
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of N-heteroaromatic N-termini in Cu(II) ATCUN metallopeptides on their biorelevant redox activity†
Jannis
Barrera
ab,
Haleh
H. Haeri
c,
Julian
Heinrich
a,
Matthias
Stein
 d,
Dariush
Hinderberger
c and
Nora
Kulak
*a
d,
Dariush
Hinderberger
c and
Nora
Kulak
*a
aInstitute of Chemistry, Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: nora.kulak@ovgu.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, 12489 Berlin, Germany
cInstitute of Chemistry, Martin-Luther-Universität Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle, Germany
dMax Planck Institute for Dynamics of Complex Technical Systems, Molecular Simulations and Design Group, Sandtorstrasse 1, 39106 Magdeburg, Germany
First published on 22nd December 2022
Abstract
Cu(II) complexes with ATCUN peptide ligands have been investigated for their ROS (reactive oxygen species) generation and oxidative DNA degradation abilities. The biological activity of most ATCUN complexes such as Cu-GGH (Gly-Gly-His) is, however, low. Tuning the redox chemistry by incorporation of N-heteroaromatics reinstates ROS production which leads to efficient DNA cleavage.
Introduction
Cu(II) complexes have been thoroughly investigated for their capability to promote DNA degradation through an oxidative pathway in the presence of molecular oxygen and reducing agents. Due to their intrinsic redox properties, also modulated by the choice of ligands, ROS formation during the redox process induces the cleavage of the biomolecule.1–4 The amino terminal Cu(II)- and Ni(II)-binding (ATCUN) motif refers to a distinctive site in peptides and proteins like albumins and histatins.5–7 The N-terminus, two deprotonated amide N atoms of the peptide bonds, and a histidine residue constitute the binding sites with the metal from which a complex with square planar coordination geometry is formed.8 In the last decades, various applications for ATCUN metallopeptides have been suggested in the literature ranging from catalysis to medicine.9–16 One of the very first examples, however, already dates back from 1983: the simplest Cu(II)-ATCUN complex, Cu-GGH, was evaluated for antitumor activity toward Ehrlich ascites tumor cells inoculated in mice in the presence of ascorbate. The capability of killing the malignant cells by generating ROS was demonstrated,9 and thus suggested the therapeutic application of Cu(II)-ATCUN complexes. However, the 4N coordination mode of the ATCUN ligand towards Cu(II) results in high complex stability. In consequence, redox activity is (partially) suppressed causing very low ROS generation.10 A modification of the original ligand scaffold with alternative amino acids or additional organic moieties may enhance the potential of these Cu(II) species as artificial nucleases. Jin and Cowan have previously developed Cu-KGH and Cu-KGHK complexes with a higher nuclease activity than Cu-GGH which was assigned to positively charged lysine residues at physiological pH that allows stronger interaction with the negatively charged DNA target.12 In addition, the substitution of the α-amino acid glycine (Gly) by the β-amino acid β-alanine (β-Ala) in the ATCUN peptide sequence has been investigated.11,17,18 As we have shown previously, such a modification results in a more flexible peptide scaffold with more facile access to Cu(II)/Cu(I) redox cycling due to the easiness of switching between square planar (Cu(II)) and tetrahedral (Cu(I)) coordination. This flexibility was, however, accompanied by a decrease of complex stability.19 As a consequence of enhanced Cu(II)/Cu(I) redox cycling, ROS generation and DNA cleavage was significantly enhanced in the presence of ascorbate (ascH−). Increased DNA affinity by the introduction of positively charged Lys in the amino acid sequence lead to further enhancement of DNA cleavage activity.19 A contrary approach has been followed by Kritzer et al.: cyclization of the ATCUN motif led to enhanced Cu(II)/Cu(III) redox cycling due to the applied constraint and was concomitant with high DNA cleavage activity under oxidative conditions in the presence of hydrogen peroxide (H2O2).20,21Within this work we are examining whether the incorporation of N-terminal modifications of the ATCUN-like peptide through heteroaromatic systems may increase the nuclease activity of complexes in view of a higher DNA affinity through π–π stacking interactions with the nucleobases. Inspired by the ATCUN motif, we have now designed a series of Cu(II) complexes containing not only Gly and β-Ala in the 1st and 2nd position of the peptide sequence, but also N-heteroaromatics. As occurred with the introduction of β-Ala, the coordination of the aromatic N-donor groups results in successive increase of the chelate ring size (5,5,6 → 5,6,6 → 6,6,6) (Chart 1), which in turn, should have a direct impact on the complex stability,22 and consequently redox chemistry.19,23
 | ||
| Chart 1 Cu(II) complexes Cu-AA1–Cu-AA4 and Cu-CA1–Cu-CA5 incorporating β-Ala and N-heteroaromatics. | ||
Results and discussion
Synthesis of peptides and complexes
N-Heteroaromatics (shown in green in Chart 1) were synthesized following previously described procedures (ESI, S2†). Incorporation of these derivatives by Fmoc-SPPS protocols24,25 at the N-terminus resulted in tetrapeptides CA1–CA5 (N-heteroaromatic-β-Ala-His-Lys, ESI, S0†). Gly and β-Ala were interchanged between 1st and 2nd positions to afford peptides AA1–AA4. All peptides were purified by RP-HPLC and characterized via ESI-MS and analytical RP-HPLC (ESI, S3†).Cu(II) complexes were prepared in situ by mixing aqueous solutions of CuCl2·2H2O and tetrapeptides in the presence of MOPS buffer (pH 7.4) (ESI, S0 and S4†).
