
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A database of molecular properties integrated in the Materials Project†
Evan Walter Clark
Spotte-Smith
 *ab,
Orion Archer
Cohen
c,
Samuel M.
Blau
d,
Jason M.
Munro
a,
Ruoxi
Yang
a,
Rishabh D.
Guha
a,
Hetal D.
Patel
ab,
Sudarshan
Vijay‡
b,
Patrick
Huck
a,
Ryan
Kingsbury
e,
Matthew K.
Horton
f and
Kristin A.
Persson
*bg
*ab,
Orion Archer
Cohen
c,
Samuel M.
Blau
d,
Jason M.
Munro
a,
Ruoxi
Yang
a,
Rishabh D.
Guha
a,
Hetal D.
Patel
ab,
Sudarshan
Vijay‡
b,
Patrick
Huck
a,
Ryan
Kingsbury
e,
Matthew K.
Horton
f and
Kristin A.
Persson
*bg
aMaterials Science Division, Lawrence Berkeley National Laboratory (LBNL), 1 Cyclotron Road, Berkeley, CA 94720, USA. E-mail: espottesmith@gmail.com
bDepartment of Materials Science and Engineering, University of California, Berkeley (UC Berkeley), 210 Hearst Memorial Mining Building, Berkeley, CA 94720, USA. E-mail: kapersson@lbl.gov
cDepartment of Chemistry, UC Berkeley, 419 Latimer Hall, Berkeley, CA 94720, USA
dEnergy Storage and Distributed Resources, LBNL, 1 Cyclotron Road, Berkeley, CA 94720, USA
eDepartment of Civil and Environmental Engineering, Princeton University, E209A Engineering Quadrangle, Princeton, New Jersey 08544, USA
fMicrosoft Research, Microsoft Building 99, 14820 NE 36th Street, Redmond, Washington 98052, USA
gMolecular Foundry, LBNL, 1 Cyclotron Road, Berkeley, CA 94720, USA
First published on 13th October 2023
Abstract
Advanced chemical research is increasingly reliant on large computed datasets of molecules and reactions to discover new functional molecules, understand chemical trends, train machine learning models, and more. To be of greatest use to the scientific community, such datasets should follow FAIR principles (i.e. be findable, accessible, interoperable, and reusable). In this work, we present a FAIR expansion of the Materials Project database (“MPcules”) that adds more than 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules studied using density functional theory (DFT) to the existing data, which comprises crystalline solids. MPcules is a diverse collection of DFT-calculated molecular properties, with an emphasis on reactive, open-shell, and charged species—relevant for studying reaction pathways—and a wide array of structural, electronic, vibrational, and thermodynamic properties. This database can be queried through an OpenAPI-compliant application programming interface and a featureful web application. We continue to expand the data available on MPcules and encourage contributions from the community.
000 molecules studied using density functional theory (DFT) to the existing data, which comprises crystalline solids. MPcules is a diverse collection of DFT-calculated molecular properties, with an emphasis on reactive, open-shell, and charged species—relevant for studying reaction pathways—and a wide array of structural, electronic, vibrational, and thermodynamic properties. This database can be queried through an OpenAPI-compliant application programming interface and a featureful web application. We continue to expand the data available on MPcules and encourage contributions from the community.
Introduction
With advances in scientific workflow automation and high-performance computing, it has become increasingly facile to generate large datasets of molecules,1 materials,2 and reactions,3,4 as well as their computed and predicted properties. Such datasets, which commonly rely on atomistic simulations using some combination of classical or ab initio molecular dynamics, semi-empirical quantum chemistry, density functional theory (DFT), and wavefunction methods, have ushered in a new paradigm of data-driven chemical research, enabling in-depth understanding of complex chemical domains5–7 and the discovery and design of new materials and molecules with desirable properties in various applications.8–12 Well-designed, systematic, and diverse datasets are also essential for virtually all machine learning (ML) tasks in chemistry and materials science.13–22While the wealth of data available to researchers is a boon, not all data is equally useful. It is increasingly recognized that for data to maximally benefit the scientific community, they should follow FAIR principles:23 they should be findable (the data can be easily searched using rich metadata and unique identifiers or IDs); accessible (the data are as open to the public as possible and can be reached using standard communication protocols); interoperable (the data can be readily combined with other data or used with a wide range of tools); and reusable (the data contain many useful attributes relevant to the domain of interest, have provenance allowing for verification of their accuracy, and are licensed in such a way as to allow others to employ them in their own work).
Since the advent of the Materials Genome Initiative in the United States,24,25 a number of databases of materials and their computed properties have been developed. Many of these databases, including the Open Quantum Materials Database (OQMD),26,27 Novel Materials Discovery (NOMAD) repository,28 and the Materials Project,29 aspire to follow FAIR principles. Though they vary in scale, the types of materials contained, and the properties reported, these repositories are all alike in that they have web interfaces where users can easily search and visualize data as well as application programming interfaces (APIs) that allow programmatic access to a wide range of data and metadata – enabling individual users with knowledge of computer programming to more easily navigate large collections of materials properties and allowing these databases to be integrated into other applications.
In contrast, few FAIR databases of calculated molecular properties exist. It remains common for computational chemistry data to be presented as a single unit (for instance, a zipped file that cannot be easily searched), or worse, not be publicly shared at all. The Molecular Sciences Software Institute's QCArchive30 and the Public Computational Chemistry Database Project (PCCDB)31 are noteworthy and laudable examples of quantum chemical databases approaching FAIR standards.
QCArchive hosts large collections of internally generated and user-submitted data, including the popular QM932 and ANI-1 datasets.33 The data on QCArchive can be downloaded in HDF5 format from their web site or can be accessed through a representational state transfer (REST) API with a high-level Python client, making it accessible and interoperable. QCArchive data is also reasonably findable and reusable. Molecules in QCArchive are given unique IDs. However, at the time of writing, it is not possible to search for specific molecules in the datasets listed on the web interface. Moreover, data visualization tools are either limited or nonexistent, making it difficult for users to discover or digest the data without downloading and sifting through large collections. In terms of reusability, QCArchive boasts an enormous collection of molecules and datapoints with provenance based on over 10 million calculations, but the available data are often limited in scope and applicability. Many of the datasets included in QCArchive contain relatively few properties (for instance, only structures and electronic energies), meaning that the data can only easily be applied to very specific tasks, e.g. training ML force-fields for molecular dynamics.
PCCDB hosts data from PubChemQC, a collection of electronic structure properties for more than 2 million molecules taken from the PubChem database.34 PCCDB has a web app that allows users to search for molecules with particular properties and then visualize those molecules, their absorption spectra, and their molecular orbitals. Calculation inputs are available through the web interface, providing users with some means to access (meta)data about e.g. calculation parameters. An API is also available, and the standard is specified in the web site's documentation. However, no client for this API has been released, which nontrivially increases the burden for end users to interact with the data and especially to download large collections of data for e.g. high-throughput screening or ML applications. Like QCArchive, data in PCCDB is limited in scope, with a strong emphasis on excited state and optical absorption properties. In our assessment, data in PCCDB is findable and interoperable but is somewhat lacking in accessibility and reusability.
In order to continue the advancement of data-driven chemical research, new platforms are needed that emphasize ease of access and diversity of data and data attributes. Here, in an effort to fill this need and support computational chemistry and chemical ML research, we report an extension of the Materials Project for calculated molecular data which we call the “Materials Project for Molecules”, or “MPcules” for short. We have developed a database schema and modular data processing pipeline that allows molecular DFT calculations to be converted into rich molecule and molecular property documents with unique, robust, and chemically meaningful IDs. This data pipeline can be used to add data to MPcules or to develop bespoke datasets. As a means to access the data in MPcules, we have expanded the Materials Project API and associated Python API client. Further, we have developed and released a new application (app) on the Materials Project web site allowing users to visualize the data in MPcules without any programming knowledge. MPCules currently contains more than 170000 molecules assembled from more than half a million DFT calculations. It is envisioned as a dynamic database that will continue to grow both in terms of the number of molecules as well as the number and types of properties included. In this paper, we describe the methods used to construct MPcules and report on the current status of the database.
Quantum chemical methods
All data currently included in MPcules are directly calculated or derived from DFT calculations. Specifically, all calculations were performed with the Q-Chem electronic structure code, using either version 5 or 6.35 Calculation automation and initial processing of DFT inputs and outputs relied on the fireworks,36custodian,37,38 and atomate39 Python libraries.At present, the calculations that make up MPcules use a small set of DFT methods. Specifically, calculations have been performed using three exchange-correlation functionals—the range-separated hybrid generalized gradient approximation (GGA) functionals ωB97X-D40 and ωB97X-V41 and the range-separated hybrid meta-generalized gradient approximation (meta-GGA) functional ωB97M-V42—as well as three basis sets from the def2 family with polarization and diffuse functions added: def2-SVPD, def2-TZVPPD, and def2-QZVPPD.43 Many calculations were performed in vacuum, but calculations using the polarizable continuum model (PCM)44 and the solvent model with density (SMD)45 implicit solvent methods are also included. We note that while these calculation methods reflect the data currently in MPcules, the database can easily accept calculations applying any functional and basis set included in Q-Chem.
Database construction
The MPcules database is constructed using the emmet Python packages. emmet-core defines “data models” or “documents” (using the pydantic data validation framework) that represent everything from the output of a DFT calculation to a molecule or a specific property; emmet-builders describes how raw calculation outputs can be converted into molecule and molecule property documents (defined in emmet-core) and how these documents should be inserted as entries in a database (MPcules, like most of the Materials Project, uses a MongoDB NoSQL architecture). Lastly, emmet-api defines how MPcules can be queried to obtain the data that has been built. Here, we elaborate on the structure of MPcules and how the database is constructed.Assigning priority to calculations
As discussed above (“Quantum chemical methods”), MPcules can accommodate calculations that use many different of levels of theory, where we define “level of theory” as the combination of a density functional, basis set, and solvent method. Therefore, when a particular property has been calculated using multiple levels of theory, we must rank them in order to retain and report only the “best” property available.Each component of the level of theory—the functional, basis set, and solvent method—is assigned a score. Because the accuracy and appropriateness of computational methods depends sensitively on the property of interest and the types of molecules being considered, these scores are inherently arbitrary and heuristic in nature and are based on e.g. previous benchmark studies and simple rules. Further details regarding the scoring of the components of level of theory are provided in the ESI.†
While one solvent method may be considered more accurate or reliable than another, the same cannot be said of solvents themselves. That is, a calculation using PCM parameterized with ε = 80 (roughly approximating an aqueous medium) is no more or less accurate than one parameterized with ε = 7 (approximating the dielectric of e.g. tetrahydrofuran). Rather, calculations performed with different solvent media are better or worse suited for particular applications. When tabulating molecular properties, we therefore select the best level of theory available for each solvent medium. We note that calculations conducted in vacuum are ranked below those performed using implicit solvent methods, but vacuum properties are still reported when available, as we treat vacuum as a distinct solvent medium.
Tasks
A single DFT workflow may correspond to one calculation (e.g. a single-point energy calculation or geometry optimization) or may be a collection of related calculations (e.g. a geometry optimization followed by a vibrational frequency analysis to confirm that the optimized structure is a local minimum of the potential energy surface or PES). In either case, the metadata, input parameters, and results of the calculation(s) are parsed by atomate and stored in a MongoDB database in a single “task document” (represented in emmet-core as a TaskDocument object). Tasks/TaskDocuments are the most fundamental collections of data used to construct MPcules, corresponding almost directly to the parameters and raw outputs of DFT calculations.Molecules
“Molecules” are central to MPcules. Most data in MPcules are conceptually organized and grouped by molecule. How we define the term “molecule” therefore affects how users will access and interact with data. Although chemists and physicists have intuitive understandings of what a molecule is, we must be careful in defining the term and consider how best to represent a molecule in a database.A molecule can be minimally described by its chemical composition, charge, and spin multiplicity. This is in line with common written nomenclature for molecules and ions. As a small example, diatomic oxygen in the triplet ground state (3O2) is differentiated by composition from the oxygen atom (O1), by charge from a peroxide anion (O2−2), and by spin from the singlet excited state (1O2). Notably, additional information may be needed to distinguish between ground and excited states. To specify beyond this starting point, there are two natural definitions: one based on PES, and another based on the idea of chemical bonding (Fig. 1).
