
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorescent cyclometalated platinum(II) complexes with phenyldiazine N^C ligands†
Mariia
Hruzd
a,
Samia
Kahlal
a,
Nicolas
le Poul
 b,
Laurianne
Wojcik
b,
Marie
Cordier
a,
Jean-Yves
Saillard
a,
Julián
Rodríguez-López
c,
Françoise
Robin-le Guen
a,
Sébastien
Gauthier
*a and
Sylvain
Achelle
*a
b,
Laurianne
Wojcik
b,
Marie
Cordier
a,
Jean-Yves
Saillard
a,
Julián
Rodríguez-López
c,
Françoise
Robin-le Guen
a,
Sébastien
Gauthier
*a and
Sylvain
Achelle
*a
aUniv. Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35000 Rennes, France. E-mail: sebastien.gauthier@univ-rennes1.fr; sylvain.achelle@univ-rennes1.Fr
bUniversité de Bretagne Occidentale, Laboratoire de Chimie, Electrochimie Moléculaires et Chimie Analytique, UMR CNRS 6521, UFR Science et Techniques, 6 Avenue Victor le Gorgeu, CS 93837, Brest Cedex 3, France
cUniversidad de Castilla-La Mancha, Área de Química Orgánica, Facultad de Ciencias y Tecnologías Químicas, Avda. Camilo José Cela 10, 13071, Ciudad Real, Spain
First published on 12th January 2023
Abstract
A series of phosphorescent platinum(II) complexes containing various phenyldiazine-type bidentate N^C ligands have been successfully synthesized and characterized. Structural modifications have been made to bidentate cyclometalating ligands regarding the nature of the diazine ring (pyrimidine, pyrazine and quinazoline), the substituent groups at the C4 position of the pyrimidine ring (OCH3, CF3) and the EDGs at the para position of the Pt atom (OCH3, Ph, NPh2, carbazol). In addition, the electronic properties of the azaheterocyclic ancillary ligand have been modulated in this series of complexes (pyridine, 4-methoxy-pyridine or pyrimidine). X-ray diffraction studies have been performed on three complexes, revealing Pt(II) ions in a distorted square-planar geometrical environment with no Pt(II)⋯Pt(II) interactions but with moderate π–π interactions in the solid-state structure. Electrochemical and computational studies suggest a ligand-centered reduction on the diazine ligands with, in some cases, additional contribution from the azaheterocyclic ancillary ligand, whereas oxidation occurs on the Pt-phenyl ring substituent moieties. All complexes exhibit phosphorescence emission ranging from green to red/near-infrared, both in solution and in the solid state. Complexes bearing a 2-(3-methoxyphenyl)pyrimidine ligand show the best PLQY of the series, up to 52% in a CH2Cl2 solution and 20% in the solid state. Furthermore, the solid state PLQY of one of the near-infrared emitting phenylquinazoline complex has been found to be 6%.
Introduction
Organic light emitting diodes (OLEDs), initially developed by Tang and VanSlyke,1 have been a subject of intensive research over the last two decades because of their attractive application in flexible displays.2 Due to the spin statistics, excitons are produced with a triplet![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :singlet ratio of 3:1, thus limiting the internal quantum efficiency (IQE) of fluorescent OLEDs to 25%.3 A second generation of OLEDs, discovered in 1998,4 based on phosphorescent emitters (PhOLEDs) has been extensively developed.5 These materials can harvest both singlet and triplet excitons through direct trapping or an energy transfer process (intersystem crossing), leading to a theoretical IQE of 100%.6 In this context, transition metal-based materials have been widely studied as phosphorescent emitters for OLEDs due to the strong spin–orbit coupling exhibited by the metal. Cyclometalated iridium(II) complexes are the benchmark emitters for PhOLEDs7 but cyclometalated Pt(II) complexes have also been developed for this application.8 The distinctive square-planar geometry of the Pt(II) complexes allows for intimate Pt(II)⋯Pt(II) contacts and intermolecular π–π stacking interactions, enabling metal–metal-to-ligand charge transfer (MMLCT) and excimer π–π* excited states.9 This can have detrimental effects on photoluminescence properties, but can also be used for red or near-infrared emission.10 Cyclometalated Pt(II) complexes have also been described for their thermally activated delayed fluorescence (TADF).11
:singlet ratio of 3:1, thus limiting the internal quantum efficiency (IQE) of fluorescent OLEDs to 25%.3 A second generation of OLEDs, discovered in 1998,4 based on phosphorescent emitters (PhOLEDs) has been extensively developed.5 These materials can harvest both singlet and triplet excitons through direct trapping or an energy transfer process (intersystem crossing), leading to a theoretical IQE of 100%.6 In this context, transition metal-based materials have been widely studied as phosphorescent emitters for OLEDs due to the strong spin–orbit coupling exhibited by the metal. Cyclometalated iridium(II) complexes are the benchmark emitters for PhOLEDs7 but cyclometalated Pt(II) complexes have also been developed for this application.8 The distinctive square-planar geometry of the Pt(II) complexes allows for intimate Pt(II)⋯Pt(II) contacts and intermolecular π–π stacking interactions, enabling metal–metal-to-ligand charge transfer (MMLCT) and excimer π–π* excited states.9 This can have detrimental effects on photoluminescence properties, but can also be used for red or near-infrared emission.10 Cyclometalated Pt(II) complexes have also been described for their thermally activated delayed fluorescence (TADF).11
The main classes of phosphorescent Pt(II) complexes are based on tridentate N^C^N or N^N^C ligands, with halogen and pseudo-halogen or acetylide ancillary ligands.12 Platinum complexes with the bidentate N^C ligand are less in number and are often based on 2-phenylpyridine (ppy)-type ligands. They generally contain bidentate coligands such as acetylacetonate (acac).13 Platinum(II) complexes bearing ppy-type N^C ligands with pyridine and chloride ancillary monodentate ligands have been frequently used as precursors for other luminescent ppy complexes.14a,b Recently, Zhao et al. have studied the photophysical properties of such complexes.14c The free rotation of the coordinated pyridine ligand reduced the efficiency of intersystem crossing and the radiative decay of the first triplet excited state (T1) in solution, leading to low emission in solution. Nevertheless, the phosphorescence ability of these complexes was restored in a solid matrix, where the rotation of the pyridine ring could be restricted. Aggregation-induced phosphorescence has been demonstrated in these complexes and analogues bearing an acetylide ligand instead of chloride.15 The non-planar conformation of the pyridine ring may prevent excimer formation in the solid state.
