
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium and ruthenium complexes of methylene-bridged, P-stereogenic, unsymmetrical diphosphanes†‡
Javier
Eusamio
 ab,
Yaiza M.
Medina
ab,
Javier C.
Córdoba
a,
Anton
Vidal-Ferran
abc,
Daniel
Sainz
ab,
Albert
Gutiérrez
ab,
Mercè
Font-Bardia
d and
Arnald
Grabulosa
*ab
ab,
Yaiza M.
Medina
ab,
Javier C.
Córdoba
a,
Anton
Vidal-Ferran
abc,
Daniel
Sainz
ab,
Albert
Gutiérrez
ab,
Mercè
Font-Bardia
d and
Arnald
Grabulosa
*ab
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès, 1-11, E-08028, Barcelona, Spain
bInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, E-08028, Barcelona, Spain. E-mail: arnald.grabulosa@qi.ub.es
cInstitució Catalana de Rercerca i Estudis Avançats (ICREA), Passeig Lluís Companys 23, E-08010, Barcelona, Spain
dUnitat de Difracció de Raigs X, Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB), Solé i Sabarís 1-3, E-08028, Barcelona, Spain
First published on 23rd January 2023
Abstract
Enantiopure P-stereogenic methylphosphane-boranes (SP)-P(BH3)PhArMe (ArMe; Ar = 1-naphthyl (NpMe), and 2-biphenylyl (BiphMe)) have been used to prepare diphosphanes of the type ArPhPCH2PR2 (R = Ph, iPr or tBu; ArR). The ligands have been reacted with [Rh(COD)2]BF4 to furnish the corresponding six monochelated [Rh(COD)(ArR)]BF4 organometallic compounds (RhArR) or, depending on the reaction conditions, the bis(chelated) coordination compound [Rh(BiphiPr)2]BF4 as a mixture of cis and trans isomers. The crystal structure of cis-[Rh(BiphiPr)2]BF4 was obtained. The coordination of the BiphR with [RuCl(μ-Cl)(η6-p-cymene)2]2 under different conditions produced cationic chelated complexes of the type [RuCl(η6-p-cymene)(κ2-BiphR)]PF6 (RuBiphR) and the neutral monocoordinated complex [RuCl2(η6-p-cymene)(κ1-BiphPh)] (RuBiphPh′) with the uncoordinated P-stereogenic moiety. The Rh(I) complexes were used in the catalytic hydrogenation of functionalized olefins and the Ru(II) complexes were tested in the transfer hydrogenation of acetophenone. Both precursors displayed good activities with moderate enantioselectivities.
Introduction
Throughout the history of organometallic homogeneous catalysis, phosphorus has been and remains the most important donor group in ancillary ligands of catalytic precursors, due to its ability to coordinate to most of the catalytically more capable metals.1 Among the many phosphorus-based ligands that have been synthesized, diphosphanes are still the most popular, especially in enantioselective catalysis.2,3 The relatively high inversion energy of phosphorus allows for the enantioselective preparation of P-stereogenic ligands,4,5 which have been very successful in several enantioselective reactions. These P-stereogenic diphosphanes shone especially bright in the early days of asymmetric catalysis specifically in enantioselective hydrogenation,6–9 to the point that one of the awardees of the 2001 Nobel Prize in Chemistry was William S. Knowles10 for his research on the synthesis of L-DOPA —a drug used to treat Parkinson's disease— by enantioselective hydrogenation using the DIPAMP ligand, a P-stereogenic diphosphane.After a long period with low activity, many more successful P-stereogenic diphosphanes have been developed,5,11–15 affording excellent catalytic results. Some of the most successful have been those with a single atom linker between the two phosphorus atoms,16 like the MiniPHOS family,17,18 TriChickenFootPHOS (TCPF),19 or MaxPHOS.20–22 These short-bridged ligands (Fig. 1), despite their simple structures, have excelled in enantioselective hydrogenation.23
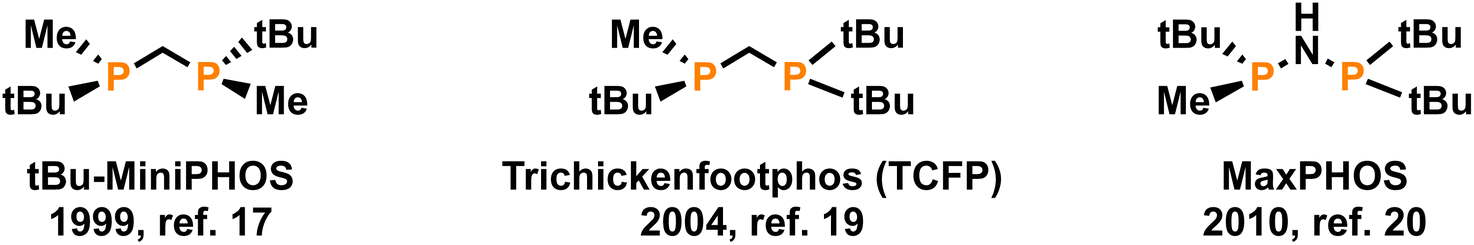 | ||
| Fig. 1 Structures of MiniPHOS, TCFP and MaxPHOS. | ||
For these reasons, following our own research on P-stereogenic monophosphanes,24–28 we recently reported an array of modular P-stereogenic diphosphanes with a methylene bridge and their Pd(II) complexes.29 The ligands are rare examples of unsymmetrical (C1-symmetric) diphosphanes, which have previously proved to be amongst the most efficient in metal-catalysed asymmetric hydrogenation.17–19,23,30–34
In the present work, we present two new ligands bearing a P(tBu)2 moiety on the non-stereogenic phosphorus, and the study of the coordination of this kind of ligands to Rh(I) and Ru(II), to further explore the coordinative variability they display. In addition, catalytic results in the hydrogenation and transfer hydrogenation reactions are presented.
Results and discussion
Unsymmetrical diphosphanes
The methylene-bridged diphosphanes ArPhP1CH2P2R2 (ArR; Ar = 2-biphenylyl (Biph) and 1-naphthyl (Np); R = iPr, tBu or Ph) contain an enantiopure P-stereogenic moiety (denoted as P1) and a non-P-stereogenic moiety (denoted as P2). The ligands were prepared from optically pure methylphosphane–boranes (ArMe) by deprotonation of the methyl group and phosphination of the resulting carbanion with the corresponding chlorophosphane29 (Fig. 2). The diphosphanes could be conveniently stored as their bis(borane) adducts (ArR·BH3), which can be easily deprotected when needed with morpholine. | ||
| Fig. 2 Synthesis of the unsymmetrical diphosphanes. NptBu was synthesized by a variation of method A, and BiphtBu with a variation of method B. | ||
Given the good results that several P-stereogenic diphosphanes containing the bis(tert-butyl)phosphino group have produced in enantioselective catalysis,13,22 the same procedure was followed to prepare the protected diphosphane BiphtBu·BH3 (Fig. 2). Unexpectedly, it was found that this compound could not be isolated, but the monoboronated adduct BiphtBu·2-BH3 —with only the P2 phosphorus protected— was instead obtained (Fig. 3), a compound that was found to be stable in air for a few days.
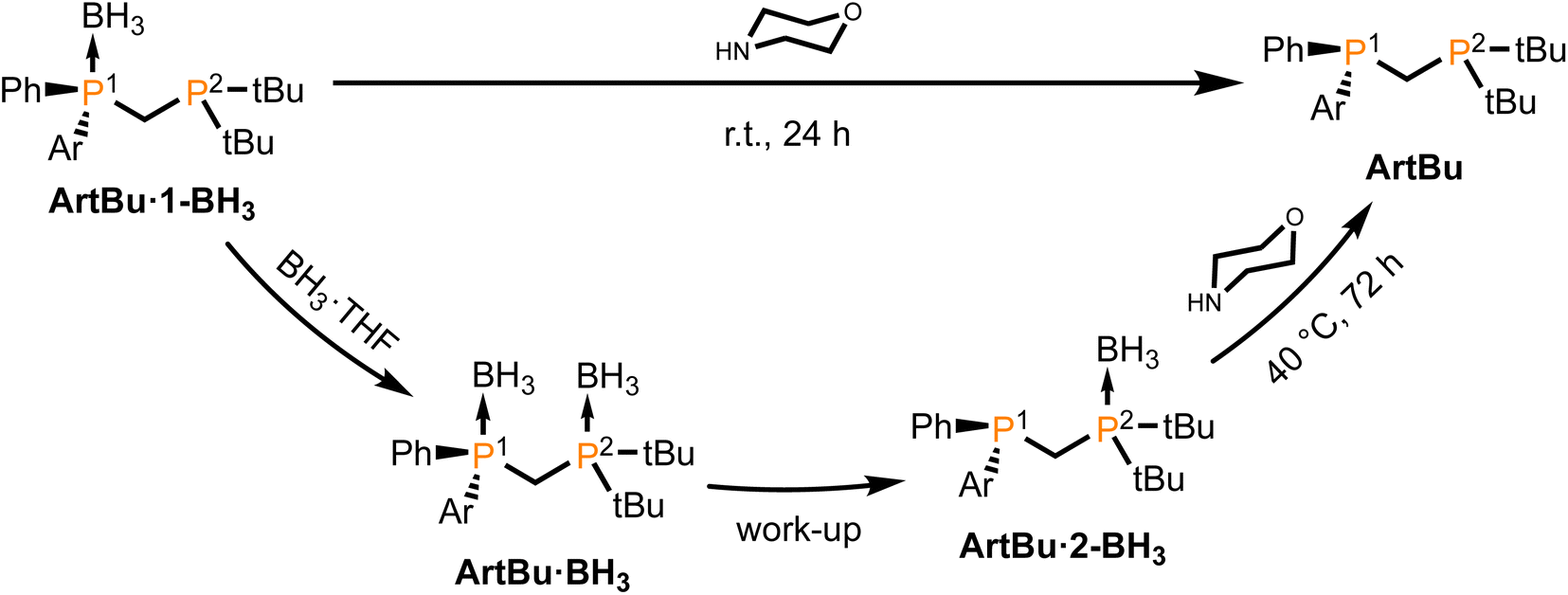 | ||
| Fig. 3 Synthesis of ArtBu diphosphanes. | ||
In the 31P{1H} NMR spectrum of the reaction mixture after workup, BiphtBu·2-BH3 was observed as a broad singlet at δP = +45.1 ppm and a sharp doublet at −30.2 ppm (JP1–P2 = 46.1 Hz), corresponding to P2 and P1 respectively. The formation of BiphtBu·BH3 (with both phosphorus boronated) could only be observed by 31P{1H} NMR spectroscopy in solution in the presence of an excess of BH3·THF as two broad signals at δP +50.3 and +26.3 ppm, corresponding to P2 and P1, respectively. After following the aqueous work-up as in the previously reported procedure,29 the species BiphtBu·2-BH3 was obtained, meaning that P1 had suffered a spontaneous deboronation reaction. This result can be explained first by the reduced basicity of the diarylphosphane P1 compared to the trialkylphosphane P2 and, second, by the steric hindrance exerted by the bis(tert-butyl)phosphane moiety of P2, weakening the B–P1 bond. It has to be noted that these two factors are necessary for the spontaneous cleavage of the B–P bond, since borane adducts of both alkyl,18,19 and aryldiphosphanes29,35,36 with a methylene bridge are known to be perfectly stable compounds.
The deprotection of BiphtBu·2-BH3 with morpholine29,37 was found to be very slow, requiring 72 h at 40 °C (Fig. 3). The synthesis was therefore adapted to furnish BiphtBu in a one-pot procedure, treating without isolation the intermediate compound BiphtBu·1-BH3 —with only P1 protected— with morpholine at room temperature for 24 h (Fig. 3). Following this same procedure, the ligand NptBu was also synthesized and fully characterized.
