
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineered aldolases catalyzing stereoselective aldol reactions between aryl-substituted ketones and aldehydes†
Eugenia Chukwu
Cornelius‡
,
Michael
Bartl‡
 ,
Louise J.
Persson
,
Ruisheng
Xiong
,
Daniela
Cederfelt
,
Farshid Mashayekhy
Rad
,
Thomas
Norberg
,
Sarah
Engel
,
Erik G.
Marklund
,
Doreen
Dobritzsch
and
Mikael
Widersten
*
,
Louise J.
Persson
,
Ruisheng
Xiong
,
Daniela
Cederfelt
,
Farshid Mashayekhy
Rad
,
Thomas
Norberg
,
Sarah
Engel
,
Erik G.
Marklund
,
Doreen
Dobritzsch
and
Mikael
Widersten
*
Department of Chemistry – BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden. E-mail: mikael.widersten@kemi.uu.se
First published on 26th July 2023
Abstract
An A129G/R134V/S166G triple mutant of fructose 6-phosphate aldolase (FSA) from Escherichia coli was further engineered with the goal to generate new enzyme variants capable of catalyzing aldol reactions between aryl substituted ketones and aldehydes. Residues L107 and L163 were subjected to saturation mutagenesis and the resulting library of FSA variants was screened for catalytic activity with 2-hydroxyacetophenone and phenylacetaldehyde as substrates. A selection of aldolase variants was identified that catalyze the synthesis of 2,3-dihydroxy-1,4-diphenylbutanone. The most active enzyme variants contained an L163C substitution. An L107C/L163C variant was further tested for activity with substituted phenylacetaldehydes, and was shown to afford the production of the corresponding diphenyl substituted butanones with good diastereoselectivities (anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn dr of 10 to 30) and reasonable to good enantioselectivities of syn enantiomers (er of 5 to 25).
:syn dr of 10 to 30) and reasonable to good enantioselectivities of syn enantiomers (er of 5 to 25).
Introduction
Generation of new carbon–carbon bonds forms the foundation of synthetic chemistry. The diverse aldol reaction is one of the most important routes since it provides the facile formation of a new C–C bond between two prochiral centers.1,2 Aldol reactions can be facilitated by organo- and metallo-organic catalysts offering electrophilic groups stabilizing the intermediate enolate, or by forming a transient covalent enamine intermediate.1,3,4The enzyme fructose 6-phosphate aldolase A (FSA) from Escherichia coli5 is of interest as a biocatalyst in carboligation reactions towards chiral ketols.6–14 The enzyme structure is that of a homodecamer of α8β8 (TIM) barrel-folded subunits, arranged in two pentameric rings15 (see Fig. S1 in the ESI†). FSA is a class I type aldolase and catalysis depends on the stabilization of a reactive enamine with an active-site lysine residue. The catalytic cycle in the aldol reaction can be described to encompass five steps (Fig. 1): ① nucleophilic attack of the catalytic lysine residue (K85 in FSA) on a donor carbonyl substrate. Proton transfer from the ammonium nitrogen to the ketone-derived oxyanion generates an unstable carbinolamine intermediate. ② Protonation of the hydroxyl group of the carbinolamine, presumably with a water molecule in the active site acting as proton donor,16 facilitates expulsion of a water, forming the iminium. ③ Deprotonation of the alpha carbon of the iminium, again involving the mentioned active-site water molecule as proton acceptor (presumably assisted by Y131), forming a nucleophilic enamine. ④ Nucleophilic attack by the enamine on bound aldehyde acceptor substrate and protonation of the formed alcoholate, forms the new iminium intermediate. ⑤ Subsequent hydrolysis of the iminium releases the aldol product and concludes the catalytic cycle. Rate limiting step(s) of the reaction pathway has not been firmly established but can be assumed to be either the formation of the carbinolamine17 or the deprotonation of the iminium carbon acid.
