
Zeolite-based catalysts for oxidative upgrading of methane: design and control of active sites
Mizuho
Yabushita
 *a,
Ryota
Osuga
*b,
Toshiyuki
Yokoi
c and
Atsushi
Muramatsu
*de
*a,
Ryota
Osuga
*b,
Toshiyuki
Yokoi
c and
Atsushi
Muramatsu
*de
aDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: m.yabushita@tohoku.ac.jp
bInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: osuga@cat.hokudai.ac.jp
cInstitute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan
dInternational Center for Synchrotron Radiation Innovation Smart, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: mura@tohoku.ac.jp
eInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
First published on 6th June 2023
Abstract
Oxidative upgrading of methane into carbon monoxide, methanol, lower hydrocarbons (such as ethane and ethylene), and other oxygenates has been achieved at relatively low temperatures over zeolite-based catalysts. The high tunability afforded by the framework structures of zeolites offers a unique and precious opportunity for the design of active species, resulting in outstanding catalytic performance. Thus, we categorize the various reported active species in zeolite-based catalysts into three groups in terms of their origin, viz., active species originated from intra-framework heteroatoms, active species introduced by ion-exchange, and active species prepared by impregnation. We present an overview of the performance, structure, and catalysis of each category and comment on the general outlook.
1. Introduction
Methane, the simplest hydrocarbon, is the main component of natural gas and shale gas. This abundant gaseous compound is currently used as a fuel, while its transformation into platform chemicals in industry has been central to C1 chemistry. The exemplified reactions for methane upgrading are dry/steam reforming into a gaseous mixture of CO and H2 (i.e., synthesis gas, also called syngas),1–3 partial oxidation into syngas,4 partial oxidation to methanol and other oxygenates,5,6 oxidative/non-oxidative coupling of methane into lower hydrocarbons such as ethane and ethylene,7 and dehydroaromatization.8,9 Among these reactions, oxidative upgrading is an attractive approach to synthesizing useful compounds directly from methane by skipping the reforming steps, which are endothermic reactions and thus require very high reaction temperatures to achieve high conversion of methane. The extremely high chemical stability of methane, as evidenced by the C–H dissociation energy of ca. 440 kJ mol−1, is a common obstacle and typically necessitates high reaction temperatures for the activation of methane.10 In addition to the requirement of high energy input, such harsh conditions in the presence of oxidant(s) have another drawback, namely, low selectivity for the desired product(s) because the products are more reactive than methane and readily undergo complete oxidation to CO2. Thus, the development of catalysts that can greatly activate methane has been desired and investigated extensively for these decades. Furthermore, the shale gas boom (also called the shale gas revolution) that commenced in the USA in the late 2000s has enhanced the research activity for catalytic upgrading of methane worldwide.10,11 Because of the ease of continuous operation by flow reactors and separation from unreacted methane and products as well as the high thermal stability, heterogeneous catalysts have attracted much attention.Among the variety of heterogeneous catalysts reported, such as metal-oxide-based ones,10,11 zeolite-based materials, which consist of crystalline and porous frameworks, have great potential to be active, selective, and durable catalysts for methane upgrading. Indeed, the number of publications and citations on methane conversion over zeolite-based catalysts has gradually increased since the 1990s, and as described above, the shale gas boom has boosted this trend (Fig. 1).12 The unique tunability of zeolites arises from the replacement of intra-framework Si4+ by heteroatoms (i.e., isomorphous substitution). In particular, the isomorphous substitution by trivalent heteroatoms such as Al3+ generates negatively charged frameworks, which require charge-compensating cations. Various cations such as proton and metal cations can be introduced as active sites onto the thus-generated ion-exchange sites. For supported metal nanoparticles, negatively charged frameworks are beneficial for fixation and isolation of the supported species through the anchoring effect.13,14 Likewise, isomorphously introduced species themselves are able to function as active sites. Another important and attractive feature imparting tunability to zeolites is their well-defined porous structures. In some cases, relatively wide nanospaces such as cages and channel intersections are also present. Such nanospaces of zeolites offer not only high surface area but also the confinement effect,13–15 both of which lead to the high dispersion and anti-aggregation nature of supported metal (or metal oxide) species that act as active sites for methane conversion.
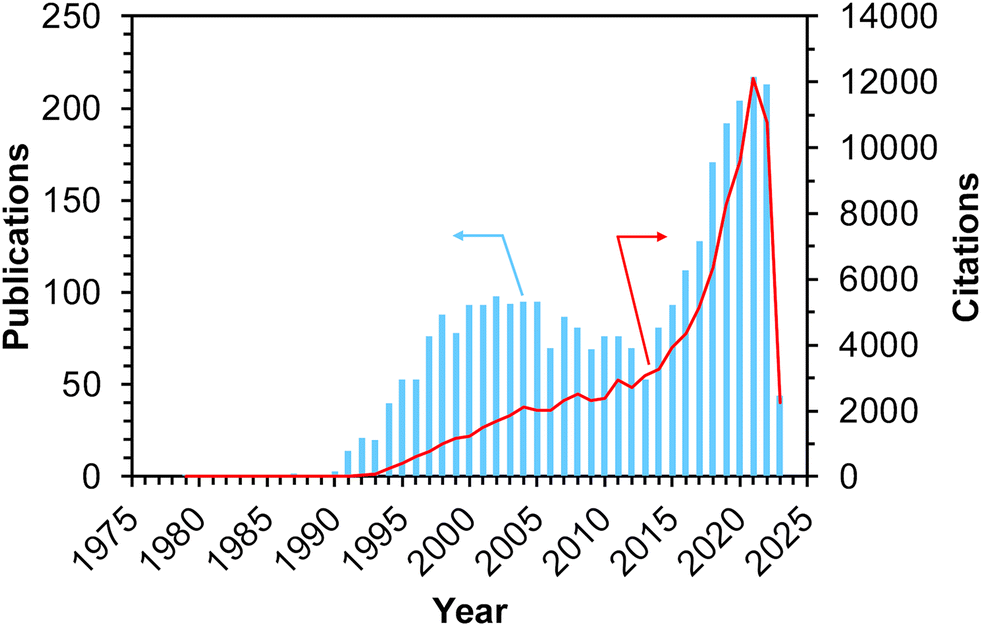 | ||
| Fig. 1 Number of publications and citations searched with the keywords of “methane + zeolite + catalyst” on Web of Science™ (accessed and searched on April 1st, 2023).12 | ||
In light of these insights, we have categorized the zeolite-based catalysts reported for oxidative upgrading of methane into three groups (Fig. 2): active sites originated from intra-framework heteroatoms (section 2); active sites created in zeolite micropores via ion-exchange (section 3); and active sites prepared via impregnation on zeolite surfaces (section 4). For each category, we have summarized the performance, structure, and catalysis of representative materials.
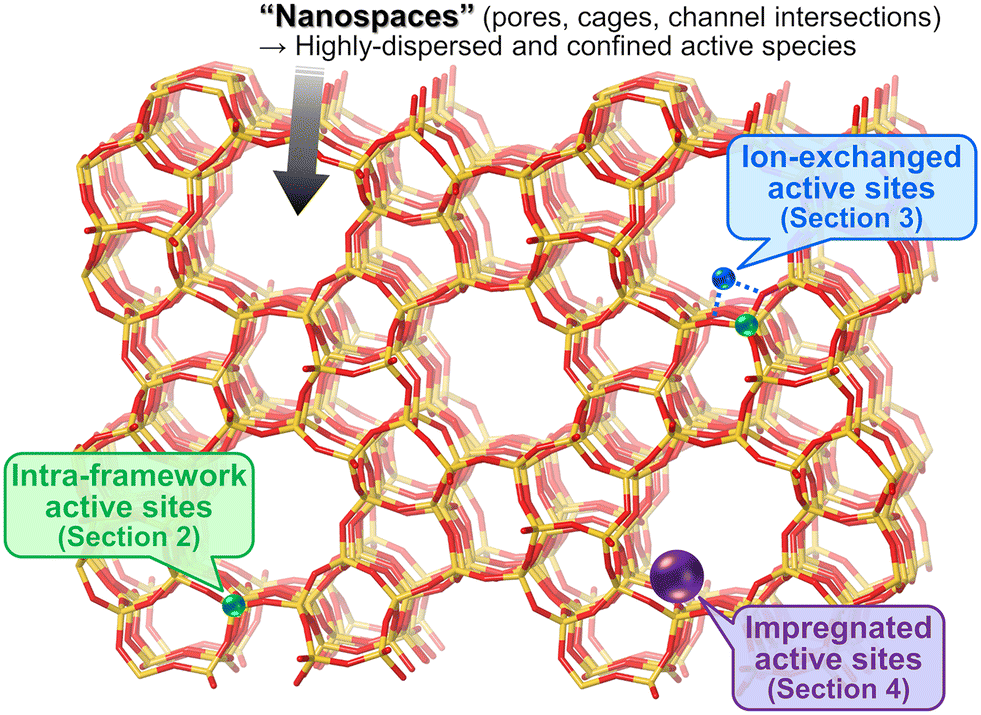 | ||
| Fig. 2 Approaches to designing zeolite-based catalysts. | ||
2. Active sites originated from intra-framework heteroatoms
A variety of heteroatoms such as Al3+, Ga3+, Fe3+, and Ti4+ are known to be introduced into zeolite frameworks via isomorphous substitution of Si4+, as mentioned above. Such isomorphously substituting heteroatoms function as catalytic active sites themselves, and cationic species introduced via ion-exchange sites also contribute to catalytic reactions (details are described in section 3, except for the case of H+). In this section, we discuss active sites originated from intra-framework heteroatoms for oxidative upgrading of methane. The exemplified catalysts introduced in this section are summarized in Table 1.| Entry | Catalyst | Framework topology | Plausible active site | Oxidant | Reactor type | Solvent | T /K | t /h | SVd/mL h−1 gcat−1 | P /MPa | Conv./% | Target C-based product(s) | Target product | By-product(s) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield/% | Sel./% | |||||||||||||||
| a Abbreviations in this table: MeOH = methanol; MeOOH = methyl hydroperoxide. b Reaction temperature. c Reaction time (only for batch-type reaction). d Space velocity (only for flow-type reaction). e Reaction pressure. f The catalytic performance at various temperatures is shown in Fig. 3. g Proposed catalytic cycle consisting of W-, Mn-, and Ti-including species is shown in Fig. 6. h These values include both C2 and C3 compounds. | ||||||||||||||||
| 1 | Fluorinated H-MOR | MOR | H+ (superacid site) | N2O | Flow | — | 623 | — | 4.2 × 104 | n.d. | 0.18 | C2 compounds | 0.17 | 93 | MeOH | 18 |
| 2 | [Ga]-MFIMCf | MFI | Ga species | O2 | Flow | — | 923 | — | 7.5 × 103 | 0.1 | 10 | CO | 8.0 | 80 | CO2 | 22 |
| 3 | [Ga]-MFIHTf | MFI | Ga species | O2 | Flow | — | 923 | — | 7.5 × 103 | 0.1 | 6.7 | CO2 | 6.3 | 94 | CO | 22 |
| 4 | Fe/ZSM-5 | MFI | Fe dinuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.05 | 0.7 | MeOH | 0.084 | 12 | HCOOH, MeOOH, CO2 | 30 |
| 5 | Cu–Fe/ZSM-5 | MFI | Fe dinuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.05 | 0.7 | MeOH | 0.68 | 85 | HCOOH, MeOOH, CO2 | 30 |
| 6 | Fe-silicalite-1 | MFI | Fe dinuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.05 | 0.3 | MeOH | 0.051 | 17 | HCOOH, MeOOH, CO2 | 30 |
| 7 | 1.5% Cu–1.5% Fe/ZSM-5 | MFI | Fe dinuclear complex | H2O2 | Flow | H2O | 323 | — | 4.0 × 102 | 2.0 | 0.5 | MeOH | 0.46 | 92 | CO2 | 32 |
| 8 | Fe-MFI | MFI | Fe species | H2O2 | Batch | Sulfolane/H2O | 323 | 2 | — | 3 | n.d. | MeOH | n.d. | 85 | HCOOH, HCHO | 35 |
| 9 | Mn–Na2WO4/Ti-MWW | MWW | W6+ speciesg | O2 | Flow | — | 993 | — | 8.0 × 103 | 0.1 | 26 | C2 + C3 compounds | 20h | 76h | CO, CO2 | 36 |
Upon the isomorphous substitution of Si4+ by trivalent Al3+ and the introduction of H+ as a charge-compensating cation, Brønsted acid sites are generated in zeolites. Although the activation of methane via typical acids is extremely difficult owing to its low proton affinity (544 kJ mol−1) and weak acidity (pKa ∼ 40),11 superacids, which exhibit stronger acidity than 100% sulfuric acid (the Hammett acidity function H0 of sulfuric acid = −12), have been reported to activate methane to a pentacoordinated carbocation, i.e., CH4 + H+ (superacid) → CH5+ → CH3+ + H2.16 The typical heterogeneous superacid catalyst for methane conversion is sulfated zirconia,17 but zeolite-based superacid catalysts have also been investigated. In the presence of N2O as an oxidant, a sample of commercially available H-MOR provided a methane conversion of at most 0.2% at 698 K, possibly owing to its insufficient Brønsted acidity for methane activation (H0 = −10.6), while the fluorination of H-MOR enhanced its acidity (H0 = −13) and more than doubled the methane conversion.18 In this reaction, the major products at 623–673 K were C2–C4 compounds without the formation of CO or CO2 (entry 1 in Table 1), probably as a result of the coupling of methane with the surface-generated carbocations containing a penta-coordinated carbon atom as suggested for homogeneous catalytic systems.16 In contrast, at 698 K, the oxidation of methane and produced hydrocarbons proceeded readily to form CO and CO2. In a related study by Spivey et al., the surface of H-MFI (i.e., H-ZSM-5) was modified by HBr + AlBr3, a combination known to be a superacid,19 and the resulting catalyst converted methane into C2, C3, C4, and aromatic compounds in the absence of an oxidant at 573 K.20 However, the methane conversion was only ∼1%. These reports suggest that the activation of methane remains a great challenge even with state-of-the-art zeolite-based superacid catalysts, owing to the almost inert nature of methane against acid–base reactions. Thus, different activation approaches are much preferred.
We found that the Ga species introduced in the MFI zeolites (Si/Ga molar ratio = 41) by a two-step synthetic procedure called the mechanochemical method, which consists of the mechanochemical treatment of SiO2 and a heteroatom source to obtain a mixed metal oxide and subsequent hydrothermal treatment of the resulting mixed metal oxide to synthesize heteroatom-containing zeolites,21 exhibited a high selectivity for CO in the oxidative conversion of methane.22 This catalyst (hereafter, [Ga]-MFIMC) produced CO as the main product (80% selectivity) with CO2 (20%) at 10% conversion of methane at 923 K in the presence of O2 as an oxidant (Fig. 3A and entry 2 in Table 1). Given the lack of stoichiometry between CO and H2 (the H2/CO molar ratio was at most 1.1), this partial oxidation was assumed to proceed via an indirect pathway, i.e., the complete oxidation of methane followed by steam/dry reforming reactions and the reverse water-gas shift reaction, rather than the direct partial oxidation pathway. The fact that the Brønsted acidity of Ga-incorporated zeolites is weaker than that of Al-incorporated zeolites23,24 indicates that the methane activation over [Ga]-MFIMC occurs in a completely different manner from the cases of superacid catalysts (vide supra). Interestingly, the product distribution given by [Ga]-MFIMC was in stark contrast to that obtained using the control specimen prepared by the typical one-step hydrothermal treatment for tetraethyl orthosilicate (TEOS) and Ga(NO3)3 ([Ga]-MFIHT), which yielded CO2 as the main product (Fig. 3B and entry 3 in Table 1). This could arise from the difference in the nature of the Ga species in [Ga]-MFIMC and [Ga]-MFIHT. Our related studies on zeolites synthesized via the mechanochemical method25,26 suggest that the Ga species in [Ga]-MFIMC are distributed in the particles more uniformly than those in [Ga]-MFIHT, and these well-distributed Ga species result in the unique catalytic activity. For the aromatization of propane, Giannetto et al. demonstrated that extra-framework Ga species and Brønsted acid sites in steam-treated [Al,Ga]-MFI zeolites cooperatively functioned as active sites by altering the ratio of extra-framework Ga species to acid sites.27 Stepanov et al. revealed via isotope tracer analysis relying on solid-state 13C cross-polarization/magic angle spinning nuclear magnetic resonance (13C CP/MAS NMR) spectroscopy that the dissociative adsorption of methane to form Ga-methyl species (upper route in Fig. 4) was the major pathway over Ga2O3 supported on H-beta, and another activation pathway to generate Ga-methoxy species (lower route in Fig. 4) was also possible but minor.28 On the basis of these reports, the highly dispersed Ga species in [Ga]-MFIMC achieved by the mechanochemical method22 could enhance the formation of binary active sites, extra-framework Ga species and acid sites, leading to the unique catalytic performance of [Ga]-MFIMC for the oxidative conversion of methane.