On the other hand, diazines are six-membered aromatic heterocycles with two nitrogen atoms. Depending on the position of N atoms, pyridazine (1,2-diazine), pyrimidine (1,3-diazine) and pyrazine (1,4-diazine) can be distinguished. The quinazoline ring corresponds to benzo-annulated pyrimidine. The diazine rings exhibit a stronger π-deficient character than pyridine due to the presence of a second N atom and have been used as the attracting part in numerous push–pull structures.16
For bidentate N^C ligands, the combination of the strong σ-donor effect of phenylate and the π-accepting character of the azaheterocyclic ring has been shown to result in a high ligand-field for the coordinated metal, which increases the energy of quenching d–d states while reducing emissive metal-to-ligand charge transfer (MLCT) and ligand-centered excited states.6a In this context, the replacement of pyridine with diazine rings appears as an interesting option. Thus, it has been shown that a 2-(4-N,N-diphenylaminophenyl)pyrimidine Pt(II) complex bearing pyridine and chloride auxiliary ligands exhibits strong green phosphorescence emission in a solid matrix and promising electroluminescent properties.14c We have recently demonstrated that functionalization of 2-phenylpyrimidine with electron-donating groups (EDGs) at the meta position of the phenyl ring induces a bathochromic shift in the emission of the corresponding Pt(II) complexes associated with a significant increase in the intensity of phosphorescence in solution.17
In this contribution, we have designed a series of 24 Pt(II) complexes based on N^C phenyldiazine ligands and monodentate ancillary ligands (Chart 1). Various structural modifications have been made concerning the nature of the diazine ring (pyrimidine, pyrazine and quinazoline), the substituent groups at the C4 position of the pyrimidine ring, the EDGs at the para position of the Pt atom in the phenyldiazine ligand, and the azaheterocyclic ancillary ligand.
 | ||
| Chart 1 Chemical structures of the cyclometalated platinum-based complexes 1–24 investigated in this work. | ||
Results and discussion
Synthesis and characterization
The synthesis of the N^C ligands 25–38 was easily accomplished in good yields from commercially available chlorodiazine and the appropriate arylboronate ester derivatives. The Suzuki–Miyaura cross-coupling reaction was used according to a method previously reported in the literature (ESI, Scheme S1†).16–18 The N^C cyclometalated chloro-Pt(II) complexes 1–24 were prepared according to the well-established method of synthesis for the traditional counterparts, by treating the Pt(II) μ-chloro-bridged dimer complexes with the pyridine, 4-methoxy-pyridine or pyrimidine ancillary ligand, in low to moderate yields (Schemes 1–4).14 These air-stable compounds were isolated in high purity as solids after column chromatography on silica gel with the appropriate eluent. It should be noted that the yields of the complexes with phenylpyrazine ligands are significantly lower than those of their pyrimidine and quinazoline analogues due to the electronic effect of the second N atom in the conjugated position to the Pt atom. The N^C ligands 25–38 and chloro-platinum complexes 1–24 were characterized by NMR (1H and 13C) and high-resolution mass spectrometry (HRMS). The characterization data were found to be in complete agreement with the proposed structures as detailed in the ESI.† | ||
| Scheme 1 Synthesis of N^C cyclometalated chloro-Pt(II) complexes 1–15. | ||
 | ||
| Scheme 2 Synthesis of N^C cyclometalated chloro-Pt(II) complex 16. | ||
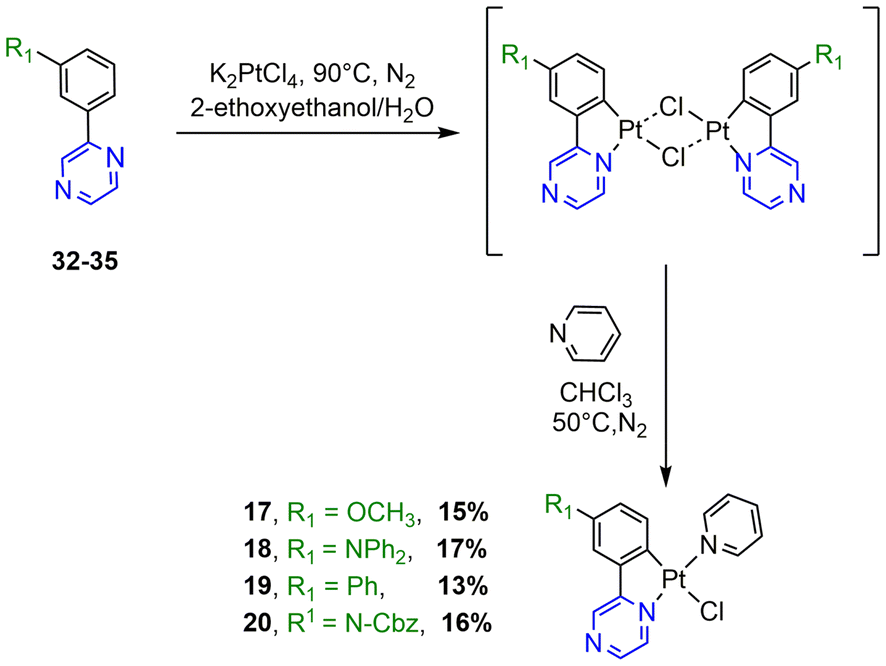 | ||
| Scheme 3 Synthesis of N^C cyclometalated chloro-Pt(II) complexes 17–20. | ||
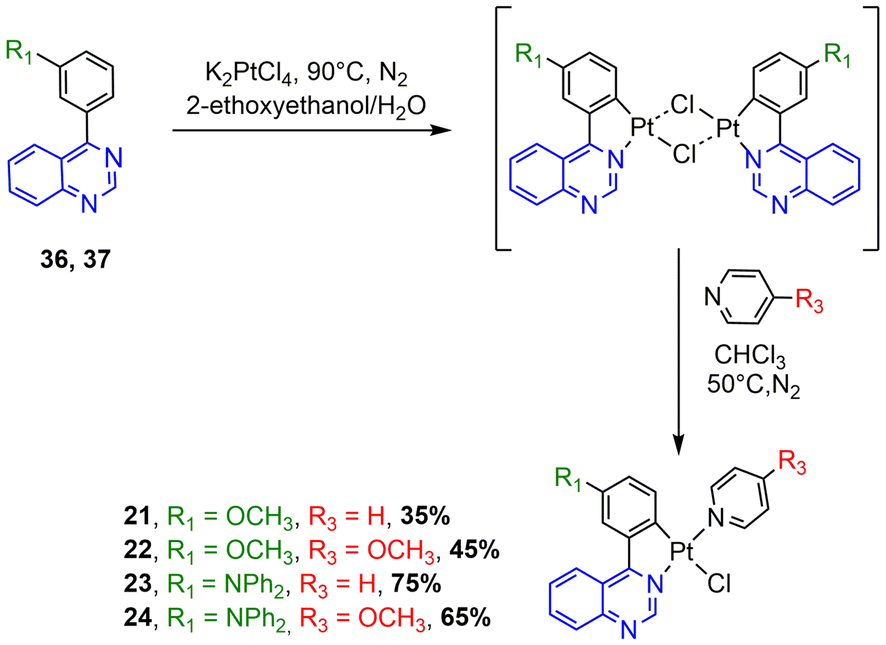 | ||
| Scheme 4 Synthesis of N^C cyclometalated chloro-Pt(II) complexes 21–24. | ||
X-ray crystal structures
Single crystals of chloro-platinum complexes 7, 12, and 14 were obtained by slow evaporation of a hexane/ethyl acetate (2/1) solution of the complex at room temperature. Their molecular structures were determined by X-ray crystallography analysis (Fig. 1 and S63, S64†). The crystal data and structure refinement details are provided in the ESI (Tables S1–S3†), and selected bond lengths and angles are listed in Table 1. As can be seen from the X-ray structures, the central platinum ion is coordinated to a bidentate cyclometalating N^C ligand and two monodentate ligands (chloride and pyridine). The coordination sphere of the Pt(II) cation in all complexes (7, 12, and 14) adopts a distorted square-planar geometry with trans-C,Cl, trans-N,N chelating disposition, characteristic of d8 metal complexes. The C1–Pt1–N2 angles (81.19–81.51°) are found to deviate from the ideal angle 90° due to the restricted bite angle of the bidentate C^N ligands. This is accompanied by a concomitant opening of the C1–Pt1–N1 (94.66–95.38°) and N1–Pt1–Cl1 (87.17–88.70°) bond angles. The bonds chelating around the platinum ion exhibit typical bond lengths compared with some similar complexes.14,15,19 | ||
