
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From ferrocene to decasubstituted enantiopure ferrocene-1,1′-disulfoxide derivatives†
Min
Wen
a,
William
Erb
 *a,
Florence
Mongin
a,
Jean-Pierre
Hurvois
a,
Yury S.
Halauko
*b,
Oleg A.
Ivashkevich
c,
Vadim E.
Matulis
b,
Marielle
Blot
a and
Thierry
Roisnel
a
*a,
Florence
Mongin
a,
Jean-Pierre
Hurvois
a,
Yury S.
Halauko
*b,
Oleg A.
Ivashkevich
c,
Vadim E.
Matulis
b,
Marielle
Blot
a and
Thierry
Roisnel
a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: william.erb@univ-rennes1.fr
bDepartment of Chemistry, Belarusian State University, 4 Nezavisimosti Av., 220030 Minsk, Belarus. E-mail: hys@tut.by
cLaboratory for Chemistry of Condensed Systems, Research Institute for Physical Chemical Problems of Belarusian State University, 14 Leningradskaya St., 220030 Minsk, Belarus
First published on 21st February 2023
Abstract
The functionalization of (R,R)-S,S′-di-tert-butylferrocene-1,1′-disulfoxide by deprotolithiation-electrophilic trapping sequences was studied towards polysubstituted, enantiopure derivatives for which the properties were determined. While the 2,2′-disubstituted ferrocene derivatives were obtained as expected, subsequent functionalization of the 2,2′-di(phenylthio) and 2,2′-bis(trimethylsilyl) derivatives occurred primarily at the 4- or 4,4′-positions. This unusual regioselectivity was discussed in detail in light of pKa values and structural data. The less sterically hindered 2,2′-difluorinated derivative yielded the expected 1,1′,2,2′,3,3′-hexasubstituted ferrocenes by the deprotometallation-trapping sequence. Further functionalization proved possible, leading to early examples of 1,1′,2,2′,3,3′,4,4′-octa, nona and even decasubstituted ferrocenes. Some of the newly prepared ferrocene-1,1′-disulfoxides were tested as ligands for enantioselective catalysis and their electrochemical properties were investigated.
Introduction
In 70 years,1,2 ferrocenes have risen to an important place among the different families of organometallic compounds. This stems from the good stabilities towards air, water, heat and light, combined with the reversible redox behaviour of these sandwich compounds.3 The specific physical and chemical properties that a ferrocene can impart to molecules have allowed applications in various fields4 such as catalysis,5–7 medicinal chemistry8–12 and materials science.13–16 Since these properties can be tuned by the rational addition of selected substituents, there is a need for synthetic strategies that allow access to functionalized ferrocenes, including enantiopure derivatives.Since the discovery of ferrocene,1,2 many mono, 1,2- and 1,1′-disubstituted derivatives have been synthesized.17–23 However, other substitution patterns and more substituted derivatives have been significantly less studied.17–22,24,25 Among the highly substituted ferrocenes already described, those containing five identical substituents on the same cyclopendadienyl (e.g. Me,26,27cPr,28 benzyl,29 ethynyl,30 Ph,31 allyl,32 F,33,34 Cl,35,36 Br,30,37–40 I,37,38 SMe,41 SPh41) are well represented although they do not offer a high degree of property tuning. Regarding decasubstituted ferrocenes, most representatives with two (e.g. Me/phosphine,42,43 Me/ketone,44,45 Me/benzyl,29 Me/alkylthio,42,46 Cl/SMe47) or three48,49 different substituents are achiral compounds. Moreover, if chiral decasubstituted ferrocenes have been reported once, it is only in the form of racemic mixtures.50
As the controlled introduction of substituents on ferrocene allows to modulate its electronic51–53 and steric54,55 properties, being able to access hetero-polysubstituted derivatives should provide an unprecedented tuning of properties. Moreover, for potential applications in catalysis, medicinal chemistry or sensing, it is necessary to develop access to enantiopure polysubstituted ferrocenes.
Diastereoselective deprotometallation of a chiral directing group (DG)-substituted ferrocene currently represents the most general approach to access enantiopure 1,2-disubstituted ferrocenes.18,20,56–58 These can be further functionalized to various tri, tetra and even pentasubstituted ferrocenes. The enantiopure 1,2,3-tri and 1,2,3,4-tetrasubstituted ferrocenes have generally been obtained by successive functionalization of the positions adjacent to the chiral DG which can be for example (R)-α-(dimethylamino)ethyl,59–61 (S)-(2-methoxymethyl-1-pyrrolidyl)methyl,62 (S)-4-isopropyloxazoline,63,64 or the acetal developed by Kagan55,65–68 (Fig. 1a).
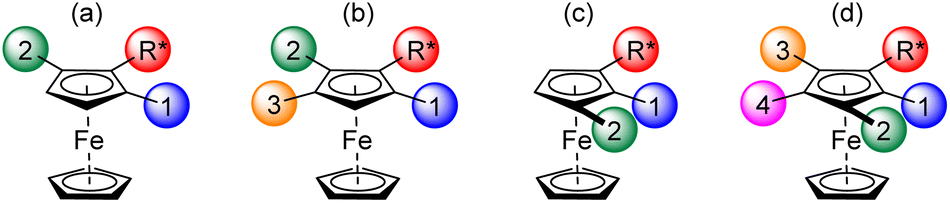 | ||
| Fig. 1 Already reported enantiopure ferrocenes with three, four or five different substituents and their introduction order. | ||
A fourth substituent can also be introduced next to the best DG, as shown by Top, Jaouen69 and Pugin60 (Fig. 1b). It should be noted that the substituents can be placed differently if the first substituent introduced is more directing than the chiral group (e.g. Br60,61,70,71 or (R)-tert-butylsulfinyl72vs. (R)-α-(dimethylamino)ethyl, and (R)-4-tolylsulfinyl69vs. Kagan's acetal; Fig. 1c). This strategy was exploited in our group to introduce successively, on the amine developed by Ugi,73 Cl, F, I and, after iodine migration,22,74 SiMe3 (Fig. 1d).75
Sulfoxides are one of the most important chiral groups able to direct a diastereoselective deprotometallation. Originally reported by Kagan and co-workers in 1993,76,77 the 4-tolyl-69,78,79 and tert-butylsulfinyl72 groups are the most employed since they can then be converted by reduction to sulfides70,78,80–82 or oxidation to sulfones,83 by sulfoxide/lithium exchange followed by trapping,69,78,79 or simply removed.72 Their use to access polyfunctionalized ferrocenes is therefore not surprising.
While Kagan and co-workers chose in 1993 to oxidize the sulfoxide to the sulfone in order to introduce a third substituent76 (Fig. 2a), we recently showed that the least activated position next to the sulfoxide could be directly functionalized84,85 (Fig. 2b). As demonstrated by Weissensteiner and co-workers,61,70 it is also possible to introduce Br and CHO in 2 and 3 position, respectively, taking advantage of the directing group properties of the former (Fig. 2c). Finally, by protecting the most reactive position next to the sulfoxide with a trimethylsilyl, we succeeded in synthesizing the first hetero-1,2,3,4,5-pentasubstituted ferrocene by successive introduction of F, Me and, after deprotection, Cl and SiMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 85 (Fig. 2d).
85 (Fig. 2d).
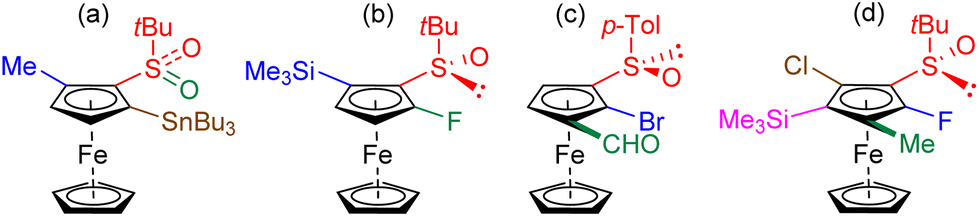 | ||
| Fig. 2 Use of a chiral sulfoxide to access enantiopure ferrocenes with three or five different substituents. The 1st, 2nd, 3rd and 4th functions introduced are shown in blue, green, brown and pink, respectively. | ||
In this article, we report how the wise choice of substituents can allow variously functionalized, especially decasubstituted, ferrocenesulfoxides to be delivered as enantiopure products.
Results and discussion
Synthesis
In order to completely decorate the ferrocene scaffold with different substituents, our idea was to identify a substrate both 1,1′-disubstituted with chiral DGs and easy to access. (R,R)-S,S′-Di-tert-butylferrocene-1,1′-disulfoxide (1) fulfilled our requirements as it has already been tested in deprotometallation-trapping by Zhang's team.86 However, we did not use their approach in two steps towards 1 (conversion of ferrocene to 1,1′-di(tert-butylthio)ferrocene followed by Kagan's asymmetric oxidation; 30% yield from 1,1′-dilithioferrocene), and preferred a one pot procedure. Thus, the disulfoxide 1 was easily prepared in 41% yield by reacting ferrocene-1,1′-dilithium87 with (R)-S-tert-butyl-tert-butanethiosulfinate88 (Scheme 1, top).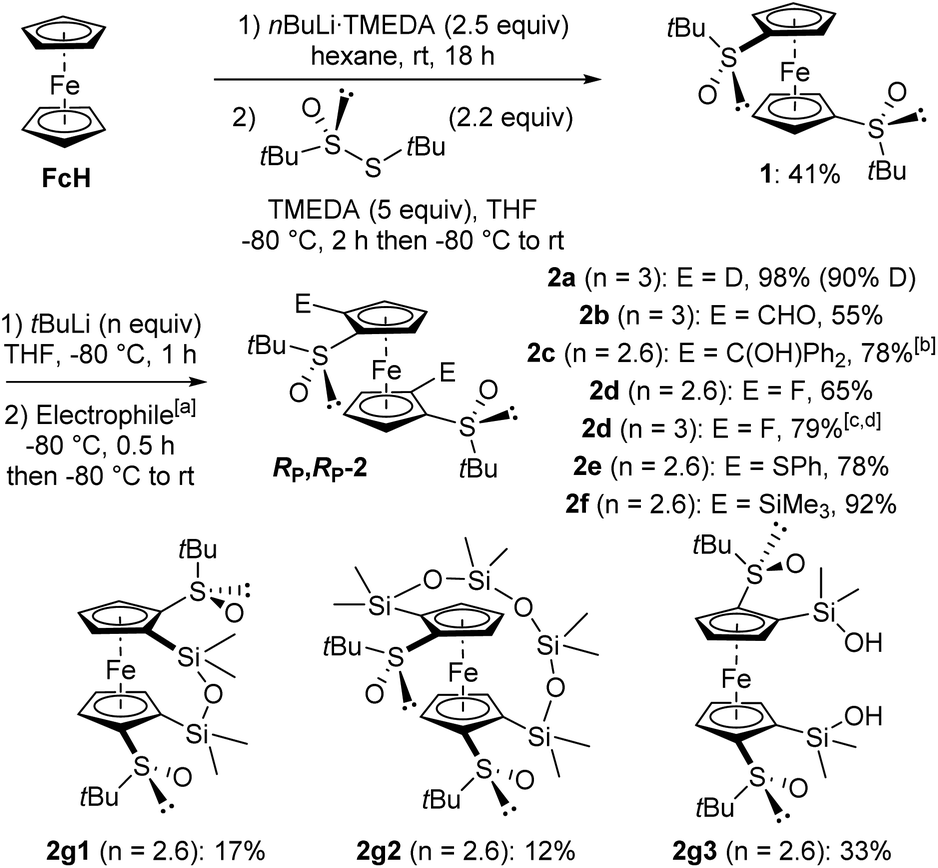 | ||
Scheme 1 Synthesis of the 2,2′-disubstituted (R,R)-S,S′-di-tert-butylferrocene-1,1′-disulfoxides RP,RP-2 from ferrocene. aElectrophiles used: D2O (2a), Me2NCHO (2b), Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (2c), (PhSO2)2NF (2d), PhSSPh (2e), ClSiMe3 (2f) and Cl2SiMe2 (2g). bAfter hydrolysis. cReaction performed at −90 °C. dMonofluoride also isolated by reducing the amount of base to 2.5 equivalents (see ESI†). O (2c), (PhSO2)2NF (2d), PhSSPh (2e), ClSiMe3 (2f) and Cl2SiMe2 (2g). bAfter hydrolysis. cReaction performed at −90 °C. dMonofluoride also isolated by reducing the amount of base to 2.5 equivalents (see ESI†). | ||
While Zhang and co-workers employed n-butyllithium in tetrahydrofuran (THF) at 0 °C to deprotolithiate the disulfoxide 1,86 we instead used tert-butyllithium at −80 °C. Under these conditions, the derivatives RP,RP-2 were isolated after electrophilic trapping with deuterated water (product 2a), dimethylformamide (2b), benzophenone (2c), N-fluorobenzenesulfonimide (NFSI; 2d), diphenyl disulfide (2e) and chlorotrimethylsilane (2f). With NFSI as the electrophile, better yield (79%) and reproducibility were achieved by working at −90 °C with 3 equivalents of tert-butyllithium (Scheme 1, middle). When dichlorodimethylsilane was used, the expected bridged silane89 was not obtained and we rather isolated the two siloxanes842g1 and 2g2, as well as the bis-silanol 2g3 (Scheme 1, bottom). While the formation of 2g1 and 2g3 probably originates from the hydrolysis of an intermediate 2,2′-bis(chlorodimethylsilyl)ferrocene-1,1′-disulfoxide, it is currently more difficult to explain the formation of the higher siloxane 2g2. However, this surprising result prompted us to take a closer look at the reactivity of these ferrocene-1,1′-disulfoxides.
