
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors controlling the structure of alkylzinc amidinates: on the role of N-substituents†
Michał
Terlecki
 a,
Iwona
Justyniak
b,
Michał K.
Leszczyński
ab,
Piotr
Bernatowicz
b and
Janusz
Lewiński
*ab
a,
Iwona
Justyniak
b,
Michał K.
Leszczyński
ab,
Piotr
Bernatowicz
b and
Janusz
Lewiński
*ab
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 3rd February 2023
Abstract
Despite various applications of alkylzinc complexes supported by N,N-bidentate ligands in chemistry and materials science, the corresponding organozinc amidinates still represent an insufficiently explored area. To gain a more in-depth understanding of factors controlling the structure and stability of alkylzinc amidinates, we selected benzamidinate and N,N′-diphenylformamidinate ligands as model N,N′-unsubstituted and N,N′-diaryl substituted ligands, respectively, to systematically modify the secondary coordination sphere of the Zn center. A series of new alkylzinc amidinates has been synthesized and their molecular structures identified in both the solid state (single-crystal X-ray crystallography) and solution (NMR and FTIR spectroscopy). The results indicate that [RZnL]x-type amidinate moieties are essentially unstable and tend to undergo Schlenk equilibria-mediated ligand scrambling leading to more thermodynamically stable non-stoichiometric [R2Zn3L4]- and [R3Zn4L5]-type complexes. This process is significantly influenced by the secondary coordination sphere noncovalent interactions as well as the steric hindrance provided by both zinc-bounded alkyl groups and the N-substituents.
Introduction
Heteroleptic alkylzinc compounds are of longstanding interest due to their important role in various stoichiometric and catalytic chemical processes1–7 and more recently as efficient predesign precursors of modern functional materials.8–11 However, the characterization of organozinc reactants and intermediate states is often challenging as they commonly exhibit complicated behavior in solutions involving multiple equilibria between various aggregated forms and non-stoichiometric species arising from Schlenk equilibria.2,12–15 The nature and reactivity of heteroleptic alkylzincs are often not only determined by the primary coordination sphere (i.e., the array of the direct metal–ligands interactions) but they can be strongly influenced by the secondary coordination sphere interactions of the metal environment,16,17 which includes organic skeletons of supporting ligands or external functionalities attached to the internal scaffolds that not only provide a specific steric environment but may participate in an array of noncovalent interactions and be very effective in the construction of supramolecular assemblies.18–21 Hence, many of the important transformations involving alkylzinc compounds are still not well understood, such as, among others, the fascinating autocatalytic Soai reaction,5,7 and the oxygenation of organozincs, which mechanism has been under debate for over 160 years.22,23 In this view, a more in-depth understanding of the multifaceted chemistry of alkylzinc species is crucial for the development of new, more efficient reaction systems.Among various supporting ligands used for the stabilization of heteroleptic organozinc complexes amidinates are particularly interesting due to a prominent tunability of steric and electronic requirements by systematic variations of the substituents at the carbon and nitrogen atoms.24–26 These ligands exhibit a vast array of coordination modes, including μ2-bridging, κ2-chelating, as well as more complex μ3-bridging and mixed bridging–chelating μ2-κ2:κ1 (Fig. 1b), which originates in a specific parallel orientation of the N donor electron pairs in the NCN anchoring groups that can be tuned by for example sterically demanding substituents or strains in macrocycle organic backbones (Fig. 1a).24,25,27 Furthermore, amidinate ligands introduce N-bonded hydrogen atoms or organic groups like aromatic rings or bulky alkyls to the proximity of the metal center that can interfere with its primary coordination sphere either by steric hindrances blocking the access to the metal ion28 or noncovalent interactions providing specific structural stabilization.29 A particularly interesting example of the influence of N-substituents on the stabilization of molecular structure of alkylzinc complexes concerns N,N′-diaryl amidinate ligands. Introduction of sterically demanding substituents to the ortho position of N-bonded aromatic rings results in the decrease of nuclearity of the respective alkylzinc systems from a trinuclear [Et2Zn3(dipf)4] in the case of ortho-unsubstituted N,N′-diphenylformamidinate (dipf) ligands to a dinuclear aggregate [Et2Zn2L2·THF] for the 2,6-dimethyl or 2-isopropyl analogs28 and a monomeric complex [MeZn{tBuC(NDipp)2}] comprising 2,6-diisopropylphenyl (Dipp) N-substituents.29 Furthermore, aromatic rings in the secondary coordination sphere are prone to participate in specific CH–π, π–π, or ion–π interactions, which may also affect the stabilization of the coordination system. For instance, ligands in [MeZn{tBuC(NDipp)2}] adopt an unusual κ1:η3 coordination mode involving efficient stabilization of the low coordinated zinc center via a Zn–π interaction (Fig. 2a).29 Theoretical calculations revealed that this type of coordination is slightly preferred, by only 1.5 kcal mol−1, over the common chelating κ2 mode, which shows that noncovalent interactions involving aromatic π systems may effectively compete with the formation of donor–acceptor bonds (this type of structural diversity has been observed for some other heteroleptic zinc alkyls30–32).
 | ||
| Fig. 1 (a) Tunable spatial orientation of the donor electron pairs in NCN anchoring groups; (b) coordination modes of amidinate ligands in zinc coordination systems. | ||
 | ||
| Fig. 2 Multifaceted role of aromatic N-substituents (a and b) and N-bonded hydrogen atoms (c and d) of N,N′-diaryl and N,N′-unsubstituted amidinate ligands, respectively, in the structural stabilization and self-assembly of zinc coordination systems. | ||
In turn, N,N′-unsubstituted amidinates involving NH units are an interesting group of ligands that can participate in the formation of intra- and intermolecular hydrogen bonds in the proximity of the coordination center. Strikingly, the chemistry of alkylzinc complexes supported by this type of ligands is highly unexplored, and, as far as we know, a series of macrocyclic trimeric group 13 benzamidinates, [Me2M(bza)]3 (M = Al, Ga, In; bza–H = benzamidine) was until now the only example of structurally characterized main group organometallics incorporating N,N′-unsubstituted amidinates.33 Very recently, we used in situ generated alkylzinc derivatives of benzamidine as efficient precursors of zinc–oxido clusters21 and zinc oxide nanostructures.11 In these studies, we found that the NH units of the benzamidinate ligands play important role in the stabilization of ZnO nanocrystals’ surface providing H-donor sites for intermolecular hydrogen bonds participating in a unique facet-specific bimodal ligands interaction, which led to the preparation of unprecedented nanoplatelets with a controlled thickness (Fig. 2d).11
Remarkably, the secondary coordination sphere of the metal environment may not only influence the stabilization of molecular coordination systems but it also is responsible for self-assembly processes mediated by noncovalent interactions. This is well exemplified by tetrahedral zinc–oxido clusters coated by carboxylate18 or amidinate ligands.19–21 For example, we observed an interesting solvatomorphism and pressure-induced dynamic behavior of an N,N′-diphenylformamidinate (dipf) stabilized zinc–oxido cluster, originated in high adaptability of its secondary coordination sphere comprising numerous N-bonded phenyl rings.20 Interestingly, in this case, the aromatic subunits not only mediated intermolecular CH–π interactions but their conformational movement gradually absorbed compression strain induced by the increasing pressure, which promoted stepwise phase transitions (Fig. 2b) and thus these observations provided a profound understanding of the multistep single-crystal-to-single crystal phase transitions at the atomic level. Other studies also found a profound effect of the introduction of NH units to the proximal secondary coordination sphere of polyhedral zinc–oxido clusters on their crystal packing.21 In this view, benzamidinate (bza) ligands provided efficient H-donor sites for intermolecular NH⋯O bonds with the solvating THF molecules leading to a honeycomb supramolecular structure with 1D open-channels (Fig. 2c).
