
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and molecular structural studies of racemic chiral-at-vanadium(V) complexes using an unsymmetric achiral phenolic bidentate ligand†
Koichi
Nagata‡
a,
Ayako
Hino
a,
Hitoshi
Ube
 a,
Hiroyasu
Sato
b and
Mitsuhiko
Shionoya
*a
a,
Hiroyasu
Sato
b and
Mitsuhiko
Shionoya
*a
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shionoya@chem.s.u-tokyo.ac.jp
bRigaku Corporation, 3-9-12 Matsubara-cho, Akishima, Tokyo 196-8666, Japan
First published on 11th January 2023
Abstract
Mononuclear oxovanadium(V) complexes [V(O)XL (1: X = Ot-Bu, 2: X = Cl)] [H2L: 2,2′-methylene bis(4,6-di-tert-butylphenol)(4′-tert-butyl-6′-(1-adamantyl)phenol)] directed towards asymmetric catalysis have been synthesised as racemic compounds using an unsymmetric and achiral phenolic bidentate ligand (H2L), and NMR and UV-vis absorption spectroscopies, single-crystal X-ray diffraction, and IR spectroscopy revealed their racemic chiral-at-vanadium structures in solution and in the crystal. In addition, theoretical calculations revealed that the HOMO–LUMO energy gap is smaller for unsymmetric ligands, which promotes d-orbital splitting of the metal centre.
Introduction
Chiral-at-metal complexes, which have chirality only at the metal centre due to the asymmetric arrangement of the achiral ligands, have attracted much attention in recent years and are widely used for asymmetric catalysis and chiroptical properties.1,2 However, most of them are related to relatively inert octahedral metal complexes, and the control of metal centre chirality of tetrahedral metal complexes has remained an open question due to their lability and rapid racemisation.3 Recently, we have reported the successful asymmetric construction of a tetrahedral chiral-at-zinc complex with high configuration stability using a designed unsymmetric tridentate ligand, opening up new possibilities for tetrahedral chiral-at-metal complexes that can be used as asymmetric catalysts.4Vanadium is known as an important element in catalytic reactions as well as bioactive systems, and there has been much research on ligand design to improve the catalytic reactivity of oxovanadium complexes in particular.5–11 Recently, enantiomeric excess (ee) measurements of chiral compounds have been reported using the absorption properties of oxovanadium complexes with aminotriphenolate-type tetradentate ligands, which are expected to serve as molecular sensors specific to their coordination structures.12
Vanadium(V) complexes can form a variety of coordination structures. For example, V(O)(OMe)3 forms an oligomeric structure in the crystal, and the vanadium centre adopts an octahedral structure.13 On the other hand, when bulky alkoxides or phenoxides are used as ligands, the complex exists as a monomer and the vanadium centre exhibits a tetrahedral structure.14–17 In general, vanadium complexes are reported to be more stable in a higher-coordinated structure than in a four-coordinate structure.18 This is because the early-transition-metal, vanadium(V), is electron-deficient, and the alkoxy bridges the central metal to the electron-rich structure. Vanadium complexes with a variety of coordination structures have been synthesised and their reactivity and optical properties have been investigated, but most of them are penta- or hexacoordinate vanadium complexes.19 Here we report the synthesis and structural characterisation of mononuclear oxovanadium(V) complexes (1 and 2) with a tetrahedral structure using an unsymmetric bidentate ligand with bulky substituents to introduce chirality to the metal centre, and the stability and electronic properties of these racemic chiral-at-vanadium complexes in solution and in the solid state. Furthermore, the d-orbital splitting of the metal centre coordinated by the unsymmetric achiral ligand was promoted, and so its effect on their electronic properties was revealed by its NMR and UV-vis absorption spectroscopies, single-crystal X-ray diffraction, IR spectroscopy and DFT calculations.