One application of chiral arylmethanols is their use as ligands in the addition of diethylzinc to benzaldehyde.90–94 This reaction was considered here to evaluate the catalytic potential of the compound RP,RP-2c, which is close to the C2-symmetric ligand of Ikeda and co-workers.95 For comparison purposes, we also prepared the diol RP,RP-3, lacking the gem-diphenyl moiety, by reduction of RP,RP-2b (Scheme 2, top left). When benzaldehyde was treated with diethylzinc in toluene containing a catalytic amount of RP,RP-3 at −10 °C overnight,95 the expected alcohol was isolated in 76% yield and 19% ee in favour of the R enantiomer (Scheme 2, bottom). Under the same reaction conditions, the diol RP,RP-2c afforded the product in quantitative yield and an ee of 15%, while working at room temperature led to a 21% ee. Finally, replacing toluene with hexane94 gave an almost similar result (92% yield, 24% ee). For comparison, we finally used the 1,2-disubstituted ferrocene RP-2c (see ESI†) in toluene at rt and obtained the alcohol with similar yield and ee. A similar trend for di- vs. tetratopic ligands has been previously reported,95 although here with lower enantioselectivity.
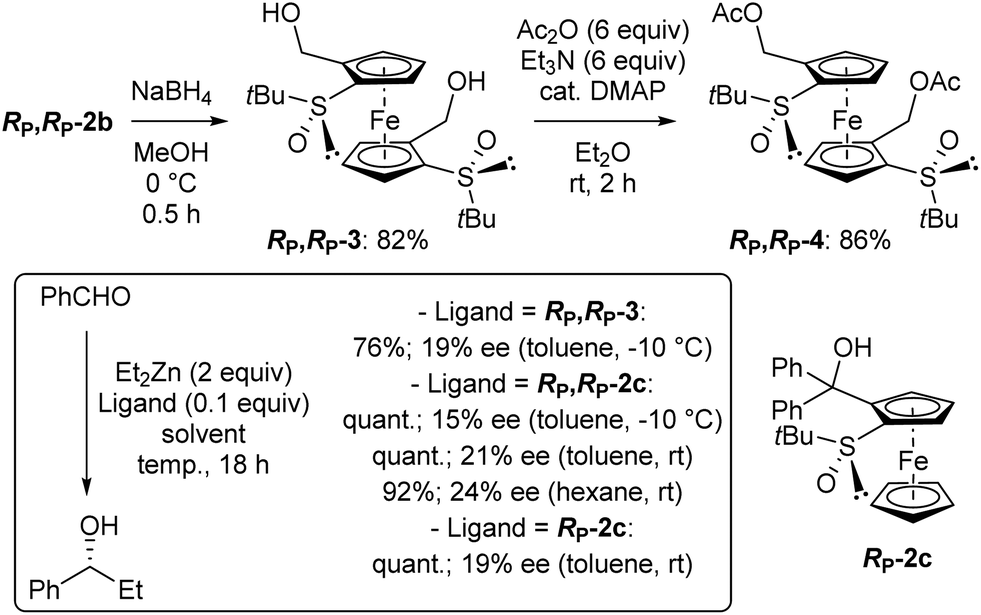 | ||
| Scheme 2 Attempts to use the alcohols RP,RP-3, RP,RP-2c and RP-2c as ligands in the diethylzinc addition to benzaldehyde. | ||
The diacetate RP,RP-4 also appeared to us as a suitable substrate to attempt the synthesis of the corresponding 2-phospha[3]ferrocenophanes. Indeed, related ferrocene-based chiral phosphines have been successfully used as organocatalysts for enantioselective transformations.96–98 This diacetate was easily prepared from the diol RP,RP-3 (Scheme 2, top right). However, whether from the diol RP,RP-3 or the diacetate RP,RP-4, no substitution occurred with phosphino-99 or (phosphinomethyl)ferrocene100 in acetic acid at room101 or at reflux96 temperature. Inspired by a previous study carried out in our group,102 we also attempted to replace the two acetates with these phosphines (as well as amines and diamines; see ESI†) in hexafluoroisopropanol at 60 °C, but without success. The recovery of the starting material in all these reactions shows the very low reactivity of this sterically hindered diacetate in substitution reactions. On the other hand, the particular behaviour of these 1,1′,2,2′-tetrasubstituted ferrocenes can also be seen as an opportunity to study original reactivities.
We next explored the possibility to reach more substituted derivatives from the 1,1′,2,2′-tetrasubstituted ferrocenes RP,RP-2e and RP,RP-2f. Indeed, their 1,2-disubstituted counterparts RP-2e and RP-2f (see Fig. 3a and b) can be functionalized at the remaining position next to the sulfoxide by deprotometallation in THF either with an excess of n-butyllithium at room temperature84 or with tert-butyllithium at −80 °C.85 Therefore, we tried to introduce two additional substituents from RP,RP-2e and RP,RP-2f in a similar way. For this purpose, RP,RP-2e was first treated with tert-butyllithium (3 equiv.) at −80 °C for 1 h before trapping with chlorotrimethylsilane. Surprisingly, the 4,4′-disilylated product RP,RP-5a was obtained in 40% yield instead of the expected 5,5′-disilylated derivative, as well as the 4-monosilylated derivative RP,RP-5′a in 11% yield (Scheme 3).
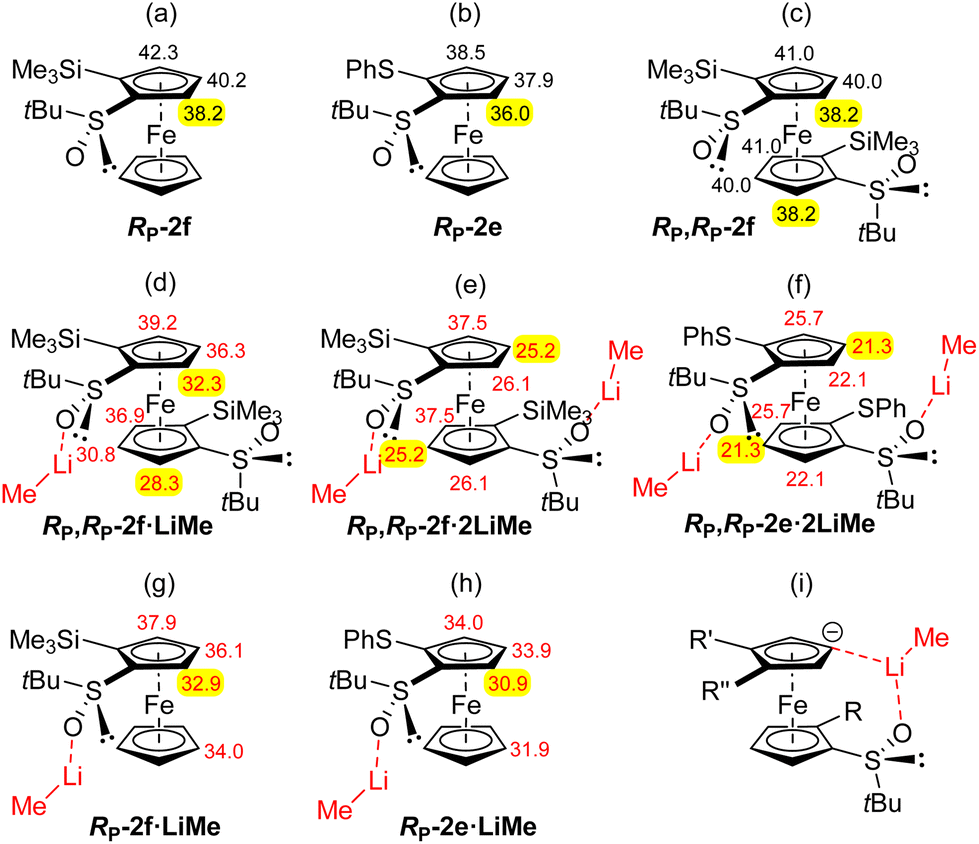 | ||
| Fig. 3 Top. Selected calculated pKa values of RP-2f (a), RP-2e (b) and RP,RP-2f (c). Middle. pKa values of RP,RP-2f (d), RP,RP-2f (e) and RP,RP-2e (f) as complexes with one, two and two LiMe, respectively. Bottom. pKa values of RP-2f (g) and RP-2e (h) as complexes with one LiMe, and a schematic of the stabilization effect (i). | ||
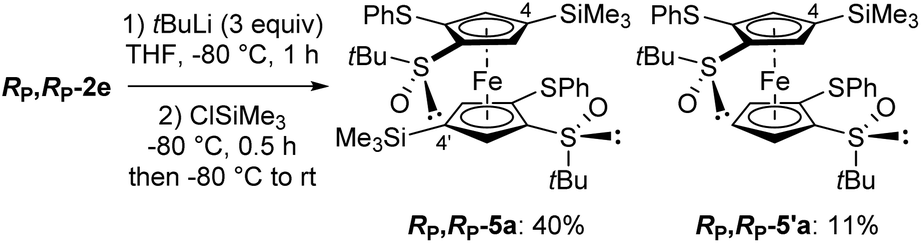 | ||
| Scheme 3 Deprotometallation-trimethylsilylation of (R,R,RP,RP)-S,S′-di-tert-butyl-2,2′-di(phenylthio)ferrocene-1,1′-disulfoxide (RP,RP-2e). | ||
In 2015, structurally-related 1,1′,2,2′,4,4′-hexasubstituted ferrocenes were synthesized by Pirio, Hierso and co-workers by double deprotolithiation of dl-1,1′-bis[(dialkylamino)methyl]-3,3′-di-tert-butylferrocenes.103 However, in our case, we benefited from the presence of the chiral sulfoxides to reach enantiopure 1,1′,2,2′,4,4′-hexasubstituted ferrocenes by double deprotolithiation-trapping.
To favour the formation of a dilithiated intermediate, the reaction was also performed at higher temperature (−50 °C). However, degradation mainly occurred (the 4,4′-diiodinated derivative RP,RP-5b was only isolated in 14% yield after iodolysis; see ESI†). This tends to suggest that the lithiated intermediates easily undergo decomposition, especially if they are not quickly trapped with an electrophile.
