
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncovering novel liquid organic hydrogen carriers: a systematic exploration of chemical compound space using cheminformatics and quantum chemical methods†
Hassan
Harb
 a,
Sarah N.
Elliott
b,
Logan
Ward
c,
Ian T.
Foster
c,
Stephen J.
Klippenstein
b,
Larry A.
Curtiss
a and
Rajeev Surendran
Assary
*a
a,
Sarah N.
Elliott
b,
Logan
Ward
c,
Ian T.
Foster
c,
Stephen J.
Klippenstein
b,
Larry A.
Curtiss
a and
Rajeev Surendran
Assary
*a
aMaterials Science Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: assary@anl.gov; Tel: +1 630-252-3536
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
cData Sciences and Learning Division, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 11th October 2023
Abstract
We present a comprehensive, in silico-based discovery approach to identifying novel liquid organic hydrogen carrier (LOHC) candidates using cheminformatics methods and quantum chemical calculations. We screened over 160 billion molecules from ZINC15 and GDB-17 chemical databases for structural similarity to known LOHCs and employed a data-driven selection criterion connecting molecular features with dehydrogenation enthalpy. This scoring criterion effectively predicts dehydrogenation enthalpies from SMILES strings, streamlining the LOHC screening process. After rigorous screening and down-selection, we compiled a database of 3000 dehydrogenation reactions for the most promising LOHC candidates, setting the stage for future selection based on kinetics and catalysis. This work demonstrates the significant impact of integrating quantum chemistry and cheminformatics in materials discovery, accelerating the selection process while reducing experimental efforts and time. By proposing new molecules as prospective LOHC candidates, our study provides a valuable resource for researchers and engineers in the development of advanced LOHC systems and showcases a successful approach for high-throughput discovery, contributing to more efficient and sustainable energy storage solutions.
1 Introduction
There is an urgent need to reduce carbon dioxide (CO2) emissions due to their role in the ongoing climate change crisis. This reduction requires a decarbonization of the world's energy systems, with a shift away from fossil fuel dependence and into clean, sustainable energy sources. The UN Glasgow climate pact highlights the need to reduce CO2 emission by 45% by 2030,1 and the European Union's Roadmap 2050 proposes to reduce CO2 emissions by 80%.2 To reach these goals, and thereby limit the extent and most negative outcomes of climate change, numerous renewable and clean energy sources are being implemented on grand scales. The most important of these sources include solar, wind, and hydroelectricity, each of which is accompanied by large scale time-dependent fluctuations in the power generated. Thus, a transformation to such decarbonized sources is only viable when accompanied by energy storage systems to obtain stable energy availability.3A wide variety of energy storage systems are currently under development, including chemical, electrochemical, electrical, mechanical, and thermal systems. Among these, chemical storage, and in particular the use of H2 as a fuel, represents a promising and efficient solution. This approach is primarily due to the high energy density of hydrogen and its potential for clean, sustainable energy.4 In a proposed hydrogen economy, energy is stored in hydrogen carrier molecules, providing a consistent delivery of energy through chemical transformations that alternately add or release hydrogen.5 The Department of Energy (DOE) Hydrogen Earthshot6 puts forth a plan to accelerate innovations and spur demand of clean hydrogen by reducing the cost of clean hydrogen by 80% to $1 per 1 kilogram in 1 decade (“111”).7
The hydrogenation of nitrogen gas to ammonia and of carbon dioxide to formic acid9,10 or methanol11 are two examples of the chemical storage of hydrogen. Both systems, however, require methods to isolate N2 or CO2 at the end stages of the catalytic dehydrogenation process and are prone to release mixtures of gases instead of pure H2.12 Various hydrogen-lean liquid organic hydrogen carriers (LOHCs)3,13,14 have been proposed as alternative, more promising carrier molecules. LOHCs are unsaturated organic molecules that can chemically trap (hydrogenation) and then release (dehydrogenation) H2 molecules, with the hydrogenation/dehydrogenation cycles mediated by a catalyst under specific reaction conditions.15 These organic liquids can, in principle, enable a fully reversible catalytic hydrogenation/dehydrogenation cycle.3,16–21 Importantly, LOHCs also provide a decarbonized energy system when they are implemented in a recyclable fashion with multiple hydrogenation/dehydrogenation cycles.
LOHCs can have a high hydrogen storage capacity and thus a high energy density.15,17,18 As a result, they can be suitable for portable fuel cells and other portable applications. Compared to H2 as a fuel, LOHCs have a much better safety profile in that they are stored in liquid form (rather than compressed gas) and are not as prone to explosion. Thus, LOHCs are an attractive option for use in transportation and other applications for which safety and portability are priorities.3,8,14,15,17,22–25
A major criterion set by DOE is that the gravimetric hydrogen capacity of LOHCs be greater than 5.5%, to ensure that the energy density is high enough for transportation applications.15,26,27 Additionally, several studies of potential LOHC systems suggest that an optimal hydrogenation/dehydrogenation enthalpy range lies between 40–70 kJ per mol of H2.15,20 This range facilitates low-temperature conversions between the hydrogenated and dehydrogenated forms. Ideally, both hydrogen-lean and hydrogen-rich species should be liquids at room temperature.15,20,27 Other critical requirements for LOHCs are that the hydrogenation/dehydrogenation cycles occur without secondary degradation reactions and that the molecules have acceptable toxicity profiles.15,18,27,28
Several classes of prospective LOHCs have been reported,3,5,20,27 with the first of these being aromatic hydrocarbons. The extensively-explored benzene/cyclohexane system, for example, has catalyst-mediated hydrogenation/dehydrogenation (ΔH = 68.8 kJ per mol H2) processes.29,30 Another example is the toluene/methyl cyclohexane system, which was seen as a promising LOHC due to its good hydrogen storage capacity (6.1%) and high heat of hydrogenation (ΔH = 68.3 kJ per mol H2).31–34 Notably, toluene/methyl cyclohexane was the system used in a 1985 hydrogen-powered prototype truck.35 Japan recently built the world's first hydrogen supply chain utilizing toluene as its LOHC.36
The N-ethyl carbazole/dodecahydro-N-ethyl carbazole (H0-NEC/H12-NEC) pair is another extensively studied LOHC system.24,37,38 This system has a dehydrogenation enthalpy of 50 kJ per mol H2 and a hydrogen capacity of 5.8%. Systems containing indoles28,39,40 and quinones41 have also been of interest, with some dehydrogenation enthalpies being around 50 kJ per mol H2. Other N-containing heterocycles studied as LOHC include phenazines, pyridines, anilines, quinoline, and pyrrole.5,15,18,20,40,42–44
LOHCs, while promising, face significant hurdles such as benzene's high toxicity, the flammability and low boiling point (81 °C) of cyclohexane, and the high melting point (68 °C) of H0-NEC. Despite these challenges, successful modification of the NEC structure produced a liquid derivative (by investigating mixtures of N-alkyl substituted carbazoles) with a considerably reduced melting point of 24 °C.45 A further limitation of N-alkyl carbazoles and their hydrogenated analogs is the propensity for the N-alkyl substituent to dissociate at temperatures significantly lower than the thermal decomposition point of the heteroaromatic ring.46 Similar problems are present with the toluene/methylcyclohexane system. Under the hydrogenation/dehydrogenation reaction conditions, degradation reactions are likely to occur.19 Additionally, both toluene and methylcyclohexane are gaseous at both hydrogenation and dehydrogenation conditions.5,15,47 Generally, key obstacles hindering the widespread adoption of LOHCs in cutting-edge energy applications include chemical degradations and side reactions that occur during hydrogenation/dehydrogenation processes.15,18,28,48 Briefly addressing the molecular intricacies, the incorporation of nitrogen within LOHC molecules has been shown to favorably influence dehydrogenation thermodynamics and kinetics. However, this same feature, while beneficial, can also introduce challenges. The presence of nitrogen tends to increase thermal lability, thereby creating opportunities for the molecule to produce undesired degradation products. Additionally, the requirement for elevated temperatures in the dehydrogenation process poses another barrier, as it complicates the efficient use of waste heat for driving the intrinsically endothermic dehydrogenation reactions.15 Moreover, another concern is the inherent inefficiency in fully saturating LOHCs with hydrogen.49 For instance, certain studies have found that the hydrogen loading of dibenzyl toluene reaches only about 90% of its maximum capacity.50 Generally, current LOHC systems also possess technological limitations, these include catalyst development, low-temperature dehydrogenation, safety concerns, H2 loading capability, alongside economic factors such as the cost of production.14,21,51
To address these drawbacks, a primary focus for LOHC development is fine-tuning their physical properties and chemical conditions, which will facilitate hydrogenation/dehydrogenation processes without triggering degradation reactions. This necessitates a careful consideration of various factors, such as operating at ambient temperature and pressure conditions for enhanced safety and practicality. The dehydrogenation enthalpy (ΔH) should range between 40–70 kJ per mol H2 to ensure efficient energy storage and release.15,27 Furthermore, the molecules of interest must exhibit acceptable toxicity profiles for safe handling and usage. Attention must also be given to other critical factors, such as adequate storage and liberation of molecular hydrogen, the capacity for multiple hydrogenation/dehydrogenation cycles, and high gravimetric and volumetric capacities. By tackling these challenges head on, LOHCs can be transformed into a compact and efficient hydrogen storage solution, revolutionizing the energy landscape.15,18,27,28
Research efforts towards discovering new LOHC systems have been focused on using NH3 or petroleum-derived hydrocarbon materials.52–55 Those that have investigated alternative classes have failed to identify materials with longer-term stability because they did not undertake a systematic exploration of chemical space with a relevant selection criteria.5,15,20,27 Recent investigations highlight the current state of the art in hydrogen storage using LOHC and stationary energy storage systems.5,56–59 Yet while feasibility of LOHC technology60 has recently been proven in commercial demonstrators using toluene,36 cost aspects demand LOHC with longer stability (>60 days). Thus, new, and improved LOHCs are needed, and there is a significant chemical space (billions of molecules) that can now be explored to identify optimal materials. The growing fields of data science, artificial intelligence, and machine learning methods allows scientists to accelerate the design and discovery of new materials.61–65 The popularity of these approaches has been reflected by successes in automated chemical synthesis, predictions of material properties, drug design, and predictions of quantum chemical properties.66–69
In this study, we employ cheminformatics methods and quantum chemical calculations to explore the chemical compound space, with the aim of identifying promising candidates for novel LOHCs as shown in Fig. 1. Our approach can be summarized in the following steps, as shown in Fig. 2: (1) screening of ZINC15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 70 (1.2 billion molecules) and GDB-1771 (166 billion molecules) chemical databases for molecules having structural similarity to known LOHCs; then down selecting the high-similarity candidates for enthalpy, based first on (2) a heuristic chemical descriptor-based selection criterion and then on (3) results of DFT calculations, and (4) further screening out impractical molecules, including by using machine-learning-models to predict molecule melting and boiling points. Our methodology successfully identifies 41 potential candidate LOHCs. Using this approach, we compiled a database of 3000 dehydrogenation reaction energies for candidate LOHC systems (see GitHub repository),72 formulated a dehydrogenation index for the prediction of dehydrogenation enthalpies of organic molecules from graph representation, provided extensive chemical and physical properties that constitute a good LOHC candidate, and finally, identified new molecules as prospective LOHC candidates. An alternative computational screening of large databases for LOHC molecules was presented by Paragian et al.73 Starting with over 1 million seed molecules from PubChem, they utilized tools like RING74–76 (to obtain candidate structures), OPERA77 (to estimate melting and boiling points), and machine-learning-methods (to predict dehydrogenation enthalpies), to identify and analyze LOHC pairs. They identified 14k feasible pairs and selected 37 promising LOHC candidate pairs based on criteria like hydrogen capacity and synthetic accessibility. The authors further explored key LOHC features using a sparse linear discriminant analysis model. Despite methodological differences in identifying candidate LOHC structures and calculations of dehydrogenation enthalpies, Paragian and coworkers' approach was successful, highlighting the importance of diverse strategies in LOHC discovery.