Results & discussion
Characterisation of oxovanadium(V) complexes 1 and 2
The reaction of V(O)X3 (X = Ot-Bu, Cl) with the unsymmetrical achiral bidentate ligand H2L in CHCl3 at room temperature gave monomeric tetrahedral oxovanadium(V) complexes rapidly and quantitatively (Scheme 1). The 1H NMR spectra of oxovanadium(V) complexes 1 and 2 showed diastereotopic methylene proton signals split into doublets [1: 4.40, 3.60 ppm (J = 13.7 Hz); 2: 4.74, 3.72 ppm (J = 14.6 Hz)]. The 51V NMR spectrum of complex 1 showed a singlet at δ = −498.1 ppm (w1/2 ≈ 198 Hz), which is comparable to the reported chemical shift values of the tetrahedral complexes.19,20 In addition, 2 showed a singlet at δ = −244.3 ppm (w1/2 ≈ 347 Hz), which was shifted further downfield than that of 1. The different chemical shifts of the signals of 1 and 2 are due to the different electron-withdrawing or electron-donating strengths of the coordinating chlorine atom or alkoxide and phenoxide, respectively. The solid-state ATR-IR spectra of 1 and 2 showed signals assigned to their V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at 1002 (νcalcd = 1101 cm−1) and 1007 cm−1 (νcalcd = 1116 cm−1), respectively.
O stretching vibrations at 1002 (νcalcd = 1101 cm−1) and 1007 cm−1 (νcalcd = 1116 cm−1), respectively.
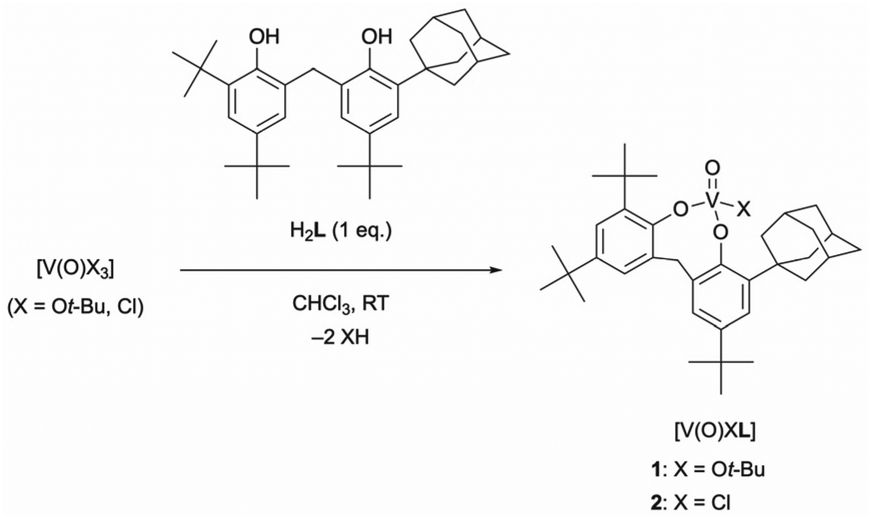 | ||
| Scheme 1 Synthesis of tetrahedral oxovanadium(V) complexes (1 and 2). | ||
The tetrahedral structures of oxovanadium(V) complexes 1 and 2 were determined by X-ray crystallographic analyses (Fig. 1), and the geometries of their V(1) centres are τ4 = 0.98 and 0.96 for 1 and 2, respectively,21 with the internal coordination sphere angles from 107.12(5)° to 111.80(5)°. Both 1 and 2 have a monomeric structure and no intermolecular contacts within ca. 2.7 Å. Their V(1)O(1) bond distances [1: 1.5862(10) Å, 2: 1.581(10) Å] are almost the same as those of the reported tetrahedral complexes (ca. 1.58 Å).14,15,19,20,22 The V–O single bonds for 1 [V(1)–O(2) 1.7902(10) and V(1)–O(3) 1.7892(10)] are slightly longer than those of 2 [V(1)–O(2) 1.771(10) and V(1)–O(3) 1.744(9)]. This difference between structures of 1 and 2 can be attributed to the strong electron-withdrawing nature of the chlorine ligand of 2, which shortens the V–O bond lengths of the coordinated oxygen atoms due to the strong donating natures of the oxygen atoms attached to the vanadium(V) centre. The eight-membered chelate ring of 2 adopts a boat–boat conformation (Fig. 1d), as does the reported complex [V(O) (ultra)Cl; ultra = 2,2′-CH2(4-Me,6-t-BuC6H2O−)2],22 while 1 adopts a boat–chair conformation (Fig. 1b).
