
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-free, oxidative C(sp3)–H arylation of amides with zeolite catalysts†
Jannick
Vercammen
 *,
Besir
Krasniqi
and
Dirk
De Vos
*
*,
Besir
Krasniqi
and
Dirk
De Vos
*
Centre For Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F P.O. box 2461, 3001 Leuven, Belgium. E-mail: Jannick.vercammen@kuleuven.be; dirk.devos@kuleuven.be
First published on 20th September 2023
Abstract
The oxidative coupling of amides and heteroarenes would allow for joining two of the most privileged scaffolds in medicinal chemistry. Here, we show that zeolite catalysts in combination with tert-butylperoxy ethylhexyl carbonate (TBEC) as oxidant can improve the formation of heterocoupled products. The exceptional selectivity and high yields are attributed to a pathway proceeding by a 2-electron oxidation of the amide to an N-acyliminium cation, followed by an electrophilic attack on the heterocycle.
Introduction
The prevalence of heteroatoms in pharmaceuticals renders the development of new reaction protocols involving such compounds of paramount importance. Important scaffolds are amide functionalities, since the amide is the most common functional group in pharmaceutical compounds,1 and N-heterocycles, as approximately 60% of unique small-molecule drugs contain at least one nitrogen heterocycle.2 An interesting class of drugs are the racetams, which contain a pyrrolidone backbone, and are often attributed with cognition-enhancing or nootropic effects.3Amides and amines can be coupled with heterocyclic aromatics using peroxides as oxidant.4 In the case of N-rich heterocycles, the reaction is thought to proceed through a radical mediated mechanism.5 Indeed, under autoxidative conditions, a large pool of radicals will be generated; and the reactions can be further accelerated by addition of redox mediators (iodide,5,6 ionic Ru,7 Fe,8,9 or Cu10). The group of Lei managed to replace the peroxide oxidants with an electrochemical oxidation setup.11
The low C–H bond dissociation energy of methylene groups in the α-position of an amide allows for an easy initial activation, e.g. by a 1-electron oxidation (Fig. 1A).12,13 If the reaction proceeds to loss of a second electron, the obtained N-acyliminium cation can subsequently react with nucleophiles, resulting in a direct C(sp3)–H functionalization of non-activated amides.14 The most notable example of this activation strategy for amides is the Shono-type anodic oxidation (Fig. 1B).15,16 The group of Kawakami identified Zr(OTf)4 as a Lewis acid catalyst in the oxidative coupling of lactams with heterocyclic arenes using oxygen as oxidant (Fig. 1C).17 Later, iron18 and copper19 catalytic systems were developed, and shown to proceed by a 2-electron oxidation of the amide. The formed N-acyliminium cation then participates in an electrophilic aromatic substitution (EAS) reaction of the N-heterocycle.
 | ||
| Fig. 1 Oxidative activation of amides. | ||
