
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure sensitivity of alumina- and zeolite-supported platinum ammonia slip catalysts†
Vasyl
Marchuk
 a,
Xiaohui
Huang
bc,
Jan-Dierk
Grunwaldt
ad and
Dmitry E.
Doronkin
*ad
a,
Xiaohui
Huang
bc,
Jan-Dierk
Grunwaldt
ad and
Dmitry E.
Doronkin
*ad
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology, Engesserstr. 20, 76131, Karlsruhe, Germany. E-mail: dmitry.doronkin@kit.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
cDepartment of Materials and Earth Sciences, Technische Universität Darmstadt, Alarich-Weiss-Straße 2, 64287, Darmstadt, Germany
dInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
First published on 6th March 2023
Abstract
The influence of the size and structure of Pt particles on the catalytic performance in selective ammonia oxidation for emission control applications is hardly understood. There is a lack of in situ structural characterization to explain the catalyst performance under atmospheric pressure and relevant reactant ratios. In this study we complemented conventional laboratory tests with operando X-ray absorption spectroscopy (XAS) to determine the activity- and selectivity-governing factors in ammonia slip catalysts with different Pt particle sizes supported on γ-alumina and a ZSM-5 zeolite. A previously reported narrow activity range of platinum catalysts was extended by using catalysts with atomically and sub-nanometre dispersed Pt. The increase in activity with particle size was mainly caused by the availability of favourable Pt ensembles on the surface, probably B5 sites. Below the required size for the existence of these ensembles (∼2 nm) Pt possessed poor activity for ammonia oxidation, while upon reaching it, the activity drastically rose and was further moderately dependent on particle size. Spectroscopic data revealed the same reaction mechanism for particles ≥∼2 nm. It included initial reduction during heating in the reaction mixture and subsequent re-oxidation at high temperatures. For Pt single sites/small clusters, the mechanism was different and involved only gradual reduction without further re-oxidation. The individual spectral components for the two types of mechanisms were resolved. Their evolution correlated with catalyst activity and selectivity change. While ammonia oxidation proceeded through the Ostwald mechanism on active catalysts with sufficiently large particles, single or low-coordinated sites were rather likely to catalyse selective catalytic reduction.
Introduction
Ammonia is regarded as a potential carbon-free energy carrier of the future.1 It has high energy density, can be easily liquefied and transported through a well-developed infrastructure, which can make it an important component of transition to sustainable economy.2–4 To regain the energy stored in ammonia, it can be split to hydrogen, used in fuel cells or directly combusted.4,5 During these processes, ammonia slip will inevitably occur. Since it is a toxic and environmentally harmful gas, ammonia abatement is required. An effective way to accomplish this is using ammonia slip catalysts (ASCs) that selectively oxidise ammonia to harmless nitrogen and water.6,7Ammonia slip catalysts are also widely used in the automotive industry to eliminate residual ammonia that is overstoichiometrically dosed for selective catalytic reduction (SCR) of NOx that are formed during fuel combustion.8,9 ASCs usually combine oxidation functionality, typically a supported Pt catalyst, with SCR functionally, often provided by zeolite-supported Fe or Cu.8,10 Due to rising noble metal prices, it is important to optimise the content of platinum in ASCs, increasing its activity while decreasing the loading. Another issue of platinum catalysts is that, despite sufficient activity at low temperatures (<200 °C), they demonstrate low selectivity to nitrogen at increased temperatures (>300 °C) producing high amounts of N2O or NOx.11–13 To solve these problems and to improve the performance of ASCs, understanding the structure of a catalyst and its relation to activity and selectivity is required.
Many studies that detect catalyst structure and directly correlate it with its performance apply model reaction conditions, such as vacuum or unsupported Pt in the form of single crystals, foil or wires.14–20 On the other hand, studies of more realistic systems with supported Pt under atmospheric pressure were conducted.13,18,21,22 These studies mostly characterise the state of Pt either before or after the reaction. It is necessary to probe the catalyst also during operation since its state might drastically vary under reaction conditions.23,24
A number of studies addressed the topic of the structure sensitivity of ammonia oxidation inspecting ASCs with different particle sizes. This approach is well-established in unravelling structure–activity relationships, since particles of different sizes often expose different groups of surface atoms with varying coordination and oxidation states and thus with different activities25 For example, earlier studies by Ostermaier et al.26,27 and van den Broek28,29 have already reported an increase of the reaction rate with growth of Pt particle size. However, the conditions applied in these studies did not completely correspond to the conditions present in emission control systems. In particular, Ostermaier et al. used a narrow temperature range (120–200 °C). In the case of van den Broek, too low O2 partial pressures were used. These points were taken into account in the following studies, and the size effect in ammonia oxidation was thoroughly examined for a Pt nanoparticle size range from 1 to 23 nm (ref. 30) and even for a size range of 1.3–200 nm.31 Interestingly, in the latter case, despite the huge size range studied and a realistic gas feed, only a moderate size effect on the activity was observed. The variation of the temperature of 50% conversion (T50%) did not exceed ∼35 °C. Hence, it is important to study the factors which could extend this activity window. According to the present literature, there is only a modest possibility to further lower Pt light-off temperatures by solely increasing its particle size.31 Moreover, it is unclear to which extent the light-off temperatures can rise for extremely dispersed Pt. This requires especially particle sizes smaller than 1 nm, which to date have hardly been studied for ASCs.
For understanding the Pt particle size dependence on ammonia oxidation, several factors need to be considered. To explain the structure sensitivity, many experimental studies focus on the varying reducibility of different Pt sites.26,27,30–34 Various studies conclude that a reduced metallic platinum surface is a prerequisite for high ammonia oxidation activity.26,27,30,32–34 At the same time, oxygen more strongly binds to step and edge Pt sites, which are more abundant on smaller particles.31,35 As a result, smaller particles are less active for ammonia oxidation. Other studies, many of which are of theoretical nature, claim that ammonia activation requires a favourable ensemble of surface Pt atoms or the presence of terrace steps for an efficient reaction.14,16,36 The presence of adsorbed oxygen on the Pt surface facilitates the activation even more.14,32,36 This is characteristic of the Ostwald process mechanism that includes stepwise hydrogen subtraction from ammonia by oxygen (O-assisted ammonia dissociation).16 A certain size of particles related to the specific Pt surface is required for these favourable conditions, typically reported to be ∼2 nm.36 In this case, the reaction rate should decline with particle size decrease with an abrupt drop at a point when the Pt particle surface is too small to host the adsorbates and intermediates of O-assisted ammonia dissociation. Despite being in line with the observed trends, concrete experimental evidence under conditions close to real applications of ASCs (i.e. atmospheric pressure, relevant NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 ratio, presence of water) is missing. Furthermore, literature reports of the size effect in ammonia oxidation are lacking operando spectroscopic data on the catalyst structure. Applying them would allow the catalyst performance to be directly correlated with structural changes of platinum catalysts with different Pt particle sizes. This would promote catalyst development based on rational design rather than on a trial and error approach.
:O2 ratio, presence of water) is missing. Furthermore, literature reports of the size effect in ammonia oxidation are lacking operando spectroscopic data on the catalyst structure. Applying them would allow the catalyst performance to be directly correlated with structural changes of platinum catalysts with different Pt particle sizes. This would promote catalyst development based on rational design rather than on a trial and error approach.
This work aims at studying factors causing Pt structure sensitivity in the selective ammonia oxidation reaction by applying operando XAS. This X-ray technique allows us to track the structure while determining the performance simultaneously at pressures and reactant concentrations close to real applications.24,37–39 In addition to a conventional catalyst series on γ-Al2O3, another series was based on ZSM-5 zeolite. Such a comparison has been reported earlier, for example, by Li and Armor22 and van den Broek.29 However, these studies did not show if the support influence remains constant with varying particle sizes of Pt. Additionally, it is unclear how big the difference is between the impacts of the support and particle size or how the presence of water might affect the structure–activity relationship. Furthermore, particular attention in this study has been devoted to the catalyst series exhibiting a broader range of reaction rates compared to earlier literature reports. This has been attempted by decreasing Pt particle size to values lower than required for the ammonia activation step of the Ostwald process. In this regard, the roles of the Pt oxidation state and the minimal surface necessary for ammonia activation have been examined.
Experimental
Catalyst preparation
Three 2 wt% Pt/Al2O3 samples were synthesised through incipient wetness impregnation (IWI). A series of samples with varying Pt particle sizes was prepared by means of thermal treatment of an initial catalyst in air flow.The PtA-IW sample was the initial catalyst from which the other two in the series were obtained. For its preparation, γ-alumina (SASOL SCFa-230, pre-calcined at 750 °C for 4 h in air) was impregnated with a solution of Pt(II) nitrate (Chempur, anhydrous, 99.95%), dried under air in a fume cupboard for 40 h at room temperature and then for 1 h at 60 °C in static air. The impregnated and dried sample was then calcined at 400 °C (ramp rate 3 °C min−1) for 4 h in static air. After calcination, the sample was reduced at 400 °C for 2 h in hydrogen flow (5% H2, N2 balance, temperature ramp rate 3 °C min−1).
To obtain alumina-supported catalysts with varying particle size, parts of PtA-IW were calcined in air flow for 4 h at 500 °C and 700 °C (ramp rate 5 °C min−1 in both cases) resulting in samples PtA-IW-500 and PtA-IW-700, respectively.
Additionally, three catalysts on zeolite support were synthesised. For this NH4-ZSM-5 zeolite was used (Si/Al = 11, Clariant). The platinum loading for the zeolite-supported series constituted 1 wt%. Before preparation, the support was dried at 150 °C for 16 h in static air.
PtZ-IW was synthesised by incipient wetness impregnation of the zeolite using platinum(II) nitrate, anhydrous (Chempur), as a precursor. The impregnated support was dried in air flow at room temperature for 16 h and then additionally at 80 °C for 4 h. The sample was calcined at 500 °C (heating rate – 4 °C min−1) in static air for 2 h.
The PtZ-IE sample was prepared via ion exchange (IE) following techniques described in the literature.40,41 For this the required amount of [Pt(NH3)2]Cl2 (Aldrich, 98%) for obtaining 1% Pt loading on 3 g of the support (51 mg of the precursor) was dissolved in 600 ml of deionized water. Ion exchange was conducted with magnetic stirring at room temperature in ambient atmosphere for 24 h. After that the sample was filtered and rinsed with 800 ml of distilled water. The filtered residue was dried in air flow at room temperature for 16 h and then for 3.5 h at 80 °C. The dried sample was calcined at 450 °C in air flow for 2 h. The temperature was reached with a slow ramp rate of 0.5 °C min−1.