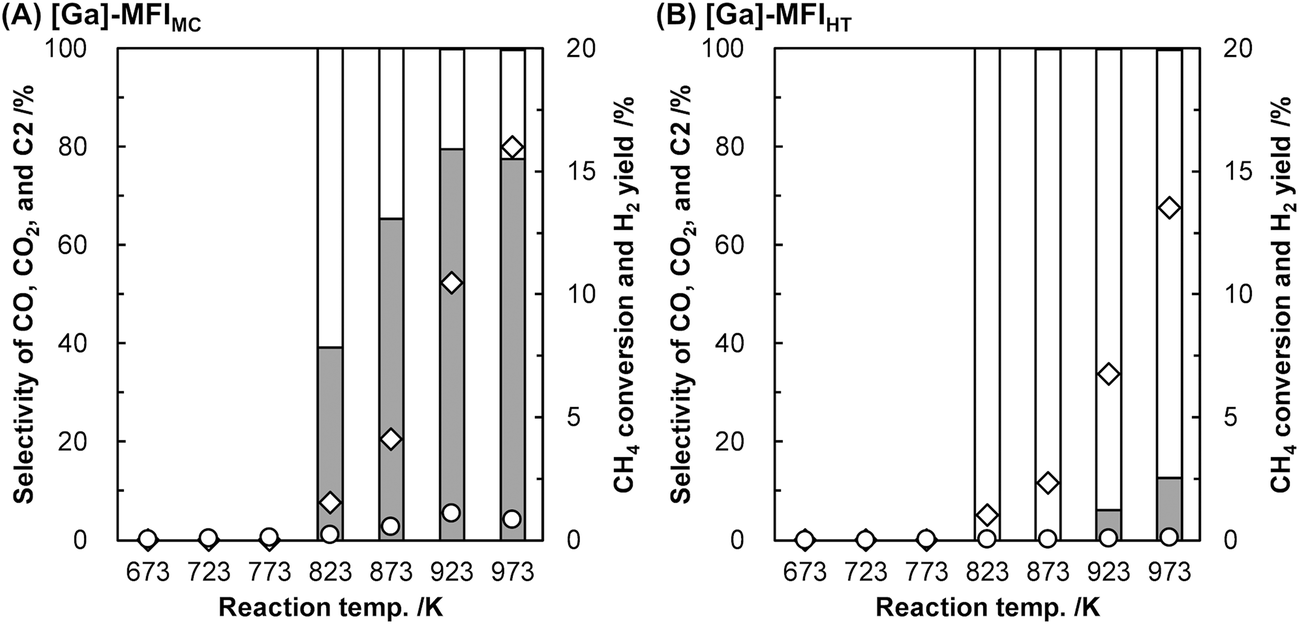 | ||
| Fig. 3 Oxidative conversion of methane over (A) [Ga]-MFIMC (Si/Ga = 41) and (B) [Ga]-MFIHT (Si/Ga = 37).22 Reaction conditions: CH4/O2/Ar = 8.0/2.0/2.5 mL min−1; SV = 7.5 × 103 mL h−1 gcat−1; 673–973 K; 0.1 MPa. Legends: grey bars = CO selectivity; white bars = CO2 selectivity; black bars = C2 selectivity; diamonds = CH4 conversion; circles = H2 yield. Although C2 hydrocarbons were detected, their selectivity was <1%. | ||
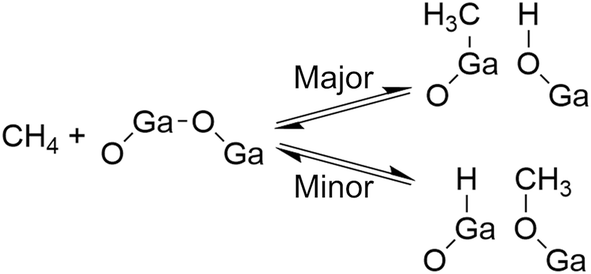 | ||
| Fig. 4 Methane activation over Ga2O3 species supported on H-beta. | ||
Fe-containing MFI zeolites were reported as catalysts for the methane-to-methanol (MTM) reaction with the oxidant H2O2 under mild conditions.29 As in the case of Ga-incorporated zeolites (vide supra), the weak acidity of Fe-incorporated zeolites23,24 rules out the possibility of acid–base-type activation of methane. Hutchings et al. found that Fe-containing ZSM-5 (Si/Al molar ratio = 15, denoted as Fe/ZSM-5) prepared via solid-state ion exchange (SSIE) produced methanol in 12% selectivity along with formic acid, methyl hydroperoxide, and CO2 in the presence of H2O2 at 323 K (entry 4 in Table 1).30 The addition of Cu species to this catalyst suppressed the over-oxidation of methanol to formic acid and CO2 and enabled the selective production of methanol (85% selectivity, entry 5 in Table 1) because Cu species suppress the formation of ˙OH radicals, which cause such undesired over-oxidation.31 From ex situ X-ray absorption spectroscopy (XAS), ultraviolet-visible (UV-vis) spectroscopy, and density functional theory (DFT) calculations, Hutchings et al. identified the dinuclear Fe complex containing antiferromagnetically coupled high-spin octahedral Fe3+ centers, [Fe2(μ2-OH)2(OH)2(H2O)2]2+, as the most likely active site and proposed the catalysis mechanism shown in Fig. 5. This active site seemed to be anchored on the divalent ion-exchange sites (so-called pair sites) of ZSM-5, which allowed it to perform constantly in a continuous operation inside a fixed-bed flow reactor at 323 K (entry 7 in Table 1).32 In the same report, the hydrothermally-synthesized Fe-containing silicalite-1 (silicalite-1 is an Al-free (i.e., pure-silica) MFI-type zeolite) was found to exhibit a catalytic activity similar to Fe/ZSM-5 (entry 6 in Table 1).30 Although the Fe atoms were present in the framework just after hydrothermal synthesis, a part of the Fe species were extracted to form the octahedral Fe species located outside the framework upon the calcination performed to remove the molecules used as an organic structure-directing agent (OSDA). This phenomenon could generate both ion-exchange sites on the remaining intra-framework Fe species and the dinuclear Fe complex [Fe2(μ2-OH)2(OH)2(H2O)2]2+. However, spectroscopic discrimination of these two Fe species is quite difficult and the local structure of actual active sites has remained unclear. Previous studies have reported intra-framework Fe species and Fe oligomers as active sites for Fenton-type oxidation of hydrocarbons with H2O2 over Fe-containing zeolites.33,34 These reports suggest that such sites also are responsible for the oxidative conversion of methane with H2O2. For the same reaction, one member of our research team investigated solvent effects with H2O2 and the hydrothermally synthesized Fe-containing MFI zeolite catalyst in a batch reactor.35 A mixed solvent of 50 vol% sulfolane/H2O yielded the highest selectivity for methanol (85%) (entry 8 in Table 1). Given that the formation of CO2 was negligible in the presence of sulfolane in the catalytic system, this solvent seems to have a similar role to the Cu additive described above (i.e., the suppression of the formation of ˙OH radicals).
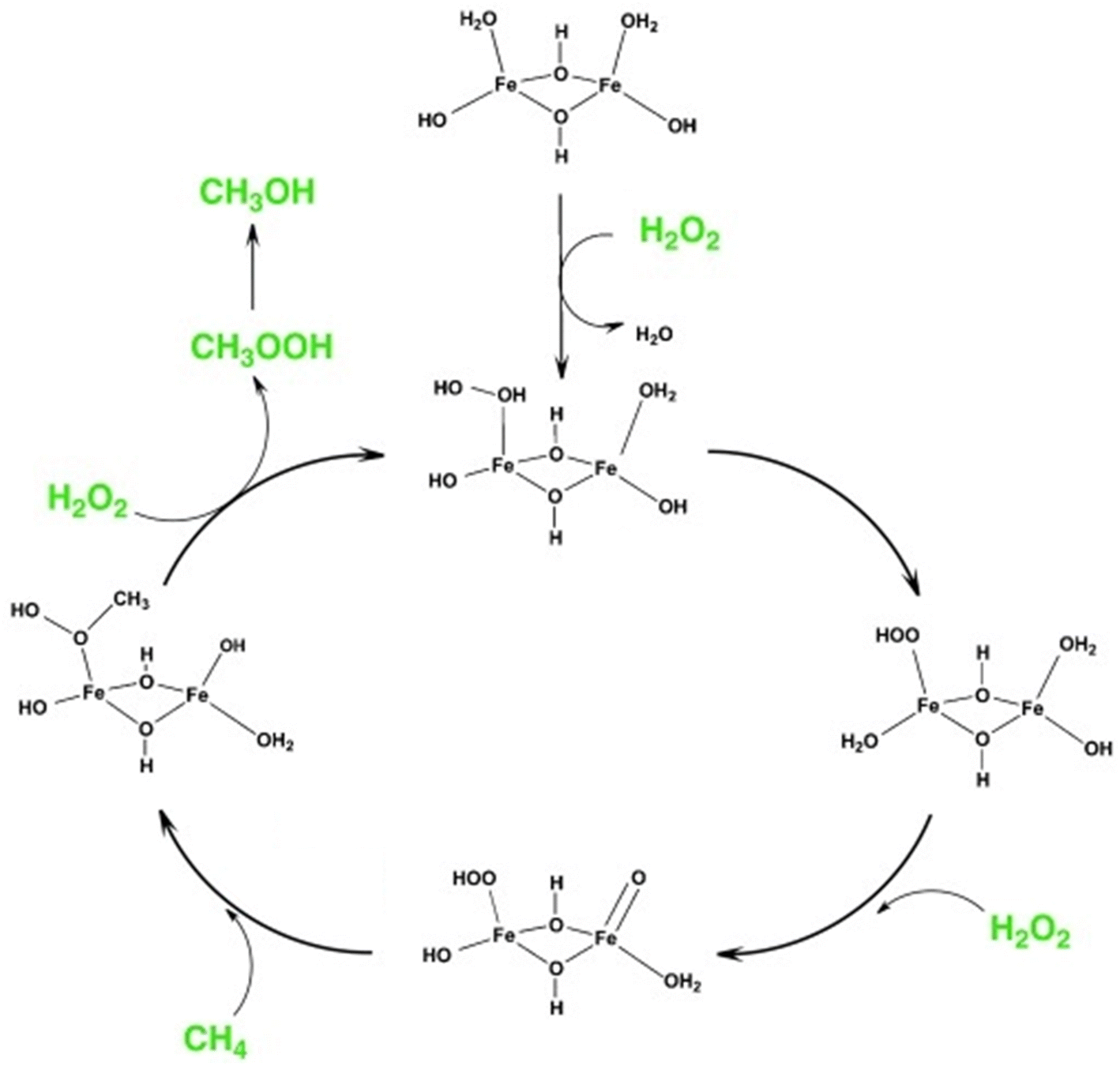 | ||
| Fig. 5 Proposed reaction mechanism of MTM reaction over the active site of [Fe2(μ2-OH)2(OH)2(H2O)2]2+ in Fe-containing ZSM-5. Reprinted from ref. 30 with permission from John Wiley and Sons, copyright 2012. | ||
As mentioned above, intra-framework heteroatoms themselves can be used as catalytic active sites or as precursors to active sites. For the latter scenario, Lu et al. employed a Ti-incorporated MWW zeolite as a support for Mn–Na2WO4 species,36 which have been known to be outstanding active sites for the oxidative coupling of methane (OCM) since their discovery.37–39 The Mn–Na2WO4/Ti-MWW catalyst exhibited a lower activation energy (80–100 kJ mol−1) than the typical Mn–Na2WO4/SiO2 (180–200 kJ mol−1), enabling the production of C2 and C3 compounds at relatively low reaction temperatures of 993–1073 K (entry 9 in Table 1). Ti-MWW behaved as a source of both SiO2 and Ti, the former being a typical support for the Mn–Na2WO4 catalyst.37–39 The Ti species derived from Ti-MWW and the Mn species supplied via the incipient wetness impregnation (IWI) method formed the MnTiO3 phase. In the presence of both CH4 and O2, this new phase triggered the redox cycle of Mn2+ ↔ Mn3+ and resulted in a high O2 activation rate (Fig. 6), leading to the low-temperature OCM activity of Mn–Na2WO4/Ti-MWW.
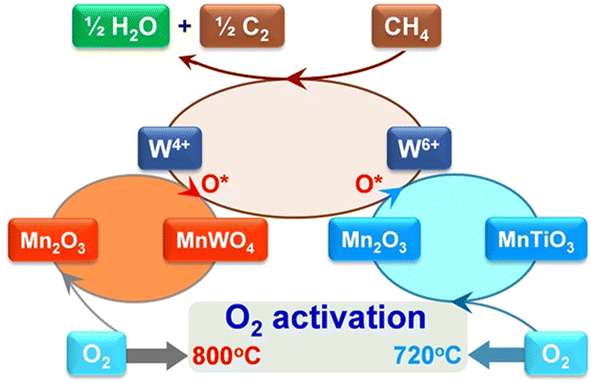 | ||
| Fig. 6 Proposed catalytic cycle for OCM reaction mediated by the redox cycle Mn2+ ↔ Mn3+ over Mn–Na2WO4/Ti-MWW. Reprinted from ref. 36 with permission from The American Association for the Advancement of Science, copyright 2017. | ||
3. Active sites created in zeolite micropores via ion exchange
Ion exchange can offer various types of chemical species in the zeolite micropores, including isolated and multi-nucleus cations, sub-nano clusters, and nano-sized metals (metal oxides). Such metal species exhibit unique and excellent catalytic performance in various catalytic reactions. Here, the active sites on the zeolite surfaces prepared via the ion exchange method are introduced and examined with some exemplified studies summarized in Tables 2 and 3.| Entry | Catalyst | Framework topology | Plausible active site | Oxidant | Reactor type | Solvent | T /K | t /h | SVd/mL h−1 gcat−1 | P /MPa | Conv./% | Target C-based product(s) | Target product | By-product(s) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield/μmol gcat−1 | Yield mmol gcat−1 h−1 | Sel./% | |||||||||||||||
| a Abbreviations in this table: MeOH = methanol; DME = dimethyl ether. b Reaction temperature. c Reaction time (only for batch-type reaction). d Space velocity (only for flow-type reaction). e Reaction pressure. f Reaction data under various conditions are shown in Fig. 11. g Water was co-flowed. The reaction data including product selectivity are shown in Fig. 12. h Reaction data for TOS of 30 h are shown in Fig. 13G. i Reaction data for TOS of 190 min are shown in Fig. 14. | |||||||||||||||||
| 1 | Cu-MFI | MFI | Cu dinuclear complex | O2 | Chemical looping | — | 398 | — | — | 0.1 | — | MeOH | 8.2 | — | 98 | CO2 | 46 |
| 2 | Cu-MOR | MOR | Cu trinuclear complex | O2 | Chemical looping | — | 473 | — | — | 0.1 | — | MeOH, DME | 160 | — | 80 | CO, CO2 | 47 |
| 3 | Cu-MOR | MOR | Cu dinuclear complex | H2O | Chemical looping | — | 473 | — | — | 0.7 | — | MeOH | 204 | — | 97 | CO2 | 49 |
| 4 | Cu-H-ZSM-5f | MFI | CuxOy | O2 | Flow | — | 483 | — | 2.4 × 103 | 0.1 | — | MeOH | — | 1.81 × 10−3 | 70 | CO2 | 50 |
| 5 | Cu-BEAg | Beta | Cu dinuclear complex | N2O | Flow | — | 593 | — | 1.2 × 104 | 0.1 | 1.0 | MeOH | — | 0.24 | 72 | CO2, C2H4, DME | 52 |
| 6 | 5Cu/AEI(Na)-850h | AEI | Cu dinuclear complex | N2O | Flow | — | 623 | — | 1.5 × 104 | 0.1 | 1.0 | MeOH | — | 1.64 | 50 | CO, CO2, DME, hydrocarbons | 62 |
| 7 | Cu–H/CHA-FAUi | CHA | Cu dinuclear complex | N2O | Flow | — | 623 | — | 1.5 × 104 | 0.1 | 0.6 | MeOH, lower olefins | — | 0.90 | 66 | CO, CO2, DME | 63 |
| 8 | Cu–H/CHA-Ami | CHA | Cu dinuclear complex | N2O | Flow | — | 623 | — | 1.5 × 104 | 0.1 | 0.2 | MeOH, lower olefins | — | 0.30 | 57 | CO, CO2, DME | 63 |
| 9 | Fe/ZSM-5SSIE | MFI | Fe mononuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | — | C1 oxygenates | — | 21.5 | 96 | CO2 | 65 |
| 10 | Fe/ZSM-5IWI | MFI | Fe mononuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | — | C1 oxygenates | — | 9.42 | 97 | CO2 | 65 |
| 11 | Fe/ZSM-5IE | MFI | Fe mononuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | — | C1 oxygenates | — | 12.5 | 98 | CO2 | 65 |
| 12 | Fe/ZSM-5(TPA) | MFI | Fe mononuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | — | C1 oxygenates | — | 25.9 | — | CO2 | 66 |
| 13 | Fe/ZSM-5(TPA + Na) | MFI | Fe mononuclear complex | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | — | C1 oxygenates | — | 200 | n.d. | CO2 | 66 |
| Entry | Catalyst | Framework topology | Plausible active site | Oxidant | Reactor type | T /K | SVb/mL h−1 gcat−1 | P /MPa | Conv./% | Target C-based product | Target product | By-product | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield/% | Sel./% | |||||||||||||
| a Reaction temperature. b Space velocity. c Reaction pressure. d Reaction data at different SV are shown in Fig. 16. | ||||||||||||||
| 1 | Rh-MOR | MOR | Rh sub-nano cluster | O2 | Flow | 873 | 1.2 × 106 | 0.1 | 84 | CO | 76 | 91 | CO2 | 71 |
| 2 | Rh-MFI[TPA]d | MFI | Rh sub-nano cluster | O2 | Flow | 873 | 1.2 × 106 | 0.1 | 86 | CO | 79 | 91 | CO2 | 72 |
| 3 | Rh-MFI[TPA,Na]d | MFI | Rh sub-nano cluster | O2 | Flow | 873 | 1.2 × 106 | 0.1 | 62 | CO | 50 | 80 | CO2 | 72 |
| 4 | Rh-Y | FAU | Rh cation | O2 | Flow | 873 | 1.2 × 106 | 0.1 | 78 | CO | 69 | 89 | CO2 | 73 |
| 5 | Rh-ASA | — | Rh oxide | O2 | Flow | 873 | 1.2 × 106 | 0.1 | 86 | CO | 78 | 90 | CO2 | 73 |
Among the numerous studies, Cu ion-exchanged zeolites are the most widely investigated zeolite catalyst for methane conversion, where Cu-oxo nanoclusters have emerged as promising active sites for the partial oxidation of methane into methanol.40,41 The design of active sites in these catalysts is based on enzymes. Methane monooxygenase (MMO), a naturally occurring enzyme, produces methanol from methane even under ambient conditions.42 The active sites of MMOs are the ionic binuclear metal centers Cu and Fe, which possess metal-oxo structures with highly active oxygens (Fig. 7).43–45 Industrial application of MMOs for methanol production, however, is not realistic from the viewpoint of cost and reaction efficiency. To overcome these issues, specific nanospaces within the zeolite micropores have attracted much attention because the confinement effect afforded by such nanospaces can stabilize metal and metal oxide clusters. Thus, the MMO-mimetic catalyst design, relying mainly on the ion exchange of Cu species on the zeolite surface, has been one of the mainstreams for the development of zeolite-based catalysts.
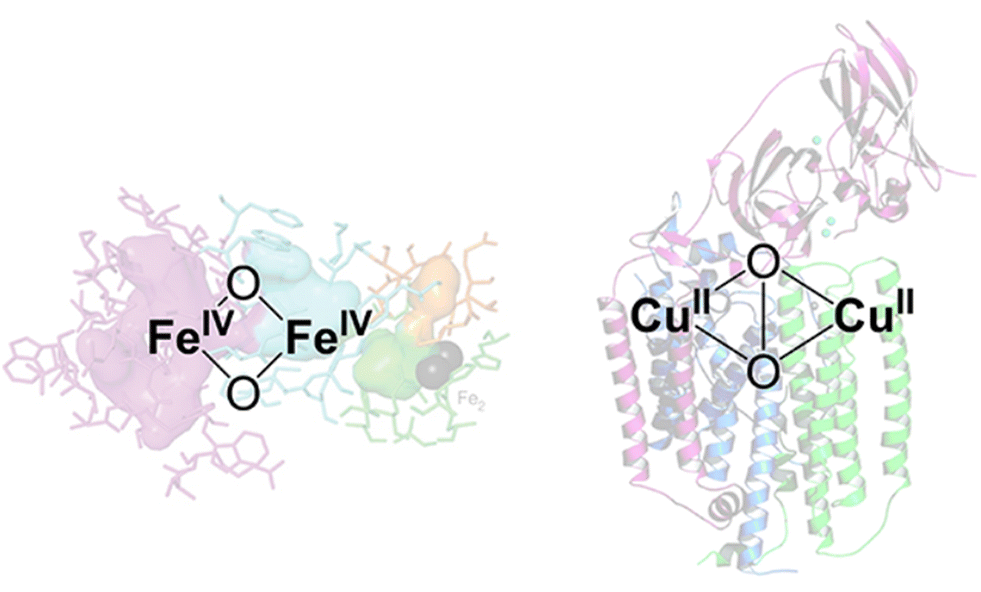 | ||
| Fig. 7 Proposed Fe- and Cu-based binuclear active sites in MMOs. Reprinted from ref. 45 with permission from American Chemical Society, copyright 2018. | ||
In 2005, Groothaert et al. reported that Cu ion-exchanged ZSM-5 (Cu-ZSM-5) enabled the selective oxidation of methane to methanol, where 8.2 μmol g−1 of methanol was obtained with 98% of selectivity (entry 1 in Table 2).46 The zeolite, which was pretreated with O2 at 573 K, converted methane to methanol at 398 K under flowing methane. In the UV-vis spectra of the O2-activated catalyst, a band attributed to the bis(μ-oxo)dicopper species was observed at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 cm−1, and the integrated intensity of the band decreased with methane flowing. This result suggests that the bis(μ-oxo)dicopper species are active sites in this reaction. On the other hand, the extraction of methoxy species generated on the zeolite surface as methanol using water is essential and inevitable for completing the reaction cycle, owing to the high adsorption enthalpy of methanol on the active sites (Fig. 8), resulting in low reaction efficiency. Such disadvantages of the chemical looping process are serious and need to be overcome for industrial applications.
