
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning-augmented docking. 1. CYP inhibition prediction†
Benjamin
Weiser
 *a,
Jérôme
Genzling
a,
Mihai
Burai-Patrascu‡
a,
Ophélie
Rostaing‡
a and
Nicolas
Moitessier
*ab
*a,
Jérôme
Genzling
a,
Mihai
Burai-Patrascu‡
a,
Ophélie
Rostaing‡
a and
Nicolas
Moitessier
*ab
aMcGill University, Montreal, Canada. E-mail: nicolas.moitessier@mcgill.ca
bMolecular Forecaster Inc., Montreal, Canada
First published on 9th October 2023
Abstract
A significant portion of the oxidative metabolism carried out by the human body is accomplished by six cytochrome P450 (CYP) enzymes. The binding of small molecules to these enzymes affects drug activity and half-life. Additionally, the inhibition or induction of a CYP isoform by a drug can lead to drug–drug interactions, which in turn can lead to toxicity. To predict CYP inhibition, a variety of computational methods have been used, with docking methods being less accurate than machine learning (ML) methods. However, the latter methods are sensitive to training data and show reduced accuracy on test sets outside of the chemical space represented in the training set. In contrast, docking methods do not have this generalization issue and allow for visual analysis. We hypothesize that combining ML methods with docking can improve CYP inhibition predictions. To test this hypothesis, we pair our in-house docking program FITTED with several ML techniques to investigate the accuracy and transferability of this hybrid methodology, which we term ML-augmented docking. We find that ML-augmented docking can significantly improve the accuracy of docking software while consistently surpassing the performance of ligand-only models. Additionally, we show that ML-augmented docking is more generalizable than machine learning models trained on ligand-only data. The open-source code created for this project can be found at https://github.com/MoitessierLab/ML-augmented-docking-CYP-inhibition.
1 Introduction
1.1 Cytochrome P450s and adverse drug reactions
The majority of administered drugs are metabolized by the liver to be more efficiently excreted from the human body. In Phase I metabolism, the molecules are modified by a set of enzymes, primarily oxidases (e.g., cytochrome P450 enzymes, or CYPs) and hydrolases. Out of this set of oxidases, six isoforms (CYP1A2, 2C9, 2C19, 2D6, 2E1, and 3A4), expressed mainly in the liver and in the gut, are responsible for more than 90% of this oxidative metabolism and represent the main focus of medicinal chemists and pharmacologists.1–3 As a result, the activity of these enzymes is key to the half-life of drugs; more active or increased concentrations of CYPs (CYP Induction) and increased metabolism can lead to lower drug half-lives and reduced drug efficacy; less active CYPs (CYP Inhibition) and decreased metabolism can lead to extended activity, accumulation, and toxicity.It is well known that adverse drug reactions (ADRs) and toxicity are major causes of the high attrition rates observed in drug discovery programs. ADRs are the 4th leading cause of death in the US.4 A common cause of ADRs are drug–drug interactions (DDIs). More specifically, the co-administration of drugs may result in DDIs due to the ability of a drug to inhibit a CYP isoform involved in the metabolism of another drug (CYP inhibition) or to induce the biosynthesis of CYPs (CYP induction).
CYP inhibition can either be reversible, quasi-irreversible or irreversible, with the most prevalent form being reversible inhibition.5 Reversible inhibition primarily occurs when a ligand (termed Type II ligand) coordinates to the heme iron of a CYP isoform. Type II ligands often contain one or more basic nitrogen atoms with an available lone pair that can coordinate with the heme.6 In docking-based methods, the proper description of heme-nitrogen coordination is paramount for identifying whether a compound can be a CYP inhibitor or not. To this end, we have recently published our efforts to accurately describe the heme-nitrogen coordination in FITTED.7
1.2 Methods to predict CYP metabolism and inhibition
While there are several medium throughput techniques available to predict CYP metabolism and inhibition, access to higher throughput techniques would enable medicinal chemists to make more informed decisions at the early stages of the drug design and development process.8–11 For instance, predicting the site of metabolism (SoM), the binding mode of small molecules to CYPs, and the inhibitory activity of drugs and their metabolites could be useful to (1) flag potential in vitro hits, (2) help prioritize experiments, (3) provide key insights enabling the design of compounds with a modulated half-life, (4) predict potentially toxic metabolites, (5) predict potential CYP inhibitors or even (6) predict the effect of CYP polymorphism (i.e., inter-individual variability).12 One approach for predicting CYP SoMs is IMPACTS (In silico Metabolism Prediction by Activated Cytochromes and Transition States),13 a fully automated program we developed, which combines molecular docking, ligand reactivity estimation and transition state (TS) structure prediction to predict the SoMs of drugs metabolized by CYPs. While IMPACTS has been shown to accurately predict the SoMs of a variety of drugs, it is unable to predict whether a drug or its metabolites, can inhibit CYPs or not.Research groups proposed to use of ML techniques to tackle the protein/ligand scoring problem in general.14,15 These efforts resulted in various ML-based scoring functions integrating neural networks (e.g., NNScore,16 DeepVS,17 DLScore and CNN-based methods18,19), Random Forest (RF-Score20), support vector machines (SVR-EP,21,22 an optimized function for eHiTS23 and PLEIC-SVM, a classifier based on protein-ligand interaction maps24) and gradient boosting (GBDT: EIC-Score25). Among the most successful approaches is KDEEP, which uses a 3D convolutional neural network (CNN) with protein–ligand complexes represented as 3D grids labelled as hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), aromatic, hydrophobic, positive ionizable, negative ionizable, metallic and excluded volumes.18 A similar approach using more atom types encoded in different channels of the 3D grids (as are the 3 colours RGB in 2D images) was also disclosed.19