We were keen to see if this deprotometallation remote from the directing group could be extended to other substrates, and therefore selected RP,RP-2f in which the phenylthio groups of RP,RP-2e are replaced with trimethylsilyls. Pleasingly, the use of TMEDA-activated sec-butyllithium (TMEDA = N,N,N′,N′-tetramethylethylenediamine; 1.5 equiv.) in THF at −80 °C for 1 h allowed the ferrocene 4-position of RP,RP-2f to be cleanly deprotolithiated, as demonstrated by subsequent trapping with iodomethane and methyl chloroformiate, leading to the products RP,RP-6a (90% yield) and RP,RP-6b (81% yield), respectively (Scheme 4). Note that trapping with concentrated DCl furnished, in addition to the expected 4-deuterated derivative (70%), the 5-deuterated one, next to the sulfoxide (20%). This confirms the presence of a major, although not exclusive, 4-lithiated derivative before the trapping step. This result is also consistent with the equilibrium constant calculated in this work for the transformation of an anion at C5 into an anion at C4, both coordinated with two LiMe (Fig. 3e), which is approximately equal to 8.
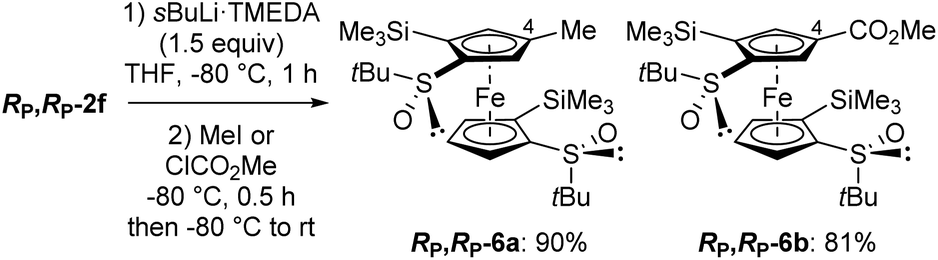 | ||
| Scheme 4 Deprotometallation-trapping of (R,R,RP,RP)-S,S′-di-tert-butyl-2,2′-bis(trimethylsilyl)ferrocene-1,1′-disulfoxide (RP,RP-2f) using sec-butyllithium·TMEDA (1.5 equiv.). | ||
However, all attempts to introduce a sixth substituent from the 4-methyl-2,2′-bis(trimethylsilyl)ferrocene-1,1′-disulfoxide RP,RP-6a by using sec-butyllithium·TMEDA and subsequent trapping failed with either Eschenmoser's salt, chlorotrimethylsilane or chlorodiphenylphosphine as the electrophile. In addition, under similar reaction conditions, RP,RP-6b was only moderately converted (∼25%) into the 4,4′-disubstituted product RP,RP-6′b (see ESI†) after trapping with methyl chloroformiate. Therefore, we came to the conclusion that, to reach 1,1′,2,2′,4,4′-hexasubstituted derivatives, it would be preferable to start from RP,RP-2f and perform a double deprotolithiation-trapping sequence.
Preliminary tests were performed by using either tert-butyllithium (2.6 equiv.) at −80 or −50 °C, or n-butyllithium (3 equiv.) at 0 °C, in THF. However, after interception with iodine, mixtures of 4-mono and polyiodinated products as well as starting material were obtained. Further, attempts to deprotometallate RP,RP-2f using lithium 2,2,6,6-tetramethylpiperidide (LiTMP; 6 or 12 equiv.) in the presence of ZnCl2·TMEDA (2 or 4 equiv.) as an in situ trap provided only inseparable mixtures of the starting material and its 4-iodinated derivative (see ESI†).
Since the 4,4′-difunctionalized product RP,RP-6′c was already present (21% yield) in a test reaction using the sec-butyllithium·TMEDA chelate (1.5 equiv.) in THF at −80 °C for 1 h and isobutyl chloroformiate as the electrophile (see ESI†), we decided to increase the amount of this base to access enantiopure 1,1′,2,2′,4,4′-hexasubstituted ferrocenes (Scheme 5). Thus, by using sec-butyllithium·TMEDA (3 equiv.), the expected product RP,RP-6′c was obtained in 57% yield while the 4-functionalized product RP,RP-6c was isolated in only 7% yield. In contrast, with benzophenone as the electrophile, the only pure product isolated, in 25% yield, was the 5-substituted product RP,RP-6′′d, from a crude also containing the 4-substituted RP,RP-6d (∼47% yield) and the 4,4′-disubstituted (∼24% yield) RP,RP-6′d derivatives. Moreover, the use of Eschenmoser's salt provided the corresponding 4-(dimethylaminomethylated) ferrocene-1,1′-disulfoxide RP,RP-6e as the main product (64% yield). With chlorodiphenylphosphine, the mono and diphosphines SP,RP-6f and SP,SP-6′f were isolated with yields of 35% and 38%, respectively. However, when diphenyl disulfide was employed, the corresponding di and monofunctionalized products SP,RP-6′g and SP,RP-6′′g were formed in low yields, 10% and 16% respectively, due to recovery of 59% of the starting material. Finally, the use of iodine led to the 4-monoiodinated product RP,RP-6h as the main product (77% yield) beside the corresponding product RP,RP-6′′h (15% yield).
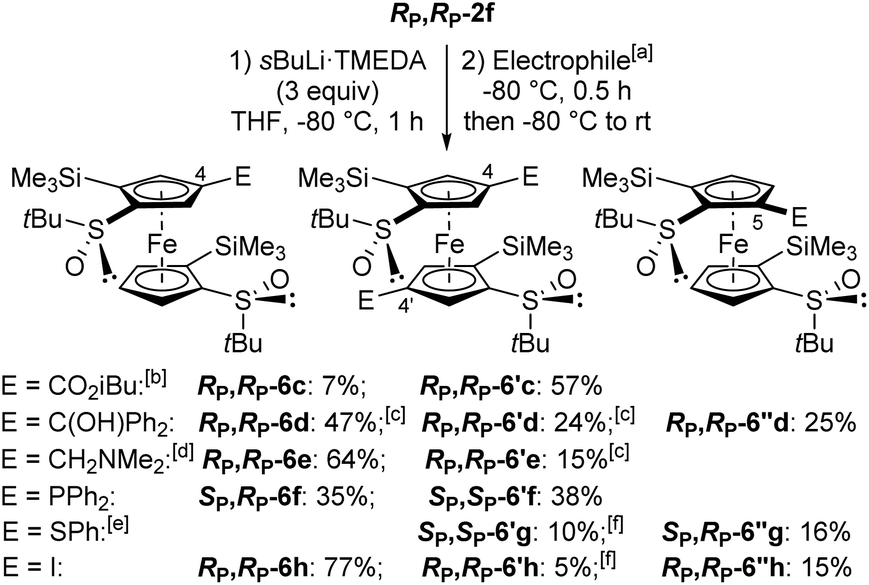 | ||
| Scheme 5 Deprotometallation-trapping of (R,R,RP,RP)-S,S′-di-tert-butyl-2,2′-bis(trimethylsilyl)ferrocene-1,1′-disulfoxide (RP,RP-2f) using sec-butyllithium·TMEDA (3 equiv.). aElectrophiles used: ClCO2iBu (6c/6′c), Ph2CO (6d/6′d/6′′d), CH2NMe2I (6e/6′e), ClPPh2 (6f/6′f), PhSSPh (6′g/6′′g) and I2 (6h/6′h/6′′h). bRecovery of 2% of the starting material. cEstimated yield. dRecovery of 18% of the starting material. eRecovery of 59% of the starting material. fYield given after cleavage of the trimethylsilyl groups. | ||
Our results concerning the functionalization of RP,RP-2e and RP,RP-2f contrast with those of Park and co-workers who converted C2-symmetric 1,1′-bis(diphenylphosphino)-2,2′-di(4-isopropyl-2-oxazolyl)ferrocene to the stereopure 3,3′-disilylated derivative using a similar deprotolithiation-trapping sequence (sBuLi-ClSiMe3).104 A more related study was reported by Brown and co-workers who claimed the deprotonation at C3 of (tert-butylthio)ferrocene using sec-butyllithium in THF.105,106 However, no detailed explanation was provided for this result. In order to compare the stabilities of the 4- and 5-lithiated derivatives of RP,RP-2e and RP,RP-2f, the equilibrium CH acidities of different positions were determined. The pKa values, calculated as before within the DFT framework (see ESI† for details),107–110 are presented in Fig. 3.
We have previously noticed that for mono and disubstituted ferrocenes with markedly distinct acidities at different positions,108,109 the pKa values are good indicators for deprotolithiation-trapping sequences. But when acidities are close and bulky substituents present,85,107 coordination to lithium should further be considered. Compared to the values previously obtained for RP-2f,85 those of RP,RP-2f are quite similar (Fig. 3c) and do not differ significantly within the molecule. As coordination to lithium significantly impacts deprotolithiations, e.g. through stabilization of transition states and lithiated products by formation of cyclic structures involving a lithium ion,85,111 pKa values were also calculated for the complexes RP,RP-2f·LiMe (Fig. 3d), RP,RP-2f·2LiMe (Fig. 3e) and RP,RP-2e·2LiMe (Fig. 3f). To this end, the various alkyllithiums used in the experiments were replaced by methyllithium in the calculations.
These data allow some comments on the regioselectivity observed in these deprotometallations. Indeed, for the 1,2-disubstituted ferrocenes RP-2f and RP-2e, coordination of the oxygen of the sulfinyl group to lithium lowers the pKa values by up to ∼5 units (Fig. 3g and h). According to the calculations, the position next to the sulfoxide remains the most acidic, in line with the observed regioselectivity during the functionalization of RP-2f.86 In contrast, the pKa values of RP,RP-2f·LiMe, RP,RP-2f·2LiMe and RP,RP-2e·2LiMe (Fig. 3d–f) tend to suggest that a ferrocenyl anion formed by deprotonation can be significantly stabilized by a remote sulfoxide through coordination to lithium. In this scenario, the stabilization probably results from the formation of a bridged structure involving a lithium ion such as the one depicted in Fig. 3i. Furthermore, one should keep in mind the small energy barrier separating the staggered and eclipsed conformations of the cyclopentadienyl rings (usually of the order of thermal energy).112 This could explain why the above mentioned stabilization is possible under deprotonation in both positions 4 and 5 (Fig. 3e and f). Furthermore, the pKa values of RP,RP-2f·2LiMe and RP,RP-2e·2LiMe (putative species, as the base is used in excess), tend to show that the 4-lithiated derivatives are slightly more stable than the 5-lithiated ones, in good agreement with our experimental results (Schemes 3–5). Finally, for the ferrocene RP,RP-2e the abovementioned stabilization (see ESI† for illustration) is possible under deprotonation in all three positions of the remote cyclopentadienyl ring while for the bulkier ferrocene RP,RP-2f in positions 4 and 5 only (Fig. 3e and f).
Since deprotometallations using alkyllithiums are generally not reversible, the most stable ferrocenyllithiums are not always produced, and conformations can also play a role. Indeed, in their related study on the double deprotometallation of 1,1′-bis(trimethylsilyl)ferrocene, Roberts, Silver and co-workers attributed the selective formation of a C2-symmetric diastereomer to the smaller steric interactions between the silyl groups in the dilithiated intermediate.113,114 Similarly, one could imagine that the ferrocene RP,RP-2f adopts a privileged conformation in which the trimethylsilyl groups move away from each other to limit the steric hindrance (conformation found in the solid state and in solution in CDCl3; see below and ESI†). Thus, the regioselectivity observed could also correspond to the least sterically hindered deprotolithiation position.
However, some discrepancies between the results obtained with different electrophiles from RP,RP-2f using 3 equivalents of base (Scheme 5) remain difficult to interpret. The main question that arises is whether a dilithiated intermediate is formed before the addition of the electrophile or whether the generated 4-functionalized product undergoes a second deprotometallation-trapping sequence.
