
Replacement of the phosphodiester backbone between canonical nucleosides with a dirhenium carbonyl “click” linker—a new class of luminescent organometallic dinucleoside phosphate mimics†
Joanna
Skiba
 a,
Aleksandra
Kowalczyk
b,
Aleksander
Gorski
c,
Natalia
Dutkiewicz
c,
Magdalena
Gapińska
d,
Józef
Stróżek
a,
Krzysztof
Woźniak
e,
Damian
Trzybiński
e and
Konrad
Kowalski
*a
a,
Aleksandra
Kowalczyk
b,
Aleksander
Gorski
c,
Natalia
Dutkiewicz
c,
Magdalena
Gapińska
d,
Józef
Stróżek
a,
Krzysztof
Woźniak
e,
Damian
Trzybiński
e and
Konrad
Kowalski
*a
aFaculty of Chemistry, Department of Organic Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: konrad.kowalski@chemia.uni.lodz.pl; kondor15@wp.pl; Tel: +48-42-635-5759
bDepartment of Molecular Microbiology, Institute of Microbiology, Biotechnology and Immunology, Faculty of Biology and Environmental Protection, University of Łódź, Banacha 12/16, 90-237 Łódź, Poland
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw, Poland
dFaculty of Biology and Environmental Protection, Laboratory of Microscopic Imaging and Specialized Biological Techniques, University of Łódź, Banacha 12/16, 90-237 Łódź, Poland
eFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089 Warszawa, Poland
First published on 11th January 2023
Abstract
The first-in-class luminescent dinucleoside phosphate analogs with a [Re2(μ-Cl)2(CO)6(μ-pyridazine)] “click” linker as a replacement for the natural phosphate group are reported together with the synthesis of luminescent adenosine and thymidine derivatives having the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] entity attached to positions 5′ and 3′, respectively. These compounds were synthesized by applying inverse-electron-demand Diels–Alder and copper(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition reactions in three or four steps. The obtained compounds exhibited orange emission (λPL ≈ 600 nm, ΦPL ≈ 0.10, and τ = 0.33–0.61 μs) and no toxicity (except for one nucleoside) to human HeLa cervical epithelioid and Ishikawa endometrial adenocarcinoma cancer cells in vitro. Furthermore, the compounds’ ability to inhibit the growth of Gram-positive Staphylococcus aureus and Gram-negative Escherichia coli bacterial strains was moderate and only observed at a high concentration of 100 μM. Confocal microscopy imaging revealed that the “dirhenium carbonyl” dinucleosides and nucleosides localized mainly in the membranous structures of HeLa cells and uniformly inside S. aureus and E. coli bacterial cells. An interesting finding was that some of the tested compounds were also found in the nuclei of HeLa cells.
Introduction
Chemical modifications of nucleic acids and their building blocks such as nucleosides and nucleotides have grown as an important field of academic and industrial studies. Many modified nucleosides and nucleotides have been marketed as anticancer and antiviral drugs.1–3 Furthermore, the discovery of novel chemically modified nucleosides and nucleotides has been beneficial for the development of RNA/DNA-targeting technologies such as antisense and short interfering RNA as well as the increasingly popular CRISPR-Cas9 gene editing technique.4–8 Structural alterations of canonical nucleic acid components pertains to the modification of the nucleobase, the (deoxy)ribose and the phosphodiester backbone.1,2,9–11 In the case of the phosphodiester backbone, three general classes of modifications can be distinguished. The first class comprises the substitution of the nonbonding oxygen atom in the phosphodiester backbone. The second deals with the replacement of the bonding oxygen atom, while the third involves the complete replacement of the phosphodiester backbone with a nonphosphodiester linker. There have been many examples of the phosphodiester backbone modifications reported in the literature. Relevant examples include phosphorothioate,12,13 phosphoroamidate,14 boranophosphate,15,16 amide,5,6,8,17,18 formacetal,19 amine,20 guanidinium,21,22 and 1,2,3-triazole23–27 internucleotide linkers.Alternatively to these organic modifications, there is growing interest in nucleic acids modified with coordination or organometallic metal complexes.28–41 The most notable examples of such modifications resulted in metallo-DNA nanostructures and nanowires.29,33 Furthermore, metal-containing oligonucleotides and nucleosides were shown to exhibit useful redox,38,42 radioactive,43 and luminescence44 properties. Literature data indicate that at least 13 metals, namely Cr, Mn, W, Tc, Fe, Re, Co, Os, Ir, Rh, Hg, Pd, and Sn, have been used for the organometallic derivatization of nucleosides.30 Four classes of organometallic nucleosides have been postulated based on the position of the metal-containing group within the nucleoside/nucleotide scaffold.30 The most common class comprises compounds in which a metal-containing entity is attached to the nucleobase part of a given nucleoside or nucleotide. On the other hand, the group of canonical dinucleoside phosphates with organometallic modifications in the phosphodiester backbone is the least prevalent.45–48 Dinucleoside phosphates A and B (Fig. 1) are examples of this least abundant class of compounds.45,46
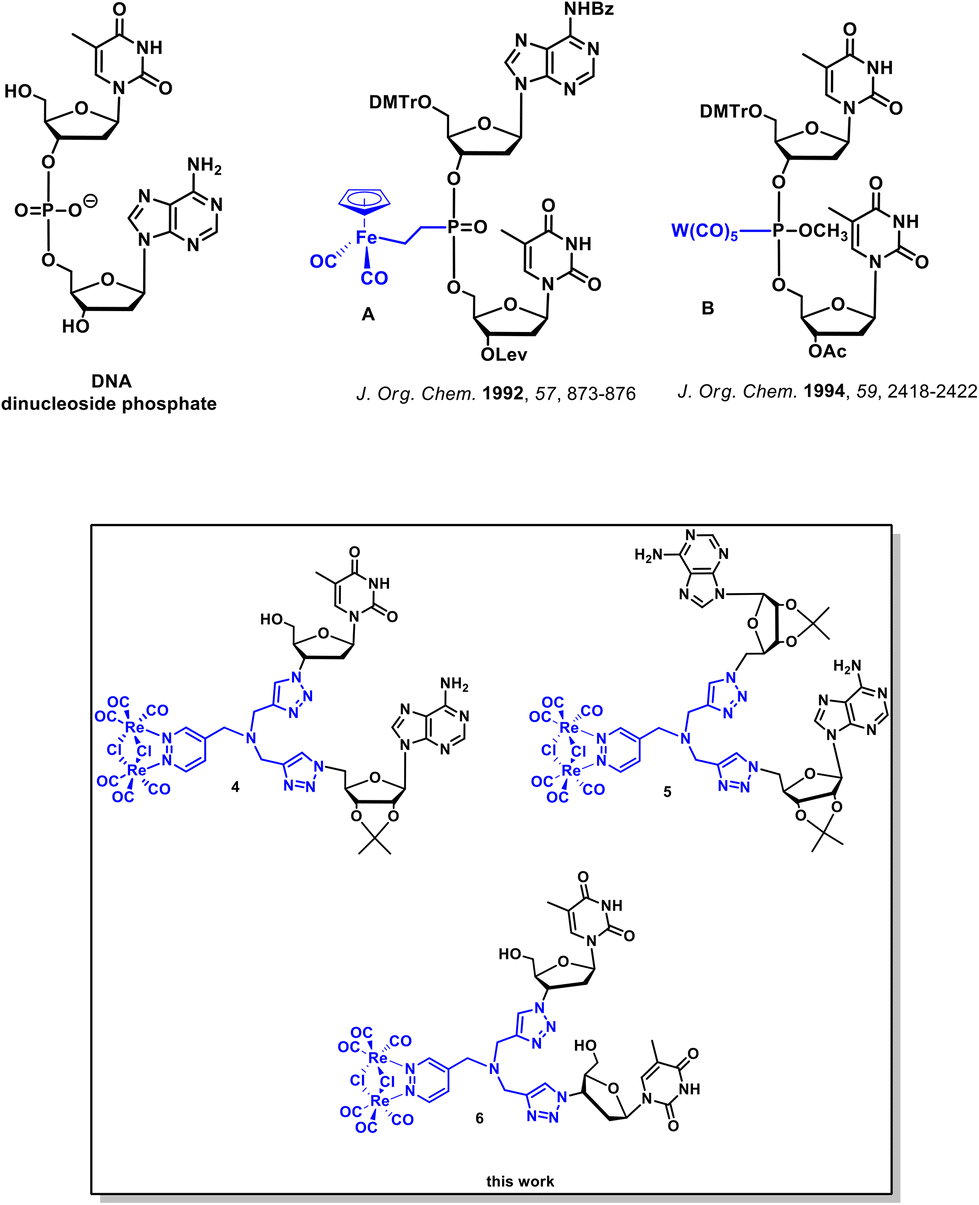 | ||
| Fig. 1 Structures of canonical DNA dinucleoside phosphate, metal–containing dinucleoside phosphates A and B and compounds 4, 5 and 6 studied in this work. Ac = acetyl; Bz = benzoyl; DMTr = 4,4′-dimethoxytrityl; and Lev = levulinyl. | ||
To the best of our knowledge, there are no literature reports on dinucleoside phosphates in which the entire phosphodiester backbone has been replaced by the luminescent organometallic linker. Therefore, we attempted to synthesize this new class of compounds as part of our research on the chemistry and biology of organometallic nucleic acid components.49,50
This paper describes the synthesis, photophysical properties, anticancer/antibacterial activity, and results of the imaging studies of the first-in-class dinucleosides 4, 5, and 6 (Fig. 1 and Scheme 1) bearing a luminescent dirhenium carbonyl [Re2(μ-Cl)2(CO)6(μ-pyridazine)] 1,2,3-triazolyl (“click”) linker in place of the canonical phosphate backbone. Furthermore, in this work, we report on luminescent dirhenium carbonyl “click” thymidine and adenosine conjugates 10 and 11 (Scheme 2). This work emphasizes on how the complex functional (luminescent) artificial “nucleosides” can be obtained by using simple cycloaddition reactions in a few synthetic steps. Furthermore we showed that the fate of these nucleosides could be followed in living cells by the luminescence confocal microscopy.