PtZ-IE-R was prepared using the same procedure as PtZ-IE, but with an additional reduction step. The reduction was conducted in 5% H2 (N2 balance) at 400 °C (heating rate 5 °C min−1) for 30 min. Pt concentration in PtZ-IE and PtZ-IE-R samples was estimated by the edge jump in the X-ray absorption spectra using the impregnated PtZ-IW sample as a reference. The estimated Pt concentration in the ion-exchange samples was 0.9%. Standard deviation was evaluated on three different zeolite-supported samples obtained through incipient wetness impregnation and constituted 0.17 abs%. The prepared samples, their abbreviations and their synthesis conditions are summarised in Table 1.
| Sample name | Preparation method | Precursor | Preparation conditions |
|---|---|---|---|
| 2 wt% Pt/γ-Al2O3 | |||
| PtA-IW | IWI | Pt(NO3)2 | Calcined at 400 °C, then reduced in 5% H2 at 400 °C |
| PtA-IW-500 | PtA-IW, additionally calcined at 500 °C in air flow | ||
| PtA-IW-700 | PtA-IW, additionally calcined at 700 °C in air flow | ||
| 1 wt% Pt/ZSM-5 | |||
|---|---|---|---|
| PtZ-IE | IE | [Pt(NH3)2]Cl2 | Calcined at 450 °C in air flow |
| PtZ-IE-R | IE | [Pt(NH3)2]Cl2 | As for PtZ-IE, additionally reduced at 400 °C in 5% H2 |
| PtZ-IW | IWI | Pt(NO3)2 | Calcined at 500 °C |
TEM
Powder samples of the synthesised catalysts were directly dispersed on copper grids coated with lacey carbon film (Quantifoil) without use of solvents. High angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) imaging was performed on a Themis300 transmission electron microscope (ThermoFisher Scientific) equipped with a probe aberration corrector, operating at 300 kV. TEM images were analysed with ImageJ software to estimate particle size.42Catalytic experiments
The catalysts were tested in a plug-flow quartz reactor in a temperature-programmed mode. Three cycles of heating from 50 °C to 400 °C and cooling to 50 °C (ramp rate 3 °C min−1, 10 min dwell times) were used. A reaction feed of 500 ppm NH3, 13% O2 in N2 was used in the first two cycles. In the third cycle, the same NH3 and O2 concentrations with additional 5% H2O and 10% CO2 balanced with N2 were applied. The first cycle served as catalyst pre-treatment, and the results of the second catalytic cycle are compared in this study for general activity and selectivity discussion. The results of the third catalytic cycle in a wet stream are discussed separately in the end of the catalytic results section to show the effect of water on activity. For alumina-supported catalysts with 2 wt% Pt loading, 25 mg of sample was taken. In the case of 1 wt% zeolite-supported catalysts, 50 mg of sample was used in order to maintain the same weight Pt loading in the reactor. Total gas flow constituted 1050 cm3 min−1. The sieved fraction of catalysts with grain size of 100–200 μm was diluted with silicon carbide (the same sieve fraction) to obtain the catalyst bed volume corresponding to a gas hourly space velocity of 63000 cm−1. The composition of the gas mixture at the outlet of the reactor was analysed on-line using a Multigas 2030 FTIR spectrometer (MKS Instruments, USA). Conversion of ammonia (XNH3) was calculated according to  , where
, where 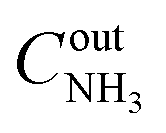 and
and  are ammonia concentrations at the reactor outlet and inlet, respectively. Selectivity to products was calculated according to the formula
are ammonia concentrations at the reactor outlet and inlet, respectively. Selectivity to products was calculated according to the formula  , where Ci is product i concentration, and ni – number of nitrogen atoms in the corresponding product molecule. The nitrogen concentration was calculated from NH3 conversion and concentrations of other products based on the mass balance.
, where Ci is product i concentration, and ni – number of nitrogen atoms in the corresponding product molecule. The nitrogen concentration was calculated from NH3 conversion and concentrations of other products based on the mass balance.
Kinetic analysis
Reaction orders of ammonia and oxygen were estimated in ammonia oxidation over alumina-supported Pt catalysts. For this purpose, integral reaction rates were measured as functions of concentrations of one of the reactants at constant temperatures. A reaction rate was calculated as an amount of converted ammonia (in mmol) per minute over a gram of Pt under certain conditions. The used catalyst loadings amounted to 25 mg. The same setup and gas flow were employed as for activity and selectivity tests. The data used in the calculations were recorded after the establishment of equilibrium, which took 30–120 min.Ex situ and operando X-ray absorption spectroscopy
To measure ex situ and operando X-ray absorption spectra (XAS), the catalysts were loaded in 1.5 mm-diameter quartz capillaries (0.02 mm wall thickness) as 100–200 μm sieved powders with the catalyst bed length of approximately 5 mm. In all cases, spectra at the Pt L3 absorption edge were recorded in transmission mode. For energy shift correction, a platinum foil spectrum was recorded simultaneously.Ex situ extended X-ray absorption fine structure (EXAFS) spectra of the alumina-supported samples were measured at the CAT-ACT beamline at the KIT light source (Karlsruhe, Germany).43Ex situ EXAFS spectra of the zeolite-supported samples were recorded at the P64 beamline of the PETRA III synchrotron radiation source (DESY, Hamburg, Germany). In both cases, the X-ray energy was selected using Si(111) monochromators with a pair of Si mirrors to eliminate higher harmonics. Energy correction and normalisation were conducted in Athena software from the IFEFFIT package.44
Next, the background-subtracted and k2-weighted spectra were Fourier-transformed (k-range of 2–10 Å−1. and 2.5–10.5 Å−1 for the alumina and zeolite based samples, respectively). The amplitude reduction factors were received by fitting a Pt foil (structural model from the Inorganic Crystal Structure Database, (ICSD, CC 243678)). They amounted to S02 = 0.68 and S02 = 0.78 for the alumina and zeolite supported samples (measured at the KIT light source and PETRA III), respectively. Fitting of EXAFS spectra was conducted in order to refine coordination numbers, interatomic distances, energy shift (δE0) and mean square deviation of interatomic distances (σ2). The misfit between the calculated and measured spectra is expressed by ρ. The refining was conducted on k1, k2, and k3-weighted data by a least squares method in Artemis software from the IFEFFIT package.44 Structural models for the fitting were based on metallic Pt and PtO2 structures, ICSD CCs 243678 and 1008935. The zeolite structure model (MFI type) that was used for fitting was described by Meier et al.45 Coordinates of cationic positions in the MFI zeolite structure were obtained from Mentzen et al.46
Operando quick-scanning extended X-ray absorption fine structure (QEXAFS) spectra were measured at the P64 beamline of the PETRA III synchrotron radiation source (DESY, Hamburg, Germany) using a continuously oscillating Si(111) channel-cut monochromator and the Si mirrors described above. Scanning speed (up and down energy scans) constituted 1 Hz. Energy correction and normalisation for the Pt foil reference were conducted in Athena software from the IFEFFIT package.44 Energy correction and normalisation for the spectra of the samples were conducted in ProQEXAFS software, v. 2.42.47 The spectral components of the recorded QEXAFS spectra were extracted using multivariate curve resolution (MCR) as implemented in the SIMPLISMA algorithm built in the ProQEXAFS software.47 Later on, these components were used to conduct linear combination analysis (LCA) of the spectra in the same program.
The in situ setup is similar to the previously described in ref. 48. Gas composition during the operando XAS experiments on Al2O3-supported samples was 500 ppm NH3, 10 vol% O2 balanced with He and N2 mixture. Gas flow constituted 75 cm3 min−1. Gas composition for the ZSM-5-supported samples was 890 ppm NH3, 10 vol% O2 balanced with He and N2 mixture. This mixture was fed with a flow rate of 70 cm3 min−1. For both series of samples, the temperature was increased from 50 °C to 400 °C with a heating rate of 5 °C min−1. Before the operando measurements, an additional heating and cooling cycle in this temperature range with the same heating rate was conducted to pre-treat the samples. For calculation of ammonia conversion and product selectivities, the same approach was used as in the catalytic experiments. A detailed explanation is given on p. S1 of the ESI.†
In situ DRIFTS
In situ diffuse reflection infrared Fourier transform spectra (DRIFTS) were measured using a VERTEX 70 Fourier transform infrared spectrometer (Bruker) with a Praying Mantis diffuse reflection optics (Harrick) and a mercury cadmium telluride (MCT) detector. 60 mg of catalyst samples with 100–200 μm grain size fraction were measured in the Harrick high temperature cell covered with a flat CaF2 window.A gas flow of 200 cm3 min−1 passed downwards through the catalyst bed. Samples were heated in Ar to 300 °C and kept at this temperature for 20 min. Then they were cooled to room temperature and flushed for 20 min. After this spectra in Ar were recorded and used as background measurements. Then the samples were exposed to 1% CO in Ar at room temperature for 30 min and subsequently flushed with pure Ar for 30 min. For the PtZ-IE sample, an additional treatment in 0.1% CO flow (balanced with Ar) for 30 min was conducted at room temperature between the treatment with 1% CO and pure Ar flushing. DRIFTS spectra were recorded for all the samples at the end of each time interval after the gas environment change.