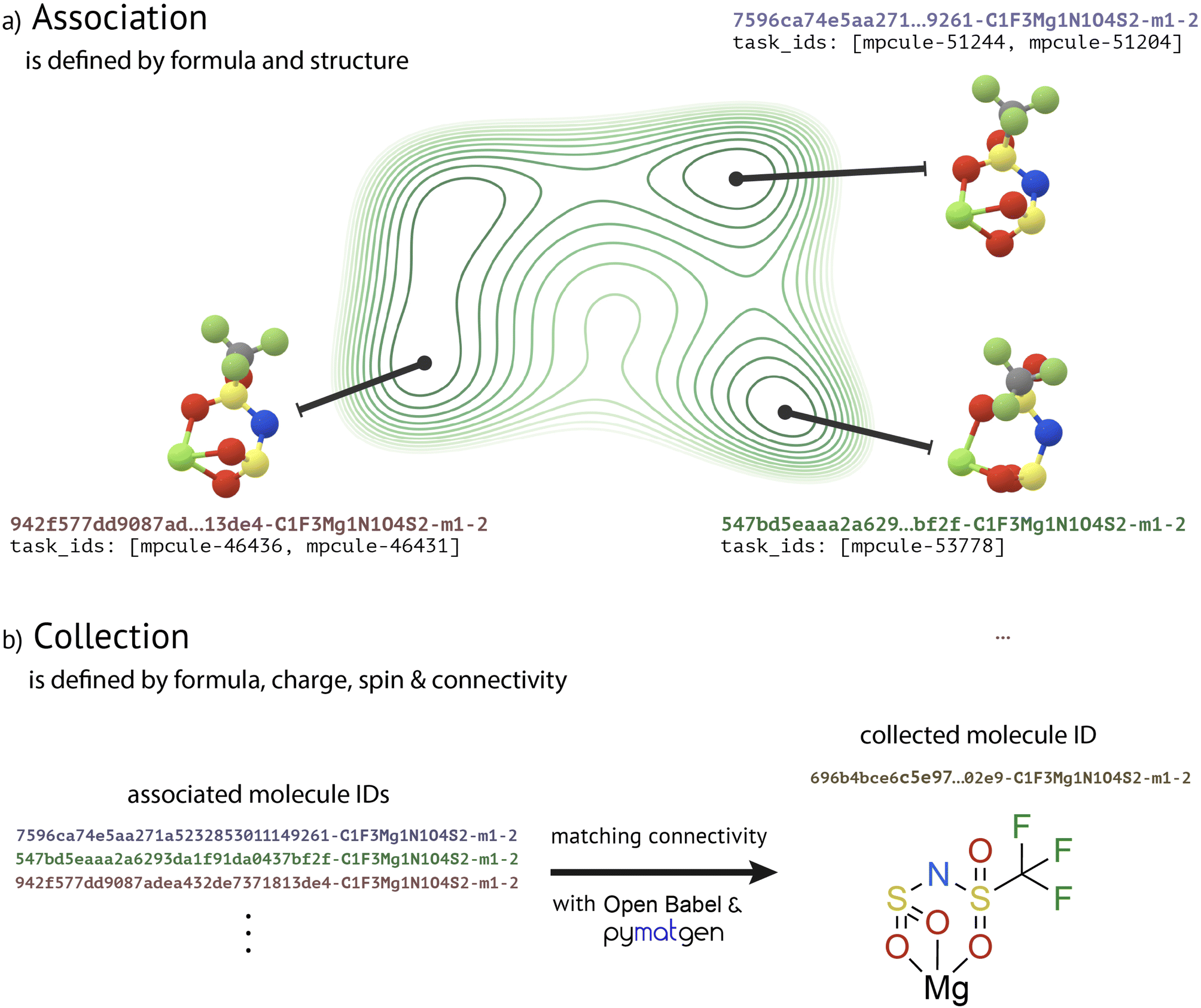 | ||
| Fig. 1 Conceptual guide to molecule definition and construction in MPcules. (a) A molecule can be defined as a unique minimum of a potential energy surface, defined by some composition and structure (e.g. interatomic distances and angles). This definition is used in the first (association) stage of molecule building. (b) Alternatively, a molecule can be defined by its composition, charge, spin multiplicity, and connectivity. Different conformers (species with distinct structures but the same connectivity) can all be thought of as the same molecule. This definition is used for the second (collection) stage of molecule building, where we use OpenBabel and pymatgen to determine bonding. | ||
In the first definition (Fig. 1a), a molecule is defined as a local minimum on a PES. The PES, in turn, is defined by the chemical composition, total number of electrons, spin multiplicity, and the DFT methods (level of theory and other calculation parameters) employed. In this definition, every unique atomic structure (in terms of interatomic distances and angles) corresponding to a local PES minimum obtained via a geometry optimization calculation is a different molecule. It is worth noting that this PES-based definition is used within the Materials Project's data for crystalline solids to define a unique “material”.
In the second definition, it is the connectivity of a molecule—the way that atoms are linked to each other through chemical bonds and other interatomic interactions—that distinguishes molecules. Different local minima on a PES may correspond to structures with different bonds, but they may also simply be different conformational isomers (conformers). This definition is somewhat more complex than the picture based on PES, as it requires additional definitions and decisions. For instance, this definition relies on the idea of a “bond” and associated criteria determining when two or more atoms are or are not bonded. We note that it is extremely challenging to rigorously define chemical bonding, and ultimately, most definitions are arbitrary.
In MPcules, we use both the PES-based and the bonding-based definitions to construct molecules, as described below (“Building Molecules”). However, as most chemical observables of interest—including various spectra, electrochemical properties, and reaction properties like thermodynamics or kinetics—are averaged over different interconverting conformers,46 we rely in most cases on the definition based on bonding.
In the second (collection) stage (Fig. 1b), the structures of associated MoleculeDocs are compared on the basis of connectivity. Though we can define bonding using several methods informed by quantum chemistry (see “Molecular Properties” below), for the purpose of collecting MoleculeDocs we need to choose a definition of bonding such that the connectivity of every molecule in MPcules can be determined regardless of what calculations have been performed. We use the bond detection algorithm included in the OpenBabel cheminformatics toolkit48 and then identify missing coordinate bonds to metals with the metal_edge_extender defined in pymatgen.37 This method is entirely based on heuristics and does not depend on any electronic structure calculations. Upon detecting bonds, we construct a molecular graph representation using the pymatgenMoleculeGraph functionality. When defining connectivity for a graph representation, we consider bonds to hydrogen atoms, which are always included explicitly in our 3D molecular structures. If there are multiple associated MoleculeDocs with the same formula, charge, spin, and connectivity, then we rank the different documents (again in terms of the level of theory used to optimize the best structure and the associated electronic energy) and choose the best to represent the group. All other associated MoleculeDocs with the same formula, charge, spin, and connectivity are linked to this representative as “similar molecules”.
Molecules are assigned unique identifiers (“MPculeIDs”) based on their chemical formulae, charges, spin multiplicities, and connectivity; further details regarding the MPculeID format are provided below (see “Unique Identifiers”). Likewise, tasks are given unique IDs defined by an (optional) prefix and some integer. MoleculeDocs store a list of the IDs of all tasks performed on the same geometry. Collected MoleculeDocs additionally store the IDs of the tasks that produced the “best” structure for each implicit solvent medium (including vacuum) for that molecule and the MPculeIDs of the “similar” associated MoleculeDocs (documents with the same connectivity, but with geometries representing different PES minima). This allows users to collect the properties of various conformers of a given molecule.
Molecular properties
MoleculeDocs and their underlying TaskDocuments contain all of the information about a molecule that is stored in MPcules. To aid in accessibility and reusability, we further process task- and molecule-level data to generate property documents. Typically, property documents are uniquely defined by the combination of MPculeID and solvent. In some cases, a property can be calculated or determined using multiple different methods; for instance, atomic partial spins can be defined using Mulliken population analysis49 or the natural atomic populations determined by the natural bonding orbital (NBO) program.50,51 For such properties, a property document is uniquely defined by MPculeID, solvent, and method.At present, we generate property documents for the following properties: natural atomic and molecular orbitals (based on NBO); atomic partial charges; atomic partial spins; bonding; thermodynamics; vibrational properties; redox and electrochemical properties; and coordination or binding of metals. Basic details for these different properties are provided below, and a schematic of how collections of tasks, molecules, and properties are connected is shown in Fig. 2.
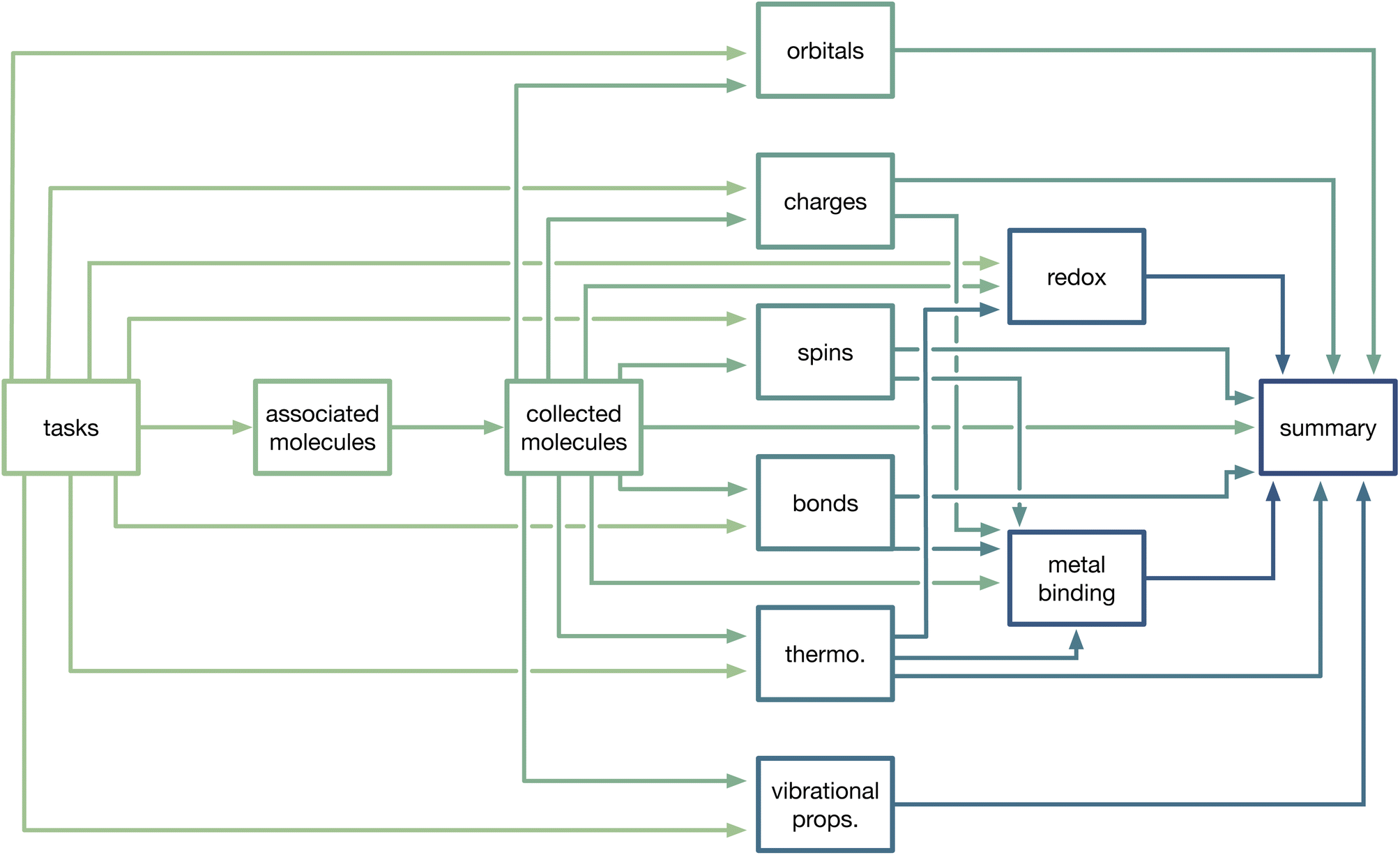 | ||
| Fig. 2 Diagram showing how different collections of tasks, (associated or collected) molecules, and properties are linked together in MPcules. An arrow from a source collection (box) to another destination collection indicates that documents in the source are used to construct documents in the destination. | ||
For molecules with multiple optimized structures for a given solvent medium (i.e., for cases where multiple associated MoleculeDocs were collapsed into a single MoleculeDoc during the collection stage), we only calculate properties based on the “best” structure. This ensures that comparable properties for the same molecule always refer to the same structure. We further note that, for atomic properties (e.g. atomic partial charges, atomic partial spins) or properties with atomic components (e.g. normal modes of vibration), we consistently use the same atomic indices as the pymatgenMolecule object for the “best” structure.
