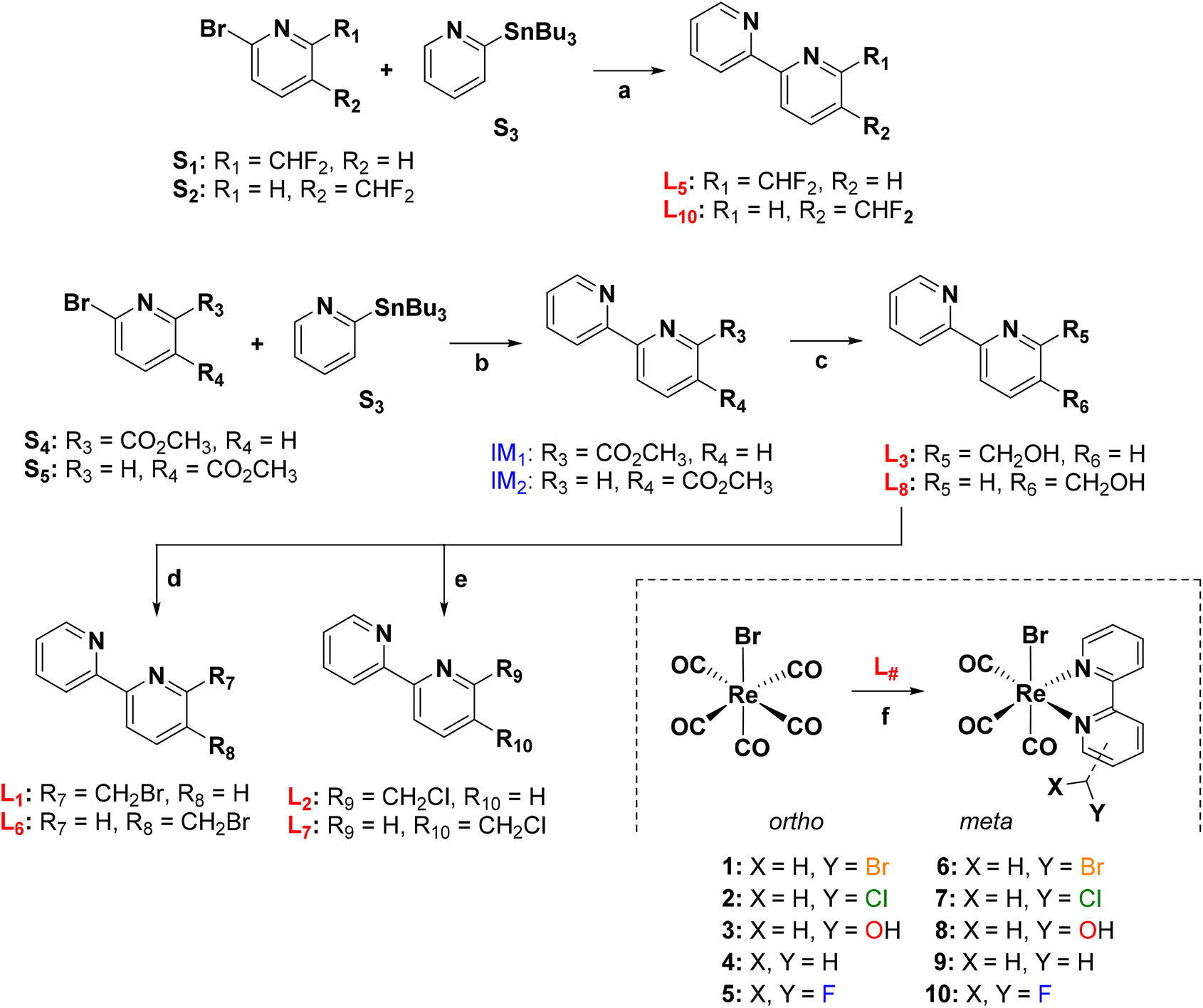 | ||
| Scheme 1 Synthesis and chemical structures of bipyridine ligands (L#) and of Re complexes (bottom left). Conditions: (a) & (b) Pd(PPh3)2(Cl)2, dry DMF, 130 °C, 10 h, under argon, 17–41%; (c) NaBH4, ethanol, H2O, 85 °C, 3 h, under argon, 76–79%; (d) PBr3, dry DCM, rt, 10 h, under argon, 77–89%; (e) SOCl2, NaHCO3, 80 °C, 2 h, 80–83%, (f) and (a) toluene, 85–100 °C, 10–12 h, 68–96%. | ||
1H NMR spectra of complexes (ESI, Fig. S1–S9†) showed pure diamagnetic compounds, according to the symmetry given by the facial-arranged CO's and low-spin d6 nature of the metal ion. IR spectroscopy analysis was in accordance with the typical tricarbonyl vibration pattern. Crystals suitable for X-ray diffraction analysis were obtained for seven of nine new species. Crystallographic details are given in ESI,† whereas Fig. 1 and 2 depict respectively the ORTEPs of the meta- and ortho-substituted derivative. Complexes 2, 6, 7, 9 and 10 all crystallized in a monoclinic lattice and space groups C2/c (2), P21/c (6, 7 and 10), Pc (9) respectively, whereas 3 and 5 were obtained in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and in the orthorhombic space group Pca21. All seven crystal structures of the complexes (Fig. 1 and 2) present a slightly distorted octahedral geometry around the central metal ion, but structural parameters are not significantly different from similar fac-[Re(CO)3]+ species (CCDC search).
and in the orthorhombic space group Pca21. All seven crystal structures of the complexes (Fig. 1 and 2) present a slightly distorted octahedral geometry around the central metal ion, but structural parameters are not significantly different from similar fac-[Re(CO)3]+ species (CCDC search).
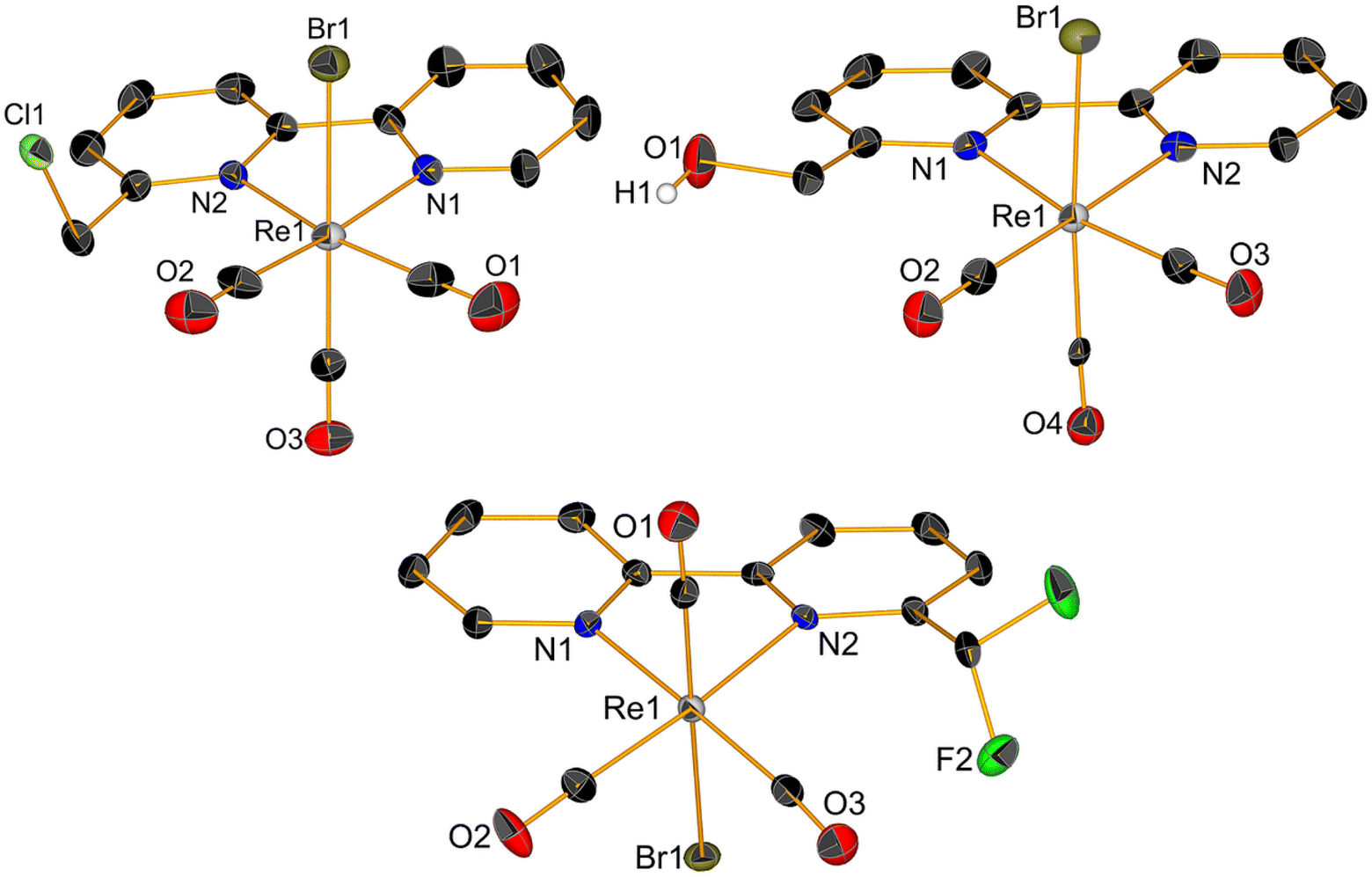 | ||
| Fig. 1 Crystal structures of compounds 2, 3 and 5. Thermal ellipsoids are at 30% probability. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 2 Crystal structures of compounds 6, 7, 9 and 10. Thermal ellipsoids are at 30% probability. Hydrogen atoms are omitted for clarity. | ||
Spectroscopically, 2–10 show the characteristic stretching bands expected for tricarbonyl complexes in the water-free region of their IR spectra. For all complexes, as predicted,44 the pattern is very similar both in terms of frequency and in intensity of the stretching vibration. The UV-Vis spectra of the compounds display two main absorptions. All complexes show similar π → π* intra-ligand transitions (LLCT) as sharp bands at 300 nm attributed to the diimine-ligand system. In addition, the spectra show a metal-to-ligand charge transfer transition (MLCT, responsible for the luminescent properties of the species) cantered at 370 nm.45
2.2. Relative lipophilicity of complexes
The activity of rhenium tricarbonyl anticancer drugs is often correlated to their lipophilicity.10,46–51 The lipophilicity of the complexes is generally defined by octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, or Kow value) referring to the ratio of the compound's concentration in the octanol phase to its concentration in the aqueous phase in the two-components system. Although previously we did not see a correlation between the lipophilicity and IC50 values of 6-(methyl-derivatized)-2,2′-bipyridine fac-[Re(CO)3]+ compounds, we decided to calculate and provide drug-likeness data of the complexes presented herein. We believe it is important to add such data in an era of computer-aided drug design of metal-based drug discovery. The drug-likeness properties of compounds 1–10 were calculated via the Molinspiration software. The results are given in Table 1. Complexes, overall, show very similar lipophilicity with the chloro and bromo derivatives (i.e. compounds 1–2 and 6–7) being the most lipophilic species and the hydroxo derivatives (3 and 8) the most hydrophilic. With the exception of their molecular weight, molecules rate well in the drug-likeness assessment (Table 1).