Results and discussion
Sample characterisation
| PtZ-IW fits | Scattering atoms | R (Å) | CN | σ 2 (10−3 Å2) | δE 0 (eV) | ρ (%) |
|---|---|---|---|---|---|---|
| 2 wt% Pt/γ-Al2O3 | ||||||
| PtA-IW | Pt–O | 2.03 ± 0.11 | 3.1 ± 0.6 | 5.7 ± 4.4 | 14.1 ± 2.2 | 3.4 |
| PtA-IW-500 | Pt–Pt | 2.76 ± 0.03 | 4.3 ± 1.7 | 1.5 ± 4.8 | 9.0 ± 2.5 | 2.7 |
| Pt–O | 1.99 ± 0.07 | 2.8 ± 0.5 | 0.1 ± 4.3 | |||
| PtA-IW-700 | Pt–Pt | 2.75 ± 0.04 | 11.5 ± 2.2 | 4.7 ± 1.9 | 11.5 ± 2.2 | 0.7 |
| Pt–O | 1.95 ± 0.03 | 0.5 ± 0.3 | ||||
| 1 wt% Pt/ZSM-5 | ||||||
|---|---|---|---|---|---|---|
| PtZ-IW | Pt–Pt | 2.75 ± 0.04 | 0.9 ± 0.7 | 2.2 ± 2.1 | 9.1 ± 1.6 | 2.0 |
| Pt–O | 1.99 ± 0.01 | 3.7 ± 0.6 | ||||
| PtZ-IE | Pt–Si | 2.73 ± 0.05 | 1.1 ± 0.7 | 3.1 ± 2.6 | 12.7 ± 1.8 | 2.3 |
| Pt–O | 2.02 ± 0.02 | 4.4 ± 0.8 | ||||
| PtZ-IE-R | Pt–Si | 2.77 ± 0.07 | 0.4 ± 0.4 | 0.8 ± 2.5 | 9.5 ± 2.5 | 9.7 |
| Pt–O | 1.99 ± 0.02 | 1.6 ± 0.4 | ||||
The results of EXAFS spectra fitting of the zeolite-supported samples are presented in Table 2, and the fits in Fig. S5–S7.† The spectra of the incipient-wetness-obtained PtZ-IW sample most reliably show platinum in the second coordination sphere (Table 2, Fig. S5†). Its average coordination number of 0.9 corresponds to very small clusters.49
The other two samples, which were synthesised using ion exchange, exhibit the most credible fitting results for the models including O and Si from zeolite as backscatterers. On the contrary, the fits including O and Pt from Pt0 result in unrealistic negative coordination numbers of Pt. A fitting attempt with the model including Pt atoms from PtO2, instead of metallic Pt, yields a similar result for PtZ-IE-R. For PtZ-IE, the coordination number of Pt from the oxide structure is positive but has too high uncertainty. Additionally, this fitting model shows a somewhat higher misfit compared to the model including O and Si as backscatterers. Thus, the spectra of the ion-exchanged samples correspond to a highly amorphous state which lacks pronounced scattering from defined Pt–Pt distances. A significant fraction of the spectra originates from Pt–O and Pt–Si contributions, typical for atomically dispersed Pt. However, the highly amorphous nature does not exclude other dispersed oxidised Pt fractions with low structural order.
Nevertheless, X-ray absorption spectroscopy is a bulk technique that gives an overview over the averaged state of a sample. If a sample is not uniform, for example several fractions of different-sized particles exist, this averaged picture may not be a true representation of any of them. To evaluate the presence of different size fractions and their contributions, EXAFS analysis was complemented with TEM and in situ DRIFTS with chemisorbed CO.
 | ||
| Fig. 1 HAADF-STEM images of the synthesized catalysts. Alumina-supported samples: A – PtA-IW, B – PtA-IW-500, C – PtA-IW-700. ZSM-5-supported samples: D – PtZ-IE, E – PtZ-IE-R, F – PtZ-IW. The insets in each image display size distributions of measurable particles for the respective samples. | ||
The PtA-IW-700 sample significantly differs from the other two. While some clusters and individual small nanoparticles are observed (see Fig. S11†), the main contribution is made by large polydisperse Pt nanoparticles with an average diameter of 13.6 nm.
As for the impregnated zeolite samples, the ones obtained with ion exchange show the most dispersed Pt species. PtZ-IE contains a great abundance of Pt clusters, maybe even single sites and only a minor fraction of small nanoparticles (>1.2 nm). Some atomically distributed Pt sites in this sample are shown in more detail in a magnified image in Fig. S11.† The reduced catalyst PtZ-IE-R displays fewer clusters and hardly any individual single sites compared to the PtZ-IE. Instead, numerous nanoparticles with an average diameter of 1.4 nm are present in the TEM images.
PtZ-IW, obtained with incipient wetness impregnation, contains the biggest Pt nanoparticles in the zeolite-supported series with an average diameter of 2.2 nm. The distribution of the nanoparticles on zeolite crystals is not homogeneous. Apparently, this is due the fact that the size of the Pt nanoparticles exceeds the zeolite pore and channel diameter. Hence, they can only be located on the outer surface of the zeolite crystals.
In general, Pt species in the zeolite-supported samples are more disperse than the alumina-supported ones and contain almost no large nanoparticles on the bigger-scale TEM images (Fig. S11†).
For the samples studied, due to the big size of support crystals and their low conductivity, part of the single atoms was not distinctly visible in the TEM images. For this reason, to complement the understanding of Pt dispersion and to evaluate contributions of different size fractions, in situ DRIFTS spectra of chemisorbed CO were measured.
 | ||
| Fig. 2 DRIFTS spectra of CO adsorbed on ZSM-5-supported catalysts: A – PtZ-IE sample; B – PtZ-IE-R sample; C – PtZ-IW sample. Black line – spectra in 1% CO flow. Dashed black line – spectrum in 0.1% CO flow. Grey line – spectra recorded during Ar flushing after switching off CO flow. Dashed grey line – spectrum of the ZSM-5 support in 1% CO flow. | ||
 | ||
| Fig. 3 DRIFTS spectra of CO adsorbed on alumina-supported catalysts: A – PtA-IW sample; B – PtA-IW-500 sample; C – PtA-IW-700 sample. Black line – spectra in 1% CO flow. Grey line – spectra during Ar flushing after switching off CO flow. | ||
On the contrary, the bands corresponding to linearly bonded CO at wavenumbers above 2000 cm−1 are very distinct. The range ∼2090–2000 cm−1 contains the bands of linearly adsorbed CO on Pt terraces in reduced nanoparticles or clusters50,52,56–60 (bands highlighted in pink in Fig. 2 and 3). The shift to higher wavenumbers indicates a decrease of platinum particle size56 down to single sites including cationic ones that were reported to show their absorption band starting from 2090 cm−1 (ref. 56, 59, 61 and 62) and above. The area at ∼2120–2090 cm−1 (highlighted in yellow in Fig. 2 and 3) is the one in which the bands of platinum single sites are most frequently reported.56–63 According to the literature, the band at 2090 cm−1 corresponds to CO adsorbed on more reduced platinum that can be either partially reduced single sites60,61 or corner and edge metal atoms which are abundant on small nanoparticles.57,60,64 In other studies,57–59,63 CO on Pt single sites shows absorption bands around 2120–2100 cm−1. This range is also typical for platinum in ionic (oxidised) state that forms a monocarbonyl complex and is not necessarily single sites.51,58,60,61 Except for the difference in oxidation state, a reason for the difference in the reported Pt single site absorption frequencies can also be the location in a different position of a zeolite network.56 Finally, the bands above ∼2120 cm−1, highlighted in green in Fig. 2, belong to polycarbonyl complexes of ionic Pt.51,57,60 They are found only on the zeolite-supported samples and are absent on the alumina-supported ones. The spectra of the pure ZSM-5 support in CO exhibit absorption bands of gaseous carbon monoxide at 2160 and 2125 cm−1 – in the same range as the polycarbonyls (Fig. 2, grey dashed line). However, these bands have low intensity compared to the main bands of the zeolite-supported spectra. Therefore, their interference with determining types of Pt sites is minor. The low intensity bands at 2170 and 2125 cm−1 on alumina-supported samples in Fig. 3 belong to gaseous CO and quickly disappear during Ar flushing.60
As it is apparent from the literature data above, only the absorption frequency may not be sufficient to unequivocally distinguish between small nanoparticles/clusters, oxidised platinum particles and singe sites. The exception is probably the polycarbonyls on isolated Ptn+ ions (n = 1–3), which exhibit their absorption bands above 2120 cm−1.51,60 For other species, a way to discriminate them from single sites is to vary the coverage of CO and follow the frequencies of the observed absorption bands. For isolated platinum atoms, the band position is supposed to be stable due to the absence of dipole–dipole interaction of adsorbate molecules. This approach has been demonstrated for absorption bands in the 2120–2090 cm−1 range56–59 (yellow in Fig. 2). To change the CO surface concentration in this study, we switched off CO flow after keeping the samples in its atmosphere so that pure Ar flushing remained. For strongly absorbed CO, certain conditions (e.g. high vacuum) may be required to change its surface coverage. Hence, even a stable band position in this region requires careful consideration before it can be assigned to atomically dispersed Pt. However, if a band changes its energy and symmetry, like it will be shown further, it allows one to exclude its origin from single site species.
The PtZ-IE sample possesses the most intense CO-bands of all the zeolite-supported catalysts. This can be indirect evidence of the highest Pt dispersion. The sample exhibits pronounced bands for polycarbonyls and monocarbonyl on ionic Pt (∼2177–2100 cm−1, areas highlighted in green and yellow respectively). A closer look at polycarbonyl bands around 2177 and 2137 cm−1 reveals their possible origin from platinum coordinating not only to CO, but also to Cl, probably remaining from the precursor.65,66 All the carbonyl bands retain their position during flushing with Ar, which is an indication of highly dispersed platinum. These absorption frequencies correspond to polycarbonyls on isolated platinum in ionic state.51,60 The less intense band at around 2080 cm−1 suggests a concurrent presence of some small nanoparticles. Light red shift by 2 cm−1 implies minor dipole–dipole interactions not found for single sites. The low intensity bridged CO bands in the 1940–1745 cm−1 region reveal the possible presence of few bigger Pt particles with a small surface area.
The PtZ-IE-R catalyst, which underwent additional reductive treatment after ion exchange, exhibits a similar profile in the polycarbonyl area, but differs in the monocarbonyl and reduced platinum wavenumber ranges below ∼2120 cm−1 (yellow and pink highlights in Fig. 2B). The most intense absorption band is now at 2094 cm−1 corresponding to reduced single sites or small clusters. Slight red shift by 2 cm−1 may indicate rather the latter type of Pt species. As for the polycarbonyl bands, they also start to exhibit a minor energy shift by 2–3 cm−1, which implies possible clustering of oxidised Pt atoms. The generally less intense absorption bands signify lower total dispersion compared to the unreduced PtZ-IE sample.