| Fig. 1 ORTEP representation of complex 7 (left) with 50% ellipsoid probability. All co-crystallized solvent molecules have been omitted for clarity. ORTEP representation of the crystal packing structure in the dimeric form: complex 7 (right) showing head-to-tail configuration. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms and all co-crystallized solvent molecules have been omitted for clarity. | ||
| 7 | 12 | 14 | |
|---|---|---|---|
| a No significant π–π interactions. | |||
| Pt1–C1 | 1.988(3) [1.983] | 1.990(3) [1.982] | 1.988(3) [1.982] |
| Pt1–N1 | 2.025(3) [2.027] | 2.021(3) [2.025] | 2.010(3) [2.025] |
| Pt1–N2 | 2.028(3) [2.018] | 2.017(3) [2.019] | 2.017(3) [2.020] |
| Pt1–Cl1 | 2.3890(9) [2.421] | 2.3952(9) [2.418] | 2.3874(9) [2.416] |
| C1–Pt1–N1 | 94.66(12) [95.6] | 95.33(13) [95.6] | 95.38(13) [95.5] |
| C1–Pt1–N2 | 81.23(12) [81.1] | 81.51(3) [81.1] | 81.19(13) [81.1] |
| N1–Pt1–N2 | 175.86(10) [176.6] | 176.84(11) [176.7] | 176.29(12) [176.6] |
| C1–Pt1–Cl1 | 176.56(10) [176.3] | 175.57(10) [176.3] | 177.26(11) [176.4] |
| N1–Pt1–Cl1 | 88.70(10) [88.1] | 88.62(9) [87.9] | 87.17(9) [88.1] |
| N2–Pt1–Cl1 | 95.41(8) [95.3] | 94.54(9) [95.3] | 96.24(9) [95.3] |
| Pt⋯Pt | 5.144 | 5.815 | 10.051 |
| π–π | ∼3.40 | ∼3.51 | —a |
Fig. 1 and S64† show that complexes 7 and 12 are packed as head-to-tail dimers in crystals. The closest Pt–Pt distances (dPt–Pt) in the crystals of 7 and 12 are 5.144 Å and 5.815 Å respectively, indicating the absence of metal–metal interactions. However, the dimers have slight overlap between the adjacent [Pt(C^N)] moieties with a vertical plane-to-plane separation (dπ–π) of ∼3.40 Å for 7 and ∼3.51 Å for 12, indicative of moderate π–π interactions for these two complexes. Notably, the molecules of complex 14 are packed as head-to-tail dimers in the crystal packing with one co-crystallized ethyl acetate solvent molecule between two Pt complexes. No significant Pt(II)⋯Pt(II) or π–π interactions could be detected, since the closest Pt–Pt distance is 10.051 Å with no direct overlap between adjacent complexes.
Electrochemical properties
Voltammetric studies were carried out in CH2Cl2 in order to decipher the redox properties of the complexes in relation with their photophysical features. Table 2 gathers the electrochemical data using a Pt working electrode. All complexes show several oxidation peaks above 0.3 V vs. Fc+/Fc and one reduction peak below ca. −1.6 V (see Fig. 2 and ESI Fig. S65–S70† for cyclic voltammograms). | ||
| Fig. 2 (A) Cyclic voltammograms of complexes 1 (black), 17 (blue), and 21 (red). (B) Cyclic voltammograms of complexes 6 (orange) and 11 (blue). CV obtained by using a Pt working electrode in 0.1 M NBu4PF6 in CH2Cl2 (E/V vs. Fc+/Fc, ν = 0.1 V s−1, C = 0.5 mM). The arrow indicates the scanning direction from the initial potential. | ||
| Complex | E°(1) | E pa(2) | E°(3) |
|---|---|---|---|
| a Irreversible peak (Epa or Epc value). b Low-intensity wave which becomes more intense after reduction at −2.3 V. c A supplementary low-intensity reversible wave is detected at −1.42 V upon reduction. | |||
| 1 | 0.78a | −2.27a | |
| 2 | 0.75a | −2.29a | |
| 3 | 0.75a | −2.39a | |
| 4 | 0.70a | −2.40a | |
| 5 | 0.83a | −2.26a | |
| 6 | 0.36 | 0.58b | −2.25a |
| 7 | 0.35 | 0.62b | −2.21a |
| 8 | 0.35 | 0.56b | −2.36a |
| 9 | 0.33 | 0.63b | −2.40a |
| 10 | 0.39 | 0.57b | −2.24a |
| 11 | 0.42 | 1.12a | −1.84 |
| 12 | 1.01a | −2.21a | |
| 13 | 0.92a | −2.27a | |
| 14 | 0.85a | −2.28a | |
| 15 | 0.75a | −2.24a | |
| 16 | 0.50 | −2.27a | |
| 17 | 0.82a | −1.97 | |
| 18 | 0.40 | 1.05a | −1.97a,c |
| 19 | 1.02a | −1.96a | |
| 20 | 0.82a | −1.94a | |
| 21 | 0.82a | −1.67 | |
| 22 | 0.77a | −1.69 | |
| 23 | 0.42 | 1.04a | −1.67 |
| 24 | 0.40 | 1.00a | −1.68 |
As a first observation, it seems clear that the nature of the diazine ring significantly affects the reduction potential of the complexes. This is best exemplified by the methoxy series (complexes 1, 17, 21), which shows reduction peaks at −2.27 V (pyrimidine), −1.97 V (pyrazine), and −1.67 V (quinazoline) against Fc+/Fc, respectively. The potential difference of 300 mV between each of these complexes (Fig. 2A) can be correlated with the electronic properties of diazine, with the most electron deficient system (quinazoline) moving the reduction peak towards the most positive values. Compared to the pyrimidine derivative, the annulated benzene ring of the quinazoline system probably favors electron delocalization upon electrochemical reduction, leading to better stabilization of the reduced species.13c Another interesting observation is that both pyrazine and quinazoline complexes exhibit a reversible reduction process, whereas the pyrimidine complex exhibits a completely irreversible reduction process. Such irreversibility may be caused by the reaction between the reduced complex and the electrolytic solution. The process could also be interpreted as a disproportionation reaction involving the reduced pyrimidine complex.20
Voltammetric studies were also carried out with complexes bearing various substituents at the 4-position of the pyrimidine ring. A striking result was obtained with the trifluoromethyl substituted complex 11. Indeed, the reduction process occurs at a much less negative potential compared to the H-substituted pyrimidine complex 6 (410 mV difference) (Fig. 2B).
