
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDigital design and 3D printing of reactionware for on demand synthesis of high value probes†
Przemyslaw
Frei
,
Philip J.
Kitson
,
Alexander X.
Jones
and
Leroy
Cronin
 *
*
School of Chemistry, The University of Glasgow, University Avenue, Glasgow G12 8QQ, UK. E-mail: lee.cronin@glasgow.ac.uk
First published on 2nd August 2023
Abstract
Performing chemical transformations in a standardised way is important for increasing the accessibility to high value reagents for specific purposes. Building on the use of 3D-printed reactionware, we present a new concept in the design of reactionware devices for generating such high value compounds. In this approach integrated reactor sequences are treated as modular, with each module able to effect a specific chemical transformation, rather than individual chemical processing steps. The initial workflow is mapped into a core set of modules, and different synthetic pathways can be selected by attaching different peripheral modules in a ‘plug and react manner’. We utilised this system to synthesise a set of diazirine based photoaffinity probes. Starting from appropriately functionalised starting materials, a core sequence of reactors furnishes a central diazirine moiety on a variable sized molecular scaffold, with exchangeable peripheral reactors facilitating the attachment of auxiliary moieties. Yields and purities range from 29–39% and 93–97%, respectively, comparable to, or exceeding literature yields for similar compounds. The activity of photoprobes produced was validated by analysis of their interaction with the peptide hormone, human Angiotensin II.
A wide array of complex reagents which are specifically tuned for particular applications, such as ligand–catalyst systems and biological markers with precise activities and physical properties. The synthesis of such specific compounds is itself a significant area of research requiring significant expertise to design and execute, meaning such reagents are often expensive if a laboratory does not have the facilities or expertise to synthesise them. To address this problem, we have developed the concept of reactionware, where 3D printed reactor cartridges embody specific reaction sequences allowing complex synthetic routes to be performed with minimal synthetic expertise required. Vital to this approach is the need to develop synthesis methods that are as reliable as possible, a non-trivial task that often poses significant challenges.1–4 Whilst the scope of reactions and reactivity control available to chemists has vastly grown during the last decades, the approach to manual chemical synthesis has largely remained unchanged.5–7
With the advent of more affordable 3D printers, this technique has moved outside of industrial modelling8 into areas of engineering and science,9 finding uses in a host of scenarios, such as manufacturing of microfluidic,10 pneumatic,11,12 and electronic devices.13 In 2012, we first investigated the concept of using a digital architectural control of the reactor system and associated unit operations, via using additive manufacturing for the design and production of laboratory-scale (active) chemical reactors.14 Our recent work challenges this status quo by developing low cost and easily accessible devices which can be manufactured by 3D printing. Previously, we have utilised this process to create a variety of devices, such as reactors for synthesis of fine chemicals and pharmaceuticals,15,16 reactors for synthesis of costly and unstable reagents from low cost precursors,17 and even to prepare a polymer electrolyte membrane (PEM) electrolyzer for the electrolysis of water.18 These devices are based on an abstraction of the chemical reaction, whereby a complete telescoped synthesis is translated into as few individual reactor modules as possible, to effect the desired reaction sequence, resulting in a bespoke reactor which is unique for any given synthesis. However, this means that each new design must be produced from the ground up, whenever a new molecule is required.
Herein, we present a new development where a modular approach is used to more closely connect the synthesis sequence to the reaction design process. This is achieved by developing reactors designed to simplify and streamline divergent synthesis by means of assigning segments of synthetic processes to dedicated sets of reactors. Any multi-step synthesis can be divided into parts that correspond to series of operations on a given functional group of the substrate compound. These may include significant alterations of chemical structure, like an addition of a new moiety, or critical functional group interconversions. This presents an opportunity for the development of standard reactor architectures to perform these operations for each such segment of a given synthesis. Moreover, one set of reactors can then be exchanged for another to facilitate a different transformation to prepare a different product. Using this technique, we are modularizing synthesis above an individual unit operation level to obtain a sequence of modules that encompass multiple unit processes, designed to achieve a specific transformation on any given substrate (Fig. 1).
