
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFTIR microscopy for direct observation of conformational changes on immobilized ω-transaminase: effect of water activity and organic solvent on biocatalyst performance†
Iulia-Ioana
Rădoi
ad,
Diana Eva
Bedolla
 bc,
Lisa
Vaccari
b,
Anamaria
Todea
a,
Federico
Zappaterra
a,
Alexey
Volkov
d and
Lucia
Gardossi
*a
bc,
Lisa
Vaccari
b,
Anamaria
Todea
a,
Federico
Zappaterra
a,
Alexey
Volkov
d and
Lucia
Gardossi
*a
aDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via L. Giorgieri 1, 34127 Trieste TS, Italy. E-mail: gardossi@units.it
bElettra-Sincrotrone Trieste S.C.p.A, Area Science Park, 34149 Basovizza TS, Italy
cAREA Science Park, Padriciano 99, IT-34149 Trieste, Italy
dEnginZyme AB, Tomtebodavägen 6, 171 65 Solna, Sweden
First published on 22nd June 2023
Abstract
Enzyme immobilization is a key strategy to expand the scope of enzyme applications and to enable the recycling of biocatalysts, resulting in greener and more cost-efficient processes. The full exploitation of the technology advantages is strictly connected to the optimal selection of the carriers and the rational development of the immobilization protocol. The present study achieved such objectives by investigating the activity of a ω-transaminase in organic solvent (toluene) upon immobilization on commercially controlled porosity glass carriers (EziG™) with diverse porosity and surface functionalization. In addition to more conventional wet-chemistry approaches and confocal microscopy, infrared microspectroscopy and imaging were exploited to highlight the enzyme distribution in a label-free manner and provide details on the immobilized enzyme's conformation with respect to the native form. Contrary to what could be expected, the highest activity of the enzyme in organic solvent was achieved for the immobilization protocol on the most hydrophilic support that more severely affects the enzyme secondary structure, promoting a beta-sheet rich folding. Experimental data show that values of water activity above 0.90 in the reaction system had a positive effect on the efficiency of the transaminase reaction. The present study represents the first example of rational development of immobilization protocols relying on direct observation of the enzyme conformation upon immobilization, shedding light on the mutual interaction between the diverse process parameters and the carrier properties.
Introduction
Enzymes are increasingly used to perform a range of chemical reactions. These catalysts from nature are in general sustainable, selective, and efficient and may offer a variety of benefits such as environmentally friendly manufacturing processes, reduced use of solvents, lower energy requirement, high atom efficiency, and reduced cost.1 Enzyme immobilization enables continuous processing, lowering production costs and waste stream generation. Moreover, the prevention of protein contamination is one further motivation that induces industry to employ immobilized biocatalysts.2–4Transaminases (TAs), also known as aminotransferases (EC 2.6.1), are enzymes that catalyze the transamination reaction between an amino acid and an α-keto acid, by using the coenzyme pyridoxal-5′-phosphate (PLP). PLP, the active form of vitamin B6, forms a Schiff base with the ε-amino residue of lysine in the active site. The amino group is transferred from an amino donor to PLP, yielding pyridoxamine-5-phosphate (PMP), while the amino donor is released as the corresponding ketone. The amino acceptor compound reacts with PMP to form the corresponding amine, thereby automatically regenerating PLP.5–7 The position of the amino group in the substrates accepted by the transaminases determines their classification as α-TAs and ω-TAs. α-TA allows the formation of only α-amino acids, while ω-TA can perform the transfer of an amino group from a substrate that has several carbon atoms between the carboxyl and the amino groups. ω-TAs are of higher interest since they can aminate a wider range of substrates, such as keto acids, aldehydes, and ketones, and they accept ketones and amines without carboxyl groups.7,8 Transaminases have enormous potential as sustainable biocatalysts in the production of chiral amines,9 representing a key priority for the pharmaceutical industry since approximately 40% of current pharmaceutical products contain an amine functional group.10 Decades of investigations of enzymes in non-aqueous media have demonstrated that the use of biocatalysts in monophasic organic solvents can lead to significant advantages when hydrophobic substrates are required, and/or the thermodynamic equilibria of reactions should be shifted. The easier recovery of both the products and the biocatalysts represents an additional advantage that motivates the adoption of organic media in biotransformation.11 Despite the enormous potential of ω-TAs, fundamental challenges associated with severe by-product inhibition and with displacing unfavorable equilibrium towards product formation have prevented the widespread application of transaminases.12 In the present study, we have investigated an ω-transaminase variant His6-ATA-117,13 which was expressed in E. coli. Some of the features of this variant are its tolerance to high concentrations of cosolvent and its acceptance of low-cost amine donors such as isopropyl amine.12,13
The nature of the materials used as carriers for enzyme immobilization is crucial for achieving process efficiency and biocatalyst reusability. Besides chemical and mechanical stability, the hydrophobic–hydrophilic balance of the carrier and its porosity play an important role in determining the ability of the material not only to interact with specific surface regions of the enzyme14 but also to adsorb water, thus creating a microenvironment beneficial for the enzyme activity. Moreover, it is known that the partition of water between the carrier and the bulk medium affects the thermodynamic equilibrium of chemical reactions.15 Therefore, a careful control of water partition and the distribution of the enzyme molecules over and across the carriers would be of major help for the optimization of biocatalysts' performances. To the best of our knowledge, the scientific literature reports a very limited number of studies describing the direct visualization of these phenomena.16
As a support material for this study, we focused the attention on EziG™ carriers, a class of hybrid controlled porosity glass (CPG) immobilization carriers developed by EnginZyme AB (Sweden) which can bind protein through affinity tags. More details on the properties of the carriers such as size, pore diameter, and coating polymer are reported in Table S1 in the ESI.† CPG provides a favorable environment for enzymes and the porous structure enhances the mass transfer of reactants and products throughout the material.17,18 CPG materials are generally stable in most organic solvents and aqueous media at pH below 10. In the case of EziG™ carriers, the CPG surface is coated with functional organic polymer bearing chelating groups suitable for the selective binding of metal ions.11,17 As the binding metal ion, Fe3+ was selected due to its high binding capacity, low environmental impact and virtually non-toxicity. Notably, the selective binding allows the immobilization to be performed directly from the cell lysate, without pre-purification steps,17 with protein loadings up to 20% w/w. The diverse polymeric coating17 provides a different degree of hydrophobicity to the carriers: EziG1-Opal – hydrophilic (trace organics and Fe3+), EziG2-Coral – hydrophobic (derivatized polystyrene and Fe3+) and EziG3-Amber – semi-hydrophobic (derivatized polystyrene and Fe3+). These carriers have been previously tested with different enzyme classes, obtaining significant improvements in stability for arylmalonate decarboxylase,19 lipase B from Candida antarctica,20 norcoclaurine synthase,21 co-immobilized dehydrogenases22 and hydroxynitrile lyase.23
In the current study, we made a more detailed characterization of an immobilized ω-transaminase by exploiting, in addition to confocal microscopy, the advantage brought by Fourier Transform Infrared (FTIR) microscopy and imaging (Fig. 1). Both FTIR and Raman vibrational spectroscopy are sensitive to chemical vibrations, in a label-free way, and they are good methods to track the conformational state of a protein. FTIR spectroscopy has some advantages over Raman microscopy for this purpose: higher sensitivity, non-radiation damage, and no fluorescence interference. Specifically, FTIR spectroscopy can provide information about the secondary structure of proteins, such as α-helices, β-sheets, and random coils,24 while Raman microscopy is less sensitive to changes in protein secondary structure and may require more sophisticated analysis to extract meaningful information about protein conformational changes.25,26 With FTIR, samples can be analyzed repeatedly without changing their structure, due to the non-ionizing nature of IR radiation. On the other hand, Raman microscopy can cause photodegradation of the sample due to the high intensity of the laser used for excitation27 and may impose a demanding data pre-processing in order to minimize background fluorescence.28 Due to these advantages, several papers have been published on the use of vibrational spectroscopy to characterize the immobilization of enzymes.16,29–31 Confocal microscopy has been used as well, and in one recent study, Santiago-Arcos et al.16 reported the distribution of an alcohol dehydrogenase from Bacillus (Geobacillus) stearothermophilus (BsADH) on the surface of EziG™ carriers exploiting fluorescently labeled enzymes.
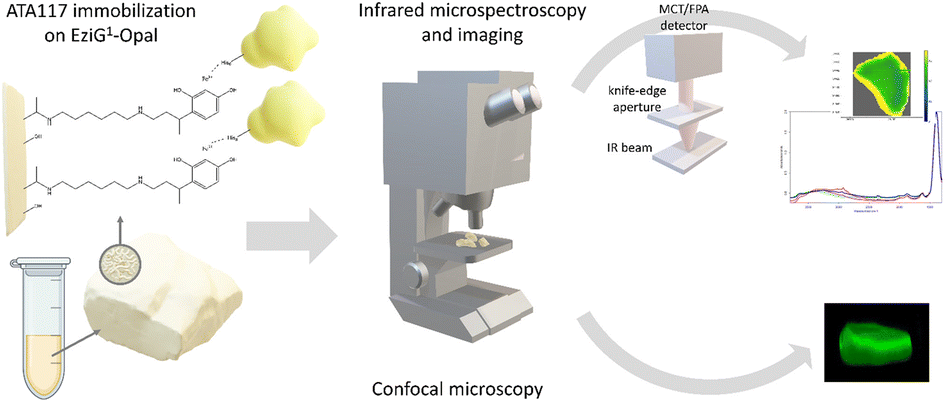 | ||
| Fig. 1 Schematic overview of the ATA-117 transaminase immobilization on EziG carriers and the microscopic characterization of the resulting biocatalysts. | ||
Here, we complemented the information on the enzyme distribution along the carrier depth by confocal microscopy with the enzyme distribution highlighted by FTIR imaging and with the enzyme conformational changes resulting from the immobilization process by exploiting FTIR microscopy. To the best of our knowledge, this is the first time that spatially-resolved IR vibrational analysis is used to study the immobilized enzyme on a glass-based carrier and to investigate the enzyme's conformational changes due to different immobilization protocols. The retrieved details have been correlated with the results on the partition of water between the reaction phases and the carriers, which allowed us to characterize the effect of the water activity (aw) of the reaction system on the enzyme efficiency in toluene and to control the equilibria taking place during the biocatalyzed transamination (which involves the formation of imines).