The 31P{1H} NMR spectra of ligands BiphtBu (δP1 = −24.1 ppm, δP2 = +15.3 ppm; JP1–P2 = 127.7 Hz) and NptBu (δP1 = −28.1 ppm, δP2 = +13.8 ppm; JP1–P2 = 143.8 Hz) consist of two doublets while the hydrogen atoms of the tert-butyl groups appear in the 1H NMR spectra as two sharp doublets (JH–P2 = 10.8 Hz) at around 0.8 ppm, in accordance with other methylene-bridged diphosphanes.29,38,39
Rhodium complexes
Initially, we focused on obtaining the rhodium(I) complexes with the general formulae [Rh(ArR)(COD)]BF4 (RhArR), which are typical hydrogenation catalytic precursors,23,40 starting from the rhodium precursor [Rh(COD)2]BF4 (Fig. 4). | ||
| Fig. 4 Synthesis of the RhArR complexes. | ||
However, upon adding one equivalent of [Rh(COD)2]BF4 to a solution of one equivalent of the deprotected diphosphane, a complex mixture of species was observed according to 31P{1H} NMR, after a standard work-up in air. At lower field (50–20 ppm) some peaks probably corresponding to the oxidized diphosphanes39,41,42 could be identified while at higher field (with a chemical shift between 0 and −40 ppm), where the signals of the Rh–P complexes tend to appear,19,43–49 more than one species was observed. From the presence of oxidized diphosphanes, it was deduced that the complexes were air-sensitive and tended to decompose when exposed to air, an uncommon feature for compounds with this structure, which are often stable.19 However, some hints in the literature suggest a certain lability of the ligands.50
The repetition of the syntheses and work-up under strictly inert atmosphere solved the oxidation issues, but a mixture of species was still observed at higher field in 31P{1H} NMR spectra. From the great variety of coordination possibilities that the ligands had shown when coordinated to Pd(II),29 it was assumed that more than one metal complex could be formed. In the literature, some examples of bischelated complexes were found for short-bridged ligands so we wondered whether mixtures of mono- and bischelated complexes could be formed (Fig. 5).17,32,33
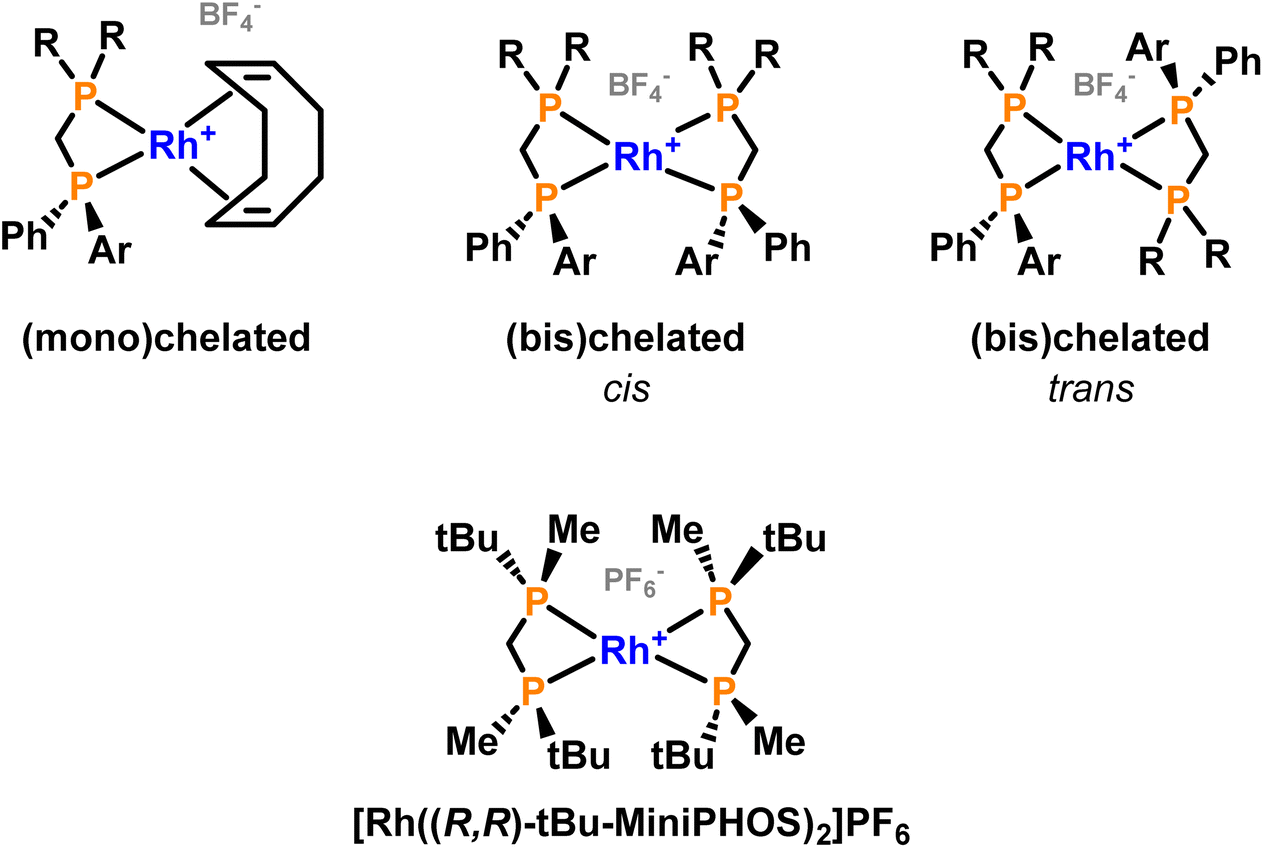 | ||
| Fig. 5 General structure of the mono(chelated) and bis(chelated) complexes (top) and an example of a reported bis(chelated) complex with a methylene-bridged diphosphorus ligand (bottom).17 | ||
To favour the formation of the mono(chelated) structure, the order of the complexation was altered, adding a solution of the diphosphane to a solution of the rhodium precursor in a dropwise manner under vigorous stirring,19,32,33,50,51 to ensure a low local concentration of the ligand and thus avoid the formation of the bis(chelated) complexes. This new procedure afforded the pure mono(chelated) structures after recrystallisation under inert atmosphere. The complexes were found to be indefinitely stable if kept under nitrogen at low temperature. In Fig. 6 the 31P{1H} NMR spectra of the complexes are displayed and the values of the chemical shifts and coupling constants are given in Table 1.
 | ||
Fig. 6
31P{1H} NMR (CD2Cl2) spectra of the mono(chelated) Rh complexes. (a) RhBiphPh, (b) RhNpPh, (c) RhBiphiPr, (d) RhNpiPr, (e) RhBiphtBu, (f) RhNptBu. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) At 162 MHz. bAt 202 MHz. At 162 MHz. bAt 202 MHz. | ||
| Complex | δ P1 | δ P2 | 2 J P1–P2 | 1 J P1–Rh | 1 J P2–Rh |
|---|---|---|---|---|---|
| a 202 MHz. b 162 MHz. c Values obtained using the software package Spinworks.52 | |||||
| RhBiphiPr | −34.9 | −13.4 | 76.1 | 129.4 | 126.9 |
| RhBiphtBu | −35.8 | −4.7 | 63.1 | 130.6 | 125.4 |
| RhBiphPh , | −29.8 | −29.1 | 75.7 | 130.6 | 126.6 |
| RhNpiPr | −39.1 | −12.3 | 77.4 | 131.7 | 126.7 |
| RhNptBu | −38.3 | −2.9 | 63.9 | 132.7 | 125.1 |
| RhNpPh | −33.1 | −28.9 | 78.0 | 130.1 | 130.2 |
Each compound features two doublets of doublets, each one corresponding to one of the phosphorus atoms, coupled to the other one and to the rhodium nucleus. As it can be seen, the chemical shift for the P-stereogenic phosphorus (P1) ranges between −40 and −33 ppm. The shift of the non-P-stereogenic phosphorus (P2) is defined by its substituents: ca. −30 ppm for PPh2, −12 for PiPr2 and −3 ppm for PtBu2. This order is given by a combination of acidity and bulkiness of the substituents, influence of which on 31P{1H} NMR shifts has been well studied and match with comparable complexes containing diphosphanes with the PPh2, PiPr2 and PtBu2 groups.19,43,46–49,51,53
In the case of RhBiphPh, the two phosphorus are so similar that they appear almost overlapped, appearing as a multiplet in the NMR spectrum due to a rather strong roofing effect on the peaks while in the case of complexes RhBiphtBu and RhNptBu the separation between the shifts of the phosphorus is more than 30 ppm and no roofing effect is observed whatsoever.
Obtaining single crystals of the complexes for X-ray diffraction measurements was found to be difficult, due to their tendency to form oily substances and their air sensitivity. Nonetheless, after many attempts we managed to obtain a single crystal of complex RhBiphtBu (Fig. 7) by vapor diffusion of hexane into a concentrated solution of the complex in tetrahydrofuran.
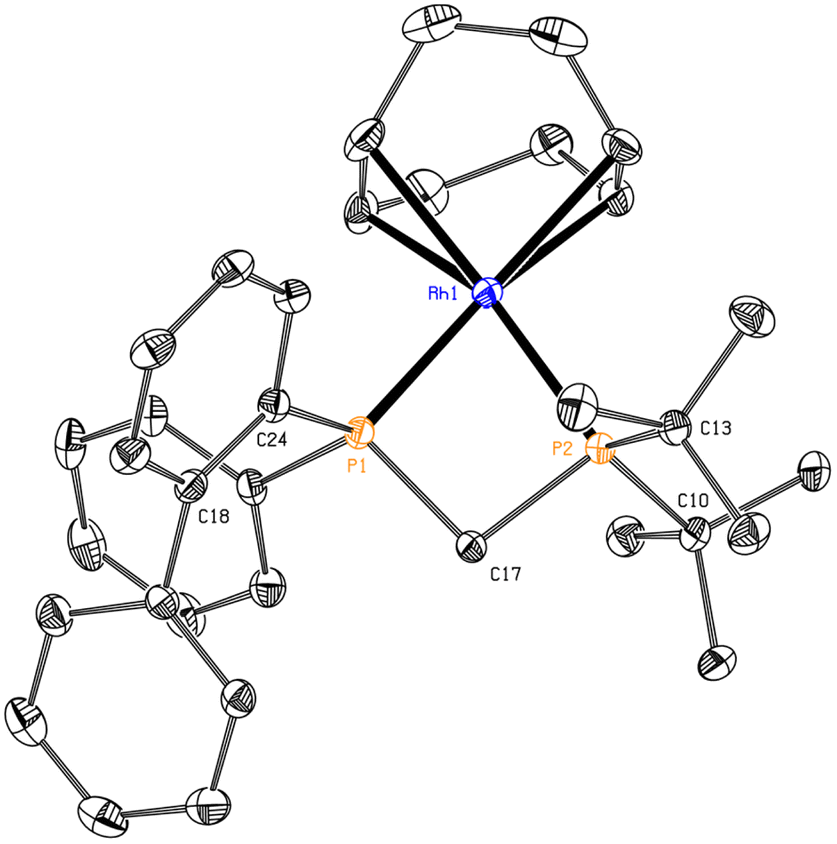 | ||
| Fig. 7 ORTEP plot of RhBiphtBu with ellipsoids drawn at 50% probability level. Hydrogen atoms and the tetrafluoroborate anion have been omitted for clarity. Selected distances (Å) and angles (°): Rh1–P1 2.2861(7), Rh1–P2 2.3356(7), P1–C17 1.837(3), P2–C17 1.857(3), P1–Rh1–P2 72.35(2). | ||
As it can be seen, the bond distance between the rhodium atom and the P-stereogenic phosphorus (P1) is shorter than that of its non-P-stereogenic counterpart (P2), probably accounting for the bulkiness of the PtBu2 moiety. The bite angle, 72.35(2)°, differs considerably from the ideal 90° that corresponds to square planar structures, but its value is within the range found for other Rh(I) complexes with diphosphane ligands with single-atom linkers.19,21,50,54–56
After having obtained the mono(chelated) complexes we focused on obtaining a pure bis(chelated) complex. To this end, a solution of slightly less than two equivalents of ligand BiphiPr was treated with one equivalent of [Rh(COD)]2BF4 in dichloromethane. The apparently simple reaction posed a greater challenge than expected since the formation of some amount of mono(chelated) complex was observed (Fig. 8a and b), even when working at a stoichiometric ratio of ligand/Rh precursor of 2:1. This has been previously observed by Vidal-Ferran and coworkers in Rh(I) complexes with narrow bite-angle chiral phosphane–phosphite ligands.32,33 Finally, adjusting the reaction conditions, it was possible to obtain the bis(chelated) complex Rh(BiphiPr)2 containing only a small amount (∼1–2%) of RhBiphiPr. High-resolution ESI-MS analysis revealed the presence of the M+ cation at m/z = 887.2698 (requires 887.2701), while the 31P{1H} NMR spectrum showed a complex spectrum (Fig. 8c).
