
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium catalysed sequence selective ring opening terpolymerisation: a mechanistic study†
Peter
Deglmann
*a,
Sara
Machleit
b,
Cesare
Gallizioli
b,
Susanne M.
Rupf
b and
Alex J.
Plajer
 *b
*b
aBASF SE, Carl-Bosch-Strasse 38, 67056 Ludwigshafen am Rhein, Germany. E-mail: peter.deglmann@basf.com
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, 14195 Berlin, Germany. E-mail: plajer@zedat.fu-berlin.de
First published on 10th April 2023
Abstract
The catalytic construction of well-defined materials from mixtures of building blocks is an important challenge in sustainable catalysis. In this regard, we have recently reported a new type of selective ring-opening terpolymerisation (ROTERP), in which three monomers (A, B, C) are selectively enchained into a (ABA′C)n sequence, but the reasons behind this unusual selectivity remained unanswered. In this study, we present a detailed investigation into the full ROTERP mechanism based on the reactivity of model intermediates, computational studies investigating >100 possible intermediates and transition states and reaction kinetics. Experimental verification of the intermediate speciation, the primary insertion steps and the side-reactions lets us show that although most insertions and side-reactions are thermodynamically viable, kinetic selection processes at the propagating chain end determine the sequence selectivity. Computational studies elucidate the special role and speciation of the lithium catalyst which during the catalytic cycle involves mono-metallic, bi-metallic and charge separated transition states comprising both coordinative activation of incoming monomers and functional groups of the polymer backbone adjacent to the propagating chain. Our study not only deciphers the mechanism of a rare selective terpolymerisation but also helps answering open questions relevant to ring-opening copolymerisation (ROCOP) and alkali-metal catalysis in general, thus guiding the design of future polymerisation catalysis for degradable materials.
Introduction
Controlled ways to make degradable and chemically complex materials from mixtures of building blocks are increasingly required in order to tailor their properties to ever more demanding technological requirements.1–8 One popular methodology to achieve this is the alternating ring-opening copolymerisation (ROCOP) of a strained heterocycle with a heteroallene (yielding e.g. polycarbonates from CO2 and epoxides) or a second heterocycle (yielding e.g. polyesters from cyclic anhydrides and epoxides).9–16 Sustainable catalysts featuring main-group metals are enjoying increasing popularity, but the associated reaction pathways are only partially understood and it is often unclear which mono- or multimetallic, cooperative or uncooperative and neutral or zwitterionic routes occur.17–25 Polymerising more complex ROCOP monomer mixtures with such catalysts can achieve the selective one-pot construction of higher order polymer architectures. For example, ternary mixtures comprising cyclic anhydrides, CO2 and epoxides can be selectively polymerised into polyester-b-polycarbonate blockpolymers, i.e. CO2/epoxide ROCOP only occurring once all anhydride is consumed and similar selectivities are observed for related monomer combinations.4,26–34 Both insertion kinetics and intermediate thermodynamics have been proposed to be responsible for this selectivity which still remains under debate.26,27,35–37 Moving down the periodic table, sulfur containing ROCOP monomer combinations likewise exist which mostly remain to be explored in terpolymerisation reactions.33,38–45 Phthalic thioanhydride (PTA)/epoxide ROCOP for example yields poly[ester-alt-thioesters] while CS2/epoxide ROCOP can yield poly(monothio-alt-trithiocarbonate) as reported by Werner and co-workers although the formal product of alternating ROCOP would be a poly(dithiocarbonate).41,46–48 Combining these ROCOPs, we recently reported a lithium catalysed PTA/CS2/epoxide ring-opening terpolymerisation (ROTERP, with propylene oxide (PO) or butylene oxide (BO)) and observed the formation of poly(ester-alt-ester-alt-trithiocarbonates) with a few erroneous thioester links (Fig. 1).49–51 The ROTERP polymers were produced in an unusual (head-to-head)-alt-(tail-to-tail) regioselectivity meaning that the trithiocarbonate links sit adjacent to secondary CH2 groups while the ester links sit adjacent to tertiary CH groups. This regioselectivity pointed towards a close mechanistic relation to Werner's CS2/epoxide ROCOP (ESI† Fig. S1) which led us to propose the reaction mechanism displayed in Fig. 1. Starting from a lithium alkoxide intermediate ATT insertion of PTA generates a lithium thiocarboxylate TC in propagation step (1). The intermediate TC then selectively attacks at the CH2 position of another epoxide in reaction step (2) which is commonly termed a “tail” selective attack. This forms a secondary lithium alkoxide ATE that sits adjacent to a thioester link. The lithium alkoxide ATE intramolecularly attacks the adjacent thioester group in step (3) to form a tetrahedral intermediate I. This intermediate then collapses in reaction step (4) into an ester appended primary lithium thiolate TE and the overall transformation of an alkoxide into a thiolate chain end has been termed O/S exchange. Propagation steps (2)–(4) also generates a link resulting from an isomerised epoxide (akin a virtual thiirane; A′ in Fig. 1) and an ortho-terephthalate link in which both esters sit adjacent to tertiary CH “head” groups of the ring-opened monosubstituted epoxides. TE then inserts into CS2 in reaction step (5) to generate a primary lithium trithiocarbonate TT. In the final propagation step, which closes the catalytic cycle, the primary TT ring opens another epoxide in reaction step (6) which regenerates the alkoxide ATT from which the cycle started. Importantly as the ring opening event is again tail selective, it constructs a trithiocarbonate link in “tail-to-tail” stereoselectivity with two CH2 groups