Building on the mentioned above results and our previous experience in studies on the structure and self-assembly of amidinate metal complexes,11,20,21,34,35 we selected dipf and bza ligands as a model N,N′-diaryl substituted and N,N′-unsubstituted NCN scaffold to gain a more in-depth understanding of factors controlling the structure and stability of alkylzinc amidinates. To this aim, we investigated the reactions of dipf–H and bza–H proligands with ZnR2 compounds (R = Me, Et, tBu) and characterized products containing a systematically modified secondary coordination sphere of the Zn center in both the solid state and solution.
Results and discussion
Alkylzincs stabilized by N,N′-diaryl amidinate ligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which indicates subsequent transformations of the initially formed [RZn(dipf)]-type species. Thus, in order to better understand the ZnR2/dipf-H reaction system and the formation of 1R-type compounds, we performed Diffusion-Ordered NMR spectroscopy (DOSY NMR) experiments on the equimolar reactions of dipf–H with ZnR2 (R = Me, Et) in d8-toluene (for details, see ESI†). Molecular weights of examined species (MWest) were estimated utilizing an external calibration curve (ECC) approach with normalized diffusion coefficients, exploiting 1,2,3,4-tetraphenylnaphtalene (TPhN) as an internal reference,36 and compared with the molecular weights of respective compounds including correction due to the presence of heavy elements (MWcorr).37 Analysis revealed two components in the post-reaction mixtures with the MWest of 762 and 66 g mol−1, and 761 and 117 g mol−1 for methyl- and ethylzinc derivatives, respectively, which indicates the equimolar formation of [R2Zn3(dipf)4]-type moieties (MWcorr = 746 and 776 g mol−1 for R = Me, Et, respectively) and ZnR2 (MWcorr = 49 and 79 g mol−1 for R = Me, Et, respectively), irrespective of the character of Zn-bonded alkyl group (Fig. S13–S18, and Tables S1–S4†). This suggests that the initially formed [RZn(dipf)]x-type species are relatively unstable and undergo the Schlenk equilibria-mediated ligand scrambling leading to the more thermodynamically stable non-stoichiometric [R2Zn3(dipf)4]-type clusters (Fig. 3). Interestingly, we observed a similar ligand rearrangement in our previous studies concerning alkylzinc pyrazolates.13 However, contrary to the dipf derivatives, the pyrazolate [EtZnL]x-type species formed during the synthesis were stable in solution and undergo ligand scrambling only during the crystallization leading to non-stoichiometric [Et2Zn3L4]- or [Et2Zn4L6]-type complexes that preserved their structure upon subsequent dissolution.
:1, which indicates subsequent transformations of the initially formed [RZn(dipf)]-type species. Thus, in order to better understand the ZnR2/dipf-H reaction system and the formation of 1R-type compounds, we performed Diffusion-Ordered NMR spectroscopy (DOSY NMR) experiments on the equimolar reactions of dipf–H with ZnR2 (R = Me, Et) in d8-toluene (for details, see ESI†). Molecular weights of examined species (MWest) were estimated utilizing an external calibration curve (ECC) approach with normalized diffusion coefficients, exploiting 1,2,3,4-tetraphenylnaphtalene (TPhN) as an internal reference,36 and compared with the molecular weights of respective compounds including correction due to the presence of heavy elements (MWcorr).37 Analysis revealed two components in the post-reaction mixtures with the MWest of 762 and 66 g mol−1, and 761 and 117 g mol−1 for methyl- and ethylzinc derivatives, respectively, which indicates the equimolar formation of [R2Zn3(dipf)4]-type moieties (MWcorr = 746 and 776 g mol−1 for R = Me, Et, respectively) and ZnR2 (MWcorr = 49 and 79 g mol−1 for R = Me, Et, respectively), irrespective of the character of Zn-bonded alkyl group (Fig. S13–S18, and Tables S1–S4†). This suggests that the initially formed [RZn(dipf)]x-type species are relatively unstable and undergo the Schlenk equilibria-mediated ligand scrambling leading to the more thermodynamically stable non-stoichiometric [R2Zn3(dipf)4]-type clusters (Fig. 3). Interestingly, we observed a similar ligand rearrangement in our previous studies concerning alkylzinc pyrazolates.13 However, contrary to the dipf derivatives, the pyrazolate [EtZnL]x-type species formed during the synthesis were stable in solution and undergo ligand scrambling only during the crystallization leading to non-stoichiometric [Et2Zn3L4]- or [Et2Zn4L6]-type complexes that preserved their structure upon subsequent dissolution.