Overall, the integration of the experimental studies provided detailed information to identify suitable conditions for the employment of the immobilized enzyme in organic media and on the hydration necessary to preserve the enzyme activity.
Results and discussion
Immobilization and dehydration of the transaminase
The ω-transaminase variant His6-ATA-11732 was immobilized on three different EziG™ carriers: EziG1-Opal hydrophilic, EziG2-Coral hydrophobic and EziG3-Amber semi-hydrophilic33 (see Table S1 and Fig. S1 in the ESI† for more details on the carriers and their spectroscopic profile). The organic polymer bears Fe3+, the binding metal ion that allows for the selective binding of the histidine-tagged enzyme. Consequently, the immobilization was performed directly from the cell free extract, without the need to pre-purify the enzyme.17Fig. 2A illustrates the secondary and tertiary structure of the enzyme.32 From the literature, it is known that the use of polyhistidine affinity tags (His6) allows for an efficient purification process (in this case immobilization, since the EziG technology serves both as a purification and immobilization technique) but can also negatively affect the activity and/or the structure of the enzyme.34 Notably, Fig. 2B indicates the presence of several hydrophobic regions on the surface of the ω-transaminase, which are expected to interact differently with the carriers depending on their hydrophobic or hydrophilic nature. It should be underlined that the enzyme was expressed in E. coli and, conversely, does not bear glycans that might shield such hydrophobic regions on the surface35 in aqueous media.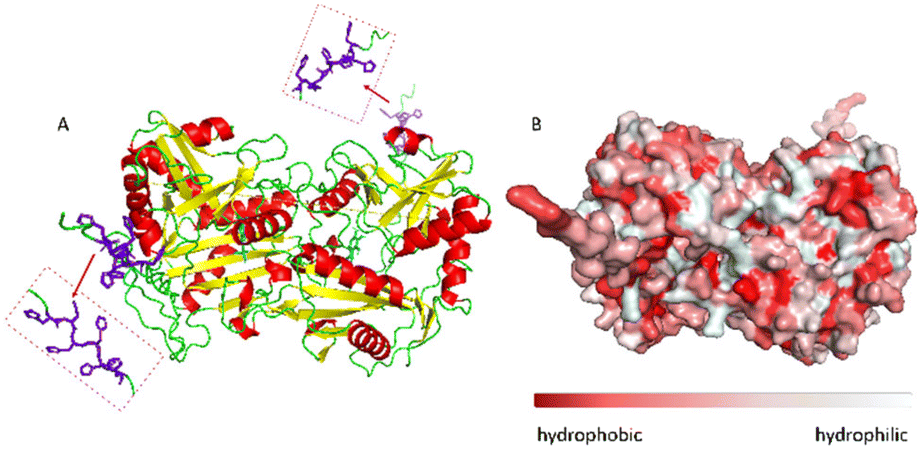 | ||
| Fig. 2 A: Visualization of the secondary structure of N-terminal His-tagged (R)-selective ω-transaminase from Arthrobacter sp. (His6-ATA-117). In purple the histidine tag and in cyan the PLP cofactor, enzyme diameter: 7.17 nm. B: Surface hydrophobicity: in red the hydrophobic areas and in light-grey the hydrophilic areas. The enzyme structure was predicted using AlphaFold software, figures and the enzyme diameter was obtained using PyMOL software. | ||
However, the solvation of the enzyme surface or even its conformational features might change when the protein is exposed to different media or interacts with carriers of different chemical nature, as in the case of the three different EziG™ CPG matrixes.33,36
To date, no study has been reported to describe the distribution of transaminases upon immobilization on EziG™ and, more importantly, no method has been used so far for describing the structural changes imposed on the enzyme upon the immobilization process nor upon exposure to organic solvent. The present study aims to fill this gap, investigating the effects of hydration, dehydration, and solvation on the dynamic and catalytic properties of the immobilized protein.37 For this purpose, various samples of immobilized His6-ATA-117 were prepared following a number of protocols. In order to prepare the enzymatic formulations for their application in organic solvent, the biocatalysts were rinsed with different aqueous solutions and then dried under vacuum to remove the water in excess. The purpose was to evaluate these protocols aiming to preserve the activity of the enzyme when exposed to the dehydration step. As a matter of fact, removing the water in excess is mandatory to prevent the agglomeration of the wet particles of the biocatalyst when placed in hydrophobic organic media. As described in detail in the Experimental section, the immobilized His6-ATA-117 samples were treated with four different buffer solutions (see Table 1, “Pretreatment” column) supplemented with the co-enzyme or sucrose. In the latter case, polyol was used to promote the binding of extra water able to protect the protein from denaturation.38,39 As a matter of fact, it is known that a harsh dehydration procedure (drying at 100 °C) can strip essential water molecules from the enzyme structure, thus jeopardizing its activity. The immobilized His6-ATA-117 samples were also rinsed with iPrOH to remove the water in excess and then with toluene for removing the residual water-miscible alcohol, which has a detrimental effect on protein stability40 and might also interfere with the reaction chemistry. After these pretreatments, the different formulations of immobilized His6-ATA-117 were also dehydrated in a desiccator at 25 °C at 100 mbar for 24 h, to verify the effect of an extensive dehydration treatment on the biocatalyst.
| Entry | Pre-treatment | EziG1-Opal hydrophilic (pd: 500 ± 50 Å) | EziG2-Coral hydrophobic (pd: 300 ± 50 Å) | EziG3-Amber semi-hydrophobic (pd: 300 ± 50 Å) | |||
|---|---|---|---|---|---|---|---|
| Water adsorbed by the carrier [% w/w] | Weight variation after drying (+His6-ATA-117) [% w/w] | Water adsorbed by the carrier [% w/w] | Weight variation after drying (+His6-ATA-117) [% w/w] | Water adsorbed by the carrier [% w/w] | Weight variation after drying (+His6-ATA-117) [% w/w] | ||
| pd: pore diameter; buffer – 25 mM NaP pH 9.0; 0.3 mM PLP; 20% w/v sucrose.a The weight variation can be ascribed to evaporation of both water and residual organic solvent. | |||||||
| 1 | Buffer | 3.4 ± 1.1 | −4.1 ± 0.6 | 3.3 ± 0.2 | −0.2 ± 0.0 | 3.3 ± 0.1 | −1.2 ± 0.2 |
| 2 | Buffer + PLP | 2.7 ± 0.2 | −2.4 ± 0.4 | 3.0 ± 0.3 | +1.1 ± 0.1 | 2.7 ± 0.2 | +0.3 ± 0.1 |
| 3 | Buffer + sucrose | 3.6 ± 0.1 | +15.3 ± 2.1 | 4.4 ± 0.1 | +22.1 ± 0.2 | 4.1 ± 0.0 | +19.6 ± 0.8 |
| 4 | Buffer + sucrose + PLP | 3.4 ± 0.1 | +17.1 ± 1.1 | 4.2 ± 0.1 | +22.8 ± 1.2 | 3.8 ± 0.2 | +21.3 ± 1.2 |
| 5 | iPrOH followed by toluene | 1.6 ± 0.3a | −2.0 ± 1.4a | 2.3 ± 0.3a | −1.4 ± 0.0a | 2.0 ± 0.6a | −3.9 ± 1.4a |
Table 1 reports a schematic overview of the water retained by the enzyme-free carriers (first column for each type of carrier) and by the carriers containing the immobilized enzyme (second column for each type of carrier). The values were determined by the difference in weight before and after extensive drying at 100 °C (until constant weight) for the enzyme-free carriers and drying under vacuum at 25 °C (100 mbar, 24 h) for the immobilized enzyme.
Data in Table 1 indicate that EziG carriers have a modest water adsorption capacity, ranging from 2.7 to 4.7 (% w/w). This is most probably a consequence of the organic polymeric coating, since previous studies on the absorption of water on porous siliceous materials showed that it heavily depends on the polarity of the material and on the size of the pores.41,42 For instance, calcined porous celite,43 a silica-based matrix consisting of diatomaceous earth broken up and subsequently recalcined to create porous particles with controlled pores, has the ability to retain water as much as 90% of its weight. When the carriers were washed with aqueous buffer, the dehydration procedure at 25 °C and 100 mbar for 24 h was able to remove most of the residual water. However, the hygroscopic nature of polyol makes the drying process in the desiccator at 25 °C under vacuum ineffective, but rather the samples adsorb extra water from the environment, up to more than 20% w/w. It is worth mentioning that all the samples were placed in the same desiccator, and it was opened only once, namely at the end of the 24 hour drying process. The samples rinsed with iPrOH and toluene display the lowest differences in weight after the drying treatment (entry 5, Table 1), which can be ascribed to the evaporation of the residual water and also traces of organic solvents.
In conclusion, all different formulations of immobilized His6-ATA-117 retain <5% of water when rinsed with all different aqueous buffers and the dehydration of the immobilized enzymes at 25 °C under vacuum removes most of this residual water, except in samples containing the hygroscopic polyol. Finally, the polar iPrOH is effective in stripping most of the water bound on the carrier.