Characterization of the complexes
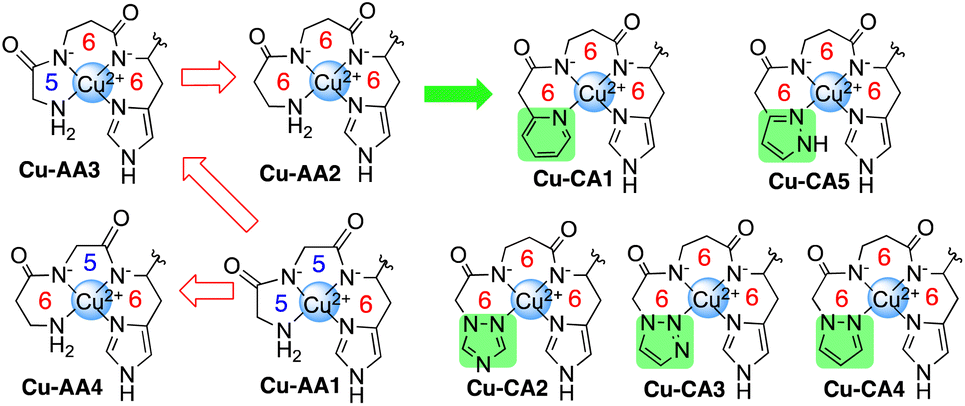
As already known, the formation of a single absorption band at 525 nm in the visible spectrum and at physiological pH is a distinctive feature of the d–d transition of Cu-GGH.5,8 According to this, the d–d transition band of complex Cu-AA1 (Gly-Gly-His-Lys) is observed at 527 nm (Fig. 2). In contrast, the absorption bands of complexes containing β-Ala in the second position of the peptide sequence (Cu-AA2, Cu-AA3 and Cu-CA1–CA5) are redshifted at longer wavelengths between 555 nm and 670 nm (Fig. 1).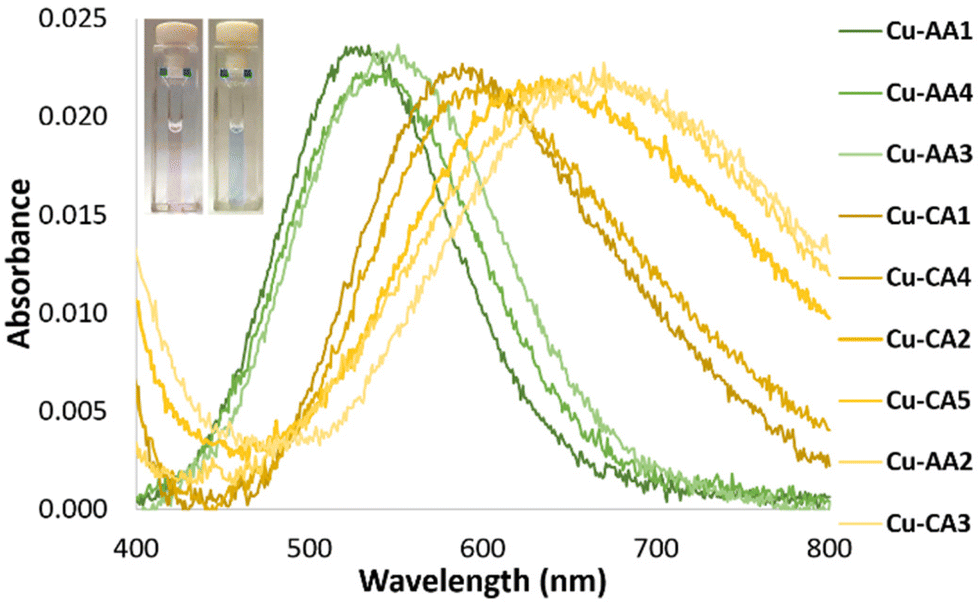 | ||
| Fig. 1 d–d absorption bands of Cu(II) complexes: Cu-AA1, Cu-AA3 and Cu-AA4 (green dash-dotted lines, pink solution), Cu-AA2 and Cu-CA1–Cu-CA5 (yellow dash-dotted lines, blue solution) (λmax d–d transition band see Table S3.2†). | ||
CD spectroscopic measurements of the complexes can provide a better understanding of the active Cu(II) species, which initiate DNA cleavage at pH 7.4. As evidenced in Fig. 2, the CD spectrum of complex Cu-AA1 shows d–d bands with maximal and minimal values at 508 and 600 nm, respectively. This is in agreement with results for complexes of the type Cu-GGH.19,26 Unlike Cu-AA1, Cu-AA3 and Cu-AA4 (ESI, S4†), Cu-AA2, is the only one of the type AA that does not show a d–d band in its CD spectrum (Fig. 2). On the other hand, Cu-CA1 (Fig. 2) and Cu-CA4 (ESI, S4†) show distinctive d–d bands with maxima and minima in the range of 550 and 700 nm, but complexes Cu-CA2, Cu-CA3, and Cu-CA5 do not (ESI, S4†). To conclude, since only chiral complexes are observable in the CD spectra, the Cu(II) complexes Cu-AA1, Cu-AA3, Cu-AA4, Cu-CA1 and Cu-CA4 should be 3N- or 4N-coordinated species, whereas those without d–d bands Cu-AA2, Cu-CA2, Cu-CA3 and Cu-CA5‡ are expected to be 2N species.
These results are consistent with UV/VIS spectroscopic data (Fig. 1): complexes with no bands in the CD spectra show higher λmax for the d–d transition band in the VIS spectra, which is known for ATCUN-like complexes when coordination mode is changed from 4N → 3N → 2N.27 The wavelength shift is consistent with a loss in complex stability due to formation of 6-membered, thus more flexible, chelate rings in the coordination sphere.20,21
 | ||
| Fig. 2 CD spectra of Cu(II) complexes Cu-AA1, Cu-AA2 and Cu-CA1 in MOPS buffer (20 mM) pH 7.4 (prepared in situ from 1.0 mM peptides and 0.8 mM CuCl2). | ||
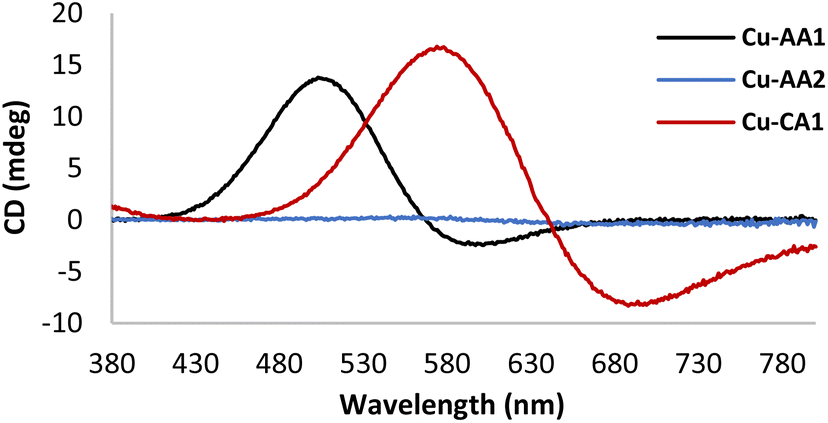
Furthermore, cyclic voltammetry experiments were carried out with the purpose of understanding the ability of Cu(II) complexes to generate ROS in accordance with their redox chemistry. Fig. 3 exemplarily illustrates voltammograms obtained for complexes Cu-AA1, Cu-AA2 and Cu-CA1 (ESI, S8† for other complexes, cathodic and anodic scanning directions). For the sake of comparison with relevant literature for Cu(II) ATCUN complexes,23 the conditions for these experiments were initially chosen accordingly (KNO3/HNO3 electrolyte).
 | ||
| Fig. 3 Cyclic voltammograms of Cu-AA1, Cu-AA2 and Cu-CA1 (0.5 mM) recorded in KNO3/HNO3 (96 mM/4 mM) at pH 7.4. The arrow indicates the direction of the potential scanning (scan rate 50 mV s−1). | ||
Whereas for Cu-AA2 and Cu-CA1, the reduction from Cu(II) to Cu(I) is the only process observed (Ered Cu(II) → Cu(I) −0.15 V and −0.02 V vs. Ag/AgCl, respectively), Cu-AA1 does not show a Cu(II)/Cu(I) reduction process (Fig. 3). When the potential was scanned towards the anodic direction, oxidation of Cu(II) to Cu(III) in Cu-AA1 takes place instead as similarly reported by Wawrzyniak et al.23 (Eox Cu(II) → Cu(III) +0.84 V vs. Ag/AgCl, ESI, Fig. S8.1a†). As expected, complexes Cu-CA2–Cu-CA4 exhibit a similar behaviour to the one observed for Cu-AA2 and Cu-CA1 (6,6,6 chelate rings) with reduction potentials in the range of −0.02 V and −0.08 V vs. Ag/AgCl (ESI, S8†). In the case of Cu-AA3, the reduction to Cu(I) is visible when the potential for the oxidation to Cu(III) is reached first (anodic scanning direction), as it has been already demonstrated by Faller, Bal and coworkers.23,27 Remarkably, complexes Cu-CA1–Cu-CA4 and Cu-AA2 show a Cu(II)/Cu(I) redox process independent of the oxidation to Cu(III), which is a key feature in the redox chemistry of these species compared with the classic Cu-GGH and other previously reported complexes carrying Gly and β-Ala in the peptide sequence (vide supra). This fact serves as a basis for explaining the higher ROS generation and consequently the increased nuclease activity of these complexes towards DNA.