 | ||
| Fig. 1 Schematic representation of the catalytic mechanism of class I aldolases. | ||
The native enzyme accepts a broad range of acceptors, including alkyl and aryl-substituted aldehydes, and variants have been engineered to further widen the aldehyde acceptance.11,18 A limiting factor, however, has been the more narrow scope of accepted donor ketone substrates. The native enzyme can only make efficient use of short-chain aliphatic ketones such as hydroxyacetone (1 in Chart 1) or dihydroxyacetone although also cyclic ketones can be conjugated to glyceraldehyde 3-phosphate.19 Again, enzyme engineering has created variants that exhibit wider scopes.20,21
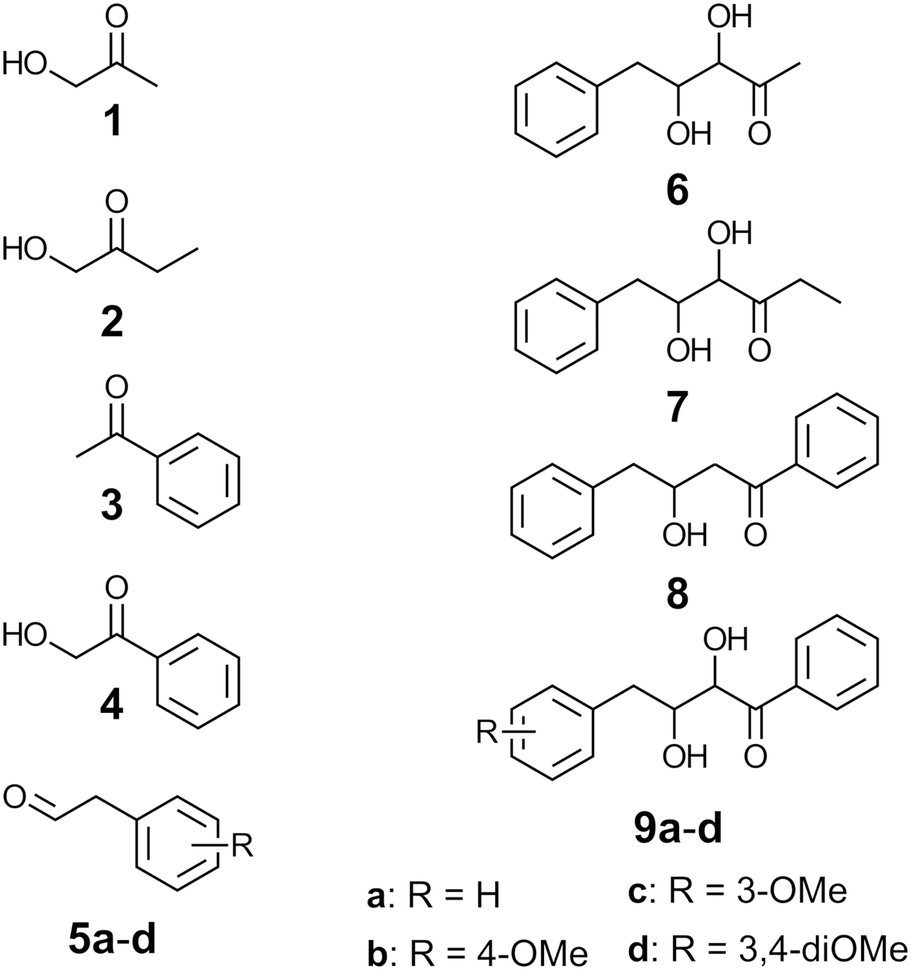 | ||
| Chart 1 Compounds studied. | ||
We set out to engineer FSA into accepting also aryl-substituted ketones such as acetophenone (3) or 2-hydroxyacetophenone (4). Such enzymes would open up additional avenues for biocatalytic synthesis of a more diverse range of polyhydroxylated ketols than currently available. Natural products built upon the phenone moiety have been isolated22–25 (Chart 2) that exhibit interesting biological activities such as cytotoxicity to tumor cells or anti-inflammatory activities. We have in an earlier study widened the aldehyde acceptance towards a range of substituted phenylacetaldehydes (5)11 and used the variant enzymes for synthesis of chiral di- and trihydroxypentanone derivatives.12 One of these engineered FSA variants contains two substitutions in the active site, R134V and S166G (dubbed ‘VG’). The rational for substituting R134 and S166, which in the native enzyme are assumed to interact with phosphorylated substrates, was to introduce amino acid residues that could better accept non-phosphorylated, aryl substituted acceptor aldehyde substrates.11
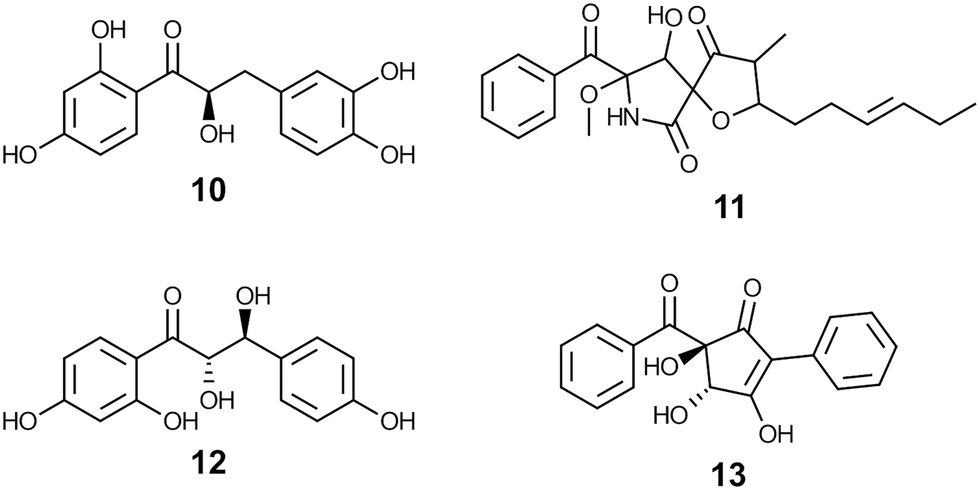 | ||
| Chart 2 Examples of natural products sharing core elements with aldols synthesized in this work. Compound 10 is a precursor to insecticidal glycosides,22 cephalimysin C (11) is cytotoxic to cultured tumor cell lines,23 compound 12 elicits growth inhibition in cultured tumor cells and displays antiinflammatory properties,24 and compound 13 has been tested for antifungal activity.25 | ||
We have solved the crystal structure of this VG variant to provide a detailed structural basis for further active-site engineering. Examination of its structure together with modeling and results from others26,27 led us to perform targeted mutagenesis in a step-wise manner, beginning with an A129G substitution that was expected to enlarge the active site cavity to accommodate a bulky ketone substituent, such as phenyl (Fig. 2). This new variant, ‘VGG’, was subsequently subjected to saturation mutagenesis of L107 and L163, creating an enzyme library (‘VGGX107X163’) in theory consisting of 399 additional variants. Fig. 2 shows a model structure that illustrates the hypothesis and starting point for the construction of the aldolase VGGX107X163 library. The two leucines are located on opposite sides of the phenyl substituent in the modeled active site cavity. Thus, alterations in the active-site structure by residue replacements were hypothesized to influence productive ketone binding and facilitate the formation of the transient carbinolamine, and the resulting iminium. It is noteworthy that the same leucine residues were mutated in the study of Güclü et al. (2016), who demonstrated that a double L107A/L163A variant exhibits a wider ketone scope.21 In our work, the goal was to further widen the ketone acceptance and to identify FSA variants that catalyze aldol additions between arylated ketones and aldehydes.