For each atom, we store the number of core, valence, and Rydberg electrons, as well as the total number of electrons assigned to that atom. Lone pair information includes the orbital character of the lone pair (fraction of the lone pair made up of s, p, d, and f natural atomic orbitals), as well as its total occupancy. Similarly, for bonds we include the orbital character of each atom's contribution and the total occupancy, as well as information regarding the bond polarization. We also store information regarding orbital interactions, including the perturbation energy, the energy difference between the donor and acceptor orbitals, and the Fock matrix element for the interaction. For each type of hybrid orbital (e.g. long pair or bond) or orbital interaction, we retain information regarding the atoms involved in the hybrid orbital(s) as well as the orbital type(s), using the codes from NBO outputs. For instance, lone pair orbitals are labeled “LP”, while antibonding orbitals are labeled as “BD*”.
For molecules with unpaired electrons, NBO separates its analysis for α and β electrons. Accordingly, the orbital data on MPcules is structured differently for open-shell and closed-shell molecules. Closed-shell molecules have singular collections of populations, lone pairs, bonds, and interactions, while open-shell molecules have one collection of each type of property for α electrons and one for β electrons.
NBO version 7 significantly improved the bond-detection algorithm over version 5.51 As a result, we currently only allow NBO 7 calculations to be included in MPcules. Users wishing to adopt our methodology should note that Q-Chem is packaged with NBO version 5 and uses this version by default, meaning that configuration of an external NBO application is necessary to benefit from the improvements and produce data that can be incorporated into MPcules.
In addition to the heuristic method of defining bonds using OpenBabel and pymatgen, we can determine bonding in two ways: (1) with the method of Spotte-Smith, Blau, et al.,57 in which OpenBabel/pymatgen heuristic bonding is augmented with bonds identified by analyzing the critical points of the total electron density (using critic2) and (2) via natural bonding orbitals identified with NBO.
NBO reports bonds that form hybrid orbitals based on the sharing of electrons between atoms—in other words, covalent bonds. Ionic bonds, coordinate bonds, and other electrostatic interatomic interactions are not captured directly as bonds in the NBO output. However, such non-covalent bonds and interactions can be inferred from other NBO-reported quantities. Specifically, to identify metal coordinate bonds, we examine NBO's second-order perturbation theory analysis of the Fock matrix. If there is an interaction between a lone pair (“LP”) orbital on a nonmetal (donor) and a lone vacant (“LV”) or anti-Rydberg orbital (“RY*“) on a metal (acceptor) where the metal is within 3.0 Å of the nonmetal and the perturbation energy is greater than or equal to 3.0 kcal mol−1, then we determine the metal and nonmetal to be electrostatically bonded. These cutoff values were determined by manual inspection of set of metal-containing complexes and are, like most definitions of bonding, arbitrary.
In order to obtain optimized geometries and calculate free energies at reduced cost, it is common for computational chemists to optimize structures at a relatively inexpensive level of theory (e.g. using a small basis set, or ignoring solvent effects) and then re-calculate the electronic energy with a single-point calculation using a more accurate and expensive level of theory (e.g. using a larger basis set or including an implicit solvent model). There are therefore two natural ways to calculate molecular thermodynamics: one in which all thermodynamic quantities of interest (electronic energy, enthalpy, entropy, etc.) are calculated from a single vibrational frequency analysis calculation, and another in which most properties are obtained from a vibrational frequency analysis but the electronic energy is instead obtained from a single-point energy calculation performed on the same structure at a higher level of theory.
We construct thermodynamic property documents (MoleculeThermoDocs in emmet-core) using both methods. If, for a given solvent, one can produce a MoleculeThermoDoc both with and without a single-point energy correction, then the scores for the “best” uncorrected document (based on the level of theory used and the electronic energy) and “best” corrected document (based on the average of the scores of the levels of theory used for the vibrational frequency analysis and the single-point energy calculation, and the electronic energy) are compared, and the document with the better (lower) score is selected.
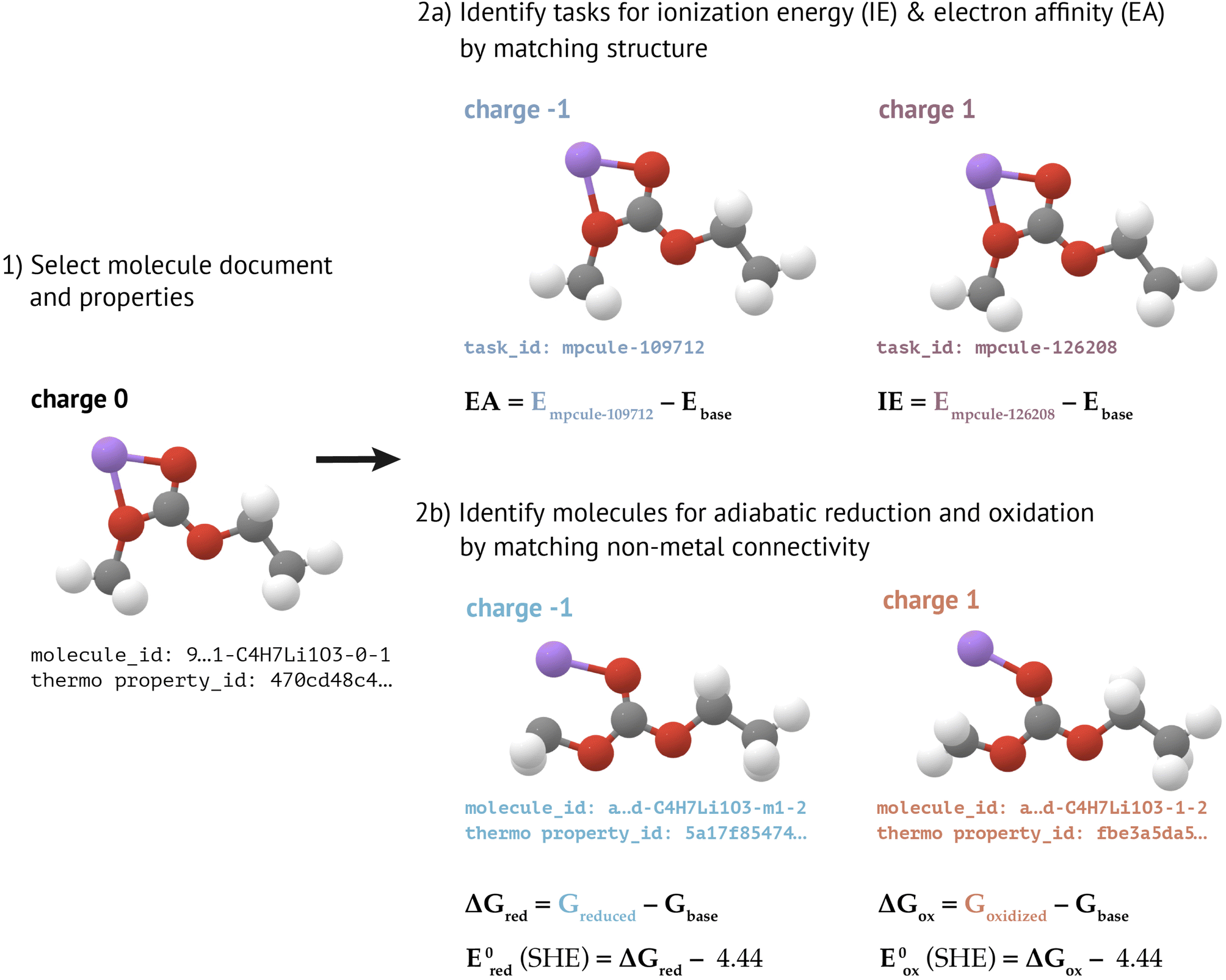 | ||
| Fig. 3 Calculation of redox and electrochemical properties in MPcules. (1) For a given molecule, the molecule document and related thermodynamics properties are needed. (2a) Vertical ionization energy (IE) and electron affinity (EA) can be calculated by identifying tasks with the same structure as the molecule of interest, but with charge shifted by +1 (for ionization energy) or −1 (for electron affinity). (2b) Adiabatic reduction and oxidation properties can be calculated by identifying molecules with the same connectivity (ignoring metal coordination) but with charge shifted by −1 (for reduction) or +1 (for oxidation). For clarity, the charge of each molecule and task is shown next to its structure. | ||
In the adiabatic approximation, one allows a reduced or oxidized molecule to fully relax. Calculating an adiabatic reduction or oxidation (free) energy therefore requires two optimized structures (and therefore two MoleculeDocs) at different charge states. Because it can be challenging to predict a priori how a molecule may decompose upon charge transfer, we neglect the possibility of dissociative redox reactions. Instead, when calculating adiabatic redox properties for a given molecule, we search for MoleculeDocs that have the same connectivity as that molecule (not including bonds involving metals), but with charge shifted by −1 (for reduction) or +1 (for oxidation). Reaction free energies are calculated using previously-constructed MoleculeThermoDocs (see “Molecular Thermodynamics”). If the oxidized and/or reduced MoleculeDocs can be identified, we also calculate reduction or oxidation potentials, referenced to the standard hydrogen electrode (SHE) using the relative potentials reported by Trasatti.58
To determine metal binding properties (Fig. 4), we must first ascertain the charge and spin state of each metal in a given molecule. To do this, we round the atomic partial charge and the atomic partial spin of the metal atom in the molecule to the nearest integer. These atomic partial charges and spins are obtained from previously-constructed collections in MPcules (see “Atomic Partial Charges” and “Atomic Partial Spins” above). If the rounded charge and spin are incompatible—for instance, if a Mg atom were assigned a charge of +1 and a spin multiplicity of 1 (spin 0)—then the charge is shifted by +1 or −1 (whichever produces a charge which is closer to the calculated atomic partial charge). We shift the charge, rather than the spin multiplicity, because we have found that the spin state of metals is more often well described by DFT than metal charge states (see “Comparison of Atomic Partial Charges and Spin” in the ESI†).