 | ||
| Fig. 3 Schematic representation of equimolar reactions of the model dipf–H and bza–H proligands with the representative homoleptic organozincs ZnR2 (R = Me, Et, tBu). | ||
Complexes 1Me and 1Et can be also easily obtained with almost quantitative yields by the reaction of dipf–H with ZnR2 in a 4:3 molar ratio and as received products were characterized by single-crystal X-ray diffraction (SC-XRD), FTIR and NMR spectroscopy, and elemental analysis. Crystal structures examination showed that the molecules of 1Me and 1Et are essentially isostructural but differ in the conformation of the N-bonded aromatic rings in the secondary coordination sphere. Their molecular structure can be described as a combination of two [RZn(dipf)] and one [Zn(dipf)2] moieties. The core of 1R comprises three Zn centers with a right triangle geometry (the largest Zn–Zn–Zn angles within the individual complexes are in the range of 88.81(2)–92.55(2)°) (Fig. 4a). All zinc centers exhibit a similar distorted tetrahedral geometry of the coordination sphere, however, the two of them comprised alkyl groups while the last one is bonded only by four amidinate ligands. The {Zn3} core is stabilized by four dipf ligands, two adopting μ2 coordination and the other two acting as μ3 bridges (Fig. 4d and e). In the μ2 coordination mode, both N–Zn bonds are of similar length (1.988(2)–2.055(3) Å) and coplanar with the NCN plane. The μ3 coordination mode comprises two similar N–Zn distances essentially coplanar with the NCN unit and the perpendicular third one that is significantly longer (the Zn–N lengths of two bonds coplanar with the NCN units and the third perpendicular bond are in the ranges of 2.045(2)–2.073(2) Å and 2.261(2)–2.346(2) Å, respectively). The different character of both donor N centers in μ3 dipf ligands introduces significant asymmetry to the amidinate group (the C–N distances to mono- and bi-dentate N centers of the μ3 ligands are in the ranges of 1.292(4)–1.306(4) Å and 1.348(3)–1.366(4) Å, respectively, while all the C–N distances in the μ2 ligands are in the range of 1.313(4)–1.328(4) Å). Interestingly, the eight phenyl rings in the secondary coordination sphere of 1Me and 1Et complexes are arranged indicating the presence of numerous specific noncovalent interactions. Especially, in 1Me the phenyl rings seem to form two CH–π:π–π:CH–π systems of cooperative interactions on top and bottom of the triangular core (Fig. 4b; the distances between the ring planes in π–π interactions are about 3.12–3.67 Å and the distances of H atoms to respective ring planes in CH–π interactions are about 2.68–2.94 Å). In turn, 1Et differs in the conformation of two aromatic rings on the bottom side of the {Zn3} core resulting in CH–π:CH–π:CH–π system of cooperative noncovalent interactions (Fig. 4c; the distance between the ring planes in π–π interaction is about 3.51 Å and the distances of H atoms to respective ring planes in CH–π interactions are about 2.43–2.91 Å). Notably, in solution, the conformation of aromatic rings in both 1Me and 1Et is similar to that observed in the crystal structure of 1Me (vide infra) indicating that the system involving two CH–π:π–π:CH–π sets of interactions is likely more thermodynamically favored. Thus, the different conformation of phenyl rings observed in the crystal structure of 1Et is probably a combined effect of the ethyl group steric hindrance and a specific environment of the close-packed molecules.
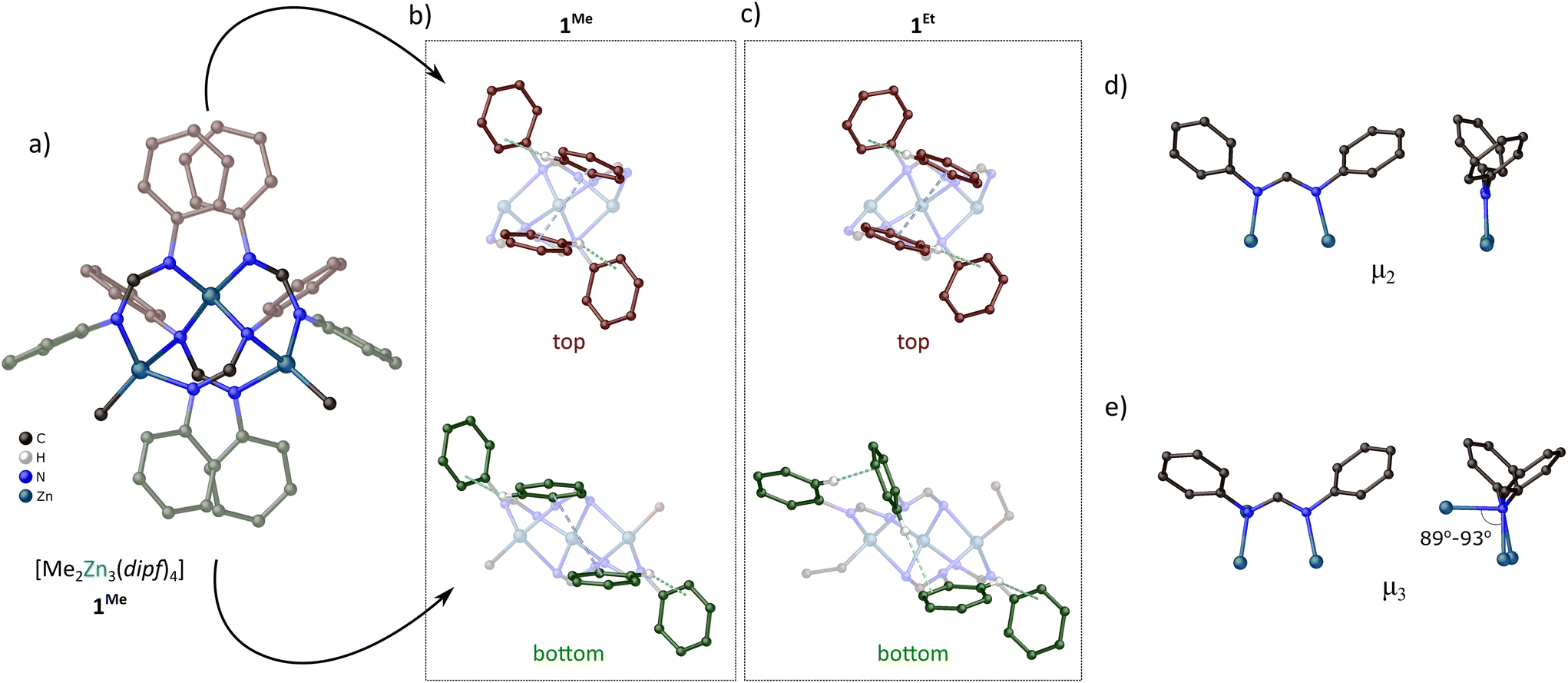 | ||
| Fig. 4 Molecular structure of 1Me (a); view on the configuration of aromatic rings on the top and bottom of triangular core in 1Me (b) and 1Et (c); μ2 (d) and μ3 (e) coordination modes of dipf ligands in 1R. | ||
Analysis of the NMR spectra indicates that the solid-state molecular structures of 1Me and 1Et are essentially preserved in solution. Especially, the 1H NMR spectrum of 1Me in C6D6 shows two singlets with the same intensity at 8.24 and 7.85 ppm and a complicated set of resonances from the aromatic hydrogen atoms in the range 8.4–5.7 ppm, which confirm the presence of two different forms of dipf ligands (Fig. 5). Furthermore, one of the aromatic resonances, with the relative intensity of 4H, is highly upfield shifted to about 5.7 ppm, which is likely related to the four specific CH–π contacts. This indicates a similar configuration of the aromatic rings to that observed in the crystal structure of 1Me, involving four CH–π interactions. The 1H NMR spectrum of 1Et (Fig. S3†) is essentially identical to that of 1Me indicating the same conformation of aromatic rings in the secondary coordination sphere (four specific CH–π contacts based on 1H NMR signal intensity).