700 cm−1, and the integrated intensity of the band decreased with methane flowing. This result suggests that the bis(μ-oxo)dicopper species are active sites in this reaction. On the other hand, the extraction of methoxy species generated on the zeolite surface as methanol using water is essential and inevitable for completing the reaction cycle, owing to the high adsorption enthalpy of methanol on the active sites (Fig. 8), resulting in low reaction efficiency. Such disadvantages of the chemical looping process are serious and need to be overcome for industrial applications.
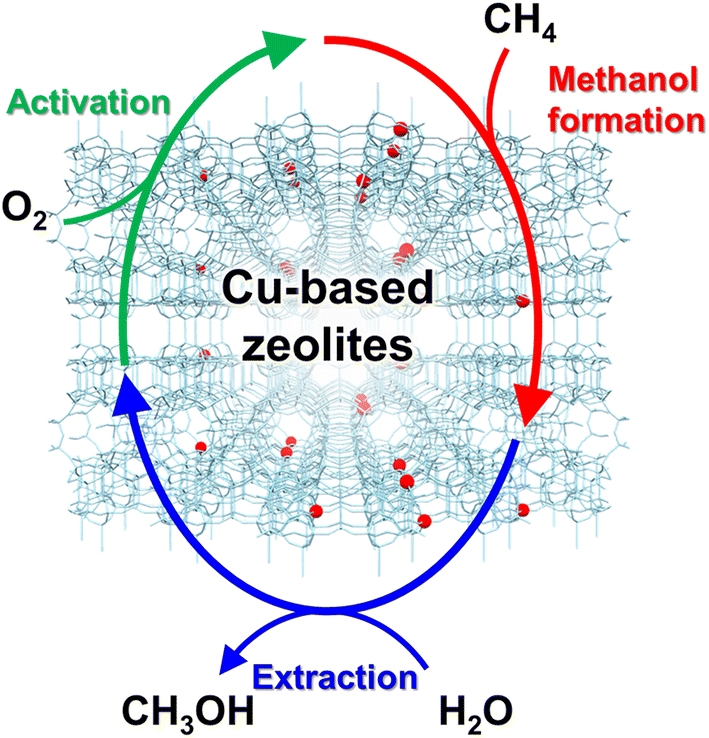 | ||
| Fig. 8 Schematics of chemical looping process for partial oxidation of methane to methanol over Cu-based zeolites. | ||
Dicopper oxygen clusters have been regarded as primary active species for some time now, but trinuclear copper oxygen clusters in MOR-type zeolites have also been found to behave as active species for the partial oxidation of methane to methanol.47 The study cited as ref. 47 employed the chemical looping process, which is almost identical to the process used in ref. 46. In the case of ref. 47, methanol and dimethyl ether were detected as reaction products, and the total yield of methanol and dimethyl ether reached 160 μmol g−1 (entry 2 in Table 2), an order of magnitude higher than that of related studies reported at the time.46,48 Despite the different Si/Al ratios, the total yield of methanol and dimethyl ether was proportional to the Cu concentration (Fig. 9A), suggesting that only one type of active site was formed regardless of the different Cu concentrations. Some spectroscopic and computational approaches proposed that [Cu3(μ-O)3]2+ clusters functioned as active sites and were located at the 8-ring (8R) side pocket of the MOR-type structure (Fig. 9B and C). The authors inferred that the hydrophobic environment provided by the 8R side pockets enhanced the activity because of the presence of a similar hydrophobic environment in MMOs. In this respect, highly active zeolites are thought to require a high local concentration of framework Al atoms at the 8R side pockets. Thus, these results suggest the importance of the location of the framework Al atoms in zeolite frameworks. In 2017, a breakthrough study in the partial oxidation of methane to methanol over Cu ion-exchanged zeolites with a chemical looping process was reported by Sushkevich and van Bokhoven et al.49 Notably, their study used water molecules as an oxidant for methane. The chemical looping process used for the partial oxidation of methane to methanol consisted of three steps as mentioned above (see also Fig. 8): (i) activation with O2, (ii) reaction of methane, and (iii) extraction of methoxy species as methanol from the catalyst surface with water. In contrast, the direct stepwise method was successfully used for converting methane into methanol with 204 μmol g−1 of productivity and 97% of selectivity in the study cited as ref. 49 (entry 3 in Table 2). The active site was determined to be the “mono”(μ-oxo)dicopper species by using XAS and Fourier transform infrared (FT-IR) measurements combined with DFT calculations. In the first step of the reaction, methane reacted with the mono(μ-oxo)dicopper species at 473 K, resulting in the formation of methoxy species and Brønsted acid sites, accompanied by the reduction of Cu2+ to Cu+ (Fig. 10). After the reaction, methanol and hydrogen were formed with the regeneration of mono(μ-oxo)dicopper species by flowing water vapor at the same temperature as the flowing methane. In addition, the zeolite was reusable with high activity for at least five cycles.
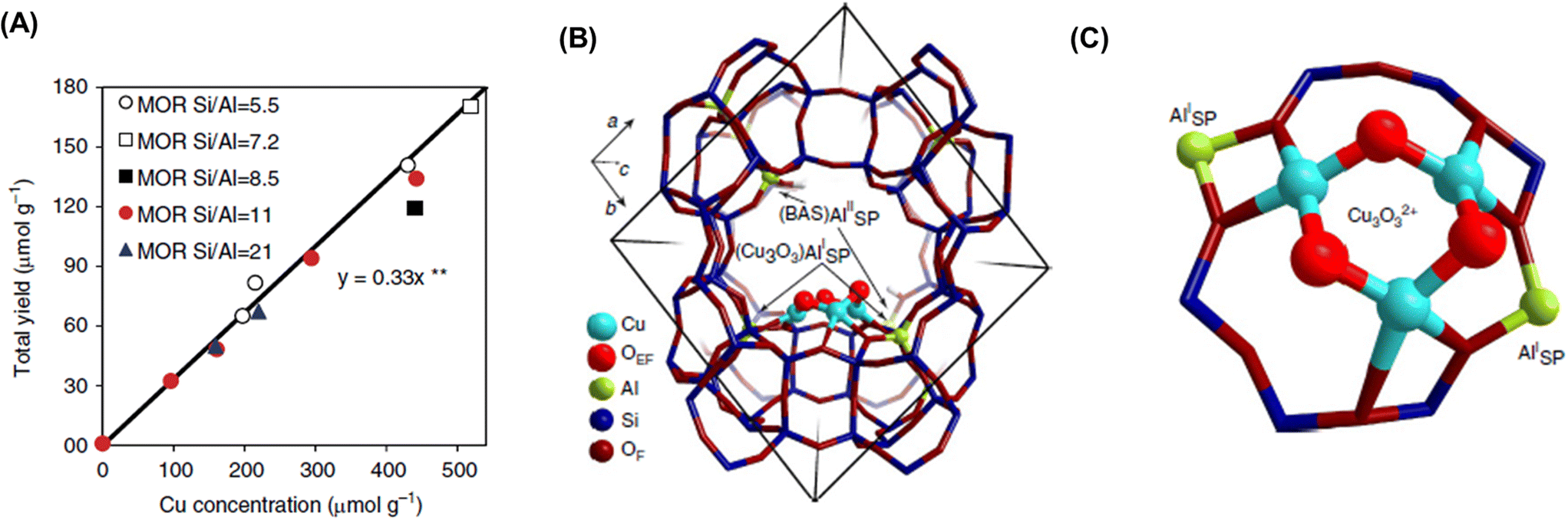 | ||
| Fig. 9 (A) Total yield of methanol over Cu-MOR with different Si/Al ratios and Cu concentrations. (B) Location and (C) structure of [Cu3(μ-O)3]2+ cluster in MOR determined by DFT calculations. Reproduced from ref. 47 with permission from Springer Nature, copyright 2015. | ||
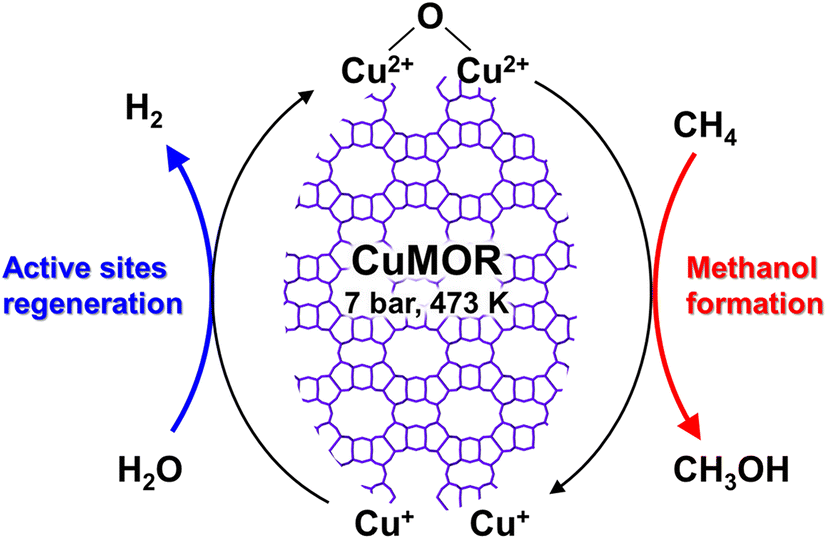 | ||
| Fig. 10 Plausible reaction scheme for partial oxidation of methane to methanol over Cu-MOR via the chemical looping process, using water as an oxidant. | ||
As mentioned above, the chemical looping process is a powerful method for the partial oxidation of methane to methanol. However, the establishment of a continuous-flow system has been highly desired to realize the MTM reaction in the chemical industry. Román-Leshkov and his co-workers reported the first demonstration of the partial oxidation of methane to methanol using O2 as an oxidant over Cu ion-exchanged zeolites at low reaction temperatures (483–498 K).50 This continuous system was achieved by co-feeding methane, O2, and water, where Cu ion-exchanged ZSM-5 (Cu-H-ZSM-5) showed high catalytic activity (entry 4 in Table 2). The catalytic activity depended on the framework types of zeolites, and the catalytic yield for methanol reached 491 μmol g−1 (Fig. 11). This was the highest value reported by any study at the time, owing to the continuous-flow system. In the study by Román-Leshkov et al., UV-vis spectra revealed the generation of copper oxide species during the reaction; however, the catalytically active sites were not identified. Starting with this report, the partial oxidation of methane to methanol via the continuous-flow system has been actively investigated by several research groups.41,51,52 O2 and water have been widely accepted as effective oxidants for the partial oxidation of methane in both the chemical looping and the continuous-flow processes. In addition, stronger oxidants such as N2O were used for the continuous-flow system. Liu et al. reported the continuous methane oxidation to methanol over Cu ion-exchanged beta zeolite (Cu-BEA).52 This reaction was boosted by the presence of water; methanol productivity was much higher in the presence of water than in the absence of water (Fig. 12A and B and entry 5 in Table 2). The authors demonstrated that water molecules contributed to the reaction via a high-speed proton transfer bridge between the generated CH3− and OH− moieties (Fig. 12C). The catalytically active sites for this reaction were identified by XAS as the mono(μ-oxo)dicopper species. The methanol productivity was recorded as 242.9 μmol gcat−1 h−1 with 71.6% selectivity for methanol. Moreover, this catalyst showed a long catalyst lifetime (70 h). This excellent catalytic performance achieved in ref. 52 can be considered as one of the benchmarks for analogous systems.
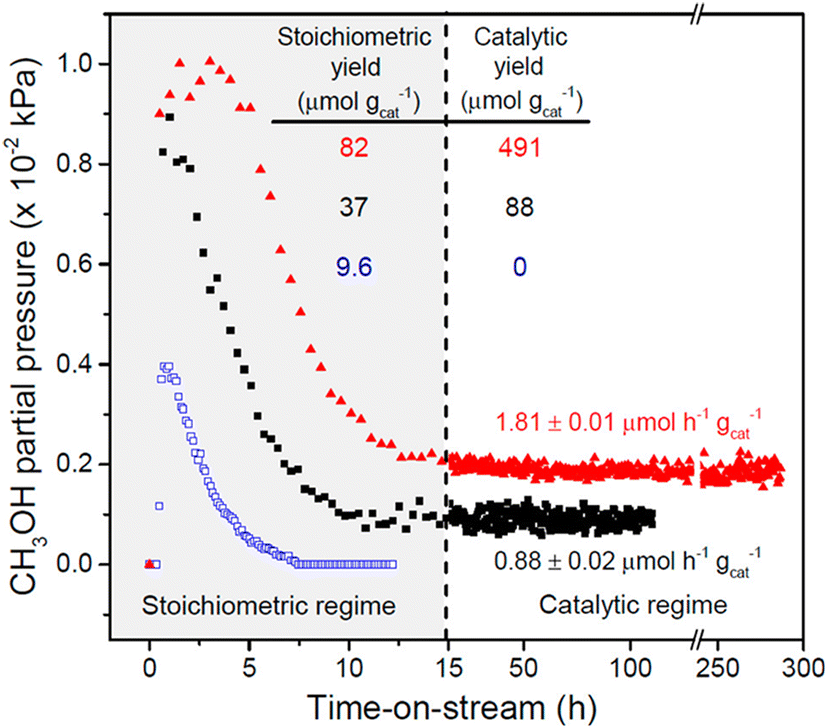 | ||
| Fig. 11 Catalytic performance of Cu-ZSM-5 in the partial oxidation of methane. Pretreatment conditions: O2 flow at 823 K for 5 h. Reaction conditions (chemical looping) for Cu-Na-ZSM-5 (blue open squares, Cu/Al = 0.37, Na/Al = 0.26): reaction with methane, WHSV = 2400 mL h−1 gcat−1, 483 K, 0.5 h; extraction of methanol, WHSV = 2400 mL h−1 gcat−1, 483 K, PHe = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa. Reaction conditions (continuous-flow) for Cu-Na-ZSM-5 (black filled squares) and Cu-H-ZSM-5 (red filled triangles, Cu/Al = 0.31): WHSV = 2400 mL h−1 gcat−1, 483 K, PCH4 = 98.1 kPa, PH2O = 3.2 kPa, PO2 = 0.0025 kPa. Reprinted from ref. 50 with permission from American Chemical Society, copyright 2016. | ||
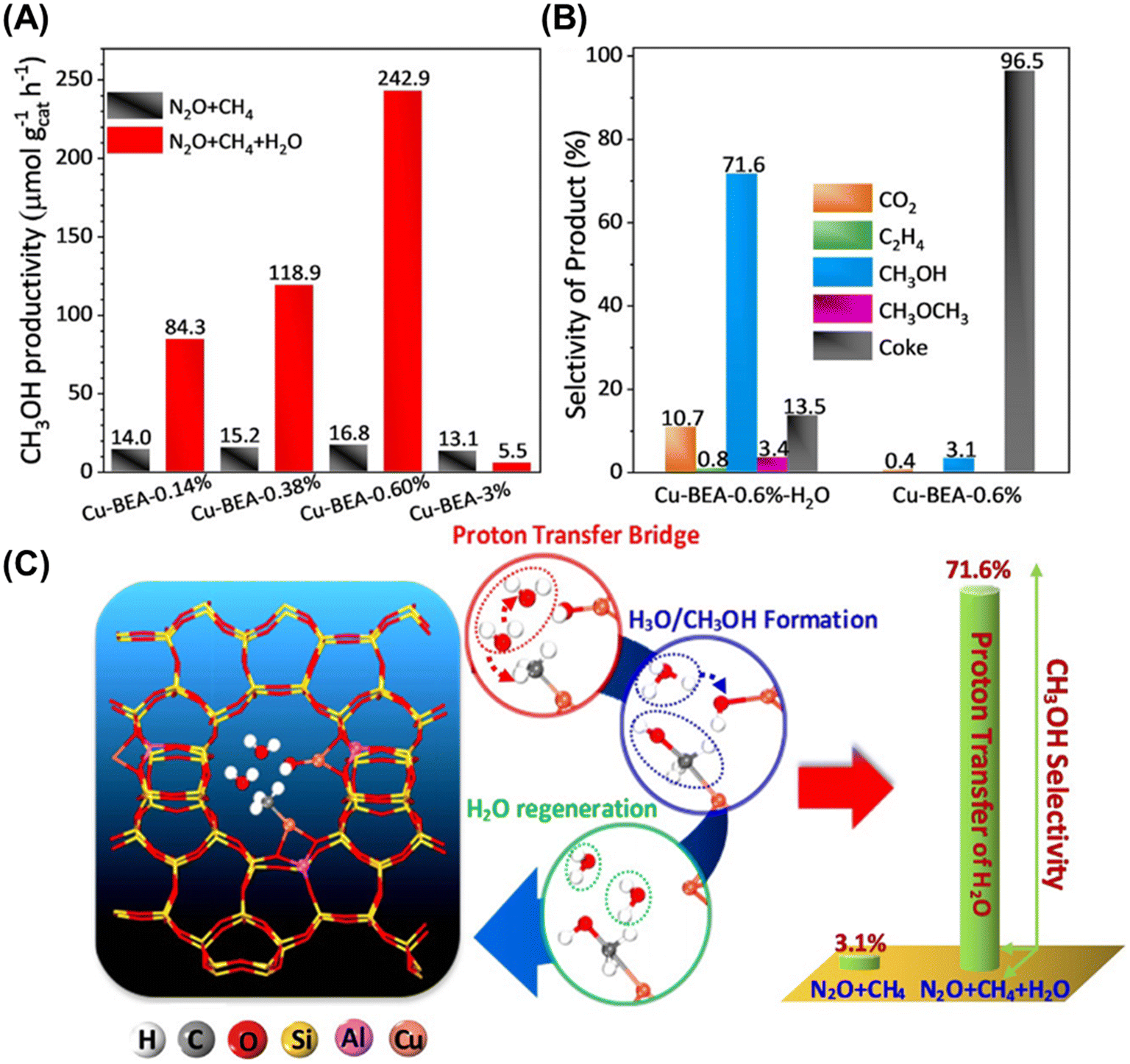 | ||
| Fig. 12 (A) Methanol productivity of Cu-BEAs for methane to methanol reaction in the presence (red) and absence (black) of H2O. Reaction conditions: N2O:CH4:H2O:He = 30:15:10(0):45(55); GHSV = 12000 h−1; 593 K; TOS = 6 h. (B) Selectivity of products over Cu-BEA under the same reaction conditions as figure A, TOS = 70 h. (C) Schematic of high-speed proton transfer bridge between the generated CH3− and OH− moieties. Reprinted from ref. 52 with permission from John Wiley and Sons, copyright 2021. | ||
Enormous efforts have been dedicated to achieving a high productivity for the partial oxidation of methane to methanol. The general consensus is that the catalytic active sites are cationic species such as [Cu2(μ-O)]2+, [Cu2(μ-O)2]2+, and [Cu3(μ-O)3]2+. To selectively form these clusters in the zeolite micropores, a precise materials design, in particular the location of the framework Al atoms and the framework structure of the zeolites, is important. Therefore, control of the location of framework Al atoms has been a hot topic in zeolite chemistry over the past several decades.53–56 Because the capture of divalent Cu clusters is crucial for the preparation of MMO-mimetic active sites in zeolite micropores, it is essential to construct the framework Al atoms in close proximity to each other, which has been the goal of many studies including some of our own.57–61 Recently, we developed a highly active catalyst for the partial oxidation of methane into methanol using N2O as an oxidant, which was achieved by controlling the position of the framework Al atoms.62 OSDAs with/without Na cations strongly influence the position of the framework Al atoms in AEI-type structures; the AEI-type zeolite synthesized with Na cations (denoted as AEI(Na)) had a greater number of paired Al sites compared to that synthesized without Na cations (AEI(Na-free)). Such a paired Al-rich environment generated a relatively larger number of dicopper species ([Cu2(μ-O)]2+) via ion exchange, resulting in higher catalytic performance (Fig. 13G). Furthermore, the activity and stability of Cu ion-exchanged AEI-type zeolites (5Cu/AEI(Na) and 5Cu/AEI(Na-free)) were controlled by calcination accompanied by partial dealumination. As a result, 5Cu/AEI(Na) calcined at 1123 K (5Cu/AEI(Na)-850) exhibited excellent catalytic performance, a methanol formation rate of 1638 μmol gcat−1 h−1 and a selectivity for methanol of 50% at 623 K (entry 6 in Table 2), with a long catalyst lifetime. After this finding, our group further improved these catalytic systems with a novel approach very recently.63 As already mentioned, methanol is easily sequentially oxidized, owing to its higher reactivity compared to methane. In order to overcome this issue, we attempted to develop a novel catalytic system for the rapid conversion of the produced methanol to lower olefins via the methanol-to-olefins (MTO) reaction. Here again, control of the location of the framework Al atoms is crucial because active copper species and Brønsted acid sites need to be placed in juxtaposition for efficient consecutive reactions consisting of MTM and MTO reactions. In this case, CHA-type zeolites were synthesized using amorphous silica (CHA-Am) or FAU-type zeolite (CHA-FAU) as starting materials under OSDA-free conditions. For these samples, different Al distributions were observed: CHA-FAU showed a higher proportion of paired Al sites than CHA-Am. The unique distribution of the framework Al atoms in CHA-FAU facilitated the pairing of active Cu and Brønsted acid sites, which led to a higher selectivity for lower olefins than in the case of CHA-Am (Fig. 14 and entries 7 and 8 in Table 2). This approach based on the distribution of framework Al atoms would open a new door for the development of zeolite-based catalysts for the oxidative upgrading of methane.