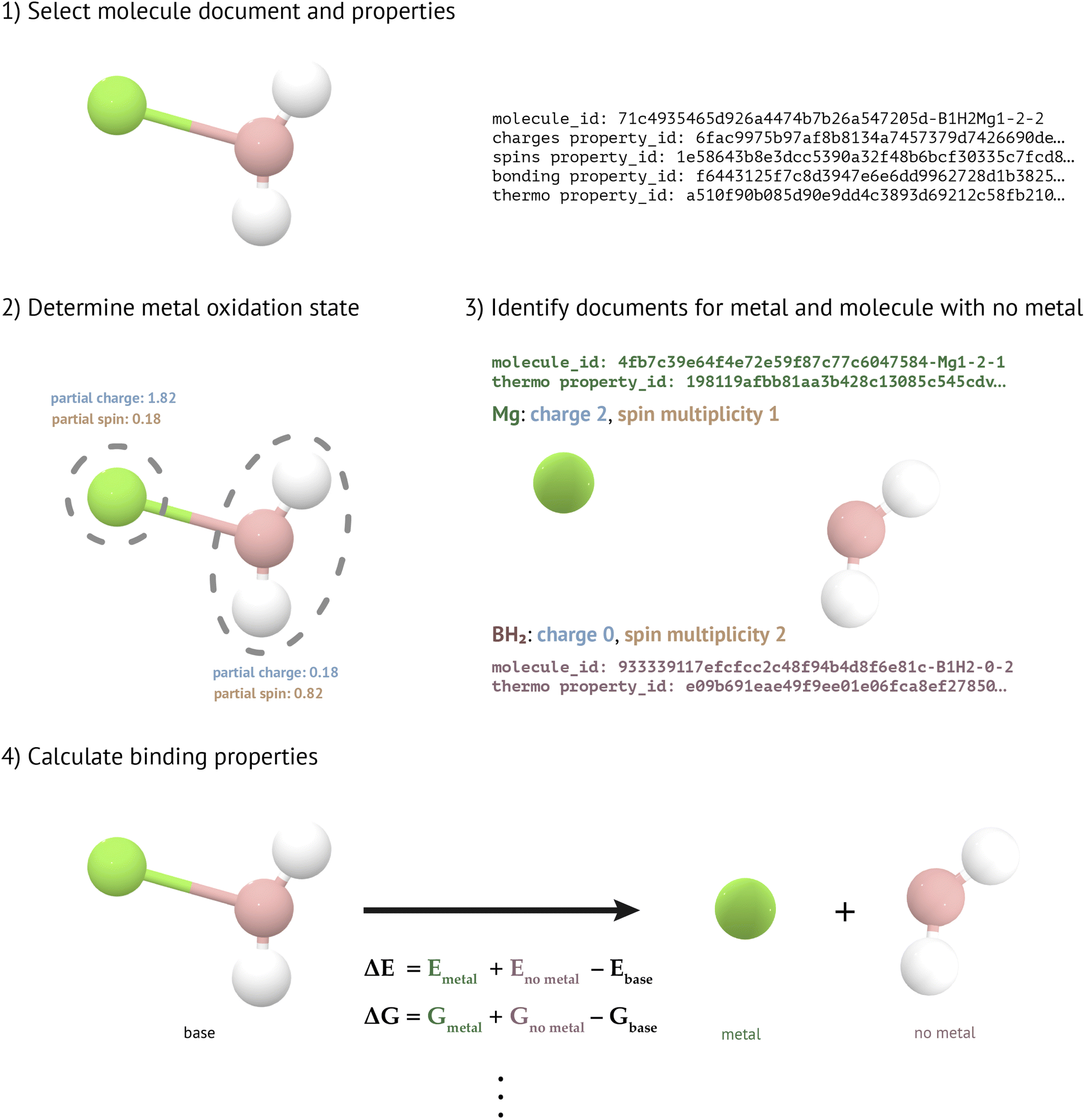 | ||
| Fig. 4 Calculation of metal binding properties in MPcules. (1) For a given molecule, the molecule document, along with atomic partial charge, atomic partial spin (if the molecule is open-shell), bonding, and thermodynamics information, must be available. (2) For each metal in the molecule, the atomic partial charges and spins (if applicable) are used to determine the metal's oxidation state. This specifies what non-bound species will be searched for. (3) Documents for the unbound metal and the molecule without that metal attached (no metal), along with their thermodynamic information, are sought. (4) Metal binding properties can be calculated using the thermodynamics of the base molecule, metal, and no metal. | ||
After the oxidation and spin state of each metal have been determined, the bonding environment around the metal atoms are characterized using previously calculated bonding information (see “Bonding”). For each metal, we then construct a MoleculeGraph of the molecule with that metal removed. Using this graph, we search for molecules with the same connectivity and the appropriate charge and spin multiplicity. If appropriate MoleculeDocs can be located for both the metal (M in the previous chemical equation) and the molecule without the metal (A), then we calculate the reaction thermodynamics using previously-constructed MoleculeThermoDocs (see “Molecular Thermodynamics”).
Atomic partial charges, atomic partial spins, and bonding can all be determined in multiple ways, which means that there are numerous possible combinations that could determine the metal-binding properties of a molecule. However, with the aim of ensuring that the various methods used are as consistent as possible, we currently only allow two combinations of methods to be used. In the first, atomic partial charges, atomic partial spins, and binding are all determined using NBO. In the second, the atomic partial charges and atomic partial spins are both calculated using the Mulliken method, and the bonding is determined using the default method combining OpenBabel with pymatgen.
We would like to point out that DFT can struggle to accurately predict the thermochemical properties of single atoms and ions, whether in vacuum or in implicit solvent. This may affect the accuracy of computed binding (free) energies.
Summary documents
After all property documents have been constructed, we compile a subset of calculated properties into a single document called a MoleculeSummaryDoc. Whereas property documents are uniquely defined by MPculeID, solvent medium, and sometimes method, the summary document is uniquely defined only by the MPculeID. Properties in the summary document that are not method-dependent are represented as key-value pairs, where the keys are the names of solvents used to calculate the property and the values are the properties calculated in those implicit solvent media. For properties that are method-dependent, the values are instead key-value pairs, with the keys being various methods and the values being the properties calculated using specific combinations of solvent and method.Unique identifiers
The principles of findability and accessibility require that data be given IDs which can be used to search for and reference specific information. In addition to being unique and persistent, it is desirable (though less essential) for these IDs to carry chemical information and to be interpretable by human users.The most widely used representations for molecules which could be used as IDs are the simplified molecular-input line-entry system (SMILES)62 and the International Chemical Identifier (InChI).63 SMILES has numerous pitfalls which make it inappropriate for use as a database ID. Most importantly, SMILES strings are not unique, and there can be several valid SMILES for the same structure. Though it is possible to generate unique “canonical” SMILES,64 this fundamental lack of uniqueness makes searching for molecules by SMILES problematic. SMILES is also designed primarily for organic molecules and struggles to robustly represent metals and coordination complexes. As many of the molecules in MPcules contain coordinated metal atoms or ions, this is a severe limitation. The self-referencing embedded strings (SELFIES) devices by Krenn, Aspuru-Guzik, and colleagues,65,66 significantly improve on SMILES—most notably, by ensuring that all possible SELFIES strings represent chemically valid molecules—and can in principle support arbitrary metal bonding. However, at present, SELFIES can only be generated via SMILES, which ultimately means that many of the same pitfalls persist. InChIs are guaranteed to be unique—for a given molecular structure, there can be only one InChI—but the InChI generation algorithm explicitly ignores metal bonding, again meaning that metal-coordinated molecules with different coordination environments cannot be distinguished by InChI.
To overcome the limitations of existing IDs and molecular representations, we have devised a new ID format: the MPculeID (Fig. 5). The basic ID has four parts that are separated by hyphens; these four parts represent the connectivity, composition, charge, and spin multiplicity of the molecule. For connectivity, we generate a graph representation of the molecule (see “Building molecules”) and hash it using the Weisfeiler-Lehman graph hashing algorithm67 originally implemented in networkx.68 This hash can be augmented with features of the nodes (atoms) or edges (bonds). In the association stage of molecules building, where MoleculeDocs are differentiated by their exact structure, we augment the graph with the Cartesian (XYZ) coordinates of the atoms. In the collection stage, where MoleculeDocs are distinguished by connectivity only (without concern for exact bond lengths, angles, etc.), we instead augment only with the string representation of the element (e.g. “Li” for lithium). To ensure consistency, when representing the composition, we always use the alphabetized chemical formula (e.g. “C1Li2O3” for lithium carbonate or Li2CO3). For molecules with negative charge, we prefix the charge with “m” instead of a minus sign “-” to distinguish from the hyphen separators.
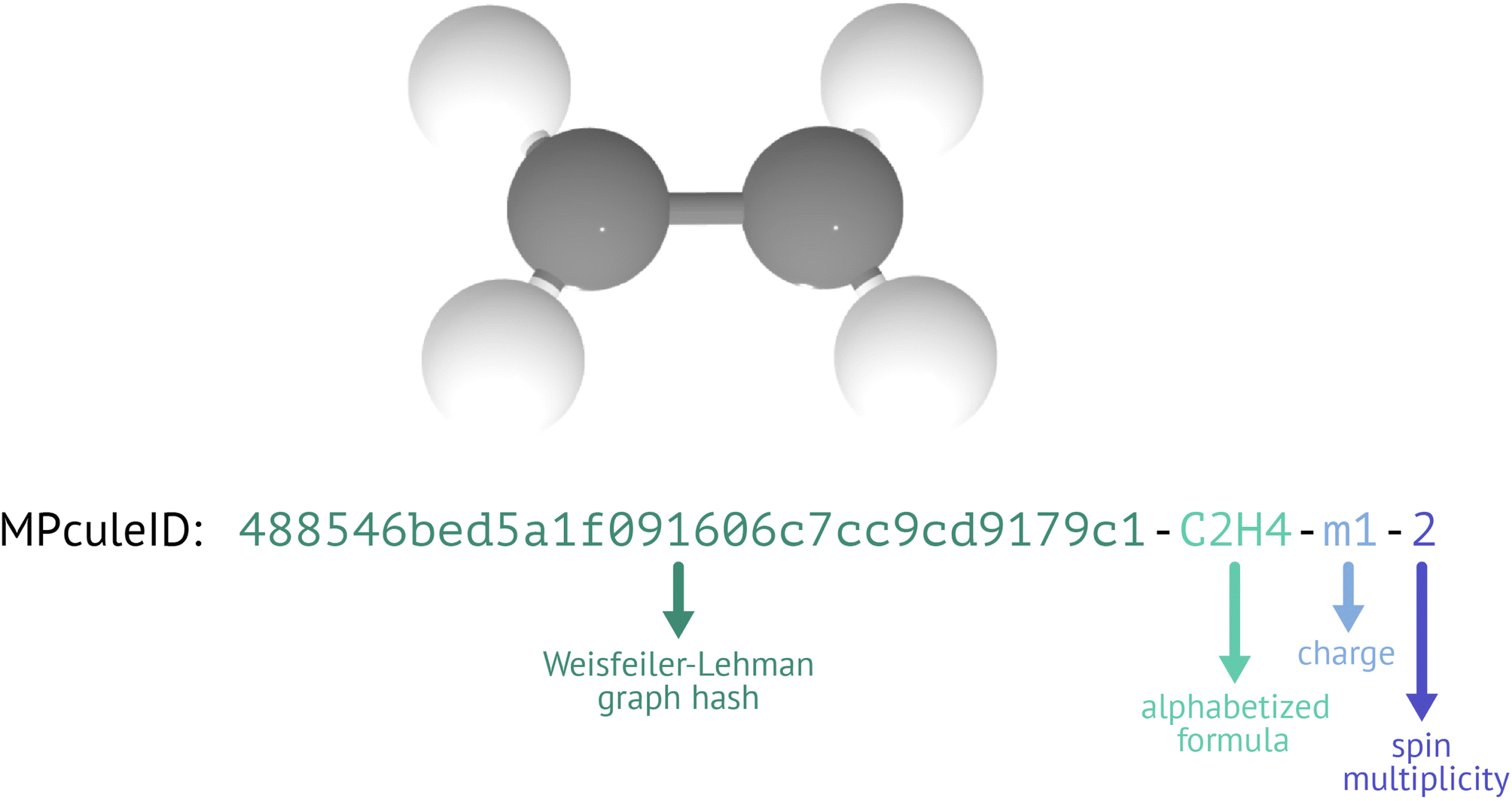 | ||
| Fig. 5 Explanation of the sections of an MPculeID, using doublet C2H4− as an example. | ||
The MPculeID comes closer to simultaneously meeting the goals of uniqueness, persistence, and interpretability. Though hash collisions—in which multiple distinct inputs are converted to the same hashed output—are essentially unavoidable with the Weisfeiler-Lehman hash or any other hashing method, it is exceptionally unlikely that any two molecules with different connectivities will nonetheless have the same hash, formula, charge, and spin. In practice, the MPculeID should therefore always be unique. Because the hashing algorithm is deterministic, the same graph input will always receive the same hash, meaning that MPculeIDs will not change over time. The Weisfeiler-Lehman algorithm further guarantees that graphs that are isomorphic produce the same hash, which means that these hashes can be used to compare molecular structures (acknowledging the possibility of hash collisions). Finally, though graph hashes are not human-interpretable, they do carry chemical information, and as the formula, charge, and spin information in the MPculeID are easily understood, users reading an MPculeID should be able to obtain a basic understanding of the underlying data.