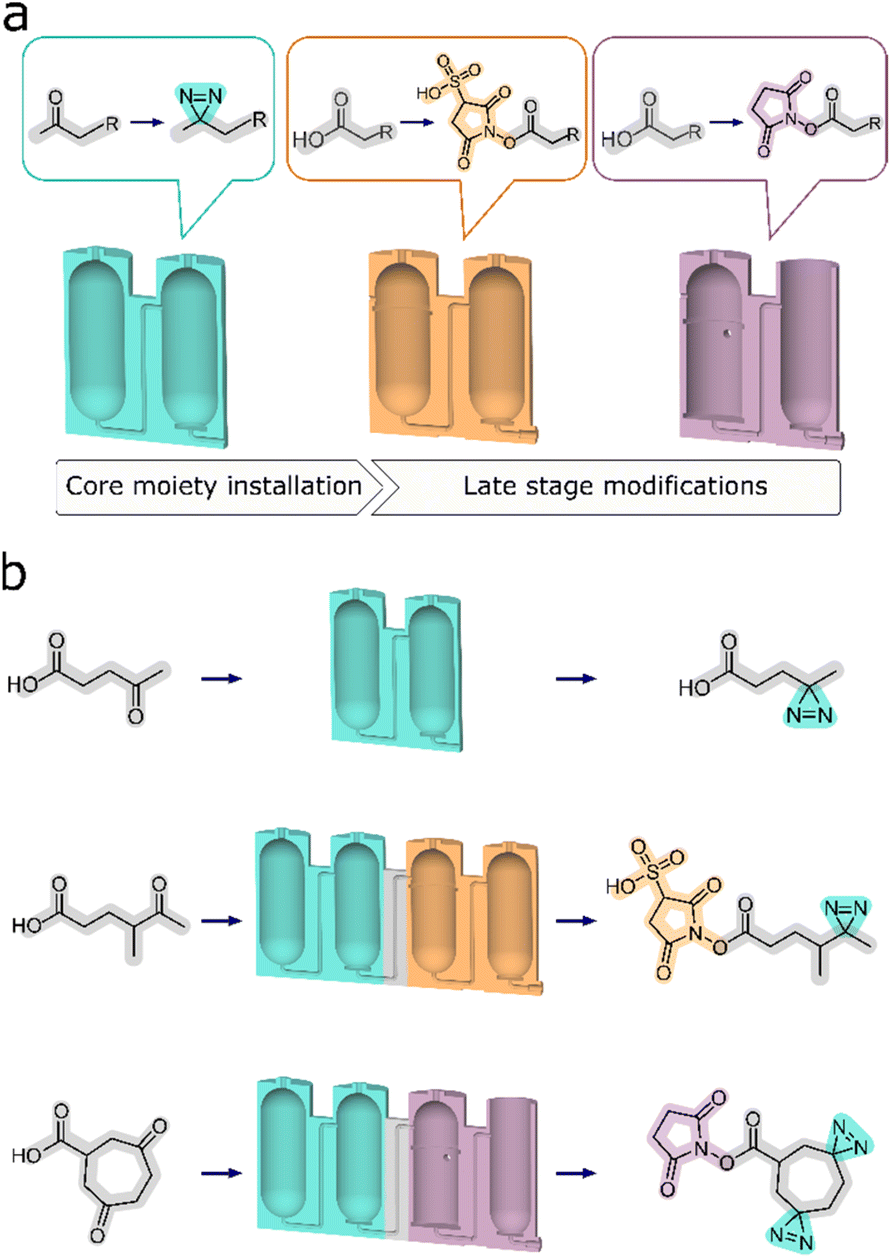 | ||
| Fig. 1 (a) Each individual set of reactors functions exclusively in providing access to one specific moiety. (b) Top – an instance of core moiety installation to a molecular backbone. Middle and bottom – instances of both core and peripheral moieties, furnished by the combined sets of reactors. | ||
We demonstrate that the operation of reactors organized in this way, coupled with precise instructions to carry out the associated synthetic procedures, gives reliable synthetic outcomes. Each of the specific target synthesis was carried out multiple times (usually no less than five), to ensure good reproducibility. With relative simplicity of operation, this novel approach provides a convenient gateway to high value, difficult to synthesize compounds for end users. Following the selection of a synthetic target, suitable modular sets of reactors need to be assembled into a single monolith, in accordance with the underlying chemical transformation. After manufacturing, the complete reactor can be used for the synthesis of structurally related compounds. In our case we substantiate this by synthesizing two distinct types of diazirine-based photo-probes. Each type can be obtained by utilizing a single set of operations in a corresponding monolithic reactor. The possibility of synthesising other analogous photoprobes also exists, provided that their physical properties remain alike and therefore compatible with the pre-designated function of each of the reactors. Similarly, the peripheral sets of modules can also be designated to facilitate the intended chemical transformations, but on a different set of starting materials.
Naturally, this is also subject to physical compatibility of the resultant products with the subsequent purification method. Diazirine based photo-probes have grown increasingly popular since their discovery,19,20 and gradually found use in a host of applications, ranging from polymer crosslinking21 to surface functionalization,22–24 with a higher impact in the domain of cell biology research,25,26 and biological sciences.27,28 The exceptional momentum in research involving diazirines can certainly continue to proliferate further if granted convenient and inexpensive access to a wide range of these useful molecular tools.
Diazirine based photo-probes tend to be extremely expensive when purchased from a commercial vendor, often costing in the Millions of US dollars per mole while the combined cost of the starting materials required to synthesize the compounds is several orders of magnitude less. This implies that preparation of small batch quantities, as needed, makes strong economic sense and is often the preferred method of obtaining these materials for researchers. Besides cost, issues related to storage and stock keeping are significant. Most, if not all diazirine based photo-affinity probes have a propensity to decompose over time, even if stored in the dark and under inert atmosphere. In our experience, even when stored under desiccator conditions and excluding light, the probes degrade at a steady rate and after six months, approximately 50% of the material will have deteriorated rendering the batch unusable. When stored less carefully, or if the container is repeatedly opened to obtain a small portion of material each time, the useable life of a batch is shortened to just a few days. This is one of the key reasons for the retail cost as they must be made on demand in very small batch sizes.
Carrying out a systematic study often requires a few structurally related photoaffinity probes, to find the best candidate for a particular use case.25,29 Being able to prepare a range of related diazirine analogues with varying length of the backbone or with different solubility profiles, provides a gateway to more comprehensive and tailored research, independently of the aspect of commercial availability. To demonstrate our design approach, it was decided to synthesize a set of four related diazirine based compounds which could act as photolinking probes originating from two starting materials, each of which can produce two potential final products depending on the configuration of the reactionware cartridge in which they are processed.