next to it. However, the mechanism mostly remained a mere hypothesis in our previous report and many questions remained unanswered. Gratifyingly though, all proposed intermediates are synthetically accessible lithium salts which allows to study their reactivity. This represents a rare opportunity to synthetically study the reactivity of intermediates in such co/terpolymerisation processes as corresponding model intermediates in related catalyses are not easily synthetically accessible.9 Combined with kinetics and detailed computational studies we here present an investigation into the full ROTERP mechanism. We reveal the origins of (i) the observed monomer selection processes at the chain end, (ii) the suppression and assistance of side processes leading to O/S exchange and erroneous links as well as (iii) the role of the Li catalyst and (iv) the energetic profile of the main propagation cycle. | ||
| Fig. 1 PTA/CS2/epoxide ROTERP and postulated propagation mechanism. R = Me, Et; Rn = polymer chain. | ||
Results and discussion
Monomer selection by ATT: step (1)
Our investigation starts from the lithium alkoxide A that serves as a model for a secondary lithium alkoxide at the propagating chain end (ESI† section S3). In step (1) of Fig. 1, ATT selectively reacts with PTA over CS2 or epoxides which are also present in the mixture and accordingly we attempted reaction of A with those in d8-THF at room temperature. As shown in Fig. 2, A instantaneously reacts with PTA to yield the lithium thiocarboxylate TC (δ [R(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)SLi] = 216.0 ppm, δ [R(CO)OR] = 169.2 ppm,
O)SLi] = 216.0 ppm, δ [R(CO)OR] = 169.2 ppm, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CO) = 1701.5 cm−1) and with CS2 generating lithium dithiocarbonate DT (δ [RO(CS)SLi] = 244.6 ppm, (CS) = 1037.1 cm−1) as confirmed by multinuclear NMR, IR and a yellow discolouration due to formation of the (CS) chromophore. In contrast no reaction is observed between A and BO (which we employed as the model epoxide throughout the study due to its higher boiling point compared to PO) showing that epoxide insertion by the lithium alkoxide is associated with significantly higher barriers explaining the absence of ether linkages during ROTERP. However, this leaves the question unanswered why PTA is selected over CS2 by A in ROTERP and this could either be due to insertion kinetics or intermediate thermodynamics. In terms of the latter, we found that CS2 insertion into A to form DT is at least partially reversible. Heating of DT under dynamic vacuum at 80 °C for 1 min results in formation of A as visually indicated by a vanishing of the yellow colour and confirmed by 1H and 7Li NMR. However, some insoluble decomposition products also form. Analogous attempts to revert PTA insertion into A starting from TC at high temperatures failed. If CS2 into A is reversible but PTA insertion isn't, we hypothesized that transformation of DT into TC should be feasible. Addition of PTA to DT in d8-THF initially results in no reaction at room temperature but heating to 80 °C overnight results in complete transformation to TC. This clearly indicates that PTA insertion from ATT is the thermodynamically favoured propagation event. However, as ROTERP also occurs at room temperature with identical sequence selectivity and DT cannot be converted to TC at room temperature intermediate thermodynamics cannot be the only reason behind the insertion selectivity of ATT. To determine the kinetically favoured propagation step, we conducted a competition experiment in which we reacted A with a 10
(CO) = 1701.5 cm−1) and with CS2 generating lithium dithiocarbonate DT (δ [RO(CS)SLi] = 244.6 ppm, (CS) = 1037.1 cm−1) as confirmed by multinuclear NMR, IR and a yellow discolouration due to formation of the (CS) chromophore. In contrast no reaction is observed between A and BO (which we employed as the model epoxide throughout the study due to its higher boiling point compared to PO) showing that epoxide insertion by the lithium alkoxide is associated with significantly higher barriers explaining the absence of ether linkages during ROTERP. However, this leaves the question unanswered why PTA is selected over CS2 by A in ROTERP and this could either be due to insertion kinetics or intermediate thermodynamics. In terms of the latter, we found that CS2 insertion into A to form DT is at least partially reversible. Heating of DT under dynamic vacuum at 80 °C for 1 min results in formation of A as visually indicated by a vanishing of the yellow colour and confirmed by 1H and 7Li NMR. However, some insoluble decomposition products also form. Analogous attempts to revert PTA insertion into A starting from TC at high temperatures failed. If CS2 into A is reversible but PTA insertion isn't, we hypothesized that transformation of DT into TC should be feasible. Addition of PTA to DT in d8-THF initially results in no reaction at room temperature but heating to 80 °C overnight results in complete transformation to TC. This clearly indicates that PTA insertion from ATT is the thermodynamically favoured propagation event. However, as ROTERP also occurs at room temperature with identical sequence selectivity and DT cannot be converted to TC at room temperature intermediate thermodynamics cannot be the only reason behind the insertion selectivity of ATT. To determine the kinetically favoured propagation step, we conducted a competition experiment in which we reacted A with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CS2 and PTA at room temperature in d8-THF (note that a 10 CS2:1 PTA ratio is also employed in the required monomer mixtures of ROTERP). Interestingly we see 96% selectivity for PTA insertion yielding TC and only 4% CS2 insertion yielding DT although an excess of CS2 was provided. We hence propose that lithium alkoxide ATT selects PTA on kinetic grounds. Nevertheless, the same insertion is likewise favoured thermodynamically, and CS2-PTA exchange could play a role at increased reaction temperatures.