The activity of all immobilized His6-ATA-117 formulations pretreated according to the protocols of Table 1 was finally assessed in aqueous media in order to verify the effect of all pre-treatments on the activity of the biocatalyst using a standard activity assay. The formation of acetophenone, starting from α-methylbenzylamine and using sodium pyruvate as an amine acceptor (see Scheme S1 in the ESI†), was followed for 15 minutes (the results are presented in Fig. 3). Compared to the activity of the native enzyme, the activity after the immobilization was 2.9 times lower.
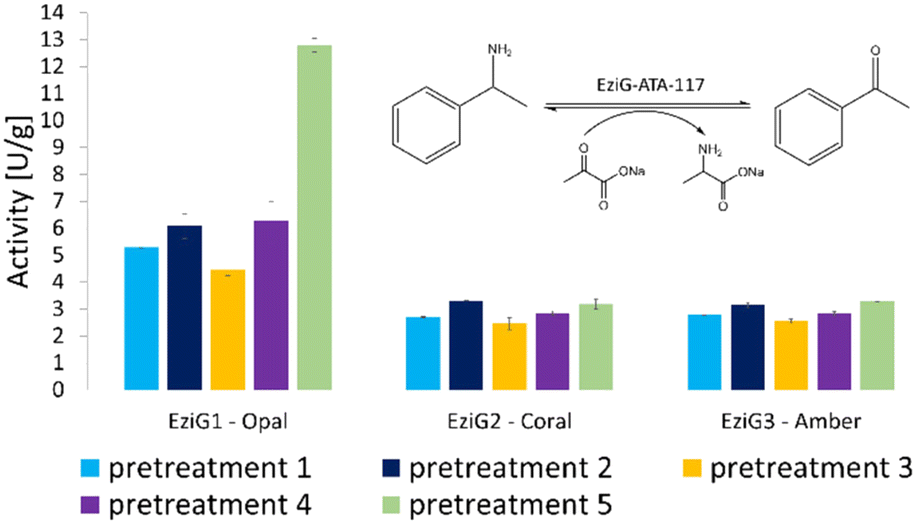 | ||
| Fig. 3 Enzymatic activity (expressed in enzymatic unit per g of immobilized enzyme) of the immobilized His6-ATA-117 formulations on EziG1-Opal, EziG2-Coral, and EziG3-Amber pre-treated according to the protocols of Table 1. Reaction conditions: 100 mM HEPES pH 9.0, 10 mM sodium pyruvate, 20 mM α-MBA, 25 °C, 15 rpm, 15 min. The formation of acetophenone was monitored by measuring the increase in absorbance at 245 nm (see the scheme in the inset of the figure). Immobilization yields: 92.4% EziG1-Opal, 92.5% EziG2-Coral and 92.2% EziG3-Amber. | ||
Results in Fig. 3 indicate that EziG1-Opal performs better (∼50% higher activity) than EziG2-Coral and EziG3-Amber for the reported model reaction in aqueous media. Notably, the surface of EziG1-Opal has the highest hydrophilicity, and the matrix has the widest pores (∼500 Å compared to 300 Å of EziG2-Coral and EziG3-Amber): the combination of these properties appears to be beneficial for this specific reaction. Independent from the carrier, there is no remarkable effect of the pretreatment protocols except for the case of pretreatment 5 (iPrOH + toluene rinsing) of the EziG1-Opal formulation, which demonstrates by far the highest activity. Therefore, starting from these experimental observations, detailed spectroscopic analyses were planned in order to gather direct observations of the behavior of the His6-ATA-117 enzyme once immobilized on the three EziG carriers and exposed to the different washing and dehydration procedures.
Spectroscopic characterization of immobilized His6-ATA-117 enzyme
Infrared vibrational microspectroscopy and imaging44 were used for the localization and structural characterization of the immobilized biocatalysts. As a matter of fact, FTIR spectroscopy is sensitive to the secondary structure of proteins. In an FTIR spectrum of a protein, two prominent features can be identified, the Amide I band (∼1650 cm−1) which comes mainly from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the peptide backbone, and the Amide II band (∼1540 cm−1) attributed to the N–H bending and C–N stretching vibration of the peptide backbone.45,46 Based on the different hydrogen-bonding environments (α-helix, β-sheet, turn, unordered conformations) the spectral shape of Amide I band changes, and this can be related to the protein secondary structure.47 Indeed, most proteins have a mixture of secondary structural elements, with the Amide I band being a combination of these components. In general, α-helices and β-sheets have Amide I vibrational frequencies around 1655 and 1630 cm−1, respectively.48,49
O stretching vibration of the peptide backbone, and the Amide II band (∼1540 cm−1) attributed to the N–H bending and C–N stretching vibration of the peptide backbone.45,46 Based on the different hydrogen-bonding environments (α-helix, β-sheet, turn, unordered conformations) the spectral shape of Amide I band changes, and this can be related to the protein secondary structure.47 Indeed, most proteins have a mixture of secondary structural elements, with the Amide I band being a combination of these components. In general, α-helices and β-sheets have Amide I vibrational frequencies around 1655 and 1630 cm−1, respectively.48,49
FTIR measurements were performed after the five different pretreatments (Table 1) as described in the Material and methods section. For all the prepared samples, both single-point spectroscopy and infrared hyperspectral images were collected. Even if the analyzed carriers are made of glass (a very strong IR absorber) and they are thick in the millimeter range, due to their high porosity it was possible to analyze the samples in transmission without any other treatment, except for the saturation of the Si–O stretching region (see Fig. S1 in the ESI†). At this stage, it is relevant to highlight that the spectral region characteristic of proteins is substantially free of carrier spectral features that may hinder the vibrational analysis.
FTIR hyperspectral images are particularly useful for correlating chemical information with the sample's morphology. Fig. 4 reports the analysis of representative chemical images of the transaminase immobilized on EziG1-Opal and washed with organic solvent as described in Table 1, entry 5 (a complete overview of all carriers and pre-treatments can be found in ESI,† Fig. S2). The hyperspectral images, obtained by plotting the intensity of Amide I and II bands normalized to the glass carrier thickness at each point, prove that the enzyme is accumulated at the border regions of the carriers. Nevertheless, the analysis pointed out a significant penetration of the protein also in the inner areas.