The trend of decreasing absorption band intensities and, hence, lower platinum dispersion further continues for the incipient-wetness-impregnation-obtained PtZ-IW catalyst. The absorption band corresponding to reduced Pt species (pink-highlighted range in Fig. 2C) is at lower energy of 2077 cm−1 typical for small Pt nanoparticles. A higher red shift of 6 cm−1 during Ar flushing also evidences larger particle size compared to the PtZ-IE catalysts. The profile of carbonyl bands on ionic Pt, which are visible above ∼2090 cm−1 (ranges of green and yellow colours in Fig. 2C), is also slightly different for this sample. It has a new band at 2112 cm−1 that may correspond to Pt2+ monocarbonyl species or, together with the other two bands at 2170 and 2137 cm−1, signify tricarbonyl Pt+ species.51
Unlike the zeolite-supported samples, the alumina-based catalysts exhibit absorption bands which correspond mainly to metallic platinum nanoparticles and clusters without the bands of polycarbonyls. Thus, only two zones containing information about Pt dispersion can be distinguished for these samples. The first one, around 2090–2080 cm−1 (yellow in Fig. 3), corresponds to platinum clusters or small nanoparticles. Assigning these bands to clusters rather than reduced single sites is based on a relatively significant energy shift of 4–6 cm−1, which is not typical for atomically dispersed platinum. The second energy range from 2090 to 2000 cm−1 (pink in Fig. 3) contains bands that originate from platinum nanoparticles. The energy of the main band in this region decreases with the increase of the temperature of catalyst treatment. It has the highest value of ∼2071 cm−1 for the untreated sample PtA-IW, a lower value of ∼2060 cm−1 for the sample PtA-IW-500, treated at 500 °C, and the lowest with the value of 2040 cm−1 for the sample Pt-IW-700 treated at 700 °C. This decrease of the absorption energy is an indication of particle size increase.56 The higher intensity of the absorption bands below 2090 cm−1 (pink in Fig. 3) for PtA-IW and PtA-IW-500 indicates a higher fraction of nanoparticles compared to small clusters. A higher relative intensity of the band at 2092 cm−1 does not necessarily mean the inverse ratio for PtA-700, because the intensity of all the bands is in general significantly lower for this sample hinting at a lower Pt surface area and bigger particle size. In general, the spectra of the alumina-supported catalysts tentatively reveal the presence of two types of platinum sites, one of which being small Pt clusters and another, dominating one, being nanoparticles. The size of the nanoparticles grows with the increase of temperature of catalyst treatment.
| Sample | EXAFS | TEM | DRIFTS |
|---|---|---|---|
| 2 wt% Pt/γ-Al2O3 | |||
| PtA-IW | High dispersion, disordered structure with significant contribution of atomically distributed Pt | Single atoms and clusters | Clusters are predominant |
| 1.7 nm nanoparticles | |||
| PtA-IW-500 | Clusters of 0.5–1 nm size | Single atoms and clusters | Small nanoparticles are predominant |
| 1.8 nm nanoparticles | |||
| PtA-IW-700 | Nanoparticles of ≥5 nm size | 13.6 nm nanoparticles | Nanoparticles are predominant |
| Individual clusters | |||
| 1 wt.% Pt/ZSM-5 | |||
|---|---|---|---|
| PtZ-IE | High dispersion, disordered structure with significant contribution of atomic Pt | Single atoms and clusters | Single atoms and clusters are predominant |
| 1.2 nm nanoparticles | |||
| PtZ-IE-R | High dispersion, disordered structure with significant contribution of atomically distributed Pt | Clusters, individual single sites | Clusters are predominant |
| 1.4 nm nanoparticles | |||
| PtZ-IW | Small clusters | 2.2 nm nanoparticles | Nanoparticles are predominant |
| A substantial number of small clusters | |||
For the PtA-IW sample, TEM data reveals the presence of two main fractions – platinum single atoms and clusters together with small nanoparticles with a diameter of 1.7 nm. In situ DRIFTS demonstrates the predominance of clusters. This is confirmed with ex situ EXAFS results showing the average coordination number typical for single atoms and lack of ordered Pt structure. As EXAFS alone usually cannot fully prove single site structure,67 its combination with other techniques, like TEM and DRIFTS, is needed.38
Although the fractions of Pt particles in PtA-IW-500 are practically the same as in PtA-IW, the general platinum dispersion is smaller. Because of the additional treatment at 500 °C, the average nanoparticle size slightly increases to 1.8 nm. In situ DRIFTS results reveal that this also leads to redistribution of Pt atoms between the fractions, and nanoparticles with a diameter of 1.8 nm prevail over clusters and single sites. Ex situ EXAFS analysis shows the average coordination number of platinum reaching the values typical for 0.5–1 nm clusters. Together with the appearance of Pt in the coordination environment, it signifies the increase in Pt particle size.
Treatment of PtA-IW at 700 °C leads to a considerable decrease in the number of clusters and single sites and sintering of nanoparticles which increase their average size to 13.6 nm. In situ DRIFTS spectra contain absorption bands of low intensity implying the general decrease of dispersion. The absorption bands originate from a small number of single sites or clusters and bigger nanoparticles with a small surface area. Ex situ EXAFS results are in line with these observations. They exhibit the average coordination number almost as high as in the case of bulk platinum suggesting a platinum particle size of ≥5 nm.
Zeolite-supported catalysts generally show higher dispersion for ion-exchanged samples compared to the incipient-wetness-impregnated one. Thus, TEM images of PtZ-IE reveal a high number of small clusters and even single sites together with small nanoparticles of 1.2 nm diameter. The DRIFTS results of this sample demonstrate the prevalence of isolated Pt ions in it. The ion-exchanged sample PtZ-IE-R that underwent reductive treatment contains Pt with lower dispersion. As revealed by TEM, it contains 1.4 nm nanoparticles together with some Pt clusters. According to DRIFTS, it also contains a number of Pt single sites, but the main fraction is Pt in a state of reduced clusters. Finally, the in situ DRIFTS spectra of the PtZ-IW sample display low intensity absorption bands signifying its low dispersion. The bands correspond to small oxidised nanoparticles and a low number of Pt clusters. It is in line with the TEM results that reveal mainly nanoparticles of 2.2 nm average diameter. Ex situ EXAFS confirms the trend in the increase of Pt particle size for the IWI sample compared to the IE-ones. However, it overestimates the sample dispersion compared to in situ DRIFTS. This may be caused by lower average particle size for the zeolite-supported samples compared to the alumina-supported ones, higher polydispersity of the former ones and Pt being in a more amorphous state. In this case, a higher number of smaller particles or disordered crystallites lower the average coordination number and backscattering on neighbouring Pt atoms in the Pt coordination environment that are observed in ex situ EXAFS.
Catalytic tests
 | ||
| Fig. 4 Catalytic tests results for the alumina-supported catalysts during the second light-off in the reaction mixture. A – Conversion; B – selectivity to N2; C – selectivity to N2O; D – selectivity to NOx. Red line – PtA-IW-700; orange line – PtA-IW-500; blue line – PtA-IW. Reaction feed – 500 ppm NH3, 13% O2 in N2. Flow −1050 cm3 min−1. GHSV = 63000 cm−1. Loading – 25 mg of 2 wt% Pt catalyst. Heating rate of 3 °C min−1. | ||
The variation in platinum size on the nanometre scale does not affect catalyst performance as strongly as the change on the subnanometre (cluster) scale. A narrow T50% range for Pt nanoparticles of different sizes at high, close to real-application oxygen concentration, was reported earlier.30,31 Expanding this range to the lower activity zone for smaller particles hints at the important role of particle size in addition to the Pt oxidative behaviour widely described in the literature.26,27,31–33 It was postulated in previous studies that specific ensembles of Pt atoms are required for ammonia activation through oxygen-assisted dissociation. The ensembles that are reported to be optimal for this step and to have the lowest energy barriers are step-edge B5-sites, for example Pt(211) facets.14,36,68–70 According to the literature, Pt nanoparticles of less than 5 nm have spherical shape in air with no distinct crystal faces on their surface.71 At the same time, Pt(211) facets result in the lowest surface energy for small nanoparticles below 5 nm in oxidative media.72 It is also reported that these facets have a very low probability to be present on transition metal particles with size below ∼2 nm.36 The reason for this is a too low number of atoms to form a step site on a small particle retaining an energy-efficient spherical shape.36,73,74 Thus, the fraction of B5 sites on particles below ∼2 nm diameter is close to 0 and abruptly increases around 2 nm constituting a maximum surface concentration that further gradually diminishes with particle size increase in favour of terrace sites.73,74 In this study, we observe a sharp increase in ammonia oxidation activity in the size range of ∼2 nm at which surface B5-sites are reported to emerge. The role of particle size in determining Pt activity appears to be more crucial at realistic NH3:O2 ratios and pressures than a commonly reported Pt oxidation state. A number of studies highlight the superior activity of reduced platinum compared to the oxidised one in the ammonia oxidation reaction.26,27,30–34 However, it was shown that this factor becomes less important at high oxygen concentrations at atmospheric pressure.30 Although pre-reduced and pre-oxidised catalysts with similar Pt dispersions performed significantly differently at 4% O2 concentration, they had almost equal activity in a reaction mixture containing 13% O2 already after the first catalytic cycle. This study also evidences the less significant influence of the oxidation state on activity and a major role of particle size which may be linked to the oxidation state as the smaller particles/clusters oxidise easier. Comparing the ex situ X-ray absorption near-edge structure (XANES) spectra of the catalysts tested (Fig. S13†), one can see that PtA-IW-500 is more active than PtA-IW although the first one is more oxidised and prepared from the latter one. The sign of a more reduced state of PtA-IW is a lower absorption intensity of its XANES spectrum near the adsorption edge, which is also called white line, compared to the white line for PtA-IW-500. In general, the PtA-IW spectrum is also closer to the Pt foil reference than that of PtA-IW-500. Evidently, the bigger particle size of the latter catalyst plays a more important role in its activity than the oxidation state. It is also interesting to compare the performance of PtA-IW, after reductive treatment, with the same catalyst before reduction (Fig. S14†). The catalytic performance of this sample before and after reductive treatment is almost identical despite a great difference in oxidation state and the unreduced sample almost reaching the spectrum of PtO2 in its white line intensity (Fig. S13†). Since the difference in Pt oxidation state in PtA-IW before and after reduction is drastic (significantly higher than the change of oxidation state under reaction conditions, cf. later), its almost unchanged performance points out that it is the particle size that is at the origin of the structure sensitivity. Note that this may still also be coupled to the oxidation state if the catalyst structure changes under reaction conditions. Nevertheless, favourable ensembles of surface Pt atoms seem to be present for large particles and decisive for activity under the studied conditions.