Although the MPculeID format meets the basic requirements for a database ID format and overcomes certain key limitations of previous chemical identifiers, MPculeIDs have limitations of their own. For example, similar graphs do not in general produce similar Weisfeiler-Lehman hashes; these hashes therefore cannot be used to search for similar molecules, including molecules containing a particular substructure or functional group. There are also limits to the current implementation of MPculeIDs in the MPcules database that are not limitations of the basic format. As we have explained, when generating graph hashes for use in MPculeIDs, the graphs can be augmented with atom or bond features. Depending on how the graphs are augmented, different hashes will be produced, which can change if and how species are distinguished. As an example, consider chiral molecules. Different enantiomers have the same connectivity but are thought of as distinct because of their optical, structural, and (in some cases) reactive properties. Because they are by definition non-superimposable, enantiomers can be distinguished by their MPculeIDs in the association stage (where the graphs are augmented with Cartesian coordinates). However, MPculeIDs in the collection stage cannot distinguish between enantiomers because we do not augment the graphs with any information about chirality.
Although existing identifiers like InChI are not sufficient for use as a unique identifier in the MPcules database, they are widely used and supported. As such, to improve interoperability with other databases, we associate InChIs and InChI-key hashes with each molecule and molecule summary document in MPcules. We intend for users to be able to search for molecules based on their InChI strings in the future.
Provenance
As we have noted, data provenance is vital to allow users to verify the accuracy of a calculation and to trace processed data back to the raw data that they are based on. Throughout the construction of MPcules, we include provenance information, allowing users to trace back to individual DFT calculations/tasks.Already, we have mentioned how provenance information is stored during the construction of associated and collected MoleculeDocs (see “Molecules”). In addition, each property document stores the IDs of the other documents used to calculate the relevant properties. For data obtained from a single task—for instance, atomic partial charges—the task ID for that property is stored. For data obtained from other property documents, the property ID is stored. When data for a particular document comes from other documents with different MPculeIDs, then the MPculeIDs of those other documents are also stored. Finally, MoleculeSummaryDocs store the property IDs of all of the documents used to construct them, linked to the relevant solvent and (where relevant) method through key-value pairs.
Accessing molecular data
The data in MPcules can be accessed in three ways: (1) directly via the Materials Project API; (2) using the high-level Python interface to the Materials Project API, mp-api; or (3) via a web app on the Materials Project web site. Here, we briefly describe these means of accessing MPcules data. Further documentation can be found online (see https://api.materialsproject.org/docs and https://docs.materialsproject.org).The Materials Project API
Upon making an account (https://profile.materialsproject.org/), users of the Materials Project gain access to an API key (https://https://next-gen.materialsproject.org/api). This allows users to interact with the Materials Project API.A Materials Project API request begins with a uniform prefix (https://api.materialsproject.org/). All data in MPcules can be accessed via an API endpoint under the/molecules/root endpoint; for instance, molecules summary documents can be obtained from the endpoint/molecules/summary/. Following these endpoints, query parameters can be provided to limit the results of the search.
Because the Materials Project API provides an OpenAPI-compliant specification, it is facile to incorporate this API into software using a variety of web frameworks and programming languages. However, to avoid having to interface with this specification directly, users can also apply the MPRester class implemented in the mp-api Python package. MPRester includes straightforward Python interfaces to each of the MPcules API endpoints. For example, to search for a molecule summary document with charge +1 and formula C2H4, one can write the following Python code:

In the MPcules database at the time of this writing, there is exactly one molecule with charge +1 and formula C2H4, so query_output will contain a list with one entry. Due to the quantity of data included in the MPcules summary collection, we will not show the entire output, but it is worth illustrating how one can interact with the results of a query:

This yields the following output:

We note that, in addition to obtaining complete task, molecule, property, and summary documents, we have also provided API endpoints that extract more targeted information. For instance, using the /molecules/tasks/trajectory/endpoint, it is possible to extract information from a task related to a geometry optimization trajectory, including the structures, energies, and forces along that trajectory. This off-equilibrium data could be used, among other purposes, to train ML interatomic potentials.22,70
The Molecules Explorer
The new Molecules Explorer web app is built using the Crystal Toolkit Python framework for data visualization,71 as well as suites of custom React JavaScript components (mp-react-components) and Plotly Dash components (dash-mp-components). The root of the Molecules Explorer presents a search interface for discovering new molecules.The Molecule Details Page visualizes data from the summary document of each molecule. It allows users to explore key computed properties under different solvent media and bonding schemes. At the top (Fig. 6), solvent-invariant properties (e.g. number of atoms, charge, and spin multiplicity) are shown alongside a 3D molecular visualization rendered with Crystal Toolkit. The solvent medium and bonding scheme can be selected from two drop-down boxes that determine the computed properties displayed on the rest of the page. Below this, a set of property sections are shown that closely map onto the MPcules database schema. Namely, we have created sections for bonding, thermodynamic stability (containing molecular thermodynamics data), partial charges and spins (containing data on atomic partial charges and atomic partial spins), vibrations (containing information on vibrational properties), and redox stability (containing redox and electrochemical properties). We plan to add sections describing orbital information from NBO and metal binding properties.
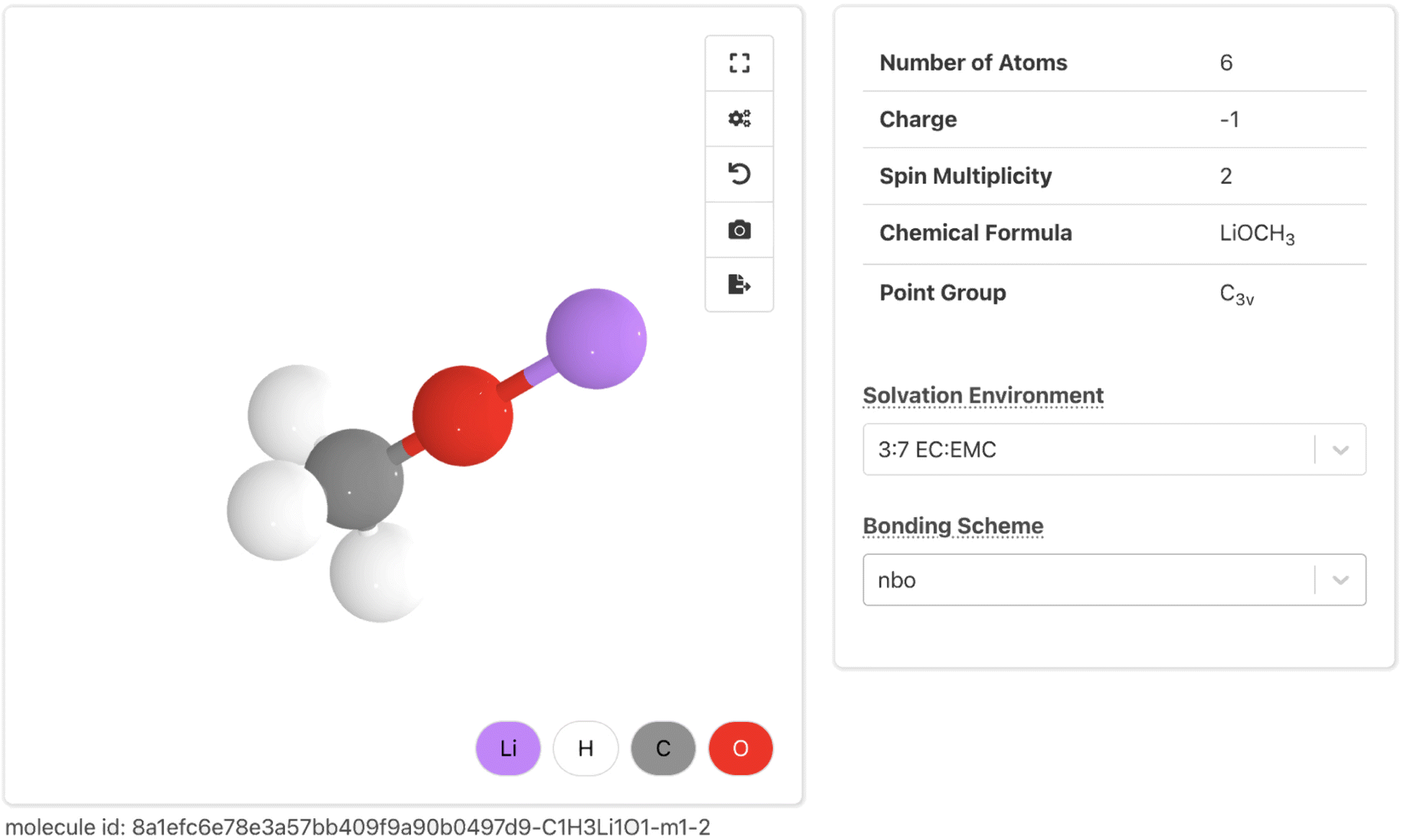 | ||
| Fig. 6 The summary section of the Molecule Details Page. | ||
Each property section consists of a data tab including the processed data from the summary document, a methods tab describing how the data was obtained from DFT and post-processing, and an API tab describing how users can programmatically access the data. For example, the data tab for the Partial Charges and Spins section (Fig. 7) consists of two drop-down menus to select the method for calculating charges and spins, a table of atomic charge and spin values, and a 2D molecular visualization of the molecule. Selecting rows in the table highlights the corresponding atoms in the molecular visualization and shows the total charge and spin of the selected atoms. This provides a map between atoms in the table and their position in the molecular topology. The other property sections each contain unique user interface elements. The Bonding section contains an interactive 2D visualization of bond distances, angles, and dihedrals; the Thermodynamic Stability and Redox Stability sections present tables of properties; and the Vibrations sections contains an interactive simulated IR spectra. At the bottom of the page, after the property sections, the parameters for the selected solvent method are shown.