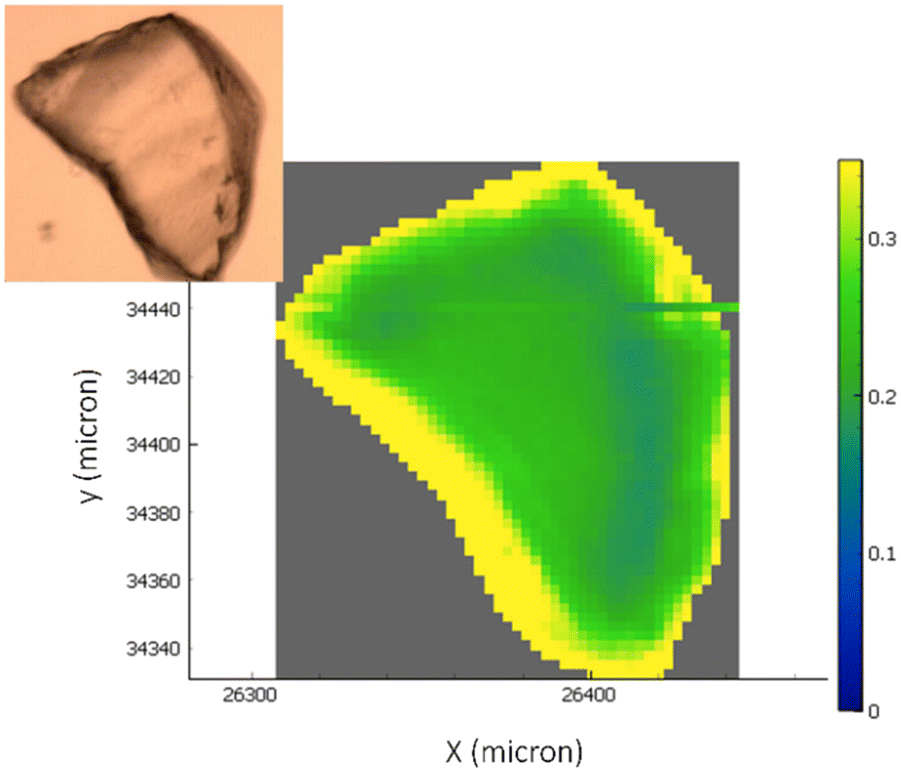 | ||
| Fig. 4 The optical image of the carriers with the overlap of the chemical infrared image generated by the ratio of the integration in the specific region of amide I and amide II bands (∼1710–1480 cm−1) and the band identifying the glass carrier (∼1500–1300 cm−1). The colour scale bar ranges from higher biocatalyst concentrations (yellow areas) to not-detectable concentrations (blue areas). | ||
Analysis of the spatial distribution of His6-ATA-117 across the EziG carriers by fluorescence confocal microscopy
FTIR imaging, being a non-confocal microscopy technique, does not allow to distinguish between surface and core penetration of the enzyme. So, to have a second confirmation on the distribution of the enzyme on the surface of the carriers, and also to get information about its infiltration along the carrier depth, we used confocal laser scanning microscopy (CLSM) imaging. To this aim, we chemically labelled His6-ATA-117 with FITC (more details in the “Material and methods” section) and then immobilized it to perform the studies (the immobilized enzyme was treated according to entry 1 of Table 1). Fig. 5 shows different “virtual” layers of each of the carriers and, on the lower row, the 3D reconstruction of all these layers (the distance between the layers is 2 μm). We found that His6-ATA-117 was mainly located in the outer regions, with all three carriers following a similar distribution pattern to what it was observed with FTIR imaging. The distribution and accumulation of the enzyme differs from one bead to another, as well as from one carrier type (EziG1-Opal, EziG2-Coral or EziG3-Amber) to another, because they do not have a regular shape or size. In all three carriers the enzyme infiltrated towards the core of the carriers at different average depths: EziG1-Opal 45.8 ± 0.1 μm, EziG2-Coral 43.7 ± 0.1 μm and EziG3-Amber 38.7 ± 0.1 μm respectively, as described in the Experimental section. The radial cross-section fluorescence profile of these EziG carriers is presented in Fig. S3.†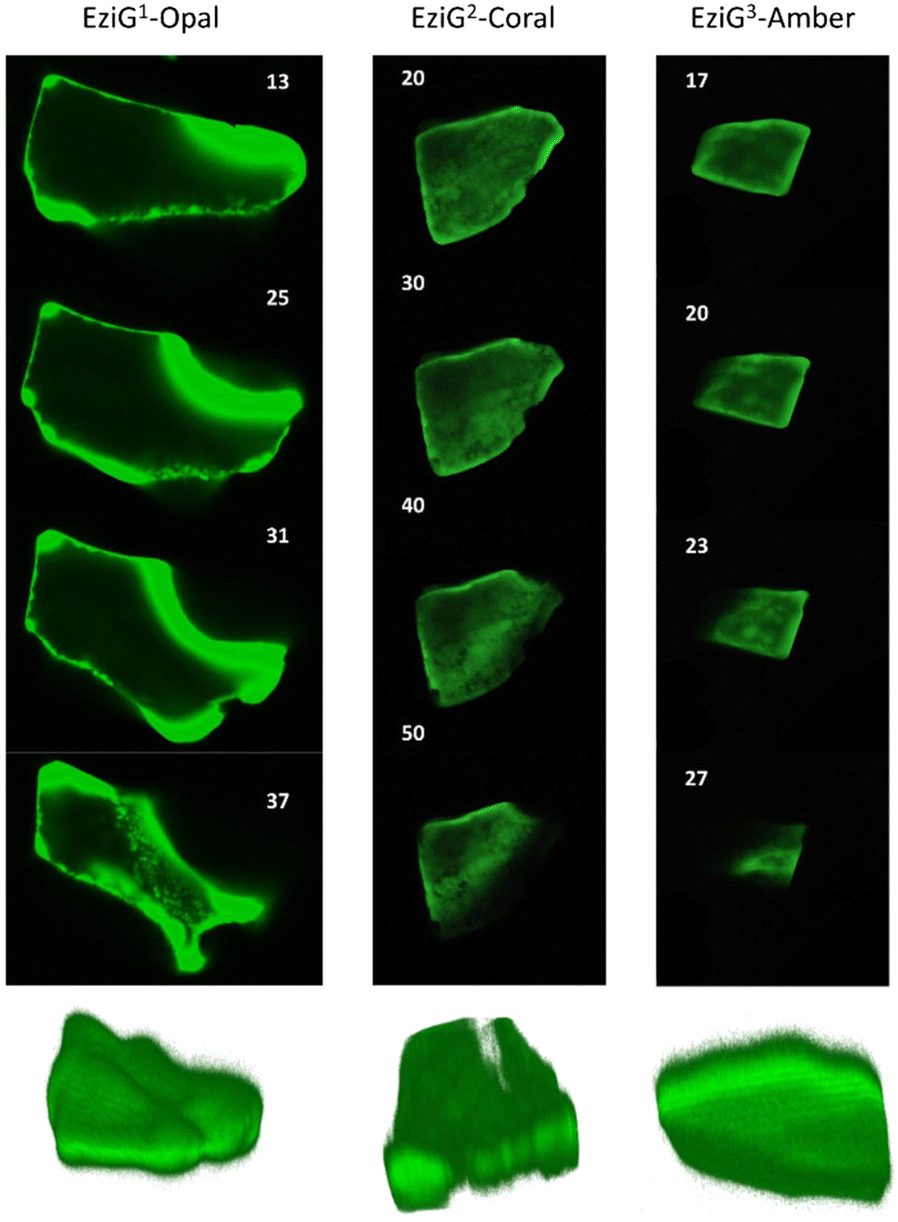 | ||
| Fig. 5 2D images (40×) of different layers of EziG1, EziG2 and EziG3 carriers obtained by acquiring 3D image stacks with scanning confocal microscopy showing the FITC labelled His6-ATA-117 distribution (samples were pretreated with NaP buffer before drying – pretreatment 1). Row below: 3D reconstruction of the shown carriers. | ||
FTIR investigation of the secondary structure of immobilized and pure His6-ATA-117
To characterize the conformation of the immobilized enzyme, we measured the infrared absorption for the pure enzyme and used it as a reference. For this analysis, the second derivatives of the spectra in the Amide I–Amide II spectral range, for all types of pretreatments used before the drying step, were compared (see Fig. S4 in the ESI†). Spectra were acquired averaging 2048 scans on a border region of the carrier, setting the lateral resolution at 50 × 50 μm2, in order to enhance the spectral quality while sacrifying the spatial resolution.In Fig. 6, an informative comparison is shown between samples pretreated with simple buffer (in blue) or with organic solvents (in green) as described in Table 1 entry 1 and 5, respectively.
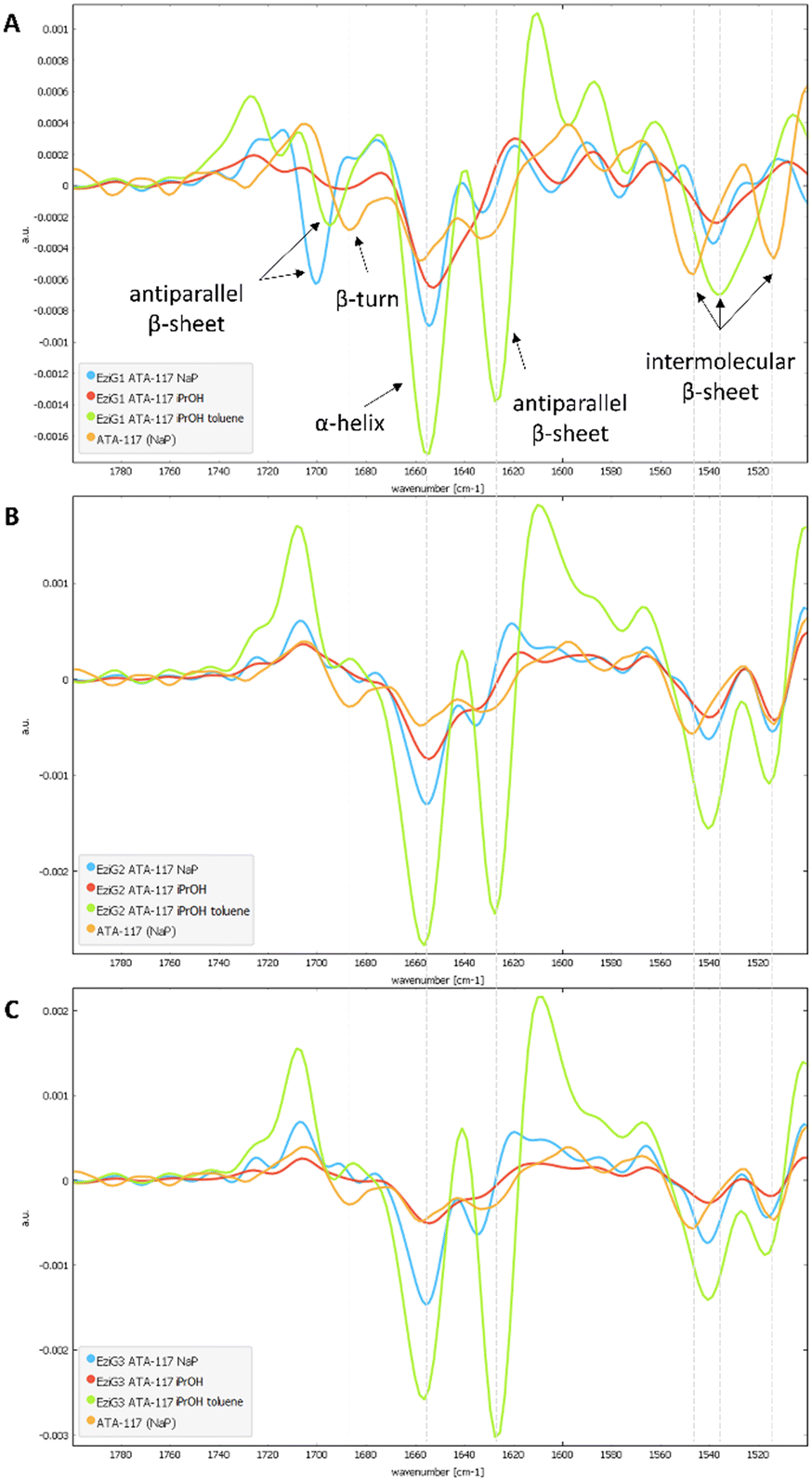 | ||
| Fig. 6 Comparison of the second derivative spectra of NaP (blue), iPrOH (red) and iPrOH + dry toluene (green) washings for all EziG carriers (A. EziG1, B. EziG2, C. EziG3) containing the immobilized His6-ATA-117. For each group, the comparison was made between the immobilized enzyme (washed and dried under vacuum at 100 mbar, 25 °C for 24h) and the native enzyme (resuspended in NaP buffer; orange). | ||
The second derivative spectrum of the immobilized enzyme displayed a main peak at ∼1654 cm−1, along with the peak at ∼1635 cm−1, which is assigned to the α-helix and β-sheet structures of the protein, respectively. The minor component at ∼1601 cm−1 occurred in the spectral region of intermolecular β-sheet absorption. As can be appreciated from the figure, the second derivative spectra for all the preparations washed with buffer solutions (blue) are similar to that of the pure free enzyme (in orange). Upon analysis of the samples pretreated with the organic solvents (iPrOH followed by toluene, entry 5 Table 1), an increase in the intensity of the band at about 1630 cm−1 was observed for all three carriers. When the enzyme was exposed to the organic solvent a conformational rearrangement occurred, probably because the protein tends to rearrange to reach a stable conformation between a hydrophilic and a hydrophobic portion. To verify whether the rearrangement was caused by the treatment with the polar and hygroscopic isopropyl alcohol, a new sample of immobilized His6-ATA-117–EziG1-Opal was prepared, which was pre-treated only with iPrOH before drying. The comparison of the new spectrum (red line in spectra of Fig. 6) with those registered using pre-treatment 1 and 5 confirms that protein misfolding is observable only when the enzyme is exposed to non-polar solvents such as toluene. Most probably, the hydrophobic organic solvent promotes the orientation of a hydrophobic part of the protein to the hydrophobic reaction medium, in this case to toluene.