70 (1.2 billion molecules) and GDB-1771 (166 billion molecules) chemical databases for molecules having structural similarity to known LOHCs; then down selecting the high-similarity candidates for enthalpy, based first on (2) a heuristic chemical descriptor-based selection criterion and then on (3) results of DFT calculations, and (4) further screening out impractical molecules, including by using machine-learning-models to predict molecule melting and boiling points. Our methodology successfully identifies 41 potential candidate LOHCs. Using this approach, we compiled a database of 3000 dehydrogenation reaction energies for candidate LOHC systems (see GitHub repository),72 formulated a dehydrogenation index for the prediction of dehydrogenation enthalpies of organic molecules from graph representation, provided extensive chemical and physical properties that constitute a good LOHC candidate, and finally, identified new molecules as prospective LOHC candidates. An alternative computational screening of large databases for LOHC molecules was presented by Paragian et al.73 Starting with over 1 million seed molecules from PubChem, they utilized tools like RING74–76 (to obtain candidate structures), OPERA77 (to estimate melting and boiling points), and machine-learning-methods (to predict dehydrogenation enthalpies), to identify and analyze LOHC pairs. They identified 14k feasible pairs and selected 37 promising LOHC candidate pairs based on criteria like hydrogen capacity and synthetic accessibility. The authors further explored key LOHC features using a sparse linear discriminant analysis model. Despite methodological differences in identifying candidate LOHC structures and calculations of dehydrogenation enthalpies, Paragian and coworkers' approach was successful, highlighting the importance of diverse strategies in LOHC discovery.
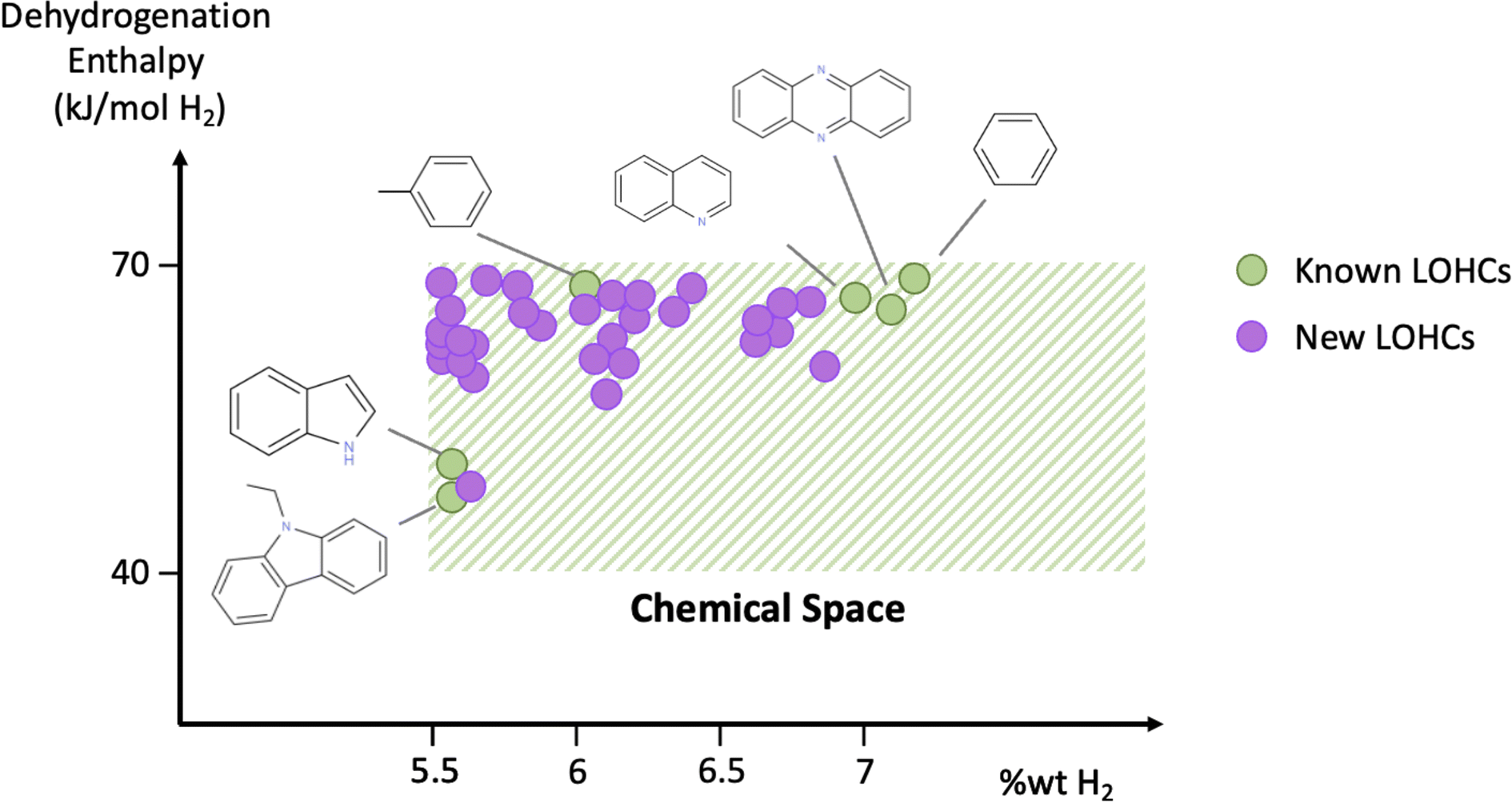 | ||
| Fig. 1 Schematic overview of the studied chemical space. Shaded area indicates the subspace of interest that contains molecules exhibiting gravimetric capacities of >5.5% wt H2 and dehydrogenation enthalpies in the 40–70 kJ per mol per H2 range. Green circles indicate known LOHCs, and purple circles refer to LOHCs that are identified in this study. A comprehensive list of known LOHCs, along with corresponding references, is shown in Table S2 of the ESI.† | ||
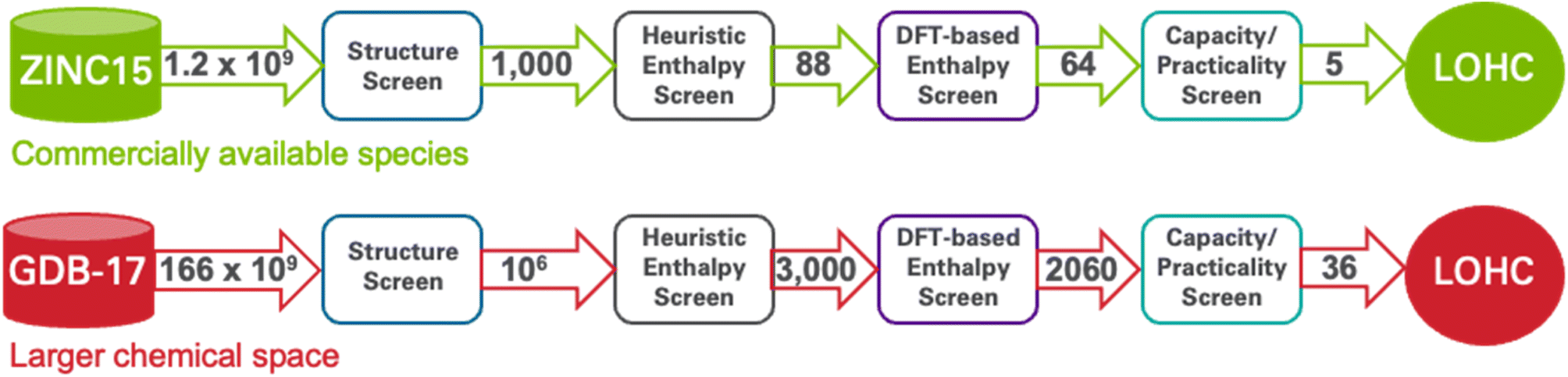 | ||
| Fig. 2 The figure presents the application of our four-step workflow for identifying candidate LOHC molecules to the ZINC15 (above) and GDB-17 (below) chemical databases. (1) The structure screen uses the Tanimoto similarity index to select molecules with high structural similarity to known LOHCs. (2) The heuristic enthalpy screen then down-selects those candidates based on a set of chemical descriptor-based selection criteria (what we refer to in the manuscript text as the provisional scoring criteria). (3) The DFT-based enthalpy screen performs DFT calculations on the resulting candidates and eliminates those for which DFT results indicate dehydrogenation enthalpies outside the desired range of 40–70 kJ mol−1. (4) The capacity/practicality screen eliminates molecules with low H2 capacity, that a machine learning melting point model predict will not be liquid at room temperature, or that a synthesizability model predicts are not easily synthesizable. The result is 41 new candidate LOHCs – 5 from ZINC15 and 36 from GDB-17. The numbers on the arrows represent the size of the molecular set involved at each step, illustrating the winnowing process from the initial database to the final selection of candidate LOHCs. | ||
2 Computational details
The dehydrogenation energy is key to predicting the utility of LOHC molecules. Given the complexity of many LOHC candidates, it is not practical to use the highest accuracy quantum chemistry methods. As a result, a more affordable Density Functional Theory (DFT) approach is adopted. For this, a set of 31 experimental LOHC molecules (Exp-31, Table S2†) is collected to validate the DFT approach. Molecules that pass the screening process (as outlined in Fig. 2) have their SMILES strings converted to 3D structures using the RDKIT package.78 Subsequently, minimum energy 3D conformers were predicted using the universal force field (UFF) method,79 and were used as initial guesses for electronic structure calculations. Density functional theory (DFT) calculations were carried out using the Gaussian 16 program80 and the ωB97X-D hybrid functional, chosen for its balance between cost-efficiency, accuracy, and its capability in addressing the long-range interactions often encountered in aromatic species in the dataset.81,82 Additionally, a comparative analysis against other popular density functionals like B3LYP and M06, as discussed in the ESI,† further justifies the choice of ωB97X-D, for a range of organic molecules.83 The 6-31G(2df,p) basis set83,84 was used for all first, second, and third row elements, and the Dunning style aug-cc-pVTZ basis set85 was used for molecules containing elements beyond the third row. Performance assessment of this model chemistry is presented in the ESI86–88 (Fig. S2 and S3).† Geometry optimizations were carried out and stationary points were verified using frequency calculations.89All reaction enthalpies (ΔHrxn) are reported as enthalpies of dehydrogenation per mole of H2 in units of kJ per mol H2 and are calculated according to eqn (1):
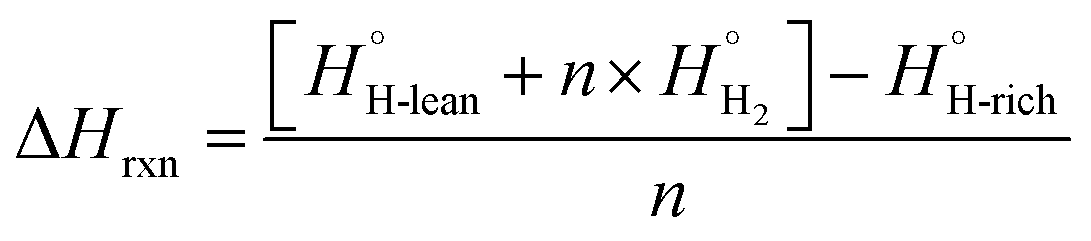 | (1) |
Predictions of physical properties, i.e. melting and boiling points, were performed using Leruli,90 which employs the OPERA77 model to predict quantitative structural activity and property relationships. OPERA is a tool that employs QSAR/QSPR (quantitative structure activity/property relationship) modeling to correlate molecular structure with physical properties. Drawing from extensive datasets containing over 40000 chemicals, OPERA was previously used to predict the physical states of organic molecules, including new potential LOHC molecules.73,91
The gravimetric capacity, denoted as % wt H2, is calculated using eqn (2):
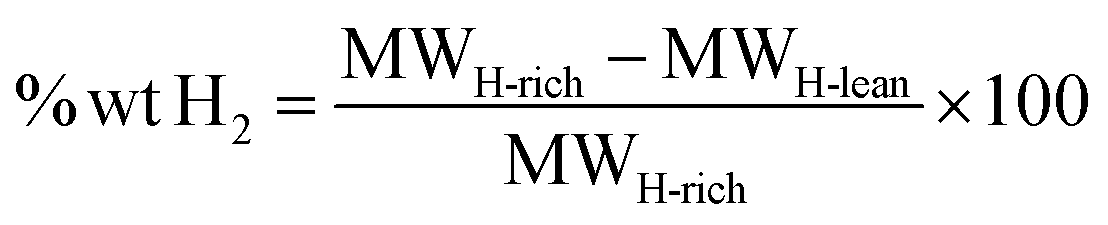 | (2) |
To identify molecules in the chemical databases that are structurally similar to the benchmark set, we employ the Tanimoto similarity index (T),92,93 shown in eqn (3):
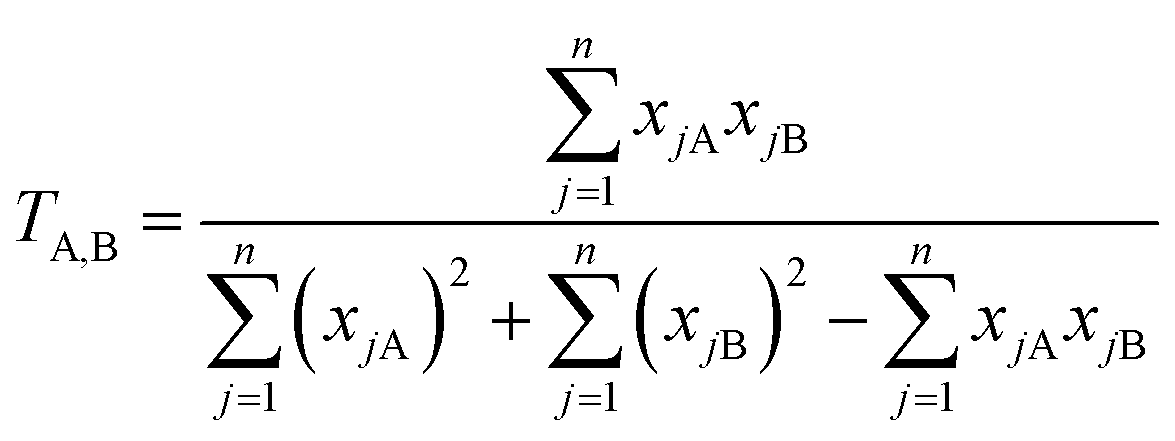 | (3) |
3 Results and discussions
3.1. Examining LOHC structure–property relationships
Large-scale screening of LOHC candidates necessitates an understanding of the relationship between desired dehydrogenation enthalpies and structural/molecular properties. Our approach integrates experimental data and literature knowledge on optimal LOHCs. Firstly, we collate available experimental data and identify common properties of leading LOHCs from the literature. Next, we expand our dataset by applying various permutations to the functional groups of known LOHC molecules, yielding a benchmark set. This set, rich in diversity, enables us to conduct effective Tanimoto similarity comparisons on large molecular databases, facilitating the discovery of novel LOHC candidates.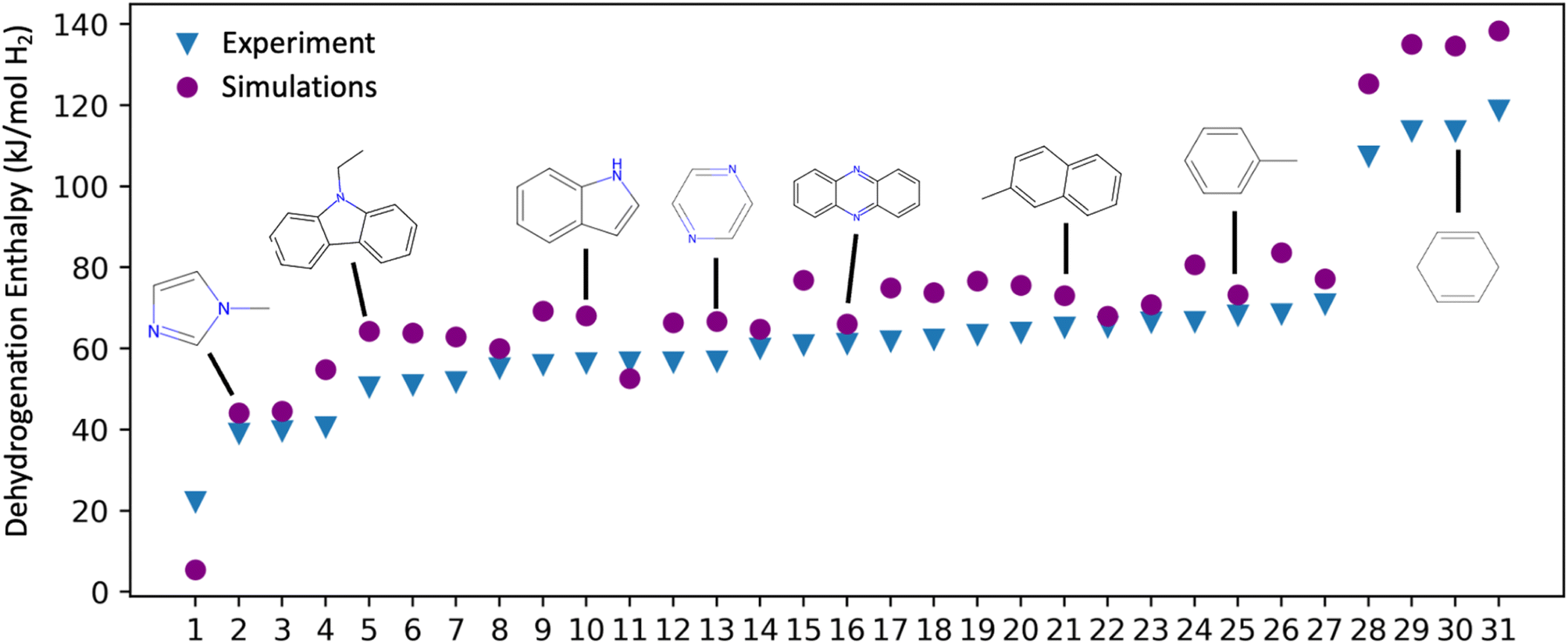 | ||