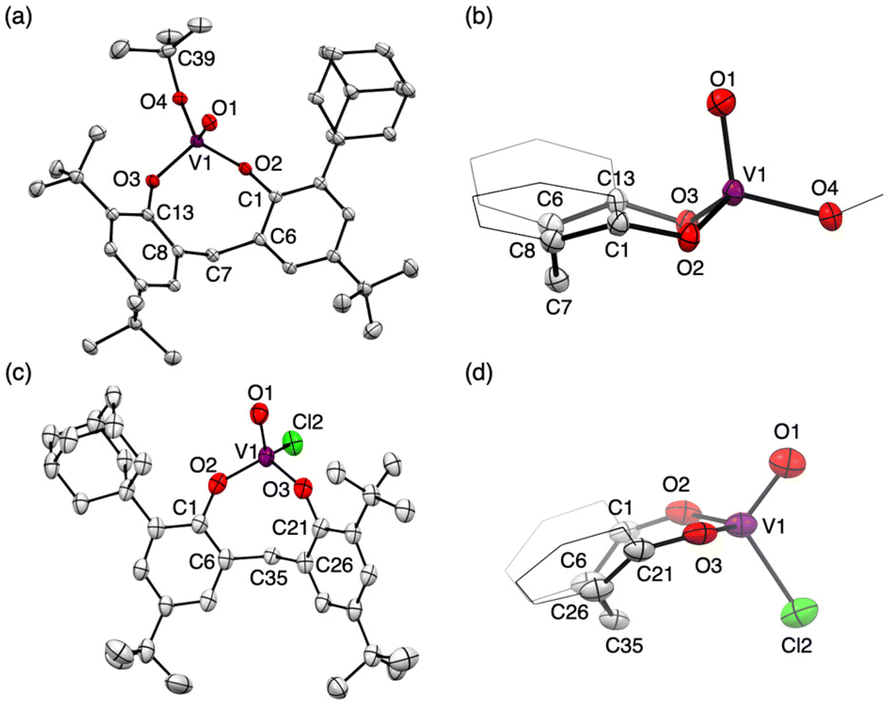 | ||
| Fig. 1 Molecular structures of 1 (a) and 2 (c). Side views of the eight-membered chelate ring (b: 1, d: 2). Thermal displacement ellipsoids are set at the 50% probability level; hydrogen atoms have been omitted for clarity. Two crystallographically independent molecules of 2 were found in the unit cell. Only one of them is shown here (for details, see Fig. S35 and S36 in the ESI†). | ||
DFT calculations
To obtain further information with respect to their geometry, density functional theory (DFT) calculations at the B3LYP23/Def2SVP24 theoretical level were performed for the complete molecular structures of oxovanadium(V) complexes 1 and 2. The molecular structures of 1 and 2 in the crystalline state were reproduced in the DFT calculations, as can be seen by comparing the selected bond lengths and angles of the crystal structures with the optimised geometries of I and IV (Table 1). Among the structures calculated for the oxovanadium complexes [V(O)XL], boat–boat conformations I and II and boat–chair conformations III and IV were found as the energy equilibrium structures on the potential energy hypersurface in the ground state. For the hydrocarbon cyclooctane, the boat–chair conformation is more stable than the boat–boat structure due to steric hindrance.25–27 Furthermore, the bulky substituents tend to be located in the equatorial positions. In contrast, for the oxovanadium complexes, the DFT calculations suggest that form I is the most stable structure in the cases of 1 and 2. The distance (d) between the vanadium and carbon atoms of the CH2 moiety in the boat–boat structure (I) is shorter than the sum of their van der Waals radii (3.75 Å),28 suggesting electronic interactions between them. Even if an equilibrium exists among these structures, the low equilibrium constant suggests that structure I is predominant in solution.| ● = C or CH2 |
|
|
||
|---|---|---|---|---|
| I | II | III | IV | |
| a Calculated at the B3LYP/Def2SVP theoretical level. b At 298.15 K, in kcal mol−1. c XRD results. Substituents of the bidentate ligand have been omitted for clarity. | ||||
| X = Ot-Bu | ||||
| θ [°] | 147.2 | 143.6 | 130.1 | 131.7 [127.67(5)]c |
| 142.6 | 137.2 | 129.3 | 134.8 [129.91(5)]c | |
| d [Å] | 3.22 | 3.26 | 3.37 | 3.45 [3.419(5)]c |