Docking methods and ML techniques have been used to predict CYP inhibition. However, docking scores are still poorly correlated with the binding affinity to proteins and this is especially true for CYP enzymes due to the heme group and shape. In general, the lack of high-quality CYP ligand data has been a major hurdle.14 Docking to CYPs with available programs (AutoDock, FlexX and GOLD) was evaluated for pose prediction in terms of root-mean-square deviation, but only 19 crystallized compounds and only pose prediction was assessed.7,26
Several machine learning (ML) methods have also been reported for the prediction of CYP inhibition, including CYPlebrity by Kirchmair and co-workers.27 While these models are developed from inhibitors and non-inhibitors of CYPs, distinct isoform sets enable the implicit incorporation of the isoform under evaluation. CYPlebrity, as well as other ML-based methods,28–31 have been trained using the same datasets for five major CYP isoforms (CYP1A2, 2C9, 2C19, 2D6 and 3A4). These sets are extracted from the AID1851 bioassay (PubChem assay identifier), which contains CYP inhibition data for ≈17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules. For these models, molecules were encoded with various molecular descriptors and fingerprints including Morgan3,27 GraphOnly,31 Klekota-Roth (PaDEL32),31 fingerprints made of pairs of Extended Connectivity Fingerprint (ECFP), ECFP8,33,34 MACCS, and PubChem35 fingerprints,31 MOE31,36 and PaDEL32 descriptors.30 ML techniques including random forests (RF),28,31 naïve Bayesian method,33 eXtreme gradient boosting (XGB),31 support vector machines,29 to multitask deep neural networks (DNN)30 and k-nearest neighbours (KNN)37 have been used. All of these reports resulted in good accuracy on their respective test sets, but as pass/fail classifiers, they provide little information to medicinal chemists on the possible modifications that would reduce the inhibition. While docking would provide such information, no docking study has been found to outperform the CYP ligand-based ML models.38
000 molecules. For these models, molecules were encoded with various molecular descriptors and fingerprints including Morgan3,27 GraphOnly,31 Klekota-Roth (PaDEL32),31 fingerprints made of pairs of Extended Connectivity Fingerprint (ECFP), ECFP8,33,34 MACCS, and PubChem35 fingerprints,31 MOE31,36 and PaDEL32 descriptors.30 ML techniques including random forests (RF),28,31 naïve Bayesian method,33 eXtreme gradient boosting (XGB),31 support vector machines,29 to multitask deep neural networks (DNN)30 and k-nearest neighbours (KNN)37 have been used. All of these reports resulted in good accuracy on their respective test sets, but as pass/fail classifiers, they provide little information to medicinal chemists on the possible modifications that would reduce the inhibition. While docking would provide such information, no docking study has been found to outperform the CYP ligand-based ML models.38
Whether ML models can generalize well to compounds outside the training set is often minimally investigated. When it is, a significant decrease in accuracy is observed when the methods are tested on compounds in different regions of the chemical space than what is included in the training set.27,39 Here we present our investigations of docking in combination with ML (ML-augmented docking) and its potential to yield accurate, and generalizable results while maintaining the ability to visualize docking poses, which can be used as an informative tool for medicinal chemists in their workflow.
2 Methodology
We first clustered our datasets using two methods as seen in Fig. 1, one investigating analogues (Fig. 9), and the other investigating the generalizability of models by using max train-test Tanimoto similarity (Fig. 5, and 6). We docked each inhibitor and non-inhibitor to their respective CYP450 isoform (3A4,40 2C19,41 2C9,42 2D6,43 and 1A244) using our docking software FITTED. We repeated docking five times due to the stochastic nature of the docking algorithm and completing multiple runs has been shown to yield more consistent results.7 We then collected ligand–residue interaction data (hydrogen bonds, van der Waals and electrostatic interactions with either side-chain or backbone of each residue in the binding site), which we term docking data, and ligand data (e.g., molecular weight, logP, Fsp3, complete list provided as ESI). Together, the ligand and docking data combined are termed all features. Subsequently, we developed an RF model which predicts CYP activity to determine a feature's importance to select key features alongside ones highlighted in the literature, visual analysis, or medicinal chemistry knowledge. Finally, we tuned hyperparameters and evaluated various ML techniques. Last, using the highest performing model, eXtreme Gradient Boosting (XGB), we investigated (1) various featurization techniques, (2) the implication of similarity between testing and training sets as well as the effect of analogues in the construction of train and test sets, and (3) the difference in the generalizability of ML models trained with ligand-only data, docking data, and combination of two.