However, the work of Kawakami produced rather low yields and used a homogeneous catalyst.17 On the other hand, the reaction pathway involving radical mediated aromatic substitution limits the substrate scope to N-rich heterocycles, and unavoidably also produces homocoupling side-products.20 Additionally, the latter research direction focused on the development of catalysts for the autoxidation of the amide reactant. Here, we focus on the functionalization of heterocyclic reactants, containing fewer heteroatoms. To facilitate the electrophilic attack of the formed cation onto these less-reactive heterocycles, we targeted acidic zeolites as catalyst, due to their use in the petrochemical industry as alkylation catalysts.21
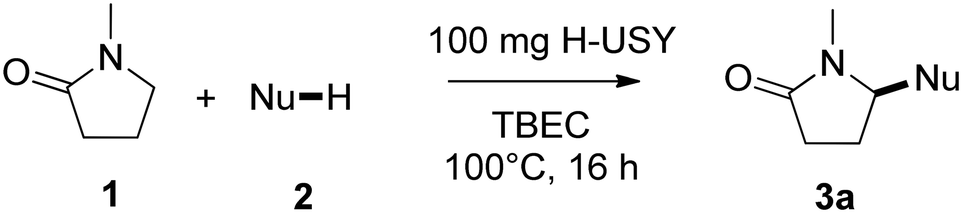
Results and discussion
The oxidative heterocoupling of N-methylpyrrolidone (1) and N-methylpyrrole (2) was investigated with different additives (Table 1). Reaction conditions were initially adapted from Kawakami et al.,17 who used 1 as a solvent. A slightly milder reaction temperature of 100 °C was employed rather than 130 °C, and reactions were performed overnight instead of over only 5 hours. We aimed to replace and improve the homogeneous Zr(OTf)4 catalyst in this study.|
|
||||
|---|---|---|---|---|
| Entry | Solid acid | Conv.d [%] | Yieldd [%] |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3b:3c :3b:3c |
| a N-Methylpyrrole (1.5 mmol), N-methylpyrrolidone (2 ml), additive (100 mg), O2 (balloon), 100 °C, 16 hours. b No O2 balloon was used, and TBEC (1.65 mmol) was added instead. c N-Methylpyrrole (1.5 mmol), N-methylpyrrolidone (3 mmol), TBEC (2.25 mmol), propylene carbonate (2 ml), H-USY (100 mg), 100 °C, 16 hours. d GC yield and conversion based on N-methylpyrrole. e Reaction conducted under N2, instead of O2. f The zeolite was dried at 200 °C, and activated molecular sieves (4 Å, 0.5 g) were added. | ||||
| 1e | — | <1 | <1 | — |
| 2 | — | 40 | 6 | 40:51:10 |
| 3 | Sulphated ZrO2 | 26 | 16 | 39:57:5 |
| 4 | Amberlyst 15 | 21 | 21 | 37:60:3 |
| 5 | H-ZSM-5 (Si/Al = 25) | 68 | 17 | 40:54:6 |
| 6 | H-Beta (Si/Al = 75) | 38 | 24 | 52:46:3 |
| 7 | H-MOR (Si/Al = 100) | 52 | 23 | 27:67:7 |
| 8 | H-MCM-22 (Si/Al = 14) | 75 | 27 | 28:69:3 |
| 9 | H-USY (Si/Al = 40) | 54 | 54 | 37:57:6 |
| 10 | Na-USY (Si/Al = 40) | <1 | <1 | — |
| 11f | H-USY (Si/Al = 40) | <1 | <1 | — |
| 12b | H-USY (Si/Al = 40) | 84 | 84 | 38:58:4 |
| 13c | H-USY (Si/Al = 40) | 59 | 57 | 34:65:1 |
No product was formed under N2 (entry 1), which shows the need for an oxidant, while barely any product is obtained without additive or catalyst (entry 2). The reaction could be boosted strongly by addition of solid acids, such as sulfated zirconia or sulfonic acid containing resins (entries 3–4). Acidic zeolites could be employed as well to improve the yield of this reaction (entries 5–9) with yields ranging from 17 to 54%; H-USY, an ultrastabilized zeolite which imposes few steric constraints, showed the best performance. The requirement of acidity was confirmed by conducting the reaction with a non-acidic Na-exchanged USY, which did not yield any product (entry 10). Furthermore, the reaction was conducted in anhydrous conditions by addition of activated molecular sieves (entry 11); no product was formed under these conditions, which highlights the importance of traces of water. Interestingly, product distributions deviating from those obtained with non-microporous acids were obtained, with variations of the position (2- or 3-) at which the N-methylpyrrole is functionalized. While typically a 40:60 ratio between 3a and 3b was observed, this could be slightly tuned to more 3a product formation with zeolite H-Beta, or more 3b product formation with H-MOR or H-MCM-22.