In addition, when looking at random spots in FTIR images at the border and at the center of the carrier for all preparations (see Fig. S4 in the ESI†), it was found that for pretreatment 5 with toluene the conformational change seems to happen everywhere in the carrier, while it is not the same for pretreatment 1, where some variability on the conformation of the protein is observed on the external parts of the carrier (see Fig. S5†).
In conclusion, the spectroscopic studies highlighted a conformational rearrangement favoring β-sheet conformation for all immobilized His6-ATA-117 formulations washed with iPrOH followed by toluene and verified that the modification is promoted by the exposure to toluene. The conformational change was observed for the enzyme immobilized on all three EziG carriers. The experimental data on enzymatic activity collected in aqueous solution in Fig. 3 indicate that this conformational modification is not detrimental for the activity of the immobilized His6-ATA-117 on EziG carriers, but rather an hyperactivation might occur as observed for His6-ATA-117–EziG1-Opal where the activity is almost doubled as compared to the formulation treated with a simple buffer solution. For this reason, the study continued with the investigation of the performance of His6-ATA-117–EziG1-Opal in a transamination reaction in organic solvent. Toluene was selected as the reaction medium in order to reduce the variables that might affect the conformation and activity of the immobilized enzyme.
EziG1-Opal-immobilized ATA 117: activity in organic solvent at different aw values
The activity of EziG1-Opal–His6-ATA-117 pretreated with iPrOH and toluene was assessed in a transamination reaction between 1-phenoxypropan-2-one and isopropyl amine as an amine donor using toluene as the reaction medium (Scheme S2 in the ESI†). Because the hydration of the enzyme and the partition of the water are key parameters for enzymatically catalyzed reactions,50,51 and our experimental design comprises a drying step after immobilization, we evaluated the efficiency of the enzymatic formulation in similar reaction systems characterized by different water activity (aw) values. The direct measurement of aw allows one to compare the performance of enzymes in different media, with the hydration state of an enzyme being always fixed by the aw value regardless of the chosen solvent.52 For instance, the reaction system employing the biocatalyst washed with iPrOH and toluene and then dried under vacuum led to aw = 0.50.As expected, the ketone disappears also in the absence of the enzyme since, as already known, it reacts spontaneously with the amine donor53 (isopropyl amine) establishing an equilibrium with the corresponding imine (N-isopropyl-1-phenoxypropan-2-imine), which was confirmed by means of GC–MS. Therefore, a clearer view of the behavior of the biocatalyzed transamination is shown in Fig. 7, which compares the amount of N-isopropyl-1-phenoxypropan-2-imine and the desired 1-phenoxypropan-2-amine product. The GC–MS data is expressed as the ratio between the area of the peaks of the molecules and an internal standard (dodecane).
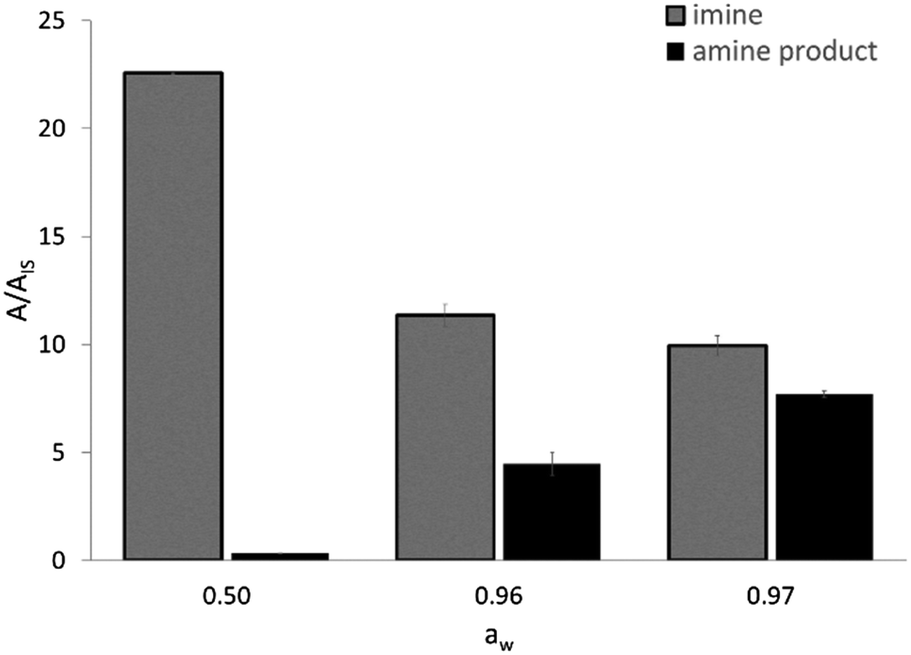 | ||
| Fig. 7 GC-MS results of the transamination of 1-phenoxypropan-2-one in the presence of isopropyl amine as an amine donor, catalyzed by His6-ATA-117 immobilized on EziG1-Opal at different aw values. Reaction conditions: 1 mL toluene, 250 mM isopropyl amine, 100 mM 1-phenoxypropan-2-one, 35 °C, 150 rpm, overnight. The aw values were obtained and measured as described in the Experimental section. | ||
The formation of N-isopropyl-1-phenoxypropan-2-imine was observed in all reaction mixtures as the main component (Fig. 7). At aw as low as 0.50 the formation of the amine product is negligible and an increase of aw at values >0.95 leads to a significant increase in the formation of the desired amine, accompanied by a decrease in the concentration of the imine. Most probably, at aw > 0.95 there are two positive effects which are synergic. Firstly, the highly hydrated biocatalyst is more efficient and secondly the equilibrium between the imine and the free amine donor is shifted towards the second chemical species, which can be converted by a more active enzyme.
In order to increase the aw values, the dehydration step was omitted, thus achieving aw = 0.96. The employment of water saturated toluene both in the rinsing step and as the reaction medium led to a measured aw = 0.97. All measurements were carried out after 24 h of incubation and it must be noted that differences in aw values close to aw = 1.0 are not informative and they simply indicate that the system is close to water saturation. In all experiments only one single liquid phase was observed.
Table 2 reports the calculated conversions based on GC–MS analysis collected for the samples obtained after 24 h of incubation of 1-phenoxypropan-2-one with isopropyl amine as an amine donor with and without the biocatalyst at the corresponding measured aw values. Notably, the aw values of the reactions performed in the absence of the biocatalyst are different since the contribution of the carrier, either in terms of subtracting or adding water to the system, is missing. The aw values of the reference reaction mixtures (i.e. dry toluene and water saturated toluene) are available in Table S2 of the ESI.†
| Reaction solvent | Pretreatment of EziG1-Opal–His6-ATA-117 | a w | Conversion of ketone [%] | |
|---|---|---|---|---|
| Rinse with | Drying | |||
| Control reactions | ||||
| Dry toluene | — | — | 0.63 ± 0.01 | 46 ± 0.5 |
| Water-saturated toluene | — | — | 0.73 ± 0.01 | 39 ± 0.2 |
| After 24 h of biocatalyzed transamination | ||||
| Dry toluene | iPrOH followed by dry toluene | Under vacuum 100 mbar, 25 °C, 24 h | 0.50 ± 0.01 | 65 ± 2.1 |
| Dry toluene | iPrOH followed by dry toluene | No drying | 0.96 ± 0.01 | 44 ± 2.7 |
| Water-saturated toluene | iPrOH followed by water-saturated toluene | No drying | 0.97 ± 0.01 | 52 ± 6.2 |
Enzyme reactivity in organic solvents can be affected positively by an increase of aw, as also reported previously for other enzymes, such as amidases50 and proteases.15 The causes can include the increase in conformational flexibility and also the importance of water molecules for the reaction mechanism itself. For sure the aw affects the thermodynamic equilibrium of the reversible reaction between the ketone and the amine donor, leading to the imine. This is evident from the percentage of conversion of the ketone towards the imine in the absence of the biocatalyst (Table 2), which increases from 39% to 46% when the aw decreases from 0.73 to 0.63. At aw close to 1.0 the system is saturated with water, even when a second liquid phase is not visible, and further addition of water would not affect the equilibrium of the reaction. Therefore, the observed differences between the reactions conducted at aw > 0.90 are most probably ascribable to effects at the level of the enzymatic activity since the use of water saturated toluene in the washing procedure appears to be beneficial.
Overall, the data here reported shed light on the effect of aw on the biocatalyzed transamination in toluene and the importance of the protocols for the formulation of immobilized transaminases to maintain the activity of transaminases.54 The reported protocols do not make use of hydrated salts for adjusting the hydration of the reaction system11 and more importantly, provide a direct confirmation of reaching the equilibrium in the distribution of water between the different phases.
