
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding W/H-ZSM-5 catalysts for the dehydroaromatization of methane†
Mustafa
Çağlayan
 a,
Abdallah
Nassereddine
b,
Stefan-Adrian F.
Nastase
c,
Antonio
Aguilar-Tapia
d,
Alla
Dikhtiarenko
a,
Sang-Ho
Chung
e,
Genrikh
Shterk
a,
Tuiana
Shoinkhorova
a,
Jean-Louis
Hazemann
b,
Javier
Ruiz-Martinez
e,
Luigi
Cavallo
c,
Samy
Ould-Chikh
c and
Jorge
Gascon
*a
a,
Abdallah
Nassereddine
b,
Stefan-Adrian F.
Nastase
c,
Antonio
Aguilar-Tapia
d,
Alla
Dikhtiarenko
a,
Sang-Ho
Chung
e,
Genrikh
Shterk
a,
Tuiana
Shoinkhorova
a,
Jean-Louis
Hazemann
b,
Javier
Ruiz-Martinez
e,
Luigi
Cavallo
c,
Samy
Ould-Chikh
c and
Jorge
Gascon
*a
aKAUST Catalysis Center (KCC), Advanced Catalytic Materials, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bInstitut Néel, UPR 2940 CNRS – Université Grenoble Alpes, Grenoble F-38000, France
cKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia
dInstitut de Chimie Moléculaire de Grenoble, UAR2607 CNRS Université Grenoble Alpes, Grenoble F-38000, France
eKAUST Catalysis Center (KCC), Catalysis Nanomaterials and Spectroscopy, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia
First published on 22nd March 2023
Abstract
Tungsten is the most interesting and promising metal to replace molybdenum in methane dehydroaromatization (MDA) catalysis. Located in the same column of the periodic table, tungsten displays similar chemical features to molybdenum (i.e., formation and stability of oxidation states, acidity of trioxides, tendency toward formation of polynuclear species, atomic radii, ionic radii, etc.) but shows higher thermal stability. The latter could be an advantage during high-temperature reaction–regeneration cycles. However, the MDA activity of W–ZSM-5 catalysts is much lower than the activity obtained with their Mo counterpart. In order to gain a further understanding of such differences in catalytic activity, we present a thorough investigation of the effect of dispersion and distribution of W sites on the zeolite, their relation with catalytic activity, and the temporal evolution of dispersion with reaction–regeneration cycles. The structure of W sites is elucidated with advanced and detailed characterization techniques, including operando X-ray absorption spectroscopy (XAS). The information obtained can help the catalysis community to design better W catalysts for MDA and other reactions (i.e., metathesis, hydrocarbon cracking, hydrodesulfurization, isomerization, etc.) where this is the metal of choice.
Introduction
In today's world, increasing methane emissions1 and high transportation-storage costs of natural gas2 (compared to other fossil fuels) make on-site methane conversion into condensed phase chemicals a prominent topic in catalysis science. Despite the commercialized indirect technologies (i.e., through steam-reforming reaction to methanol synthesis or Fischer–Tropsch synthesis) to incorporate natural gas into the existing petrochemical infrastructure, there is still an intense desire to develop catalytic processes that would convert methane to value-added chemicals and fuels in one single step. In this spirit, the non-oxidative methane dehydroaromatization (MDA, 6CH4 = C6H6 + 9H2, ΔH998K = 598.4 kJ mol−1) over metal-incorporated zeolite catalysts remains of high interest among catalysis science researchers, who are trying to gain insight into the direct methane valorization and further improve the process despite its inherent challenges (i.e., thermodynamic limitations, high coke selectivity, quick catalyst deactivation).Since the discovery of MDA activity of Mo-loaded ZSM-5 catalyst,3 several different transition metals (W, Fe, Cr, V, Re, etc.)4 and zeolites (MCM-41, ZSM-8, TNU-9, MOR, SSZ-13, etc.)5 were utilized and investigated. Yet, the best combination for high MDA activity is still Mo/H-ZSM-5. Consequently, many researchers – including us – have focused on Mo/H-ZSM-5 catalysts intensively to understand the formation of active Mo sites and the reaction mechanism.6–10 Since the aromatization feature of MFI topology is well-known for many zeolite-catalyzed reactions (i.e., methanol-to-hydrocarbons,11 aliphatics aromatization,12etc.), further investigations on the zeolite fragment (topologies) of the catalyst may not be the prioritized research topic. However, there is still a gap of understanding for other metallozeolite catalysts; highlighting the importance of more research to address unresolved fundamental issues (i.e., nature of active catalytic sites) and applied aspects (i.e., enhancing aromatics yield, inhibiting coke formation). Among all the metals tested for MDA, the most interesting and promising one to replace Mo would be W, a well-known metathesis catalyst.13,14 It is known that Mo and W differ from Cr which is also a VIB group element in terms of oxidation state stability, reactivity of oxides, ionic radii, etc. features.15,16 Besides having similar chemical features to Mo (Table 1), W has certain advantages for the MDA process. One of them is its high thermal stability (Table 1). Since MoO3-based materials may suffer from sublimation at high temperatures, the high thermal stability of WO3 could be an advantage during high-temperature MDA reaction–regeneration cycles. Also, the interaction between Al3+ sites of zeolite and metal (Mo or W) is critical. In several studies, MDA-inactive extra-framework Al2(MoO4)3 (or Anderson-type aluminum polyoxymolybdates, Al(OH)6Mo6O18) species were reported on zeolites upon Mo introduction and thermal treatments.17–19 However, in the case of W, aluminum tungstate formation could not be observed. The detailed solid-state NMR study (27Al and 1H–27Al cross-polarization (CP) magic angle spinning (MAS) NMR) focusing on W/ZSM-5 catalysts from Feng Deng and colleagues20 revealed that the introduction of W may lead to distortion and dealumination of the zeolite framework, but no formation of aluminum tungstate species.
| Mo | W | |
|---|---|---|
| a Molybdenum is one of the exceptional elements from the diagonal rule in the periodic table. | ||
| Group | VIB | VIB |
| Electron configurationa | [Kr]5s14d5 | [Xe]6s24f145d4 |
| Atomic radius (vdW) | 209 pm | 210 pm |
| Atomic mass | 95.95 | 183.84 |
| Maximum oxidation state | 6+ | 6+ |
| Ionization energy of metal | 7.092 eV | 7.980 eV |
| Ionization energy of MO3 | 11.8 eV | 12.5 eV |
| Electron affinity of metal | 0.749 eV | 0.816 eV |
| Electron affinity of MO3 | 3.17 eV | 3.33 eV |
| MO3 melting point | 795 °C | 1472 °C |
On the other hand, the previous catalytic results20–25 indubitably show that the MDA activity of W is much lower than the activity of Mo. For instance, Iglesia et al.21 related the lower methane conversion and aromatics selectivity over W/H-ZSM-5 catalyst to the difficult carburization of WOx sites via CH4 compared to MoOx counterparts. They suggested WCx clusters as the main methane activation sites (contrary to some other studies discussed below), which they detected in situ after a 1 h MDA reaction at 700 °C via XANES. Lunsford et al.24 also showed that W/H-ZSM-5 catalysts bear very low methane conversion activity at 750 °C unless they are pre-treated with CO before the reaction. In their follow-up study,26 where they investigated various metals (V, Cr, Fe, W, and Mo) on ZSM-5 via XPS after CH4 treatment at 750 °C, they could observe only sub-oxides except Mo/H-ZSM-5 for which metal-carbide (Mo2C) was detected as well.13C MAS NMR studies from Deng et al.20,23 also corroborated the absence of tungsten carbide formation during MDA reaction at 627–727 °C. Although no tungsten carbide phases were detected in their case, they observed the formation of benzene, ethylene, and ethane on the catalyst. Thus, they suggested that WOx (x < 3) species formed during the initial stages (i.e., WO3 + CH4 → WOx + CO + CO2 + H2O) can activate methane. This was in line with the theoretical findings of Goddard et al.,27 who studied various Mo, W, and Cr molecular oxide structures for methane activation via DFT calculations. They found that molecular WO2 is the most active structure for the oxidative methane addition leading to methyl metal hydride (i.e., H–M(O2)–CH3). In contrast to these, Kozlov et al.,28 who applied high-resolution transmission electron microscopy on post-reacted W/ZSM-5 catalysts, found the presence of WCx(Oy) clusters inside the zeolite pores and proposed them as the main active sites for methane conversion. Besides these, Zhang and colleagues29 studied how catalyst preparation affects the W sites and MDA activity by regulating the pH of the aqueous (NH4)2WO2 impregnation solution. They observed the formation of (WO6)n− polytungstate ions in the precursor solution prepared at low pH values (ca. 2–3), which was different from (WO4)2− ions in the base (i.e., pH 8–9) solutions. Since easier W reduction and higher MDA activity were also detected with the catalyst prepared with an acidic precursor in their experiments, they related these to the W coordination in the precursor solution. In general, although the differences and gaps between the abovementioned previous studies can be explained with catalysts (i.e., preparation method, W amount, zeolite acidity), process conditions (i.e., contact time, reactor structure), and inherent challenges of the characterization techniques they used, we believe that further investigations would be helpful to understand the evolution and structure of active W sites during MDA reaction.
In the present study, we studied the MDA activity of W/ZSM-5 catalysts from a different perspective. Besides comparing W catalyst to Mo one in terms of catalytic performance, we investigated how the dispersion–distribution of W sites on zeolites affects the catalytic activity via changing W loading and applying high-temperature reaction–regeneration cycles. In addition to a battery of advanced ex situ characterization techniques (i.e., 27Al MAS NMR, XRD-refinement, DRIFT, pyridine-IR, micro-Raman, ICP-OES, etc.), we attempted to shed light on the activity of tungsten catalysts via operando XAS and DFT-based thermodynamic analyses.