 | ||
| Fig. 8
31P{1H} NMR spectra (CD2Cl2) of the mono- and bis(chelated) complexes. (a) Pure RhBiphiPr; (b) crude mixture of RhBiphiPr and Rh(BiphiPr)2; (c) ∼99% pure Rh(BiphiPr)2 as a mixture of cis- and trans isomers; (d) pure cis-Rh(BiphiPr)2 from a monocrystal. aAt 202 MHz. bAt 162 MHz. | ||
The compound features a complex 31P{1H} NMR spectrum (Fig. 8c) comprising six highly symmetric multiplets with chemical shifts between 0 and −28 ppm. The high-field signals in the 31P{1H} NMR spectrum are in the range expected for methylene-bridged diphosphanes (for dppm43 and dcypm48δP = −23.3 and −16.9 ppm, respectively) and the complexity of is not unexpected since a mixture of the cis and trans isomers (Fig. 5) could be present and both constitute second order AMM′XX′ spin systems32,57 (M and M′ represent P2, X and X′ P1 and A, Rh). To confirm that an isomeric mixture was present, a bidimensional homonuclear COSY 31P{1H}–31P{1H} NMR experiment (Fig. 9) and also a HMBC 1H–31P{1H} (see ESI‡) were carried out.
 | ||
| Fig. 9 COSY 31P{1H}–31P{1H} NMR spectrum (202 MHz, CD2Cl2) of Rh(BiphiPr)2. | ||
The COSY spectrum clearly shows a correlation between the two central multiplets and the four external multiplets, but not between these two groups of signals. Therefore, it was deduced that the complex was present as a mixture of cis- and trans-isomers in an approximate 1:1.4 ratio.
Several attempts to separate the isomers by crystallization failed, but changes in the relative integrations of the two mentioned groups of signals in the recrystallized solids and the mother liquors were observed. This confirmed that two species were present and indicating that their separation could be possible. Luckily, it was possible to grow single crystals of the complex (Fig. 10), which corresponded to cis-Rh(BiphiPr)2. The 31P{1H} NMR spectrum of a crystal after its dissolution in CDCl3 was recorded (Fig. 8d). From the figure it can be seen that the multiplets between −24 and −28 ppm and between 0 and −4 ppm correspond to cis-Rh(BiphiPr)2. The structure of cis-Rh(BiphiPr)2, obtained by X-ray diffraction, is to our knowledge the first reported example of a Rh(I) bis(chelated) complex with an unsymmetrical diphosphane (Fig. 10).
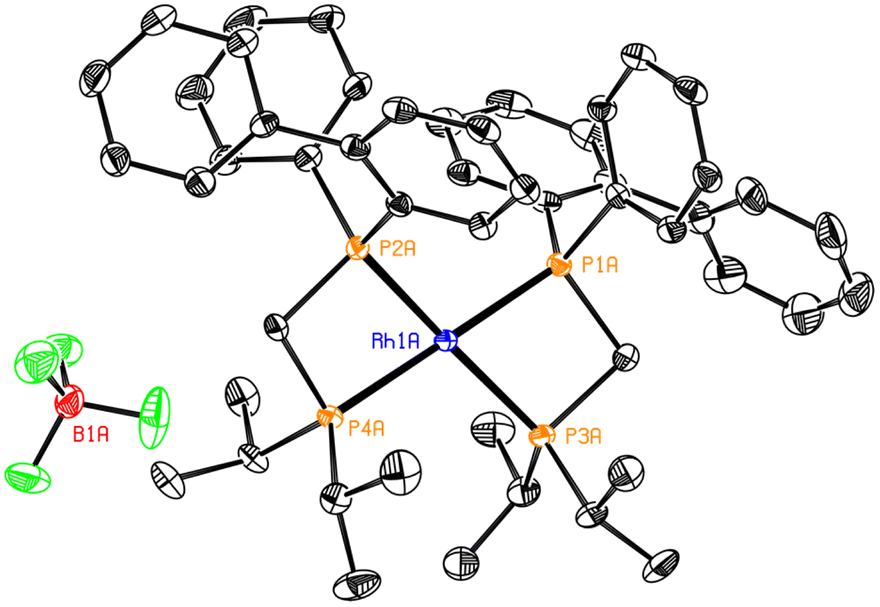 | ||
| Fig. 10 ORTEP plot of the crystal structure of cis-Rh(BiphiPr)2 with ellipsoids drawn at 50% probability level. Hydrogen atoms have been omitted for clarity. Selected distances (Å) and angles (°): P1A–Rh1A 2.295(1), P2A–Rh1A 2.328(1), P3A–Rh1A 2.292(1), P4A–Rh1A 2.297(1), P1A–Rh1A–P3A 72.60(4), P2A–Rh1A–P4A 72.72(4), P1A–P2A–P3A–P4A −1.40(3). | ||
Interestingly, the isomer that crystallized was cis-Rh(BiphiPr)2, exactly the same that had been observed with Pd(II).29 As both isomers are formed in approximately equal ratio, it is reasonable to think that the cis isomer is more insoluble and easier to precipitate.
In the crystal structure, the bite angle matches that of the mono(chelated) structure with the same ligand and of other reported bis(chelated) complexes with similar ligands.17,58 The near-zero torsion angle indicates that the Rh centre adopts a nearly perfect square-planar configuration, even though the bite angle differs greatly from the ideal 90°.
Despite many attempts, it was not possible to obtain, in a synthetically useful way, the pure isomers although the obtention of a crystal of cis-Rh(BiphiPr)2 shows that it should be possible by simple recrystallisation.
Ruthenium complexes
After the study of the coordination chemistry of the ligands in the square-planar environments of d8 palladium(II)29 and rhodium(I) we decided to study their behaviour with the well-known ruthenium(II)-p-cymene unit, to form octahedral, d6 Ru(II) complexes. Two ligands with a 2-biphenylyl moiety (BiphiPr and BiphPh) were chosen because it seems to lead to more stable and crystalline complexes.Following previous studies,59 2 eq. of the BiphR ligand were added to a dichloromethane solution of [RuCl(μ-Cl)(η6-p-cymene)]2 containing 5 equivalents of NH4PF6 in order to form the cationic complexes [RuCl(η6-p-cymene)(BiphR)]PF6, (RuBiphR, Fig. 11, top). However, similarly to what had been observed with Rh(I) and Pd(II),29 the reactions yielded several types of complexes, shown in Fig. 11 (bottom). It has to be noted that the complexes tend to form oils that had to be triturated with diethyl ether or pentane to give the final compounds as orange or reddish solids, a fact that has been mentioned in the literature for similar complexes.60
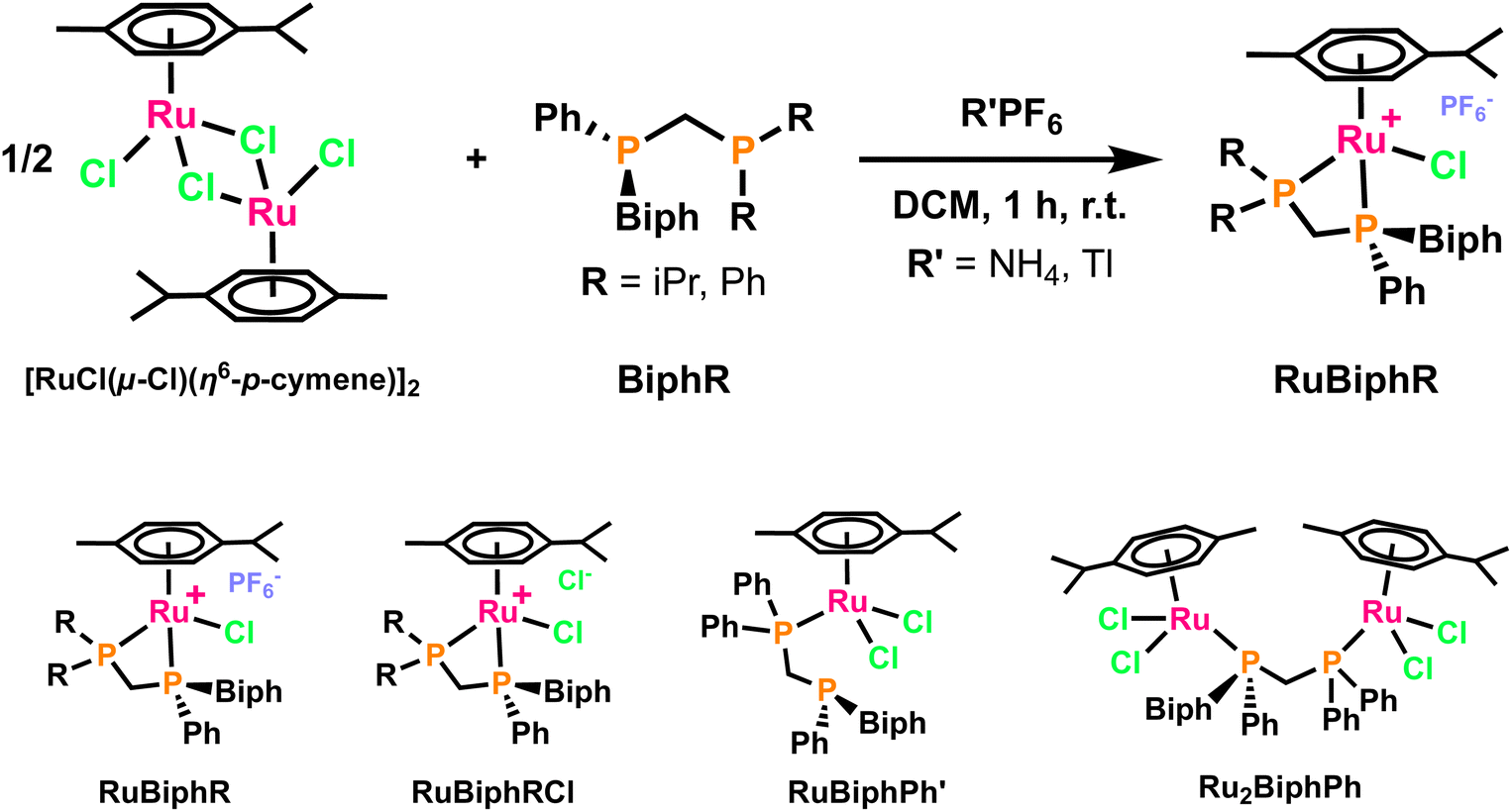 | ||
| Fig. 11 Top: reaction scheme for the complexation of the ligands to Ru(II) complexes. Bottom: structures of the different complexes. | ||
Interestingly, the coordination behaviour is determined by the substituents of the non-P-stereogenic atom, P2. The ligand BiphiPr, with the relatively bulky iPr groups preferentially forms the chelated structures,61 regardless of the addition of PF6 as a counterion or not. Hence, RuBiphiPr was isolated with the standard procedure (Fig. 11, top) and when NH4PF6 was not added, RuBiphiPrCl was obtained instead, with chloride as a counterion.