Experimental
Materials
Kanamycin monosulfate, Auto Induction Media Terrific Broth Base, 3-(morpholin-4-yl)propane-1-sulfonic acid (MOPS), sodium chloride, pyridoxal-5′-phosphate, sodium phosphate, toluene, isopropyl alcohol, sodium pyruvate, α-methylbenzylamine, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethane-1-sulfonic acid (HEPES), Bradford reagent, fluorescein isothiocyanate isomer I (FITC), acetophenone, sucrose and dimethylformamide were purchased from Sigma-Aldrich. The EziG™ carriers (EziG1-Opal, EziG2-Coral, EziG3-Amber) and the His6-ATA-117 ω-transaminase (round 11 variant originating from Arthrobacter sp. expressed using Escherichia coli BL21 (DE3) as a host organism) were provided by EnginZyme AB (Stockholm, Sweden). Relative humidity and water activity were measured using a humidity sensor resistant to the detrimental effects of organic solvent vapors (DARAI Instruments, Trieste, Italy).Expression and of ω-transaminases
N-Terminal His-tagged (R)-selective ω-transaminase from Arthrobacter sp. (His6-ATA-117, pET-28a) was previously developed by Savile et al. by performing 11 rounds of mutagenesis.55 The ω-TA variant His6-ATA-117 was expressed using Escherichia coli BL21 (DE3) as a host organism: 500 mL of TB AIM medium supplemented with kanamycin (final concentration of 50 μg mL−1) was inoculated with 50 μL cell stock. Protein expression was conducted overnight at 30 °C, 220 rpm and thereafter terminated by centrifugation of the culture (4 °C, 4000 rpm, 20 min). The cell pellet was resuspended in buffer solution (50 mM MOPS, 150 mM NaCl, pH 7) and disrupted by sonication. Lysed cells were centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 1 hour. The clarified cell-free extract solution was lyophilized in the freeze dryer for 48 hours. The activity of the obtained protein was evaluated by using the standard assay described in the activity assay section and the protein concentration was determined by using the Protein A280 assay (protein content 6.362 mg mL−1, specific activity 0.031 U mg−1).
000 rpm at 4 °C for 1 hour. The clarified cell-free extract solution was lyophilized in the freeze dryer for 48 hours. The activity of the obtained protein was evaluated by using the standard assay described in the activity assay section and the protein concentration was determined by using the Protein A280 assay (protein content 6.362 mg mL−1, specific activity 0.031 U mg−1).
Enzyme immobilization onto EziG carriers and dehydration protocols
The immobilization of His6-ATA-117 onto EziG carriers was performed using the cell free extract (CFE) obtained after cell disruption and clarification procedures. The CFE solution used for the immobilization was prepared by resuspending 1.5 g of freeze-dried CFE in 150 mL buffer (25 mM sodium phosphate and 0.1 mM pyridoxal-5′-phosfhate, pH 9.0) and incubated for two hours. 50 mL of the obtained solution was further incubated for two more hours in the presence of 50 mg of carriers at 25 °C covered by aluminum foil (the procedure was done for each of the carriers). In order to determine the total protein loading, the Bradford assay was performed before (A595 initial) and after the immobilization process (A595 final) for calculating the amount of total protein bound to the beads.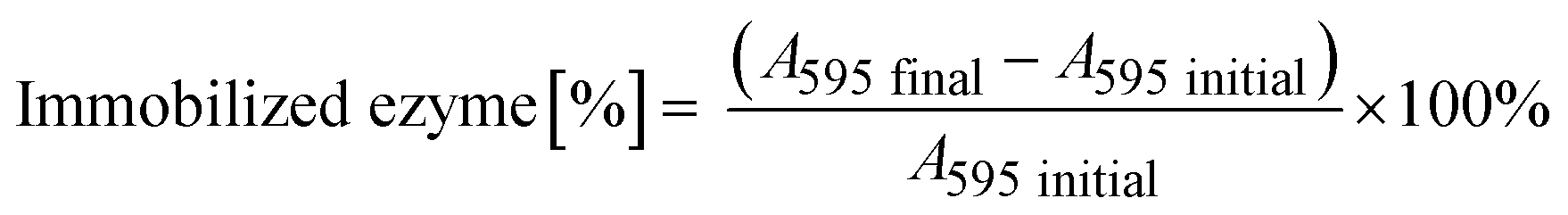 | (1) |
The immobilized enzyme was left to sediment before removing the liquid phase by pipetting. In order to prepare the enzymatic formulations for their application in organic solvent, the biocatalysts were rinsed with different aqueous solutions and then dried under vacuum to remove the water in excess. Different samples (50 mg) of immobilized His6-ATA-117 were washed 3 times with 5 mL of each of the following solutions: a) 25 mM sodium phosphate buffer (pH = 9); b) 25 mM sodium phosphate buffer (pH = 9) + 0.3 mM pyridoxal-5′-phosphate; c) 25 mM sodium phosphate buffer (pH = 9) + 20% w/v sucrose; d) 25 mM sodium phosphate buffer (pH = 9) + 0.3 mM pyridoxal-5′-phosphate + 20% w/v sucrose; e) isopropyl alcohol followed by dry toluene. The first washing solution (25 mM NaP, also used as a buffer for immobilization) is usually used after immobilization to remove the non-immobilized enzyme from the carrier surface. This step is mandatory especially when the cell free extract (CFE) is used. Pyridoxal-5′-phosphate acts as a coenzyme for all transamination reactions and it was demonstrated that it improves the stability of the enzyme.5
Finally, samples of immobilized His6-ATA-117 were also washed (3 times with 5 mL) with a polar solvent, namely iPrOH (logP −0.16) followed by rinsing (3 times with 5 mL) with toluene (logP 2.7). While iPrOH is used for removing the water in excess, the chemically inert and hydrophobic toluene is necessary for removing the water-miscible alcohol, which has a detrimental effect on protein potential inhibition and might also interfere with the reaction chemistry.
After the washing protocols described above, the immobilized formulations were also dried in a desiccator under vacuum at 25 °C, 100 mbar for 24 h. The temperature was carefully controlled in order to prevent the inactivation of the enzyme during the drying process.
Determination of the water adsorption capacity of the carriers
The washing procedures described above were also performed for the enzyme-free carriers, which were finally dried in an oven at 100 °C for 24 hours. In order to determine the water retained by the carrier after each washing treatment, each sample (washed enzyme-free EziG carrier) was dried until constant weight and then the difference in weight between the wet and the dried sample was determined. The drying process was monitored for 24 h, with the samples being weighed five times during this time. It was experimentally verified that the presence of the enzyme does not affect the water adsorption capacity of each of the three EziG carriers.Determination of the enzymatic activity of the immobilized enzyme using microplates
To determine the activity of the immobilized enzyme, 10 mg of EziG formulations were mixed with 1 mL reaction mixture (10 mM sodium pyruvate and 20 mM α-methylbenzylamine were dissolved in 100 mM HEPES pH 9.0). The reaction was run for 15 minutes (25 °C, 15 rpm) and the formation of acetophenone was followed in a microplate reader (Infinite M1000 pro reader) by measuring the increase in absorbance at 245 nm (ε = 12 mM−1 cm−1).56,57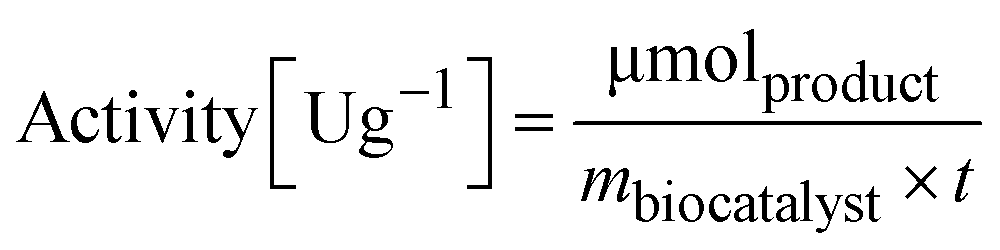 | (2) |
m biocatalyst = amount of dry biocatalyst [g]
t = time of the enzymatic assay [min]
Measurement of the water activity (aw) of the reaction systems
The water activity, aw, of the sealed and equilibrated reaction systems was determined according to eqn (3), by measuring the relative humidity (ERH) of the headspace of the sealed reaction vessel, assuming that the reaction system had reached the equilibrium and that the aw is the same in all phases after the partition of the water molecules between the different components of the system (solid, liquid and gas).51,58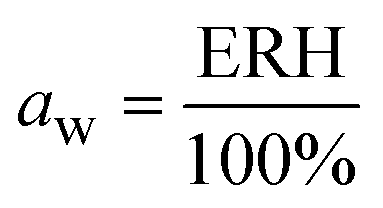 | (3) |
In a 4 mL vial, 10 mg of the sample containing the immobilized enzyme (EziG carrier + His6-ATA-117) were stored in the presence of 1 mL of dry toluene and kept for 24 h in the closed vial at 25 °C to ensure the equilibrium of the system. Measurements were conducted by carefully sealing the sensor inside the vial until constant reading to verify the achievement of the equilibrium. The sensor did not get in contact with the sample.
The calibration of the sensor was made at 25 °C using standard salt solutions at four different aw values (0.12, 0.43, 0.75, 0.84). The saturated solutions were prepared at 25 °C using the following salts (analytical grade): NaOH (aw = 0.12), K2CO3 (aw = 0.43), NaCl (aw = 0.75), and KCl (aw = 0.84). Notably, the humidity-temperature sensor can measure differences in ERH values in the range of 10–90%. Values above 90% indicate that the system is close to water-saturation (i.e. ERH 100%) but differences are not informative.
Biocatalyzed transamination in toluene
To perform the reaction 10 mg of immobilized His6-ATA-117 on the EziG1-Opal carrier were used with 1 mL reaction mixture in an orbital shaker at 35 °C, 150 rpm. The aw of the reaction systems was tuned by using the toluene either water saturated or dry. Moreover, the hydration of the biocatalyst EziG1-Opal–His6-ATA-117 was tuned by modifying slightly the rinsing step: i) in one case the biocatalyst was used after the pretreatment as described in Table 1 entry 5 (washing with iPrOH and dry toluene and then dried); ii) in the second case the drying step was omitted; iii) a third biocatalyst sample was rinsed with iPrOH and water-saturated toluene, without any drying step, in order to maintain a higher hydration level.The reaction mixtures were prepared by dissolving isopropyl amine (250 mM) and 1-phenoxypropan-2-one (100 mM) in the selected medium (either water-saturated toluene or dry toluene). The reaction was carried out for 24 hours and monitored by GC chromatography by analyzing 40 μL withdrawals of the reaction mixture diluted with 460 μL of ethyl acetate containing 40 mM dodecane as an internal standard. The progress of the transamination was followed by evaluating the formation of the amine product (1-phenoxypropan-2-amine), the disappearance of the ketone substrates and also the presence of N-isopropyl-1-phenoxypropan-2-imine derived from the spontaneous reaction between isopropyl amine and the ketone. The product 1-phenoxypropan-2-amine was detected as a racemate since the priority of the current work was the control of the activity of the enzyme under different experimental conditions, whereas the enantioselectivity was out of the scope of the investigation. The quantitative analysis of the chemical species was carried out on a SHIMADZU GC Chromatograph GC-2010 Plus with an AOC-20i autosampler equipped with a Supelco SLB™-5 ms 30 m × 0.25 mm × 0.5 μm fused silica capillary column, using the following conditions: oven temperature: 120–200 °C (5 °C min−1) and 200–295 °C (10 °C min−1), injector temperature 295 °C, carrier gas (helium) flow 1.0 mL min−1. Retention time: 1-phenoxypropan-2-one 8.17 min, 1-phenoxypropan-2-amine 9.09 min, N-isopropyl-1-phenoxypropan-2-imine 12.17 min. Mass spectra were obtained from a SHIMADZU GCMS-QP2010 SE Gas Chromatograph Mass Spectrometer (mass range 33–700 Daltons). The method will be referred to as GC–MS. No other product was observed in the reaction mixtures.
FITC labelling
The coupling reaction of ω-TA with FITC was started by the dropwise addition of FITC solution (10 mgFITC mLdimethylformamide−1) into the ω-TA solution (10 mg mL−1 in sodium phosphate buffer 0.1 M, pH 8). The mixture was incubated for 1 h at 25 °C. The protocol previously used by Todea et al.59 is based on the PIERCE EZ-Label TM FITC labeling kit. The amount of FITC required for the reaction depends on the amount of protein to be labeled.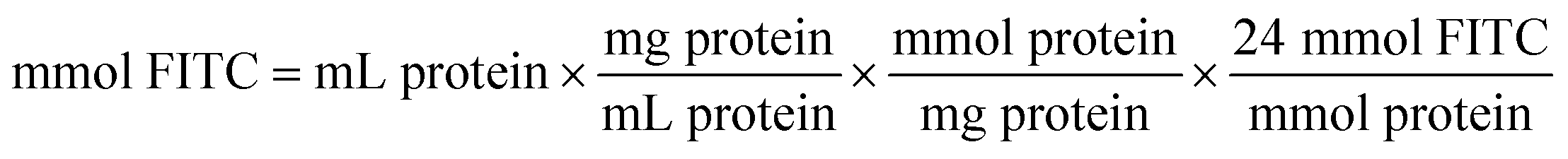 | (4) |
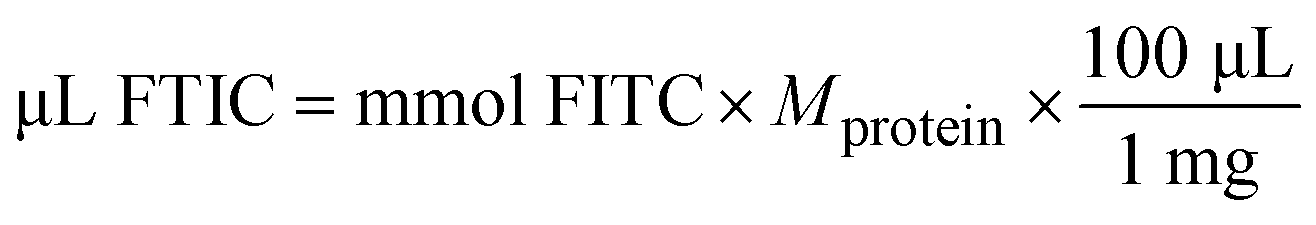 | (5) |
24 = recommended molar ratio of FITC to protein
100 = μL of solvent in which 1 mg FITC is dissolved
The labeled ω-TA was separated from unreacted FITC by several washes with sodium phosphate buffer (0.1 M, pH 8) using a centrifugal filter (50 KDa cutoff). UV-vis spectra were collected after each washing step, until the absorbance at 490 nm (characteristic value for FITC) reached 0.1 absorbance unit. To determine the protein concentration a Bradford assay (A595) was performed.
Confocal laser scanning microscopy (CLSM) imaging
The localization and distribution of the FITC-labelled ω-TA immobilized on the EziG carriers were visualized by means of a Nikon Eclipse TE2000-E Inverted Fluorescence Microscope. Confocal imaging was performed using 20× and 40× objectives and the resulting micrographs were analyzed by means of FIJI software.60 From the collected confocal images, it was possible to obtain an average and normalized fluorescence radius using the FIJI plugin for the generation of the radial profile. A Gaussian fit was applied, after which the fitted data point corresponding to 50% of the maximum normalized fluorescence fitted peak (yFWHM) was searched. FWHM (full width half maximum), the infiltration distance of the enzyme in the inner surface of the analyzed beads, was calculated by subtracting the coordinates corresponding to the FWHM radius (xFWHM) from the particle radius (R).FTIR microspectroscopy and imaging
FTIR analysis was carried out at the Chemical and Life Sciences branch of the SISSI beamline at Elettra Sincrotrone Trieste.61,62 Measurements were performed with a Bruker Hyperion 3000 microscope (Bruker Optics, Ettlingen, Germany) coupled to a Bruker Vertex 70v interferometer equipped with both a single-point liquid-nitrogen-cooled MCT (Mercury Cadmium Telluride) detector for FTIR microspectroscopy and a bidimensional nitrogen cooled Focal Plane Array detector (FPA) for FTIR imaging. Samples were placed on a CaF2 window (13 mm diameter × 1 mm thickness) transparent to both visible and infrared light. The carrier surface was brought into focus prior to the collection of the sample spectrum. A reference spectrum was collected in a clean spot near the sample. For microspectroscopy analysis, 2048 scans were averaged with a spectral resolution of 4 cm−1 in the 3800–750 cm−1 spectral range with a lateral resolution of 50 μm × 50 μm aperture to define the precise area of analysis. The samples analyzed with this technique were the EziG carriers with and without immobilized protein (washed with different solutions as described in the immobilization section) and the pure His6-ATA-117 enzyme suspended in sodium phosphate buffer (50 mM, pH = 7.0 supplemented with 5 mM pyridoxal-5′-phosphate) was left to dry to acquire the spectrum. On these same types of samples, FTIR imaging was performed with the FPA detector by acquiring 512 scans with a spectral resolution of 4 cm−1 in the spectral range from 4000–900 cm−1.FTIR data analysis
All the collected spectra were pre-processed with Bruker software OPUS 7.5 and Quasar 1.6.0 Miniconda software.63,64 Pre-processing consisted of atmospheric compensation, rubber band baseline correction in the 3800–800 cm−1 spectral range and cut in the regions of interest (ROIs), such as: C–H stretching region, 3080–2800 cm−1 characteristic for symmetric and asymmetric stretching of methyl and methylene groups (diagnostic for carrier functionalization) and 1700–1500 cm−1 characteristic for Amide I and Amide II peaks (diagnostic for the enzyme). Second derivatives of the spectra were computed by applying the Savitzky–Golay algorithm using the third order polynomial and 13 points of smoothing. The integral from baseline of Amide I and Amide II at 1710–1485 cm−1 and for the glass carrier at 1500–1300 cm−1 was calculated and rationed. In Fig. 4 just for clarity reasons, the empty space was removed by using a basic Hierarchical Cluster Analysis (HCA) that identified the empty space from the carrier.Conclusions
The integration of different experimental studies provided information necessary to develop rational protocols for preserving the activity of an ω-transaminase immobilized on EziG™ carriers when applied in hydrophobic organic solvent (toluene). Infrared microspectroscopy and imaging, coupled with confocal microscopy, not only allowed the direct observation of the distribution of the immobilized transaminase on the carriers but also disclosed a specific conformational change in the β-sheet domain of the immobilized transaminase as a consequence of the exposure to toluene. Unexpectedly, the conformational change resulted in activity increase for the enzyme immobilized on the most hydrophilic EziG™ carrier. When such formulation was applied in toluene at different aw values the best reaction conditions were characterized with aw values close to water saturation, which ensured the shift of the thermodynamic equilibrium between the amine donor and its imine in favor of the first species. Overall, careful preservation of the hydration of the biocatalyst during the whole post-immobilization procedures appears crucial for the enzymatic activity and simple preparation protocols are reported herein.The behavior of EziG1-Opal–His6-ATA-117 when exposed to toluene provides new hints for future investigations of the conformational behavior of transaminases in non-aqueous media by means of spectroscopic analysis and molecular dynamics simulations. As a matter of fact, the structural analysis of the enzyme highlighted various hydrophobic regions on its surface. Conformational rearrangements cannot be excluded when the protein is exposed to more hydrophobic environments, as a consequence the differences in solvation of the enzyme surface and the chemical nature of the carrier might play an active role in such phenomena.