 | ||
| Fig. 2 Model of putative iminium formed following reaction of the VGG variant of FSA with ketone 4 and subsequent addition of glyceraldehyde. The model is based on the crystal structure of the VG variant of FSA11 with the A129G mutation introduced in silico. The phenyl subsituent (yellow) has also been modeled onto the trihydroxyhexanone linked to the active-site lysine (Fig. S1†). Albeit a crude model, it provides information of the expected spatial requirements for (productive) binding of the iminium intermediate generated from reaction with an arylated ketone such as 4. Residue 129, that has been mutated into glycine in the VGG variant is shown together with the two residues subjected to saturation mutagenesis (L107 and L163) are shown. The surface represents available space in the active-site cavity. Image created in Pymol ver. 2.5 (ref. 28) from the atomic coordinates in PDB entry 7QXF. | ||
Initially, the constructed enzyme library was screened via medium-throughput methods based on microtiter (96-wells) format for clone cultivation and crude enzyme sample production. The assay was based on coupling FSA catalyzed retroaldol cleavage of 9a with an engineered aldehyde reductase consuming NADH.29 Because of insufficient signal-to-noise ratios the library screening outcome was inconclusive and we instead turned to a low-throughput approach when searching the protein library; every aldolase variant was expressed and purified, and carboligation activity was assayed by chromatographic analysis of the reaction mixtures. Here we present the collected data on structure–activity relationships of >70 VGG L107X/L163X variants. We have identified specific enzymes that are capable of catalyzing asymmetric synthesis of aldols from 4 with a series of substituted phenylacetaldehydes (5a–d).
Results
Crystal structure of FSA R134V/S166G
The structure was solved to a resolution of 2.6 Å. Comparison of the overall structure with wild-type FSA15 reveals very small changes in the structure (RMSD of 0.96 Å3 over 8990 atoms). The two substitutions are clearly visible in the electron density map (Fig. S1D†) and result in a widening of the active-site cavity, presumably facilitating the binding of the bulkier 5 series of aldehydes. The VG variant crystallized with 15 subunits in the unit cell (Fig. S1C†) where the extra pentameric ring is a part of an adjacent decamer.The enzyme was co-crystallized with ketone 1 with the aim to obtain a crystal structure of the corresponding iminium. However, the electron density extending from the nucleophilic K85 rather suggests an adduct consisting of six carbons (Fig. S1D†). The relatively low resolution of the structure obscures a straightforward assignment of the bound molecule but it has been modeled as a proposed adduct between the iminium of 1 bound to K85 that has subsequently reacted with glycerol from the crystallization liquor.
Library distribution and general characteristics of mutants
The constructed VGG L107X/L163X (‘VGGX107X163’) library was sampled by sequencing 96 randomly picked variant E. coli clones. The distribution of amino acids found at the two mutation sites L107 and L163 differed, but all 20 possible proteinogenic amino acids were observed at least once, resulting in a reasonable cross section of the 400 possible combinations (Fig. 3). While alanine and cysteine were over-represented at position L107, lysine or isoleucine were not observed at all. Position L163 also displayed an uneven distribution with an over-representation of cysteine and glycine. Phenylalanine, proline and asparagine were not spotted at this position. Therefore, some limitations regarding a fully representative cross section had to be accepted. The gene sequencing and subsequent expression and purification of 96 proteins resulted in a total of 66 VGGX107X163 mutants, which were analyzed further in the context of a possible structure–function relationship. Some aldolase variants showed additional, not pre-intended point-mutations and were compared to their closest VGGX107X163 relatives when present (vide infra). Proteins encoded by genes carrying frame-shift mutations were excluded from characterization. During expression and purification of the different variants, links between amino acid substitutions and protein behavior in solution became evident.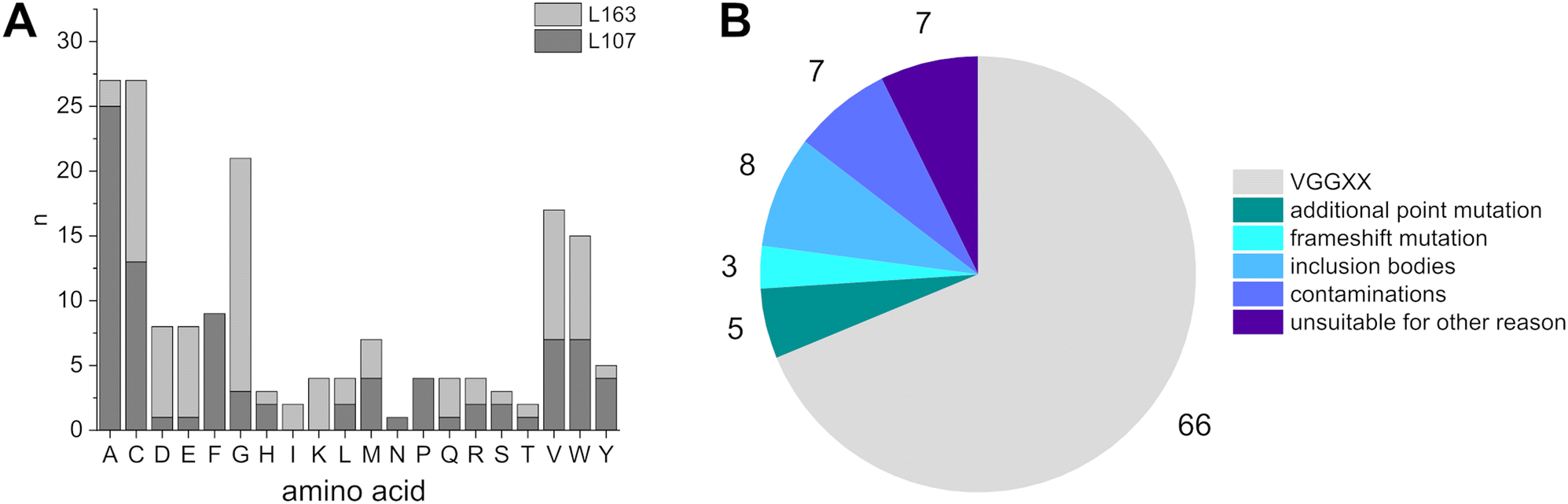 | ||
| Fig. 3 (A) Total count of amino acids at the two mutation sites of all sequenced samples. Residues of frame-shift mutations were not included in the histogram if the frame-shift occurred upstream of the targeted position. (B) Overview of the results of members of the VGGX107X163-library following gene sequencing and enzyme purification. Mutants with multiple characteristics were assigned to only one category, resulting in a total number of 96 initial samples. The VGG parent enzyme was not included in this statistics. | ||
Mutations to proline, arginine, aspartate and tryptophan resulted in direct effects on the solubility of the proteins with substitution to proline in all cases resulting in the formation of inclusion bodies. Variants that had acquired substitutions to arginine, aspartate or tryptophan generally were soluble, i.e. present in the supernatant fraction of the cleared lysates, but displayed varying tendencies to aggregate after purification. L163R mutants remained soluble, whereas an L107R mutant was not. VGGW107W163 formed inclusion bodies as well. Aldolases with an L163W or L163D mutation formed precipitates when exposed to high imidazole concentrations but remained soluble in other buffers. L107W and L107E mutants precipitated only occasionally at high imidazole concentrations. Some variants were not obtained at adequate enough yields to allow for conclusive characterization. Out of the 96 isolates, 66 VGGX107X163 aldolase derived proteins were obtained. 13 mutants were obtained multiple times. Including the VGG parent, 48 unique variants were analyzed in full, equivalent to a library coverage of 12% (Fig. 3).