For the two complexes, very clean ESI-MS spectra with a single peak at m/z = 663.2 were obtained, corresponding to the parent [RuCl(p-cymene)(BiphiPr)]+ cation. In the 31P{1H} spectra NMR, both complexes featured two pairs of doublets at δP of approximately +21 and 0 ppm with a coupling constant of ca. 91 Hz. The chemical shifts can be compared to those of the related complexes with the simple κ2-coordinated ligand dppm, reported to occur at δP of +2.7 (PF6),60 +0.4 ppm (Cl)62 and similarly for other anions63,64 in accordance with the expected upfield shift due to the ring effect of a four-membered metallacycle.60,65–67 In addition, the spectrum of complex RuBiphiPr contains the diagnostic septet centred at −142.0 ppm of the hexafluorophosphate anion. It is interesting to note that a single set of signals was observed in 31P{1H}, 13C{1H} and 1H spectra, suggesting that the complexes are present as single species in solution despite the fact that the ruthenium atom becomes a stereogenic centre and two configurations are possible. The rigidity of the rings probably accounts for this fact, as it happened with the short-bridge ligand MaxPHOS.59
In the case of the ligand BiphPh, the standard conditions (Fig. 11, top) did not provide the expected chelated complex RuBiphPh, but the monocoordinated complex RuBiphPh′, with the BiphPh ligands coordinated in a κ1 fashion (Fig. 11, bottom). Obviously, the very same complex was obtained if NH4PF6 was not added. There is only one related complex described in the literature, bearing the dppm ligand coordinated in κ1 fashion.68–70
The identity of RuBiphPh′ was confirmed by the ESI-MS spectrum, with the base peak at m/z = 767.1 corresponding to the [M + H]+ ion. Interestingly, a minor peak at m/z = 731.1 can also be detected, which can be assigned to the [M − Cl]+ cation, in which the ligand presumably acts as bidentate, most probably formed during the ionization process in the mass spectrometer chamber. The 31P{1H} showed the absence of the hexafluorophosphate anion and the presence of two sharp doublets at δP of +20.9 and −36.9 ppm (in CDCl3), with a coupling constant of 31.0 Hz. These values can be conveniently compared to those reported for the related complex with a κ1-coordinated dppm (δP = +26.1, −27.6 ppm; JPP = 31.8 Hz)68,69 and for the free BiphPh (δP1 = −29.7 ppm, δP2 = −22.6 ppm; JP1–P2 = 126.8 Hz). This comparison reveals that the BiphPh ligand is coordinated to the Ru(II) atom by the non-P-stereogenic phosphorus P2, probably due to steric effects, preventing the coordination of the more crowded P-stereogenic atom P1 bearing the 2-biphenylyl substituent. Interestingly, when the 31P{1H} was recorded in CD3OD the spectrum looked completely different (δP = +6.9, −2.4 ppm; JP1–P2 = 99.5 Hz) because it corresponded to the chelated complex RuBiphPhCl, highlighting the subtle effects that govern the chelation of the methylene-bridged diphosphanes. A similar solvent effect was observed by Dyson and coworkers71 for dppm. After performing kinetic studies, they concluded that methanol favours ring-closing due to hydrogen bond formation with the chloride ligands, helping in their activation.
The synthesis and isolation of RuBiphPh was finally achieved using TlPF6 instead of NH4PF6, the former being a much stronger halide scavenger.28,72,73 This reaction was effective if the starting reagents were either [RuCl(μ-Cl)(η6-p-cymene)]2 and the free BiphPh ligand, or the monodentate RuBiphPh′ complex, yielding the cationic complex after filtration of thallium chloride. The ESI-MS spectrum featured the expected peak centred at m/z = 731.1, corresponding to the organometallic cation. The 31P{1H} NMR spectrum confirmed the identity of RuBiphPh, showing the expected coupled pair of doublets (δP1 = −2.4 ppm, δP2 = +6.9, JP1–P2 = 99.6 Hz) apart from the septet of the hexafluorophosphate anion. The chemical shift values are very similar to those of the analogous complex with dppm.60 It is interesting to note that the 2JP1–P2 in the chelated complexes is around 100 Hz but only 30 Hz in the complexes with the monocoordinated ligands.
Finally, BiphPh was treated with an excess of [RuCl(μ-Cl)(η6-p-cymene)]2 in order to obtain the bimetallic complex Ru2BiphPh, as described in the literature a long time ago with dppm by Atwood, Dixneuf, Bonnet and coworkers74,75 and by Lahuerta and coworkers76 and much more recently by Blom and coworkers77 using η6-benzene instead of η6-p-cymene. However, after a few attempts including heating a dichloromethane solution of the Ru dimer and the BiphPh ligand to reflux for several hours,77 the desired complex was not formed, and only RuBiphPh′, impurified with RuBiphPhCl, free p-cymene and other species due to decomposition78 was obtained. This unsuccessful synthesis shows that the steric crowding of the BiphPh ligand makes it rather different compared to dppm, despite having very similar electronic characteristics.
The crystal structure of RuBiphiPr was obtained (Fig. 12) after slow diffusion of hexane onto a saturated solution of the complex in dichloromethane. It appears that the complexes bearing the BiphiPr ligand are more prone to crystallize, since it is the only ligand for which it has been possible to elucidate at least one crystal structure for all the metals to which it has been coordinated (Pd,29 Rh, and Ru), and also the only one to have an elucidated crystal structure in its boronated form.29
 | ||
| Fig. 12 ORTEP plot of the crystal structure of RuBiphiPr with ellipsoids drawn at 50% probability level. Hydrogen atoms and the hexafluorophosphate anion have been omitted for clarity. Selected distances (Å) and angles (°): Ru1–centroid 1.760, Ru1–Cl1 2.3825(6), Ru1–P1 2.3492(5), Ru1–P2 2.3399(5), P1–C11 1.8341(19), P2–C11 1.8369(19); P1–Ru1–P2 70.997(17), Cl1–Ru1–P1 83.886(17), Cl1–Ru1–P2 85.467(18), centroid–Ru1–Cl1 122.51, centroid–Ru1–P1 139.43, centroid–Ru1–P2 134.66, P1–C11–P2 95.76(9). | ||
As it can be seen in the figure, the complex forms a pseudo-tetrahedral structure, widely known as three-legged piano-stool, typical for this kind of complexes. The distance Ru-centroid (1.760 Å) is similar to other [RuCl(η6-p-cymene)(κ2-diphosphane)]+ complexes.60 The bite angle of the diphosphane in the complex (71.00(2)°) is much smaller than that observed with the same ligand in a Pd(II) complex (74.266(18)°)29 and in the Rh complex Rh(BiphiPr)2 (72.60(4)° and 72.72(4)°) discussed before, but very similar to the bite angle of dppm62,63,79 and a related ligand66 in the few crystal structures reported for Ru(II) compounds containing the cation [RuCl(η6-p-cymene)(κ2-dppm)]. The smaller bite angle with Ru(II) is probably due to the octahedral coordination geometry enforced by a d6 metal, that allows the methylene-bridged diphosphane to reach its “natural” bite angle of 72°.16,80,81 The absolute configuration of the Ru centre is SRu according to the priority sequence η6-p-cymene > Cl > P(Biph)Ph > P(iPr)2 and the Cahn–Ingold–Prelog Rules.82–84
Rh-catalysed hydrogenation
The rhodium complexes described in a previous section were explored as catalytic precursors in the reaction of asymmetric hydrogenation.23 Two typically used benchmark substrates, Z-MAC ((Z)-methyl α-acetamido cinnamate) and DMI (dimethyl itaconate) were subjected to hydrogenation using 1% of Rh precursor at 20 bar of hydrogen pressure (Table 2).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Substrate | Conversion (%) | ee (%) |
| 1 | RhBiphiPr | Z-MAC | >99 | 16 (R) |
| 2 | DMI | >99 | 29 (S) | |
| 3 | RhBiphtBu | Z-MAC | 61 | rac |
| 4 | DMI | >99 | rac | |
| 5 | RhBiphPh | Z-MAC | >99 | 24 (R) |
| 6 | DMI | >99 | 6 (S) | |
| 7 | RhNpiPr | Z-MAC | >99 | 29 (R) |
| 8 | DMI | >99 | 23 (S) | |
| 9 | RhNptBu | Z-MAC | >99 | 50 (R) |
| 10 | DMI | >99 | 9 (S) | |
| 11 | RhNpPh | Z-MAC | >99 | 16 (R) |
| 12 | DMI | >99 | rac | |
| 13 | Rh(BiphiPr)2 | Z-MAC | 12 | rac |
| 14 | DMI | 59 | rac | |

The precursors are active in the reaction, leading to full conversions except for the hydrogenation of Z-MAC with RhBiphiPr (entry 3) and the two catalytic runs with Rh(BiphiPr)2 (entries 13 and 14). The lower activity of these precursors can be understood by the steric hindrance of the ligand and by the fact that the latter complex does not contain a labile ligand. Indeed, Imamoto and coworkers found a similar behaviour when studying their mono- and bis(chelated) complexes with MiniPHOS in hydrogenation.17,18 Regarding the enantioselectivities, they are moderate, without a clear pattern and reaching a maximum value of 50% (entry 9), with Z-MAC usually giving slightly better enantioselectivities, as it is usually found.5,23,40 As expected, the sense of enantioinduction is the same.85
The moderate enantioselectivities obtained with the ligands of this manuscript and the availability of the crystal structures of RhBiphtBu (Fig. 7) and the analogous complex with the related ligand TriChickenFootPhos (TCFP, a highly enantioselective ligand in enantioselective hydrogenation),86 reported by Hoge and coworkers,19 prompted us to evaluate the steric properties of these two ligands by calculation of the percent buried volume (%Vbur) parameter87 and the topographic steric maps using the SambVca 2.1 web application88–90 (Fig. 13).