On the oxidation side, the presence of a diphenylamino group on the Pt-bound phenyl moiety (complexes 6–11) induces the appearance of a reversible wave at ca. E°(1) = 0.35 V, which can be ascribed to the oxidation of the amine.14c The different substituents at the para position of the phenyl ring in relation to the Pt metal center affect the oxidation potential according to their electron donating properties. Therefore, the lowest oxidation potential value was obtained for the strongest electron-donating NPh2 group: phenyl (12: 1.07 V); 9H-carbazol-9-yl (14: 0.90 V); methoxy (1: 0.78 V), and diphenylamino (6: 0.58 V).
The redox properties of the present complexes can be compared with those already reported by Zhao and co-workers14c for an analogous 2-phenylpyrimidine complex with the diphenylamino group at the meta position of the Pt atom. Although the voltammetric behavior is similar, the first two oxidation peaks for complex 6 are found at much less positive values (approx. 300 mV). This discrepancy may be ascribed to the difference in the position of the diphenylamino group on the phenyl ring, which may affect both the amine and Pt(II) oxidation processes. In the cathodic part of the voltammogram, both complexes show irreversible reduction peaks at around −2.2 V (170 mV difference). Accordingly, this process was proposed to occur primarily at the pyrimidine and ancillary pyridine groups. Further comparison can be made with tridentate C^N^N–Pt complexes. For example, complexes 1 and 5 display oxidation peaks at more positive potential values than the reported N^C^N–Pt chloro analogue, where N = pyrimidine and C = methoxybenzene.12f Conversely, the reduction occurs at more negative values. Even if the comparison is not strictly correct, this difference suggests that the tridentate structure stabilizes the oxidized and reduced states and lowers the HOMO–LUMO gap compared to the bidentate case.
Photophysical properties
The UV/vis absorption and photoluminescence properties of complexes were measured in degassed CH2Cl2 solution and in potassium bromide pellets (2 wt%). The resulting data are summarized in Table 3.| CH2Cl2a |
KBr (2 wt%) | |||||||
|---|---|---|---|---|---|---|---|---|
| UV/Vis | PL | Stokes shiftc, cm−1 | PL | |||||
| λ max, nm (ε, mM−1 cm−1) | λ max, nm | τ, μs | Φ PL , % | λ max, nm | τ, μs | Φ PL , % | ||
| a All spectra were recorded at room temperature at c ∼ 1.0 × 10−5 M. The solutions were deoxygenated by bubbling N2. b Photoluminescence quantum yield (±10%) determined in relation to 9,10-bisphenylethynylanthracene in cyclohexane (ΦPL = 1.00).21 c Calculated using the less energetic absorption and the more energetic emission bands. d Absolute value measured with an integrating sphere (powder). e Emission too low to permit accurate measurement. | ||||||||
| 1 | 254 (42.3), 294 (14.0), 347 (6.22), 427 (2.95) | 556, 582 | 12.9 | 45 | 5434 | 531, 593 | 14.8 | 4 |
| 2 | 254 (40.9), 289 (13.9), 351 (5.55), 430 (2.55) | 556, 582 | 9.7 | 54 | 5553 | 551, 589 | 11.6 | 20 |
| 3 | 253 (46.0), 297 (10.8), 341 (5.89), 416 (4.57) | 545, 575 | 12.0 | 47 | 5404 | 547, 577 | 13.2 | 3 |
| 4 | 252 (38.5), 288 (18.4), 338 (5.04), 415 (2.99) | 545, 575 | 7.7 | 15 | 5632 | 562, 594 | 9.6 | 2 |
| 5 | 251 (28.8), 285 (14.3), 345 (4.69), 413 (2.49) | 543, 571 | 4.3 | 9 | 5797 | 587 | 9.6 | <1 |
| 6 | 258 (24.4), 293 (35.2), 449 (2.92) | 643 | 8.9 | 5 | 6744 | 635 | 9.7 | 2 |
| 7 | 258 (25.0), 294 (35.0), 451 (3.13) | 643 | 7.4 | 4 | 6474 | 630 | 15.6 | 8 |
| 8 | 259 (30.6), 293 (38.5), (5.71), 437 (1.67) | 630 | 6.8 | 8 | 6523 | 625 | 13.6 | 5 |
| 9 | 262 (33.6), 293 (50.0), 350 (8.24), 433 (2.17) | 630 | 6.1 | 6 | 6422 | 604 | 10.9 | 3 |
| 10 | 259 (19.9), 292 (28.7), 342 (6.53), 442 (1.74) | 630 | 6.0 | 3 | 6498 | 598, 627 | 13.4 | 5 |
| 11 | 255 (24.1), 299 (44.4), 345 (3.57), 383 (1.45) 477 (0.84) | 642 | —e | <1 | —e | 691 | 9.9 | 2 |
| 12 | 277 (41.3), 346 (3.94), 404 (1.27) | 508, 538 | 8.5 | 5 | 4705 | 495, 531, 566 | 12.0 | 7 |
| 13 | 277 (35.1), 348 (4.06), 402 (1.79) | 508, 538 | 4.4 | 7 | 6253 | 517sh, 555, 590 | 11.5 | 3 |
| 14 | 241 (68.9), 261 (52.5), 291 (38.7), 341 (10.6), 412 (2.29) | 547 | 4.7 | 12 | 5990 | 517, 568 | 12.2 | 5 |
| 15 | 270 (36.4), 330 (9.11), 342 (10.0), 415 (2.46) | 543 | 3.9 | 15 | 6094 | 567 | 13.2 | 14 |
| 16 | 271 (26.7), 298 (12.0), 341 (14.1), 379 (8.33), 455 (1.84) | 583 | 7.3 | 22 | 5040 | 630 | 14.5 | 1 |
| 17 | 256 (33.0), 319 (13.1), 357 (7.82), 466 (2.12) | 630 | 6.3 | 12 | 5725 | 634, 699 | 17.4 | 4 |
| 18 | 264 (53.5), 283 (60.0), 372 (4.31), 490 (1.57) | —e | —e | —e | —e | 684 | 7.4 | 2 |
| 19 | 270 (61.2), 321 (19.6), 347 (11.9), 445 (2.55) | 583 | 7.9 | 30 | 6275 | 652 | 14.6 | 3 |
| 20 | 243 (56.8), 267 (53.6), 328 (13.2), 343 (10.6), 452 (1.02) | 608 | 5.6 | 9 | 5928 | 638 | 10.5 | 2 |
| 21 | 274 (29.4), 351 (10.5), 401 (4.53), 504 (3.32) | 671 | 5.4 | 26 | 4975 | 728 | 9.2 | 3 |
| 22 | 246 (21.3), 259 (23.1), 276 (22.4), 351 (8.05), 367 (7.53), 408 (3.98), 511 (3.13) | 677 | 5.1 | 16 | 4776 | 720 | 6.3 | 6 |