:1 mixture of CS2 and PTA at room temperature in d8-THF (note that a 10 CS2:1 PTA ratio is also employed in the required monomer mixtures of ROTERP). Interestingly we see 96% selectivity for PTA insertion yielding TC and only 4% CS2 insertion yielding DT although an excess of CS2 was provided. We hence propose that lithium alkoxide ATT selects PTA on kinetic grounds. Nevertheless, the same insertion is likewise favoured thermodynamically, and CS2-PTA exchange could play a role at increased reaction temperatures.
 | ||
| Fig. 2 (a) Reactivity and solid-state structure of model alkoxide A; 1H–13C HMBC (d8-THF, 25 °C, 500 MHz) NMR spectra of TC and DT; (b) 1H NMR spectrum (d8-THF, 25 °C, 400 MHz) of products obtained from the reaction of A with a 10:1 mixture of CS2 and PTA. | ||
O/S exchange: step (2)–(4)
Next up in the mechanism are steps (2)–(4) leading from TC to TEvia O/S exchange and these could be verified as thermodynamically favoured events in our previous report. Reacting TC with BO under a range of conditions always exclusively led to ester containing products and no thioesters from incomplete O/S exchange could be detected.49 In order to then study reaction step (5) from species similar to TE, we prepared the primary lithium thiolate T adjacent to a tertiary carbon centre (ESI† section S4).Monomer selection by TE: step (5)
As shown in Fig. 3, T instantaneously reacts with PTA to yield the lithium thiocarboxylate TC′ (δ [R(CO)SLi] = 216.0 ppm, = 1689.1 cm−1 and δ [R(CO)SR] = 193.7 ppm, = 1556.3 cm−1) and with CS2 generating the lithium trithiocarbonate TT (δ [RS(CS)SLi] = 244.6 ppm, = 996.5 cm−1) as confirmed by multinuclear NMR, IR and a yellow discolouration due to the CS chromophore. Since both CS2 and PTA insertion are again possible from T this raises the question whether monomer selection in this step during ROTERP occurs on a thermodynamic or kinetic basis. Again, we found CS2 insertion to be reversible. Heating TT at 80 °C under vacuum cleanly regenerates T as observed by 1H and 7Li NMR which is also visible by the disappearance of the yellow (CS) chromophore. Reversible CS2 insertion could again provide a pathway for CS2 to PTA exchange. However, in contrast to DT, this reaction does not occur cleanly for TT and results in complex product mixtures. Although some PTA ring opening is observed indicating this insertion to be thermodynamically favoured over CS2 insertion, monomer exchange pathways are unlikely to play a role during ROTERP and we thus investigated the potential for kinetic selection. Reacting T with a 10:1 mixture of CS2 to PTA, which is the monomer ratio present during ROTERP, results in 99% selective CS2 insertion yielding TT. We hence suggest the monomer selection in step (5) of Fig. 1 to occur on a kinetic basis; note that this is the opposite chemoselectivity that A shows. Surprisingly we observe ring opening of BO by T to form thioethers in d8-THF at room temperature. However only trace amounts of thioether links are formed during ROTERP and this is likely because of faster CS2 than BO insertion. Accordingly reacting T with a 2:1 mix of CS2 and BO resulted in no thioether and only heterocarbonate formation. Another aspect concerning TE is the origin of erroneous thioester links (Fig. 1) which either form from TE if it inserts PTA or if ATE propagates without O/S exchange. However, we previously found O/S exchange from TC to always occur quantitatively after BO insertion, making this thioester formation pathway unlikely.49 On the other hand, we observed that lower CS2 loadings in the starting monomer feed result in more thioester errors making PTA over CS2 insertion from TE the more likely mechanistic origin of errors. Seeking to confirm this hypothesis we conducted are series of competition experiments like those depicted in Fig. 3b in which T was combined with mixtures of 1 eq. PTA and 5 eq., 2 eq. or 1 eq. CS2 in d8-THF. In contrast to the competition experiment featuring a 1:10 PTA:CS2 ratio, we observe PTA ring opening to an increasing extent as the amount of CS2 is lowered. Hence kinetic CS2 over PTA selection by the thiolate T is not achieved at lower relative CS2 concentrations making erroneous PTA insertion by the thiolate intermediate T the likely origin of thioester errors.
 | ||
| Fig. 3 (a) Reactivity and solid-state structure of model thiolate T; 1H–13C HMBC (d8-THF, 25 °C) NMR spectra of TT and TC′; (b) 1H NMR spectrum (d8-THF, 25 °C, 400 MHz) of the reaction products of T with a 10:1 mixture of CS2 and PTA. | ||
Propagation from TT: step (6)
Closing the loop, we confirmed reaction step (6) by reacting TT with BO leading to organic heterocarbonate formation (Fig. 4b, ESI† section S5). This however leads to O/S scrambled heterocarbonate links rather than to clean formation of a trithiocarbonate appended lithium alkoxide like ATT demonstrating that O/S exchange from ATT (Fig. 4a) is thermodynamically feasible at room temperature. Accordingly, suppression of O/S exchange from ATT must be a kinetic effect which we confirmed by reaction of TT with 1 eq. BO in presence of 1 eq. PTA cleanly generating the lithium thiocarboxylate TC* (Fig. 4c). NMR and IR analysis of the obtained product clearly shows the generation of an organic trithiocarbonate (δ [RSC(S)SR] = 224.1 ppm, = 1054.0 cm−1) and an ester link (δ [RC(O)OR] = 168.5 ppm, = 1700.0 cm−1) as well as a lithium thiocarboxylate chain end (δ [RC(O)SLi] = 217.1 ppm) and tail-selectively ring-opened BO. Going one step further, all selection processes occurring in steps (1) to (5) (Fig. 1) can be coupled to each other as shown in Fig. 5. Here, we reacted T with a mixture of 10 eq. CS2, 1 eq. PTA and 1 eq. BO to likewise form the thiocarboxylate TC* while preserving the unscrambled trithiocarbonate link.