 | ||
| Fig. 13 Topographic steric maps and %Vbur values (for each quadrant and average value) of BiphtBu using the crystal structure of RhBiphtBu (Fig. 7) (left) and TCFP using the crystal structure of [Rh(cod)2TCFP]BF419 (right). | ||
The averaged %Vbur values of the two ligands is indeed very similar with BiphtBu being slightly bulkier than TCFP. The spatial distribution of this bulkiness, however is rather different for the two ligands because the normalised %Vbur(NW + SW):%Vbur(NE + SE) ratios are 100:94 and 100:91 for BiphtBu and TCFP respectively. This means that in TCFP the steric differentiation between the two phosphane moieties (i.e. between the eastern and western halves in the topographic maps of Fig. 13) is more pronounced than for BiphtBu. In addition, the NE quadrant is more sterically unhindered in TCFP than the comparable SE quadrant for BiphtBu.91 It can be concluded that the phenyl/2-biphenylyl substituents of P1 are probably not distinct enough and hence the introduction at this phosphorus of a bulkier aromatic group instead of 2-biphenylyl could be a strategy to explore future generations of the ligands.92
Ru-catalysed transfer hydrogenation
Ruthenium–arene complexes with P-stereogenic monophosphanes have been used by us26–28,93,94 and others95 in the catalytic reduction of ketones under transfer hydrogenation reactions. Following these studies, ruthenium complexes RuBiphiPr, RuBiphiPrCl and RuBiphPh′ were studied as catalytic precursors, at 1% catalyst loading, in the transfer hydrogenation of acetophenone in refluxing isopropanol in the presence of base (Fig. 14).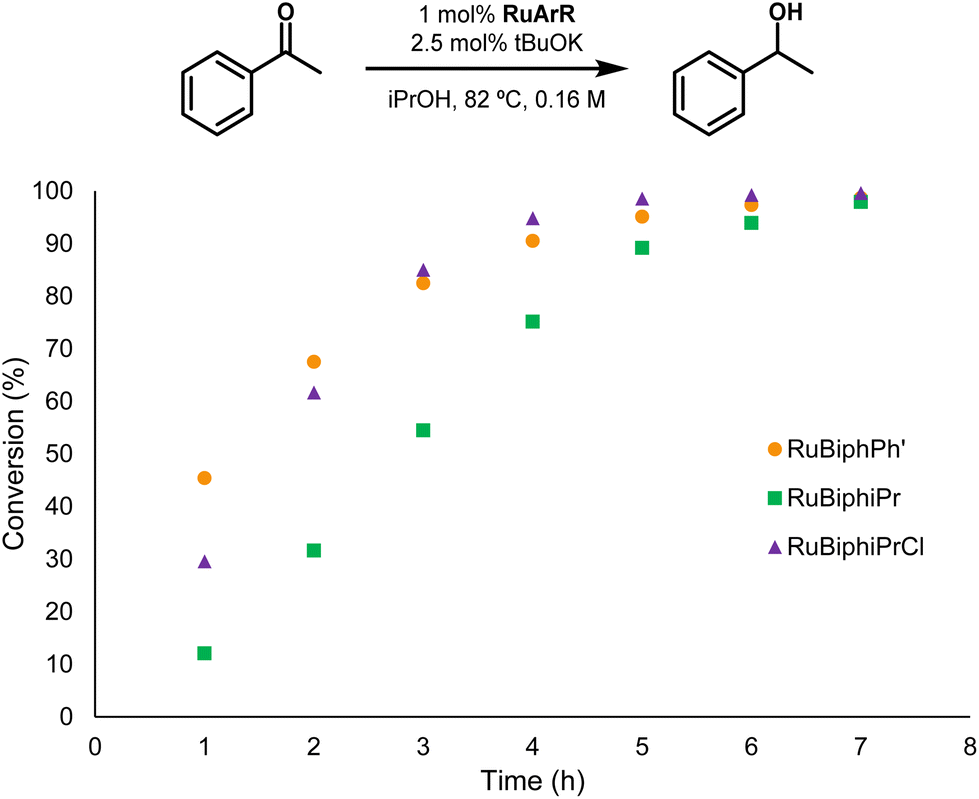 | ||
| Fig. 14 Conversion of acetophenone vs. time with complexes RuBiphiPr, RuBiphiPrCl, and RuBiphPh′. | ||
The three complexes were found to be effective catalysts in the reaction with complexes RuBiphPh′ and RuBiphiPrCl being equally active furnishing over 90% conversion after 4 h, while RuBiphiPr is somewhat less active. The activity of the complexes is similar to the previously reported neutral complexes of the type [Ru(η6-p-cymene)Cl2(P*)] (P* = P-stereogenic monophosphane, including BiphMe26–28,93–95) but in contrast to the moderate enantioselectivity obtained with some of the reported complexes with monophosphanes,28 the enantioselectivity obtained with the complexes of Fig. 14 was very low (ee < 10%).
Conclusions
In this paper the rich coordination chemistry towards Rh(I) and Ru(II) of a group of six P-stereogenic, methylene-bridged, unsymmetrical diphosphanes has been studied. Two out of these six ligands, bearing two tert-butyl moieties on the non-P-stereogenic phosphorus, are described for the first time in this paper.For Rh, mono(chelated) and bis(chelated) structures have been synthesized and isolated. The bis(chelated) complexes present the cis- and the trans-isomers, and it has been observed that the former crystallizes preferentially, which has allowed us to obtain its crystal structure by X-ray diffraction. This selectivity is in accordance with the previous results of the group when coordinating the same ligand to Pd.
For Ru, two different structures have been obtained for the BiphR ligands, namely a chelated (κ2-P1,P2), and a monocoordinated (κ1-P2) one, the latter with the ligand coordinated through the non-P-stereogenic phosphorus atom. The ligand BiphiPr shows a preference for the chelated structure, while BiphPh also forms the monocoordinated and the chelated complexes. The clean formation of the monocoordinated complex is very interesting, since it is an obvious entryway to heterobimetallic complexes with a variety of methylene-bridged diphosphanes, which up to now have only been studied with parent dppm, but already showing promising biological activities, especially the Ru(II)–Au(I) and Ru(II)–Fe(II) complexes.64,77,96–98
Lastly, the applicability of the different complex structures has been tested in asymmetric catalysis. For the Rh complexes, asymmetric hydrogenation has been studied, while for the Ru ones the chosen reaction has been asymmetric transfer hydrogenation. The conversion has been excellent in both cases, although the enantioselectivity has ranged between moderate and low.
Experimental part
Generalities
All compounds were prepared under a purified nitrogen atmosphere using standard Schlenk and vacuum-line techniques or inside a glovebox. The solvents were purified by a solvent purification system or by standard procedures99 and kept under nitrogen. 1H, 13C{1H}, 31P{1H}, and bidimensional NMR spectra were recorded at room temperature with 400 or 500 MHz spectrometers. The fields are 400 or 500 MHz (1H), 101 or 126 MHz (13C{1H}) and 162 or 202 MHz (31P{1H}). Chemical shifts are reported downfield from standards (SiMe4 for 1H and 13C and H3PO4 for 31P NMR) and the coupling constants are given in Hz. IR spectra were recorded with an ATR and the main absorption bands are expressed in cm−1. High-resolution mass analyses (HRMS) were carried out in a time-of-flight instrument using electrospray ionisation. Diphosphanes BiphiPr, NpiPr, BiphPh and NpPh were prepared following the reported procedure.29Ligands
The next day, 1 mL of methanol was added, and the mixture was brought to dryness. After that, it was dissolved in 5 mL of morpholine and stirred for 24 h at room temperature. The solvent was evaporated in vacuo, and the residue was subjected to column chromatography purification (alumina, toluene) under nitrogen to yield the desired product as a viscous white oil. Yield: 90% (379 mg).
1 H NMR (400 MHz, C 6 D 6 ): 7.48–7.41 (m, 1H, Ar), 7.37–7.28 (m, 2H, Ar), 7.26–7.19 (m, 2H, Ar), 7.10–6.79 (m, 9H, Ar), 1.89–1.81 (m, 2H, CH2(bridge)), 0.84 (d, J = 10.8, 9H, CH3(tBu)), 0.76 (d, J = 10.5, 9H, CH3(tBu)). 13C{1H} NMR (101 MHz, C6D6): 147.8–122.1 (m, 18C, Ar), 30.7 (m, 2C, CtBu), 28.7 (m, 6C, CH3(tBu)), 20.1 (dd, J = 36.1, 24.1, 1C, CH2(bridge)). 31P{1H} NMR (162 MHz, C6D6): +13.8 (d, J = 129.8, P2), −25.1 (d, J = 129.8, P1).
1 H NMR (400 MHz, C 6 D 6 ): 8.66 (m, 1H, Ar), 7.64 (ddd, J = 7.1, 4.1, 1.3, 1H, Ar), 7.49–7.36 (m, 3H, Ar), 7.20–7.10 (m, 1H, Ar), 7.07–6.87 (m, 3H, Ar), 6.88–6.78 (m, 3H, Ar), 2.36–2.28 (m, 1H, CHbridge), 2.06–1.96 (m, 1H, CHbridge), 0.93 (d, J = 10.8, 9H, CH3(tBu)), 0.80 (d, J = 10.8, 9H, CH3(tBu)). 13C{1H} NMR (101 MHz, C6D6): 133.3–123.1 (m, 16C, Ar), 30.8 (m, 2C, CtBu), 29.6–27.8 (m, 6C, CH3(tBu)), 20.6–18.7 (m, 1C, CH2(bridge)). 31P{1H} NMR (162 MHz, C6D6): +13.8 (d, 2JP1–P2 = 144.0, P2), −28.1 (d, 2JP1–P2 = 144.0, P1).
Rhodium complexes
1 H NMR (400 MHz, CD 2 Cl 2 ): 9.22–9.10 (m, 1H, Ar), 8.52–8.32 (m, 3H, Ar), 8.31–8.01 (m, 5H, Ar), 7.98–7.85 (m, 4H, Ar), 7.60–7.52 (m, 1H, Ar), 6.46 (s, br, 1H, CHCOD), 6.21 (s, br, 1H, CHCOD), 5.91 (s, br, 1H, CHCOD), 5.67 (s, br, 1H, CHCOD), 4.41–4.29 (m, 1H, CHbridge), 3.98–3.86 (m, 1H, CHbridge), 3.39–3.08 (m, 8H, CH2(COD)), 3.07–2.96 (m, 1H, CHiPr), 2.89–2.80 (m, 1H, CHiPr), 2.12 (dd, J = 17.8, 7.2, 3H, CH3(iPr)), 2.00–1.74 (m, 6H, CH3(iPr)). 13C{1H} NMR (101 MHz, CD2Cl2): 142.2–123.3 (m, 18C, CHAr), 99.5 (s, 1C, CHCOD), 95.0 (s, 1C, CHCOD), 93.8 (s, 1C, CHCOD), 93.6 (s, 1C, CHCOD), 27.8 (dd, J = 24.6, 19.0, 1C, CH2(bridge)), 26.5–24.7 (m, 4C, CH2(COD)), 21.2–20.4 (m, 2C, CHiPr), 14.6 (s, 1C, CH3(iPr)), 14.0 (s, 1C, CH3(iPr)), 13.8 (s, 1C, CH3(iPr)), 12.6 (s, 1C, CH3(iPr)). 31P{1H} NMR (202 MHz, CD2Cl2): −13.4 (dd, 1JP2–Rh = 126.9, 2JP1–P2 = 76.1, P2), −34.9 (dd, 1JP1–Rh = 129.4, 2JP1–P2 = 76.1, P1). HRMS: calcd for [M − BF4 + 2O]+ 635.1715, found 635.1703. IR: 2962, 1629, 1435, 1258, 1037, 879, 795, 702.
1 H NMR (400 MHz, CD 2 Cl 2 ): 7.63–7.56 (m, 2H, Ar), 7.47–7.14 (m, 6H, Ar), 7.04–7.01 (m, 2H, Ar), 6.96–6.91 (m, 2H, Ar), 6.59–6.57 (m, 2H, Ar), 5.91 (s, br, 1H, CHCOD), 5.49 (s, br, 1H, CHCOD), 4.86 (s, br, 1H, CHCOD), 4.64 (s, br, 1H, CHCOD), 3.56–3.42 (m, 1H, CHbridge), 3.21–3.07 (m, 1H, CHbridge), 2.61–1.98 (m, 8H, CH2(COD)), 1.30 (d, J = 14.5, 9H, CH3(tBu)), 1.11 (d, J = 14.8, 9H, CH3(tBu)). 13C{1H} NMR (101 MHz, CD2Cl2): 146.7–126.2 (m, 18C, CHAr), 103.4–103.0 (m, 1C, CHCOD), 101.5–101.0 (m, 1C, CHCOD), 99.3–98.9 (m, 1C, CHCOD), 91.7–91.3 (m, 1C, CHCOD), 37.1 (dd, J = 7.5, 3.6, 1C, C(tBu)), 36.0 (d, J = 7.2, 1C, C(tBu)), 33.3 (dd, J = 23.6, 13.7, 1C, CH2(bridge)), 31.5 (s, 1C, CH2(COD)), 30.7 (s, 1C, CH2(COD)), 29.4 (d, J = 4.9, 3C, CH3(tBu)), 29.3 (d, J = 5.6, 3C, CH3(tBu)), 29.1 (s, 1C, CH2(COD)), 28.9 (s, 1C, CH2(COD)). 31P{1H} NMR (202 MHz, CD2Cl2): −4.7 (dd, 1JP2–Rh = 125.4, 2JP1–P2 = 63.1, P2), −35.8 (dd, 1JP1–Rh = 130.6, 2JP1–P2 = 63.1, P1). HRMS: calcd for [M − COD − BF4]+ 523.1191, found 523.1183. IR: 2951, 2081, 2031, 1466, 1435, 1062, 736, 704.