Regarding selectivity to nitrogen, its trend appears to be inverse to activity – PtA-IW with the smallest particle size is the most selective to N2 in the series. PtA-IW-500 and PtA-IW-700 are less selective, but no clear trend can be seen between the two of them. They demonstrate a similar relatively high N2 selectivity at low temperatures. In the intermediate temperature range, PtA-IW-500 shows the lowest N2 selectivity and is the most selective to N2O of all three catalysts. At high temperature, especially during light-out (Fig. S12†), PtA-IW-500 exceeds PtA-IW-700 in its selectivity to nitrogen. For all three samples, NOx selectivity rises monotonously starting from ∼240 °C and is the lowest for PtA-IW-500 mainly in favour of N2O. Higher selectivity for less active catalysts was already described in the literature.30 T. Hansen reported31 the opposite trend but notably only for low temperatures. Literature studies suggest different Pt surface coverages of Nads, Oads or NOads as the reason for different product yields in this system.75–78 The observation that the selectivity to N2 decreases as soon as conversion reaches 100% in this and other studies30,32 complies with this explanation. Under these conditions, the catalysts with bigger particles on which ammonia is oxidised faster yield surfaces poorer in ammonia-derived species, which decreases selectivity to nitrogen.
 | ||
| Fig. 5 Catalytic test results for the ZSM-5-supported catalysts during the second light-off in the reaction mixture. A – Conversion; B – selectivity to N2; C – selectivity to N2O; D – selectivity to NOx. Red line – PtZ-IW; orange line – PtZ-IE-R; blue line – PtZ-IE. Reaction feed – 500 ppm NH3, 13% O2 in N2. Flow −1050 cm3 min−1. GHSV = 63000 cm−1. Loading – 50 mg of 1 wt% Pt catalyst. Heating rate of 3 °C min−1. | ||
T 50% values for the nanoparticle-dominated sample PtZ-IW (210 °C) and cluster-dominated sample PtZ-IE-R (229 °C) resemble those of the corresponding alumina-supported samples with particle sizes in a similar range – PtA-IW-700 and PtA-IW-500. Remarkably, the sample PtZ-IW with 2.2 nm nanoparticles has practically the same T50% as PtA-IW-700 with a much bigger average particle diameter of 13.6 nm. At the same time, the sample PtA-IW-500 with predominant nanoparticles of 1.8 nm is less active than the PtZ-IW.
The activity of the PtZ-IE sample with mainly Pt single sites/small clusters is drastically poorer. Its T50% during light-off constitutes 384 °C which is significantly higher than the range of light-off temperatures reported in the literature for similar reaction conditions.30,31 PtZ-IE also stands out in its hysteresis. While for PtZ-IW and PtZ-IE-R hysteresis is slightly larger than that for the alumina-supported samples with Pt particles in a similar size range (PtA-IW-700 and PtA-IW-500 respectively), for PtZ-IE it is as big as 143 °C (cf.Fig. 5 and S16†). This cannot be explained only with the change of the structure of the sample under the reaction conditions. Different activities in the first and the second catalytic cycle shown in Fig. S17† indicate that some structural changes do occur. However, the effect from them is smaller than the observed hysteresis in both cycles. Such a distinct behaviour of the PtZ-IE sample, together with its low activity, is a strong hint for a different reaction mechanism. Selectivity to N2 for all three zeolite-supported samples increases during the light-out with decreasing particle size (Fig. S16†). This trend, with the exception of the single-site-dominated sample (PtZ-IE), is also observed during the light-off. Samples PtZ-IE-R and PtZ-IW show similar selectivity to N2 at low temperatures, i.e. below ∼315 °C during the light-off and ∼280 °C during the light-out. At higher temperatures, selectivity to nitrogen increases from ∼40% to 50% in the case of PtZ-IE-R and decreases from the same initial level to 30% in the case of PtZ-IW. In the case of the single-site-dominated sample PtZ-IE, selectivity to nitrogen is the highest among all zeolite-supported samples during cooling. It decreases from ∼80% at 200 °C to ∼50% at 400 °C. During the light-off this sample shows a completely different behaviour producing almost exclusively NOx (mostly NO) at low conversions. With conversions above 10%, the selectivity of this sample becomes similar to that of PtZ-IW with ∼60% NOx and ∼30% N2 yields. In general, selectivity to nitrogen is higher for zeolite-supported samples than for alumina-supported samples. This can be explained with the higher acidity and ability to store more ammonia in zeolites which ensures higher coverage of surface N-intermediates and improves N2 yield.79,80
The peculiar performance of the sample containing Pt single sites and small clusters may be explained with unavailable Pt ensembles for the mechanism of the Ostwald process as the activation step of O-assisted ammonia dissociation on single sites/small clusters is hardly possible. An additional factor contributing to the low activity of single sites/small clusters may be their coordination to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or –OH-groups of the zeolite which limits adsorption of the reactants. As in the case of the alumina-supported catalysts, the oxidation state of this sample does not seem to be an essential factor for its activity. According to the ex situ XANES spectrum of PtZ-IE (Fig. S19†) this catalyst is oxidised with an intermediate oxidation state between Pt(0) and Pt(IV). This can be deduced from the white line height almost in the middle between the white lines of the two references. In contrast, the spectrum of a more active PtZ-IE-R catalyst has a less intense maximum, which points to its more reduced state. Nevertheless, the lowest activity of PtZ-IE cannot be explained with Pt being more oxidised, because the most active sample in the series, PtZ-IW, is even more oxidised than the least active PtZ-IE. Hence, also for the zeolite-supported catalysts, the explanation of activity difference should be mainly based on the particle size which enables or prevents the existence of B5 Pt sites for efficient ammonia activation.
O or –OH-groups of the zeolite which limits adsorption of the reactants. As in the case of the alumina-supported catalysts, the oxidation state of this sample does not seem to be an essential factor for its activity. According to the ex situ XANES spectrum of PtZ-IE (Fig. S19†) this catalyst is oxidised with an intermediate oxidation state between Pt(0) and Pt(IV). This can be deduced from the white line height almost in the middle between the white lines of the two references. In contrast, the spectrum of a more active PtZ-IE-R catalyst has a less intense maximum, which points to its more reduced state. Nevertheless, the lowest activity of PtZ-IE cannot be explained with Pt being more oxidised, because the most active sample in the series, PtZ-IW, is even more oxidised than the least active PtZ-IE. Hence, also for the zeolite-supported catalysts, the explanation of activity difference should be mainly based on the particle size which enables or prevents the existence of B5 Pt sites for efficient ammonia activation.
Given the constraints for the Ostwald mechanism at critically small Pt single sites and clusters, a more likely option remains the selective catalytic reduction (SCR) of NOx with NH3 that under these conditions also occurs on Pt.81 In this case, NOx can be produced on less abundant Pt nanoparticles or clusters that are also present in PtZ-IW, according to DRIFTS. Higher selectivity to N2 at high temperatures during cooling supports the SCR mechanism hypothesis. Significantly lower light-off activity and lower selectivity may result from inhibition of the SCR mechanism while the catalyst is saturated with some of the adsorbates that are not on the catalyst surface during cooling down. A common adsorbate that poisons the SCR Cu and Fe catalysts is ammonia which prevents re-oxidation of active sites.82 Another adsorbate that is abundant on the surface during catalyst heating and absent during cooling is water. Its presence in the case of SCR on Cu was shown to keep the catalyst in the oxidised state and its desorption – to dynamically alter catalyst activity.83,84
The normalised reaction rates per surface atom of Pt allow for a better comparison of the catalytic activity of Pt on different supports. For this purpose, TOFs of catalysts on alumina and zeolite with similar particle sizes are presented in Fig. S25.† Among the catalysts with a particle size of about 2 nm (1.8 nm for PtA-IW-500 and 2.2 nm for PtZ-IW) the zeolite-supported one was about 1.4 times more active. This can be explained with higher acidic properties of ZSM-5 compared to alumina. Such behaviour is in line with the studies of Wang et al.85 who highlighted increased activity of ASCs containing acid sites. They reported that the rate of ammonia oxidation positively correlated with the number of acid sites for catalysts containing Pt with particles in the nanometre size range. Nonetheless, in this study, the samples with Pt dispersed in a sub-nanometre range showed the opposite activity trend. Specifically, among the samples containing a high number of Pt clusters, the alumina-supported one was 1.4 times more active (PtA-IW and PtZ-IE-R in Fig. S25†). This might be another indication of a different mechanism of ammonia activation on sub-nanometre Pt particles. However, this observation needs to be treated with caution and in the context of other results, since the particles in PtA-IW and PtZ-IE-R are not monodisperse with some larger particles.
In spite of the evident influence of the catalyst support on activity, the effect of particle size turns out prevailing and determining the overall catalyst activity. While the support variation changed TOF by about 1.4, the particle size increase from single sites to clusters and from clusters to nanoparticles changed the reaction rate by orders of magnitude (see Fig. S23†). Therefore, all the studied catalysts on the two supports form the same activity trend in the TOF dependence on particle diameter. It appears that after reaching a certain size around 2 nm, the activity of ammonia oxidation, normalised by the Pt surface, starts to depend on Pt particle size relatively modestly. Before this threshold, the rate dependence on particle size is exponential, while after it still increases but with a linear trend. As discussed above, Pt B5 sites, which were shown to facilitate O-assisted ammonia dissociation, can appear on Pt particles starting from this size.
To analyse the impact of the support on selectivity, alumina- and zeolite-based samples with similar light-off profiles are compared in Fig. S26.† Zeolite-supported catalysts demonstrated higher N2 yield at elevated temperatures when ammonia conversion was full. This was true for both heating and cooling in the reaction mixture. Zeolite-supported catalysts were still generally more selective at low temperatures and under incomplete ammonia conversions during cooling. However, during heating in the reaction mixture alumina-supported samples generally produced more N2. As shown in the literature by Wang et al.85 and Lin et al.,86 the overall higher selectivity of the zeolite-supported samples is explained with the higher acidity of the support and, particularly, with the higher number of Brønsted acid sites. The latter are much more abundant on the zeolite than on the alumina and, together with Lewis acid sites, act to form the ammonia pool on the surface of the support enabling constant supply of ammonia to active Pt species.