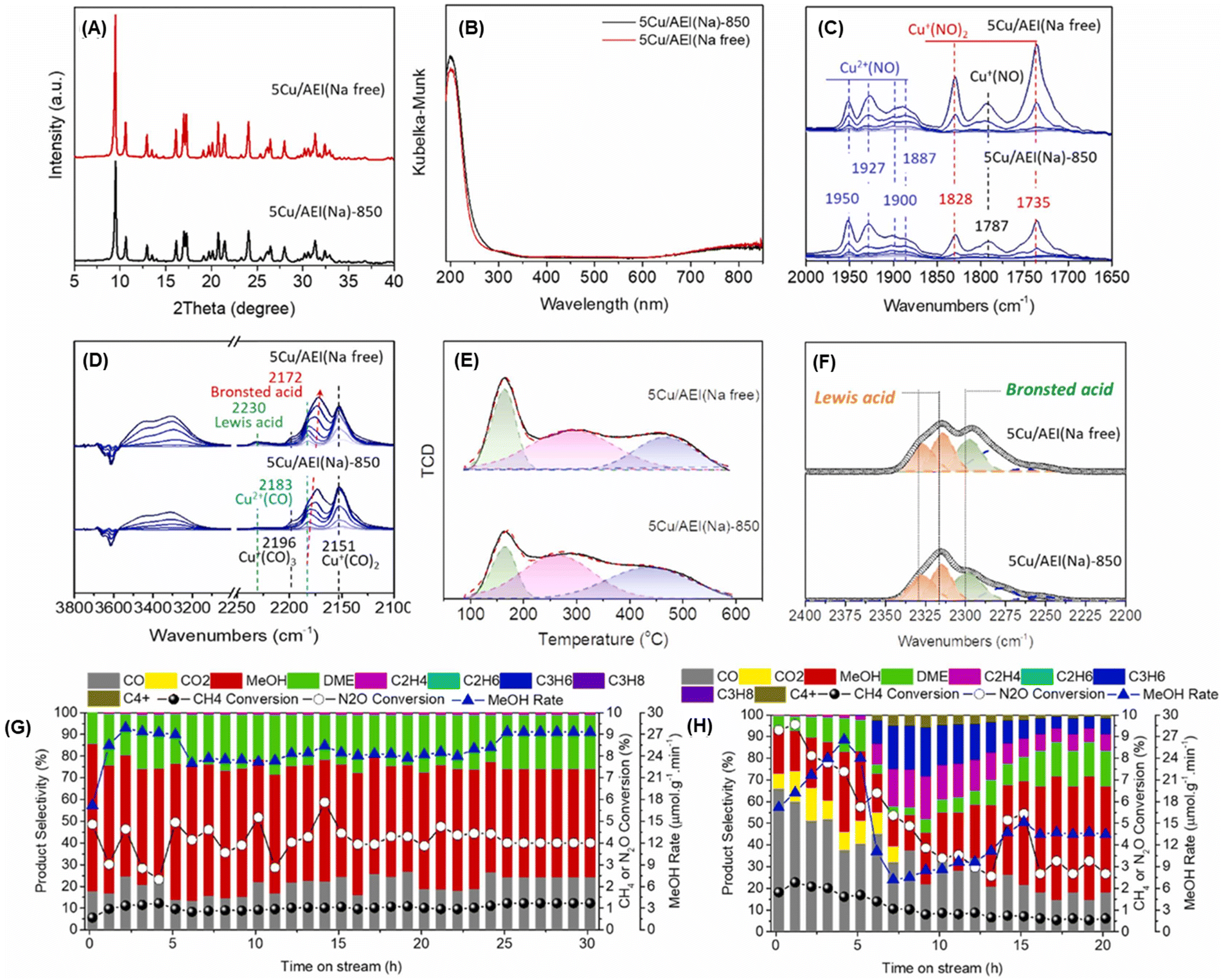 | ||
| Fig. 13 (A) XRD patterns and (B) UV-vis spectra of 5Cu/AEI(Na)-850 and 5Cu/AEI(Na free). In situ FT-IR spectra of adsorbed (C) NO (5–120 Pa) and (D) CO (5–1000 Pa) on Cu ion-exchanged AEI-type zeolites at 153 K. (E) NH3-TPD profiles. (F) In situ FT-IR spectra of adsorbed CD3CN species on Cu ion-exchanged AEI-type zeolites at room temperature. Catalytic performance of (G) 5Cu/AEI(Na)-850 and (H) 5Cu/AEI(Na free) in the MTM reaction. Reaction conditions: 100 mg catalysts; CH4/N2O/H2O/Ar = 10/10/2/3 mL min−1; WHSV = 15000 mL h−1 gcat−1; 623 K. Reprinted from ref. 62 with permission from Elsevier, copyright 2023. | ||
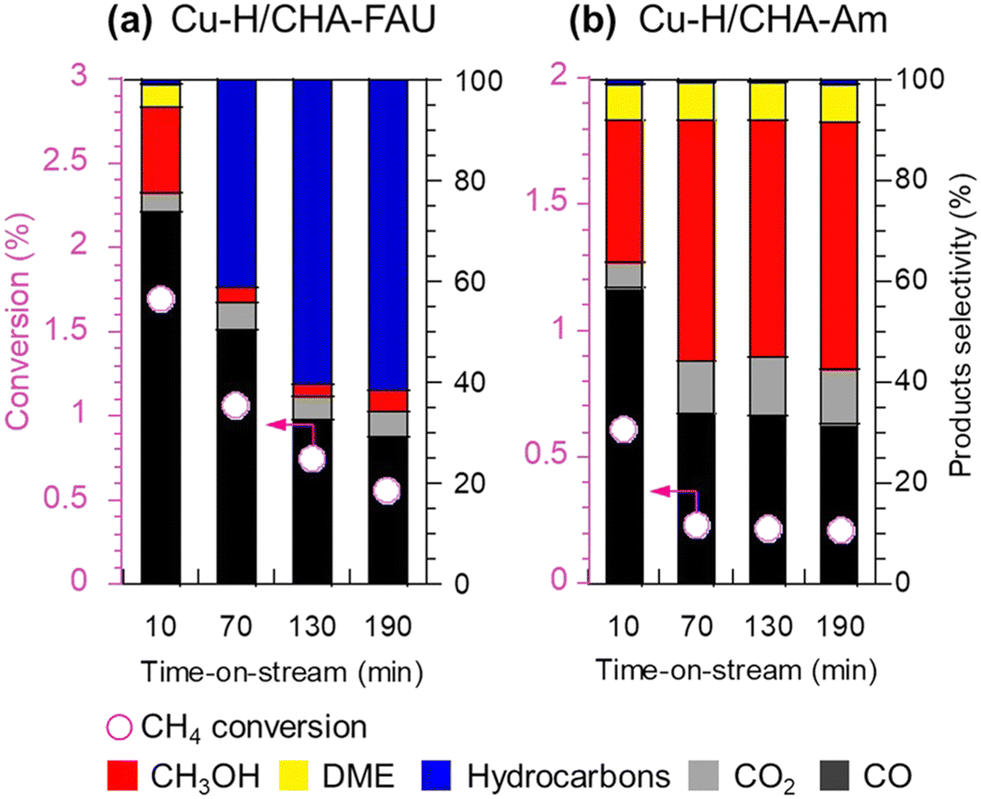 | ||
| Fig. 14 Catalytic performance of (a) Cu-H/CHA-FAU and (b) Cu-H/CHA-Am in the partial oxidation of methane. Reaction conditions: 100 mg catalyst; PCH4:PN2O:PH2O:PAr = 0.4:0.4:0.08:balance; total pressure 0.1 MPa; SV = 15000 mL h−1 gcat−1; W/F = 3.7 gcat h molCH4−1; 623 K. Reprinted from ref. 63 with permission from Royal Society of Chemistry, copyright 2023. | ||
Cu ion-exchanged zeolites are widely recognized as promising catalysts for the MTM reaction, while Fe species, which are also found in MMOs,43,45 are also promising candidates as catalytically active sites. Yoshizawa et al. proposed that Fe, Co, and Ni species are candidates for the MTM reaction on the basis of DFT calculations.64 According to these calculation results, the orders of increasing reactivity to C–H bond dissociation and of the selectivity for methanol were estimated to be CoO+–ZSM-5 < NiO+–ZSM-5 < FeO+–ZSM-5 < CuO+–ZSM-5 and FeO+–ZSM-5 < CoO+–ZSM-5 < NiO+–ZSM-5 < CuO+–ZSM-5, respectively. Weckhuysen and Luo et al. reported Fe ion-exchanged ZSM-5 to be an efficient catalyst for the low-temperature oxidation of methane.65 In that study, Fe-containing ZSM-5 samples were prepared by one of three methods: IWI, liquid-phase ion exchange (IE), and SSIE. In the cases of Fe/ZSM-5IWI and Fe/ZSM-5IE, Fe-derived particles were observed in the scanning transmission electron microscope (STEM) images, while Fe/ZSM-5SSIE did not contain such visible Fe-derived particles (Fig. 15A). In addition, UV-vis and Mössbauer spectroscopies revealed that a larger number of monomeric Fe3+ species were present in Fe/ZSM-5SSIE compared to the others (Fig. 15B and C). Fe/ZSM-5SSIE exhibited a remarkably high total yield for the target products (CH3OH, CH3OOH, HOCH2OOH, and HCOOH), as compared with Fe/ZSM-5IWI and Fe/ZSM-5IE (entries 9, 10, and 11 in Table 2). Recently, more highly active Fe ion-exchanged MFI-type zeolites (Fe/ZSM-5) have been reported. In these catalysts, the local environment of the framework Al atoms is the key to creating active single Fe species and suppressing the oligomerization of Fe species.66 The MFI-type structure has straight and sinusoidal 10R channels, and the widths of both channels are ca. 5.5 Å. In contrast to these channels, a relatively larger spherical space (ca. 10 Å) is located at the intersection at these channels. In general, tetrapropylammonium (TPA) hydroxide is used as an OSDA for the synthesis of MFI-type zeolites, and TPA+ can sit only at the intersection in the MFI-type framework since TPA+ is too large for the 10R channels. Thus, owing to the electrostatic interaction between TPA+ and negatively charged building blocks for the zeolite framework, the framework Al atoms are selectively located at the channel intersections when only TPA cations are used for zeolite synthesis.67 The catalyst prepared via Fe-ion exchange adopted to the ZSM-5 synthesized by using TPA only (denoted as Fe/ZSM-5(TPA)) showed higher catalytic activity than the catalyst prepared via Fe-ion exchange for ZSM-5 synthesized by using both TPA and Na cations (Fe/ZSM-5(TPA + Na)) (entries 12 and 13 in Table 2). This clearly indicates the importance of the location of the framework Al atoms (i.e., ion-exchange sites). In addition, the reactive Fe center was confirmed by Fe K-edge XAS measurement, where no oligomeric Fe species were detected. Thus, it was concluded that the active sites of Fe/ZSM-5(TPA) were single Fe species anchored on the ion-exchange sites.
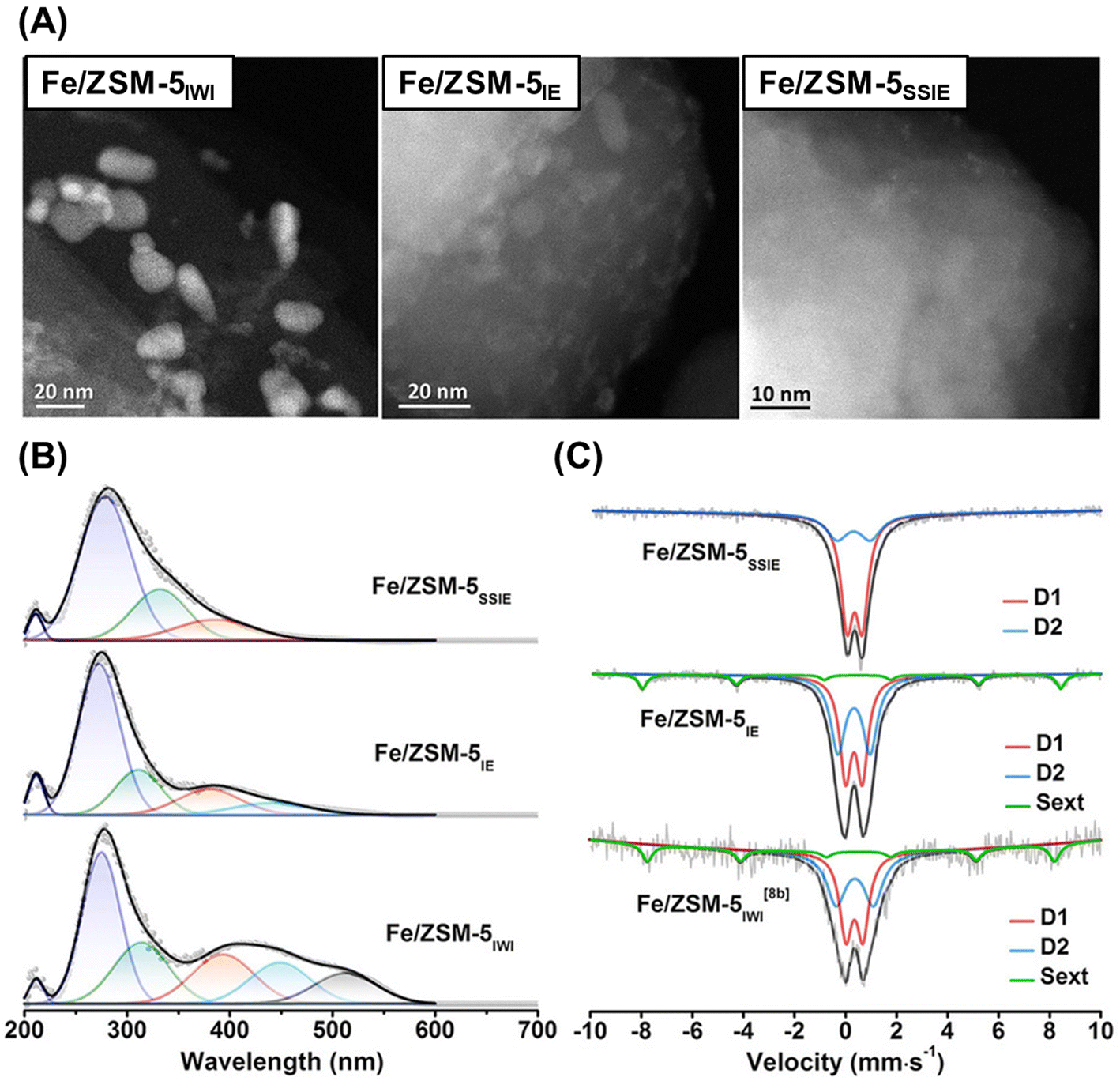 | ||
| Fig. 15 (A) STEM images, (B) UV-vis spectra, and (C) 57Fe Mössbauer spectra of Fe/ZSM-5s. Reprinted from ref. 65 with permission from John Wiley and Sons, copyright 2021. | ||
Ion-exchanged zeolites are applicable to the oxidation of methane into not only methanol but also other chemicals.68–70 Here, we introduce syngas production over Rh ion-exchanged zeolites. Direct steam reforming of methane is a common means of syngas production but requires a high reaction temperature because the reaction is endothermic. By contrast, the partial oxidation of methane into syngas has great potential in terms of achieving a catalytic system that can be operated at low reaction temperatures since the partial oxidation of methane is exothermic. Hou et al. prepared a MOR-type zeolite-supported Rh sub-nano cluster (Rh-MOR) via ion exchange as a highly active catalyst for the low-temperature selective methane oxidation to syngas.71 This catalyst lowered the reaction temperature, generally 1073 K or higher, to 873 K. Moreover, almost the equilibrium CO yield was obtained even under high space velocity (SV) conditions (entry 1 in Table 3). In this reaction, the Rh sub-nano clusters with a size of ca. 0.6 nm, formed via ion exchange, served as active sites. Inspired by this research, our team has tried to selectively form such active Rh sub-nano clusters in the zeolite micropore on the basis of the location of the framework Al atoms.72 In the case of MFI[TPA], which is analogous to ZSM-5(TPA), the framework Al atoms are located selectively at the channel intersection. The ion exchange sites, derived from such site-controlled Al species, provide enough space for the formation of Rh sub-nano clusters, because the sizes of the channel intersections and the Rh sub-nano clusters are ca. 1.0 and 0.6 nm, respectively, as mentioned above. In addition, no clusters were expected to form in the channels (ca. 0.55 nm) owing to the size limitation. In the study cited as ref. 72, the chemical state of the Rh species was characterized by UV-vis and in situ FT-IR spectroscopies. The isolated Rh cations were observed for Rh-MFI[TPA,Na], in which the ion-exchange sites were present both in the channels and at the intersections, while Rh oxide clusters were found in Rh-MFI[TPA] (Fig. 16A and B). A significant difference in catalytic activity was found between Rh-MFI[TPA] and Rh-MFI[TPA,Na] even at almost the same Rh loading (ca. 0.2 wt%) (Fig. 16C and D, entries 2 and 3 in Table 3): Rh-MFI[TPA] showed greater catalytic performance and higher stability compared to Rh-MFI[TPA,Na], indicating that active Rh clusters of a suitable size were successfully created in the channel intersections owing to the selective sitting of the framework Al atoms in MFI[TPA]. Furthermore, we investigated the role of the zeolite framework in this reaction by using model catalysts.73 To this end, we prepared two kinds of model catalysts: Rh ion-exchanged Y zeolite (Rh-Y) and amorphous silica-alumina (Rh-ASA). The Rh species were characterized by UV-vis and in situ FT-IR spectroscopies, transmission electron microscopy (TEM), and H2 temperature-programmed reduction (H2-TPR) measurements. Owing to the abundance of ion-exchange sites of Y zeolites, isolated Rh cations were preferentially formed on Rh-Y, while Rh oxide particles (or clusters) were generated on Rh-ASA by the reaction. In the comparison of the catalytic performance of these model catalysts, Rh-ASA showed a slightly higher CO yield than Rh-Y (entries 4 and 5 in Table 3), which is consistent with the results for the Rh-MFI zeolites described above. On the other hand, the stability and catalytic performance of Rh-Y were higher than those of Rh-ASA. In TEM images of the spent catalysts, some aggregated Rh species were observed for Rh-ASA, indicating that the zeolite framework suppressed the aggregation of metal species, owing to the electrostatic interaction between the cations and the anionic field of the zeolite framework (Fig. 17). These results suggest that the active Rh clusters in Rh-MFI[TPA] were preserved by the zeolite framework. In other words, the zeolite framework has two main roles: the creation and preservation of catalytic active sites during the reaction.