Author contributions
All the authors planned the experiments and I. I. R. performed the experimental work. I. I. R. together with D. E. B. and L. V. performed the IR data analysis and wrote the relative discussion. All authors contributed with manuscript revisions and discussed the data prior to submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 860414, “INTERfaces” project. We acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities (proposal number: 20210308) for using the beamline “Chemical and Life Sciences branchline (SISSI-Bio)”. The authors also wish to thank Prof. Enrico Tongiorgi and Dr. Gabriele Baj from University of Trieste, Department of Life Sciences for the help with the confocal microscopy analysis.Notes and references
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- R. di Cosimo, J. Mc Auliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- K. E. Cassimjee, M. S. Humble, V. Miceli, C. G. Colomina and P. Berglund, ACS Catal., 2011, 1, 1051–1055 CrossRef CAS.
- K. E. Cassimjee, B. Manta and F. Himo, Org. Biomol. Chem., 2015, 13, 8453–8465 RSC.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- M. S. Malik, E. S. Park and J. S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS PubMed.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS PubMed.
- W. Böhmer, A. Volkov, K. E. Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef PubMed.
- M. D. Truppo, J. D. Rozzell and N. J. Turner, Org. Process Res. Dev., 2010, 14, 234–237 CrossRef CAS.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- V. Ferrario, C. Ebert, L. Knapic, D. Fattor, A. Basso, P. Spizzo and L. Gardossi, Adv. Synth. Catal., 2011, 353, 2466–2480 CrossRef CAS.
- L. de Martin, C. Ebert, L. Gardossi and P. Linda, Tetrahedron Lett., 2001, 42, 3395–3397 CrossRef CAS.
- J. Santiago-Arcos, S. Velasco-Lozano, E. Diamanti, A. L. Cortajarena and F. López-Gallego, Front. Catal., 2021, 1, 715075 CrossRef.
- K. E. Cassimjee, M. Kadow, Y. Wikmark, M. S. Humble, M. L. Rothstein, D. M. Rothstein and J. E. Bäckvall, Chem. Commun., 2014, 50, 9134–9137 RSC.
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS.
- M. Aßmann, C. Mügge, S. K. Gaßmeyer, J. Enoki, L. Hilterhaus, R. Kourist, A. Liese and S. Kara, Front. Microbiol., 2017, 8, 448 Search PubMed.
- K. E. Cassimjee, P. Hendil-Forssell, A. Volkov, A. Krog, J. Malmo, T. E. V. Aune, W. Knecht, I. R. Miskelly, T. S. Moody and M. S. Humble, ACS Omega, 2017, 2, 8674–8677 CrossRef CAS PubMed.
- H. Lechner, P. Soriano, R. Poschner, H. C. Hailes, J. M. Ward and W. Kroutil, Biotechnol. J., 2018, 13, 1700542 CrossRef PubMed.
- W. Böhmer, T. Knaus and F. G. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed.
- J. Coloma, T. Lugtenburg, M. Afendi, M. Lazzarotto, P. Bracco, P. L. Hagedoorn, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 1–14 CrossRef.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- M. Sezer, P. Kielb, U. Kuhlmann, H. Mohrmann, C. Schulz, D. Heinrich, R. Schlesinger, J. Heberle and I. M. Weidinger, J. Phys. Chem. B, 2015, 119, 9586–9591 CrossRef CAS PubMed.
- N. Kuhar, S. Sil, T. Verma and S. Umapathy, RSC Adv., 2018, 8, 25888–25908 RSC.
- K. Eberhardt, C. Stiebing, C. Matthaüs, M. Schmitt and J. Popp, Expert Rev. Mol. Diagn., 2015, 15, 773–787 CrossRef CAS PubMed.
- Z. M. Zhang, S. Chen, Y. Z. Liang, Z. X. Liu, Q. M. Zhang, L. X. Ding, F. Ye and H. Zhou, J. Raman Spectrosc., 2010, 41, 659–669 CrossRef CAS.
- A. Deep, U. Tiwari, P. Kumar, V. Mishra, S. C. Jain, N. Singh, P. Kapur and L. M. Bharadwaj, Biosens. Bioelectron., 2012, 33, 190–195 CrossRef CAS PubMed.
- S. F. Prazeres, F. Zapata, N. Canilho, A. Pasc, C. García-Ruiz and G. Montalvo, Microporous Mesoporous Mater., 2019, 278, 149–155 CrossRef CAS.
- Y. Mei, L. Miller, W. Gao and R. A. Gross, Biomacromolecules, 2003, 4, 70–74 CrossRef CAS PubMed.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- K. E. Cassimjee and J.-E. Bäckvall, Immobilized proteins and use thereof, US20210139881A1, 2021.
- K. A. Majorek, M. L. Kuhn, M. Chruszcz, W. F. Anderson and W. Minor, Protein Sci., 2014, 23, 1359–1368 CrossRef CAS PubMed.
- A. Basso, P. Braiuca, S. Cantone, C. Ebert, P. Linda, P. Spizzo, P. Caimi, U. Hanefeld, G. Degrassi and L. Gardossi, Adv. Synth. Catal., 2007, 349, 877–886 CrossRef CAS.
- W. Böhmer, T. Knaus, A. Volkov, T. K. Slot, N. R. Shiju, K. E. Cassimjee and F. G. Mutti, J. Biotechnol., 2019, 291, 52–60 CrossRef PubMed.
- A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 8017–8021 CrossRef CAS PubMed.
- A. Sellami-Kamoun, A. Haddar, N. E. H. Ali, B. Ghorbel-Frikha, S. Kanoun and M. Nasri, Microbiol. Res., 2008, 163, 299–306 CrossRef CAS PubMed.
- L. M. Miller, M. W. Bourassa and R. J. Smith, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2339–2346 CrossRef CAS PubMed.
- H. Yun, Y.-H. Yang, B.-K. Cho, B.-Y. Hwang and B.-G. Kim, Simultaneous synthesis of enantiomerically pure (R)-1-phenylethanol and (R)-α-methylbenzylamine from racemic α-methylbenzylamine using ω-transaminase/alcohol dehydrogenase/glucose dehydrogenase coupling reaction, Biotechnol. Lett., 2003, 25, 809–814 CrossRef CAS PubMed.
- S. G. Allen, P. C. L. Stephenson and J. H. Strange, J. Chem. Phys., 1998, 108, 8195–8198 CrossRef CAS.
- T. Takamuku, M. Yamagami, H. Wakita, Y. Masuda and T. Yamaguchi, J. Phys. Chem. A, 1997, 101, 5730–5739 CrossRef CAS.
- A. Basso, L. de Martin, C. Ebert, L. Gardossi and P. Linda, J. Mol. Catal. B: Enzym., 2000, 8, 245–253 CrossRef CAS.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed.
- P. I. Haris and F. Severcan, J. Mol. Catal. B: Enzym., 1999, 7, 207–221 CrossRef CAS.
- L. M. Miller, M. W. Bourassa and R. J. Smith, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2339–2346 CrossRef CAS PubMed.
- M. Maglione, L. Vaccari, L. Mancini, R. Ciancio, D. Bedolla, L. Bevilacqua and P. Tonellato, Int. J. Oral Maxillofac. Implants, 2019, 34, 631–641 CrossRef PubMed.
- D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469–487 CrossRef CAS PubMed.
- E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biophys. J., 2006, 90, 2946–2957 CrossRef CAS PubMed.
- A. Basso, L. de Martin, C. Ebert, L. Gardossi, P. Linda and V. Zlatev, J. Mol. Catal. B: Enzym., 2001, 11, 851–855 CrossRef CAS.
- E. N. Vulfson, P. J. Halling and H. L. Holland, Enzymes in Nonaqueous Solvents, 2001 Search PubMed.
- R. Lortie, Biotechnol. Adv., 1997, 15, 1–15 CrossRef CAS PubMed.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 2015, 6965–6982 CrossRef CAS PubMed.
- F. G. Mutti and W. Kroutil, Adv. Synth. Catal., 2012, 354, 3409–3413 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 303–305 CrossRef PubMed.
- K. E. Cassimjee, M. S. Humble, H. Land, V. Abedi and P. Berglund, Org. Biomol. Chem., 2012, 10, 5466–5470 RSC.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81, 8244–8248 CrossRef PubMed.
- G. V. Barbosa-Cánovas, Water activity in foods : fundamentals and applications, 2nd edn, 2020, vol. 2 Search PubMed.
- A. Todea, A. Hiseni, L. G. Otten, I. W. C. E. Arends, F. Peter and C. G. Boeriu, J. Mol. Catal. B: Enzym., 2015, 119, 40–47 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- G. Birarda, D. Bedolla, F. Piccirilli, C. Stani, H. Vondracek and L. Vaccari, in Conference Paper, SPIE-Intl Soc Optical Eng, 2022, p. 31 Search PubMed.
- D. J. Nagle, G. A. George, L. Rintoul and P. M. Fredericks, Vib. Spectrosc., 2010, 53, 24–27 CrossRef CAS.
- M. Toplak, G. Birarda, S. Read, C. Sandt, S. M. Rosendahl, L. Vaccari, J. Demšar and F. Borondics, Synchrotron Radiat. News, 2017, 30, 40–45 CrossRef.
- M. Toplak, S. T. Read, C. Sandt and F. Borondics, Cells, 2021, 10, 2300 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01949c |
| This journal is © The Royal Society of Chemistry 2023 |