Synthetic approach and reactor design
We have previously presented a general approach to the design and manufacture of reactionware devices for such multi-step syntheses, including software for a parametric generation of reactor modules such as those described below.8–10,16 Before any work on reactor design commences, we review the scientific literature to determine the most suitable procedure for the synthesis of our target compounds. Particular attention is paid to the number of synthetic steps, solvent compatibility, reagent handling and physicochemical aspects of a candidate synthetic pathway. Synthetic procedures with the least number of steps, or steps that could be performed in succession in the same reactor module without purification are favored. Moreover, having smaller number of individual reactors comprising a monolith, shortens the physical path between the first and the last reactor, which in turn reduces the risk of progressive loss of material to a relatively larger surface area of an otherwise longer path. Similarly, difficult to handle reagents, either for their reactivity or physical state, are generally avoided. Liquid ammonia serves as a good example here, as it is sometimes used in the formation of one of the intermediates during formation of diazirine moiety.30,31 Once a suitable synthetic route is selected, it is then validated by performing a glassware synthesis to ensure that yield and purity of a final product was meeting our expectations and was in line or exceeding the data reported in the literature. Testing and optimization of the reactions in glassware often proves to be particularly time-consuming part, as our aim is to obtain robust, consistent results over a number of repeated synthetic runs. Fig. 2 illustrates the reaction sequence chosen for the synthesis of diazirine compounds 1a, 1b, 2a and 2c along with the sequence of reactionware modules designed to accomplish each necessary set of transformations. Diazirines 2a and 2b produced in this work are not commercially available, whereas 2b has not been previously reported to the best of our knowledge.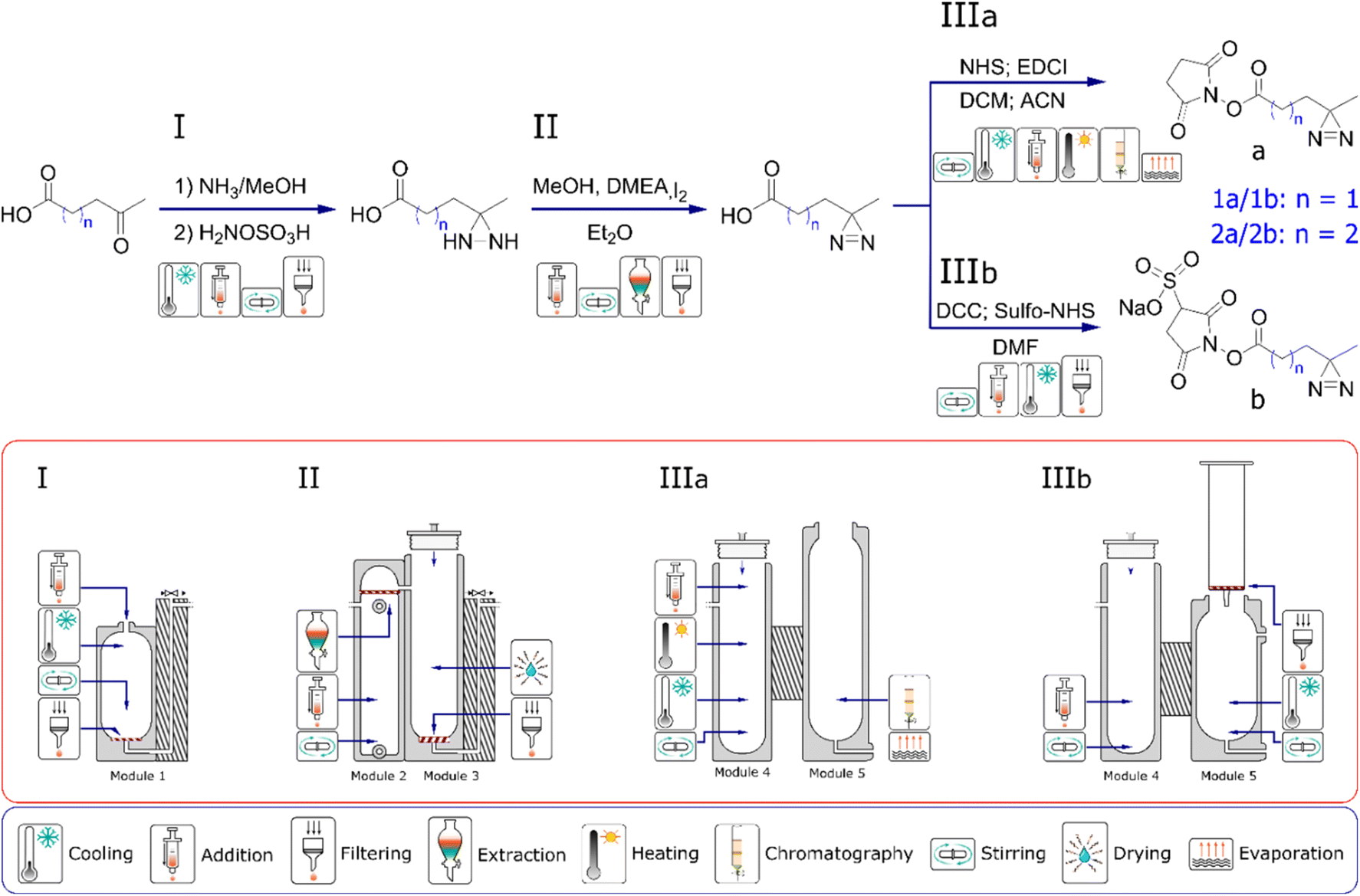 | ||
| Fig. 2 Schematic representation for the synthesis of two different types of diazirines in single monolithic assembly of reactors. In the first part I–II, all of the molecular targets follow an identical synthetic route. This is reflected in the common architecture of the reactors. In the second part IIIa and b, procedures diverge to give two different species of photo-probes. | ||
In the first step in the syntheses of all four diazirines, the starting materials, either 3-acetylpropionic acid or 4-acetylbutyric acid, are combined with ammonia solution in methanol in the presence of a desiccant to form the intermediate imines. Following this step, hydroxylamine-O-sulfonic acid is added as a solution in anhydrous methanol to obtain the next intermediate; diaziridine. In the next step, the solvent along with ammonia are removed by evaporation and the diaziridine is then dissolved in fresh anhydrous methanol. The solution is chilled and N,N-dimethylethylamine is added as catalyst for the next transformation. Solid iodine is then added progressively, to oxidize the diaziridine to diazirine. Addition of iodine is terminated when it stops being consumed in the reaction. The end point is indicated by dark coloration of the reaction mixture, lasting for more than ten minutes after addition of the last portion of iodine. Subsequently, an aqueous solution of potassium iodide is introduced into the reaction mixture to generate a biphasic mixture. A solution of ascorbic acid is then added to neutralize any unreacted molecular iodine, and the mixture is then acidified with a small amount of 3M hydrochloric acid. Next, diethyl ether is introduced into the lower chamber to extract the product into the organic phase. Following the drying process, the extract is pushed into reactor module four where the solvent is evaporated, before the next synthetic steps can take place.