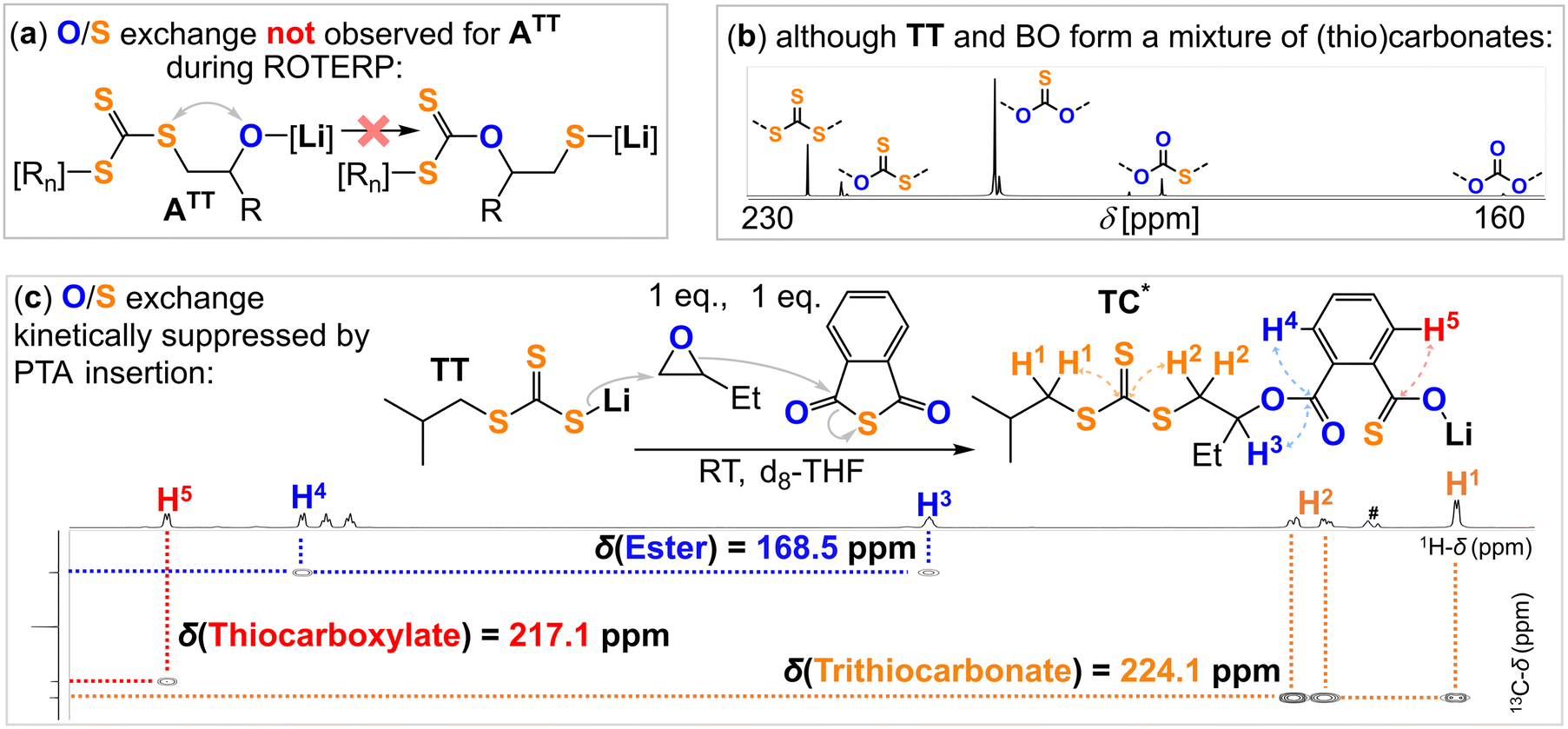 | ||
| Fig. 4 (a) Formal O/S exchange from ATT not observed during ROTERP; (b) reaction of TT with 1 eq. BO in d8-THF leads to O/S scrambled organic heterocarbonates (13C (126 MHz, d8-THF, 25 °C) NMR spectra of the reaction mixture displayed) showing that O/S exchange is thermodynamically feasible; (c) consecutive enchainment of PTA, following BO insertion by T suppresses O/S exchange; 1H–13C HMBC spectrum (d8-THF, 25 °C) and corresponding 1H and 13C NMR spectra of TC*. #Denotes residual THF and d8-THF signals. | ||
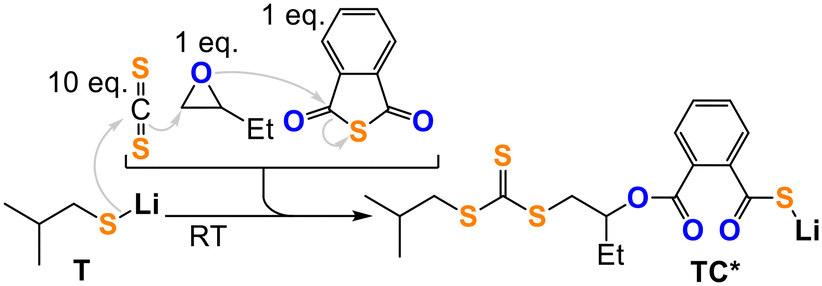 | ||
| Fig. 5 Four component cascade reaction generating TC* from T, CS2, BO and PTA. | ||
The rate determining step of ROTERP
Finally, we were wondering what the rate determining step of ROTERP is and at which type of chain end the lithium catalyst hence rests (ESI† section S6). Following the PTA consumption during ROTERP by 1H NMR of aliquots removed at regular time intervals (Fig. 6(a)) reveals a linear dependence of PTA consumption versus time. This supports a 0th order dependence of the reaction rate regarding PTA further supporting that lithium alkoxides like ATT are unlikely to be the resting states of ROTERP. We previously showed experimentally that ester appended lithium thiolates like TE are unstable due to rapid thiirane elimination and that thioester appended lithium alkoxides like ATE are unstable intermediates due to rapid O/S exchange.49 Hence there remains the question whether BO insertion from TC in step (2) or from TT in step (6) is faster. We therefore performed a competition experiment in which we reacted a 1:1 mixture of TC and TT with 1 eq. BO in d8-THF/CS2 at room temperature. Under these conditions negligible reaction of TC occurs whereas TT reacts to form organic heterocarbonates (Fig. 6(b)) by BO insertion and initiation of CS2/BO coupling. We then compared the reaction kinetics of PTA/CS2/BO ROTERP with that of the parent PTA/BO and CS2/BO ROCOP reactions which complemented our findings (Fig. 6(c)). Here we tracked the reaction progress of Li catalysed PTA/CS2/BO ROTERP (at 1 eq. LiOBn: 100 eq. PTA: 800 eq. BO: 1600 eq. CS2), CS2/BO ROCOP (at 1 eq. LiOBn: 800 eq. BO: 1600 eq. CS2) and PTA/BO ROCOP (at 1 eq. LiOBn: 100 eq. PTA: 800 eq. BO) employing an equal volume of toluene in place of CS2 to reach comparable reactant concentrations. The reaction kinetics at room temperature assessed by 1H NMR aliquot analysis as shown in Fig. 6(c) reveal that ROTERP (TOF ca. 59 h−1) is slower than CS2/BO ROCOP (ca. 234 h−1) but faster than PTA/BO ROCOP (ca. 13 h−1) under comparable reaction conditions with regards to BO turnover. This suggests that reaction steps during ROTERP like PTA/BO ROCOP (i.e. PTA insertion and BO insertion from TC) are slower than the ones like CS2/BO ROCOP (i.e. CS2 insertion and BO insertion from TT). As ROTERP is 0th order in PTA this leaves BO insertion from TC as the slowest reaction step as indicated by our prior model intermediate studies. We therefore propose that the intermediate TC is the likely resting state of ROTERP. Furthermore, the experiment outlined in Fig. 5 can be viewed as a ROTERP at high Li-initiator loading (i.e. 100 mol%) as all monomer are present which are enchained to the TC* oligomer. Hence this provides direct spectroscopic evidence for the thiocarboxylate chain-end in the resting state of the ROTERP which confirms the conclusions made in this paragraph.
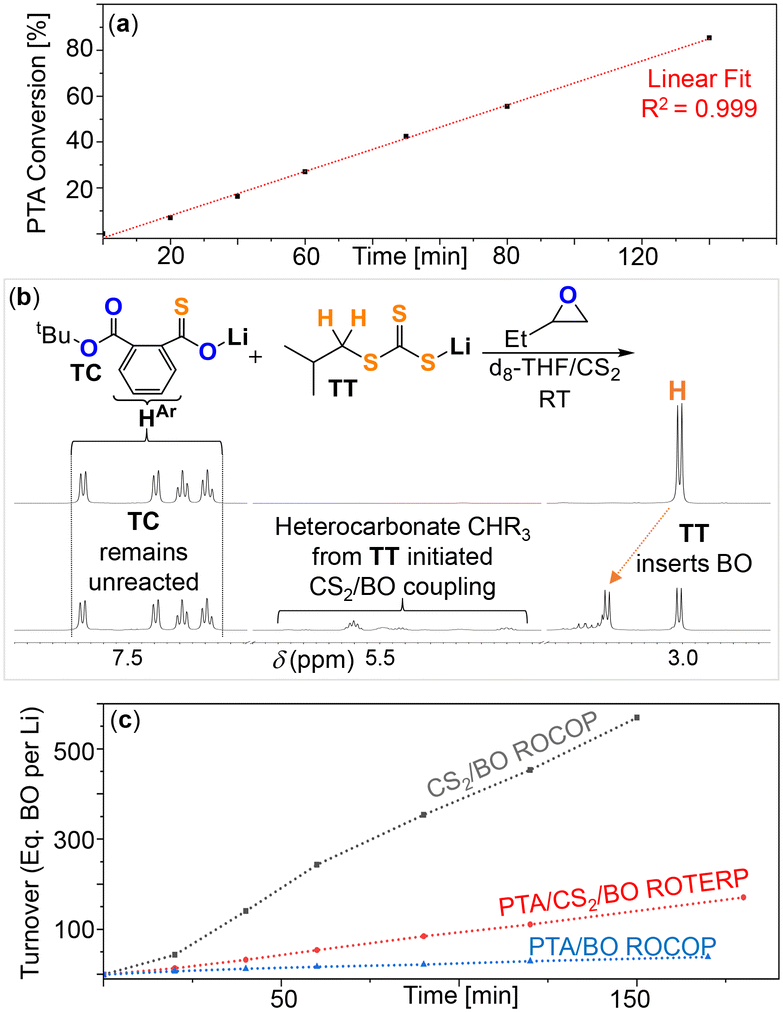 | ||
| Fig. 6 (a) Linear fit of the PTA conversion vs. time at 1 eq. LiOBn: 100 eq. PTA: 800 eq. BO: 1600 eq. CS2 at RT. (b) Competition experiment reacting a 1:1 mixture of TC and TT with 1 eq. BO at room temperature in d8-THF (overlaid 400 MHz 1H NMR shown); note that the previously reported tBu derivative of TC was employed to avoid signal overlap.49 (c) Comparison of the BO turnover versus time for ROTERP and the two parent ROCOP reactions as outlined in the main text; TOF as discussed in the main text obtained by linear fitting over the entire displayed range. | ||