Isolation of FSA-VGGX107X163 variants able to synthesize di-phenyl substituted aldol 9a
It has been established earlier that ketone 1 is a well-accepted donor substrate for FSA VG, combined with aldehyde 5a to generate aldol 6.11 We confirmed that also the VGG variant catalyzed this reaction with comparable efficiency. Thus, the reaction with these substrates was assayed as a control of basic catalytic function in parallel to the sought-after aldol addition activity with ketone 4 and aldehyde 5a to generate 9a. The respective reactions were analyzed for product formation by reversed phase HPLC where the product peaks could be identified after comparisons with reference compounds comprising mixtures of anti- and syn-isomers of 6 and 9a.12 The assay also allowed us to judge the relative amounts of product formation and to estimate the distereomeric ratios directly from the HPLC chromatograms, using standardized conditions and enzyme concentrations.Others were scored as inactive and had apparently lost the ability to utilize this ketone.
 | ||
| Fig. 4 Bar chart of scored activities in FSA VGGX107X163 catalyzed formation of syn or anti-9a following incubation of ketone and aldehyde substrates in the presence of 10 μM (2 nmol) of respective FSA variants. Reaction mixtures were analyzed by reversed phase HPLC. The FSA VGGL107L163 parent data are shown as white bars with the anti-9a data to the left of syn-9a. The estimated error was ±13% (see the ESI† for experimental details, numerical values of integrated peak areas (Table S2†) and error estimation). | ||
The diastereomeric ratios between anti and syn isomers of 9a were estimated from the respective integrated peak areas of the diastereomers (Fig. 4, Table S2†). Again, enzymes exhibiting higher degree of selectivity were found in the L163C lineage of variants. The highest diastereoselectivity was observed with VGGV107C163 and VGGC107C163, but also with VGGN107C163.
The VGGC107C163 variant was further employed in the synthesis of 9a (10 μmol of 5a and 4, 0.2 μmol FSA VGGC107C163, purified yield: approximately 35%) to allow for NMR analysis of the aldol product.
The diastereomer ratio of 10:90 of anti:syn was estimated from the integrals of the C-2 and C-3 protons12 (Table 1). The obtained ratio of the two syn enantiomers was estimated after chiral HPLC to be 71:29 (Table 1, Fig. S3†). The same enzyme variant was also tested for the ability to accept methoxy substituted aldehydes 5b–d together with 4, and indeed this enzyme produced aldols 9b–d as judged by mass spectrometry analysis (Fig. S12†). Aldol 9b was produced at good diastereomer ratio, favoring the formation of the syn diastereomer by >90% (Table 1). The enantiomeric ratios were 3 to 25 between the syn enantiomers of 9a and 9b, respectively (Fig. S3†).
| Compound | dr anti:syn |
er of syn enantiomersa |
|---|---|---|
| a Estimated from integrated peak areas following chiral HPLC (Chiralpak AS-H). b Estimated from integrated peak areas following reversed-phase HPLC (C-18). c Estimated from integrated NMR peaks of protons at positions 2 and 3. | ||
| 9a | 7:93,b 10:90c |
71:29 |
| 9b | 6:94b |
96:4 |
Certain variants showed an increased formation of anti-9a relative to VGG. A combination of a hydrophobic and somewhat bulky residue at position 107 (V/M/F) with Gly at position 163 appears to favor formation of the anti diastereomer (Fig. 4). It is conceivable that the extra free volume introduced by the L163G substitution together with sterical limitations by a bulky residue at position 107 steers binding of the phenylacetaldehyde substrate to adapt a conformation favoring addition to the re face of the aldehyde, thereby preferentially forming the anti diastereomer. Reactions catalyzed by VGGV107G163 and VGGY107M163 gave the highest amounts of anti-9a, although also being promiscuous regarding stereoselectivities. These FSA variants thus appear to allow for less selective binding of aldehyde 5a, thereby allowing for attack from both the re and si faces of the carbonyl.
