
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sewage remediation using solar energy and a triply fused Zn-porphyrin dimer molecular graphene photocatalytic agent†
Fei
Cheng
a,
Taotao
Qiang
 *a,
Mingli
Li
a and
Tony D.
James
*bc
*a,
Mingli
Li
a and
Tony D.
James
*bc
aCollege of Bioresources and Materials Engineering, Shaanxi Collaborative Innovation Center of Industrial Auxiliary Chemistry & Technology, Shaanxi University of Science & Technology, Xi'an, 710021, China
bDepartment of Chemistry, University of Bath, Bath, BA27AY, UK
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 5th October 2023
Abstract
Photocatalysis plays a pivotal role in achieving clean energy and a pollution-free environment. However, its practical application is limited by the complexity of photoreactors and the fragility of external light-emitting diode (LED) sources. In this study, a novel zinc diporphyrin molecular graphene Oligo-Zn-Por photocatalyst was synthesized using the intermolecular edge-matching effect of porphyrins and oxidative fusion using a high-valence metal (Sc3+). The photocatalyst displayed Cr(VI) reduction ability using direct solar energy. The stacking of graphene lamellae and a unique void structure were observed. Oligo-Zn-Por exhibited excellent photogenerated electron capacity (∼60 μA cm−2) and reasonable photogenerated electron lifetimes at multiple wavelengths (5.4, 5.5 and 21.2 ps at absorption wavelengths of 413, 576 and 1040 nm, respectively), thus enhancing photovoltaic conversion efficiency. Oligo-Zn-Por exhibited high Cr(VI) reduction efficiency (over 90% reduction at Cr(VI) concentrations below 60 mg L−1) when a low dose (5 mg) of the catalyst was used in an aqueous environment under the irradiation of sunlight. Furthermore, the catalyst exhibited a high Cr(VI) reduction rate of 90% under weak alkaline conditions, demonstrating high environmental adaptability. Additionally, after ten cycles, the catalyst exhibited a high cycling ability and retained 99% of its initial Cr(VI) reduction rate. Thus, the excellent catalytic performance of Oligo-Zn-Por makes it an ideal material for constructing inexpensive, portable, and non-external power driven photocatalytic reactors.
Introduction
As a revolutionary technology for treating environmental pollution, photocatalytic reactions play a pivotal role in maintaining a healthy environment and ecological balance.1–11 However, the utilization and effectiveness of photocatalytic technology are significantly hindered by the intricate nature of photoreactors and the construction of external LED light sources, which consequently necessitates a fixed working environment, expensive instrument upkeep and maintenance, and a large energy expenditure.12–17 Solar energy, a renewable energy source, can effectively replace external electrical LED light sources in photocatalytic systems owing to its various advantages, such as the synergy between energy development and environmental treatment, portability and ease of operation of the instrument, low cost, and low energy consumption.18–21 To replace an external electrical LED light source with a solar energy source, the photocatalyst must exhibit the following three properties: 1) a small energy band gap to ensure a large light absorption range, 2) a low photogenerated charge carrier complexation rate to ensure a high photogenerated electron lifetime, and 3) a large specific surface area to ensure enhanced adsorption sites for a complete photocatalytic process.22–24 However, due to the high compounding rate of the photogenerated charge carriers, the photocatalytic activity of low bandgap semiconductors is greatly inhibited, thus limiting the application of solar energy in replacing external electrical LED light sources in photocatalytic systems.Polyporphyrin fused molecular graphene is a new material that combines the robust rigid planar structure of traditional hydrocarbon graphene with good stability and high molar extinction coefficients.25–27 Meanwhile, the molecular structure can be precisely tuned by altering the degree of molecular conjugation, and introducing heteroatoms and metal ions. In addition, a low singlet (S1)/triplet (T1) energy level difference as well as spin–orbit coupling strength of S1/T1 enables intersystem conversion (ISC: the process by which electrons in an excited molecule undergo a spin reversal to a non-radiative transition that changes the multiplicity of the molecule) and a reduced rate of photogenerated charge carrier recombination.28–30
With this research, we developed a novel zinc complex porphyrin fused molecular graphene photocatalyst, Oligo-Zn-Por, with a large optical absorption range (300–1184 nm) and small HOMO–LUMO band gap (Eg = 1.36 eV), which exhibits a graphene lamellar stacking structure. The photocatalyst exhibits a low photogenerated electron/hole recombination rate, as evidenced by its small Nyquist arc radius, high photocurrent intensity (∼60 μA cm−2) and picosecond photogenerated electron lifetimes (5.4 ps, 5.5 ps and 21.2 ps at 413 nm, 576 nm and 1040 nm wavelengths, respectively). Furthermore, solar photocatalytic reduction of Cr(VI) showed that a low dose of Oligo-Zn-Por (5 mg) could maintain a high reduction efficiency of Cr(VI) (up to 90% when the concentration of Cr(VI) was lower than 60 mg L−1), with excellent pH stability (90% reduction rate of Cr(VI) under weak alkaline conditions) and cycling stability (less than 1% loss of Cr(VI) reduction rate after ten cycles). This result shows that Oligo-Zn-Por material can be used to construct a low-cost, portable, LED light source photocatalytic reactor without external power.