Density functional theory
With a mechanistic idea of monomer incorporation selectivity, side reactions and intermediate speciation at hand we turned to DFT modelling to shed light on the catalytic cycle and the nature of transition states as well as the exact nature and role of the Li centres which is otherwise hard to elucidate (ESI† section S8). The fact that heavier alkali metals Na and K show worse or no appreciable activity/selectivity in ROTERP implies that Li acts as a true catalyst rather than a mere spectator counter cation. Quantum chemical computations were performed with the program package TURBOMOLE at a meta-hybrid DFT level of theory employing conformational searches and structure optimizations at the TPSSH/def-TZVP level (using the solvation model COSMO and assuming a dielectric constant of infinity) followed by M06-2X/def2-TZVPD single-point calculations.52–58 For the final Gibbs free energies G given here, the solvation model COSMO-RS was used assuming as solvent a 2:1 mixture (by molarity) of CS2 and PO; a temperature of 80 °C was assumed for this solvation treatment as well as for computing the statistic thermodynamics contributions to allow for an optimal comparison with our initial work.49,59 For all transition states, it was checked which combination of reactants and products they actually link together by slight distortions (corresponding to 30 K) along both directions of the imaginary vibrational mode and subsequent structural relaxation. Before entering into the discussion of our computational results, we would like to point out that identification of reaction intermediates and transition states is non-trivial due to the manifold of possible ligands for coordination to the Li centres, which can be chain-ends, functional groups of the polymer chain adjacent to the chain-ends and the monomers. With respect to monomer coordination, it could be shown that PO (as a model alkylene oxide) binds more strongly than CS2 as well as PTA, so that in the results shown in the following always PO was used to saturate available coordination sites at Li. It became also obvious that phthalic diester groups of the product are in spite of their chelating nature not superior as ligands compared to two PO, so that coordination to remote functional groups of the propagating chain or groups of other polymer chains is not expected (see ESI†). Another complication represents the tendency of lithium salts to aggregate into dimers and even more aggregated structures in solution. The most favoured intermediates and transition states from a Gibbs free energy perspective were identified assessing over 100 potential species for all of which searches for the lowest energy conformer at the level of structure optimization were performed; the reader is referred to the ESI† for a summary of the structures and their associated Gibbs free energies. Despite of these efforts, our investigations cannot deliver an exhaustive list of all possible lithium complexes involved in the ROTERP cycle, but rather deliver one possible reactive pathway which nevertheless allows to draw some general conclusions about the transition state geometries, intermediate speciation, and the role of the lithium catalyst. A summary of the computed catalytic cycle is given in Fig. 7. The starting point represents the suspected lithium thiocarboxylate resting state in its monomeric form Im for which coordination of three additional epoxides results in the most stable monomeric structure. We used thiobenzoate as a model polymer chain end and propylene oxide as model alkylene oxide throughout this study for reasons of computational simplicity. Aggregation to Id results in a slightly more stable dimeric structure with a Li2O2 core bridged by the thiocarboxylate oxygens. It should be mentioned here, that for all aggregation equilibria, lithium thiocarboxylates were inferred as reagent, as these represent the suspected chain ends in the resting state of the catalytic cycle and must therefore be primarily available in the reaction