| Fig. 3 Comparison of experimental and DFT-calculated (ωb97X-D/6-31G(2df,p)) dehydrogenation enthalpies for molecules in the Exp-31 set. Each molecule is represented by a blue triangle (experimental value) and a purple circle (DFT value). The specific molecules depicted above the points were chosen to illustrate the range and diversity within the Exp-31 set. A comprehensive list of these molecules, along with corresponding references is shown in Table S2 of the ESI.† | ||
The Exp-31 experimental dataset, while highly useful for DFT method validation, is not large and diverse enough to determine structure–property relationships and trends. Consequently, we expanded our dataset by constructing a larger test set, Bench-93, primarily for a more extensive range of Tanimoto similarity comparisons (vide infra). The Bench-93 molecules, which resulted from applying various permutations to the functional groups of Exp-31 molecules (changed position of the substituted group on five- and six-membered rings, changed positions of heteroatoms in aromatic rings, and modifying sizes of aliphatic chains), were not initially conceived as LOHC candidates. Rather, their role was to broaden the spectrum of molecular structures in our dataset, with the primary scoring system remaining grounded in literature reports on good LOHC candidates.15,20,27 We note that all the molecules in the Exp-31 set are included in Bench-93.
We combine the 11 descriptors into a single numeric score via this equation:
 | (4) |
We intend s, as defined by eqn (4) to provide a coarse representation of how well the dehydrogenation enthalpy of molecules fit within the desired 40–70 kJ mol−1 range; a higher value of s indicates that the molecule is more likely to have a dehydrogenation enthalpy within that range. In practice, we found that molecules with an s score of around 4 did not consistently align within the desired dehydrogenation enthalpy range of 40–70 kJ mol−1, while molecules that scored between 5 and 7 demonstrated a more consistent alignment within this optimal range. Thus, we conclude that this scoring system has utility in identifying suitable LOHC candidates, albeit in a coarse manner (see Fig. S1 of the ESI†).
3.2. Molecular database screening
In our systematic exploration of the chemical compound space to identify novel LOHC molecules, we employed a targeted approach to navigate large molecular libraries efficiently. Fig. 4 shows a schematic overview of the chemical compound space and the subspaces that are of interest in our study. Since the core concept of LOHC technology is the chemical storage of hydrogen molecules in unsaturated bonds, our search strategically concentrated on the subspace of unsaturated molecules which hold significant potential for LOHC discovery. We classified the search space into four subspaces based on specific criteria, shown in Fig. 4 as the four-circle Venn diagram: | ||
| Fig. 4 Visual representation of the search space and approach for identifying suitable LOHCs. The large outer circle represents the entire chemical compound space, estimated to contain around 1011 molecules. The smaller circle within this represents the subset of these molecules that are unsaturated. Within this subset, a four-circle Venn diagram represents additional selection criteria, labeled as Structure, Practicality, Capacity, and Enthalpy, and they correspond to structural similarity to known LOHCs, desirable physical properties and molecule synthesizability, a gravimetric capacity of 5.5% or higher, and a dehydrogenation enthalpy in the 40–70 kJ per mol H2 range, respectively. We hypothesize that the intersection of these four criteria indicates the most promising candidates for LOHCs. | ||
(1) Structure: high structural similarity to known LOHCs, as determined by the Tanimoto similarity coefficient.
(2) Practicality: melting points for both hydrogen-rich and hydrogen-poor forms of less than 40 °C, as estimated by ML models to guarantee that the candidate LOHCs are liquid at room temperature, high synthetic accessibility (low synthesis accessibility score), and listed in PubChem.
(3) Capacity: gravimetric capacity for hydrogen storage of 5.5% wt H2 or more, calculated by comparing the molar mass of hydrogenated and dehydrogenated forms (eqn (2)).
(4) Enthalpy: hydrogenation enthalpy values in the range 40–70 kJ per mol H2, as first estimated via our provisional scoring criteria and then confirmed, for promising molecules, by using DFT calculations.
To scrutinize the intersection of these subspaces and identify promising LOHC candidates, we adopted a stepwise approach, starting with the structural similarity subspace (thereby ensuring that our focus was on the most closely related structures) and progressively incorporating additional criteria. This targeted strategy facilitated an efficient exploration of the chemical compound space, revealing the potential for novel LOHC discovery. One of the crucial bottlenecks in screening large chemical databases (billions of molecules) is data management and the innovation we adopted to address this is described below.
The massive scale of the GDB-17 database, containing 1.66 × 1011 organic molecules, created significant challenges in identifying structurally similar compounds based on Tanimoto similarity coefficients. Our matching algorithm divides large sets of candidate molecules into smaller chunks and calculates similarity scores between target and candidate molecules using molecular fingerprints. Maintaining a priority queue for storing the best molecules found during the screening process, the algorithm updates the list as new candidates are processed. This parallelized and distributed approach expedites the screening process, reduces computational resource demands, and facilitates accurate identification of structurally similar compounds, ultimately enabling efficient exploration of the chemical compound space and contributing to a deeper understanding of its potential for novel LOHC discovery.
The size of GDB-17 at 4.7 TB makes it too large to store in memory for most computers and the sheer number of molecules requires distributed computing – 1 ms per molecule is more than 5 years of compute time. We created a highly-scalable molecular screening application built on the Colmena98 and Parsl99 parallel computing libraries to enable screening in reasonable timescales. We ran the workflow on 8192 cores of Argonne Leadership Computing Facility (ALCF) Theta supercomputer and achieved an aggregate screening rate of 3 million molecules per second.