 | ||
| Fig. 5 1H NMR spectrum of 1Me in C6D6 solution. | ||
We note that 1Et was obtained previously by Zhao and coworkers, who studied the effect of steric hindrances in the aromatic rings on the structure of ethylzinc N,N′-diarylformamidinates.28 However, the authors didn't consider the role of secondary coordination sphere interactions on its structural stabilization neither their solution behavior. Still, they reviled that introduction of substituents in the ortho-position of N-bonded aromatic subunits of amidinate ligands resulted in the stabilization of THF solvated dimers [EtZnL]2·THF, instead of non-stoichiometric trinuclear [Et2Zn3L4]-type complexes isolated for ortho-unsubstituted ligands. Notably, these ortho-substituents not only introduce steric hindrances to the amidinate ligands but also prevent the formation of CH–π interactions described above.
 | ||
| Fig. 6 Molecular structure of 2 (a), and view along Zn–Zn line on the Δ/Λ conformers (b). | ||
The 1H and 13C NMR spectra of 2 in C6D6 solution are consistent with the structure observed in solid-state indicating the presence of only one form of dipf ligand (Fig. S5 and S6†). The 1H NMR spectrum shows a singlet at 7.68 ppm, three multiplets in the range of 6.8–7.0 ppm, and an intensive singlet at 1.47 ppm, associated with amidinate, aromatic, and tBu hydrogen atoms, respectively. In turn, the 13C NMR spectrum shows single resonances at 161.6 and 148.5 ppm associated with amidinate and tertiary aromatic carbon atoms of the single dipf ligand. Further three signals at 129.9, 124.3, 122.0 ppm, and two signals at 33.4 and 24.8 ppm are associated with the rest of the phenyl group carbon atoms and tBu group, respectively.
The above results indicate that the steric hindrances in the proximity of metal centers play a significant role in the stabilization of [RZn(L)]x-type amidinate species. Thus, while the methyl and ethyl dipf derivatives selectively stabilized non-stoichiometric [Et2Zn3L4]-type coordination systems, the more sterically demanding tert-butylzinc analogue selectively leads to the dimeric [(tBu)Zn(dipf)]2 complex (Fig. 3). This is in line with results obtained by Zhao and coworkers, who demonstrated that the dimeric [EtZn(L)]2·THF-type structures were favored over the non-stoichiometric [Et2Zn3(L)4]-type complexes in the case of amidinate ligands with sterically demanding N-bonded aryl subunits.28 This emphasizes that the steric hindrance of both organic ligands and alkylzinc groups are important factors competing with secondary coordination sphere noncovalent interactions in the stabilization of the molecular structure of organometallic complexes.
Alkylzincs stabilized by N,N′-unsubstituted amidinate ligands
:0.3 and 1:0.4 for methyl- and ethylzinc derivatives, respectively, which suggests the formation of non-stoichiometric complexes with an excess of bza ligands compared to alkylzinc groups.
 | ||
| Fig. 7 1H NMR spectrum from the equimolar reaction of bza–H with ZnMe2 in d8-toluene solution. | ||
To gain more insights into the bza–H/ZnR2 reaction system, we performed DOSY NMR analysis of the post-reaction mixtures in d8-toluene (for details, see ESI†). Analysis revealed the presence of two main components with the MWest of 609 and 86 g mol−1, and 653 and 124 g mol−1 for methyl- and ethylzinc derivatives, respectively, which indicates the formation of [R3Zn4(bza)5]-type moieties (MWcorr = 610 and 655 g mol−1 for R = Me, Et, respectively) and ZnR2 (MWcorr = 49 and 79 g mol−1 for R = Me, Et, respectively) (Fig. S19–S24, and Tables S5–S8†). Furthermore, the 1H NMR spectra show several additional signals in the aliphatic region that suggest the formation of minor products, which in the case of methylzinc derivative can be assigned to other non-stoichiometric complexes like [Me4Zn5(bza)6] (MWest = 735, MWcorr = 741 g mol−1) and [MeZn3(bza)6] (MWest = 567, MWcorr = 561 g mol−1) moieties (Fig. 7). Thus, the data indicates a low selectivity of bza ligands in the stabilization of specific alkylzinc complexes. As in the case of methyl- and ethylzinc derivatives of dipf–H, the initially formed benzamidinate [RZn(bza)]x-type species are essentially unstable and undergo fast Schlenk equilibria-mediated ligand scrambling leading to more thermodynamically stable non-stoichiometric coordination systems (Fig. 3). However, the bza ligand provide less specific stabilization than dipf one resulting in a mixture of various non-stoichiometric complexes instead of one dominant form. Notably, bza ligands create little steric hindrance, thus in this case, the structure of non-stoichiometric alkylzinc systems is likely stabilized by noncovalent interactions involving the NH groups in the secondary coordination sphere.
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry built by four fused by edges Zn–N–Zn–NCN–metallamacrocycles (Fig. 8a). The phenyl rings of bza ligands are directed above and below the barrel-like core, while the zinc-bonded alkyl groups occupy the side positions around it. All Zn centers are symmetrically equivalent adopting the same slightly distorted geometry of the coordination sphere. Four bza ligands exhibit the same μ3 coordination mode resulting in the diversification of the N–C bonds within the amidinate functionality (the N–C distances are 1.298(2) and 1.360(2) Å to the monodentate and bridging N centers, respectively). This μ3 coordination mode of bza ligands differs significantly from that observed for dipf ligands in the complexes 1R (vide supra). Particularly, in the μ3-bza ligands both Zn–N bonds to the bridging N center substantially deviate from the NCN plane of amidinate group and the angle between them is 105.25(7)° (Fig. 8b), while in the μ3-dipf ligands, two of the Zn centers are essentially coplanar with the NCN plane and the third Zn center is coordinated almost perpendicular to them (Fig. 4e). Moreover, the Zn–N distances in 3 are 2.000(2), 2.083(1), and 2.165(2) Å to the monodentate and bridging N centers, respectively, exhibiting much smaller diversification than in 1R.
symmetry built by four fused by edges Zn–N–Zn–NCN–metallamacrocycles (Fig. 8a). The phenyl rings of bza ligands are directed above and below the barrel-like core, while the zinc-bonded alkyl groups occupy the side positions around it. All Zn centers are symmetrically equivalent adopting the same slightly distorted geometry of the coordination sphere. Four bza ligands exhibit the same μ3 coordination mode resulting in the diversification of the N–C bonds within the amidinate functionality (the N–C distances are 1.298(2) and 1.360(2) Å to the monodentate and bridging N centers, respectively). This μ3 coordination mode of bza ligands differs significantly from that observed for dipf ligands in the complexes 1R (vide supra). Particularly, in the μ3-bza ligands both Zn–N bonds to the bridging N center substantially deviate from the NCN plane of amidinate group and the angle between them is 105.25(7)° (Fig. 8b), while in the μ3-dipf ligands, two of the Zn centers are essentially coplanar with the NCN plane and the third Zn center is coordinated almost perpendicular to them (Fig. 4e). Moreover, the Zn–N distances in 3 are 2.000(2), 2.083(1), and 2.165(2) Å to the monodentate and bridging N centers, respectively, exhibiting much smaller diversification than in 1R.