Effects of additional mutations
Out of a total of five aldolases that had acquired additional, not pre-designed point mutations (considered as artifacts introduced during gene syntheses by polymerase chain reactions), two were suitable for direct comparison to their closest VGGX107X163 relatives, VGGA107C163 with an additional T185N-mutation (referred to as VGGA107C163N185) and VGGV107V163 with a P187Q-mutation (VGGV107V163Q187, Fig. 5). While VGGA107C163 showed better catalytic properties regarding the reaction of 4 and 5a, as compared to VGG, the additional T185N-mutation impaired this activity approximately five-fold (Table S2†), and the activity with 1 and 5a was even further reduced (Fig. 5). VGGV107V163Q187 also displayed a lower degree of formation of 6 as compared to VGG or its closest relative VGGV107V163. The overall catalytic efficiency towards the 9a product was comparable to the one of VGGV107V163, but the anti-9a:syn-9a diastereomeric ratio dropped dramatically from 1:40 to 1:1.5, mainly due to increased formation of the anti diastereomer. P187 is not part of the active site but is situated at the C-terminal end of β8 contributing to the loop connecting to α8. The substitution for glutamine could affect the local folding and thereby influence the active-site structure via β8.
 | ||
| Fig. 5 (A) Localization of non-targeted T185N and P187Q mutations in the FSA VG monomer (a chain of the FSA VG decamer (PDB entry 7QXF)). (B) Effect of additional, unplanned mutations on the catalytic properties of the resulting variants. The FSA variants containing the additional T185N and P187Q-mutations, named VGGA107C163N185 (light green) and VGGV107V163Q187 (dark blue), respectively. This allowed a comparison to their closest relatives (VGGA107C163 in darker green and VGGV107V163 in light blue, respectively) as well as to VGG as parent reference (gray). Bars represent percentage of product formation as compared to the most active variant in this comparison. Product amounts were determined from the integrated peak areas detected at 212 (aldol 6) or 244 (aldol 9a) nm after separation of reaction mixtures by reversed phase HPLC. See Tables S1 and S2† for absolute values of peak integrals and the ESI† for experimental details and error estimations. | ||
Molecular dynamics simulation of enzyme–carbinolamine interactions in the active sites
For the aldolase to be catalytically competent, a ketone derived enzyme bound iminium must be formed. The iminium is the result of dehydration of an intermediate carbinolamine. As an approach to gain further information regarding structure-dynamics in the formation of the iminium state, via the carbinolamine intermediate state, molecular dynamics simulations of FSA were carried out. The wild-type, VGG and VGGC107C163 enzymes were modeled with both possible enantiomers of the carbinolamine intermediates in the respective active sites and simulations were run over 200 ns. t. The initial models inferred neutralization through rapid proton transfer between the nitrogen and the oxygen in the initial zwitterionic species.The simulations revealed that large conformational changes would be required to accommodate an (S)-configured carbinolamine, and hence, we consider this an improbable intermediate. Therefore, the analysis of the simulations were focused on the results obtained with the (R)-carbinolamine bound in these FSA variants (Fig. 6). The catalytically most active VGGC107C163 variant distinguished itself by favoring a shorter distance between the hydroxyls of Y131 and of the leaving group hydroxyl (Fig. 6B). The side-chains of the introduced C107 and C163 were frequently seen to stabilize the phenyl ring of the intermediate, as judged by the respective atomic distances (Fig. 6C), interactions that appear to result in a shortening of the distance between the Y131 phenol oxygen and the leaving group hydroxyl oxygen on the carbinolamine.
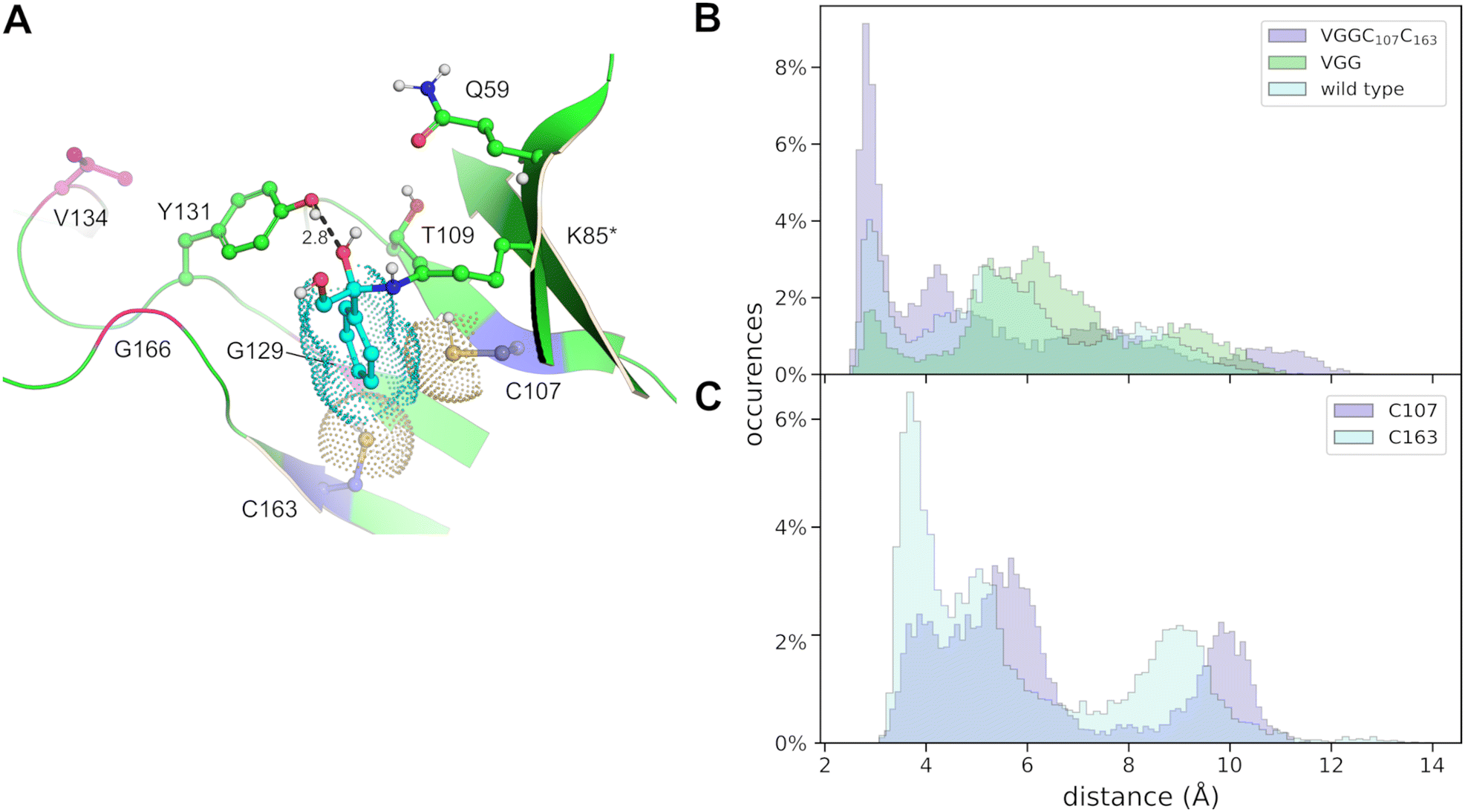 | ||
| Fig. 6 (A) Model of VGGC107C163 with the (R)-configured carbinolamine, showing a structure of the highest populated structure cluster. The two introduced C107 and C163 residues are shown together with the R134V, A129G and S166G substitutions from the VGG parent and the proposed catalytic amino acid residues Q59, T109 and Y131. The ketone derived portion of the carbinolamine (‘K85*’), is in cyan carbons connected to the K85 side-chain. The dot representation of the cysteines and the phenyl substituent illustrates a snug fit of the (R)-carbinolamine between these two residues. The atomic distance between the Y131 phenol oxygen and the leaving group hydroxyl of the carbinolamine is given in Å. (B) and (C), atomic distances observed during 100 ns molecular dynamics simulation between (B), the phenol oxygen of Y131 and the carbinol leaving group hydroxyl in either wild type (palecyan), VGG (green) or VGGC107C163 (palemagenta), or (C), between the sulfur atoms of C107 (palemagenta) or C163 (palecyan) and carbon-4 of the phenyl substituent of the (R)-carbinolamine in the VGGC107C163 variant. | ||
Discussion
The activity assays used in screening for functional variants were performed on individually purified enzymes which lowered the throughput of analyzed clones. We therefore ended the search after 96 tested enzyme expressing clones. However, the collected data provide a rich source of enzymes that exhibit altered, and in many cases non-overlapping, catalytic activities with different substrates. In this study, we limited ourselves to focus on the arylated ketone 4, but the produced enzyme library may prove to be a source of also other aldolase activities. Added benefits of performing isolation of each individual enzyme variant were also that more accurate analyses of expression levels, solubility and stability were possible, which is usually not the case when screening enzyme activity directly from cell lysates.Wild-type FSA favors low-molecular ketone substrates, and our preconception was that substitution into smaller amino acid residues such as Ala or Gly could be beneficial to allow productive binding of the bulkier ketone 4. The added accessible volume could possibly facilitate the initial imine formation since this process would be expected to include dynamic structural changes during the transition via the carbinolamine intermediate. However, somewhat surprisingly, the VGGX107G163 and VGGG107X163-variants did in general not exhibit notably increased activities with ketone 4. One could argue that Gly may destabilize the structure and cause inactivation of the enzyme, but several of the glycine containing variants did display reasonable activities with hydroxyacetone (1) as donor (Fig. S2†) thereby demonstrating that the enzymes are indeed functional but not improved for activity with the bulkier ketone 4. This underlines the elusiveness of the interplay between active-site architecture combined with structural dynamics that makes a priori structure–activity predictions challenging. The naive shotgun approach in the enzyme screening was therefore helpful in that beneficial amino acid combinations could be identified that would probably have been discarded in a more rational mutagenesis approach. The variants of highest aldol addition activities both contained cysteine at position 163 (VGGV107C163 and VGGC107C163). The mechanistic reasons for the improvement in activity with 4 are at this stage speculative, and further muddled by the lack of detailed understanding of rate limitations in catalysis. However, ab initio studies have suggested that S–π interactions between thiols and aromatic rings may contribute substantially to inter-molecular stabilization up to as much as 2.6 kcal mol−1 in relevant model systems, and with corresponding interactions between Cys thiols and aromatic rings of Phe or Tyr confirmed also in experimentally determined protein structures.30,31 It is therefore possible that the Cys substitutions indeed promote productive binding of the donor ketone and contribute to stabilization of the reactive eneaminol intermediate.
The molecular dynamics simulations provided information that aids in rationalizing the observed improvement in activity with 4 as ketone donor. The pathway to the essential enzyme bound iminium is via formation of an unstable carbinolamine intermediate (② in Fig. 7). Since the iminium is a prerequisite for the aldol reaction to proceed we hypothesized that a requirement for catalytic activity with aryl-substituted ketones would be the ability to facilitate the formation and accommodate the preceeding carbinolamine and the subsequent dehydration into the stable iminium. Thus, both possible enantiomers of the carbinolamine that could be generated following reaction between K85 and 4 were modeled in the active sites of the wild-type, the VGG and the VGGC107C163 variants.