Zeolite H-USY was chosen for further optimization of the catalytic system due to both the high yield and selectivity obtained with this zeolite. Substituting the O2 environment by the peroxide oxidant tert-butylperoxy 2-ethylhexyl carbonate (TBEC) led to a significant increase to 84% yield (entry 12, Table 1). Furthermore, this allowed for easier and safer handling of the oxidant and more reproducible experiments. Finally, to allow the use of more expensive amides, we investigated different solvents in order to reduce the amount of 1 from solvent-like to stoichiometric quantities. Although a slightly lower yield of 57% was obtained, we were able to substitute the excess of 1 with the green solvent propylene carbonate (entry 13, Table 1).
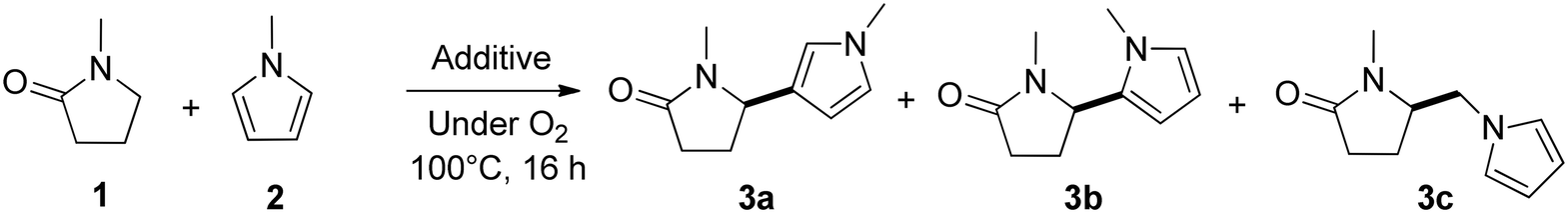
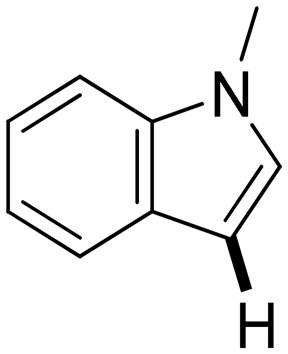
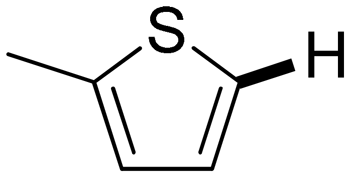
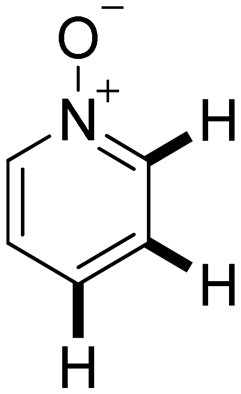
Next, the substrate scope of the C–H functionalization of amides using zeolites was investigated (Table 2). A large range of nucleophiles was assessed as we believed the model reaction to proceed by an electrophilic aromatic substitution of pyrrole with an in situ generated NMP-derived cation. A good yield was observed with the slightly larger N-methylindole; the reaction could even be performed with the non-protected indole and pyrrole (entries 1–3). Additional heteroatoms in the ring were also tolerated as could be seen with pyrazole, imidazole and triazoles (entries 4–8). Other heteroatom containing substrates such as furan and thiophenes were also tolerated, albeit with lower yields (entries 9–10). Pyridines could only be functionalized by employing pyridine N-oxide as substrate (entry 11). The reaction of the NMP-derived cation with amides and carbamates as N-nucleophilic reactants resulted in C–N coupled products (entries 12–15). Caffeine was assessed as an example of a more complex molecule, and found to be compatible with the investigated methodology (entry 16). Finally, we found that benzothiazole, a privileged motif in pharmaceuticals, was also reactive under the investigated conditions (entry 17). A similar performance could be obtained for electron-deficient heterocycles, such as benzothiazole and pyridine N-oxide, in the absence of the zeolite catalyst.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Nucleophile | Conv. [%] | Yield [%] | Entry | Nucleophile | Conv. [%] | Yield [%] |
|
a 100 mg H-USY (CBV-780, Si/Al = 40), 1.0 mmol nucleophile, 1.5 mmol TBEC, 2 ml NMP, 100 °C, 16 hours. The position of the functionalization is indicated by a bold bond.
b 1.5 mmol of nucleophile and 1.65 mmol of TBEC were used instead.
c The C5 and N-coupled product were obtained in a ratio of 1:4, respectively.
d 3 regiomers in a 77:6:17 ratio with m/z of 176.
e 0.5 mmol of caffeine was used.
f A similar conversion and yield were obtained in the absence of H-USY.
|
|||||||
| 1 |
|
87 | 67 | 10b |
|
10 | 8 |
| 2 |