From this point onwards the syntheses diverge depending on whether a diazirine with an NHS headgroup (‘IIIa’ pathway, see Fig. 2) or a Sulfo-NHS headgroup (‘IIIb’ pathway) are to be synthesized. In the case of a diazirine with NHS headgroup, following solvent evaporation, the crude diazirines, 3-(3-methyldiaziridin-3-yl) propanoic acid or 4-(3-methyldiaziridin-3-yl)butanoic acid are dissolved in anhydrous acetonitrile and combined with N-Ethyl-N′-(3-dimethylaminopropyl) carbodiimide dissolved in anhydrous DCM. To this mixture, NHS is added as solution in anhydrous acetonitrile and the reaction stirred to form the final products of either 2,5-dioxopyrrolidin-1-yl 3-(3-methyl-3H-diazirin-3-yl) propanoate or 2,5-dioxopyrrolidin-1-yl 4-(3-methyl-3H-diazirin-3-yl)butanoate. Molecular sieves are used during the coupling of NHS headgroups to remove any residual water and prevent hydrolysis of the intermediates of o-acylisourea or the desired products. Following the coupling reaction, solvents are evaporated, and the crude products purified by silica gel chromatography carried out in reaction module five. To minimize contact between the NHS-diazirines and water during purification and subsequently avoid their hydrolysis, eluent and silica gel are carefully dried before use. Average yield and purity of the 2,5-dioxopyrrolidin-1-yl-3-(3-methyl-3H-diazirin-3-yl)propanoate (1a) as synthesised in reactionware was 39% and 94%, respectively. The average yield and purity of 2,5-dioxopyrrolidin-1-yl 4-(3-methyl-3H-diazirin-3-yl) butanoate (2a) was 30% and 97% respectively. Yields and purities are reported as averages from a minimum of five consecutive synthetic runs.
In the case of diazirines with Sulfo-NHS, following the evaporation of solvent from the organic extract, the crude diazirines, 3-(3-methyldiaziridin-3-yl)propanoic acid or 4-(3-methyldiaziridin-3-yl)butanoic acid are dissolved in anhydrous DMF. N,N′-dicyclohexylmethanediimine is then added as solid, followed by sodium 1-hydroxy-2,5-dioxopyrrolidine-3-sulfonate, also added as solid. The reaction mixture is then stirred to achieve the coupling between Sulfo-NHS and a respective diazirine carboxylic acid. The coupling reaction produces a precipitate of dicyclohexylurea, characterized by its poor solubility in organic solvents. Following completion of the reaction, the mixture is thoroughly cooled to precipitate any free, uncoupled Sulfo-NHS. Subsequently, the reaction is filtered and the filtrate containing the product is collected in reactor module five, removing both precipitates of dicyclohexylurea and free Sulfo-NHS. Filtration takes place under a flow of dry nitrogen to minimise contact between the product and any airborne water, which could consequently cause hydrolysis of the ester bond between Sulfo-NHS and the rest of the molecule. Next, anhydrous ethyl acetate is added, and the solution is cooled to trigger precipitation of the final product. Finally, the white precipitate of the product is recovered by filtration under flow of dry nitrogen to once again minimize contact between the product and any airborne water. In the final stage of filtration, the precipitate is successively washed with anhydrous ethyl acetate, diethyl ether, and pentane to remove any traces of DMF which would otherwise be difficult to evaporate owing to its high boiling point. The product is then dried under vacuum to remove traces of the low boiling solvents. Average yield and purity of sodium 1-{[3-(3-methyl-3H-diazirin-3-yl)propanoyl]oxy} 2,5-dioxopyrrolidine-3-sulfonate (1b) synthesized in reactionware was 34% an 95%, respectively. Average yield of sodium 1-{[4-(3-methyl-3H-diazirin-3-yl) butanoyl]oxy}2,5-dioxopyrrolidine-3-sulfonate (2b) synthesized in reactionware was 29% and purity 93%.