We employ several innovations to scan GDB-17 within a reasonable amount of time. The core screening application itself is simple: a steering process submits tasks to score molecules from GDB-17 based on their similarity to our seed set (Bench-93) and continually collects the results into a top list of the most similar. One innovation is to progressively reduce communication overhead by only sending molecules likely to be in the top list. We implement this concept by waiting to submit a batch of molecules to be scored until previous tasks complete and supplying the lowest score of the current top list as a threshold. The cost to send results reduces from a mean of 2.3 s per task for the first 10% of tasks to 0.3 ms per task for the last 10% of tasks, which leads to higher utilization of each compute node through lower time spent on communication. Delayed submission required concurrently submitting new tasks and ranking the molecules from completed tasks, which we implemented by expressing the relationship between submission and processing using Colmena. The strategy employed is deterministic, producing the same list of top molecules regardless of their order in the search space, using a batching method for score computation and a screening step that doesn't affect the rank list but accelerates the process.
Our other innovation is to automatically offload large task inputs or outputs to the filesystem to avoid congestion of networks. Offloading to disk reduces the size of control messages sent by the workflow engine, leading to much better scaling performance.98 Storing the messages on disk also reduces the memory requirements for the steering process. The queue of molecule scores waiting to be added to our top list (which peaked at 449627 tasks during the run) resided on the petabyte-scale global filesystem rather than the small resources available to the steering process. We implemented offloading using ProxyStore,100 which required only adding the storage location and message size threshold to Colmena and no other changes to application logic. A schematic overview of the screening procedure is presented in the ESI (Fig. S4).†
(1) Structure screen: we computed the Tanimoto similarity index92,101 between each of the 1.5 billion molecules in the ZINC15 database and each of the 93 molecules in Bench-93: a total of ∼2 × 1011 similarity computations. We then selected the 1000 ZINC15 molecules with the highest Tanimoto similarity (lower bound cutoff of 0.56) to at least one molecule in Bench-93.
(2) Heuristic enthalpy screen: we applied the provisional screening criteria (eqn (4)) to identify the best 88 of these 1000 molecules, yielding what we term the ZINC-88 set.
(3) DFT-based enthalpy screen: we performed DFT calculations on these 88 molecules to determine their dehydrogenation enthalpies and rejected the 23 of the 88 molecules for which the DFT-computed dehydrogenation enthalpy lies outside the desired range of 40–70 kJ per mol H2. The effectiveness of applying provisional scoring criteria after a Tanimoto similarity search is indicated by the 65 molecules that exhibited dehydrogenation enthalpies within the desired range, successfully filtering out molecules outside this range.
(4) Capacity/practicality screen: finally we further down selected from these 65 LOHC candidates by applying two further requirements: hydrogen storage capacity of at least 5.5% by weight, and ML-predicted melting points, for both the hydrogen-rich and hydrogen-poor forms, of at most 40 °C. (LOHCs are ideally liquid at room temperature, but we considered melting temperatures up to 70 °C for hydrogen-lean LOHCs, which is similar to the melting point of N-ethyl Carbazole.) Note that all molecules in ZINC15 satisfy the synthesizability and PubChem criteria listed above.
Application of these criteria reveals five candidate LOHC molecules that fit all four requirements. Fig. 5 shows the five new candidate LOHC molecules and Table 1 provides a summary of their physicochemical properties.
 | ||
| Fig. 5 Structures of five LOHC candidates identified from the ZINC15 chemical database. Their detailed physicochemical properties are listed in Table 1. | ||
| MP(HL) | MP(HR) | BP(HL) | BP(HR) | ΔHrxn | % wt H2 | |
|---|---|---|---|---|---|---|
| ZINC15-A | 19.6 | 4.8 | 290.7 | 269.0 | 69.5 | 6.2 |
| ZINC15-B | 59.7 | 83.4 | 344.5 | 277.3 | 64.8 | 5.95 |
| ZINC15-C | 38.5 | 6.9 | 265.3 | 213.4 | 70.2 | 5.82 |
| ZINC15-D | 33.7 | 50.0 | 350.2 | 254.6 | 63.4 | 5.67 |
| ZINC15-E | 57.4 | 53.2 | 313.4 | 252.1 | 61.6 | 6.2 |
A choice of a good LOHC candidate is a tradeoff between four requirements: a molecule that can store high quantity of molecular hydrogen, be liquid at room temperature in its hydrogen rich and hydrogen lean forms, has a high boiling point, and be on the lower end of the 40–70 kJ mol−1 enthalpy range (ΔHrxn). The five chosen molecules, shown in Fig. 5, exhibit good physicochemical properties (summarized in Table 1), with ZINC15-A showing the best overall properties. As shown in Table 1, both the H-rich and H-lean forms of ZINC15-A are liquids at room temperature; the molecule can store up to 6H2 molecules, resulting in a gravimetric capacity of 6.2% H2; and its DFT-calculated dehydrogenation enthalpy is 69.5 kJ mol−1. These properties make ZINC15-A a good candidate for a new LOHC system. The four remaining molecules also show promise as candidate LOHC systems, but will require further investigation, given that their predicted melting and boiling points suggest that at least either the hydrogenated or dehydrogenated form is a solid at room temperature.
(1) Structure screen: to screen GDB-17 chemical space of 166 billion molecules, we first employed the Tanimoto similarity index (T) to identify molecules that are structurally like those in Bench-93. Given the larger size of the GDB-17 dataset, we selected the one million with the highest degree of Tanimoto similarity (lower bound cutoff of T is 0.375).
(2) Heuristic enthalpy screen: we then selected from this set the best scoring 2000 molecules based on our provisional scoring criteria. To further include the diversity of the molecular space, we also selected an additional 1000 molecules chosen at random from the top 10000 (1%) of the set of 1 million molecules. We refer to this total of 3000 molecules as the GDB-3000 dataset.
(3) DFT-based enthalpy screen: we next performed DFT computations on the 3000 molecules72 in GDB-3000 to determine their dehydrogenation enthalpies (see Fig. 6), and removed the 940 with calculated enthalpies (ΔH) outside the desired 40–70 kJ mol−1 range.
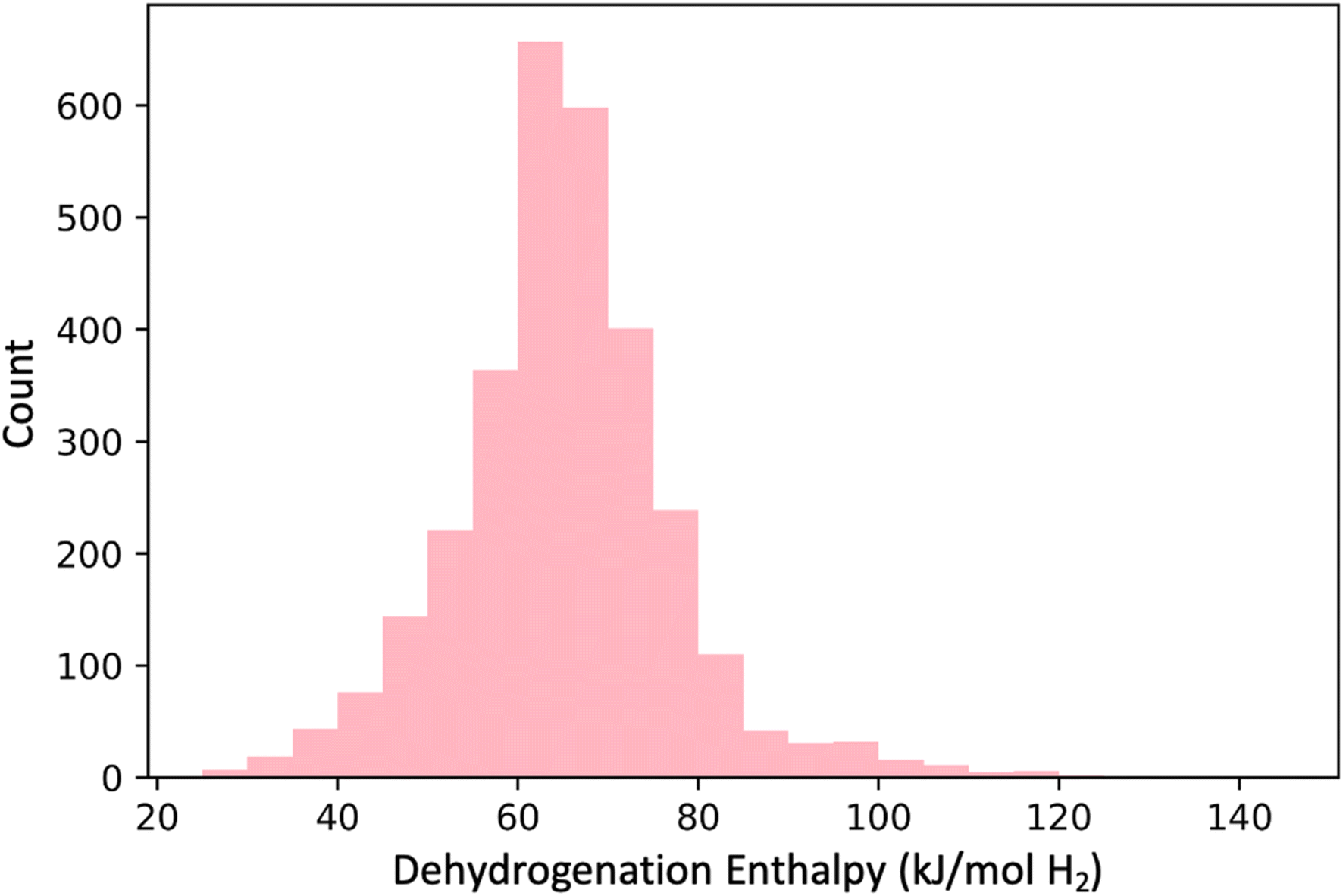 | ||
| Fig. 6 Histogram representation of DFT-calculated dehydrogenation enthalpies (ΔHrxn) for the GDB-3000 dataset. Most molecules fall within the 40–80 kJ mol−1 range, and approximately two-thirds (2060 molecules) are specifically within the desired 40–70 kJ mol−1 range. | ||
(4) Capacity/practicality screen: we remove molecules with a hydrogen storage capacity less than the DOE standard of at least 5.5% by weight, or for which either their hydrogen-rich or hydrogen-lean states has a melting point >40 °C, indicating that it is not liquid at room temperature. Synthetic Accessibility (SA) scores, as computed by the method of Ertl and Schuffenhauer,102 and PubChem database presences are reported for all molecules under investigation (Table 2). A lower SA score implies higher likelihood of a molecule to be synthesized. However, these metrics do not serve as exclusionary parameters in our screening process. This strategy enables us to bypass the conventional restriction of focusing solely on molecules that have already been synthesized or reported. By adopting such an approach, we aim to uncover promising LOHC candidates among molecules that, while theoretically feasible, have not yet been synthesized. Consequently, while SA scores and PubChem database entries serve as useful reference points, they do not limit our exploration for previously not synthesized yet promising LOHC molecules. Those latter steps are important because GDB-17, unlike ZINC15, contains many molecules that are neither commercially available nor even synthesizable. (It is an enumeration of organic molecules of up to 17 heavy atoms containing C, N, O, S, and halogens,71 generated using a combinatorial approach, considering all possible combinations of atoms, bonds, and their properties.)