P, or Kow value) referring to the ratio of the compound's concentration in the octanol phase to its concentration in the aqueous phase in the two-components system. Although previously we did not see a correlation between the lipophilicity and IC50 values of 6-(methyl-derivatized)-2,2′-bipyridine fac-[Re(CO)3]+ compounds, we decided to calculate and provide drug-likeness data of the complexes presented herein. We believe it is important to add such data in an era of computer-aided drug design of metal-based drug discovery. The drug-likeness properties of compounds 1–10 were calculated via the Molinspiration software. The results are given in Table 1. Complexes, overall, show very similar lipophilicity with the chloro and bromo derivatives (i.e. compounds 1–2 and 6–7) being the most lipophilic species and the hydroxo derivatives (3 and 8) the most hydrophilic. With the exception of their molecular weight, molecules rate well in the drug-likeness assessment (Table 1).
| Complex | miLogPa |
TPSAb |
M
Wc |
N
atomsd |
N
ONe |
N
OHNHf |
N
violg |
N
rotbh |
Vol.i |
|---|---|---|---|---|---|---|---|---|---|
|
a Octanolewater partition coefficient (logP value obtained using Molinspiration method).
b Molecular polar surface area in Å2.
c Molecular weight.
d Number of nonhydrogen atoms.
e Number of hydrogen-bond acceptors (O and N atoms).
f Number of hydrogen-bond donors (OH and NH groups).
g Number of “Rule of five” violations.
h Number of rotatable bonds.
i Molecular volume in Å3.
|
|||||||||
| 1 | 3.78 | 61.08 | 602.28 | 22 | 5 | 0 | 1 | 4 | 297.04 |
| 2 | 3.65 | 61.08 | 557.83 | 22 | 5 | 0 | 1 | 4 | 292.69 |
| 3 | 2.41 | 81.31 | 539.38 | 22 | 6 | 1 | 1 | 4 | 287.17 |
| 4 | 3.06 | 61.08 | 523.38 | 21 | 5 | 0 | 1 | 3 | 278.91 |
| 5 | 3.61 | 61.08 | 559.36 | 23 | 5 | 0 | 1 | 4 | 289.04 |
| 6 | 3.88 | 61.08 | 602.28 | 22 | 5 | 0 | 1 | 4 | 297.04 |
| 7 | 3.75 | 61.08 | 557.83 | 22 | 5 | 0 | 1 | 4 | 292.69 |
| 8 | 2.51 | 81.31 | 539.38 | 22 | 6 | 1 | 1 | 4 | 287.17 |
| 9 | 3.62 | 61.08 | 523.38 | 21 | 5 | 0 | 1 | 3 | 278.91 |
| 10 | 3.71 | 61.08 | 559.36 | 23 | 5 | 0 | 1 | 4 | 289.04 |
2.3. In vitro anticancer activity evaluation
The antiproliferative effect of compounds 2–10 was evaluated on a panel of four cancer cell lines (2 colorectal and 2 pancreatic) and compared to the previously reported activity of 1. In addition, the toxicity of all new complexes was assessed on a normal cell line (MRC-5) in order to determine the selectivity index (Si) of the molecules. Table 2 presents the results of our analysis. As evident from the data, the potentially alkylating (i.e. “reactive”) chloro- and bromomethyl complexes 2, 6 and 7 showed higher antiproliferative activity, as compared to the unreactive complexes. Of these latter species, only 4 (i.e. the 6-methyl-2,2′-bipyridine complex) revealed comparable activity to 2, 6 and 7. As it is the case for 1, the compounds are particularly effective against the colorectal HCT-116 cell line, with IC50 values ranging from ca. 5 to 10 μM. We consider this result of particular relevance since HCT-116 is a CRC cell line that rapidly acquires resistance to clinical anticancer drugs, including cisplatin, oxaliplatin, docetaxel, 5-FU, and others.52–55 As we have previously pointed out,42 it is possible that unique characteristics of HCT-116 cells, including mutation in the KRAS proto-oncogene, exhibitions of wild-type p53 expression, stem cell-like properties, low differentiation level, fast division as well as epithelial morphology,55–57 could all be factors contributing to their higher sensitivity of tumor line this class of rhenium tricarbonyl complexes. Unlike 1, however, the new compounds show in general a lower Si (Table 2) towards cancer cells, being only ca. twice as active when compared to the IC50 value against healthy MRC-5 cells. Only complex 7 showed moderately good selectivity towards HCT-116 with an Si of 3.1. Nevertheless, we decided to further investigate in vivo selected compounds with acceptable Si (here defined as Si > 1).| Cells/comp. | HT-29 | HCT-116 | MiaPaCa-2 | Panc-1 | MRC-5 | Zebrafisha |
|---|---|---|---|---|---|---|
| IC50 (Si/Ti)b | LC50 | |||||
| nd = not determined.a The LC50 values have been determined in the zebrafish model only for the compounds with the IC50 values ≤11 μM.b Selectivity index (Si) is determined as the ratio between IC50 values on the normal and tumour cell lines; therapeutic index (Ti) is determined as the ratio between corresponding LC50 and IC50 values. The most potent compounds showing the IC50 ≤ 11 μM and/or Si ≥ 3 and Ti ≥ 4 are bolded. | ||||||
| 1 | nd | 5.0 (6.8/48.9) | 10.7 (3.2/22.8) | nd | 34 | 244.4 |
| 2 | 14.9 | 10.3 | 14.6 | 13.3 | 12 | 68.9 |
| 3 | 49.5 | 27.7 | 20.3 | 20.1 | >50 | nd |
| 4 | 38.7 | 9.5 (2.4/5.1) | 39.6 | 10.1 (2.3/4.9) | 23 | 48.6 |
| 5 | 25 | 21.8 | 19.3 | 16.5 | 25.3 | nd |
| 6 | 11.6 | 7.5 (1.6/6.5) | 12 | 16.7 | 12.3 | 49.1 |