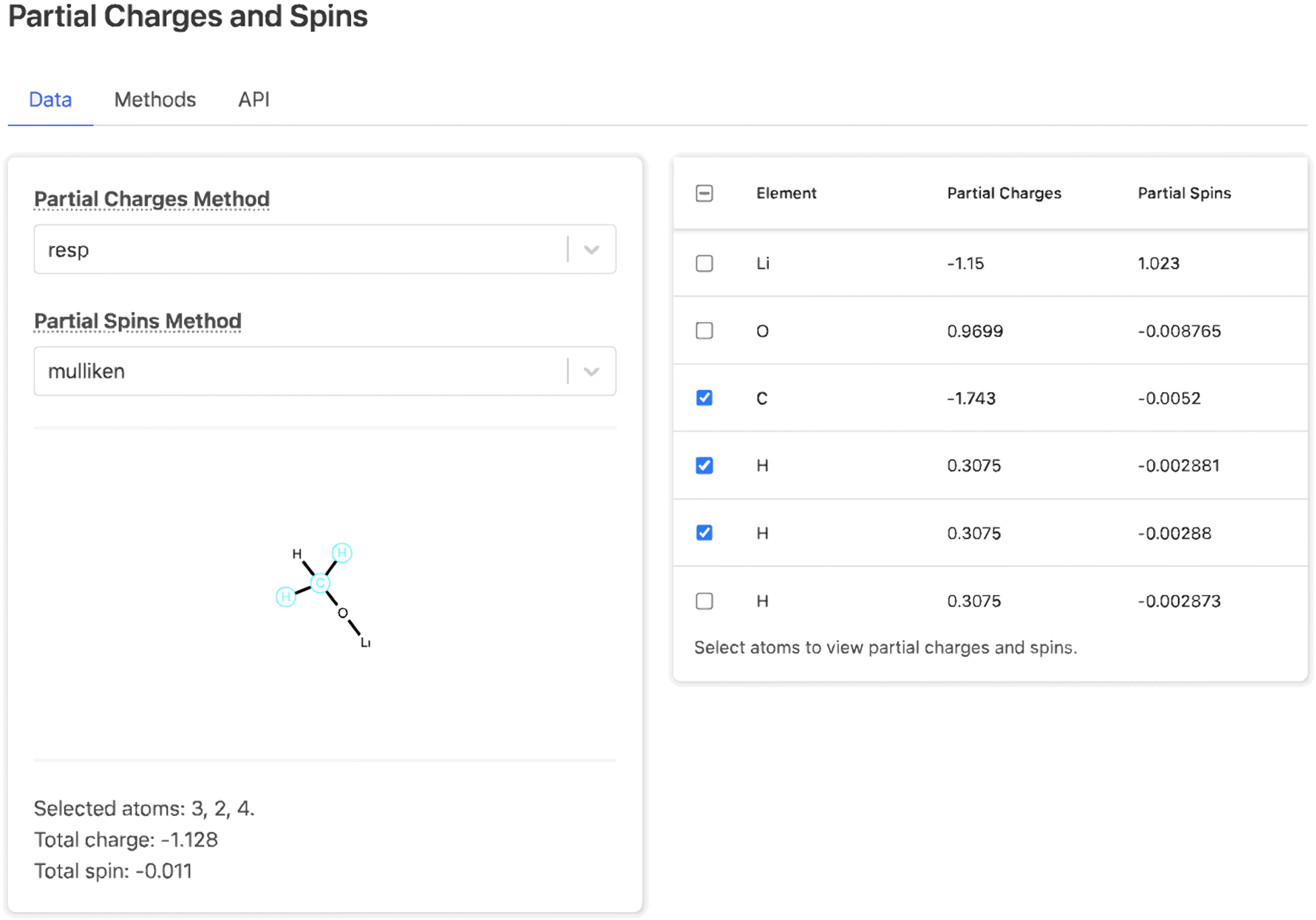 | ||
| Fig. 7 The Partial Charges and Spins section of a Molecule Detail Page on the Materials Project web site. An interactive molecule visualization allows users to select atoms and see their atomic partial charges and spins; these are also represented in tabular form. | ||
The current state of MPcules
The main focus of this work is to describe a general computational infrastructure for processing, storing, and disseminating calculated molecular properties. We expect the data stored on MPcules to change and grow over time, and specific additions to the database will be discussed in future works. Nonetheless, we here briefly discuss the scale and scope of the MPcules database as it exists at the time of this writing.MPcules currently contains data on 172874 (collected) molecules (248302 associated molecules) based on 568004 tasks (Fig. 8). Most properties are present for all relevant molecules. Because atomic partial charges and electronic energy are calculated by default for all DFT calculations in Q-Chem, there is at least one partial charge document and one molecular thermodynamics document per molecule. Likewise, there is at least one bonding document per molecule (because bonding can be assessed from an optimized geometry without any further electronic structure calculations) and at least one atomic partial spins document for each open-shell molecule. While we do not strictly require that optimized structures be validated by performing a vibrational frequency analysis, all molecules currently in MPcules that are not single atoms have been subjected to such analyses. As such, almost all molecules in MPcules have vibrational properties calculated. Other properties—namely natural atomic and molecular orbital properties, redox properties, and metal binding properties—are available only for a subset of molecules, either because these properties require specialized calculations (e.g. NBO analysis must be performed for orbital properties, and single-point calculations at different charges must be performed to calculate vertical redox properties) or because the calculation of certain properties for a given molecule require other molecules with specific structures and charges to be present (e.g. calculating a metal binding energy requires three molecules: the metal ion, the molecule-metal complex, and the same molecule not bound to the metal).
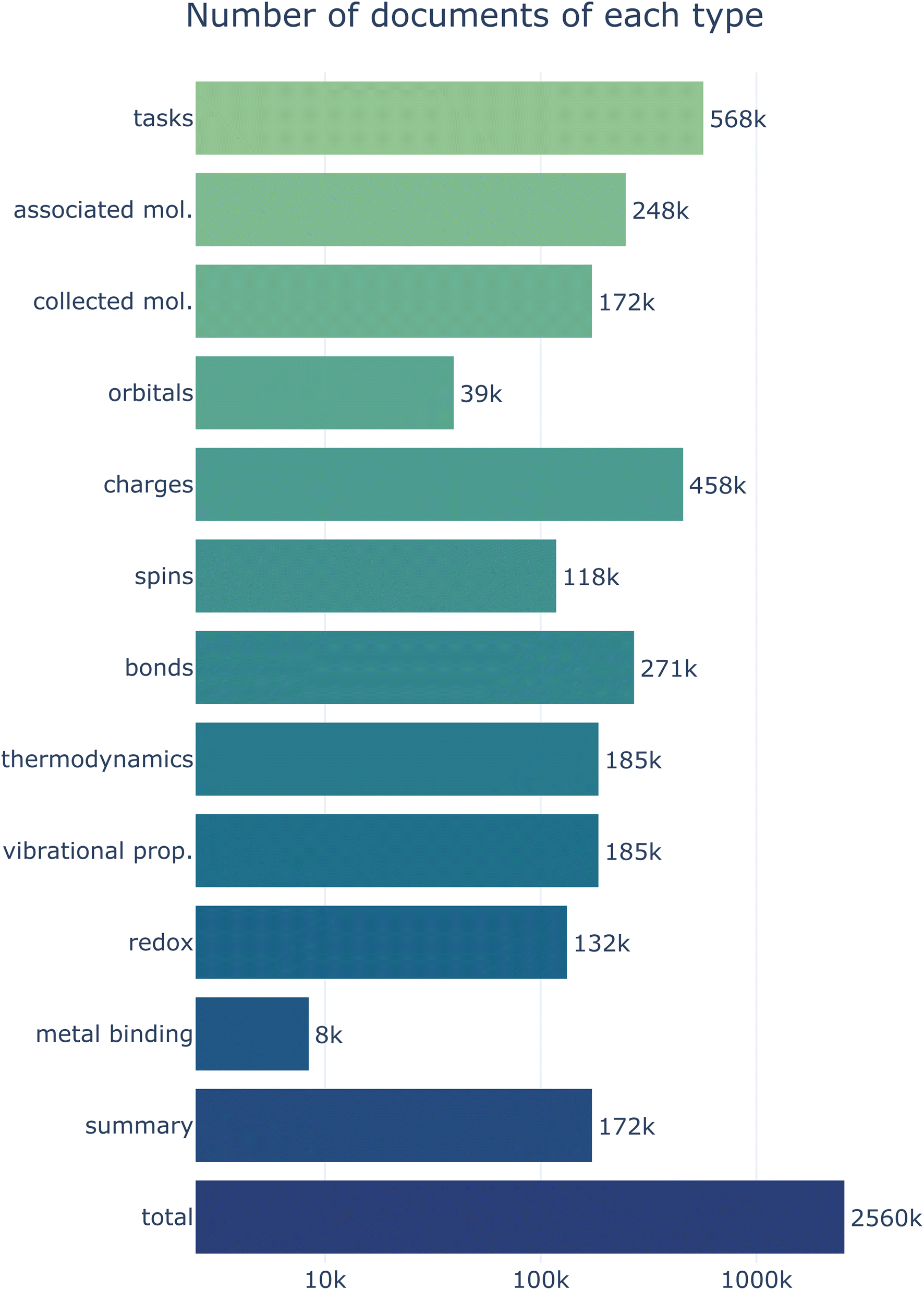 | ||
| Fig. 8 Scale of the current MPcules database in terms of number of documents, broken down by type. | ||
Molecules in the MPcules database do not come from a single source and are not selected based on any single set of criteria. We note that some of the data in MPcules has been previously released in different collections. Specifically, we previously reported the Lithium-Ion Battery Electrolyte (LIBE) dataset,57 a collection of the properties of 17190 molecules relevant to electrolyte decomposition and interphase formation in Li-ion batteries with carbonate electrolytes. More recently, we released the MAgnesium Dataset of Electrolyte and Interphase ReAgents (MADEIRA),7 containing properties of 11502 species relevant to electrolyte degradation and gas evolution in Mg-ion electrolytes consisting of magnesium bistriflimide dissolved in diglyme. Properties in LIBE and MADEIRA were calculated at the ωB97X-V/def2-TZVPPD/SMD level of theory. In addition to the molecules in LIBE and MADEIRA, MPcules contains molecules relevant to Mg-ion battery electrolytes with tetrahydrofuran electrolytes, as well as large numbers of small organic molecules, the properties of which have been calculated in vacuum and in many cases in an implicit solvent medium approximating water. As mentioned above, we intend to describe these data in further detail in future works.
Fig. 9 details the current composition of the MPcules database. In contrast to many existing molecular datasets, MPcules contains molecules with diverse charges and spin multiplicities (Fig. 9a and b). Currently, there are more ions in MPcules (98480) than neutral molecules (74394) and more radicals (89715) than closed-shell molecules (83159). Most ions have charge ±1, with nontrivial numbers of molecules with charge ±2. A very small number of ions with charge −3 (7) and +3 (6) are also included. These are all single atoms, the properties of which were studied in order to calculate redox properties. Currently, MPcules contains a relatively small number of triplets (2942); this presents a natural area for expansion.