1 H NMR (400 MHz, CD 2 Cl 2 ): 7.57–7.01 (m, 24H, Ar), 5.22 (s, br, 1H, CHCOD), 5.04 (s, br, 2H, CHCOD), 4.59 (s, br, 1H, CHCOD), 4.08–3.94 (m, 1H, CHbridge), 3.70–3.58 (m, 1H, CHbridge), 2.39–2.07 (m, 8H, CH2(COD)). 13C{1H} NMR (101 MHz, CD2Cl2): 146.9–126.3 (m, 30C, CHAr), 102.8–102.4 (m, 1C, CHCOD), 100.3–99.9 (m, 1C, CHCOD), 98.3–97.7 (m, 2C, CHCOD), 41.1 (t, 1JCP = 22.6, 1C, CH2(bridge)), 30.8 (s, 1C, CH2(COD)), 30.7 (s, 1C, CH2(COD)), 29.5 (s, 1C, CH2(COD)), 29.0 (s, 1C, CH2(COD)). 31P{1H} NMR (162 MHz, CD2Cl2): −29.1 (dd, 1JP2–Rh = 126.6, 2JP1–P2 = 75.7, P2), −29.8 (dd, 1JP1–Rh = 130.6, 2JP1–P2 = 75.7, P1). HRMS: calcd for [M − COD − BF4 + O]+ 687.1453, found 687.1453. IR: 2886, 2837, 1481, 1433, 1345, 1314, 1281, 1164, 1091, 1032, 995, 863, 829, 784, 742, 692.
1 H NMR (400 MHz, CD 2 Cl 2 ): 8.43 (ddd, J = 16.8, 6.8, 1.2, 1H, Ar), 8.18 (d, J = 8.4, 1H, Ar), 8.02 (d, J = 8.0, 1H, Ar), 7.76 (d, J = 8.4, 1H, Ar), 7.67 (td, J = 7.4, 2.0, 1H, Ar), 7.57 (td, J = 7.6, 0.8, 1H, Ar), 7.53–7.40 (m, 6H, Ar), 5.75 (s, br, 1H, CHCOD), 5.57 (s, br, 1H, CHCOD), 5.15 (s, br, 1H, CHCOD), 4.92 (s, br, 1H, CHCOD), 4.16–4.06 (m, 1H, CHbridge), 3.66–3.60 (m, 1H, CHbridge), 2.54–2.26 (m, 8H, CH2(COD)), 2.08–1.97 (m, 2H, CHiPr), 1.55 (dd, J = 15.0, 6.9, 3H, CH3(iPr)), 1.26 (dd, J = 15.0, 6.9, 3H, CH3(iPr)), 1.10 (dd, J = 17.9, 7.1, 3H, CH3(iPr)), 0.83 (dd, J = 17.3, 6.9, 3H, CH3(iPr)). 13C{1H} NMR (101 MHz, CDCl3): 138.0–124.0 (m, 16C, CHAr), 102.9–102.7 (m, 1C, CHCOD), 99.6–99.4 (m, 1C, CHCOD), 99.1–99.0 (m, 1C, CHCOD), 96.2–96.1 (m, 1C, CHCOD), 33.8–33.3 (m, 1C, CH2(bridge)), 30.8 (s, 1C, CH2(COD)), 30.1 (s, 1C, CH2(COD)), 30.0 (s, 1C, CH2(COD)), 29.4 (s, 1C, CH2(COD)), 25.5–25.2 (m, 2C, CH(iPr)), 19.4 (d, J = 3.1, 1C, CH3(iPr)), 18.3 (s, 1C, CH3(iPr)), 18.6 (d, J = 4.0, 1C, CH3(iPr)), 17.4 (s, 1C, CH3(iPr)). 31P{1H} NMR (162 MHz, CD2Cl2): −12.3 (dd, 1JPRh = 126.7, 2JPP = 77.4, P2), −39.1 (dd, 1JPRh = 131.7, 2JPP = 77.4, P1). HRMS: calcd for [M − COD − BF4 + 2CH3CN]+ 567.1201, found 567.1206. IR: 2922, 1696, 1506, 1458, 1436, 1335, 1052, 881, 805, 694, 663, 639.
1 H NMR (500 MHz, CD 2 Cl 2 ): 8.69 (ddd, J = 17.2, 7.0, 1.3, 1H, Ar), 8.20 (d, J = 8.5, 1H, Ar), 8.02 (d, J = 8.0, 1H, Ar), 7.98 (d, J = 8.0, 1H, Ar), 7.73–7.34 (m, 8H, Ar), 6.07 (s, br, 1H, CHCOD), 5.74 (s, br, 1H, CHCOD), 4.98 (s, br, 1H, CHCOD), 4.68 (s, br, 1H, CHCOD), 4.25 (ddd, J = 16.7, 10.8, 8.3, 1H, CHbridge), 3.79–3.68 (m, 1H, CHbridge), 2.70–2.60 (m, 2H, CH2(COD)), 2.57–2.46 (m, 1H, CH2(COD)), 2.35 (m, 2H, CH2(COD)), 2.28–2.18 (m, 3H, CH2(COD)), 1.59 (d, J = 14.5, 9H, CH3(tBu)), 1.07 (d, J = 14.9, 9H, CH3(tBu)). 13C{1H} NMR (126 MHz, CD2Cl2): 142.2–121.8 (m, 16C, Ar), 104.2 (s, 1C, CHCOD), 101.3 (s, 1C, CHCOD), 94.5 (s, 1C, CHCOD), 91.5 (s, 1C, CHCOD), 36.2 (m, 1C, CH2(bridge)), 31.7 (s, 1C, CH2(COD)), 31.2 (s, 1C, CH2(COD)), 30.0 (d, J = 5.1, CH3(tBu)), 29.7 (d, J = 6.0, CH3(tBu)), 29.4 (s, 1C, CtBu), 29.3 (s, 1C, CtBu). 31P{1H} NMR (202 MHz, CD2Cl2): −2.9 (dd, 1JP2–Rh = 125.1, 2JP1–P2 = 63.9, P2), −38.3 (dd, 1JP1–Rh = 132.7, 2JP1–P2 = 63.9, P1). IR: 2962, 2237, 1588, 1435, 1262, 1047, 886, 804, 775, 731, 694.
1 H NMR (500 MHz, CD 2 Cl 2 ): 8.26 (d, J = 8.5, 1H, Ar), 8.05 (d, J = 8.0, 1H, Ar), 8.00 (d, J = 8.0, 1H, Ar), 7.87 (ddd, J = 16.0, 7.0, 1.5, 1H, Ar), 7.62–7.35 (m, 18H, Ar), 5.47 (s, br, 1H, CHCOD), 5.32 (s, br, 3H, CHCOD), 4.47–4.43 (m, 2H, CHbridge), 2.42–2.21 (m, 8H, CH2(COD)). 13C{1H} NMR (126 MHz, CD2Cl2): 137.5–125.0 (m, Ar), 103.7 (s, CHCOD), 101.1 (m, CHCOD), 100.1 (s, CHCOD), 42.4 (t, J = 21.9, 1C, CH2(bridge)), 30.5 (m, CH2(COD)). 31P{1H} NMR (162 MHz, CD2Cl2): −28.9 (dd, 1JP2–Rh = 130.2, 2JP1–P2 = 78.0, P2), −33.1 (dd, 1JP1–Rh = 130.1, 2JP1–P2 = 78.0, P1). HRMS: calcd for [M − COD − BF4 + 2O]+ 677.1246, found 677.1238. IR: 2887, 2838, 1482, 1434, 1345, 1314, 1282, 1165, 1090, 1050, 992, 864, 828, 804, 776, 741, 962.
1 H NMR (500 MHz, CD 2 Cl 2 ): 7.71–6.50 (m, 28H, Ar), 3.39–3.18 (m, 2H, CHbridge), 2.77–2.64 (m, 2H, CHbridge), 1.98–1.90 (m, 1H, CHiPr), 1.82–1.72 (m, 3H, CHiPr), 1.27–0.63 (m, 24H, CH3(iPr)). 13C{1H} NMR (126 MHz, CD2Cl2): 132.9–126.2 (m, Ar), 33.9 (m, CH2(bridge)), 25.7 (m, CHiPr), 24.9 (m, CHiPr), 19.5–15.3 (m, CH3(iPr)). 31P{1H} NMR (202 MHz, CD2Cl2): 0.3−(−1.8) (m, P2cis), −1.8−(−3.9) (m, P2cis), −6.7−(−9.0) (m, P2trans), −17.3−(−19.6) (m, P1trans), −24.2−(−26.1) (m, P1cis), −26.1−(−28.2) (m, P1cis). HRMS: calcd for [M − BF4]+ 887.2701, found 887.2698. IR: 3054, 2957, 1460, 1438, 1049, 735, 694, 630.
Ruthenium complexes
1 H NMR (400 MHz, CDCl 3 ): 8.10 (ddd, J = 17.2, 8.0, 0.8, 1H, Ar), 7.88 (dt, J = 8.0, 2.0, 1H, Ar), 7.62 (dt, J = 7.2, 1.6, 1H, Ar), 7.40–7.35 (m, 1H, Ar), 7.32–7.24 (m, 5H, Ar), 7.20 (ddd, J = 7.6, 4.0, 1.6, 1H, Ar), 7.16–7.12 (m, 2H, Ar), 6.66–6.58 (m, 2H, Ar), 6.26 (d, J = 6.1, 1H, Ar(p-cymene)), 6.07 (d, J = 6.5, 2H, Ar(p-cymene)), 5.90 (d, J = 6.1, 1H, Ar(p-cymene)), 5.73 (d, J = 6.4, 1H, Ar(p-cymene)), 3.57–3.42 (m, 1H, CHbridge), 3.42–3.27 (m, 1H, CHbridge), 2.76–2.62 (m, 1H, CHiPr), 2.62–2.52 (m, 1H, CHiPr(p-cymene)), 2.35–2.22 (m, 1H, CHiPr), 2.10 (s, 3H, CH3(p-cymene)), 1.30 (dd, J = 7.2, 2.6, 3H, CH3(iPr)), 1.26 (dd, J = 7.3, 2.0, 3H, CH3(iPr)), 1.22 (d, J = 7.3, 3H, CH3(iPr, p-cymene)), 1.03 (dd, J = 13.9, 6.9, 6H, CH3(iPr)), 0.96 (dd, J = 15.7, 7.2, 3H, CH3(iPr)). 13C{1H} NMR (101 MHz, CDCl3): 145.1–128.0 (m, 18C, Ar), 120.1 (s, 1C, Ar(p-cymene)), 101.8 (s, 1C, Ar(p-cymene)), 94.0 (d, J = 7.1, 1CH, Ar(p-cymene)), 89.6 (d, J = 6.0, 1CH, Ar(p-cymene)), 89.2 (d, J = 6.3, 1CH, Ar(p-cymene)), 87.7 (s, 1CH, Ar(p-cymene)), 35.0 (dd, J = 27.3 21.2, 1C, CH2(bridge)), 31.2 (s, 1C, CHiPr(p-cymene)), 27.6 (d, J = 21.5, 1C, CHiPr), 26.1 (dd, J = 18.4, 9.9, 1C, CHiPr), 21.9 (d, J = 43.8, 2C, CH3(iPr, p-cymene)), 19.6 (s, 3C, CH3(iPr)), 19.2 (s, 1C, CH3(iPr)), 19.0 (s, 1C, CH3(p-cymene)). 31P{1H} NMR (162 MHz, CDCl3): +20.9 (d, 2JP1–P2 = 91.0, P2), −0.2 (d, 2JP1–P2 = 91.0, P1), −144.2 (hept, 1JP–F = 712.8, PF6). HRMS: calcd for [M − PF6]+ 663.16502, found 633.1643. IR: 2966, 2935, 2880, 1462, 1436, 1387, 1096, 1059, 877, 832, 758, 737, 706, 692, 556, 556.