We already observed that the conversion of a 1,1′,2,2′,4-penta into a 1,1′,2,2′,4,4′-hexasubstituted ferrocene was hardly possible. Indeed, the deprotometallation of the methyl ester RP,RP-6b using sec-butyllithium·TMEDA before interception with methyl chloroformiate only afforded 17% of RP,RP-6′b. However, when similar deprotometallation conditions were applied to the isobutyl ester RP,RP-6c before adding isobutyl chloroformate as the electrophile, the expected product RP,RP-6′c was not observed. Thus, even if it cannot be totally excluded in the case of electrophiles enough compatible with hindered alkyllithiums (e.g. chlorotrimethylsilane), all these data make an in situ deprotometallation-trapping sequence from the 4-functionalized product formed rather unlikely. Consequently, what seems most reasonable is that after its reaction with a first equivalent of electrophile, the 4,4′-dilithiated intermediate finds itself in a situation where it is more or less able (depending on the steric hindrance generated or/and the reactivity of the electrophile) to attack the second equivalent of electrophile before hydrolysis happens. For example, steric hindrance might be too high in the case of benzophenone or iodine while reactivity might be too low in the case of Eschenmoser's salt.
As some of the 4,4′-substituted ferrocenes prepared so far might behave as ligands, we were eager to evaluate them in asymmetric catalysis. Therefore, inspired by studies of Snieckus and co-workers who successfully used the enantiopure C2-symmetric N,N-diisopropyl-2,2′-bis(diphenylphosphino)ferrocene-1,1′-dicarboxamide as ligand for enantioselective palladium-catalysed allylic substitution,115 we selected for this purpose the di and monophosphines SP,SP-6′f and SP,RP-6f, as well as the corresponding desilylated counterparts SP,SP-7 and SP-7′ (Scheme 6, top).
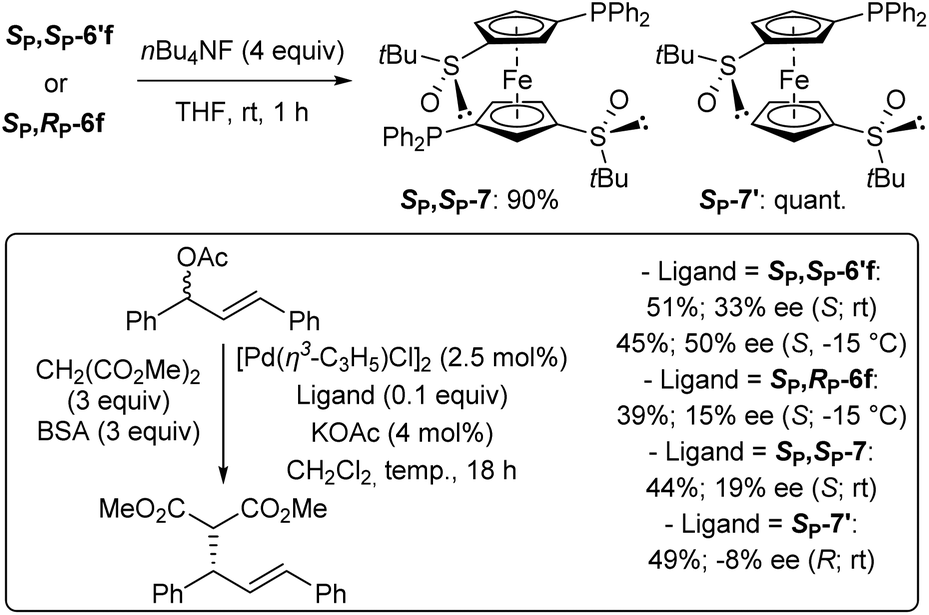 | ||
| Scheme 6 Attempts to use the phosphines SP,SP-6′f, SP,RP-6f, SP,SP-7 and SP-7′ as ligands in the palladium-catalysed allylic substitution of racemic phenylcinnamyl acetate with dimethyl malonate. | ||
They were involved in the reaction of racemic (E)-1,3-diphenyl-2-propenyl acetate with dimethyl malonate (3 equiv.) under Trost conditions using catalytic allylpalladium(II) chloride dimer, ligand and potassium acetate, and excess of N,O-bis-(trimethylsilyl)acetamide (BSA) in dichloromethane at room temperature.116 Under these conditions, the best ligand proved to be the diphosphine SP,SP-6′f, giving the expected product in 45% and a moderate enantioselectivity (50% ee) in favour of the S enantiomer (Scheme 6, bottom).
The results obtained so far (Scheme 5 and Fig. 3e) suggest that the steric hindrance generated by the two trimethylsilyl groups of the substrate RP,RP-2f interferes with the synthesis of a decasubstituted ferrocene. Hence, we decided to continue with the less crowded 2,2′-difluorinated ferrocene RP,RP-2d for which the pKa values are also promising for further functionalization, expected next to fluorine (Fig. 4).
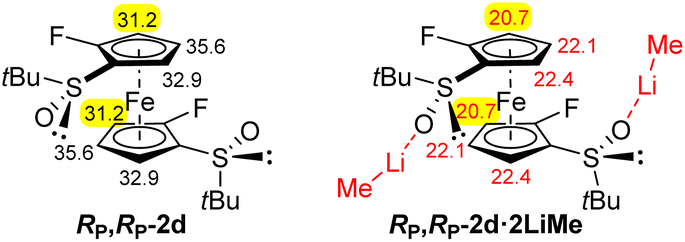 | ||
| Fig. 4 pKa values of RP,RP-2d and its complex with two LiMe. | ||
We first tested the double functionalization of RP,RP-2d by using LiTMP (2.5 equiv.) in the presence of TMEDA (1.4 equiv.) in hexane at rt for 20 min, as proposed by Butler for the double deprotolithiation of polyhalogenated ferrocenes.117 However, only starting material and degradation products were observed under these conditions. Next, we switched to sec-butyllithium in THF at −80 °C for 1 h, as used for fluoroferrocene.108 After subsequent quenching with chlorotrimethylsilane, the expected disilylated product RP,RP-8a was obtained in an encouraging 38% yield although degradation was also observed at this temperature. At −90 °C, the use of sec-butyllithium·TMEDA (3 equiv.) led to a slightly improved 44% yield. In order to progress towards more substituted derivatives, we then successively treated RP,RP-2d with sec-butyllithium (3 equiv.) and hexachloroethane, leading to the 3,3′-dichlorinated derivative SP,SP-8b in 61% yield. Finally, we similarly prepared the corresponding diiodide SP,SP-8c in 48% yield (Scheme 7, top). We attempted to isomerize this iodide by heavy halogen migration22,74 using LiTMP (3.4 equiv.) under the conditions described previously (THF, −50 °C, 2 h).107 However, after hydrolysis, deiodinated product was mainly formed (29% yield), resulting from a competitive iodine/lithium exchange.107,108,118,119 The expected 2,2′-difluoro-4,4′-diiodoferrocene-1,1′-disulfoxide was only detected in one of the fractions separated by column chromatography (12% estimated yield).
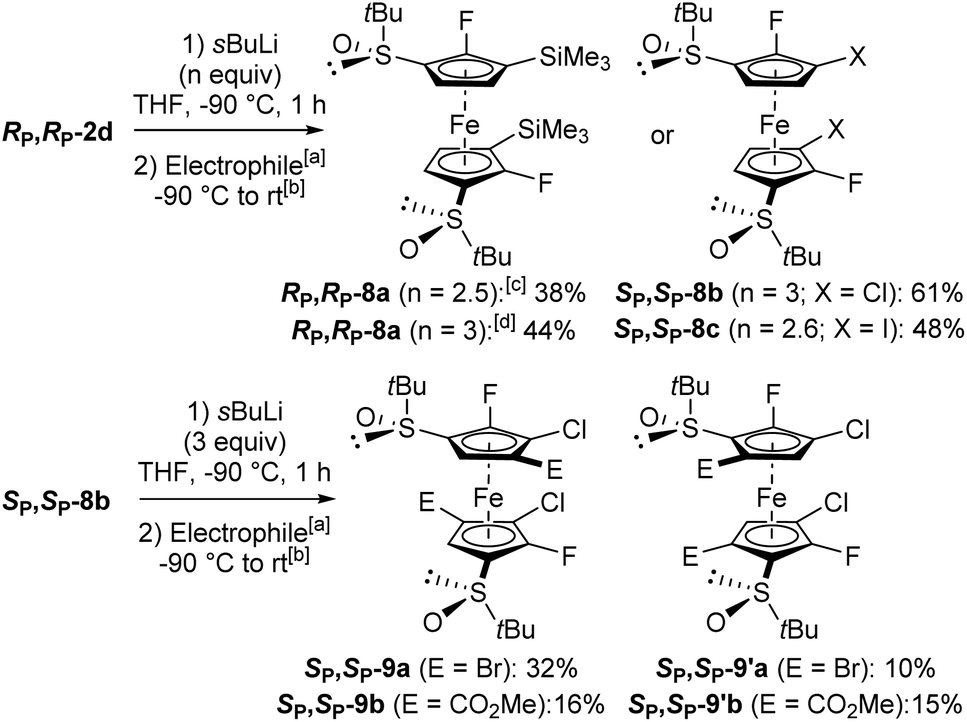 | ||
| Scheme 7 Synthesis of hexa and octasubstituted ferrocenes. aElectrophiles used: ClSiMe3 (RP,RP-8a), C2Cl6 (SP,SP-8b), I2 (SP,SP-8c), CF3CFBrCF2Br (SP,SP-9a and SP,SP-9′a), and ClCO2Me (SP,SP-9b and SP,SP-9′b). bSee ESI† for details on the trapping step. c−80 °C instead of −90 °C. dIn the presence of TMEDA (3 equiv.). | ||
In order to progress towards unprecedented octasubstituted ferrocene disulfoxides, we treated the 3,3′-dichlorinated derivative SP,SP-8b with sec-butyllithium (2.6 equiv.) in THF at −90 °C for 1 h. As subsequent deuteriolysis led to a mixture of regioisomers, we attempted to use other electrophiles. By trapping with 1,2-dibromo-1,1,2,3,3,3-hexafluoropropane, two octasubstituted ferrocenes were isolated: SP,SP-9a possessing its bromine atoms next to the chlorines (32% yield), and its regioisomer SP,SP-9′a with its bromine atoms next to the sulfoxides (10% yield). Similarly, the use of methyl chloroformiate afforded the two regioisomers SP,SP-9b and SP,SP-9′b, respectively possessing their ester functions next to the chlorines (16% yield) and next to the sulfoxides (15% yield; Scheme 7, bottom). Although the functionalization of the 5 and 5′ positions was not expected from the substrate 8b, a similar reaction was recently observed on related ferrocenesulfoxides.85
To finally reach decasubstituted ferrocenes, the regioisomers SP,SP-9a and SP,SP-9′a were subjected to the action of an excess of LiTMP (3 equiv., −90 °C, 1 h) before trapping with chlorotrimethylsilane. From SP,SP-9a, this made it possible to isolate the decasubstituted derivative SP,SP-10a as the main product (52% yield; Scheme 8, top). The use of methyl chloroformiate for the trapping step also led to the corresponding product SP,SP-10b, but in a lower 10% yield (see ESI†). By starting from SP,SP-9′a, the disilylated SP,SP-10′a coming from a double deprotolithiation-trapping sequence was also obtained as the main product (38% yield); however, it was accompanied by the regioisomer SP,SP-10′′a (16% yield; Scheme 8, bottom). For the latter, a single halogen migration took place on one cyclopentadienyl (Cp) ring before trapping. Single deprotometallation also took place in these two reactions, leading to the nonasubstituted ferrocenes SP,SP-10′′′a (13% yield; Scheme 8, top) and SP,SP-10′′′′a (14% yield).