| 7 | 16.9 | 4.9 (3.2/7) | 9.2 (1.7/3.7) | 11 (1.4/3.1) | 15.5 | 34.2 |
| 8 | 302.8 | 31.8 | 33.1 | 23.1 | 49.3 | nd |
| 9 | 173.2 | 22.1 | 21.8 | 16.4 | >50 | nd |
| 10 | 32.9 | 21.5 | 21 | 20.1 | >50 | nd |
2.4. In vivo anticancer activity in the zebrafish xenograft models
The activity of complexes 2, 4, 6, 7 and 9 was finally investigated in vivo against human pancreatic and colorectal carcinoma tumours using the zebrafish-Panc-1 and -HCT-116 xenograft models. Zebrafish xenografts are an established platform for translational research to human cancers, allowing the study of key hallmarks of cancer biology, such as tumour cells proliferation, dissemination, metastasis and angiogenesis.56,58,59 Accordingly, in two separate experiments, Panc-1 and HCT-116 cells were fluorescently labelled and injected into the yolk of Tg(fli1:EGFP) and Tg(-2.8fabp10a:EGFP) embryos, respectively (Fig. 3 and 4). At 3 days post injection (dpi), xenografts were processed for fluorescence microscopy in order to evaluate the effects of applied complexes on the tumour mass development and cancer cells dissemination and metastasis.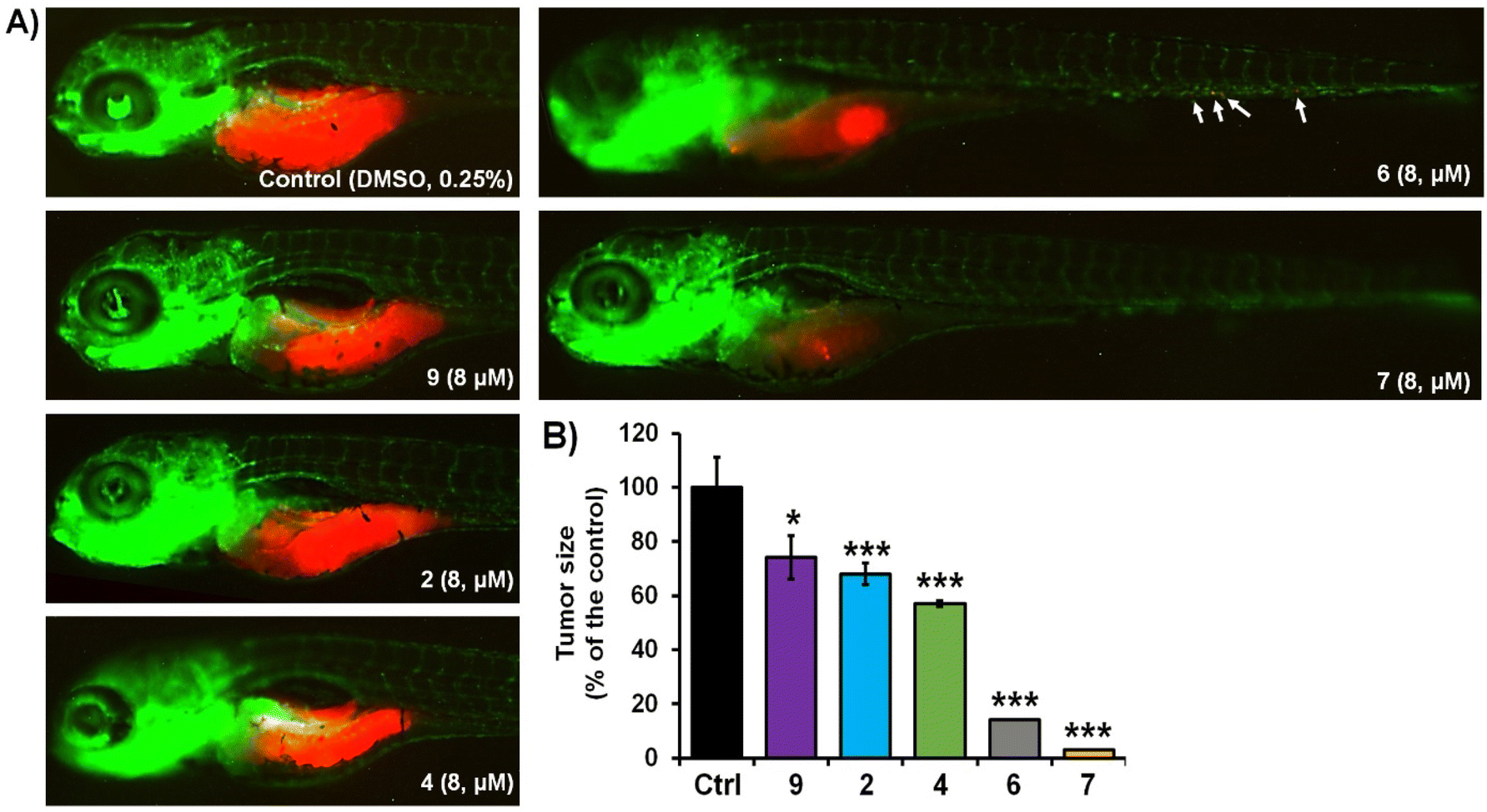 | ||
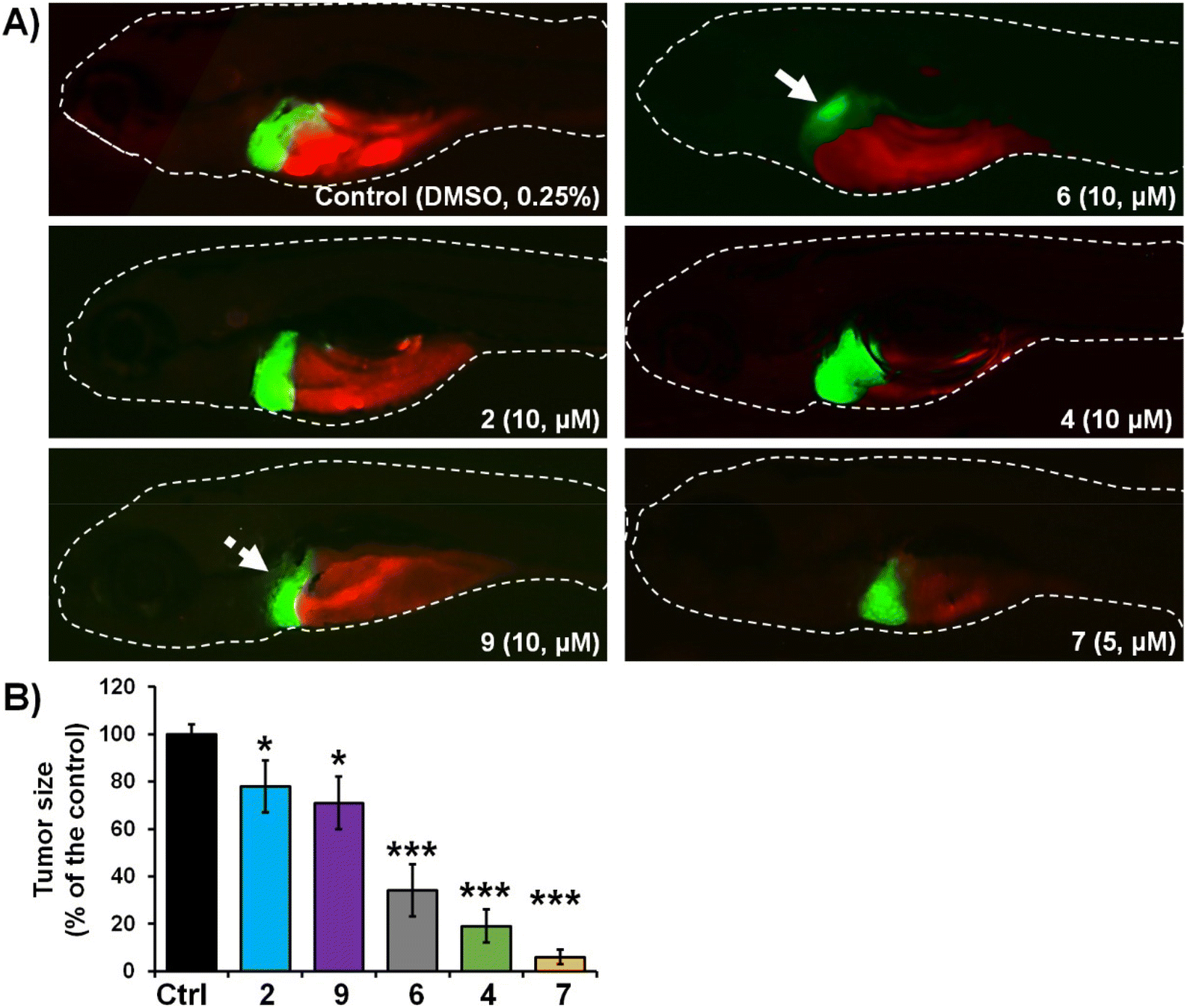
| Fig. 3 Anticancer activity of selected complexes against human Panc-1 cells in zebrafish xenografts. Tg(fli1:EGFP) xenografts (n = 20) were exposed to complexes at 8 μM doses, and then analysed after 3-days treatments for tumour progression and metastasis. Representative fluorescent microscopy images are shown (A); white solid arrows indicate disseminated cells. Only applied treatments of complexes 6 and 7 markedly reduced the tumour, growth compared to those in the control group (B). Data are normalized in relation to the control group (B). *P < 0.05; **P < 0.01; ***P < 0.001. | ||
 | ||
| Fig. 4 Anticancer activity of selected complexes against highly metastatic human HCT-116 cells in zebrafish xenografts. Tg(-2.8fabp10a:EGFP) xenografts (n = 20) were exposed to complexes at 10 μM doses, and then analysed after 3-day treatments for tumour progression and metastasis. Representative fluorescent microscopy images are shown (A); white solid arrow indicates treatment-affected liver (hepatotoxicity). Only applied treatments of complexes 4 and 7 markedly reduced the tumour growth, compared to those in the control group (B). Data are normalized in relation to the control group (B and C). *P < 0.05; **P < 0.01; ***P < 0.001. | ||
Our results of zebrafish-Panc-1 xenografts (Fig. 3) show that only treatments with 6 and 7 significantly inhibited pancreatic tumour growth in vivo (P < 0.001). Compound 7 was also effective in inhibiting cancer cells dissemination (P < 0.001). Both complexes were noticeably effective at an 8 μM concentration, lower than their respective in vitro IC50 values (Table 2). Comparison of tumour growth to untreated Panc-1 xenografts at 3 dpi (120 hpf) indicated that 6 and 7 reduced tumour mass in the treated xenografts by 86.0 ± 1.0% and 97.5 ± 0.5%, respectively (P < 0.0001, for both compounds). Compound 7 was also the most effective complex of the series in inhibiting colorectal tumour growth in zebrafish-HCT-116 xenografts (Fig. 4). However, significant cancer growth inhibition was archived at 2× in vitro IC50 value of the compound (i.e. at 10 μM). In zebrafish-HCT-116 xenografts, 6 showed much lower efficacy than in the pancreatic xenograft model (66.4 ± 11.1% vs. 86.1 ± 4.2%, P < 0.0001). Finally, in the colorectal carcinoma model, compound 4 showed good inhibition of tumour growth (80.9 ± 7.3%, P < 0.0001).
3. Conclusion
We have reported the synthesis, characterization, and in vivo evaluation of the anticancer activity of a series of 5- and 6-(halomethyl)-2,2′-bipyridine rhenium tricarbonyl complexes. On the basis of the initial identification of compound 1,42 the study was initiated in order to understand if the presence and position of a reactive halomethyl substituent on the diimine ligand system of fac-[Re(CO)3]+ species may be a key structural or molecular feature for the design of active and non-toxic anticancer agents. Overall, our study revealed that, within the series of methyl-substituted diimine complexes, only compounds potentially able of ligand-based alkylating reactions (i.e. chloro- and bromomethyl complexes 2, 6 and 7) showed significant antiproliferative activity (as compared to the unreactive complexes, which are generally inactive). Of the new species presented in this study, compound 7 showed significant inhibition of pancreatic tumour growth in vivo in zebrafish-Panc-1 xenografts. The complex is noticeably effective at 8 μM concentration, lower than its in vitro IC50 values, and it is also effective in inhibiting cancer cells dissemination, but it does not surpass the genuine potential of 1.4. Experimental protocols