 | ||
| Fig. 8 Molecular structure of 3 and μ3 coordination mode of bza ligands. | ||
Interestingly, NMR spectra analysis reviled that upon dissolution in d8-toluene complex 3 undergoes ligand scrambling with the formation of various forms of tert-buthylzinc benzamidinates (see ESI, Fig. S25 and S26†). Both 1H and 13C NMR spectra of 3 show multiple signals in the range of 1.7–0.7 ppm and 37–17 ppm, respectively, indicating the presence of tBu groups with various chemical environments (Fig. 9 and S12†). In turn, multiple signals in the ranges of 182–172 ppm and 144–136 ppm in the 13C NMR spectrum characteristic for the amidinate and tertiary aromatic carbon atoms, respectively, suggest at least four different coordination modes of bza ligands. 1H DOSY NMR analysis shows that the three most intensive resonances in the region of alkyl groups, at ca. 1.40, 1.26, and 1.07 ppm, belong to various tert-butylzinc complexes with estimated molecular weights of 696, 746, and 158 g mol−1 matching well to the parent tetramer [(tBu)Zn(bza)]4 (MWcorr = 706 g mol−1), a non-stoichiometric complex [(tBu)3Zn4(bza)5] (MWcorr = 744 g mol−1), and ZntBu2 (MWcorr = 139 g mol−1), respectively (Fig. S25, S26 and Tables S9, S10†). However, the presence of other less intensive signals in this region indicates a more complex mixture of products likely resulted in complicated equilibria in solution. Note, that several attempts to obtain crystals of [(tBu)3Zn4(bza)5] by crystallization in various conditions (temperature, different solvents) yielded selectively the tetrameric complex 3, which is likely the most thermodynamically stable (or the least soluble) form of the tert-butylzinc bza derivatives. This is in agreement with the result obtained for the analogous dipf derivatives, indicating that the sterically demanding tert-butyl substituents on zinc centers favor the formation of [RZnL]x-type species, which is also strongly affected by a combination of steric and electronic effects mediated by the N-bonded substituents.
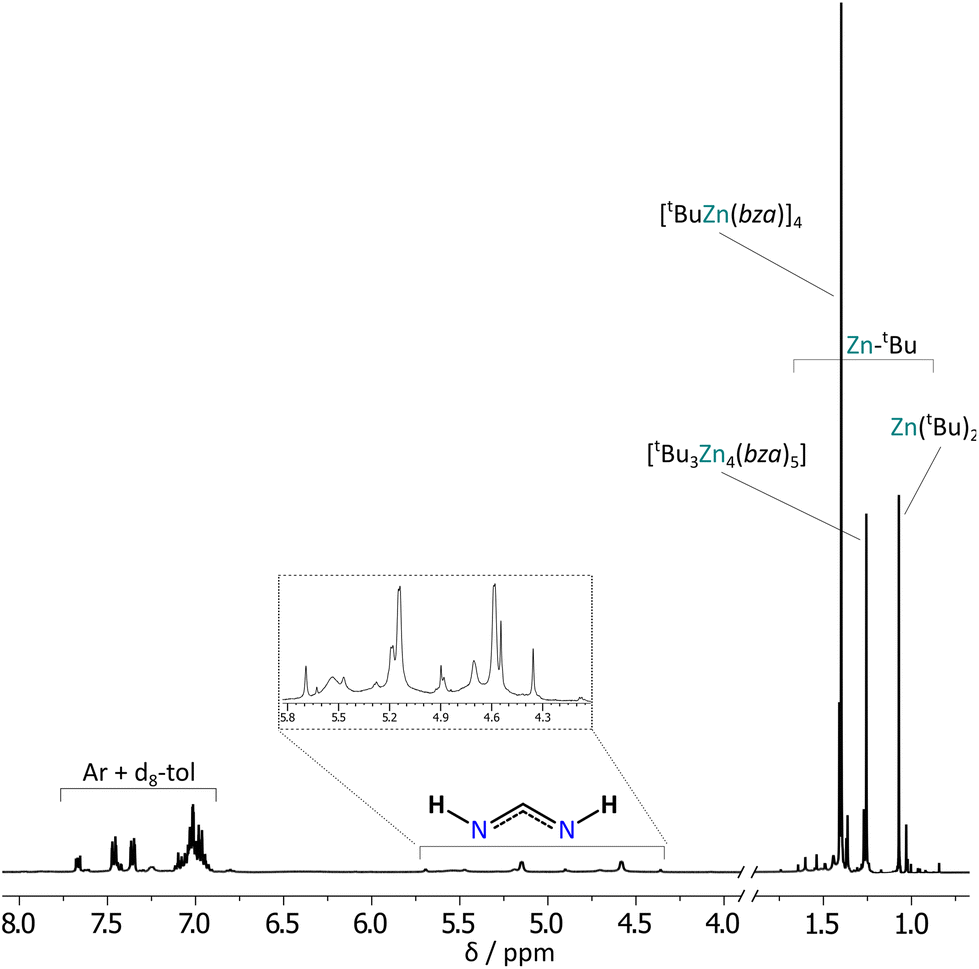 | ||
| Fig. 9 1H NMR spectrum of 3 in d8-toluene solution. | ||
The above results show the unique ability of bza ligand for the stabilization of a vast array of non-stoichiometric alkylzinc complexes, including [RZn3(bza)5]-, [R3Zn4(bza)5]-, and [R4Zn5(bza)6]-type systems. To the best of our knowledge, non-stoichiometric trinuclear and tetranuclear alkylzinc complexes were so far characterized only as [R2Zn3L4] and [R2Zn4L6]-type coordination systems (either as linear spiro-type structures,13,15 or clusters with the trigonal-, or square-planar arrangement of metal centers28,38) and [RZn3L5]- as well as [R3Zn4L5]-type structural motives remain unexplored. In turn, an interesting [R4Zn5L6]-type structure was characterized for an ethylzinc acetate, comprising a central six-coordinate inorganic Zn ion in a tetrahedral surrounding of four alkylzinc centers connected by six μ3-acetate ligands.39 A similar structure may be adapted by [R4Zn5(bza)6]-type complexes.