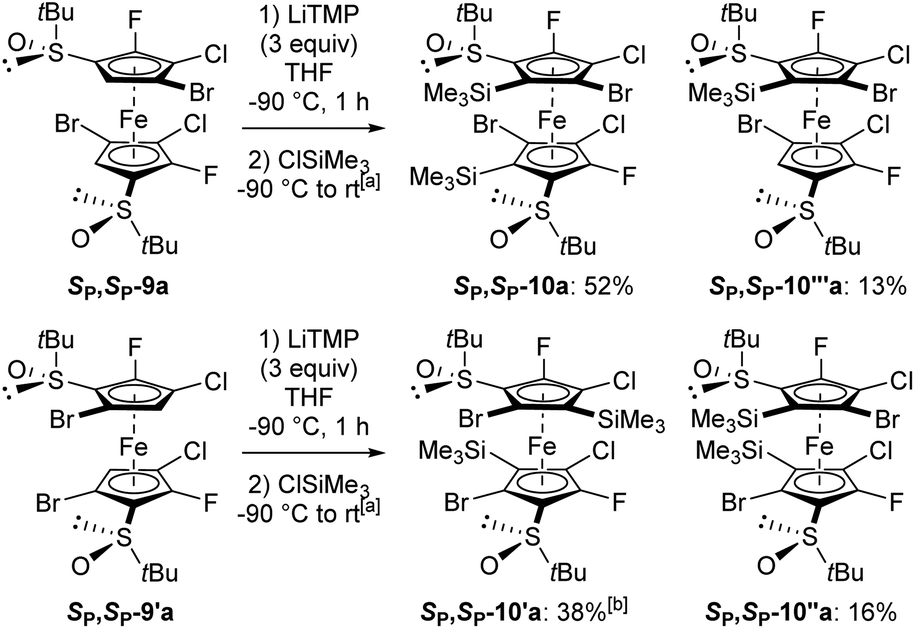 | ||
| Scheme 8 Synthesis of nona and decasubstituted ferrocenes. aSee ESI† for details on the trapping step. b(R,R,SP,SP)-2,5′-dibromo-S,S′-di-tert-butyl-4,3′-dichloro-5,2′-difluoro-3-(trimethylsilyl)ferrocene-1,1′-disulfoxide (SP,SP-10′′′′a) was also isolated in 14% yield. | ||
Thus, the first enantiopure hexa, octa, nona and decasubstituted ferrocenes could be achieved for the first time. Although none of these highly substituted ferrocenes afforded crystals suitable for X-ray diffraction analysis, high-resolution mass spectrometry analysis as well as the comparison of the NMR spectra (in particular, the 1H, 13C, NOESY and HOESY data) supported the proposed structures.
Electrochemical characterization
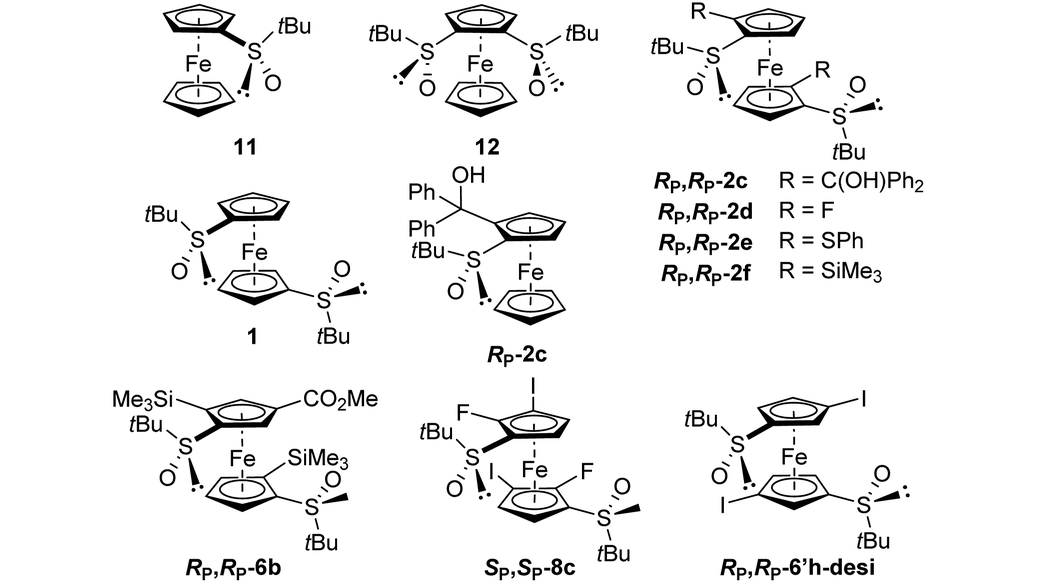
Few studies have been dedicated to the evaluation of the electrochemical behaviour of ferrocene sulfoxides. In 1997, Kagan highlighted the electron-withdrawing properties of the sulfoxide group as various alkyl- and aryl-sulfoxides raise the redox potentials from +0.25 to +0.29 V vs. FcH/FcH+.120 Except for S-butylferrocenesulfoxide, the E1/2 values of the S-arylated ferrocenesulfoxides were slightly higher than those of the S-alkylated ones. More recently, Jahn reported that the introduction of a phenylsulfinyl raises the redox potential of ferrocene by +0.30 V vs. FcH/FcH+,51 close to the +0.28 V value reported by Kagan. However, the introduction of a phenylsulfinyl group on each Cp ring raises the E1/2 value by only +0.50 V vs. FcH/FcH+. Taking advantage of the various ferrocene-1,1′-disulfoxides described in this study, we were eager to further investigate the electrochemical properties of these derivatives. This was done by performing cyclic voltammetry (CV) and differential pulse voltammetry (DPV) analyses in dry, oxygen-free dichloromethane (Table 1).| Compound |
E
pab |
E
pcb |
i
pa/ipcb |
E
1/2c |
|---|---|---|---|---|
| a Potential values referenced to Ag/AgCl and recalculated to FcH/FcH+; scan rate = 100 mV s−1. b From CV experiments. c From DPV experiments. d Not determined due to irreversible oxidation. | ||||
| 11 | 0.29 | 0.19 | 1.0 | 0.22 |
| 12 | 0.49 | 0.40 | 0.80 | 0.38 |
| 1 | 0.49 | 0.40 | 0.92 | 0.44 |
| R p-2c | 0.36 | 0.27 | 1.0 | 0.32 |
| R p,Rp-2c | 0.68 | 0.59 | 0.90 | 0.61 |
| R p,Rp-2d | 0.74 | ndd | ndd | 0.63 |
| R p,Rp-2e | 0.38 | 0.29 | 1.0 | 0.33 |
| R p,Rp-2f | 0.47 | 0.37 | 1.0 | 0.42 |
| R p,Rp-6b | 0.68 | 0.58 | 1.0 | 0.60 |
| S p,Sp-8c | 0.87 | nd | nd | 0.79 |
| R P,RP-6′h-desi | 0.77 | 0.70 | 1.0 | 0.71 |
|
||||
For comparison purposes, we also included (R,RP)-S-tert-butyl-2-[(α,α-diphenyl)hydroxymethyl]ferrocenesulfoxide (Rp-2c), and the previously reported (S)-S-tert-butylferrocenesulfoxide (11) and (R,R)-S,S′-di-tert-butylferrocene-1,2-disulfoxide (12).
The introduction of a phenylsulfinyl onto ferrocene raised the redox potential by +0.22 V, close to the +0.25 V value recorded by Kagan in different analysis conditions. The introduction of a second sulfoxide group further increases the E1/2 value from +0.18 to +0.22 V, depending on the position functionalized. Indeed, it seems that when introduced next to the first sulfoxide, the second one cannot exert its full effect (compound 12). However, when introduced on the other Cp ring (compound 1; Fig. 5), a fully additive effect was noticed which perfectly matched with the ∑σp Hammett parameters (E1/2 = 2.227 ∑σp − 0.0215, R2 = 1, Plot ESI1†). Similar results were previously reported by others who plotted the E1/2 value against the sum of different Hammett parameters, depending on the substituent studied.53,121–125 From the compound 11, the introduction of a tertiary alcohol raised the redox potential by +0.10 V with the compound Rp-2c while the introduction of two alcohol groups from the ferrocene 1 only raised the redox potential by +0.17 V (compound Rp,Rp-2c). This additivity trend was confirmed with the difluorinated compound Rp,Rp-2d (+0.10 V per fluorine atom) although an irreversible oxidation was also observed. However, while the trimethylsilyl group had almost no effect (compound Rp,Rp-2f), the introduction of two phenylthio substituents lowered the E1/2 value by −0.11 V, a surprising result considering the increase of E1/2 by +0.13 V per phenylthio substituent already observed.41,51 From the tetrasubstituted ferrocene Rp,Rp-2d, the introduction of two iodine atoms next to the fluorine raised the redox potential by +0.08 V per iodine. However, when the iodine atoms were introduced remoted from another group (compound RP,RP-6′h-desi), the redox potential increased by +0.13 V per iodine, in better agreement with the +0.15 V expected value.124 From the tetrasubstituted ferrocene Rp,Rp-2f to the pentasubstituted compound Rp,Rp-6b, an increase of +0.18 V was recorded, which is lower than the expected value (+0.22 V from the work of Jahn,51 +0.26 V from the work of Heinze53).
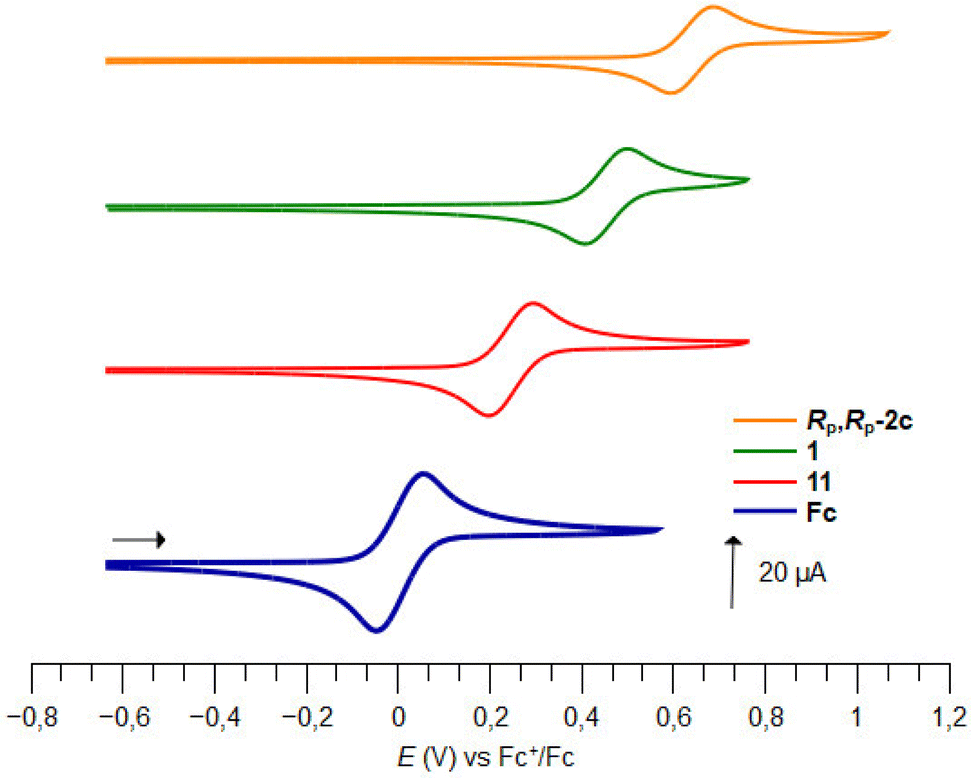 | ||
| Fig. 5 Plots of cyclic voltammograms of ferrocene (Fc) and the derivatives 11, 1 and RP,RP-2c. | ||
Therefore, taking together, these results tend to highlight the importance of the substituent, and its position, already present on a ferrocene derivative on the redox potential. To get a more general view on the substituent effect in this ferrocene-1,1′-disulfoxide series, we plotted the E1/2 values of ferrocene and the compounds 1, 2c, 2d, 2f, 8c and RP,RP-6′h-desi against either the sums of σp, σm or σp + σm (plots ESI2–4†). Given the diversity of substituents, the best correlation was obtained with the equation E1/2 = 1.751 ∑σp + 0.048, R2 = 0.9438. However, focusing our study on the effect of halogen atoms resulted in a better correlation between the redox potential value and the ∑σp (E1/2 = 1.812 ∑σp + 0.045, R2 = 0.9784, plot ESI5†).