Results and discussion
Observations on MDA performance of W/ZSM-5 catalysts
We started our investigations studying the effect of reaction temperature on catalytic performance for 2 wt% W/H-ZSM-5 (Silica/Alumina:26) catalyst (throughout the text, “XMetal-Zyy” notation was used to define the catalysts having “X wt% Metal” loading on H-ZSM-5 zeolite with “Silica/Alumina:yy”), which displays a good dispersion of W on the zeolite (see the discussions below and ESI†). As depicted in Fig. 1, the MDA activity of the W catalysts at 700 °C is almost negligible, with no production of aromatics and aliphatics. This is opposite to the Mo counterpart of the catalyst we used in our previous studies.6,17 2Mo-Z26 shows the typical MDA activity even at 675 °C under the same reaction conditions (see section S2.1 in ESI†). These results support the previous studies21,24,26 indicating that activation of W sites for MDA reaction is much more demanding than activating Mo. When the reaction temperature reached 750 °C, a significant amount of aromatics and aliphatics were detected in the effluent stream (Fig. 1b). However, this was different from the typical MDA activity observed over Mo/H-ZSM-5 catalysts. The benzene and ethylene yields at 750 °C were on the same level; the CH4 conversion and total yield were not reaching peak values and quickly decaying with time, as we observed in the case of Mo catalysts. This may imply that full activation (i.e., carburization) of W sites on 2W-Z26 was not achieved under this condition but partially reduced W sites seem to have the capability of attaining methane activation, as suggested by the previous studies.20,23,26,27 When the reactor temperature reached 800 °C (Fig. 1), a typical MDA activity from the 2W-Z26 catalyst could be observed. The methane conversion and product yields reached their maximum; most importantly, benzene became the primary product, like in the case of Mo/H-ZSM-5 catalysts. This performance at 800 °C implies the activation of W sites which lead to typical MDA activity.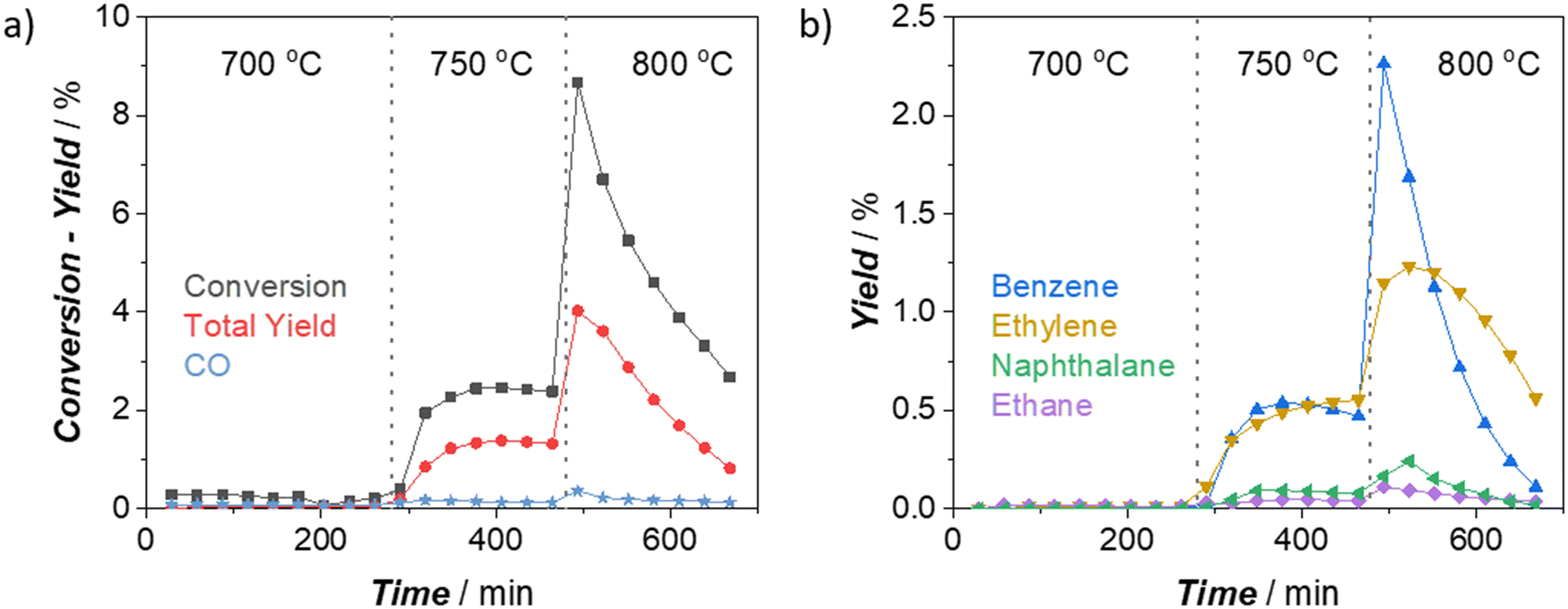 | ||
| Fig. 1 Catalytic performance of 2W-Z26 (250 mg) catalyst at different temperatures under atmospheric pressure (11.8 mol% N2 and 88.2 mol% CH4 in the inlet, total gas flow = 17 ml min−1 at STP): a) CH4 conversion, total yield (without CO and CO2), and CO yield, b) yields of major aromatic (benzene and naphthalene) and major aliphatic (ethylene and ethane) products. | ||
Furthermore, we studied how W loading affects the MDA performance at 750 and 800 °C (Fig. S3†) and examined these metal-loaded zeolites with 27Al MAS NMR, PXRD, XPS, N2 physisorption, TGA-TPO, DRIFT-IR, UV-vis DRS, and micro-Raman to understand the activity observed (see section S3 in the ESI† for detailed discussions). Catalysts having higher tungsten amounts (>2 wt%) were activated like 2Mo-Z26 catalyst at 750 °C and showed typical MDA performance (Fig. S3a–c†). This indicates that W amount and dispersion are critical parameters to initiate typical MDA activity at lower temperatures (<800 °C). Also, at both temperature levels, the overall performance (i.e., methane conversion, total product yield, and H2 production rate) was significantly enhanced, and the product yields (aliphatics and aromatics, Fig. S4a and b†) reached values comparable to 2Mo-Z26 catalyst as tungsten amount increased. However, the deactivation of the catalyst also became faster along with this enhancement in the performance related to the W amount. Similar to the previous experience17 with 2Mo-Z26; the higher the reaction temperature becomes, the faster the deactivation gets, and the higher initial activity occurs (Fig. S3 and S4†).
To critically analyze the MDA performances of these catalysts, we also calculated the product formation rates per mole of metal atom (turnover frequency, TOF) (Fig. 2 and 3). For these calculations, we used the metal amounts found by ICP-OES analysis (Table S4†). As presented in Fig. 2, the aromatic formation rates of 2Mo-Z26 are much higher than the W-loaded catalysts at both temperatures. However, this trend changes when it comes to aliphatic formation rates (Fig. 3). Whereas 2W-Z26 has the highest aliphatics TOF values (except ethane production at the early stages of 750 °C-experiment), 2Mo-Z26 and 3W-Z26 catalysts having very close metal densities (i.e., metalmmoles per gzeolite, Table S4†) show similar aliphatic performance at both temperature levels. Overall, these comparisons imply that Mo is a slightly better aromatization catalyst than W despite their similar aliphatic formation rates. Also, this activity per mole of W analyses indicates that there is no linear relation between product formation rates and the amount of metal on the zeolite. This clearly reveals that active metal site formation and accessibility are different depending on the loading. Therefore, the sample characterizations should be very carefully weighed here. When the dispersion/distribution of W was investigated with micro-Raman (Fig. S16a†) and UV-vis DRS (Fig. S20 and S21†), the spectroscopic features which could be attributed to the formation of monomeric and polymeric W sites in/on ZSM-5 particles were observed. However, in addition to the aforementioned W features, the crystalline WO3 formation was observed at 9 wt% W loading. For instance, the crystalline WO3-related vibrations (ν(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) WO) at 811 cm−1, ν(W2O6 & W3O8) at 717 cm−1, and ν(O–W–O) at 272 cm−1) were detected on the Raman spectrum of 9W-Z26 sample. However, these crystalline phase vibrations were broader than the bulk WO3 vibrations, which indicates nanoparticle formation rather than larger particles.30,31 As shown and discussed with the catalytic activity of WO3 & ZSM-5 physical mixture catalyst (2W-Z26 Phys-Mix) in the sections below (Fig. 5f and 6f), the formation of WO3 is very critical, because crystalline WO3 is not a convenient precursor for active W site formation. Thus, this points out that the primary activity on the 9W-Z26 catalyst mainly resulted from well dispersed–distributed W sites in/on the zeolite particles, not from the crystalline WO3 particles.
WO) at 811 cm−1, ν(W2O6 & W3O8) at 717 cm−1, and ν(O–W–O) at 272 cm−1) were detected on the Raman spectrum of 9W-Z26 sample. However, these crystalline phase vibrations were broader than the bulk WO3 vibrations, which indicates nanoparticle formation rather than larger particles.30,31 As shown and discussed with the catalytic activity of WO3 & ZSM-5 physical mixture catalyst (2W-Z26 Phys-Mix) in the sections below (Fig. 5f and 6f), the formation of WO3 is very critical, because crystalline WO3 is not a convenient precursor for active W site formation. Thus, this points out that the primary activity on the 9W-Z26 catalyst mainly resulted from well dispersed–distributed W sites in/on the zeolite particles, not from the crystalline WO3 particles.
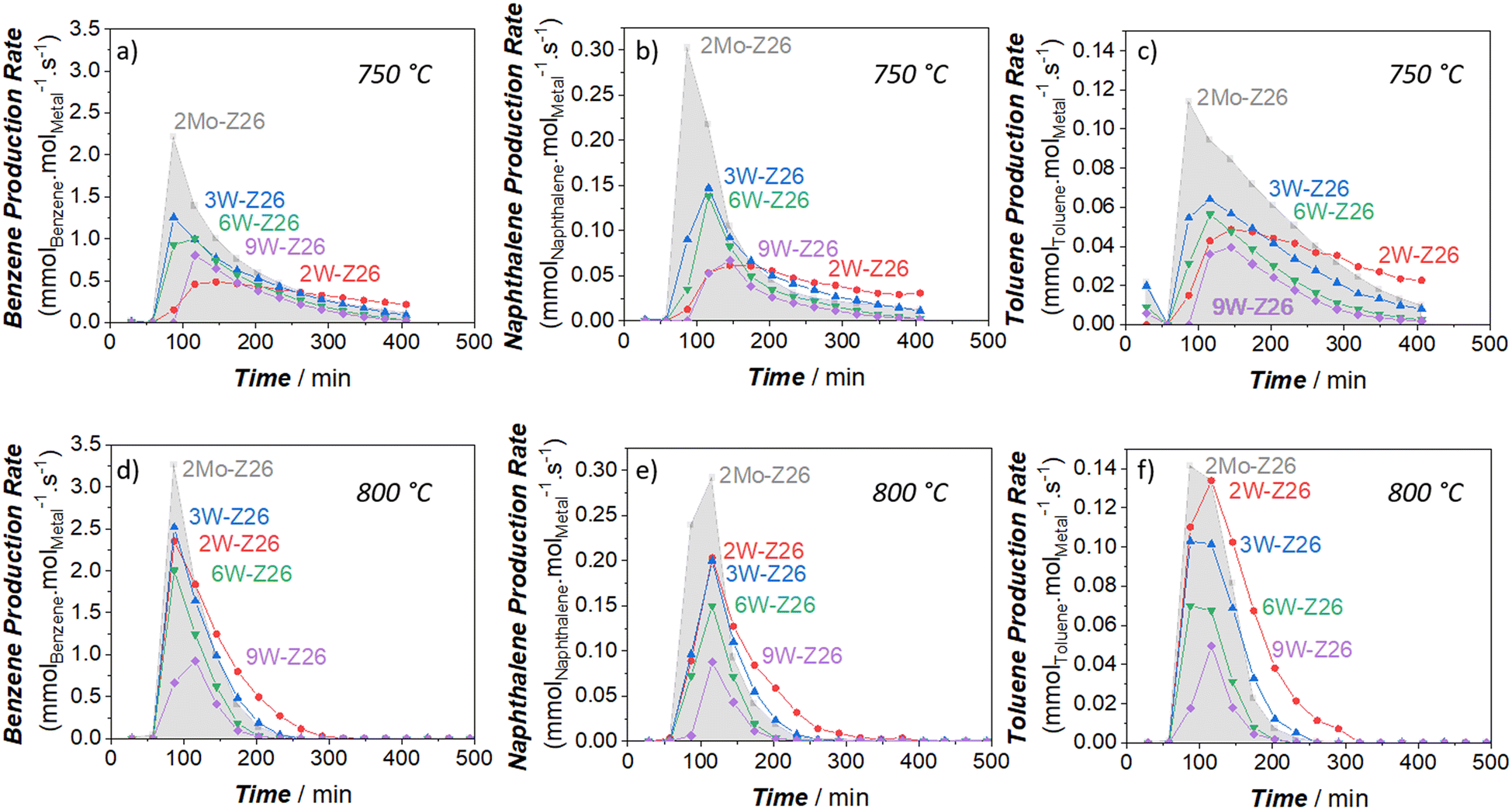 | ||
| Fig. 2 Aromatic production rates (TOFs) of the prepared W and Mo catalysts (250 mg) at 750 °C (top: a–c) and 800 °C (bottom: d–f); a and d: benzene – b and e: naphthalene – c and f: toluene (11.8 mol% N2 and 88.2 mol% CH4 in the inlet, total gas flow = 17 ml min−1 at STP). | ||
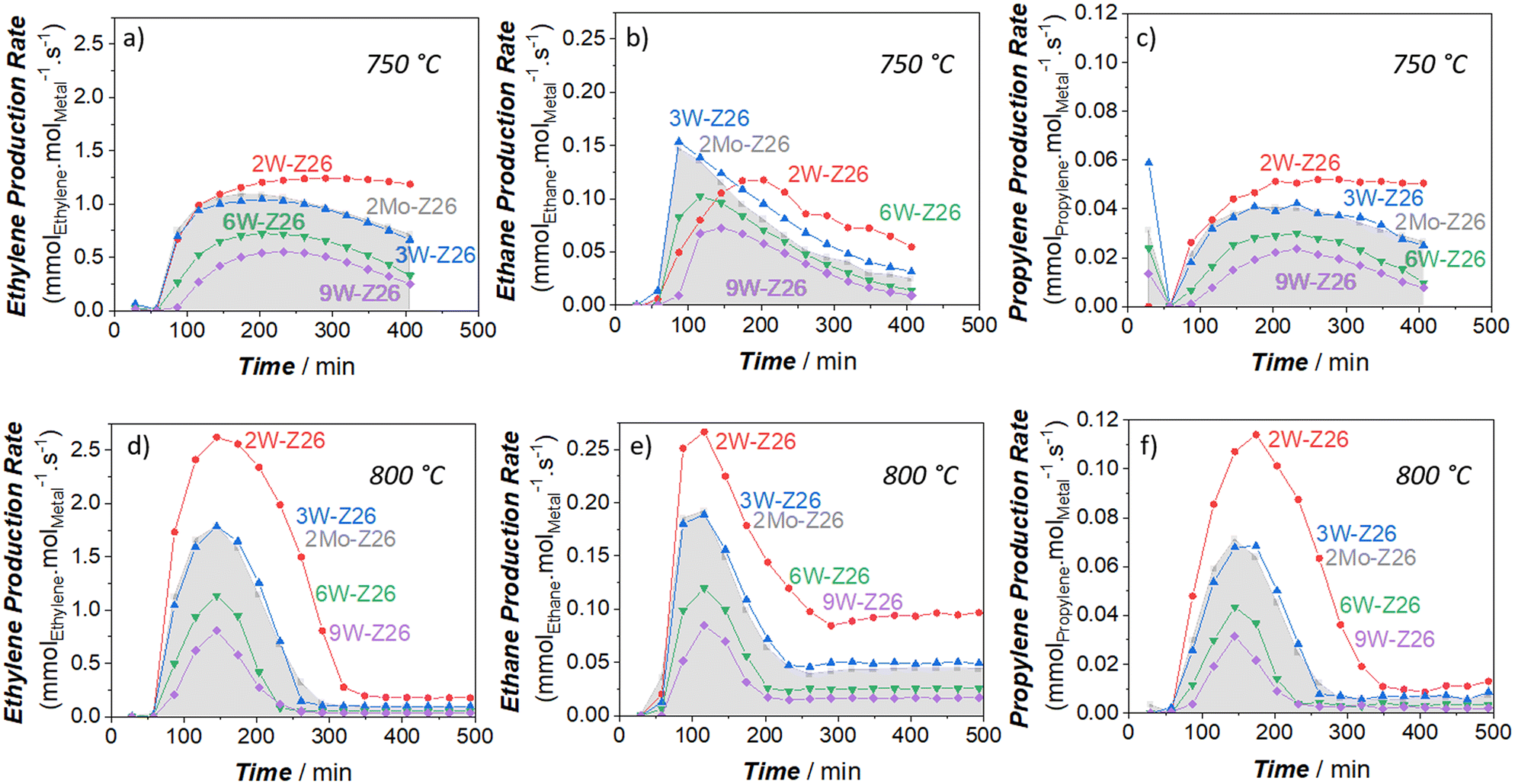 | ||
| Fig. 3 Aliphatic production rates (TOFs) of the prepared W and Mo catalysts (250 mg) at 750 °C (top: a–c) and 800 °C (bottom: d–f); a and d: ethylene – b and e: ethane – c and f: propylene (11.8 mol% N2 and 88.2 mol% CH4 in the inlet, total gas flow = 17 ml min−1 at STP). | ||