Results and discussion
Characterization
Fig. 1a shows the synthetic route for Por, Zn-Por and Oligo-Zn-Por. Details of the synthesis and the characterization of each compound are given in Fig. S1–S5.† High-resolution field emission scanning electron microscopy (SEM) images of Zn-Por and Oligo-Zn-Por are shown in Fig. 1b–d. SEM images reveal the hypodynamic planar structure of Zn-Por and the incomplete arylation of the porphyrin meso-site results in a disordered stacking state (Fig. 1b). Meanwhile, Oligo-Zn-Por can overcome its π–π stacking effect due to a stiff and rigid planar structure with multiple aryl groups perpendicular to the porphyrins, and thus exhibits an ordered lamellar stacking structure (Fig. 1c).31,32 The use of multiple perpendicular aryl groups to overcome the π–π stacking effect endowed Oligo-Zn-Por with significant intermolecular porosity, thereby enhancing the penetration and utilization of solar light (Fig. 1d).33Fig. 1e shows the dispersion of Oligo-Zn-Por in buffers of different pH values. The lower the pH value, the better the dispersion, indicating that the acidic environment improves the dispersion of Oligo-Zn-Por in aqueous solutions. Furthermore, proton nuclear magnetic resonance spectroscopy (1H NMR) and high-resolution mass spectrometry (HR-MS) were used to analyze Oligo-Zn-Por. The results are shown in Fig. 1f and g. 1H NMR of the porphyrin monomer Por exhibits porphyrin monomer peaks of four porphyrin β-hydrogens (delta = 9.46, 9.27, 9.20, and 9.16 ppm), four porphyrin meso-substituted benzene ring hydrogens (delta = 8.35, 8.32, 8.03, and 8.01 ppm), porphyrin meso-hydrogens (delta = 10.31 ppm), and porphyrin intra-ring nitrogen hydrogens (delta = −2.64 ppm).34 In contrast to the porphyrin monomer Por, the 1H NMR of the metalloporphyrin Zn-Por exhibits the disappearance of the high field −2.64 ppm peak for the intra-ring nitrogen hydrogen signals of the porphyrin, indicating that the porphyrin ring was successfully complexed with the metal ion The final molecule Oligo-Zn-Por was obtained through the oxidation of Zn-Por using a high-valence metal,35,36 which was confirmed by 1H NMR with the disappearance of the meso-hydrogen signal (delta = 10.26 ppm) of the porphyrin and the β-hydrogens (two sets of double peaks delta = 7.75 and 7.71 ppm and a single peak delta = 7.35 ppm) due to the intermolecular (β–β, meso–meso, β–β) linkage. Simultaneously, the intermolecular condensation causes the formation of a larger conjugated system, which weakens the aromaticity and shifts the porphyrin β-hydrogen and the intermediate aromatic ring substituted hydrogen signals to a higher field. High-resolution mass spectroscopic data display a molecular ion peak of m/z = 1867.0891 (calculated value of 1866.9627) for Oligo-Zn-Por, further demonstrating the successful preparation of Oligo-Zn-Por. The absorption spectra indicates that Oligo-Zn-Por with a larger π-system resulted in a significant red shift in its absorption wavelength compared with Zn-Por.37 These results are shown in Fig. 2a. Oligo-Zn-Por absorption spectrum exhibits a split peak in the Soret band and a red shift in the Q-band to 1040 nm in the near-infrared spectra compared with Zn-Por. This phenomenon confirms the feasibility of a strategy to extend the light absorption range of molecular graphene materials through oxidation by high-valence metals to form polyporphyrin.38–40 At the same time, the absorption spectrum of Oligo-Zn-Por exhibits a clear solvent dependence (Fig. S6†). With increasing solvent polarity, the solubility of Oligo-Zn-Por significantly decreased, thereby decreasing its absorption intensity. The absorption intensity was almost negligible due to the insolubility of Oligo-Zn-Por in water. These results indicated that Oligo-Zn-Por could be completely separated from the reaction system after the photocatalytic reduction of Cr(VI), resulting in 100% recovery of Oligo-Zn-Por and preventing secondary contamination of the reaction system. The results from the cyclic voltammetry tests revealed that the energy level difference between Zn-Por and Oligo-Zn-Por was 1.65 eV and 1.36 eV respectively (Fig. 2b), signifying that the high-valence metal oxidation strategy can decrease the photocatalyst energy level difference, thus resulting in the red shift of the absorption wavelength.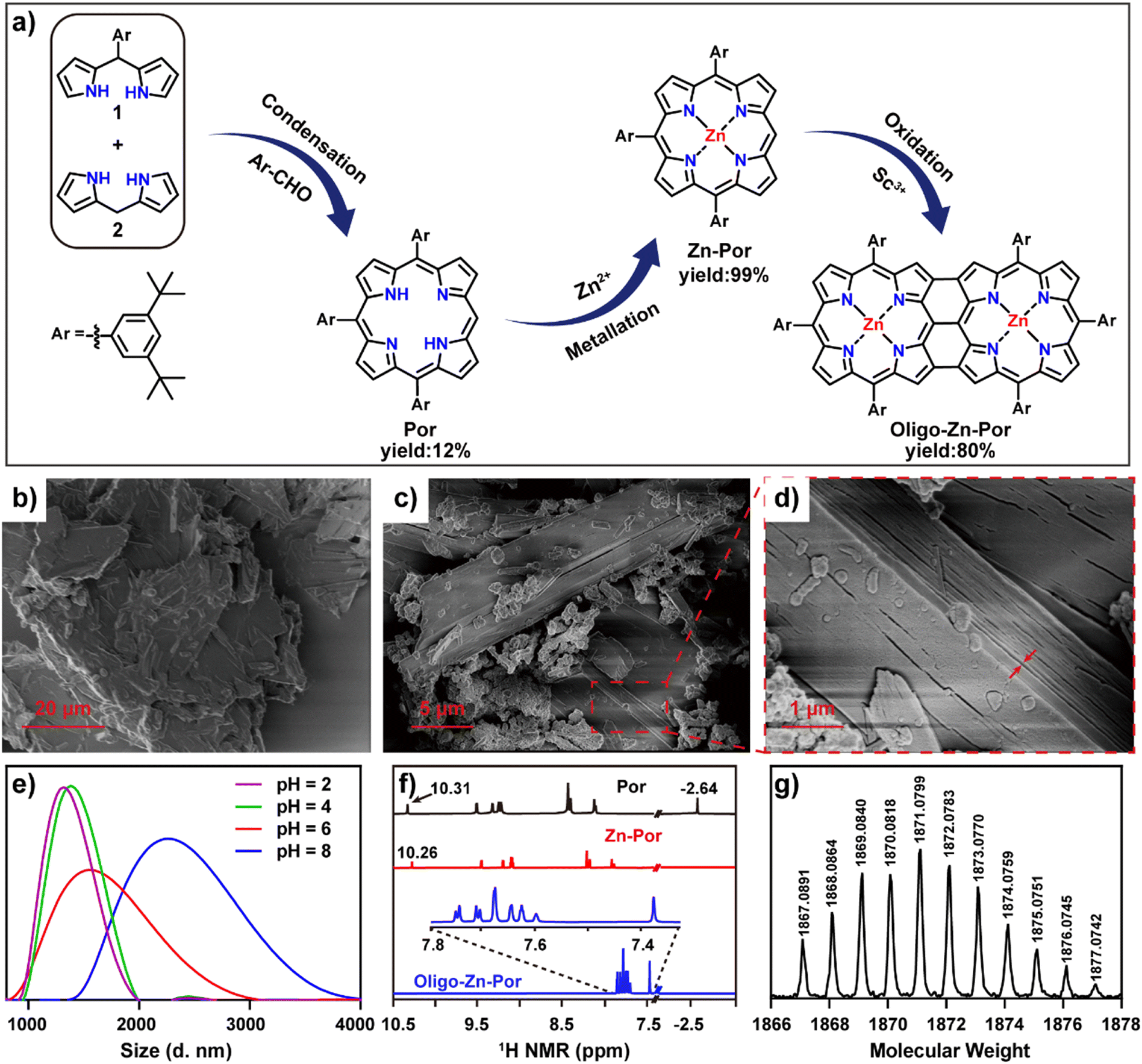 | ||
| Fig. 1 (a) Schematic diagram of the Oligo-Zn-Por synthetic route and yield; high-resolution field emission scanning electron microscopic images of Zn-Por (b) and Oligo-Zn-Por (c); (d) magnified view of (c); (e) dynamic light scattering spectra of Oligo-Zn-Por at different pH conditions; (f) 1H NMR spectra of Por, Zn-Por, and Oligo-Zn-Por; (g) high-resolution mass spectra of Oligo-Zn-Por. | ||
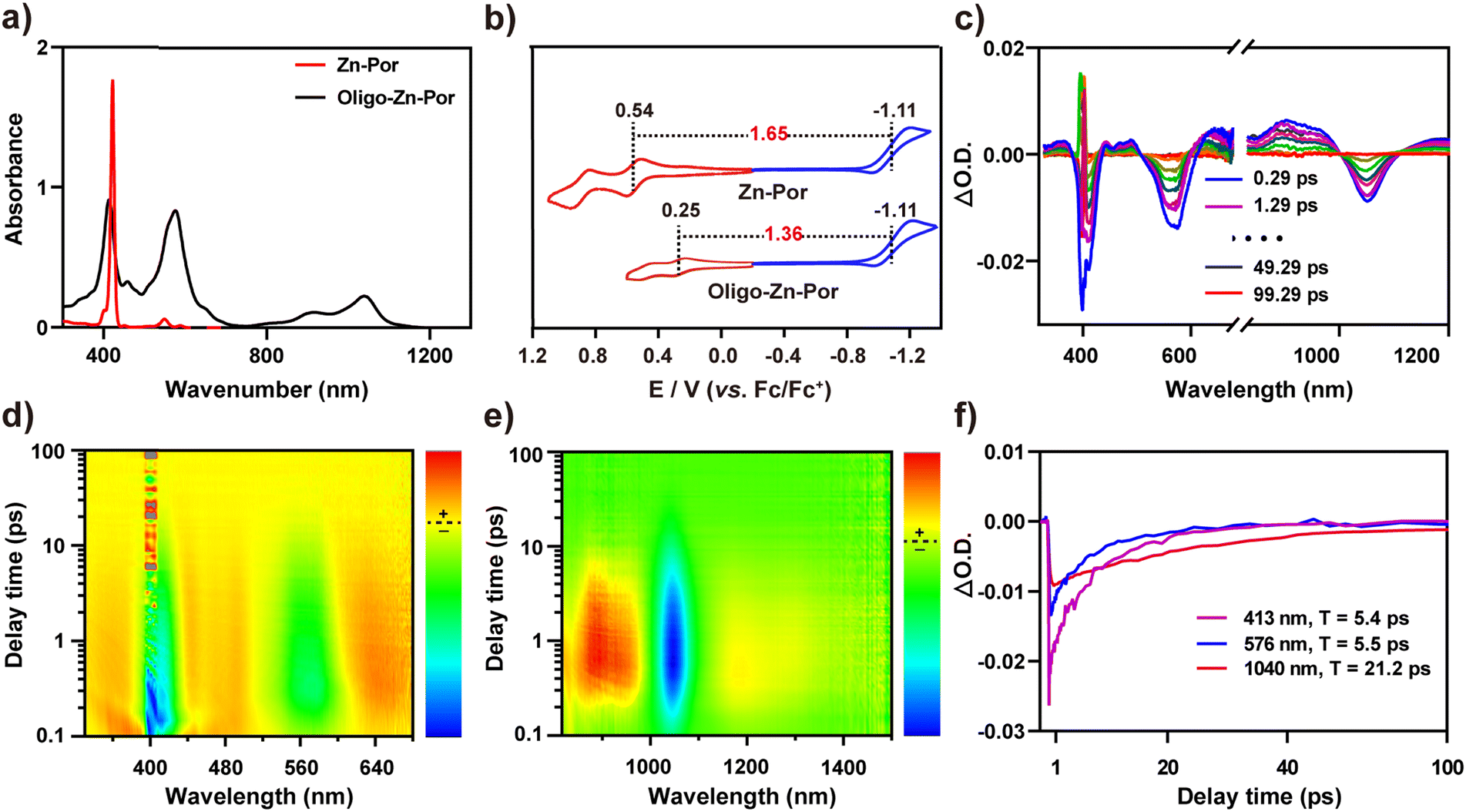 | ||
| Fig. 2 (a) Absorption spectra of Zn-Por and Oligo-Zn-Por (solvent dichloromethane, c = 10−5 M); (b) cyclic voltammetric profiles of Zn-Por and Oligo-Zn-Por (conditions: supporting electrolyte: 0.1 M nBu4NPF6 in CH2Cl2, counter electrode: Pt wire, reference electrode: Ag/AgNO3, working electrode: glassy carbon. Scan rate: 0.05 V s−1); (c) variation of the femtosecond-transient absorption spectra of Oligo-Zn-Por (0.29–99.29 ps) (dichloromethane solvent, excitation wavelength of 400 nm, c = 10−4 M); (d) top view of the 2D femtosecond-transient absorption spectrum of Oligo-Zn-Por (dichloromethane solvent, excitation wavelength 400 nm, wavelength detection range 320–680 nm, c = 10−4 M); (e) top view of the 2D femtosecond-transient absorption spectrum of Oligo-Zn-Por (dichloromethane solvent, excitation wavelength 400 nm, wavelength detection range 820–1500 nm, c = 10−4 M); (f) femtosecond-transient absorption spectral kinetics of Oligo-Zn-Por at different absorption wavelengths. | ||
To further explore the photogenerated electron capability of the Oligo-Zn-Por photocatalyst, the charge transfer kinetics and the photogenerated electron lifetime of Oligo-Zn-Por were investigated.41–43 The results are shown in Fig. 2c–e. Femtosecond-transient absorption detection of Oligo-Zn-Por over the full absorption band (320–1500 nm) was performed using 400 nm excitation, and two positive signals at 458 and 916 nm were observed from the two-dimensional top view spectra and transient absorption spectra of the transient absorption contour plots of Oligo-Zn-Por. The two positive signals were attributed to the electron generation of the S1–Sn transition (ESA, a singlet rather than a triplet transition as determined by the picosecond lifetime of the process). The three negative signals at 413, 576 and 1040 nm are attributable to ground state bleaching (GSB) or the excited emission (SE) from the S1–S0 transition of the electron. However, the absolute quantum yield of Oligo-Zn-Por was approximately 0.3%, indicating the absence of stimulated emission (SE), which implied that the three negative signals at 413, 576 and 1040 nm were all GSB signals due to the S1–S0 transition. Thus, with this research, the photogenerated electron lifetime of Oligo-Zn-Por was characterized via measuring the intensity of the three GSB absorption wavelengths over time. The results are shown in Fig. 2f. Only fast charge recombination was observed in the transient absorption spectra at absorption wavelengths (λabs) of 413 and 576 nm, which results in photogenerated electron lifetimes of 5.4 ps and 5.5 ps at specific wavelengths of 413 and 576 nm, respectively. In addition, the transient absorption spectrum at 1040 nm showed a decay curve that can be fitted using two components: a fast component (0–0.69 ps) and a slow component (0.69–100 ps). The fast component signified the population of the lowest excited states, while the slow component represented the slow charge recombination, yielding a photogenerated electron lifetime of 21.2 ps at 1040 nm. The experimental results indicated that Oligo-Zn-Por exhibited an impressive photogenerated electron lifetime and charge separation efficiency.
We then investigated the photogenerated charge generation and transfer capabilities and photogenerated charge/hole separation efficiency of the photocatalyst using electrochemical impedance spectroscopy (EIS) and transient photocurrent response tests.44 The photogenerated charge transfer capability of the photocatalyst interface was first analyzed using EIS. The size of the arc radius reflects the strength of the interfacial charge transfer resistance of the photocatalyst, i.e., the arc radius is proportional to the interfacial resistance. As shown in Fig. 3a, Zn-Por and Oligo-Zn-Por exhibited different semicircular arc trends in the Nyquist plot. Compared with Zn-Por, a significant decrease in the interfacial resistance occurred in Oligo-Zn-Por, indicating that the creation of a large π-conjugated system improves the photocatalyst charge transfer ability. The transient photocurrent responses of Zn-Por and Oligo-Zn-Por are shown in Fig. 3b. Oligo-Zn-Por exhibited a higher photocurrent response density, indicating that the π-conjugated system extended the light absorption range of the photocatalyst and enhanced the light utilization, thus improving the Oligo-Zn-Por photogenerated electron efficiency (∼60 μA cm−2). The reduced Nyquist arc radius and high photocurrent intensity demonstrate high photoconversion efficiency and good photoelectron/hole separation efficiency of Oligo-Zn-Por, thus providing a theoretical basis for the subsequent efficient catalytic reduction of Cr(VI) through solar energy absorption using Oligo-Zn-Por.
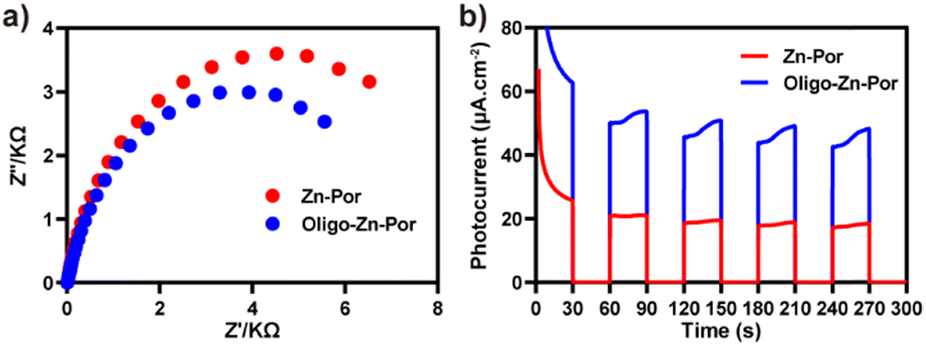 | ||
| Fig. 3 (a) Schematic electrochemical impedance spectra of Zn-Por and Oligo-Zn-Por; (b) schematic transient photocurrent response of Zn-Por and Oligo-Zn-Por. | ||
Photocatalytic performance
Given the above conclusions, a simple experimental device was evaluated for sunlight-driven heavy metal Cr(VI) reduction where the photocatalytic performance of Zn-Por and Oligo-Zn-Por were evaluated (Fig. 4a). Through comparative experiments, we found that in the absence of both Zn-Por and Oligo-Zn-Por in the control group, no significant change was observed in the Cr(VI) content with or without sunlight irradiation, indicating that Cr(VI) could not be actively reduced without the catalyst (Fig. 4b). At the same time, the free Cr(VI) concentration in the control group was significantly higher than that in the Zn-Por group (Zn-Por adsorption of Cr(VI) for 30 min) and the Oligo-Zn-Por group (Oligo-Zn-Por adsorption of Cr(VI) for 30 min), indicating that the catalysts were capable of adsorbing Cr(VI). Furthermore, by examining the Cr(VI) adsorption capacity of Zn-Por and Oligo-Zn-Por, we found that both catalysts reached Cr(VI) adsorption equilibrium within 30 min under dark conditions; afterward, the Cr(VI) concentration remained almost stable with time (Fig. S7†).45 More importantly, under sunlight irradiation, Oligo-Zn-Por had a higher reduction efficiency for Cr(VI) than Zn-Por, which was attributable to the broader light absorption range and superior photogenerated electron capacity of Oligo-Zn-Por (Fig. 4b). Simultaneously, the adsorption and catalytic capability of Oligo-Zn-Por towards Cr(VI) was investigated using a colorimetric method, the results of which are presented in Fig. 2c.46 It was observed that in both the dark and sunlight groups, the color after radiating for 0 min was significantly less than the control group, which was attributed to the adsorption of the catalyst. Afterward, with the extension of the catalytic reaction time, the color of the dark group remained almost unchanged. Meanwhile, the color of the sunlight group was notably diminished, demonstrating that Oligo-Zn-Por can rapidly result in the reduction of Cr(VI) under the influence of solar photocatalysis.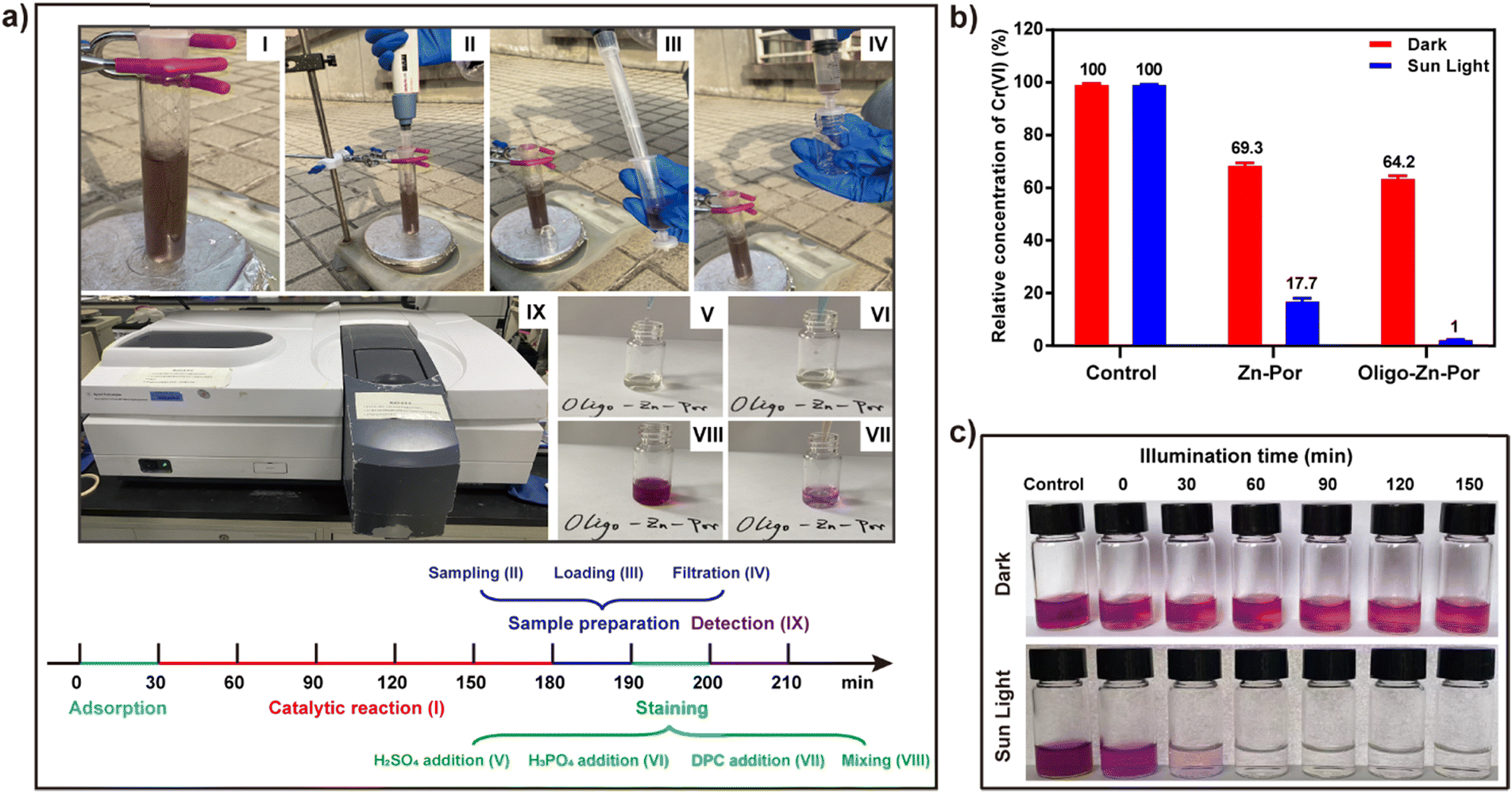 | ||
| Fig. 4 (a) Schematic diagram of the device and process for the catalytic reduction of Cr(VI) by solar energy (Sun light catalytic reaction (I), sampling (II), loading (III), filtration photocatalyst (IV), H2SO4 addition (V), H3PO4 addition (VI), diphenyl carbamide addition (VII), mixing (VIII), detection (IX)); (b) actual Cr(VI) content under different conditions (photocatalyst dosage of 5 mg, Cr(VI) concentration of 10 mg L−1); (c) schematic diagram of the color development of Cr(VI) content under different light conditions (Oligo-Zn-Por dosage of 5 mg, Cr(VI) concentration of 10 mg L−1). | ||
Then, we used the Oligo-Zn-Por photocatalyst to optimize the system conditions and study the ability of Oligo-Zn-Por to reduce Cr(VI) in real samples under optimal photocatalytic conditions. First, we evaluated the effect of the photocatalyst dose on the efficiency of the Cr(VI) reduction (Fig. S8†). The result indicated that both Zn-Por and Oligo-Zn-Por photocatalysts effectively reduced Cr(VI). The photocatalytic efficiency of Oligo-Zn-Por was significantly higher than that of Zn-Por. However, the Cr(VI) reduction rate was inversely proportional to the photocatalyst dosage, indicating that a high concentration of photocatalyst increased the chance of intermolecular collisions, thus limiting its photogenerated electron capacity and consequently reducing the effectiveness of photocatalytic reduction of Cr(VI). In addition, the above experimental results confirmed 99% reduction of Cr(VI) by Oligo-Zn-Por at a dose of 5 mg, which is a lower dose of catalyst than those used in other reported research (20–50 mg).44–46 Moreover, Oligo-Zn-Por was used for the solar photocatalytic reduction of Cr(VI) at different concentrations to further investigate the maximum efficiency of the photocatalyst. The results are shown in Fig. S9.† The Cr(VI) reduction rate decreased with increasing Cr(VI) concentration. When the Cr(VI) concentration was below 60 mg L−1, the reduction rates were above 90%, indicating that Oligo-Zn-Por could reduce high concentrations of Cr(VI). Industrial effluent discharges are usually acidic; such effluent requires a photocatalyst with high stability in acidic environments. Therefore, we evaluated the efficiency of the Oligo-Zn-Por photocatalyst for solar-catalytic reduction of Cr(VI) under different pH conditions. The results indicated that Oligo-Zn-Por exhibited high efficiency for the catalytic reduction of Cr(VI) under different pH environments (all above 80% reduction), and the reduction efficiency was enhanced in acidic environments (pH = 2–6), which were all above 84%. This outstanding reduction efficiency of the catalyst can be attributed to its higher dispersion performance in acidic environments (Fig. 1e and S10†).
We also investigated the effect of the sunlight intensity on the photocatalytic Cr(VI) reduction rate of Oligo-Zn-Por at different times of the day. As shown in Fig. S11,† the highest Cr(VI) reduction efficiency of 84.6% was obtained at 13:00 using Oligo-Zn-Por. The Oligo-Zn-Por-catalyzed Cr(VI) reduction rate under sunlight irradiation was more than 99% at 13:00. The result indicates that the Oligo-Zn-Por photocatalyst is sensitive to sunlight intensities and can effectively reduce Cr(VI) under different sunlight intensities. To further verify the effect of sunlight on the Cr(VI) reduction efficiency of Oligo-Zn-Por during the year, catalytic reduction of Cr(VI) experiments were performed under sunlight irradiation over months. The experimental results indicated that the highest Cr(VI) reduction rate of Oligo-Zn-Por (84.6%) was observed in July. However, in January, the Cr(VI) reduction rate of Oligo-Zn-Por was still more than 91% compared with July, when the sunlight intensity was extremely weak, thus indicating good adaptability of the catalyst at different intensities of sunlight (Fig. S12†).
Stability of the photocatalyst
Photocatalyst stability is an important indicator of photocatalytic performance of photocatalysts. A stable photocatalyst can contribute to an economical and efficient photocatalytic reaction.47 With this research, the photocatalytic cycle stability of Oligo-Zn-Por photocatalyst for reducing Cr(VI) under sunlight irradiation was investigated As shown in Fig. S13,†Oligo-Zn-Por retained more than 99% of the initial efficiency after ten photocatalytic cycles, indicating that the Oligo-Zn-Por photocatalyst exhibited good stability. Finally, the stability of the photocatalyst was confirmed using 1H NMR. 1H NMR analysis of the photocatalyst Oligo-Zn-Por after ten cycles of the solar photocatalytic reduction of Cr(VI) indicated no structural changes, demonstrating good stability of the Oligo-Zn-Por photocatalyst (Fig. S14†). In addition, the XPS spectrum of Oligo-Zn-Por after ten cycles of experiments show that there are two sets of single peaks, Cr 2p3/2 and Cr 2p1/2 of Cr(III), at 577.96 and 587.07 eV, respectively (Fig. S15†). This result show that the reduction of Cr(VI) to Cr(III) by Oligo-Zn-Por catalyzed by sunlight can be realized.48Mechanism of photocatalysis
Finally, we evaluated the mechanism of the solar photocatalysis of Cr(VI) reduction by Oligo-Zn-Por using radical, hole, and electron capture experiments and electron spin resonance (ESR) spectroscopy.48–50 The active components of the solar photocatalytic reduction of Cr(VI) were determined using benzoquinone (BQ), isopropanol (IPA), disodium ethylene diamine tetraacetic acid (EDTA-2Na) and potassium bromide (KBr) as trapping agents for superoxide radicals (·O2−), hydroxyl radicals (·OH), holes (h+), and electrons (e−), respectively. Fig. 5a shows the effect of adding different trapping agents on the Cr(VI) reduction rate of Oligo-Zn-Por under sunlight irradiation. The results indicated that BQ and KBr had a significant impact by decreasing the rate of Cr(VI) reduction (the reduction rates of Cr(VI) were 43.5% and 31.1%, respectively), whereas IPA and EDTA-2Na caused an increase in the reduction rate (the reduction rates of Cr(VI) were 93.4% and 94.7%, respectively). Therefore, ·O2− and ·OH were the main active species in the photocatalytic reduction process. As shown in Fig. 5b, the ESR spectrum of Oligo-Zn-Por exhibits six peaks of DMPO–·O2− and four signal peaks of DMPO–·OH, indicating that the reduction of Cr(VI) using Oligo-Zn-Por under sunlight irradiation produces ·O2− and ·OH as the two active species. It was further proved that ·O2− and ·OH could generate and participate in the photocatalytic reduction of hexavalent chromium. Specifically, when the ·O2− was captured by BQ, the continuity of the photocatalytic reaction was blocked, the production of H2O2 was reduced, and the efficiency of photocatalytic reduction of Cr(VI) was inhibited. When ·OH was trapped by IPA, the formation of H2O2 was promoted, and the efficiency of photocatalytic reduction of Cr(VI) was improved. After EDTA-2Na trapping the h+, the recombination rate of e− with the h+ was reduced, and more e− was released to participate in the Cr(VI) reduction reaction. In brief, photogenerated e− plays a dominant role in the photocatalytic reduction of Cr(VI).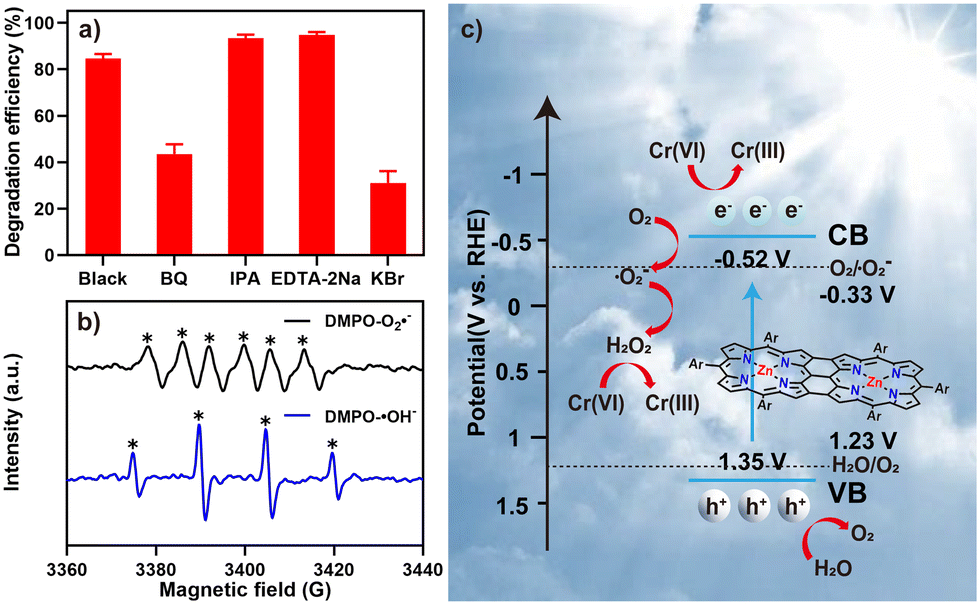 | ||
| Fig. 5 (a) Photocatalytic reduction of Cr(VI) using Oligo-Zn-Por under sunlight irradiation in the presence of different trapping agents (Oligo-Zn-Por dosage of 5 mg, trapping agent dosage of 2 mM, Cr(VI) concentration of 70 mg L−1); (b) electron spin resonance (ESR) spectra of superoxide radicals and hydroxyl radicals after visible light irradiation for 180 s; (c) sunlight-catalyzed schematic diagram of the Oligo-Zn-Por Cr(VI) reduction process. | ||
In order to further study the mechanism of the photocatalytic reduction of Cr(VI) by Oligo-Zn-Por, the energy band structure of the photocatalyst Oligo-Zn-Por was characterized (Fig. 5c). The valence band (VB) and band gap (Eg) of Oligo-Zn-Por were confirmed by valence-band X-ray photoelectron spectrum and UV-vis DRS. As shown in Fig. S16,† the VB value (EVB = 1.35 V) of Oligo-Zn-Por was obtained by linear intersection method. At the same time, the band gap (Eg = 1.87 eV) was obtained by UV-vis DRS (Fig. S17†). The CB value (ECB = −0.52 V) can be calculated according to the equation: Eg = EVB − ECB. Therefore, the electrons produced by photoexcitation of Oligo-Zn-Por can react with oxygen to form ·O2− (O2 + e− → ·O2−) because the CB value of Oligo-Zn-Por is more negative than O2/·O2− (−0.33 V vs. RHE). Meanwhile, because the VB value of Oligo-Zn-Por is corrected than that of H2O/O2 (1.23 V vs. RHE), the h+ left on VB after light excitation can oxidize H2O to O2 (2H2O + 4 h+ → O2 + 4H+). In addition, ·O2− generated by the reaction of photogenerated e− with O2 can further interact with H2O to produce H2O2 in an acid environment (H2O + ·O2− + H+ → H2O2 + ·OH). The strong reducibility of H2O2 can reduce Cr(VI) to Cr(III) in acidic environment.51
Compared with other photocatalysts
To date, few studies have reported that the catalyst can directly use sunlight for photocatalytic reduction of Cr(VI). Table S1† provides a simple comparison of the work reported in recent years on the reduction of Cr(VI) with different photocatalysts. The results show that the molecular graphene photocatalyst Oligo-Zn-Por developed in this work has the ability of photocatalytic reduction of Cr(VI) by direct use of sunlight. Compared with the reported results (Table S1†), Oligo-Zn-Por has the characteristics of using less amount and direct use of sunlight in the photocatalytic reduction of Cr(VI), which reduces the cost of photocatalytic reduction of Cr(VI) and solves the problem of energy consumption in the actual reduction of Cr(VI).Conclusion
In this study, we developed a zinc-complexed diporphyrin molecular graphene Oligo-Zn-Por photocatalyst using the intermolecular edge-matching effect of porphyrins and oxidative fusion using high-valent metals. This research was the first to explore the photocatalytic reduction performance of Oligo-Zn-Por photocatalyst for treating Cr(VI), a heavy metal ion pollutant, under sunlight irradiation. The absorption spectra revealed that Oligo-Zn-Por had a broader light absorption range than existing photocatalysts, which enhanced its sunlight absorbance capacity. Femtosecond-transient absorption spectroscopy, EIS, and transient photocurrent response experiments revealed that Oligo-Zn-Por exhibited a considerable photogenerated electron capability and lifetime, which enhanced its photovoltaic conversion efficiency. High-resolution field emission SEM experiments showed that Oligo-Zn-Por had a significant void structure, significantly increasing its ability to adsorb and reduce Cr(VI). Finally, the results of the solar-catalysed Cr(VI) reduction experiments demonstrate that the Oligo-Zn-Por photocatalyst is cost-effective, efficient, highly adaptable, and stable. Thus, this study provides a theoretical basis for constructing a low-cost, portable, and LED light source photocatalytic reactor without using external electrical energy.Conflicts of interest
TDJ acts as an academic consultant for TQ as part of a guest professorship at SUST.Acknowledgements
This work was funded by the National Natural Science Foundation of China (52273268) and the Shaanxi Province Technology Innovation Guidance Project (2023GXLH-079). TDJ wishes to thank the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01).References
- J. Li, W. F. Pan, Q. Y. Liu, Z. Q. Chen, Z. J. Chen, X. Z. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. Gordon, G. G. Wallace and D. L. Officer, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- C. D. Wang, Y. Lei, Q. Y. Lv, P. Y. Wang, W. P. Kong, F. Wan and W. G. Chen, Appl. Catal., B, 2022, 315, 121547 CrossRef CAS.
- L. H. Ai, C. H. Zhang, L. L. Li and J. Jiang, Appl. Catal., B, 2014, 148, 191–200 CrossRef.
- B. P. Zhang, P. W. Wong and A. K. An, Chem. Eng. J., 2022, 430, 133054 CrossRef CAS.
- S. Silvestri, A. R. Fajardo and B. A. Iglesias, Environ. Chem. Lett., 2022, 20, 731–771 CrossRef CAS.
- A. Call, M. Cibian, K. Yamauchi and K. Sakai, Sustainable Energy Fuels, 2022, 6, 2160–2164 RSC.
- D. S. Chen, Z. Jin and H. Z. Xing, Langmuir, 2022, 38, 12292–12299 CrossRef CAS PubMed.
- S. M. Ghoreishian, S. R. Ranjith, B. Park, S. K. Hwang, R. Hosseini, R. Behjatmanesh-Ardakani, S. M. Pourmortazavi, H. U. Lee, B. Son, S. Mirsadeghi, Y. K. Han and Y. S. Huh, Chem. Eng. J., 2021, 419, 129530 CrossRef CAS.
- S. M. Ghoreishian, S. R. Ranjith, M. Ghasemi, B. Park, S. K. Hwang, N. Irannejad, M. Norouzi, S. Y. Park, R. Behjatmanesh-Ardakani, S. M. Pourmortazavi, S. Mirsadeghi, Y. K. Han and Y. S. Huh, Chem. Eng. J., 2023, 452, 139435 CrossRef CAS.
- D. X. Wang, J. Chen, X. Gao, Y. H. Ao and P. F. Wang, Chem. Eng. J., 2022, 4331, 134105 CrossRef.
- J. G. Yu, W. G. Wang, B. Cheng and B. L. Su, J. Phys. Chem. C, 2009, 113, 6743–6750 CrossRef CAS.
- M. Shekofteh-Gohari, A. Habibi-Yangjeh, M. Abitorabi and A. Rouhi, Crit. Rev. Environ. Sci. Technol., 2018, 48, 806–857 CrossRef CAS.
- S. Zinatloo-Ajabshir and M. Salavati-Niasari, J. Mol. Liq., 2016, 216, 545–551 CrossRef CAS.
- J. A. Navío, G. Colón, M. Trillas, J. Peral, X. Domènech, J. J. Testa, J. Padrón, D. Rodríguez and M. I. Litter, Appl. Catal., B, 1998, 16, 187–196 CrossRef.