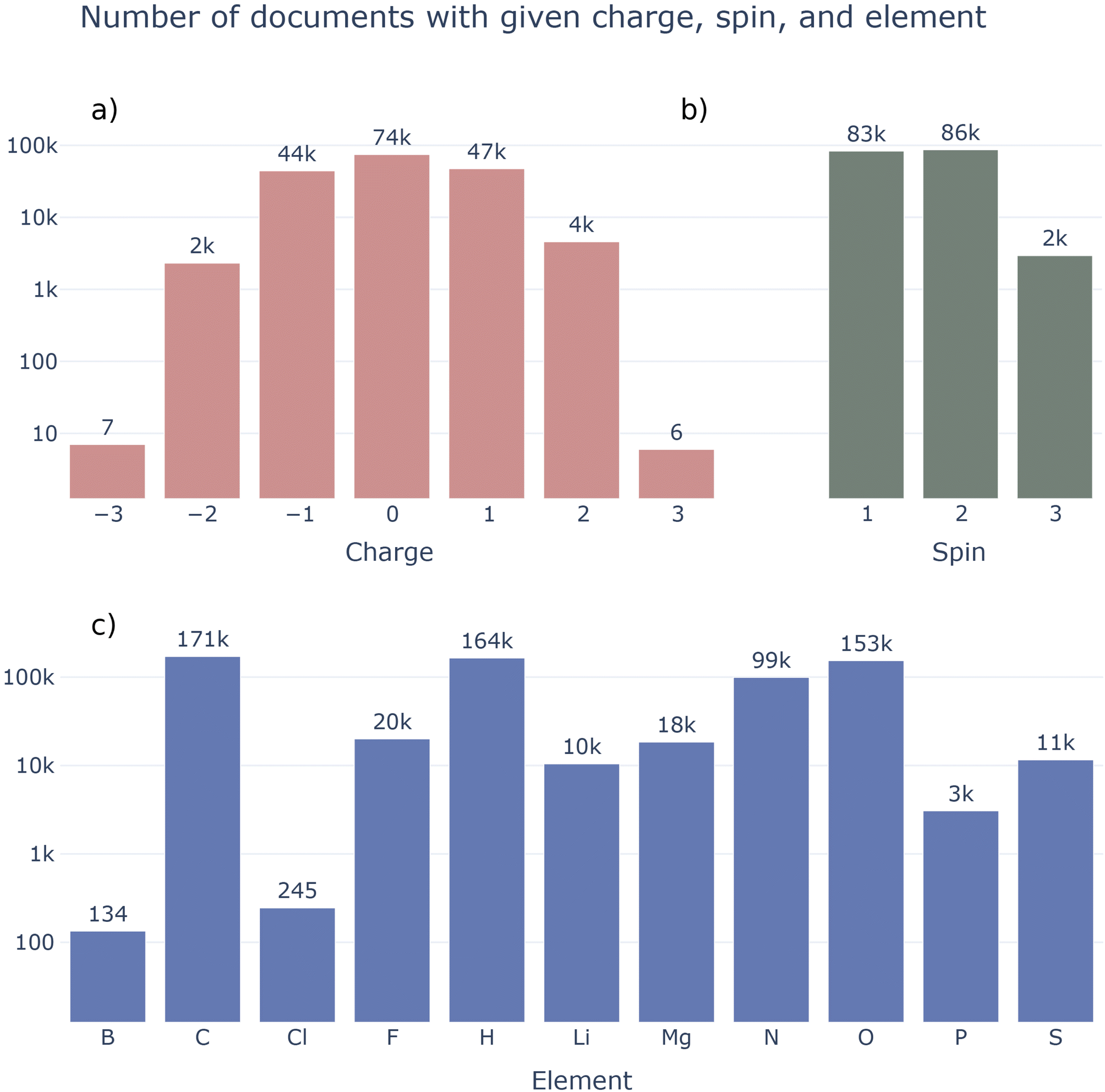 | ||
| Fig. 9 Composition of the MPcules database. (a) Number of molecules with different charges; (b) number of molecules with different spin multiplicities; (c) number of molecules including different elements. | ||
In terms of elements (Fig. 9c), MPcules is skewed towards organic molecules containing C, H, N, and O. In large part because of the previous LIBE and MADEIRA datasets, there are many (>10000) molecules containing F, Li, Mg, and S. While we do believe that MPcules is relatively diverse in terms of elements and chemical formulae, there are obviously many opportunities to expand its coverage through the addition of molecules containing B, P, halogens (e.g. Cl and Br), Si, or transition metals.
Future work
Just as the Materials Project has evolved from its initial release in 2011 to today, increasing in scale, scope, and structure, MPcules will continue to develop over time. We have already mentioned types of molecules that we intend to add to MPcules (e.g. transition-metal complexes and triplets). Here, we outline further plans to expand MPcules. We note that while we aim to internally develop the MPcules code(s) and dataset, we welcome user-submitted contributions of data as well as features (in the form of code contributions to emmet-core, emmet-builders, emmet-api, and the other Materials Project packages).Calculation methods and sources
Currently, MPcules accepts only DFT calculations from Q-Chem. This means that we are presently excluding calculations using high-quality wavefunction methods based on e.g. coupled-cluster theory and multireference methods, which might be useful for benchmarking electronic structure methods or for Δ-machine learning of electronic energy and other molecular properties. At the same time, we exclude calculations based on semiempirical quantum chemistry methods such as self-consistent extended tight-binding (e.g. GFN2-xTB),72 which have become increasingly popular for generating large datasets of molecules at low computational cost. Even within the narrower realm of DFT, the restriction to using a single electronic structure code could limit the ease with which users can contribute data to MPcules.In the future, we intend to create a more flexible interface which can parse and construct molecule and molecule property documents from calculations using a variety of DFT and non-DFT methods with multiple quantum chemistry codes (e.g.xTB,72ORCA,73,74 or NWChem).75
Molecular properties
MPcules already contains diverse atomic, molecular, and reaction properties. In the future, we aim to expand the properties available, both to aid in chemical analysis and to enable the development of ML methods. In particular, we are interested in expanding properties in two directions: spectroscopy and electronic densities. At present, the only spectra included in MPcules are IR spectra obtained via vibrational analysis. With modest modifications to our existing workflows, we should be able to additionally obtain molecular Raman spectra, including Raman activities and intensities. We also intend to incorporate nuclear magnetic resonance (NMR) spectra, including chemical shifts and J-couplings. In terms of charge densities, NBO provides detailed information regarding atomic and hybrid orbitals but does not describe the spatial extent of such orbitals or the total charge density around atoms in a molecule. Inspired by the recently developed charge density dataset included in the Materials Project,76 we intend to present total molecular charge densities to MPcules users via the Materials Project API and web site, as well as information regarding the electron densities of individual molecular orbitals.Conclusions
As chemical research grows increasingly reliant on big data and ML approaches, high-quality and open datasets of molecular properties will become vital cornerstone resources, accelerating the understanding of existing chemical systems and the design of novel functional molecules with optimized properties. MPcules, expanding on the existing Materials Project database, is a database of molecular calculations adhering to FAIR principles. The MPcules database currently contains over 170000 molecules which can be accessed through an API and featureful web app. MPcules is unique both because it grants users facile access to data and because that data is particularly diverse, containing many charged, open-shell, and metal-coordinated species as well as properties related to molecular bonding, electronic structure, thermodynamics, electrochemistry, vibrations, and reactions. Since MPcules relies on a suite of open source software, it is possible for users to add calculations to MPcules or to develop standalone datasets based on the same underlying schema. We believe that MPcules could serve as a community center for chemical datasets, with collaborative contributions of both code and data from users.
Software availability
The Materials Project software stack used to develop MPcules is publicly available on GitHub:• Pymatgen: https://github.com/materialsproject/pymatgen.
• Custodian: https://github.com/materialsproject/custodian.
• Atomate: https://github.com/hackingmaterials/atomate.
• Emmet: https://github.com/materialsproject/emmet.
• mp-api: https://github.com/materialsproject/api.
• Crystal Toolkit: https://github.com/materialsproject/crystaltoolkit.
• mp-react-components: https://github.com/materialsproject/mp-react-components.
• dash-mp-components: https://github.com/materialsproject/dash-mp-components.
Data availability
All data discussed in this work is publicly available through the Materials Project API, which can be accessed directly or viamp-api. Most data can also be accessed through the Materials Project web site (https://materialsproject.org/molecules).Author contributions
E. W. C. S.-S.: Conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, software, visualization, writing – original draft, writing – review & editing, O. A. C.: Funding acquisition, methodology, software, visualization, writing – original draft, writing – review & editing, S. M. B.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, writing – review & editing, J. M. M.: data curation, methodology, software, supervision, writing – review & editing, R. Y.: methodology, software, writing – review & editing, R. D. G.: investigation, writing – review & editing, H. D. P.: investigation, writing – review & editing, S. V.: investigation, writing – review & editing, P. H.: data curation, writing – review & editing, R. K.: methodology, writing – review & editing, M. K. H.: methodology, supervision, writing – review & editing, K. A. P.: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editingConflicts of interest
There are no conflicts to declare.Acknowledgements
E. W. C. S.-S. was supported by the Kavli Energy NanoScience Institute Philomathia Graduate Student Fellowship. O. A. C. was supported by the National Science Foundation Graduate Research Fellowships Program. Additional support was provided collaboratively by the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences; the Silicon Consortium Project directed by Brian Cunningham under the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy, Contract No. DE-AC02-05CH11231; and the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under U.S. Department of Energy Contract No. DE-AC02-05CH11231. Data described in this study were produced using computational resources provided by the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231, the Eagle and Swift high-performance computing (HPC) systems at the National Renewable Energy Laboratory (NREL), and the Lawrencium HPC cluster at Lawrence Berkeley National Laboratory.References
- M. Nakata and T. Maeda, PubChemQC B3LYP/6-31G*//PM6 dataset: the Electronic Structures of 86 Million Molecules using B3LYP/6-31G* calculations, arXiv, 2023, preprint, arXiv:2305.18454v1, DOI:10.48550/arXiv.2305.18454.
- L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick and Z. Ulissi, Open Catalyst 2020 (OC20) Dataset and Community Challenges, ACS Catal., 2021, 11, 6059–6072 CrossRef.
- C. A. Grambow, L. Pattanaik and W. H. Green, Reactants, products, and transition states of elementary chemical reactions based on quantum chemistry, Sci. Data, 2020, 7, 137 CrossRef PubMed.
- Q. Zhao, S. M. Vaddadi, M. Woulfe, L. A. Ogunfowora, S. S. Garimella, O. Isayev and B. M. Savoie, Comprehensive exploration of graphically defined reaction spaces, Sci. Data, 2023, 10, 145 CrossRef PubMed.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, To address surface reaction network complexity using scaling relations machine learning and DFT calculations, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- S. M. Blau, H. D. Patel, E. W. Clark Spotte-Smith, X. Xie, S. Dwaraknath and K. A. Persson, A chemically consistent graph architecture for massive reaction networks applied to solid-electrolyte interphase formation, Chem. Sci., 2021, 12, 4931–4939 RSC.
- E. W. C. Spotte-Smith, S. M. Blau, D. Barter, N. J. Leon, N. T. Hahn, N. S. Redkar, K. R. Zavadil, C. Liao and K. A. Persson, Chemical Reaction Networks Explain Gas Evolution Mechanisms in Mg-Ion Batteries, J. Am. Chem. Soc., 2023, 145, 12181–12192 CrossRef PubMed.
- J. Dagdelen, J. Montoya, M. de Jong and K. Persson, Computational prediction of new auxetic materials, Nat. Commun., 2017, 8, 323 CrossRef PubMed.
- P. Gorai, V. Stevanović and E. S. Toberer, Computationally guided discovery of thermoelectric materials, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- R. Berraud-Pache, F. Neese, G. Bistoni and R. Izsák, Computational Design of Near-Infrared Fluorescent Organic Dyes Using an Accurate New Wave Function Approach, J. Phys. Chem. Lett., 2019, 10, 4822–4828 CrossRef PubMed.