On the other side, we can state that 2Mo-Z26 catalyst has better metal dispersion/distribution and prevailing microporosity after Mo introduction based on our previous studies (in which we used the same catalyst)6,17 and characterizations presented here (vide infra section S3 in the ESI†). However, there are also pronounced differences compared to W-samples when it comes to metal–zeolite interaction and acidity. As depicted in our previous study17via27Al MAS NMR, Mo introduction leads to aluminum molybdate formation (i.e., Anderson-type aluminum polyoxymolybdates) whose tungsten-analogue was not observed in the W cases (Fig. S31†). Based on NH3-TPD (Table 2) and pyridine-IR (Table 3), there is a sharp loss of strong acid sites on 2Mo-Z26 catalyst upon metal introduction compared to W-Z26 samples. Pyridine-IR analyses indicate that these disappeared strong acids are mostly Brønsted acid sites (BASs) since 2Mo/Z26 has a lower BAS/LAS (Brønsted acid sites/Lewis acid site, B/L) ratio compared to bare ZSM-5 and W-loaded ones (Table 3). The well-dispersion of metals, lower acid density, and lower B/L ratio of 2Mo-Z26 catalyst clearly suggest that the high aromatization activity could be linked to the active Mo sites rather than the zeolite acidity. In the case of 2W-Z26, NH3-TPD analysis (see Table 2, Fig. S26†) revealed the loss of some acid sites upon W introduction, especially the strong ones. However, it is not to the same extent as its Mo counterpart. The loss of acidity could be explained by the anchoring of W cations on bridging oxygen sites between framework Al3+ and Si4+ atoms (see pyridine-IR data in Table 3, Fig. S28a and S29a†). However, this trend seems to be reversed at higher loadings (Table 2) since some tungsten sites provide acidity (except 9W-Z26 sample having nano-sized crystalline WO3 particles which could block pore entrances and decrease the accessibility). It is well known that supported WOx can act as an acid.32–35 The amount of W, dispersion–distribution, and support type are critical factors defining this W acidity. For instance, Wachs et al.33 found a correlation between site acidity and electron density of the bridging W–O–support bonding (i.e., as support (X) cation electronegativity increases, electron density of bridging W–O–X decreases and acidity increases), when they studied supported (Al2O3, Nb2O5, TiO2, and ZrO2) tungsten oxide catalysts for methanol-to-dimethylether-dehydration reaction. Also, W–support interaction is closely related to the dispersion of metal sites and their reducibility. For example, previous studies33,35 revealed that W–SiO2 interaction is very weak compared to W–Al2O3, and this could lead to bigger particles (i.e., polytungstates and aggregated crystalline WO3) compared to highly dispersed species (i.e., isolated tetrahedral) on alumina supports. Moreover, reducing tungsten sites on alumina were found more demanding than reducing tungsten sites on silica support via H2.35
| Samples | Peak position (°C) | Amount (μmol g−1 at STP) | |||
|---|---|---|---|---|---|
| Weak acid | Strong acid | Weak acid | Strong acid | Total acid | |
| Z26 | 201 | 361 | 282 | 427 | 709 |
| 2W-Z26 | 203 | 346 | 290 | 321 | 611 |
| 3W-Z26 | 209 | 361 | 316 | 353 | 669 |
| 6W-Z26 | 205 | 338 | 337 | 360 | 697 |
| 9W-Z26 | 205 | 320 | 338 | 282 | 620 |
| 2Mo-Z26 | 205 | 342 | 302 | 260 | 562 |
| Samples Fresh | 150 °C | 350 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| BAS (μmol g−1) | LAS (μmol g−1) | Total (μmol g−1) | B/L | BAS (μmol g−1) | LAS (μmol g−1) | Total (μmol g−1) | B/L | |
| Z26 | 409.2 | 264.6 | 673.8 | 1.55 | 251.9 | 95.7 | 347.6 | 2.63 |
| 2W-Z26 | 314.8 | 209.6 | 524.4 | 1.50 | 203.0 | 81.7 | 284.7 | 2.49 |
| 6W-Z26 | 334.6 | 202.6 | 537.2 | 1.65 | 202.5 | 97.0 | 299.5 | 2.09 |
| 2Mo-Z26 | 199.5 | 230.8 | 430.3 | 0.86 | 104.6 | 79.7 | 184.4 | 1.31 |
The MDA activity differences observed between 2W-Z26 and other W loaded catalysts at 750 °C could be explained through considering these. Apparently, there is a high probability of forming WOx sites attached to silanol groups and located on the external surface at higher loadings (>2 wt%). So, these W-sites attached to “silica phase” of zeolite could be easily activated/reduced in comparison to other tungsten sites attached to the BASs inside the pores. Eventually, this results in the activity differences (typical MDA vs. non-typical MDA) between 2W-Z26 and higher W-loaded samples at 750 °C. Similar examples showing a correlation between overall activity and W–support interaction can also be found in the literature36 (vide infra section S3 in the ESI†).
Beyond these, the pyridine-IR analyses performed to investigate the acid sites of fresh Z26, 2W-Z26, and 6W-Z26 depict that the number of BASs sharply decreases upon W introduction to zeolite compared to the decrease in the number of LASs (at 150 °C – after removing physisorbed pyridine). This indicates that W cations replace protons of some BASs. However, 6W-Z26 has slightly higher BASs than 2W-Z26 at 150 °C, which could mean there are more polymeric W sites at 6 wt% loading and results in more free zeolite BASs. When we further treated the samples at 350 °C and forced some pyridine to desorb, it was noticed that 2W-Z26 and 6W-26 have a similar number of BASs but 6W-Z26 has a higher amount of LASs. These LASs resulted from W-sites may explain the higher catalytic activity of 6W-Z26 at 750 °C. It is known that Lewis acid–base pairs can activate C–H bonds heterolytically:37 the electron-deficient/undercoordinated metal site (Lewis acid) can polarize the C–H bond and form σ-bond with CH3 moieties while pair basic site (i.e., neighbouring basic oxygen center) accepts a proton. Also, while one-oxidation state (W6+) was observed at 2 wt% loading, W5+ features were also identified to a small extent at 6 wt% loading on the XPS spectra (Fig. S30†) collected for fresh samples. The latter could imply that the formation of defective W sites becomes more probable at higher loadings. These defects could ease the activation of W sites (vide infra section S3 in the ESI†).
Understanding the MDA activity through DFT-based thermodynamic calculations: Mo vs. W
To gauge the reduction and stability of W species, we benefited from DFT-based calculations as we did in our previous publication.17 First of all, monomeric and dimeric W structures anchored on ZSM-5 were built and optimized (Fig. S54†). Then, we performed thermodynamic analysis for reduction-via-methane reactions of respective W and Mo states under a gas phase composition similar to the experimental one (for more details see section S5†).The thermodynamic analyses provided in Fig. 4 reveal that the different exergonic and endogenic steps for Mo structures are also exergonic and endogenic for the corresponding W models. In other words, they have similar trends (also see Fig. S55, S56, S58–S63 in the ESI†). However, when we compare the extent of exergonicity, we see that Mo species (both dimeric and monomeric) are significantly more prone to get reduced during the initial oxygen removal steps compared to W. This difference between the two metals could explain why it is much more demanding to activate W/ZSM-5 catalysts and attain typical MDA activity. Also, it is noteworthy that the reduction of dimeric structures is thermodynamically more spontaneous than monomeric ones. Since the formation of dimeric/polymeric structures is more possible at higher W loadings (>2 wt%), this finding can explain the observation of typical MDA activity for higher W loadings at 750 °C.