| ΔGa,b | 0.00 | +1.95 | +6.46 | +2.12 |
| X = Cl | ||||
| θ [°] | 149.8 [147.0(3)]c | 145.7 | 145.7 | 135.8 |
| 148.0 [142.2(3)]c | 136.0 | 136.1 | 131.1 | |
| d [Å] | 3.29 [3.216(3)]c | 3.27 | 3.27 | 3.47 |
| ΔGa,b | 0.00 | +2.70 | +2.78 | +2.31 |
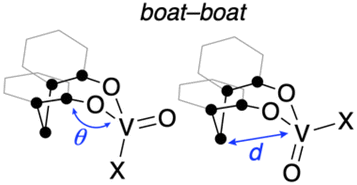
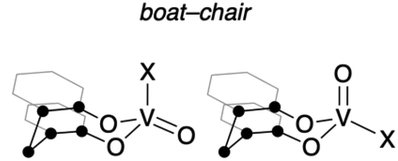
To evaluate the stabilizing effect of the boat–boat structure I, NBO calculations29 for I–IV were performed at the B3LYP/Def2SVP theoretical level. In the NBO calculations, the NBO 7.0 program was used to estimate the second-order perturbation of I–IV based on the donation of the lone pairs of O/Cl atoms to π*(VO) orbitals or vacant 3d(V) orbitals. These interaction energies were highest for the boat–boat structure I, where the VO site is equatorial (for the details; see Fig. S31 and Table S5 in ESI†). Furthermore, interactions between the π (C–H) and vacant π*(VO) orbitals of the bridged CH2 moiety were observed only in the boat–boat structure I, with the interaction energies estimated to be 1.87 (X = Ot-Bu) and 1.41 kcal mol−1 (X = Cl), respectively. These results mean that oxovanadium(V) complexes 1 and 2 are most similar to structure I in solution and are also consistent with their UV-vis spectra. The HOMO and LUMO orbitals of 1 and 2 represent the π orbitals on the aromatic rings of the bidentate ligand and the vacant 3d(V) orbitals, respectively (Fig. 2).
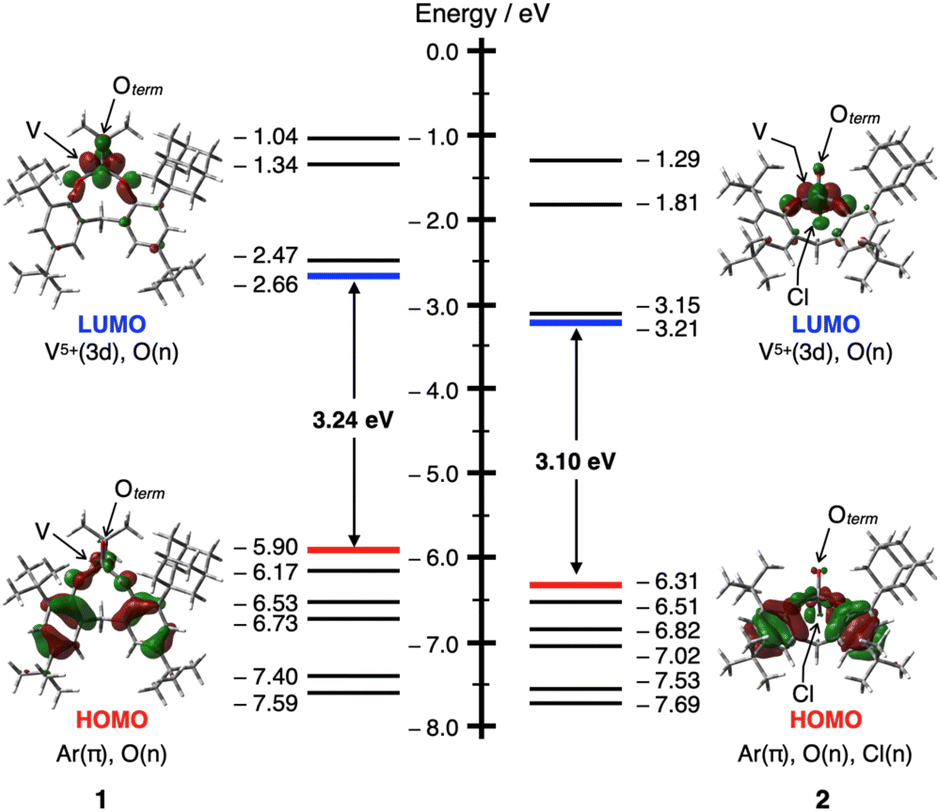 | ||
| Fig. 2 An energy diagram showing the frontier Kohn–Sham orbitals and energy values for 1 (left) and 2 (right) calculated at the B3LYP/Def2SVP theoretical level (isosurface level: 0.04 e au−3). | ||
UV-vis absorption spectra
The UV-vis absorption spectra of oxovanadium(V) complexes 1 and 2 provided further insight into their structure in solution. In CHCl3, 1 showed a broad absorption band with a maximum at 419 nm (ε = 6600). Time-dependent density functional theory (TD-DFT) calculations of 1 suggested that a broad absorption band with a maximum at 419 nm was considered to be a ligand-to-metal charge-transfer (LMCT) [π (bisphenolate ligand) → V(3d)] transition, and that the minimum energy absorption band at 330 nm (ε = 6900) was considered to be an intraligand charge transfer (ILCT) [π → π* (bisphenolate ligand)] transition (see Tables S1, S2 and Fig. S29 in the ESI†).The UV-vis