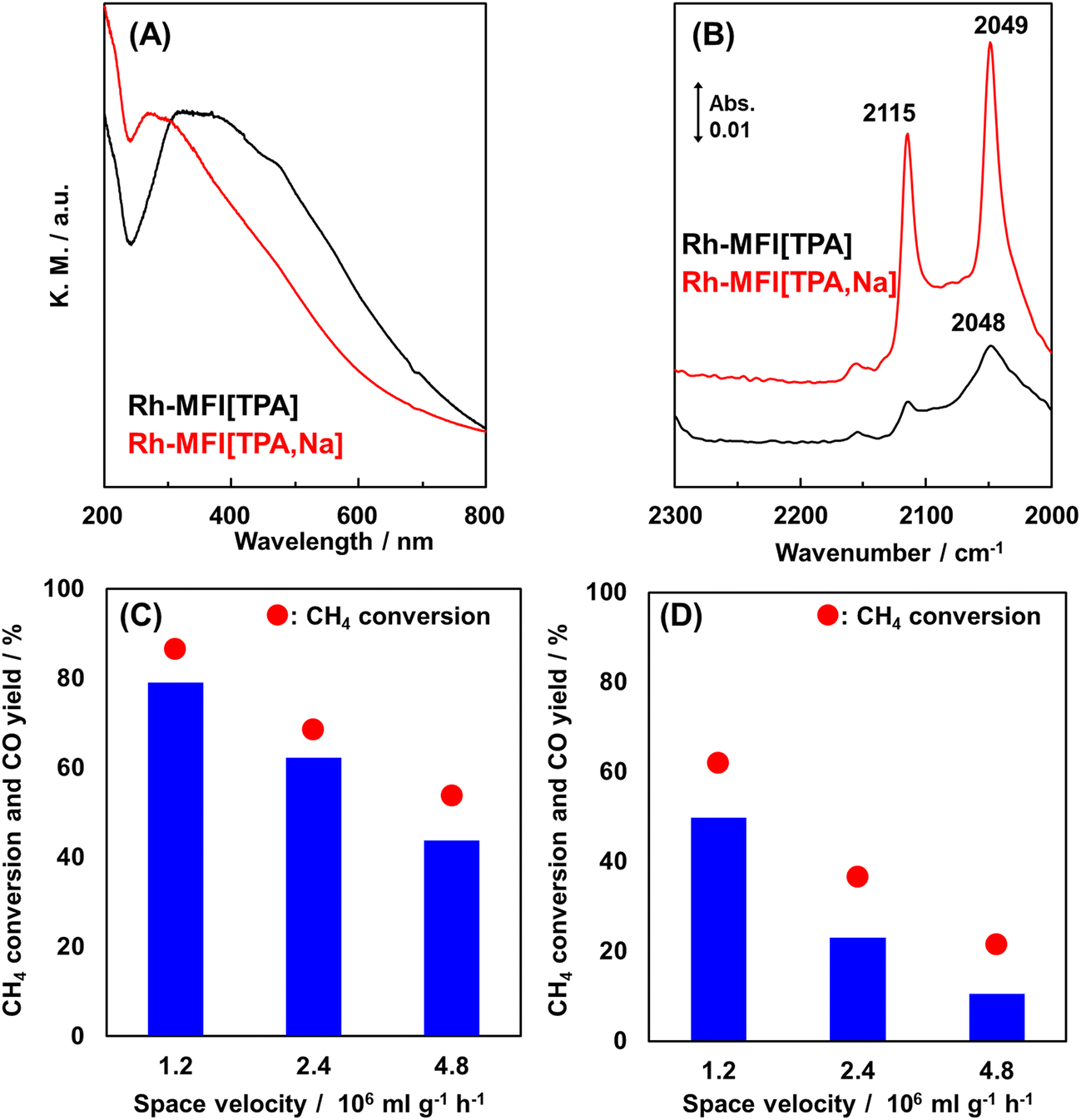 | ||
| Fig. 16 (A) UV-vis spectra of Rh ion-exchanged MFI-type zeolites. (B) In situ FT-IR spectra of adsorbed CO species on Rh-MFI[TPA] and Rh-MFI[TPA,Na] at 298 K. Catalytic performance of (C) Rh-MFI[TPA] and (D) Rh-MFI[TPA,Na] in the partial oxidation of methane at 873 K with different space velocities. Reaction conditions: 10–25 mg catalyst; PCH4:PO2:PAr = 3.33:1.67:95; total pressure 0.1 MPa. Reprinted from ref. 72 with permission from Royal Society of Chemistry, copyright 2020. | ||
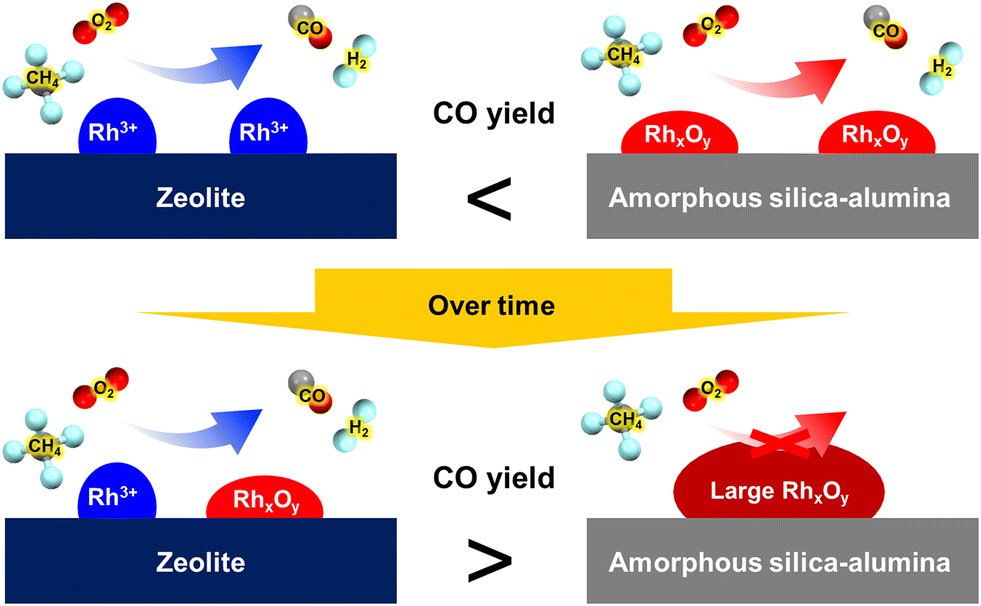 | ||
| Fig. 17 Schematics for changing behavior of both model catalysts during the reaction. Reprinted from ref. 73 with permission from American Chemical Society, copyright 2021. | ||
4. Active sites prepared via impregnation on zeolite surfaces
The impregnation technique, used commonly to prepare various supported metal (or metal oxide) catalysts, has also been widely employed in the field of zeolite-based catalysts. This section discusses zeolite-based catalysts that possess active sites prepared via impregnation technique. The zeolite-based catalysts introduced in this section and their catalytic performance for methane upgrading are summarized in Table 4.| Entry | Catalyst | Framework topology | Plausible active site | Oxidant | Reactor type | Solvent | T /K | t /h | SVd/mL h−1 gcat−1 | P /MPa | Conv./% | Target C-based product(s) | Target product | By-product(s) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield/% | Sel./% | |||||||||||||||
| a Abbreviations in this table: MeOH = methanol; MeOOH = methyl hydroperoxide; AcOH = acetic acid. b Reaction temperature. c Reaction time (only for batch-type reaction). d Space velocity (only for flow-type reaction). e Reaction pressure. f Reaction data for TOS of 8000 min are shown in Fig. 18B. g Reaction data for TOS of 50 h are shown in Fig. 19B. h Pd loading was 0.01 wt%. i CO was also used as a reactant along with methane. j Rh loading was 0.10 wt%. k Reaction data at different temperatures are shown in Fig. 22B. l Au and Pd loading amounts were 3.24 wt% and 1.76 wt%, respectively. m In situ formed H2O2 from H2 and O2 over Au–Pd alloy nanoparticles. n Au and Pd loading amounts were both 0.5 wt%. | ||||||||||||||||
| 1 | 6.4 wt% Pb/NaX | FAU | PbO species | O2 | Flow | — | 1023 | — | 3.6 × 103 | 0.1 | 8.6 | C2 compounds | 5.9 | 69 | CO2 | 74 |
| 2 | 2 wt% Ni/[Al]-CHA | CHA | Small NiO | O2 | Flow | — | 773 | — | 1.5 × 104 | 0.1 | 22 | CO | 5.7 | 26 | CO2 | 75 |
| 3 | 5 wt% Ni/MORf | MOR | Ultra-small NiO | O2 | Flow | — | 973 | — | 3.0 × 104 | 0.1 | 98 | CO | 90 | 92 | CO2 | 77 |
| 4 | 0.005 wt% Rh–3 wt% Co/MORg | MOR | Ultra-small Co | O2 | Flow | — | 923 | — | 1.2 × 106 | 0.1 | 86 | CO | 78 | 91 | CO2 | 79 |
| 5 | 0.3 wt% Rh/ZSM-5-2D | MFI | Ultra-small Rh | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.0 | 0.2 | MeOOH | 0.15 | 73.2 | MeOH, HCOOH | 80 |
| 6 | Pd1O4@ZSM-5 + 2 wt% CuOh | MFI | Pd1O4 | H2O2 | Batch | H2O | 368 | 0.5 | — | 3.0 | n.d. | MeOH | n.d. | 86 | MeOOH, CO2 | 81 |
| 7i | Rh1O5@ZSM-5j | MFI | Rh1O5 | O2 | Batch | H2O | 423 | 12 | — | 6.8 | n.d. | AcOH | n.d. | 70.1 | MeOH, HCOOH | 82 |
| 8 | 3 wt% Pd/Ce-MFIMCk | MFI | Small Ce species with Pd | O2 | Flow | — | 523 | — | 7.5 × 103 | 0.1 | 3.8 | C2H6 | 0.19 | 4.9 | CO2 | 85 |
| 9 | 1 wt% Pd/Ce-MFIMC | MFI | Small Ce species with Pd | O2 | Flow | — | 573 | — | 7.5 × 103 | 0.1 | 3.0 | C2H6 | 0.11 | 3.7 | CO2 | 88 |
| 10 | 1 wt% Pd–1 wt% Co/Ce-MFIMC | MFI | Small Ce species with Pd and Co | O2 | Flow | — | 573 | — | 7.5 × 103 | 0.1 | 4.7 | C2H6 | 0.16 | 3.4 | CO2 | 88 |
| 11 | AuPd@ZSM-5-C16l | MFI | Au–Pd alloy | H2O2m | Batch | H2O | 343 | 0.5 | — | 3.0 | 17.3 | MeOH | 16 | 92 | HCOOH, CO2 | 91 |
| 12 | AuPd/HZSM-5n | MFI | Au–Pd alloy | H2O2 | Batch | H2O | 323 | 0.5 | — | 3.05 | n.d. | MeOH | n.d. | 16.9 | HCOOH, MeOOH, CO2 | 92 |
The Pb species impregnated over NaX zeolite were reported to be active for the oxidative coupling of methane, producing the C2 compounds ethane and ethene (entry 1 in Table 4).74 The conversion of methane and C2 selectivity exhibited a volcano-type dependence on the loading amount of Pb species, the optimum loading amount being 6.4 wt%. At 1073 K, the catalyst with this Pb loading produced a 5.9% yield of C2 compounds with 69% selectivity. The X-ray diffraction (XRD) patterns of the catalyst before/after the reaction indicated that the redox cycle Pb2+ ↔ Pb0 was the key to methane coupling. Control experiments with acidic zeolites were missing in the paper cited as ref. 74, but the lattice oxygen of the basic zeolite, NaX, was proposed to act as a hydrogen acceptor in the reaction. For metal oxide catalysts employed for methane activation, namely oxidative coupling of methane, the importance of their basicity due to lattice oxygens is widely recognized.10 A part of the metal sources (typically metal cations) can be introduced onto the ion-exchange sites of zeolites even by impregnation, and these species lead to the formation of metal (or metal oxide) nanoparticles anchored on the negatively charged framework during the calcination treatment. In other words, active metal (or metal oxide) species are in proximity to basic lattice oxygens, which is beneficial for methane activation. Thus, the combination of a metal oxide that causes a redox reaction and a zeolite framework with appropriate surface properties is a promising approach to designing catalysts for methane activation.
As for the case of Rh clusters supported on MFI-type zeolite frameworks (vide supra), an anchoring effect afforded by negatively charged zeolite frameworks is also useful for isolating small metal clusters/nanoparticles and suppressing their aggregation.13,14 NiO particles were prepared on the surface of a CHA-type framework substituted by B, Al, and Ga via impregnation and subsequent calcination.75 At the same Ni loading amount of 2 wt%, the size of NiO particles was found from XRD to be dependent on a class of framework-substituting atoms, and the NiO particle size increased in the order [Al]-CHA < [Ga]-CHA < [B]-CHA. This order might be connected to the deprotonation energy (i.e., basicity) of these frameworks, given the consistent trend;76 thus, the consideration of such parameters could help researchers design and prepare zeolite-based catalysts with supported metal species of a desirable size. The smaller and more highly dispersed NiO particles on [Al]-CHA produced CO and H2 along with CO2 in the oxidative conversion of methane at lower reaction temperatures (entry 2 in Table 4), compared to those on [Ga]-CHA and [B]-CHA.75
Although it is unclear whether an anchoring effect was at play, ultra-small metal nanoparticles deposited on the MOR-type zeolite were reported to maintain their isolated and highly dispersed state during the partial oxidation of methane and show a long catalyst lifetime. In the first example introduced here, ultra-small NiO particles with a mean diameter of 1.6 nm were successfully deposited on the MOR surface via an IWI method; the TEM image for 5 wt% Ni/MOR is shown in Fig. 18A.77 This catalyst produced CO (91–92% selectivity) and H2 with a molar ratio of 2.0 at 97–98% methane conversion at 973 K for a time on stream (TOS) as long as 8000 min (entry 3 in Table 4 and Fig. 18B), in which the lattice oxygen atoms of NiO were suggested to participate (i.e., Mars-van Krevelen mechanism). The high selectivity and long lifetime of this catalyst arose from the ultra-small sized NiO particles. Particle size was demonstrated to greatly affect catalytic activity, viz., both methane conversion and selectivity for CO; NiO particles larger than 15 nm exhibited at least a 2.4-fold lower methane conversion as compared to NiO particles smaller than 3 nm and accelerated the complete oxidation of methane, yielding CO2. In addition, methane decomposition into H2 and carbon nanofibers is known to occur readily on large Ni particles in the absence of O2.78 In contrast to large Ni particles, ultra-small NiO nanoparticles on MOR did not form carbonaceous species, which resulted in a long catalyst lifetime. In the second example, Co nanoparticles with a mean diameter of 1.5 nm were loaded on the MOR surface via the standard impregnation method, as seen in the image obtained by a high-angle annular dark-field STEM (HAADF-STEM; Fig. 19A).79 This 3 wt% Co/MOR catalyst pre-reduced with H2 initially produced CO in 91% selectivity at a methane conversion of 7.6% at 873 K in the presence of O2 but lost this activity for CO production in the steady state, where CO2 was the main product. This observed alteration of product selectivity resulted from the transformation of Co0 into Co2+ species, the former of which is necessary for the partial oxidation of methane to CO. In stark contrast, the surface modification of the 3 wt% Co/MOR catalyst with a tiny amount of Rh (0.005 wt%) drastically improved the catalytic performance to yield CO with 90% selectivity at a methane conversion of 15% even in the steady state under the same reaction conditions. The Rh species were revealed by extended X-ray absorption fine structure (EXAFS) to be mono-atomically dispersed and attached on some of the Co nanoparticles. Such Rh species were proposed to cause the spillover of hydrogen atoms onto the catalyst surface, which allowed Co species to be in the zero-valence state (Fig. 19B). As a result, this 0.005 wt% Rh–3 wt% Co/MOR catalyst exhibited an outstanding lifetime and produced a mixture of CO and H2 with a molar ratio of 2.0 at an SV as high as 1.2 × 106 mL h−1 gcat−1, as seen in Fig. 19C (see also entry 4 in Table 4).