Conclusion
In conclusion, we investigated the solid state and solution structure of a series of alkylzinc complexes incorporating N,N′-diaryl substituted and N,N′-unsubstituted amidinate ligands. We found that [RZnL]x-type amidinate moieties are essentially unstable and tend to undergo ligand scrambling reactions according to Schlenk equilibria leading to more thermodynamically stable non-stoichiometric coordination systems, like [R2Zn3L4]- and [R3Zn4L5]-type complexes among others. This process is significantly influenced by the size of alkylzinc species and more sterically demanding tert-butyl groups favor the stabilization of the [RZnL]x-type system. Interestingly, while the dip-based complexes maintain their solid-state structure in solution, the dissolution of alkylzinc compounds stabilized by bza ligands leads to complicated mixtures of moieties with various stoichiometries. Importantly, the crystallographic and spectroscopic investigations indicate that the interplay between primary and secondary coordination spheres is an important factor controlling the character of resulting products. Moreover, as far as we know, we presented the first systematic characterization of alkylzinc systems stabilized by N,N′-unsubstituted amidinate ligands. The results provide a better understanding of the factors controlling the structure and stability of alkylzinc amidinates and shed new light on their complex behavior in solution, which in the future may contribute to the development of new efficient reaction systems.Experimental section
General remarks
Organometallic reagents were handled under a nitrogen atmosphere by using the standard Schlenk technique. Dimethylzinc (ABCR), diethylzinc (ABCR), bza–H (Sigma-Aldrich), and dipf–H (ABCR) were purchased from commercial vendors and used as received. Di-tert-butylzinc was synthesized according to a literature procedure40 and purified by careful sublimation in the dark. All solvents were purified and dried using MBraun Solvent Purification System (SPS). The standard NMR and FTIR spectra were acquired on Bruker AVANCE III HD (400 MHz) and Bruker-Tensor II (ATR) spectrometers, respectively. The DOSY NMR experiments were carried out on Bruker AVANCE II (300 MHz) spectrometer. Elemental analyses were performed on the Elementar VarioMicro Cube analyzer.Synthesis of 1Me
A solution of ZnMe2 in hexane (2 M, 1.5 mL, 3 mmol) was added to a suspension of dipf–H (784 mg, 4 mmol) in toluene (10 mL) cooled to −78 °C. The reaction mixture was stirred at room temperature for 4 h and then crystalized at 5 °C with the addition of hexane to give high-quality colorless crystals of 1Me (isolated yield 86%, 866 mg). Alternatively, the solvent can be removed under vacuum to give the product as a white crystalline powder in a practically quantitative yield. Note, that similar reactions with 1:1 and 2:1 ZnMe2 to dipf–H molar ratio followed by crystallization at 5 °C also give high-quality colorless crystals of 1Me (yield 76%, 765 mg, and 79%, 796 mg for 1:1 and 2:1 molar ratio, respectively). 1H NMR (C6D6, 400 MHz): δ = 8.24 (s, 2H, NC(H)N), 7.86 (s, 2H, NC(H)N), 7.52 (d, 4H, Ar), 7.34–6.72 (m, 26H, Ar), 6.47 (m, 6H, Ar), 5.75 (d, 4H, Ar), −0.27 (s, 6H, CH3); 13C NMR (C6D6, 100 MHz): δ = 169.5, 166.3, 150.8, 150.1, 149.5, 149.0, 129.8, 129.6, 129.0, 125.4, 125.2, 124.8, 124.7, 124.5, 124.4, 123.9, 123.7, −10.5; FTIR (ATR, cm−1): 3056 (w), 3029 (w), 2897 (w), 2834 (w), 1610 (m), 1595 (w), 1554 (s), 1484 (s), 1450 (w), 1366 (m), 1359 (s), 1213 (s), 1080 (m), 1027 (m), 993 (s), 772 (s), 756 (s), 694 (s), 648 (m), 526 (s). Elemental analysis (%) calcd for C54H50Zn3N8: C 64.40, H 5.00, N 11.13; found: C 64.43, H 4.91, N 11.09.
Synthesis of 1Et
The procedure was similar to that described for 1Me. Using ZnEt2 in hexane (2 M, 1.5 mL, 3 mmol) and dipf–H (784 mg, 4 mmol) compound 1Et was obtained as colorless crystals after crystallization at 5 °C (isolated yield: 921 mg, 89%). 1H NMR (C6D6, 400 MHz): δ = 8.36 (s, 2H, NC(H)N), 7.85 (s, 2H, NC(H)N), 7.51 (d, 4H, Ar), 7.25–6.75 (m, 26H, Ar), 6.45 (m, 6H, Ar), 5.72 (d, 4H, Ar), 1.19 (t, 6H, CH3), 0.50 (q, 4H, CH2); 13C NMR (C6D6, 100 MHz): δ = 168.5, 166.0, 150.6, 149.8, 149.2, 148.9, 129.4, 129.3, 128.8, 125.0, 124.6, 124.0, 123.9, 123.6, 123.3, 13.5, 2.7; FTIR (ATR, cm−1): 3056 (w), 3029 (w), 2889 (w), 2849 (w), 1609 (m), 1557 (s), 1486 (s), 1451 (w), 1352 (s), 1296 (m), 1209 (s), 1078 (w), 1028 (w), 989 (m), 770 (m), 755 (s), 692 (s), 644 (m), 608 (m), 522 (s). Elemental analysis (%) calcd for C56H54Zn3N8: C 64.97, H 5.26, N 10.82; found: C 65.04, H 5.13, N 10.75.Synthesis of 2
A solution of Zn(tBu)2 in hexane (0.8 M, 1.25 mL, 1 mmol) was added to a suspension of dipf–H (196 mg, 1 mmol) in toluene (4 mL) cooled to −15 °C. Then the reaction mixture was stirred at room temperature for 4 h and crystalized at 5 °C with the addition of hexane to give high-quality colorless crystals of 2 (isolated yield 289 mg, 91%,). 1H NMR (C6D6, 400 MHz): δ = 7.68 (s, 2H, NC(H)N), 7.09 (t, 8H, mAr), 6.92 (t, 4H, pAr), 6.84 (d, 8H, oAr), 1.47 (s, 18H, tBu); 13C NMR (C6D6, 100 MHz): δ = 161.6, 148.5, 129.9, 124.3, 122.0, 33.4, 24.8; FTIR (ATR, cm−1): 3056 (w), 3023 (w), 2931 (w), 2916 (w), 2810 (m), 1661 (m), 1604 (m), 1547 (s), 1537 (s), 1484 (s), 1406 (m), 1320 (s), 1220 (s), 1080 (m), 977 (s), 929 (m), 810 (m), 766 (s), 756 (s), 690 (s), 649 (m), 523 (m). Elemental analysis (%) calcd for C34H40Zn2N4: C 64.26, H 6.34, N 8.82; found: C 64.35, H 6.30, N 8.75.Synthesis of methyl- and ethylzinc derivatives of bza