spectrum of 2 in CHCl3 also showed characteristic absorption maxima at 472 nm (ε = 8800) and 592 nm (shoulder, ε = 4200), consistent with LMCT [π (bisphenolate ligand) → V(3d)] (see Tables S3, S4 and Fig. S30 in ESI†). The maximum absorption wavelength of 2 was red-shifted compared to that of 1, possibly due to the chlorine substituent effect, which lowers the LUMO [V(3d)] and reduces the HOMO–LUMO gap (Fig. 3).
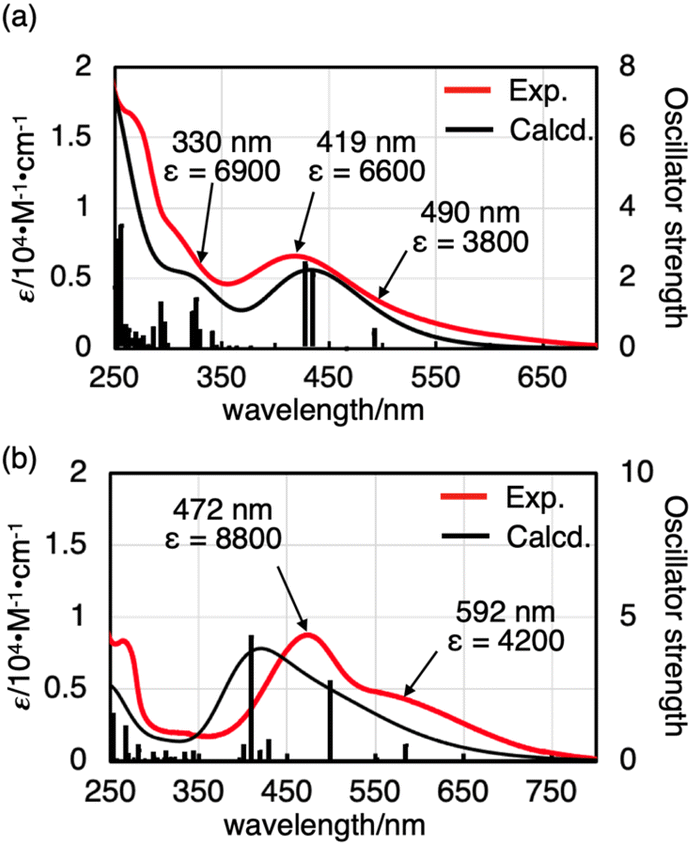 | ||
| Fig. 3 UV-vis absorption spectra (red lines) of (a) 1 (1.1 × 10−3 M) and (b) 2 (4.0 × 10−5 M) in CHCl3 (293 K), along with the computed spectrum of V(O)XL (black lines) at the B3LYP/Def2SVP (PCM: CHCl3) theoretical level. | ||
To discuss the effect of the unsymmetric bidentate ligand, we synthesised an oxovanadium(V) complex [V(O)(Ot-Bu)L′] (3) with a symmetrical bidentate ligand 2,2′-methylenebis[4,6-di-tert-butylphenol] (H2L′). The absorption coefficient of 1 was larger than that of 3 (see Fig. S28 in the ESI†), suggesting that the use of an unsymmetric ligand promotes d-orbital splitting of the metal centre of 1. DFT calculations also suggested that the LUMO energy level of 1 (−2.66 eV) was slightly lower than that of 3 (−2.46 eV), but that of the HOMO is similar (−5.90 eV for 1 and −5.91 eV for 3). Thus, the HOMO–LUMO energy gap is smaller for 1 (3.24 eV) than for 3 (3.45 eV) (Fig. S32†).
Conclusion
The synthesis, structure and photophysical properties of the racemic tetrahedral oxovanadium(V) complexes 1 and 2 with chirality at the metal centre are described above. These complexes are thermally stable solids, and their molecular structures have been clearly assigned on the basis of spectroscopic and crystallographic analyses and theoretical calculations. Since the structure of the V(O)XL synthesised in this study is chiral, the conversion of racemic compounds to diastereomeric complexes by ligand exchange with chiral alcohols is being investigated for the asymmetric induction of the metal-centered chirality.Experimental
Materials and methods