Reactor fabrication and preparation
Once a glassware procedure was well tested and delivering consistent results, we were able to design a reactor for each of the synthetic steps. The reactionware modules were printed individually and each step of a synthesis was carried out in the module it was assigned to. Polypropylene was used as the printing material for the fabrication of the reactionware modules and monoliths due to its superior chemical resistance to standard FDM 3D printing materials such as polylactic acid (PLA). Optimization of the geometries of the individual reactors happened at this stage, to ensure that each of them performed as expected as a standalone module before they were assembled into a monolithic design which would then be tested as a single unit. Each of the monolithic designs for both types of diazirines respectively, comprise three basic types of reactor modules (Fig. 3h), albeit having varying topologies. The standard module consists of a simple chamber in which a reaction or other operations such as precipitation or chromatography can take place. The filter module, similar to the standard module but equipped with a fritted filter disc at the bottom of the chamber and an outlet below the filter enables the separation of solids from liquids. A floating filter module is similar to the filter module but here the position of the filter can be adjusted in such a way that it divides the reactor chamber into two separate parts. The volumes of each of the chambers, as well as height at which the floating filter is positioned, can be defined by the user as required. Solid–liquid or liquid–liquid separations are possible with such a design when a hydrophobic filter disc is used in place of a standard fritted disk. Additionally, three different module tops (i.e., open top, closed top with inlet(s), and closed top) could be specified (Fig. 3h, right).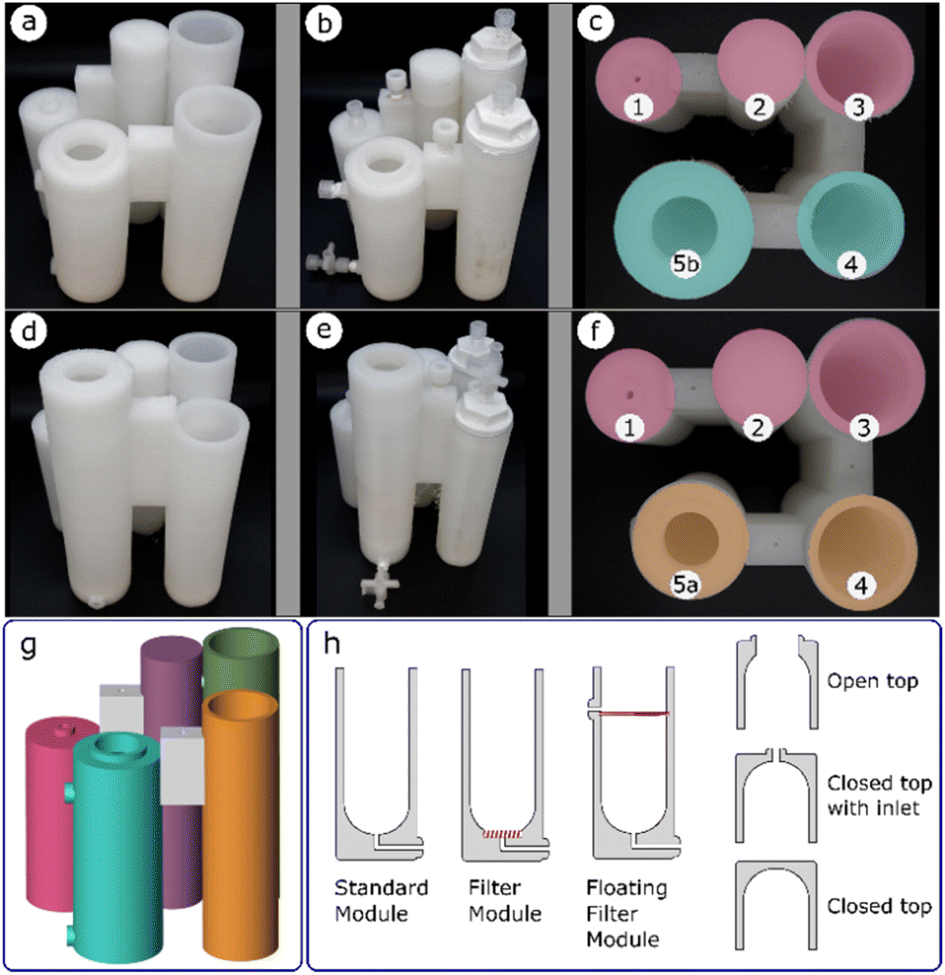 | ||
| Fig. 3 (a and d) printed modules, (b and e) fully fitted modules, (c and f) top views of the printed modules, (g) example of a digital blueprint, (h) examples of module types with customisable tops (right). | ||
Two final monoliths designs were produced, one for synthesis of diazirines with NHS (N-hydroxysuccinimide) headgroup and one for synthesis of diazirines with Sulfo-NHS headgroup. Each of the monoliths was assembled from five individual reactor modules. The formation of the diazirine moiety and subsequent drying of the crude, common to all the syntheses, share the same procedural steps, these were carried in the core modules 1, 2 and 3 (Fig. 3c and f).
Peripheral modules 4 and 5 served to install different head groups to the core backbone of the molecule. The fourth reactor module is also common to both designs as it performs a similar function in both syntheses, albeit with different procedural steps. In both designs, the terminal vessels, denoted by 5, have significant geometrical differences, but are both classified as standard modules. In the case of diazirines equipped with NHS headgroup, the terminal vessel 5a, performs the function of a chromatography column, whereas in the case of diazirines with Sulfo-NHS headgroup, the terminal vessel 5b, performs function of a precipitation chamber.
Both monolithic reactors were designed with some of the modules having a filter or a phase separator incorporated into their structure. Pauses in printing were introduced to enable insertion of the required elements before the printing process could be restarted. Upon completion (Fig. 3a and d) the reactors required fitting of other non-printed parts, such as Luer to thread adapters, Luer lock valves and caps. This was achieved by tapping of ports in the reactors, so that the fittings could be attached by screwing them into the threaded cavities. All threads were wrapped in PTFE tape, to ensure a good seal. Fitting of Luer to thread adapters allowed for connection with syringes through which an addition of solvent or a reagent could be performed. Additionally, Luer lock valves could be connected, to facilitate repetitive additions, while maintaining a leak free system. Since some of the modules were designed to have an open top to provide better access at times, during reactions they were kept sealed by means of polypropylene caps. A screw valve was inserted between modules 1 and 2, to block the channel connecting both modules and avoid unintended transfer of liquid, during reaction. The channel could be opened when required, by partially lifting the screw and enabling the flow of liquid. An example of fully fitted reactor assemblies are shown in (Fig. 3b and e).
Comparison with glassware syntheses
Glassware and reactionware procedures were carried out with as little difference in operational procedures as possible between the two regimes, resulting in reaction times which were comparable between the two modes of production, although it should be noted that the constrained nature of interactions with the reactionware modules required significantly less skilled interaction than traditional glassware synthesis. One significant process difference was cooling during precipitation of impurities, following coupling between Sulfo-NHS and the diazirine acids. In the case of glassware, 4 hours of cooling at 0 °C was entirely sufficient to precipitate and remove most of the impurities. In reactionware on the other hand, the same cooling conditions did not suffice and resulted in some of the impurities making their way to the next step and into the final products. The key cause of this is very low heat conductivity of polypropylene as compared to the thin glass walls of regular laboratory glassware resulting in hindered precipitation of impurities during the prescribed time. To mitigate this, a lower temperature (−20 °C) was applied to increase the temperature gradient between the inside and outside of the polypropylene reactors and in this way allow for complete precipitation, while keeping cooling period the same as with glassware reactors. Comparative results, detailing yields and purities of reactionware and glassware syntheses are shown in Table 1. In the first row we observe that the yield and purity of the glassware synthesis of the shorter NHS-diazirine 1a is marginally better than that of reactionware. In the syntheses of the longer NHS-diazirine 2a, the average yield for both glassware and reactionware syntheses is exactly the same at 30%, whereas the purity of the reactionware synthesis is marginally lower. The slightly lower purity of the NHS-diazirines obtained from reactionware can be attributed to the final purification step, where column chromatography is performed in printed reactor module. FDM printing of polypropylene forms corrugated surfaces at outer boundaries of the printed structures. Corrugation of the surfaces in effect constitutes a large and uneven surface area when compared to smooth glass surfaces. Passage of eluent and analyte along high surface area of the walls of printed chromatography module, is impeded and leads to uneven flow of the eluent through the column. As result the desired product may bear a larger amount of trace impurities as compared to that purified in glass chromatography column. We observed that the yield and purity of the shorter Sulfo-NHS diazirine 1b is slightly better when synthesised in reactionware. Yield of the longer analogue 2b is identical for both glassware and reactionware syntheses.| Glassware | Reactionware | |||
|---|---|---|---|---|
| Yield% | Purity% | Yield% | Purity% | |
| 1a | 43 | 97 | 39 | 94 |
| 1b | 31 | 94 | 34 | 95 |
| 2a | 30 | 99 | 30 | 97 |
Validation experiments
Bearing in mind the nature of utility of diazirines, we felt that it was important to validate the functionality of the material produced in the reactionware syntheses. Although molecules of this class can be made to interact with a wide range of substrates, in our validation experiments we have used human Angiotensin II, a relatively short peptide hormone. This allowed us to verify the outcomes of reactions, using relatively common analytical methods, such as ESI-MS and DEPTQ carbon NMR. The specific purpose of the experiments was to observe and verify interaction of diazirines with the peptide in the first instance and then compare this interaction between diazirines synthesized by traditional methods (in glassware) and those synthesized in reactionware to determine whether any significant