| 23 | 280 (48.9), 299 (49.8), 345 (24.8), 364 (16.0), 397 (6.71), 546 (2.79), 563 (3.17) | —e | —e | —e | —e | 795 | 3.5 | <1 |
| 24 | 247 (24.6), 292 (38.9), 364 (12.9), 396 (6.44), 566 (3.23) | —e | —e | —e | —e | —e | —e | —e |
Several absorption bands at below 400 nm are observed for all complexes, with the most intense one being attributed to intraligand n–π* and π–π* transitions.17 Furthermore, one or, in some cases, two significantly less intense bands appear between 400 and 570 nm. The absorption spectra of phenylpyrimidine complexes 1, 6, 12, and 14 are shown in Fig. 3 (see the ESI, Fig. S71–S76† for other complexes). These bands have metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) character. In the phenylpyrimidine series, the less energetic absorption band red-shifts with increasing the strength of the electron-donating group at the para position of the Pt atom on the phenyl moiety: from 404 nm for phenyl (12) to 412 nm for 9H-carbazol-9-yl (14), 427 nm for methoxy (1), and 449 nm for diphenylamino (6). The same trend is observed in the phenylpyrazine and phenylquinazoline series. 9H-carbazol-2-yl-pyrimidine complex 16 exhibits a less energetic absorption band that is red-shifted compared to those of all the phenylpyrimidine analogues. In the phenylpyrimidine series, the presence of an electron-donating methoxy group on the pyrimidine ring induces a slight blue-shift of up to 18 nm (1, 2, 6, and 7vs.3, 4, 8, and 9 respectively). On the other hand, a strong electron-withdrawing trifluoromethyl group on the pyrimidine ring (compound 11) induces a red-shift of 28 nm regarding 6 (Fig. S72†). Meanwhile, in the pyrimidine series, a change in the ancillary ligand (pyridine with 4-methoxypyridine or pyrimidine) does not significantly modify the position of the absorption band.
 | ||
| Fig. 3 (A) UV-Vis absorption spectra of phenylpyrimidine complexes 1, 6, 12, and 14 in CH2Cl2 solution (C ∼ 10−5 M). (B) TD-DFT-simulated UV-vis absorption spectra of complexes 1, 6, 12, and 14. | ||
Phenylpyrazine and phenylquinazoline analogues have a less energetic absorption band that is notably red-shifted as can be seen, for example, in the methoxy series: 427 nm for the phenylpyrimidine complex 1, 466 nm for the phenylpyrazine complex 17, and 504 nm for the phenylquinazoline complex 21 (Fig. S73†). It has been reported that this bathochromic shift can be explained by the presence of a second N atom in the conjugated position to the Pt atom in the pyrazine complexes,22 and by the more pronounced electron-withdrawing character of the quinazoline fragment.18a
Most of the complexes are luminescent in CH2Cl2 solutions with emission lifetimes ranging from 4 to 12 μs and Stokes shifts between 4700 and 6300 cm−1, characteristic of phosphorescence. The presence of strong electron-donating diphenylamino groups on the phenylpyrazine/phenylquinazoline ligands (complexes 18, 23, and 24) leads to non-emissive complexes. The latter may be explained by a too strong charge transfer that disfavors the radiative return to the ground state.23 The position of the emission band in the phenylpyrimidine complexes follows the same trend as that in the less energetic absorption band; a red-shift is observed as the strength of the electron-donating substituent increases: green emission for phenyl (12, 508/538 nm), yellow-green for 9H-carbazol-9-yl (14, 547 nm), yellow for methoxy (1, 556/582 nm), and red for diphenylamino group (6, 644 nm) (Fig. 4). In this series, the highest PLQY is observed for the methoxy complex 1 (45%). Complex 16 with the 9H-carbazol-2-yl-pyrimidine N^C ligand shows yellow-orange emission with a PLQY of 22%. The presence of a methoxy substituent on the pyrimidine ring induces a slight blue-shift (8–17 nm) of the emission (complexes 1, 2, 6, and 7vs.3, 4, 8, and 9, respectively) due to the reduction of the electron-withdrawing character of the heterocyclic part of the N^C ligand. In the phenylpyrimidine series, the modification of the azaheterocyclic ancillary ligand does not appreciably change the emission maxima (2, 4/5, 7, and 9/10vs.1, 3, 6, and 8, respectively), although the pyrimidine ligands (complexes 5 and 10) significantly decrease the PLQY.
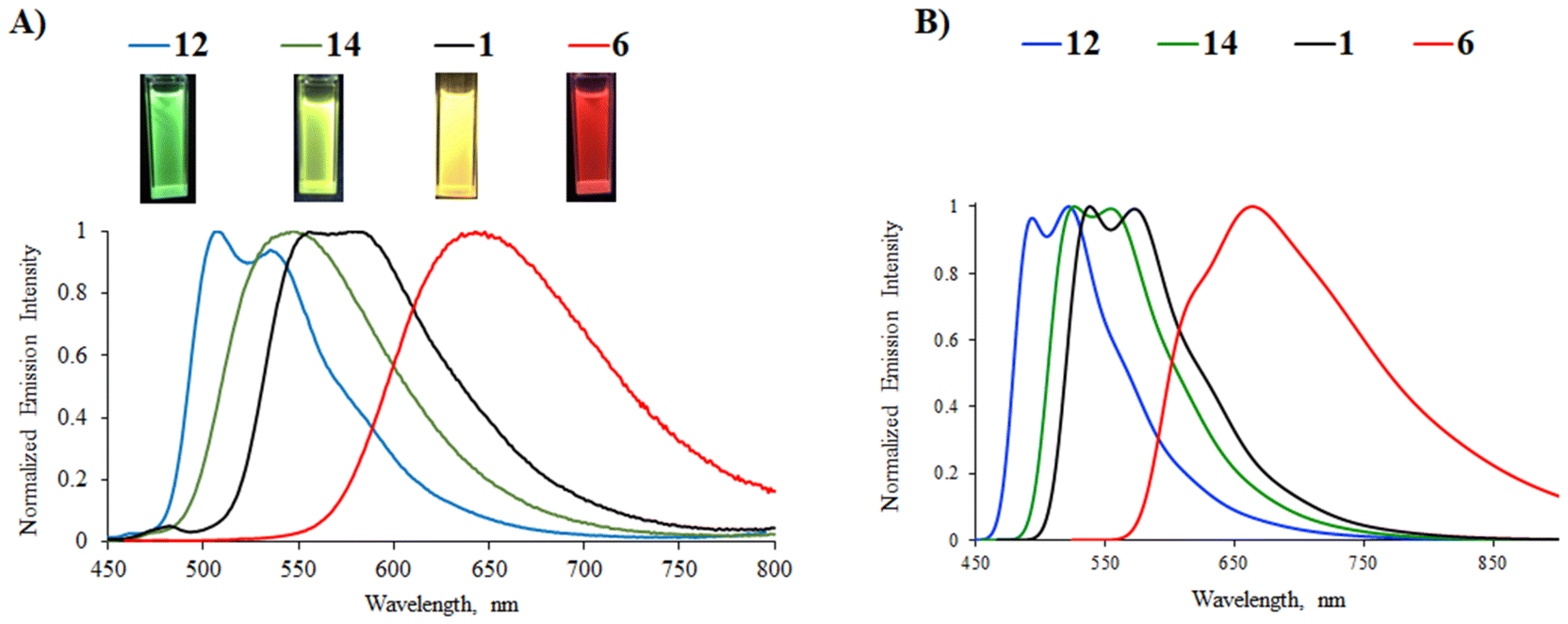 | ||
| Fig. 4 (A) Normalized emission spectra of phenylpyrimidine complexes 1, 6, 12, and 14 in deoxygenated CH2Cl2 solution (C ∼ 10−5 M). λexc = λabsmax of the lowest energy band. Inset: Pictures of the CH2Cl2 solutions taken under UV irradiation (from left to right: 12, 14, 1, and 6). (B) DFT-simulated phosphorescence spectra of complexes 12, 14, 1, and 6. | ||
Phenylpyrazine complexes 17, 19, and 20 exhibit a red-shift of around 60 nm in relation to their phenylpyrimidine analogues and exhibit lower PLQY. As for the methoxy-substituted complex 17, the emission is shifted to the orange-red region of the spectrum (Fig. 5). The emission of the methoxy-substituted phenylquinazoline complex 22 is the most red-shifted in the series (λem = 678 nm).