Although the above conditions in voltammetric experiments are not comparable to those in nuclease activity studies, the CV data of the Cu-AA series show that for larger chelate ring sizes (5,6,6/6,5,6/6,6,6) in general the Cu(II) → Cu(I) reduction process is more easily accessible than for the natural ATCUN motif (5,5,6). In case of the CA-type metallopeptides the Cu(II) → Cu(I) redox process is even accessible without prior oxidation to Cu(III), which is reflecting the special redox feature of these new N-heterocyclic metallopeptides.
Additionally, in order to mimic the conditions of ROS and DNA cleavage studies, all voltammograms were also recorded in buffer solution (2.5 mM MOPS). It is important to note that a lower buffer concentration was used since the Cu(II)/Cu(I) process was not visible at the higher concentration (10 mM MOPS as in the other experiments). In buffered solution, the trends of the non-buffered system can be corroborated, however, it is also possible in this case to explicitly compare Ered values: Ered of Cu-AA1 (−0.12 V vs. Ag/AgCl, ESI, Fig. S8.1d†) is found at more negative potential than that of Cu-AA2 (−0.02 V vs. Ag/AgCl, ESI, Fig. S8.2d†) and Cu-CA1 (+0.03 V vs. Ag/AgCl, ESI, Fig. S8.5d†) and all other 6,6,6-metallopeptides (∼+0.1 V vs. Ag/AgCl, ESI, Fig. S8.6–8d†‡).
EPR measurements were performed on copper complexes, since it is shown in literature that their redox properties are closely related to their geometrical structures.20,28,29
Several factors can affect redox properties of a metal complex; the kind of metal ion, the nature of bound ligands, interaction between metal and ligand(s) and non-covalent interactions in the coordination sphere like hydrogen bonding, hydrophobicity and π-stacking.30 One of the main information obtained by EPR is about ligand coordination sphere. Hyperfine spectroscopy methods monitoring interactions of paramagnetic metal centers with neighboring ligands (primary or secondary coordination sphere) and structural geometry constraints could be observed by g-tensor. In an axial spectral symmetry system, g∥ > g⊥ is indicative of a dx2–y2 electronic ground state which correlates to square planar or tetragonal geometries. It is known from literature31 that the g∥ is affected by change in ligand type (N- or S- or O-based). Increase in g∥ could be also correlated to the distortion degree from tetrahedral geometrical structure.20,29 On the other hand, it is demonstrated that changes in ligand structure (and therefore possible geometrical distortion) are capable of tuning redox properties (for example see ref. 32 and 33). So, it is possible to deductively correlate redox potentials to structural geometry constrains derived from EPR g-tensor.
We measured at two frequency bands; while we get highly resolved g-values at higher frequency of ∼34 GHz, the hyperfine couplings were not resolved due to the larger g-anisotropy. Therefore, we also measured copper complexes at conventional X-band frequencies (9.4 GHz) to get information about the interaction of the copper center with the surrounding nuclei (Fig. 4, for derived spin Hamiltonian parameters cf. Table S4.3†).
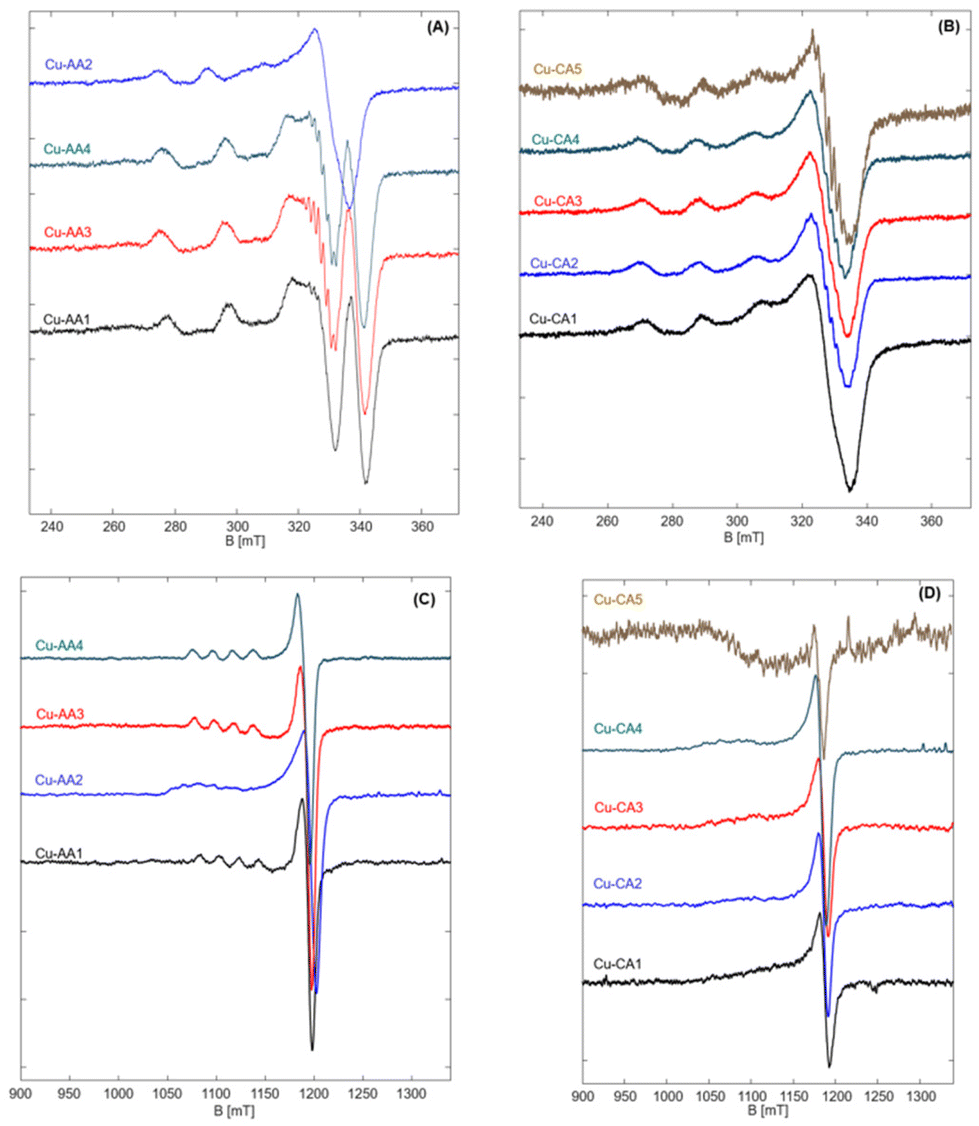 | ||
| Fig. 4 EPR spectra of Cu-AA (A) and Cu-CA (B) complexes measured at 9.4 GHz and 77 K, and EPR spectra of Cu-AA (C) and Cu-CA (D) complexes measured at 34 GHz and 50 K. | ||
EPR spectra of the Cu(II) peptide complexes reveal the typical copper (type II) with a dx2–y2 ground state, characteristic of square pyramidal, square planar or elongated hexagonal tetragonal geometries distorted to tetragonal along with generally axial g-tensor and large Azz hyperfine couplings of copper (∼10–21 mT).31,34
Most of the complexes show axial symmetry, regardless of the used frequency. In the series of β-Ala-substituted complexes, however, three complexes Cu-AA1, Cu-AA3 and Cu-AA4 show anisotropy in hyperfine couplings in addition to g-anisotropy. This indicates that spin densities in these complexes are not evenly distributed and delocalized from the dx2–y2 orbital to the nearby ligands at the equatorial plane. This fact is reflected in nitrogen splittings that were clearly observable for AA3 and AA4 complexes. At higher frequencies, however, these complexes showed axial symmetric spectra in both, g- and A-tensor (Fig. 4C). Although no explicit ligand hyperfine splitting could be resolved for the Cu-AA1 complex, the set of (Azz, gzz) fits typical CuN4-spectra with square planar coordination of equatorial ligands (Fig. S4.14A, E and Table S4.3†).34