Solid-state structures of S,S′-di-tert-butylferrocene-1,1′-disulfoxide derivatives
While Zhang and coll. already prepared S,S′-di-tert-butylferrocene-1,1′-disulfoxide and a few derivatives, their solid-state structures remained elusive up to now. In the frame of this study, we were able to grow crystals suitable for X-ray diffraction analysis for some compounds of interest. Compound 1 was found in a staggered conformation at the solid state, with the two tert-butyl moieties perpendicular to each Cp ring (Fig. 6). As a result of the R configuration of both sulfinyl groups, the two SO bonds are pointing in opposite directions and both are slightly bent towards the iron atom.
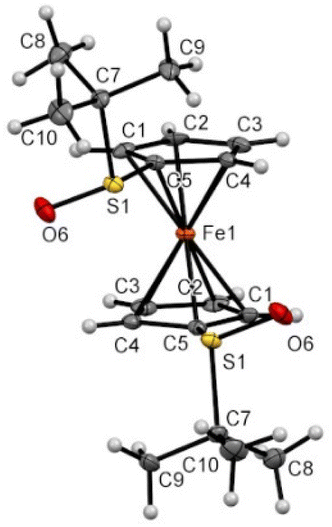 | ||
| Fig. 6 Molecular structure of the compound 1 (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C5–S1 1.758(4), C5–Cg1⋯Cg1–C5 −37.64(0.35) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring), C1–C5–S1–O6 18.5(4), C1–C5–S1–C7 −92.1(4), O6–S1⋯S1–O6 175.75(0.28). | ||
The introduction of fluorine atoms next to each sulfoxide was almost without impact on the solid-state structure. Indeed, the compound 2d also adopted a staggered conformation, with the two SO bonds bent towards the iron and opposite to each other (Fig. 7). However, replacing the small fluorine atom by a bulkier trimethylsilyl group induced more changes. Indeed, the two SO bonds in compound 2f were still observed pointing in opposite directions, but with one close to coplanarity with the Cp ring (Fig. 8). Furthermore, probably due to the bulkiness of the silyl group, the tert-butyl moieties were found more bent than in compound 1 (−98.5° and −105.9° torsion angles compared to −92.1°). The ferrocene core further adopts an eclipsed conformation with the two silyl groups pointing in opposite directions. In addition to the pKa calculation already discussed, the solid-state structure of ferrocene 2f also helps to rationalize the regioselectivity observed during deprotometallation experiments. Indeed, the positions adjacent to the trimethylsilyl groups are protected by these bulky substituents. However, the sulfinyl oxygen is well positioned to stabilize a lithiated intermediate at the 4 or 5 position of the opposite Cp ring until subsequent electrophilic trapping.
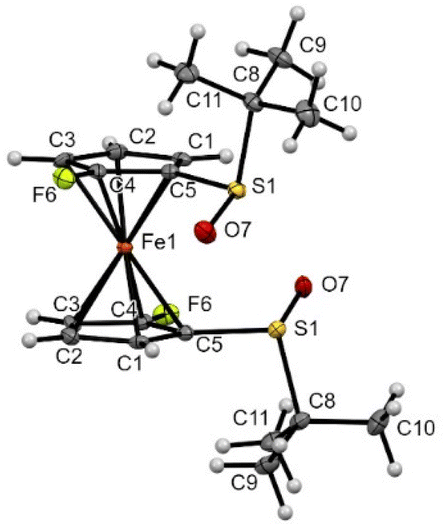 | ||
| Fig. 7 Molecular structure of the compound 2d (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C5–S1 1.776(4), C4–F6 1.342(3), C5–Cg1⋯Cg1–C5–43.56(0.33) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring), C4–C5–S1–C8 −89.6(4), O7–S1⋯S1–O7 175.6(2), F6–C4⋯C4–F6 168.3(3). | ||
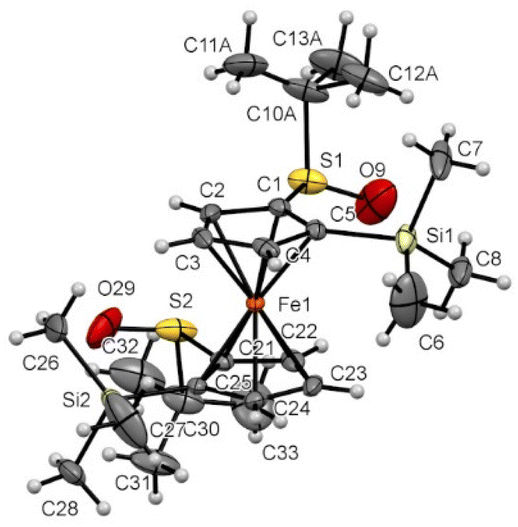 | ||
| Fig. 8 Molecular structure of the compound 2f (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C1–S1 1.770(3), C5–Si1 1.883(3), C21–S2 1.7770(3), C25–Si2 1.883(3), C1–Cg1⋯Cg2–C21 12.91(0.23) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring, Cg2 being the centroid of the C21–C22–C23–C24–C25 ring), C25–C21–S2–O29 5.7(4), C5–C1–S1–O9 15.2(4), C5–C1–S1–C10A −98.5(4), C25–C21–S2–C30 −105.9(3), O9–S1⋯S2–O29 161.51(0.28), Si1–C5⋯C25–Si2 132.73(0.18). | ||
Although not as bulky as the trimethylsilyl group, the hydroxymethyl moiety induced more changes at the solid state (compound 3, Fig. 9). Indeed, the presence of the two primary alcohols leads to the establishment of two intramolecular hydrogen bonds between a hydroxyl and the oxygen of a remote sulfinyl group acting as an acceptor. The hydrogen bond lengths and angles are within the expected range.126 As a result, an eclipsed conformation was observed, with both SO now pointing in the same direction, to lead to an O–S⋯S–O torsion angle of 35.26°. However, these bonds are also bent towards the iron atom with a torsion angle value of 19.6° while the tert-butyl groups are still perpendicular to their respective Cp rings. For comparison, the solid-state structure of the alcohol RP-2c has also been investigated and an intramolecular hydrogen bond has been similarly identified (see ESI†).
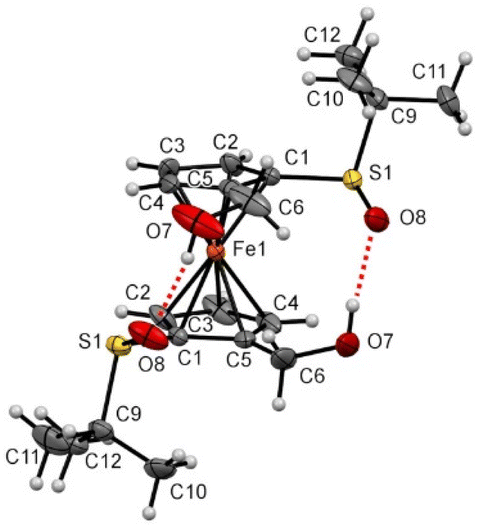 | ||
| Fig. 9 Molecular structure of the compound 3 (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C1–S1 1.759(3), C5–C6 1.500(6), C5–Cg1⋯Cg1–C5 −14.33(0.33) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring), C5–C1–S1–O8 19.6(4), C1–C5–S1–C7 −92.1(4), O8–S1⋯S1–O8 35.26(0.28), H7⋯O8 1.98(0.07), O7–H7⋯O8 171.93(7.27), H7⋯O8–S1 121.67(2.12). | ||
The hexasubstituted ferrocene 8a was also found in an almost perfectly eclipsed conformation, probably to reduce the steric hindrance between the two trimethylsilyl substituents at the 3 and 3′ positions (Fig. 10). However, the proximity of these bulky groups also induced the tilting of the Cp rings, with the C5–F6 bound inclined upwards. The two SO bonds are observed pointing in the same direction, both bent towards the iron atom, with tert-butyl groups almost perpendicular to their respective Cp rings.
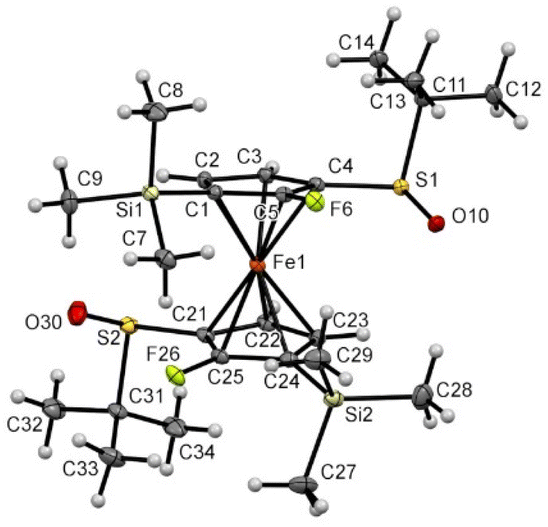 | ||
| Fig. 10 Molecular structure of the compound 8a (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C4–S1 1.772(3), C5–F6 1.346(3), C1–Si1 1.871(3), C21–S2 1.774(3), C25–F26 1.342(3), C24–Si2 1.874(3), C5–Cg1⋯Cg2–C24 −0.59(0.18) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring, Cg2 being the centroid of the C21–C22–C23–C24–C25 ring), mean plane C1–C2–C3–C4–C5⋯mean plane C21–C22–C23–C24–C25 9.31, mean plane C1–C2–C3–C4–C5⋯mean plane C4–S1C11 89.9, mean plane C21–C22–C23–C24–C25⋯mean plane C21–S2–C31 86.0, C5–C4–S1–O10 18.7(3), C25–C21–S2–O30 26.2(3), C5–C4–S1–C11 −92.6(3), C25–C21–S2–C31 −85.1(3); O10–S1⋯S2–O30 76.15(0.15), Si1–C1⋯C24–Si2 −62.33(0.18). | ||
From the ferrocene 2f, the introduction of an iodine atom at the 4 position (compound 6h) induced minor changes on the solid-state structure (Fig. 11). Indeed, the ferrocene core was also found in an eclipsed conformation with the two SO bonds pointing in opposite directions. However, both of them were almost coplanar with the Cp ring they are attached to (−2.0° and 3.2° torsion angle) and the tert-butyl groups were also more bent away from the silyl groups.