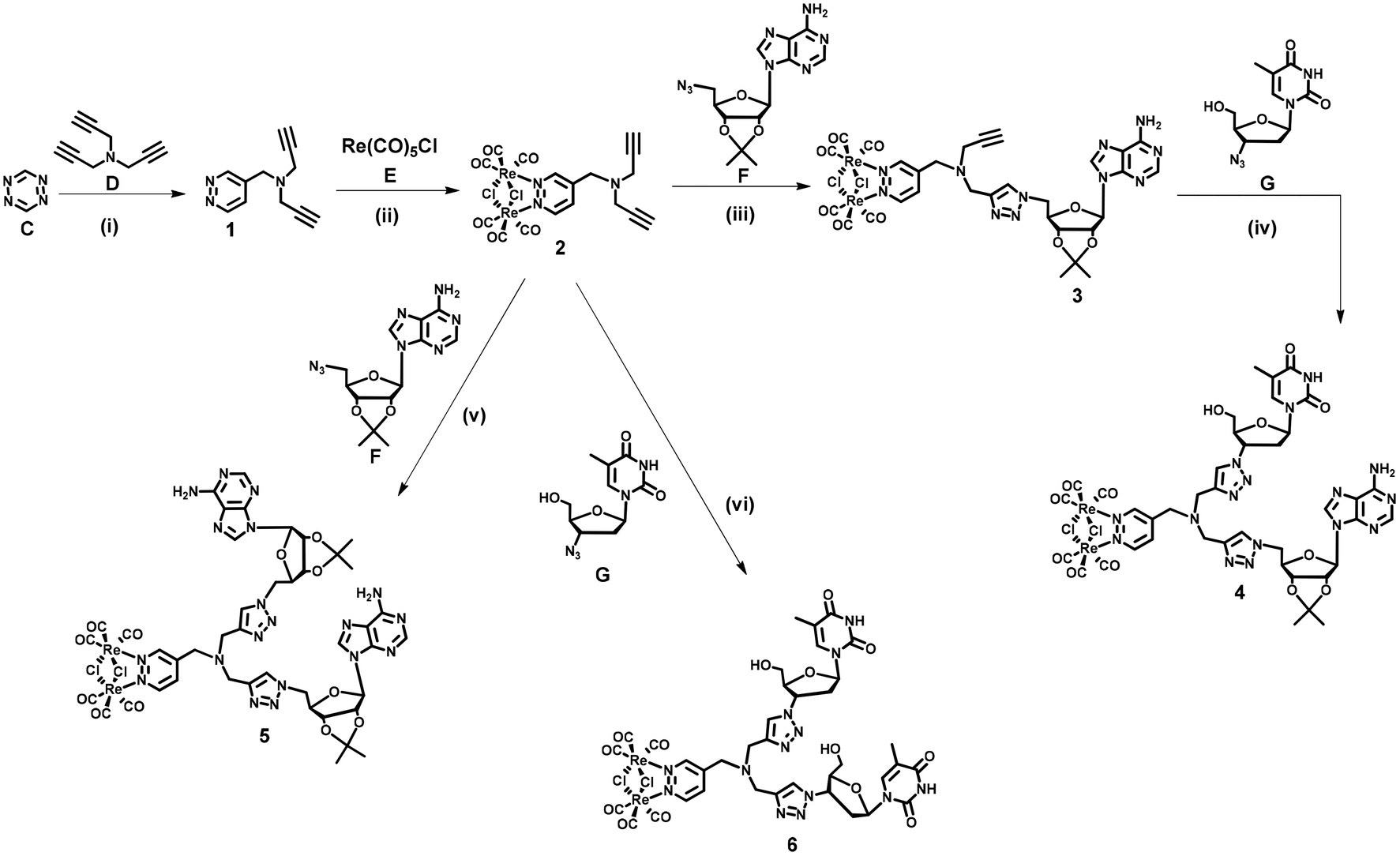 | ||
| Scheme 1 Synthetic pathway to compounds 4, 5, and 6. Reagents and conditions: (i) DCM, ambient temperature, and 18 h; (ii) toluene, reflux, and 30 min; (iii) CuSO4·5H2O, Na ascorbate, tert-BuOH, H2O, ambient temperature, and 2 h; (iv, v, vi) CuSO4·5H2O, Na ascorbate, tert-BuOH, H2O, ambient temperature, and 3 h. DCM = dichloromethane. | ||
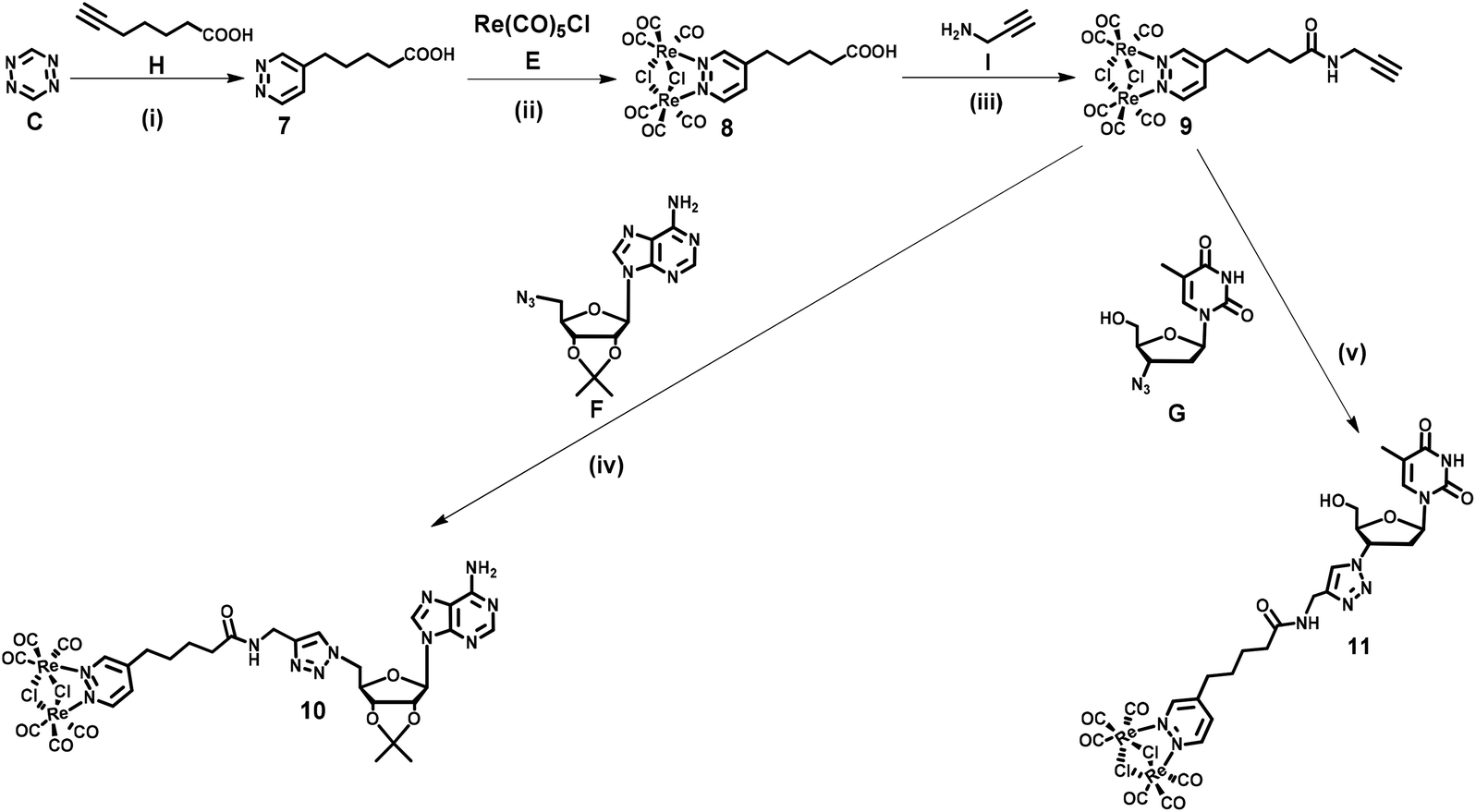 | ||
| Scheme 2 Synthetic pathway to compounds 10 and 11. Reagents and conditions: (i) DCM, ambient temperature, and 18 h; (ii) toluene, reflux, and 2 h; (iii) DCC, DMAP, DCM, ambient temperature, and 1.5 h; (iv, v) CuSO4·5H2O, Na ascorbate, tert-BuOH, H2O, ambient temperature, and 3 h. DCM = dichloromethane; DMAP = 4-(dimethyl)aminopyridine; and DCC = N,N′-dicyclohexylcarbodiimide. | ||
The 1,2,3-triazole scaffold was chosen as the primary component of the internucleotide linker because it can be easily synthesized via the copper(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC, “click”) reaction which is being widely utilized in nucleic acid chemistry.11,49,51 Re(I)-carbonyl, on the other hand, was chosen as the metal-containing motif due to its inherent photoluminescence properties.52,53 Luminescent Re(I) conjugates of biologically relevant compounds such as sugars, drugs and so forth have been used as imaging probes and phototoxic agents.54–58 Furthermore Re(I)-carbonyl complexes have been used as IR probes59 and as “cold” isostructural counterparts to the 99mTc radionuclide used in positron emission tomography.60 In this study, we used dinuclear [Re2(μ-Cl)2(CO)6(-μ-1,2-diazine)] rhenium derivatives. Dinuclear rhenium 1,2-diazine complexes have generally received significantly less attention than mononuclear Re(I)-carbonyl complexes.61–67 Like the majority of their mononuclear Re(I)-carbonyl counterparts, they exhibit strong phosphorescence in the range of ca. 550–620 nm. This emission is assigned to the lowest triplet state of the metal-to-ligand charge transfer (3MLCT) character resulting from a d(Re2(μ-Cl)2(CO)6) → π*(pyridazine) electronic transition.65,67 Luminescent [Re2(μ-Cl)2(CO)6(μ-1,2-pyridazine)] conjugates with estradiol,65 glycodendrons,63,64 and peptide nucleic acids (PNA)61,62 have been reported in the literature e.g., as probes for cell imaging which encouraged us to use them as confocal microscopy-detectable labels in the course of the studies reported here.
Results and discussion
Synthesis
Dinucleoside 4 was synthesized in four steps, while the synthesis of dinucleosides 5 and 6 required three synthetic steps each (Scheme 1). The key for all procedures 1,2,4,5-tetrazine (C) was obtained according to the literature.68 The first step of the synthetic sequence was an inverse-electron-demand Diels–Alder (IEDDA) [4 + 2]-cycloaddition reaction69,70 of 1,2,4,5-tetrazine (C) with tripropargylamine (D) which afforded ligand 1 in 44% yield. Refluxing of 1 with Re(CO)5Cl in a toluene solution resulted in the [Re2(μ-Cl)2(CO)6] complex 2 in 61% yield. In the subsequent step, complex 2 was reacted with 5′-azido-2′,3′-O-isopropylidene adenosine71 (F) under CuAAC reaction conditions to afford an intermediate 3 in 21% yield. In the last step, the CuAAC reaction of 3 with 3′-azido-3′-deoxythymidine (G) resulted in “click” dinucleoside 4 in 40% yield. Compounds 5 and 6 were synthesized in 38 and 42% yields via a CuAAC reaction of complex 2 and (F) or (G) in 38 and 42% yield, respectively.Nucleosides 11 and 10 were synthesized in four steps according to Scheme 2. In the first step, an IEDDA reaction of 1,2,4,5-tetrazine (C) with 6-heptynoic acid (H) afforded 5-(pyridazine-4-yl)pentanoic acid 7 in 31% yield. In the second step, the rhenium complex E was reacted with ligand 7 which afforded acid 8 in 87% yield. In the following step, acid 8 was transformed into the corresponding amide 9 (87% yield) by conjugation with propargylamine (I) under Steglich reaction conditions.72 The last step involved a CuAAC reaction of amide 9 with azidonucleoside (F) or (G) which afforded target conjugates 10 and 11 in 47 and 45% yield, respectively.
At this point it should be emphasised that the synthetic approach for 4–6, 10 and 11 compounds vividly shows how different types of cycloaddition reactions together with simple coordination and amide bond formation reactions might be utilized for the engineering of structurally complex and functional (in this case luminescent) artificial nucleic acid components.
With the exception of 3, all compounds were characterized by 1H NMR, 13C NMR and FTIR spectroscopy, high-resolution mass spectrometry, and elemental analysis. The resulting analytical data confirmed the proposed structures. The 1H-NMR spectra are shown in Fig. S1–S10 (ESI†). Also, the structures of compounds 2 and 8 in the solid state were determined by single-crystal X-ray structural analysis. All compounds were found to be stable in the solid state and could be stored as such for months without decomposition. On the other hand, synthetic intermediate 3 decomposes in organic solvents. We also noticed gradual decomposition of other complexes in DMSO solution when left overnight at room temperature. This did not, however, interfere with their structural assignment and spectroscopic characterization.
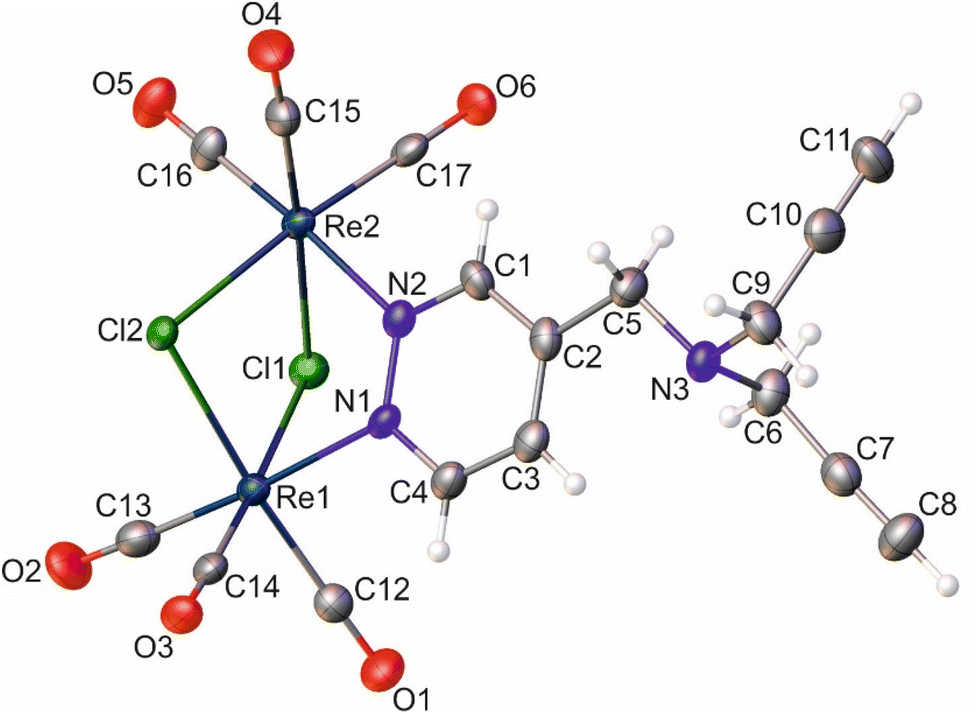 | ||
| Fig. 2 Molecular diagram of 2 with atomic displacement ellipsoids at the 50% probability level. The H-atoms are shown as small spheres of arbitrary radius. Selected bond lengths, distances [Å], and angles [°]: Re1⋯Re2, 3.5459(3); Re1–Cl1, 2.4830(12); Re1–Cl2, 2.4978(12); Re2–Cl1, 2.4925(13); Re2–Cl2, 2.5034(12); Re1–N1, 2.204(5); Re2–N2, 2.204(4); Re1–C12, 1.918(6); Re1–C13, 1.942(7); Re1–C14, 1.899(6); Re2–C15, 1.905(6); Re2–C16, 1.914(6); Re2–C17, 1.907(6); N1–N2, 1.354(6); C7–C8, 1.196(10); C10–C11, 1.180(10); Re1–N1–N2, 120.1(3); Re2–N2–N1, 119.5(3); Re1⋯Re2–C16, 115.88(19); Re2⋯Re1–C13, 115.04(18). | ||
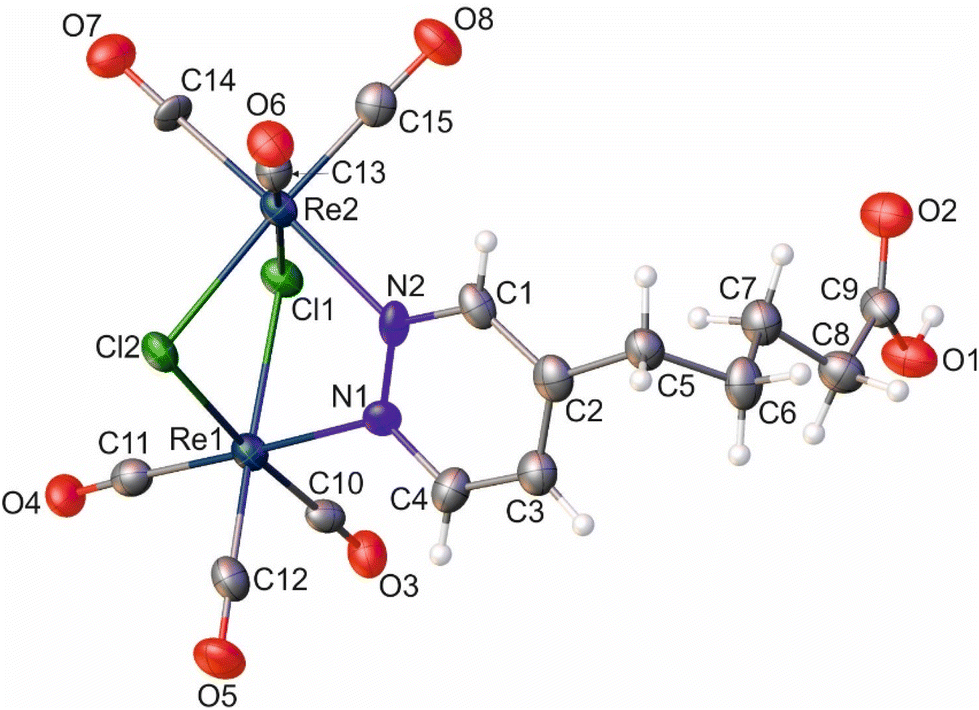 | ||
| Fig. 3 Molecular diagram of 8 with atomic displacement ellipsoids at the 50% probability level. The H-atoms are shown as small spheres of arbitrary radius. Selected bond lengths, distances [Å], and angles [°]: Re1⋯Re2, 3.5564(6); Re1–Cl1, 2.5033(19); Re1–Cl2, 2.503(2); Re2–Cl1, 2.4921(19); Re2–Cl2, 2.4883(19); Re1–N1, 2.204(7); Re2–N2, 2.182(7); Re1–C10, 1.905(9); Re1–C11, 1.935(9); Re1–C12, 1.892(10); Re2–C13, 1.889(10); Re2–C14, 1.922(7); Re2–C15, 1.897(9); N1–N2, 1.356(10); C9–O1, 1.306(11); C9–O2, 1.227(11); Re1–N1–N2, 119.4(5); Re2–N2–N1, 120.7(5); Re1⋯Re2–C14, 115.6(3); Re2⋯Re1–C11, 118.4(3). | ||
Similar to the literature data,67 the X-ray analysis revealed that the coordination sphere around the Re(I) atoms of the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] moiety in complexes 2 and 8 was distorted octahedral. The Re⋯Re distance in these two complexes was 3.5459(3) and 3.5564(6) Å, respectively. Within the coordination spheres of Re1 and Re2, the planes defined by equatorial C-atoms and bridging Cl atoms were inclined to themselves by 135.92(11) (2) and 134.90(16)° (8) (Fig. S11 [ESI†]). In both compounds, the pyridazine moiety was planar, with deviation from planarity of 0.008 (2) and 0.007 Å (8). In compound 2, the triple bond lengths were 1.180(10) and 1.196(10) Å for C10–C11 and C7–C8, respectively. In the crystal lattice of 8, the adjacent molecules of the complex formed dimers, where individual molecules were held together by O–H⋯O hydrogen bonds arranged in an R22(8) motif (Fig. S12 [ESI†]) (d(D⋯A) = 2.607(10) Å; <(D–H⋯A) = 172(5)°). In turn, in the crystal lattice of 2, there were only weak intermolecular hydrogen bonds found.