Possessing a rather flexible ligand AA2, the copper complex has a very broad spectrum as expected. It has a completely different spectral shape when compared to other complexes and deviates profoundly from the suggested square planar to a rather distorted tetragonal geometry. The measured Q-band EPR spectrum is still axial but reveals a clear fifth splitting originating from a second component, which could not be resolved at X band (Fig. S4.14B and F†). This is a common feature of type II copper complexes that can manifest itself in dependence of the type and charge of the surrounding ligands.34,35 It is shown that large local concentration of Cu(II) leads to broadening of EPR spectra, as well.36 The presence of two different components, which results in change of the local geometry, can explain the observed inconsistency in CD spectra of Cu-AA2, in comparison to other Cu-AA complexes. Also, this deviation from square planar geometry suggests a higher redox activity of this complex compared to the other ones in the β-Ala substituted series.
Spectral simulations of the Cu-AA3 complex reveal a 2N complex with nitrogen splittings of about 0.5 mT (Fig. S4.14C†). Nitrogen hyperfine splittings were not resolved for other complexes, indicating that these most probably are remote nuclei weakly interacting with the copper center.
The Cu-CA complexes, no matter of measured frequency, are typically broad and rather unstructured at the parallel position, as a result of β-Ala and N-heteroaromatic ring substitutions. The Cu-CA1 complex was chosen for quantitative simulation, since it outperformed the other regarding its DNA cleavage activity (vide infra). We found an approximately axial symmetry with a large Azz coupling (no matter of measured frequency, Fig. S4.14D and G†). The value of gzz is increased compared to the Cu-AA1 complex upon β-Ala and N-heteroaromatic ring substitutions, but still smaller than that of the Cu-AA2 complex. We also found that the g-tensor is tilted against the molecular frame by about 15° towards the y-direction. The broadness of the spectrum is due to the presence of species with slightly different g-values, reflected as a g-strain at the parallel position of the spectrum.
As we have shown before,19 addition of β-Ala does not lead to a significant change of gzz of the complex, so it must be the steric effect of the added N-heteroaromatic ring which distorts the structure (to some extent) from square planar (increased gzz and tilted gyy) while preserving the local environment around copper as shown by Azz.
Computational studies
In order investigate the structural flexibility and Cu(II)/Cu(I) redox chemistry of the different chelates, efficient computational methods were used. Since the His-Lys C-terminus is identical to all complexes, focus was on the complex core with 4N coordination (Lys-truncated). As a measure of intramolecular flexibility, the number of unique conformers accessible at room temperature were calculated with CREST/xTB-GFN2 using several refinement steps.37,38 Upon successive substitution of Gly by β-Ala, the number of conformers increases significantly. Introduction of N-heteroaromatics again reduces the number of possible conformers (Cu-CA1-Cu-CA5). When reducing Cu(II) to Cu(I), the number of accessible conformers significantly increases (Fig. 5).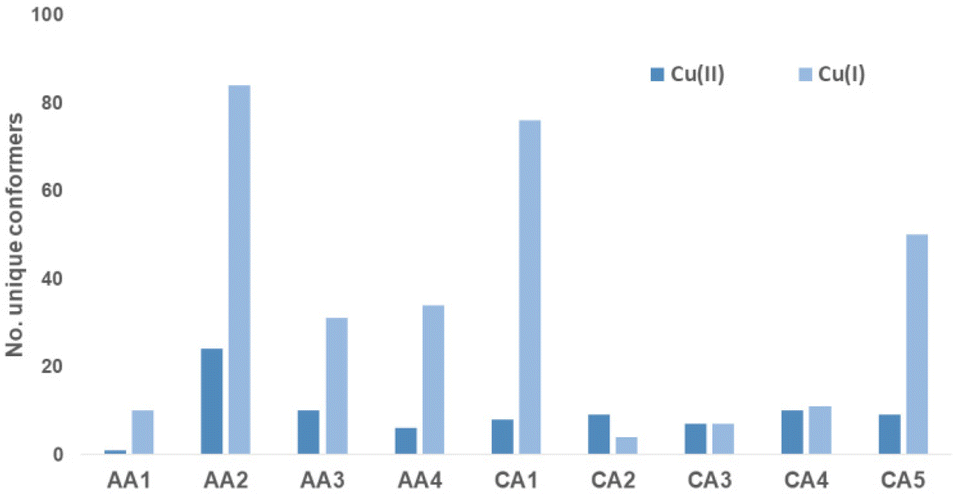 | ||
| Fig. 5 Number of unique conformers for Cu(II)/Cu(I) ATCUN complexes. | ||
Upon reduction, the copper β-Ala-His coordination is maintained, while the bond between N(donor at N-terminus)-Cu(I) breaks affording 3N-coordinated Cu(I) species. This leads to a larger number of distinct conformers (except for Cu-CA2). In Fig. 6, the global minimum structures of Cu-CA1 are shown with Cu–N(pyridine) distances of 2.1 Å in Cu(II) and 5.4 Å in Cu(I). It should be noted that the Cu(I) species as calculated bound to two amide functionalities is probably elusive, since we did not find experimental evidence for such a compound, neither during our investigation nor in the literature. Nevertheless, this also supports the assumption of facile reoxidation to Cu(II) necessary for the ROS generation.