|
71 | 42 | 11f |
|
80 | 31d |
| 3 |

|
97 | 83 | 12 |
|
90 | 62 |
| 4b |

|
55 | 27 | 13 |

|
82 | 55 |
| 5 |

|
85 | 85c | 14 |

|
64 | 53 |
| 6b |

|
34 | 34 | 15b |

|
56 | 29 |
| 7b |

|
100 | 61 | 16 |

|
44 | 29e |
| 8b |

|
37 | 17 | 17f |

|
34 | 34 |
| 9 |

|
64 | 46 | 18 |

|
37 | 15 |
Next, the amide reactant in the oxidative heterocoupling with N-methylpyrrole was varied (Table 3). The use of the N–H amide, 2-pyrrolidone, and different N-alkyl substituted variants resulted in acceptable yields. For the latter reactions, 3 regiomers were observed as with NMP, of which the distribution was strongly affected by the steric bulk of the N-substituent. In the case of the non-substituted 2-pyrrolidone, only one regiomer was observed.
|
|
|||
|---|---|---|---|
| Entry | Substituent R | Conv. [%] | Yield [%] |
|
a 100 mg H-USY (CBV-780, Si/Al = 40), 1.5 mmol N-methylpyrrole, 1.65 mmol TBEC, 2 ml amide, 100 °C, 16 hours.
b 3 regiomers in a ratio of 81:12:6.
c 3 regiomers in a ratio of 13:58:29.
d 3 regiomers in a ratio of 11:52:37.
|
|||
| 1 | H | 48 | 48 |
| 2 | Cyclohexyl | 69 | 48b |
| 3 | n-Octyl | 86 | 49c |
| 4 | n-Butyl | 79 | 46d |