mixture. From Id, ring opening occurs through a bimetallic transition state TSI (ΔG‡ = 125.4 kJ mol−1) in which the reacting thiocarboxylate is coordinated by one Li while the inserted PO ring is coordinatively activated by the second Li centre. It should be noted that tail-selective PO ring-opening at the CH2 position is energetically most favourable as it represents the SN2 reaction at the less substituted carbon atom. Monometallic PO ring-opening transition states starting from Im result in significantly higher ΔG‡ as a consequence of the angular constraints around the attacked C-atom at which in SN2 reactions nucleophile and leaving group optimally assume an angle of 180°. Further bimetallic transition states in which two monomeric units are not linked by an anionic bridging ligand are likewise less favoured as a consequence of the fact that either reactant or product have to be of zwitterionic nature. TSI leads to a dimeric intermediate IId with the thiocarboxylate bridging through the O and S centres (generally, μ2- or κ2-bridging by either O only or O and S, respectively, was found to be energetically quite close for thiocarboxylate links between two Li) as well as a bridging alkoxide. Notably the carbonyl oxygen of the adjacent thioester link coordinates in this most stable form of IId to one of the Li centres and this already hints the subsequent activation of the thioester group for nucleophilic attack in the O/S exchange step. Intramolecular attack (ΔG‡ = 48.4 kJ mol−1) of the thioester by the alkoxide viaTSII transforms the alkoxide into a thiolate and this exchange step is orders of magnitude-faster than the PO ring-opening before (and expected to be also faster than any reaction with CS2 or PTA, when comparing the analogous differences in Gibbs free energies between Vd and transition states TSV or TSV*). TSII actually represents the initial alkoxide attack to form as primary product a tetrahedral intermediate in which the two O-atoms as in the transition state are linked to Li; however, any attempt of optimizing analogous species with the negatively charged O and S coordinated to Li – as a prerequisite for thiolate formation – led to spontaneous C–S bond cleavage, so that it can be concluded that this carbonyl addition–elimination reaction is limited by the addition step and that elimination is fast and does not require more than dissociation of a neutral donor atom from Li and coordination of another. Notably the thiolate IIId is 24.4 kJ mol−1 more stable than its alkoxide counterpart IId demonstrating that O/S exchange at the chain-end is a thermodynamically favourable process. Monometallic O/S exchange transition states starting from monomeric lithium alkoxides lead to significantly higher barriers, which fits to the general observation that the strongly coordinating alkoxide anion has a high preference for dinuclear species which is much less pronounced for less coordinating anions like thiolate or anions with negative charge delocalization like thiocarboxylates or trithiocarbonates. Moreover, transition states similar to TSII for which the thiocarboxylate spectator ligand acts as a μ2-bridge via O only (instead of κ2-bridging via O and S) also lie energetically higher, which results from the fact that the corresponding structures contain too many small rings annulated with each other, showing that a modulation of the Li⋯Li separation through different bridging modes of the thiocarboxylates plays a role in the ROTERP mechanism. From IIId CS2 insertion has to occur in order to continue propagation, however in this case bimetallic transition states are less favoured than monometallic ones. We infer that this is partially due to steric encumbrance of the thiolates by the two flanking Li centres which also withdraw electron density from the nucleophilic S. In order to achieve monometallic CS2 insertion, fragmentation of the dimers IIId into their monomeric form IIIm under coordination of an additional equivalent of PO per Li occurs, overall requiring 21.7 kJ mol−1. The four-membered transition state TSIII (ΔG‡ = 45.1 kJ mol−1 with respect to IIIm) that leads to CS2 insertion also lies from a free energy perspective lower than the prior PO ring-opening. Importantly CS2 insertion is only associated with a slightly lower barrier than PTA insertion (TSIII*, ΔG‡ = 46.5 kJ mol−1) confirming that although CS2 insertion is kinetically favoured, monomer selection by the lithium