differences in reactivity could be observed.In the first instance Angiotensin II was stirred in solution with each of the photo-probes at room temperature. The resulting solution was irradiated under UV light, to activate the diazirine moiety. Subsequently the mixtures were probed by DEPTQ NMR. This data was compared against control experiments, which comprised of experimental runs without either Angiotensin II or a photo-probe. An example comparison of stacked spectra is presented in Fig. 4 below. Each of the two spectra (c and d) on the bottom of the figure reveal areas (highlighted in red) where more peaks were found when compared against the sum of the two top spectra (a and b). It is expected that a chemical modification, as illustrated in (Fig. 4e) should produce a peak shift for the carbon centre (encircled). Whereas peak shift and inversion would be expected in DEPTQ NMR. Both effects can be clearly observed on spectra c and d.
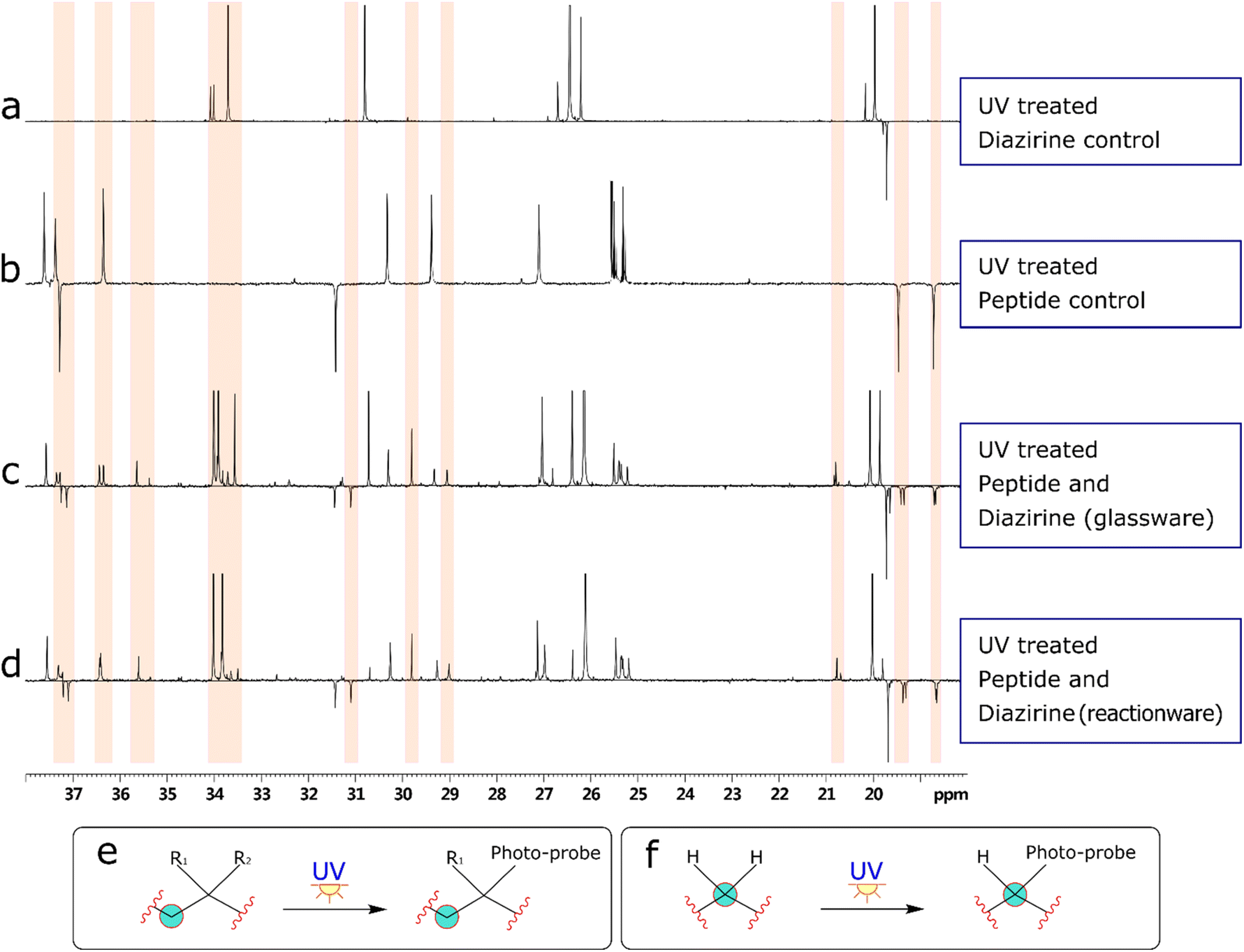 | ||
| Fig. 4 (a) and (b) show spectra from control experiments. Spectra (c) and (d) depict spectra obtained from the validation experiments. Areas highlighted in red indicate parts of spectra (c) and (d) where additional peaks are found when correlated with spectra (a) and (b). (e) and (f) depict examples of chemical modifications affecting the observed changes in spectra (c) and (d). Illustration (e) is not representative of coupling between any specific groups. | ||
Based on the NMR data alone it was of course difficult to infer the identities of any new species present in the crude mixtures. For this reason, ESI-MS experiments were carried out to verify that the observed masses corresponded to the anticipated products of reaction between the peptide and diazirine compounds. As expected, a number of species were identified, however those common to all of the experiments, matched with products a, b and c presented in Fig. 5. Both diazirines obtained from reactionware as well as those synthetized in glassware produced very similar results, therefore suggesting equal potency. The detailed procedure for the validation experiments, including complete analytical data can be found in the ESI† as well as full details to reproduce the work including the fabrication of the 3D printed reactionware.
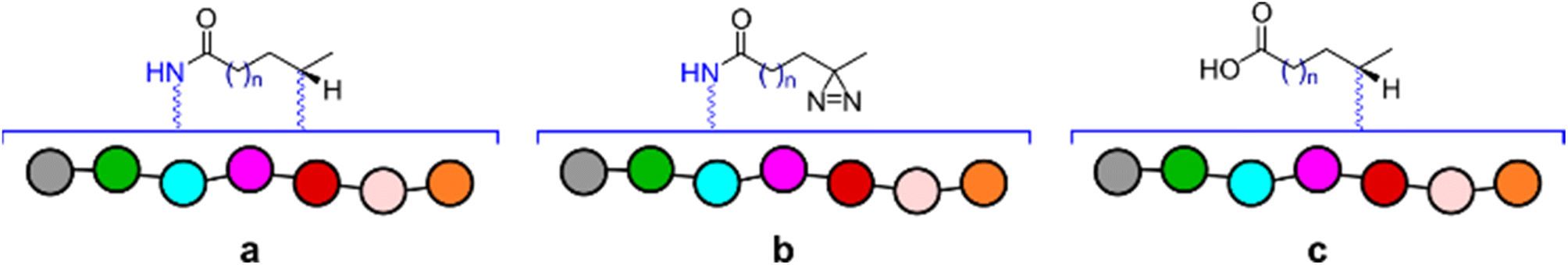 | ||
| Fig. 5 Depiction of species common to all validation experiments for which peaks with corresponding masses were identified. | ||
In previous works, we introduced the concept of 3D printed chemical reactors1 and demonstrated how their use may benefit those who wish to carry out a small-scale synthesis of high value compounds.2 Moreover, previously our reactors were designed to serve targeted synthesis of specific molecules and were not designed to accommodate any bifurcation of the synthesis to obtain analogue compounds, when required. This work demonstrates a step change in the area of reactionware design, where parts of synthetic process take place in designated sets of reactor modules which are interchangeable with other sets in such way, as to enable synthesis of analogues. One reactor set serves to install a central moiety to the backbone of a molecule, whereas other reactor sets serve to install peripheral moieties to the already completed molecular backbone. Furthermore, one set of instructions can be used to obtain at least two or potentially more structurally related and high value compounds, allowing for widening of the spectrum of one's research, whenever their requirements for analogue substances increases. We believe this approach is safer, more accessible, reduces waste, and should help both non-expert synthetic chemists and molecular biologists gain access to important labels that are both expensive and have extremely short-lived shelf life.