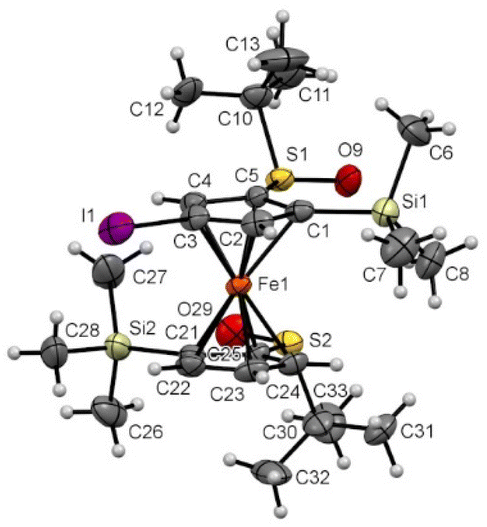 | ||
| Fig. 11 Molecular structure of the compound 6h (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C5–S1 1.776(6), C1–Si1 1.853(7), C3–I1 2.070(7), C25–S2 1.778(6), C21–Si2 1.895(7), C5–Cg1⋯Cg2–C25 4.82(0.43) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring, Cg2 being the centroid of the C21–C22–C23–C24–C25 ring), C1–C5–S1–O9 −2.0(6), C21–C25–C2–O29 3.2(6), C1–C5–S1–C10 −113.1(6), C21–C25–S2–C30 −108.9 (6), O9–S1⋯S2–O29 −142.82(0.34), Si1–C1⋯C21–Si2 −147.09(0.39). | ||
A similar solid-state structure was finally determined for the compound 6′c having one isopropyl ester at the 4 position of each Cp ring (Fig. 12). It features an eclipsed conformation for the ferrocene, two SO bonds pointing in opposite directions, the silyl groups almost opposite (Si1–C1⋯ C25–Si2 torsion angle of −169.18(0.35)°) and two slightly bent downwards. A molecule of chloroform co-crystallized with our compound and a weak interaction between a chlorine atom and one of the esters was also observed.
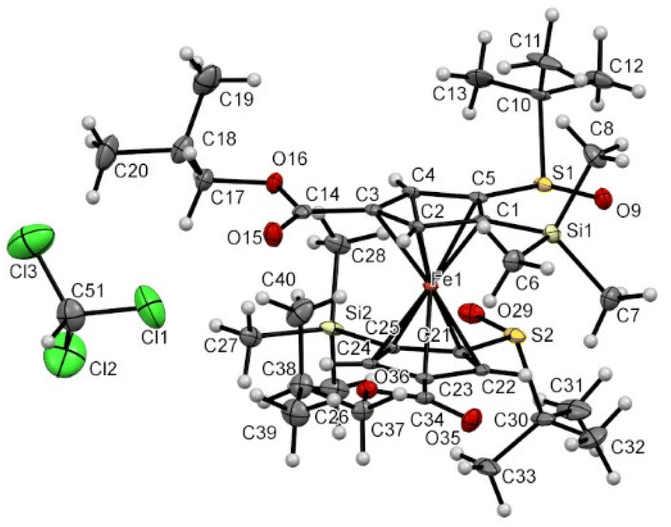 | ||
| Fig. 12 Molecular structure of the compound 6′c (thermal ellipsoids shown at the 30% probability level). Selected lengths [Å] and angles (°): C5–S1 1.780(4), C1–Si1 1.884(6), C3–C14 1.469(8), C21–S2 1.771 (5), C25–Si2 1.887 (6), C23–C34 1.477(8), C5–Cg1⋯Cg2–C21 −13.40(0.45) (Cg1 being the centroid of the C1–C2–C3–C4–C5 ring, Cg2 being the centroid of the C21–C22–C23–C24–C25 ring), C1–C5–S1–O9 −1.7(6), C25–C21–S2–O29 3.0(6), C1–C5–S1–C10 −112.9(6), C25–C21–S2–C30 −108.5(6), O9–S1⋯S2–O29 −160.34(0.27), Si1–C1⋯C25–Si2 −169.18(0.35), Cl1⋯O15 3.078(0.005), C51–Cl1⋯O15 138.34(0.38), Cl1⋯O15–C14 145.71(0.45). | ||
Conclusions
Although many ferrocenesulfoxide derivatives have been prepared over the last decades, the behaviour of (R,R)-S,S′-di-tert-butylferrocene-1,1′-disulfoxide and its derivatives in deprotolithiation have never been studied in details. Here, we found that the trends of regioselectivity observed with the 1,2- and 1,1′-disubstituted ferrocenes do not consistently apply to the functionalization of more hindered substrates such as 1,1′,2,2′-tetrasubstituted derivatives. Thus, while a 2,2′-difluorinated derivative could be 3,3′-difunctionalized, the corresponding 2,2′-di(phenylthio) and 2,2′-bis(trimethylsilyl) substrates were instead 4,4′-difunctionalized.While structural data were informative in explaining these results, additional pKa calculations helped us gain a more detailed understanding of reactivity trends. Further functionalization by deprotolithiation-electrophilic trapping sequences was also possible, affording the first enantiopure 1,1′,2,2′,4,4′-hexa, 1,1′,2,2′,3,3′-hexa, 1,1′,2,2′,3,3′,4,4′-octa, nona and even decasubstituted ferrocenes. The understanding of the key parameters allowing the synthesis and the control of the properties of these original compounds is fundamental for the development of molecules tailored for applications. Therefore, we hope that this first general study will be valuable in achieving this goal.
Author contributions
Investigation, M. W., W. E., J.-P. H., M. B., T. R., Y. S. H. and V. E. M.; funding acquisition, project administration and supervision, W. E.; writing – original draft preparation, W. E., F. M., M. W., J.-P. H. and Y. S. H.; writing – review and editing, W. E., F. M., M. W., J.-P. H., Y. S. H., T. R., V. E. M. and O. A. I.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded, in part, by the Agence Nationale de la Recherche (Ferrodance project). A CC-BY public copyright licence has been applied by the authors to present document and will be applied to all subsequent versions up to the Authors Accepted Manuscript arising from this submission, in accordance with the grant's open access conditions. This work was also supported by the Université de Rennes 1 and CNRS. We gratefully acknowledge the Fonds Européen de Développement Régional (FEDER; D8 VENTURE Bruker AXS diffractometer) and Thermofisher (generous gift of 2,2,6,6-tetramethylpiperidine).References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632 RSC.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6 CrossRef CAS.
- A. Togni and T. Hayashi, Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, Wiley-VCH, Weinheim, Germany, 1995 Search PubMed.
- T. J. Colacot, Chem. Rev., 2003, 103, 3101 CrossRef CAS PubMed.
- R. Gómez-Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed.
- N. A. Butt, D. Liu and W. Zhang, Synlett, 2014, 615 CAS.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802 RSC.
- Y. C. Ong, S. Roy, P. C. Andrews and G. Gasser, Chem. Rev., 2019, 119, 730 CrossRef CAS PubMed.
- Y. Lin, H. Betts, S. Keller, K. Cariou and G. Gasser, Chem. Soc. Rev., 2021, 50, 10346 RSC.
- B. Sharma and V. Kumar, J. Med. Chem., 2021, 64, 16865 CrossRef CAS PubMed.
- B. Zhou, L. Liu, P. Cai, G. Zeng, X. Li, Z. Wen and L. Chen, J. Mater. Chem. A, 2017, 5, 22163 RSC.
- M.-L. Hu, M. Abbasi-Azad, B. Habibi, F. Rouhani, H. Moghanni-Bavil-Olyaei, K.-G. Liu and A. Morsali, ChemPlusChem, 2020, 85, 2397 CrossRef CAS.
- Z. Wei, D. Wang, Y. Liu, X. Guo, Y. Zhu, Z. Meng, Z.-Q. Yu and W.-Y. Wong, J. Mater. Chem. C, 2020, 8, 10774 RSC.
- L. Li, J. Zhang, C. Yang, L. Huang, J. Zhang, J. Bai, C. Redshaw, X. Feng, C. Cao, N. Huo, J. Li and B. Z. Tang, Small, 2021, 17, 2103125 CrossRef CAS PubMed.
- G. G. A. Balavoine, J. C. Daran, G. Iftime, E. Manoury and C. Moreau-Bossuet, J. Organomet. Chem., 1998, 567, 191 CrossRef CAS.
- R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC.
- I. R. Butler, Eur. J. Inorg. Chem., 2012, 4387 CrossRef CAS.
- D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668 CrossRef CAS.
- H. Butenschön, Synthesis, 2018, 3787 CrossRef.
- M. Korb and H. Lang, Eur. J. Inorg. Chem., 2022, e202100946 CAS.
- W. Erb and F. Mongin, Synthesis, 2019, 146 CAS.
- E. Lerayer, L. Radal, T. A. Nguyen, N. Dwadnia, H. Cattey, R. Amardeil, N. Pirio, J. Roger and J.-C. Hierso, Eur. J. Inorg. Chem., 2020, 419 CrossRef CAS.
- G. Werner and H. Butenschön, Organometallics, 2013, 32, 5798 CrossRef CAS.
- E. E. Bunel, L. Valle and J. M. Manriquez, Organometallics, 1985, 4, 1680 CrossRef CAS.
- M. Malischewski, K. Seppelt, J. Sutter, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2018, 57, 14597 CrossRef CAS PubMed.
- I. Emme, S. Redlich, T. Labahn, J. Magull and A. De Meijere, Angew. Chem., Int. Ed., 2002, 41, 786 CrossRef CAS PubMed.
- M. D. Rausch, W. M. Tsai, J. W. Chambers, R. D. Rogers and H. G. Alt, Organometallics, 1989, 8, 816 CrossRef CAS.
- K. Sünkel and S. Bernhartzeder, J. Organomet. Chem., 2011, 696, 1536 CrossRef.
- D. W. Slocum, S. Duraj, M. Matusz, J. L. Cmarik, K. M. Simpson and D. A. Owen, Met.-Containing Polym. Syst., 1985, 59 CAS.
- P. Jutzi, C. Batz, B. Neumann and H.-G. Stammler, Angew. Chem., Int. Ed. Engl., 1996, 35, 2118 CrossRef CAS.
- K. Sünkel, S. Weigand, A. Hoffmann, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy and N. W. Mitzel, J. Am. Chem. Soc., 2015, 137, 126 CrossRef PubMed.
- W. Erb, N. Richy, J.-P. Hurvois, P. J. Low and F. Mongin, Dalton Trans., 2021, 50, 16933 RSC.
- S. Bernhartzeder and K. Sünkel, J. Organomet. Chem., 2012, 716, 146 CrossRef CAS.
- F. L. Hedberg and H. Rosenberg, J. Am. Chem. Soc., 1973, 95, 870 CrossRef CAS.
- V. I. Boev and A. V. Dombrovskii, Zh. Obshch. Khim., 1977, 47, 727 CAS.
- Y.-H. Han, M. J. Heeg and C. H. Winter, Organometallics, 1994, 13, 3009 CrossRef CAS.
- I. R. Butler, Inorg. Chem. Commun., 2008, 11, 484 CrossRef CAS.
- S. M. Rupf, I. S. Dimitrova, G. Schroeder and M. Malischewski, Organometallics, 2022, 41, 1261 CrossRef CAS.
- T. Blockhaus, C. Klein-Heβling, P. M. Zehetmaier, F. L. Zott, H. Jangra, K. Karaghiosoff and K. Sünkel, Chem. – Eur. J., 2019, 25, 12684 CrossRef CAS PubMed.
- R. Broussier, S. Ninoreille, C. Bourdon, O. Blacque, C. Ninoreille, M. M. Kubicki and B. Gautheron, J. Organomet. Chem., 1998, 561, 85 CrossRef CAS.
- G. Trouve, R. Broussier, B. Gautheron and M. M. Kubicki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1966 CrossRef.
- M. Roemer, B. W. Skelton, M. J. Piggott and G. A. Koutsantonis, Dalton Trans., 2016, 45, 18817 RSC.
- K. H. Taylor, S. E. Kalman, T. B. Gunnoe and M. Sabat, Organometallics, 2016, 35, 1978 CrossRef CAS.
- S. Zürcher, V. Gramlich, D. von Arx and A. Togni, Inorg. Chem., 1998, 37, 4015 CrossRef PubMed.
- T. Blockhaus, S. Bernhartzeder, W. Kempinger, C. Klein-Hessling, S. Weigand and K. Sünkel, Eur. J. Org. Chem., 2020, 6576 CrossRef CAS.
- T. Hatanaka, Y. Ohki and K. Tatsumi, Angew. Chem., Int. Ed., 2014, 53, 2727 CrossRef CAS PubMed.