4.1. Materials and methods
All chemical reagents were purchased as reagent or analytical grade from commercial suppliers (Sigma-Aldrich, Alfa Aesar, TCI, Fluorochem) and used without further purification. Solvents were either used as received or dried over molecular sieves prior to use. 1H and 13C NMR spectra were recorded on a Bruker Avance III 400 MHz using residual solvent peaks as internal references. The following abbreviations are used: singlet (s), doublet (d), doublet of doublets (dd), triplet (t), doublet of triplets (td), quintuplet (quint), sextuplet (sext), and multiplet (m). HPLC analysis was performed on a Merck-Hitachi L7000. The analytical separations were conducted on a Machereye Nagel Nucleodur PolarTec column (5 μm particle size, 110 Å pore size, 250/3). The preparative separations were conducted on a Machereye Nagel Nucleodur C18 HTec column (5 μm particle size, 110 Å pore size, 250/21). The flow rate was set to 0.5 mL min−1 for analytical separations and 5 mL min−1 for the preparative ones. Eluting solvents and gradient are as previously described.60 The eluting bands were detected at 250 nm. Analytical thin-layer chromatography (TLC) was performed on commercial silica plates (Merck 60-F 254, 0.25 mm thickness); compounds were visualized by UV light (254 nm and 366 nm). Preparative flash chromatography was performed with Merck silica gel (Si 60, 63–200 mesh). IR spectra were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer. The UV-Vis spectra were recorded on a Jasco V-730 and the emission on a spectrofluorometer FS5 (Edinburgh Instruments Ltd). Single crystal diffraction data collection was performed on a Stoe IPDS2 diffractometer (CuKα1 (λ = 1.5406 Å)) equipped with a cryostat from Oxford Cryosystems. The structures were solved with the ShelXT structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using Least Squares minimization.61,62 All crystal structures are deposited at the Cambridge Crystallographic Data Centre. CCDC numbers 2090791–2090797 contain the supplementary crystallographic data for this paper.†4.2. Synthesis and characterization of ligands and complexes
Preparation of 6-(difluoromethyl)-2,2′-bipyridine (L5) and 5-(difluoromethyl)-2,2′-bipyridine (L10). 2-Bromo-6-(difluoromethyl)pyridine (S1, or 2-bromo-5-(difluoromethyl)pyridine S2, for L5 and L10 respectively, 1.0 equiv.) was added to a suspension of 2-(tributylstannyl)pyridine (S3, 1.2 equiv.) in dry DMF. Pd(PPh3)2(Cl)2 (cat.) was added under inert conditions, and then the reaction mixture was heated to 130 °C and stirred overnight. After completion of the reaction, the mixture was cooled to room temperature, and filtered over Celite. The residue was washed with DMF until the filtrate was colourless. The filtrate was evaporated and residue was dissolved in water. The solution was extracted 3 times with DCM, washed with brine, the combined organic phase was dried with MgSO4 and then solvent was evaporated. The crude product was purified by flash column chromatography using DCM/MeOH 98
:2 (v:v) to give a dark solid. This was dissolved in a small amount of diethyl ether followed by the addition of pentane. The solution was stirred overnight, then filtered and the residue washed with cold pentane. In order to increase the yield, the filtrate was evaporated and then precipitated again in pentane. The combined products were dried to give the desired compounds. Yields for L5 and L10 were 17% and 38% respectively. L5: 1H NMR (400 MHz, CDCl3) δ ppm 6.58–6.88 (m, 1 H), 7.35 (ddd, J = 7.46, 4.77, 1.22 Hz, 1 H), 7.67 (d, J = 7.70 Hz, 1 H), 7.85 (td, J = 7.76, 1.83 Hz, 1 H), 7.98 (t, J = 7.83 Hz, 1 H), 8.47 (dt, J = 7.95, 1.04 Hz, 1 H), 8.55 (dd, J = 7.95, 0.98 Hz, 1 H), 8.69–8.73 (m, 1 H). L10: 1H NMR (400 MHz, CDCl3) δ ppm 6.62–6.94 (m, 1 H), 7.36 (ddd, J = 7.46, 4.77, 1.10 Hz, 1 H), 7.85 (td, J = 7.73, 1.77 Hz, 1 H), 7.97 (dt, J = 8.25, 0.95 Hz, 1 H), 8.45 (d, J = 7.95 Hz, 1 H), 8.53 (d, J = 8.19 Hz, 1 H), 8.71 (dd, J = 4.71, 0.67 Hz, 1 H), 8.81 (d, J = 0.73 Hz, 1 H).