| Molecule | MP(HL) | MP(HR) | BP(HL) | BP(HR) | SA(HL) | SA(HR) | PC(HL) | PC(HR) | ΔH | % wt H2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Subset A | ||||||||||
| A1 | −39.9 | 12.8 | 197.5 | 181.6 | 1.72 | 3.48 | Yes | Yes | 69.6 | 5.75 |
| A2 | 15.9 | −10.5 | n/a | 252.5 | 2.92 | 2.52 | Yes | Yes | 67.6 | 6.75 |
| A3 | 16.8 | −10.5 | 239.1 | 226.6 | 4.11 | 4.07 | No | No | 63.1 | 6.60 |
| A4 | 23.7 | 25.8 | 211.4 | 160.7 | 2.40 | 4.01 | Yes | Yes | 68.2 | 6.34 |
|
||||||||||
| Subset B | ||||||||||
| B1 | −1.7 | −4.7 | 259.1 | 197.0 | 1.58 | 3.45 | Yes | Yes | 64.1 | 6.58 |
| B2 | 28.8 | 9.2 | 285.2 | 250.1 | 1.92 | 3.35 | Yes | No | 63.0 | 5.63 |
| B3 | 28.8 | 9.1 | 285.2 | 250.1 | 1.95 | 3.35 | Yes | No | 66.1 | 5.63 |
| B4 | 28.8 | 9.1 | n/a | 250.1 | 1.91 | 3.44 | Yes | No | 66.5 | 5.63 |
| B5 | 28.8 | 5.8 | 285.2 | 250.1 | 1.96 | 3.44 | Yes | No | 62.2 | 5.63 |
| B6 | 28.8 | 5.6 | n/a | 250.2 | 1.77 | 3.37 | Yes | No | 66.0 | 5.63 |
| B7 | 28.8 | 5.5 | 287.2 | 250.2 | 1.97 | 3.45 | Yes | No | 67.1 | 5.63 |
| B8 | 28.9 | 5.5 | 287.1 | 250.2 | 1.79 | 3.51 | Yes | No | 65.9 | 5.63 |
| B9 | 28.9 | 5.5 | 287.1 | 250.2 | 1.81 | 3.41 | Yes | No | 66.8 | 5.63 |
| B10 | 28.9 | 14.4 | 287.1 | 250.2 | 1.79 | 3.34 | Yes | No | 61.6 | 5.63 |
|
||||||||||
| Subset C | ||||||||||
| C1 | 17.6 | 2.3 | 307.1 | 261.4 | 1.60 | 3.50 | Yes | No | 64.3 | 6.20 |
| C2 | 17.8 | −2.2 | n/a | 261.4 | 1.71 | 3.64 | Yes | No | 65.4 | 6.20 |
| C3 | 18.4 | 1.8 | 308.0 | n/a | 1.61 | 3.53 | Yes | No | 63.7 | 6.20 |
| C4 | 19.7 | 4.6 | 290.7 | 269.0 | 1.62 | 3.15 | Yes | Yes | 68.5 | 6.20 |
| C5 | 37.7 | 0.4 | 284.8 | 257.5 | 1.48 | 3.31 | Yes | Yes | 67.6 | 6.67 |
|
||||||||||
| Subset D | ||||||||||
| D1 | 20.5 | 176.6 | 207.5 | n/a | 1.59 | 3.41 | Yes | Yes | 49.3 | 5.56 |
| D2 | 78.5 | 10.1 | 342.2 | 261.5 | 2.00 | 4.43 | Yes | No | 58.9 | 6.20 |
| D3 | 74.6 | 2.6 | 320.0 | 241.0 | 1.88 | 3.70 | Yes | No | 59.7 | 6.78 |
| D4 | 52.9 | 13.3 | 291.3 | 247.6 | 1.70 | 3.88 | Yes | Yes | 60.8 | 6.23 |
| D5 | 47.6 | 1.9 | 268.5 | 248.5 | 1.75 | 3.93 | Yes | No | 60.9 | 5.56 |
| D6 | 70.4 | 7.0 | 305.4 | 268.7 | 2.69 | 3.75 | Yes | No | 61.6 | 5.56 |
| D7 | 46.2 | −0.5 | n/a | 257.5 | 1.67 | 3.52 | Yes | No | 61.8 | 5.56 |
| D8 | 74.6 | 3.5 | n/a | 241.1 | 1.96 | 3.56 | Yes | No | 62.2 | 6.78 |
| D9 | 3.6 | 80.7 | 276.2 | 214.8 | 2.66 | 3.22 | Yes | Yes | 62.3 | 6.39 |
| D10 | 44.5 | 6.6 | 292.1 | 258.7 | 1.73 | 3.99 | Yes | No | 66.4 | 6.07 |
| D11 | 44.5 | 6.8 | 303.4 | 262.1 | 1.74 | 3.93 | Yes | No | 66.5 | 5.62 |
| D12 | 44.6 | 9.8 | 304.3 | 263.9 | 1.69 | 3.56 | Yes | No | 66.7 | 5.62 |
| D13 | 57.8 | 0.2 | 262.8 | 223.6 | 1.77 | 3.92 | Yes | Yes | 67.1 | 6.03 |
| D14 | 3.6 | 81.8 | 276.2 | 212.1 | 2.68 | 2.77 | Yes | Yes | 67.2 | 6.39 |
| D15 | 44.2 | 4.9 | 266.7 | 212.1 | 1.74 | 4.13 | Yes | No | 67.3 | 5.89 |
| D16 | 49.8 | 4.0 | 296.5 | 258.6 | 1.66 | 3.81 | Yes | No | 69.0 | 6.07 |
| D17 | 78.1 | 6.4 | 361.9 | 272.6 | 2.11 | 4.03 | No | No | 70.0 | 6.27 |
To identify the best LOHC candidates, we began with the results from 2060 DFT calculations, employing the OPERA model to predict the melting points of the hydrogen-lean molecules. Of these, only 203 molecules displayed melting points below 80 °C, and further, a smaller subset, 113 molecules, demonstrated melting points less than or equal to that of N-ethyl carbazole (70 °C). It is significant to note that these temperatures, albeit lower, still exceed the typical range expected for room temperature liquids. We proceeded with our analysis by employing OPERA to estimate the melting points of the hydrogen-rich counterparts of these 203 molecules. This step aimed to give us a comprehensive picture of the melting point distribution across both hydrogen-rich and -lean molecular pairs. Further, we introduced a stringent threshold of 40 °C, eliminating those pairs where either the hydrogen-lean or -rich molecule exceeded this value. This criterion led to the identification of 19 promising candidate molecules, which we categorized into groups A, B, and C for further discussion. Following this, we revisited the initial pool of 203 molecules, seeking pairs where one molecule (either hydrogen-rich or -lean) met our 40 °C threshold, while its counterpart showed a melting point above 40 °C but below 80 °C. From this reevaluation, we have also identified 17 intriguing molecules that, while not individually satisfying the melting point criteria, hold substantial promise. These molecules, though not ideally tailored for standalone use, can potentially shine in mixed-LOHC systems. This is due to the unique property of mixed materials, where they may exhibit a lower melting point than either of the individual components, provided the experimentally determined molar ratios are optimal. This additional set of 17 molecules expand our pool of promising LOHC candidates to a total of 36. These molecules have been designated as category D, and characteristics of which will be discussed in detail below.
We group the down selected candidate LOHC molecules into four groups, as shown in Fig. 7. Group A consists of new molecules (A1 to A4) that are not like any molecule in the Exp-31 set; group B (B1 to B11) includes substituted quinolines; group C (C1–C5) contains phenyl pyridines; and group D (D1–D17) comprises molecules that do not meet all requirements but could be useful. Selected molecules from each group and their calculated properties (melting points, boiling points, synthetic accessibility) are listed in Table 2. Entries marked with ‘n/a’ signify instances where the OPERA machine learning model was unable to predict the melting and/or boiling point of a molecule. We note, moreover, that even when predictions are made, current machine learning models can exhibit errors of up to 50 °C in estimating melting and boiling points.77,103 In the next sections we discuss these candidate LOHC molecules identified from the GDB-17 database.
 | ||
| Fig. 7 Structural representations of identified LOHC candidates from the GDB-17 database, grouped into four subsets: A (A1–A4), B (B1–B10), C (C1–C5), and D (D1–D17). Each molecule shown here exhibits properties that qualify it as a promising LOHC candidate. | ||
3.2.2.1. New molecules (subset A). We present the first group of molecules (group A) in Fig. 7 and Table 2. All four molecules show excellent LOHC-like physicochemical properties: all are liquid at room temperature, both in their hydrogen-rich and hydrogen-lean states. They all exhibit desired dehydrogenation enthalpy and high gravimetric capacity. The A1 and A4 structures belong to the benzofuran and benzoxazole groups, respectively. They are closed-shell aromatic molecules with minimal geometrical strains. Conversely, the unsaturated carbon radical on A3 and the strained, cumulated double bonds in one of the rings in A2 can both give rise to potential stability issues, making these molecules more prone to side reactions. For these reasons, A1 and A4 are better LOHC candidates from this set of molecules.
3.2.2.2. Substituted quinolines (subset B). The second group of molecules, subset B, shown in Fig. 7, are quinoline derivatives. Note that the methyl quinolines have recently gained attention as potential LOHC molecules due to their good physicochemical properties.104 Results from GDB-17 screening show that a new family of substituted quinolines can make a good LOHC system. Several monosubstituted cyclopropyl quinones were identified with good LOHC-like properties. All shown cyclopropyl groups have good gravimetric capacities (5.6%) and the position of the substituted group has minimal effect on the physicochemical properties of the molecule.
3.2.2.3. Phenyl pyridines (subset C). The Fig. 7 shows a group of substituted phenyl pyridine molecules, denoted as C1–C5. Separately, phenyl groups and pyridine groups were shown to be good LOHC candidate molecules (toluene, N-ethyl carbazole, quinoline). The reported phenyl pyridines show good dehydrogenation enthalpies and have melting points less than 20 °C, except for C5. They also exhibit high gravimetric capacity (>6% wt H2) and high boiling points. The position of the substituted methyl groups has little effect on the molecule's properties but results in Table 2 show that molecules with two substituted methyl groups (or in case of C4 a linker CH2 group) have lower melting points than the singly substituted molecule (C5).
3.2.2.4. Molecules that partially meet criteria (subset D). In this section, we briefly mention some LOHC candidates that partially meet the criteria. These molecules are represented by subset D (D1–D17). We report molecules where their hydrogen-rich and hydrogen-lean molecules have different physical states at room temperature. Fig. 7 shows some of these molecules. All these structures have good hydrogen storage capacity (5.6–6.8%), desired dehydrogenation enthalpies (49.3–70.0 kJ per mol H2), and all except D17 are reported as synthesizable molecules in PubChem. Stark and coworkers45 have successfully demonstrated that modifying the alkyl chain on N-alkyl-carbazoles significantly lowered the melting point of the molecule. They also report that binary and multinary mixtures of different N-alkyl carbazoles could lower the melting point from 68 °C to 12.5 °C. Following up on this successful approach, molecules from subset D can have utility in mixtures or serve as the basis for future candidates, which can attain lower the melting point by modifying the structures through addition of alkyl groups. Designing systems comprising binary or multiple organic molecules necessitates investigating the phase equilibria of the mixtures. The objective is to determine the optimal mole ratio between the components, which leads to a reduced melting point for the integrated system.
The LOHCs identified by Paragian and coworkers73 differ significantly from ours, with no overlap in the molecules discovered. Their 37 identified molecules are mostly benzene or pyridine substituents, or five- and six-membered rings with substituents, highlighting the value of diverse approaches in exploring the chemical compound space. In contrast to our methodology, their approach, which does not use Tanimoto similarity, allowed for the discovery of new and unique molecules. These compounds, predominantly containing N atoms as exocyclic substituents, can be found in the PubChem database. The dehydrogenation pathways and enthalpies further emphasize the unique characteristics of these LOHCs, including the favorable impact of N incorporation on the thermochemistry. This divergence in discovery paths underscores that there is no one way to discover LOHCs; different approaches can lead to different and valuable insights, enriching our collective understanding and expanding the horizons of LOHC research.