 | ||
| Fig. 7 Formation of enzyme iminium in the VGGC107C163 as suggested by molecular dynamics simulations. ① Nucleophilic attack by the amino group of K85 on the si face of bound 4. ② Rapid proton exchange to generate an unstable carbinolamine intermediate. ③ Protonation of leaving group hydroxyl leading to expulsion of a water equivalent. ④ Formation of stable iminium. The iminium is subsequently deprotonated at the α-carbon to yield the nucleophilic eneaminol that may react with an incoming aldehyde. | ||
The simulation results suggest the following: (1) the sulfur atoms of the two introduced Cys residues in the VGGC107C163 variant are too far apart to form a disulfide bond (average distance ∼6 Å, Table S7†). (2) The only carbinolamine enantiomer that can lead to the iminium state has the (R)-configuration. This conclusion is based on the necessary geometry for possible protonation of the leaving group hydroxyl by a proposed catalytic water molecule hydrogen bonded to Q59, T109 and Y131 (Fig. S15†).15,16 Furthermore, to generate the (S)-carbinolamine would require binding of the ketone in a conformation allowing for attack of K85 on the re face of the carbonyl. This would require extensive re-shaping of the active site, which is unlikely due to the expected unfavorable energy involved. To generate the (R) enantiomer, the initial attack by the K85 amino group has to occur on the si face of bound ketone 4 substrate. (3) Accomodation of the phenyl substituent appears to be favored in the VGGC107C163 variant, as judged by the atomic distances in the highest populated structure clusters of wild-type FSA, and the VGG and VGGC107C163 variants (Fig. 6). Two additional results from the simulations are worth noting: firstly, the sulfur of C163 is in contact with the phenyl substituent of the carbinolamine to a larger extent compared to the sulfur of C107 (Fig. 6C). This may explain the fact that the cysteine at position 163 also appears to be the main determinant for activity with ketone 4 as judged by the activity profile in Fig. 4, where also VGGA107C163, VGGN107C163 and VGGV107C163, all show improved activities with 4, compared to the VGG parent. Secondly, the average distance between the phenol oxygen of Y131 and the leaving group hydroxyl oxygen is shortened in the highest populated structure cluster of the VGGC107C163 variant, compared to the VGG parent and the wild type (Fig. 6B and S15†). This may suggest direct proton transfer from Y131 to the leaving group, circumventing a water proton relay. If this observation is linked to the increased activity with ketone 4 is unlikely – rates of proton transfer reactions between electronegative atoms such as oxygen and nitrogen are expected to be many orders of magnitude faster than the catalytic rates determined here. Thus, the possible change in active site geometry may just be an accommodation to allow productive binding of the bulkier ketone substrate. It may, however, result in that the hydroxyl of Y131 becomes better positioned in space for protonation of the leaving group hydroxyl, thereby increasing the frequency of iminium formation. Furthermore, when analyzing the abilities of these three FSA variants to accommodate a water molecule during molecular dynamics simulations, positioned for possible proton transfer to the carbinolamine leaving group, the wild-type enzyme displays the highest frequencies (Fig. S6†). However, further studies are clearly needed to establish the exact mechanism in this step of the catalytic cycle.
We have demonstrated, albeit at yet small scale and without any efforts of optimizing the product work-up, that the VGGC107C163 enzyme may be considered as a starting enzyme scaffold for development of useful biocatalysts affording asymmetric synthesis of bis-arylated ketols. The produced diphenyl substituted compounds share structural features with isolated natural products with biological activities, such as compounds 10 and 12 in Chart 2. The obtained isomeric ratios in the aldol products formed with methoxy substituted aldehyde 5b by far exceed those that have been obtained with the organocatalyst cinchonine in the synthesis of this class of compounds.12 The products are enriched enantiomers of the syn diasteromer configuration, in line with previously described reactions with short-chain ketones.7,11,12,18 The somewhat mediocre enantiomeric ratio between the syn-9a aldols, synthesized by the VGGC107C163 variant, will require further refinement to yield similar discrimination as obtained with the methoxy substituted aldehydes. However, differences in the respective proportions of enantiomer ‘B’ between aldols 9a and 9b (Fig. S3†), implies stereo-restricting mechanisms that depend on the different structures of the acceptor aldehydes. In order to shift the configuration around carbon-2, rotation of the nucleophilic enamine (or the preceding iminium) has to occur. If such rotations indeed are present it should be independent of the aldehyde since the enamine is supposedly generated before aldehyde binding. Thus, the higher degree of the enantiomeric ratios in aldol 9b would then suggest restrictions in aldehyde binding that favor productive complexes preferentially leading to the ‘syn-A’ isomer. These attempts to explain the observed differences in stereoselectivity are still speculative and demand further investigations for clarification of the mechanisms.
Experimental
Experiments and methods are summarized here with detailed descriptions in the ESI.† A C-terminally His-tagged A129G/R134V/S166G triple mutant of FSA, dubbed ‘VGG’ was employed as starting scaffold in the aldolase library construction. Its gene was employed to template codon unbiased saturation mutagenesis32 of codon positions L107 and L163 achieved by overlapping polymerase chain reactions (Fig. S2†). Library DNA was transformed into E. coli BL-21 AI [pREP4-GroEL-ES] allowing for co-expression of chaperonins GroEL/ES.33FSA variants were expressed at 100 ml culture scale and purified by immobilized nickel ion chromatography. Purified proteins were subsequently challenged for aldolase activity with either hydroxyacetone (1) or 2-hydroxyacetophenone (4) as donor ketones and with phenylacetaldehyde (5a) as acceptor aldehyde. Reaction mixtures were analyzed by reversed phase (C-18) HPLC. A selection of isolated enzyme variants was also tested for activity with ketones 2 or 3 together with 5a. Products were identified from their chromatographic properties after comparing with reference compounds, and in cases also by NMR spectroscopy and ESI-MS/MS spectrometry. Diastereomeric ratios of obtained aldol products were analyzed by reversed phase HPLC and 1H-NMR (H2–H3 integrals in the case of aldol 9a, Fig. S7†) and enantiomeric ratios by chiral HPLC.
Steady state kinetic parameters of the reaction between 4 and 5a, catalyzed by VGGC107C163 were determined.
The FSA variant R134V/S166G (‘VG’) was crystallized and the 3-D structure solved by molecular replacement with wild type FSA15 as search model (Fig. S5†).
Molecular dynamics simulations were carried out with the Gromacs simulation package (https://doi.org/10.1016/j.softx.2015.06.001) for decameric wild-type FSA as well as the VGGC107C163 and VGG variants. In each monomer, K85 had been remodeled as the carbinolamine intermediate. Details are provided in the ESI.†
Conclusions
We have isolated new FSA derived enzyme variants that catalyze stereoselective synthesis of biphenyl substituted aldols. The enzymes provide protein scaffolds for refinement of (bio)catalytic properties and as models for studies of fundamental enzymological studies. The structure–activity data, points towards amino acid position 163 to be a determinant for improved activity with a bulky ketone.Author contributions
ECC, MB, RX, DC, FMR, TN, SE and DD contributed different parts of the experimental work. LJP and EGM conducted the molecular dynamics simulations. MW planned the work together with all co-authors. All authors contributed to writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The contributions of MSc Noha Saad are gratefully acknowledged. This work was supported by Olle Engkvist Byggmästare foundation (MW, #194-0638 and #218-0061). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC, partially funded by the Swedish Research Council through grant agreement no. 2018-05973.Notes and references
- B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC.
- Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC.
- B. List, Synlett, 2001, 11, 1675–1686 CrossRef.
- W. Notz, F. Tanaka and C. F. Barbas, III, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed.
- M. Schürmann and G. A. Sprenger, J. Biol. Chem., 2001, 276, 11055–11061 CrossRef PubMed.
- A. K. Samland and G. A. Sprenger, Appl. Microbiol. Biotechnol., 2006, 71, 253–264 CrossRef CAS PubMed.
- A. L. Concia, C. Lozano, J. A. Castillo, T. Parella, J. Joglar and P. Clapés, Chem. – Eur. J., 2009, 15, 3808–3816 CrossRef CAS PubMed.
- P. Clapés, W.-D. Fessner, G. A. Sprenger and A. K. Samland, Curr. Opin. Chem. Biol., 2010, 14, 154–167 CrossRef PubMed.
- P. Clapés and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263–2283 CrossRef.
- K. Fesko and M. Gruber-Khadjawi, ChemCatChem, 2013, 5, 1248–1272 CrossRef CAS.
- H. Ma, S. Engel, T. R. Enugala, D. Al-Smadi, C. Gautier and M. Widersten, Biochemistry, 2018, 57, 5877–5885 CrossRef CAS PubMed.
- D. Al-Smadi, T. R. Enugala, V. Kessler, A. R. Mhasal, S. C. L. Kamerlin, J. Kihlberg, T. Norberg and M. Widersten, J. Org. Chem., 2019, 84, 6982–6991 CrossRef CAS PubMed.
- M. Widersten, Methods Enzymol., 2020, 644, 149–167 CAS.
- V. Hélaine, C. Gastaldi, M. Lemaire, P. Clapés and C. Guérard-Hélaine, ACS Catal., 2022, 12, 733–761 CrossRef.
- S. Thorell, M. Schürmann, G. A. Sprenger and G. Schneider, J. Mol. Biol., 2002, 319, 161–171 CrossRef CAS PubMed.
- L. Stellmacher, T. Sandalova, S. Lepthin, G. Schneider, G. A. Sprenger and A. K. Samland, ChemCatChem, 2015, 7, 3140–3151 CrossRef CAS.
- I. A. Rose, E. L. O'Connell and A. H. Mehler, J. Biol. Chem., 1965, 240, 1758–1765 CrossRef CAS PubMed.
- A. Soler, M. L. Gutiérrez, J. Bujons, T. Parella, C. Minguillon, J. Joglar and P. Clapés, Adv. Synth. Catal., 2015, 357, 1787–1807 CrossRef CAS.
- R. Roldán, I. Sanchez-Moreno, T. Scheidt, V. Hélaine, M. Lemaire, T. Parella, P. Clapés, W.-D. Fessner and C. Guérard-Helaine, Chem. – Eur. J., 2017, 23, 5005–5009 CrossRef PubMed.
- R. Roldán, K. Hernandez, J. Joglar, J. Bujons, T. Parella, I. Sánches-Moreno, V. Hélaine, M. Lemaire, C. Guérard-Hélaine, W.-D. Fessner and P. Clapés, ACS Catal., 2018, 8, 8804–8809 CrossRef PubMed.
- D. Güclü, A. Szekrenyi, X. Garrabou, M. Kickstein, S. Juncker, P. Clapés and W.-D. Fessner, ACS Catal., 2016, 6, 1848–1852 CrossRef.
- L. Alvarez and G. Delgado, Phytochemistry, 1999, 50, 681–687 CrossRef CAS.
- T. Yamada, E. Imai, K. Nakatuji, A. Numata and R. Tanaka, Tetrahedron Lett., 2007, 48, 6294–6296 CrossRef CAS.
- K. H. Kim, E. Moon, S. U. Choi, S. Y. Kim and K. R. Lee, Phytochemistry, 2013, 92, 113–121 CrossRef CAS PubMed.
- D. Kälvö, A. Menkis and A. Broberg, Molecules, 2018, 23, 1417–1427 CrossRef PubMed.
- J. A. Castillo, C. Guérard-Hélaine, M. Gutiérrez, X. Garrabou, M. Sancelme, M. Schürmann, T. Inoue, V. Hélaine, F. Charmantray, T. Gefflaut, L. Hecquet, J. Joglar, P. Clapés, G. A. Sprenger and M. A. Lemaire, Adv. Synth. Catal., 2010, 352, 1039–1046 CrossRef CAS.
- A. Szekrenyi, A. Soler, X. Garrabou, C. Guérard-Helaine, T. Parella, J. Joglar, M. Lemaire, J. Bujons and P. Clapés, Chem. – Eur. J., 2014, 20, 12572–12583 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC Search PubMed.
- H. Ma, T. R. Enugala and M. Widersten, ChemBioChem, 2015, 16, 2595–2598 CrossRef CAS PubMed.
- G. Duan, V. H. Smith Jr and D. F. Weaver, Mol. Phys., 2001, 99, 1689–1699 CrossRef CAS.
- A. L. Ringer, A. Seneneko and C. D. Sherrill, Protein Sci, 2007, 16, 2216–2223 CrossRef CAS PubMed.
- L. Tang, H. Gao, X. Zhu, X. Wang, M. Zhou and R. Jiang, BioTechniques, 2012, 52, 149 CrossRef CAS PubMed.
- G. E. Dale, H.-J. Schönfeld, H. Langen and M. Stieger, Protein Eng., 1994, 7, 925–931 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00181d |
| ‡ Have provided equal contributions to the presented work. |
| This journal is © The Royal Society of Chemistry 2023 |