Preparation of methyl [2,2′-bipyridine]-6-carboxylate (IM1) and methyl [2,2′-bipyridine]-5-carboxylate (IM2). The same procedure described above for the preparation of L5 and L10 was used starting with methyl 6-bromopicolinate (S4, or methyl 6-bromonicotinate S5, for IM1 and IM2 respectively, 1.0 equiv.) and S3 (1.2 equiv.). Yields for IM1 and IM2 were 47% and 41% respectively. Analytical data are in agreement with data reported in literature for IM163,64 and IM2.65
Preparation of [2,2′-bipyridin]-6-ylmethanol (L3) and [2,2′-bipyridin]-5-ylmethanol (L8). Under argon, IM1 (or IM2, for L3 and L8 respectively, 1.0 equiv.) was dissolved in ethanol followed by the portion wise addition of NaBH4 (3.0 equiv.). At the end of the addition, the reaction mixture was heated to 85 °C and stirred for 3 h. After completion of the reaction, the solution was cooled down to room temperature and quenched by the addition of H2O. The residual ethanol was evaporated, and the solution acidified by the addition of sulphuric acid and then washed 3 times with DCM. The aqueous phase was basified with NaOH 30% and extracted 3 times with DCM. The combined organic phase was then dried with MgSO4. Evaporation of the solvent gave the pure products. Yields for L3 and L8 were 79% and 76% respectively. Analytical data are in agreement with data reported in literature for L3
66 and L8.67
Preparation of 6-(bromomethyl)-2,2′-bipyridine (L1) and 5-(bromomethyl)-2,2′-bipyridine (L6). L3 (or L8, for L1 and L6 respectively, 1.0 equiv.) was dissolved in dry DCM under inert conditions while being cooled in an ice bath, followed by a slow addition of PBr3 (3.0 equiv.). The ice bath was then removed, and the reaction mixture was stirred overnight at room temperature. After completion of the reaction, the solution was cooled again in an ice bath, and the reaction was quenched by the slow addition of cold water. Residual DCM was evaporated, and the residue was basified by the addition of NaOH 30%. For L1, the solution was filtered, and the residue was washed with cold water. For L6, the mixture was extracted 3 times with DCM, washed with water and dried with MgSO4. The residual solvent was evaporated to give the desired product. Yields for L1 and L6 were 77% and 89% respectively. Analytical data are in agreement with data reported in literature for L1 and L6.68
Preparation of 6-(chloromethyl)-2,2′-bipyridine (L2) and 5-(chloromethyl)-2,2′-bipyridine (L7). L3 (or L8, for L2 and L7 respectively, 1.0 equiv.) was placed in a beaker and the solid was cooled in an ice bath before the dropwise addition of thionyl chloride (17.0 equiv.). Then the reaction mixture was heated to 80 °C and stirred for 2 h. After completion of the reaction, the solution was cooled to room temperature and excess thionyl chloride was evaporated. The orange residue was then gently added to a saturated NaHCO3 solution. The mixture was extracted 3 times with ethyl acetate, washed with brine, and dried with MgSO4. The solvent was evaporated and the residue was purified by normal phase column chromatography using pentane/ethyl acetate 5
:1 (v:v) to give the desired product. Yields for L2 and L7 were 83% and 80% respectively. Analytical data are in agreement with data reported in literature for L769 and L2.70
fac-[Re(CO)3(L2)Br] (2). Yellow powder, yield 96%. ESI-MS analysis (positive mode) m/z = 474.9 [M − Br]+ measured; calculated for [C14H9ClN2O3Re]+ 475.0. IR (solid, νCO cm−1): 2015.43, 1882.86. UV-Vis (CH3CN, nm): 301, 372. 1H NMR (400 MHz, acetonitrile-d3) δ ppm 5.11–5.23 (m, 2 H) 7.65 (ddd, J = 7.64, 5.56, 1.10 Hz, 1 H) 7.89–8.01 (m, 1 H) 8.14–8.28 (m, 2 H) 8.35–8.49 (m, 2 H) 9.06–9.15 (m, 1 H). 13C NMR (126 MHz, acetonitrile-d3) δ ppm 48.64–51.40 (m, 1 C) 123.48–126.23 (m, 3 C) 126.65–128.85 (m, 2 C) 139.05–142.77 (m, 3 C) 153.49 (d, J = 4.54 Hz, 2 C). Crystals suitable for X-ray diffraction were obtained from layering of hexane on DCM.
fac-[Re(CO)3(L3)Br] (3). Yellow powder, yield 96%. ESI-MS analysis (positive mode) m/z = 456.9 [M − Br]+ measured; calculated for [C14H10N2O4Re]+ 457.0. IR (solid, νCO cm−1): 2015.70, 1919.28, 1889.42. UV-Vis (CH3CN, nm): 299.5, 367.5. 1H NMR (400 MHz, DMSO-d6) δ ppm 4.81–4.96 (m, 2 H) 6.20 (br. s., 1 H) 7.72–7.78 (m, 1 H) 8.00 (d, J = 7.93 Hz, 1 H) 8.28–8.37 (m, 2 H) 8.65 (d, J = 7.93 Hz, 1 H) 8.76 (d, J = 8.24 Hz, 1 H) 9.07 (d, J = 5.34 Hz, 1 H). 13C NMR (126 MHz, DMSO-d6) δ ppm 67.53 (s, 1 C) 122.55 (s, 1 C) 123.87 (s, 1 C) 124.55 (s, 1 C) 127.74 (s, 1 C) 140.49 (d, J = 14.53 Hz, 1 C) 152.77 (s, 1 C) 155.53 (s, 1 C) 156.14 (s, 1 C) 164.80 (s, 1 C) 188.44 (s, 1 C) 197.05 (s, 1 C). Crystals suitable for X-ray diffraction were obtained from layering of hexane on DCM.
fac-[Re(CO)3(L4)Br] (4). Yellow powder, yield 91%. ESI-MS analysis (positive mode) m/z = 440.9 [M − Br]+ measured; calculated for [C14H10N2O3Re]+ 441.0. IR (solid, νCO cm−1): 2017.70, 1896.71. UV-Vis (CH3CN, nm): 299, 369. 1H NMR (400 MHz, acetonitrile-d3) δ ppm 3.01 (s, 3 H) 7.57–7.69 (m, 2 H) 8.04 (t, J = 7.89 Hz, 1 H) 8.12–8.20 (m, 1 H) 8.26 (d, J = 7.95 Hz, 1 H) 8.35–8.41 (m, 1 H) 9.04–9.12 (m, 1 H). 13C NMR (126 MHz, acetonitrile-d3) d ppm 17.67 (s, 1 C) 123.73 (d, J = 17.26 Hz, 1 C) 127.29 (s, 1 C) 138.26 (s, 1 C) 139.20–141.08 (m, 2 C) 151.49–153.45 (m, 2 C) 155.21 (s, 1 C).
