
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning catalysis by surface-deposition of elements on oxidation catalysts via atomic layer deposition†
Frederik
Rüther
 a,
Robert
Baumgarten
a,
Fabian
Ebert
a,
Esteban
Gioria
a,
Raoul
Naumann d'Alnoncourt
*a,
Annette
Trunschke
b and
Frank
Rosowski
ac
a,
Robert
Baumgarten
a,
Fabian
Ebert
a,
Esteban
Gioria
a,
Raoul
Naumann d'Alnoncourt
*a,
Annette
Trunschke
b and
Frank
Rosowski
ac
aBasCat - UniCat BASF JointLab, Technische Universität Berlin, D-10623 Berlin, Germany. E-mail: r.naumann@bascat.tu-berlin.de
bDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, D-14195 Berlin, Germany
cCatalysis Research, BASF SE, D-67065 Ludwigshafen, Germany
First published on 22nd March 2023
Abstract
Based on the concept that most reaction steps proceed only at the surface layer of a bulk catalyst, the catalytic impact of the surface-modification with POx, BOx, and MnOx in the selective oxidation of ethane, propane, and n-butane is systematically studied. Three different promoter elements are deposited as a sub-monolayer on the surface of oxidation catalysts with high dispersion by sequential and self-limiting reactions at the solid–gas interface, using atomic layer deposition. Oxygenate and olefin selectivities are tuned by the surface deposition of POx and BOx, leading to improved product yields. The mixed metal oxide MoVTeNbOx is used as a case study to demonstrate the effect of the modification in different reactions with yield improvements of up to 24% in the propane oxidation towards acrylic acid. It is shown that the beneficial performance is related to a change in surface composition, a modification in the electronic properties of the redox active element vanadium, and a decrease in acidity. A comparative study considering several bulk catalysts and deposited elements revealed further promoting effects for different oxidation catalysts. In particular, the deposition of POx on V-containing oxides suppresses COx formation. Precisely adjusted surface modifications leading to enhanced product yields demonstrate the potential of atomic layer deposition as a powerful tool for tuning catalytic properties of bulk catalysts.
Introduction
On the way towards a greener chemical industry existing processes must be optimized, which depends strongly on advances in heterogeneous catalysis.1–3 Every chemical conversion step is accompanied by a loss in resources and energy. The goal of sustainability can only be achieved by minimizing this loss, either by reducing the overall number of transformations or tuning overall product yields. Partial oxidations of light alkanes have been studied for decades as promising reaction routes converting feedstocks directly into valuable chemicals.4 Preventing consecutive and parallel reactions, leading to the formation of combustion products, is still a difficult task. However, in the case of highly optimized selective oxidation reactions like the n-butane (C4) oxidation to maleic anhydride (MAN) on vanadyl(IV) pyrophosphate (VPP)5–9 or the direct oxidation of propane (C3) to acrylic acid (AA) using MoVTeNbOx,10–12 two promising catalysts were identified.5,13 The oxidative dehydrogenation (ODH) of ethane (C2) is a prospective reaction route for the on-demand production of ethylene using similar catalysts.14,15 One of the main challenges in understanding the underlying catalytic principles is the dynamic behaviour and self-adapting nature of the catalysts under reaction conditions.16–19 In literature, this phenomenon is described as ‘flexible surfaces’, where the chemical potential of the gas phase affects the surface state and changes the surface composition of the bulk catalyst.20–23 However, structural changes are actually proceeding only at the surface itself or within a thin subsurface layer (0.5–1 nm determined for VPP24 or MoVTeNbOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18), while the bulk functions e.g. as an electron or oxygen carrier.20,25
18), while the bulk functions e.g. as an electron or oxygen carrier.20,25
In situ characterization revealed a shift in the oxidation state of the redox active element vanadium(V) on the surface of VPP.17,26–28 While it is fixed to V4+ based on the stoichiometry of the bulk, the detection of V5+-species supports the hypothesis that the actual active phase of a catalyst can be very different from the bulk.8,29–32 Similarly, the crucial influence of the gas phase is studied for MoVTeNbOx.13,18,33,34 The active and selective surface layer is formed under reaction conditions with a strong influence of added steam, inducing an enrichment of V5+ and depletion of Mo6+ sites.12,19,20,35,36 Recently, a data driven approach37 was applied to a clean catalytic data set with the goal to connect the materials properties and underlying physical principles to the performance of different catalysts in selective oxidations.38,39 The targeted reduction of total oxidation can be related to surface composition, surface area, redox activity, and isolation of active sites responsible for the formation of olefins (C2, C3). With oxygenates as the target product (C4, C3) the electronic properties of the bulk and the surface, as well as the adsorption properties of the catalyst are crucial.40
Hence, the catalytic functionality of bulk catalysts is determined by the properties of the termination layer, which depends on the bulk composition and is formed under reaction conditions.24,26,36,41–43 However, the surface can additionally be modified using synthetic methods like atomic layer deposition (ALD). The deposition of elements via volatile precursors using self-limiting surface reactions during ALD is utilized to tune the properties of the catalyst precursor at the atomic level.44–50 Most commonly it is used for surface decoration or promotion of bulk catalysts51–53 or advanced catalyst synthesis.54–56 Here, ALD provides a high dispersion of the deposited elements, to improve activity, selectivity, or stability. Additionally, ALD has the advantage of a solvent-free deposition of promotors, so that the mechanical properties of the bulk catalyst are sustained. This enables to elucidate on reaction mechanisms or property performance relationships.52
This work features the surface modification of oxidation catalysts to optimize catalytic (surface-)properties. First, the catalytic functionality of six different oxidation catalysts and their potential to selectively activate alkanes is discussed. Then, oxides (POx, BOx, MnOx) with potentially promoting effects are deposited on the catalyst surface in sub-monolayers using ALD and the catalytic impact is studied in the selective oxidation of ethane, propane, and n-butane. As the deposition of POx on V2O5 showed promising results in n-butane oxidation,51,52 this concept is transferred to reactions of propane and ethane and more selective oxidation catalysts. The mixed metal oxide MoVTeNbOx is tuned by the deposition of POx and BOx, leading to improved product yields. With this study we demonstrate that ALD is as a versatile tool to promote oxidation catalysts, while the basic catalytic features of the host structures are maintained.
Results and discussion
Comparative functionality analysis of base catalysts
A collection of six oxide and phosphate catalysts were selected for their broad range in catalytic behaviour and studied in the oxidation of ethane, propane, and n-butane. From a variety of elements, V or Mo-based materials show the most promising performance in selective oxidations. However, binary or monometallic oxides V2O551,57 or MoO358,59 show either a very low alkane conversion or product selectivity. In contrast, the combination of these elements, preferably with more complex structural features, e.g. MoVOx with M1-structure type,60–62 have a much higher capability to selectively oxidize the reactant. By introducing Te and Nb into the M1 structure, MoVTeNbOx was obtained as an excellent catalyst for the oxidative dehydrogenation of ethane (ODHE) and the direct oxidation of propane to AA.15,63–65 For an efficient oxidation of n-butane to MAN, vanadium phosphate (VPO) catalysts or more specifically VPP, are industrially applied.9,66 As an alternative for V, as redox active element, Mn is studied for the activation of light alkanes. Here, the perovskite structured SmMnO3 is included, which was applied in the oxidative dehydrogenation of propane67,68 (see Fig. SI1† for XRPD patterns of all catalysts). While these catalysts were mostly studied in literature in one specific reaction, a comparative study under different reaction feeds is way less prominent.63,69 The functionalization of different alkanes (see desired reaction pathways eqn (1)–(3)) follows very similar steps, namely the activation of a C–H bond, the abstraction of two hydrogen atoms and, in case an oxygenate is formed, the insertion of oxygen into the molecule. Also, in all three reactions the water formation as couple-product is an important driving force.42 However, for each reaction a different amount of electron transfers needs to be performed and the amount of side products increases with the alkane chain-length.| C2H6 + 0.5O2 → C2H4 + H2O | (1) |
| C3H8 + 2O2 → C3H4O2 + 2H2O | (2) |
| C4H10 + 3.5O2 → C4H2O3 + 4H2O | (3) |
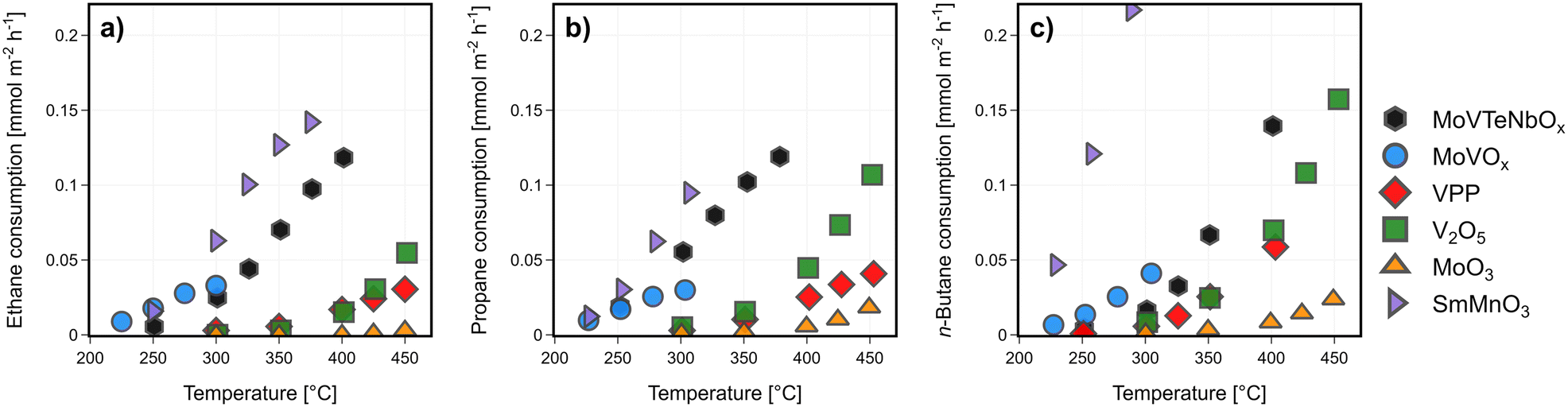 | ||
| Fig. 1 Catalytic activity of MoVTeNbOx. MoVOx, MoO3, V2O5, VPP and SmMnO3: alkane consumption rates normalized to the surface area [mmol m−2 h−1] as a function of the reaction temperature in (a) ethane, (b) propane, and (c) n-butane oxidation; the corresponding alkane conversions and consumption rates normalized to weight can be found in Fig. SI3;† oxygen consumption rates are shown in Fig. SI4;† C2H6/O2/H2O = 3/9/0 % vol, C3H8/O2/H2O = 3/9/20 % vol, C4H10/O2/H2O = 2/20/3 % vol, 1 atm, 1000 h−1 (C2, C3)/2000 h−1 (C4). | ||
In general, the rate of alkanes consumption is not only a function of C–H activation. Fig. 2 shows the product distribution obtained with the catalysts normalized to a fixed alkane conversion level. The formation of the various products requires additional centres for C–C bond cleavage or oxygen insertion. In the case of propane and n-butane, oxygenates are formed in much higher selectivity on the catalysts surface compared to ethane oxidation (see Fig. 2 and contact time variation study in Fig. SI5 and SI6†). Therefore, the reaction rate is not only determined by the alkane activation, as the constant rates on MoVTeNbOx and MoVOx suggest. A re-adsorption of already formed products also might play a role.
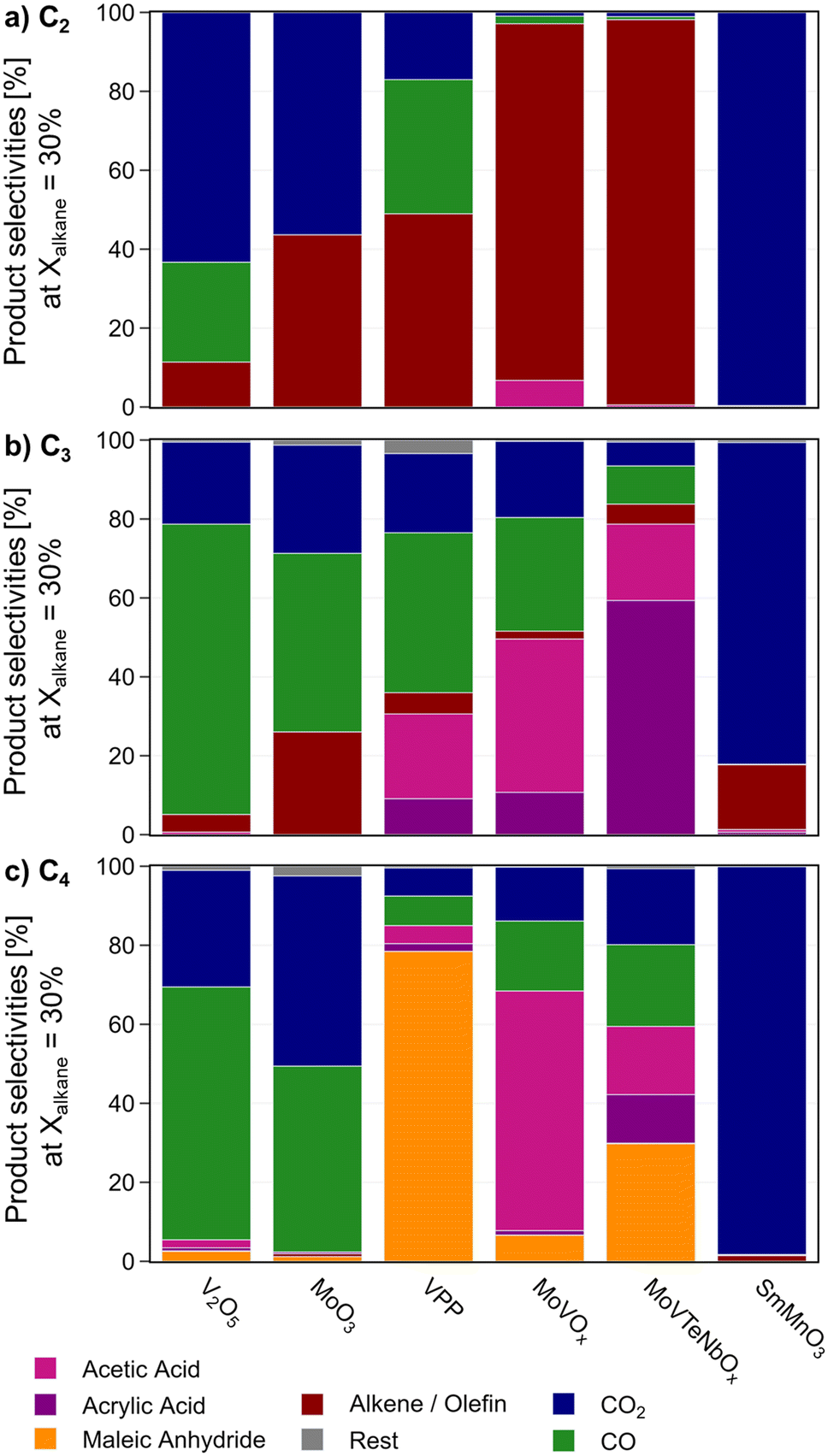 | ||
| Fig. 2 Product selectivity in a) ethane, b) propane, and c) n-butane oxidation, interpolated to a fixed alkane conversion of Xalkane = 30% during the temperature variation study; in case of MoO3 the maximum temperature setpoint of 450 °C is used, due to overall low conversions Xalkane < 30%; C2H6/O2/H2O = 3/9/0 % vol, C3H8/O2/H2O = 3/9/20 % vol, C4H10/O2/H2O = 2/20/3 % vol, 1 atm, 1000 h−1 (C2, C3)/2000 h−1 (C4). | ||
In all three reactions, the perovskite SmMnO3 produces CO2 as the main product. As a minor product the respective olefin is formed. V2O5 mostly generates CO2 from ethane, while CO is formed as main product from propane and n-butane. Also, in C4-oxidation MAN and acetic acid can be detected as reaction products, especially at a low conversion level. MoO3 shows a similar product distribution, while the overall alkane conversion is very low (<5%). MoVTeNbOx, MoVOx, and VPP are by far the most selective catalysts. While VPP suffers from consecutive oxidation of the desired products in C3 and C2 oxidation (see contact time variation study in Fig. SI6†), it shows the maximal yield towards MAN from n-butane. The mixed metal oxides are more prone to break the C–C bond in this reaction leading to the formation of lower oxygenates like acrylic and acetic acid. On the other hand, higher oxygenate selectivity is achieved by the oxide catalysts from propane with MoVTeNbOx producing AA as main product. Based on a comparison between the consumption rate of the alkane and oxygen (see Fig. SI4†), the superior performance of MoVTeNbOx in ODHE and C3-oxidation is related to a comparably low oxygen consumption at high alkane conversion levels. In comparison, MoVOx is more active in C–C bond splitting and forms lower oxygenates from the feedstock. The oxidative dehydrogenation of ethane proceeds very selectively on both mixed metal oxides with M1 structure, while the formation of small amounts of acetic acid can only be attributed to MoVOx.
The studied catalysts clearly differ in the capability to activate the different alkanes, based on the change in performance (see Fig. 1). The apparent activation energies of both reactants (ethane, propane, or n-butane, and oxygen) are plotted to compare the potential of the respective catalyst to partially oxidize the feedstock (yield 1-COx), visualized in Fig. SI7.† In case of oxidative dehydrogenation of ethane, the highest ethylene selectivity is measured for medium apparent activation energies. In case of SmMnO3 apparent activation energies are very low, which corresponds to the high ethane consumption rate with CO2 as main product. The low capability of V2O5 to activate ethane is clearly visible, indicated by very high activation barriers. The samples producing ethylene in medium (VPP) or even high yields (MoVTeNbOx, MoVOx) show both an intermediate apparent activation energy of the alkane and the gas phase oxygen, which is assumed to mostly act to re-oxidize the oxygen vacancy of the bulk following the Mars–van Krevelen mechanism.26,71,72 Compared to the oxidative dehydrogenation, the selective oxidation of propane and n-butane forming oxygenates is more complex. Therefore, the trends between the alkane activation and the product yield are not following linear relationships (C3, C4), as multiple reaction pathways are proceeding in parallel. As a result, certainly different catalytic properties are required to either proceed the initial dehydrogenation step of the alkane forming olefins than the consecutive oxidation towards oxygenates like AA or MAN.40 In general, the measured apparent activation energies with the respect to the oxygen consumption rate for all catalysts depend on the alkane, which clearly shows that the chemical potential of the gas phase has an impact on the surface composition, defect concentration, and the electronic properties of the catalysts.
Surface modification by deposition of elements via ALD
Based on the functionality analysis of the bulk catalysts in the literature and in the present work, it is found that the combination of different elements within a phosphate or oxide structure is favourable for catalysis (e.g. the incorporation of Nb and Te into a Mo–V oxide structure73). Accordingly, the six base catalysts are modified by three different promoting elements, namely phosphorus, boron and manganese, introduced to the surface, ultimately as oxides (details can be found in the Experimental section). Within the entire periodic table, a large number of elements have been identified in the past that are active or beneficial in catalytic conversion of hydrocarbons.74 Many elements have already been deposited on oxides by ALD, but only a minority of the materials thus prepared have been studied in catalysis.50 Phosphates are discussed to play a crucial role in the oxidation of n-butane.47,52,75 While most of the redox steps are attributed to the V, phosphorus itself or the ratio between V and P is often discussed as one important factor for a selective oxidation of the alkane.5,75–78 Additionally, P has a positive effect as doping element in mixed metal oxide catalysts and P–Mo–V compounds were found to selectively oxidize isobutane.79,80 It was proposed that additional acidic properties correlate with increased performance. Similarly, boron is discussed to promote the partial oxidation of alkanes.81,82 Bulk boron containing catalysts are discussed to be highly selective in oxidative dehydrogenation reactions e.g. of propane.83 Surface-stabilized BOx species are proposed as active site,84,85 although it has been shown that the function of the boron oxide can at most be the initiation of radical chain reactions in the gas phase.81,86–88 While P mostly shows promoting effects in combination with a transition metal, the deposition of Mn introduces another redox active element on the surface of the catalysts. Mn-Tungstates are among the rare cases of V-free oxidation catalysts for the dehydrogenation of propane to propylene.89 In summary, it can be expected that Mn introduced to the system predominantly contributes to the activity, while P promotes selectivity. Added surface sites of BOx potentially accelerate the dehydrogenation of the alkanes leading to higher product yields.The impact of the surface modification on the surface properties and the catalytic performance is discussed exemplary for MoVTeNbOx, as it shows unique performance in the selective oxidation of C2, C3 and C4. The deposition of the element precursors, the subsequent treatment (ligand-removal under O2 or H2O feed diluted with N2) and the final calcination of the modified catalyst maintain the structural integrity of the bulk. Comparison of the XRPD patterns (Fig. SI11†) and N2-physisorption measurements (Fig. SI12†) before and after the modification confirms that no additional phases are formed and that the morphology, the specific surface area, and the pore volume remain constant (see Table SI2†). The amount of deposited POx, BOx and MnOx is quantified by the overall mass gain throughout the surface deposition (e.g., Δw = +1.8 wt% of POx on MoVTeNbOx, see Fig. SI13†). Additionally, ICP-OES is used to analyse the overall element loading. With regard to the total catalyst mass, it is found that only 0.9 wt% (B) or 0.8 wt% (P) are deposited (Table SI4†). The deviation between the quantification methods is related to the introduction of the additive as oxide species to the surface, adding weight in the balance. The modified surface composition and therefore the amount of the deposited ALD-element is determined by the properties of both, the base catalyst (e.g., OH-group density, surface area) and the utilized ALD-precursor (reactivity, ligand-molecule size) and therefore varies between the modified samples. A homogeneous dispersion is confirmed by STEM-EDX mapping of POx on MoVTeNbOx (Fig. 3). Moreover, the dispersion of POx on MoVTeNbOx can be maintained during the reaction in propane oxidation under the rather harsh conditions (cSteam = 20%), as the mappings of the spent catalyst indicate.
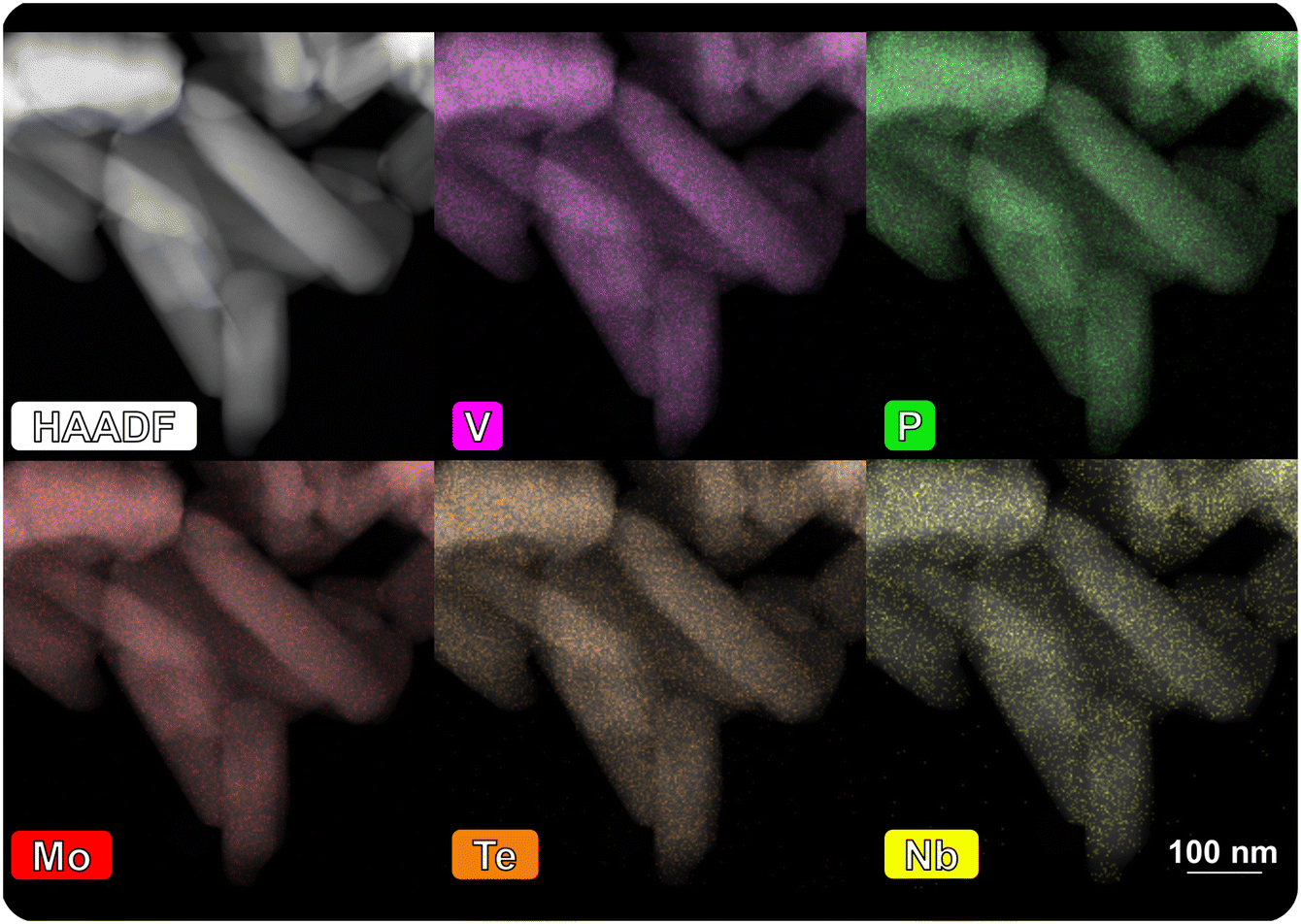 | ||
| Fig. 3 STEM-HAADF images of POx surface-modified MoVTeNbOx after catalysis (spent sample): EDX-mappings show element dispersion of both bulk elements (Mo, V, Te, Nb) and the additionally deposited phosphorus; the images of the fresh sample are shown in Fig. SI8,† the mapping-spectra of both samples are attached in Fig. SI9 and SI10.† | ||
In contrast to the bulk characteristics, the introduction of the elements shows a significant effect on the near-surface properties. In order to describe these changes in detail, the chemical state of the samples at the near-surface is investigated by XPS (details can be found in the Experimental section). Fig. 4a illustrates the variation in composition of near-surface elements by the deposition of BOx, POx and MnOx on MoVTeNbOx. Interestingly, the addition of a fifth element does not always lead to a similar change in the surface concentrations of the bulk elements. While the deposition of Mn lowers the amount of the other elements evenly, the addition of BOx shows specifically a depletion of Mo6+ on the surface and, at the same time, a slight increase in V availability. Furthermore, the combination of ICP-OES and XPS measurements is used to specify the location of the elements after calcination. Therefore, the B seems to migrate directly into the bulk of the catalyst, as a higher content of B can be detected in the bulk (ICP-OES) compared to the near-surface (XPS, see Fig. 4b). In contrast POx is enriched on the surface (Fig. 4c).
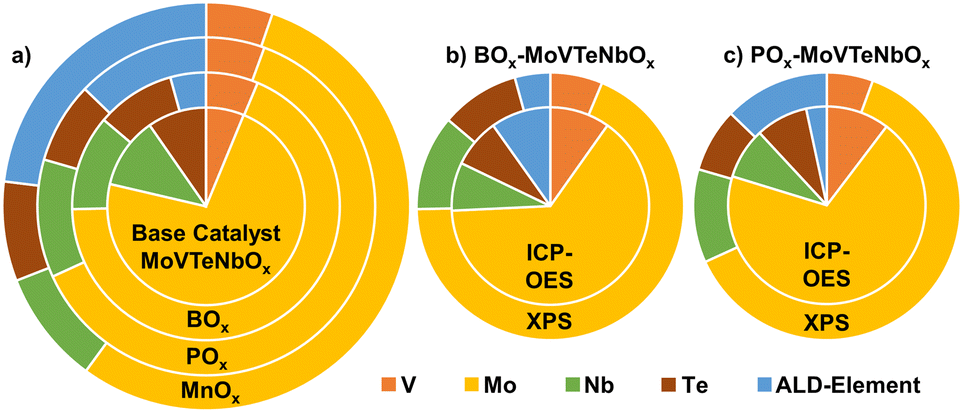 | ||
| Fig. 4 (a) Impact on near-surface composition (at%), while excluding the oxygen content, by surface modification (BOx, POx, MnOx) of the MoVTeNbOx catalyst before catalysis determined by XPS measurements; comparison between near-surface (XPS) and overall bulk (ICP-OES) composition of (b) BOx-modified and (c) POx-modified MoVTeNbOx. The raw data of the measurements is summed up in Tables SI3 and SI4† and the composition of the spent samples is visualized in Fig. SI14 and SI15.† | ||
Tuning surface properties of oxidation catalysts
The deposition of additional elements on the catalysts results in a modification of the near-surface composition, with respect to the added ALD-elements, but also with respect to the original bulk elements. To elaborate further on this effect, the chemical state of the (redox active) elements at the near-surface is studied by XPS. While most of the transition metals only show one specific oxidation state in the spectra (Mo6+, Nb5+, Te4+), which is maintained during the modification, a change in the average oxidation state of V is observed, presented after catalysis in Fig. 5. In all MoVTeNbOx-samples surface-species with two different oxidation states (V4+ and V5+) are detected (XPS spectra can be found in Fig. SI16†). To characterize a more oxidized or reduced overall surface state, the ratio RXPS between V4+ and the sum of V4+ and V5+ is calculated (see Experimental section for details). The unmodified MoVTeNbOx shows a slightly reduced surface state with more V4+ present at the surface. The deposition of the elements by ALD leads to a change in the oxidation state of the redox active element after catalysis. While the Mn modified sample shows an even more reduced surface state with respect to V, the POx- and BOx-modified surfaces show a higher average oxidation state of V. An enrichment of V4+ or V5+ at the surface is often correlated to catalytic performance based on both in situ and ex situ analytics in case of MoVTeNbOx.12,34,59 In a previous work, Knemeyer et al. show that the deposition of a phosphorus precursor has the potential to reduce surface V5+ to V4+ starting from a V2O5, which mostly contains V5+ in its bulk structure and at the surface.52 However, the calcination of the sample in the second ALD half-cycle (POx on V2O5), resulted in a re-oxidation of the V-species at the surface. In the present case of MoVTeNbOx, the decoration with POx and BOx induces an oxidized surface state after catalyst testing.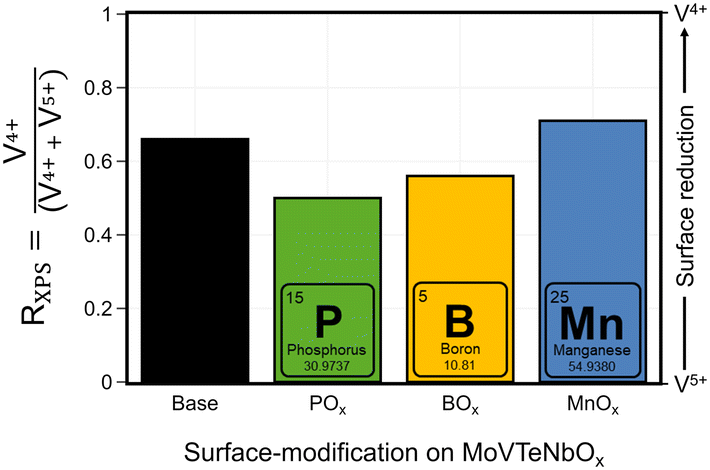 | ||
| Fig. 5 Effect of surface deposition of BOx, POx, MnOx on the oxidation state of V in the near-surface region of MoVTeNbOx after catalysis (spent); the ratio RXPS between reduced V4+ species and the sum of V4+ and V5+ calculated from the XPS spectra in the of V 2p3/2 region (see Fig. SI16 and SI17† for more details) is shown. | ||
In addition to the electronic properties of the redox-active element V, the selectivity of heterogeneous catalysts in oxidation reactions is determined by multiple phenomena, that need to collaborate to activate adsorbed reactants and convert them into the desired product. Many of these processes are strongly influenced by the interaction between the surface elements and the gas phase molecules. As a result, the surface-acidity of a catalyst often correlates with performance.90–93 Acidity can contribute to the C–H activation,94 while strong acid sites can lower the product selectivity by undesired parallel or consecutive reactions.92 As a result, the acidity of mixed metal oxides and especially of Mo and V based catalysts are studied in much detail in literature with the goal to correlate it with the performance in partial oxidation reactions.69,73,95–97
While both Brønsted and Lewis acid sites are discussed to play a role in catalysis, their presence is also crucial for the described deposition of atomic layers on the oxide surface. During ALD, the element precursors are interacting e.g. with surface hydroxyl-groups, which can exhibit Brønsted acidity, and thus chemisorb onto the surface.50,53 After the surface is saturated with the precursor, in a second step using O2 or H2O new hydroxyl groups both on the deposited elements and bulk-species are created. The determination of the surface acidity is complicated, as multiple surface properties of a heterogeneous catalyst are mutually depending on each other (e.g. oxidation state or redox activity and acidity93). In this work, NH3-pulse studies are used as a tool to describe the impact on the surface characteristics by the surface deposition of different elements. NH3-desorption experiments at temperatures above the calcination temperature of the catalysts can lead to an undesired formation of gaseous species other than ammonia, which impedes correct quantification of desorbed ammonia. POx introduced to the surface of MoVTeNbOx by ALD clearly decreases the overall amount of adsorbed ammonia, while for BOx a slight reduction can be observed, which indicates a lower availability of acidic sites on the surface of the modified catalysts, as shown in Fig. 6. The lower acidity is in line with the composition change on the surface of the catalyst, as the boron mostly migrates to the bulk of the catalysts (see Fig. 4). Although the overall amount of deposited BOx surpasses the deposition of POx (ICP-OES), on the surface of the modified catalysts the opposite effect can be observed, with more POx accumulated at the surface (see Tables SI3 and SI4†). This is an indicator why the POx-deposition has a more pronounced effect on the surface properties like the acidity. However, the addition of P and B is discussed to add acidic features as dopants,79 increased P-content in Mo-phosphate lead to higher acidity,98 and surface P–OH groups were shown to contribute to the Brønsted-acidity in VPP.99 On the contrary, in case of supported alumina100 or zeolite101 catalysts, the addition of phosphates was utilized to reduce acidity. In this study, the deposition of the surface groups has two effects. First, surface OH-groups are exchanged and saturated by the added ALD-groups. This introduces additional sites, where ammonia can adsorb, but also potentially blocks acid sites of the base catalysts. A similar decrease in adsorbed NH3 or acidity of the catalyst was observed for doping experiments of MoVTeNbOx with Bi, leading to improved performance in the synthesis of acrylonitrile.102 The sample modified by MnOx shows that the surface treatment can also result in an increased NH3 adsorption capacity. In this case, the introduction of new surface groups creates more adsorption centres for ammonia than blocking existing ones.
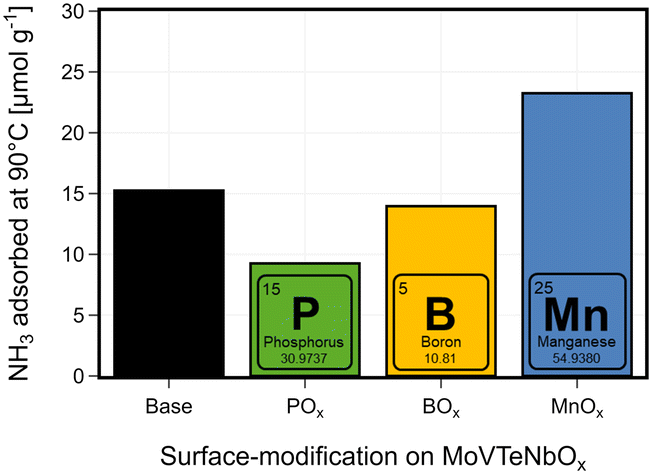 | ||
| Fig. 6 Quantification of adsorbed ammonia in the NH3-pulsing study: total amount of NH3 adsorbed on the base and surface-modified (BOx–, POx–, MnOx–) MoVTeNbOx catalyst before catalysis. The samples were dried at 150 °C under He flow for 1 h; 20 NH3-pulses were performed until saturation of the samples at 90 °C; the overall amount of NH3 adsorbed is calculated from the differences between the first 15 pulses and the mean signal of saturation (peaks 16–20); see Fig. SI18† for more detail. | ||
The influence of the surface modification on catalysis
All samples were tested in the oxidation of ethane, propane, and n-butane. The base mixed metal oxide MoVTeNbOx shows high product yields in all the three reactions, forming ethylene, AA, or MAN from the respective alkane. At 375 °C, alkane conversions of >50% can be achieved on the unmodified catalyst (see Fig. 7a; see Fig. SI19a† for exemplary error bars). Fig. 7b shows the effect on the selectivity towards the desired products at a fixed interpolated conversion level of Xalkane = 70%.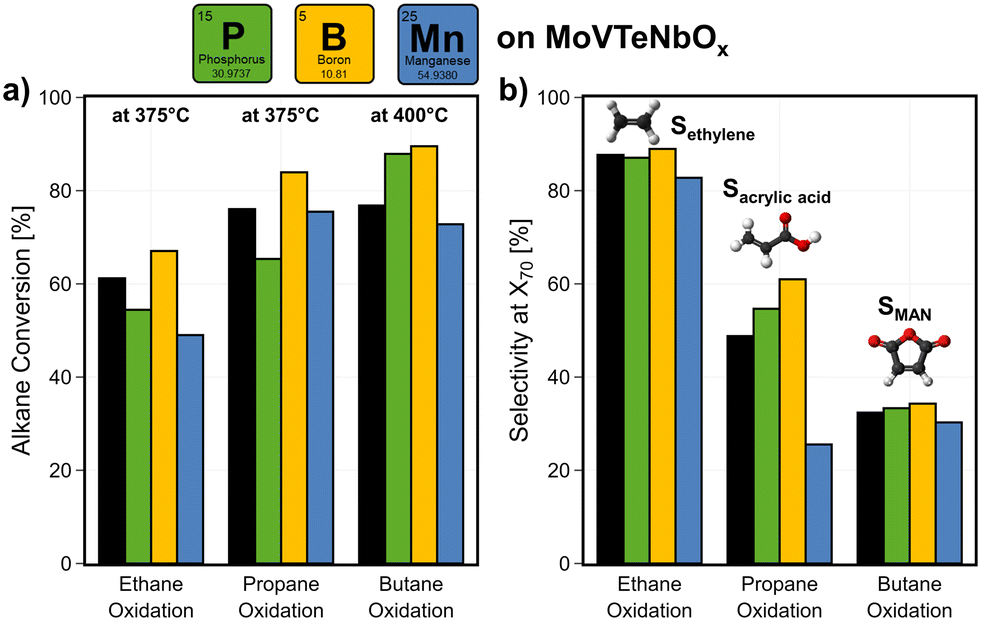 | ||
| Fig. 7 Impact of surface modification with BOx, POx, MnOx on the catalyst performance of MoVTeNbOx: (a) change in alkane conversion at a fixed temperature setpoint of 375 and 400 °C and (b) target-product selectivity (C2: ethylene, C3: AA, C4: MAN) at a fixed conversion level of Xalkane = 70% (interpolated) in ethane, propane, and n-butane oxidation. C2H6/O2/H2O = 3/9/0 % vol, C3H8/O2/H2O = 3/9/20 % vol, C4H10/O2/H2O = 2/20/3 % vol, 1 atm, 1000 h−1 (C2, C3), 2000 h−1 (C4). | ||
BOx increases both activity and selectivity over MoVTeNbOx in all three reactions. As MoVTeNbOx already shows very high ethylene selectivity of Sethylene > 80%, a surface modification with BOx shows a small improvement. Together with the increased activity, the yield is improved by 7%. Especially in propane oxidation a boost in conversion is achieved, as the conversion is increased from Xpropane = 76% to 84% at 375 °C (see Fig. SI19b† for change in alkane consumption rates). The selectivity to AA increases by 25% at a fixed conversion. Thus, the overall yield is improved by up to 24% in the selective oxidation of propane at a fixed temperature of 375 °C. Slight improvements of the selectivity towards MAN are achieved in n-butane oxidation. However, BOx disappears from the near-surface of the catalyst under reaction conditions (Fig. SI14†). This behaviour is similar to what is observed in the industrially used method for moderating temperature profiles within catalyst beds of VPP, in which a volatile phosphorus compound is dosed during operation.5,103 The deposition of BOx changes the surface composition of MoVTeNbOx, but partly migrates into its bulk (see Fig. 4b). With that a blocking of active sites cannot be observed in the performance data, while additionally the modified surface (e.g. change in electronic properties of V, surface-acidity) helps to desorb the formed products leading to higher selectivity. Furthermore, an increased TOF of all three alkanes seems to be induced by the deposition of BOx (Fig. SI19c†). Similarly, the incorporation of Nb and Te into the Mo- and V-based oxide structure significantly improves the catalytic performance and lowers the potential for C–C bond cleavage in all reactions (see Fig. 2). In literature, this effect is attributed to the changing surface compositions under reaction conditions.19,73 Therefore, an enrichment of Te leads to a reduced abundance of V5+, while on MoVOx a strong surface-enrichment with V is observed. Thus, the improved performance of BOx-modified MoVTeNbOx is attributed to the lowered availability of V on the surface.19
The introduction of POx lowers the catalytic activity of MoVTeNbOx in ethane and propane oxidation, but shows a beneficial effect in n-butane oxidation. A slight increase in product selectivity is observed in propane and n-butane oxidation. This effect could be attributed to the blocking of active, but also unselective sites on the surface. Calculated turn over frequencies (TOFs) of the respective alkane (see Fig. SI19b†) show that POx deposition has either no or even a positive effect on the alkane consumption rate normalized to V available on the surface.
In contrast to this, the presence of MnOx leads either to a small decrease (C2, C4) or a drop in the selectivity to AA in propane oxidation. In this case, the catalyst is not only less active, but also unselective sites are introduced into the system. This observation is in agreement with the catalytic performance of the perovskite SmMnO3 comprising Mn as redox active element, mostly forming CO2 from the feedstock. Thus, the addition of MnOx to the surface of MoVTeNbOx appears to promote the consecutive combustion of the formed products to COx.
In summary, the surface modification of MoVTeNbOx has both positive and negative effects on the catalytic performance. The analysis of the spent samples shows that the surface deposition of elements does not prevent the surface from adapting to the reaction feed and therefore from restructuring. A comparison before and after catalysis by ICP-OES and XPS indicates a drastic loss in both P and B during the whole catalytic study with >160 h time on stream (see Fig. SI14 and SI15†). This behaviour is known for phosphorus in VPO catalysts, where a loss in surface phosphorus leads to an activation of the catalyst under reaction condition and a change in the V:P ratio.5,76,103 Nonetheless, a formation of the samples or a change in performance during the study could not be observed, after the samples were exposed to the maximum reaction temperature. The demonstrated tuned performance by the surface modification is therefore maintained and does not follow the loss of the ALD-elements. Additionally, the impact on the surface properties like the oxidation state of V by the surface-modification is still present after the reaction (Fig. 5). While the surface of the base catalyst MoVTeNbOx is noticeably reduced after catalysis, the XPS spectra of the spent POx- and BOx-modified samples reveal an enrichment of V5+ in the near-surface region. Recently, a doping study on MoVTeNbOx with Bi showed a similar effect of increased AA selectivity, which was correlated to a more oxidized surface state and also an increased Lewis acidity.104 In contrast, the MnOx-modified sample shows an even more reduced oxidation state of V compared to the base catalyst, which is maintained after reaction. Thus, the low AA selectivity measured on this catalyst is consistent with the discussion above. Therefore, a certain flexibility of V at the near-surface appears to be another factor that determines selective oxidation.105
Apart from the selected example MoVTeNbOx, the effect of the surface modification on the catalytic performance is investigated systematically for MoVOx, VPP, V2O5, MoO3, and SmMnO3 (Fig. SI20–SI24†). In previous studies V2O5 was used as a model catalyst to study the role of P in VPO-catalysts.52 A promoting effect was found in n-butane oxidation, which is confirmed for the oxidation of C2 and C3 in the present study. In all reactions, the formation of COx is reduced, leading to higher product selectivity. In particular, the POx-deposition in sub-monolayers (1 ALD-cycle) also leads to an improved catalytic activity. The BOx-modification, however, does not lead to significant improvements on V2O5 (see Fig. SI22†). On the other hand, the deposition of POx or BOx, has a beneficial effect on activity and selectivity in some cases, especially for catalysts that already show certain performance in the reactions under investigation (VPP, MoVOx). At the same time, the surface deposition of MnOx in most cases leads to a reduced performance in partial oxidations. An exception to this trend is MoO3, which shows very low capability to activate the three alkane feedstocks. Here, the surface modification with BOx or MnOx leads to slightly elevated alkane conversions and improved selectivity. Especially, the deposited sub-monolayer of MnOx on MoO3 promotes the oxidative dehydrogenation of propane and a selectivity above 50% is observed (see Fig. SI23†). This result reveals parallels with a study on MnWO4,106 in which an isolation of MnOx-species on the surface resulted in improved performance.89 However, the Mn-based perovskite catalyst acts like a total combustion catalyst forming mostly CO2. Here, the selectivity cannot be tuned by the surface-modification, while a surface-enrichment with Mn clearly leads to a higher catalytic activity.
In addition to temperature, the contact time was varied in catalytic testing. The temperature during this study is fixed to achieve a medium conversion of the base catalysts, while the modified samples are tested under the same conditions. Based on this data, an overview of the different effects of the surface modification is shown in Fig. SI25.† The COx formation is often promoted by the deposition of MnOxe.g., on catalysts, that already show high selectivity (e.g. VPP in C4-oxidation, see Fig. SI26†). While a surface-enrichment of POx on the studied phosphate catalyst (VPP), shows no significant change, the addition of POx on V2O5, MoVOx, and MoVTeNbOx leads to an improvement in product selectivity (Fig. 8). Apparently, the interaction between the V-containing bulk and well dispersed POx species on the surface of the catalyst reduces the total oxidation of the reactants. It seems that unselective V-sites at the surface are blocked, which in case of MoVTeNbOx leads to an increased concentration of oxidized V-species in the near-surface region compared to the unmodified catalyst (Fig. 5). Furthermore, the POx modification reduces the surface acidity, which could be the reason for the lower activity and the increased selectivity. On the contrary, the deposition of POx on the surface of catalysts with another redox active bulk element than V shows no beneficial effect. In comparison, the deposition of BOx promotes the selectivity in partial oxidations only on specific base catalysts like MoVTeNbOx, while it shows only minor effects on other materials. The mobility of the deposited species and consecutively the migration to the bulk of catalysts, as shown for BOx–MoVTeNbOx, could be the reason for only slight changes in performance on catalysts like V2O5 or MoVOx (see Fig. SI27†). In contrast to this, in case of MoVTeNbOx the most notable improvements are achieved by the BOx-modification. The fact that this catalyst is already very selective in the studied reactions might play a role here, as the element deposition could also lead to blocking of active sites. Therefore, with the addition of BOx on the surface most of the features of the base catalyst are sustained, while surface properties can be tuned. In summary, the surface modification by ALD enables an optimized potential of the catalysts to partially oxidize the alkane feedstocks, as shown Fig. 8.
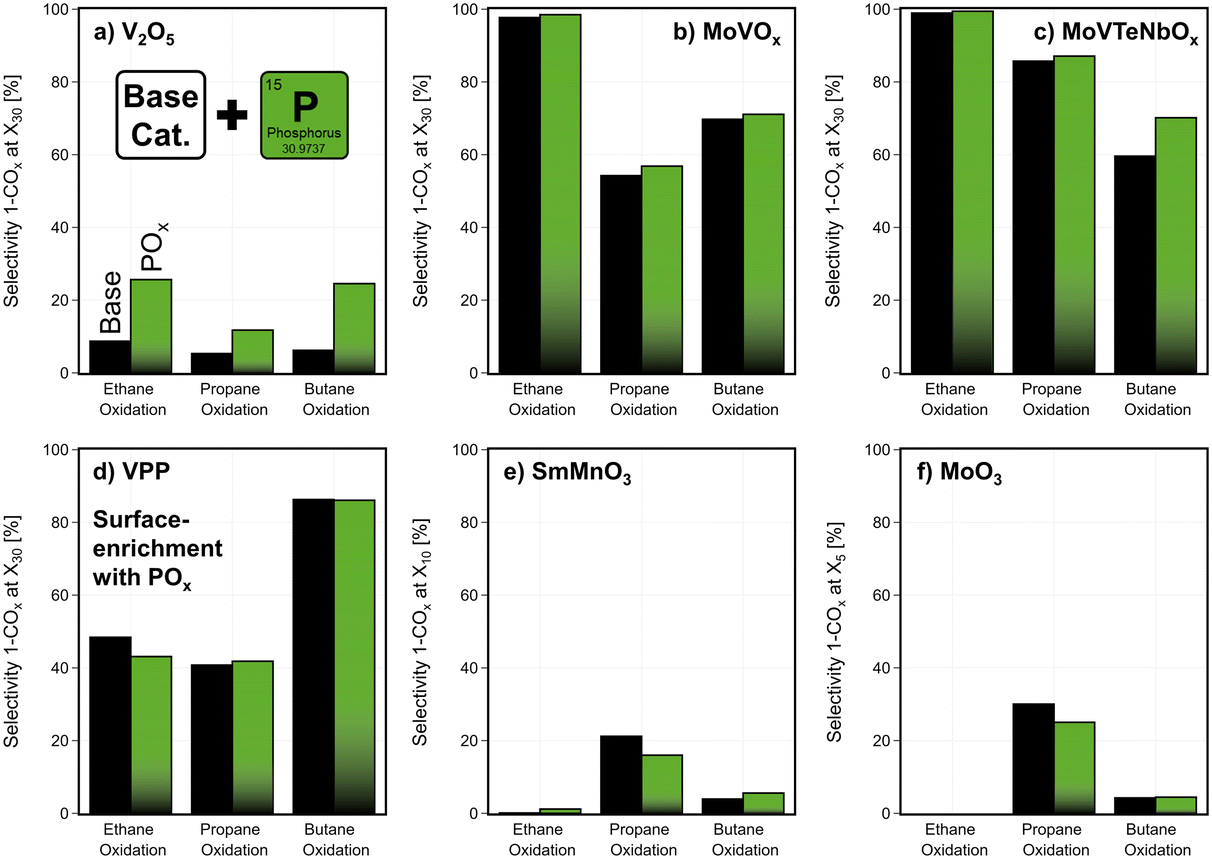 | ||
| Fig. 8 Change in product formation by the deposition of POx on the surface of the six studied catalysts (a) V2O5, (b) MoVOx, (c) MoVTeNbOx, (d) VPP, (e) SmMnO3, (f) MoO3: selectivity towards the sum of partial oxidation products (1-COx) shown in ethane, propane, n-butane oxidation at fixed temperature setpoint for each reaction and catalyst (see contact time variation study Fig. SI5 and SI6†); the product selectivity is shown at a fixed alkane conversion level by varying the contact time in a range of 500–5000 h−1; the complete contact time variation study can be found in the data set provided in the ESI.† C2H6/O2/H2O = 3/9/0 % vol, C3H8/O2/H2O = 3/9/20 % vol, C4H10/O2/H2O = 2/20/3 % vol, 1 atm. | ||
According to these results the surface modification has both beneficial and unfavourable effects on selectivity, depending on the catalyst and the reaction. However, the overall product distribution (see Fig. 2) can only be adjusted without completely changing in selectivity. One reason for this is the deposition of only one ALD-cycle on the catalysts, which correspond to a sub-monolayer.53 In previous studies it could be shown that the deposition of multiple cycles can intensify the observed effects.52 Beyond a certain amount of precursor deposited on the surface of the catalyst, however, the formation of new (crystalline-) phases is possible. With the deposition of one ALD-cycle resulting in a homogeneous dispersion, the effect of oxide species (POx, BOx, MnOx) as promotors is studied rather than forming new active phases on the surface. On the other hand, the catalytic performance of the samples modified with a sub-monolayer is still dominated by the properties of the base catalyst. Furthermore, the catalyst surfaces still undergo a restructuring process, when exposed to the reaction feeds, as shown by the comparison of fresh and spent samples (see Fig. SI15 and SI17†). Therefore, the formation of the actual active surface layer is supported by stable bulk characteristics. This is in line with the fact that the bulk has an important function in catalysis,107,108 while most of the elemental steps of the actual reaction are proceeding at the termination layer. As a result, in particular the interplay between the bulk and the dynamic surface of a bulk catalyst seems to be the key for high performance in selective oxidation reactions.41,42,109
Conclusion
An efficient selective oxidation of different alkanes requires catalysts with the ability to activate the feedstock while the total oxidation of the products or intermediates by parallel or consecutive reactions is suppressed. The best performance with high yields of the desired products (MAN, AA, ethylene) is achieved by catalysts from two different families, namely the mixed metal oxide MoVTeNbOx, and the vanadium phosphate VPP. A common feature of both materials is the formation of a surface layer under reaction conditions, that significantly differs from its bulk. The surface modification with the promoting elements POx, BOx, and MnOx is used to tune the catalytic properties of oxidation catalysts. A homogeneous element dispersion on the surface is ensured by the utilization of ALD, while maintaining the mechanical properties of the base catalysts. The deposition of POx on V-based oxides is demonstrated to enhance the oxygenate or olefin formation in alkane oxidation. Similarly, BOx could be identified as another promotor of the studied reactions, resulting in increased product selectivity and catalytic activity. The systematic study of six base catalyst with three deposition-elements revealed a broad range of effects, of which certain modified surface properties can be attributed to improved performance. In case of the mixed metal oxide MoVTeNbOx, the deposition of POx and BOx yields in an increase in productivity of up to 24%, for instance in the oxidation of propane to AA. In this case, XPS studies reveal that the beneficial effect in catalysis is connected to an oxidized surface state with the accumulation of V5+ in the near-surface region. Furthermore, a reduced acidity is shown to be another common characteristic of the surface modification of MoVTeNbOx with POx and BOx. On the other hand, the deposition of another redox element like Mn induces a reduction of the V-species on the surface and furthermore to additional acidic sites. These changes in surface properties come along with an increased formation of COx and a reduction in catalytic activity. However, the introduction of Mn to a mostly inert oxide like MoO3 or an enrichment of Mn on the SmMnO3 perovskite shows an increase in performance. The elements are deposited as partial monolayers by ALD on the oxidation catalysts without changing their basic features. Thus, the change in surface-properties like the acidity or the surface state of the termination groups shows a good agreement with the observed change in performance. This study, including 18 combinations of base catalysts and promoting elements studied in three reactions, suggests that the catalytic performance of a bulk catalyst is tuneable by varying its surface properties with synthetic means. Here, it is crucial to preserve the important bulk properties of the catalysts by using solvent free techniques like ALD. This method proves to be a powerful tool for precise addition of promotor compounds to bulk catalysts. Based on this study, the product formation of V-containing oxide catalysts can be tuned by the introduction of finely dispersed POx. For future applications, the deposition of more than one sub-monolayer of potentially beneficial elements in multiple ALD-cycles could further enhance product formation. Additionally, a variety of different elements can be deposited by ALD, potentially leading to further improved catalytic performance.Experimental section
Base catalysts
Six bulk oxidation catalysts are selected from a collection of oxides or phosphates with a well-known catalytic activity in selective oxidations.38 A preferably broad range of catalytic behaviours is covered with highly active and less active catalysts, full combustion catalysts, and very selective catalysts. First, binary or monometallic oxides comprising the most commonly used elements in selective oxidation reactions are included, namely V2O5 and MoO3. The combination of both elements as multi-metal oxides with M1 structure type, represented by MoVOx and MoVTeNbOx, enables improved activity in the respective reactions. While the state-of-the-art performance in C2 and C3 oxidation can be achieved by these metal oxide catalysts, for an efficient n-butane oxidation to MAN especially VPO catalysts, like VPP, are used. Finally, the perovskite catalyst SmMnO3 is selected, which could be shown to activate C–H bonds very efficiently but shows rather low selectivity in partial oxidation reactions. The bulk catalysts were prepared in sufficient batch size (20 g) and as phase pure samples, according to synthesis-protocols, that are described in detail elsewhere.40 Linked references are listed in the ESI,† from which the synthesis protocols are adapted. The samples are pressed (1 t, 1 min, dpellet = 14 mm), crushed, and sieved to a particle size fraction of 100–200 μm, for both the performance studies and the surface modification.Atomic layer deposition
Sub-monolayers of phosphorus-, boron-, and manganese-oxide are deposited on the surface of the base catalysts using atomic layer deposition (ALD).44–46,50 During the first ALD half cycle, the gaseous precursors of the respective element P (tris-(dimethylamino)-phosphate, DMAP, Sigma-Aldrich, 97%), B (trimethylborate, TMB, Sigma-Aldrich, ≥98%), Mn (bis(ethyl-cyclopentadienyl)manganese, MnCpEt, Strem, ≥98%) diluted with argon (99.999%) is introduced in a quartz tube fixed bed reactor containing 5 ml of the catalyst until saturation of the surface sites (100 ml min−1, 1 atm) is achieved. The reactor is heated to 70 °C for the deposition of the phosphorus precursor and 150 °C in the case of boron and manganese. The precursor dosing units were kept at 25 °C (DMA, TMB) or 100 °C (MnCpEt). The point of precursor or reactant saturation was determined by online mass spectrometry. The mass gain or ALD growth during the whole process is monitored by an in situ magnetic suspension balance44 (see example in Fig. SI13†). In the second half cycle, water or oxygen is used as a reactant to remove the ligands, leaving the surface with POx and BOx, or MnOx-groups, respectively. In between, before and after the half-cycles the sample is purged with argon. Details about the experimental setup and the ALD process are described elsewhere.47,53 In this study one ALD-cycle is applied to the catalysts, which yields in sub-monolayers of the elements on the catalysts. After a fixed pre-treatment in dry synthetic air (20% O2 in N2) at 275 °C (mixed metal oxides) or 450 °C for 4 h, the modified samples are cooled down and sieved for catalyst testing.Catalyst testing
Base catalysts and modified samples are studied in the selective oxidation of short-chain alkanes (n-butane, propane, ethane). The feed composition is fixed based on industrial or academic standards (C4-oxidation,24 C3-oxidation11) balanced by nitrogen (C4H10/O2/H2O: 2/20/3; C3H8/O2/H2O: 3/9/20; C2H6/O2/H2O: 3/9/0 % vol) at a gas hourly space velocity (GHSV) of 2000 h−1 (C4) or 1000 h−1 (C2, C3) and atmospheric pressure. The performance studies are performed in commercial parallel test setups (hte GmbH, ILS GmbH) with sample volumes of V = 0.7/1.0 ml in stainless steel reactors with a diameter of d = 7/10 mm. The reaction temperature is individually controlled in the range of 225 to 450 °C, while an alkane or oxygen conversion of Xalkane = 85% must not be exceeded. Outlet concentrations are monitored using an online gas chromatography system. Argon is used as an internal standard to eliminate possible gas expansion in the calculation of conversion and selectivity and a blank reactor is used for online measurements of the input gas feed. More details on the technical setup and the evidence of independence of mass and heat transfer limitations are described elsewhere.9,110The design of experiments (DoE) is fixed for all tested samples and includes a temperature and GHSV variation study. The reaction temperature is gradually increased in 25 K steps until 450 °C max or until a reactant conversion Xalkane < 85% is achieved. After the temperature is decreased again, it is fixed to a setpoint of about Xalkane = 15%. The GHSV is varied at four different setpoints (500 to 5000 h−1). Raw-data evaluation was performed using the commercial software tool myhte™. The calculation of the presented performance parameters (X, S) is done product-based (see eqn (4)–(8)); the whole set of raw data, can be found in the ESI.†
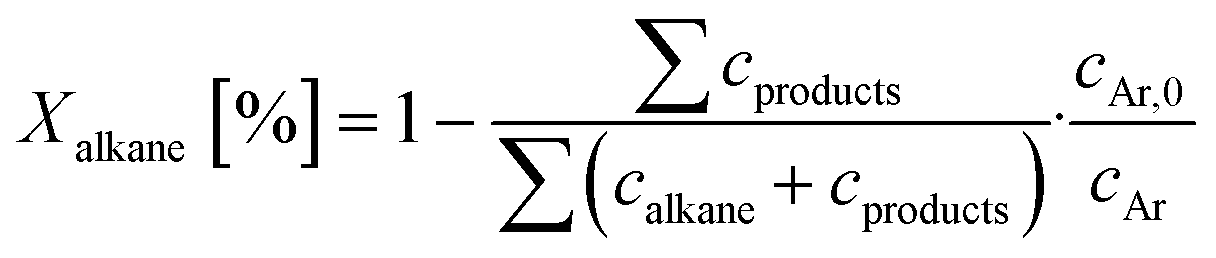 | (4) |
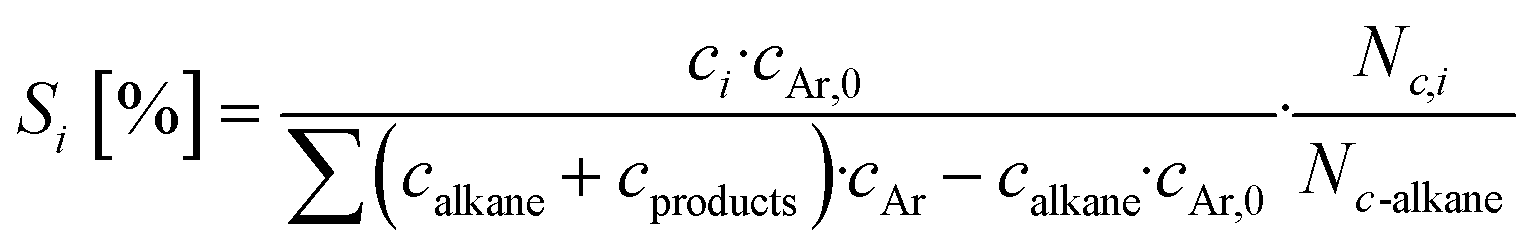 | (5) |
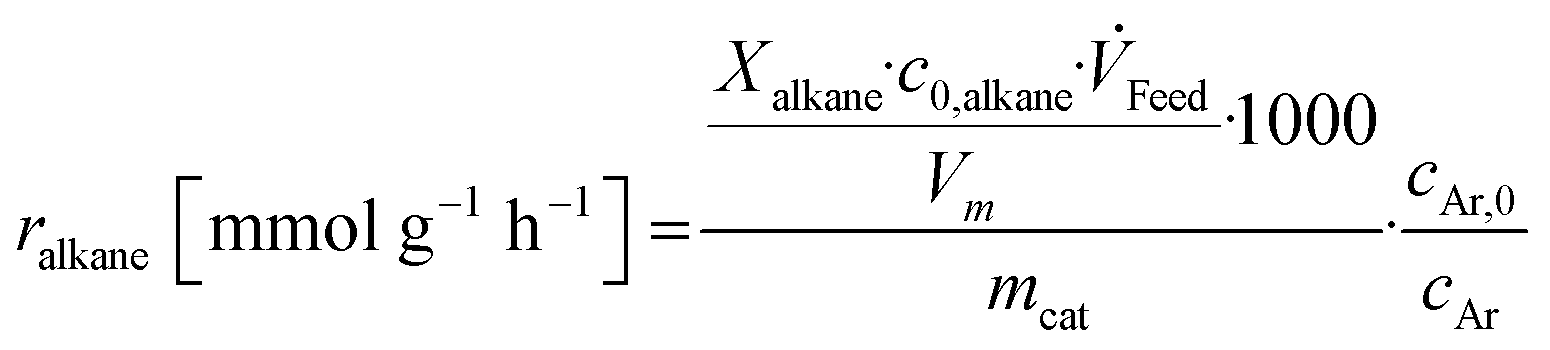 | (6) |
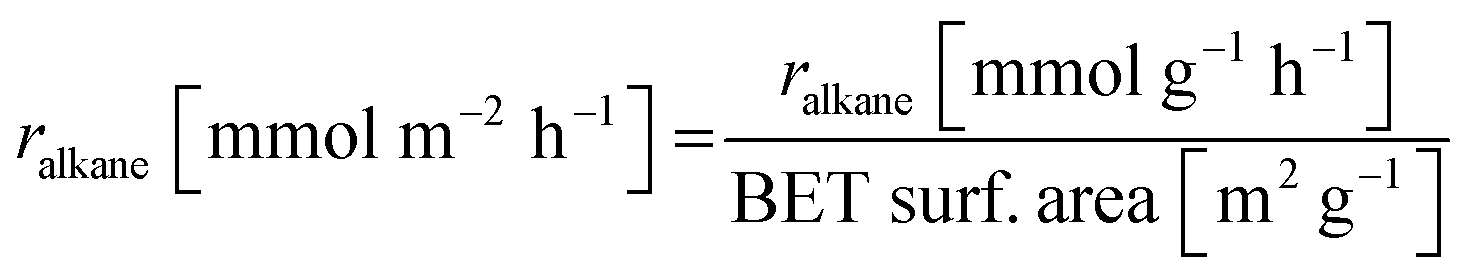 | (7) |
| ri [mmol g−1 h−1] = ralkane [mmol g−1 h−1]·Si | (8) |
Characterization
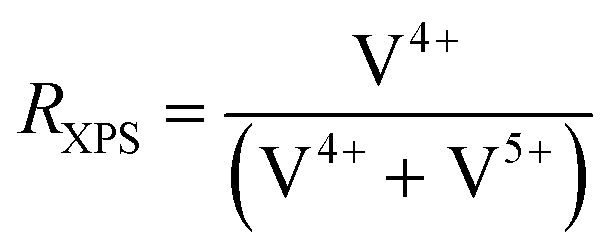 | (9) |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was conducted in the framework of the BasCat – UniCat BASF JointLab between BASF SE, Technische Universität Berlin (TU Berlin), and Fritz-Haber-Institut (FHI) der Max-Planck-Gesellschaft. Funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat is acknowledged. We would like to thank Dr. Michael Geske (TU Berlin) for scientific discussions, Jan Meißner (TU Berlin), Stephen Lohr (BASF SE), Sven Richter (FHI), Dr. Gregor Koch (FHI) for technical assistance in the laboratory and the synthesis work, Christina Eichenauer (TU Berlin) for BET measurements, Dr. Johannes Schmidt (TU Berlin) for the support in the XPS measurements, Kevin Profita (TU Berlin), Sophie Hund (TU Berlin) and Dr. Frank Girgsdies (FHI) for the XRPD measurements and Dr. Majd Al-Naji (TU Berlin) for the support in the NH3 adsorption experiments.References
- Research needs towards sustainable production of fuels and chemicals, ed. J. K. Nørskov, A. Latimer and C. F. Dickens, 2019, Accessed Nov. 2022 Search PubMed.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2003, 77, 287–297 CrossRef CAS.
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS PubMed.
- G. Mestl, D. Lesser and T. Turek, Top. Catal., 2016, 59, 1533–1544 CrossRef CAS.
- N. Ballarini, F. Cavani, C. Cortelli, S. Ligi, F. Pierelli, F. Trifirò, C. Fumagalli, G. Mazzoni and T. Monti, Top. Catal., 2006, 38, 147–156 CrossRef CAS.
- F. Trifirò and R. K. Grasselli, Top. Catal., 2014, 57, 1188–1195 CrossRef.
- N. F. Dummer, J. K. Bartley and G. J. Hutchings, Adv. Catal., 2011, 54, 189–247 CAS.
- C. Schulz, F. Pohl, M. Driess, R. Glaum, F. Rosowski and B. Frank, Ind. Eng. Chem. Res., 2019, 58, 2492–2502 CrossRef CAS.
- G. Mestl, J. L. Margitfalvi, L. Végvári, G. P. Szijjártó and A. Tompos, Appl. Catal., A, 2014, 474, 3–9 CrossRef CAS.
- R. Naumann d'Alnoncourt, L. I. Csepei, M. Hävecker, F. Girgsdies, M. E. Schuster, R. Schlögl and A. Trunschke, J. Catal., 2014, 311, 369–385 CrossRef.
- M. Hävecker, S. Wrabetz, J. Kröhnert, L. I. Csepei, R. Naumann d'Alnoncourt, Y. V. Kolen'Ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48–60 CrossRef.
- R. Schlögl, Top. Catal., 2011, 54, 627–638 CrossRef.
- D. Melzer, G. Mestl, K. Wanninger, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Top. Catal., 2020, 63, 1754–1764 CrossRef CAS.
- D. Melzer, G. Mestl, K. Wanninger, Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 4012 CrossRef PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
- J. C. Volta, Catal. Today, 1996, 32, 29–36 CrossRef CAS.
- A. C. Sanfiz, T. W. Hansen, D. Teschner, P. Schnörch, F. Girgsdies, A. Trunschke, R. Schlögl, M. H. Looi and S. B. Abd Hamid, J. Phys. Chem. C, 2010, 114, 1912–1921 CrossRef CAS.
- A. Trunschke, J. Noack, S. Trojanov, F. Girgsdies, T. Lunkenbein, V. Pfeifer, M. Hävecker, P. Kube, C. Sprung, F. Rosowski and R. Schlögl, ACS Catal., 2017, 7, 3061–3071 CrossRef CAS.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- T. Zambelli, J. Wintterlin, J. Trost and G. Ertl, Science, 1996, 273, 1688–1690 CrossRef CAS.
- G. A. Somorjai, Langmuir, 1991, 7, 3176–3182 CrossRef CAS.
- G. Ertl, Angew. Chem., 1990, 102, 1258–1266 CrossRef CAS.
- C. Schulz, R. Kraehnert, F. Rosowski and B. Frank, ChemCatChem, 2018, 10, 5523–5532 CrossRef CAS.
- E. W. Arnold and S. Sundaresan, Chem. Eng. Commun., 2007, 58, 213–230 CrossRef.
- C. Heine, M. Hävecker, E. Stotz, F. Rosowski, A. Knop-Gericke, A. Trunschke, M. Eichelbaum and R. Schlögl, J. Phys. Chem. C, 2014, 118, 20405–20412 CrossRef CAS.
- M. Eichelbaum, M. Hävecker, C. Heine, A. Karpov, C.-K. Dobner, F. Rosowski, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2012, 51, 6246–6250 CrossRef CAS PubMed.
- M. A. Carreon and V. V. Guliants, Catal. Today, 2003, 78, 303–310 CrossRef CAS.
- M. Abon, K. E. Bere, A. Tuel and P. Delichere, J. Catal., 1995, 156, 28–36 CrossRef CAS.
- G. Busca and G. Centi, J. Am. Chem. Soc., 1989, 111, 46–54 CrossRef CAS.
- F. Ben Abdelouahab, R. Olier, N. Guilhaume, F. Lefebvre and J. C. Volta, J. Catal., 1992, 134, 151–167 CrossRef.
- E. A. Mamedov, Appl. Catal., A, 1994, 116, 49–70 CrossRef CAS.
- V. V. Guliants, R. Bhandari, B. Swaminathan, V. K. Vasudevan, H. H. Brongersma, A. Knoester, A. M. Gaffney and S. Han, J. Phys. Chem. B, 2005, 109(50), 24046–24055 CrossRef CAS PubMed.
- V. V. Guliants, H. H. Brongersma, A. Knoester, A. M. Gaffney and S. Han, Top. Catal., 2006, 38, 41–50 CrossRef CAS.
- T. V. Andrushkevich and E. V. Ovchinnikova, Mol. Catal., 2020, 484, 110734 CrossRef CAS.
- C. Heine, M. Hävecker, A. Trunschke, R. Schlögl and M. Eichelbaum, Phys. Chem. Chem. Phys., 2015, 17, 8983–8993 RSC.
- A. Trunschke, Catal. Sci. Technol., 2022, 12, 3650–3669 RSC.
- A. Trunschke, G. Bellini, M. Boniface, S. J. Carey, J. Dong, E. Erdem, L. Foppa, W. Frandsen, M. Geske, L. M. Ghiringhelli, F. Girgsdies, R. Hanna, M. Hashagen, M. Hävecker, G. Huff, A. Knop-Gericke, G. Koch, P. Kraus, J. Kröhnert, P. Kube, S. Lohr, T. Lunkenbein, L. Masliuk, R. Naumann d'Alnoncourt, T. Omojola, C. Pratsch, S. Richter, C. Rohner, F. Rosowski, F. Rüther, M. Scheffler, R. Schlögl, A. Tarasov, D. Teschner, O. Timpe, P. Trunschke, Y. Wang and S. Wrabetz, Top. Catal., 2020, 63, 1683–1699 CrossRef CAS.
- L. Foppa, L. M. Ghiringhelli, F. Girgsdies, M. Hashagen, P. Kube, M. Hävecker, S. J. Carey, A. Tarasov, P. Kraus, F. Rosowski, R. Schlögl, A. Trunschke and M. Scheffler, MRS Bull., 2021, 46, 1016–1026 CrossRef CAS PubMed.
- L. Foppa, F. Rüther, M. Geske, G. Koch, F. Girgsdies, P. Kube, S. Carey, M. Hävecker, O. Timpe, A. Tarasov, M. Scheffler, F. Rosowski, R. Schlögl and A. Trunschke, J. Am. Chem. Soc., 2023, 145(6), 3427–3442 CrossRef CAS PubMed.
- S. V. Merzlikin, N. N. Tolkachev, L. E. Briand, T. Strunskus, C. Wöll, I. E. Wachs, W. Grünert, S. V. Merzlikin, W. Grünert, N. N. Tolkachev, N. D. Zelinsky, L. E. Briand, T. Strunskus and C. Wöll, Angew. Chem., Int. Ed., 2010, 49, 8037–8041 CrossRef CAS PubMed.
- R. Schlögl, Top. Catal., 2016, 59, 1461–1476 CrossRef.
- M. Eichelbaum, M. Hävecker, C. Heine, A. M. Wernbacher, F. Rosowski, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 2922–2926 CrossRef CAS PubMed.
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS.
- B. J. Oneill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef CAS.
- H. Zhang and C. L. Marshall, Chin. J. Catal., 2019, 40, 1311–1323 CrossRef CAS.
- V. E. Strempel, R. Naumann D'Alnoncourt, M. Driess and F. Rosowski, Rev. Sci. Instrum., 2017, 88, 74102 CrossRef CAS PubMed.
- K. Knemeyer, R. Baumgarten, P. Ingale, R. Naumann D'Alnoncourt, M. Driess and F. Rosowski, Rev. Sci. Instrum., 2021, 92, 25115 CrossRef CAS PubMed.
- R. Baumgarten, P. Ingale, K. Knemeyer, R. Naumann d'Alnoncourt, M. Driess and F. Rosowski, Nanomaterials, 2022, 12, 1458 CrossRef CAS PubMed.
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, J. Appl. Phys., 2013, 113, 021301 CrossRef.
- V. E. Strempel, D. Löffler, J. Kröhnert, K. Skorupska, B. Johnson, R. Naumann d'Alnoncourt, M. Driess and F. Rosowski, J. Vac. Sci. Technol., A, 2015, 34, 01A135 CrossRef.
- K. Knemeyer, J. D. Epping, F. Rüther, C. Schulz, B. Frank, P. Müller, R. Naumann d'Alnoncourt, M. Driess and F. Rosowski, ChemCatChem, 2021, 13, 4201 CrossRef CAS.
- K. Knemeyer, M. P. Hermida, P. Ingale, J. Schmidt, J. Kröhnert, R. Naumann d'Alnoncourt, M. Driess and F. Rosowski, Phys. Chem. Chem. Phys., 2020, 22, 17999–18006 RSC.
- P. Ingale, K. Knemeyer, P. Preikschas, M. Ye, M. Geske, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Catal. Sci. Technol., 2021, 11, 484–493 RSC.
- P. Ingale, K. Knemeyer, M. P. Hermida, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Nanomaterials, 2020, 10, 981 CrossRef CAS PubMed.
- R. Baumgarten, R. Naumann d'Alnoncourt, S. Lohr, E. Gioria, E. Frei, E. Fako, S. De, C. Boscagli, M. Drieß, S. Schunk and F. Rosowski, Chem. Ing. Tech., 2022, 94, 1765–1775 CrossRef CAS.
- S. T. Oyama and G. A. Somorjai, J. Phys. Chem., 1990, 94, 5022–5028 CrossRef CAS.
- T. Ressler, J. Wienold, R. E. Jentoft and F. Girgsdies, Eur. J. Inorg. Chem., 2003, 2003, 301–312 CrossRef.
- C.-C. Liao, C.-C. Chang, Y. M. Choi and M.-K. Tsai, Surf. Sci., 2018, 674, 45–50 CrossRef CAS.
- T. Katou, D. Vitry and W. Ueda, Catal. Today, 2004, 91–92, 237–240 CrossRef CAS.
- W. Ueda, D. Vitry and T. Katou, Catal. Today, 2005, 99, 43–49 CrossRef CAS.
- A. M. Wernbacher, P. Kube, M. Hävecker, R. Schlögl and A. Trunschke, J. Phys. Chem. C, 2019, 123, 13269–13282 CrossRef CAS.
- J. M. López Nieto, B. Solsona, P. Concepción, F. Ivars, A. Dejoz and M. I. Vázquez, Catal. Today, 2010, 157, 291–296 CrossRef.
- J. M. López Nieto, P. Botella, M. I. Vázquez and A. Dejoz, Chem. Commun., 2002, 1906–1907 RSC.
- C. Heine, M. Hävecker, M. Sanchez-Sanchez, A. Trunschke, R. Schlögl and M. Eichelbaum, J. Phys. Chem. C, 2013, 117, 26988–26997 CrossRef CAS.
- G. J. Hutchings, Catal. Today, 1993, 16, 139–146 CrossRef CAS.
- P. Ciambelli, S. Cimino, S. De Rossi, M. Faticanti, L. Lisi, G. Minelli, I. Pettiti, P. Porta, G. Russo and M. Turco, Appl. Catal., B, 2000, 24, 243–253 CrossRef CAS.
- G. Koch, M. Hävecker, D. Teschner, S. J. Carey, Y. Wang, P. Kube, W. Hetaba, T. Lunkenbein, G. Auffermann, O. Timpe, F. Rosowski, R. Schlögl and A. Trunschke, ACS Catal., 2020, 10, 7007–7020 CrossRef CAS.
- F. Ivars, P. Botella, A. Dejoz, J. M. López Nieto, P. Concepción and M. I. Vázquez, Top. Catal., 2006, 38, 59–67 CrossRef CAS.
- J. T. Herron and R. E. Huie, J. Phys. Chem., 1969, 73, 3327–3337 CrossRef CAS.
- J. Haber, Handbook of Heterogeneous Catalysis, 2008 Search PubMed.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- A. de Arriba, B. Solsona, E. García-González, P. Concepción and J. M. López Nieto, Appl. Catal., A, 2022, 643, 118780 CrossRef CAS.
- M. Jaber Darabi Mahboub, J. L. Dubois, F. Cavani, M. Rostamizadeh and G. S. Patience, Chem. Soc. Rev., 2018, 47, 7703–7738 RSC.
- M. J. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2013, 135, 4600–4603 CrossRef CAS PubMed.
- B. K. Hodnett and B. Delmon, J. Catal., 1984, 88, 43–53 CrossRef CAS.
- Y. H. Taufiq-Yap, C. K. Goh, G. J. Hutchings, N. Dummer and J. Bartley, Catal. Lett., 2009, 130, 327–334 CrossRef CAS.
- W. C. O'Leary, W. A. Goddard and M. J. Cheng, J. Phys. Chem. C, 2017, 121, 24069–24076 CrossRef.
- R. K. Grasselli, C. G. Lugmair and A. F. Volpe, Top. Catal., 2008, 50, 66–73 CrossRef CAS.
- T. Ushikubo, Catal. Today, 2003, 78, 79–84 CrossRef CAS.
- O. V. Buyevskaya, D. Müller, I. Pitsch and M. Baerns, in Natural gas conversion V, ed. A. Parmaliana, Elsevier, Amsterdam, New York, 1998, pp. 671–676 Search PubMed.
- P. Kube, J. Dong, N. S. Bastardo, H. Ruland, R. Schlögl, J. T. Margraf, K. Reuter and A. Trunschke, Nat. Commun., 2022, 13, 7504 CrossRef CAS PubMed.
- W. P. McDermott, M. C. Cendejas and I. Hermans, Top. Catal., 2020, 63, 1700–1707 CrossRef CAS.
- J. M. Venegas, Z. Zhang, T. O. Agbi, W. P. McDermott, A. Alexandrova and I. Hermans, Angew. Chem., Int. Ed., 2020, 59, 16527 CrossRef CAS PubMed.
- J. T. Grant, W. P. McDermott, J. M. Venegas, S. P. Burt, J. Micka, S. P. Phivilay, C. A. Carrero and I. Hermans, ChemCatChem, 2017, 9, 3623–3626 CrossRef CAS.
- J. A. Loiland, Z. Zhao, A. Patel and P. Hazin, Ind. Eng. Chem. Res., 2019, 58, 2170–2180 CrossRef CAS.
- P. Kraus and R. P. Lindstedt, J. Phys. Chem. C, 2021, 125, 5623–5634 CrossRef CAS.
- X. Zhang, R. You, Z. Wei, X. Jiang, J. Yang, Y. Pan, P. Wu, Q. Jia, Z. Bao, L. Bai, M. Jin, B. Sumpter, V. Fung, W. Huang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 8042–8046 CrossRef CAS PubMed.
- X. Li, T. Lunkenbein, V. Pfeifer, M. Jastak, P. K. Nielsen, F. Girgsdies, A. Knop-Gericke, F. Rosowski, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2016, 55, 4092–4096 CrossRef CAS PubMed.
- K. Amakawa, Y. Wang, J. Kröhnert, R. Schlögl and A. Trunschke, Mol. Catal., 2019, 478, 110580 CrossRef CAS.
- V. V. Guliants, J. B. Benziger, S. Sundaresan and I. E. Wachs, Stud. Surf. Sci. Catal., 2000, 130, 1721–1726 CrossRef.
- F. Cavani and F. Trifirò, Stud. Surf. Sci. Catal., 1997, 110, 19–34 CrossRef CAS.
- M. Niwa, Y. Habuta, K. Okumura and N. Katada, Catal. Today, 2003, 87, 213–218 CrossRef CAS.
- Y. Moro-oka, Appl. Catal., A, 1999, 181, 323–329 CrossRef CAS.
- P. Concepción, P. Botella and J. M. López Nieto, Appl. Catal., A, 2004, 278, 45–56 CrossRef.
- M. Baca, A. Pigamo, J. L. Dubois and J. Millet, Catal. Commun., 2005, 6, 215–220 CrossRef CAS.
- F. Ivars-Barceló, J. Millet, T. Blasco, P. Concepción, J. S. Valente and J. M. López Nieto, Catal. Today, 2014, 238, 41–48 CrossRef.
- K. Bhaduri, A. Auroux, A. Bhaumik and B. Chowdhury, Appl. Organomet. Chem., 2022, 36(12), e6904 CrossRef CAS.
- G. Busca, G. Centi, F. Trifiro and V. Lorenzelli, J. Phys. Chem., 1986, 90, 1337–1344 CrossRef CAS.
- A. Stanislaus, M. Absi-Halabi and K. Al-Doloma, Appl. Catal., 1988, 39, 239–253 CrossRef CAS.
- D. C. Upham, M. Orazov and T. F. Jaramillo, J. Catal., 2021, 399, 132–141 CrossRef CAS.
- T. V. Andrushkevich, G. Y. Popova, Y. A. Chesalov, E. V. Ischenko, M. I. Khramov and V. V. Kaichev, Appl. Catal., A, 2015, 506, 109–117 CrossRef CAS.
- F. Garbassi, J. Catal., 1986, 98, 317–325 CrossRef CAS.
- R. Quintana-Solórzano, I. Mejía-Centeno, H. Armendáriz-Herrera, J. Ramírez-Salgado, A. Rodríguez-Hernandez, M. D. L. Guzmán-Castillo, J. M. López Nieto and J. S. Valente, ACS Omega, 2021, 6, 15279–15291 CrossRef PubMed.
- J. Ramírez-Salgado, R. Quintana-Solórzano, I. Mejía-Centeno, H. Armendáriz-Herrera, A. Rodríguez-Hernández, M. de Lourdes Guzmán-Castillo and J. S. Valente, Appl. Surf. Sci., 2022, 573, 151428 CrossRef.
- X. Li, T. Lunkenbein, J. Kröhnert, V. Pfeifer, F. Girgsdies, F. Rosowski, R. Schlögl and A. Trunschke, Faraday Discuss., 2016, 188, 99–113 RSC.
- C. Zhao and I. E. Wachs, J. Catal., 2008, 257, 181–189 CrossRef CAS.
- R. K. Grasselli, Top. Catal., 2002, 21, 79–88 CrossRef CAS.
- M. Eichelbaum, R. Glaum, M. Hävecker, K. Wittich, C. Heine, H. Schwarz, C.-K. Dobner, C. Welker-Nieuwoudt, A. Trunschke and R. Schlögl, ChemCatChem, 2013, 5, 2318–2329 CrossRef CAS.
- C. Schulz, S. C. Roy, K. Wittich, R. Naumann d'Alnoncourt, S. Linke, V. E. Strempel, B. Frank, R. Glaum and F. Rosowski, Catal. Today, 2019, 333, 113–119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimentation, details on experiments and characterisation (PDF) and complete catalytic data set (XLSX). See DOI: https://doi.org/10.1039/d2cy02184f |
| This journal is © The Royal Society of Chemistry 2023 |