A recycling test was executed on the zeolite catalyst in the coupling reaction between indole and NMP under the standard reaction conditions (Table 4). Surprisingly, the yield of NMP-coupled product increased throughout the consecutive runs from 83% to 94%, whereas the selectivity improved significantly. Possibly, reagents or products accumulated inside the zeolite catalyst throughout the runs. A thermogravimetric analysis on the fresh and spent catalyst (Fig. S2†) revealed an additional mass of approximately 11 wt% compared to the fresh zeolite. The increase in performance can be explained by a selective adsorption of indole during the first run. Finally, powder X-ray diffraction patterns were recorded on both the fresh and spent catalyst (Fig. S3†), which revealed no significant loss in crystallinity after the reaction.
| Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | |
|---|---|---|---|---|---|
| a 100 mg H-USY (CBV-780, Si/Al = 40), 1.0 mmol indole, 1.5 mmol TBEC, 2 ml NMP, 100 °C, 16 hours. The catalyst was washed with 2 ml ethanol and subsequently dried for 5 hours in between runs. | |||||
| Conversion | 98% | 90% | 92% | 94% | 94% |
| Yield | 81% | 87% | 92% | 94% | 93% |
The oxidative coupling of amides and heterocycles can either proceed through a cationic or radical amide-derived intermediate, depending on the structure of the investigated amide and heterocycle, as was proposed by Singh et al.5 For example, in the case of amides with solely primary C(sp3)–H bonds in the α-position, such as N,N-dimethylacetamide (DMA), a 2-electron oxidation of such compounds is strongly disfavored as a primary carbocation would be formed. The formation of C–C or C–N bonds was therefore proposed to proceed by the recombination of radical species formed by 1-electron oxidations of both the amide and heterocycle reactant. Azole-based radicals were proposed as key intermediates for the heterocycle in this pathway; the high N-content of the azoles was deemed beneficial to their generation.
To evaluate the possibility that the zeolite-promoted reaction would exclusively proceed through radical intermediates, we investigated the oxidative coupling of N,N-dimethylacetamide (DMA) and benzimidazole with TBHP as oxidant (Table 5). This specific reaction was shown by Singh et al. to proceed exclusively by a radical recombination as discussed before.5 Other studies have shown that mediators of one-electron redox processes such as Ru, Fe and Cu can promote this particular reaction, probably involving Fenton-like chemistry.5,7,8,10 However, the addition of the zeolite catalyst did not result in an improved yield in these conditions. These results indicate the zeolite catalyst is not capable of activating TBHP for the radical-mediated oxidation of DMA.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Conv. [%] | Yield [%] |
| a Reaction conditions: 1.5 mmol benzimidazole, 1.65 mmol oxidant, 2 ml DMA, 100 °C, 16 hours. b TBAI = Tetra-n-butylammonium iodide. | ||||
| 1 | TBHP | — | 9 | 1 |
| 2 | 75 μmol RuCl3·3H2O | 29 | 15 | |
| 3 | 375 μmol FeCl3·6H2O | 49 | 42 | |
| 4 | 150 μmol TBAIb | 45 | 42 | |
| 5 | 100 mg H-USY | 7 | 3 | |
| 6 | TBEC | — | 45 | 14 |
| 7 | 75 μmol RuCl3·3H2O | 50 | 24 | |
| 8 | 375 μmol FeCl3·6H2O | 57 | 26 | |
| 9 | 150 μmol TBAI | 55 | 26 | |
| 10 | 100 mg H-USY | 27 | 27 | |
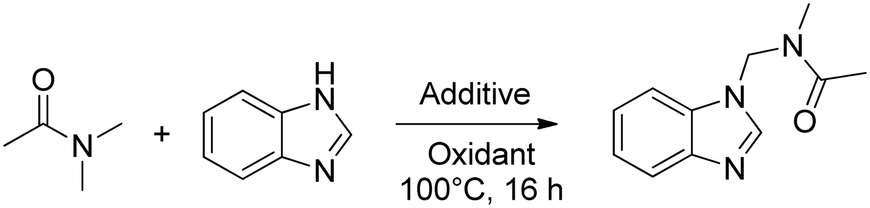
Next, we investigated whether the use of TBEC as oxidant, rather than TBHP, could alter the outcome of the previous experiment (Table 5, entries 6–10). Without any additive (entry 6), we already observed a significant amount of product, indicating that TBEC can be activated thermally without the involvement of redox mediators. While the redox mediators could improve the selectivity towards the heterocoupled product (entries 7–9), the zeolite catalyst was found to be the optimal additive when using TBEC as oxidant, as evidenced by an excellent selectivity for the heterocoupled product (entry 10). The reaction systems with TBHP and redox mediators are however strongly preferred for the oxidative coupling of DMA and benzimidazole as could be expected for reactions proceeding by a radical pathway.
Subsequently, the previous reaction systems with either TBEC or TBHP as oxidant, and using either one-electron redox mediators or the zeolite catalyst as additive were compared in our model reaction of N-methylpyrrole and N-methylpyrrolidone (Table 6). Like in the coupling of DMA and benzimidazole (Table 5), no conversion was observed when using TBHP, either without additive or with zeolite catalyst. Contrarily, in reactions with TBEC as oxidant, already a slight conversion is noticed without additive, which could be strongly boosted in combination with the zeolite catalyst. Although redox mediators can enhance the conversion of 2, they hardly promote the formation of heterocoupled products. These results suggest that although the zeolite catalyst can promote the activation of TBEC, its main role involves promoting the selective coupling of the heterocyclic and amide reactant.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Conv. [%] | Yield [%] |
| a Reaction conditions: 1.5 mmol N-methyl pyrrole, 1.65 mmol oxidant, 2 ml NMP, 100 °C, 16 hours. | ||||
| 1 | TBHP | — | <1 | <1 |
| 2 | 75 μmol RuCl3·3H2O | 75 | 26 | |
| 3 | 375 μmol FeCl3·6H2O | 69 | 35 | |
| 4 | 150 μmol TBAI | 32 | 13 | |
| 5 | 100 mg H-USY | <1 | <1 | |
| 6 | TBEC | — | 18 | 2 |
| 7 | 75 μmol RuCl3·3H2O | 72 | 14 | |
| 8 | 375 μmol FeCl3·6H2O | 44 | 18 | |
| 9 | 150 μmol TBAI | 26 | 6 | |
| 10 | 100 mg H-USY | 84 | 84 | |