fac-[Re(CO)3(L5)Br] (5). Yellow powder, yield 95%. ESI-MS analysis (positive mode) m/z = 476.9 [M − Br]+ measured; calculated for [C14H8F2N2O3Re]+ 477.0. IR (solid, νCO cm−1): 2017.49 1889.73. UV-Vis (CH3CN, nm): 296, 377. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.17–7.42 (m, 1 H) 7.80 (t, J = 6.41 Hz, 1 H) 7.91 (d, J = 5.65 Hz, 1 H) 8.35 (t, J = 7.86 Hz, 1 H) 8.91 (d, J = 8.09 Hz, 1 H) 8.94 (s, 1 H) 9.07 (s, 1 H) 9.20 (d, J = 5.65 Hz, 1 H). 13C NMR (126 MHz, DMSO-d6) d ppm 110.58 (s, 1 C) 112.49 (s, 1 C) 114.40 (s, 1 C) 120.43–121.34 (m, 1 C) 123.03–124.59 (m, 1 C) 124.84 (s, 1 C) 128.18 (s, 1 C) 140.19 (s, 1 C) 143.85–145.74 (m, 1 C) 153.07 (s, 1 C) 153.67–154.97 (m, 1 C) 156.26 (s, 1 C) 188.91 (s, 1 C) 196.99 (d, J = 16.35 Hz, 1 C). Crystals suitable for X-ray diffraction were obtained from layering of hexane on DCM.
fac-[Re(CO)3(L6)Br] (6). Yellow powder, yield 68%. ESI-MS analysis (positive mode) m/z = 519.1 [M − Br]+ measured; calculated for [C14H9BrN2O3Re]+ 518.9. IR (solid, νCO cm−1): 2016.66, 1882.55. UV-Vis (CH3CN, nm): 298, 384. 1H NMR (400 MHz, DMSO-d6) δ ppm 4.96 (s, 2 H) 7.71–7.80 (m, 1 H) 8.33 (t, J = 7.86 Hz, 1 H) 8.40 (d, J = 8.39 Hz, 1 H) 8.76 (t, J = 8.70 Hz, 2 H) 9.04 (d, J = 5.34 Hz, 1 H) 9.12 (s, 1 H). 13C NMR (126 MHz, DMSO-d6) δ ppm 14.04 (s, 2 C) 22.33 (s, 1 C) 27.04 (s, 1 C) 34.12 (s, 1 C) 54.55 (s, 1 C) 121.76–124.24 (m, 1 C) 127.25 (s, 1 C) 136.74–140.99 (m, 2 C) 151.71–156.38 (m, 1 C). Crystals suitable for X-ray diffraction were obtained from layering of pentane on chloroform.
fac-[Re(CO)3(L7)Br] (7). Yellow powder, yield 81%. ESI-MS analysis (positive mode) m/z = 474.9 [M − Br]+ measured; calculated for [C14H9ClN2O3Re]+ 475.0. IR (solid, νCO cm−1): 2017.48, 1882.97. UV-Vis (CH3CN, nm): 296, 276. 1H NMR (400 MHz, DMSO-d6) δ ppm 5.05 (s, 2 H) 7.77 (br. s., 1 H) 8.34 (s, 1 H) 8.41 (d, J = 7.17 Hz, 1 H) 8.79 (dd, J = 15.87, 8.24 Hz, 2 H) 9.05 (d, J = 5.49 Hz, 1 H) 9.12 (s, 1 H). 13C NMR (126 MHz, DMSO-d6) δ ppm 41.53 (s, 1 C) 123.27–125.47 (m, 2 C) 127.97 (s, 1 C) 137.83 (s, 1 C) 139.26–141.18 (m, 2 C) 150.56–154.00 (m, 2 C) 154.00–155.79 (m, 2 C) 189.29 (s, 1 C) 195.62–197.55 (m, 2 C). Crystals suitable for X-ray diffraction were obtained from layering of hexane on DCM.
fac-[Re(CO)3(L8)Br] (8). Yellow powder, yield 68%. ESI-MS analysis (positive mode) m/z = 456.9 [M − Br]+ measured; calculated for [C14H10N2O4Re]+ 457.0. IR (solid, νCO cm−1): 2016.90, 1869.77. UV-Vis (CH3CN, nm): 294, 371. 1H NMR (400 MHz, DMSO-d6) δ ppm 4.74 (d, J = 5.65 Hz, 2 H) 5.73 (t, J = 5.72 Hz, 1 H) 7.71–7.77 (m, 1 H) 8.22 (d, J = 8.24 Hz, 1 H) 8.32 (t, J = 7.86 Hz, 1 H) 8.73 (d, J = 8.39 Hz, 2 H) 8.96 (s, 1 H) 9.02 (d, J = 5.49 Hz, 1 H). 13C NMR (126 MHz, DMSO-d6) δ ppm 59.69 (s, 1 C) 122.58–124.92 (m, 2 C) 127.58 (s, 1 C) 137.89 (s, 1 C) 140.18 (s, 1 C) 142.51 (s, 1 C) 150.48 (s, 1 C) 152.35–154.28 (m, 2 C) 155.16 (s, 1 C) 189.45 (s, 1 C) 197.38 (s, 1 C) 201.66 (s, 1 C).
fac-[Re(CO)3(L9)Br] (9). Yellow powder, yield 74%. ESI-MS analysis (positive mode) m/z = 440.9 [M − Br]+ measured; calculated for [C14H10N2O3Re]+ 441.0. IR (solid, νCO cm−1): 2017.70, 1896.71. UV-Vis (CH3CN, nm): 295, 368. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.50 (s, 3 H) 7.68–7.76 (m, 1 H) 8.16 (d, J = 8.39 Hz, 1 H) 8.30 (t, J = 7.86 Hz, 1 H) 8.62–8.73 (m, 2 H) 8.86 (s, 1 H) 9.01 (d, J = 5.34 Hz, 1 H). 13C NMR (126 MHz, DMSO-d6) d ppm 17.67 (s, 1 C) 123.79 (s, 2 C) 127.29 (s, 1 C) 138.26 (s, 1 C) 140.00 (s, 1 C) 140.51 (s, 1 C) 151.94–153.15 (m, 2 C) 155.21 (s, 1 C) 189.41 (s, 1 C) 197.12 (s, 2 C) 206.33 (s, 1 C). Crystals suitable for X-ray diffraction were obtained from layering of pentane on DCM.
fac-[Re(CO)3(L10)Br] (10). Yellow powder, yield 71%. ESI-MS analysis (positive mode) m/z = 476.9 [M − Br]+ measured; calculated for [C14H8F2N2O3Re]+ 477.0. IR (solid, νCO cm−1): 2020.67, 1887.81. UV-Vis (CH3CN, nm): 295, 378. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.26–7.51 (m, 1 H) 7.82 (t, J = 6.56 Hz, 1 H) 8.37 (t, J = 7.86 Hz, 1 H) 8.57 (d, J = 8.55 Hz, 1 H) 8.82–8.94 (m, 2 H) 9.08 (d, J = 5.34 Hz, 1 H) 9.19 (s, 1 H). 13C NMR (126 MHz, DMSO-d6) d ppm 20.89 (s, 1 C) 110.28 (s, 1 C) 112.17 (s, 1 C) 114.07 (s, 1 C) 122.35–126.48 (m, 2 C) 127.17–129.93 (m, 2 C) 140.24 (s, 1 C) 150.39 (t, J = 7.72 Hz, 1 C) 153.17 (s, 2 C) 188.90 (s, 1 C) 196.87 (d, J = 18.17 Hz, 1 C). Crystals suitable for X-ray diffraction were obtained from layering of hexane on DCM.