 | ||
| Fig. 5 Normalized emission spectra of methoxy-substituted complexes 1, 17, and 21 in deoxygenated CH2Cl2 solution (C ∼ 10−5 M). λexc = λabsmax of the lowest energy band. Inset: Picture of the CH2Cl2 solutions taken under UV irradiation (from left to right: 1, 17, and 21). | ||
Emission can also be detected for all complexes in the solid state (2 wt% in the KBr matrix), except for 24. Emission lifetimes are higher than those in CH2Cl2 solution (up to 17.4 μs for 17). As an example, the emission spectra of complexes 1, 6, 13, and 14 are shown in Fig. 6. As in solution, the solid-state emission depends on the substituents on the phenyl ring and the nature of the diazine ring. No particular red-shift or broadening of the emission band is observed, which could indicate aggregation or excimer formation, probably because the out-of-plane azaheterocyclic ancillary ligand hinders Pt(II)⋯Pt(II) and π–π interactions. Nevertheless, the emission from the pyrazine and quinazoline complexes 17–22 is red-shifted for up to 80 nm in relation to the emission in solution.
 | ||
| Fig. 6 Solid state emission (2 wt% in the KBr matrix) of complexes 1, 6, 12, and 14. λexc = λabsmax of the lowest energy band in CH2Cl2 solution. Inset: Pictures of the KBr pellets taken under UV irradiation (from left to right: 12, 14, 1, and 6). | ||
The position of emission maxima globally follows the same rules as that in CH2Cl2 solution. The highest PLQYs are measured for complexes 2 (20%), 15 (14%), and 7 (8%), all of them bearing the 4-methoxypyrimidine ancillary ligand. Phenylquinazoline complexes 21–23 exhibit emission in the near-infrared region and the PLQY of complex 22 (6%) is particularly appealing for such emission.
Computational investigations
Density functional theory (DFT) calculations at the PBE0/Def2-TZVP level were performed to optimize the geometries of compounds 1–24, except for complex 16. Solvent (CH2Cl2) effect corrections were included (see the Computational details in the ESI†). The optimized structures are shown in Fig. S91 and S92.† The optimized geometries of 6, 12, and 14 are in a very good agreement with their experimental X-ray results (see Table 1 and compare Fig. 1 with Fig. S91 and S92†). Relevant computed data are provided in Table 4. The largest HOMO–LUMO gaps (ΔEH–L = 4.02 eV) are found for complexes with phenyl-substituted pyrimidine ligands 12 and 13, whereas the smallest HOMO–LUMO gap (3.05 eV) corresponds to 11, which bears both donor (diphenylamino) and acceptor (trifluoromethyl) substituents. The HOMOs of all the complexes are similar (see selected examples in Fig. 7). They show little or no involvement of the pyridine and pyrimidine heterocycles. Assuming that the C–Pt–Cl axis is the x axis and z is perpendicular to the Pt coordination plane, they all consist of some 5dxz(Pt) atomic orbitals (AOs) (typically between 7 and 30%) antibondingly mixed with π-type orbitals of the ligands coordinated along the x axis. This ligand contribution to the HOMOs is predominant. The HOMOs of the complexes that bear a diphenylamino substituent on the phenyl ring show a large participation of this substituent, which explains their peculiar oxidation behavior (see above). For all complexes, there is only one occupied orbital with dominant metal participation. It is 5dz2 in nature and, depending on the complexes, corresponds to the HOMO−1, HOMO−2 or HOMO−3 (see the selected MO diagrams in Fig. S100–S104†). The formally occupied 5dxy, 5dxz and 5dyz AOs are in fact diluted in a large number of the occupied molecular orbitals (down to the HOMO−10/HOMO−14), in which the ligand character always dominates. The LUMOs of all the complexes are derived from the LUMOs of the diazine ligands, with a small percentage of metal participation, and, in some cases, additional contribution from the azaheterocyclic ancillary ligands (Fig. 7). | ||
| Fig. 7 Plots of the HOMO (below) and LUMO (above) of complexes 1, 6, 12, 14, and 17. | ||
| Complex | μ (D) | ΔEH–L (eV) | IEa (eV) | EAb (eV) | λ max (nm) absorption | λ max (nm) emission |
|---|---|---|---|---|---|---|
| a Electron affinity. b Ionization energy. | ||||||
| 1 | 5.94 | 3.86 | 5.54 | −2.30 | 265, 372, 418 | 538 |
| 2 | 6.90 | 3.87 | 5.51 | −2.27 | 304, 275, 418 | 540 |
| 3 | 7.04 | 3.95 | 5.50 | −2.24 | 249, 306, 406 | 533 |
| 4 | 7.45 | 3.95 | 5.47 | −2.21 | 263, 312, 408 | 571 |
| 5 | 7.99 | 3.86 | 5.56 | −2.29 | 246, 309, 408 | 567 |
| 6 | 4.07 | 3.36 | 5.10 | −2.35 | 306, 417, 472 | 664 |
| 7 | 5.82 | 3.38 | 5.08 | −2.33 | 305, 409, 472 | 651 |
| 8 | 5.18 | 3.47 | 5.09 | −2.29 | 269, 306, 459 | 692 |
| 9 | 6.56 | 3.48 | 5.07 | −2.27 | 307, 356, 457 | 607 |
| 10 | 6.44 | 3.36 | 5.13 | −2.35 | 309, 353, 465 | 678 |
| 11 | 6.99 | 3.05 | 5.16 | −2.74 | 376, 461, 526 | 682 |
| 12 | 4.11 | 4.02 | 5.75 | −2.33 | 275, 355, 400 | 522 |
| 13 | 5.29 | 4.02 | 5.72 | −2.30 | 279, 353, 402 | 496 |
| 14 | 3.58 | 3.76 | 5.56 | −2.41 | 240, 270, 413 | 526 |
| 15 | 5.48 | 3.79 | 5.55 | −2.39 | 308, 355, 412 | 550 |
| 17 | 9.04 | 3.60 | 5.57 | −2.59 | 249, 294, 465 | 590 |
| 18 | 8.85 | 3.21 | 5.16 | −2.56 | 305, 370, 503 | 713 |
| 19 | 7.26 | 3.76 | 6.20 | −2.62 | 283, 441 | 538 |
| 20 | 6.33 | 3.50 | 5.60 | −2.69 | 241, 279, 452 | 571 |
| 21 | 10.87 | 3.28 | 5.58 | −2.92 | 274, 326, 513 | 672 |
| 22 | 10.95 | 3.28 | 5.55 | −2.90 | 327, 357, 515 | 729 |
| 23 | 9.36 | 2.80 | 5.15 | −2.95 | 353, 410, 595 | 805 |
| 24 | 10.22 | 2.82 | 5.14 | −2.93 | 317, 412, 592 | 951 |