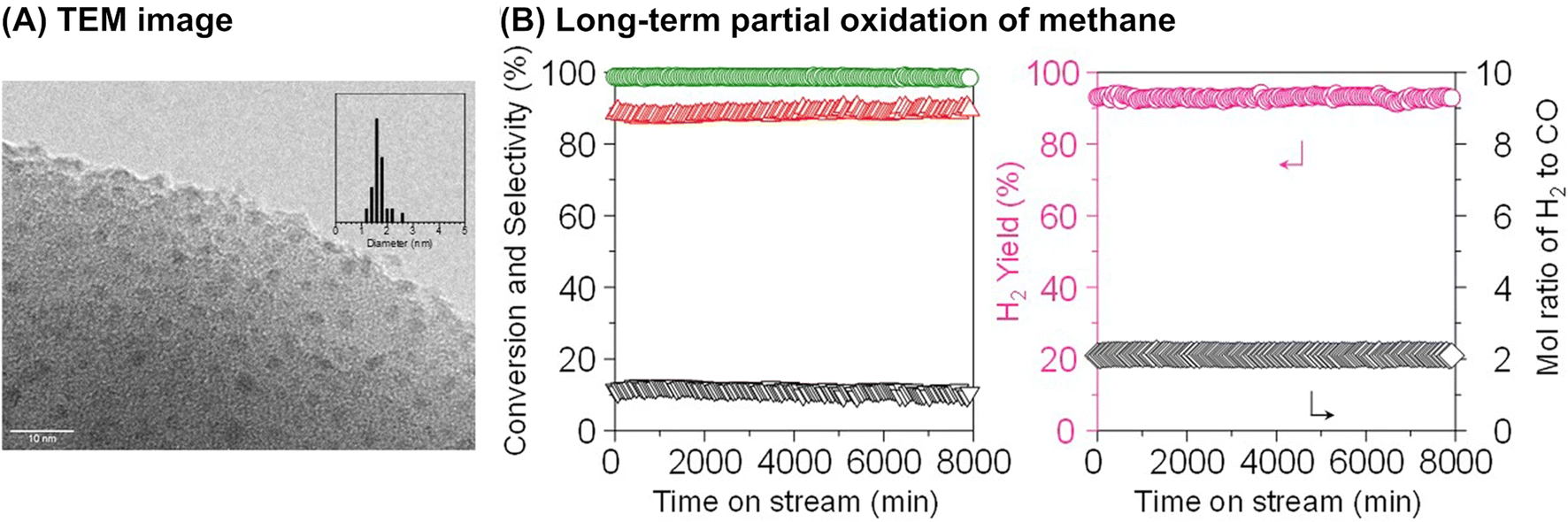 | ||
| Fig. 18 (A) TEM image of 5 wt% Ni/MOR catalyst and (B) long-term partial oxidation of methane over the Ni/MOR catalyst. Reaction conditions: CH4/O2/Ar = 0.06/0.03/0.91 atm; SV = 3.0 × 104 mL h−1 gcat−1; 973 K. Reproduced from ref. 77 with permission from Springer Nature, copyright 2020. | ||
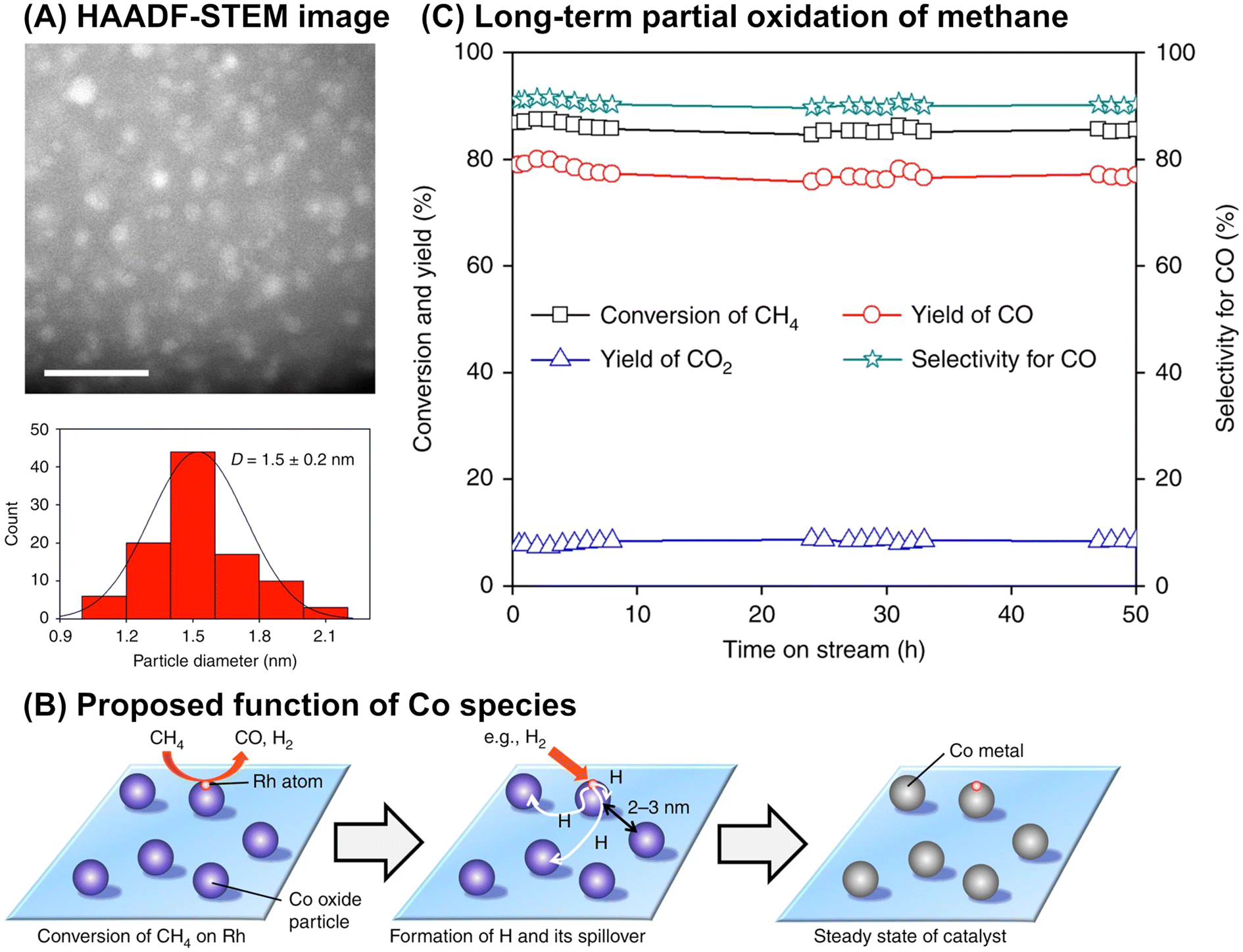 | ||
| Fig. 19 (A) HAADF-STEM image of 0.005 wt% Rh–3 wt% Co/MOR catalyst, (B) proposed function of Co species for reducing Rh species via hydrogen spillover, and (C) long-term partial oxidation of methane over the Rh–Co/MOR catalyst. Reaction conditions: CH4/O2/N2 = 3.33/1.67/95; SV = 1.2 × 106 mL h−1 gcat−1; 923 K; 0.1 MPa. Reproduced from ref. 79 with permission from Springer Nature, copyright 2018. | ||
Along with this anchoring effect, confinement of supported particles in nanospaces consisting of micropores, cages, and/or channel intersections is another attractive approach to keeping supported metal particles isolated and highly dispersed.13–15 Subnanometric Rh particles were successfully deposited in the micropores of two-dimensional ZSM-5 nanosheets (ZSM-5-2D).80 Interestingly, their size could be reversibly controlled by heat-treatment in H2 or CO, which was confirmed by FT-IR after CO adsorption and HAADF-STEM (Fig. 20). The catalytic performance for methane oxidation was dependent on the size of Rh nanoparticles; the subnanometer-sized Rh particles (∼0.9 nm) prepared via two-step heat treatment in H2 at 773 K and in CO at 773 K exhibited the highest productivity for methyl hydroperoxide (CH3OOH) (entry 5 in Table 4).
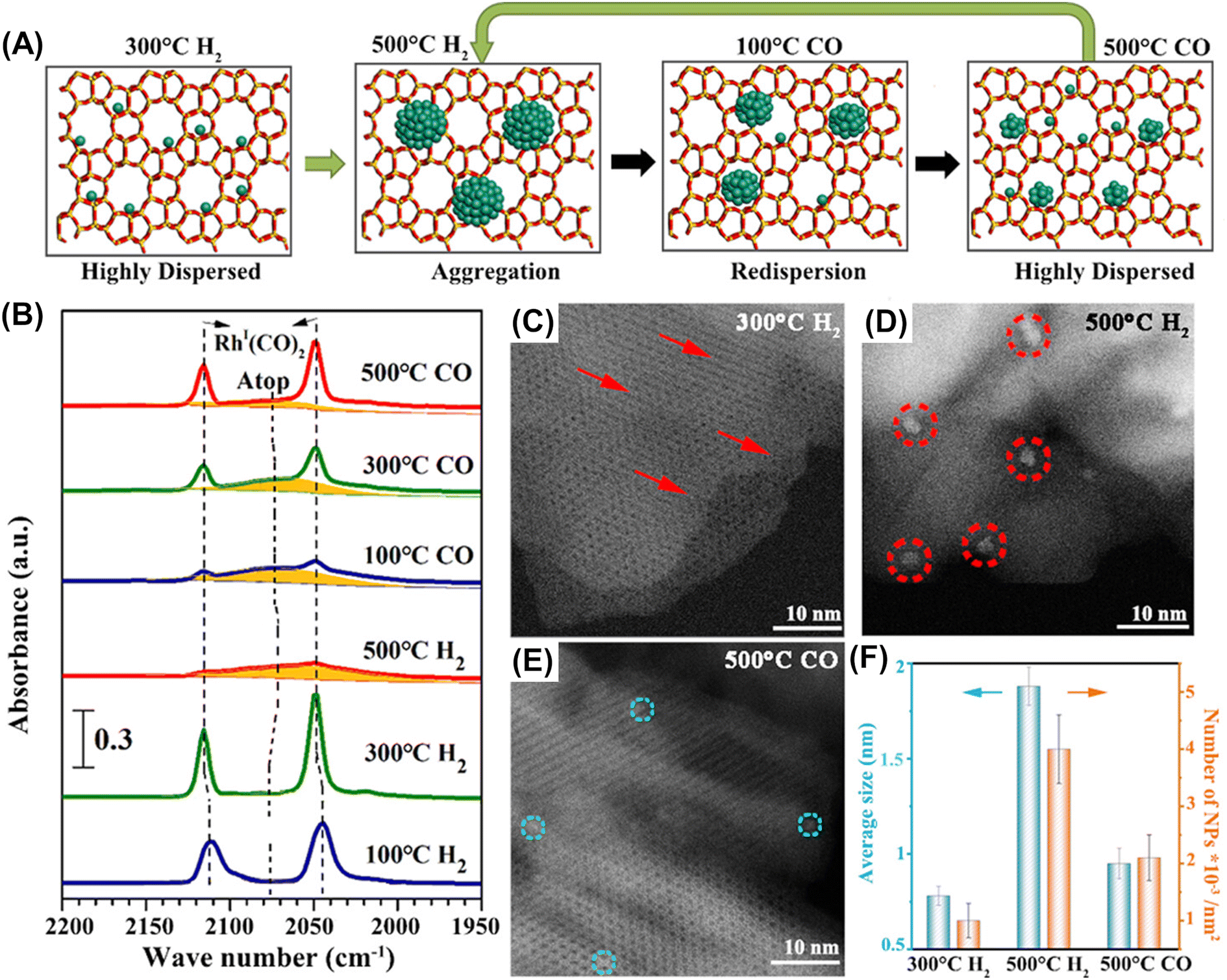 | ||
| Fig. 20 Reversible size control of Rh nanoparticles ZSM-5-2D. (A) Schematic of reversible size control of Rh particles upon heat treatment in H2 or CO. (B) CO FT-IR spectra of Rh/ZSM-5-2D heat-treated in H2 and CO step-by-step. (C–E) HAADF-STEM images of Rh/ZSM-5-2D heat-treated under different conditions. (F) Average particle size and density of Rh nanoparticles on ZSM-5-2D heat-treated under different conditions. Reprinted from ref. 80 with permission from American Chemical Society, copyright 2023. | ||
Ultimately, so-called single-site catalysts can be developed by an appropriate technique for introducing metal species. Tao et al. developed two different zeolite-supported single-site catalysts: Pd1O4 anchored on the internal surface of ZSM-5 (Pd1O4@ZSM-5)81 and Rh1O5 on ZSM-5 (Rh1O5@ZSM-5).82 In both cases, the pores of the ZSM-5 support were evacuated by a vacuum pump, and then an aqueous solution containing a metal nitrate was introduced. As in the case of a variety of single-site catalysts,83,84 the absence of the second shell of Pd–Pd (or Rh–Rh) in all EXAFS spectra revealed the local structure of the supported Pd and Rh species (Fig. 21). Pd1O4@ZSM-5 with CuO and H2O2, which were the co-catalyst and oxidant, respectively, converted methane to methanol with 86% selectivity (entry 6 in Table 4). Meanwhile, Rh1O5@ZSM-5 accelerated the coupling of methane, CO, and O2 to produce acetic acid (entry 7 in Table 4).
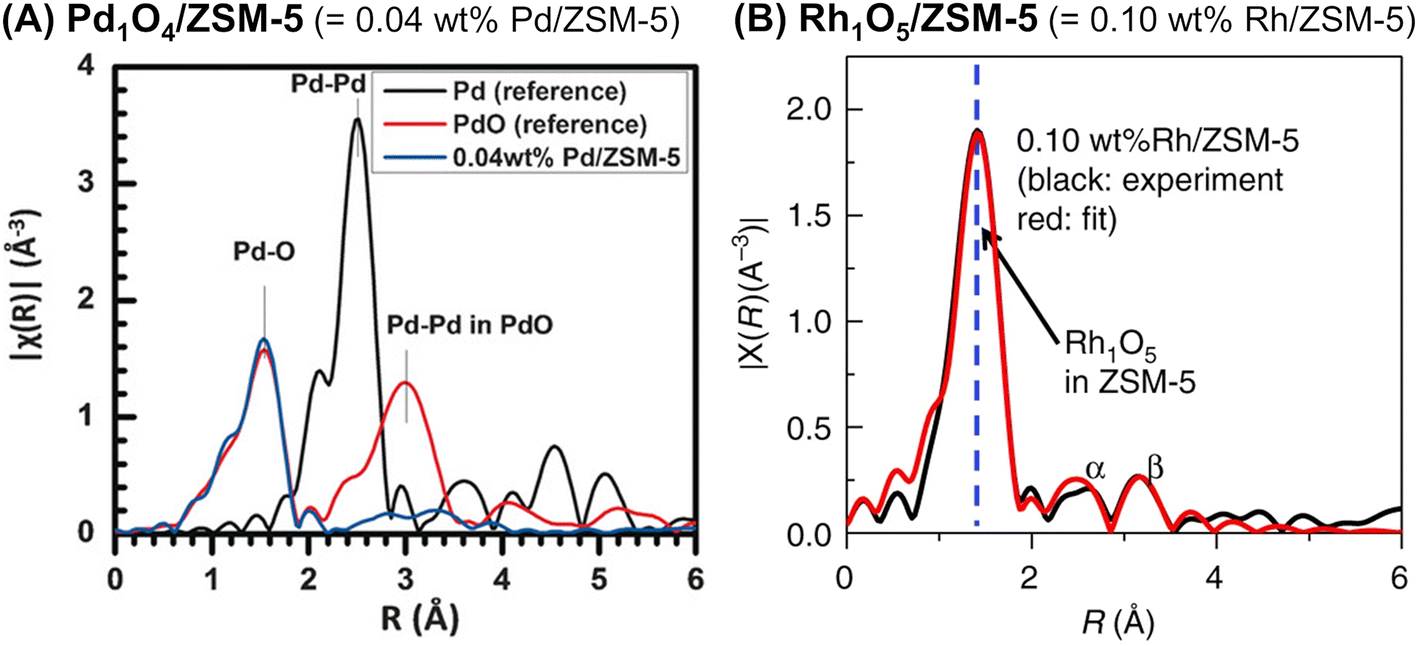 | ||
| Fig. 21 Fourier transform of EXAFS spectra of (A) Pd1O4@ZSM-5 (k2-weighted Pd K-edge) and (B) Rh1O5@ZSM-5 (k2-weighted Rh K-edge). Reprinted from (A) ref. 81 with permission from John Wiley and Sons, copyright 2016, and (B) ref. 82 with permission from Springer Nature, copyright 2018. | ||
As mentioned above, zeolite frameworks offer a unique opportunity to form highly dispersed and small species that can activate methane. We recently demonstrated that the combination of the Ce-containing MFI-type zeolite prepared via the two-step mechanochemical method and Pd nanoparticles (denoted as Pd/Ce-MFIMC) showed low-temperature activity for oxidative coupling of methane.85 The 3 wt% Pd/Ce-MFIMC produced ethane in 0.19% yield at 523 K (entry 8 in Table 4). The ethane production was probably induced by the redox cycle of CeOx species mediated by supported Pd, as suggested by H2-TPR measurements as well as a previous report where Pd/ZSM-5 without Ce species accelerated the complete oxidation of methane to CO2 and H2O.86 As seen in the EXAFS spectra in Fig. 22A, Ce-MFIMC did not provide the second shell corresponding to Ce–Ce, while the other Ce-containing supports—Ce-MFIHT (Ce-containing zeolite synthesized via typical one-step hydrothermal synthesis), Ce/SiO2 (SiO2-supported Ce, prepared by impregnation), Ce/silicalite-1 (silicalite-1-supported Ce, prepared by impregnation), and bulk CeO2—showed a peak to some extent. These spectra indicated that the size of Ce species increased in the order Ce-MFIMC < Ce-MFIHT < Ce/SiO2 < Ce/silicalite-1 < CeO2. Among these supports, Ce-MFIMC and Ce-MFIHT combined with Pd yielded ethane at low reaction temperatures below 573 K (Fig. 22B), indicating the requirement of highly dispersed and small Ce species for the low-temperature oxidative coupling of methane. Owing to the high affinity between Ce and Pd,87 Pd species were in proximity to Ce species (STEM images with elemental mapping by energy-dispersive X-ray spectroscopy (EDS) are shown in Fig. 22C) and facilitated the completion of the redox cycle of Ce species for activating methane. Such highly dispersed Ce species with Pd, i.e. Pd/Ce-MFIMC, improved catalytic activity through further surface modification by third species. When Co was loaded on Pd/Ce-MFIMC, new active sites, i.e., concerted Ce, Pd, and Co species, were formed (Fig. 23A) and gave a 1.5-fold higher ethane yield than Pd/Ce-MFIMC (entries 9 and 10 in Table 4).88 Owing to the poorer dispersion of Ce species in Ce-MFIHT than in Ce-MFIMC, such triad sites were difficult to form (Fig. 23B) and the catalytic activity was not improved.