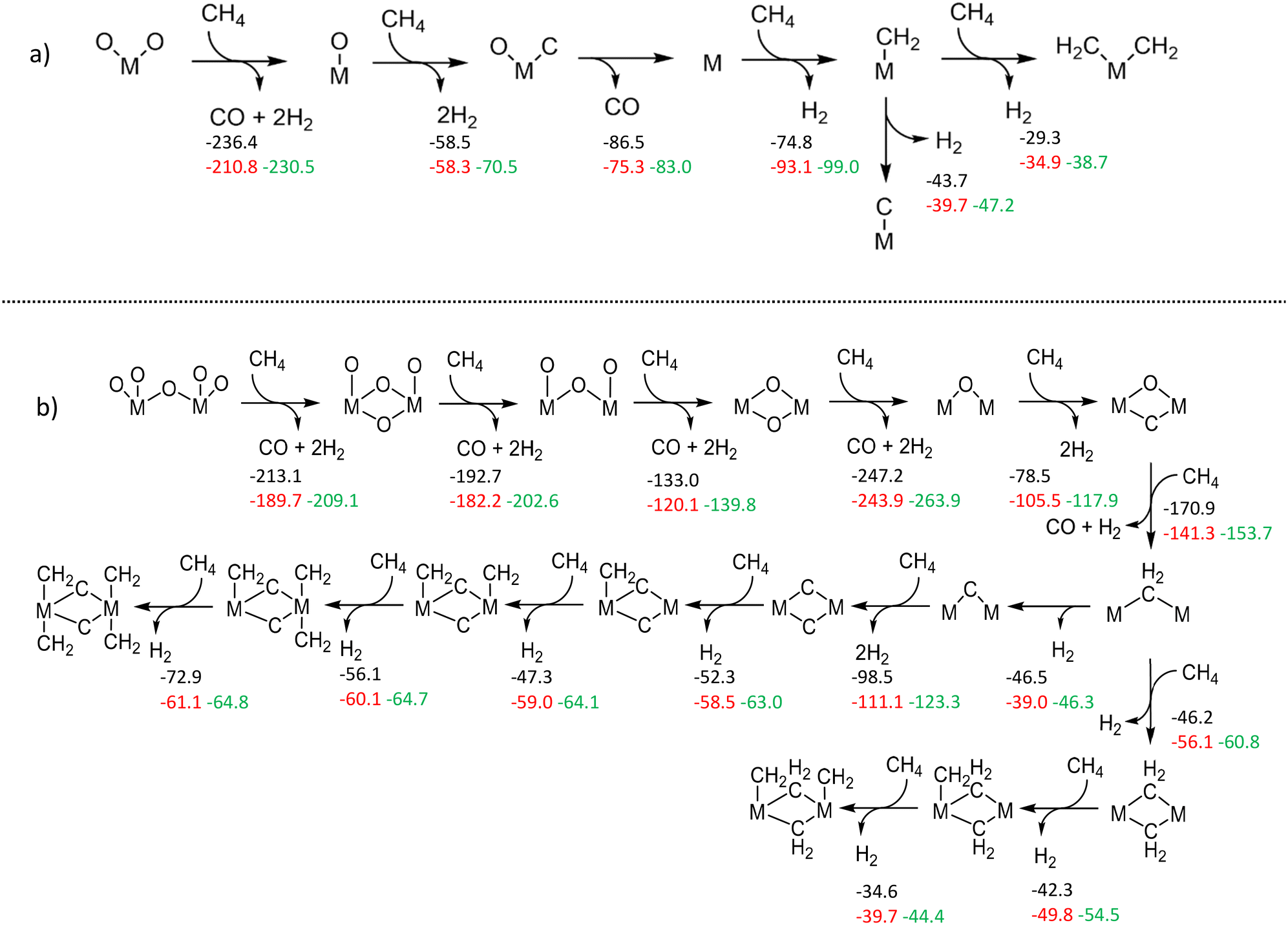 | ||
| Fig. 4 Illustration of methane activation over monomeric (a) and dimeric (b) oxide species of Mo and W, with Gibbs reaction free energies provided in kcal mol−1 at 725 °C (Mo – black, W – red) and 800 °C (W – green) and partial pressures as PCO: 10−15 atm, PH2: 10−15 atm, PN2: 0.11 atm, and PCH4: 1 − PCO − PH2 − PN2 atm. | ||
Another notable difference between Mo and W structures is the addition of carbenes (:CH2) to reduced/carbidic structures (Fig. 4). In the case of W structures, the reaction steps leading to the formation of carbene moieties are mostly more exergonic than Mo. This relative thermodynamic difference could help us in understanding the activity differences between Mo and W catalysts that we observed. Considering the arguments on carbenes which are considered as key intermediates for coupling and further oligomerization steps,38 we studied the thermodynamic feasibility of ethylene formation through the dimeric and monomeric structures in this study (vide infra Fig. S57†). It was found that carbene-dimerization-to-ethylene is relatively more thermodynamically favorable on Mo than on W. This could explain the activity difference between the two metals.
In general, our computational analysis corroborates our experimental findings, but one should remember the clusters we studied are simplified models. The real sites on zeolites are much more complex and dynamic, as we discussed throughout this study.
High temperature reaction–regeneration cycles of W/ZSM-5 catalysts
In the next phase of this study, to gain more in-depth insight into W activity, we applied high-temperature reaction–regeneration cycles on all catalysts mentioned in the previous sections (i.e., reaction at 750 °C–regeneration at 700 °C). This type of cyclic MDA performance evaluations for W-loaded catalysts was never performed before in previous studies. Here we brought benzene and ethylene TOFs (Fig. 5 and 6) to the fore since they are the major aromatic and aliphatic products, respectively (see Fig. S5–S9 in ESI† for other products). It is clear that the performance of 2Mo-Z26 catalysts deteriorated after each regeneration step (Fig. 5a and 6a), especially aromatics formation capability. However, in the case of its W counterpart (2W-Z26), the performance – particularly initial activity – was getting enhanced after each cycle (Fig. 5b and 6b); surprisingly, all production rates were significantly higher. One may relate the difference between these two catalysts to possible loss (i.e., sublimation) of Mo, but when we checked both catalysts after 4 regeneration cycles with ICP-OES, loss of metals was not detected (Table S4†). On the other hand, provocatively, as the W amount increases (>2 wt%), the enhancement tendency is no longer there. In contrast, the product formation rates (especially benzene) decreased, and much quicker deactivation was observed (Fig. 5c–e and 6c–e). So, this implies that 2W-Z26 catalyst, which was not fully activated in the first reaction run like the others (see the previous sections), experienced some structural changes leading to improved activity while others were degrading. To confirm that this enhancement tendency is dependent on W loading, we prepared another 2W-Z26 catalyst by simply mixing bulk WO3 and H-ZSM-5 physically (2W-Z26 Phys-Mix). The prepared catalyst showed almost negligible activity in the first run (Fig. 5f and 6f), especially for aromatics. This demonstrates that bulk WO3 is not a convenient precursor for active W site formation. On the other hand, 2W-Z26 Phys-Mix behaved like its equivalent after the regeneration steps; showed enhanced activity in a similar trend. This indicates there should be an optimal W loading range for the enhancement behavior. Furthermore, we investigated the effect of the zeolite's silica/alumina ratio (SAR) since it affects the distribution–dispersion of metal sites. As shown in Fig. S10a and b,† we applied the same reaction–regeneration cycles to the 2W-Z371 catalyst, which has higher SAR (371), and we did not observe a considerable enhancement in the production rates compared to 2W-Z26 catalyst. However, it also did not degrade either. So, it can be interpreted the aluminum content of the zeolite also plays a crucial role during the reaction–regeneration cycles.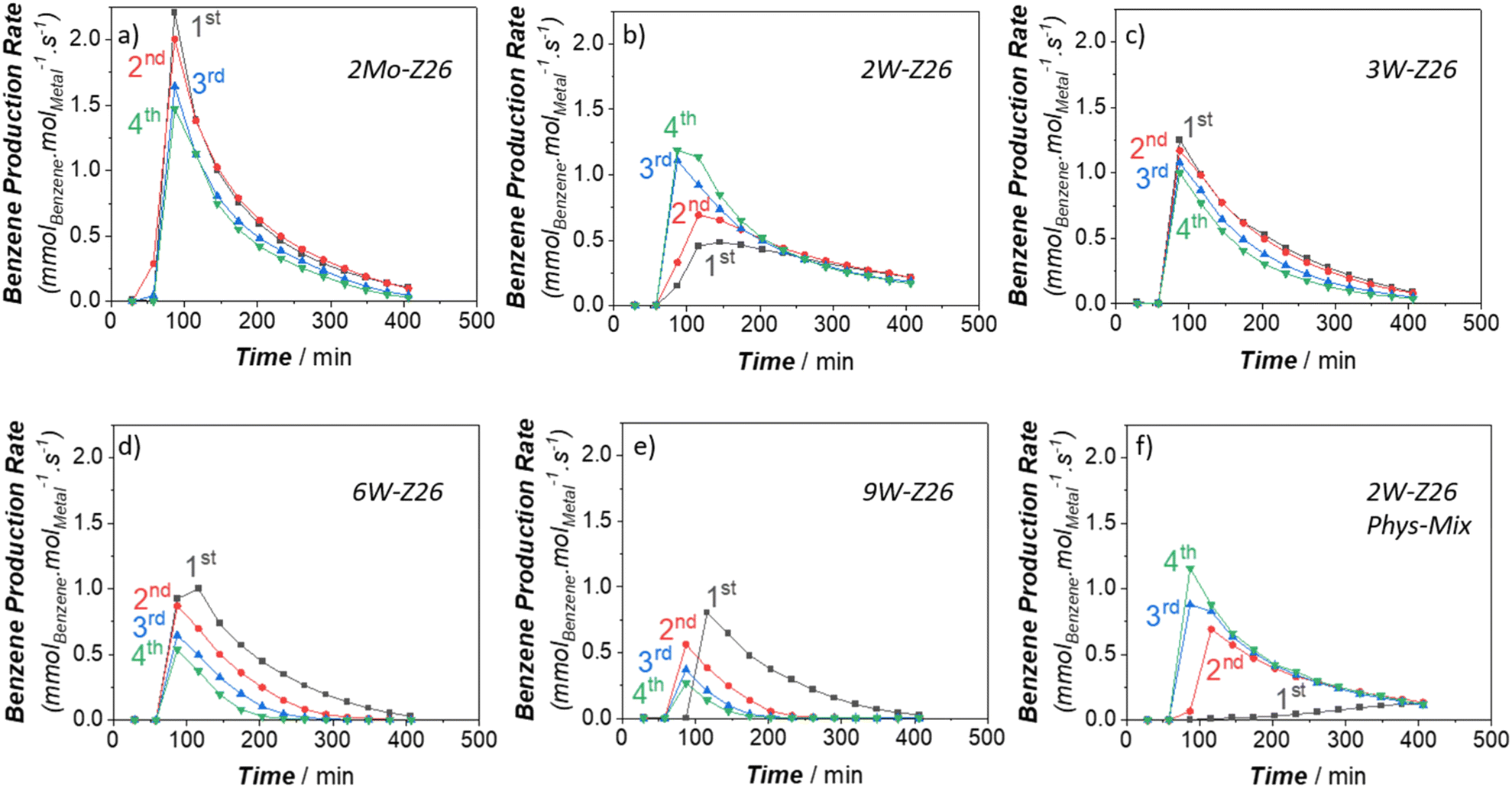 | ||
| Fig. 5 Benzene production rates (TOFs) of the prepared W and Mo catalysts (250 mg) during the reaction–regeneration cycles: a) 2Mo-Z26, b) 2W-Z26, c) 3W-Z26, d) 6W-Z26, e) 9W-Z26, and f) 2W-Z26 Phys-Mix (reaction at 750 °C: 11.8 mol% N2 and 88.2 mol% CH4 in the inlet, total gas flow = 17 ml min−1 at STP – regeneration at 700 °C: dry air flow = 15 ml min−1 at STP). | ||
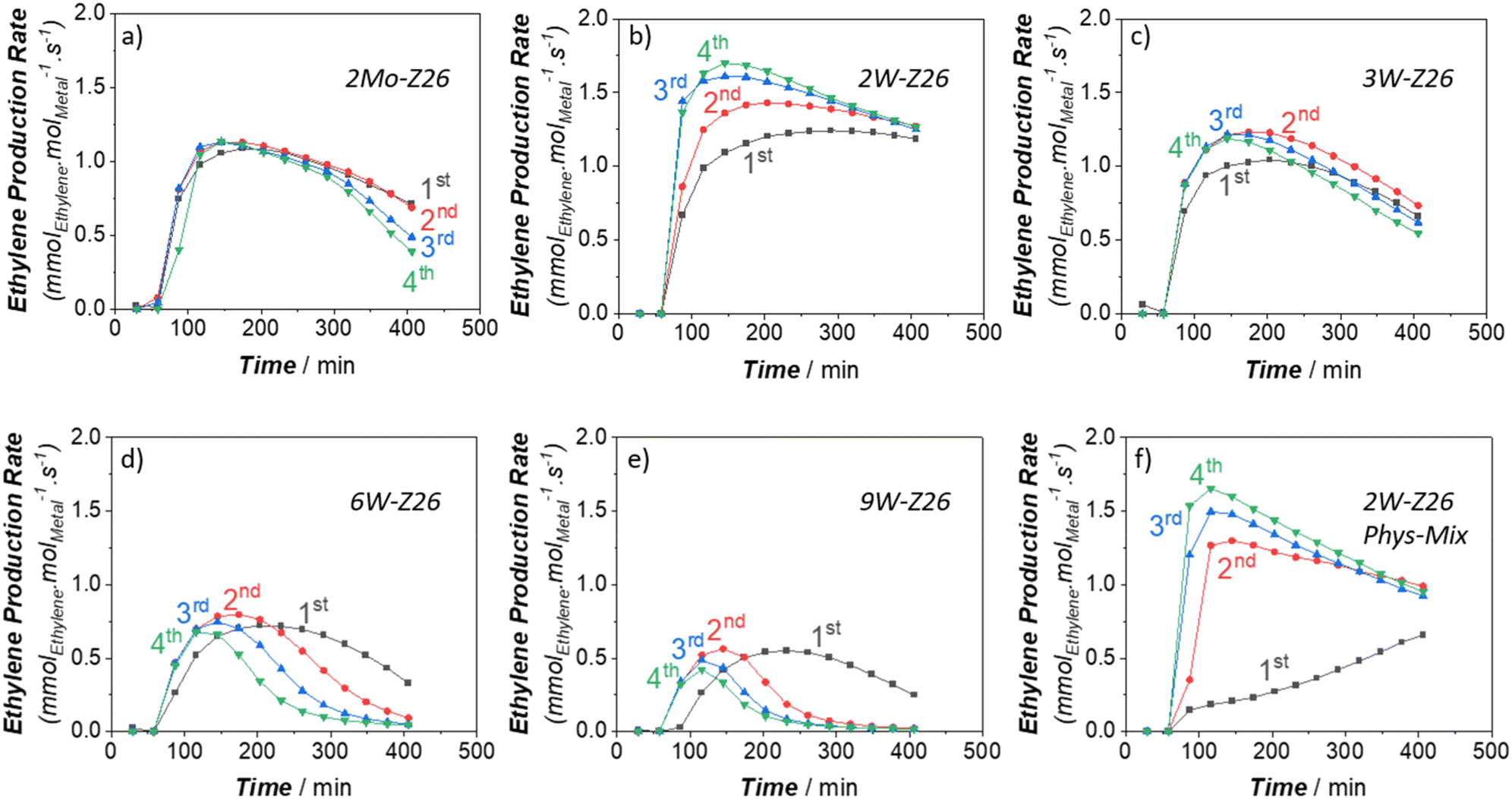 | ||
| Fig. 6 Ethylene production rates (TOFs) of the prepared W and Mo catalysts (250 mg) during the reaction–regeneration cycles: a) 2Mo-Z26, b) 2W-Z26, c) 3W-Z26, d) 6W-Z26, e) 9W-Z26, and f) 2W-Z26 Phys-Mix (reaction at 750 °C: 11.8 mol% N2 and 88.2 mol% CH4 in the inlet, total gas flow = 17 ml min−1 at STP – regeneration at 700 °C: dry air flow = 15 ml min−1 at STP). | ||
Besides these, one may also question the effect of regeneration during all these investigations. As we mentioned before, the regeneration process occurs at 700 °C under continuous air-flow. However, when we prepared the fresh catalysts, they were calcined at 550 °C in a muffle furnace (i.e., static conditions). To check how the regeneration conditions affect the fresh 2W-Z26 catalyst, we decided to pre-calcine it at 700 °C under continuous flow for 7 days prior to the reaction test at 750 °C. The obtained performance (Fig. S10c and d†) showed no enhancement in the activity; higher calcination temperature under dynamic conditions is not helpful in re-distributing/re-dispersing the W sites. To get the enhancement, a spent version of the catalyst (i.e., reduced W sites and coke deposition) is required. Meanwhile, we also looked into the effects of reaction–regeneration cycle parameters (i.e., temperature level, experimental duration, number of cycles). The cyclic pathways followed during the catalysis experiments are depicted in the ESI† (see section S2.5). In one of these pathways, when we regenerated the 2W-Z26 catalysts at 700 °C for 16 h upon the first reaction test at 750 °C, we noticed faster aging for the second reaction run, which led to the highest activity we observed in the original pathway (Fig. S13c and f†). Also, we realized this enhancement in the activity has a limit; the activity started declining after a few more cycles.
Effects of reaction–regeneration cycles on catalysts