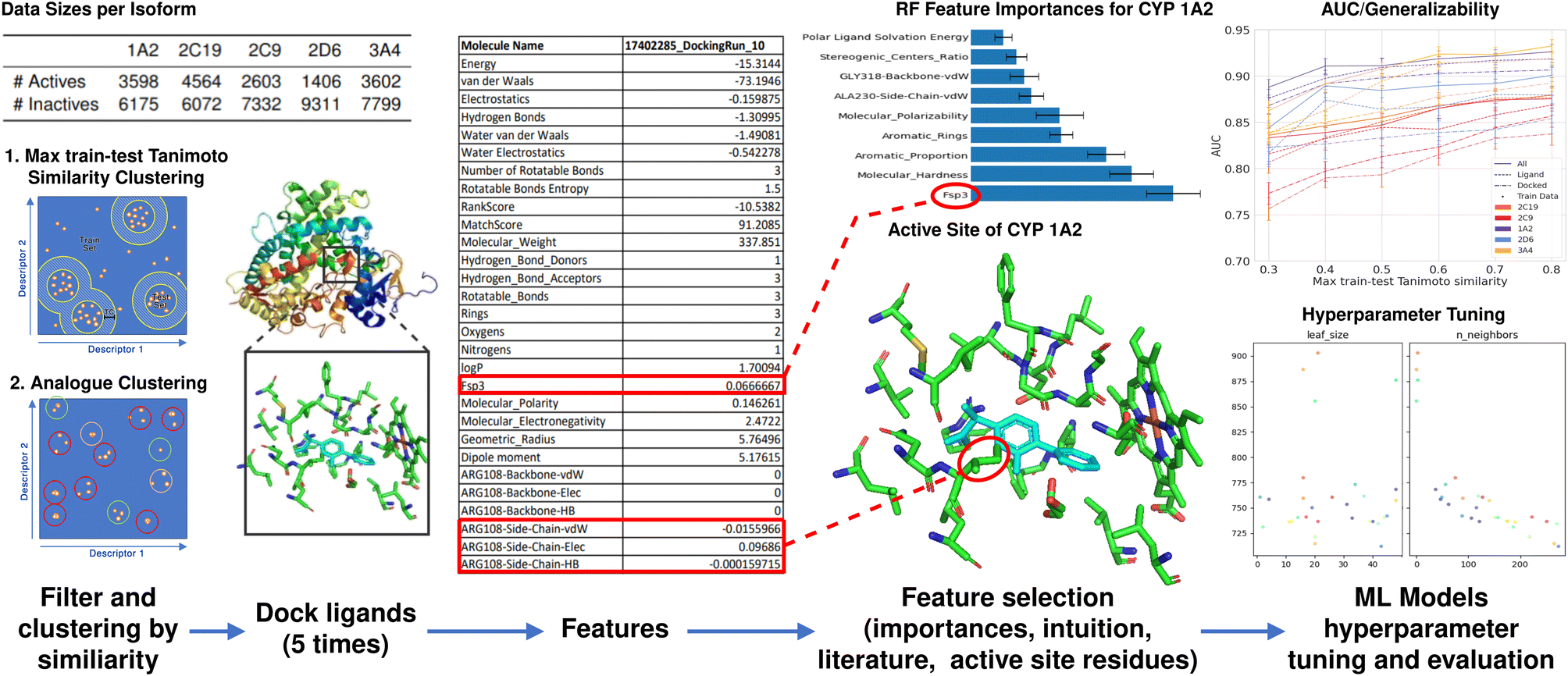 | ||
| Fig. 1 ML-augmented docking workflow. Filtering and clustering; 5 datasets for each of the CYP P450 proteins with experimentally determined activities are clustered using two methods concerning Tanimoto similarity. Max train-test Tanimoto similarity clustering is used to evaluate the generalization of ML-augmented docking. Analogue clustering is used to investigate the correlation between analogue molecules in the data set and accuracy. Dock ligands; each molecule is then docked using FITTED 5 times and a list of ligand and docking features is extracted. Feature selection; the most important feature is then selected by considering the active site, literature, and intuition. ML Models hyperparameter tuning and evaluation; the following ML classifier models were then constructed; LR, RF, GB, XGB, KNN, and DNN. Hyperparameters tuned by using hyperopt. XGB models were then created for each varying training set to evaluate generalizability. | ||
2.1 Datasets
Developing an accurate ML model requires a very careful selection of the training and test sets (and validation sets with some techniques). While some CYP inhibitor sets are available, careful use of these sets is recommended. For this work, we used the AID curated set developed by Pei and co-workers, which was also used by Hou and coworkers to derive an XGB model.31 The set excludes (1) entries containing mixtures, (2) noncovalent complexes, and (3) compounds with atoms other than C, H, O, N, P, S, F, Cl, Br.30 The compounds were labelled active or inactive according to their half-maximal effective concentration, EC50, concentration–response curves, as well as PubChem activity score which is based on the half-maximal inhibitory concentration, IC50. Any compounds which could not be definitively classified were removed.30 We deleted a number of duplicates and kept all compounds which were docked successfully (≈85%). The generalizability of these models remains an issue as it has been shown that as the test set becomes more dissimilar to molecules in the training set, the accuracy of prediction decreases.27 When using datasets, the additional challenges are:(1) To ensure that the training and testing sets are sufficiently different to evaluate the accuracy of these models on new, diverse molecules. In particular, training and testing datasets may include analogs of the same chemical series which would lead to an overestimation of the true accuracy of the models in prospective studies.
(2) To ensure that the set is diverse enough and without excessive chemical space over-representation.
(3) To ensure that the dataset is large enough.
First, to access the generalizability of the ML models (challenge #1), we curated the original dataset such that the molecules in the training and testing sets differ by a pre-determined Tanimoto similarity coefficient (Fig. 2). We term this method the max train-test similarity, where a high Tanimoto coefficient between training and testing sets represents a high level of similarity. Here the Tanimoto coefficient was calculated using Morgan fingerprints with a radius of 2, which is very similar to ECFP4. To generate the testing set, 100 compounds were randomly selected from the unclustered dataset and, for each of these, the 9 most similar molecules from the dataset were selected, creating a testing set of 1000 molecules. The remaining molecules compose the training set. The training set was filtered using the max train-test similarity to ensure that no analogues to compounds in the testing set were found in the training set. We created 8 different training sets by selecting molecules that are within a Tanimoto coefficient ranging from 0.2 to 0.9 (with increments of 0.1) of the molecules in the testing set. Molecules with a Tanimoto coefficient greater than the pre-determined threshold are automatically excluded from the training set. This allows us to generate a training set that is dissimilar in chemical space to the testing set and thus allows for better evaluation of the accuracy and generalization of our ML models.
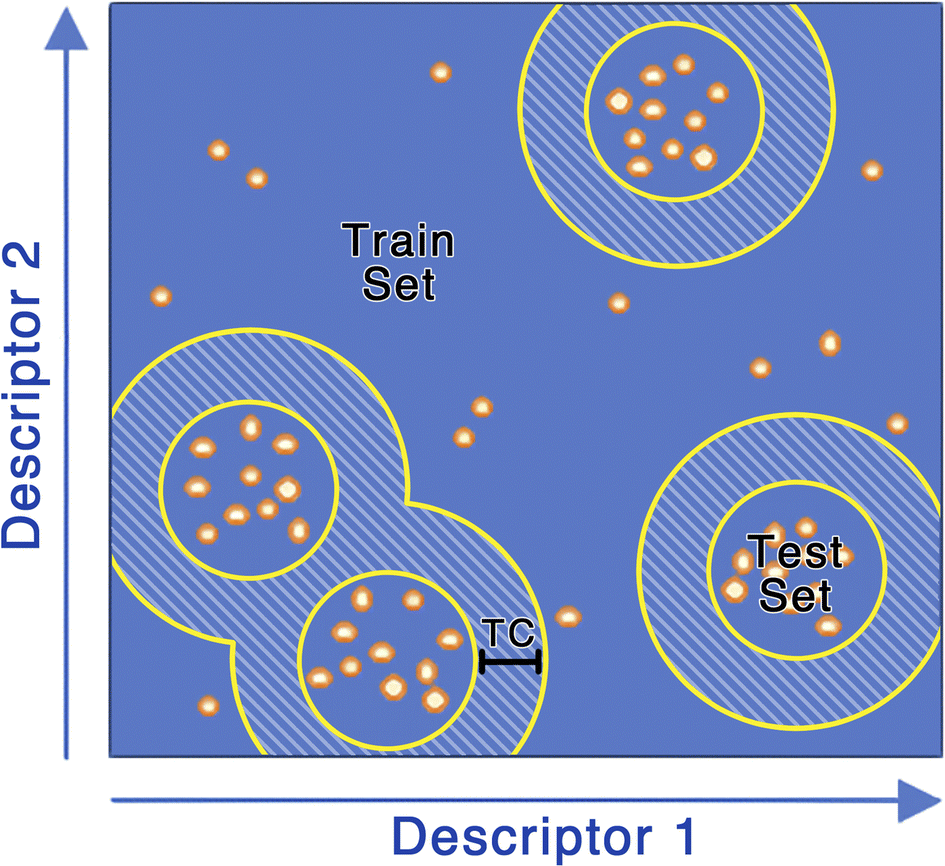 | ||
| Fig. 2 Developing the max train-test Tanimoto similarity clustering data sets. (1) 100 compounds were randomly selected from the unclustered dataset, (2) the 9 most similar molecules to each selected molecule are added to the test set, (3) Molecules with a Tanimoto coefficient (shown as TC) greater than the predetermined threshold for any molecules in the test set are deleted. The remaining molecules make up the train set. | ||
Alongside this, we investigated a clustering method to better assess the impact of analogues. We developed the protocol illustrated in Fig. 3. First, all the molecules were clustered by similarity using ECFP4 with a Tanimoto coefficient from 0.2 to 0.9 (with increments of 0.1) as implemented in SELECT, a program of our drug discovery platform FORECASTER.45 Molecules were then removed from clusters that contained more than a given number of molecules (e.g., molecules from the same chemical space). These steps ensure that chemical analogues are not overrepresented in the set (challenge #2). Data augmentation was next envisioned to increase the dataset size (challenge #3). For this purpose, in clusters with less than a given number of molecules, duplicates were added. Since our docking program is based on some stochastic methods, docking duplicates will lead to slightly (if convergent) or very different (if poorly convergent) results. Finally, the splitting into testing and training sets will be applied to the clusters, hence ensuring minimal overlap between testing and training sets (challenge #1).