A control reaction was performed in which the oxidation of NMP and the subsequent electrophilic attack were performed in two different steps (Fig. 2). The short lifetime of radicals would then preclude their presence during the C–C bond formation, hence excluding the possibility of a radical adduct intermediate. To this aim, NMP was pre-oxidized to 5-hydroxy-N-methyl-2-pyrrolidone (5-OH–NMP) according to a procedure reported by Wang and co-workers (Fig. 2A).22 The use of oxygen as oxidant allows to perform the subsequent C–C bond formation in the absence of oxidants. Approximately 1% of the solvent was converted in this way to 5-OH–NMP, which corresponds to approximately 0.2 mmol of 5-OH–NMP in 2 ml of this solvent.
 | ||
| Fig. 2 Sequential reaction. A) 20 ml NMP, 1 ml H2O, bubbling O2, 160 °C, 3 hours; B) 100 mg H-USY (Si/Al = 40), 1 mmol indole, 2 ml of the pre-oxidized solution of part A, 100 °C, 2 hours, flushed with N2. | ||
The subsequent reaction with 1 mmol of indole under N2 at 100 °C and 100 mg CBV-780 resulted in a quantitative yield of 0.2 mmol of NMP-coupled product (Fig. 2B). No product was formed in the absence of the zeolite catalyst. Through this test reaction, the role of the zeolite also becomes evident: it catalyzes the Friedel–Crafts alkylation of indole with oxidized NMP intermediates.
Based on the foregoing, we propose that the reaction proceeds by a 2-electron oxidation of the cyclic amide (Fig. 3), which can proceed without additive in the case of TBEC as oxidant, but requires redox mediators when employing TBHP. More specifically, TBEC is expected to dissociate into a tert-butoxy (tBu–O°) and carbonate radical (ROC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O°) by thermolysis. The tert-butoxy radical initiates the oxidation of the amide by a hydrogen atom transfer (HAT), driven by the thermodynamic stability of the formed tert-butanol. The carbonate-based radical then further oxidizes the amidyl radical to an N-acyliminium cation by a single electron transfer (SET), due to a kinetic preference over a HAT mechanism.23 The formed cation is possibly stabilized by the zeolite, which is negatively charged due to a proton abstraction by an alkoxide stemming from the carbonate anion. An electrophilic attack of the formed cation on the heterocyclic reactant is proposed, likely proceeding by EAS-like chemistry. By promoting the conversion of the cationic intermediates, the zeolite catalysts enable improved selectivities for the heterocoupling reaction.
O)–O°) by thermolysis. The tert-butoxy radical initiates the oxidation of the amide by a hydrogen atom transfer (HAT), driven by the thermodynamic stability of the formed tert-butanol. The carbonate-based radical then further oxidizes the amidyl radical to an N-acyliminium cation by a single electron transfer (SET), due to a kinetic preference over a HAT mechanism.23 The formed cation is possibly stabilized by the zeolite, which is negatively charged due to a proton abstraction by an alkoxide stemming from the carbonate anion. An electrophilic attack of the formed cation on the heterocyclic reactant is proposed, likely proceeding by EAS-like chemistry. By promoting the conversion of the cationic intermediates, the zeolite catalysts enable improved selectivities for the heterocoupling reaction.
 | ||
| Fig. 3 Proposed mechanism for the oxidative coupling 1 and 2. | ||
Conclusion
In this study, we have shown the possibility to functionalize amides with pyrroles via a direct C(sp3)–H functionalization. Acidic zeolites were used as benign, easily handleable catalysts in combination with the organic peroxide TBEC as oxidant. Excellent yields and selectivities for the heterocoupled product were obtained after optimization of the catalytic system. Other heterocycles such as furans and thiophenes were also reactive. Furthermore, a range of nucleophiles such as unprotected pyrroles, amides and carbamates were shown to be compatible with the technology, thereby leading to the elusive formation of C–N bonds.Methods