4.3. Cytotoxicity evaluation
Antiproliferative activity of the compounds was tested in a panel against tumour cells lines HCT-116 (colorectal carcinoma cells), HT-29 (colorectal adenocarcinoma cells) Mia PaCa-2 (pancreatic carcinoma cells) and Panc-1 (epithelial pancreatic carcinoma cells), as well as on normal human lung fibroblasts (MRC-5), all from ATCC collection. Compounds were freshly dissolved in DMSO and used for the bioactivity assessments. Cytotoxicity in terms of antiproliferative effects was tested by the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.71 The assay was carried out as previously described.424.4. In vivo toxicity assessment
Toxicity evaluation of the complexes was carried in the zebrafish (Danio rerio) model according to the general rules of the OECD Guidelines for the Testing of Chemicals (OECD, 2013, Test No. 236).72 All experiments involving zebrafish were performed in compliance with the European directive 2010/63/EU and the ethical guidelines of the Guide for Care and Use of Laboratory Animals of the Institute of Molecular Genetics and Genetic Engineering, University of Belgrade. Wild type (AB) zebrafish were kindly provided by Dr Ana Cvejić (Wellcome Trust Sanger Institute, Cambridge, UK). Experiments were performed as previously reported.424.5. Anticancer activity evaluation in human CRC-zebrafish xenografts
Cancer cells were cultured in RPMI-1640 supplemented with 10% FBS, 100 μg mL−1 streptomycin and 100 U mL−1 penicillin, and grown as a monolayer in humidified atmosphere of 95% air and 5% CO2 at 37 °C. Prior to microinjection, the cells were washed once with PBS and trypsinized (0.25% trypsin/0.53 mM EDTA) to obtain a single cell suspension. After centrifugation at 1200 rpm for 5 min, the cells were resuspended in serum-free RPMI medium and labelled with 2 μM CellTracker™ RedCMTPX (Thermofisher Scientific) according to the manufacturer's instructions.4.6. Zebrafish xenografts injection and treatment
The zebrafish xenografts with human HCT-116 cells were established according to the previously described procedure.73 Before the microinjections, Tg(fli1:EGFP) and Tg(-2.8fabp10a:EGFP) embryos were kept at 28 °C and manually dechorionated few hours before the injection. At 48 hpf, 5 nL of cells suspension containing 150 labelled cells was microinjected into the yolk of anesthetized embryos by a pneumatic picopump (PV820, World Precision Instruments, USA). Exact number of cells was confirmed by dispensing the injected volume onto a microscope slide and by visual counting. After injection, embryos were incubated to recover for at least one hour at 28 °C, dead embryos were removed, and alive embryos were transferred into 24-well plates containing 1 mL of embryo water and 10 embryos per well. The injected xenografts were treated with different doses of complex 1 and 4 (1/2, 1/4 and 1/8 of IC50 values), and maintained at 33 °C by 120 hpf. DMSO (0.25%) was used as a negative control. The survival and development of the xenografted embryos was recorded every day until the end of experiment. At 3 days post injection (dpi), anesthetized xenografts were processed by fluorescent microscopy. The tumour size was determined by the fluorescent images using ImageJ programme. The experiment was repeated two times.4.7. Statistical analysis
The experimental results were expressed as mean values ± SD. The differences in anti-angiogenic phenotypes between the untreated and treated groups were determined according to χ2 test. In other tests, the differences between the untreated and treated groups, as well as between the treatments were evaluated using the one-way ANOVA followed by a comparison of the means by Bonferroni test (P = 0.05). All analyses were performed using SPSS 20 (SPSS Inc., Chicago, IL) software package.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support from the Swiss National Science Foundation (project# 200021_196967, K. S. and F. Z.), the University of Fribourg and the Institute of Molecular Genetics and Genetic Engineering from the University of Belgrade (Ministry of Education, Science and Technological Development of the Republic of Serbia, 451-03-68/2022-14/200042) are gratefully acknowledged.References
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- S. Su, Y. Chen, P. Zhang, R. Ma, W. Zhang, J. Liu, T. Li, H. Niu, Y. Cao, B. Hu, J. Gao, H. Sun, D. Fang, J. Wang, P. G. Wang, S. Xie, C. Wang and J. Ma, Eur. J. Med. Chem., 2022, 243, 114680 CrossRef CAS PubMed.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- R. Paprocka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Coord. Chem. Rev., 2022, 452, 214307 CrossRef CAS.
- Z.-Y. Li, Q.-H. Shen, Z.-W. Mao and C.-P. Tan, Chem. – Asian J., 2022, 17, e202200270 CAS.
- K. Schindler and F. Zobi, Molecules, 2022, 27, 539 CrossRef CAS PubMed.
- Z. Huang and J. J. Wilson, Eur. J. Inorg. Chem., 2021, 2021, 1312–1324 CrossRef CAS.
- M. Mkhatshwa, J. M. Moremi, K. Makgopa and A.-L. E. Manicum, Int. J. Mol. Sci., 2021, 22, 6546 CrossRef CAS PubMed.
- H. S. Liew, C.-W. Mai, M. Zulkefeli, T. Madheswaran, L. V. Kiew, N. Delsuc and M. L. Low, Molecules, 2020, 25, 4176 CrossRef CAS PubMed.
- P. Collery, D. Desmaele and V. Vijaykumar, Curr. Pharm. Des., 2019, 25, 1–17 CrossRef PubMed.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- L. C. Lee, K. K. Leung and K. K. Lo, Dalton Trans., 2017, 46, 16357–16380 RSC.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- S. Ajay Sharma, N. Vaibhavi, B. Kar, U. Das and P. Paira, RSC Adv., 2022, 12, 20264–20295 RSC.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066–3068, 10.1039/B706657K.
- M.-W. Louie, M. Ho-Chuen Lam and K. Kam-Wing Lo, Eur. J. Inorg. Chem., 2009, 2009, 4265–4273 CrossRef.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- R. R. Ye, C. P. Tan, M. H. Chen, L. Hao, L. N. Ji and Z. W. Mao, Chem. – Eur. J., 2016, 22, 7800–7809 CrossRef CAS.
- S. Clede, F. Lambert, R. Saint-Fort, M. A. Plamont, H. Bertrand, A. Vessieres and C. Policar, Chem. – Eur. J., 2014, 20, 8714–8722 CrossRef CAS PubMed.
- I. Kitanovic, S. Z. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wolfl and G. Gasser, Chem. – Eur. J., 2014, 20, 2496–2507 CrossRef CAS PubMed.
- K. M. Knopf, B. L. Murphy, S. N. MacMillan, J. M. Baskin, M. P. Barr, E. Boros and J. J. Wilson, J. Am. Chem. Soc., 2017, 139, 14302–14314 CrossRef CAS PubMed.
- M. König, D. Siegmund, L. J. Raszeja, A. Prokop and N. Metzler-Nolte, Med. Chem. Commun., 2018, 9, 173–180 RSC.
- C. C. Konkankit, A. P. King, K. M. Knopf, T. L. Southard and J. J. Wilson, ACS Med. Chem. Lett., 2019, 10, 822–827 CrossRef CAS PubMed.
- F. X. Wang, J. H. Liang, H. Zhang, Z. H. Wang, Q. Wan, C. P. Tan, L. N. Ji and Z. W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133 CrossRef CAS PubMed.
- M. Munoz-Osses, F. Godoy, A. Fierro, A. Gomez and N. Metzler-Nolte, Dalton Trans., 2018, 47, 1233–1242 RSC.
- C.-C. Pagoni, V.-S. Xylouri, G. C. Kaiafas, M. Lazou, G. Bompola, E. Tsoukas, L. C. Papadopoulou, G. Psomas and D. Papagiannopoulou, J. Biol. Inorg. Chem., 2019, 24, 609–619 CrossRef CAS PubMed.
- M. Kaplanis, G. Stamatakis, V. D. Papakonstantinou, M. Paravatou-Petsotas, C. A. Demopoulos and C. A. Mitsopoulou, J. Inorg. Biochem., 2014, 135, 1–9 CrossRef CAS.
- G. Balakrishnan, T. Rajendran, K. Senthil Murugan, M. Sathish Kumar, V. K. Sivasubramanian, M. Ganesan, A. Mahesh, T. Thirunalasundari and S. Rajagopal, Inorg. Chim. Acta, 2015, 434, 51–59 CrossRef CAS.
- F. Zobi, B. Spingler and R. Alberto, ChemBioChem, 2005, 6, 1397–1405 CrossRef CAS PubMed.