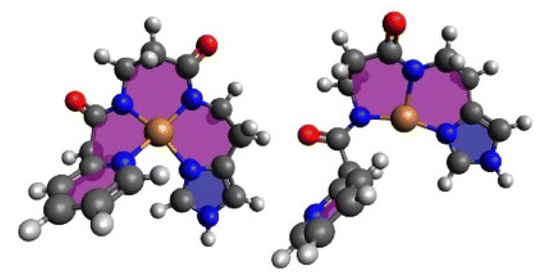 | ||
| Fig. 6 Global minimum structures of equatorially 4N-coordinate oxidized Cu(II)-CA1 and reduced Cu(I)-CA1. | ||
The lability of the Cu–N bond can also be seen from the coupled-cluster calculated Cu(II) complexation energies of compounds Cu-CA1–Cu-CA5 which are lower than those of Gly-Gly-His (−864 kJ mol−1) and Gly-β-Ala-His (−860 kJ mol−1). This enables a facile reduction of Cu(II) complexes containing N-heteroaromatics in their ligand system. As can be derived from the experimental as well as computed reduction potentials, the reduction process is indeed easier for these complexes (Table S9.3†).
Cu(II)-peptide interactions were additionally evaluated by calculating spin densities at the metal center and the peptide (for previous results cf. ref. 19). Calculations for the Cu-CA1 system confirm these previous results (Fig. 7): the spin is mostly copper-centred but partially delocalized onto the coordinating N atoms.
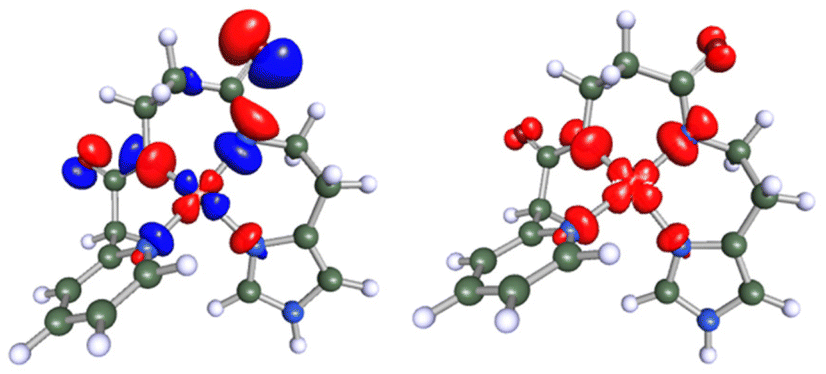 | ||
| Fig. 7 Left: the SOMO of the Cu(II)-CA1 complex at an isosurface value of 0.05 e Å−3. This orbital is an anti-linear combination of the Cu dx2–y2 and the p orbitals of the equatorial nitrogen atoms. Right: unpaired spin density plot of Cu(II)-CA1 at an isocontour value of 0.005 e Å−3. | ||
DNA cleavage
The activity of complexes Cu-AA1–Cu-AA4 and Cu-CA1–Cu-CA5 as metallonucleases in the cleavage of plasmid DNA was evaluated by agarose gel electrophoresis in the presence of ascorbate and MOPS buffer (pH 7.4). Complexes were initially tested at different concentrations from which we determined that all Cu(II) species including β-Ala were able to cleave DNA to form II (nicked DNA) from a concentration of 30 μM on. At a concentration of 35 μM, all these complexes, with the exception of Cu-AA4, promoted DNA scission to form III (linear) (Fig. 8). Complete degradation of DNA into form II and III was achieved at 40 μM for all complexes except for Cu-AA1 (ESI, S5†).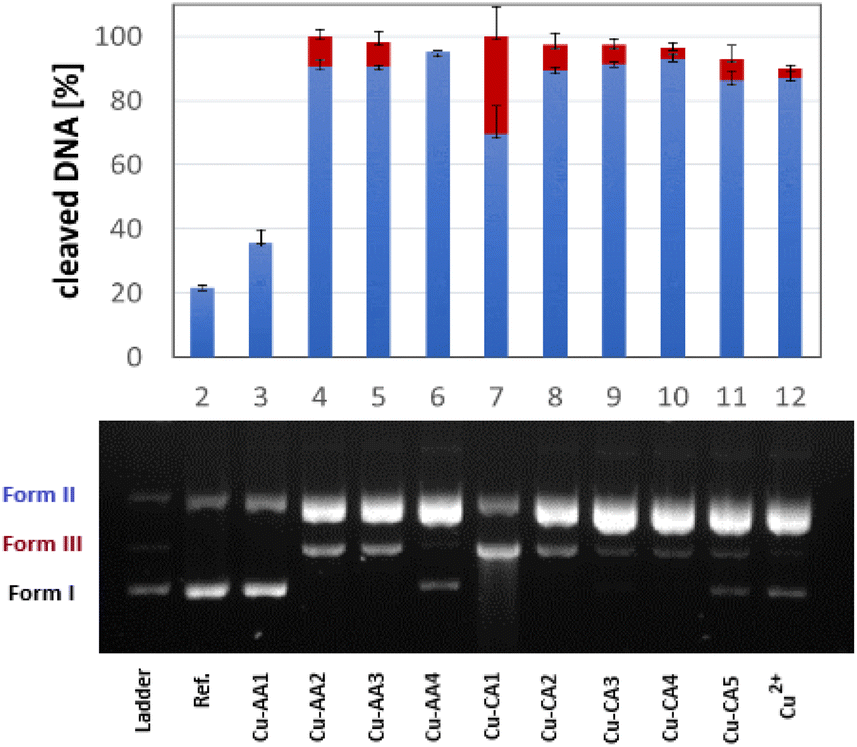 | ||
| Fig. 8 DNA cleavage activity of complexes Cu-AA1–CuAA4, Cu-CA1–Cu-CA5 and CuCl2 at 35 μM (agarose gel). Incubation was done using 0.025 μg μL−1 plasmid DNA pBR322, 50 mM MOPS buffer (pH 7.4) and 1 mM ascH− for 1 h at 37 °C and 500 rpm (same conditions for the reference without addition of a complex). Error bars for the standard deviation were obtained with at least three experiments. Blue bars indicate percentage of form II, red bars percentage of form III. The ladder contains plasmid DNA form I, II and III enzymatically prepared for band referencing. | ||
It can be concluded that the most active metallonucleases are those containing β-Ala in position 2 of the peptide as reported before by us.19 Complexes carrying N-heteroaromatics, having also β-Ala in the 2nd position, display comparable cleavage activity. Outstandingly, Cu-CA1 (pyridine at the N-terminus) is by far the most efficient cleaver among complexes of this series, also if compared to complexes Cu-AA1–Cu-AA4. Structural differences within the 5-membered heterocycles in Cu-CA2–Cu-CA5 do not exert significant influence in cutting plasmid DNA.