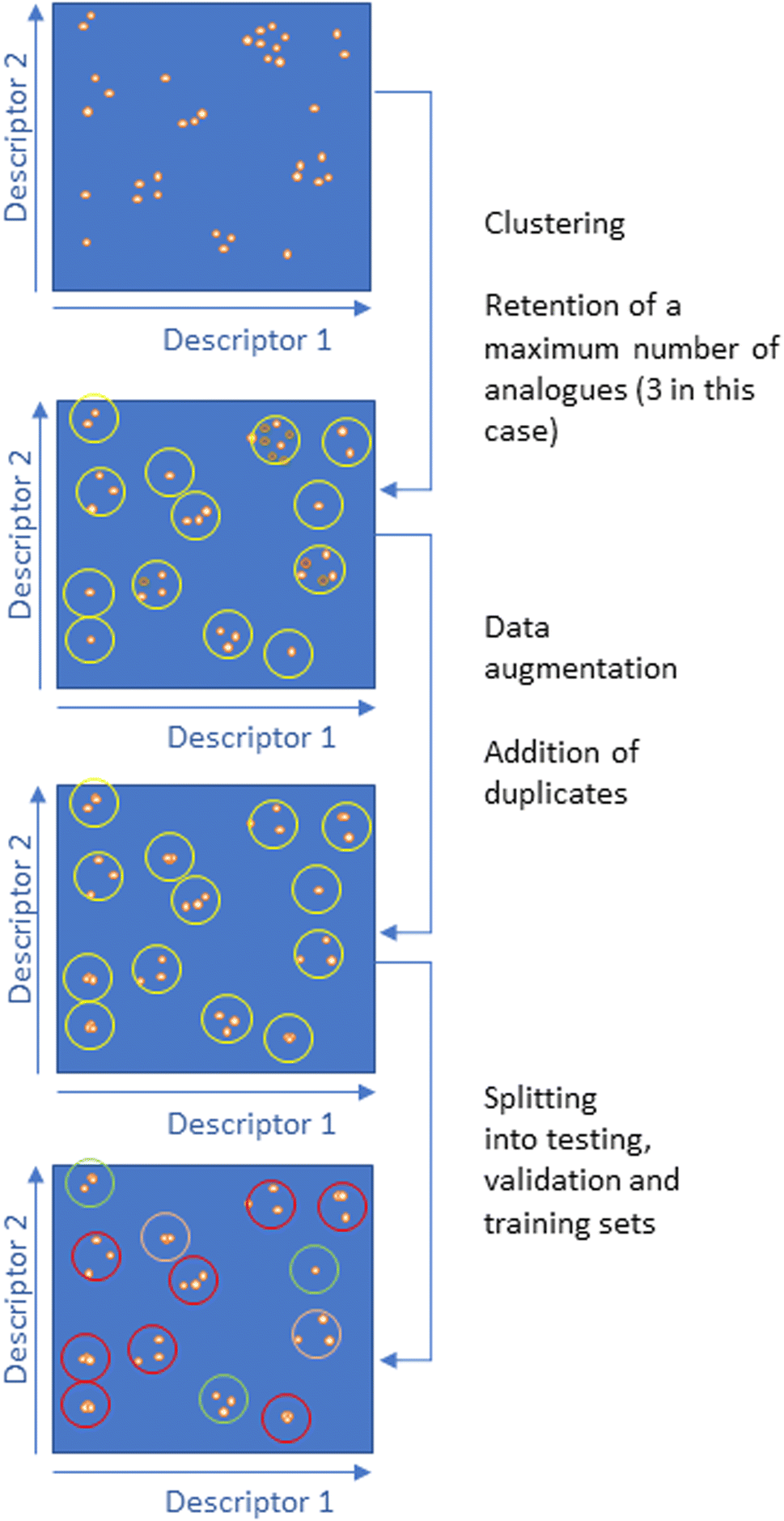 | ||
| Fig. 3 Developing the training and test sets by clustering by Tanimoto coefficient to create analogue datasets. | ||
2.2 Encoding protein ligands and/or protein–ligand complexes
At the core of several ML-based techniques is the method used to encode the objects to be predicted. Several flavours of interaction fingerprints (IFPs) have been proposed to model protein–ligand complexes including Protein–Ligand Extended Connectivity (PLEC) which rely on ECFP,34 one for the ligand, one for the protein group in close proximity,46 Simple Ligand–Receptor Interaction Descriptor (SILIRID),47 Structural Protein–Ligand Interaction Fingerprint (SPLIF) which also uses ECFP descriptors,48 Structural Interaction Fingerprints (SiFTs),49 python-based protein-ligand interaction fingerprinting (PyPLIF).50 With these approaches, 3D information is converted into a 1D string and can be used as a post-docking re-scoring strategy. Other common approaches are to use ligand atoms or groups and encode their interactions with the protein residues as proposed in DeepVS17 and PLEIC-SV24 and/or to include ligand information.51 These techniques rely on structural information.We investigate encoding protein ligands energy terms, calculated by structural information, such as electrostatic, van der Waals and hydrogen bond interaction energies. To do so, our docking program FITTED7 has been modified to output the interactions of the small molecules with each of the protein residues in 6 bits (HBD, van der Waals and electrostatic for both the side chain and the backbone) into a comma separated file (CSV). In addition, some molecular descriptors implemented into the FORECASTER program SMART (molecular weight, molecular shapes, …) were added resulting in a preliminary list of approximately 500 features for each molecule docked to each isoform. Feature selection was then performed using two approaches:
(1) A RF model was developed using all of these features and feature importances were calculated. This was done 10 times and the mean was taken. The features which showed little to no importance were discarded.