Reaction procedure under atmospheric oxygen pressure
Generally, NMP (2 ml) was added to a 10 ml crimp neck vial containing the solid acid additive and a PTFE stirring rod. Subsequently, N-methylpyrrole (1.5 mmol, 133 μl) and anisole (50 μl, internal standard for GC-analysis) were added. The glass vials were sealed with crimp caps containing a rubber septum. A balloon, pre-filled with oxygen gas, was subsequently connected to the vial by a needle. Reactions were conducted in aluminium heating blocks with magnetic stirring. After the designated reaction time, 3 ml of acetone was added to quench the reaction. Solids were separated by centrifugation, and a liquid sample was taken for GC-analysis.Reaction procedure with organic oxidants
Generally, NMP (2 ml) was added to a 3 ml glass liner containing the solid acid additive and a PTFE stirring rod. Subsequently, N-methylpyrrole (1.5 mmol, 133 μl) and anisole (50 μl, internal standard for GC-analysis) were added. The liner was sealed in a homemade, stainless steel autoclave. Reactions were conducted in aluminium heating blocks with magnetic stirring. The internal temperature dependence was calibrated beforehand, and the set temperature of the heating block was adjusted to obtain 100 °C internally. After the designated reaction time, 2 ml of acetone was added to quench the reaction. Solids were separated by centrifugation, and a liquid sample was taken for GC-analysis.Reaction analysis
Reaction mixtures were analysed quantitatively using a Shimadzu GC-2014 equipped with a CP-SIL 5 CB column (Agilent, 100% PDMS, 60 m, 0.25 μm film thickness, 0.32 mm i.d.). Samples of 1 μl were injected automatically using an AOC-20s autosampler and AOC-20i auto-injector aided by the GCsolution software bundle (version 2.44.00). Products were identified using an Agilent 6890 gas chromatograph equipped with an HP-1 MS column and coupled to a 5973 MSD mass spectrometer or by comparison with commercially obtained or synthesized reference samples. 1H NMR spectra of liquid samples were recorded using a Bruker Avance III HD 400 console at 400 MHz, equipped with a 5 mm PABBO BB/19F–1H/D probe and the data were analysed using the MestReNova 12.0.2 software package.Author contributions
JV was responsible for the conception, design and interpretation of the experiments under the supervision of DDV. BK contributed to the purification of reaction products. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Mathys Evens and Galahad O'Rourke for assistance with the experimental work and the thermogravimetric measurements respectively. JV is grateful to Research Foundation Flanders (FWO) for funding (1263522N).References
- P. Ertl, E. Altmann and J. M. McKenna, The Most Common Functional Groups in Bioactive Molecules and How Their Popularity Has Evolved over Time, J. Med. Chem., 2020, 63(15), 8408–8418 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- A. H. Gouliaev and A. Senning, Piracetam and other structurally related nootropics, Brain Res. Rev., 1994, 19, 180–222 CrossRef CAS PubMed.
- K. Moriyama, Recent advances in oxidative C–C coupling reaction of amides with carbon nucleophiles, Tetrahedron Lett., 2017, 58(50), 4655–4662 CrossRef CAS.
- H. Aruri, U. Singh, M. Kumar, S. Sharma, S. Kumar, V. K. Gupta, S. Mignani, R. A. Vishwakarma and P. Pal Singh, Metal-free Cross-Dehydrogenative Coupling of HN-azoles with α-C(sp3)-H Amides via C–H Activation and Its Mechanistic and Application Studies, J. Org. Chem., 2017, 82(2), 1000–1012 CrossRef CAS PubMed.
- H. Aruri, U. Singh, S. Sharma, S. Gudup, M. Bhogal, S. Kumar, D. Singh, V. K. Gupta, R. Kant, R. A. Vishwakarma and P. Pal Singh, Cross-Dehydrogenative Coupling of Azoles with α-C(sp3)–H of Ethers and Thioethers under Metal-Free Conditions: Functionalization of H–N Azoles via C–H Activation, J. Org. Chem., 2015, 80(3), 1929–1936 CrossRef CAS PubMed.