The first thing we noticed on all samples after four regeneration cycles, the sample color was becoming light brownish (Fig. S17†). Initially, we thought that this color change might be related to tungsten sites (i.e., partially reduced W oxides39), but we observed the same color change in the case of 2Mo-Z26 catalyst and bare ZSM-5 when we applied the same reaction–regeneration cycles. Also, XPS measurements on 2W-Z26 and 6W-Z26 confirm that oxidation states of W sites are recoverable even after 4 regeneration cycles (Fig. S30†). While investigating these samples with micro-Raman, we noticed that these brownish parts were localized, and they were very sensitive to laser irradiation under air (see Fig. S18†). This brought to mind that they might be coke species, although we didn't observe any coke-related mass loss when we performed TPO with regenerated samples (Fig. S24†). However, when we further analyzed these regenerated samples with UV-vis DRS (Fig. S21b†), we confirmed the presence of some persistent coke-type species (i.e., poly-aromatics, alkylated-naphthalenes)40,41 even after the high-temperature regeneration of spent catalysts.Moreover, the N2 physisorption analysis (Tables S1–S3, and Fig. S14 and S15†) demonstrates that after four reaction–regeneration cycles, there is a noticeable decrease in the surface area and the pore volume for all samples (especially the ones having higher W loading). This degradation in the porous structure can be explained by the trapped carbon species – which we discussed above – and the deformations in the catalyst structure. When we analyzed 2W-Z26 and 6W-Z26 samples via solid-state 27Al MAS NMR to probe the local atomic environment related to Al, significant changes were noticed after the reaction–regeneration cycles (27Al and 29Si MAS NMR: Fig. S31a and b,† respectively). In the regenerated samples, the dealumination of the zeolite framework was observed, showing broad distribution 27Al resonances with penta-coordinated 27Al species at ca. 30 ppm. We could also observe the formation of extra framework Al species (EFAl) after regeneration cycles, in line with the DRIFT results (Fig. S23†). Meanwhile, neither on fresh nor on regenerated samples, any 27Al resonances related to aluminum tungstate species were not observed. This corroborated the absence of these species in W-incorporated ZSM-5 catalysts reported in earlier studies.20,28 Besides this, we further identified the formation of defects via UV-vis DRS for all samples – including 2Mo-Z26 – as well (see Fig. S22† caption for the details). Furthermore, we investigated the crystallographic features in all these samples via PXRD analysis (Fig. S32 and Table S5†). The crystallinity, as well as the coherent domain size of the regenerated samples, does not change much compared to their fresh counterparts. To support this observation, there are many examples showing that after high-temperature steam treatment – the crystallinity of zeolites does not degrade despite the dealumination of framework and formation of defects.42–45 Moreover, the event of dealumination after the catalytic cycle can be evidenced from PXRD data, comparing the relative intensities of (011) or (200) and (501) diffraction peaks, I(011)/I(501) or I(200)/I(501), (see Fig. S33–S35, Table S5† and its caption for further details).46
On the other hand, the ex situ characterizations performed to unveil the changes in tungsten and molybdenum sites are delicate and complicated. To compare the fresh and regenerated samples (4 cycles), first, we performed micro-Raman measurements under ambient conditions (Fig. S16 and S19†). We noticed W-related Raman shifts on fresh samples (i.e., 973 cm−1: ν(OWO) from surface WO4 species, 828 cm−1: ν(W–O–W)) disappeared after several regenerations (except the bulk WO3 vibrations on 9W-Z26 catalyst). Instead of these, we observe a vibration band at ca. 1015 cm−1 which could be assigned to mono WO stretching of surface WO5/WO6 polytungstates and WO5 monotungstates species.47 A similar situation was also observed on 2Mo-Z26 catalyst after regeneration cycles (Fig. S19†). In the case of UV-vis spectra, it is complicated to identify and analyze the W and Mo-related absorptions since zeolite defects and coke-related absorptions also exist (Fig. S20 and S21†). The absorptions at ca. 209 and ca. 260 nm could be attributed to mono- and polytungstates different than the ones observed on fresh catalysts. Beyond any doubt, the dispersion and distribution of metal on regenerated samples change sharply compared to the sites on fresh catalysts. At the same time, it is clear that coke combustion during the regeneration is changing the zeolite structure (i.e., defects, dealumination), and this affects the metal dispersion–distribution.
To gain more insight into the catalyst during these regeneration cycles, we performed acidity analysis (pyridine-IR) on four times regenerated Z26, 2W-Z26, 6W-Z26 and 2Mo-Z26 samples. First of all, there was a sharp decrease in the total number of acid sites for all four samples compared to their fresh versions (Table 4). Considering the values upon treatment at 150 °C, we could state that the decrease in BASs is much more significant while the number of LASs increases slightly in the case of ZSM-5 and 2W-Z26. This result clearly suggests that we are forming defects (i.e., EFAl) that induce Lewis acidity. It is also perceptible that 2W-Z26 catalyst has the highest BAS/LAS ratio compared to the other three. This may explain the increasing trend in 2W-Z26's activity during the high-temperature reaction–regeneration cycles. While re-dispersing and re-distributing tungstens, preserving a certain amount of BASs that can contribute to aromatization and hydrocarbon-pool mechanism might be very critical. On the other hand, four times regenerated 2Mo-Z26 catalyst has the lowest B/L ratio – like we observed in the case of fresh catalysts. Again, this corroborates the aromatization capability of active Mo sites over active W sites.
| Samples 4× regenerated | 150 °C | 350 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| BAS (μmol g−1) | LAS (μmol g−1) | Total (μmol g−1) | B/L | BAS (μmol g−1) | LAS (μmol g−1) | Total (μmol g−1) | B/L | |
| Z26 | 109.2 | 274.3 | 383.5 | 0.40 | 36.2 | 70.4 | 106.6 | 0.51 |
| 2W-Z26 | 128.0 | 210.4 | 338.4 | 0.61 | 31.9 | 50.9 | 82.8 | 0.63 |
| 6W-Z26 | 96.7 | 184.8 | 281.5 | 0.53 | 20.6 | 38.0 | 58.6 | 0.54 |
| 2Mo-Z26 | 96.7 | 197.6 | 294.3 | 0.49 | 26.3 | 78.5 | 104.9 | 0.34 |
Besides these, we investigated the spent 2W-Z26, 6W-Z26 and 2Mo-Z26 catalysts after the first and third reaction steps via TPO and N2 physisorption techniques (Table 5, Fig. S14b and S25†). Based on the data obtained via TPO, 6W-Z26 catalysts have a slightly higher amount of coke compared to 2W-Z26 and 2Mo-Z26 after both steps, but the difference is not so high. Also, the coke amount decreases as these catalysts were further regenerated and tested. This trend could be attributed to the BAS degradation and re-dispersion/re-distribution of tungsten and molybdenum sites. Although the thermal gravimetric analysis does not show sharp differences between the three samples, N2 physisorption analyses set forth the differences between them during the deactivation. Despite having close coke amounts after both reaction steps, 2W-Z26 and 2Mo-Z26 spent samples have still considerable free porosity whereas the pores of 6W-Z26 are almost fully blocked. This is indicating that the coke distribution behavior in/on zeolite particles differs between the samples. From the previous studies with Mo/ZSM-5 catalysts, it is well-known that Mo species inside the pores are carburized by detaching from the zeolite pores and forming clusters on the outer surface that leads to coke accumulation and sintering of the active molybdenum carbide sites.48 Eventually, the same behavior can be expected for the tungsten version of the catalyst. And this can explain why the porous structure of 6W-Z26 was heavily blocked compared to 2W-Z26 since it has a higher amount of W. In the TPO profile of 6W-Z26 (Fig. S25b†), the oxidation of big WCx clusters could be seen around 510 °C. The unexpected result here is that while 2W-Z26 catalyst gives better performance and 6W-Z26 gets worse after each regeneration cycle, both have decreasing coke content trends. These might be attributed to the loss of BASs (Table 4) which could lead to coking-related deactivation in zeolite catalysis and/or different kinds of active tungsten sites on these two catalysts (which we discussed in the next section).