(2) Features associated with known key residue interactions within the active site of the enzyme were chosen, such as GLY318 backbone van der Waals and ALA230 side chain van der Waals interaction energy.4 Additionally, features which, through our chemical knowledge, should be useful for predicting binding such as ligand molecular size, and ligand surface areas were also selected.
2.3 ML models
Experimental data from Pei30 included the activity/inactivity of molecules for the associated isoform resulting in 5 datasets being used. Each of the molecules in the 5 datasets was docked to the corresponding CYP isoforms and the CSV files containing the fingerprints were used to train ML models. The Keras Python Library52 and scikit-learn53 were used to build and train these models. The classifiers that were built are logistic regression (LR), RF, GB, XGB, KNN, and DNN. Model hyperparameter selection was performed using the Tree of Parzen Estimator (TPE) optimizing the 5-fold cross-validation accuracy. TPE outperforms random search with significantly fewer trials.54 The DNN was tunned using the Keras tuner, Hyperband. For the XGB models, the hyperparameter search space had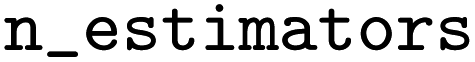 between 5 and 300, a
between 5 and 300, a 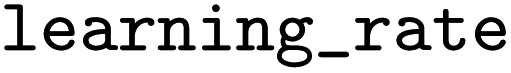 of 0.03, 0.06, 0.12, 0.25, 0.5, a
of 0.03, 0.06, 0.12, 0.25, 0.5, a  between 5 and 20, a
between 5 and 20, a  between 1 and 20, and a
between 1 and 20, and a  between 0 and 9. 15 evaluation were made before selecting the best hyperparameters. The models were evaluated based on the area under the receiver operating characteristic (AUC) as well as the Matthews correlation coefficient (MCC), sensitivity, and specificity on the testing sets. Error bars were determined using the standard deviation of bootstrapped test predictions.
between 0 and 9. 15 evaluation were made before selecting the best hyperparameters. The models were evaluated based on the area under the receiver operating characteristic (AUC) as well as the Matthews correlation coefficient (MCC), sensitivity, and specificity on the testing sets. Error bars were determined using the standard deviation of bootstrapped test predictions.
2.4 Limitations
A limitation of ML models for CYP inhibition is that prediction is made for the unmodified compounds and does not include inhibition by metabolites, or secondary compounds. Therefore, inhibition mechanisms such as catalysis-dependent inhibition whereby the molecule first reacts with the enzyme and the product of the reaction inhibits the enzyme, are not accounted for. As we dock our molecules to the CYP active sites, we assume competitive active site inhibition. Other means of inhibition due to binding to other sites are possible and are not considered in our methodology. Another consideration is that enzyme variability due to genetic heterogeneity in the global population affects the accuracy of results. For example, 5% of Caucasians and 20% of Asians have reduced or no enzyme function.28 The isoform used for data collection may be more representative of the enzymes produced by a certain population than another. Due to this, the application of these models and similar models should consider biases which arise from the data set which would, in turn, lead to varying accuracy regarding certain populations. As with all ML models, generalization is one of the most crucial, yet difficult limitations to solve.3 Results and discussion
3.1 ML-augmented docking for CYP inhibition
We propose ML-augmented docking to enable better assessment of results by medicinal chemists, improve activity prediction and investigate whether using docking energy interaction featurization improves the generalizability of ML models. As we do not replace the docking software with an ML model, medicinal chemists retain the ability to investigate docking by visual analyses and determine key residue interactions, which is crucial for the computer-accelerated drug design process.55When the most important features were analyzed, all features identified to be correlated to the isoforms' activity by Beck4 were ranked as important (above mean importance) by our RF model. First, known key residues to binding were consistently ranked as the most important features. Second, other known important ligand factors were ranked as important by the RF model such as aromaticity and polarity for isoform 2C9. This data suggests that ML-augmented docking could be used for insights into key residues and ligand properties for binding and that our RF approach to identify essential features is robust.
With these datasets and selected features, we benchmarked various ML models. Results showed that GB and XGB models performed the best (Fig. 4). However, at this stage, we wondered what role the docking data played in achieving this accuracy. To address this question, we evaluated the effect of using only ligand features and using only interaction features calculated by docking, and the respective AUC of each model (Table 1). ML-augmented docking can consistently improve the accuracy of CYP activity prediction of FITTED using both feature types. Using docking features only, therefore simply adjusting the scoring function of FITTED, consistently increased the AUC. Due to the iron-containing heme, FITTED's docking AUC is significantly lower with respect to its average AUC for other proteins.7
 | ||
| Fig. 4 AUC of ML models for max train-test similarity of 0.6 using docked features for isoform 3A4 and 2C19. The dotted line is FITTED AUC without ML-augmented docking. | ||
| Isoform | Similarity | All (%) | Docked (%) | Ligand (%) | |||
|---|---|---|---|---|---|---|---|
| AUC | Diff | AUC | Diff | AUC | Diff | ||
| 1A2 | 0.4 | 91 | 23 | 88 | 21 | 90 | 22 |
| 0.8 | 92 | 24 | 90 | 23 | 92 | 24 | |
| 2C19 | 0.4 | 85 | 24 | 79 | 18 | 83 | 22 |
| 0.8 | 87 | 26 | 83 | 22 | 87 | 25 | |
| 2C9 | 0.4 | 84 | 15 | 80 | 11 | 83 | 14 |
| 0.8 | 87 | 18 | 84 | 15 | 86 | 17 | |
| 2D6 | 0.4 | 88 | 33 | 84 | 29 | 88 | 33 |
| 0.8 | 90 | 35 | 85 | 30 | 89 | 34 | |
| 3A4 | 0.4 | 88 | 14 | 85 | 11 | 86 | 12 |
| 0.8 | 92 | 18 | 88 | 14 | 91 | 17 | |
Using docking features with ligand features consistently achieved an increase in AUC over ligand features alone. This is with the one exception of when using similar training and testing sets for isoform 2C19 which the inclusion of docking feature did not increase the AUC. This increase is of larger magnitude when looking at the generalizable regime (similarity 0.4). In addition, ML models looking at just the ligands performed better than those using the docking features alone.
For the case of 2C19, the scoring functions currently implemented in FITTED, RankScore and FittedScore, yield poorly predictive results, AUC of 61.5%, however, using our ML using docking interactions, we achieved an AUC of 83% and 79% when trained and tested on max train-test similarity of 0.8 and 0.4 data respectively.