thiolate chain end is also concentration driven and requires a large excess of CS2 as found in our experimental studies in order to achieve highly regular product structures. These findings also explain that erroneous PTA insertion from lithium thiolates viaTSIII* cause the aforementioned thioester errors. Progressing in the catalytic cycle, TSIII leads to the lithium trithiocarbonate IVm which then undergoes the thermodynamically slightly favourable dimerization to IVd with a thiocarboxylate Im. Surprisingly from here the energetically most favourable PO ring-opening pathway involves initial dissociation of the trithiocarbonate ligand under additional PO coordination to form the zwitterionic intermediate IVz (the given free energy refers to then solvent separated ion pair) which lies energetically uphill by 87.7 kJ mol−1 compared to IVd. This ligand exchange at Li effectively swaps a μ2-S for a μ2-O donor and could be facilitated by the high oxophilicity of Li. PO ring-opening in TSIV from here occurs with a very small additional activation barrier (ΔG‡ = 24.4 kJ mol−1) so that the overall barrier for PO ring-opening starting from IVd that needs to be overcome amounts to 112.1 kJ mol−1. PO ring-opening by the lithium trithiocarbonate from IVd to Vd is therefore predicted to occur more easily by 13.3 kJ mol−1 than PO ring-opening by lithium thiocarboxylates, confirming TSI to be the rate-determining step of the reaction. The zwitterionic nature of the most favourable TSIV can be understood as a consequence of the generally weaker binding between Li and trithiocarbonate anions compared to thiocarboxylates (as can be seen in the ESI by comparing the speciation of I and IV). From here, several possible propagation pathways with PTA are possible; in case of the monometallic ones some of the most favourable ones are actually barrierless, i.e. are spontaneous reactions once the corresponding monometallic precursor with a free coordination site has been generated by the energetically uphill (ΔG = 40.0 kJ mol−1) dissociation of dimers Vd into monomers Vm (see ESI† for more details) and this is in line with the experimentally determined 0th reaction order in PTA concentration; in cases with more steric crowding around Li, PTA inserts in a four-membered transition state TSV, which however does not seem the most favoured option. In contrast, CS2 insertion occurs most easily starting directly from Vd with an insertion barrier to TSV* of ΔG‡ = 69.2 kJ mol−1 confirming that PTA insertion is in fact kinetically favoured from this alkoxide intermediate that does – because of the more different barriers involved – less depend on monomer concentration than the competition of TSIII and TSIII*. One final question concerns the circumstance why no O/S exchange occurs from Vm (corresponding to ATT in Fig. 1). In this regard a cyclised variant Vc observed without explicitly searching for it upon a conformational scan of Vm suggests that cyclization initiating the O/S exchange process is a possible (and obviously reversible) process that occurs with a negligible barrier. In two cases it was found, that releasing a dialkyl dithiocarbonate and a thiolate chain end from this is actually slightly exergonic with respect to dialkyl trithiocarbonate and alkoxide chain end. However, from the activation barriers computed for TSIII, one would expect the corresponding transition states of CS2 addition around 20 kJ mol−1 higher (see ESI† for V) than the most favourable TSV. As PTA insertion is irreversible (vide supra), any Vm is thus in the presence of PTA rapidly scavenged which suppresses any O/S scrambling as predicted by our model intermediate studies.