A solution of the corresponding ZnR2 in hexane (2 M, 1 mL, 2 mmol) was added to a solution of bza–H (240 mg, 2 mmol) in toluene (7 mL) cooled to −78 °C. The reaction mixture was stirred at room temperature for 4 h. Crystallization at 5 °C with the addition of hexane afforded white precipitates that were characterized by 1H and 13C NMR, and FTIR spectroscopy. Methylzinc derivative: 1H NMR (d8-tol, 400 MHz): δ = 7.90–6.66 (m, Ar), 6.28–3.96 (m, 2H, NH), −0.07–(−0.65) (m, 0.9H, MeZn); 13C NMR (d8-tol, 100 MHz): δ = 184.4, 179.4, 147.0, 144.8, 136.1–127.9, −10.1; FTIR (ATR, cm−1): 3053 (w), 3020 (w), 2886 (w), 2813 (w), 1590 (s), 1543 (s), 1539 (s), 1510 (s), 1470 (s), 1437 (s), 1269 (s), 1073 (m), 1027 (m), 922 (m), 785 (m), 732 (m), 693 (s), 671 (s), 614 (s), 482 (s). Ethylzinc derivative: 1H NMR (d8-tol, 400 MHz): δ = 7.74–6.87 (m, Ar), 6.28–4.29 (m, 2H, NH), 1.97–1.48 (m, 1.2H, CH3), 0.77–0.40 (m, 0.8H, CH2Zn); 13C NMR (d8-tol, 100 MHz): δ = 180.5, 176.0, 174.3, 148.6, 143.0, 140.8, 132.0–123.9, 14.6, −0.3; FTIR (ATR, cm−1): 3053 (w), 3023 (w), 2916 (w), 2876 (m), 2838 (m), 1590 (s), 1540 (s), 1509 (s), 1467 (s), 1437 (s), 1266 (s), 1073 (m), 1027 (m), 922 (m), 786 (m), 732 (m), 693 (s), 671 (s), 589 (m), 487 (s).Synthesis of 3
A solution of Zn(tBu)2 in hexane (0.8 M, 1.25 mL, 1 mmol) was added to a solution of bza–H (120 mg, 1 mmol) in toluene (5 mL) cooled to −15 °C. The yellowish reaction mixture was stirred at room temperature for 4 h and then crystalized at 5 °C with the addition of hexane to give high-quality colorless crystals of 3 (yield 64%, 155 mg). 1H NMR (d8-tol, 400 MHz): δ = 7.71–6.87 (m, 5H, Ar), 5.75–4.25 (m, 2H, NH), 1.48–0.96 (m, 9H, tBu); 13C NMR (d8-tol, 100 MHz): δ = 181.6, 180.8, 175.9, 142.5, 139.61, 138.5, 137.4, 131.7–124.19, 35.5–34.1, 21.9–19.4; FTIR (ATR, cm−1): 3056 (w), 3023 (w), 2909 (m), 2850 (m), 2793 (s), 1592 (s), 1552 (s), 1508 (s), 1455 (s), 1432 (s), 1266 (s), 1219 (m), 1102 (m), 1027 (m), 842 (m), 809 (m), 788 (m), 747 (m), 697 (s), 671 (s), 488 (m). Elemental analysis (%) calcd for C44H64Zn4N8: C 54.68, H 6.67, N 11.59; found: C 54.70, H 6.72, N 11.69.NMR experiments
In all cases, a solution of the corresponding ZnR2 in d8-toluene (1 M, 0.2 mL, 0.2 mmol) was added to a solution of dipf–H or bza–H (0.2 mmol) in d8-toluene (1 mL) cooled to −78 °C. The reaction mixture was stirred at room temperature for 4 h and moved to NMR tubes equipped with J. Young valve for analysis. To the samples for DOSY NMR 1,2,3,4-tetraphenylnaphthalene (10 mg, 0.02 mmol) was added as an internal standard.X-Ray structure determination
The crystals were selected under Paratone–N oil, mounted on the nylon loops and positioned in the cold stream on the diffractometer. The X-ray data for complexes 1Me, 2, and 3 were collected at 100(2) K on a SuperNova Agilent diffractometer using MoKα radiation (λ = 0.71073 Å). The data were processed with CrysAlisPro.41 The X-ray data for complex 1Et were collected at 100(2) K on a Nonius KappaCCD diffractometer using MoKα radiation (λ = 0.71073 Å). The data were processed with HKL2000.42 Structures were solved by direct methods using the SHELXT program and were refined by full-matrix least–squares on F2 using the program SHELXL.43 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were added to the structure model at geometrically idealized coordinates and refined as riding atoms.Crystal data for 1Me (CCDC 2220050†), C54H50N8Zn3: M = 1007.13, monoclinic, space group P21/n (no. 14), a = 18.5634(3) Å, b = 24.5535(4) Å, c = 22.3642(4) Å, β = 107.018(2)°, U = 9747.2(3) Å3, Z = 8, F(000) = 4160, Dc = 1.373 g cm−3, μ(Mo-Kα) = 1.509 mm−1, θmax = 27.000°, 21042 unique reflections. Refinement converged at R1 = 0.0640, wR2 = 0.0844 for all data (R1 = 0.0407, wR2 = 0.0763 for 16019 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.044.
Crystal data for 1Et (CCDC 2220051†), C56H54N16Zn6: M = 1035.18, monoclinic, space group P21/n (no. 14), a = 11.7300(2) Å, b = 13.4060(3) Å, c = 31.6940(7) Å, β = 100.5050(10) °, U = 4900.42(18) Å3, Z = 4, F(000) = 2144, Dc = 1.403 g cm−3, μ(Mo-Kα) = 1.503 mm−1, θmax = 27.419°, 11110 unique reflections. Refinement converged at R1 = 0.0789, wR2 = 0.0957 for all data (R1 = 0.0474, wR2 = 0.0883 for 8048 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.039.
Crystal data for 2 (CCDC 2220052†), C34H40N4Zn2: M = 635.44, monoclinic, space group P21 (no. 4), a = 15.2257(2) Å, b = 22.1226(2) Å, c = 19.5545(2) Å, β = 104.4370(10)°, U = 6378.59(12) Å3, Z = 8, F(000) = 2656, Dc = 1.323 g cm−3, μ(Mo-Kα) = 1.532 mm−1, θmax = 25.981°, 18795 unique reflections. Refinement converged at R1 = 0.0469, wR2 = 0.1148 for all data (R1 = 0.0434, wR2 = 0.1111 for 17490 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.025.
Crystal data for 3 (CCDC 2220053†), C44H64N8Zn4: M = 966.51, tetragonal, space group I41/a (no. 88), a = 19.5833(3) Å, b = 19.5833(3) Å, c = 12.0916(3) Å, U = 4637.20(18) Å3, Z = 4, F(000) = 2016, Dc = 1.384 g cm−3, μ(Mo-Kα) = 2.083 mm−1, θmax = 29.148°, 2758 unique reflections. Refinement converged at R1 = 0.0360, wR2 = 0.0682 for all data (R1 = 0.0286, wR2 = 0.0651 for 2399 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.064.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Foundation for Polish Science Team Programme co-financed by the European Union under the European Regional Development Fund POIR.04.04.00-00-20C6/16-00 (TEAM/2016-2/14) and the National Science Centre (Grant MAESTRO 11, No. 2019/34/A/ST5/00416) for financial support.References
- K. Soai and S. Niwa, Chem. Rev., 1992, 92, 833–856 CrossRef CAS.