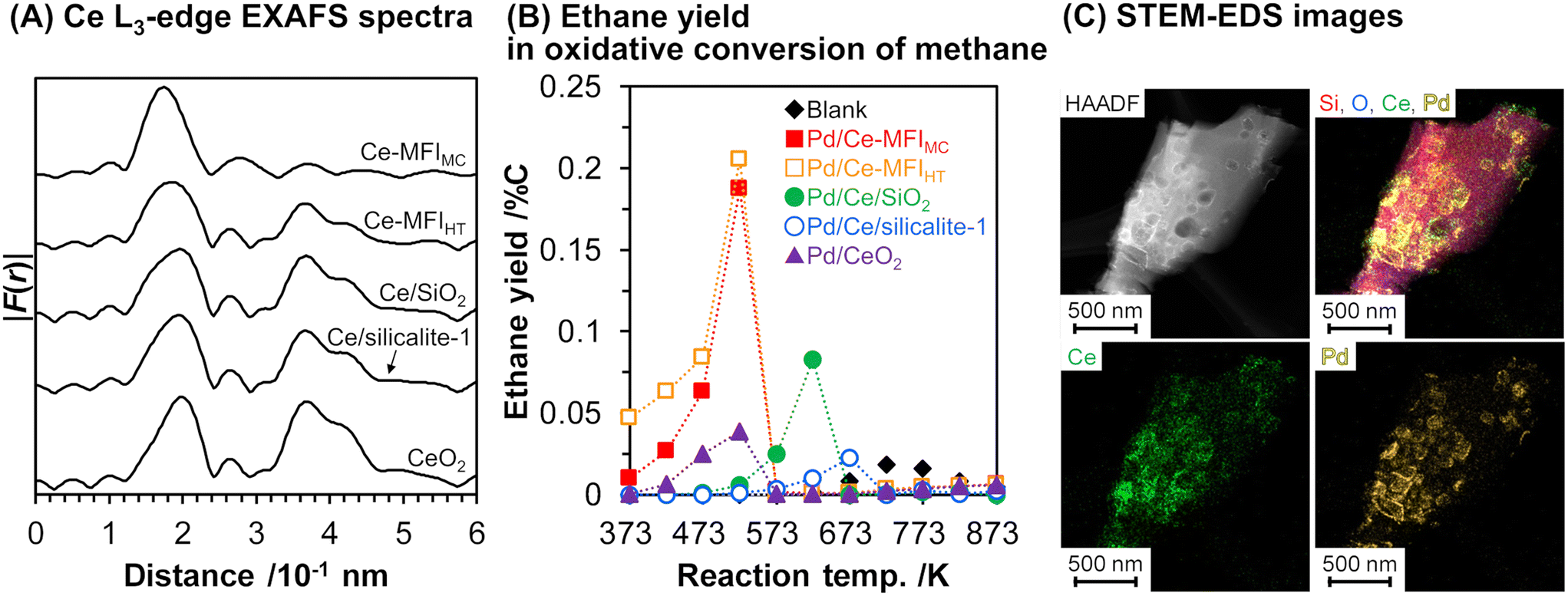 | ||
| Fig. 22 (A) Fourier transform of k3-weighted Ce L3-edge EXAFS spectra for Ce-containing supports, (B) ethane yield in oxidative conversion of methane over 3 wt% Pd/support, and (C) STEM-EDS images of 3 wt% Pd/Ce-MFIMC. Reaction conditions: CH4/O2/Ar2 = 8.0/2.0/2.5 mL min−1; SV = 7.5 × 103 mL gcat−1 h−1; 373–873 K; 0.1 MPa. Reproduced from ref. 85 with permission from American Chemical Society, copyright 2021. | ||
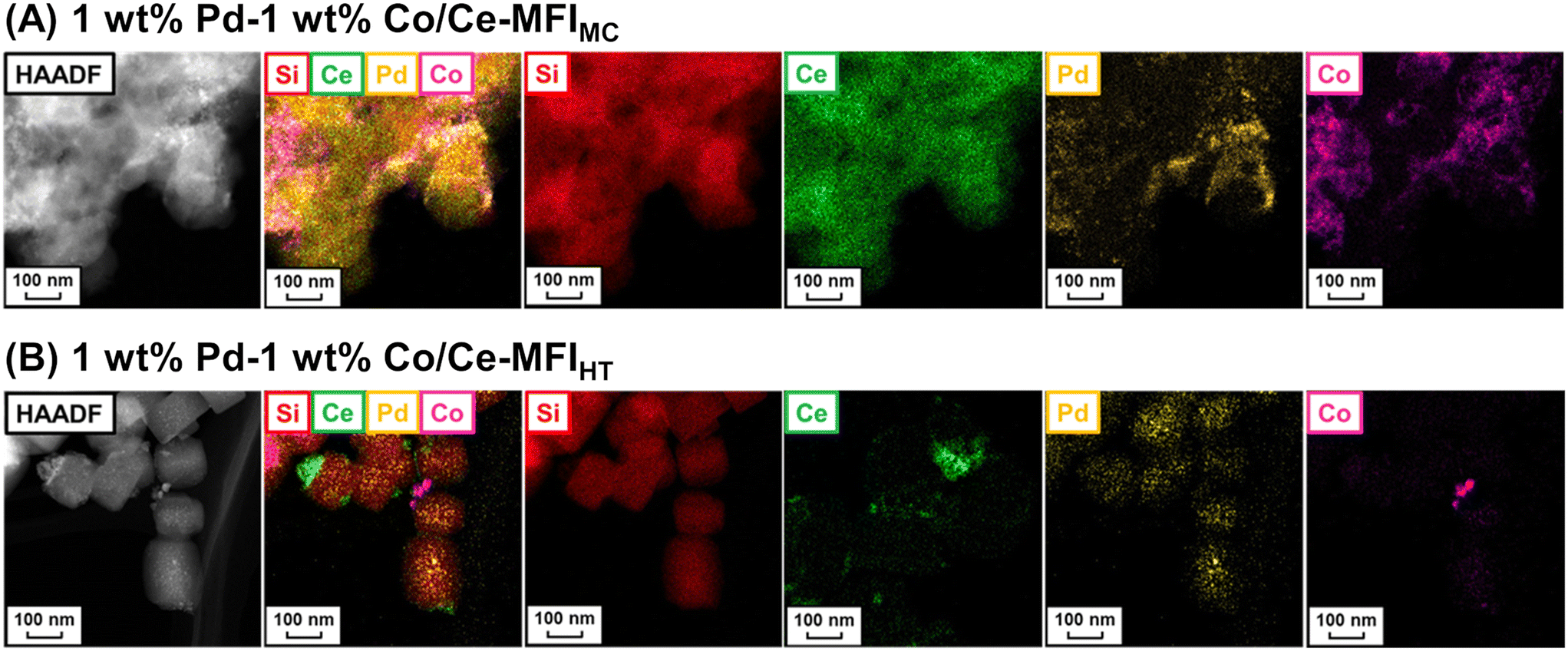 | ||
| Fig. 23 STEM-EDS images of (A) 1 wt% Pd–1 wt% Co/Ce-MFIMC and (B) 1 wt% Pd–1 wt% Co/Ce-MFIHT. Reproduced from ref. 88 with permission from American Chemical Society, copyright 2022. | ||
Encapsulation of active nanoparticles is effective in controlling and maintaining their size and performance.89,90 Xiao et al. fixed AuPd alloy nanoparticles in ZSM-5 crystals via a two-step synthetic method.91 A mixture of AuPd colloids and TEOS was hydrothermally treated to prepare AuPd nanoparticles coated with amorphous silica. The resulting material was further treated in the presence of TPA hydroxide and boehmite to obtain AuPd nanoparticles encapsulated in ZSM-5 crystals, whose external surface was further modified with organosilanes (AuPd@ZSM-5-C16). The structure of this catalyst differed completely from the control materials prepared via the conventional impregnation (AuPd/ZSM-5). This well-designed AuPd@ZSM-5-C16 catalyst converted methane to methanol in the presence of in situ generated H2O2 from H2 and O2 (entry 11 in Table 4). This catalyst was the analogue of the catalyst reported by Hutchings et al.,92 but the latter was prepared by a conventional co-impregnation method and used only for methane conversion with H2O2, not with H2 and O2 (entry 12 in Table 4). AuPd alloy nanoparticles had a dual function in this reaction system: the in situ formation of H2O2 from H2 and O2 and methane activation.92 Meanwhile, the organosilane modification of ZSM-5 provides hydrophobic external surfaces, which impede the outward diffusion of in situ formed H2O2 from inside the ZSM-5 crystals. This greatly enhanced the local concentration of H2O2 within the crystals, promoting methane conversion. Indeed, methanol productivity with AuPd@ZSM-5 (without surface modification) became 2.8-fold lower than that with AuPd@ZSM-5-C16 (with modification). Metal nanoparticles encapsulated in zeolites have also been employed for other classes of methane conversion such as combustion and dry reforming.93,94
5. Summary of advantages and disadvantages of each preparation method
Based on the exemplified catalysts and discussion in the former sections, the advantages and disadvantages of the three preparation methods—incorporation of heteroatoms into frameworks, ion-exchange, and impregnation—are overviewed in this section. These insights are expected to be guidelines for preparation of desired structures and active sites in zeolite-based catalysts for not only oxidative upgrading of methane but also other reactions.The incorporation of heteroatoms into zeolite frameworks as active sites easily prepares single-site catalysts due to the complete isolation of each introduced heteroatom by inert siliceous frameworks. Meanwhile, metal elements that can be employed in this method are severely limited. Trivalent cations consisting of Al3+ and Ga3+ are known to be introduced into zeolite frameworks relatively easily, while the complete incorporation of other metal cations into frameworks is quite challenging even in the case of widely investigated Fe3+ regardless of the same valence as the trivalent cations described above (typically, intra- and extra-framework Fe species are generated).26 The limited element scope makes the freedom of catalyst design narrower, compared to the other two preparation methods. Besides, one-pot hydrothermal synthesis of heteroatom-incorporated zeolites is sometimes impossible depending on heteroatoms, framework topologies, and molar ratios of Si to heteroatoms. In such situations, post-synthetic incorporation is necessary, making the overall catalyst preparation relatively complicated. Furthermore, the formation of binuclear heteroatom species in the framework-incorporation method is impossible due to the violation of the widely-accepted Loewenstein's rule, which omits the possibility of the formation of M–O–M species (M = heteroatom) in zeolite frameworks due to their thermodynamic instability.95 In this respect, this approach is not appropriate for creating binuclear active sites that mimic MMOs.
In contrast to the framework incorporation method, the ion-exchange method is a powerful means of preparing MMO-mimetic binuclear active sites, as introduced in section 3. This method is also useful for preparing highly dispersed metal species since each cationic metal ions are introduced via electrostatic interaction with negatively-charged sites originated from isomorphously substituting trivalent heteroatoms. Such electrostatic interaction is beneficial to anchoring introduced ions tightly, leading to the suppression of aggregation of active metal species. Yet, for the introduction of multivalent cationic species via the ion-exchange method, the presence of paired heteroatom sites in zeolite frameworks, which can compensate two positive charges, is the prerequisite; otherwise, undesired oxygen-containing ligands inevitably attach cationic metal ions for charge compensation, hampering the formation of well-designed active sites or rather triggering undesired side reactions.
The impregnation method has the broadest scope of metal elements among the three approaches introduced here; in principle, all metal elements are applicable. However, the introduction of highly-dispersed active metal species precisely into zeolite micropores requires a carefully-handled impregnation method combined with a vacuum technique. Otherwise, metal species are present on outer surfaces of zeolites and the confinement effect by zeolite micropores cannot be expected in such cases.
6. Conclusions and outlook
Zeolite-based materials have a great potential to be highly active and selective catalysts for the oxidative conversion of methane into platform chemicals such as carbon monoxide, methanol, lower hydrocarbons, and other oxygenates, owing to their high tunability. Active sites of zeolite-based catalysts consist of intra-framework heteroatoms, ion-exchanged species (H+ and metal ions), supported metal or metal oxide nanoparticles, single sites, and combinations thereof. For the preparation and stabilization of these species that function as catalytic active sites, their environment, typically nanospaces (i.e., pores, cages, and channel intersections) of siliceous frameworks, plays a significant role and needs to be tuned properly. In most cases, catalyst regeneration is quite difficult once active sites are aggregated. Therefore, isolation and fixation by both anchoring and confinement effects of nanospaces, which are inaccessible to typical catalyst supports, are among the characteristic and attractive features of zeolite-based materials. To this end, a thorough understanding of the precise design and preparation of zeolite-based catalysts is required.The conventional a posteriori approach relying on screening of each parameter (e.g., catalyst component, content, and preparation technique) is undoubtedly still important and necessary for the development of effective catalysts; however, recent advances in computational chemistry, including DFT calculations and machine learning, provide useful guidelines for catalyst design.96–99 For example, previous MMO-mimetic zeolites rely on the preparation of binuclear (or trinuclear) Cu and Fe species on the zeolite surfaces via the ion-exchange method as described in section 3. The resulting Cu- and Fe-based active sites successfully convert methane to methoxy species, which is an important intermediate of MTM reaction but also exhibit too high affinity with methoxy species, leading to the requirement of the extraction step of methoxy species with water (Fig. 8). In other words, the chemical looping system or co-feeding of water is inevitably required to complete the reaction cycle over these active sites; namely, in the case of chemical looping, the impossibility of exceeding the turnover number of one is a drawback (i.e., reactions with chemical looping are not catalytic). To establish more simplified and efficient catalytic system, suitable active sites that enable the spontaneous desorption of methoxy species need to be devised. According to computational calculations by Mahyuddin and Yoshizawa, Ni-based binuclear and trinuclear sites (i.e., [Ni2(μ-O)2]2+ and [Ni3(μ-O)3]2+) on ZSM-5 have such potential along with the methane activation;64,100 however, such sites have not yet been developed. Such computational-chemistry-driven catalyst design is a promising approach but has remained a grand challenge.
In addition to advanced computational chemistry, state-of-the-art atomic-level characterization techniques help researchers to gain a deeper understanding of the structure–activity relationship for each synthesized catalyst. One of the techniques introduced in this manuscript is high-resolution microscopy. Spherical-aberration-corrected STEM using annular dark field and low angle annular dark field (abbreviated as Cs-corrected STEM ADF and Cs-corrected STEM LAADF, respectively) have been employed to identify the locations (i.e., tetrahedral site (T-site)) of framework-substituting heteroatoms, e.g., Fe atoms in MFI101 and Ti atoms in YNU-2 (Fig. 24A).102 High-resolution NMR spectroscopy is another powerful tool used for the same objective, as described in many review papers.103–105 Integrated differential phase contrast STEM (iDPC-STEM) also enables the imaging of metal species anchored on the inner surfaces of zeolites, as exemplified by Mo species in a ZSM-5 pore (Fig. 24B).106 Furthermore, dynamic atomic-level imaging by iDPC-STEM directly visualizes organic molecules confined in nanospaces; for example, the dynamic adsorption and desorption of pyridine in a ZSM-5 pore have been directly observed.107 These “seeing-is-believing” techniques are powerful tools for understanding both catalytic active sites and the adsorption states of substrate molecules.
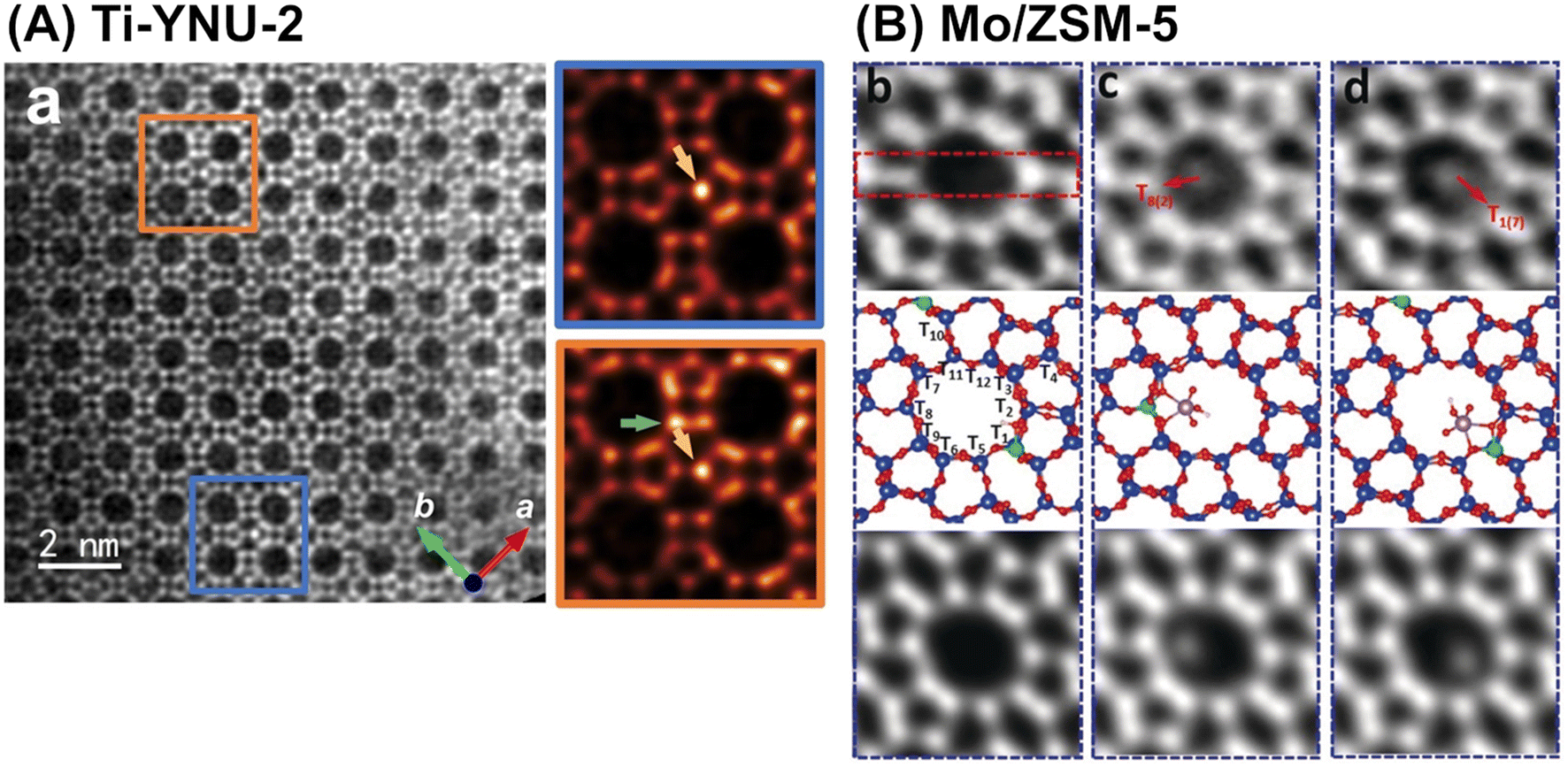 | ||
| Fig. 24 (A) Cs-corrected STEM LAADF image along (001) incidence of Ti-YNU-2. Two regions colored by blue and orange rectangles with distinct contrast differences are enlarged on the right. Green and golden arrows in the expanded figures represent Ti atoms at the T1/T2 and T7/T8 sites, respectively. Reprinted from ref. 102 with permission from John Wiley and Sons, copyright 2022. (B) iDPC-STEM images of Mo/ZSM-5, where (b–d) are different fields of view for the same sample: top = raw images; middle = calculated structural models; and bottom = simulated projected electrostatic potential. Reprinted from ref. 106 with permission from John Wiley and Sons, copyright 2019. | ||
Both microscopy and NMR spectroscopy can provide atomic-level structural information for zeolites as described above, yet these methods are relatively difficult to apply to in situ/operando modes, as compared to other characterization techniques. The static structural information of each catalyst is surely important, but dynamic structural analyses offer more direct and detailed insight into catalysis. To reveal the working states of active metal or metal oxide species over zeolites, UV-vis spectroscopy, FT-IR spectroscopy, Raman spectroscopy, and XAS have been employed in in situ/operando modes.108 FT-IR spectroscopy is also useful in terms of characterizing surface-adsorbed species. Among these techniques, in situ/operando XAS is the most popular and attractive since it provides both the valence state and local structure of active metal or metal oxide species. For example, Beato et al. successfully tracked the gradual changes in Cu active sites over SSZ-13 (i.e., Cu-SSZ-13), a typical catalyst for the MTM reaction (see section 3), by operando XAS, monitoring the step-by-step alteration of the environment and temperature necessary to complete the entire reaction process, i.e., activation of Cu species by O2, reaction with methane, and subsequent water-mediated extraction of the produced methoxy species as methanol (see Fig. 25).109 They found that the Cu2+ species, after activation by O2 and flushing with He, partially underwent reduction to Cu+, which was successively re-oxidized to Cu2+ during the water-assisted extraction of methoxy species. We should note that in the oxidative coupling of methane to lower hydrocarbons, the selectivity for target product(s) is typically low, except when the methane conversion is carefully controlled to be low,7,10 and CO and CO2 are major products. In such situations, in situ/operando spectroscopy should provide catalytic insights into partial/complete oxidation of methane, not oxidative coupling. Similar situation of low selectivity to target product(s) is also possible in other types of oxidative upgrading of methane due to the ease of over-oxidation of product(s). Therefore, the operation conditions need to be fine-tuned to gain precise and reliable data for each catalytic reaction as well as in situ/operando analysis.