The computed ionization energies correlate linearly with the first recorded oxidation peak (see above), except for compounds 12, 14, and 19, for which the match is more approximate (see Fig. S93†). In the case of these three compounds, a peculiarly explicit role of the electrolyte or solvent in the cationic form, not considered in the calculations, could be tentatively predicted. There is also a slightly lower correlation between the computed electron affinities and the recorded E°(3) potentials (Fig. S94†). It should be noted that most of the electrochemical potentials considered in Fig. S93 and S94† correspond to the irreversible peaks (see Table 2), which could be the reason for the appearance of some (small) deviations away from the root-mean-square line.
TD-DFT calculations were performed for complexes 1–24 (except for 16) to better understand their optical behavior. The simulated spectra (singlet → singlet transitions) satisfactorily reproduce the shape of the experimental spectra and their λmax values (Table 4) match reasonably well with their recorded counterparts (Table 3). The simulated spectra of 1, 6, 12, and 14 are illustrated in Fig. 3 and those of the other complexes are shown in Fig. S95–S97.† For all of them, the weak band of the lowest energy with λmax > 400 nm is associated with a HOMO–LUMO transition. The higher energy bands are majorly LLCT in nature (mainly with L = substituted diazine) with some MLCT contribution, owing to the (minor but significant) metallic admixture of most of the highest occupied molecular orbitals (MOs). It should be noted that the occupied MO, mostly of 5dz2 in nature, is not involved in these transitions.
It was found that the fully optimized lowest triplet state simply results from a HOMO → LUMO excitation. The vibrationally resolved simulated phosphorescence spectra were computed by assuming emission from this excited state (see the Computational details). The simulated spectra of complexes 1, 6, 12, and 14 match quite well with their experimental counterparts recorded in solution (Fig. 4). The λmax values obtained from the simulated spectra are listed in Table 4 where a fairly good agreement can be observed between most of these values and the corresponding experimental ones (Table 3 and Fig. S105†). It can also be noted that the computed values for the experimentally non-emissive complexes 19, 23, and 24 fall in the near-infrared region. The value corresponding to 11 (also non-emissive) is slightly smaller (682 nm).
The charge transfers associated with the triplet → singlet emission were calculated from TD-DFT calculations by using the method developed by Adamo and coworkers24 (see the Computational details). They are quantified by their corresponding transferred fractions of electron qCT over associated spatial extends dCT. They are illustrated as the plots of the differences between the densities of the two states involved in the emission process, which are shown along with their associated qCT and dCT in Fig. S98 and S99.† Their main features indicate a charge transfer from the substituted phenyl ring to the diazine heterocycle. The non-emissive compounds 11, 18, 23, and 24 are still different from the other complexes with their larger qCT and dCT values. It is noteworthy that the complexes having the largest PLQY in CH2Cl2 solution (namely 1, 2, and 3) are among those with the lowest qCT and dCT, which is surprising at the first glance. Nevertheless, it should be considered that important charge reorganisations are also associated with non-negligible structural changes upon de-excitation, which in turn tend to favor non-radiative processes. Compounds 1–3 appear to satisfy the best balance between such conflicting parameters.
Conclusion
In summary, we have designed a series of 24 Pt(II) complexes bearing phenyldiazine N^C ligands as well as chloride and azaheterocyclic ancillary ligands. The remarkable feature of the here presented material is that a rather simple structural motif of the Pt(II) complex was successfully modified, yielding interesting luminophores. Various structural modifications have been made in this series of complexes: the substituent on the phenyl ring at the para position to the metal center (methoxy, diphenylamino, phenyl, and 9H-carbazol-9-yl), the nature of the diazine fragment (pyrimidine, pyrazine, and quinazoline), the substituents at the C4 position of the pyrimidine ring (OMe and CF3), and the nature of the azaheterocyclic ancillary ligand (pyridine, 4-methoxypyridine and pyrimidine). The structural modifications have a significant effect on the optical properties of the complexes, which exhibit green to red/near-infrared phosphorescence emission both in CH2Cl2 solution and in the solid state. The following structure–property relationships can be highlighted:• The strength of the EDG on the phenyl ring tunes the oxidation potential, the HOMO energy and emission wavelength. The stronger the EDG, the more red-shifted the emission.
• The nature of the diazine fragment affects the reduction potential and the emission wavelength, with a red-shift in the following order: pyrimidine < pyrazine < quinazoline. For pyrimidine complexes, both the reduction potential and the emission wavelength can be modulated by introducing an EDG/EWG at the C4 position of the pyrimidine ring.
• The ancillary ligand has a moderate influence on the electronic properties but plays a significant role in the PLQY. In the solid state, the 4-methoxypyridine ligand has a beneficial effect.
Complexes 1–3 with a 2-(3-methoxyphenyl)pyrimidine ligand exhibit the best compromise in terms of charge transfer, leading to the best PLQY (up to 52% in CH2Cl2 solution). On the other hand, phenylquinazoline complexes 21–23 exhibit emissions in the near-infrared region. The solid-state PLQY of complex 22 (6%) is particularly appealing for such red-shifted emissions. In the near future, more attractive complexes will be incorporated in PhOLED devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. H. acknowledges the Région Bretagne, France and Conseil Départemental des Côtes d'Armor, France for her PhD funding (MMLum project).References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef.