Photophysical characterization and DFT calculations
The absorption and emission properties of compounds 2, 4–6, and 8–11 were almost identical and dominated by the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] group (Table 1).| Compound | λ abs [nm] (ε [×103 M−1 cm−1]) | λ PL [nm] |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) * [nm] * [nm] |
λ
PL* [nm] |
Φ PL(±10%) | τ [μs] | τ* [μs] |
|---|---|---|---|---|---|---|---|
| * data in air-equilibrated DMSO at ambient temperature. | |||||||
| 2 | 370 (10.0) | 612 | 335 | 630 | 0.08 | 0.53 | 0.04 |
| 4 | 370 (8.0) | 600 | 340 | 614 | 0.06 | 0.49 | 0.31 |
| 5 | 365 (8.9) | 596 | 342 | 625 | 0.07 | 0.33 | 0.29 |
| 6 | 371 (6.3) | 605 | 347 | 617 | 0.08 | 0.53 | 0.30 |
| 8 | 363 (8.5) | 589 | 330 | 640 | 0.11 | 0.61 | 0.41 |
| 9 | 359 (5.9) | 590 | 332 | 650 | 0.11 | 0.60 | 0.41 |
| 10 | 360 (7.4) | 588 | 340 | 616 | 0.08 | 0.61 | 0.24 |
| 11 | 363 (6.9) | 589 | 335 | 620 | 0.12 | 0.60 | 0.40 |
The UV/Vis absorption spectra of the studied complexes were characterized by the presence of a broad unstructured band at ca. 370 nm owing to metal-to-ligand (dπ(Re)–π*(pyridiazine)) charge transfer states (3MLCT).61,65 At higher energies (ca. 260 nm) an intense band assigned to the pyridazine intraligand π–π* transitions was present. All compounds display luminescence with a broad emission band in a range of 588–612 nm in air-equilibrated DCM at ambient temperature (Table 1, and Fig. 4). In DMSO solution, a slight red shift of the emission was observed (Fig. 4 and S13†). This broad, unstructured emission was assigned to phosphorescence from the lowest 3MLCT.61,65,66 The long decay times typical of triplet emissions further substantiate this assignment.
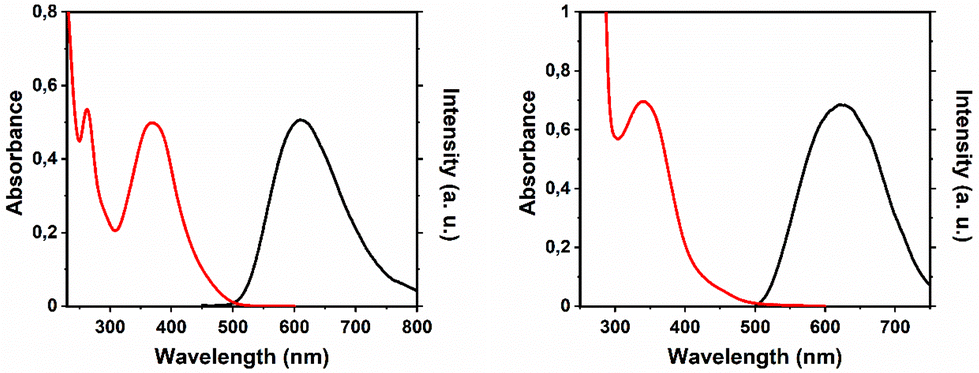 | ||
| Fig. 4 Electronic absorption (red line) and luminescence (black line) spectra of 2 in DCM (left) and 5 in DMSO (right). | ||
For a deeper understanding of the electronic structure of the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] emitting part common to all the examined compounds, DFT and TD-DFT computations were conducted on the model complex 2. According to the TD-DFT calculations, the four first excited states S1, S2, and T1, T2 of 2 involve the two highest occupied molecular orbitals (HOMO and HOMO−1) and one lowest unoccupied molecular orbital (LUMO). The orbital localization analysis showed that the HOMO, HOMO−1, and LUMO were localized on the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] moiety (Fig. 5). The HOMO and HOMO−1 had major contribution from metal centers, whereas the LUMO was mainly localized on pyridazine. Thus the calculated first excited states were characterized by dominant MLCT transitions.
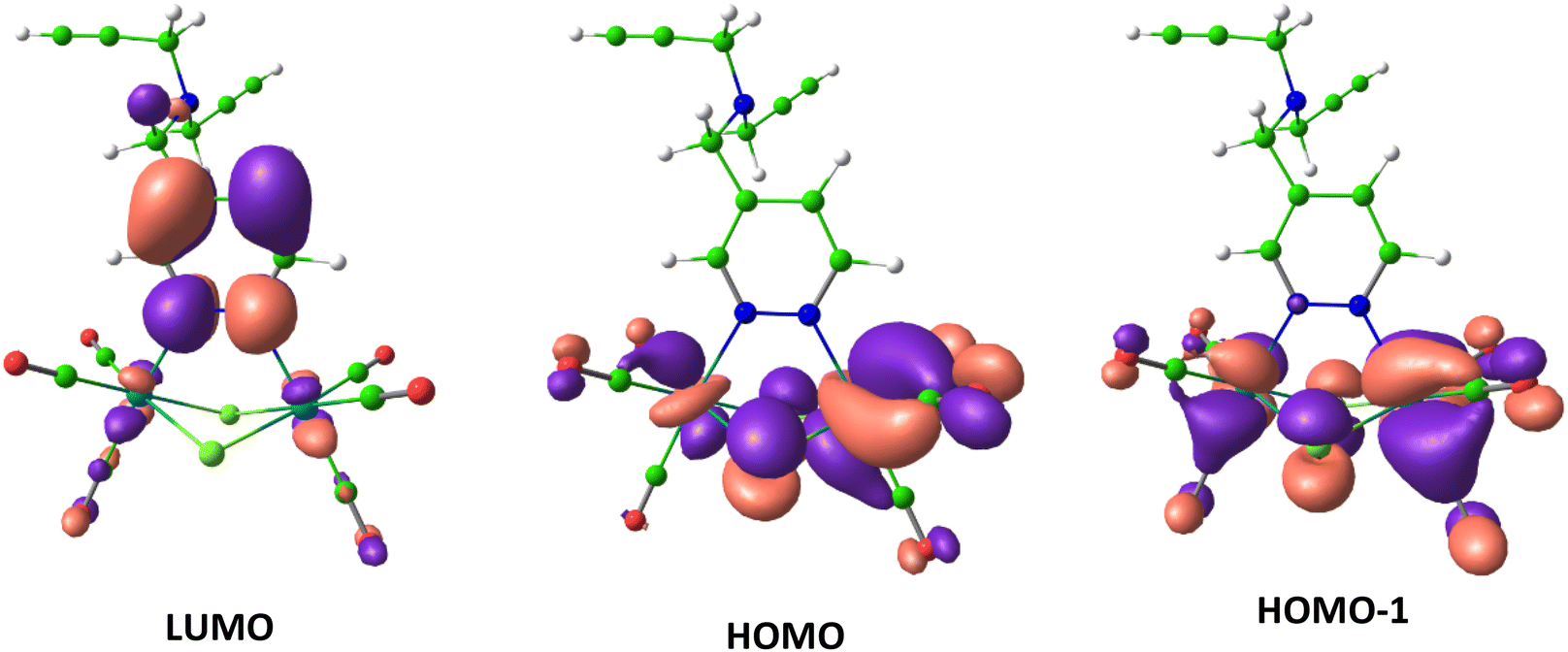 | ||
| Fig. 5 DFT-calculated iso-surface contour plots (iso-value = 0.035) of the lowest unoccupied molecular orbital (LUMO) and the highest and second highest occupied molecular orbitals (HOMO and HOMO−1) of the model compound 2. | ||
Imaging of cells with luminescent probes or compounds has several limitations. One of these is a cell's ability to show autofluorescence which occurs in the range of 350 to 550 nm and is related to the presence of aromatic cellular components (e.g., aromatic amino acids, NADPH, and riboflavin). Autofluorescence is a short-lived process typically characterized by emission decay times of several nanoseconds. The photophysical characteristics of compounds 2, 4–6, and 8–11, which include their ability to emit in the region lacking cell autofluorescence and exhibiting long emission decay times, support the use of these rhenium complexes in luminescence confocal microscopy imaging of live cells.
| IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 4 | 5 | 6 | 8 | 9 | 10 | 11 | CisPt | |
| a IC50 value was higher than the maximal compound concentration tested. | |||||||||
| L929 | 7.3 ± 0.3 | >100a | >100 | >100 | 76.2 ± 13 | 18.7 ± 0.9 | 56.8 ± 11.4 | >100 | 24.4 ± 2.0 |
| HeLa | 6.4 ± 0.6 | >100 | >100 | >100 | 48.0 ± 3.4 | 9.2 ± 1.0 | 42.7 ± 4.4 | >100 | 4.5 ± 0.3 |
| Ishikawa | 10.6 ± 0.7 | >100 | >100 | >100 | 57.3 ± 2.9 | 21.1 ± 1.6 | 42.6 ± 2.6 | >100 | 10.4 ± 1.6 |
In the MTT assay, dinucleosides 4, 5 and 6 did not cause any cytotoxicity in the tested cell lines (IC50 > 100 μM). However, their precursor, i.e., complex 2, showed the highest cytotoxicity against all the examined cell lines. The activity of complex 2 against HeLa and Ishikawa cells was comparable to that of cisplatin (CisPt). Unfortunately, complex 2 displayed higher toxicity than CisPt against nontumorigenic L929 cells which likely decreased its value from the standpoint of real anticancer applications. In the case of nucleosides 10 and 11, only the former compound displayed a moderate level of activity, with IC50 values of 42.7 ± 4.4 and 42.6 ± 2.6 μM against HeLa and Ishikawa cells, respectively. The activity of compound 10 was comparable to that of carboxylic acid 8. In contrast, amide 9, a propargyl amine derivative of 8, exhibited strong cytotoxicity toward HeLa and Ishikawa cancer cells as well as unfavorable activity against nontumorigenic L929 cells. The two most effective anticancer complexes 2 and 9 were intermediates in the synthesis of target nucleoside compounds. Also both these intermediates showed highly undesirable toxicity against nontumorigenic L929 cells. Driven by these reasons, more in-depth anticancer activity studies of 2 and 9 were not carried out. Because some anticancer nucleosides may show their activity after a longer treatment time,73 the MTT experiment was also carried out after 72 h of exposure; however, no changes in the IC50 values were noticed (data not shown). In summary, the MTT assay showed that nucleosides 4–6, 11 and 10 are appropriate for imaging in live HeLa cells with luminescence confocal microscopy due to their low cytotoxic activity. In the following stage of biological studies we used the broth microdilution technique to test the antibacterial activity of dinucleosides 4–6, 10, and 11 against Gram-positive Staphylococcus aureus ATCC 6538 and Gram-negative Escherichia coli NCTC 8196 bacterial strains. The 24 h treatment period and the same concentration range as for the MTT assay were employed. The minimal inhibitory concentration (MIC) values for the widely used antibiotics ciprofloxacin (CIP) and streptomycin (STR) were determined in order to compare the activities of the tested compounds. For CIP, the MIC values were 0.04 and 0.8 μM for E. coli and S. aureus, respectively, while in the case of STR the value was 1.4 μM for both bacterial strains. The antibacterial activity of rhenium compounds was so weak that under the applied conditions their MIC values could not be determined. However, moderate suppression of bacterial growth was seen at a high concentration of 100 μM (Fig. 6).