- M. K. Singh, H. K. Akula, S. Satishkumar, L. Stahl and M. K. Lakshman, Ruthenium-Catalyzed C–H Bond Activation Approach to Azolyl Aminals and Hemiaminal Ethers, Mechanistic Evaluations, and Isomer Interconversion, ACS Catal., 2016, 6(3), 1921–1928 CrossRef CAS PubMed.
- Q. Xia and W. Chen, Iron-Catalyzed N-Alkylation of Azoles via Cleavage of an sp3 C–H Bond Adjacent to a Nitrogen Atom, J. Org. Chem., 2012, 77, 9366–9373 CrossRef CAS PubMed.
- M. Ghobrial, K. Harhammer, M. D. Mihovilovic and M. Schnürch, Facile, solvent and ligand free iron catalyzed direct functionalization of N-protected tetrahydroisoquinolines and isochroman, Chem. Commun., 2010, 46, 8836–8838 RSC.
- S. Priyadarshini, P. J. A. Joseph and M. L. Kantam, Copper catalyzed oxidative cross-coupling of aromatic amines with 2-pyrrolidinone: a facile synthesis of N-aryl-γ-amino-γ-lactams, Tetrahedron, 2014, 70, 6068–6074 CrossRef CAS.
- Z. Wan, D. Wang, Z. Yang, H. Zhang, S. Wang and A. Lei, Electrochemical oxidative C(sp3)–H azolation of lactams under mild conditions, Green Chem., 2020, 22, 3742–3747 RSC.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, The Essential Role of Bond Energetics in C-H Activation/Functionalization, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed.
- S. M. Aschmann and R. Atkinson, Atmospheric chemistry of 1-methyl-2-pyrrolidinone, Atmos. Environ., 1999, 33, 591–599 CrossRef CAS.
- S. Murahashi, T. Naota, T. Kuwabara, T. Saito, H. Kumobayashi and S. Akutagawa, Ruthenium-catalyzed oxidation of amides and lactams with peroxides, J. Am. Chem. Soc., 1990, 112, 7820–7822 CrossRef CAS.
- T. Shono, H. Tamaguchi and Y. Matsumura, Electroorganic chemistry. XX. Anodic oxidation of carbamates, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS.
- A. M. Jones and C. E. Banks, The Shono-type electroorganic oxidation of unfunctionalised amides. Carbon–carbon bond formation via electrogenerated N-acyliminium ions, Beilstein J. Org. Chem., 2014, 10, 3056–3072 CrossRef PubMed.
- T. Tsuchimoto, Y. Ozawa, R. Negoro, E. Shirakawa and Y. Kawakami, Zirconium Triflate Catalyzed Direct Coupling Reaction of Lactams with Heterocyclic Arenes under Atmospheric Oxygen, Angew. Chem., Int. Ed., 2004, 43, 4231–4233 CrossRef CAS PubMed.
- E. Shirakawa, N. Uchiyama and T. Hayashi, Iron-Catalyzed Oxidative Coupling of Alkylamides with Arenes through Oxidation of Alkylamides Followed by Friedel-Crafts Alkylation, J. Org. Chem., 2011, 76, 25–34 CrossRef CAS PubMed.
- M. Ghobrial, M. Schnürch and M. D. Mihovilovic, Direct Functionalization of (Un)protected Tetrahydroisoquinoline and Isochroman under Iron and Copper Catalysis: Two Metals, Two Mechanisms, J. Org. Chem., 2011, 76(21), 8781–8793 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent Advances in Radical C−H Activation/Radical Cross-Coupling, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- S. Van Minnebruggen, T. De Baerdemaeker, K. Y. Cheung, A.-N. Parvulescu, U. Müller, P. Tomkins, R. de Oliveira-Silva, X. Meng, F.-S. Xiao, T. Yokoi, W. Zhang, D. Sakellariou and D. De Vos, Alkylation of isobutane with butenes using OSDA-free zeolite beta, J. Catal., 2022, 406, 206–212 CrossRef.
- S.-H. Jeon, P. Xu, N. H. Mack, L. Y. Chiang, L. Brown and H.-L. Wang, Understanding and Controlled Growth of Silver Nanoparticles Using Oxidized N-Methyl-pyrrolidone as a Reducing Agent, J. Phys. Chem. C, 2010, 114(1), 36–40 CrossRef CAS.
- O. Augusto, M. G. Bonini, A. M. Amanso, E. Linares, C. C. X. Santos and S. L. De Menezes, Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology, Free Radicals, 2002, 32(9), 841–859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00880k |
| This journal is © The Royal Society of Chemistry 2023 |