All the manipulations were performed under a nitrogen atmosphere, either by the Schlenk technique or in a glove box. All solvents were purified by standard methods, and trace amounts of oxygen and water remaining in the solvents were thoroughly removed by the freeze–pump–thaw method and activated MS4A, respectively. 1H, 13C, 51V and 2D NMR spectra were measured on a Bruker AVANCE III-500 (500 MHz) spectrometer. Tetramethylsilane was used as the internal standard for 1H NMR measurements when chloroform-d was used as the solvent. When chloroform-d (δ 77.16 ppm) was used as the solvent, the residual solvent signal was used to calibrate the 13C NMR measurements. 51V (132 MHz) NMR spectra were recorded on a Bruker AVANCE III-500 spectrometer using V(O)Cl3 in chloroform-d (δ 0 ppm) as an external standard. ESI-TOF mass data were recorded on a Micromass LCT Premier XE mass spectrometer. Unless otherwise noted, experimental conditions were as follows (ion mode, positive; capillary voltage, 3000 V; sample cone voltage, 30 V; desolvation temperature, 150 °C; source temperature, 80 °C). UV-vis absorption spectra in CHCl3 were recorded on a JASCO V-770 UV-vis spectrophotometer. IR spectra were recorded on a Jasco FT/IR 4200 with an ATR equipment. Elemental analysis was performed at the Microanalytical Laboratory of the University of Tokyo. 6,6′-Methylenebis(2,4-di-tert-butylphenol),30 2,4-di-tert-butyl-6-(5-tert-butyl-2-hydroxybenzyl)-methylphenol30 and oxovanadium(V) tri-tert-butoxide [V(O)(Ot-Bu)3]31 were prepared according to a literature.Synthetic procedures
Author contributions
K.N. conceived the development of oxovanadium(V) complexes and performed DFT calculations. A.H. performed the experiments and characterization. K.N. drafted the manuscript. M.S. directed the project and completed the manuscript. H.S. performed the X-ray crystallographic measurements and structure determination. All authors contributed to the analysis and interpretation of the data and commented on the final draft of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21H05022 (M. S.) and JP18K14236 (K. N.), MEXT KAKENHI Grant Number JP16H06509 (M. S.) and a Sasagawa Scientific Research Grant from the Japan Science Society (K. N.). A. H. acknowledges Materials Education program for the future leaders in Research, Industry, and Technology (MERIT). Calculations for complexes 1 and 2 were performed using the Research Centre for Computational Science, Okazaki, Japan (Project: 20-IMS-C100).References
- E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153–3167 RSC.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed.