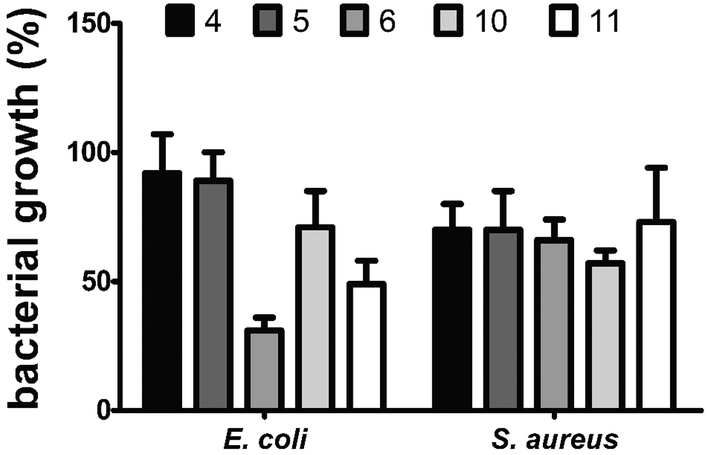 | ||
| Fig. 6 Growth inhibition of E. coli NCTC 8196 and S. aureus ATCC 6538 cells after 24 h treatment with compounds 4–6, 10, and 11 at a concentration of 100 μM. Percentage of bacterial growth was calculated in comparison with untreated control (100% of growth), and the results are presented as mean ± SEM from two independent experiments. | ||
Dinucleoside 6 (69% growth inhibition) and nucleoside 11 (51% growth inhibition) caused the highest growth inhibition of E. coli cells. The remaining compounds were significantly less effective in inhibiting the growth of E. coli. Notably, 6 and 11 are both derivatives of 3′-azido-3′-deoxythymidine, which may have an influence on their antibacterial activity. On the other hand, compounds 6 and 11 as well as other examined compounds caused less than 50% growth inhibition of S. aureus cells. This finding is interesting because it contradicts the stronger luminescence stemming from the interiors of S. aureus cells stained with the studied compounds in comparison with the E. coli cells (described in the Confocal luminescence microscopy section). It can be speculated that the higher antibacterial activity and lower luminescence intensity of compounds observed in E. coli cells were caused by the degradation of these compounds accompanied by the production of toxic degradation products. Studies on antibacterial activity revealed that the examined compounds 4–6, 10, and 11 are largely nontoxic to bacteria, which is a valuable finding to proceed with imaging by luminescence confocal microscopy.
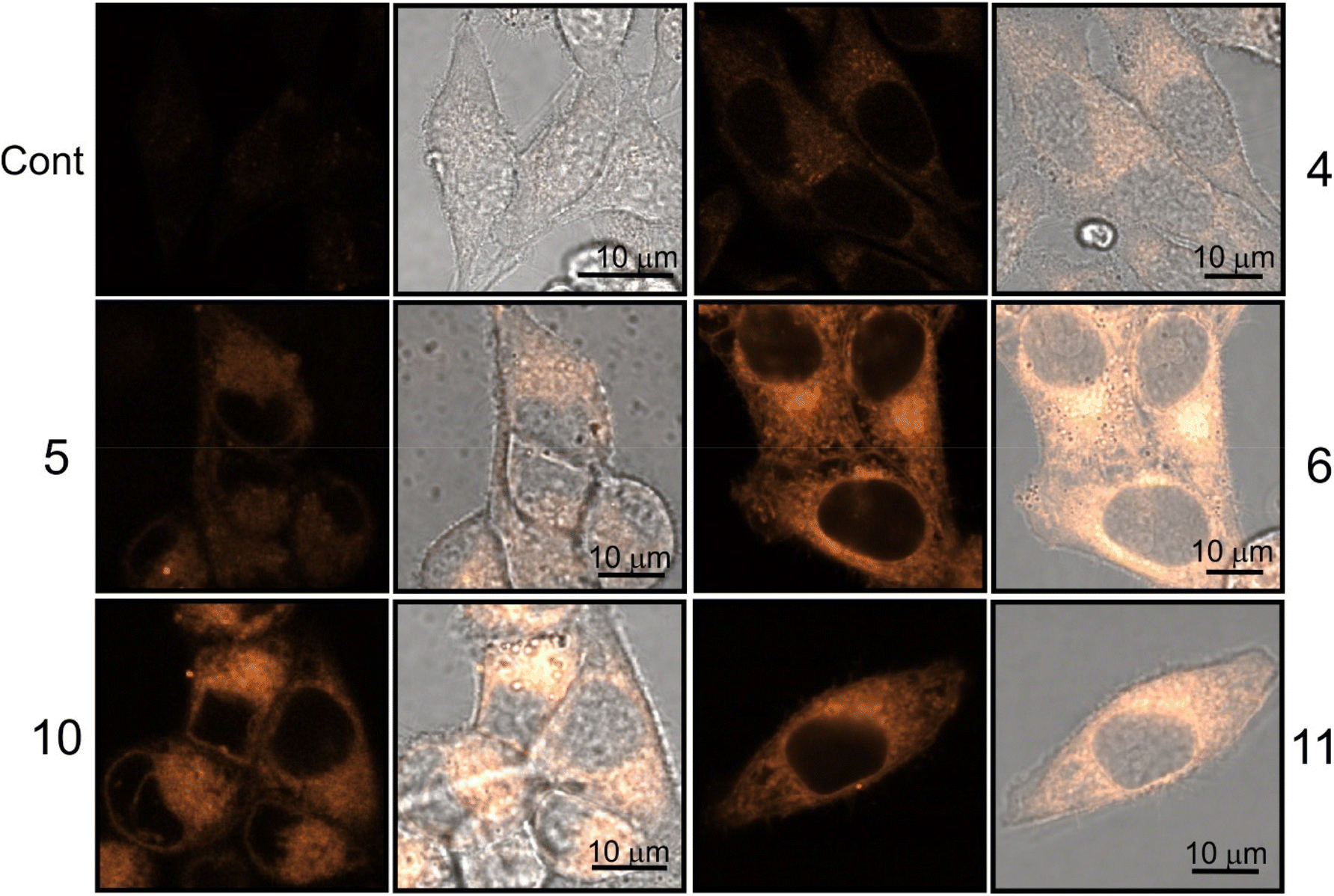 | ||
| Fig. 7 Confocal luminescence images of viable HeLa cells stained with dinucleosides 4–6 and nucleosides 10 and 11. The images were taken after 30 min treatment time. | ||
The staining pattern of HeLa cells was very similar for compounds 6, 10, and 11. The example photographs in Fig. 8 show details of dinucleoside 6 localization in the viable HeLa cell.
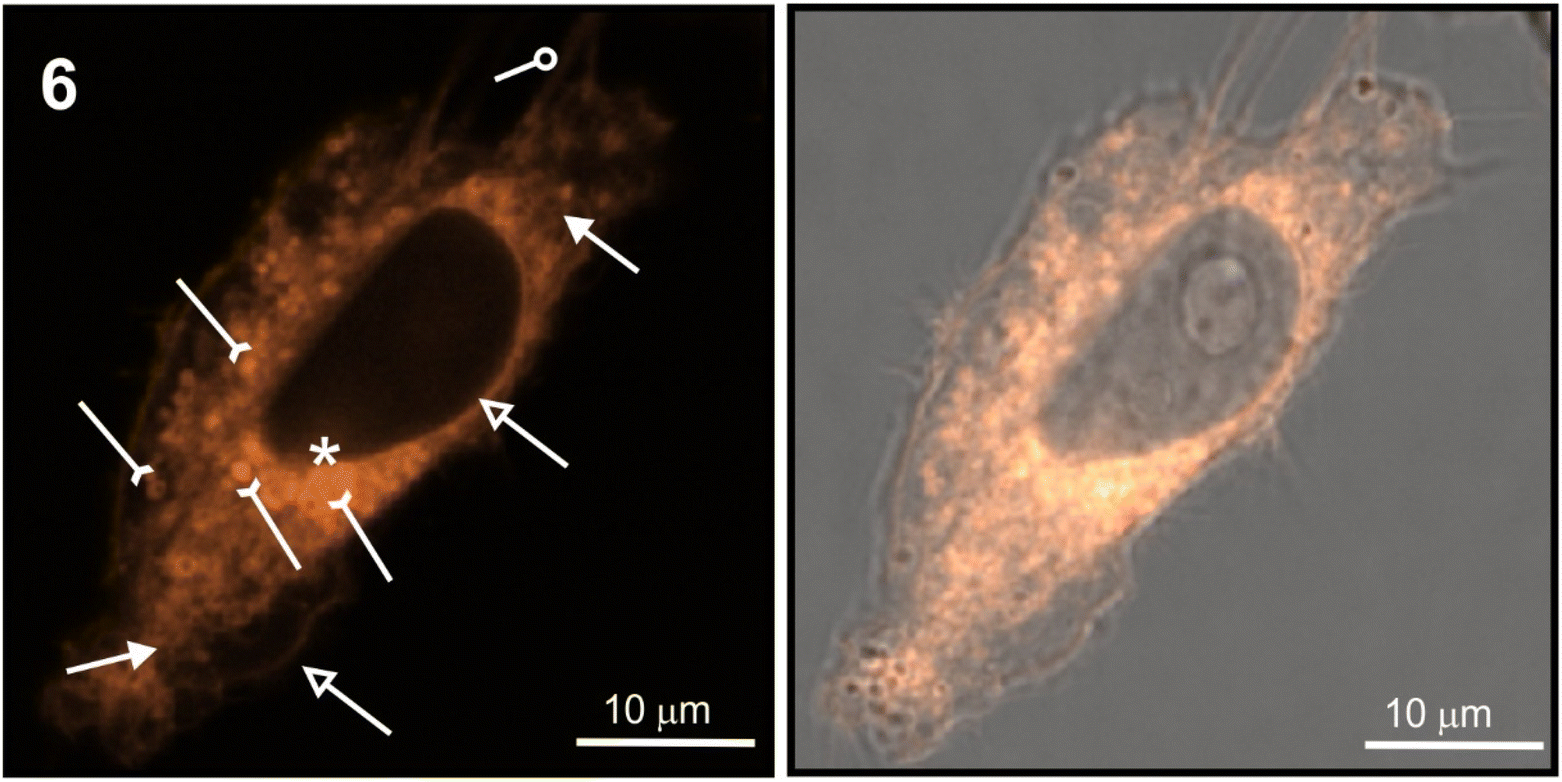 | ||
| Fig. 8 Cellular distribution of dinucleoside 6 in viable HeLa cells. The images were taken after 30 min treatment time. Left column—luminescence image, right column—merged images of luminescence and transmitted light. Lysosomes/vesicles are denoted by inverted arrows; plasmalemma and nuclear envelope are denoted by hollow arrows; filopodia are denoted by the loop; mitochondria are denoted by full-tip arrows. The compound in the nucleus is denoted by asterisk. | ||
Localization of compound 6 in the plasmalemma, nuclear envelope (hollow arrows in Fig. 8), and filopodia (loop in Fig. 8) was unambiguously confirmed. In addition, a granularly diffused pattern of emission was observed in the area of the cytoplasm which surrounds the nucleus. Within this space, bright circular structures were seen which are interpreted as lysosomes or vesicles (inverted arrows in Fig. 8). Furthermore, compound 6 was localized in the elongated structures scattered throughout the cell (arrows in Fig. 8) which were classified as mitochondria. Due to its DNA-protective role, it is difficult for many xenobiotics to cross the nuclear envelope. However, dinucleoside 6 crossed the nuclear envelope barrier and was localized in the nucleus (asterisk in Fig. 8). It is worth noting that the nuclear localization of compounds 10 and 11 was also unambiguously confirmed. In the case of dinucleosides 4 and 5, a more granular and less diffuse staining pattern of the cytoplasm was observed. Compounds 4 and 5 were not found in the nuclei of HeLa cells. Overall, the examined compounds showed a non-selective pattern of staining. They were mainly localized in lipidic membrane compartments and organelles of the cells. In comparison with luminescence imaging of human HeLa cells, the imaging of prokaryotic bacterial cells is a more challenging task. This is because it is difficult to penetrate the bacterial cell wall combined with efflux proteins which represent an evolutionary design to protect bacteria from external toxins. The low antibacterial activity of 4–6, 10, and 11 against E. coli NCTC 8196 and S. aureus ATCC 6538 strains prompted us to conduct luminescence confocal microscopy imaging in these bacteria (Fig. 9).