| Samples | S BET (m2 g−1) | S meso/ext (m2 g−1) | S micro (m2 g−1) | V total (ml g−1) | V micro (ml g−1) | Coke %d (masscoke per masscatalyst) |
|---|---|---|---|---|---|---|
| a S meso/ext = SBET − Smicro. b From N2 adsorption isotherm using the t-plot method. c Single point adsorption total pore volume at p/po = 0.95. d Calculated from TG-TPO data. | ||||||
| 2W-Z26 fresh | 397 | 104 | 293 | 0.23 | 0.11 | — |
| 2W-Z26 4× regenerated | 372 | 79 | 293 | 0.22 | 0.11 | — |
| 2W-Z26 1× reaction | 129 | 35 | 94 | 0.09 | 0.04 | 20.3% |
| 2W-Z26 3× reaction | 221 | 40 | 181 | 0.13 | 0.07 | 17.0% |
| 6W-Z26 fresh | 372 | 100 | 272 | 0.21 | 0.11 | — |
| 6W-Z26 4× regenerated | 321 | 81 | 240 | 0.20 | 0.09 | — |
| 6W-Z26 1× reaction | 31 | 22 | 9 | 0.04 | 0.00 | 22.3% |
| 6W-Z26 3× reaction | 30 | 21 | 9 | 0.04 | 0.00 | 19.0% |
| 2Mo-Z26 fresh | 384 | 98 | 286 | 0.22 | 0.11 | — |
| 2Mo-Z26 4× regenerated | 352 | 89 | 264 | 0.21 | 0.10 | — |
| 2Mo-Z26 1× reaction | 142 | 32 | 110 | 0.09 | 0.04 | 20.1% |
| 2Mo-Z26 3× reaction | 162 | 130 | 32 | 0.10 | 0.05 | 16.9% |
Operando X-ray absorption spectroscopy: active tungsten-species
We performed operando X-ray absorption spectroscopy (XAS) analysis to shed light on the W sites and how they behave during the reaction–regeneration cycles. The characterization of catalysts under high-temperature reaction/regeneration conditions was performed using a dedicated cell designed as a plug-flow reactor for operando XAS at the Neel Institute in Grenoble, France (see the further details in section S1.4 and S4†).49 In order to identify the nature of active W species of the 2W-Z26 and 6W-Z26 catalysts during MDA reaction, W L3-edge EXAFS spectra of the fresh catalysts were collected during the different stages of reaction–regeneration cycles with the same space velocity. Based on the effluent gas analysis performed with the mass spectrometer (Fig. S45†), we confirmed the reproducibility of the catalytic tests obtained with PID fixed-bed reactor. Also, we noticed changes in the initial activation-induction period during the operando experiments (see Fig. S48† for details).
Fig. 7 shows the W L3-edge XANES spectra of the two 2W-Z26 and 6W-Z26 fresh catalysts and reference samples including tungsten trioxide WO3, sodium monotungstate Na2WO4 (aq) obtained from the Solid Spectroscopy Hosting Architecture of Databases and Expertise (SSHADE),50 and ammonium metatungstate (NH4)6H2W12O40. All the spectra contain a white line at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 213.5 eV, which is characteristic of tungsten oxide W(VI) species.51,52 However, each sample has a differently shaped white line, which can be attributed, according to Yamazoe et al.,53 to the difference in structure. For example, the WO3 reference sample having a white line of lower intensity, is known to have a distorted octahedral [WO6] symmetry structure.53–55 In contrast, the increase in the intensity of the white line for the monotungstate reference sample WO42− (aq) is related to the presence of W atoms in a tetrahedral environment [WO4].53–56 For both 2W-Z26 and 6W-Z26 fresh catalysts, their XANES spectra is similar to that of ammonium metatungstate (NH4)6H2W12O40, which indicates that the catalysts are deposited as polytungstate species on the support after impregnation. This is consistent with the white line intensity of both catalysts located between those of the WO3 and WO42− (aq) references, thus confirming the presence of mixed W(VI) tetrahedral and octahedral clusters. The higher intensity of the white line of the 2W catalyst compared to that of the 6W may be due to a better dispersion of the particles after impregnation, as shown in the high-resolution HAADF-STEM images of the two fresh catalysts (Fig. S36 and S37†).
213.5 eV, which is characteristic of tungsten oxide W(VI) species.51,52 However, each sample has a differently shaped white line, which can be attributed, according to Yamazoe et al.,53 to the difference in structure. For example, the WO3 reference sample having a white line of lower intensity, is known to have a distorted octahedral [WO6] symmetry structure.53–55 In contrast, the increase in the intensity of the white line for the monotungstate reference sample WO42− (aq) is related to the presence of W atoms in a tetrahedral environment [WO4].53–56 For both 2W-Z26 and 6W-Z26 fresh catalysts, their XANES spectra is similar to that of ammonium metatungstate (NH4)6H2W12O40, which indicates that the catalysts are deposited as polytungstate species on the support after impregnation. This is consistent with the white line intensity of both catalysts located between those of the WO3 and WO42− (aq) references, thus confirming the presence of mixed W(VI) tetrahedral and octahedral clusters. The higher intensity of the white line of the 2W catalyst compared to that of the 6W may be due to a better dispersion of the particles after impregnation, as shown in the high-resolution HAADF-STEM images of the two fresh catalysts (Fig. S36 and S37†).
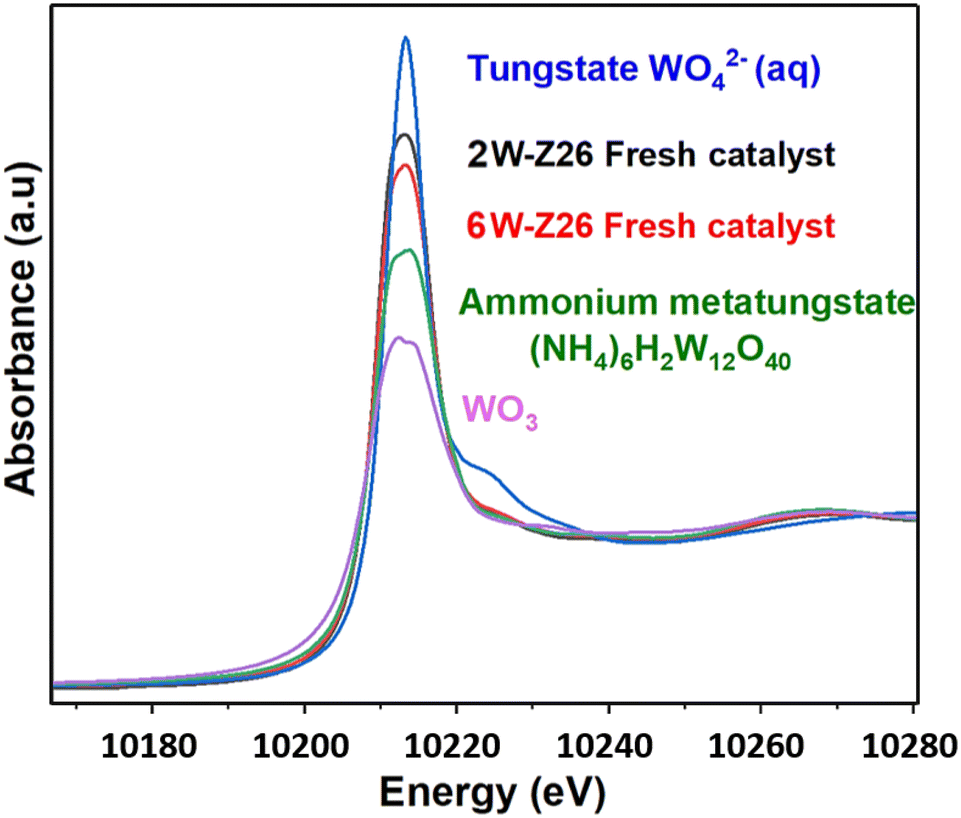 | ||
| Fig. 7 W L3-edge XANES spectra of the 2W-Z26 and 6W-Z26 fresh catalysts compared with tungsten trioxide (WO3), ammonium metatungstate (NH4)6H2W12O40 and sodium monotungstate Na2WO4 (aq) reference compounds. | ||
In order to understand the structural evolution of the W catalysts during the different reaction/regeneration cycles, we compared their XANES spectra obtained operando to those of the fresh catalysts and the Na2WO4 (aq) reference. Fig. 8 shows the W L3-edge XANES spectra of the 2W-Z26 and 6W-Z26 catalysts during the reaction cycles (under CH4/He, 750 °C) and after regeneration with air at 700 °C. In both cases, a clear evolution of the fresh 2W and 6W catalysts is observed during the reaction, with the same behavior obtained during the three cycles. Indeed, the appearance of a shoulder at 10224 eV is revealed. This type of oscillation was only observed (greatly) in the case of WO42− monotungstate reference (Fig. 8),51,57 and may then indicate the dispersion of the W species (polytungstates) initially present in monotungstates species during the reaction. This can be confirmed by the aberration-corrected HAADF-STEM images (Fig. S36 and S37†), showing the presence of highly dispersed single tungsten atoms, corresponding to surface monotungstate species, and polytungstates clusters, as previously observed in literature.58–60 Furthermore, the absorption edge of the XANES spectra shifts to lower energy during the reaction for both 2W-Z26 and 6W-Z26 catalysts (Fig. 8), which can be attributed to the chemical shift effect. Generally, the shift in edge position can be attributed to a change in the oxidation state of an element.61 An overall shift of the XANES spectra to higher energies is observed as the oxidation state increases. Indeed, when the global charge of the atom is more positive at higher degrees of oxidation, more energy is required to excite an electron out of an orbital. Conversely, the XANES spectrum shifts to lower energy when the metal element is more reduced, i.e. when there is less positive charge on the metal. Therefore, to get an idea of the oxidation state of tungsten in the two catalysts during the MDA reaction, a linear relationship between the W L3-edge energy and the W oxidation state was established, using the W references (Fig. 9). The W L3-edge absorption energies of the reference materials were 10209.7 eV for W foil (W0), 10212.1 eV for WO2 (W4+), and 10213.5 eV for WO3 (W6+). The absorption edges of the two catalysts are very close to those of WO3, indicating that both catalysts have a W6+ oxidation state before the MDA reaction. During the reaction, the absorption edges of the 2W and 6W catalysts were lower than those of the fresh counterparts and were particularly localized between those of the W foil and WO2 references. This is an indication that both catalysts exhibit a reduction in their oxidation state during the reaction, and that the W absorber shifts from a W6+ oxidation state to a mixture of oxidation states, namely between W2+ and W4+. However, the overall reduction of W sites in the case of 6W-Z26 was much sharper compared to the ones in 2W-Z26 catalyst during the reaction, which may indicate the difference in the active W site structures and oxidation states.