- (a) R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116 RSC; (b) A. Salehi, X. Fu, D.-H. Shin and F. So, Adv. Funct. Mater., 2019, 29, 1808803 CrossRef; (c) N. T. Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696 CrossRef; (d) G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- H. Sasabe and J. Kido, Eur. J. Org. Chem., 2013, 7653 CrossRef CAS.
- (a) C. Adachi, M. A. Balto, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS; (b) P. T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380 CrossRef CAS PubMed.
- (a) Y. Yo and S. Y. Park, Dalton Trans., 2009, 1267 RSC; (b) E. Baranoff, J.-H. Yum, M. Graetzel and Md. K. Nazeeruddin, J. Organomet. Chem., 2009, 694, 2661 CrossRef CAS; (c) M. G. Helander, Z. B. Wang, J. Qiu, M. T. Greiner, D. P. Puzoo, Z. W. Liu and Z. H. Lu, Science, 2011, 332, 944 CrossRef CAS PubMed.
- (a) J. Kalinnowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401 CrossRef; (b) C. Cebrián and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459 CrossRef PubMed.
- (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed; (b) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed; (c) P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084 RSC.
- (a) M. Ibrahim-Ouali and F. Dumur, Molecules, 2019, 24, 1412 CrossRef CAS PubMed; (b) S. F. Wang, L.-W. Fu, Y.-C. Wei, S.-H. Liu, J.-A. Lin, G.-H. Lee, P.-T. Chou, J.-Z. Huang, C.-I. Huang, C.-I. Wu, Y. Yuang, C.-S. Lee and Y. Chi, Inorg. Chem., 2019, 58, 13892 CrossRef CAS PubMed; (c) S. F. Wang, Y. Yuan, Y.-C. Wei, W.-H. Chan, L.-W. Fu, B.-K. Su, I.-Y. Chen, K.-J. Chou, P.-T. Chen, H.-F. Hsu, C.-L. Ko, W.-Y. Hung, C.-S. Lee, P.-T. Chou and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002173 CrossRef CAS.
- (a) P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, Chem. Sci., 2021, 12, 6172 RSC; (b) P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, J. Mater. Chem. C, 2022, 10, 4851 RSC; (c) P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, P.-H. Lanoe, V. N. Kozhevnikov and F. B. Dias, J. Mater. Chem. C, 2021, 9, 10276 RSC.
- (a) M. Hruzd, S. Gauthier, J. Boixel, S. Kahlal, N. le Poul, J.-Y. Saillard, S. Achelle and F. Robin-le Guen, Dyes Pigm., 2021, 194, 109622 CrossRef CAS; (b) A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547 RSC; (c) P. K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084 CrossRef CAS PubMed; (d) F. Nisic, A. Colombo, C. Dragonetti, D. Roberto, A. Valore, J. M. Malicka, M. Cocchi, G. R. Freeman and J. A. G. Williams, J. Mater. Chem. C, 2014, 2, 1791 RSC; (e) Z. Wang, E. Turner, V. Mahoney, S. Madakuni, T. Groy and J. Li, Inorg. Chem., 2010, 49, 11276 CrossRef CAS PubMed; (f) M. Hruzd, N. le Poul, M. Cordier, S. Kahlal, J.-Y. Saillard, S. Achelle, S. Gauthier and F. Robin-le Guen, Dalton Trans., 2022, 51, 5546 RSC.
- (a) M. Z. Shafikov, R. Daniels, P. Pander, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, ACS Appl. Mater. Interfaces, 2019, 11, 8182 CrossRef CAS PubMed; (b) G. Zhou, Q. Wang, X. Wang, C.-L. Ho, W.-Y. Wong, D. Ma, L. Wang and Z. Lin, J. Mater. Chem., 2010, 20, 7472 RSC; (c) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS PubMed.
- (a) Y. Sun, C. Zhu, S. Liu, W. Wang, X. Chen, G. Zhou, X. Yang and W.-Y. Wong, Chem. Eng. J., 2022, 449, 137457 CrossRef CAS; (b) M. Chaaban, S. Lee, J. S. R. Vellore Winfred, X. Lin and B. Ma, Small Struct., 2022, 3, 2200043 CrossRef CAS; (c) J. Zhao, F. Dang, Z. Feng, B. Liu, X. Yang, Y. Wu, G. Zhou, Z. Wu and W.-Y. Wong, Chem. Commun., 2017, 53, 7581 RSC.
- (a) J. Zhao, Z. Feng, D. Zhong, X. Yang, Y. Wu, G. Zhou and Z. Wu, Chem. Mater., 2018, 30, 929 CrossRef CAS; (b) X. Yang, L. Yue, Y. Yu, B. Liu, J. Dang, Y. Sun, G. Zhou, Z. Wu and W.-Y. Wong, Adv. Opt. Mater., 2020, 8, 2000079 CrossRef CAS; (c) H. Yang, H. Li, L. Yue, X. Chen, D. Song, X. Yang, Y. Sun, G. Zhou and Z. Wu, J. Mater. Chem. C, 2021, 9, 2334 RSC.
- (a) S. Achelle, J. Rodríguez-López and F. Robin-le Guen, ChemistrySelect, 2018, 3, 1852 CrossRef CAS; (b) P. Meti, H.-H. Park and Y.-D. Gong, J. Mater. Chem. C, 2020, 8, 352 RSC; (c) R. Plaza-Pedroche, D. Georgiou, M. Fakis, A. Fihey, C. Katan, F. Robin-le Guen, S. Achelle and J. Rodríguez-López, Dyes Pigm., 2021, 185, 108948 CrossRef CAS.
- M. Fecková, S. Kahlal, T. Roisnel, J.-Y. Saillard, J. Boixel, M. Hruzd, P. le Poul, S. Gauthier, F. Robin-le Guen, F. Bureš and S. Achelle, Eur. J. Inorg. Chem., 2021, 1592 CrossRef.
- (a) S. Achelle, J. Rodríguez-López and F. Robin-le Guen, J. Org. Chem., 2014, 79, 7564 CrossRef CAS PubMed; (b) K. Hoffert, R. J. Durand, S. Gauthier, F. Robin-le Guen and S. Achelle, Eur. J. Org. Chem., 2017, 523 CrossRef CAS.
- H. Fukuda, Y. Yamada, D. Hashizume, T. Takayama and M. Watabe, Appl. Organomet. Chem., 2009, 23, 154 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197 CrossRef CAS.
- M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2018, 94, 290 CrossRef CAS PubMed.
- S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef CAS PubMed.
- Y. Chen, K. Li, W. Lu, S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909 CrossRef CAS PubMed.
- (a) T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498 CrossRef CAS PubMed; (b) I. Ciofini, T. Le Bahers, C. Adamo, F. Odobel and D. Jacquemin, J. Phys. Chem. C, 2012, 116, 11946 CrossRef CAS; (c) D. Jacquemin, T. Le Bahers, C. Adamo and I. Ciofini, Phys. Chem. Chem. Phys., 2012, 14, 5383 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2211426–2211428. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03690h |
| This journal is © The Royal Society of Chemistry 2023 |