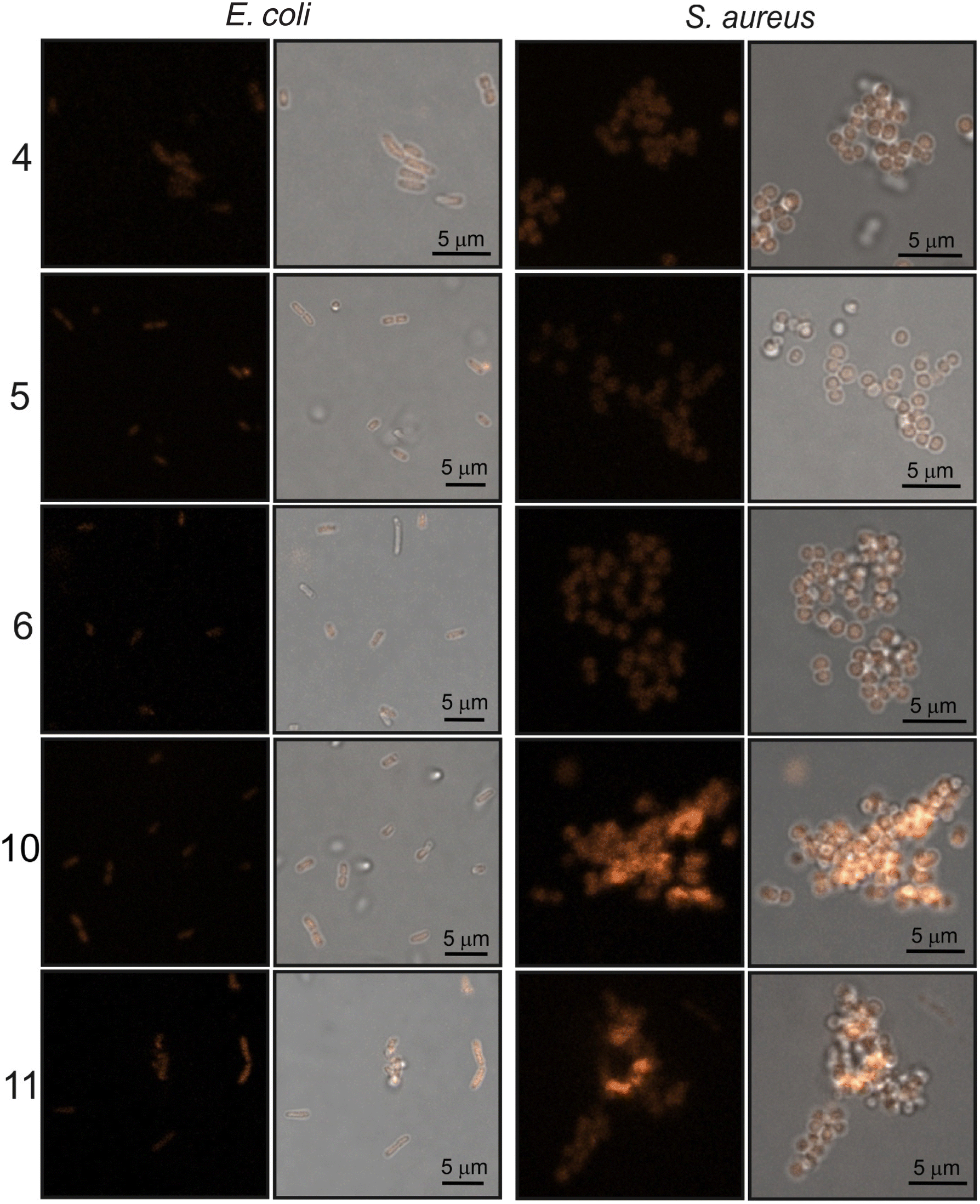 | ||
| Fig. 9 Confocal luminescence images of E. coli and S. aureus bacteria stained with dinucleosides 4–6 and nucleosides 10 and 11. The images were taken after 1 hour treatment time. Left column—luminescence images, right column—merged images of luminescence and transmitted light. | ||
After one hour of incubation, each of the tested compounds penetrated into E. coli and S. aureus bacterial cells (Fig. 9). However, it should be emphasized that less intense luminescence was observed for Gram-negative E. coli than for Gram-positive S. aureus cells. This feature can likely be explained by the weakened penetration of the assayed compounds through the E. coli bacterial envelope. Luminescence confocal microscopy enabled recognition of the characteristic stick-like morphology of E. coli cells as well as the sphere-shaped S. aureus cells. However, due to the small sizes of the bacteria and technical limitations further intracellular localization studies were not performed.
Conclusions
In this account, first-in-class luminescent organometallic analogs of canonical dinucleoside phosphates are reported. These contain the [Re2(μ-Cl)2(CO)6(μ-pyridazine)] “click” linker in place of the phosphate linker present in canonical dinucleosides. The paper also reports the synthesis of adenosine and thymidine derivatives with a dirhenium carbonyl “click” entity attached at positions 5′ and 3′. The dinucleosides were synthesized in three- (5, 6) or four-step (4) procedures by applying IEDDA and CuAAC “click” reactions as key synthetic approaches. Dirhenium nucleosides and their synthetic precursors displayed orange phosphorescence with ΦPL ≈ 0.10 and τ = 0.33–0.61 μs. With the exception of nucleoside 10, the synthesized nucleosides did not exhibit any toxic effect against human cancer HeLa and Ishikawa cells, as confirmed by biological activity assays. Additionally, the compounds were not toxic to S. aureus and E. coli bacterial cells. Favorable photophysical properties together with the low toxicity of 4–6, 10 and 11 enabled their confocal luminescence microscopy imaging in HeLa cells (as the eukaryotic model) as well as in S. aureus and E. coli (as the prokaryotic model) cells. Confocal imaging results showed that the studied compounds localized mainly in the membranous structures and compartments of HeLa cells. Additionally, compounds 6, 10, and 11 were detected in the nuclear region which indicates some interactions with nuclear components. The tested bacterial cells were more uniformly stained. In conclusion, a novel class of artificial nucleic acid components with luminescence properties were synthesized and biologically characterized. These compounds may serve as attractive building blocks for the synthesis of luminescent nucleic acid analogs for biological and other applications.Materials and methods
Reagents and general procedures
All preparations were carried out using standard Schlenk techniques. Chromatographic separations involved the use of silica gel 60 (Merck, 230–400 mesh ASTM). Toluene was distilled and purged with argon prior to use. Other solvents were of reagent grade and used without prior purification. Tripropargylamine, propargylamine, 4-(dimethyl)aminopyridine, N,N′-dicyclohexylcarbodiimide, Re(CO)5Cl, 3′-azido-3′-deoxythymidine, 6-heptynoic acid and Na ascorbate were purchased from a commercial supplier and used without further purification. 1H NMR (600 MHz) and 13C{H} NMR (150 MHz) spectra were recorded on a Bruker Avance III 600 spectrometer operating at 298 K in Fourier transform mode. Chemical shifts are reported in δ units (ppm) using the residual signal of DMSO (1H δ 2.50 ppm, 13C δ 39.5) or CD3OD (13C δ 49.00) as reference. All the mass spectra were recorded using a Synapt G2-Si mass spectrometer (Waters) equipped with an ESI source and quadrupole-TOF mass analyzer. The mass spectrometer was operated in negative ion detection mode. The measurements were performed with the capillary voltage set to 2.7 kV and sampling cone voltage to 20 V. The source temperature was 110 °C. To ensure the accuracy of mass measurements, data were collected in centroid mode and mass was corrected during acquisition using leucine enkephalin solution as an external reference (Lock-Spray). The results of the measurements were processed using MassLynx 4.1 software (Waters) incorporated with the instrument. IR spectra (KBr pellets) were measured on a FTIR Nexus Nicolet apparatus. Microanalyses were carried out by the Analytical Services of the Polish Academy of the Sciences (Łódź). HPLC separations were performed with a Shimadzu Prominence system equipped with a PDA detector, using an analytical Phenomenex Lux 5 μm Silica (2) 100 A (150 × 21.2 mm) column. Detection was accomplished at λ = 254 nm and a flow rate of 8 mL min−1 was applied. Pyridazine = pydz, adenine = ade, thymine = thym, triazole = triaz, and isoprop = isopropylidene.Synthesis
:3 v/v). Compound 1 was obtained as a pale yellow solid in 44% (500 mg) yield.
(1) 1H NMR (600 MHz, DMSO-d6): δ 9.17–9.16 (bs, 2H, H-pydz), 7.59 (q, JH,H = 4.8, 2.4 Hz, 1H, H-pydz), 3.71 (s, 2H, CH2N), 3.38 (d, JH,H = 2.4 Hz, 4H, CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 3.26 (t, JH,H = 2.4 Hz, 2H, CCH) ppm. 13C{1H} NMR (150 MHz, DMSO-d6): δ = 152.0, 151.3, 137.8, 126.1, 78.6, 76.3, 52.9, 41.6. MS (TOF ESI+): m/z = 186.1038 (M + H+) (calcd for C11H12N3: 186.1031). FTIR (KBr ν [cm−1]): 3190, 2823, 2112, 2105, 1591, 1437, 1119, 927, 670. Anal. calcd for C11H11N3: C, 71.33; H, 5.99; N, 22.69%. Found: C, 71.15; H, 5.82; N, 22.54%.
C), 3.26 (t, JH,H = 2.4 Hz, 2H, CCH) ppm. 13C{1H} NMR (150 MHz, DMSO-d6): δ = 152.0, 151.3, 137.8, 126.1, 78.6, 76.3, 52.9, 41.6. MS (TOF ESI+): m/z = 186.1038 (M + H+) (calcd for C11H12N3: 186.1031). FTIR (KBr ν [cm−1]): 3190, 2823, 2112, 2105, 1591, 1437, 1119, 927, 670. Anal. calcd for C11H11N3: C, 71.33; H, 5.99; N, 22.69%. Found: C, 71.15; H, 5.82; N, 22.54%.
1H NMR (600 MHz, DMSO-d6): δ = 10.06 (d, JH,H = 6.0 Hz, 1H, H-pydz), 10.04 (s, 1H, H-pydz), 8.20 (d, JH,H = 4.2 Hz, 1H, H-pydz), 4.00 (s, 2H, CH2N), 3.53 (s, 4H, CH2CC), 3.27 (s, 2H, CCH). 13C NMR (150 MHz, CD3OD): δ = 195.6, 195.5, 191.7, 191.6, 164.2, 163.3, 148.8, 133.2, 79.1, 75.5, 54.3, 43.6. MS (TOF ESI−): m/z = 795.9045 (M − H+) (calcd for C17H10N3O6Cl2Re2: 795.9062). FTIR (KBr ν [cm−1]): 3307, 3103, 2922, 2129, 2110, 2036, 1942, 1925, 1905, 1597, 1042. Anal. calcd for C17H11N3O6Cl2Re2: C, 25.63; H, 1.39; N, 5.27%. Found: C, 25.55; H, 1.64; N, 5.07%.
1H NMR (600 MHz, DMSO-d6): 1H NMR (600 MHz, DMSO-d6): δ = 11.32 (s, 2H, NH thym), 10.04 (d, JH,H = 6.0 Hz, 1H, H-pydz), 10.01 (bs, 1H, H-pydz), 8.28 (s, 1.5H, 1,2,3-triaz and ½H-pydz), 8.27 (bs, 0.5 H, ½H-pydz), 8.20 (s, 1H, 1,2,3-triaz), 8.19 (s, 1H, ade), 7.94 (s, 1H, ade), 7.81 (d, JH,H = 0.6 Hz, 1H, thym), 7.32 (s, 2H, NH2 ade), 6.42 (t, JH,H = 6.6 Hz, 1H, H1′ deoxyribose), 6.23 (d, JH,H = 1.8 Hz, 1H, H1′ ribose), 5.46 (dd, JH,H = 6.3, 1.8 Hz, 1H, H2′ ribose), 5.33 (dt, JH,H = 8.4, 5.4 Hz, 1H, H3′ deoxyribose), 5.27 (t, JH,H = 5.4 Hz, 1H, OH), 5.15 (dd, JH,H = 6.3, 3.6 Hz, 1H, H3′ ribose), 4.76 (dd, JH,H = 14.4, 5.4 Hz, 1H, H5′ ribose), 4.66 (dd, JH,H = 14.4, 7.8 Hz, 1H, H5′ ribose), 4.55 (m, 1H, H4′ ribose), 4.19 (app q, JH,H = 3.6 Hz, 1H, H4′ deoxyribose), 3.96 (s, 2H, CH2), 3.80 (s, 2H, CH2), 3.78 (s, 2H, CH2), 3.70 (m, 1H, H5′ deoxyribose), 3.62 (m, 1H, H5′ deoxyribose), 2.71 (m, 1H, H2′ deoxyribose), 2.64 (m, 1H, H2′ deoxyribose), 1.81 (d, JH,H = 0.6 Hz, 3H, CH3 thym), 1.51 (s, 3H, isoprop), 1.30 (s, 3H, isoprop).
13C NMR (150 MHz, DMSO-d6): δ = 191.8, 191.7, 166.3, 157.4, 154.1, 152.3, 150.1, 149.7, 145.0, 142.1, 138.2, 133.0, 130.9, 126.4, 125.0, 120.7, 115.9, 108.1, 91.4, 86.7, 86.6, 86.3, 85.2, 83.1, 62.2, 61.2, 54.9, 52.7, 50.0, 49.9, 49.5, 39.0, 27.4, 25.5, 12.4. MS (TOF ESI−): m/z = 1395.1367 (M − H+) (calcd for C40H39N16O13Cl2Re2: 1395.1375). FTIR (KBr ν [cm−1]): 3396, 2955, 2923, 2048, 2029, 1939, 1911, 1694, 1642, 1476, 1212, 855. Anal. calcd for C40H40N16O13Cl2Re2: C, 34.41; H, 2.89; N, 16.05%. Found: C, 34.58; H, 3.04; N, 15.87%.
1H NMR (600 MHz, DMSO-d6): δ = 10.02 (dd, JH,H = 5.7, 0.6 Hz, 1H, H-pydz), 9.98 (bd, JH,H = 1.2 Hz, 1H, H-pydz), 8.27 (s, 2H, 1,2,3-triaz), 8.24 (dd, JH,H = 5.7, 2.4 Hz, 1H, H-pydz), 8.18 (s, 2H, ade), 7.91 (s, 2H, ade), 7.32 (bs, 4H, NH2 ade), 6.22 (d, JH,H = 2.4 Hz, 1H, H1′ ribose), 5.45 (dd, JH,H = 6.3, 2.4 Hz, 2H, H2′ ribose), 5.14 (dd, JH,H = 6.3, 3.0 Hz, 2H, H3′ ribose), 4.74 (dd, JH,H = 14.0, 5.4 Hz, 2H, H5′ ribose), 4.65 (dd, JH,H = 14.0, 7.8 Hz, 2H, H5′ ribose), 4.53 (m, 2H, H4′ ribose), 3.89 (s, 2H, CH2), 3.73 (app d, JH,H = 1.8 Hz, 4H, CH2), 1.50 (s, 6H, isoprop), 1.29 (s, 6H, isoprop). 13C NMR (150 MHz, CD3OD): δ = 195.6, 195.4, 191.7, 157.3, 154.1, 150.1, 149.6, 144.9, 142.0, 133.0, 126.2, 120.7, 115.9, 91.4, 86.6, 85.2, 83.1, 54.8, 52.7, 49.8, 27.4, 25.5. MS (TOF ESI−): m/z = 1460.1731 (M − H+) (calcd for C43H42N19O12Cl2Re2: 1460.1752). FTIR (KBr ν [cm−1]): 3334, 2955, 2925, 2048, 2030, 1942, 1912, 1723, 1640, 1597, 1474, 1236, 870, 647. Anal. Calcd for C43H43N19O12Cl2Re2: C, 35.34; H, 2.97; N, 18.21%. Found: C, 35.34; H, 3.17; N, 18.00%.