3.3. Chemical design principles of LOHCs
To identify chemical design principles, we undertook two distinct data analyses using the GDB-3000 dataset. Initially, we focus on a subset of 700 molecules from the GDB-3000 dataset, specifically those that scored highest (s score of 4.5 or above) according to our provisional scoring criteria. This ranking, or ‘scoring’, reflects the energy changes associated with the reactions, with ‘top-scoring’ reactions possessing enthalpies most aligned with optimal LOHC behavior. By comparing these calculated enthalpies to the Tanimoto similarity, we identified that our preliminary scoring criteria provided a closer correlation with the desired enthalpy range (see Fig. S1†). In this context, ‘trend’ refers to the relationship between calculated enthalpies, Tanimoto similarity, and the suitability of a molecule as an LOHC. Our provisional criteria effectively captured this trend. Subsequently, we extended our focus to include a larger subset of the top-scoring 950 molecules from GDB-3000 based on our provisional scoring criteria. The goal of this expanded analysis was to further refine our understanding of these trends or ‘design principles’, with an aim of enhancing our scoring criteria and developing a comprehensive dehydrogenation index.We do so in three steps: first, we reformulate eqn (4) to calculate  , the dehydrogenation index, shown in eqn (5). Unlike the coarse provisional scoring criteria discussed in earlier section, the dehydrogenation index offers a descriptor-based estimation of a molecule's dehydrogenation enthalpy (eqn (5) and (6)). Second, we optimize the weights of our provisional 11 descriptor set against the 950 dehydrogenation energies, where their
, the dehydrogenation index, shown in eqn (5). Unlike the coarse provisional scoring criteria discussed in earlier section, the dehydrogenation index offers a descriptor-based estimation of a molecule's dehydrogenation enthalpy (eqn (5) and (6)). Second, we optimize the weights of our provisional 11 descriptor set against the 950 dehydrogenation energies, where their 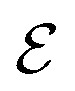 is the DFT-calculated ΔH value scaled by 0.1. By this, multiplying
is the DFT-calculated ΔH value scaled by 0.1. By this, multiplying 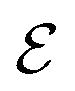 by 10 gives us an approximate value for dehydrogenation enthalpy. Molecules with
by 10 gives us an approximate value for dehydrogenation enthalpy. Molecules with 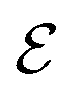 score that fall within the range of 4–7 are considered as good LOHC candidates. Third, we visualize all outliers to identify common molecular features amongst them. These features are molecular structures that affect the destabilization energy and were not captured in the provisional descriptor set. Analyzing the various molecular descriptors versus dehydrogenation energies allows us to identify 21 descriptors (details provided in Table S4†). The weights of the new descriptor set were optimized based on DFT results of 950 dehydrogenation energies scaled by a factor of 0.1.
score that fall within the range of 4–7 are considered as good LOHC candidates. Third, we visualize all outliers to identify common molecular features amongst them. These features are molecular structures that affect the destabilization energy and were not captured in the provisional descriptor set. Analyzing the various molecular descriptors versus dehydrogenation energies allows us to identify 21 descriptors (details provided in Table S4†). The weights of the new descriptor set were optimized based on DFT results of 950 dehydrogenation energies scaled by a factor of 0.1.
 | (5) |
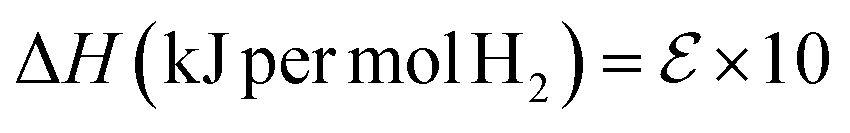 | (6) |
Fig. 8a shows the scaled values of dehydrogenation enthalpies (ΔH × 0.1), labeled as QM Score on the x-axis, versus the computed dehydrogenation index. We see that the proposed dehydrogenation index agrees with the DFT computed QM Score (0.1 × ΔH). This dehydrogenation index clearly allows us to explore a vast chemical space and identify potential LOHCs candidates by providing an approximation to their dehydrogenation energies. Additionally, the chemical descriptors that we used to construct our dehydrogenation index, such as hybridizations of carbons, numbers of carbocyclic and heterocyclic rings, and positions and count of heteroatoms provide key insight to further understand the properties of LOHCs.
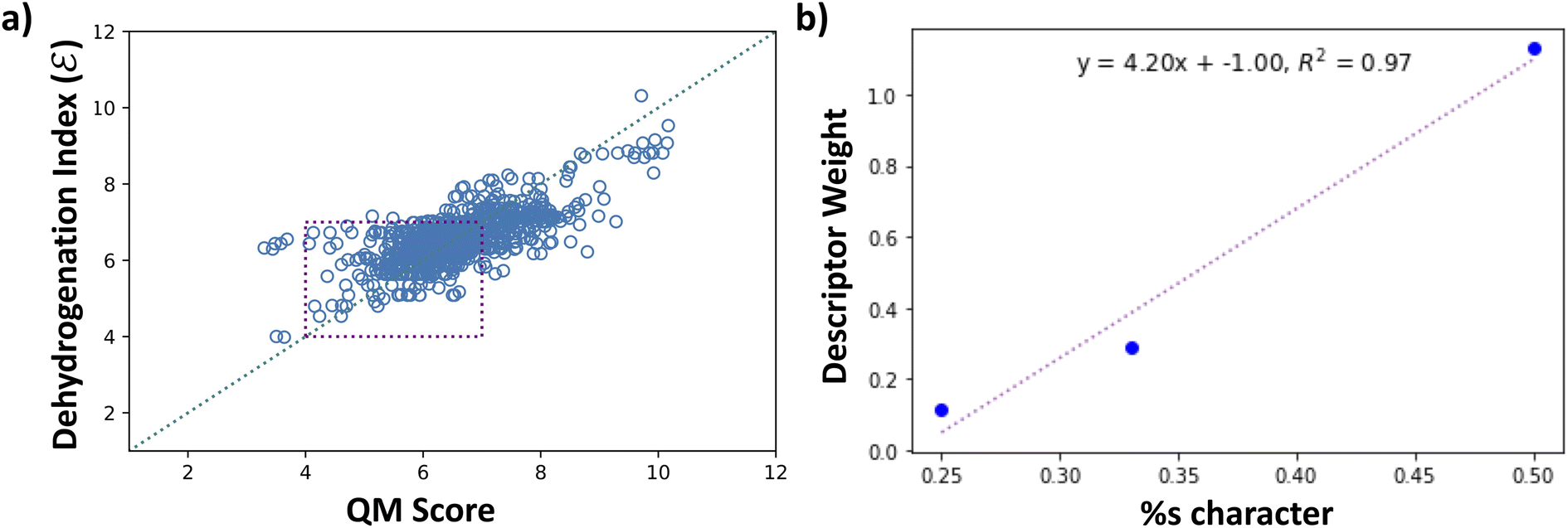 | ||
Fig. 8 (a) Plot of dehydrogenation index 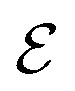 vs. QM score (left). QM scores are the DFT calculated dehydrogenation enthalpies scaled by 0.1. The purple square indicates values that are within the desired range of 4–7. (b) Plot of descriptor weights of sp3, sp2, and sp carbons versus percent s characters, showing a linear correlation between descriptor weight and percent s character of the hybrid orbital. vs. QM score (left). QM scores are the DFT calculated dehydrogenation enthalpies scaled by 0.1. The purple square indicates values that are within the desired range of 4–7. (b) Plot of descriptor weights of sp3, sp2, and sp carbons versus percent s characters, showing a linear correlation between descriptor weight and percent s character of the hybrid orbital. | ||
To understand what makes a good LOHC, we further discuss the relationships between the descriptor weights in Table S4† and the dehydrogenation energies of molecules in our dataset. From Fig. 8a, the computed dehydrogenation index shows good agreement with DFT-computed dehydrogenation enthalpies, with a mean absolute deviation of 0.49 and root-mean squared error of 0.66. These correspond to 4.9 kJ per mol H2 and 6.6 kJ per mol H2, respectively. While the Tanimoto similarity search was vital in exploring the chemical space, it is also essential to implement additional levels of chemical intuition that will serve as metrics to explore larger areas of the chemical space and provide a more fundamental understanding of the properties that characterize a good LOHC, as we will discuss in detail in this section.
A central property in organic molecules is the hybridization of its carbon atoms. The hybridization of the carbon dictates its 3D structure, and the presence of multiple bonds affects the electronic structure and electron density around the carbon centers. The dehydrogenation scoring weights of sp3, sp2, and sp carbons show a positive linear correlation with the percent-s character of the hybridized orbitals, with an R-squared value of 0.97, as shown in Fig. 8b. While sp3 carbons are saturated centers and cannot undergo further hydrogenation, their non-zero weight indicates that the presence of sp3 carbon has an indirect effect on the hydrogenation/dehydrogenation energy. Additionally, both sp2 and sp carbons involve the presence of one or more unsaturated bonds, both of which are potential sites of hydrogen addition. However, due to the weaker bond strength of π bonds relative to σ bonds, the presence of multiple π bonds on the same carbon site implies higher degrees of destabilization. This concept is well established for hydrocarbons: alkynes are thermodynamically less stable than alkenes than the latter is to alkanes – the higher energy product will have too high a dehydrogenation energy. As reflected by the difference in scoring weights of sp and sp2 carbons (1.13 vs. 0.29, respectively), while storing hydrogens in a triple bond implies higher gravimetric capacity, the major disadvantage arises from obtaining a dehydrogenation enthalpy higher than the desired 40–70 kJ per mol H2 range.
For molecules containing one or more monocyclic rings, our scoring system shows significant differences in their scoring weights. Specifically, we have identified four chemical descriptors in this arena: non-5-membered monocycles, 5-membered-containing monocycles, nitrogen-containing monocycles, and sulfur/oxygen containing monocycles. Of these four, the sulfur/oxygen containing ring is the sole descriptor with a positive weight. Five-membered rings that contain N, S, and O (pyrroles, thiophenes, and furans, respectively) are all aromatic heterocycles. However, while aromaticity in all three rings involves lone pairs on the hetero atoms, thiophenes and furans bear an additional lone pair on the sulfur and oxygen atoms, respectively. The presence of the additional lone pair switches the direction of the dipole moment on these rings, where instead of having the positive end lying on the heteroatom as in pyrroles, the sulfur and oxygen centers on thiophenes and furans hold the negative end of the dipole. From this, we conclude that the position and orientation of the dipole moment in relation to the heteroatom on 5-membered rings is significant in determining the impact of five-membered substructures on the enthalpy of dehydrogenation of the target molecule.
The effect of 5- versus 6-membered rings on dehydrogenation enthalpy can be understood through the perspective of angular strains. In both rings, the aromatic carbons are sp2 hybridized. The simplest 5-membered carbocycle, cyclopentadiene, has a C–C–C angle of 103°. On the other hand, the well-known 6-membered carbocycle, benzene, has a perfect hexagonal shape with bond angles of 120°. This shows that the angular strains in 5-membered rings are higher than those in 6-membered rings, where the “ideal” bond angle for sp2 carbons is 120°. Hydrogenation of the double bonds in both structures will permit the relaxation of these angles and the breaking of planarity of the rings. Thus, the reasoning behind obtaining a higher “negative” score for 5-membered rings may be the higher relative stability of their hydrogenated forms than the 6-membered rings analogues.