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario, M. S. Sigman and A. Aspuru-Guzik, A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef PubMed.
- A. S. Rosen, S. Vijay and K. A. Persson, Free-atom-like d states beyond the dilute limit of single-atom alloys, Chem. Sci., 2023, 14, 1503–1511 RSC.
- H. Huo, Z. Rong, O. Kononova, W. Sun, T. Botari, T. He, V. Tshitoyan and G. Ceder, Semi-supervised machine-learning classification of materials synthesis procedures, npj Comput. Mater., 2019, 5, 1–7 CrossRef.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef PubMed.
- A. R. Singh, B. A. Rohr, J. A. Gauthier and J. K. Nørskov, Predicting Chemical Reaction Barriers with a Machine Learning Model, Catal. Lett., 2019, 149, 2347–2354 CrossRef.
- S. Kim, J. Noh, G. H. Gu, A. Aspuru-Guzik and Y. Jung, Generative Adversarial Networks for Crystal Structure Prediction, ACS Cent. Sci., 2020, 6, 1412–1420 CrossRef PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. P. Turban, A. L. Gaunt, J. S. Spencer, A. G. D. G. Matthews, A. Obika, L. Thiry, M. Fortunato, D. Pfau, L. R. Castellanos, S. Petersen, A. W. R. Nelson, P. Kohli, P. Mori-Sánchez, D. Hassabis and A. J. Cohen, Pushing the frontiers of density functionals by solving the fractional electron problem, Science, 2021, 374, 1385–1389 CrossRef PubMed.
- S. Vargas, M. R. Hennefarth, Z. Liu and A. N. Alexandrova, Machine Learning to Predict Diels–Alder Reaction Barriers from the Reactant State Electron Density, J. Chem. Theory Comput., 2021, 17, 6203–6213 CrossRef PubMed.
- M. Wen, S. M. Blau, E. W. Clark Spotte-Smith, S. Dwaraknath and K. A. Persson, BonDNet: a graph neural network for the prediction of bond dissociation energies for charged molecules, Chem. Sci., 2021, 12, 1858–1868 RSC.
- S. Wengert, G. Csányi, K. Reuter and J. T. Margraf, Data-efficient machine learning for molecular crystal structure prediction, Chem. Sci., 2021, 12, 4536–4546 RSC.
- C. Chen and S. P. Ong, A universal graph deep learning interatomic potential for the periodic table, Nat. Comput. Sci., 2022, 2, 718–728 CrossRef.
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Learning local equivariant representations for large-scale atomistic dynamics, Nat. Commun., 2023, 14, 579 CrossRef PubMed.
- M. D. Wilkinson, M. Dumontier, I. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. da Silva Santos, P. E. Bourne, J. Bouwman, A. J. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. T. Evelo, R. Finkers, A. Gonzalez-Beltran, A. J. G. Gray, P. Groth, C. Goble, J. S. Grethe, J. Heringa, P. A. C. ’t Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. J. Lusher, M. E. Martone, A. Mons, A. L. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. A. Swertz, M. Thompson, J. van der Lei, E. van Mulligen, J. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao and B. Mons, The FAIR Guiding Principles for scientific data management and stewardship, Sci. Data, 2016, 3, 160018 CrossRef PubMed.
- J. J. de Pablo, B. Jones, C. L. Kovacs, V. Ozolins and A. P. Ramirez, The Materials Genome Initiative, the interplay of experiment, theory and computation, Curr. Opin. Solid State Mater. Sci., 2014, 18, 99–117 CrossRef.
- J. J. de Pablo, N. E. Jackson, M. A. Webb, L.-Q. Chen, J. E. Moore, D. Morgan, R. Jacobs, T. Pollock, D. G. Schlom, E. S. Toberer, J. Analytis, I. Dabo, D. M. DeLongchamp, G. A. Fiete, G. M. Grason, G. Hautier, Y. Mo, K. Rajan, E. J. Reed, E. Rodriguez, V. Stevanovic, J. Suntivich, K. Thornton and J.-C. Zhao, New frontiers for the materials genome initiative, npj Comput. Mater., 2019, 5, 1–23 CrossRef.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD), JOM, 2013, 65, 1501–1509 CrossRef.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies, npj Comput. Mater., 2015, 1, 1–15 Search PubMed.
- C. Draxl and M. Scheffler, NOMAD: The FAIR concept for big data-driven materials science, MRS Bull., 2018, 43, 676–682 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- D. G. A. Smith, D. Altarawy, L. A. Burns, M. Welborn, L. N. Naden, L. Ward, S. Ellis, B. P. Pritchard and T. D. Crawford, The MolSSI QCArchive project: An open-source platform to compute, organize, and share quantum chemistry data, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11, e1491 CrossRef.
- M. Nakata and T. Shimazaki, PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry, J. Chem. Inf. Model., 2017, 57, 1300–1308 CrossRef PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1, 140022 CrossRef PubMed.
- J. S. Smith, O. Isayev and A. E. Roitberg, ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules, Sci. Data, 2017, 4, 170193 CrossRef PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He and B. A. Shoemaker, et al., PubChem substance and compound databases, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, d. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, d. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, III, R. A. DiStasio, Jr., A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, III, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, III, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 CrossRef PubMed.
- A. Jain, S. P. Ong, W. Chen, B. Medasani, X. Qu, M. Kocher, M. Brafman, G. Petretto, G.-M. Rignanese, G. Hautier, D. Gunter and K. A. Persson, FireWorks: a dynamic workflow system designed for high-throughput applications, Concurr. Comput., 2015, 27, 5037–5059 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef.
- S. Blau, E. W. C. Spotte-Smith, B. Wood, S. Dwaraknath and K. PerssonAccurate, Automated Density Functional Theory for Complex Molecules Using On-the-fly Error Correction. ChemRxiv, 2020, preprint, DOI:10.26434/chemrxiv.13076030.v1.
- K. Mathew, J. H. Montoya, A. Faghaninia, S. Dwarakanath, M. Aykol, H. Tang, I.-h. Chu, T. Smidt, B. Bocklund, M. Horton, J. Dagdelen, B. Wood, Z.-K. Liu, J. Neaton, S. P. Ong, K. Persson and A. A. Jain, A high-level interface to generate, execute, and analyze computational materials science workflows, Comput. Mater. Sci., 2017, 139, 140–152 CrossRef.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- N. Mardirossian and M. Head-Gordon, ω B97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- N. Mardirossian and M. Head-Gordon, ω B97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- D. Rappoport and F. Furche, Property-optimized Gaussian basis sets for molecular response calculations, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- B. Mennucci, Polarizable continuum model, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 386–404 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- P. C. D. Hawkins, Conformation Generation: The State of the Art, J. Chem. Inf. Model., 2017, 57, 1747–1756 CrossRef PubMed.
- S. P. Ong and G. Ceder, Investigation of the Effect of Functional Group Substitutions on the Gas-Phase Electron Affinities and Ionization Energies of Room-Temperature Ionic Liquids Ions using Density Functional Theory, Electrochim. Acta, 2010, 55, 3804–3811 CrossRef.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminformatics, 2011, 3, 33 CrossRef PubMed.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 2004, 23, 1833–1840 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural bond orbital methods, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 1–42 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI, 2018 Search PubMed.
- J. Meister and W. H. E. Schwarz, Principal Components of Ionicity, J. Phys. Chem., 1994, 98, 8245–8252 CrossRef.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed.
- A. Otero-de-la Roza, E. R. Johnson and V. Luaña, Critic2: A program for real-space analysis of quantum chemical interactions in solids, Comput. Phys. Commun., 2014, 185, 1007–1018 CrossRef.
- S. Lehtola and H. Jónsson, Pipek–Mezey Orbital Localization Using Various Partial Charge Estimates, J. Chem. Theory Comput., 2014, 10, 642–649 CrossRef PubMed.
- E. W. C. Spotte-Smith, S. M. Blau, X. Xie, H. D. Patel, M. Wen, B. Wood, S. Dwaraknath and K. A. Persson, Quantum chemical calculations of lithium-ion battery electrolyte and interphase species, Sci. Data, 2021, 8, 203 CrossRef PubMed.
- S. Trasatti, The absolute electrode potential: an explanatory note (Recommendations 1986), Pure Appl. Chem., 1986, 58, 955–966 CrossRef.
- R. Custelcean and B. A. Moyer, Anion Separation with Metal–Organic Frameworks, Eur. J. Inorg. Chem., 2007, 2007, 1321–1340 CrossRef.
- G. J. Kubas, Hydrogen activation on organometallic complexes and H2 production, utilization, and storage for future energy, J. Organomet. Chem., 2009, 694, 2648–2653 CrossRef.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, High-Voltage Lithium-Metal Batteries Enabled by Localized High-Concentration Electrolytes, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, InChI, the IUPAC International Chemical Identifier, J. Cheminformatics, 2015, 7, 23 CrossRef PubMed.
- D. Weininger, A. Weininger and J. L. Weininger, SMILES. 2. Algorithm for generation of unique SMILES notation, J. Chem. Inf. Comput. Sci., 1989, 29, 97–101 CrossRef.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation, Mach. learn.: sci. technol., 2020, 1, 045024 Search PubMed.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. Falk von Rudorff, A. Wang, A. D. White, A. Young, R. Yu and A. Aspuru-Guzik, SELFIES and the future of molecular string representations, Patterns, 2022, 3, 100588 CrossRef PubMed.
- N. Shervashidze, P. Schweitzer, E. J. Van Leeuwen, K. Mehlhorn and K. M. Borgwardt, Weisfeiler-Lehman Graph Kernels, J. Mach. Learn. Res., 2011, 12 Search PubMed.
- A. Hagberg, P. Swart and D. S Chult, Exploring network structure, dynamics, and function using NetworkX, 2008 Search PubMed.
- J.-P. Aumasson, S. Neves, Z. Wilcox-O’Hearn and C. Winnerlein, BLAKE2: Simpler, Smaller, Fast as MD5, Applied Cryptography and Network Security, Berlin, Heidelberg, 2013, pp. 119–135 Search PubMed.
- Y.-L. Liao and T. Smidt, Equivariant graph attention transformer for 3d atomistic graphs, arXiv, 2022, preprint, arXiv:2206.11990v2, DOI:10.48550/arXiv.2206.11990.
- M. Horton, J.-X. Shen, J. Burns, O. Cohen, F. Chabbey, A. M. Ganose, R. Guha, P. Huck, H. H. Li, M. McDermott, J. Montoya, G. Moore, J. Munro, C. O'Donnell, C. Ophus, G. Petretto, J. Riebesell, S. Wetizner, B. Wander, D. Winston, R. Yang, S. Zeltmann, A. Jain and K. A. Persson, Crystal Toolkit: A Web App Framework to Improve Usability and Accessibility of Materials Science Research Algorithms, arXiv, 2023, preprint, arXiv:2302.06147v2, DOI:10.48550/arXiv.2302.06147.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef.
- J.-X. Shen, J. M. Munro, M. K. Horton, P. Huck, S. Dwaraknath and K. A. Persson, A representation-independent electronic charge density database for crystalline materials, Sci. Data, 2022, 9, 661 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further details on ranking of levels of theory; composition of MPcules in terms of level of theory; comparison of Mulliken and NBO atomic partial charges and atomic partial spins. Ref. 40–45. See DOI: https://doi.org/10.1039/d3dd00153a |
| ‡ Present address: VASP Software GmbH, Sensengasse 8, A-1090 Vienna, Austria. |
| This journal is © The Royal Society of Chemistry 2023 |