1H NMR (600 MHz, DMSO-d6): δ = 11.36 (s, 2H, NH thym), 10.07 (d, JH,H = 6.0 Hz, 1H, H-pydz), 10.05 (bs, 1H, H-pydz), 8.32 (dd, JH,H = 6.0, 1.8 Hz, 1H, H-pydz), 8.26 (s, 2H, 1,2,3-triaz), 7.82 (d, JH,H = 0.6 Hz, 2H, thym), 6.43 (t, JH,H = 6.6 Hz, 2H, H1′ deoxyribose), 5.35 (dt, JH,H = 9.0, 4.8 Hz, 2H, H3′ deoxyribose), 5.30 (t, JH,H = 5.4 Hz, 2H, OH), 4.20 (app q, JH,H = 3.6 Hz, 2H, H4′ deoxyribose), 4.01 (s, 2H, CH2), 3.85 (s, 4H, CH2), 3.70 (m, 2H, H5′ deoxyribose), 3.62 (m, 2H, H5′ deoxyribose), 2.71 (m, 2H, H2′ deoxyribose), 2.63 (m, 2H, H2′ deoxyribose), 1.81 (s, 6H, CH3 thym). 13C NMR (150 MHz, CD3OD): δ = 195.5, 195.4, 191.9, 191.7, 166.3, 164.1, 163.1, 152.3, 149.9, 145.3, 138.1, 133.0, 130.6, 125.1, 111.7, 86.7, 86.4, 62.3, 61.3, 55.0, 50.1, 39.0, 12.4. MS (TOF ESI−): m/z = 1330.1001 (M − H+) (calcd for C37H36N13O14Cl2Re2: 1330.0997). FTIR (KBr ν [cm−1]): 3434, 3068, 2930, 2048, 2030, 1938, 1913, 1693, 1469, 1273, 1103, 1051, 771. Anal. calcd for C37H37N13O14Cl2Re2 and 1.333 n-hexane: C, 37.38; H, 3.88; N, 12.59%. Found: C, 37.54; H, 3.64; N, 11.77%.
1H NMR (600 MHz, DMSO-d6): δ = 12.04 (s, 1H, COOH), 9.12 (bs, 1H, H-pydz), 9.08 (dd, JH,H = 5.4, 1.2 Hz, 1H, H-pydz), 7.53 (dd, JH,H = 4.8, 2.4 Hz, 1H, H-pydz), 2.62 (t, JH,H = 7.8 Hz, 2H, CH2), 2.24 (t, JH,H = 7.8 Hz, 2H, CH2), 1.60 (qui, JH,H = 7.8 Hz, 2H, CH2), 1.49 (qui, JH,H = 7.8 Hz, 2H, CH2). 13C{1H} NMR (150 MHz, DMSO-d6): δ = 174.2, 152.7, 151.0, 141.4, 126.1, 33.2, 31.1, 28.5, 23.8. MS (TOF ESI+): m/z = 181.0973 (M + H+) (calcd for C9H13N2O2: 181.0977). FTIR (KBr ν [cm−1]): 3073, 2930, 2503, 1934, 1708, 1590, 1032, 673, 492. Anal. calcd for C9H12N2O2: C, 59.99; H, 6.71; N, 15.55%. Found: C, 59.98; H, 6.45; N, 15.46%.
1H NMR (600 MHz, DMSO-d6): δ 12.05 (s, 1H, COOH), 9.12 (s, 1H, H-pydz), 9.08 (d, JH,H = 4.8 Hz, 1H, H-pydz), 7.53 (dd, JH,H = 4.8, 1.8 Hz, 1H, H-pydz), 2.62 (t, JH,H = 7.2 Hz, 2H, CH2), 2.24 (t, JH,H = 7.2 Hz, 2H, CH2), 1.61 (qui, JH,H = 7.2 Hz, 2H, CH2), 1.49 (qui, JH,H = 7.2 Hz, 2H, CH2). 13C NMR (150 MHz, CD3OD): δ = 195.7, 195.6, 191.8, 191.7, 177.0, 164.9, 163.0, 151.7, 133.4, 34.2, 33.0, 297, 25.4. MS (TOF ESI−): m/z = 790.8998 (M − H+) (calcd for C15H11N2O8Cl2Re2: 790.9007) FTIR (KBr ν [cm−1]): 2049 (CO), 2028 (CO), 1930 (CO), 1900 (CO), 1701 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Anal. calcd for C15H12N2O8Cl2Re2: C, 22.76; H, 1.53; N, 3.54%. Found: C, 22.65; H, 1.68; N, 3.69%.
O). Anal. calcd for C15H12N2O8Cl2Re2: C, 22.76; H, 1.53; N, 3.54%. Found: C, 22.65; H, 1.68; N, 3.69%.
1H NMR (600 MHz, DMSO-d6): δ = 10.16 (d, JH,H = 1.2 Hz, 1H, H-pydz), 10.05 (dd, JH,H = 6.0, 1.2 Hz, 1H, H-pydz), 8.27 (t, JH,H = 5.4 Hz, 1H, NH), 8.19 (q, JH,H = 6.0, 1.8 Hz, 1H, H-pydz), 3.84 (dd, JH,H = 5.4, 2.4 Hz, 2H, CH2CC), 3.08 (t, JH,H = 2.4 Hz, 1H, CCH), 2.91 (t, JH,H = 7.2 Hz, 2H, CH2), 2.15 (t, JH,H = 7.2 Hz, 2H, CH2), 1.66 (qui, JH,H = 7.2 Hz, 2H, CH2), 1.59 (qui, JH,H = 7.2 Hz, 2H, CH2). 13C NMR (150 MHz, DMSO-d6): δ = 195.6, 195.5, 191.8, 191.7, 175.1, 164.9, 163.0, 151.6, 133.4, 80.6, 72.0, 36.0, 34.7, 32.9, 29.8, 26.0. MS (TOF ESI−): m/z = 827.9303 (M − H+) (calcd for C18H14N3O7Cl2Re2: 827.9324). FTIR (KBr ν [cm−1]): 3301, 3069, 2933, 2121, 2048, 2028, 1904, 1659, 1523, 1456. Anal. calcd for C18H15N3O7Cl2Re2: C, 26.09; H, 1.82; N, 5.07%. Found: C, 26.01; H, 2.08; N, 4.98%.
1H NMR (600 MHz, DMSO-d6): δ = 10.16 (d, JH,H = 1.8 Hz, 1H, H-pydz), 10.04 (dd, JH,H = 6.0, 0.6 Hz, 1H, H-pydz), 8.28 (s, 1H, 1,2,3-triaz), 8.27 (t, JH,H = 5.4 Hz, 1H, CONH), 8.19 (d, JH,H = 2.4 Hz, 1H, H-pydz), 8.18 (s, 1H, ade), 7.77 (s, 1H, ade), 7.38 (bs, 2H, NH2 ade), 6.22 (d, JH,H = 2.4 Hz, 1H, H1′ ribose), 5.45 (dd, JH,H = 6.0, 2.4 Hz, 1H, H2′ ribose), 5.13 (dd, JH,H = 6.0, 3.0 Hz, 1H, H3′ ribose), 4.73 (dd, JH,H = 14.1, 5.4 Hz, 1H, H5′ ribose), 4.65 (dd, JH,H = 14.1, 7.8 Hz, 1H, H5′ ribose), 4.53 (m, 1H, H4′ ribose), 4.23 (d, JH,H = 5.4 Hz, 2H, NHCH2), 2.89 (t, JH,H = 7.2 Hz, 2H, CH2), 2.14 (t, JH,H = 7.2 Hz, 2H, CH2), 1.65 (qui, JH,H = 6.6 Hz, 2H, CH2), 1.58 (qui, JH,H = 7.2 Hz, 2H, CH2), 1.52 (s, 3H, isoprop), 1.31 (s, 3H, isoprop). 13C NMR (150 MHz, CD3OD): δ = 195.6, 195.5, 191.7, 164.9, 163.1, 151.6, 133.4, 115.9, 91.5, 86.4, 85.2, 83.1, 54.7, 52.7, 49.5, 36.1, 35.5, 32.9, 32.7, 29.7, 27.4, 26.0, 25.5, 23.6, 14.3. MS (TOF ESI−): m/z = 1160.0671 (M − H+) (calcd for C31H30N11O10Cl2Re2: 1160.0669). FTIR (KBr ν [cm−1]): 3326, 2954, 2924, 2048, 2029, 1909, 1640, 1597, 1457, 1376, 1212, 1081, 854, 479. Anal. calcd for C31H31N11O10Cl2Re2: C, 32.07; H, 2.69; N, 13.27%. Found: C, 32.21; H, 2.90; N, 13.32%.
1H NMR (600 MHz, DMSO-d6): δ = 11.35 (s, 1H, NH thym), 10.16 (d, JH,H = 1.2 Hz, 1H, H-pydz), 10.04 (dd, JH,H = 5.4, 1.2 Hz, 1H, H-pydz), 8.34 (t, JH,H = 6.0 Hz, 1H, CONH), 8.20 (dd, JH,H = 6.0, 1.8 Hz, 1H, H-pydz), 8.10 (s, 1H, 1,2,3-triaz), 7.80 (d, JH,H = 1.2 Hz, 1H, thym), 6.40 (t, JH,H = 6.6 Hz, 1H, H1′ deoxyribose), 5.33 (dt, JH,H = 8.4, 5.4 Hz, 1H, H3′ deoxyribose), 5.27 (t, JH,H = 5.4 Hz, 1H, OH), 4.30 (d, JH,H = 6.0 Hz, 2H, NHCH2), 4.18 (app q, JH,H = 3.6 Hz, 1H, H4′ deoxyribose), 3.68 (m, 1H, H5′ deoxyribose), 3.59 (m, 1H, H5′ deoxyribose), 2.91 (t, JH,H = 7.2 Hz, 2H, CH2), 2.68 (m, 1H, H2′ deoxyribose), 2.63 (m, 1H, H2′ deoxyribose), 2.18 (t, JH,H = 7.2 Hz, 2H, CH2), 1.80 (d, JH,H = 0.6 Hz, 3H, CH3 thym), 1.68 (qui, JH,H = 7.2 Hz, 2H, CH2), 1.62 (qui, JH,H = 7.2 Hz, 2H, CH2) ppm. 13C NMR (150 MHz, CD3OD): δ = 191.7, 175.5, 163.1, 152.3, 151.6, 146.5, 138.1, 133.4, 123.8, 111.7, 86.7, 86.3, 62.2, 61.1, 39.0, 36.1, 35.5, 32.9, 32.7, 29.7, 26.0, 12.4 ppm. MS (TOF ESI−): m/z = 1095.0292 (M − H+) (calcd for C28H27N8O11Cl2Re2: 1095.0291). FTIR (KBr ν [cm−1]): 3331, 3067, 2956, 2927, 2047, 2028, 1909, 1688, 1529, 1458, 1272, 1101, 774, 520. Anal. calcd for C28H28N8O11Cl2Re2: C, 30.69; H, 2.58; N, 10.23%. Found: C, 30.82; H, 2.86; N, 10.17%.