Compared to monocyclic fragments, bicyclic compounds have higher scoring weight. While monocyclic fragments decrease the dehydrogenation enthalpy by around 12 kJ mol−1 (for non-5-membered rings) and 16 kJ mol−1 (for 5-membered rings), bicyclic fragments have an opposite effect. Our scoring system shows that bicyclic structures increase the enthalpy by around 50 kJ mol−1. In comparing these scoring weights, we can see that the number of rings plays a major role in predicting the dehydrogenation enthalpies of organic molecules. Going further beyond bicyclic rings, our scoring system shows that molecules containing 3 or more rings have smaller weights than bicyclic molecules, with polycyclic rings containing a 5-membered ring adding around 35 kJ mol−1 to the dehydrogenation enthalpy, while non-5 membered ring polycycles only contribute 10 kJ mol−1 to the dehydrogenation enthalpy. Nevertheless, molecules containing one bicyclic or polycyclic fragment can better constitute a good LOHC than a monocyclic-containing molecules.
As seen with monocyclic rings, the presence of nitrogen, sulfur, and oxygen rings also results in interesting effects on dehydrogenation enthalpies in bicyclic and polycyclic rings. Nitrogen-containing bicyclic compounds decrease the dehydrogenation enthalpy by ∼3 kJ mol−1 while sulfur- and oxygen-containing bicycles have little effect on the enthalpy. In contrast, nitrogen-containing polycyclic rings have an opposite effect on the molecule's dehydrogenation enthalpy. Based on our models, a presence of a nitrogen atom in a polycyclic ring increases the enthalpy of dehydrogenation by 5 kcal mol−1. The presence of sulfur and oxygen atoms has little effect on the dehydrogenation enthalpies of polycycles.
While the number of sulfur and oxygen atoms has little or no effect on the dehydrogenation enthalpies, their major contribution arises from scoring weights for their atomic positions within bicyclic and polycyclic rings. The sulfur and oxygen atoms have two lone pairs, only one of which participates in the aromatic stabilization via delocalization, while the additional lone pair affects the polarity of the molecule. The position of this lone pair relative to the ring has the capacity to affect the strength and direction of the dipole moment of the whole polycyclic molecular fragment. This effect is reflected in our scoring criteria as the weights of sulfur- and oxygen-containing rings are dependent on the positions of the heteroatoms within the rings: entries “SObadpos” and “SO_adjrng” in Table S4.†
Our scoring criteria also detected additional properties. Aromatic ring substituents in general have little effect on dehydrogenation enthalpy, with the highest effect being less than 3 kJ mol−1 for substituents at 1,3 positions. This result can be interpreted as a balanced trade-off between the electronic effects that these substituents have on dehydrogenation enthalpies, on the one hand, and their incapability of binding hydrogens due to their saturated structures, on the other. Another property that we identified is the presence of a 3-membered ring fused to another ring, with our scoring criteria predicting that it increases the dehydrogenation enthalpy by around 40 kJ mol−1. This property is due to the high reactivity of these fragments arising from their angular strains where the fully hydrogenated carbons will then have an angle closer to the desired sp3 bonds.
Analysis of the scoring criteria provides us with a deeper understanding of the chemical properties that shape good LOHCs. The initial step of our screening process, which utilizes the Tanimoto similarity index on ZINC15 and GDB-17 databases, assumes that structural similarity correlates with dehydrogenation energy. However, the scoring system focuses in on specific interactions that influence the dehydrogenation enthalpies of organic molecules. An understanding of the structural–property relationships in LOHC systems provides the means to effectively evaluate potential candidate molecules. The values assigned to the molecular groups in the scoring system, based on DFT calculations, represent a broad range of electronic interactions, including electron delocalization, atom hybridizations, molecular polarity, ring aromaticity, and geometric strains.
The chemical design principles developed in this study are grounded in thermodynamic data derived from DFT calculations. While this approach provides valuable insights into the properties and behavior of LOHCs, it is important to recognize that these principles are not comprehensive.14,51,59,60 Future work in this area could benefit from a more nuanced exploration that includes the development of scoring methods for reactivity and reaction pathways, kinetic analysis, examination of catalysis, stability assessments of intermediates, and careful consideration of side reactions. For example, while activated –CH2 groups may be thermodynamically favorable, they could be prone to oxidation side reactions.105 Similarly, halogens, though viable in some contexts, may be susceptible to side reactions (e.g. elimination reactions) under high heat and pressure conditions.105 For these reasons, careful consideration should be taken with further studies on molecules B2–B10 to prevent degradation of the cyclopropyl substituent. Additionally, given these complexities and potential susceptibilities to side reactions, we can also exclude halogen-substituted molecules, specifically D10, D11, D12, D15, and D17 from GDB-3000, as well as ZINC15-C and ZINC15-D from ZINC-88, from further consideration in future studies. This aligns with our aim to identify only the most promising and robust LOHC candidates. Future work may further investigate these exclusions with detailed kinetic studies and stability assessments, ensuring that our approach remains aligned with practical applications and real-world conditions. These complexities underline the need for a multi-faceted approach that considers not just thermodynamics but also the broader chemical behavior and interactions within the system. By embracing these additional dimensions, we can further refine our understanding of LOHCs and create more robust and reliable design guidelines that account for not only thermodynamic constraints, but also kinetic constraints, environmental conditions, and the interplay of various chemical reactions. By integrating these diverse factors, we can craft LOHCs that not only meet theoretical expectations but also demonstrate practical viability and resilience in real-world applications.
In the quest to identify prime LOHC candidates, it is vital to address the known drawbacks of existing LOHCs, some of which are susceptible to chemical degradation and side reactions, particularly dealkylation during the hydrogenation/dehydrogenation stepwise reactions. One approach to avoid this issue is to promote candidate LOHCs that do not possess long chains. From our selection, three molecules emerged as the most promising candidates: ZINC15-E (9-hydroxycarbazole), A1 (2-methyl-1-benzofuran), and A4 (2,1-benzoxazole). The appeal of these candidates is further augmented by the fact that they are readily available for purchase from chemical vendors. While there has been prior research into the hydrogenation of some of these compounds, such as 2-methyl-1-benzofuran,106,107 the methods have yet to be perfected. Our work strives to build upon these foundations to advance the development of these promising LOHCs. The synthesis, scalability, and catalytic processes of these identified LOHC candidates remain areas for further investigation. Analyzing the feasibility and reversibility of these compounds will be essential for their successful deployment in practical applications. Such analyses will involve assessing potential methods for synthesizing the compounds on a large scale, as well as evaluating the reversibility of the hydrogenation/dehydrogenation processes.
4 Conclusion
In this manuscript, we have successfully utilized in silico-based discovery approaches to accelerate the search for suitable LOHC candidates from billions of molecular space. Our approach utilizes prior experimental studies and data-driven approaches to identify new LOHC candidates. Importantly, our rigorous screening and down selecting process identified 5 promising molecules from the ZINC15 dataset (1.5 billion molecules) and 36 within the GDB-17 dataset (166 billion molecules) dataset that have potential to operate effectively as LOHC systems.The screening protocol steps through four considerations. The first searches for Tanimoto similarity, a structural similarity evaluation, to a set of 31 molecules that are known. To evaluate this similarity across both ZINC15 and GDB-17 databases, we develop a highly parallelized screening application, which utilized ALCF's Theta supercomputer, and has been made readily available on GitHub. As a second step in the screening protocol, we develop a LOHC selection criteria that connects the molecular features with the dehydrogenation enthalpy, a critical parameter for LOHC performance. We validate, with DFT computations, that this scoring criterion can predict if dehydrogenation enthalpies of 3000 organic molecules will fall within the accepted 40–70 kJ per mol H2 range. Moreover, because it requires only SMILES strings as inputs, the scoring criterion provides a more streamlined and efficient screening process for LOHC selection. The third step of the screening protocol directly computes, with DFT methods, the dehydrogenation energy of each molecule that has passed through the previous screening steps. For 2125 molecules with dehydrogenation energies within the accepted range, we calculated their SA scores to predict synthetic accessibility and utilized the OPERA model to determine their melting and boiling points.
This investigation demonstrates a successful approach for the discovery of new materials with tailored properties, enabling researchers to achieve high-throughput discovery in a wide range of applications. Our future investigations will investigate catalysis including estimating activation barriers and identifying suitable catalysts for both the hydrogenation and dehydrogenation reactions. In this scope, future work can make use of newly developed machine learning models, such as ALFABET, to efficiently predict energetics of elementary reaction steps and implementation of machine learning models for catalysis screening.108–110 This research highlights the power of computational approaches in navigating the vast chemical compound space for viable LOHC molecules. We innovatively tackled the enormity of the GDB-17 database by employing a targeted strategy, high-performance molecular screening application, and distributed computing. These tactics facilitated a comprehensive understanding of the compound space, unearthing novel LOHC candidates and exemplifying the potential of integrating quantum chemistry and cheminformatics in materials discovery. This study will serve as a valuable resource for researchers and engineers working to develop advanced LOHC systems. Additionally, we anticipate that our results will inspire further innovation in the field of materials discovery by combining cutting-edge technologies to address major challenges in the quest for more efficient and sustainable energy storage solutions.
Data availability
DFT-calculated dehydrogenation enthalpies of GDB-3000 and ZINC-88 are publicly available on GitHub: https://github.com/HydrogenStorage/LOHC. Details on the different datasets generated in this study and descriptors involved in the dehydrogenation scoring criteria, and experimental and benchmark datasets are provided in the ESI.† Python scripts for finding similar molecules within GDB-17 or other large datasets can be found on GitHub: https://github.com/HydrogenStorage/screening-large-databases.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This material is based upon work supported by Laboratory Directed Research and Development (LDRD) funding from Argonne National Laboratory, provided by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-06CH11357. We acknowledge the computing resources provided on “BEBOP”, a computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory (ANL). This research used resources of the Argonne Leadership Computing Facility (ALCF); a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357.References
- Glasgow Climate Pact, https://unfccc.int/process-and-meetings/the-paris-agreement/the-glasgow-climate-pact-key-outcomes-from-cop26, accessed 7 November 2022 Search PubMed.
- Roadmap, 2050, https://www.roadmap2050.eu/, accessed 7 November 2022 Search PubMed.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 CrossRef CAS.
- J. Meng, F. Zhou, H. Ma, X. Yuan, Y. Wang and J. Zhang, Top. Catal., 2021, 64, 509–520 CrossRef CAS.
- D. Wei, X. Shi, R. Qu, K. Junge, H. Junge and M. Beller, ACS Energy Lett., 2022, 7, 3734–3752 CrossRef CAS.
- Energy Earthshots Initiative, https://www.energy.gov/policy/energy-earthshots-initiative, accessed 4 January 2023 Search PubMed.
- Hydrogen Shot, https://www.energy.gov/eere/fuelcells/hydrogen-shot, accessed 3 April 2023 Search PubMed.
- J. W. Makepeace, T. He, C. Weidenthaler, T. R. Jensen, F. Chang, T. Vegge, P. Ngene, Y. Kojima, P. E. de Jongh, P. Chen and W. I. F. David, Int. J. Hydrogen Energy, 2019, 44, 7746–7767 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- M. Markiewicz, Y. Q. Zhang, A. Bösmann, N. Brückner, J. Thöming, P. Wasserscheid and S. Stolte, Energy Environ. Sci., 2015, 8, 1035–1045 RSC.
- T. M. Narayanan, G. He, E. Gençer, Y. Shao-Horn and D. S. Mallapragada, ACS Sustain. Chem. Eng., 2022, 10, 10768–10780 CrossRef CAS.
- P. Modisha and D. Bessarabov, in Industrial Arene Chemistry, ed. J. Mortier, Wiley, 1st edn, 2023, pp. 1037–1065 Search PubMed.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC.
- M. Niermann, S. Timmerberg, S. Drünert and M. Kaltschmitt, Renewable Sustainable Energy Rev., 2021, 135, 110171 CrossRef CAS.
- T. He, Q. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- F. Valentini, A. Marrocchi and L. Vaccaro, Adv. Energy Mater., 2022, 12, 2103362 CrossRef CAS.
- P. T. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef CAS.
- P. Modisha and D. Bessarabov, Curr. Opin. Green Sustainable Chem., 2023, 42, 100820 CrossRef CAS.