- V. Desvergnes-Breuil, V. Hebbe, C. Dietrich-Buchecker, J. P. Sauvage and J. Lacour, Inorg. Chem., 2003, 42, 255–257 CrossRef CAS PubMed.
- K. Endo, Y. Liu, H. Ube, K. Nagata and M. Shionoya, Nat. Commun., 2020, 11, 6263 CrossRef CAS PubMed.
- E. Amadio, J. González-Fabra, D. Carraro, W. Denis, B. Gjoka, C. Zonta, K. Bartik, F. Cavani, S. Solmi, C. Bo and G. Licini, Adv. Synth. Catal., 2018, 360, 3286–3296 CrossRef CAS.
- J. A. L. da Silva, J. J. R. F. da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 2232–2248 CrossRef CAS.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- C. Bolm, Coord. Chem. Rev., 2003, 237, 245–256 CrossRef CAS.
- S. Takizawa, H. Gröger and H. Sasai, Chem. – Eur. J., 2015, 21, 8992–8997 CrossRef CAS PubMed.
- H. Pellissier, Coord. Chem. Rev., 2015, 284, 93–110 CrossRef CAS.
- H. Pellissier, Coord. Chem. Rev., 2020, 418, 213385 CrossRef.
- P. Zardi, K. Wurst, G. Licini and C. Zonta, J. Am. Chem. Soc., 2017, 139, 15616–15619 CrossRef CAS PubMed.
- H. M. S. Charles, N. Caughlan and K. Watenpaugh, Inorg. Chem., 1966, 5, 2131–2134 CrossRef.
- F. J. Feher and J. F. Walzer, Inorg. Chem., 1991, 30, 1689–1694 CrossRef CAS.
- J. Spandl, I. Brüdgam and H. Hartl, Z. Anorg. Allg. Chem., 2000, 626, 2125–2132 CrossRef CAS.
- D. C. Crans, H. J. Chen and R. A. Felty, J. Am. Chem. Soc., 1992, 114, 4543–4550 CrossRef CAS.
- D. L. H. Richard, A. Henderson, Z. Janas, R. L. Richards, P. Sobota and S. Szafert, J. Organomet. Chem., 1998, 554, 195–201 CrossRef.
- J. Y. Kempf, B. Maigret and D. C. Crans, Inorg. Chem., 1996, 35, 6485–6494 CrossRef CAS PubMed.
- C. Redshaw, M. J. Walton, M. R. J. Elsegood, T. J. Prior and K. Michiue, RSC Adv., 2015, 5, 89783–89796 RSC.
- F. Ge, Y. Dan, Y. Al-Khafaji, T. J. Prior, L. Jiang, M. R. J. Elsegood and C. Redshaw, RSC Adv., 2016, 6, 4792–4802 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- P. J. Toscano, E. J. Schermerhorn, C. Dettelbacher, D. Macherone and J. Zubieta, J. Chem. Soc., Chem. Commun., 1991, 933–934 RSC.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. B. Hendrickson, J. Am. Chem. Soc., 1967, 89, 7036–7043 CrossRef CAS.
- K. B. Wiberg, J. Org. Chem., 2003, 68, 9322–9329 CrossRef CAS PubMed.
- P. Khalili, C. B. Barnett and K. J. Naidoo, J. Chem. Phys., 2013, 138, 184110 CrossRef PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- G. Sartori, F. Bigi, R. Maggi and C. Porta, Tetrahedron Lett., 1994, 35, 7073–7076 CrossRef CAS.
- C. Redshaw, M.-J. Walton, D.-S. Lee, C. Jiang, M.-R. J. Elsegood and K. Michiue, Chem. – Eur. J., 2015, 21, 5199–5210 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation, single X-ray diffraction data, selected spectra and theoretical calculation details. Cartesian coordinates for the calculated structures (xyz file). CCDC 2210585 and 2210598. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03436k |
| ‡ Present Address: Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |