
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the intimate mechanism of NAD+ hydrogenation with phosphonic acid catalysed by Cp*Ir(pyridine-2-sulfonamidate) complexes†
Leonardo
Tensi
 ab,
Luca
Rocchigiani
*a,
Gabriel
Menendez Rodriguez
a,
Edoardo
Mosconi
c,
Cristiano
Zuccaccia
a,
Filippo
De Angelis
*a and
Alceo
Macchioni
*a
ab,
Luca
Rocchigiani
*a,
Gabriel
Menendez Rodriguez
a,
Edoardo
Mosconi
c,
Cristiano
Zuccaccia
a,
Filippo
De Angelis
*a and
Alceo
Macchioni
*a
aDepartment of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di sotto 8, 06123, Perugia, Italy. E-mail: luca.rocchigiani@unipg.it; filippo.deangelis@unipg.it; alceo.macchioni@unipg.it
bDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123, Perugia, Italy
cComputational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), Istituto CNR di Scienze e Tecnologie Chimiche “Giulio Natta” (CNR-SCITEC), 06123 Perugia, Italy
First published on 30th October 2023
Abstract
The reaction mechanism of nicotine amide dinucleotide hydrogenation (NAD+ to NADH) catalysed by Cp*Ir(pyridine-2-sulfonamidate) complexes in the presence of phosphonic acid has been elucidated. Multivariate kinetic experiments and NMR spectroscopy revealed that the enhanced performance of this class of catalysts arises from the hemilability of the pyridine ligand, which is displaced during P–H bond activation and facilitates the generation of the metal-hydride intermediate. Experimental results are backed by DFT calculations showcasing the importance of hydrogen bonding interactions in the activation of phosphite anions. Direct comparison between the prototypical unsubstituted catalyst and the 6-aminopyridine-2-sulfonamidate derivative allowed tracing of the molecular origin of the superior performance of the latter, paving the way for the intelligent design of better performing catalysts for NADH regeneration.
Introduction
Nicotine amide dinucleotide(phosphate) enzymatic cofactors [NAD(P)H] are key actors in cellular metabolism and take part in a plethora of biologically relevant redox reactions.1 Most oxidoreductase enzymes depend, indeed, on exploiting NAD(P)H as an electron transfer and proton carrier for performing their target transformations.2 With the development of modern biocatalysis, the interest in the chemistry of NAD(P)H cofactors has also been steadily growing for their application in selective preparative biotransformations.3–7 Nevertheless, the high cost of such cofactors ($70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mol−1 NADPH and $2600 mol−1 NADH) is a major drawback and makes their application as stoichiometric reagents unfeasible, especially in large-scale reactions. As a direct consequence, the industrial implementation of oxidoreductases is tightly bound to the possibility of regenerating the cofactor in situ from the oxidised form. Ideally, the regenerative method should be affordable and not interfere with the enzymatic process.8,9
000 mol−1 NADPH and $2600 mol−1 NADH) is a major drawback and makes their application as stoichiometric reagents unfeasible, especially in large-scale reactions. As a direct consequence, the industrial implementation of oxidoreductases is tightly bound to the possibility of regenerating the cofactor in situ from the oxidised form. Ideally, the regenerative method should be affordable and not interfere with the enzymatic process.8,9
In the case of NADH regeneration from NAD+, multiple chemical,10,11 photochemical,12,13 enzymatic,14,15 or electrochemical16–18 protocols have been developed.19 An appealing route is to use cheap reducing agents, such as formic or phosphonic acids, in combination with transition metal catalysts. Over the past two decades, half-metallocenes of Rh,20,21 Ru,22,23 and Ir24–27 emerged as a competent class of NAD+ hydrogenation catalysts, even though their performance strongly depends on the metal–ligand combination employed.28 Seminal work by Sadler et al. showed, for instance, that the tethered Ru–arene complexes A (Scheme 1) mediate NAD+ hydrogenation by formate salts with modest performance (turnover frequency, TOF ≈10 h−1).29
 | ||
| Scheme 1 Relevant organometallic catalysts for NAD+ hydrogenation. | ||
On the other hand, the Rh phenanthroline complex B (Scheme 1) reported by Štěpnička et al. shows much higher activity, approaching a TOF of 2000 h−1, again using HCOO− as reducing agent.30 A very similar bipyridine derivative was also used by Hollmann and co-workers to reduce NAD+ in combination with phosphonic acid and alcohols, albeit with sensibly inferior performance.31,32
More recently, we have showed that [Cp*Ir(R–pysa)NO3] complexes (pysa = κ2-pyridine-2-sulfonamidate; R = H, 1; R = 6-NH2, 2) (Scheme 1, Cp* = pentamethylcyclopentadienyl anion) are very active in NADH regeneration using phosphonic acid as reducing agent.33 Upon catalyst screening, we observed that the structure of the ancillary ligand is key in determining the rate of NADH regeneration. In particular, the presence of an amino group in position 6 of the pyridine ring proved extremely beneficial. Catalyst 2 shows, indeed, a maximum TOF value exceeding 3700 h−1, which is one order of magnitude higher than that of the unsubstituted derivative 1. According to the reaction mechanism we proposed in our earlier work33 and the corresponding rate law, the improved performance of catalyst 2 was ascribed to (i) a lower tendency to generate out of cycle adducts with NAD+ and (ii) a higher tendency to form Ir hydride intermediates.
While these findings can be qualitatively correlated with the steric and electronic properties of the ancillary ligand, a clear explanation of the origin of these effects is still not available. This is an aspect that deserves attention, as a full mechanistic understanding would provide an enlightened guide in the design of better performing catalysts for this specific application. In this work, we have performed an extensive integrated experimental/computational study on the intimate catalytic mechanism of NAD+ hydrogenation with phosphonic acid, setting the specific objective of tracing the molecular origin of the enhanced performance of catalyst 2. Combining the information derived from multivariate kinetics, NMR spectroscopy and DFT calculations, we aimed at refining the proposed catalytic mechanism to a much better level of detail and pinpointing the role of the amino substituent in each step of the cycle, with the goal of providing a full rationale for the effect of the ligand substitution on reaction rates.
Results and discussion
Kinetic experiments
To explore a wide range of conditions suitable for a compelling mechanistic investigation, we performed kinetic hydrogenation experiments by using catalysts 1 (1–10 μM) under different phosphite buffer (0.1–1 M, pH = 6.58) or enzymatic cofactor (0.25–10 mM) concentration conditions. NADH production was monitored spectroscopically by following the formation of the diagnostic UV-vis absorption band due to the 1-4-β-NADH isomer (λ = 340 nm, ε340 = 6220 M−1 cm−1). Initial rates (rNADH) were determined by the linear fit of [NADH] versus time trends up to 10% conversion. The results are summarized in Table 1 and Fig. 1.| Entry | [Cat] (μM) | [H2PO3− + HPO32−] (M) | [NAD+] (mM) | TOFini (h−1) | r NADH (10−7 M s−1) |
|---|---|---|---|---|---|
| 1 | 5 | 0.4 | 0.25 | 357 ± 23 | 5.0 ± 0.3 |
| 2 | 5 | 0.4 | 0.5 | 531 ± 35 | 7.4 ± 0.5 |
| 3 | 1 | 0.4 | 1.00 | 578 ± 38 | 1.6 ± 0.1 |
| 4 | 2.5 | 0.4 | 1.00 | 560 ± 36 | 3.9 ± 0.3 |
| 5 | 5 | 0.4 | 1.00 | 558 ± 36 | 7.7 ± 0.5 |
| 6 | 10 | 0.4 | 1.00 | 544 ± 35 | 15 ± 1 |
| 7 | 5 | 0.4 | 1.50 | 518 ± 34 | 7.2 ± 0.5 |
| 8 | 5 | 0.4 | 3.00 | 411 ± 27 | 5.7 ± 0.4 |
| 9 | 5 | 0.4 | 6.00 | 292 ± 19 | 4.1 ± 0.3 |
| 10 | 5 | 0.4 | 10.00 | 195 ± 13 | 2.7 ± 0.2 |
| 11 | 5 | 0.8 | 0.25 | 440 ± 29 | 6.1 ± 0.4 |
| 12 | 5 | 0.8 | 0.50 | 607 ± 39 | 8.4 ± 0.5 |
| 13 | 5 | 0.8 | 0.77 | 701 ± 46 | 9.7 ± 0.6 |
| 14 | 5 | 0.8 | 1.25 | 659 ± 43 | 9.1 ± 0.6 |
| 15 | 5 | 0.8 | 1.50 | 613 ± 40 | 8.5 ± 0.6 |
| 16 | 5 | 0.8 | 3.00 | 501 ± 33 | 7.0 ± 0.5 |
| 17 | 1 | 0.8 | 6.00 | 350 ± 23 | 0.97 ± 0.06 |
| 18 | 2.5 | 0.8 | 6.00 | 343 ± 23 | 2.4 ± 0.2 |
| 19 | 5 | 0.8 | 6.00 | 333 ± 22 | 4.6 ± 0.3 |
| 20 | 10 | 0.8 | 6.00 | 321 ± 21 | 8.9 ± 0.6 |
| 21 | 5 | 0.8 | 10.00 | 211 ± 14 | 2.9 ± 0.2 |
| 22 | 5 | 0.1 | 6.00 | 189 ± 12 | 2.7 ± 0.2 |
| 23 | 5 | 0.2 | 6.00 | 265 ± 17 | 3.7 ± 0.2 |
| 24 | 5 | 0.6 | 6.00 | 312 ± 20 | 4.3 ± 0.3 |
| 25 | 5 | 1.0 | 6.00 | 342 ± 22 | 4.8 ± 0.3 |
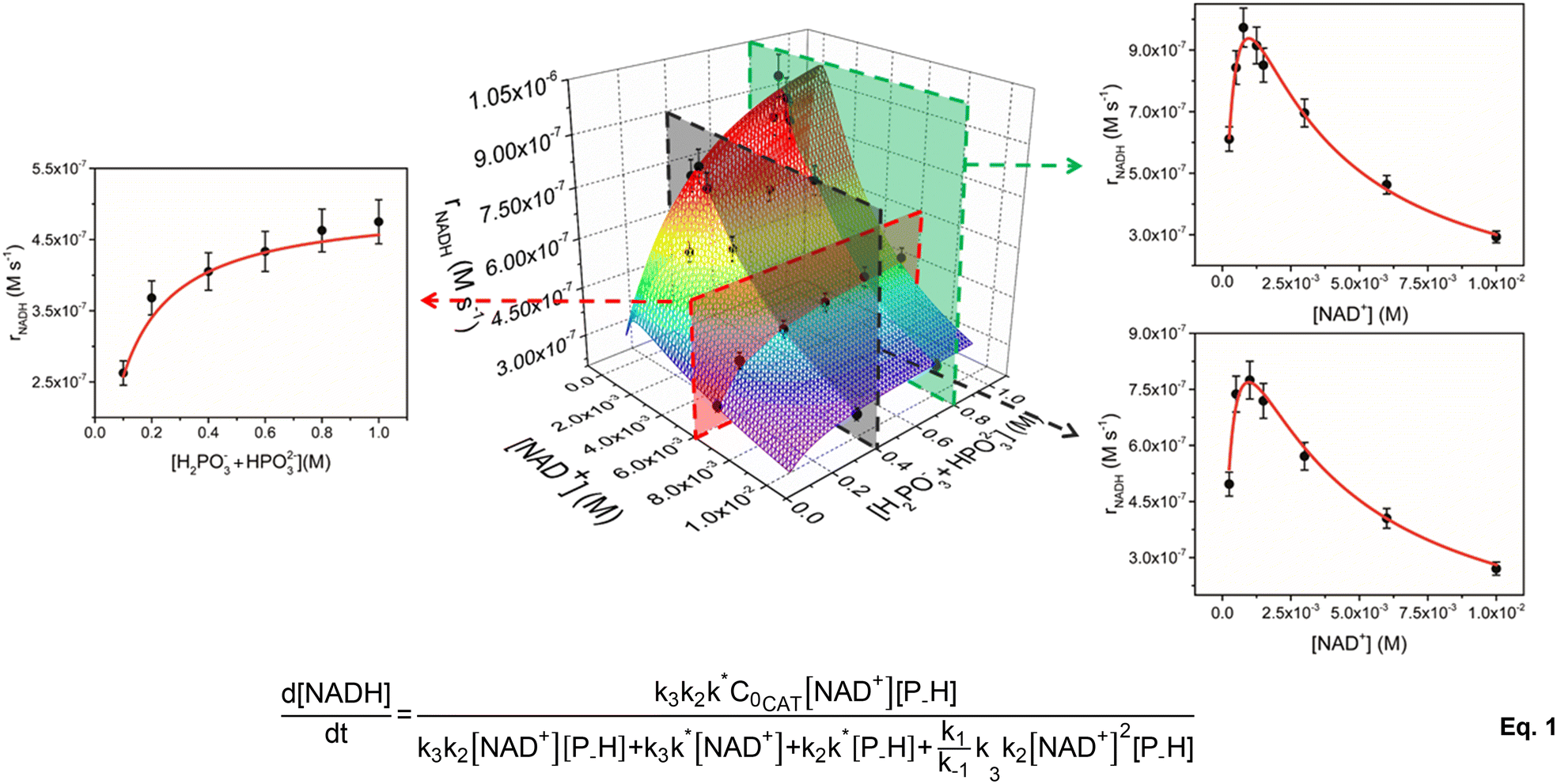 | ||
| Fig. 1 Global fitting of 3D and 2D trends of initial rate of reaction (rNADH, M s−1) vs. concentration of reactants (NAD+ on the right and phosphonic acid on the left) for the NADH regeneration with phosphonic acid mediated by 1 (experimental conditions: T = 313 K, [1] = 5 μM, pH = 6.58). The fitting of the data was made with eqn (1). | ||
To account for all variables at the same time, kinetic data were plotted in a 3D graph (Fig. 1) in which the rate of NADH formation is simultaneously a function of [NAD+] and [H2PO3− + HPO32−] at a given catalyst concentration. In analogy to our previous results,33 the 2D slices of the graph corresponding to rNADHversus [NAD+] show a positively skewed bell trend. For instance, at 5 μM catalyst concentration and [H2PO3− + HPO32−] = 0.4 M, rNADH increases to a peak value of 7.7 × 10−7 M s−1 at [NAD+] = 1 mM and then drops below 3 × 10−7 M s−1 when [NAD+] is increased to 10 mM (see Table 1). The same trend is active at higher concentration of the reducing agent ([H2PO3− + HPO32−] = 0.8 M), for which a slightly faster peak rate is observed (9.7 × 10−7 at [NAD+] = 0.77 mM). rNADH is also not linear with [H2PO3− + HPO32−] and increases to reach a plateau at buffer concentration values ≥0.8 M. TOFini values are instead substantially independent of [1] (entries 3–6 and 17–20, Table 1), suggesting that the reaction is first order with respect to the catalyst.
At first glance, the observed dependence of rNADH on [NAD+] is in line with our early mechanistic proposal, in which the catalytically active species equilibrates with an out of cycle catalyst–NAD+ adduct prior to P–H bond activation and hydrogenation of the cofactor.33 The concentration of the out of cycle intermediate is likely negligible at low [NAD+] and, in a scenario where hydride transfer to NAD+ is the turnover limiting step, it is expected to observe a rate increase on increasing cofactor concentration. At higher [NAD+], the relative amount of the out of cycle adduct becomes more relevant and the number of active sites decreases, with a detrimental effect on rNADH.
The peculiar dependence of rNADH on buffer concentration, though, is not compatible with the previously formulated mechanism and suggests that a more complex reaction pathway may be active. In agreement with this hypothesis, we found that our original rate law does not fit the current extended kinetic data and fails particularly in reproducing the [H2PO3− + HPO32−] dimension (see the ESI†). Therefore, we explored the feasibility of alternative reaction mechanisms that would explain a phosphite buffer concentration dependence. We initially considered two alternatives: (i) catalyst inhibition by phosphite and (ii) Michaelis–Menten type mechanism (see the ESI† for details). By applying the corresponding rate laws to the 3D plot, we found that neither mechanism (i) nor mechanism (ii) is a good model for the kinetic data. This seems to suggest that the observed trends are not simply accounted for by the presence of an inhibitory interaction between the reducing agent (or phosphates) and the active species. This differs from other reports on similar Rh and Ir systems, for which a Michaelis–Menten type kinetics is usually observed.20,31
Therefore, we also tested whether a mechanism including a third slow step, which would occur before phosphite activation, would better reproduce the data. In this scenario, the catalyst C, while in equilibrium with the out of cycle C_NAD+ adduct, undergoes an activation step to afford C*. The latter would engage in phosphite activation to give the hydride species C_H that, in turn, hydrogenates NAD+ to give NADH and regenerate C (Scheme 2).
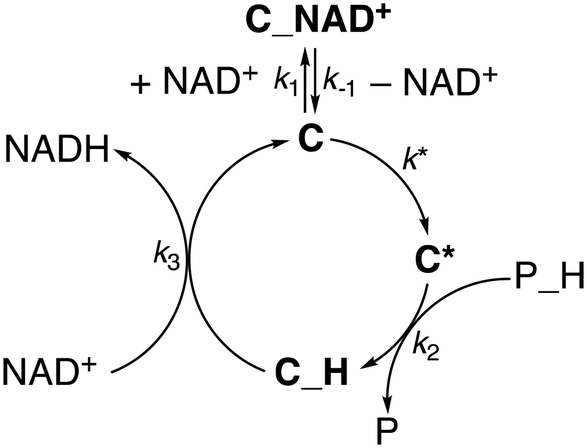 | ||
| Scheme 2 Proposed kinetic model for NAD+ hydrogenation by 1. | ||
Gratifyingly, the rate law derived from the latter model (eqn (1), see the ESI† for details) perfectly fits the data in all dimensions (R2 = 0.98) and affords kinetic parameters with low relative uncertainty (Fig. 1). The comparison between the obtained rate constants indicates that k* (0.7 ± 0.1 s−1) and k2 (1.0 ± 0.1 M−1 s−1) have quite similar values, while k3 (780 ± 50 M−1 s−1) is much larger. This is consistent with the presence of a slow catalyst activation step that is competitive with the reaction with phosphonic acid at higher [NAD+], while cofactor hydrogenation is by far the fastest reaction in the cycle. An appreciable catalyst–cofactor association constant (KM = k1/k−1 = 900 ± 200 M−1) was obtained, suggesting that 1 has a high tendency to bind NAD+ and rests mostly as a dormant cofactor adduct.
Considering the similarity between k* and k2 values, the observed dependence of rNADH on buffer concentration may be explained assuming two regimes. At low buffer concentration, the slowest reaction is phosphite activation and the reaction rate increases upon increasing the concentration of phosphonic acid (first order in P–H). At higher buffer concentration, instead, the slowest reaction is the activation step, whose intrinsic rate is independent of the buffer concentration (zero order in P–H), thus explaining the observation of a plateau in rNADH.
With a compelling kinetic model in hand, we also explored the behaviour of the better performing catalyst 2. As in the case of 1, we investigated a large range of variables by performing kinetic experiments with 2 (5 μM) at different cofactor (0.25–10.0 mM) and phosphite buffer (0.1–1.0 M) concentrations (see Table S1 in the ESI†). As observed for 1, trends of rNADHvs. buffer concentration are not linear and a plateau is observed at higher [H2PO3− + HPO32−] values (Fig. 2). rNADHvs. [NAD+] trends are again not linear, even though the detrimental effect of increasing NAD+ concentration seems to be less important in comparison with that observed for 1 (Fig. 2). For instance, a peak rNADH value of 5.5 × 10−6 M s−1 is obtained at [NAD+] = 3 mM when [H2PO3− + HPO32−] = 0.8 M, while a drop by only 22% was observed when [NAD+] was increased to 10 mM.
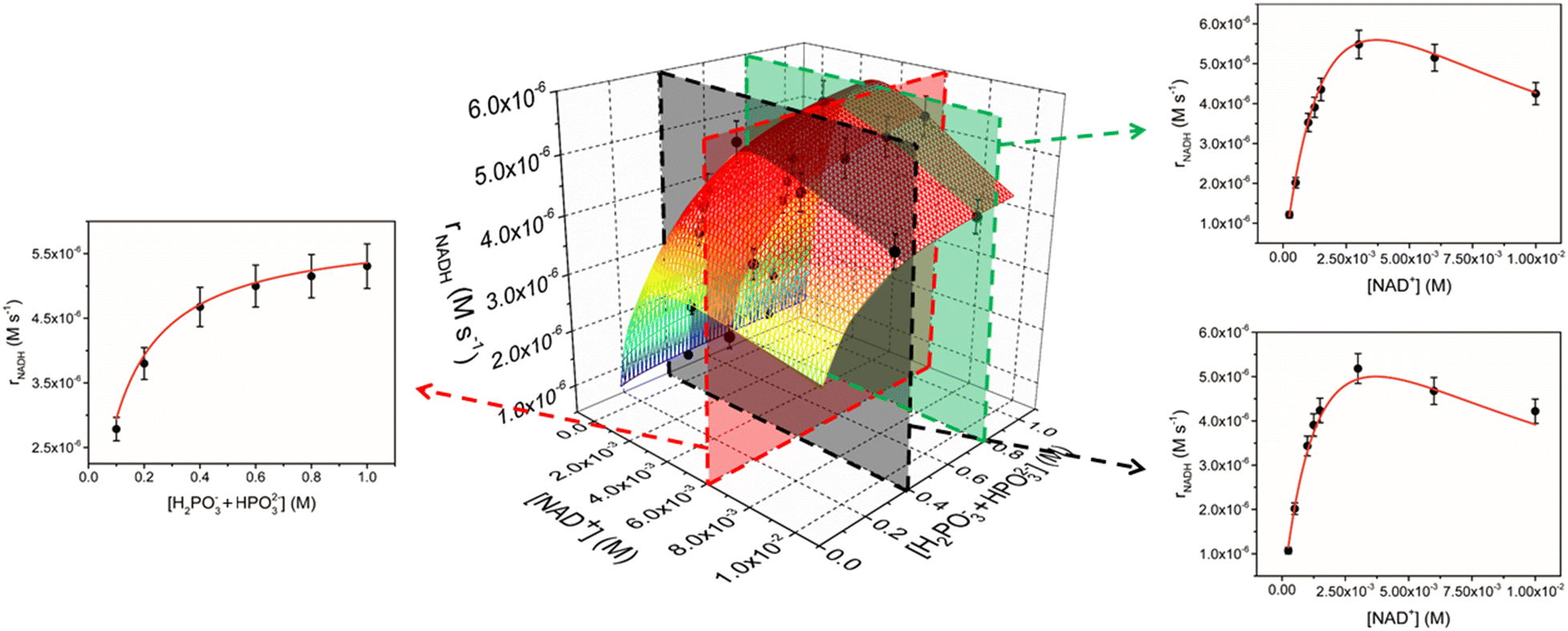 | ||
| Fig. 2 Global fitting of 3D and 2D trends of initial rate of reaction (rNADH, M s−1) vs. concentrations of reactants (NAD+ on the right and phosphonic acid on the left) for the NADH regeneration with phosphonic acid mediated by 2 (experimental conditions: T = 313 K, [2] = 5 μM, pH = 6.58). The fitting of the data was made with eqn (1). | ||
The 3D data for 2 are perfectly reproduced by eqn (1) (R2 = 0.990) in all dimensions (Fig. 2), indicating that rate law and catalytic mechanism are unaffected by the subtle modification in the structure of the catalyst. Analysis of the rate constants, though, shows a significant increase in both k* (3.7 ± 0.6 s−1) and k2 values (12 ± 1 M−1 s−1) for 2 with respect to the unsubstituted analogue. On the other hand, k3 (1040 ± 60 M−1 s−1) is only marginally affected, while the association constant between the catalyst and NAD+ is appreciably smaller (KM = 260 ± 60 M−1).
These results clearly show that the improved performance of catalyst 2 with respect to 1 arises from two constructive independent effects (see also Table 2 for a direct comparison). First, the 5-fold increase in k* and the 12-fold increase in k2 suggest that complex 2 is intrinsically more reactive than 1 in both activation and phosphite activation steps. Evidently, the presence of an NH2 group in proximity to the catalyst's active site facilitates the interaction with the reducing agent and P–H bond activation. Second, the reduced tendency of 2 to interact with NAD+, which is corroborated by the 3.5-fold decrease in KM, indicates that a higher fraction of iridium complex can engage in catalytically productive reactions. Overall, these effects combine, leading to rNADH values for 2 that are one order of magnitude higher than those of 1.
| Parameter | 1 | 2 |
|---|---|---|
| k 1/k−1 | 900 ± 200 M−1 | 260 ± 60 M−1 |
| k* | 0.7 ± 0.1 M−1 s−1 | 3.7 ± 0.6 M−1 s−1 |
| k 2 | 1.0 ± 0.1 M−1 s−1 | 12 ± 1 M−1 s−1 |
| k 3 | 780 ± 50 M−1 s−1 | 1040 ± 60 M−1 s−1 |
NMR studies
With the aim of intercepting reaction intermediates relevant to the proposed mechanism, we investigated the speciation in solution and reactivity with phosphonic acid of complexes 1 and 2 by NMR spectroscopy.Initially, we looked at elucidating the nature of the entry point of the catalyst in the cycle. As we reported earlier, this family of complexes undergoes dimerization phenomena when their nitrate salts are dissolved in H2O or phosphate buffer solutions.34 Such a process is driven by the formation of Ir(μ-N)2Ir moieties, in which the amide nitrogen of a pyridylsulfonamidate ligand binds two metal centres at the same time. Depending on the reaction medium and pH conditions, bridging amide groups can be protonated or not, potentially affording multiple active species in solution and affecting catalytic activity. In the case of complex 1, we showed that it forms an approximately 70:30 monomeric aquo complex–dimer mixture in aqueous solution at C = 16.0 mM.34 Here we found that catalyst 2 (C = 12.0 mM) also undergoes monomer–dimer equilibrium upon dissolution in unbuffered H2O, even though the tendency to dimerize is somewhat reduced with respect to 1 (monomer/dimer ratio = 86:14). A van't Hoff analysis of the equilibrium constant values derived by 1H NMR spectra recorded at different temperatures afforded a ΔH0 = −0.85 ± 0.04 kcal mol−1 and a ΔS0 = +2.8 ± 0.1 cal mol−1 k−1 for complex 2 (ΔG0298K = −1.68 ± 0.07 kcal mol−1). These values are only marginally different from those obtained for 1 (ΔH0 = −1.79 ± 0.08 kcal mol−1 and ΔS0 = +1.7 ± 0.3 cal mol−1 k−1, ΔG0298K = −2.3 ± 0.2 kcal mol−1), indicating that dimerization of 2 is just slightly less exothermic than that of 1. Most likely, the steric demand of the NH2 groups makes the association of two Ir monomers slightly more difficult with respect to the unsubstituted analogue.
It is quite reasonable, though, that the striking difference in catalytic performance between 1 and 2 is not related to this phenomenon. Under the low concentration conditions used in the catalytic experiments, the relative amount of dimeric species present in solution is indeed negligible. For example, at 5 μM concentration, the total amount of dimer expected in the case of 1 would be 1 nM based on the equilibrium constant at 313 K. Moreover, we noted previously that TOFini values are basically independent of catalyst concentration (entries 3–6 and 17–20, Table 1) and that a first-order reaction in the Ir complex is active. Therefore, it can be inferred that dimerization is not kinetically relevant for the catalytic reaction.
These iridium complexes are found to be very reactive with phosphite. Upon addition of a few drops of buffer solution to D2O samples containing 1 or 2, an immediate disappearance of the starting materials was observed. In the case of 1, an intractable mixture composed of a series of unstable hydridic species was obtained, while the formation of a precipitate was observed in the case of 2. Redissolution of the solid in DMSO-d6 indicated the formation of a single hydridic species, whose full characterisation was unfortunately hampered by exchange reactions with residual excess D2O.
Strikingly, if the reaction between 2 and phosphonic acid is directly performed in DMSO-d6, no hydride transfer to Ir takes place. On the other hand, when HCOOK is used as hydride source, 1H NMR reveals the clean formation of a single hydride species 2–H. The latter features a terminal Ir–H moiety appearing at δH = −13.56 ppm and a single Cp* resonance at δH = 1.67 ppm. A close inspection of the map of dipolar contacts in the 1H NOESY NMR spectrum of 2–H (Fig. 3) revealed the presence of strong interactions between the Cp* protons and all the aromatic hydrogen atoms of the PYSA ligand. Moreover, the hydride signal was found to be dipolarly coupled with the hydrogen atom in position 5 of the ancillary ligand, while no interaction was observed with the amino group 9. This is consistent with a structural rearrangement of the catalyst, in which the NH2 group is re-oriented far away from the metal centre. Displacement of the pyridine ligand by a DMSO solvent molecule may account for the observed structural rearrangement upon hydride generation. A similar pyridine detachment upon hydride formation was observed by Pizarro and co-workers on tethered Cp*–pyridine derivatives.35 Unfortunately, 2–H proved to be rather unstable and all attempts to grow single crystals resulted in decomposition.
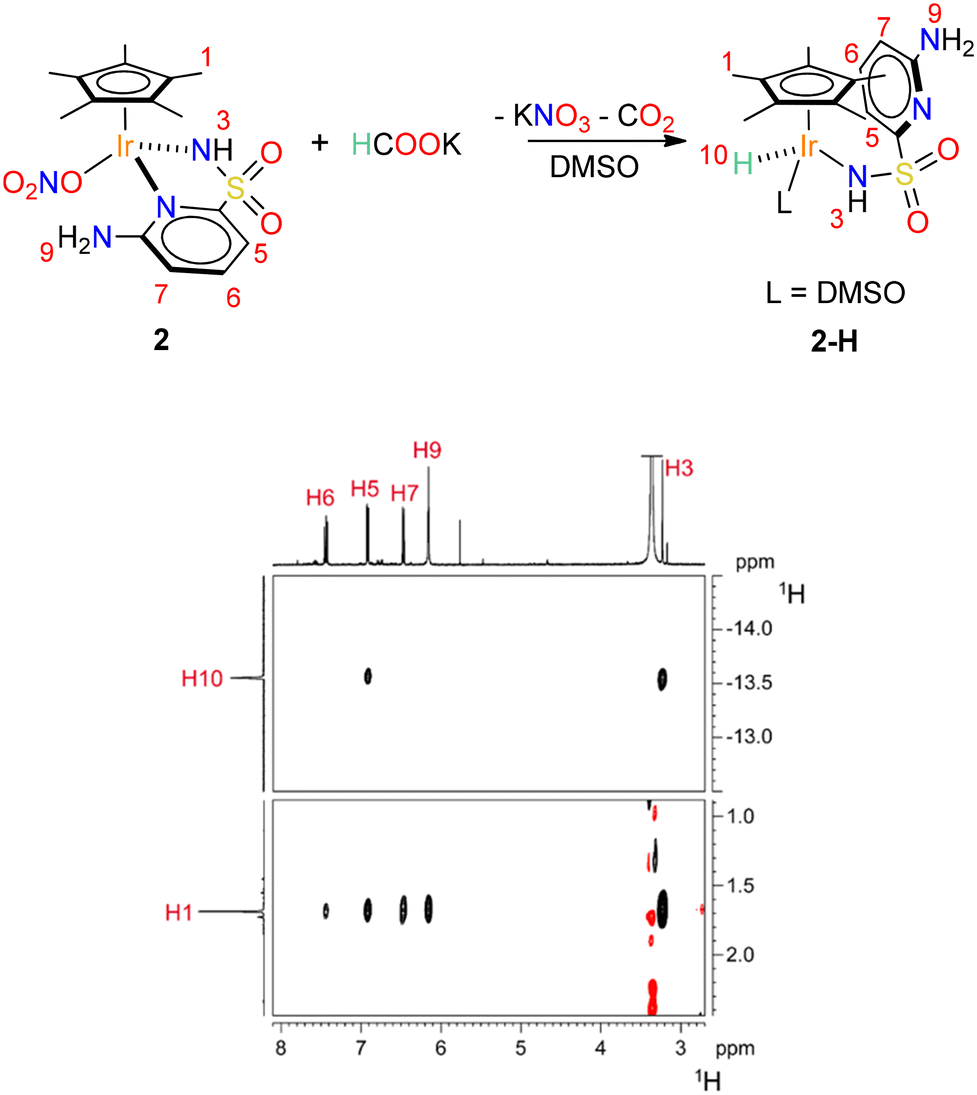 | ||
| Fig. 3 Reaction of formation of complex 2–H and sections of the 1H NOESY spectrum (298 K, DMSO-d6) relative to 2–H showing the dipolar correlations discussed in the main text. | ||
DFT calculations
From the NMR results, it appears clear that the pathway to the formation of Ir hydride species may be more complex than simple ligand substitution. With the aim of shedding some light on this step, the route to intermediate 2–H was also investigated by means of DFT calculations. For the sake of simplicity, studies were performed on catalyst 1 at the B3LYP/6-311++g**/sdd (for core electrons of Ir only) level of theory. The reaction solvent (H2O) was simulated with five explicit water molecules complemented by an implicit solvation model (SCRF = CPCM). The results are depicted in Scheme 3. | ||
| Scheme 3 Reaction profile of phosphonic acid activation by 1–H2O. ΔE and ΔE‡ values are given in kcal mol−1. | ||
The entry point of the energy profile is the hydrogen bonded adduct between the aquo complex 1–H2O and a HPO32− anion, which was set as a zero energy reference structure. Detachment of the pyridine ring from the metal center of 1–H2O occurs via1–TS1 (ΔE‡ = 11.0 kcal mol−1) to give the intermediate 1TP–OH (ΔE0 = 0.3 kcal mol−1), where a proton of the coordinated water molecule is transferred to the phosphite anion and the Ir(III) center is formally neutral. The latter assumes a pseudo-trigonal planar coordination geometry, in which the calculated Ir–N bond distance (2.04 Å) is significantly shorter than that found in 1–H2O (2.15 Å) and the Ir–N–H angle approaches 115°.
This feature indicates that the amide portion of the pyridylsulfonamidate ligand switches from an X to an LX type coordination mode in 1TP–OH upon pyridine detachment, similarly to what was proposed for analogous picolinamide complexes.36
After pyridine detachment, the H2PO3− anion coordinates the Ir center via bridging interaction with the P–H bond, assisted by P–O⋯H–O and P–O⋯H–N hydrogen bonding, leading to the high energy intermediate 1–HPO3− (ΔE0 = 11.5 kcal mol−1). To stabilize such a product, the Ir–OH moiety is protonated back by the phosphite anion to give a formally cationic Ir(III) center. There is a significant energy barrier to the generation of 1–HPO3− (ΔE‡ = 14.5 kcal mol−1), suggesting that phosphite activation by the pseudo-tricoordinated Ir complex is a slow reaction.
From this stage, the pathway goes downhill: first, displacement of the PO3− anion (ΔE‡ = 6.3 kcal mol−1 above 1–HPO3−) and its subsequent reaction with the coordinated H2O leads to the formation of H2PO4− and the stable piano-stool hydride complex 1–H (ΔE0 = −12 kcal mol−1).
Overall, the reaction profile leading from 1–H2O to 1–H shows the presence of two activated processes that may be turnover limiting in the catalytic reaction. The first is the generation of a coordinative vacancy on the metal upon pyridine detachment and switching of the amide coordination mode. This process can be reasonably associated to what we described as the catalyst activation process (Scheme 2); thus k* is directly related to the energy barrier leading to 1–TSI and the intermediate C* can be associated with 1TP–OH. The second activated process is the activation of the P–H bond, so k2 is correlated with the energy of either 1–TSII or 1–TSIII. Consistently, no other intermediate is required by the kinetic model prior to hydride generation, as species such as 1–HPO3− are too high in energy and would not accumulate.
The calculated mechanism for P–H bond activation is also consistent with the observation that an excess of water is required to favour the activation of the reducing agent. The coordinated water molecule, indeed, acts as a proton donor, stabilizing all the reaction intermediates/transition states via hydrogen bonding and is a reactive fragment in the generation of H2PO4−. Clearly, these steps are likely to be disfavoured in non-aqueous solvents such as DMSO.
Reaction mechanism
By merging all the pieces of information obtained experimentally and theoretically, a compelling detailed reaction mechanism for the hydrogenation of NAD+ catalysed by 1 in the presence of phosphonic acid was formulated (Scheme 4).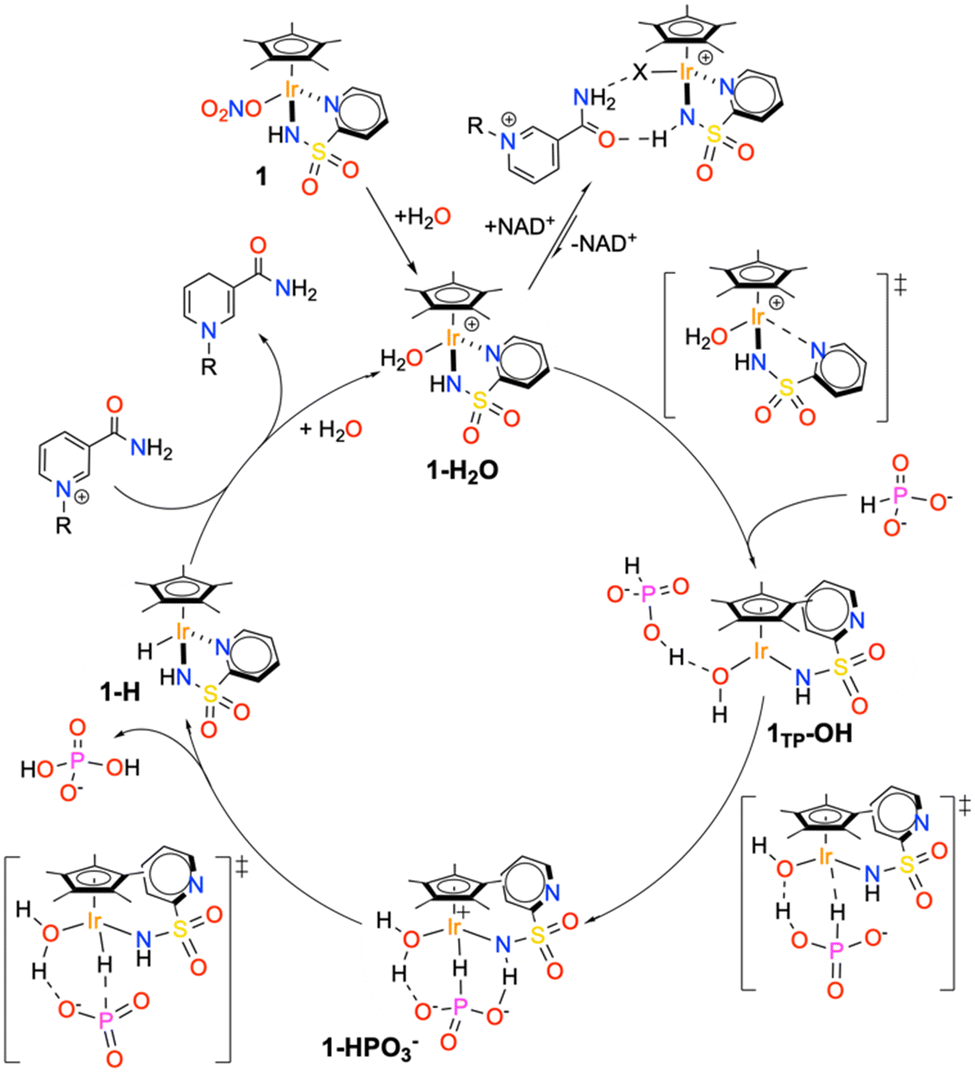 | ||
| Scheme 4 Proposed reaction mechanism based on theoretical, kinetic and spectroscopic data. | ||
The active species entering the cycle is the monomeric aquo complex 1–H2O, which is immediately generated upon dissolving the nitrate precatalyst in water or phosphonic acid buffer. In the presence of the reactants, the catalyst can either form an out of cycle adduct with NAD+, held together by multiple hydrogen bonding interactions, or generate a coordinative vacancy upon pyridine detachment and deprotonation of the coordinated water. This step generates the pseudo-tricoordinated species 1TP–OH, which, in turn, activates the phosphite anion, affording the high-energy intermediate 1–HPO3−. PO3− release and reaction with the coordinated H2O leads to the hydridic intermediate 1–H and the ortho-dihydrogenophosphate anion. In the final step, the iridium hydride intermediate reduces the enzymatic cofactor through hydride migration, thus regenerating the catalyst.
Considering the proposed cycle, we can fully rationalize the origin of the improved performance of catalyst 2 with respect to catalyst 1. First of all, the presence of the amino group close to the metal centre of 2 inhibits the association between the latter and NAD+, with the important consequence of reducing the amount of catalyst resting as out of cycle species. It is reasonable to assume that the NH2 in position 6 can destabilize the Ir–NAD+ adduct because of its steric pressure and/or disruption of the hydrogen bonding interactions holding together the catalyst–cofactor adduct. Moreover, the presence of the NH2 has a clear beneficial effect on the activation process (k* step), possibly by enhancing the tendency of the pyridine fragment to leave the metal centre upon reaction with the hydride donor. Also, the hydrogen bonding accepting character of the amine may contribute to the network of interactions leading to phosphite activation, thus explaining the rate increase we observed also for the k2 step. It cannot be excluded that the enhanced tendency to interact with phosphites is directly related to the decreased ability of 2 to bind NAD+. On the other hand, no significant effect is observed in the final hydrogenation step, suggesting that the hydricity of 1–H and 2–H is basically comparable.
Conclusions
In summary, in-depth experimental and computational mechanistic investigations allowed us to draw a compelling reaction pathway for the hydrogenation of nicotine amide dinucleotide (NAD+) catalysed by the highly performing [Cp*Ir(R–pysa)NO3] complexes (pysa = κ2-pyridine-2-sulfonamidate; R = H, 1; R = 6-NH2, 2) in the presence of phosphonic acid. Multiple catalytic experiments with 1 and 2 revealed non-linear trends of the initial rate of NADH formation (rNADH) with the concentration of the substrates, which were modelled with an ad hoc kinetic law. Integration of the kinetic results with NMR spectroscopic and theoretical studies suggested the presence of two slow steps, namely generation of a coordinative vacancy on the metal and P–H bond activation, that may be turnover limiting depending on the relative concentration of phosphonic acid used. Remarkably, the coordinative vacancy is generated via pyridine decoordination from Ir, affording a pseudo-tricoordinated species that activates phosphite anions more favourably. The establishment of a hydrogen bonding network between the phosphonic acid and both coordinated H2O and amide moieties is functional for P–H bond activation, which would not occur otherwise.Such a fine level of understanding of the catalytic mechanism gave us the opportunity to trace the molecular origin of the enhanced performance of catalyst 2 with respect to 1. In particular, the amino substituent has a dual beneficial effect as (i) it reduces the tendency of the catalyst to form out of cycle adducts with NAD+ and (ii) it makes pyridine detachment and P–H bond activation steps kinetically more efficient.