 | ||
| Fig. 7 Energy profile for the ROTERP propagation cycle computed on the M06-2X/def2-TZVPD‖TPSSH/def-TZVP/COSMO(ε = ∞) level of theory. | ||
Conclusions
In conclusion, we have presented a thorough experimental verification of the intermediate speciation, the propagation events, and the side processes of the ROTERP reaction mechanism. We found that although most insertions and side processes are thermodynamically feasible a series of kinetic selection processes at the propagating chain-end determine the ROTERP sequence selectivity. Nevertheless, as CS2 insertion is reversible, erroneous insertions can be corrected at elevated temperatures. Although O/S scrambling pathways are thermodynamically favoured for alkoxide chain ends adjacent to sulfurated links, these are kinetically suppressed for alkoxides adjacent to trithiocarbonate links by propagation. Furthermore, we identified epoxide ring opening following PTA insertion as the rate-determining step. Our computational investigations helped to elucidate the special role of the Li catalyst. ROTERP is enabled by fluxional aggregation at the chain end allowing for mono and multimetallic insertion steps and is furthermore facilitated by a certain degree of plasticity of the intermetallic distance through different bridging ligands and adaptable bridging modes of those. The high oxophilicity of lithium leads to coordinative activation of carbonyl groups adjacent to the chain-end as well as formation of zwitterionic intermediates enabling the associated transition states. All these factors collaboratively enable the polymerisation process which has been oversimplified in the past. Our results demonstrate how exactly sustainable main-group metals enable the selective catalytic synthesis of degradable heteroatom-containing polymers and provide guidance for future catalyst selection and unprecedented polymer sequence and architecture design.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The VCI is acknowledged for a Liebig Fellowship for A. J. P. and a Ph.D. scholarship for C. G. Prof. Dr Christian Müller and Prof. Dr Rainer Haag are thanked for continuous support and valuable discussions.References
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- C. Hu, X. Pang and X. Chen, Macromolecules, 2022, 55, 1879–1893 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, e202104495 CAS.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, C. Lu, H. Qi, L. Yang, Y. Wang and G.-P. Wu, Nat. Synth., 2022, 1–10 Search PubMed.
- Y. Popowski, Y. Lu, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2022, 144, 8362–8370 CrossRef CAS PubMed.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
- Y. Yu, B. Gao, Y. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2022, e202204492 CAS.
- A. J. Plajer and C. K. Williams, ACS Catal., 2021, 11, 14819–14828 CrossRef CAS.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- J. Xu and N. Hadjichristidis, Angew. Chem., Int. Ed., 2021, 60, 6949–6954 CrossRef CAS PubMed.
- X. Xia, R. Suzuki, K. Takojima, D.-H. Jiang, T. Isono and T. Satoh, ACS Catal., 2021, 11, 5999–6009 CrossRef CAS.
- X. Xia, R. Suzuki, T. Gao, T. Isono and T. Satoh, Nat. Commun., 2022, 13, 163 CrossRef CAS PubMed.
- X. Xia, T. Gao, F. Li, R. Suzuki, T. Isono and T. Satoh, J. Am. Chem. Soc., 2022, 144, 17905–17915 CrossRef CAS PubMed.
- Y. Reddi and C. J. Cramer, ACS Catal., 2021, 11, 15244–15251 CrossRef CAS.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- L. Song, M. Liu, D. You, W. Wei and H. Xiong, Macromolecules, 2021, 54, 10529–10536 CrossRef CAS.
- D. Zhang, H. Zhang, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2016, 49, 2484–2492 CrossRef CAS.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS PubMed.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- Y. Liu, J.-Z. Guo, H.-W. Lu, H.-B. Wang and X.-B. Lu, Macromolecules, 2018, 51, 771–778 CrossRef CAS.
- Z. Yang, C. Hu, F. Cui, X. Pang, Y. Huang, Y. Zhou and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117533 CAS.
- J. Xu, X. Wang and N. Hadjichristidis, Nat. Commun., 2021, 12, 7124 CrossRef CAS PubMed.
- G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC.
- J. Tang, M. Li, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, e202115465 CAS.
- X. Wang and R. Tong, J. Am. Chem. Soc., 2022, 144, 20687–20698 CrossRef CAS PubMed.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., Int. Ed., 2022, e202201785 CAS.
- Y. Liu, H. Zhou, J.-Z. Guo, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed.
- J.-L. Yang, H.-L. Wu, Y. Li, X.-H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., 2019, 131, 628–633 CrossRef.
- W.-M. Ren, J.-Y. Chao, T.-J. Yue, B.-H. Ren, G.-G. Gu and X.-B. Lu, Angew. Chem., Int. Ed., 2022, 61, e2021159 Search PubMed.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- Y. Wang, Y. Zhu, W. Lv, X. Wang and Y. Tao, J. Am. Chem. Soc., 2023, 145(3), 1877–1885 CrossRef CAS PubMed.
- T.-J. Yue, G. A. Bhat, W.-J. Zhang, W.-M. Ren, X.-B. Lu and D. J. Darensbourg, Angew. Chem., Int. Ed., 2020, 59, 13633–13637 CrossRef CAS PubMed.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 618–623 CrossRef CAS PubMed.
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, e202115189 CAS.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Macromolecules, 2019, 52, 2439–2445 CrossRef CAS.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- D. Silbernagl, H. Sturm and A. J. Plajer, Polym. Chem., 2022, 13, 3981–3985 RSC.
- A. J. Plajer, ChemCatChem, 2022, 14, e2022008 CrossRef.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, WIREs Comput. Mol. Sci., 2014, 4, 91 CrossRef CAS.
- J. Tao, J. Perdew, V. Staroverov and G. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. Staroverov, G. Scuseria, J. Tao and J. Perdew, J. Chem. Phys., 2003, 119, 12129 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2236905 and 2236906. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00301a |
| This journal is © The Royal Society of Chemistry 2023 |