X-ray structure determination
Good quality single-crystals of 2 and 8 were selected for the X-ray diffraction experiments at T = 100(2) K. Diffraction data were collected on an Agilent Technologies SuperNova Dual Source diffractometer with CuKα radiation (λ = 1.54184 Å) using CrysAlis Pro software.74 The analytical numerical absorption correction using a multifaceted crystal model based on expressions derived by R.C. Clark & J.S. Reid (2) and numerical absorption correction based on Gaussian integration over a multifaceted crystal model (8) were applied.75 The structural determination procedure was carried out using the SHELX package.76 The structures were solved with the intrinsic phasing method and then successive least-square refinement was carried out based on the full-matrix least-squares method on F2 using the SHELXL program.76 The hydroxyl H-atom in the case of 8 was located on the Fourier difference electron density map and refined with the O–H bond length restrained to 0.82 Å, with Uiso(H) = 1.5Ueq(O). The remaining H-atoms were positioned geometrically with the C–H bond length equal to 0.93 and 0.97 Å for the aromatic and methylene H-atoms, respectively, and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(C). The figures for this publication were prepared using the Olex2 program.77 CCDC 2224012 (2) and 2224011 (8) contain the supplementary crystallographic data for this paper.†Photophysical characterization and DFT calculations
Electronic absorption spectra were measured with a Shimadzu UV 2700 spectrophotometer. Stationary emission was obtained using a Fluorolog-3 spectrofluorometer (model FL3-22, Horiba Jobin Yvon). The spectra were corrected for the spectral sensitivity of the instrument. Photoluminescence curves, recorded as a function of wavelength (λ), were subsequently multiplied by λ2 in order to convert counts per wavelength into counts per wavenumber. The quantum yield of luminescence, Φ, was determined by a reference method, using 0.05 mol dm−3 quinine sulfate in sulfuric acid (Φf = 0.51) as the standard.78 Photoluminescence decay lifetimes in a nanosecond range were determined by time-resolved single photon counting (SPC) on an Edinburgh FL 900 CDT spectrofluorometer (excitation by an IBH Nanoled emitting at 370 nm, pulse width <1.5 ns was used). Photoluminescence decays were analyzed using FAST Advanced Analysis of Fluorescence Kinetics software (Edinburgh Instruments, version 3). The DFT calculations were performed with Gaussian 16 (C.01)79 quantum-chemical software packages at the M11L80,81/def2-SVP82 theory level. The C-PCM83 polarizable continuum model was applied with the solvent parameters of dichloromethane. Ground-state (S0) and first excited triplet state (T1) geometries were found to possess no imaginary frequencies. The reported computational results refer to the optimized T1 state geometry, as it is most relevant in terms of the discussed photophysics.Cell viability studies
The effect of compounds 2, 4, 5, 6, 8, 9, 10, 11 and cisplatin (CisPt) on the viability of mammalian cells was determined using the MTT reduction assay. The MTT assay enables one to assess the number of metabolically active (viable) cells in a given cell population. The MTT assay protocol is based on the conversion of water soluble MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to an insoluble formazan product by NAD(P)H-dependent oxidoreductase enzymes, present in viable cells. Human cervical cancer HeLa cells (ATTC®—CCL-2™), endometrial adenocarcinoma Ishikawa cells (ECACC 99040201) and murine fibroblast L929 cells (ATTC®—CCL-1) were plated in 96-well microplates with a density of 1 × 104 cells per well, according to the international standard ISO 10993–5:2009(E) (American National Standard 2009), in Dulbecco's modified Eagle's medium F12 (DMEM-F12, Biowest), supplemented with 10% fetal bovine serum (FBS, Biowest) and 100 units per ml of penicillin G with 100 μg ml−1 of streptomycin (Biowest). After overnight incubation of the cells, at 37 °C, under a humidified atmosphere containing 5% CO2, the culture medium was removed and replaced with a freshly prepared solution of the compounds in culture medium or medium alone (control). Stock solutions (10 mM) of each compound were prepared in DMSO and diluted with growth medium, supplemented with 10% FBS, to obtain concentrations ranging from 1.56 to 100 μM. Cisplatin (Sigma-Aldrich P4394) was used as the control anticancer drug. The final content of DMSO in the solutions did not exceed 1% and an additional control group with 1% DMSO was included to rule out solvent effects. Cells were incubated with the compounds at 37 °C in 5% CO2, for 24 h. After incubation, the medium was removed and MTT was added at a concentration of 50 μg per well. The plates were incubated for the next 2 h at 37 °C in 5% CO2. The formazan crystals were then solubilized in 150 μl of DMSO and quantified by spectrophotometric measurements at 570 nm using the SpectraMax i3 Multi-Mode Platform (Molecular Devices). Each concentration was tested in triplicate with two independent experiments and the results were calculated as a percentage of viable cells of the control group. The IC50 values (compound concentration that inhibits cell viability by 50%) were determined using a nonlinear regression, from the plot of % viability against the log dose of the compounds, using GraphPad Prism 5.01 software.Antibacterial activity
The antibacterial activity of dinucleosides 4, 5, and 6 and nucleosides 10 and 11 was evaluated using E. coli NCTC 8196 and S. aureus ATCC 6538 strains. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of the compound preventing the visible growth of the microorganism using the microdilution method according to EUCAST guidelines [ISO 20776-1 (2006)]. Each compound was pipetted into wells of 96-well microplates as a series of two-fold dilutions in Mueller–Hinton broth in the concentration range from 1.56 to 100 μM. All compounds were dissolved in DMSO. The final concentration of DMSO in the medium was 1% which had no influence on the growth of the microorganisms. Ciprofloxacin hydrochloride and streptomycin sulfate salt were purchased from a commercial supplier and were used as reference antibacterial agents. Then bacteria were added with an inoculum of approximately 5 × 105 CFU mL−1. The plates were incubated at 37 °C for 24 h and the optical density at 600 nm was measured using the SpectraMax i3 Multi-Mode Platform (Molecular Devices). Due to the weak activity of the tested compounds, MIC determination was impossible and the percentage of bacterial growth was calculated for each concentration, and the untreated control was taken as 100% of bacterial growth.Confocal fluorescence microscopy
For confocal microscopy analysis E. coli NCTC 8196 and S. aureus ATCC 6538 strains were first grown on an L-agar plate to obtain single colonies. The day before microscopic observation, single colonies of each strain were transferred to Mueller–Hinton broth and were cultured for 24 h at 37 °C using an automated shaking incubator (140 rpm). After incubation, the tested dinucleosides (4, 5, and 6) and nucleosides (10, 11) were added to bacteria at a concentration of 10 μM and treated for 1 hour. Then 10 μl of each sample was put on microscopic slides and immediately imaged using a confocal fluorescence microscope. In order to compare the uptake of the tested compounds by Gram-negative and Gram-positive microorganisms, the excitation and fluorescence detection parameters were initially unified, but due to the significant differences in fluorescence intensity between species observed during the experiment, the UV diode laser (405 nm) power for E. coli was set at 5% and for S. aureus at 2% of the nominal potency. The detection parameters remained the same for both species. The emission was collected by the PMT detector in the range of 500–700 nm (gain: 750, offset: 0, scan acquisition speed: 600 Hz, and line average – 6×).HeLa cells (ATCC Cat. no CCL-2™, human epithelial cells) were cultivated in Dulbecco's modified Eagle's medium F12 (DMEM-F12, Biowest), free of fluorescent markers supplemented with 10% fetal bovine serum (FBS, Biowest) at 37 °C under a humidified atmosphere with 5% CO2. Twenty-four hours before the experiment, HeLa cells at a density of 1 × 105 mL−1 were plated in a chamber culture slide with a glass bottom and cultured under the same conditions. After that, the cells were incubated for 30 min or 2 h with a freshly prepared 10 μM solution of tested dinucleosides (4, 5, and 6), nucleosides (10 and 11) and their precursors (2 and 9), then rinsed with phosphate buffered saline (PBS) (3×) and immediately imaged under a fluorescence confocal microscope. Similarly, as for prokaryotic cells in the experiment on live HeLa cells to compare the fluorescence intensities of all tested nucleosides and dinucleosides and their precursors as well as the control, the excitation and fluorescence detection parameters were unified. The compounds were excited with a UV diode laser of 405 nm wavelength and 5% power. The emission was collected by the PMT detector in the range of 500–700 nm (gain: 750, offset: 0, scan acquisition speed: 400 Hz, and line average – 3×). In the experiment demonstrating the intracellular localization of the compounds, the analyses were performed after 30 min and 2 hours of incubation with tested compounds. In order to best visualize the cells, the laser power (2–5%) and detection parameters were adjusted to the signal intensity.
All imaging was performed using a DMI 600 SC inverted microscope (HC PL APO CS2 63×/1.40 oil immersion objective or 100×/1.40 oil immersion objective) and a scanning confocal system TCS SP8 (Leica Microsystem, Wetzlar, Germany) operated using LAS 2.0.2.15022 software.
Statistical analysis
All data in the text and figures are provided as mean ± SEM from two independent experiments. The results were analyzed using GraphPad Prism version 5.01, 2007. P < 0.05 was considered as the level of statistical significance.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
K. K. thanks the National Science Center in Cracow, Poland (grant OPUS UMO-2018/29/B/ST5/00055) for financial support. The X-Ray diffractometric measurements were performed at the Core Facility for Crystallographic and Biophysical Research to support the development of medicinal products sponsored by the Foundation for Polish Science (FNP).References
- J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, and base analogs, Chem. Rev., 2016, 116, 14379–14455 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Synthesis of nucleoside phosphate and phosphonate prodrugs, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- E. De Clercq and G. Li, Approved antiviral drugs over the past 50 years, Clin. Microbiol. Rev., 2019, 29, 695–747 CrossRef PubMed.
- W. B. Wan and P. P. Seth, The medicinal chemistry of therapeutic oligonucleotides, J. Med. Chem., 2016, 59, 9645–9667 CrossRef CAS PubMed.
- V. Kotikam and E. Rozners, Amide-Modified RNA: using protein backbone to modulate function of short interfering RNAs, Acc. Chem. Res., 2020, 53, 1782–1790 CrossRef CAS PubMed.
- T. Hardcastle, I. Novosjolova, V. Kotikam, S. K. Cheruiyot, D. Mutisya, S. D. Kennedy, M. Egli, M. L. Kelley, A. van Brabant Smith and E. Rozners, A single amide linkage in the passenger strand suppresses its activity and enhances guide strand targeting of siRNAs, ACS Chem. Biol., 2018, 13, 533–536 CrossRef CAS PubMed.
- M. L. Hammill, K. Tsubaki, L. Salim, A. J. Varley, I. Giorgees, M. Kitamura, T. Okauchi and J.-P. Desaulniers, SiRNAs with neutral phosphate triester hydrocarbon tails exhibit carrier-free gene-silencing activity, ACS Med. Chem. Lett., 2022, 13, 695–700 CrossRef CAS PubMed.
- V. Kotikam, P. K. Gajula, L. Coyle and E. Rozners, Amide internucleoside linkages are well tolerated in protospacer adjacent motif-distal region of CRISPR RNAs, ACS Chem. Biol., 2022, 17, 509–512 CrossRef CAS PubMed.
- V. Verma, J. Maity, V. K. Maikhuri, R. Sharma, H. K. Ganguly and A. K. Prasad, Double-headed nucleosides: synthesis and applications, Beilstein J. Org. Chem., 2021, 17, 1392–1439 CrossRef CAS PubMed.
- C. Liczner, K. Duke, G. Juneau, M. Egli and C. J. Wilds, Beyond ribose and phosphate: selected nucleic acid modifications for structure–function investigations and therapeutic applications, Beilstein J. Org. Chem., 2021, 17, 908–931 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, A Hitchhiker's guide to click-chemistry with nucleic acids, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
- N.-S. Li, J. K. Frederiksen and J. A. Piccirilli, Automated solid-phase synthesis of RNA oligonucleotides containing a nonbridging phosphorodithioate linkage via phosphorothioamidites, J. Org. Chem., 2012, 77, 9889–9892 CrossRef CAS PubMed.
- S. Sakamuri, L. Eltepu, D. Liu, S. Lam, B. R. Meade, B. Liu, G. D. Iacono, A. Kabakibi, L. Luukkonen, T. Leedom, M. Foster and C. W. Bradshaw, Impact of phosphorothioate chirality on double–stranded siRNAs: a systematic evaluation of stereopure siRNA designs, ChemBioChem, 2020, 21, 1304–1308 CrossRef CAS PubMed.
- H. Nguyen, M. Abramov, E. Eremeeva and P. Herdewijn, In vivo expression of genetic information from phosphoramidate DNA, ChemBioChem, 2020, 21, 272–278 CrossRef CAS PubMed.
- K. Sato, H. Imai, T. Shuto, R. I. Hara and T. Wada, Solid-phase synthesis of phosphate/boranophosphate chimeric DNAs using the H–phosphonate–H–boranophosphonate method, J. Org. Chem., 2019, 84, 15032–15041 CrossRef CAS.
- R. I. Hara, T. Saito, T. Kogure, Y. Hamamura, N. Uchiyama, Y. Nukaga, N. Iwamoto and T. Wada, Stereocontrolled Synthesis of Boranophosphate DNA by an Oxazaphospholidine Approach and Evaluation of Its Properties, J. Org. Chem., 2019, 84, 7971–7983 CrossRef CAS PubMed.
- D. Mutisya, T. Hardcastle, S. K. Cheruiyot, P. S. Pallan, S. D. Kennedy, M. Egli, M. L. Kelley, A. van Brabant Smith and E. Rozners, Amide linkages mimic phosphates in RNA interactions with proteins and are well tolerated in the guide strand of short interfering RNAs, Nucleic Acids Res., 2017, 45, 8142–8155 CrossRef CAS PubMed.
- C. Selvam, S. Thomas, J. Abbott, S. D. Kennedy and E. Rozners, Amides as excellent mimics of phosphate linkages in RNA, Angew. Chem., Int. Ed., 2011, 50, 2068–2070 CrossRef CAS PubMed.
- A. Kolarovic, E. Schweizer, E. Greene, M. Gironda, P. S. Pallan, M. Egli and E. Rozners, Interplay of structure, hydration and thermal stability in formacetal modified oligonucleotides: RNA may tolerate nonionic modifications better than DNA, J. Am. Chem. Soc., 2009, 131, 14932–14937 CrossRef CAS PubMed.
- M. Kuwahara, H. Takeshima, J.-i. Nagashima, S. Minezaki, H. Ozaki and H. Sawai, Transcription and reverse transcription of artificial nucleic acids involving backbone modification by template–directed DNA polymerase reactions, Bioorg. Med. Chem., 2009, 17, 3782–3788 CrossRef CAS PubMed.
- M. Park and T. C. Bruice, Binding properties of positively charged deoxynucleic guanidine (DNG), AgTgAgTgAgT and DNG/DNA chimeras to DNA, Bioorg. Med. Chem., 2008, 18, 3488–3491 CrossRef CAS PubMed.
- B. A. Linkletter, I. E. Szabo and T. C. Bruice, Solid-Phase Synthesis of Deoxynucleic Guanidine (DNG) Oligomers and Melting Point and Circular Dichroism Analysis of Binding Fidelity of Octameric Thymidyl Oligomers with DNA Oligomers, J. Am. Chem. Soc., 1999, 121, 3888–3896 CrossRef CAS.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Triazole–linked analogue of deoxyribonucleic acid (TLDNA): design, synthesis, and double-strand formation with natural DNA, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed.
- C. N. Birts, A. P. Sanzone, A. H. El-Sagheer, J. P. Blaydes, T. Brown and A. Tavassoli, Transcription of click-linked DNA in Human cells, Angew. Chem., Int. Ed., 2014, 53, 2362–2365 CrossRef CAS PubMed.
- M. Kukwikila, N. Gale, A. H. El-Sagheer, T. Brown and A. Tavassoli, Assembly of a biocompatible triazole–linked gene by one–pot click–DNA ligation, Nat. Chem., 2017, 9, 1089–1098 CrossRef CAS PubMed.
- L. Taemaitree, A. Shivalingam, A. H. El-Sagheer and T. Brown, An artificial triazole backbone linkage provides a split-and-click strategy to bioactive chemicallymodified CRISPR sgRNA, Nat. Commun., 2019, 10, 1610 CrossRef PubMed.
- S. Epple, A. Modi, Y. R. Baker, E. Węgrzyn, D. Traoré, P. Wanat, A. E. S. Tyburn, A. Shivalingam, L. Taemaitree, A. H. El-Sagheer and T. Brown, A new 1,5−disubstituted triazole DNA backbone mimic with enhanced polymerase compatibility, J. Am. Chem. Soc., 2021, 143, 16292–16301 CrossRef PubMed.