 | ||
| Fig. 8 W L3-edge XANES spectra of a) 2W-Z26 and b) 6W-Z26 catalysts before and during different reaction/regeneration cycles. The experimental spectra of the two catalysts are compared to the sodium monotungstate Na2WO4 (aq) reference spectra. | ||
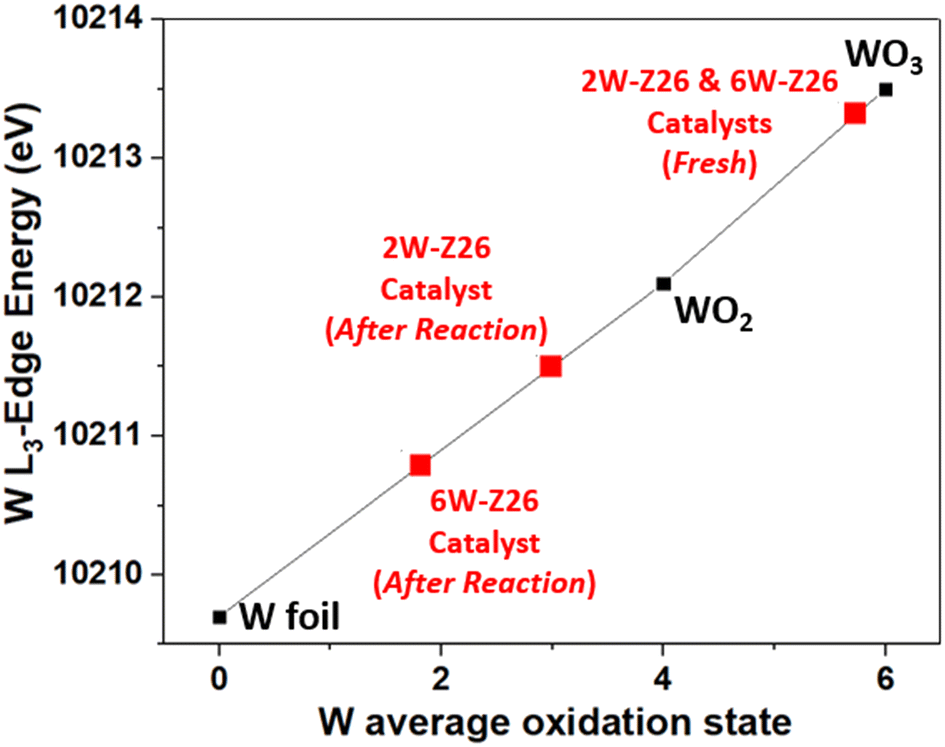 | ||
| Fig. 9 Average oxidation states of tungsten obtained from W L3-edge XANES for the references (W foil, WO2, and WO3) and the 2W-Z26 and 6W-Z26 catalysts before reaction (fresh catalysts) and during MDA reaction. | ||
After regeneration in air at 700 °C (Fig. 8), we observe a remarkable increase of the shoulder present at 10224 eV. Indeed, the XANES spectra of the two catalysts after regeneration become very similar to that of the Na2WO4 (aq) reference containing monotungstate WO42− species. This indicates that the state of the catalyst obtained after regeneration is different from its initial state just before the first reaction, with the presence of monotungstates species better dispersed on the support than those of polytungstates present in the 2W and 6W fresh catalysts. This well dispersed–distributed W sites after regeneration can explain the better catalytic activity of the 2W-Z26 catalyst during the second reaction cycle. However, what is surprising is that the 6W-Z26 catalyst shows a lower catalytic activity during the second reaction cycle, probably due to a difference in structure, which cannot be distinguished in XANES analysis.
To have a clearer idea of the structure of the catalysts and the nature of the tungsten species present during the reaction, as well as that formed after regeneration, the radial distribution functions of the EXAFS spectra (FT-EXAFS) of each catalyst during the different reaction/regeneration cycles are showed in Fig. 10 (the corresponding k2-weighted EXAFS spectra are shown in Fig. S49 in ESI†). For a more comprehensive comparison, the experimental FT-EXAFS spectra and their corresponding Cauchy wavelet transforms are compared to those of known reference spectra WO3, Na2WO4 (aq), ammonium meta tungstate ((NH4)6H2W12O40) and WC obtained at room temperature (see Fig. S50–S53 in ESI†). Both catalysts exhibit one large feature around 1.7 Å during MDA reaction and regeneration under air at 700 °C. This peak corresponds to the first W-coordination shell in which the anion of the oxygen atoms surrounds the central atom.62,63 Peaks less than 1 Å generally do not correspond to actual coordination spheres and may appear from atomic XAFS and/or multi-electron excitations.64 The difference in peak intensity at 1.7 Å between the reaction and the regeneration cycles is due to a modification of the structure of the W species. In general, the disordered octahedral structure of the W(VI) species results in octahedral W–O shells showing much lower intensities than ordered tetrahedral tungstates. Obtaining a higher intensity during regeneration can then explain the formation of well-dispersed monotungstates WO42− species, which is consistent with XANES analyses (Fig. 8). On the other hand, the decrease in intensity during the reaction indicates that a mixture of W(VI) tetrahedral and octahedral clusters is formed (mono/polytungstates), and some sites are reduced. The noticeable difference between the two catalysts is the emergence of a peak around 2.3 Å for 6W-Z26 (Fig. 10). The comparison of the continuous Cauchy wavelet transforms (WT-EXAFS) applied on the 6W-Z26 EXAFS spectra obtained during the reaction to the known WC reference seems to show that this contribution at 2.3 Å is probably associated with the formation of tungsten carbide bonds W–C during the reaction. The presence of both tungsten-oxo and carburized tungsten species may indicate that the tungsten-oxo particles (W6+) present in the fresh catalyst are partially carburized after contact with CH4 during the reaction. Considering the previous studies performed on Mo/HZSM-5 catalysts,65–67 it could be stated that the formation of tungsten carbide species may occur on 6W-Z26 samples during the first stages of the reaction, and these serve as an active phase giving the typical MDA activity which we discussed in previous sections. Overall, these analyses corroborate that there might be different active W sites in terms of structure and oxidation state depending on the tungsten loading. Also, it is possible that structural changes in ZSM-5 support during reaction–regeneration cycles play key roles in the re-dispersion and re-orientation of W sites, which leads to enhanced/declined overall MDA activity depending on the initial W loading (see discussions in previous sections).
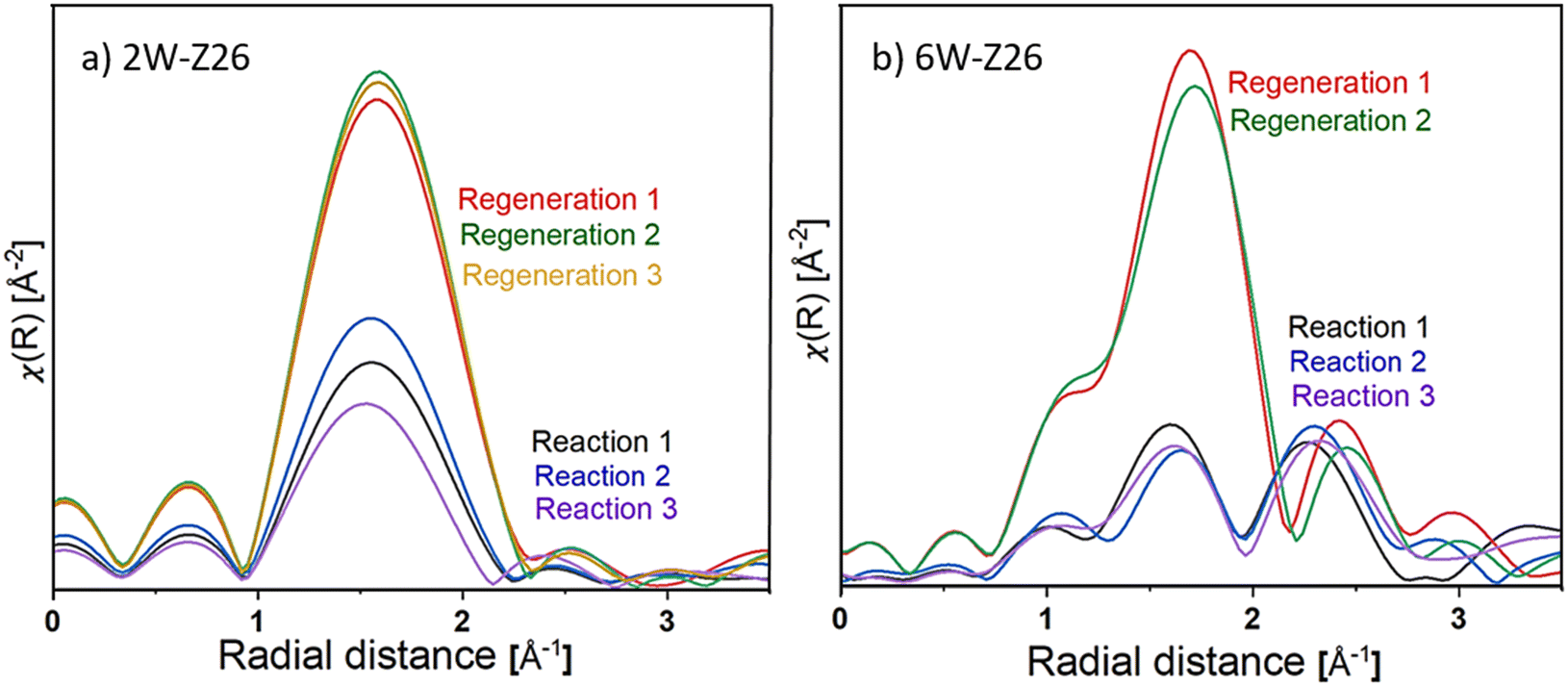 | ||
| Fig. 10 W L3-edge FT-EXAFS spectra for the 2W-Z26 and 6W-Z26 catalysts after different reaction (88.2% CH4/11.8% He flow at 750 °C) and regeneration (dry air flow at 700 °C) cycles. The curves are obtained in the 2.3–7.8 Å−1k-space using a Hanning window. | ||
Conclusions
In this study, we examined the W-loaded ZSM-5 catalysts from a different perspective than the previous studies in the literature. Besides showing that W activation is more demanding than activation of Mo, and that Mo is a better aromatization catalyst in general; we revealed that W dispersion/distribution on the zeolite is very critical through high-temperature reaction–regeneration cycles. Our results clearly indicate that if W–zeolite interaction and zeolite acidity can be tuned, it would be a promising alternative to replace Mo for MDA reactions. In addition to this, via operando XAS analyses, we found the existence of two different active tungsten species (partially reduced tungsten sites and carburized tungsten sites) on W loaded ZSM-5 catalysts. As discussed in the introduction, there was no consensus on the oxidation state of active W species in the MDA literature up to this study. Future endeavours in this field should be fully understanding the structure of these active W species and unravelling the zeolite–tungsten interaction thoroughly.Experimental methods
Catalyst preparation and characterization
All W and Mo catalysts were prepared via incipient wetness impregnation unless otherwise stated, using (NH4)6H2W12O40·xH2O and (NH4)6Mo7O24·4H2O as precursors, respectively. The details about catalyst preparation and catalyst characterization are presented in ESI.†Catalytic activity tests
All the catalytic performance tests (reaction–regeneration cycles) were conducted in a fixed-bed reactor setup provided by PID Eng & Tech at atmospheric pressure and corresponding temperatures (please see the further details in ESI†). Methane conversion-% (1), product yield-% (2), total yield-% (not including CO and CO2) (3), H2 flow at the outlet (ml min−1 at STP) (4), and product formation rate per metal atom (turnover frequencies, TOFs) (5) were calculated as follows: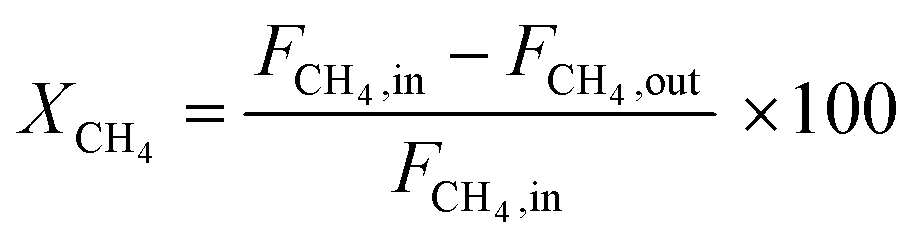 | (1) |
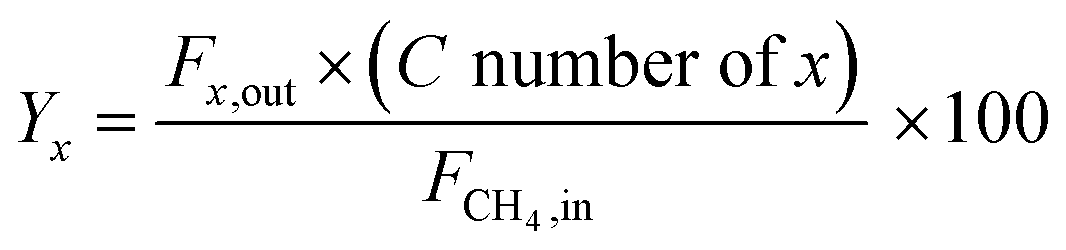 | (2) |
 | (3) |
| FH2,out = yH2,out × Ftotal,out | (4) |
 | (5) |
Author contributions
J. G. conceived, designed and supervised the project together with M. Ç. who prepared the catalysts and performed the catalytic experiments, and most of the characterizations. A. N., A. A. T., J. L. H., and S. O. C. were responsible for the XAS characterization, data analyses and interpretations. S. A. F. N. and L. C. performed the advanced DFT calculations. Solid-state NMR experiments and analyses were carried out by S. C. and J. R. M., A. D., G. S., and T. S. were responsible for the XRD-refinement analysis, XPS experiments, and NH3-TPD data acquisition, respectively. The manuscript was drafted by M. Ç. and J. G. with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by King Abdullah University of Science and Technology (KAUST). Also, we would like to thank Söhrab Askarli, Qingxiao Wang, Eganathan Kaliyamoorthy and Büşra Dereli for their supports in zeolite-steaming experiments, HAADF-STEM imaging, ICP-OES analysis, and DFT-calculations, respectively. We acknowledge the European Synchrotron Radiation Facility for the provision of synchrotron radiation facilities, and we would like to thank Eric Lahera for assistance in using beamline BM30.References
- M. Saunois, A. R. Stavert, B. Poulter, P. Bousquet, J. G. Canadell, R. B. Jackson, P. A. Raymond, E. J. Dlugokencky and S. Houweling, Earth Syst. Sci. Data, 2020, 12, 1561–1623 CrossRef
.