These results provide unprecedented insights on the action mode of this family of catalysts and the elucidation of the intimate hydrogenation mechanism represents a cornerstone in this area of research. Indeed, our findings provide clear and rational strategies for optimizing both catalyst structure and reaction conditions. Further work on exploring such strategies is ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality, Progetti Fondo Ricerca di Ateneo 2021 and PON – Ricerca e Innovazione – DM 1062 (J91B21003250006). We also thank the Fondazione Perugia (project 21101, 2022.0405) for supporting our research.Notes and references
- S. K. Spaans, R. A. Weusthuis, J. van der Oost and S. W. M. Kengen, Front. Microbiol., 2015, 6, 742 CrossRef PubMed.
- L. Sellés Vidal, C. L. Kelly, P. M. Mordaka and J. T. Heap, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 327–347 CrossRef PubMed.
- D. Monti, G. Ottolina, G. Carrea and S. Riva, Chem. Rev., 2011, 111, 4111–4140 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- J. S. Rowbotham, M. A. Ramirez, O. Lenz, H. A. Reeve and K. A. Vincent, Nat. Commun., 2020, 11, 1–7 CrossRef PubMed.
- M. Hambourger, G. F. Moore, D. M. Kramer, D. Gust, A. L. Moore and T. A. Moore, Chem. Soc. Rev., 2009, 38, 25–35 RSC.
- T. Quinto, V. Köhler and T. R. Ward, Top. Catal., 2014, 57, 321–331 CrossRef CAS.
- J. W. H. Burnett, Z. Sun, J. Li, X. Wang and X. Wang, Green Chem., 2021, 23, 7162–7169 RSC.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880–1886 CrossRef CAS.
- M. Wang, X. Ren, M. Guo, J. Liu, H. Li and Q. Yang, ACS Sustainable Chem. Eng., 2021, 9, 6499–6506 CrossRef CAS.
- Q. Pan, H. Liu, Y. Zhao, S. Chen, B. Xue, X. Kan, X. Huang, J. Liu and Z. Li, ACS Appl. Mater. Interfaces, 2019, 11, 2740–2744 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- J. M. Vrtis, A. K. White, W. W. Metcalf and W. A. Van Der Donk, Angew. Chem., Int. Ed., 2002, 41, 3257–3259 CrossRef CAS PubMed.
- H. Slusarczyk, S. Felber, M. R. Kula and M. Pohl, Eur. J. Biochem., 2000, 267, 1280–1289 CrossRef CAS PubMed.
- N. Ullah, I. Ali and S. Omanovic, Mater. Chem. Phys., 2015, 149, 413–417 CrossRef.
- L. Zhang, M. Etienne, N. Vilà, T. X. H. Le, G. W. Kohring and A. Walcarius, ChemCatChem, 2018, 10, 4067–4073 CrossRef CAS.
- R. Barin, S. Rashid-Nadimi, D. Biria and M. A. Asadollahi, Electrochim. Acta, 2017, 247, 1095–1102 CrossRef CAS.
- V. K. Sharma, J. M. Hutchinson and A. M. Allgeier, ChemSusChem, 2022, 15, e202200888 CrossRef CAS PubMed.
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS.
- V. Ganesan, D. Sivanesan and S. Yoon, Inorg. Chem., 2017, 56, 1366–1374 CrossRef CAS PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS PubMed.
- F. Chen, I. Romero-Canelón, J. J. Soldevila-Barreda, J. I. Song, J. P. C. Coverdale, G. J. Clarkson, J. Kasparkova, A. Habtemariam, M. Wills, V. Brabec and P. J. Sadler, Organometallics, 2018, 37, 1555–1566 CrossRef CAS PubMed.
- Z. Liu, R. J. Deeth, J. S. Butler, A. Habtemariam, M. E. Newton and P. J. Sadler, Angew. Chem., Int. Ed., 2013, 52, 4194–4197 CrossRef CAS PubMed.
- A. Bucci, S. Dunn, G. Bellachioma, G. Menendez Rodriguez, C. Zuccaccia, C. Nervi and A. Macchioni, ACS Catal., 2017, 7, 7788–7796 CrossRef CAS.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367–374 CrossRef CAS PubMed.
- L.-J. Zhao, Z. Yin, Y. Shi, W. Sun, L. Sun, H. Su, X. Sun, W. Zhang, L. Xia and C. Qi, Catal. Sci. Technol., 2021, 11, 7982–7991 RSC.
- S. Fukuzumi, Y.-M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, P. C. A. Bruijnincx, A. Habtemariam, G. J. Clarkson, R. J. Deeth and P. J. Sadler, Organometallics, 2012, 31, 5958–5967 CrossRef CAS.
- J. Canivet, G. Süss-Fink and P. Štěpnička, Eur. J. Inorg. Chem., 2007, 30, 4736–4742 CrossRef.
- M. M. Grau, M. Poizat, I. W. C. E. Arends and F. Hollmann, Appl. Organomet. Chem., 2010, 24, 380–385 CrossRef CAS.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19–20, 167–176 CrossRef CAS.
- L. Tensi and A. Macchioni, ACS Catal., 2020, 10, 7945–7949 CrossRef CAS.
- C. Trotta, G. Menendez Rodriguez, L. Tensi, A. V. Yakimov, L. Rocchigiani, A. Donnadio, C. Zuccaccia, C. Copéret and A. Macchioni, Eur. J. Inorg. Chem., 2023, e202300211 CrossRef CAS.
- A. C. Carrasco, V. Rodriguez-Fanjul and A. M. Pizarro, Inorg. Chem., 2020, 59, 16454–16466 CrossRef CAS PubMed.
- G. Menendez Rodriguez, F. Zaccaria, L. Tensi, C. Zuccaccia, P. Belanzoni and A. Macchioni, Chem. – Eur. J., 2021, 27, 2050–2064 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on materials and methods, NMR spectra, kinetic data, computational details and coordinates of optimized geometries. See DOI: https://doi.org/10.1039/d3cy01048a |
| This journal is © The Royal Society of Chemistry 2023 |