Nevertheless, the general trend of rate dependence on particle size was still the same as in the dry feed with an inflection point at about 2 nm (Fig. S27†). This shows that the reaction mechanism does not change in the presence of water. Therefore, further XAS experiments were conducted in the dry feed.
Operando QEXAFS studies
The contour plot for PtA-IW-500 is shown in Fig. 6C. This plot can be imagined as a “top view” of a series of XANES spectra measured at different temperatures. The bottom right graph in Fig. 6 represents a temperature profile of normalized absorption of the spectra around their maxima (11568.9 eV). The top left graph in the figure shows selected XANES spectra at constant temperatures at which the white line shapes differ most. It is evident from the plot that for PtA-IW-500, all most distinct white line shapes corresponded to partially oxidized Pt. Their intensity was between Pt foil and PtO2 references (Fig. S30A†).
 | ||
| Fig. 6 Contour plot of PtA-IW-500 operando Pt L3-edge XANES spectra measured during the light-off in the reaction mixture (500 ppm NH3 and 10% O2 in inert). A – Spectra with three most distinct white line shapes (energy profiles from the contour plot). B – The contour plot. C – Absorption profile at the energy close to maximum absorption (11568.9 eV). Flow – 75 cm3 min−1 (corresponds to a GHSV of about 127000 h−1). Heating rate – 5 °C min−1. 2 wt% Pt catalyst of 100–200 μm sieved fraction was loaded into a 1.5 mm-diameter quartz capillary to form a bed length of ∼5 mm. | ||
The slight but distinct changes in the Pt white line of these spectral components indicate that the main changes in Pt state occur on its surface with, to a great extent, an unchanged core. Initially, the white line intensity decreases during heating in the reaction mixture and reaches its minimum around 240 °C. Then it becomes more intense again with further heating. Following the white line maximum along the temperature, one can see that it shifts to lower energies by ∼0.5 eV. These tendencies in changing white line intensity indicate reduction in the reaction mixture with the highest reduction extent at ∼240 °C and then re-oxidation at even higher temperatures.87,88 The absorption maximum shift to lower energies indicates that the oxidised states at high and low temperatures have different coordination environments and, thus, are not equal.87,89
Catalytic data for the sample PtA-IW-500 recorded during operando QEXAFS measurements is shown in Fig. 7A. The trends are the same as in laboratory tests. Rapid light-off starts at ∼200 °C and is accompanied with the highest selectivity to N2. Then, around full conversion, N2O becomes the main product and NOx starts to form. With further heating, the N2O yield drops and the NOx yield gradually increases.
 | ||
| Fig. 7 Results of operando QEXAFS analysis of PtA-IW-500 measured during a light-off in the reaction feed (500 ppm NH3 and 10% O2 in inert). A – Catalytic data; B – MCR-resolved spectral components from the QEXAFS dataset; C – evolution of relative fractions of the spectral components in the course of the light-off. | ||
XANES regions of the QEXAFS spectra were analysed using multivariate curve resolution (MCR), and three main spectral components were identified (Fig. 7B), in line with the analysis of absorption of the white line shape (cf.Fig. 6). The MCR-derived spectral components correspond to partially oxidized Pt with its white line intensity between Pt foil and PtO2 (cf. Fig. S30B†). The obtained components were then used to perform the linear combination analysis (LCA) of the recorded spectra to track the interchange of Pt states in the course of the light-off.
The catalytic data acquired during the QEXAFS study was analysed along with the LCA results to correlate the catalyst performance to observed spectral features. Thus, the Pt L3 spectra recorded at temperatures below the light-off correspond to the first oxidized state, or the “low temperature component” of LCA in Fig. 7B and C. The relative fraction of this component gradually declines with heating in the reaction mixture. The second, more reduced Pt state evolves with rise in temperature, marked as “middle temperature component” (Fig. 7B and C). As soon as its concentration exceeds the initial oxidized state concentration, light-off starts. The more reduced middle temperature state reaches its maximum around 100% conversion and then decreases. The predominance of this component can be correlated to the high conversion and the highest selectivity to N2. Shortly after the complete conversion, platinum re-oxidises and another, high temperature oxidised component becomes predominant. It is denoted as “high temperature component” in Fig. 7B and C. The prevalence of the oxidised high-temperature component over the reduced one is accompanied by N2O becoming the main product. From this point, up to the maximum temperature tested, the NOx content in the products gradually grows. Thus, the three spectral components detected with MCR correspond to three main states of Pt during light-off that clearly correlate with its activity and selectivity. The surface species of Pt, from which the above-mentioned principal spectral components originate, should be further identified in future. This may help to deepen the understanding of the activity- and selectivity-governing factors and requires operando techniques complementary to XAS.
The spectra obtained for PtA-IW-700 were treated analogously to PtA-IW-500 and showed similar, though less pronounced, trends. Like for PtA-IW-500, three components with different white line shapes are observed in the operando QEXAFS spectra and shown in the contour plot in Fig. S31.† The white line intensity initially decreased with heating and then increased again at higher temperatures. The absorption maximum, as in the case of PtA-IW-500, slightly drifted to lower energies as the temperature grew. MCR analysis of PtA-IW-700 resolved three spectral components (Fig. 8B), the LCA-derived concentrations of which behave analogously to PtA-IW-500. The low-temperature component prevailing during the inactive phase of the catalytic cycle was again transformed to a more reduced middle-temperature component reaching its highest fraction with the highest N2 yield. Finally, the third, high-temperature component was mostly observed during pronounced formation of the undesired N2O and NOx products.
 | ||
| Fig. 8 Results of operando QEXAFS analysis of PtA-IW-700 measured during a light-off in the reaction feed (500 ppm NH3 and 10% O2 in inert). A – Catalytic data; B – MCR-resolved spectral components from the QEXAFS dataset; C – evolution of relative fractions of the spectral components in the course of the light-off. | ||
With all the similarities mentioned above, the difference between PtA-IW-700 and PtA-IW-500 is that the changes of the white line in the former catalyst are less pronounced. For example, the change in the maximum absorption intensity in the MCR-resolved spectral components is ∼0.15 (varies between ∼1.6 and 1.75) for PtA-IW-500 containing Pt clusters, while this difference for the sample PtA-IW-700 with big nanoparticles constitutes only ∼0.1 (varies between 1.3 and 1.4). The smaller white line differences also result in smaller concentration variations of the components. For PtA-IW-500 they are ∼0.1–0.8, while for PtA-IW-700 – only ∼0.2–0.5. These less distinct changes in the case of the sample with bigger particles can be explained with the fact that mainly surface Pt atoms participate in the reaction with bulk atoms being not much affected. In this way, the sample with smaller particles, that has a higher fraction of surface atoms, exhibits more pronounced changes in the average state of platinum atoms that result in the final XAS spectra. However, regardless of the different proportions of surface platinum atoms involved in the catalytic process, the similar white line changes evidence that the mechanism is the same for larger and smaller particles.
 | ||
| Fig. 9 Results of operando QEXAFS analysis of PtZ-IW measured during a light-off in the reaction feed (890 ppm NH3 and 10% O2 in inert). A – Catalytic data; B – spectral components for the QEXAFS dataset; C – evolution of relative fractions of the spectral components in the course of the light-off. | ||
In contrast, PtZ-IE containing mostly single Pt sites and small clusters demonstrated remarkably different spectral trends. Fig. 10 shows that the white line intensity of PtZ-IE decreased monotonously in the whole tested temperature range and did not undergo any combined reduction and reoxidation in the course of the light-off. Thus, only two different spectra can be resolved – the most oxidised catalyst at low temperature and the most reduced catalyst at high temperature. An absorption maximum shift to lower energies was also observed, as for other samples.
 | ||
| Fig. 10 Contour plot of PtZ-IE or operando XANES spectra measured during the light-off in the reaction mixture (890 ppm NH3 and 10% O2 in inert). A – Spectra with two most distinct white line shapes (energy profiles from the contour plot). B – The contour plot. C – Absorption profile at the energy close to maximum absorption (11567.9 eV). Flow −70 cm3 min−1 (corresponds to a GHSV of about 120000 h−1). Heating rate – 5 °C min−1. 1 wt% Pt catalyst of 100–200 μm sieved fraction was loaded into a 1.5-mm-diameter quartz capillary to form a bed length of ∼5 mm. | ||
The main spectral components for operando QEXAFS data of PtZ-IE were found with MCR and their concentrations are juxtaposed with the catalytic data along the temperature axis in Fig. 11. One spectral component prevails at low temperature with ∼80% concentration and decreases with temperature rise. The Pt state associated with it appears to be inactive, because the light-off starts very close to the point where the low-temperature component falls to less than half of the total content. The appearance of the high-temperature component was accompanied with an activity increase and also, to a large extent, a monotonous growth of yields of N2 and NOx (predominantly NO), which are the main products. This selectivity pattern is different from that of nanoparticle/cluster-containing samples that include the stage of high N2O formation. This observation, together with a different spectral behaviour, strongly evidences a different mechanism of ammonia oxidation on platinum single sites and small clusters.
 | ||
| Fig. 11 Results of operando QEXAFS analysis of PtZ-IE measured during a light-off in the reaction feed (890 ppm NH3 and 10% O2 in inert). A – Catalytic data; B – MCR-resolved spectral components from the QEXAFS dataset; C – evolution of relative fractions of the spectral components in the course of the light-off. | ||
In the section Catalytic tests we already speculated that platinum single sites/small clusters are more likely to catalyse ammonia SCR than ammonia oxidation as the probability of ammonia dissociation on them is low. Based on our laboratory tests which show a very pronounced hysteresis, we assume that the presence of adsorbed species on the Pt sites inhibits the catalytic activity at low temperatures. According to literature data, the inhibiting species could be either ammonia, which is a known poison for Fe and Cu SCR catalysts, or water which can compete for adsorption sites and alter the activity.83,84 From the reduction in the course of the light-off, it can be deduced that the low-temperature spectral component does not originate from Pt-adsorbed ammonia, but rather from water which desorbs in reaction mixture leaving the Pt surface more reduced and available for ammonia adsorption. Hence, water desorption may initiate ammonia oxidation on Pt nanoparticles in the PtZ-IE catalyst, since platinum reduction is known to benefit its activity.30,33 In this case the reaction is initiated by ammonia oxidation that produces some NOx which further reacts with NH3 by SCR on single sites and clusters.