- J. Kondo, T. Yamada, I. Okamoto, Y. Tanaka and A. Ono, Crystal structure of metallo DNA duplex containing consecutive Watson–Crick-like T–HgII–T base pairs, Angew. Chem., Int. Ed., 2014, 53, 2385–2388 CrossRef CAS PubMed.
- J. Kondo, Y. Tada, T. Dairaku, Y. Hattori, H. Saneyoshi, A. Ono and Y. Tanaka, A metallo–DNA nanowire with uninterrupted one–dimensional silver array, Nat. Chem., 2017, 9, 956–960 CrossRef CAS PubMed.
- K. Kowalski, Organometallic nucleosides–synthesis, transformations, and applications, Coord. Chem. Rev., 2021, 432, 213705 CrossRef CAS.
- B. Jash and J. Müller, Metal–mediated base pairs: from characterization to application, Chem. – Eur. J., 2017, 23, 17166–17178 CrossRef CAS PubMed.
- J. Müller, Nucleic acid duplexes with metal–mediated base pairs and their structures, Coord. Chem. Rev., 2019, 393, 37–47 CrossRef.
- H. Yang, K. L. Metera and H. F. Sleiman, DNA modified with metal complexes: applications in the construction of higher order metal–DNA nanostructures, Coord. Chem. Rev., 2010, 254, 2403–2415 CrossRef CAS.
- K. Kowalski, Ferrocenyl–nucleobase complexes: synthesis, chemistry and applications, Coord. Chem. Rev., 2016, 317, 132–156 CrossRef CAS.
- T. A. Lönnberg, M. A. Hande and D. U. Ukale, Oligonucleotide Complexes in Bioorganometallic Chemistry, in Compr. Organomet. Chem. IV, 2022, vol. 15, pp. 146–182 Search PubMed.
- D. Ukale and T. Lönnberg, Organomercury nucleic acids: past, present and future, ChemBioChem, 2021, 22, 1733–1739 CrossRef CAS PubMed.
- Y. Takezawa, J. Müller and M. Shionoya, Artificial DNA base pairing mediated by diverse metal ions, Chem. Lett., 2017, 46, 622–633 CrossRef CAS.
- G. Chatelain, A. Meyer, F. Morvan, J.-J. Vasseur and C. Chaix, Electrochemical detection of nucleic acids using pentaferrocenyl phosphoramidate α-oligonucleotides, New J. Chem., 2011, 35, 893–901 RSC.
- H. V. Nguyen, Z. Zhao, A. Sallustrau, S. L. Horswell, L. Male, A. Mulas and J. H. R. Tucker, A ferrocene nucleic acid oligomer as an organometallic structural mimic of DNA, Chem. Commun., 2012, 48, 12165–12167 RSC.
- E. Alberico, D. Dewaele, T. Kiss and G. Micera, Oxovanadium(IV) complexation by adenosine 5′-di- and -tri-phosphate and nucleotide building blocks, J. Chem. Soc., Dalton Trans., 1995, 425–430 RSC.
- H. Deng, J. W. Burgner and R. H. Callender, Structure of the ribonuclease uridine – vanadate transition state analogue complex by Raman difference spectroscopy: mechanistic implications, J. Am. Chem. Soc., 1998, 120, 4717–4722 CrossRef CAS.
- A. Simonova, I. Magriñá, V. Sykorová, R. Pohl, M. Ortiz, L. Havran, M. Fojta, C. K. O'Sullivan and M. Hocek, Tuning of oxidation potential of ferrocene for ratiometric redox labeling and coding of nucleotides and DNA, Chem. – Eur. J., 2020, 26, 1286–1291 CrossRef CAS PubMed.
- H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, “Click-to-Chelate”: Design and Incorporation of Triazole-Containing Metal-Chelating Systems into Biomolecules of Diagnostic and Therapeutic Interest, Chem. – Eur. J., 2008, 14, 6173–6183 CrossRef CAS PubMed.
- C. Lorenzo-Aparicio, M. G. Gallego, C. R. de Arellano and M. A. Sierra, Phosphorescent Ir(III) complexes derived from purine nucleobases, Dalton Trans., 2022, 51, 5138–5150 RSC.
- D. Bergstrom and T. Schmaltz, Organoiron–mediated alkylation of phosphite esters: synthesis of (dicarbonyl)(η5−cyclopentadienyl)iron–derived nucleoside phosphonate esters, J. Org. Chem., 1992, 57, 873–876 CrossRef CAS.
- J. M. D. R. Toma and D. E. Bergstrom, Transition metal labeling of oligodeoxyribonucleotides: synthesis and characterization of (pentacarbonyl)tungsten(0) nucleoside phosphites, J. Org. Chem., 1994, 59, 2418–2422 CrossRef.
- D. Bergstrom and T. Schmaltz, Synthesis of (Dicarbonyl)(η5−cyclopentadienyl)iron–derived nucleoside phosphonate esters, Nucleosides Nucleotides, 1989, 8, 1057–1059 CrossRef.
- D. Bergstrom, P. Beal and R. Lind, Synthesis of (Dicarbonyl)(η5−cyclopentadienyl)manganese complex stabilized nucleoside phosphite esters, Nucleosides Nucleotides, 1989, 8, 1061–1063 CrossRef.
- P. Biegański, E. Kovalski, N. Israel, E. Dmitrieva, D. Trzybiński, K. Woźniak, V. Vrček, M. Godel, C. Riganti, J. Kopecka, H. Lang and K. Kowalski, Electronic coupling in 1,2,3-triazole bridged ferrocenes and its impact on reactive oxygen species generation and deleterious activity in cancer cells, Inorg. Chem., 2022, 61, 9650–9666 CrossRef PubMed.
- M. Piotrowicz, A. Kowalczyk, D. Trzybiński, K. Woźniak and K. Kowalski, Redox–active glycol nucleic acid (GNA) components: synthesis and properties of the ferrocenyl–GNA nucleoside, phosphoramidite, and semicanonical dinucleoside phosphate, Organometallics, 2020, 39, 813–823 CrossRef CAS.
- F. Seela, H. Xiong and S. Budow, Synthesis and ‘double click’ density functionalization of 8−aza–7−deazaguanine DNA bearing branched side chains with terminal triple bonds, Tetrahedron, 2010, 66, 3930–3943 CrossRef CAS.
- J. G. Vaughan, B. L. Reid, P. J. Wright, S. Ramchandani, B. W. Skelton, P. Raiteri, S. Muzzioli, D. H. Brown, S. Stagni and M. Massi, Photophysical and Photochemical Trends in Tricarbonyl Rhenium(I) N-Heterocyclic Carbene Complexes, Inorg. Chem., 2014, 53, 3629–3641 CrossRef CAS PubMed.
- R. Czerwieniec, A. Kapturkiewicz, J. Lipkowski and J. Nowacki, Re(I)(tricarbonyl)+ complexes with the 2-(2-pyridyl)-N-methyl-benzimidazole2-(2-pyridyl)benzoxazole and 2-(2-pyridyl)benzothiazole ligands – syntheses, structures, electrochemical and spectroscopic studies, Inorg. Chim. Acta, 2005, 358, 2701–2710 CrossRef CAS.
- K. K.-W. Lo, Molecular design of biorthogonal probes and imaging reagents derived from photofunctional transition metal complexes, Acc. Chem. Res., 2020, 53, 32–44 CrossRef CAS PubMed.
- A. M.-H. Yip and K. K.-W. Lo, Luminescent rhenium(I)ruthenium(II), and iridium(III) polypyridine complexes containing a poly(ethylene glycol) pendant or bioorthogonal reaction group as biological probes and photocytotoxic agents, Coord. Chem. Rev., 2018, 361, 138–163 CrossRef CAS.
- M.-W. Louie, H.-W. Liu, M. H.-C. Lam, Y.-W. Lam and K. K.-W. Lo, Luminescent Rhenium(I) Polypyridine Complexes Appended with an α-D-Glucose Moiety as Novel Biomolecular and Cellular Probes, Chem. – Eur. J., 2011, 17, 8304–8308 CrossRef CAS PubMed.
- J. Skiba, A. Kowalczyk, P. Stączek, T. Bernaś, D. Trzybiński, K. Woźniak, U. Schatzschneider, R. Czerwieniec and K. Kowalski, Luminescent fac–[Re(CO)3(phen)] carboxylato complexes with non-steroidal anti-inflammatory drugs: synthesis and mechanistic insights into the in vitroanticancer activity of fac–[Re(CO)3(phen)(aspirin)], New J. Chem., 2019, 43, 573–583 RSC.
- J. Skiba, T. Bernaś, D. Trzybiński, K. Woźniak, G. Ferraro, D. Marasco, A. Merlino, M. Z. Shafikov, R. Czerwieniec and K. Kowalski, Mitochondria targeting with luminescent rhenium(I) complexes, Molecules, 2017, 22, 809–825 CrossRef PubMed.
- S. Clède and C. Policar, Metal–Carbonyl Units for Vibrational and Luminescence Imaging: Towards Multimodality, Chem. – Eur. J., 2015, 21, 942–958 CrossRef PubMed.
- K. A. Stephenson, S. R. Banerjee, T. Besanger, O. O. Sogbein, M. K. Levadala, N. McFarlane, J. A. Lemon, D. R. Borenham, K. P. Maresca, J. D. Brennan, J. W. Babich, J. Zubieta and J. F. Valliant, Bridging the Gap between in Vitro and in Vivo Imaging: Isostructural Re and 99mTc Complexes for Correlating Fluorescence and Radioimaging Studies, J. Am. Chem. Soc., 2004, 126, 8598–8599 CrossRef CAS PubMed.
- E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Luminescent conjugates between dinuclear rhenium(I) complexes and peptide nucleic acids (PNA) for cell imaging and DNA targeting, Chem. Commun., 2010, 46, 6255–6257 RSC.
- C. Mari, M. Panigati, L. D'Alfonso, I. Zanoni, D. Donghi, L. Sironi, M. Collini, S. Maiorana, C. Baldoli, G. D'Alfonso and E. Licandro, Luminescent conjugates between dinuclear rhenium complexes and peptide nucleic acids (PNA): synthesis, photophysical characterization, and cell uptake, Organometallics, 2012, 31, 5918–5928 CrossRef CAS.
- A. Palmioli, M. Panigati and A. Bernardi, Glycodendron–rhenium complexes as luminescent probes for lectin sensing, Org. Biomol. Chem., 2018, 16, 7035–7041 RSC.
- A. Palmioli, A. Aliprandi, D. Septiadi, M. Mauro, A. Bernardi, L. De Cola and M. Panigati, Glyco–functionalized dinuclear rhenium(I) complexes for cell imaging, Org. Biomol. Chem., 2017, 15, 1686–1699 RSC.
- M. Proverbio, E. Q. Procopio, M. Panigati, S. Mercurio, R. Pennati, M. Ascagni, R. Leone, C. La Porta and M. Sugni, Luminescent conjugates between dinuclear rhenium complexes and 17α–ethynylestradiol: synthesis, photophysical characterization, and cell imaging, Org. Biomol. Chem., 2019, 17, 509–518 RSC.
- M. Panigati, M. Mauro, D. Donghi, P. Mercandelli, P. Mussini, L. De Cola and G. D'Alfonso, Luminescent dinuclear rhenium(I) complexes containing bridging 1,2-diazine ligands: Photophysical properties and application, Coord. Chem. Rev., 2012, 256, 1621–1643 CrossRef CAS.
- D. Donghi, G. D'Alfonso, M. Mauro, M. Panigati, P. Mercandelli, A. Sironi, P. Mussini and L. D'Alfonso, A new class of luminescent tricarbonyl rhenium(I) complexes containing bridging 1,2-diazine ligands: electrochemical, photophysical, and computational characterization, Inorg. Chem., 2008, 47, 4243–4255 CrossRef CAS PubMed.
- A. T. M. Marcelis and H. C. van der Plas, Cycloaddition reactions of cyclic ketene–N,S–acetals with 1,2,4,5−tetrazines, J. Heterocycl. Chem., 1987, 24, 545–548 CrossRef CAS.
- J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, 1,2,4,5−Tetrazine: synthesis and reactivity in [4 + 2] cycloadditions, Eur. J. Org. Chem., 1998, 2885–2896 CrossRef CAS.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels–Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- Y. Luan, L. L. Blazer, H. Hu, T. Hajian, J. Zhang, H. Wu, S. Houliston, C. H. Arrowsmith, M. Vedadi and Y. G. Zheng, Design of a fluorescent ligand targeting the S–adenosylmethionine binding site of the histone methyltransferase MLL1, Org. Biomol. Chem., 2016, 14, 631–638 RSC.
- B. Neises and W. Steglich, Simple method for the esterification of carboxylic acids, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- A. Kowalczyk, M. Piotrowicz, M. Gapińska, D. Trzybiński, K. Woźniak, T. M. Golding, T. Stringer, G. S. Smith, R. Czerwieniec and K. Kowalski, Chemistry of glycol nucleic acid (GNA): Synthesis, photophysical characterization and insight into the biological activity of phenanthrenyl GNA constituents, Bioorg. Chem., 2022, 125, 105847 CrossRef CAS PubMed.
- CrysAlis CCD and CrysAlis RED, Oxford Diffraction, Oxford Diffraction Ltd, Yarnton, 2008 Search PubMed.
- R. C. Clark and J. S. Reid, The analytical calculation of absorption in multifaceted crystals, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. A. Velapoldi and H. H. Tonnesen, Corrected emission spectra and quantum yields for a series of fluorescent compounds in the visible spectral region, J. Fluoresc., 2004, 14, 465–472 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford, Connecticut, USA, 2016 Search PubMed.
- R. Peverati and D. G. Truhlar, M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics, J. Phys. Chem. Lett., 2012, 3, 117–124 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Performance of the M11 and M11-L density functionals for calculations of electronic excitation energies by adiabatic time-dependent density functional theory, Phys. Chem. Chem. Phys., 2012, 14, 11363–11370 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2224012 and 2224011. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03995h |
| This journal is © The Royal Society of Chemistry 2023 |