- X. J. Liu, L. K. Pan, Q. F. Zhao, T. Lv, G. Zhu, T. Q. Chen, T. Lu, Z. Sun and C. Q. Sun, Chem. Eng. J., 2012, 183, 238–243 CrossRef CAS.
- C. R. Dong, Y. L. Yang, X. M. Hu, Y. Cho, G. Jang, Y. H. Ao, L. Y. Wang, J. Y. Shen, J. H. Park and K. Zhang, Nat. Commun., 2022, 13, 4982 CrossRef CAS PubMed.
- C. Tudisco, L. Pulvirenti, P. Cool and G. G. Condorelli, Dalton Trans., 2020, 49, 8652–8660 RSC.
- Q. Li, Y. Liu, S. H. Guo and H. S. Zhou, Nano Today, 2017, 16, 46–60 CrossRef CAS.
- R. R. Hernandez, A. Armstrong, J. Burney, G. Ryan, K. Moore-óLeary, I. Diédhiou, S. M. Grodsky, L. Saul-Gershenz, R. Davis, J. Macknick, D. Mulvaney, G. A. Heath, S. B. Easter, M. K. Hoffacker, M. F. Allen and D. M. Kammen, Nat. Sustain., 2019, 2, 560–568 CrossRef.
- X. Z. Zheng and L. W. Zhang, Energy Environ. Sci., 2016, 9, 2511–2532 RSC.
- E. S. Shremzer, D. A. Polivanovskaia, K. P. Birin, G. Yulia, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2023, 210, 110935 CrossRef CAS.
- F. Wu, Y. F. Sun, H. Gao, X. Zhi, Y. Zhao and Z. Shen, Sci. China: Chem., 2023, 66, 164–173 CrossRef CAS.
- Q. Zhang, J. Chen, X. Gao, H. N. Che, P. F. Wang and Y. H. Ao, Appl. Catal., B, 2022, 313, 121443 CrossRef CAS.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- L. M. Mateo, Q. Sun, S. X. Liu, J. J. Bergkamp, K. Eimre, C. A. Pignedoli, P. Ruffieux, S. Decurtins, G. Bottari, R. Fasel and T. Torres, Angew. Chem., Int. Ed., 2020, 59, 1334–1339 CrossRef CAS PubMed.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS PubMed.
- Y. Kuramochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed.
- F. Ema, M. Tanabe, S. Saito, T. Yoneda, K. Sugisaki, T. Tachikawa, S. Akimoto, S. Yamauchi, K. Sato, A. Osuka, T. Takui and Y. Kobori, J. Phys. Chem. Lett., 2018, 9, 2685–2690 CrossRef CAS PubMed.
- Y. Hong, J. Oh, Y. M. Sung, Y. Tanaka, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2017, 56, 2932–2936 CrossRef CAS PubMed.
- N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945–3947 CrossRef CAS PubMed.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124–2127 CrossRef CAS PubMed.
- Y. Y. Li, Y. Q. Wang, J. Lu, W. W. Wang and D. Wang, Chem. Eng. J., 2023, 457, 141299 CrossRef CAS.
- M. Umetani, T. Tanaka, T. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 8095–8099 CrossRef CAS PubMed.
- N. Fukui, W. Cha, D. Shimizu, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2017, 8, 189–199 RSC.
- I. M. Blake, A. Krivokapic, M. Katterle and H. L. Anderson, Chem. Commun., 2002, 1662–1663 RSC.
- Y. Nakamura, S. Y. Jang, T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem. – Eur. J., 2008, 14, 8279–8289 CrossRef CAS PubMed.
- A. K. Sahoo, Y. Nakamura, N. Aratani, S. K. Kim, S. B. Noh, H. Shinokubo, D. Kim and A. Osuka, Org. Lett., 2006, 8, 4141–4144 CrossRef CAS PubMed.
- H. Mori, T. Tanaka, S. Lee, J. M. Lim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 2097–2106 CrossRef CAS PubMed.
- T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem. Sci., 2011, 2, 1414–1418 RSC.
- W. B. Zhao, D. D. Chen, K. K. Liu, Y. Wang, R. Zhou, S. Y. Song, F. K. Li, L. Z. Sui, Q. Lou, L. Hou and C. X. Shan, Chem. Eng. J., 2023, 452, 139231 CrossRef CAS.
- J. Li, W. F. Pan, Q. Y. Liu, Z. Q. Chen, Z. J. Chen, X. Z. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
- H. Wang, D. Y. Yong, S. C. Chen, S. L. Jiang, X. D. Zhang, W. Shao, Q. Zhang, W. S. Yan, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2018, 140, 1760–1766 CrossRef CAS PubMed.
- T. T. Qiang and Y. J. Xia, J. Alloys Compd., 2020, 845, 156155 CrossRef CAS.
- F. P. Zhao, Y. P. Liud, S. B. Hammouda, B. Doshi, N. Guijarro, X. B. Min, C. J. Tang, M. Sillanpää, K. Sivula and S. B. Wang, Appl. Catal., B, 2020, 272, 119033 CrossRef CAS.
- T. T. Qiang, L. Chen and X. T. Qin, Catal. Sci. Technol., 2021, 11, 4931–4943 RSC.
- T. T. Qiang, S. T. Wang, L. F. Ren and X. D. Gao, J. Environ. Chem. Eng., 2022, 10, 108784 CrossRef CAS.
- Q. L. Huang, J. Q. Hu, Y. F. Hu, J. C. Liu, J. L. He, G. B. Zhou, N. Hu, Z. Yang, Y. C. Zhang, Y. Zhou and Z. G. Zou, Environ. Sci.: Nano, 2022, 9, 4433–4444 RSC.
- J. L. He, J. Q. Hu, Y. F. Hu, S. E. Guo, Q. L. Huang, Y. Q. Li, G. B. Zhou, T. Gui, N. Hu and X. S. Chen, Inorg. Chem., 2022, 61, 19961–19973 CrossRef CAS PubMed.
- J. Lin, J. L. He, J. Q. Hu, J. Q. Dong, A. Y. Liu, Y. Yang, L. Q. Tang, L. Li, Y. Zhou and Z. G. Zou, Catal. Sci. Technol., 2021, 11, 3885–3893 RSC.
- X. Y. Yuan, X. Wu, Z. J. Feng, W. Jia, X. X. Zheng and C. Q. Li, Catalysts, 2019, 9, 624 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation and materials/synthesis and characterization/Fig. S1–S13. See DOI: https://doi.org/10.1039/d3cy01131c |
| This journal is © The Royal Society of Chemistry 2023 |