
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expedient tandem dehydrogenative alkylation and cyclization reactions under Mn(I)-catalysis†
Reshma
Babu
,
Subarna S.
Padhy
,
Ganesan
Sivakumar
and
Ekambaram
Balaraman
 *
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Tirupati, Tirupati-517507, India. E-mail: eb.raman@iisertirupati.ac.in
First published on 10th February 2023
Abstract
Sustainable chemical production requires fundamentally new types of catalysts and catalytic technologies. The development of coherent and robust catalytic systems based on earth-abundant transition metals is essential; however, it is extremely challenging. Herein, an expedient divergence strategy for tandem dehydrogenative alkylation and cyclization reactions of hydrazides is reported under manganese catalysis. Using abundantly available alcohols (including MeOH, EtOH) and earth-abundant metal catalysts with water as a sole by-product makes the present strategy more sustainable and affordable. Interestingly, unsymmetrical selective deuterium incorporation is also presented for the first time. Several selective bond activation/formation reactions occur sequentially via amine–amide metal–ligand cooperation.
Introduction
N,N-Disubstituted acyl hydrazides and their derivatives are exceptional synthetic scaffolds that have been widely used for the synthesis of agrochemicals, polymers, pharmaceuticals, and bioactive functional materials.1 In particular, a significant number of N,N-disubstituted acyl hydrazide analogues having biological activities have been discovered, including antibacterials, antifungals, anthelmintics, antitumors, PG12 agonists, papillomavirus inhibitors, and pesticides (Scheme 1).2 Notably, cyclic hydrazide organic moieties are highly prominent building blocks and play a significant role in drug synthesis.3 Consequently, the progress on synthetic strategies that can lead to N,N-disubstituted and cyclic hydrazide derivatives under an eco-benign approach has gained much more attention in medicinal science. | ||
| Scheme 1 Biologically active N,N-disubstituted hydrazide derivatives (selected examples). | ||
Typically, the N,N-disubstitution of acyl hydrazides has been achieved by the direct treatment of acyl hydrazides with alkyl halides in the presence of a stoichiometric amount of base.4 Substituted acyl hydrazides can also be prepared by the direct reaction of acyl halides with disubstituted hydrazines.5 In the recent past, N-alkylation of hydrazides and N-acyl-N,N-disubstituted hydrazines has also been reported.6 This approach normally requires multistep synthesis, harsh reaction conditions, and harmful reagents, and generates copious amounts of waste. Thus, the development of an eco-benign, sustainable and affordable method for the synthesis of substituted and cyclized hydrazides is highly desirable.
Recently, the hydrogen auto-transfer (HA) strategy utilizing abundantly available renewable feedstock alcohols for selective C–C or C–N bond formation has been highly sought after.7 The transition metal-catalyzed C–N coupling of carbonyl compounds via alcohol activation through the HA strategy overcomes the limitations of the classical approach.8,9 Overall, the HA strategy is redox neutral and eliminates water as the only by-product. However, N,N-dialkylation via double borrowing hydrogenation is rarely reported. In 2017, the research group of Zhou reported the nickel/bis(dicyclohexylphosphine)propane (dcpp) and nickel/(S)-binapine complex catalyzed simple N-alkylation of acyl hydrazides using secondary alcohols under acidic conditions.10 In 2020, Gunanathan and co-workers developed the first noble-metal catalyzed N,N-dialkylation of acyl hydrazides with alcohols using a Ru-complex.11 Very recently, the research group of Renaud and Poater reported a diaminocyclopentadienone ruthenium tricarbonyl complex-catalyzed synthesis of mono- or dialkylated acyl hydrazide compounds using the borrowing hydrogen strategy.12
Of late, there have been many massive benefits for studying the progress of catalytic approaches based on earth-abundant base-metal catalysts in sustainable catalysis. Manganese is known to be the third most earth-abundant transition element and found to have manifold applications in dehydrogenation and hydrogenation reactions.13 To the best of our knowledge, first-row 3d-transition metal catalyzed N,N-dialkylation (symmetrical and unsymmetrical) and cyclization of acyl hydrazides using alcohols and diols, respectively, have been rarely explored. Herein, we report the first earth-abundant base-metal catalyzed direct one-pot N,N-dialkylation and cyclization of acyl hydrazides using abundantly available alcohols as potential alkylating agents. The reaction is catalyzed by a well-defined Mn(I)-based PNP-pincer complex and it operates via the HA strategy. Interestingly, the more challenging intermolecular cyclization of acyl hydrazides with diols was successfully demonstrated. Additionally, diversification of N,N-dialkylated acyl hydrazides and a study of the kinetics and mechanistic insights into the present Mn-catalysis were performed (Scheme 2).
 | ||
| Scheme 2 Various approaches for the synthesis of N,N-disubstituted acyl hydrazides. | ||
Results and discussion
Initially, we systematically studied various reaction parameters for the N,N-dialkylation and cyclization of acyl hydrazides using alcohols as an alkylating agent under Mn-catalysis. Hence, benzohydrazide (1a) and 1-butanol (2a) were chosen as benchmark substrates. The reaction of 1a (0.5 mmol) with 2a (1.25 mmol) in the presence of a catalytic amount of Mn-catalyst ([Mn]-1 at 5 mol%) and KOtBu (50 mol%) at 130 °C in toluene for 24 h afforded the desired N,N-dialkylated product 3a in 83% isolated yield (Table 1, entry 1). Interestingly, the reaction in the presence of 2.2 equiv. of alcohol (2a) slightly increased the yield of 3a (Table 1, entry 2). An excellent outcome was obtained with 3 mol% of the catalyst (Mn-1) (Table 1, entry 3). Further changing the catalyst systems to [Mn]-2 and [Mn]-3 yielded the desired product 3a in moderate yield (Table 1, entries 4 and 5; up to 71%). Also, there is no product formation in the absence of either a base or the Mn-catalyst (Table 1, entries 6 and 7). This confirmed that the combination of the base and the present Mn-1 catalytic system is essential for this N,N-dialkylation strategy. In particular, decreasing the mole percentage of the base ended up in a low yield of 3a (Table 1, entry 8). Lowering the temperature from 130 °C to 110 °C afforded 84% 3a (Table 1, entry 9). Notably, various bases such as NaOtBu, LiOtBu, KOH, and NaHCO3 resulted in a lower yield of 3a under the optimal conditions (Table 1, entry 10, and Table S5†). Under identical conditions, changing the solvents to m-xylene, n-octane, THF, 1,4-dioxane, and acetonitrile ended with a low yield of 3a (Table 1, entry 11).
With the optimal reaction conditions in hand, we applied the present catalytic protocol for the N,N-dialkylation of different acyl hydrazides with alcohols. Initially, we explored the substrate scope of symmetrical N,N-dialkylation of acyl hydrazides using diverse unactivated alcohols (Table 2). A range of unactivated aliphatic alcohols such as 1-butanol, 1-pentanol, 1-hexanol, and 1-heptanol with acyl hydrazides afforded the corresponding products 3a–3e and 3g–3k in excellent yields. Interestingly, more demanding diethylation and dimethylation reactions of benzohydrazides were performed using an excess amount of ethanol and methanol and 3l and 3y were obtained in 93% and 92% yield, respectively, under solventless conditions. Similarly, p-toluic hydrazide also smoothly underwent dimethylation with methanol and led to the product 3z in 95% yield. Notably, 3-phenylpropanol was also reactive under the optimized conditions and yielded 3f in 88% isolated yield. Next, a series of benzohydrazides having different substituents on the aryl ring was studied for the N,N-diethylation reactions. Indeed, acyl hydrazides with electron-donating substituents such as methyl, methoxy, and ethyl groups afforded 3m–3p in excellent yields.

Similarly, halide (–Cl, –Br, and –F) substituent containing benzohydrazides successfully underwent N,N-diethylation reaction with the optimized reaction conditions and yielded the corresponding products 3q–3w in good to excellent yields (80–89% yield). These results suggest that the electronic nature of substituents does not play any significant role in the present Mn-catalysis. Interestingly, 2-thiophenecarboxylic acid hydrazide was well-tolerated and gave 3x in 85% isolated yield. Further, we extended our synthetic strategy to achieve the challenging unsymmetrical N,N-dialkylation of acyl hydrazides using two different alcohols (Table 3).
| a Reaction conditions: 1 (0.5 mmol, 1 equiv), 2 (0.55 mmol, 1.1 equiv), Mn-1 (3 mol%), KOtBu (0.5 mmol), and 1 mL of toluene heated at 130 °C under argon flow for 4 h. After 12 h, different alcohol (0.55 mmol, 1.1 equiv), Mn-1 (3 mol%), base (0.5 mmol), and 1 mL of toluene were added into the reaction mixture and heating continued for another 4 h. All the represented yields are isolated yields of the compounds. For products 4g–4j: 5 equiv of ethanol was used. For products 4k–4n: 5 equiv of methanol was used. |
|---|

|
In our initial set of experiments, benzohydrazides were treated with 1.1 equiv. of aliphatic primary alcohols to achieve a monoalkylated product. Further, the reaction was carried out using a different primary aliphatic alcohol to yield the unsymmetrical N,N-dialkylation products 4a–4s with good to excellent yields (up to 98% isolated yield). Interestingly, more challenging and demanding aliphatic alcohols such as ethanol and methanol underwent highly selective N,N-dialkylation reactions and gave the corresponding products 4g–4n in excellent yields (92–98% yield). Interestingly, performing the reaction with deuterated methanol (CD3OD) instead of CH3OH afforded deuterium incorporation in the methyl α-position and yielded the products 4o–4s in excellent yields. Impressively, such a kind of unsymmetrical selective deuterium incorporation is unprecedented.
Delightfully, we have explored the intermolecular cyclization of acyl hydrazides using diols. As shown in Table 4, a range of neutral and electron-rich benzohydrazides (p-Me, p-OMe, m-OEt, and m-OMe) readily reacted with 1,4-butanediol under optimal conditions and afforded the corresponding five-membered ring acyl hydrazides in excellent yields (products 5a–5e, up to 98% isolated yield). Under the optimized catalytic conditions, halide (–F, –Cl, and –Br) substituted benzohydrazides yielded the expected products 5f–5k in good to excellent yields. Further, we extended this strategy for six-membered ring cyclization with 1,5-pentanediol. Under optimal reaction conditions, alkyl, alkoxy, and halide (–Cl, –F, and –Br) substituted acyl hydrazides underwent cyclization reaction via double borrowing hydrogenation and gave the respective six-membered ring cyclized products 5m–5r in 81–94% isolated yields (Table 4). More interestingly, 2-thiophenecarboxylic acid hydrazide also endured the cyclization with 1,5-pentanediol and yielded 5s in 77% isolated yield.
| a Reaction conditions: 1 (0.5 mmol), 4 (1.1 mmol), Mn-1 (3 mol%), KOtBu (0.5 mmol), and 1 mL of toluene heated at 130 °C under argon flow for 4 h. All the represented yields are isolated yields of the compounds. |
|---|

|
Several control experiments were conducted to gain insights into the reaction mechanism (Scheme 3). The formation of H2 gas was detected from the reaction of 1a with 2a under optimal conditions (by using gas chromatography (GC) as well as gasometer analysis; Scheme 3a; see the ESI†). Next, N-butylidenebenzohydrazide (3a′) was subjected to the present Mn-catalytic alkylation reaction with 1-butanol 2a and the expected product 3a was obtained in 92% yield (Scheme 3b). The in situ generated N-alkylidenebenzohydrazide intermediate undergoes catalytic transfer hydrogenation reaction and subsequent N-alkylation leads to the desired N,N-dialkylated product 3a. This result shows that the reaction proceeds via the N-alkylidenebenzohydrazide intermediate. We have also performed a tandem one-pot reaction to directly access N,N-dialkylated products from the corresponding ester derivatives. The reaction of ethyl benzoate with hydrazine hydrate to (in situ) generate benzohydrazide, which further reacted with 1-heptanol 2d in a one-pot manner under the optimal catalytic conditions, afforded the corresponding N,N-dialkylated product (3d) in 94% isolated yield (Scheme 3c). Then, three independent deuterium labeling experiments were carried out; almost 40–80% deuterium incorporated was observed at the α-methylene carbon of the N,N-dialkylation products 3y-D, 3l-D, and 3d-D under the present catalytic conditions (Scheme 3d). This experiment revealed that the present manganese catalytic cycle follows the borrowing hydrogen strategy. The lower deuterium incorporation may be due to hydrogen–deuterium (H–D) exchange by the presence of formed water molecules during the catalytic process. Finally, to determine the nature of the present Mn-catalysis, we have performed two independent experiments in the presence of mercury and radical scavengers such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and butylated hydroxytoluene (BHT). Indeed, performing the reaction in the presence of mercury didn't affect the yield of 3a. However, performing the reaction in the presence of dibenzo[a,e]cyclooctatetraene (DCT) significantly affected the yield of 3a (section 4.5, ESI†). These results confirmed the homogeneous character of the present Mn-catalyst and excluded the involvement of a SET (radical) mechanism (Scheme 3e and section 4.5, ESI†). The kinetic experiments reveal that the consumption of acyl hydrazide follows an exponential decay.14
 | ||
| Scheme 3 Control experiments (a–e). | ||
Next, we have successfully demonstrated the diversification of the acquired N,N-dialkylated product 2d (Scheme 4a). The reaction of 2d with lithium aluminum hydride yielded the corresponding reduced product 6a in 93% isolated yield (Scheme 4a). Then, the reaction of 2d with methanolic ammonia gave the corresponding ester product 6b in 32% isolated yield (Scheme 4). Performing Pd-catalyzed hydrogenation reaction in the presence of a catalytic amount of pyridine resulted in the corresponding products 6c and 6c′ in moderate yields.
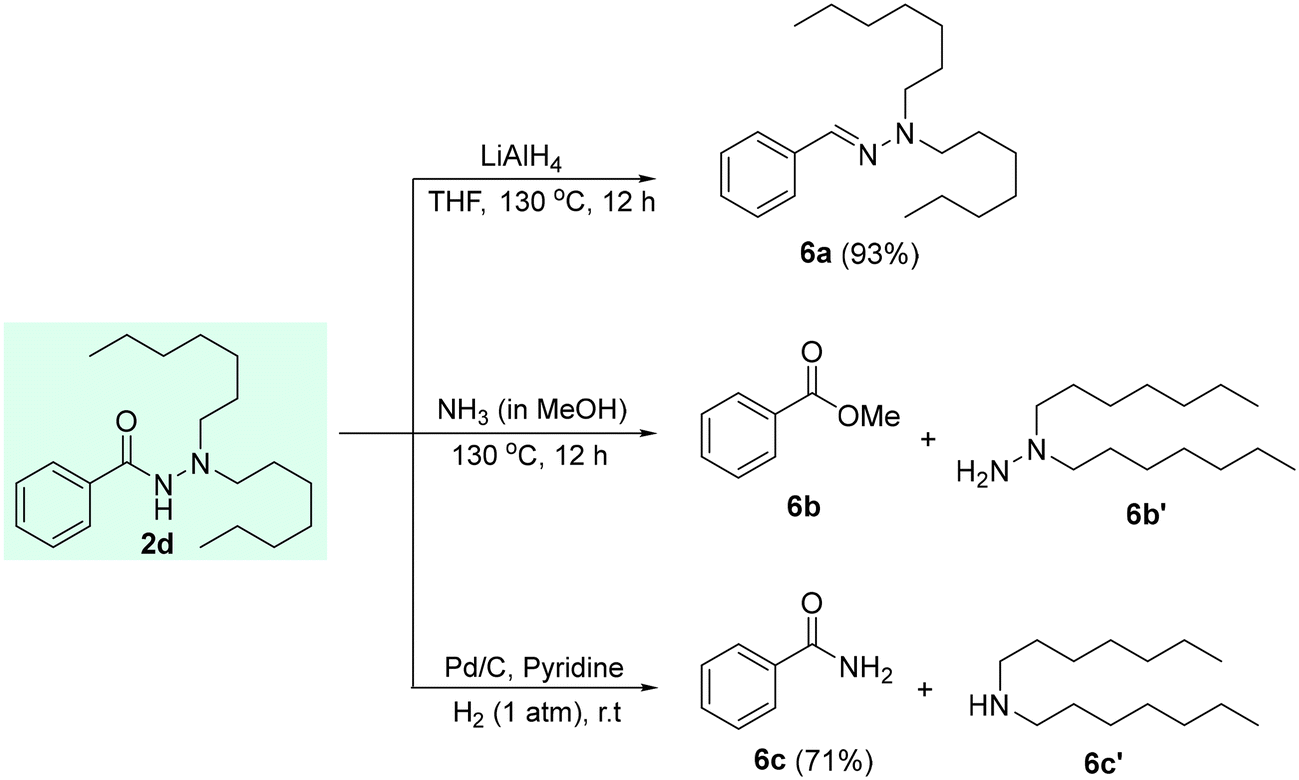 | ||
| Scheme 4 Diversification of 2d. | ||
Based on the experimental results and the literature,15 a plausible mechanism for the manganese-catalyzed N,N-dialkylation of acyl hydrazides with alcohols is proposed in Scheme 5. Initially, treatment of the precatalyst I with a base leads to the active catalyst II, followed by alcohol 2 activation via MLC leading to an alkoxy species III. Then, the dehydrogenation (of 2) occurs via β-hydride elimination, yielding the corresponding carbonyl compound, and the manganese hydride complex IV.15a,b However, recent DFT computational studies suggested that the initial dehydrogenation reaction proceeds via an outer-sphere mechanism.15c Thus, we believe that a blended mechanism may be operative. Further, the base-catalyzed condensation reaction of the in situ generated carbonyl compound with acyl hydrazide 1 leads to a hydrazone intermediate 3′, which undergoes hydrogenation by complex IV, yielding monoalkyl acyl hydrazide 3′′ with the regeneration of the active catalyst II. Subsequently, the monoalkyl acyl hydrazide again undergoes a second condensation with the in situ generated aldehyde 2′, which provides iminium intermediate V. Finally, hydrogenation of the iminium intermediate by the manganese complex IV yields the expected N,N-dialkylacylhydrazide 3 with the regeneration of II.
 | ||
| Scheme 5 A plausible mechanism for the manganese-catalyzed N,N-dialkylation of acyl hydrazides using alcohols. | ||
In summary, we have disclosed an earth-abundant, Mn-catalyzed, and efficient protocol for the one-pot direct coupling of acyl hydrazides with alcohols to N,N-dialkylacylhydrazides under mild and benign conditions. The present work was extended to the formation of more challenging five and six-membered heterocyclic acyl hydrazides. Interestingly, demanding diethylation and dimethylation of acyl hydrazides were demonstrated using ethanol and methanol, respectively, as potential alkylating agents. Control and labeling experiments suggest that the reaction proceeds via double borrowing hydrogenation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the CSIR (Project No: 01/(3030)/21/EMR-II). E. B. acknowledges funding from the Swarnajayanti Fellowship (SERB/F/5892/2020-2021). R. B. acknowledges the SERB-PMRF, India for a fellowship. G. S. thanks IISER-Tirupati for a fellowship.Notes and references
- K. Othmer, Hydrazine and its Derivatives in Encyclopedia of Chemical Technology, Wiley, New York, 4th edn, 1995, vol. 13 Search PubMed.
- P. Majumdar, A. Pati, M. Patra, R. K. Behera and A. K. Behera, Chem. Rev., 2014, 114, 2942 CrossRef CAS PubMed.
- M. Slusarczyk, W. M. De Borggraeve, G. Hoornaert, F. Deroose and J. T. M. Linders, Eur. J. Org. Chem., 2008, 8, 1350 CrossRef.
- A. S. S. Peter and W. K. Norman, J. Org. Chem., 1958, 23, 1599 CrossRef.
- A. N. Butkevich, M. L. Bossi, G. Lukinavičius and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 981 CrossRef CAS PubMed.
- (a) Y. Kawase, T. Yamagishi, J. Kato, T. Kutsuma, T. Kataoka, T. Iwakuma and T. Yokomatsu, Synthesis, 2014, 46, 455 CrossRef; (b) X. D. Xiong, Y. W. Jiang and D. W. Ma, Org. Lett., 2012, 14, 2552 CrossRef CAS PubMed; (c) J. Q. Zhang, G. B. Huang, J. Weng, G. Lu and A. S. C. Chan, Org. Biomol. Chem., 2015, 13, 2055 RSC.
- (a) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed; (b) Y. Iuchi, Y. Obora and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 2536 CrossRef CAS PubMed; (c) Y. Cai, F. Li, Y. Q. Li, W. B. Zhang, F. H. Liu and S. L. Shi, Tetrahedron Lett., 2018, 59, 1073 CrossRef CAS; (d) Y. Li, H. Q. Li, H. Junge and M. Beller, Chem. Commun., 2014, 50, 14991 RSC.
- (a) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410 CrossRef CAS PubMed; (b) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC; (c) A. Nandakumar, S. P. Midya, V. G. Landge and E. Balaraman, Angew. Chem., Int. Ed., 2015, 54, 11022 CrossRef CAS PubMed; (d) T. D. Obora and Y. Ishii, Synlett, 2011, 11, 30 CrossRef; (e) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853 CrossRef; (f) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed.
- (a) M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., Int. Ed., 2005, 44, 6913 CrossRef CAS PubMed; (b) X. Liu, R. S. Ding, L. He, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, ChemSusChem, 2013, 6, 604 CrossRef CAS PubMed.
- P. Yang, C. Zhang, Y. Ma, C. Zhang, A. Li, B. Tang and B. S. Zhou, Angew. Chem., Int. Ed., 2017, 56, 14702 CrossRef CAS PubMed.
- S. Thiyagarajan and C. Gunanathan, Org. Lett., 2020, 22, 6617 CrossRef CAS PubMed.
- (a) N. Joly, L. Bettoni, S. Gaillard, A. Poater and J.-L. Renaud, J. Org. Chem., 2021, 86, 6813 CrossRef CAS PubMed; (b) L. Bettoni, N. Joly, L. Jean-François, S. Gaillard, A. Poater and J.-L. Renaud, Adv. Synth. Catal., 2021, 363, 4009 CrossRef CAS.
- For selected recent examples of Mn-catalyzed AD and BH reactions, see: (a) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344 CrossRef CAS; (b) B. Maji and M. Barman, Synthesis, 2017, 49, 3377 CrossRef CAS; (c) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459 RSC; (d) S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (e) F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261 CrossRef CAS PubMed; (f) B. G. Reed-Berendt and L. C. Morrill, J. Org. Chem., 2019, 84, 3715 CrossRef CAS PubMed; (g) T. Liu, L. Wang, K. Wu and Z. Yu, ACS Catal., 2018, 8, 7201 CrossRef CAS; (h) J. Rana, V. Gupta and E. Balaraman, Dalton Trans., 2019, 48, 7094 RSC; (i) P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734 CrossRef CAS PubMed; (j) V. G. Landge, A. Mondal, V. Kumar, A. Nandakumar and E. Balaraman, Org. Biomol. Chem., 2018, 16, 8175 RSC; (k) Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180 CrossRef CAS; (l) K. Das, S. Waiba, A. Janaa and B. Maji, Chem. Soc. Rev., 2022, 51, 4386 RSC.
- For further details, see the ESI.†.
- (a) M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967 CrossRef PubMed; (b) G. Sivakumar, M. Subaramanian and E. Balaraman, ACS Sustainable Chem. Eng., 2022, 10(22), 7362 CrossRef CAS; (c) A. E. Owen, A. Preiss, A. McLuskie, C. Gao, G. Peters, M. Bühl and A. Kumar, ACS Catal., 2022, 12(12), 6923 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data. See DOI: https://doi.org/10.1039/d3cy00009e |
| This journal is © The Royal Society of Chemistry 2023 |