1 H NMR (400 MHz, CDCl 3 ): 8.36 (dd, J = 15.6, 7.2, 1H, Ar), 8.11 (t, J = 6.8, 1H, Ar), 7.61 (t, J = 6.0, 1H, Ar), 7.39–7.08 (m, 9H, Ar), 6.65–6.57 (m, 2H, Ar), 6.47 (s, 1H, Ar(p-cymene)), 6.43 (s, 1H, Ar(p-cymene)), 6.32 (d, J = 6.5, 1H, Ar(p-cymene)), 5.77 (d, J = 6.5, 1H, Ar(p-cymene)), 3.46 (ddd, J = 15.2, 12.8, 11.6, 1H, CHbridge), 3.30 (ddd, J = 15.3, 10.5, 9.1, 1H, CHbridge), 2.75–2.63 (m, 1H, CHiPr), 2.65–2.53 (m, 1H, CHiPr(p-cymene)), 2.37 (m, 1H, CHiPr), 2.20 (s, 3H, CH3(p-cymene)), 1.33–1.14 (m, 9H, CH3(iPr)), 1.03 (d, J = 6.1, 6H, 2CH3(p-cymene)), 0.93 (dd, J = 15.9, 7.2, 3H, CH3(iPr)). 13C{1H} NMR (101 MHz, CDCl3): 144.8–127.9 (m, 18C, Ar), 117.8 (d, 1C, Ar(p-cymene)), 102.4 (s, 1C, Cp-cymene), 94.4 (d, 1CH, Ar(p-cymene)), 90.4 (d, 1CH, Ar(p-cymene)), 89.9 (d, 1CH, Ar(p-cymene)), 88.6 (s, 1CH, Ar(p-cymene)), 34.7 (dd, JCP = 27.1, 20.9, 1C, CH2(bridge)), 31.4 (s, 1C, CH(iPr, p-cymene)), 27.6 (d, J = 21.3, 1C, CH(iPr)), 26.2 (dd, J = 18.3, 9.9, 1C, CH(iPr)), 22.2 (s, 1C, CH3(iPr, p-cymene)), 22.1 (s, 1C, CH3(iPr, p-cymene)), 19.8 (s, 1C, CH3(p-cymene)), 19.8 (s, 1C, CH3(iPr)), 19.6–19.3 (m, 3C, CH3(iPr)). 31P{1H} NMR (162 MHz, CDCl3): +21.5 (d, 2JP1–P2 = 91.4, P2), +0.1 (d, 2JP1–P2 = 91.4, P1). HRMS: calcd for [M − Cl]+ 663.1650, found 663.1645. IR: 3357, 3051, 2961, 1625, 1459, 1434, 1259, 1090, 1008, 877, 799, 690, 603.
1 H NMR (400 MHz, CDCl 3 ): 8.36 (dd, J = 17.2, 7.6, 1H, Ar), 7.92 (t, J = 7.6, 1H, Ar), 7.63 (t, J = 6.0, 1H, Ar), 7.42–7.01 (m, 17H, Ar), 7.79 (d, J = 8.4, 1H, Ar), 7.76 (d, J = 7.2, 1H, Ar), 6.58 (d, J = 5.6, 1H, Ar(p-cymene)), 6.44–6.42 (m, br, 2H, Ar), 6.17 (d, J = 6.3, 1H, Ar(p-cymene)), 5.75 (d, J = 6.3, 1H, Ar(p-cymene)), 5.70 (d, J = 5.7, 1H, Ar(p-cymene)), 4.06 (dt, J = 15.0, 12.8, 1H, CHbridge), 3.49 (dt, J = 15.2, 10.1, 1H, CHbridge), 2.40–2.26 (m, 1H, CHiPr(p-cymene)), 1.37 (s, 3H, CH3(p-cymene)), 0.95 (dd, J = 7.1, 2.3, 6H, CH3(iPr, p-cymene)). 13C{1H} NMR (101 MHz, CDCl3): 143.8–123.3 (m, 30C, Ar), 117.3 (s, 1C, Arp-cymene), 101.5 (s, 1C, Arp-cymene), 95.4 (d, 1C, Ar(p-cymene)), 89.8 (d, 1C, Ar(p-cymene)), 89.3 (d, 1C, Ar(p-cymene)), 87.3 (s, 1C, Ar(p-cymene)), 39.5 (t, J = 27.3, 1C, CH2(bridge)), 30.0 (s, 1C, CH3(p-cymene)), 22.0 (s, 1C, CHiPr(p-cymene)), 19.7 (s, 1C, CH3(p-cymene)), 16.8 (s, 1C, CH3(p-cymene)). 31P{1H} NMR (162 MHz, CDCl3): +6.5 (d, 2JP1–P2 = 98.6, P2), −2.8 (d, 2JP1–P2 = 98.6, P1), −144.12 (hept, 1JPF = 713.0, PF6). HRMS: calcd for [M − PF6]+ 731.1337, found 731.1326. IR: 3057, 2966, 1436, 1302, 1098, 1058, 911, 832, 723, 690, 556.
1 H NMR (400 MHz, CDCl 3 ): 7.80–7.71 (m, 2H, Ar), 7.59–7.45 (m, 2H, Ar), 7.38–7.15 (m, 12H, Ar), 7.09–6.95 (m, 4H, Ar), 6.94–6.81 (m, 4H, Ar), 5.28 (d, J = 6.2, 1H, Ar(p-cymene)), 5.16 (d, J = 5.8, 2H, Ar(p-cymene)), 4.93 (d, J = 6.0, 1H, Ar(p-cymene)), 3.52 (dd, J = 16.4, 8.4, 1H, CHbridge), 3.13 (dd, J = 16.0, 8.8, 1H, CHbridge), 2.45 (hept, J = 7.2, 1H, CHiPr(p-cymene)), 1.82 (s, 3H, CH3(p-cymene)), 0.87 (d, J = 6.8, 3H, CH3(iPr, p-cymene)), 0.70 (d, J = 6.9, 3H, CH3(iPr, p-cymene)). 13C{1H} NMR (126 MHz, CDCl3): 135.1–124.4 (m, Ar), 106.8 (s, Ar(p-cymene)), 93.9–79.1 (m, Ar(p-cymene)), 28.9 (s, CHiPr(p-cymene)), 22.6 (s, CH3(p-cymene)), 20.7 (s, CH2(bridge)), 19.8 (s, CH3(p-cymene)). 31P{1H} NMR (162 MHz, CDCl3): +26.0 (d, 2JP1–P2 = 30.8, P2), −36.9 (d, 2JP1–P2 = 31.0, P1). 1H NMR (400 MHz, CD3OD): 8.36–6.85 (m, 24H, Ar), 6.53 (d, J = 6.4, 1H, Ar(p-cymene)), 6.43 (d, J = 6.4, 1H, Ar(p-cymene)), 5.87 (d, J = 6.4, 1H, Ar(p-cymene)), 5.79 (d, J = 6.0, 1H, Ar(p-cymene)), 4.27 (dt, J = 15.3, 12.8, 1H, CHbridge), 3.93 (dt, J = 15.4, 10.1, 1H, CHbridge), 2.34 (septet, J = 6.7, 1H, CHiPr(p-cymene)), 1.42 (s, 3H, CH3(p-cymene)), 0.97 (d, J = 7.0, 3H, CH3(iPr, p-cymene)), 0.76 (d, J = 6.9, 3H, CH3(iPr, p-cymene)). 13C{1H} NMR (101 MHz, CD3OD): 146.8–125.3 (m, Ar), 94.8 (d, J = 7.5, Ar(p-cymene)), 91.8 (d, J = 6.4, Ar(p-cymene)), 89.5 (d, J = 6.5, Ar(p-cymene)), 88.9 (s, Ar(p-cymene)), 40.7 (t, J = 27.7, CH2(bridge)), 30.9 (s, CHiPr(p-cymene)), 21.9 (s, CH3(iPr, p-cymene)), 19.7 (s, CH3(iPr, p-cymene)), 16.4 (s, CH3(p-cymene)). 31P{1H} NMR (162 MHz, CD3OD): +5.8 (d, 2JP1–P2 = 100.4, P2), −0.78 (d, 2JP1–P2 = 100.4, P1). HRMS: calcd for [M + H]+ 767.1103, found 767.1097. IR: 3057, 2966, 1436, 1302, 1098, 1058, 910, 832, 723, 690, 556.
Catalytic runs
Author contributions
J. E.: methodology, formal analysis, investigation, validation, data curation, writing-original draft, visualisation. Y. M. M.: investigation, validation, writing-original draft. J. C. C.: investigation. A. V.-F.: methodology, resources, writing-review & editing, supervision, funding acquisition. A. G.: investigation, validation. D. S.: resources, funding acquisition. M. F.-B.: investigation, formal analysis. A. G.: methodology, resources, writing-original draft, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the MICINN (PID2020-115658GB-I00) and AGAUR (2021-SGR-01107) for financial support. Y. M. M. thanks IN2UB for a Master fellowship.References
- Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley, 2012 Search PubMed.
- Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, Wiley, Hoboken, NJ, 2022 Search PubMed.
- K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375–1411 CrossRef CAS.
- A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, Royal Society of Chemistry, Cambridge, 2011 Search PubMed.
- L. Horner, H. Siegel and H. Büthe, Angew. Chem., Int. Ed. Engl., 1968, 7, 942–942 CrossRef CAS.
- W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445–1446 RSC.
- W. S. Knowles, M. J. Sabacky and B. D. Vineyard, J. Chem. Soc., Chem. Commun., 1972, 10–11 RSC.
- W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567–2568 CrossRef CAS.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS.
- O. I. Kolodiazhnyi, in Phosphorus Chemistry I: Asymmetric Synthesis and Bioactive Compounds, ed. J.-L. Montchamp, Springer International Publishing, Cham, 2015, pp. 161–236 Search PubMed.
- M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
- T. Imamoto, Chem. Rec., 2016, 16, 2659–2673 CrossRef PubMed.
- X. Ye, L. Peng, X. Bao, C.-H. Tan and H. Wang, Green Synth. Catal., 2021, 2, 6–18 CrossRef.
- D. S. Glueck, Synlett, 2021, 31, 875–884 CrossRef.
- S. M. Mansell, Dalton Trans., 2017, 46, 15157–15174 RSC.
- Y. Yamanoi and T. Imamoto, J. Org. Chem., 1999, 64, 2988–2989 CrossRef CAS PubMed.
- I. D. Gridnev, Y. Yamamoi, N. Higashi, H. Tsuruta, M. Yasutake and T. Imamoto, Adv. Synth. Catal., 2001, 343, 118–136 CrossRef CAS.
- G. Hoge, H. Wu, W. S. Kissel, D. A. Pflum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS PubMed.
- M. Revés, C. Ferrer, T. León, S. Doran, P. Etayo, A. Vidal-Ferran, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2010, 49, 9452–9455 CrossRef PubMed.
- E. Cristóbal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martín-Gago, A. Grabulosa, A. R. Constantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795–804 CrossRef.
- A. Cabré, A. Riera and X. Verdaguer, Acc. Chem. Res., 2020, 53, 676–689 CrossRef PubMed.
- A. Vidal-Ferran, A. Grabulosa, X. Verdaguer and A. Riera, in Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, Wiley, Hoboken, NJ, 2022, ch. 15, pp. 561–616 Search PubMed.
- A. Grabulosa, G. Muller, J. I. Ordinas, A. Mezzetti, M. A. Maestro, M. Font-Bardia and X. Solans, Organometallics, 2005, 24, 4961–4973 CrossRef CAS.
- L. Rodríguez, O. Rossell, M. Seco, A. Grabulosa, G. Muller and M. Rocamora, Organometallics, 2006, 25, 1368–1376 CrossRef.
- R. Aznar, A. Grabulosa, A. Mannu, G. Muller, D. Sainz, V. Moreno, M. Font-Bardia, T. Calvet and J. Lorenzo, Organometallics, 2013, 32, 2344–2362 CrossRef CAS.
- M. Navarro, D. Vidal, P. Clavero, A. Grabulosa and G. Muller, Organometallics, 2015, 34, 973–994 CrossRef CAS.