Conclusions
To reveal factors governing the structural sensitivity in selective ammonia oxidation, laboratory tests and operando QEXAFS studies were conducted on well-characterised γ-Al2O3 and zeolite supported platinum catalysts with different particle sizes. The rate of ammonia activation rose with increasing particle size growing exponentially in the range of the smallest particles from single sites up to ∼2 nm (Fig. 12). In the nanometre size range, the activity rose more moderately demonstrating an almost linear but small gain with size increase. Hence, for an effective ammonia oxidation, particles of sufficient diameter are required that contain surface ensembles of Pt atoms needed for the activation step through oxygen-assisted ammonia dissociation, typical for the mechanism of ammonia oxidation in the Ostwald process. These ensembles are most probably B5 sites of Pt that can occur with a sufficient probability only on nanoparticles larger than ∼2 nm. For larger particles, their fraction grows linearly, which corresponds to the observed linear increment of the reaction rate for particles larger than 2 nm (see Fig. 12). In turn, Pt single sites and clusters, being below this threshold, exhibit lower activity due to apparent constraints for the activation step. It is proposed that Pt particles in this size range rather catalyse SCR than ammonia oxidation. These conclusions are schematically summarised in Fig. 12. The depicted trend was predicted theoretically36 but has not been experimentally proven on supported catalysts under atmospheric pressures so far. The spectroscopic results further suggest a common mechanism for nanoparticle-containing samples, regardless of the support. Three surface Pt states correlating with catalyst performance were found for these catalysts. An initial oxidized state dominated at low temperatures while the catalyst was inactive. At intermediate temperatures the second more reduced state (tentatively, clean nanoparticle surface) was prevalent and corresponded to the light-off and the highest nitrogen yield. At high temperatures, the third state emerged that was oxidised again and was accompanied by high selectivity to N2O and NOx. Selectivity to nitrogen was generally higher for the less active samples, presumably due to slower removal of N-containing species from the surface. Zeolite-supported catalysts were generally more selective to N2 because of their higher acidity and ammonia storage properties. | ||
| Fig. 12 The dependence of the TOF of ammonia oxidation as well as of the fraction of B5 sites on the size of supported Pt particles. The fraction of B5 sites on the Pt surface was plotted based on the results of G. A. Tritsaris et al.72 and K. Honkala et al.74 The number of B5 sites was estimated based on the relation of dispersion with particle size described on page S14 of the ESI† and a dependence of number of atoms on particle size for a spherical particle of Pt described by A. Jentys.49 | ||
Constraints for the mechanism of ammonia oxidation on critically small Pt particles were confirmed spectroscopically for Pt single sites/small clusters on zeolite. This sample demonstrated only two main states with a gradual change between them during the light-off, instead of a consecutive reduction and re-oxidation, as in the case of Pt nanoparticles.
In summary, the varying activity of Pt particles with their size increase can be used for tuning catalytic performance. Specifically, the use of catalysts with about 2 nm Pt particle size can ensure high concentration of active surface species necessary for efficient ammonia conversion. The impact of particle size on activity is substantially more significant than the impact of the studied supports. However, a support can be used as an additional variable to adjust catalytic properties. Thus, the zeolite support increases catalyst selectivity and activity in the dry feed; however, it leads to stronger deactivation in the presence of water compared to alumina. Further identification of the states found in the platinum-containing catalysts and the surface species from which they originate is expected to deepen the understanding of the mechanism of this reaction and enable a more rational catalyst design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by Helmholtz Initiative and Networking Fund (HRSF-0046). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III, and we would like to thank Dr. Vadim Murzin (DESY), Dr. Wolfgang Caliebe (DESY) and Dr. Benjamin Bornmann (BU Wuppertal, Germany) for assistance in using P64 beamline. Beamtimes were allocated for proposals I-20200270 and I-20181000. A further part of the XAS spectra for this research was measured at the CAT-ACT beamline of the KIT light source (Karlsruhe, Germany). We thank Dr. Anna Zimina for her support during the measurements. The authors express their gratitude to Dr. Paolo Dolcet and Simon Barth (ITCP/IKFT, KIT) for their help during synchrotron measurements. We also thank Dr. Andrey I. Stadnichenko, Dr. Lidiya S. Kibis, Dr. Dmitry A. Svintsitskiy, and Dr. Andrei I. Boronin (Boreskov Institute of Catalysis) for their help during beamtimes, project planning and for valuable discussions. The authors also acknowledge China Scholarship Council (CSC) for funding the PhD studies of Xiaohui Huang at the Karlsruhe Nano Micro Facility (KNMF) where the TEM data was recorded. We finally thank Dr. Di Wang (Institute of Nanotechnology, KIT) who supervised and assisted the TEM measurements. The authors are thankful to Peter Nossier for his help with the design of the cover image. We gratefully acknowledge Rye Terrell and Speck atomic system visualiser which was used for the design of the cover image.References
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- Y. Kojima, Int. J. Hydrogen Energy, 2019, 44, 18179–18192 CrossRef CAS.
- D. Miura and T. Tezuka, Energy, 2014, 68, 428–436 CrossRef CAS.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- N. Morlanes, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef CAS.
- T. Lan, Y. Zhao, J. Deng, J. Zhang, L. Shi and D. Zhang, Catal. Sci. Technol., 2020, 10, 5792–5810 RSC.
- F. Gao, Y. Liu, Z. Sani, X. Tang, H. Yi, S. Zhao, Q. Yu and Y. Zhou, J. Environ. Chem. Eng., 2021, 9, 104575 CrossRef CAS.
- L. Chmielarz and M. Jabłońska, RSC Adv., 2015, 5, 43408–43431 RSC.
- M. Colombo, I. Nova and E. Tronconi, Catal. Today, 2012, 197, 243–255 CrossRef CAS.
- P. S. Dhillon, M. P. Harold, D. Wang, A. Kumar and S. Y. Joshi, Chem. Eng. J., 2019, 377, 119734 CrossRef CAS.
- A. Scheuer, A. Drochner, J. Gieshoff, H. Vogel and M. Votsmeier, Catal. Today, 2012, 188, 70–79 CrossRef CAS.
- A. Scheuer, W. Hauptmann, A. Drochner, J. Gieshoff, H. Vogel and M. Votsmeier, Appl. Catal., B, 2012, 111–112, 445–455 CrossRef CAS.
- P. S. Dhillon, M. P. Harold, D. Wang, A. Kumar and S. Joshi, Catal. Today, 2019, 320, 20–29 CrossRef CAS.
- R. Imbihl, A. Scheibe, Y. F. Zeng, S. Günther, R. Kraehnert, V. A. Kondratenko, M. Baerns, W. K. Offermans, A. P. J. Jansen and R. A. van Santen, Phys. Chem. Chem. Phys., 2007, 9, 3522–3540 RSC.
- A. Scheibe, U. Lins and R. Imbihl, Surf. Sci., 2005, 577, 1–14 CrossRef CAS.
- J. Pérez-Ramírez, E. V. Kondratenko, G. Novell-Leruth and J. M. Ricart, J. Catal., 2009, 261, 217–223 CrossRef.
- R. Kraehnert and M. Baerns, Appl. Catal., A, 2007, 327, 73–81 CrossRef CAS.
- M. Baerns, R. Imbihl, V. A. Kondratenko, R. Kraehnert, W. K. Offermans, R. A. van Santen and A. Scheibe, J. Catal., 2005, 232, 226–238 CrossRef CAS.
- J. Schäffer, V. A. Kondratenko, N. Steinfeldt, M. Sebek and E. V. Kondratenko, J. Catal., 2013, 301, 210–216 CrossRef.
- Y. F. Zeng and R. Imbihl, J. Catal., 2009, 261, 129–136 CrossRef CAS.
- D. P. Sobczyk, E. J. M. Hensen, A. M. de Jong and R. A. van Santen, Top. Catal., 2003, 23, 109–117 CrossRef CAS.
- Y. Li and J. N. Armor, Appl. Catal., B, 1997, 13, 131–139 CrossRef CAS.
- A. M. Gänzler, B. Betz, S. Baier-Stegmaier, S. Belin, V. Briois, M. Votsmeier and M. Casapu, J. Phys. Chem. C, 2020, 124, 20090–20100 CrossRef.
- D. Decarolis, A. H. Clark, T. Pellegrinelli, M. Nachtegaal, E. W. Lynch, C. R. A. Catlow, E. K. Gibson, A. Goguet and P. P. Wells, ACS Catal., 2021, 11, 2141–2149 CrossRef CAS PubMed.
- G. A. Somorjai, K. R. McCrea and J. Zhu, Top. Catal., 2002, 18, 157–166 CrossRef CAS.
- J. J. Ostermaier, J. R. Katzer and W. H. Manogue, J. Catal., 1974, 33, 457–473 CrossRef CAS.
- J. J. Ostermaier, J. R. Katzer and W. H. Manogue, J. Catal., 1976, 41, 277–292 CrossRef CAS.
- A. C. M. van den Broek, J. van Grondelle and R. A. van Santen, J. Catal., 1999, 185, 297–306 CrossRef CAS.
- A. C. M. van den Broek, PhD Thesis, Eindhoven University of Technology, 1998, DOI:10.6100/IR517149.
- E. M. Slavinskaya, L. S. Kibis, O. A. Stonkus, D. A. Svintsitskiy, A. I. Stadnichenko, E. A. Fedorova, A. V. Romanenko, V. Marchuk, D. E. Doronkin and A. I. Boronin, ChemCatChem, 2021, 13, 313–327 CrossRef CAS.
- T. K. Hansen, PhD thesis, Technical University of Denmark, 2017, https://orbit.dtu.dk/en/publications/development-of-new-diesel-oxidation-and-nhsub3sub-slip-catalysts Search PubMed.
- L. S. Kibis, D. A. Svintsitskiy, A. I. Stadnichenko, E. M. Slavinskaya, A. V. Romanenko, E. A. Fedorova, O. A. Stonkus, V. A. Svetlichnyi, E. D. Fakhrutdinova, M. Vorokhta, B. Smid, D. E. Doronkin, V. Marchuk, J.-D. Grunwaldt and A. I. Boronin, Catal. Sci. Technol., 2021, 11, 250–263 RSC.