ROS formation
In order to investigate the mechanism of DNA strand breakage reaction through an oxidative mechanism, ROS formation mediated by complexes Cu-AA2 and Cu-CA1 in the presence of ascorbate was monitored employing the fluorogenic probes terephthalate (TPA) and pentafluorobenzenesulfonyl fluorescein (PBSF) (ESI, S6†).39–41Generated hydroxyl radicals (˙OH) are trapped by TPA, which gives rise to the strongly fluorescent 2-hydroxyterephthalate. Similarly, perhydrolysis of PBSF results in the formation of fluorescein, which indicates the presence of H2O2. Both species are indeed generated by the system Cu(II) complex/ascH−, and DNA degradation is taking place as a result of the reaction with ROS. As a further proof, DMSO and pyruvate are able to quench the emission intensity of both dyes (ESI, S6†).41 A kinetic experiment for the production of H2O2 was assessed for comparing the inactive complex Cu-AA1 and a CuCl2 control with the moderately active complex Cu-AA2 and the most active complex Cu-CA1 (Fig. 9, for the other complexes cf. ESI, S6†).
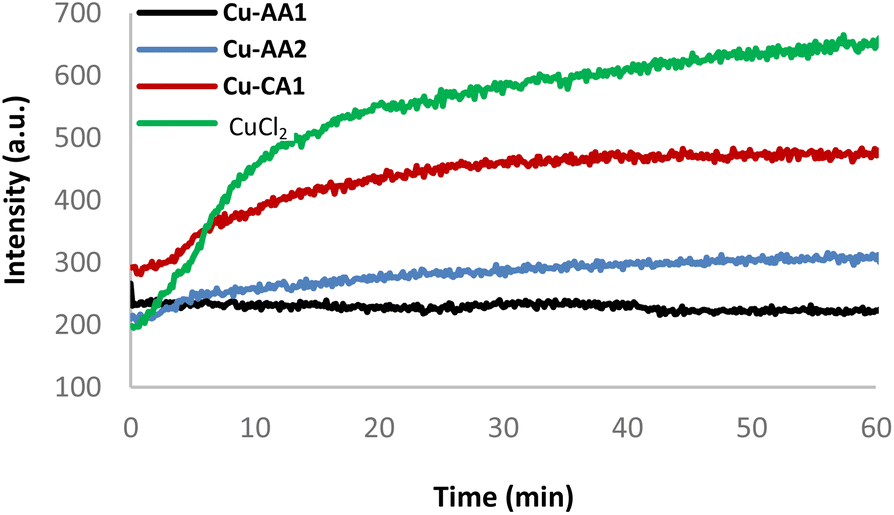 | ||
| Fig. 9 Kinetics of H2O2 evolution: fluorescence emission based on fluorescein formation by PBSF perhydrolysis (25 μM, λex = 485 nm, λem = 513 nm) with complexes Cu-AA1, Cu-AA2, Cu-CA1 and CuCl2 (35 μM) in the presence of ascH− (1 mM) and MOPS buffer (50 mM, pH 7.4) at room temperature. | ||
The formation of H2O2 was certainly enhanced with complexes Cu-AA2 and Cu-CA1, the latter with a considerable increase, compared to the poor ROS generation of complex Cu-AA1, thus explaining its lacking nuclease activity (Fig. 8) and confirming previous studies with Gly-containing Cu(II) complexes.10 The kinetics remarkably reveals faster H2O2 formation for complex Cu-CA1 in comparison to the others. Additionally, Cu-CA1 surpasses previous examples from our lab.§![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 ROS generation of Cu-CA1 and Cu-AA2 with ascorbate was also assessed in a quenching assay by gel electrophoresis. We could verify this way, firstly, an oxidative pathway of the DNA damage since there was no cleavage in the absence of ascorbate. Secondly, inhibition of nuclease activity was corroborated in the presence of scavengers DMSO and pyruvate selective for reactive species ˙OH and H2O2, respectively, but was also observed when using scavengers NaN3 for 1O2 and SOD for O2˙− (Fig. 10 for Cu-CA1, ESI, S5† for Cu-AA2).
19 ROS generation of Cu-CA1 and Cu-AA2 with ascorbate was also assessed in a quenching assay by gel electrophoresis. We could verify this way, firstly, an oxidative pathway of the DNA damage since there was no cleavage in the absence of ascorbate. Secondly, inhibition of nuclease activity was corroborated in the presence of scavengers DMSO and pyruvate selective for reactive species ˙OH and H2O2, respectively, but was also observed when using scavengers NaN3 for 1O2 and SOD for O2˙− (Fig. 10 for Cu-CA1, ESI, S5† for Cu-AA2).
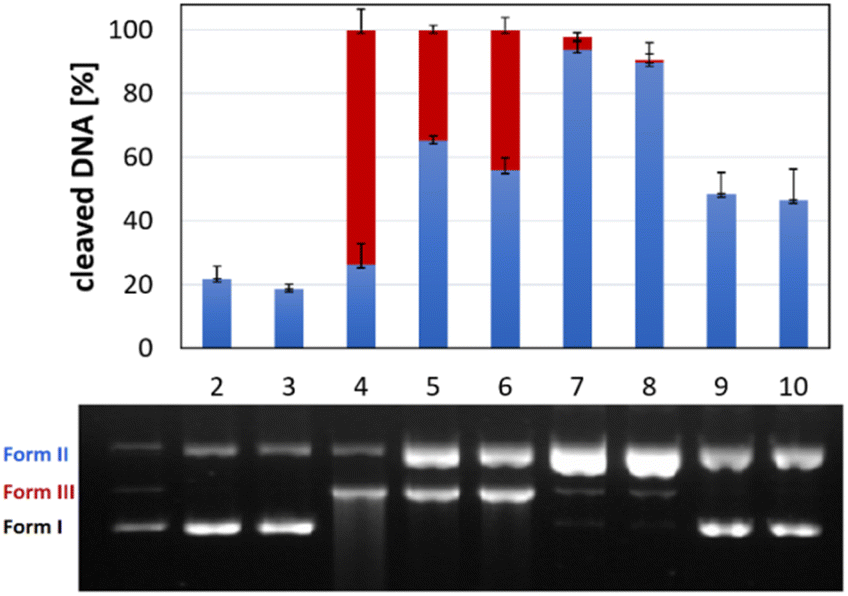 | ||
| Fig. 10 Reactive oxygen species (ROS) assay by gel electrophoresis. Incubation with complex Cu-CA1 at a concentration of 35 μM at 37 °C and pH 7.4 for 1 h. Lane 1: DNA ladder (forms I, II and III), lane 2: DNA reference, lane 3: complex without ascH−, lane 4–10: complex with ascH− and the following scavengers: lane 4: none, lane 5: DMSO (400 mM), lane 6: NaN3 (10 mM), lane 7: pyruvate (2.5 mM), lane 8: SOD (625 U mL−1), lane 9: pyruvate (2.5 mM) and SOD (625 U mL−1), lane 10: DMSO (400 mM), NaN3 (10 mM), pyruvate (2.5 mM), SOD (625 U mL−1). | ||
In order to exclude that ROS were formed by released Cu(I) upon reduction, Cu(I) stability constants with peptides AA1, AA2 and CA1 were determined by a UV/VIS competition experiment. Kapp values were 20 × 106 M−1 for the 5,5,6 chelator AA1, and below 0.4 × 106 M−1 for the 6,6,6 chelators AA2 and CA1, respectively (ESI, S4†). It could have been expected that the stability would be even lower for 6,6,6-chelating peptides (vide infra, CD spectroscopy), however, the calculated values are in the same order of magnitude of previously reported Cu(I) complexes with amyloid-β peptides.42
DNA binding
Further experiments were directed to evaluate possible DNA binding modes of the complexes in order to assess their impact on the DNA cleavage activity. The melting temperature (Tm) of calf thymus DNA (CT-DNA) in the presence of all Cu(II) complexes herein reported was monitored via absorbance at 260 nm as a function of the temperature. An increase of Tm (1–1.3 °C) was observed in the presence of Cu-CA1-CA5 and Cu-AA2 (ESI, S7†). These results suggest electrostatic and/or partial groove binding interactions with DNA.43 Additionally, CD spectral changes of CT-DNA in the presence of the metallopeptides were considered. Perturbations of the helical structure induced by Cu(II) complexes result in changes of the positive and negative band of the B-DNA conformation.44,45 The Cu(II) complexes generate relatively weak distortions, which is indicative for electrostatic interactions as the predominant mode of DNA interaction caused by the Lys tail (ESI, S7,† exemplarily Cu-AA1, Fig. 11, top).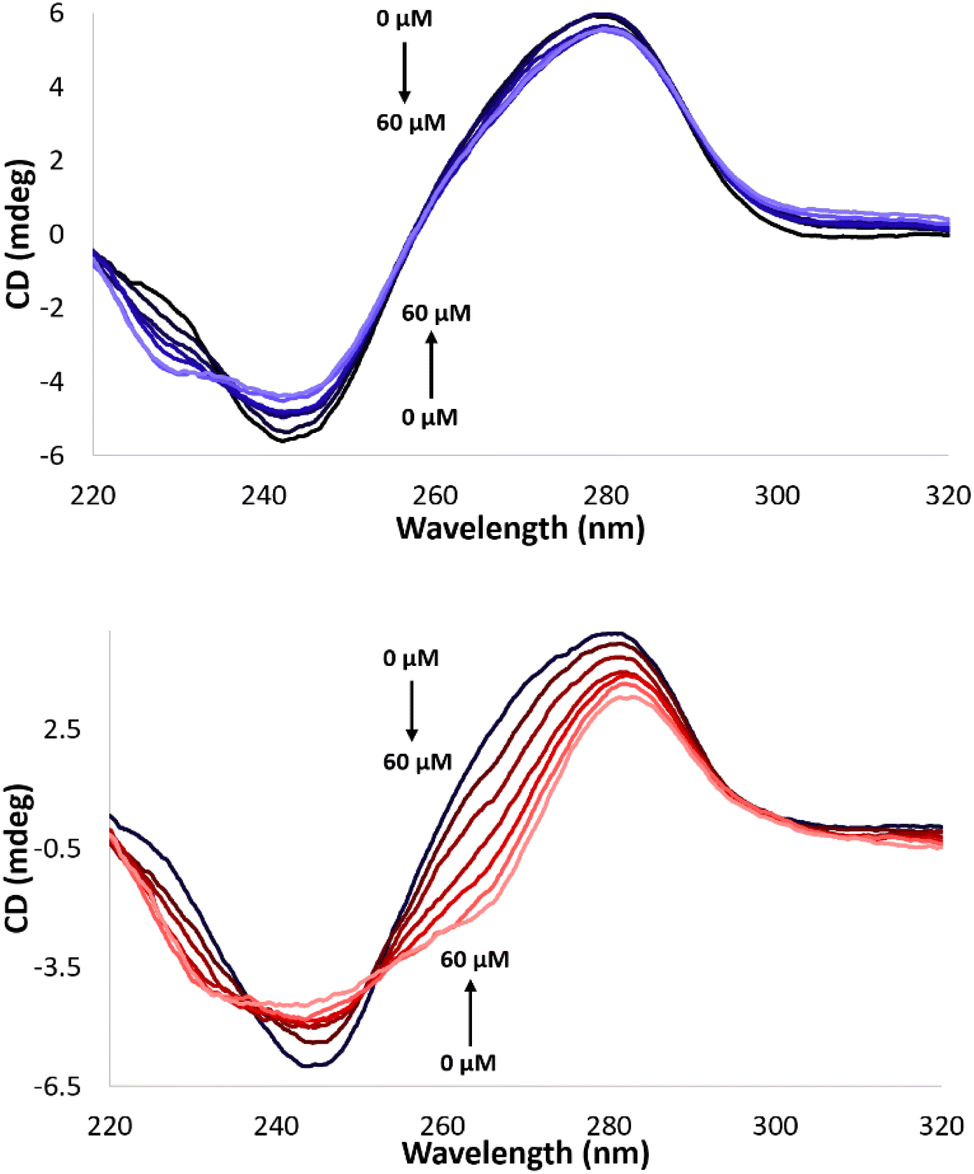 | ||
| Fig. 11 CD spectra of CT-DNA (100 μM) in MOPS buffer (50 mM, pH 7.4) with 0 to 60 μM complex Cu-AA1 (top) and Cu-CA1 (bottom), respectively. | ||
Cu-CA1 is undoubtedly the complex that exhibits the most significant changes in the CD spectrum, possibly associated with groove binding interactions (Fig. 11, bottom). The outstanding nuclease activity observed for Cu-CA1 is thus also explained in terms of its interaction with DNA besides its considerably higher ROS generation.
Conclusions
In conclusion, the experimental results herein presented show the enhanced oxidative nuclease activity towards DNA of ATCUN-based Cu(II) complexes with β-Ala and N-heteroaromatics (especially pyridine) replacing Gly in the well-studied Cu-GGH model. Through structural changes in the peptide sequence and the subsequent formation of larger chelate rings (5,5,6- → 5,6,6- → 6,6,6-chelates) when coordinating to the metal, i.e. increasing the intramolecular flexibility, we could prove that catalytic ROS generation is considerably increased compared to Cu-GGH (in our studies Cu-AA1, Gly-Gly-His-Lys, 5,5,6-chelate). A more flexible ligand like in Cu-AA2 (β-Ala-β-Ala-His-Lys, 6,6,6-chelate) ensures that Cu(II) species are indeed able to support Cu(II)/Cu(I) redox processes accompanied by a switch from a 4N to a 3N coordination. Moreover, the introduction of an N-terminal heteroaromatic moiety in the ATCUN-type peptide to form a weakly coordinated six-membered chelate ring promotes not only Cu(II) reduction, and thus enhanced ROS production, but also a better interaction with the double helix and consequently, a higher nuclease activity. This is shown with Cu-CA1, the best DNA cleaver in the presented series of compounds bearing an N-terminal pyridine moiety. This feature brings to light that Cu(II)-ATCUN complexes, initially considered poor catalysts for ROS generation, could actually display an outstanding performance as oxidative metallonucleases.Author contributions
Jannis Barrera: investigation, validation, visualization, writing – original draft. Haleh H. Haeri: investigation, validation, visualization, writing – original draft. Julian Heinrich: investigation, supervision, validation, visualization. Matthias Stein: investigation, validation, visualization, writing – original draft. Dariush Hinderberger: supervision, validation, writing – reviewing & editing. Nora Kulak: conceptualization, supervision, validation, writing – reviewing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M.S. and N.K. are thankful for support by the state of Saxony-Anhalt (European Regional Development Fund–ERDF grant ZS/2016/04/78155) within the OVGU Research Center Dynamic Systems.References
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- T. J. P. McGivern, S. Afsharpour and C. J. Marmion, Inorg. Chim. Acta, 2018, 472, 12–39 CrossRef CAS.