- P. Clavero, A. Grabulosa, M. Rocamora, G. Muller and M. Font-Bardia, Dalton Trans., 2016, 45, 8513–8531 RSC.
- J. C. Córdoba, A. Vidal-Ferran, M. Font-Bardia and A. Grabulosa, Organometallics, 2020, 39, 2511–2525 CrossRef.
- I. D. Gridnev, M. Yasutake, N. Higashi and T. Imamoto, J. Am. Chem. Soc., 2001, 123, 5268–5276 CrossRef CAS PubMed.
- K. V. L. Crépy and T. Imamoto, Adv. Synth. Catal., 2003, 345, 79–101 CrossRef.
- H. Fernández-Pérez, S. M. A. Donald, I. J. Munslow, J. Benet-Buchholz, F. Maseras and A. Vidal-Ferran, Chem. – Eur. J., 2010, 16, 6495–6508 CrossRef PubMed.
- H. Fernández-Pérez, J. Benet-Buchholz and A. Vidal-Ferran, Chem. – Eur. J., 2014, 20, 15375–15384 CrossRef PubMed.
- S. Chakrabortty, K. Konieczny, B. H. Mueller, A. Spannenberg, P. Kamer and J. G. de Vries, Catal. Sci. Technol., 2022, 12, 1392–1399 RSC.
- C. Salomon, S. Dal Molin, D. Fortin, Y. Mugnier, R. T. Boere, S. Jugé and P. D. Harvey, Dalton Trans., 2010, 39, 10068–10075 RSC.
- C. Salomon, D. Fortin, N. Khiri, S. Jugé and P. D. Harvey, Eur. J. Inorg. Chem., 2011, 2597–2609 CrossRef CAS.
- R. M. Stoop, A. Mezzetti and F. Spindler, Organometallics, 1998, 17, 668–675 CrossRef CAS.
- S. O. Grim, P. H. Smith, I. J. Colquhoun and W. McFarlane, Inorg. Chem., 1980, 19, 3195–3198 CrossRef CAS.
- F. Eisenträger, A. Göthlich, I. Gruber, H. Heiss, C. A. Kiener, C. Krüger, J. Ulrich Notheis, F. Rominger, G. Scherhag, M. Schultz, B. F. Straub, M. A. O. Volland and P. Hofmann, New J. Chem., 2003, 27, 540–550 RSC.
- P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC.
- T. L. Gianetti, R. E. Rodríguez-Lugo, J. R. Harmer, M. Trincado, M. Vogt, G. Santiso-Quinones and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 15323–15328 CrossRef CAS PubMed.
- V. P. Morgalyuk, T. Y. V. Strelkova, E. E. Nifant'ev and V. K. Brel, Mendeleev Commun., 2016, 26, 397–398 CrossRef CAS.
- J. M. Ernsting, C. J. Elsevier, W. G. J. De Lange and K. Timmer, Magn. Reson. Chem., 1991, 29, S118–S124 CrossRef CAS.
- J. Browning, G. W. Bushnell, K. R. Dixon and R. W. Hilts, J. Organomet. Chem., 1993, 452, 205–218 CrossRef CAS.
- P. Hofmann, C. Meier, W. Hiller, M. Heckel, J. Riede and M. U. Schmidt, J. Organomet. Chem., 1995, 490, 51–70 CrossRef CAS.
- M. Manger, J. Wolf, M. Teichert, D. Stalke and H. Werner, Organometallics, 1998, 17, 3210–3221 CrossRef CAS.
- J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC.
- S. K. Murphy, A. Bruch and V. M. Dong, Chem. Sci., 2015, 6, 174–180 RSC.
- A. J. Martínez-Martínez, N. H. Rees and A. S. Weller, Angew. Chem., Int. Ed., 2019, 58, 16873–16877 CrossRef PubMed.
- T. Imamoto, Y. Horiuchi, E. Hamanishi, S. Takeshita, K. Tamura, M. Sugiya and K. Yoshida, Tetrahedron, 2015, 71, 6471–6480 CrossRef CAS.
- H. El-Amouri, A. A. Bahsoun and J. A. Osborn, Polyhedron, 1988, 7, 2035–2038 CrossRef.
- K. Marat, Spinworks 4.2.0, University of Manitoba, 2015 Search PubMed.
- G. Fries, J. Wolf, K. Ilg, B. Walfort, D. Stalke and H. Werner, Dalton Trans., 2004, 1873–1881 RSC.
- P. G. Pringle, E. L. Hazeland, A. Chapman and H. Sparkes, Chem. Commun., 2015, 51, 10206–10209 RSC.
- A. L. Colebatch, A. I. McKay, N. A. Beattie, S. A. Macgregor and A. S. Weller, Eur. J. Inorg. Chem., 2017, 4533–4540 CrossRef CAS.
- Y. Sawatsugawa, K. Tamura, N. Sano and T. Imamoto, Org. Lett., 2019, 21, 8874–8878 CrossRef CAS PubMed.
- A. F. M. J. Van Der Ploeg and G. Van Koten, Inorg. Chim. Acta, 1981, 51, 225–239 CrossRef CAS.
- R. Fornika, C. Six, H. Görls, M. Kessler, C. Krüger and W. Leitner, Can. J. Chem., 2001, 79, 642–648 CAS.
- J. Tellez, A. Gallen, J. Ferrer, F. J. Lahoz, P. Garcia-Orduna, A. Riera, X. Verdaguer, D. Carmona and A. Grabulosa, Dalton Trans., 2017, 46, 15865–15874 RSC.
- S. B. Jensen, S. J. Rodger and M. D. Spicer, J. Organomet. Chem., 1998, 556, 151–158 CrossRef CAS.
- A. C. Marr, M. Nieuwenhuyzen, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 2659–2671 CrossRef CAS.
- C. Daguenet, R. Scopelliti and P. J. Dyson, Organometallics, 2004, 23, 4849–4857 CrossRef CAS.
- N. Aldeghi, D. Romano, C. Marschner, S. Biswas, S. Chakraborty, S. Prince, S. Ngubane and B. Blom, J. Organomet. Chem., 2020, 916, 121214 CrossRef CAS.
- A. Benamrane, B. Herry, V. Vieru, S. Chakraborty, S. Biswas, S. Prince, C. Marschner and B. Blom, J. Organomet. Chem., 2021, 934, 121659 CrossRef CAS.
- P. E. Garrou, Chem. Rev., 1981, 81, 229–266 CrossRef CAS.
- L. J. Hounjet, M. Bierenstiel, M. J. Ferguson, R. McDonald and M. Cowie, Inorg. Chem., 2010, 49, 4288–4300 CrossRef CAS PubMed.
- O. A. Lenis-Rojas, M. P. Robalo, A. I. Tomaz, A. R. Fernandes, C. Roma-Rodrigues, R. G. Teixeira, F. Marques, M. Folgueira, J. Yáñez, A. A. Gonzalez, M. Salamini-Montemurri, D. Pech-Puch, D. Vázquez-García, M. L. Torres, A. Fernández and J. J. Fernández, Inorg. Chem., 2021, 60, 2914–2930 CrossRef CAS PubMed.
- D. K. Gupta, A. N. Sahay, D. S. Pandey, N. K. Jha, P. Sharma, G. Espinosa, A. Cabrera, M. C. Puerta and P. Valerga, J. Organomet. Chem., 1998, 568, 13–20 CrossRef CAS.
- A. B. Chaplin, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2005, 4762–4774 CrossRef CAS.
- P. Chuklin, V. Chalermpanaphan, T. Nhukeaw, S. Saithong, K. Chainok, S. Phongpaichit, A. Ratanaphan and N. Leesakul, J. Organomet. Chem., 2017, 846, 242–250 CrossRef CAS.
- A. B. Chaplin, C. Fellay, G. Laurenczy and P. J. Dyson, Organometallics, 2006, 26, 586–593 CrossRef.
- J. Ruiz, M. P. Gonzalo, M. Vivanco, R. Quesada and M. E. G. Mosquera, Dalton Trans., 2009, 9280–9290 RSC.
- L. Rafols, D. Josa, D. Aguilà, L. A. Barrios, O. Roubeau, J. Cirera, V. Soto-Cerrato, R. Pérez-Tomás, M. Martínez, A. Grabulosa and P. Gamez, Inorg. Chem., 2021, 60, 7974–7990 CrossRef CAS PubMed.
- A. W. Coleman, D. F. Jones, P. H. Dixneuf, C. Brisson, J. J. Bonnet and G. Lavigne, Inorg. Chem., 1984, 23, 952–956 CrossRef CAS.
- A. W. Coleman, H. Zhang, S. G. Bott, J. L. Atwood and P. H. Dixneuf, J. Coord. Chem., 1987, 16, 9–17 CrossRef CAS.
- F. Estevan, P. Lahuerta, J. Latorre, A. Sanchez and C. Sieiro, Polyhedron, 1987, 6, 473–478 CrossRef CAS.
- B. Herry, L. K. Batchelor, B. Roufosse, D. Romano, J. Baumgartner, M. Borzova, T. Reifenstahl, T. Collins, A. Benamrane, J. Weggelaar, M. C. Correia, P. J. Dyson and B. Blom, J. Organomet. Chem., 2019, 901, 120934 CrossRef CAS.
- J. Popp, S. Hanf and E. Hey-Hawkins, ACS Omega, 2020, 4, 22540–22548 CrossRef PubMed.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed.
- R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl., 1966, 5, 385–415 CrossRef CAS.
- V. Prelog and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1982, 21, 567–583 CrossRef.
- C. Lecomte, Y. Dusausoy, J. Protas, J. Tirouflet and A. Dormond, J. Organomet. Chem., 1974, 73, 67–76 CrossRef CAS.
- The different absolute configurations of the products stems from the Cahn–Ingold–Prelog priority rules.
- In the two crystal structures the most relevant distances and angles are very similar. Selected parameters in RhBiphtBuvs. [Rh(COD)TCFP]BF4: Rh–P1 (2.2861(7) vs. 2.2867(18); Rh–P2 (2.3356(7) vs. 2.3377(18); P1–Rh–P2 (72.35(2) vs. 72.55(6).
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- The absolute configurations of the stereogenic phosphorus are different in the crystal structures (S for BiphtBu and R for TCFP, as free ligands) and for this reason the unhindered quadrants are different in the two steric maps of Fig. 13.
- P. Rojo, A. Riera and X. Verdaguer, Org. Lett., 2021, 23, 4802–4806 CrossRef CAS PubMed.
- A. Grabulosa, A. Mannu, A. Mezzetti and G. Muller, J. Organomet. Chem., 2012, 696, 4221–4228 CrossRef CAS.
- P. Clavero, A. Grabulosa, M. Font-Bardia and G. Muller, J. Mol. Catal. A: Chem., 2014, 391, 183–190 CrossRef CAS.
- J. Popp, A.-M. Caminade and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2020, 1654–1669 CrossRef CAS.
- L. Massai, J. Fernandez-Gallardo, A. Guerri, A. Arcangeli, S. Pillozzi, M. Contel and L. Messori, Dalton Trans., 2015, 44, 11067–11076 RSC.
- J. Fernández-Gallardo, B. T. Elie, M. Sanaú and M. Contel, Chem. Commun., 2016, 52, 3155–3158 RSC.
- B. T. Elie, Y. Pechenyy, F. Uddin and M. Contel, JBIC, J. Biol. Inorg. Chem., 2018, 23, 399–411 CrossRef CAS PubMed.
- W. L. F. Armarego, Purification of Laboratory Chemicals, Butterworth Heinemann, Oxford, Eight edn., 2017 Search PubMed.
Footnotes |
| † In memoriam of Prof. Paul Kamer (1960–2020). |
| ‡ Electronic supplementary information (ESI) available: NMR spectra of the new compounds. CCDC 2223261–2223263 for complexes RhBiphtBu, Rh(BiphiPr)2 and RuBiphiPr. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04026c |
| This journal is © The Royal Society of Chemistry 2023 |