- A. P. King, S. C. Marker, R. V. Swanda, J. J. Woods, S.-B. Qian and J. J. Wilson, Chem. – Eur. J., 2019, 25, 9206–9210 CrossRef CAS PubMed.
- C. C. Konkankit, J. Lovett, H. H. Harris and J. J. Wilson, Chem. Commun., 2020, 56, 6515–6518 RSC.
- S. C. Marker, A. P. King, S. Granja, B. Vaughn, J. J. Woods, E. Boros and J. J. Wilson, Inorg. Chem., 2020, 59, 10285–10303 CrossRef CAS PubMed.
- P. Collery, G. Bastian, F. Santoni, A. Mohsen, M. Wei, T. Collery, A. Tomas, D. Desmaele and J. D'Angelo, Anticancer Res., 2014, 34, 1679–1689 CAS.
- P. Collery, A. Mohsen, A. Kermagoret, S. Corre, G. Bastian, A. Tomas, M. Wei, F. Santoni, N. Guerra, D. Desmaele and J. d'Angelo, Invest. New Drugs, 2015, 33, 848–860 CrossRef CAS PubMed.
- V. Veena, A. Harikrishnan, B. Lakshmi, S. Khanna, D. Desmaele and P. Collery, Anticancer Res., 2020, 40, 1915–1920 CrossRef CAS PubMed.
- P. Collery, V. Veena, A. Harikrishnan and D. Desmaele, Invest. New Drugs, 2019, 37, 973–983 CrossRef CAS PubMed.
- P. V. Simpson, I. Casari, S. Paternoster, B. W. Skelton, M. Falasca and M. Massi, Chem. – Eur. J., 2017, 23, 6518–6521 CrossRef CAS PubMed.
- A. Domenichini, I. Casari, P. V. Simpson, N. M. Desai, L. Chen, C. Dustin, J. S. Edmands, A. van der Vliet, M. Mohammadi, M. Massi and M. Falasca, J. Exp. Clin. Cancer Res., 2020, 39, 276 CrossRef CAS.
- S. N. Sovari, N. Radakovic, P. Roch, A. Crochet, A. Pavic and F. Zobi, Eur. J. Med. Chem., 2021, 226, 113858 CrossRef CAS PubMed.
- J. Rossier, D. Hauser, E. Kottelat, B. Rothen-Rutishauser and F. Zobi, Dalton Trans., 2017, 46, 2159–2164 RSC.
- J. Delasoie, A. Pavic, N. Voutier, S. Vojnovic, A. Crochet, J. Nikodinovic-Runic and F. Zobi, Eur. J. Med. Chem., 2020, 204, 112583 CrossRef CAS.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- F. Zobi, Inorg. Chem., 2009, 48, 10845–10855 CrossRef CAS PubMed.
- E. Kottelat, F. Lucarini, A. Crochet, A. Ruggi and F. Zobi, Eur. J. Inorg. Chem., 2019, 2019, 3758–3768 CrossRef CAS.
- G. Schanne, L. Henry, H. C. Ong, A. Somogyi, K. Medjoubi, N. Delsuc, C. Policar, F. García and H. C. Bertrand, Inorg. Chem. Front., 2021, 8, 3905–3915 RSC.
- L. D. Ramos, G. Cerchiaro and K. P. Morelli Frin, Inorg. Chim. Acta, 2020, 501, 119329 CrossRef CAS.
- S. Clede, C. Sandt, P. Dumas and C. Policar, Appl. Spectrosc., 2020, 74, 63–71 CrossRef CAS PubMed.
- C. C. Konkankit, B. A. Vaughn, Z. Huang, E. Boros and J. J. Wilson, Dalton Trans., 2020, 49, 16062–16066 RSC.
- L. D. Ramos, L. H. de Macedo, N. R. S. Gobo, K. T. de Oliveira, G. Cerchiaro and K. P. Morelli Frin, Dalton Trans., 2020, 49, 16154–16165 RSC.
- P. T. Wilder, D. J. Weber, A. Winstead, S. Parnell, T. V. Hinton, M. Stevenson, D. Giri, S. Azemati, P. Olczak, B. V. Powell, T. Odebode, S. Tadesse, Y. Zhang, S. K. Pramanik, J. M. Wachira, S. Ghimire, P. McCarthy, A. Barfield, H. N. Banerjee, C. Chen, J. A. Golen, A. L. Rheingold, J. A. Krause, D. M. Ho, P. Y. Zavalij, R. Shaw and S. K. Mandal, Mol. Cell. Biochem., 2018, 441, 151–163 CrossRef CAS PubMed.
- P. A. Clarke, T. Roe, K. Swabey, S. M. Hobbs, C. McAndrew, K. Tomlin, I. Westwood, R. Burke, R. van Montfort and P. Workman, Oncogene, 2019, 38, 5076–5090 CrossRef CAS PubMed.
- H. Shen, R. E. Perez, B. Davaadelger and C. G. Maki, PLoS One, 2013, 8, e59848 CrossRef CAS PubMed.
- A. A. Untereiner, A. Pavlidou, N. Druzhyna, A. Papapetropoulos, M. R. Hellmich and C. Szabo, Biochem. Pharmacol., 2018, 149, 174–185 CrossRef CAS PubMed.
- P. M. De Angelis, K. L. Kravik, S. H. Tunheim, T. Haug and W. H. Reichelt, Mol. Cancer, 2004, 3, 11 CrossRef PubMed.
- R. Fior, V. Póvoa, R. V. Mendes, T. Carvalho, A. Gomes, N. Figueiredo and M. G. Ferreira, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8234–E8243 CrossRef CAS PubMed.
- K. Kai, O. Nagano, E. Sugihara, Y. Arima, O. Sampetrean, T. Ishimoto, M. Nakanishi, N. T. Ueno, H. Iwase and H. Saya, Cancer Sci., 2009, 100, 2275–2282 CrossRef CAS PubMed.
- M. Tavares Barroso, B. Costa, C. Rebelo de Almeida, M. Castillo Martin, N. Couto, T. Carvalho and R. Fior, Cells, 2021, 10, 2077 CrossRef CAS PubMed.
- R. White, K. Rose and L. Zon, Nat. Rev. Cancer, 2013, 13, 624–636 CrossRef CAS PubMed.
- F. Zobi, B. Spingler and R. Alberto, Dalton Trans., 2005, 2859–2865, 10.1039/b503874j.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. Gütz and A. Lützen, Synthesis, 2010, 85–90 Search PubMed.
- T. Norrby, A. Borje, L. Zhang and B. Akermark, Acta Chem. Scand., 1998, 52, 77–85 CrossRef CAS.
- N. C. Fletcher, M. Nieuwenhuyzen and S. Rainey, J. Chem. Soc., Dalton Trans., 2001, 2641–2648, 10.1039/B104365J.
- L.-Y. Liao, X.-R. Kong and X.-F. Duan, J. Org. Chem., 2014, 79, 777–782 CrossRef CAS PubMed.
- B. Imperiali, T. J. Prins and S. L. Fisher, J. Org. Chem., 1993, 58, 1613–1616 CrossRef CAS.
- J. Uenishi, T. Tanaka, K. Nishiwaki, S. Wakabayashi, S. Oae and H. Tsukube, J. Org. Chem., 1993, 58, 4382–4388 CrossRef CAS.
- S. A. Savage, A. P. Smith and C. L. Fraser, J. Org. Chem., 1998, 63, 10048–10051 CrossRef CAS.
- G. R. Newkome, W. E. Puckett, G. E. Kiefer, V. D. Gupta, Y. Xia, M. Coreil and M. A. Hackney, J. Org. Chem., 1982, 47, 4116–4120 CrossRef CAS.
- M. B. Hansen, S. E. Nielsen and K. Berg, J. Immunol. Methods, 1989, 119, 203–210 CrossRef CAS PubMed.
- OECD, OECD, 2013, DOI:10.1787/9789264203709-en.
- C. Zhao, X. Wang, Y. Zhao, Z. Li, S. Lin, Y. Wei and H. Yang, PLoS One, 2011, 6, e21768–e21768 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2090791–2090797. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04041g |
| This journal is © The Royal Society of Chemistry 2023 |