- D. Teichmann, K. Stark, K. Müller, G. Zöttl, P. Wasserscheid and W. Arlt, Energy Environ. Sci., 2012, 5, 9044 RSC.
- D. Teichmann, W. Arlt and P. Wasserscheid, Int. J. Hydrogen Energy, 2012, 37, 18118–18132 CrossRef CAS.
- E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231–2233 RSC.
- H. W. Langmi, N. Engelbrecht, P. M. Modisha and D. Bessarabov, in Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2022, pp. 455–486 Search PubMed.
- DOE Technical Targets for Onboard Hydrogen Storage for Light-Duty Vehicles, https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles, accessed 27 March 2023 Search PubMed.
- P. C. Rao and M. Yoon, Energies, 2020, 13, 6040 CrossRef CAS.
- D. Dean, B. Davis and P. G. Jessop, New J. Chem., 2011, 35, 417–422 RSC.
- L. Schlapbach and A. Züttel, in Materials for Sustainable Energy, Co-Published with Macmillan Publishers Ltd, UK, 2010, pp. 265–270 Search PubMed.
- N. Kariya, A. Fukuoka and M. Ichikawa, Appl. Catal., A, 2002, 233, 91–102 CrossRef CAS.
- Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef CAS.
- G. Zhu, B. Yang and S. Wang, Int. J. Hydrogen Energy, 2011, 36, 13603–13613 CrossRef CAS.
- A. A. Shukla, P. V. Gosavi, J. V. Pande, V. P. Kumar, K. V. R. Chary and R. B. Biniwale, Int. J. Hydrogen Energy, 2010, 35, 4020–4026 CrossRef CAS.
- A. Gora, D. A. P. Tanaka, F. Mizukami and T. M. Suzuki, Chem. Lett., 2006, 35, 1372–1373 CrossRef CAS.
- M. Taube, D. Rippin, W. Knecht, D. Hakimifard, B. Milisavljevic and N. Gruenenfelder, Int. J. Hydrogen Energy, 1985, 10, 595–599 CrossRef CAS.
- What is “SPERA HYDROGEN” system?, https://www.chiyodacorp.com/en/service/spera-hydrogen/innovations/, accessed 5 January 2023 Search PubMed.
- R. H. Crabtree, ACS Sustain. Chem. Eng., 2017, 5, 4491–4498 CrossRef CAS.
- Hydrogen storage by reversible hydrogenation of pi-conjugated substrates (Patent) | OSTI.GOV, https://www.osti.gov/biblio/1531566, accessed 5 January 2023 Search PubMed.
- A. Søgaard, M. Scheuermeyer, A. Bösmann, P. Wasserscheid and A. Riisager, Chem. Commun., 2019, 55, 2046–2049 RSC.
- H. Adkins and H. L. Coonradt, J. Am. Chem. Soc., 1941, 63, 1563–1570 CrossRef CAS.
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed.
- M. Niermann, A. Beckendorff, M. Kaltschmitt and K. Bonhoff, Int. J. Hydrogen Energy, 2019, 44, 6631–6654 CrossRef CAS.
- P. Linstrom, 1997.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134 RSC.
- K. Stark, P. Keil, S. Schug, K. Müller, P. Wasserscheid and W. Arlt, J. Chem. Eng. Data, 2016, 61, 1441–1448 CrossRef CAS.
- C. Gleichweit, M. Amende, S. Schernich, W. Zhao, M. P. A. Lorenz, O. Höfert, N. Brückner, P. Wasserscheid, J. Libuda, H.-P. Steinrück and C. Papp, ChemSusChem, 2013, 6, 974–977 CrossRef CAS PubMed.
- M. Markiewicz, Y.-Q. Zhang, M. T. Empl, M. Lykaki, J. Thöming, P. Steinberg and S. Stolte, Energy Environ. Sci., 2019, 12, 366–383 RSC.
- M. Yang, C. Han, G. Ni, J. Wu and H. Cheng, Int. J. Hydrogen Energy, 2012, 37, 12839–12845 CrossRef CAS.
- J. P. Viteri, S. Viteri, C. Alvarez-Vasco and F. Henao, Int. J. Hydrogen Energy, 2023, 48, 19751–19771 CrossRef CAS.
- A. X. Yee Mah, W. S. Ho, M. H. Hassim, H. Hashim, P. Y. Liew and Z. A. Muis, Energy, 2021, 218, 119475 CrossRef CAS.
- P. Perreault, L. Van Hoecke, H. Pourfallah, N. B. Kummamuru, C.-R. Boruntea and P. Preuster, Curr. Opin. Green Sustainable Chem., 2023, 41, 100836 CrossRef CAS.
- T. Q. Hua and R. K. Ahluwalia, Int. J. Hydrogen Energy, 2012, 37, 14382–14392 CrossRef CAS.
- T. Kobayashi and H. Takahashi, Energy Fuels, 2004, 18, 285–286 CrossRef CAS.
- S. Saxena, S. Kumar and V. Drozd, Int. J. Hydrogen Energy, 2011, 36, 4366–4369 CrossRef CAS.
- B. Loges, A. Boddien, F. Gärtner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS.
- G. Vishwakarma and J. Hachmann, Liquid Organic Hydrogen Carriers: High-throughput Screening of Homogeneous Catalysts, ChemRxiv., Cambridge Open Engage, Cambridge, 2023, DOI:10.26434/chemrxiv-2023-s8pkf.
- T. Zhang, J. Uratani, Y. Huang, L. Xu, S. Griffiths and Y. Ding, Renewable Sustainable Energy Rev., 2023, 176, 113204 CrossRef CAS.
- O. Lebedeva, D. Kultin, A. Kalenchuk and L. Kustov, Curr. Opin. Electrochem., 2023, 38, 101207 CrossRef CAS.
- C. Chu, K. Wu, B. Luo, Q. Cao and H. Zhang, Carbon Resour. Convers., 2023, 6(6), 334–351 CrossRef CAS.
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen, Z. Xiao, S. Lu, Y. Li and K. Sun, Sci. Adv., 2019, 5, eaay4275 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- Y. Cao, C. T. Ser, M. Skreta, K. Jorner, N. Kusanda and A. Aspuru-Guzik, Nat. Mach. Intell., 2022, 4, 667–668 CrossRef.
- M. Seifrid, R. Pollice, A. Aguilar-Granda, Z. Morgan Chan, K. Hotta, C. T. Ser, J. Vestfrid, T. C. Wu and A. Aspuru-Guzik, Acc. Chem. Res., 2022, 55, 2454–2466 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, J. Chem. Theory Comput., 2015, 11, 2087–2096 CrossRef CAS PubMed.
- N. Dandu, L. Ward, R. S. Assary, P. C. Redfern, B. Narayanan, I. T. Foster and L. A. Curtiss, J. Phys. Chem. A, 2020, 124, 5804–5811 CrossRef CAS PubMed.
- B. Huang and O. A. von Lilienfeld, Chem. Rev., 2021, 121, 10001–10036 CrossRef CAS PubMed.
- A. C. Mater and M. L. Coote, J. Chem. Inf. Model., 2019, 59, 2545–2559 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed.
- Data available at: https://github.com/HydrogenStorage/LOHC.
- K. Paragian, B. Li, M. Massino and S. Rangarajan, Mol. Syst. Des. Eng., 2020, 5, 1658–1670 RSC.
- S. Rangarajan, A. Bhan and P. Daoutidis, Comput. Chem. Eng., 2012, 45, 114–123 CrossRef CAS.
- S. Rangarajan, A. Bhan and P. Daoutidis, Comput. Chem. Eng., 2012, 46, 141–152 CrossRef CAS.
- S. Rangarajan, T. Kaminski, E. Van Wyk, A. Bhan and P. Daoutidis, Comput. Chem. Eng., 2014, 64, 124–137 CrossRef CAS.
- K. Mansouri, C. M. Grulke, R. S. Judson and A. J. Williams, J. Cheminf., 2018, 10, 10 Search PubMed.
- RDKit, https://www.rdkit.org/, accessed 5 January 2023 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- J. Pedersen and K. V. Mikkelsen, RSC Adv., 2022, 12, 2830–2842 RSC.
- B. Narayanan, P. C. Redfern, R. S. Assary and L. A. Curtiss, Chem. Sci., 2019, 10, 7449–7455 RSC.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. Bursch, J. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61(42), e202205735 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, in Theory and Applications of Computational Chemistry, Elsevier, 2005, pp. 195–249 Search PubMed.
- Leruli, https://www.leruli.com/, accessed 13 March 2023 Search PubMed.
- US EPA, EPI Suite™-Estimation Program Interface, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface, accessed 16 August 2023 Search PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- T. T. Tanimoto, An Elementary Mathematical Theory of Classification and Prediction, 1958 Search PubMed.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 CrossRef CAS.
- R. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33, 360–365 CrossRef CAS.
- M. E. Konnova, S. Li, A. Bösmann, K. Müller, P. Wasserscheid, I. V. Andreeva, V. V. Turovtzev, D. H. Zaitsau, A. A. Pimerzin and S. P. Verevkin, Ind. Eng. Chem. Res., 2020, 59, 20539–20550 CrossRef CAS.
- Y. Cui, S. Kwok, A. Bucholtz, B. Davis, R. A. Whitney and P. G. Jessop, New J. Chem., 2008, 32, 1027 RSC.
- L. Ward, G. Sivaraman, J. G. Pauloski, Y. Babuji, R. Chard, N. Dandu, P. C. Redfern, R. S. Assary, K. Chard, L. A. Curtiss, R. Thakur and I. Foster, in 2021 IEEE/ACM Workshop on Machine Learning in High Performance Computing Environments (MLHPC), IEEE, St. Louis, MO, USA, 2021, pp. 9–20 Search PubMed.
- Y. Babuji, A. Woodard, Z. Li, D. S. Katz, B. Clifford, R. Kumar, L. Lacinski, R. Chard, J. M. Wozniak, I. Foster, M. Wilde and K. Chard, in Proceedings of the 28th International Symposium on High-Performance Parallel and Distributed Computing, ACM, Phoenix AZ USA, 2019, pp. 25–36 Search PubMed.
- J. G. Pauloski, V. Hayot-Sasson, L. Ward, N. Hudson, C. Sabino, M. Baughman, K. Chard and I. Foster, Accelerating Communications in Federated Applications with Transparent Object Proxies, arXiv, 2023, preprint, arXiv:2305.09593, DOI:10.48550/arXiv.2305.09593.
- X. Chen and C. H. Reynolds, J. Chem. Inf. Comput. Sci., 2002, 42, 1407–1414 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
- G. Sivaraman, N. E. Jackson, B. Sanchez-Lengeling, Á. Vázquez-Mayagoitia, A. Aspuru-Guzik, V. Vishwanath and J. J. de Pablo, Mach. Learn.: Sci. Technol., 2020, 1, 025015 Search PubMed.
- S. P. Safronov, S. V. Vostrikov, A. A. Samarov and S. P. Verevkin, Fuel, 2022, 317, 123501 CrossRef CAS.
- D. R. Klein, Organic chemistry, Wiley, Hoboken, NJ, 4th edn, 2021 Search PubMed.
- É. A. Karakhanov and E. A. Viktorova, Chem. Heterocycl. Compd., 1976, 12, 367–375 CrossRef.
- P. C. St. John, Y. Guan, Y. Kim, S. Kim and R. S. Paton, Nat. Commun., 2020, 11, 2328 CrossRef PubMed.
- L. Davy, J. Castro, R. Wessels, M. Rey-Bayle, I. Merdrignac and B. Celse, Ind. Eng. Chem. Res., 2020, 59, 21133–21143 CrossRef CAS.
- W. Yang, T. T. Fidelis and W.-H. Sun, ACS Omega, 2020, 5, 83–88 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00123g |
| This journal is © The Royal Society of Chemistry 2023 |