Data availability
The STL files used to 3D print the reactors are available with the publication as ESI.†Author contributions
L. C. invented the concept, devised the project and helped run the team with P. J. K. P. F. identified the synthetic targets, carried out all associated synthesis, designed, optimised and manufactured the reactionware. P. F wrote the manuscript and associated ESI† with help and guidance from P. J. K and L. C. P. J. K acted as a mentor and helped to devise specific design solutions for the reactionware devices. A. J. contributed the initial idea for the synthetic targets and carried out some preliminary synthesis and optimisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the EPSRC (Grant no. EP/L023652/1, EP/R020914/1, EP/S030603/1, EP/R01308X/1, EP/S017046/1, and EP/S019472/1), the ERC (Project no. 670467 SMART-POM), the EC (Project no. 766975 MADONNA), The John Templeton Foundation (Project no. 60625 and 61184), and DARPA (Project no. W911NF-18-2-0036, W911NF-17-1-0316, and HR001119S0003).References
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS PubMed
.
-
National Research Council, Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering, The National Academies Press, Washington, DC, 2003 Search PubMed
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed
- C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 17114–17116 CrossRef CAS PubMed
- Glassware: The protagonist of the laboratory|Science Museum. https://www.sciencemuseum.org.uk/objects-and-stories/chemistry/glassware-protagonist-laboratory.
- A. F. Donnelly, Lab. Med., 1970, 1, 28–33 CrossRef
-
K. Espahangizi, M. Grote, M. Stadler and L. Otis, Membranes, Surfaces and Boundaries: Interstices in the History of Science, Technology and Culture, 2011, pp. 17–33 Search PubMed
- H. Kodama, Rev. Sci. Instrum., 1981, 52, 1770–1773 CrossRef
- A. J. Capel, R. P. Rimington, M. P. Lewis and S. D. Christie, Nat. Rev. Chem., 2018, 2, 422–436 CrossRef
- D. Therriault, S. R. White and J. A. Lewis, Nat. Mater., 2003, 2, 265–271 CrossRef CAS PubMed
- T. Hasegawa, K. Nakashima, F. Omatsu and K. Ikuta, Sens. Actuators, A, 2008, 143, 390–398 CrossRef CAS
- F. Ilievski, A. D. Mazzeo, R. F. Shepherd, X. Chen and G. M. Whitesides, Angew. Chem., Int. Ed., 2011, 50, 1890–1895 CrossRef CAS PubMed
- B. Y. Ahn, E. B. Duoss, M. J. Motala, X. Y. Guo, S. I. Park, Y. J. Xiong, J. Yoon, R. G. Nuzzo, J. A. Rogers and J. A. Lewis, Science, 2009, 323, 1590–1593 CrossRef CAS PubMed
- M. D. Symes, P. J. Kitson, J. Yan, C. J. Richmond, G. J. T. Cooper, R. W. Bowman, T. Vilbrandt and L. Cronin, Nat. Chem., 2012, 4, 349–354 CrossRef CAS PubMed
- P. J. Kitson, G. Marie, J.-P. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS PubMed
- W. Hou, A. Bubliauskas, P. J. Kitson, J.-P. Francoia, H. Powell-Davies, J. M. P. Gutierrez, P. Frei, J. S. Manzano and L. Cronin, ACS Cent. Sci., 2021, 7, 212–218 CrossRef CAS PubMed
- S. S. Zalesskiy, P. J. Kitson, P. Frei, A. Bubliauskas and L. Cronin, Nat. Commun., 2019, 10, 5496 CrossRef CAS PubMed
- G. Chisholm, P. J. Kitson, N. D. Kirkaldy, L. G. Bloor and L. Cronin, Energy Environ. Sci., 2014, 7, 3026–3032 RSC
- S. Paulsen, Angew. Chem., 1960, 72, 781–782 CrossRef CAS
- E. Schmitz and R. Ohme, Chem. Ber., 1961, 94, 2166–2173 CrossRef CAS
- M. L. Lepage, C. Simhadri, C. Liu, M. Takaffoli, L. Bi, B. Crawford, A. S. Milani and J. E. Wulff, Science, 2019, 366, 875–878 CrossRef CAS PubMed
- Y. Chevolot, O. Bucher, D. Léonard, H. J. Mathieu and H. Sigrist, Bioconjugate Chem., 1999, 10, 169–175 CrossRef CAS PubMed
- A. Blencowe, K. Cosstick and W. Hayes, New J. Chem., 2006, 30, 53–58 RSC
- R. Sun, L. Yin, S. Zhang, L. He, X. Cheng, A. Wang, H. Xia and H. Shi, Chem.–Eur. J., 2017, 23, 13893–13896 CrossRef CAS PubMed
- T. Tamura, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 2216–2226 CrossRef CAS PubMed
- D. C. McCutcheon, G. Lee, A. Carlos, J. E. Montgomery and R. E. Moellering, J. Am. Chem. Soc., 2019, 142, 146–153 CrossRef PubMed
- Y. Yang, M. He, T. Wei, J. Sun, S. Wu, T. Gao and Z. Guo, Anal. Chim. Acta, 2020, 1107, 164–171 CrossRef CAS PubMed
- Y. Agapkina, D. Agapkin, A. Zagorodnikov, Y. I. Alekseev, G. Korshunova and M. Gottikh, Russ. J. Bioorg. Chem., 2002, 28, 293–299 CrossRef CAS PubMed
- A. L. MacKinnon and J. Taunton, Curr. Protoc. Chem. Biol., 2009, 1, 55–73 CrossRef PubMed
- L. Wang, A. Ishida, Y. Hashidoko and M. Hashimoto, Angew. Chem., 2017, 129, 888–891 CrossRef
- L. Wang, Z. P. Tachrim, N. Kurokawa, F. Ohashi, Y. Sakihama, Y. Hashidoko and M. Hashimoto, Molecules, 2017, 22, 1389 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00108c |
| This journal is © The Royal Society of Chemistry 2023 |