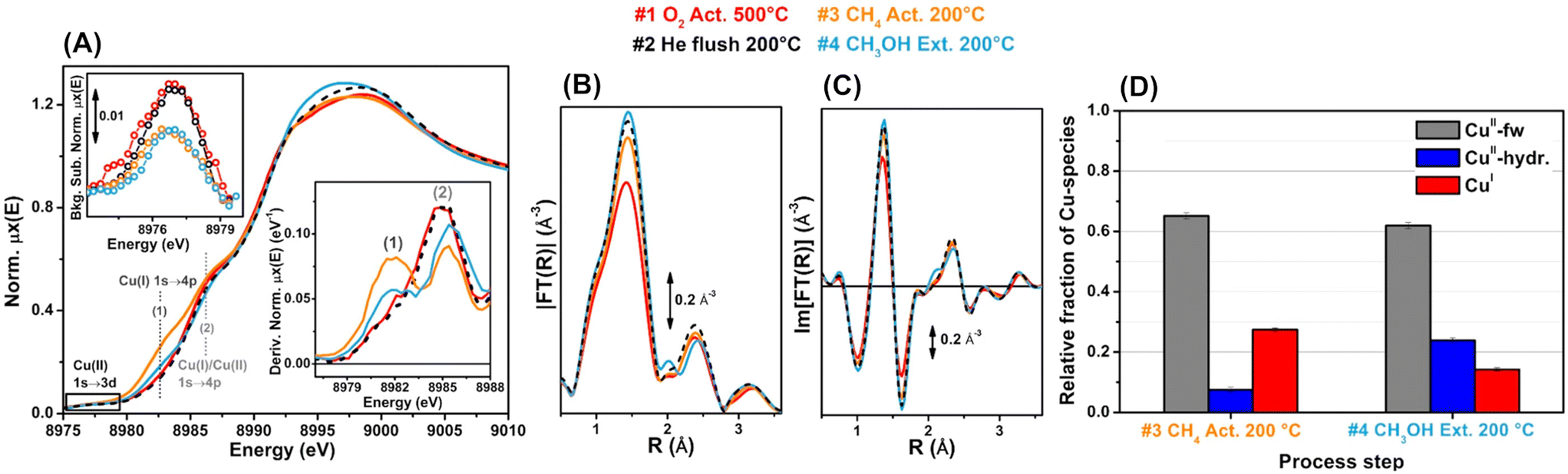 | ||
| Fig. 25 Operando Cu K-edge XAS spectra of Cu-SSZ-13 after sequential treatments: step 1 (red) = O2 activation at 773 K; step 2 (black) = He flushing after cooling to 473 K in O2; step 3 (orange) = CH4 treatment at 473 K; step 4 (blue) = H2O-assisted extraction of methoxy species produced on the catalyst. (A) X-ray absorption near edge structure (XANES) spectra, where labels (1) and (2) represent the rising-edge from 1s to 4p peaks that are characteristic of Cu+ and Cu+/Cu2+. (B) Magnitude part of Fourier transform of EXAFS spectra. (C) Imaginary part of Fourier transform of EXAFS spectra. (D) Relative fractions of Cu species estimated via linear combination fitting (LCF) of XANES spectra: CuII-fw = Cu2+ species that interact with the framework; CuII-hyd. = mobile Cu2+ aquo complexes. Reproduced from ref. 109 with permission from American Chemical Society, copyright 2017. | ||
Zeolitic materials such as metal–organic frameworks (MOFs) also offer opportunities to tune active sites precisely via design of nodes and linkers as well as modifiers.110 Similar to the case of zeolite-based catalysts introduced in section 3, the creation of MMO-mimetic active sites has also been investigated for MOF-based catalysts.111 Higher surface area of MOFs compared to zeolites112 enables the preparation of highly dispersed metal nanoparticles even by using typical impregnation technique, which could lead to better productivity per loading amount of metals. Meanwhile, due to the presence of organic linkers, thermal stability of MOFs is inferior to that of zeolites. This limitation of MOFs cannot allow reaction operation at high temperatures, and thus, reaction rates are apt to be low. This fact also points out that fine-tuned sites that are capable of highly activating C–H bond(s) of methane even at low reaction temperatures are always necessary and also that careful selection of reaction conditions is required to suppress thermal degradation of MOFs completely. With respect to the latter issue, isotope tracer analysis using 13C-labeled methane as a substrate could be helpful for clearly confirming and distinguishing methane-derived and MOF-derived products. Besides, high selectivity toward target product(s) is mandatory for MOF-based catalysts since even if carbonaceous materials are deposited onto the catalyst surfaces, these catalysts cannot be regenerated via simple calcination. In this respect, zeolites and zeolitic materials need to be carefully chosen for one's objective.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was supported financially by the following grants: Core Research for Evolutional Science and Technology of the Japan Science and Technology Agency (JST CREST, JPMJCR16P3); a Grant-in-Aid for Scientific Research (S) (KAKENHI, 21H05011), a Grant-in-Aid for Scientific Research (C) (23K04494), and a Grant-in-Aid for Early-Career Scientists (21K14459) from the Japan Society for the Promotion of Science (JSPS); a research grant from The Japan Petroleum Institute; Kurita Water and Environment Foundation (21A023 and 22K002); Nippon Life Insurance Foundation; and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials in Network Joint Research Center for Materials and Devices.References
- D. Li, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2011, 408, 1–24 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- M. Usman, W. M. A. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 45, 710–744 CrossRef CAS.
- B. C. Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef CAS.
- P. Tomkins, M. Ranocchiari and J. A. van Bokhoven, Acc. Chem. Res., 2017, 50, 418–425 CrossRef CAS PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- A. Galadima and O. Muraza, J. Ind. Eng. Chem., 2016, 37, 1–13 CrossRef CAS.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- T. Zhang, Chem. Sci., 2021, 12, 12529–12545 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- Web of Science, https://www.webofscience.com/, (accessed April 2023).
- Y. Wang, C. Wang, L. Wang, L. Wang and F.-S. Xiao, Acc. Chem. Res., 2021, 54, 2579–2590 CrossRef CAS PubMed.
- Q. Zhang, S. Gao and J. Yu, Chem. Rev., 2023, 123, 6039–6106 CrossRef CAS PubMed.
- S.-M. Wu, X.-Y. Yang and C. Janiak, Angew. Chem., Int. Ed., 2019, 58, 12340–12354 CrossRef CAS PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- P. Batamack, I. Bucsi, Á. Molnár and G. A. Olah, Catal. Lett., 1994, 25, 11–19 CrossRef CAS.
- S. Kowalak and J. B. Moffat, Appl. Catal., 1988, 36, 139–145 CrossRef CAS.
- D. Farcasiu, S. L. Fisk, M. T. Melchior and K. D. Rose, J. Org. Chem., 1982, 47, 453–457 CrossRef CAS.
- S. Kanitkar, J. H. Carter, G. J. Hutchings, K. Ding and J. J. Spivey, ChemCatChem, 2018, 10, 5019–5024 CrossRef.
- K. Yamamoto, G. S. E. Borjas, F. Saito and A. Muramatsu, Chem. Lett., 2006, 35, 570–571 CrossRef CAS.
- M. Yabushita, M. Yoshida, F. Muto, M. Horie, Y. Kunitake, T. Nishitoba, S. Maki, K. Kanie, T. Yokoi and A. Muramatsu, Mol. Catal., 2019, 478, 110579 CrossRef CAS.
- C. T. W. Chu and C. D. Chang, J. Phys. Chem., 1985, 89, 1569–1571 CrossRef CAS.
- M. S. Stave and J. B. Nicholas, J. Phys. Chem., 1995, 99, 15046–15061 CrossRef CAS.
- M. Yabushita, H. Kobayashi, R. Osuga, M. Nakaya, M. Matsubara, S. Maki, K. Kanie and A. Muramatsu, Ind. Eng. Chem. Res., 2021, 60, 2079–2088 CrossRef CAS.
- R. Osuga, G. Tanaka, M. Yabushita, K. Ninomiya, S. Maki, M. Nishibori, K. Kanie and A. Muramatsu, Sekiyu Gakkaishi, 2022, 65, 67–77 CrossRef CAS.
- A. Montes and G. Giannetto, Appl. Catal., A, 2000, 197, 31–39 CrossRef CAS.
- M. V. Luzgin, A. A. Gabrienko, V. A. Rogov, A. V. Toktarev, V. N. Parmon and A. G. Stepanov, J. Phys. Chem. C, 2010, 114, 21555–21561 CrossRef CAS.
- Y. Liu, L. Wang and F.-S. Xiao, Chem. Res. Chin. Univ., 2022, 38, 671–676 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- J. Xu, R. D. Armstrong, G. Shaw, N. F. Dummer, S. J. Freakley, S. H. Taylor and G. J. Hutchings, Catal. Today, 2016, 270, 93–100 CrossRef CAS.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- P. Xiao, Y. Wang, J. N. Kondo and T. Yokoi, Appl. Catal., A, 2019, 579, 159–167 CrossRef CAS.
- P. Xiao, Y. Wang, T. Nishitoba, J. N. Kondo and T. Yokoi, Chem. Commun., 2019, 55, 2896–2899 RSC.
- P. Wang, G. Zhao, Y. Wang and Y. Lu, Sci. Adv., 2017, 3, e1603180 CrossRef PubMed.
- S. L. X. Fang, J. Lin, J. Gu and D. Yan, J. Mol. Catal., 1992, 6, 255–262 Search PubMed.
- S. L. X. Fang, J. Lin and Y. Chu, J. Mol. Catal., 1992, 6, 427–433 Search PubMed.
- S. Arndt, T. Otremba, U. Simon, M. Yildiz, H. Schubert and R. Schomäcker, Appl. Catal., A, 2012, 425–426, 53–61 CrossRef CAS.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- L. Tao, I. Lee and M. Sanchez-Sanchez, Catal. Sci. Technol., 2020, 10, 7124–7141 RSC.
- R. S. Hanson and T. E. Hanson, Microbiol. Rev., 1996, 60, 439–471 CrossRef CAS PubMed.
- S.-P. Huang, Y. Shiota and K. Yoshizawa, Dalton Trans., 2013, 42, 1011–1023 RSC.
- J. C. S. Da Silva, R. C. R. Pennifold, J. N. Harvey and W. R. Rocha, Dalton Trans., 2016, 45, 2492–2504 RSC.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef.
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131–136 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed.
- R. Xu, N. Liu, C. Dai, Y. Li, J. Zhang, B. Wu, G. Yu and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 16634–16640 CrossRef CAS PubMed.
- M. Yabushita, R. Osuga and A. Muramatsu, CrystEngComm, 2021, 23, 6226–6233 RSC.
- R. Osuga and T. Yokoi, in Crystalline Metal Oxide Catalysts, ed. W. Ueda, Springer Nature Singapore, Singapore, 2022, pp. 167–196 Search PubMed.
- J. Bae and M. Dusselier, Chem. Commun., 2023, 59, 852–867 RSC.
- J. Li, M. Gao, W. Yan and J. Yu, Chem. Sci., 2023, 14, 1935–1959 RSC.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev.: Sci. Eng., 2012, 54, 135–223 CrossRef.
- J. R. Di Iorio and R. Gounder, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- T. Nishitoba, N. Yoshida, J. N. Kondo and T. Yokoi, Ind. Eng. Chem. Res., 2018, 57, 3914–3922 CrossRef CAS.
- J. R. Di Iorio, S. Li, C. B. Jones, C. T. Nimlos, Y. Wang, E. Kunkes, V. Vattipalli, S. Prasad, A. Moini, W. F. Schneider and R. Gounder, J. Am. Chem. Soc., 2020, 142, 4807–4819 CrossRef CAS PubMed.
- M. Yabushita, Y. Imanishi, T. Xiao, R. Osuga, T. Nishitoba, S. Maki, K. Kanie, W. Cao, T. Yokoi and A. Muramatsu, Chem. Commun., 2021, 57, 13301–13304 RSC.
- P. Xiao, Y. Wang, Y. Lu, T. De Baerdemaeker, A.-N. Parvulescu, U. Müller, D. De Vos, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, U. Kolb, H. Gies and T. Yokoi, Appl. Catal., B, 2023, 325, 122395 CrossRef CAS.
- K. Nakamura, P. Xiao, R. Osuga, Y. Wang, S. Yasuda, T. Matsumoto, J. N. Kondo, M. Yabushita, A. Muramatsu, H. Gies and T. Yokoi, Catal. Sci. Technol., 2023, 13, 2648–2651 RSC.
- M. H. Mahyuddin, A. Staykov, Y. Shiota and K. Yoshizawa, ACS Catal., 2016, 6, 8321–8331 CrossRef CAS.
- T. Yu, Y. Su, A. Wang, B. M. Weckhuysen and W. Luo, ChemCatChem, 2021, 13, 2766–2770 CrossRef CAS.
- A. Oda, K. Aono, N. Murata, K. Murata, M. Yasumoto, N. Tsunoji, K. Sawabe and A. Satsuma, Catal. Sci. Technol., 2022, 12, 542–550 RSC.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS PubMed.
- A. M. Rabie, M. A. Betiha and S.-E. Park, Appl. Catal., B, 2017, 215, 50–59 CrossRef CAS.
- Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- Y. Hou, S. Ogasawara, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2017, 7, 6132–6139 RSC.
- R. Osuga, S. Bayarsaikhan, S. Yasuda, R. Manabe, H. Shima, S. Tsutsuminai, A. Fukuoka, H. Kobayashi and T. Yokoi, Chem. Commun., 2020, 56, 5913–5916 RSC.
- R. Osuga, S. Yasuda, M. Sawada, R. Manabe, H. Shima, S. Tsutsuminai, A. Fukuoka, H. Kobayashi, A. Muramatsu and T. Yokoi, Ind. Eng. Chem. Res., 2021, 60, 8696–8704 CrossRef CAS.
- N. Davidova, P. Kovacheva and A. H. Weiss, Catal. Today, 1992, 13, 625–626 CrossRef CAS.
- S. Yasuda, Y. Kunitake, R. Osuga, K. Nakamura, T. Matsumoto, K. Sago, J. N. Kondo, M. Yabushita, A. Muramatsu and T. Yokoi, Chem. Lett., 2022, 51, 46–49 CrossRef CAS.
- R. A. Schoonheydt, P. Geerlings, E. A. Pidko and R. A. van Santen, J. Mater. Chem., 2012, 22, 18705–18717 RSC.
- S. Yasuda, R. Osuga, Y. Kunitake, K. Kato, A. Fukuoka, H. Kobayashi, M. Gao, J. Hasegawa, R. Manabe, H. Shima, S. Tsutsuminai and T. Yokoi, Commun. Chem., 2020, 3, 129 CrossRef CAS PubMed.
- S. Takenaka, S. Kobayashi, H. Ogihara and K. Otsuka, J. Catal., 2003, 217, 79–87 CAS.
- Y. Hou, S. Nagamatsu, K. Asakura, A. Fukuoka and H. Kobayashi, Commun. Chem., 2018, 1, 41 CrossRef.
- H. Li, Y. Shen, X. Xiao, H. Jiang, Q. Gu, Y. Zhang, L. Lin, W. Luo, S. Zhou, J. Zhao, A. Wang, T. Zhang and B. Yang, ACS Catal., 2023, 13, 1197–1206 CrossRef CAS.
- W. Huang, S. Zhang, Y. Tang, Y. Li, L. Nguyen, Y. Li, J. Shan, D. Xiao, R. Gagne, A. I. Frenkel and F. Tao, Angew. Chem., Int. Ed., 2016, 55, 13441–13445 CrossRef CAS PubMed.
- Y. Tang, Y. Li, V. Fung, D.-e. Jiang, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. I. Frenkel and F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- T. Zhang, Z. Chen, A. G. Walsh, Y. Li and P. Zhang, Adv. Mater., 2020, 32, 2002910 CrossRef CAS PubMed.
- L. Fang, S. Seifert, R. E. Winans and T. Li, Small Methods, 2021, 5, 2001194 CrossRef CAS PubMed.
- M. Yabushita, M. Yoshida, R. Osuga, F. Muto, S. Iguchi, S. Yasuda, A. Neya, M. Horie, S. Maki, K. Kanie, I. Yamanaka, T. Yokoi and A. Muramatsu, Ind. Eng. Chem. Res., 2021, 60, 10101–10111 CrossRef CAS.
- A. W. Petrov, D. Ferri, M. Tarik, O. Kröcher and J. A. van Bokhoven, Top. Catal., 2017, 60, 123–130 CrossRef CAS.
- S. Hosokawa, M. Taniguchi, K. Utani, H. Kanai and S. Imamura, Appl. Catal., A, 2005, 289, 115–120 CrossRef CAS.
- R. Osuga, A. Neya, M. Yoshida, M. Yabushita, S. Yasuda, S. Maki, K. Kanie, T. Yokoi and A. Muramatsu, Ind. Eng. Chem. Res., 2022, 61, 9686–9694 CrossRef CAS.
- H. Wang, L. Wang and F.-S. Xiao, ACS Cent. Sci., 2020, 6, 1685–1697 CrossRef CAS PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- R. J. Lewis, A. Bara-Estaun, N. Agarwal, S. J. Freakley, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2019, 149, 3066–3075 CrossRef CAS.
- R. L. Mortensen, H.-D. Noack, K. Pedersen, S. Mossin and J. Mielby, ChemCatChem, 2022, 14, e202101924 CrossRef CAS.
- S. Ma, X. Li, Z. Yang and H. Li, J. Catal., 2023, 417, 368–378 CrossRef CAS.
- W. Loewenstein, Am. Mineral., 1954, 39, 92–96 CAS.
- T. Toyao, K. Suzuki, S. Kikuchi, S. Takakusagi, K. Shimizu and I. Takigawa, J. Phys. Chem. C, 2018, 122, 8315–8326 CrossRef CAS.
- J. Ohyama, A. Hirayama, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Hirai, I. Miyazato and K. Takahashi, Sci. Rep., 2021, 11, 2067 CrossRef CAS PubMed.
- S. Ma and Z.-P. Liu, Top. Catal., 2022, 65, 59–68 CrossRef CAS.
- S. Ma and Z.-P. Liu, Chem. Sci., 2022, 13, 5055–5068 RSC.
- M. H. Mahyuddin and K. Yoshizawa, Catal. Sci. Technol., 2018, 8, 5875–5885 RSC.
- A. Mayoral, Q. Zhang, Y. Zhou, P. Chen, Y. Ma, T. Monji, P. Losch, W. Schmidt, F. Schüth, H. Hirao, J. Yu and O. Terasaki, Angew. Chem., Int. Ed., 2020, 59, 19510–19517 CrossRef CAS PubMed.
- J. Li, A. Mayoral, Y. Kubota, S. Inagaki, J. Yu and O. Terasaki, Angew. Chem., Int. Ed., 2022, 61, e202211196 CAS.
- Y. Jiang, J. Huang, W. Dai and M. Hunger, Solid State Nucl. Magn. Reson., 2011, 39, 116–141 CrossRef CAS PubMed.
- S. Li, O. Lafon, W. Wang, Q. Wang, X. Wang, Y. Li, J. Xu and F. Deng, Adv. Mater., 2020, 32, 2002879 CrossRef CAS PubMed.
- X. Zhao, J. Xu and F. Deng, Front. Chem. Sci. Eng., 2020, 14, 159–187 CrossRef CAS.
- L. Liu, N. Wang, C. Zhu, X. Liu, Y. Zhu, P. Guo, L. Alfilfil, X. Dong, D. Zhang and Y. Han, Angew. Chem., Int. Ed., 2020, 59, 819–825 CrossRef CAS PubMed.
- B. Shen, H. Wang, H. Xiong, X. Chen, E. G. T. Bosch, I. Lazić, W. Qian and F. Wei, Nature, 2022, 607, 703–707 CrossRef CAS PubMed.
- D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Chem. Soc. Rev., 2021, 50, 1251–1268 RSC.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- W.-G. Cui, G.-Y. Zhang, T.-L. Hu and X.-H. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- L. S. Andrade, H. H. L. B. Lima, C. T. P. Silva, W. L. N. Amorim, J. G. R. Poço, A. López-Castillo, M. V. Kirillova, W. A. Carvalho, A. M. Kirillov and D. Mandelli, Coord. Chem. Rev., 2023, 481, 215042 CrossRef CAS.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |