
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-functionality of rhodium-loaded MOR zeolite: production of H2via the water gas shift reaction and its use in the formation of NH3†
Shunsaku
Yasumura
 a,
Ken
Nagai
a,
Yucheng
Qian
a,
Takashi
Toyao
a,
Zen
Maeno
b and
Ken-ichi
Shimizu
*a
a,
Ken
Nagai
a,
Yucheng
Qian
a,
Takashi
Toyao
a,
Zen
Maeno
b and
Ken-ichi
Shimizu
*a
aInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. E-mail: kshimizu@cat.hokudai.ac.jp
bSchool of Advanced Engineering, Kogakuin University, 2665-1 Nakano-cho, Hachioji, Tokyo 192-0015, Japan
First published on 21st April 2023
Abstract
Rh-loaded mordenite (RhMOR) zeolite was investigated as a catalyst that can use CO + H2O as a H2 source for NO reduction. Operando IR measurements showed that CO was captured in the form of Rh dicarbonyl species ([Rh(CO)2]+) in zeolite, which reacts with H2O to form H2in situ via the water gas shift (WGS) reaction (CO + H2O → CO2 + H2) at >350 °C. Temperature-programmed surface reaction (TPSR) measurements under a flow of NO + CO + H2O monitored both the surface ad-species and outlet gas components. At >250 °C, the decrease in the outlet NO and CO as well as the increase in the IR intensity corresponding to NH4+ in zeolite were observed, indicating that the formed H2 was directly used for the reduction of NO into NH3, which was stored at the Brønsted acid sites (BAS). The mechanism of the WGS reaction was theoretically investigated; the predicted rate-determining step was the dissociation of H2O on [Rh(CO)2]+ to give [Rh(CO)(COOH)(H)]+ and its activation barrier was 176 kJ mol−1. The present study demonstrates the effective use of the multi-functionality of the isolated active metal anchored in zeolite as well as the potential utilization of CO + H2O as a H2 source.
1. Introduction
Atomically dispersed noble metals on solid supports, known as single-atom catalysts (SACs), have attracted a significant amount of attention because they provide the maximum utilization of expensive metals.1–3 Among the various supporting materials used for isolated metal catalysts, crystalline supports such as zeolites offer nearly uniform anchoring sites for the isolated metals, resulting in the formation of nearly uniform supported metal complexes.4–8 Much research effort has been devoted to obtaining molecular-level insight into the structure and reactivity of the metal complexes in zeolites.9–16 Rhodium complexes anchored in zeolites, as representative examples of well-defined active species,17–22 have been studied using in situ/operando spectroscopic techniques and density functional theory (DFT) calculations in order to identify the changes in their coordination environment during their reaction with small molecules.4–8,23–27 The group of Amiridis conducted X-ray absorption spectroscopy (XAS), high-resolution scanning transmission electron microscopy (HRSTEM), infrared (IR), and density function theory (DFT) calculations to clarify the detailed structure of the isolated [Rh(NO)2]+ complexes in zeolite and their catalytic activity toward the hydrogenation and dimerization of hydrocarbons.21 Although previous reports have studied model reactions over the Rh complexes in zeolites, only a few studies have focused on practical catalytic reactions under unsteady-state conditions driven by the reversible structural changes in the Rh complexes.23The use of CO + H2O as reductant to reduce NO to NH3 was proposed in the previous papers.28–44 The group of Christopher45 and that of Wang46 independently reported that atomically dispersed (single atom) Rh sites on Al2O3 and CeO2 exhibit high selectivity toward NH3 in the NO + CO + H2O reaction. These studies suggest that single-atom Rh sites catalyze the formation of NH3via the reduction of NO using H2 produced by the water gas shift (WGS) reaction (CO + H2O → H2 + CO2). Inspired by the pioneering works by Nakatsuji and co-workers,47 we have reported an unsteady-state NOx reduction system using a Rh-loaded zeolite catalyst. The method is based on a two-step cyclic operation, in which the NOx trapped on the catalyst during the lean period is reduced by H2 to form NH3 during rich period (fuel-rich condition). Based on the operando spectroscopic and DFT results, it has been found that the reduction of the [Rh(NO)2]+ complexes by H2 results in NH3 as an intermediate, which then reduces NOx to give N2.48,49
In this study, we have conducted IR experiments and DFT calculations for Rh-loaded mordenite zeolite (Rh-MOR) with a low Rh loading. Operando IR experiments were carried out using transient or temperature-programed surface reaction (TPSR) measurements. The results obtained under a flow of CO + H2O show that CO was captured in the form of [Rh(CO)2]+ complex, which reacts with H2O to produce CO2 + H2via the WGS reaction at >350 °C. In the presence of NO, the H2 formed in situ (via the WGS reaction) was directly used for the reduction of NO to form NH3 at >250 °C. Transition state calculations confirmed that the Rh cation was practical as the active site for the WGS reaction.
2. Results and discussion
2.1. TPSR and operando IR studies on the WGS reaction and NH3 formation
To assess the effect of temperature on the WGS reaction, CO + H2O was fed to 0.6 wt% Rh-loaded MOR zeolite (RhMOR) under temperature-ramping conditions. RhMOR was prepared via the aqueous ion-exchange method using NH4+ − MOR (Si/Al = 10) with the aqueous solution of RhCl3 (see ESI†). The catalyst was subjected to CO + H2O at room temperature and heated to 500 °C at a ramping rate of 20 °C min−1. The concentration of CO and CO2 in the outlet gas was analyzed using IR (gas cell) and the relative concentration of H2 was estimated using mass spectrometry (MS). The outlet gas profile (Fig. 1) shows that CO2 was observed at >250 °C and its formation rate increases at >400 °C. H2 evolution was observed at >350 °C. Since the fed H2O is the only source of hydrogen atoms, these results indicate that the WGS reaction was catalyzed by RhMOR at >350 °C. The consecutive mechanism of the WGS on RhMOR will be discussed based on our DFT calculations. | ||
| Fig. 1 MS intensity of H2 (arbitrary unit) and the concentration of CO and CO2 (ppm) during ramping the temperature (20 °C min−1) under a flow of 0.5% CO + 1% H2O over RhMOR. | ||
The mechanism of the WGS reaction over RhMOR was examined by TPSR measurements using in situ IR measurements of the adsorbed species combined with online IR and MS analyses of the effluent gas (operando IR). A schematic representation of the setup used is shown in Fig. S1.† The catalyst powder was pelletized into a self-supported IR disk (40 mg) and set in the in situ IR cell. The concentration of CO2 in the outlet gas was monitored using another IR equipped with an IR gas cell, while the evolution of H2 was monitored by MS. The IR disk was first pretreated under a flow of 2% O2 at 350 °C and then subjected to a flow of 0.5% CO/He at 200 °C. Fig. 2(left) shows the IR spectra of CO adsorbed on RhMOR. According to the literature,7,19,23 the two characteristic IR peaks observed at 2044 cm−1 and 2108 cm−1 can be ascribed to the isolated dicarbonyl Rh+ complexes ([Rh(CO)2]+) stabilized at the Al sites in the zeolite. After purging with He, the disk was further subjected to a flow of 1% H2O/He at 350 °C. Upon exposure to H2O, the intensity of the IR peaks corresponding to [Rh(CO)2]+ decreased and were scarcely observed after 600 s of exposure. Note that the formation of [Rh(CO)]+ was not observed here as discussed in the previous report which revealed that the desorption of the second CO is rapid at high temperatures using in situ IR spectroscopy.50 The time course of the concentration of CO2 as well as the MS intensity of H2 in the outlet gas are plotted in Fig. 2(right) together with the IR intensity at 2044 cm−1 for [Rh(CO)2]+. When H2O was fed to the CO adsorbed RhMOR, the IR intensity of the [Rh(CO)2]+ complex decreased, and the concentration of CO2 and MS intensity of H2 in the outlet gas increased. This indicates that CO coordinated to the Rh+ sites ([Rh(CO)2]+ complex) react with H2O to give CO2 and H2 (i.e. the WGS reaction).
 | ||
| Fig. 2 The changes in the in situ IR spectra obtained for [Rh(CO)2]+ in RhMOR under a flow of 1% H2O at 350 °C (left), the time course of the height of the IR peak for [Rh(CO)2]+, and MS intensity (arbitrary unit) of H2 and CO2 (right). The IR disk was pre-exposed to 0.5% CO at 200 °C (600 s), followed by purging with He (600 s). | ||
Recently, we reported that RhMOR catalyzed the selective formation of NH3 from NO + H2 and the subsequent capture of in situ formed NH3 at the Brønsted acid sites (BASs) in the zeolite. Aiming at H2 formation from CO and H2O, followed by NH3 formation from NO + H2, we conducted TPSR in combination with operando IR measurements under a flow of NO + CO + H2O over RhMOR. The TPSR profile shows the formation of CO2 at >250 °C (Fig. 3). The concentration of CO2 (i.e. relative rate of CO2 formation) in a temperature range of 300–500 °C under NO + CO + H2O was higher than that observed under CO + H2O (Fig. 1) due to the competitive NO + CO reaction. In the IR spectra obtained for the adsorbed species, the NH3 adsorbed on the BASs in MOR (NH4+) was observed at >250 °C (Fig. S2†), while gaseous NH3 was observed in outlet gas at >350 °C. Compared to the formation of gaseous H2 in NO + H2O flow (Fig. 1), the temperature of NH3 formation is lower (<250 °C vs. <350 °C). These results suggest that the H2 formed in situ (via the WGS reaction over RhMOR) acts as a reductant to reduce NO to form NH3.
 | ||
| Fig. 3 The MS intensity of H2 (arbitrary units), concentrations of NO, CO, and CO2, and IR intensity of NH4+ during TPSR (20 °C min−1) under a flow of 0.1% NO + 0.5% CO + 1% H2O over RhMOR. | ||
2.2. DFT study on the WGS mechanism over the Rh+ cation in MOR zeolite
Reaction route mapping, as implemented in the GRRM17 program,51 based on DFT calculations within periodic boundary condition was performed to investigate the mechanism of the WGS reaction over the RhMOR catalyst. The isolated [Rh(CO)2]+ species, which were observed under a flow of CO using in situ IR (Fig. 2), was used as the initial structure. Among the distinct T sites in MOR zeolite, the T4 site was applied as the Al replacement site (Fig. 4a). Note that MOR framework comprises four crystallographically inequivalent T sites; among them, T4 site is known as the energetically preferred Al replacement site. Assuming that one-fourth of the Al sites are T4 sites, the Rh/AlT4 ratio is calculated to be 0.14, which suggests that there are ample T4 sites available to accommodate Rh cations.52,53Fig. 4b shows the reaction pathway for the Rh(CO)2 species with a H2O molecule weakly bound to the zeolite framework. In the calculated reaction, two pathways were investigated as the initial steps: (i) The adsorption of H2O onto the Rh(CO)2 species to give Rh(CO)2(H2O) and (ii) the dissociative adsorption of H2O to give Rh(CO)(COOH)(H) species. Although the former pathway proceeds via a low activation barrier (38 kJ mol−1) as the first step, the following reaction requires a very high activation energy (370 kJ mol−1) to produce the Rh(CO)2(OH) species and BAS. The latter pathway, which forms the Rh(CO)(COOH)(H) species via an activation barrier (176 kJ mol−1) as the first step, subsequently forms a Rh(CO)(H)2 species and CO2 molecule. The formation of Rh(CO)(H)2 species is previously reported over Rh-exchanged Y zeolite17 and Rh-loaded Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 by IR measurement under H2 flow near room temperature. Fig. 5 shows the energy profile along with the reaction coordinate for the reaction of the Rh(CO)2 species with H2O. The result indicates that although the activation barrier of the first step was higher than that forming the BAS (38 vs. 176 kJ mol−1), the second step requires an activation energy of only 87 kJ mol−1, and the relative energy of the products (Rh(CO)(H)2 + CO2) was lower (72 kJ mol−1) when compared to Rh(CO)2(OH) + BAS (125 kJ mol−1). Therefore, the formation of the Rh(CO)(H)2 complex and CO2 molecule was determined as the plausible pathway for the reaction of Rh(CO)2 with H2O. The formed CO2 was then easily desorbed with a desorption energy of only 23 kJ mol−1 (Fig. S3†).
54 by IR measurement under H2 flow near room temperature. Fig. 5 shows the energy profile along with the reaction coordinate for the reaction of the Rh(CO)2 species with H2O. The result indicates that although the activation barrier of the first step was higher than that forming the BAS (38 vs. 176 kJ mol−1), the second step requires an activation energy of only 87 kJ mol−1, and the relative energy of the products (Rh(CO)(H)2 + CO2) was lower (72 kJ mol−1) when compared to Rh(CO)2(OH) + BAS (125 kJ mol−1). Therefore, the formation of the Rh(CO)(H)2 complex and CO2 molecule was determined as the plausible pathway for the reaction of Rh(CO)2 with H2O. The formed CO2 was then easily desorbed with a desorption energy of only 23 kJ mol−1 (Fig. S3†).
 | ||
| Fig. 4 (a) The periodic model used for MOR zeolite. (b) The calculated reaction pathway for Rh(CO)2 and H2O together with the relative energy (ΔE) values. The values written in dark red show the activation barrier. The relative energies are provided under each structure (units: kJ mol−1). | ||
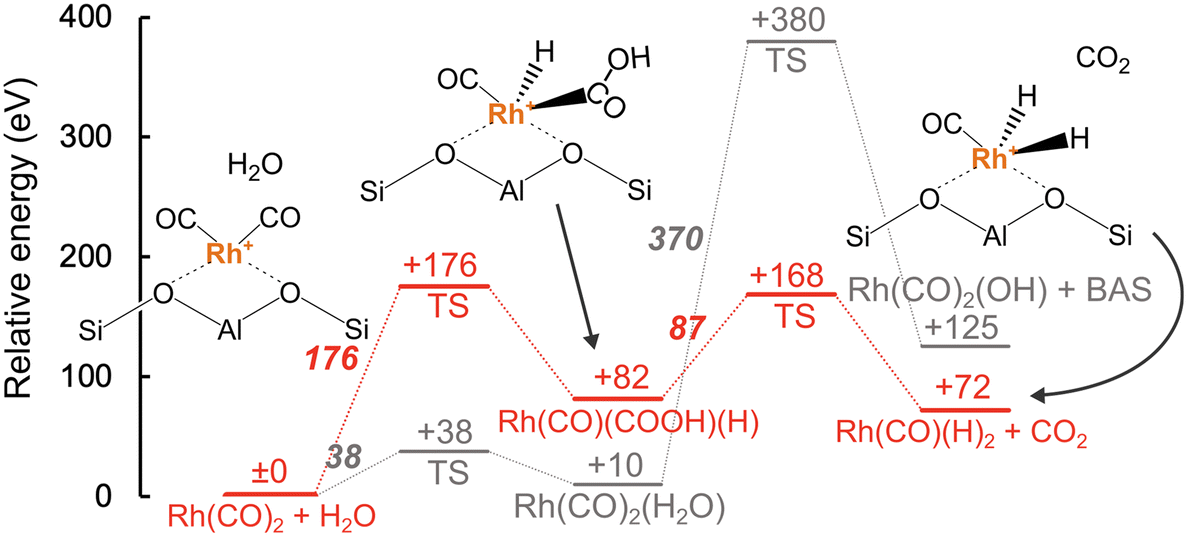 | ||
| Fig. 5 Energy profile for the reaction of Rh(CO)2 with water. The reaction pathway indicated by the red line is the most plausible pathway for CO2 formation. The relative energies are provided under each bar and the activation barriers are shown using bold italic style (units: kJ mol−1). | ||
2.3. Operando IR measurements under periodic lean (NO + O2 + H2O)/rich (NO + CO + H2O) conditions
Considering the presence of CO and water in the automotive exhaust gas and that Rh species can catalyze WGS reaction, we designed a two-stage de-NOx method with RhMOR, where NH3, formed via WGS followed by NO + H2 reaction in rich (NO + CO + H2O) period, is utilized as a reductant of NOx in the subsequent lean (NO + O2 + H2O) period. Thus, operando IR measurements were carried out to observe the ad-species on the catalyst under periodic lean/rich conditions. The rich gas (0.1% NO + 0.5% CO + 1% H2O; 300 s) and lean gas (0.1% NO + 2% O2 + 1% H2O; 600 s) were repeatedly fed into an in situ IR cell at 300 °C. Fig. 6 shows the representative IR spectra (left) and time course of outlet gases (NO, N2O, CO, and CO2) and IR intensities (B–NH3 and NOad; right). Under lean conditions, the NO species adsorbed on the Rh cations (NOad) were observed at 1950 cm−1,55,56 indicating that NO was captured by the catalyst. Under the subsequent rich conditions, the [Rh(CO)2]+ species and B–NH3 appear in the IR spectra, while the peak corresponding to NOad was hardly detected. This observation shows that Rh species in the zeolite are repeatably reduced back to isolated Rh species under the rich conditions while they were oxidized under the rich condition. We have confirmed that this process can be repeated at least 10 times (up to 9000 seconds) without significant changes in performance. Note that the adsorbed NO (e.g. Rh(NO)2) was not observed under rich conditions because the hydrogenation of NO required only 87 kJ mol−1 of the activation barrier as shown in our previous report.49 The evaluated NO conversion and N2 selectivity are 18% and 94%, respectively while gaseous NH3 was not detected from the outlet gas (the products from the NO + CO reaction were possibly included). Combined with the TPSR measurements (Fig. 3), the H2 formed in situ (via the WGS reaction) was used to reduce NO to form NH3, which was then stored at the BASs in zeolite (B–NH3). | ||
| Fig. 6 (a) Representative IR spectra and (b) time course of outlet gases (NO, N2O, CO, and CO2) and IR intensities (B–NH3 and NOad) obtained for the periodic rich (0.1% NO + 0.5% CO + 1% H2O 300 s)/lean (0.1% NO + 2% O2 + 1% H2O; 600 s) conditions at 300 °C over Rh-MOR. | ||
Assuming that both the WGS reaction and NH3 formation take place at the isolated Rh cations, their reaction mechanism can be described (Fig. 7). The WGS reaction occurs from [Rh(CO)2]+ + H2O to form [Rh(CO)(H)2]+ + CO2via activation barriers of 176 and 87 kJ mol−1, respectively. After the desorption of CO2, the resulting [Rh(CO)(H)2]+ complex follows two possible pathways: (i) the [Rh(CO)2]+ complex and adsorbed H2O are recovered by the adsorption of CO and H2O as well as the desorption of H2, and (ii) the [Rh(CO)(H)2(NO)]+ complex was formed by the adsorption of NO. In the latter case, the adsorbed NO was reduced into a N atom and H2O ([Rh(CO)(H2O)(N)]+), and then hydrogenated to form NH3, as discussed in our previous study.49,57–59 The consecutive stoichiometry is described as follows.
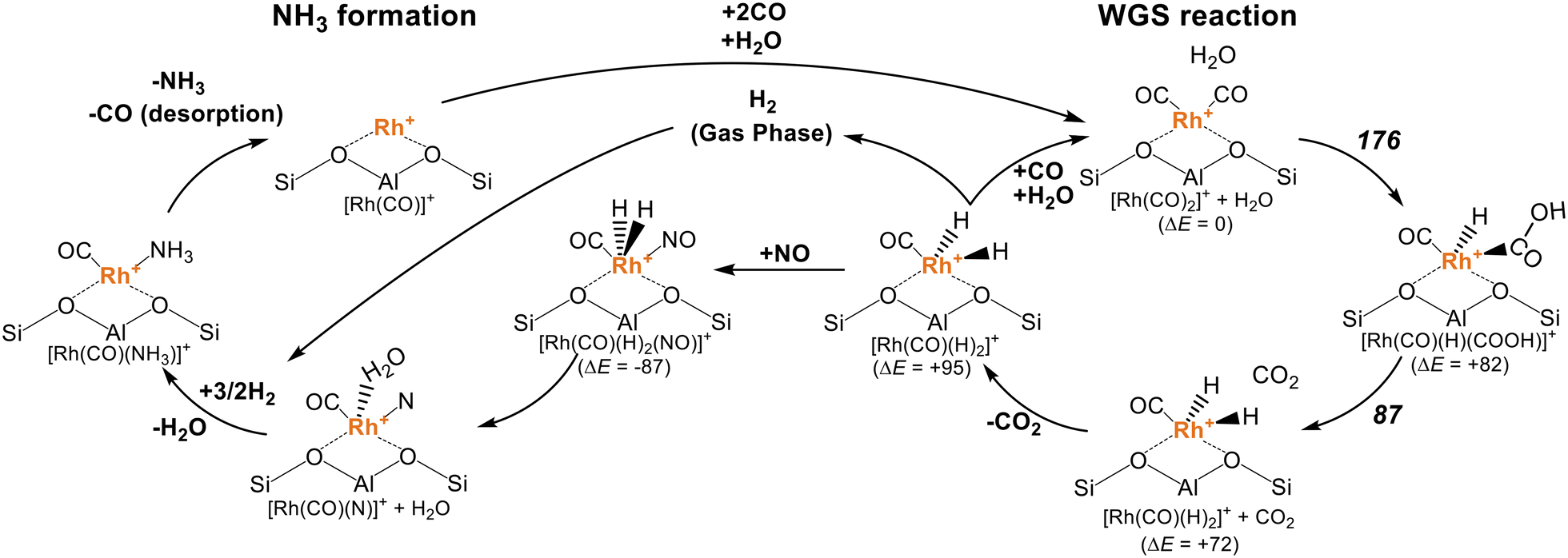 | ||
| Fig. 7 The mechanism of the de-NOx reaction using CO + H2O with the RhMOR catalyst proposed in this study. The relative energies of each structure are shown together (ΔE). The activation barriers of the WGS reaction are shown in bold italic style. The reaction pathway for NH3 formation was discussed in our previous report (unit: kJ mol−1).49 | ||
Rich condition:
| 2NO + 5CO + 3H2O → 2NH3 + 5CO2 | (1) |
Lean condition (SCR reaction):
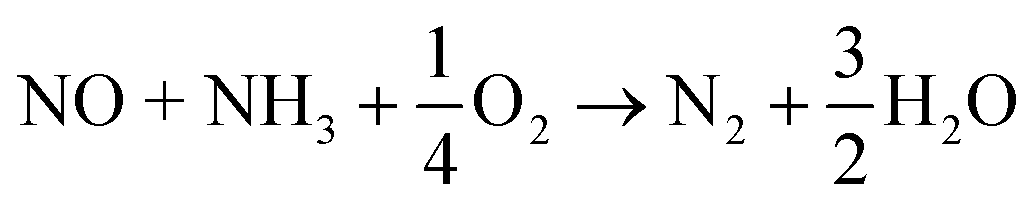 | (2) |
3. Conclusions
The NO reduction over RhMOR using CO + H2O as reductants have been investigated by in situ/operando techniques and DFT calculations. TPSR measurements showed that the Rh species in zeolite can catalyze the WGS reaction (CO + H2O → CO2 + H2O) via [Rh(CO)2]+ complex at >350 °C. Other TPSR measurements under 0.1% NO + 0.5% CO + 1% H2O demonstrated the simultaneous formation of CO2 and B–NH3 at >250 °C, indicating that in the presence of NO, the H2 formed in situ (via the WGS reaction) is directly used for the reduction of NO into NH3 (B–NH3). Our DFT calculations indicate that the rate-determining step of the WGS reaction is the dissociative adsorption of H2O on [Rh(CO)2]+ to give a [Rh(CO)(COOH)(H)]+ complex (175.6 kJ mol−1). The present study has proposed the effective use of the multi-functionality of active metals anchored in zeolite as well as the potential utilization of CO + H2O as reductants in the de-NOx reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by JSPS KAKENHI (21H04626). Some of the calculations were conducted employing the supercomputing resources at the Cyberscience Center of Tohoku University. This project was supported by the Joint Usage/Research Center for Catalysis. The authors would like to sincerely thank the Technical Division of the Institute for Catalysis at Hokkaido University.References
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- A. K. Datye and M. Votsmeier, Nat. Mater., 2021, 20, 1049–1059 CrossRef CAS PubMed.
- I. Ogino and B. C. Gates, J. Phys. Chem. C, 2010, 114, 8405–8413 CrossRef CAS.
- P. Serna and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 4714–4717 CrossRef CAS PubMed.
- P. Serna and B. C. Gates, Angew. Chem., Int. Ed., 2011, 50, 5528–5531 CrossRef CAS PubMed.
- C. Y. Fang, S. Zhang, Y. Hu, M. Vasiliu, J. E. Perez-Aguilar, E. T. Conley, D. A. Dixon, C. Y. Chen and B. C. Gates, ACS Catal., 2019, 9, 3311–3321 CrossRef CAS.
- J. E. Perez-Aguilar, C. Y. Chen, J. T. Hughes, C. Y. Fang and B. C. Gates, J. Am. Chem. Soc., 2020, 142, 11474–11485 CrossRef CAS PubMed.
- P. Rubio-Marqués, M. A. Rivero-Crespo, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2015, 137, 11832–11837 CrossRef PubMed.
- J. Cejka, A. Corma and S. Zones, Zeolites and Catalysis, Wiley, 2010 Search PubMed.
- S. Yasumura, H. Ide, T. Ueda, Y. Jing, C. Liu, K. Kon, T. Toyao, Z. Maeno and K. Shimizu, JACS Au, 2021, 1, 201–211 CrossRef CAS PubMed.
- S. Yasumura, T. Ueda, H. Ide, K. Otsubo, C. Liu, N. Tsunoji, T. Toyao, Z. Maeno and K. Shimizu, Phys. Chem. Chem. Phys., 2021, 23, 22273–22282 RSC.
- N. Kosinov, C. Liu, E. J. M. Hensen and E. A. Pidko, Chem. Mater., 2018, 30, 3177–3198 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota, A. Staykov and K. Yoshizawa, Acc. Chem. Res., 2018, 51, 2382–2390 CrossRef CAS PubMed.
- Z. Maeno, S. Yasumura, X. Wu, M. Huang, C. Liu, T. Toyao and K. Shimizu, J. Am. Chem. Soc., 2020, 142, 4820–4832 CrossRef CAS PubMed.
- G. Li and E. A. Pidko, ChemCatChem, 2019, 11, 134–156 CrossRef CAS.
- K. Khivantsev, A. Vityuk, H. A. Aleksandrov, G. N. Vayssilov, O. S. Alexeev and M. D. Amiridis, J. Phys. Chem. C, 2015, 119, 17166–17181 CrossRef CAS.
- T. T. T. Wong, A. Y. Stakheev and W. M. H. Sachtler, J. Phys. Chem., 1992, 96, 7733–7740 CrossRef CAS.
- E. Ivanova, M. Mihaylov, F. Thibault-Starzyk, M. Daturi and K. Hadjiivanov, J. Catal., 2005, 236, 168–171 CrossRef CAS.
- V. Markova, G. Rugg, A. Govindasamy, A. Genest and N. Rösch, J. Phys. Chem. C, 2018, 122, 2783–2795 CrossRef CAS.
- K. Khivantsev, A. Vityuk, H. A. Aleksandrov, G. N. Vayssilov, D. Blom, O. S. Alexeev and M. D. Amiridis, ACS Catal., 2017, 7, 5965–5982 CrossRef CAS.
- W. Shang, M. Gao, Y. Chai, G. Wu, N. Guan and L. Li, ACS Catal., 2021, 7249–7256 CrossRef CAS.
- C. Y. Fang, J. Valecillos, E. T. Conley, C. Y. Chen, P. Castaño and B. C. Gates, J. Phys. Chem. C, 2020, 124, 2513–2520 CrossRef CAS.
- W. A. Weber and B. C. Gates, J. Phys. Chem. B, 1997, 101, 10423–10434 CrossRef CAS.
- A. J. Dent, J. Evans, S. G. Fiddy, B. Jyoti, M. A. Newton and M. Tromp, Angew. Chem., Int. Ed., 2007, 46, 5356–5358 CrossRef CAS PubMed.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- S. Yasumura, T. Kato, T. Toyao, Z. Maeno and K. I. Shimizu, Phys. Chem. Chem. Phys., 2023, 8524–8531 RSC.
- K. Kobayashi, S. Ogawa, H. Matsumoto, K. Matsuda and T. Nanba, J. Jpn. Pet. Inst., 2020, 63, 28–37 CrossRef CAS.
- C. J. Yoo, A. Getsoian and A. Bhan, Appl. Catal., B, 2021, 286, 119893 CrossRef CAS.
- K. Kobayashi, R. Atsumi, Y. Manaka, H. Matsumoto and T. Nanba, Catal. Sci. Technol., 2019, 9, 2898–2905 RSC.
- E. C. Corbos, M. Haneda, X. Courtois, P. Marecot, D. Duprez and H. Hamada, Catal. Commun., 2008, 10, 137–141 CrossRef CAS.
- M. Sakai, T. Hamaguchi and T. Tanaka, Appl. Catal., A, 2019, 582, 117105 CrossRef CAS.
- J. R. González-Velasco, B. Pereda-Ayo, U. De-La-Torre, M. Urrutxua and R. López-Fonseca, ChemCatChem, 2018, 10, 2928–2940 CrossRef.
- L. Castoldi, R. Bonzi, L. Lietti, P. Forzatti, S. Morandi, G. Ghiotti and S. Dzwigaj, J. Catal., 2011, 282, 128–144 CrossRef CAS.
- M. Kim, J. Choi and M. Crocker, Catal. Today, 2014, 231, 90–98 CrossRef CAS.
- F. Can, X. Courtois, S. Royer, G. Blanchard, S. Rousseau and D. Duprez, Catal. Today, 2012, 197, 144–154 CrossRef CAS.
- M. Urrutxua, B. Pereda-Ayo, L. V. Trandafilovic, L. Olsson and J. R. González-Velasco, Ind. Eng. Chem. Res., 2019, 58, 7001–7013 CrossRef CAS.
- M. Li, V. G. Easterling and M. P. Harold, Appl. Catal., B, 2016, 184, 364–380 CrossRef CAS.
- S. H. Oh and T. Triplett, Catal. Today, 2014, 231, 22–32 CrossRef CAS.
- V. Y. Prikhodko, J. E. Parks, J. A. Pihl and T. J. Toops, Catal. Today, 2016, 267, 202–209 CrossRef CAS.
- K. Ramanathan, C. S. Sharma and C. H. Kim, Ind. Eng. Chem. Res., 2012, 51, 1198–1208 CrossRef CAS.
- C. D. Digiulio, J. A. Pihl, J. E. Parks, M. D. Amiridis and T. J. Toops, Catal. Today, 2014, 231, 33–45 CrossRef CAS.
- M. Li, S. A. Malamis, W. Epling and M. P. Harold, Appl. Catal., B, 2019, 242, 469–484 CrossRef CAS.
- C. R. Thomas, J. A. Pihl, V. Y. Prikhodko, M. K. Kidder, J. A. Lauterbach and T. J. Toops, Catal. Commun., 2021, 156, 106308 CrossRef CAS.
- C. Asokan, Y. Yang, A. Dang, A. B. Getsoian and P. Christopher, ACS Catal., 2020, 10, 5217–5222 CrossRef CAS.
- K. Khivantsev, C. G. Vargas, J. Tian, L. Kovarik, N. R. Jaegers, J. Szanyi and Y. Wang, Am. Ethnol., 2021, 133, 395–402 Search PubMed.
- T. Nakatsuji, M. Matsubara, J. Rouistenmäki, N. Sato and H. Ohno, Appl. Catal., B, 2007, 77, 190–201 CrossRef CAS.
- S. Yasumura, T. Toyao, Z. Maeno and K. Shimizu, ACS Catal., 2021, 12293–12300 CrossRef CAS.
- S. Yasumura, T. Kato, Y. Qian, T. Toyao, Z. Maeno and K. Shimizu, J. Phys. Chem. C, 2022, 126, 19147–19158 CrossRef CAS.
- E. Ivanova and K. Hadjiivanov, Phys. Chem. Chem. Phys., 2003, 5, 655–661 RSC.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233–250 CrossRef CAS PubMed.
- M. Brändle and J. Sauer, J. Am. Chem. Soc., 1998, 120, 1556–1570 CrossRef.
- H. Xia, ACS Omega, 2020, 5, 9707–9713 CrossRef CAS PubMed.
- J. T. Yates, S. D. Worley, T. M. Duncan and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1225–1230 CrossRef CAS.
- B. E. Hayden, A. King, M. A. Newton and N. Yoshikawa, J. Mol. Catal. A: Chem., 2001, 167, 33–46 CrossRef CAS.
- K. A. Almusaiteer and S. S. C. Chuang, J. Phys. Chem. B, 2000, 104, 2265–2272 CrossRef CAS.
- S. Yasumura, Y. Qian, T. Kato, S. Mine, T. Toyao, Z. Maeno and K. Shimizu, ACS Catal., 2022, 12, 9983–9993 CrossRef CAS.
- S. Yasumura, Y. Qian, T. Toyao, Z. Maeno and K. Shimizu, J. Phys. Chem. C, 2022, 126, 11082–11090 CrossRef CAS.
- S. Yasumura, C. Liu, T. Toyao, Z. Maeno and K. Shimizu, J. Phys. Chem. C, 2021, 125, 1913–1922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental and theoretical details as well as the additional data of IR spectra and DFT calculation are included. See DOI: https://doi.org/10.1039/d3cy00043e |
| This journal is © The Royal Society of Chemistry 2023 |