- H. Bauer, J. Weismann, D. Saurenz, C. Färber, M. Schär, W. Gidt, I. Schädlich, G. Wolmershäuser, Y. Sun, S. Harder and H. Sitzmann, J. Organomet. Chem., 2016, 809, 63 CrossRef CAS.
- C. J. Miller and D. O'Hare, J. Mater. Chem., 2005, 15, 5070 RSC.
- D. A. Khobragade, S. G. Mahamulkar, L. Pospíšil, I. Císařová, L. Rulíšek and U. Jahn, Chem. – Eur. J., 2012, 18, 12267 CrossRef CAS.
- S. Toma and R. Šebesta, Synthesis, 2015, 1683 CrossRef CAS.
- S. D. Waniek, J. Klett, C. Förster and K. Heinze, Beilstein J. Org. Chem., 2018, 14, 1004 CrossRef CAS PubMed.
- F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 14264 CrossRef CAS PubMed.
- X. Wang, R. Fröhlich, C. G. Daniliuc, B. Rieger, A. Jonovic, G. Kehr and G. Erker, Organometallics, 2012, 31, 6741 CrossRef CAS.
- C. J. Richards and A. J. Locke, Tetrahedron: Asymmetry, 1998, 9, 2377 CrossRef CAS.
- W.-P. Deng, V. Snieckus and C. Metallinos, in Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications, ed. L.-X. Dai and X.-L. Hou, Wiley-VCH, Weinheim, 2010, ch. 2, pp. 15–53 Search PubMed.
- B. F. Bonini, M. Fochi and A. Ricci, Synlett, 2007, 360 CrossRef CAS.
- T. Hayashi, T. Mise, M. Fukushima, M. Kagotani, N. Nagashima, Y. Hamada, A. Matsumoto, S. Kawakami, M. Konishi, K. Yamamoto and M. Kumada, Bull. Chem. Soc. Jpn., 1980, 53, 1138 CrossRef CAS.
- B. Pugin and X. Feng, WO2006-EP61861,2006114438, 2006.
- M. Steurer, Y. Wang, K. Mereiter and W. Weissensteiner, Organometallics, 2007, 26, 3850 CrossRef CAS.
- C. Ganter and T. Wagner, Chem. Ber., 1995, 128, 1157 CrossRef CAS.
- C. J. Richards and A. W. Mulvaney, Tetrahedron: Asymmetry, 1996, 7, 1419 CrossRef CAS.
- K. H. Ahn, C.-W. Cho, H.-H. Baek, J. Park and S. Lee, J. Org. Chem., 1996, 61, 4937 CrossRef CAS.
- O. Riant, O. Samuel and H. B. Kagan, J. Am. Chem. Soc., 1993, 115, 5835 CrossRef CAS.
- H. Plenio, J. Hermann and A. Sehring, Chem. – Eur. J., 2000, 6, 1820 CrossRef CAS.
- J. Chiffre, Y. Coppel, G. G. A. Balavoine, J.-C. Daran and E. Manoury, Organometallics, 2002, 21, 4552 CrossRef CAS.
- J. Niemeyer, G. Kehr, R. Fröhlich and G. Erker, Dalton Trans., 2009, 38, 3716 RSC.
- B. Ferber, S. Top, R. Welter and G. Jaouen, Chem. – Eur. J., 2006, 12, 2081 CrossRef CAS PubMed.
- M. Steurer, K. Tiedl, Y. Wang and W. Weissensteiner, Chem. Commun., 2005, 41, 4929 RSC.
- X. Feng, B. Pugin, B. Gschwend, F. Spindler, M. Paas and H.-U. Blaser, ChemCatChem, 2009, 1, 85 CrossRef CAS.
- N. D'Antona, D. Lambusta, R. Morrone, G. Nicolosi and F. Secundo, Tetrahedron: Asymmetry, 2004, 15, 3835 CrossRef.
- D. Marquarding, H. Klusacek, G. Gokel, P. Hoffmann and I. Ugi, J. Am. Chem. Soc., 1970, 92, 5389 CrossRef CAS.
- W. Erb and F. Mongin, Tetrahedron, 2016, 72, 4973 CrossRef CAS.
- W. Erb and T. Roisnel, Chem. Commun., 2019, 55, 9132 RSC.
- F. Rebière, O. Riant, L. Ricard and H. B. Kagan, Angew. Chem., Int. Ed. Engl., 1993, 32, 568 CrossRef.
- B. Ferber and H. B. Kagan, Adv. Synth. Catal., 2007, 349, 493 CrossRef CAS.
- D. H. Hua, N. M. Lagneau, Y. Chen, P. M. Robben, G. Clapham and P. D. Robinson, J. Org. Chem., 1996, 61, 4508 CrossRef CAS PubMed.
- O. Riant, G. Argouarch, D. Guillaneux, O. Samuel and H. B. Kagan, J. Org. Chem., 1998, 63, 3511 CrossRef CAS.
- J. Priego, O. García Mancheño, S. Cabrera, R. Gómez Arrayás, T. Llamas and J. C. Carretero, Chem. Commun., 2002, 38, 2512 RSC.
- T. Sasamori, M. Sakagami, M. Niwa, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. Commun., 2012, 48, 8562 RSC.
- U. Caniparoli, I. Escofet and A. M. Echavarren, ACS Catal., 2022, 12, 3317 CrossRef CAS PubMed.
- N. M. Lagneau, Y. Chen, P. M. Robben, H.-S. Sin, K. Takasu, J.-S. Chen, P. D. Robinson and D. H. Hua, Tetrahedron, 1998, 54, 7301 CrossRef CAS.
- M. Wen, W. Erb, F. Mongin, M. Blot and T. Roisnel, Chem. Commun., 2022, 58, 2002 RSC.
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel, Molecules, 2022, 27, 1798 CrossRef CAS PubMed.
- M. Raghunath, W. Gao and X. Zhang, Tetrahedron: Asymmetry, 2005, 16, 3676 CrossRef CAS.
- M. D. Rausch and D. Ciappenelli, J. Organomet. Chem., 1967, 10, 127 CrossRef CAS.
- D. J. Weix and J. A. Ellman, Org. Lett., 2003, 5, 1317 CrossRef CAS PubMed.
- A. B. Fischer, J. B. Kinney, R. H. Staley and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 6501 CrossRef CAS.
- N. Oguni and T. Omi, Tetrahedron Lett., 1984, 25, 2823 CrossRef CAS.
- M. Watanabe, N. Hashimoto, S. Araki and Y. Butsugan, J. Org. Chem., 1992, 57, 742 CrossRef CAS.
- H. Wally, M. Widhalm, W. Weissensteiner and K. Schlögl, Tetrahedron: Asymmetry, 1993, 4, 285 CrossRef CAS.
- C. Bolm, N. Hermanns, J. P. Hildebrand and K. Muniz, Angew. Chem., Int. Ed., 2000, 39, 3465 CrossRef CAS.
- T. Ahern, H. Müller-Bunz and P. J. Guiry, J. Org. Chem., 2006, 71, 7596 CrossRef CAS PubMed.
- W. Zhang, H. Yoshinaga, Y. Imai, T. Kida, Y. Nakatsuji and I. Ikeda, Synlett, 2000, 1512 CAS.
- A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CrossRef CAS PubMed.
- A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, Adv. Synth. Catal., 2009, 351, 1968 CrossRef CAS.
- M. Neel, A. Panossian, A. Voituriez and A. Marinetti, J. Organomet. Chem., 2012, 716, 187 CrossRef CAS.
- B. A. Surgenor, L. J. Taylor, A. Nordheider, A. M. Z. Slawin, K. S. Athukorala Arachchige, J. D. Woollins and P. Kilian, RSC Adv., 2016, 6, 5973 RSC.
- F. Horký, I. Císařová and P. Štěpnička, Chem. – Eur. J., 2021, 27, 1282 CrossRef PubMed.
- W. Chen, W. Mbafor, S. M. Roberts and J. Whittall, J. Am. Chem. Soc., 2006, 128, 3922 CrossRef CAS PubMed.
- W. Erb, V. Carré and T. Roisnel, Eur. J. Org. Chem., 2021, 5702 CrossRef CAS.
- F. Allouch, N. Dwadnia, N. V. Vologdin, Y. V. Svyaschenko, H. Cattey, M.-J. Penouilh, J. Roger, D. Naoufal, R. Ben Salem, N. Pirio and J.-C. Hierso, Organometallics, 2015, 34, 5015 CrossRef CAS.
- S. Lee, J. H. Koh and J. Park, J. Organomet. Chem., 2001, 637–639, 99 CrossRef CAS.
- C. Pichon, B. Odell and J. M. Brown, Chem. Commun., 2004, 40, 598 RSC.
- B. Marsh, C. Frost and D. Pearce, WO Pat., WO2013190328, 2013 Search PubMed.
- M. Tazi, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet and F. Mongin, Organometallics, 2017, 36, 4770 CrossRef CAS.
- M. Tazi, M. Hedidi, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet, G. Bentabed-Ababsa and F. Mongin, Organometallics, 2018, 37, 2207 CrossRef CAS.
- W. Erb, M. Wen, J.-P. Hurvois, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel, Eur. J. Inorg. Chem., 2021, 3165 CrossRef CAS.
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel and V. Dorcet, Organometallics, 2021, 40, 1129 CrossRef CAS.
- A. Škvorcová and R. Šebesta, Org. Biomol. Chem., 2014, 12, 132 RSC.
- N. Mohammadi, A. Ganesan, C. T. Chantler and F. Wang, J. Organomet. Chem., 2012, 713, 51 CrossRef CAS.
- R. A. Brown, A. Houlton, R. M. G. Roberts, J. Silver and C. S. Frampton, Polyhedron, 1992, 11, 2611 CrossRef CAS.
- T. J. Peckham, D. A. Foucher, A. J. Lough and I. Manners, Can. J. Chem., 1995, 73, 2069 CrossRef CAS.
- R. S. Laufer, U. Veith, N. J. Taylor and V. Snieckus, Org. Lett., 2000, 2, 629 CrossRef CAS PubMed.
- B. M. Trost and D. J. Murphy, Organometallics, 1985, 4, 1143 CrossRef CAS.
- I. R. Butler, Organometallics, 2021, 40, 3240 CrossRef CAS.
- M. Tazi, W. Erb, T. Roisnel, V. Dorcet, F. Mongin and P. J. Low, Org. Biomol. Chem., 2019, 17, 9352 RSC.
- F. Mongin, E. Marzi and M. Schlosser, Eur. J. Org. Chem., 2001, 2771 CrossRef CAS.
- A. Gref, P. Diter, D. Guillaneux and H. B. Kagan, New J. Chem., 1997, 21, 1353 CAS.
- W. F. Little, C. N. Reilley, J. D. Johnson and A. P. Sanders, J. Am. Chem. Soc., 1964, 86, 1382 CrossRef CAS.
- X. You, C. Li, J. Chen, Q. Chao, X. Pen and A. Dai, Kexue Tongbao, 1987, 32, 1410 CAS.
- A. Hildebrandt, K. Al Khalyfeh, D. Schaarschmidt and M. Korb, J. Organomet. Chem., 2016, 804, 87 CrossRef CAS.
- M. S. Inkpen, S. Du, M. Hildebrand, A. J. P. White, N. M. Harrison, T. Albrecht and N. J. Long, Organometallics, 2015, 34, 5461 CrossRef CAS.
- D. M. Evans, D. D. Hughes, P. J. Murphy, P. N. Horton, S. J. Coles, F. F. de Biani, M. Corsini and I. R. Butler, Organometallics, 2021, 40, 2496 CrossRef CAS.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Compound synthesis and analysis, NMR spectra, selected NMR NOESY correlations, HPLC data, computational details, lithium stabilization effect, Cartesian coordinates of DFT optimized structures, additional pKa values, voltammograms, additional plots. CCDC 2204517–2204523 and 2236043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03456e |
| This journal is © The Royal Society of Chemistry 2023 |