- D. A. Svintsitskiy, L. S. Kibis, A. I. Stadnichenko, E. M. Slavinskaya, A. V. Romanenko, E. A. Fedorova, O. A. Stonkus, D. E. Doronkin, V. Marchuk, A. Zimina, M. Casapu, J.-D. Grunwaldt and A. I. Boronin, ChemCatChem, 2020, 12, 867–880 CrossRef CAS.
- M. Machida, Y. Tokudome, A. Maeda, Y. Kuzuhara, T. Hirakawa, T. Sato, H. Yoshida, J. Ohyama, K. Fujii and N. Ishikawa, ACS Catal., 2020, 10, 4677–4685 CrossRef CAS.
- M. Casapu, A. Fischer, A. M. Gänzler, R. Popescu, M. Crone, D. Gerthsen, M. Türk and J.-D. Grunwaldt, ACS Catal., 2017, 7, 343–355 CrossRef CAS.
- R. A. van Santen, Acc. Chem. Res., 2009, 42, 57–66 CrossRef CAS PubMed.
- G. T. Whiting, F. Meirer and B. M. Weckhuysen, in XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer International Publishing, Cham, 2017, pp. 167–191, DOI:10.1007/978-3-319-43866-5_13.
- B. B. Sarma, F. Maurer, D. E. Doronkin and J.-D. Grunwaldt, Chem. Rev., 2023, 123(1), 379–444 CrossRef CAS PubMed.
- A. Boubnov, S. Dahl, E. Johnson, A. P. Molina, S. B. Simonsen, F. M. Cano, S. Helveg, L. J. Lemus-Yegres and J.-D. Grunwaldt, Appl. Catal., B, 2012, 126, 315–325 CrossRef CAS.
- A. Philippaerts, S. Paulussen, S. Turner, O. I. Lebedev, G. van Tendeloo, H. Poelman, M. Bulut, F. de Clippel, P. Smeets, B. Sels and P. Jacobs, J. Catal., 2010, 270, 172–184 CrossRef CAS.
- A. C. M. van den Broek, J. van Grondelle and R. A. van Santen, J. Catal., 1997, 167, 417–424 CrossRef CAS.
- M. Abramoff, P. Magalhães and S. J. Ram, Biophotonics Int., 2003, 11, 36–42 Search PubMed.
- A. Zimina, K. Dardenne, M. A. Denecke, D. E. Doronkin, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe, T. Spangenberg, R. Steininger, T. Vitova, H. Geckeis and J.-D. Grunwaldt, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. Baerlocher, W. M. Meier and D. H. Olson, in Atlas of Zeolite Framework Types, Elsevier, Amsterdam, The Netherlands, 5th edn, 2001, pp. 184–185 Search PubMed.
- B. F. Mentzen and G. Bergeret, J. Phys. Chem. C, 2007, 111, 12512–12516 CrossRef CAS.
- A. H. Clark, J. Imbao, R. Frahm and M. Nachtegaal, J. Synchrotron Radiat., 2020, 27, 551–557 CrossRef PubMed.
- J.-D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
- P. V. Menacherry and G. L. Haller, Catal. Lett., 1997, 44, 135–144 CrossRef CAS.
- K. Chakarova, M. Mihaylov and K. Hadjiivanov, Microporous Mesoporous Mater., 2005, 81, 305–312 CrossRef CAS.
- Y. Li, M. Kottwitz, J. L. Vincent, M. J. Enright, Z. Liu, L. Zhang, J. Huang, S. D. Senanayake, W.-C. D. Yang, P. A. Crozier, R. G. Nuzzo and A. I. Frenkel, Nat. Commun., 2021, 12, 914 CrossRef CAS PubMed.
- M. A. Newton, D. Ferri, G. Smolentsev, V. Marchionni and M. Nachtegaal, J. Am. Chem. Soc., 2016, 138, 13930–13940 CrossRef CAS PubMed.
- L. M. Kustov, D. Ostgard and W. M. H. Sachtler, Catal. Lett., 1991, 9, 121–126 CrossRef CAS.
- T. J. Toops, D. B. Smith, W. S. Epling, J. E. Parks and W. P. Partridge, Appl. Catal., B, 2005, 58, 255–264 CrossRef CAS.
- A. Y. Stakheev, E. S. Shpiro, O. P. Tkachenko, N. I. Jaeger and G. Schulz-Ekloff, J. Catal., 1997, 169, 382–388 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189 CrossRef CAS PubMed.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150 CrossRef CAS PubMed.
- F. C. Meunier, J. Phys. Chem. C, 2021, 125, 21810–21823 CrossRef CAS.
- F. Maurer, J. Jelic, J. Wang, A. Gänzler, P. Dolcet, C. Wöll, Y. Wang, F. Studt, M. Casapu and J.-D. Grunwaldt, Nat. Catal., 2020, 3, 824–833 CrossRef CAS.
- H. Wang, J.-X. Liu, L. F. Allard, S. Lee, J. Liu, H. Li, J. Wang, J. Wang, S. H. Oh, W. Li, M. Flytzani-Stephanopoulos, M. Shen, B. R. Goldsmith and M. Yang, Nat. Commun., 2019, 10, 3808 CrossRef PubMed.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C.-Y. Chen, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2014, 53, 8904–8907 CrossRef CAS PubMed.
- A. Kaftan, F. Kollhoff, T.-S. Nguyen, L. Piccolo, M. Laurin and J. Libuda, Catal. Sci. Technol., 2016, 6, 818–828 RSC.
- Q. Xu, B. T. Heaton, C. Jacob, K. Mogi, Y. Ichihashi, Y. Souma, K. Kanamori and T. Eguchi, J. Am. Chem. Soc., 2000, 122, 6862–6870 CrossRef CAS.
- B. P. Andreini, D. Belli Dell'Amico, F. Calderazzo, M. G. Venturi, G. Pelizzi and A. Serge, J. Organomet. Chem., 1988, 354, 357–368 CrossRef CAS.
- K. Feng, H. Z. Zhang, J. Gao, J. B. Xu, Y. M. Dong, Z. H. Kang and J. Zhong, Appl. Phys. Lett., 2020, 116, 191903 CrossRef CAS.
- G. Novell-Leruth, A. Valcárcel, A. Clotet, J. M. Ricart and J. Pérez-Ramírez, J. Phys. Chem. B, 2005, 109, 18061–18069 CrossRef CAS PubMed.
- R. S. Ghosh, P. S. Dhillon, M. P. Harold and D. Wang, Chem. Eng. J., 2021, 417, 128273 CrossRef CAS.
- H. Ma and W. F. Schneider, ACS Catal., 2019, 9, 2407–2414 CrossRef CAS.
- A. S. Ramachandran, S. L. Anderson and A. K. Datye, Ultramicroscopy, 1993, 51, 282–297 CrossRef CAS.
- G. A. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141, 909–913 CrossRef CAS.
- R. van Hardeveld and A. van Montfoort, Surf. Sci., 1966, 4, 396–430 CrossRef CAS.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- J. Pérez-Ramírez, E. V. Kondratenko, V. A. Kondratenko and M. Baerns, J. Catal., 2005, 229, 303–313 CrossRef.
- J. Pérez-Ramírez, E. V. Kondratenko, V. A. Kondratenko and M. Baerns, J. Catal., 2004, 227, 90–100 CrossRef.
- O. Ivashenko, N. Johansson, C. Pettersen, M. Jensen, J. Zheng, J. Schnadt and A. O. Sjastad, ACS Catal., 2021, 11, 8261–8273 CrossRef CAS.
- J. D. Gonzalez, K. Shojaee, B. S. Haynes and A. Montoya, Phys. Chem. Chem. Phys., 2018, 20, 25314–25323 RSC.
- C. Wang, D. Ren, G. Harle, Q. Qin, L. Guo, T. Zheng, X. Yin, J. Du and Y. Zhao, J. Hazard. Mater., 2021, 416, 125782 CrossRef CAS PubMed.
- M. M. Sun, S. N. Wang, Y. S. Li, Q. Wang, H. D. Xu and Y. Q. Chen, J. Taiwan Inst. Chem. Eng., 2017, 78, 401–408 CrossRef CAS.
- T. Yu, M. Xu, Y. Huang, J. Wang, J. Wang, L. Lv, G. Qi, W. Li and M. Shen, Appl. Catal., B, 2017, 204, 525–536 CrossRef CAS.
- A. Boubnov, H. W. P. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J.-D. Grunwaldt, J. Am. Chem. Soc., 2014, 136, 13006–13015 CrossRef CAS PubMed.
- B. Kerkeni, D. Berthout, D. Berthomieu, D. E. Doronkin, M. Casapu, J.-D. Grunwaldt and C. Chizallet, J. Phys. Chem. C, 2018, 122, 16741–16755 CrossRef CAS.
- A. R. Fahami, T. Gunter, D. E. Doronkin, M. Casapu, D. Zengel, T. H. Vuong, M. Simon, F. Breher, A. V. Kucherov, A. Brückner and J.-D. Grunwaldt, React. Chem. Eng., 2019, 4, 1000–1018 RSC.
- H. Wang, M. Lin, T. Murayama, S. Feng, M. Haruta, H. Miura and T. Shishido, J. Catal., 2021, 402, 101–113 CrossRef CAS.
- M. Y. Lin, B. X. An, T. Takei, T. Shishido, T. Ishida, M. Haruta and T. Murayama, J. Catal., 2020, 389, 366–374 CrossRef CAS.
- J. Singh, E. M. C. Alayon, M. Tromp, O. V. Safonova, P. Glatzel, M. Nachtegaal, R. Frahm and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2008, 47, 9260–9264 CrossRef CAS PubMed.
- H. Yoshida, S. Nonoyama, Y. Yazawa and T. Hattori, Phys. Scr., 2005, 115, 813–815 CrossRef.
- Y. Lei, J. Jelic, L. C. Nitsche, R. Meyer and J. Miller, Top. Catal., 2011, 54, 334–348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Ex situ XANES and EXAFS spectra, and the corresponding fits, HAADF-STEM images, additional catalytic data, Arrhenius plots and TOF calculations, and additional operando XANES data. See DOI: https://doi.org/10.1039/d2cy02095e |
| This journal is © The Royal Society of Chemistry 2023 |