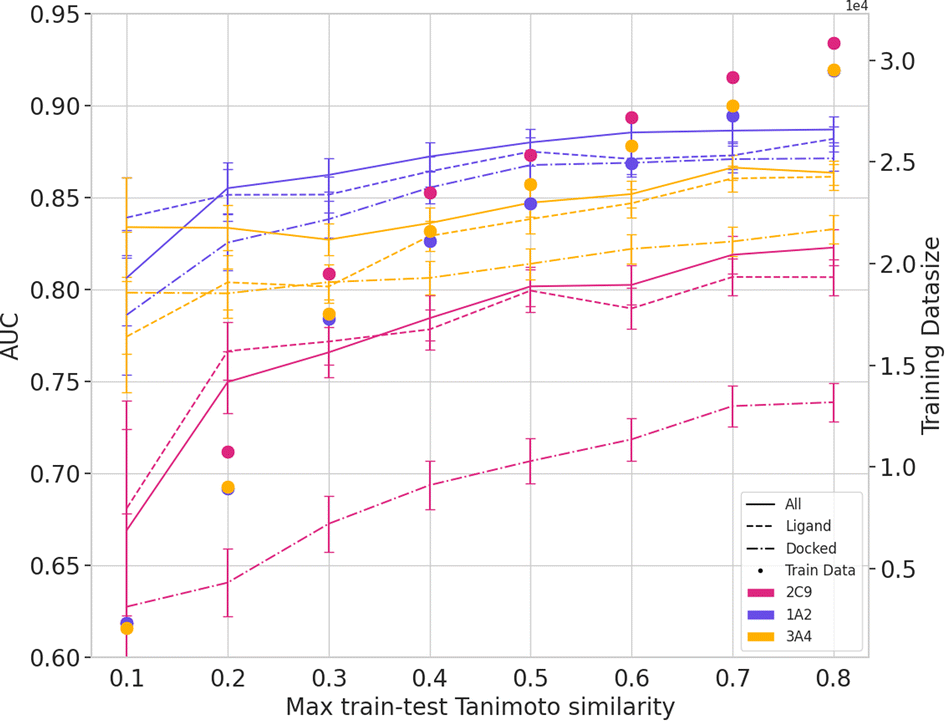
To investigate the effect of generalizability of prediction of using ML-augmented docking versus ML alone, we consider the effects of increasing the dissimilarity between the testing and training sets (Fig. 5 and 6). Using the docking features resulted in equal AUC values at max train-test Tanimoto similarity of 0.3 for three of the five isoforms. The improvement in percent AUC by using docking and ligand features over simply ligand features for each isoform is 2.4, 2.9, 1.7, 2.7, and 1.2 for isoforms 3A4, 2C19, 2C9, 2D6, and 1A2 respectively. Additionally, the slope when using all features is less steep than ligand features between max train-test similarity of 0.3 and 0.4. Our findings suggest that a combination of ligand features and docking features improves the generalizability of ML model prediction if the accuracy of the docking software is sufficiently high.
 | ||
| Fig. 5 AUC of ML-augmented docking using ligand-only features, docking features, and all features over max train-test Tanimoto similarity for isoform 1A2, 2C19 and 3A4. Training data decreases as max train-test similarity decreases. See ESI† for 2C9, and 2D6 results. | ||
 | ||
| Fig. 6 AUC of ML-augmented docking using ligand-only features, docking features, and all features over max train-test Tanimoto similarity using same data sizes per training set for isoform 1A2, 2C19, and 3A4. Train data size capped at data size of 0.3 (1A2: 2406 ligands, 2C19: 2698 ligands, 3A4: 2649 ligands). Using all features becomes more predictive than ligand featurization at low Tanimoto similarity. See ESI† for 2C9, and 2D6 results. | ||
Subsequently, weighted ensemble models, a combination of the already trained XGB models, were created using the validation set AUC − 0.75 as the weight of each of the model's predictions. Therefore, a model with poor performance (AUC < 0.75) does not contribute to the ensemble. This was done using models made with all training sets (Fig. 7) as well as only training sets with a max train-test Tanimoto similarity equal to or less than 0.4 (Fig. 8, Table 1).
 | ||
| Fig. 7 AUC of an ensemble of models for ML-augmented docking using ligand-only features, docking features, and all features. 7 models are combined for each feature for each isoform. Ensemble of all models is an ensemble using all the models of each one of the features combining 21 models per isoform. | ||
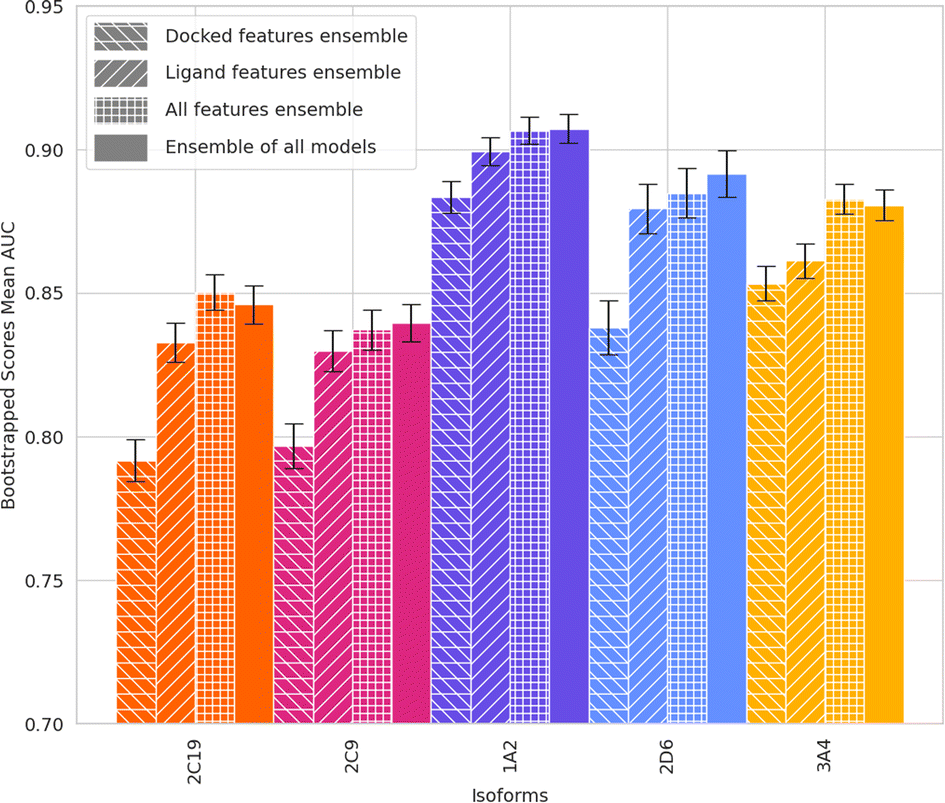 | ||
| Fig. 8 AUC of an ensemble of models for ML-augmented docking using ligand-only features, docking features, and all features. 3 models, with max train-test similarity of 0.4, 0.3, and 0.2, are combined for each feature for each isoform. Ensemble of all models is an ensemble using all the models of each one of the features combining 9 models per isoform. For isoforms 2D6, 2C19 and one of 2C9, the 0.2 max train-test Tanimoto similarity models are below 0.75 AUC and therefore do not contribute to the ensemble. | ||
The results using analog datasets (Fig. 9) show that using fewer data with more varied chemical structures can lead to similar accuracy. This shows that the breadth of chemical space represented in the data is an essential aspect of creating accurate ML models for chemistry.
 | ||
| Fig. 9 AUC of ML-augmented docking using ligand-only features, docking features, and all features over max train-test Tanimoto similarity using analog datasets for isoform 1A2, 2C9, 3A4. Training data size on the right y-axis. A minimal decrease in accuracy was observed as data size decreased substantially. See ESI† for 2C19, and 2D6 results. | ||
As additional experiments, the threshold of prediction used to classify each prediction as active or inactive can be tuned. The threshold can be tuned to get ≈95% active accuracy by taking very modest active predictions as active. We show the results for the highest, 1A2, and lowest, 2C9, predictive models. Notably, we can get a high active accuracy while maintaining between 46-71% inactive accuracy depending on the case (Table 2). This suggests notable applicability for large-scale drug candidate searches.
| Isoform | Similarity | All (%) | Ligand (%) | ||
|---|---|---|---|---|---|
| A | I | A | I | ||
| 1A2 | 0.4 | 93 | 69 | 95 | 61 |
| 0.8 | 94 | 71 | 95 | 63 | |
| 2C9 | 0.4 | 93 | 49 | 93 | 46 |
| 0.8 | 93 | 55 | 91 | 60 | |
3.2 ML-augmented docking
ML-augmented docking provides a better scoring function than what is currently implemented into FITTED. The ML model can learn to fit a parameter to adjust the scoring function to account for a combination of factors. (i) The ML models may be able to detect when the FITTED original scoring function may make a wrong prediction from the docking interaction features. (ii) The ML models may be able to infer relationships between the docking interaction features and physical effects which are not well accounted for in FITTED Score. For example, if interactions with a certain residue influence the entropy of the complex, this may not be accurately calculated by the approximations of the FITTED Score. However, this unaccounted entropy effect could be approximated by the model. (iii) The model may learn to account for the error in the conformation of the protein due to its flexibility and change in conformation when docking small molecules resulting in certain side chains being closer or farther to these ligands. (iv) The model may be learning when the crystal structure is inaccurate for experimental conditions. The crystal structure used for docking is often acquired at low temperatures, in their non-native biological environments and pH levels, and is subject to assumptions made by the crystallographers, especially regarding poorly resolved side chains (see ESI†). Thus, the side chains' position in the crystal structure may disagree with the protein structure in solution and under biological experimental conditions. The models can then account for which residues have over- or underrepresented interaction due to imperfect crystallographic data.4 Conclusions
We created various ML models using different algorithms to predict CYP inhibition. We found that XGB yielded the greatest accuracy. These models were made with various feature constructions to evaluate the effect of ML-augmented docking. We conclude that ML models used to rescore docking results can enhance the prediction power of the docking software. Even when simply using docking residue interaction features, ML augmentation provided significant improvement. ML using ligand-only feature models provides similar accuracy to docking and ligand feature models when test and training sets are similar. Combining ligand and docking results consistently increases the accuracy of our model for CYP inhibition. In the evaluation of the generalization of our models, ligand-only models proved less generalizable than using docking interaction features along with ligand features. The AID set may not be large and/or diverse enough for ML models and more high-quality data is needed. We also predict that other models in the literature should be investigated for their generalizability as their accuracy will likely be decreased as predictions are made on the chemical space outside the training set.Data availability
The open-source code created for this project can be found at https://github.com/MoitessierLab/ML-augmented-docking-CYP-inhibition. The data (models, optimization of hyperparameters, input data) and code are available as ESI.†Author contributions
Benjamin Weiser: conceptualization, methodology, software, formal analysis, investigation, writing – original draft, visualization. Jérôme Genzling: conceptualization, writing – review & editing. Mihai Burai-Patrascu: conceptualization, writing – review & editing. Ophélie Rostaing: conceptualization, writing – review & editing. Nicolas Moitessier: funding acquisition, supervision, conceptualization, writing – review & editing, Software.Conflicts of interest
FITTED is distributed by MFI (NM co-founder and CSO, MBP and OR research scientists), free for academic research.Acknowledgements
Thank you to Ziling Luo, Agathe Fayet for their contributions to this project. We thank NSERC (Discovery Grant) and CIHR (Project Grant) for funding to NM.Notes and references
- D. Dalvie, A. S. Kalgutkar and W. Chen, Drug Metab. Rev., 2015, 47, 56–70 CrossRef CAS PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2008, 21, 70–83 Search PubMed.
- E. Stjernschantz, N. P. E. Vermeulen and C. Oostenbrink, Expert Opin. Drug Metab., 2008, 4, 513–527 Search PubMed.
- T. C. Beck, K. R. Beck, J. Morningstar, M. M. Benjamin and R. A. Norris, Pharmaceuticals, 2021, 14, 472 Search PubMed.
- J. H. Lin and A. Y. Lu, Clin. Pharmacokinet., 1998, 35, 361–390 CrossRef CAS PubMed.
- M. M. Ahlström and I. Zamora, J. Med. Chem., 2008, 51, 1755–1763 CrossRef PubMed.
- A. Labarre, J. K. Stille, M. B. Patrascu, A. Martins, J. Pottel and N. Moitessier, J. Chem. Inf. Model., 2022, 62, 1061–1077 CrossRef CAS PubMed.
- H. Kato, Drug Metab. Pharmacokinet., 2020, 35, 30–44 CrossRef CAS PubMed.
- M. Hennemann, A. Friedl, M. Lobell, J. Keldenich, A. Hillisch, T. Clark and A. H. Göller, ChemMedChem, 2009, 4, 657–669 CrossRef CAS PubMed.
- J. P. Jones, M. Mysinger and K. R. Korzekwa, Drug Metab. Dispos., 2002, 30, 7 CrossRef CAS PubMed.
- R. T. Naven and S. Louise-May, Hum. Exp. Toxicol., 2015, 34, 1304–1309 CrossRef CAS PubMed.
- S. M. He, Z. W. Zhou, X. T. Li and S. F. Zhou, Curr. Med. Chem., 2011, 18, 667–713 CrossRef CAS PubMed.
- V. Campagna-Slater, J. Pottel, E. Therrien, L.-D. Cantin and N. Moitessier, J. Chem. Inf. Model., 2012, 52, 2471–2483 CrossRef CAS PubMed.
- H. Li, K.-H. Sze, G. Lu and P. J. Ballester, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10, e1465 CrossRef CAS.
- C. Shen, J. Ding, Z. Wang, D. Cao, X. Ding and T. Hou, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10, e1429 CrossRef CAS.
- J. D. Durrant and J. A. McCammon, J. Chem. Inf. Model., 2011, 51, 2897–2903 CrossRef CAS PubMed.
- J. C. Pereira, E. R. Caffarena and C. N. dos Santos, J. Chem. Inf. Model., 2016, 56, 2495–2506 CrossRef CAS PubMed.
- J. Jiménez, M. Škalič, G. Martínez-Rosell and G. De Fabritiis, J. Chem. Inf. Model., 2018, 58, 287–296 CrossRef PubMed.
- M. Ragoza, J. Hochuli, E. Idrobo, J. Sunseri and D. R. Koes, J. Chem. Inf. Model., 2017, 57, 942–957 CrossRef CAS PubMed.
- P. J. Ballester and J. B. O. Mitchell, Bioinformatics, 2010, 26, 1169–1175 CrossRef CAS PubMed.
- L. Li, B. Wang and S. O. Meroueh, J. Chem. Inf. Model., 2011, 51, 2132–2138 CrossRef CAS PubMed.
- M. S. Nogueira and O. Koch, J. Chem. Inf. Model., 2019, 59, 1238–1252 CrossRef CAS PubMed.
- S. L. Kinnings, N. Liu, P. J. Tonge, R. M. Jackson, L. Xie and P. E. Bourne, J. Chem. Inf. Model., 2011, 51, 408–419 CrossRef CAS PubMed.
- Y. Yan, W. Wang, Z. Sun, J. Z. H. Zhang and C. Ji, J. Chem. Inf. Model., 2017, 57, 1793–1806 CrossRef CAS PubMed.
- D. D. Nguyen and G.-W. Wei, Int. J. Numer. Method Biomed. Eng., 2019, 35, e3179 CrossRef PubMed.
- C. de Graaf, P. Pospisil, W. Pos, G. Folkers and N. P. E. Vermeulen, J. Med. Chem., 2005, 48, 2308–2318 CrossRef CAS PubMed.
- W. Plonka, C. Stork, M. Šícho and J. Kirchmair, Bioorg. Med. Chem., 2021, 46, 116388 CrossRef CAS PubMed.
- P. Banerjee, M. Dunkel, E. Kemmler and R. Preissner, Nucleic Acids Res., 2020, 48, W580–W585 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- X. Li, Y. Xu, L. Lai and J. Pei, Mol. Pharm., 2018, 15, 4336–4345 CrossRef CAS PubMed.
- Z. Wu, T. Lei, C. Shen, Z. Wang, D. Cao and T. Hou, J. Chem. Inf. Model., 2019, 59, 4587–4601 CrossRef CAS PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- J. H. Lee, S. Basith, M. Cui, B. Kim and S. Choi, SAR QSAR Environ. Res., 2017, 28, 863–874 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- PubChem, PubChem Substructure Fingerprint, https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt Search PubMed.
- Molecular Operating Environment (MOE), 2019.1, Chemical Computing Group, ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021 Search PubMed.
- S. Nembri, F. Grisoni, V. Consonni and R. Todeschini, Int. J. Mol. Sci., 2016, 17, 914 CrossRef PubMed.
- K. A. Feenstra, C. De Graaf and N. P. E. Vermeulen, Cytochrome P450 Protein Modeling and Ligand Docking, CRC Press, 2008 Search PubMed.
- N. N. Wang, X. G. Wang, G. L. Xiong, Z. Y. Yang, A. P. Lu, X. Chen, S. Liu, T. J. Hou and D. S. Cao, J. Cheminf., 2022, 14, 23 CAS.
- I. F. Sevrioukova and T. L. Poulos, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 18422–18427 CrossRef CAS PubMed.
- R. L. Reynald, S. Sansen, C. D. Stout and E. F. Johnson, J. Biophys. Chem., 2012, 287, 44581–44591 CAS.
- M. R. Wester, J. K. Yano, G. A. Schoch, C. Yang, K. J. Griffin, C. D. Stout and E. F. Johnson, J. Biophys. Chem., 2004, 279, 35630–35637 CAS.
- A. Wang, U. Savas, M. H. Hsu, C. D. Stout and E. F. Johnson, J. Biophys. Chem., 2012, 287, 10834–10843 CAS.
- S. Sansen, J. K. Yano, R. L. Reynald, G. A. Schoch, K. J. Griffin, C. D. Stout and E. F. Johnson, J. Biophys. Chem., 2007, 282, 14348–14355 CAS.
- E. Therrien, P. Englebienne, A. G. Arrowsmith, R. Mendoza-Sanchez, C. R. Corbeil, N. Weill, V. Campagna-Slater and N. Moitessier, J. Chem. Inf. Model., 2012, 52, 210–224 CrossRef CAS PubMed.
- M. Wójcikowski, M. Kukiełka, M. M. Stepniewska-Dziubinska and P. Siedlecki, Bioinformatics, 2018, 35, 1334–1341 CrossRef PubMed.
- V. Chupakhin, G. Marcou, H. Gaspar and A. Varnek, Comput. Struct. Biotechnol. J., 2014, 10, 33–37 CrossRef PubMed.
- C. Da and D. Kireev, J. Chem. Inf. Model., 2014, 54, 2555–2561 CrossRef CAS PubMed.
- Z. Deng, C. Chuaqui and J. Singh, J. Med. Chem., 2004, 47, 337–344 CrossRef CAS PubMed.
- M. Radifar, N. Yuniarti and E. P. Istyastono, Bioinformation, 2013, 9, 325–328 CrossRef PubMed.
- J. Li, W. Liu, Y. Song and J. Xia, RSC Adv., 2020, 10, 7609–7618 RSC.
- F. Chollet et al. , Keras, 2015, https://github.com/fchollet/keras Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- J. Bergstra, D. Yamins and D. Cox, Proceedings of the 30th International Conference on Machine Learning, pp. , pp. 115–123 Search PubMed.
- A. Fischer, M. Smiesko, M. Sellner and M. A. Lill, J. Med. Chem., 2021, 64, 2489–2500 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.1org/10.1039/d3dd00110e |
| ‡ Current address: Molecular Forecaster Inc., Montreal, Canada. |
| This journal is © The Royal Society of Chemistry 2023 |