- G. Zichittella and J. Pérez-Ramírez, Chem. Soc. Rev., 2021, 50, 2984–3012 RSC
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS
- D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Chem. Soc. Rev., 2021, 50, 1251–1268 RSC
- S. Ma, X. Guo, L. Zhao, S. Scott and X. Bao, J. Energy Chem., 2013, 22, 1–20 CrossRef CAS
- M. Çağlayan, A. Lucini Paioni, E. Abou-Hamad, G. Shterk, A. Pustovarenko, M. Baldus, A. D. Chowdhury and J. Gascon, Angew. Chem., Int. Ed., 2020, 59, 16741–16746 CrossRef PubMed
- I. Vollmer, N. Kosinov, Á. Szécsényi, G. Li, I. Yarulina, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, A. Aguilar-Tapia, J. L. Hazemann, E. Pidko, E. Hensen, F. Kapteijn and J. Gascon, J. Catal., 2019, 370, 321–331 CrossRef CAS
- M. Agote-Arán, R. E. Fletcher, M. Briceno, A. B. Kroner, I. V. Sazanovich, B. Slater, M. E. Rivas, A. W. J. Smith, P. Collier, I. Lezcano-González and A. M. Beale, ChemCatChem, 2020, 12, 294–304 CrossRef
- N. K. Razdan, A. Kumar, B. L. Foley and A. Bhan, J. Catal., 2020, 381, 261–270 CrossRef CAS
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2017, 7, 520–529 CrossRef CAS
- T. Shoinkhorova, T. Cordero-Lanzac, A. Ramirez, S. H. Chung, A. Dokania, J. Ruiz-Martinez and J. Gascon, ACS Catal., 2021, 11, 3602–3613 CrossRef CAS
- M. Guisnet, N. S. Gnep, D. Aittaleb and Y. J. Doyemet, Appl. Catal., A, 1992, 87, 255–270 CrossRef CAS
- E. Le Roux, M. Taoufik, C. Copéret, A. De Mallmann, J. Thivolle-Cazat, J. M. Basset, B. M. Maunders and G. J. Sunley, Angew. Chem., Int. Ed., 2005, 44, 6755–6758 CrossRef CAS PubMed
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2016, 6, 3061–3071 CrossRef CAS
- I. Dellien, F. M. Hall and L. G. Hepler, Chem. Rev., 1976, 76, 283–310 CrossRef CAS
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2nd edn, 1997 Search PubMed
- M. Çaǧlayan, A. L. Paioni, B. Dereli, G. Shterk, I. Hita, E. Abou-Hamad, A. Pustovarenko, A.-H. Emwas, A. Dikhtiarenko, P. Castaño, L. Cavallo, M. Baldus, A. D. Chowdhury and J. Gascon, ACS Catal., 2021, 11, 11671–11684 CrossRef
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, J. Catal., 2017, 346, 125–133 CrossRef CAS
- G. Plazenet, E. Payen, J. Lynch and B. Rebours, J. Phys. Chem. B, 2002, 106, 7013–7028 CrossRef CAS
- J. Yang, D. Ma, F. Deng, Q. Luo, M. Zhang, X. Bao and C. Ye, Chem. Commun., 2002, 3046–3047 RSC
- W. Ding, G. D. Meitzner, D. O. Marler and E. Iglesia, J. Phys. Chem. B, 2001, 105, 3928–3936 CrossRef CAS
- M. A. Abedin, S. Kanitkar and J. J. Spivey, MRS Adv., 2020, 3407–3417 CrossRef CAS
- J. Yang, F. Deng, M. Zhang, Q. Luo and C. Ye, J. Mol. Catal. A: Chem., 2003, 202, 239–246 CrossRef CAS
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338–346 CrossRef CAS
- S.-T. Wong, Y. Xu, L. Wang, S. Liu, G. Li, M. Xie and X. Guo, Catal. Lett., 1996, 38, 39–43 CrossRef CAS
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 347–351 CrossRef CAS
- X. Xu, F. Faglioni and W. A. Goddard, J. Phys. Chem. A, 2002, 106, 7171–7176 CrossRef CAS
- V. V. Kozlov, V. I. Zaikovskii, A. V. Vosmerikov, L. L. Korobitsyna and G. V. Echevskii, Kinet. Catal., 2008, 49, 117–121 CrossRef
- J. L. Zeng, Z. T. Xiong, H. Bin Zhang, G. D. Lin and K. R. Tsai, Catal. Lett., 1998, 53, 119–124 CrossRef CAS
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2016, 6, 3061–3071 CrossRef CAS
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089–15099 CrossRef CAS
- M. A. Alvarez-Merino, F. Carrasco-Marín, J. L. G. Fierro and C. Moreno-Castilla, J. Catal., 2000, 192, 363–373 CrossRef CAS
- T. Kim, A. Burrows, C. J. Kiely and I. E. Wachs, J. Catal., 2007, 246, 370–381 CrossRef CAS
- A. A. Costa, P. R. S. Braga, J. L. De MacEdo, J. A. Dias and S. C. L. Dias, Microporous Mesoporous Mater., 2012, 147, 142–148 CrossRef
- A. Guntida, K. Suriye, J. Panpranot and P. Praserthdam, Catal. Today, 2020, 358, 354–369 CrossRef CAS
- N. Liu, S. Ding, Y. Cui, N. Xue, L. Peng, X. Guo and W. Ding, Chem. Eng. Res. Des., 2013, 91, 573–580 CrossRef CAS
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS
- X. Gong, M. Çağlayan, Y. Ye, K. Liu, J. Gascon and A. Dutta Chowdhury, Chem. Rev., 2022, 122, 14275–14345 CrossRef CAS PubMed
-
M. Weil and W. D. Schubert, The Beautiful Colours of Tungsten Oxides, London, 2013 Search PubMed
- J. Goetze, F. Meirer, I. Yarulina, J. Gascon, F. Kapteijn, J. Ruiz-Martínez and B. M. Weckhuysen, ACS Catal., 2017, 7, 4033–4046 CrossRef CAS PubMed
- S. V. Konnov, V. S. Pavlov, P. A. Kots, V. B. Zaytsev and I. I. Ivanova, Catal. Sci. Technol., 2018, 8, 1564–1577 RSC
- S. Kumar, A. K. Sinha, S. G. Hegde and S. Sivasanker, J. Mol. Catal. A: Chem., 2000, 154, 115–120 CrossRef CAS
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 32, 1–29 CrossRef
- A. R. Maag, G. A. Tompsett, J. Tam, C. A. Ang, G. Azimi, A. D. Carl, X. Huang, L. J. Smith, R. L. Grimm, J. Q. Bond and M. T. Timko, Phys. Chem. Chem. Phys., 2019, 21, 17880–17892 RSC
- A. S. Al-Dughaither and H. De Lasa, Ind. Eng. Chem. Res., 2014, 53, 15303–15316 CrossRef CAS
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089–15099 CrossRef CAS
- C. H. L. Tempelman and E. J. M. Hensen, Appl. Catal., B, 2015, 176–177, 731–739 CrossRef CAS
- A. Aguilar-Tapia, S. Ould-Chikh, E. Lahera, A. Prat, W. Delnet, O. Proux, I. Kieffer, J. M. Basset, K. Takanabe and J. L. Hazemann, Rev. Sci. Instrum., 2018, 89, 035109 CrossRef PubMed
- Solid Spectroscopy Hosting Architecture of Databases and Expertise | SSHADE, https://www.sshade.eu/, (accessed 18 January 2023).
- B. C. Bostick, J. Sun, J. D. Landis and J. L. Clausen, Environ. Sci. Technol., 2018, 52, 1045–1053 CrossRef CAS PubMed
- J. L. Clausen, B. C. Bostick, A. Bednar, J. Sun and J. D. Landis, Tungsten Speciation in Firing Range Soils, U.S. Army Environmental Command, 5179 Hoadley Road, Aberdeen Proving Ground, MD, 2021, https://apps.dtic.mil/sti/pdfs/ADA535133.pdf Search PubMed.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2008, 112, 6869–6879 CrossRef CAS
- F. Hilbrig, H. E. Göbel, H. Knözinger, H. Schmelz and B. Lengeler, J. Phys. Chem., 1991, 95, 6973–6978 CrossRef CAS
- X. Carrier, E. Marceau, H. Carabineiro, V. Rodríguez-González and M. Che, Phys. Chem. Chem. Phys., 2009, 11, 7527–7539 RSC
- T. Witoon, P. Kidkhunthod, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2018, 348, 713–722 CrossRef CAS
- C. R. VanderSchee, D. Kuter, A. M. Bolt, F. C. Lo, R. Feng, J. Thieme, Y. Chen, K. Chen-Wiegart, G. Williams, K. K. Mann and D. S. Bohle, Commun. Chem., 2018, 1, 8 CrossRef
- W. Zhou, E. I. Ross-Medgaarden, W. v. Knowles, M. S. Wong, I. E. Wachs and C. J. Kiely, Nat. Chem., 2009, 1, 722–728 CrossRef CAS PubMed
- S. Li, H. Zhou, C. Jin, N. Feng, F. Liu, F. Deng, J. Q. Wang, W. Huang, L. Xiao and J. Fan, J. Phys. Chem. C, 2014, 118, 6283–6290 CrossRef CAS
- W. Zhou, I. E. Wachs and C. J. Kiely, Curr. Opin. Solid State Mater. Sci., 2012, 16 Search PubMed
- J. Kowalska and S. DeBeer, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1406–1415 CrossRef CAS PubMed
- Y. Kou, B. Zhang, J. Z. Niu, S. Ben Li, H. L. Wang, T. Tanaka and S. Yoshida, J. Catal., 1998, 173, 399–408 CrossRef CAS
- A. Kuzmin and J. Purans, Radiat. Meas., 2001, 33, 583–586 CrossRef CAS
- L. Gracia, V. M. Longo, L. S. Cavalcante, A. Beltrn, W. Avansi, M. S. Li, V. R. Mastelaro, J. A. Varela, E. Longo and J. Andrés, J. Appl. Phys., 2011, 110, 043501 CrossRef
- N. Wang, X. Dong, L. Liu, D. Cai, Q. Cheng, J. Wang, Y. Hou, A. H. Emwas, J. Gascon and Y. Han, Cell Rep. Phys. Sci., 2021, 2, 100309 CrossRef CAS
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS PubMed
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed
- NIST Chemistry WebBook, https://webbook.nist.gov/chemistry/, (accessed 13 June 2022).
- PubChem - National Library of Medicine, https://pubchem.ncbi.nlm.nih.gov/, (accessed 13 June 2022).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00103b |
| This journal is © The Royal Society of Chemistry 2023 |