- J. T. B. H. Jastrzebski, J. Boersma and G. van Koten, in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewiński, Chem. Sci., 2015, 6, 3102–3108 RSC.
- A. Raheem Keeri, I. Justyniak, J. Jurczak and J. Lewiński, Adv. Synth. Catal., 2016, 358, 864–868 CrossRef CAS.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem., 2020, 12, 412–423 CrossRef CAS PubMed.
- H. Pellissier, Coord. Chem. Rev., 2021, 439, 213926 CrossRef CAS.
- Y. Geiger, Chem. Soc. Rev., 2022, 51, 1206–1211 RSC.
- M. Terlecki, M. Wolska-Pietkiewicz and J. Lewiński, in Nanomaterials Via Single-Source Precursors, ed. A. W. Apblett, A. R. Barron and A. F. Hepp, Elsevier, 2022, pp. 245–280 Search PubMed.
- K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443–2448 CrossRef CAS.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewiński and G. De Paëpe, Angew. Chem., Int. Ed., 2019, 58, 17163–17168 CrossRef CAS PubMed.
- M. Terlecki, S. Badoni, M. K. Leszczyński, S. Gierlotka, I. Justyniak, H. Okuno, M. Wolska-Pietkiewicz, D. Lee, G. De Paëpe and J. Lewiński, Adv. Funct. Mater., 2021, 31, 2105318 CrossRef CAS.
- J. Rio, L. Perrin and P.-A. Payard, Eur. J. Org. Chem., 2022, e202200906 CAS.
- S. Komorski, M. K. Leszczyński, I. Justyniak and J. Lewiński, Dalton Trans., 2020, 49, 17388–17394 RSC.
- E. M. Hanada, K. Jess and S. A. Blum, Chem. – Eur. J., 2020, 26, 15094–15098 CrossRef CAS PubMed.
- Ł. Mąkolski, V. Szejko, K. Zelga, A. Tulewicz, P. Bernatowicz, I. Justyniak and J. Lewiński, Chem. – Eur. J., 2021, 27, 5666–5674 CrossRef PubMed.
- M. Dochnahl, K. Löhnwitz, J. W. Pissarek, M. Biyikal, S. R. Schulz, S. Schön, N. Meyer, P. W. Roesky and S. Blechert, Chem. – Eur. J., 2007, 13, 6654–6666 CrossRef CAS PubMed.
- Ł. Mąkolski, K. Zelga, R. Petrus, D. Kubicki, P. Zarzycki, P. Sobota and J. Lewiński, Chem. – Eur. J., 2014, 20, 14790–14799 CrossRef PubMed.
- W. Bury, I. Justyniak, D. Prochowicz, A. Rola-Noworyta and J. Lewiński, Inorg. Chem., 2012, 51, 7410–7414 CrossRef CAS PubMed.
- W. Bury, A. M. Walczak, M. K. Leszczyński and J. A. R. Navarro, J. Am. Chem. Soc., 2018, 140, 15031–15037 CrossRef CAS PubMed.
- M. Terlecki, S. Sobczak, M. K. Leszczyński, A. Katrusiak and J. Lewiński, Chem. – Eur. J., 2021, 27, 13757–13764 CrossRef CAS PubMed.
- M. Terlecki, I. Justyniak, M. K. Leszczyński and J. Lewiński, Commun. Chem., 2021, 4, 133 CrossRef CAS PubMed.
- J. Lewiński, W. Sliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826–4829 CrossRef PubMed.
- T. Pietrzak, I. Justyniak, M. Kubisiak, E. Bojarski and J. Lewiński, Angew. Chem., Int. Ed., 2019, 58, 8526–8530 CrossRef CAS PubMed.
- T. Chlupatý and A. Růžička, Coord. Chem. Rev., 2016, 314, 103–113 CrossRef.
- F. T. Edelmann, in Advances in Organometallic Chemistry, 2008, vol. 57, pp. 183–352 Search PubMed.
- A. Baishya, L. Kumar, M. K. Barman, H. S. Biswal and S. Nembenna, Inorg. Chem., 2017, 56, 9535–9546 CrossRef CAS PubMed.
- M. S. Khalaf, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2008, 4288–4295 RSC.
- Y. J. Tsai, W. Lo and Q. Zhao, Polyhedron, 2015, 97, 39–46 CrossRef CAS.
- S. Schmidt, S. Schulz, D. Bläser, R. Boese and M. Bolte, Organometallics, 2010, 29, 6097–6103 CrossRef CAS.
- J. Lewiński, M. Dranka, I. Kraszewska, W. Sliwiński and I. Justyniak, Chem. Commun., 2005, 4935–4937 RSC.
- Z. Wróbel, I. Justyniak, I. Dranka and J. Lewiński, Dalton Trans., 2016, 45, 7240–7243 RSC.
- M. A. Bhide, J. A. Manzi, C. E. Knapp and C. J. Carmalt, Molecules, 2021, 26, 3165 CrossRef CAS PubMed.
- J. Barker, D. R. Aris, N. C. Blacker, W. Errington, P. R. Phillips and M. G. H. Wallbridge, J. Organomet. Chem., 1999, 586, 138–144 CrossRef CAS.
- K. Korona, M. Terlecki, I. Justyniak, M. Magott, J. Żukrowski, A. Kornowicz, D. Pinkowicz, A. Kubas and J. Lewiński, Chem. – Eur. J., 2022, 28, e202200620 CrossRef CAS PubMed.
- K. Korona, A. Kornowicz, I. Justyniak, M. Terlecki, A. Błachowski and J. Lewiński, Dalton Trans., 2022, 51, 16557–16564 RSC.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- A. Kreyenschmidt, S. Bachmann, T. Niklas and D. Stalke, ChemistrySelect, 2017, 2, 6957–6960 CrossRef CAS.
- S. J. Birch, S. R. Boss, S. C. Cole, M. P. Coles, R. Haigh, P. B. Hitchcock and A. E. H. Wheatley, Dalton Trans., 2004, 3568–3574 RSC.
- K. L. Orchard, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2009, 28, 5828–5832 CrossRef CAS.
- J. Lewiński, M. Dranka, W. Bury, W. Śliwiński, I. Justyniak and J. Lipkowski, J. Am. Chem. Soc., 2007, 129, 3096–3098 CrossRef PubMed.
- CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, 1997, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2220050–2220053. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03722j |
| This journal is © The Royal Society of Chemistry 2023 |