- C. Duncan and A. R. White, Metallomics, 2012, 4, 127–138 CrossRef CAS PubMed.
- S. Tardito and L. Marchiò, Curr. Med. Chem., 2009, 16, 1325–1348 CrossRef CAS PubMed.
- T. Peters Jr. and F. A. Blumenstock, J. Biol. Chem., 1967, 242, 1574–1578 CrossRef CAS PubMed.
- C. Harford and B. Sarkar, Acc. Chem. Res., 1997, 30, 123–130 CrossRef CAS.
- G. Gasmi, A. Singer, J. Forman-Kay and B. Sarkar, J. Pept. Res., 1997, 49, 500–509 CrossRef CAS PubMed.
- N. Camerman, A. Camerman and B. Sarkar, Can. J. Chem., 1976, 54, 1309–1316 CrossRef CAS.
- E. Kimoto, H. Tanaka, J. Gyotoku, F. Morishige and L. Pauling, Cancer Res., 1983, 43, 824–828 CAS.
- A. Santoro, G. Walke, B. Vileno, P. P. Kulkarni, L. Raibaut and P. Faller, Chem. Commun., 2018, 54, 11945–11948 RSC.
- A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef CAS.
- Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 127, 8408–8415 CrossRef CAS PubMed.
- B. K. Maiti, N. Govil, T. Kundu and J. J. G. Moura, iScience, 2020, 23, 101792 CrossRef CAS PubMed.
- B. Kandemir, L. Kubie, Y. Guo, B. Sheldon and K. L. Bren, Inorg. Chem., 2016, 55, 1355–1357 CrossRef CAS PubMed.
- S. Chakraborty, E. H. Edwards, B. Kandemir and K. L. Bren, Inorg. Chem., 2019, 58, 16402–16410 CrossRef CAS PubMed.
- J. Domergue, P. Guinard, M. Douillard, J. Pécaut, O. Proux, C. Lebrun, A. Le Goff, P. Maldivi, P. Delangle and C. Duboc, Inorg. Chem., 2021, 60, 12772–12780 CrossRef CAS PubMed.
- J. Nagaj, K. Stokowa-Sołtys, I. Zawisza, M. Jeżowska-Bojczuk, A. Bonna and W. Bal, J. Inorg. Biochem., 2013, 119, 85–89 CrossRef CAS PubMed.
- C. Wende and N. Kulak, Chem. Commun., 2015, 51, 12395–12398 RSC.
- J. Heinrich, K. Bossak-Ahmad, M. Riisom, H. H. Haeri, T. R. Steel, V. Hergl, A. Langhans, C. Schattschneider, J. Barrera, S. M. F. Jamieson, M. Stein, D. Hinderberger, C. G. Hartinger, W. Bal and N. Kulak, Chem. Eur. J., 2021, 27, 18093–18102 CrossRef CAS PubMed.
- K. P. Neupane, A. R. Aldous and J. A. Kritzer, J. Inorg. Biochem., 2014, 139, 65–76 CrossRef CAS PubMed.
- K. P. Neupane, A. R. Aldous and J. A. Kritzer, Inorg. Chem., 2013, 52, 2729–2735 CrossRef CAS PubMed.
- J. Nagaj, K. Stokowa-Sołtys, E. Kurowska, T. Frączyk, M. Jeżowska-Bojczuk and W. Bal, Inorg. Chem., 2013, 52, 13927–13933 CrossRef CAS PubMed.
- M. Z. Wiloch, I. Ufnalska, A. Bonna, W. Bal, W. Wróblewski and U. E. Wawrzyniak, J. Electrochem. Soc., 2017, 164, G77–G81 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed.
- C. Conato, H. Kozłowski, P. Młynarz, F. Pulidori and M. Remelli, Polyhedron, 2002, 21, 1469–1474 CrossRef CAS.
- P. Gonzalez, K. Bossak, E. Stefaniak, C. Hureau, L. Raibaut, W. Bal and P. Faller, Chem. Eur. J., 2018, 24, 8029–8041 CrossRef CAS PubMed.
- B. Bennett and J. M. Kowalski, Methods Enzymol., 2015, 563, 341–361 CAS.
- P. Comba, N. F. Curtis, G. A. Lawrance, A. M. Sargeson, B. W. Skelton and A. H. White, Inorg. Chem., 1986, 25, 4260–4267 CrossRef CAS.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, 2016, 1857, 557–581 CrossRef CAS PubMed.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed.
- H. B. Lee and T. Agapie, Inorg. Chem., 2019, 58, 14998–15003 CrossRef CAS PubMed.
- J. D. Cope, H. U. Valle, R. S. Hall, K. M. Riley, E. Goel, S. Biswas, M. P. Hendrich, D. O. Wipf, S. L. Stokes and J. P. Emerson, Eur. J. Inorg. Chem., 2020, 1278–1285 CrossRef CAS PubMed.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- G. M. Cereghetti, A. Schweiger, R. Glockshuber and S. Van Doorslaer, Biophys. J., 2001, 81, 516–525 CrossRef CAS PubMed.
- P. Brüggeller and C. Mayer, Nature, 1980, 288, 569–571 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- X. Fang, G. Mark and C. von Sonntag, Ultrason. Sonochem., 1996, 3, 57–63 CrossRef CAS.
- H. Maeda, Y. Fukuyasu, S. Yoshida, M. Fukuda, K. Saeki, H. Matsuno, Y. Yamauchi, K. Yoshida, K. Hirata and K. Miyamoto, Angew. Chem. Int. Ed., 2004, 43, 2389–2391 CrossRef CAS PubMed.
- S. Leichnitz, J. Heinrich and N. Kulak, Chem. Commun., 2018, 54, 13411–13414 RSC.
- B. Alies, B. Badei, P. Faller and C. Hureau, Chem. Eur. J., 2012, 18, 1161–1167 CrossRef CAS PubMed.
- S. Roy, A. K. Patra, S. Dhar and A. R. Chakravarty, Inorg. Chem., 2008, 47, 5625–5633 CrossRef CAS PubMed.
- N. Shahabadi, S. Kashanian and A. Fatahi, Bioinorg. Chem. Appl., 2011, 2011, 687571 Search PubMed.
- S. Ramakrishnan and M. Palaniandavar, J. Chem. Sci., 2005, 117, 179–186 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization, DNA cleavage and interaction studies, ROS detection, cyclic voltammetry, computational details and data. See DOI: https://doi.org/10.1039/d2dt02044k |
| ‡ Complex Cu-CA5 has shown irregular results in some of the measurements (UV/VIS, CD, CV, EPR) with large deviations with respect to the other complexes. |
| § Peptides in the previous work19 contained a Ser-Ser tail. Compound Cu-AA3 corresponds to compound 3 therein |
| This journal is © The Royal Society of Chemistry 2023 |