
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, performance evaluation, and economic assessment of tailored Pt/TiO2 catalysts for selective biomass vapour upgrading via a scalable flame spray pyrolysis route†
Anurag
Kumar
a,
Rongyue
Wang
b,
Kellene A.
Orton
a,
Kurt M.
Van Allsburg
 a,
Calvin
Mukarakate
a,
Evan C.
Wegener
c,
Qiyuan
Wu
a,
Susan E.
Habas
a,
Krzystof Z.
Pupek
b,
Joseph A.
Libera
b and
Frederick G.
Baddour
*a
a,
Calvin
Mukarakate
a,
Evan C.
Wegener
c,
Qiyuan
Wu
a,
Susan E.
Habas
a,
Krzystof Z.
Pupek
b,
Joseph A.
Libera
b and
Frederick G.
Baddour
*a
aCatalytic Carbon Transformation and Scale-up Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401-3305, USA. E-mail: Frederick.Baddour@nrel.gov
bEnergy Systems Division, Argonne National Laboratory, Argonne, Illinois 60439, USA
cChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL, 60439 USA
First published on 18th July 2023
Abstract
Commercialization of next-generation nanostructured catalysts for biomass conversion is largely limited by the unique challenges associated with their large scale manufacture. In this work, we present the scalable, continuous synthesis of nanostructured Pt/TiO2 catalysts in a single step via flame-spray pyrolysis (FSP) and demonstrate their utility in the upgrading of biomass vapours via catalytic fast pyrolysis (CFP). Spectroscopic and microscopic characterization of the catalysts prepared via FSP and conventional incipient wetness impregnation (IW) demonstrated that the FSP method enabled scalable production of Pt/TiO2 with distinct Pt speciation (single atoms, clusters, and nanoparticles) that could be tuned by varying the Pt loading. This compositional control by FSP resulted in catalytic activity for upgrading of pine pyrolysis vapours to aromatics, oxygenates, and alkenes that was comparable to the IW analogues' performance, with the addition of more control over the product slate by modifying the FSP synthesis conditions. Specifically, the FSP-synthesized catalysts exhibited decreased hydrogenation performance and greater selectivity towards partially deoxygenated products, aromatics, (cyclo)alkanes, and (cyclo)alkenes with total hydrocarbon yields similar to IW-synthesized catalysts at equivalent Pt loadings. A preliminary cost analysis comparing the FSP and IW processes identified the major cost drivers, including solvent usage, process gas requirements, and Pt utilization, that can be addressed to improve the economic viability of the FSP synthesis developed herein.
Introduction
Catalytic upgrading of biomass to drop-in liquid fuels represents a significant research challenge. In particular, it requires the removal of oxygen from a complex mixture of oxygenated molecules (e.g., phenols, alcohols, syringols, ketones, and aldehydes) due to the detrimental effects of oxygenated species on fuel properties such as a low heating value, high acidity, and chemical instability.1–3Ex situ catalytic fast pyrolysis (CFP) is a promising conversion technology wherein the vapours of pyrolyzed biomass are passed over a catalyst at high temperature and near-ambient pressure (450 °C, 1 atm) to generate a stabilized bio-oil with lower oxygen content that is more suitable for down-stream upgrading to hydrocarbon fuels or high value chemicals. However, techno-economic analyses of this process have revealed the need for further development of next-generation catalysts if the goal of sustainable, low-cost CFP-derived biofuels is to be realized.3 Notably, several key catalytic properties have been identified, including the importance of bifunctionality (i.e., combination of acidic and metallic sites), such that H2 can be activated at the low-pressure-high-temperature conditions of CFP with selectivity that favours cleavage of C–O bonds over C–C bonds.1Bifunctional catalysts possessing metallic and acidic sites such as noble metals (e.g., Pt, Pd, Ru)3–7 supported on acidic reducible oxide supports (e.g., TiO2, ZrO2) have been identified as effective catalysts for biomass pyrolysis vapour upgrading at lab-scale because of their ability to activate hydrogen and perform deoxygenation reactions.3–7 Traditionally, these catalysts are synthesized by incipient wetness impregnation (IW), co-precipitation, sol–gel, or hydrothermal methods for their ease of preparation.8–10 However, recent work has highlighted the performance enhancements that are attainable in the catalytic upgrading of bio-derived feedstocks by employing advanced synthetic methods such as solution phase synthesis11,12 and strong electrostatic adsorption3,13,14 that afford more control over catalyst physical properties (e.g., particle size, dispersion, acid-site strength, and density).15,16 However, the realization and deployment of any biomass conversion technology requires not only tailored high-performance catalysts, but methods to sustainably produce them at industrial scales. A commercial biorefinery for the conversion of biomass via CFP sized to compete with incumbent fuel production technologies would require catalyst loads of 50 tons or more, which poses a significant scaling challenge for the production of tailored catalysts by advanced synthetic methods.17
There are few chemical manufacturing techniques deployed at commercial scales that are amenable to the production of catalysts with targeted physical properties at quantities suitable for the needs of a conceptual biorefinery. For example, high surface area catalyst supports exhibiting the aforementioned physical and chemical properties are commonly produced using flame synthesis methods such as vapour-fed aerosol flame synthesis, in which precursor vapours are continuously combusted in a controlled stream. The versatility18–20 and scalability21–23 of this process is highlighted by its industrial maturity, with flame-synthesized materials manufactured on the scale of millions of tons per year to produce a wide variety of engineered materials including activated carbons and fumed metal oxides (e.g., SiO2, TiO2, Al2O3). A particularly promising method for advanced catalyst synthesis is flame spray pyrolysis (FSP), in which a continuously fed solution of metal or metal oxide precursor(s) in organic solvent is combusted to form nanoparticles (NPs) from the aerosolized precursors. This methodology has been adapted to prepare multi-component heterogeneous catalysts in a single step (i.e., metal active phase and a metal oxide support) by co-feeding a judicious choice of metal and metal oxide precursor solutions,24–26 including single-atom and nanostructured Pt on TiO2.27,28 This approach offers substantial benefits for green synthesis of catalytic materials, as it combines two conventional steps—flame synthesis of the catalyst support and loading of an active phase—into a single FSP step. FSP is particularly promising in the application of catalyst synthesis because it is a continuous, readily scalable process that can significantly decrease the number of processing steps involved in catalyst manufacturing compared to traditional solution-based routes (Scheme 1). Furthermore, it offers precise control over catalytically relevant physical properties (e.g., spatial distribution of active components) that may not be as readily controlled through traditional synthetic methods.24 However, despite the maturity of flame synthesis technologies and the development of “green manufacturing” technologies for NPs afforded by FSP, the viability and potential for widespread market adoption of this technology for catalytic applications is uncertain without a detailed economic assessment.
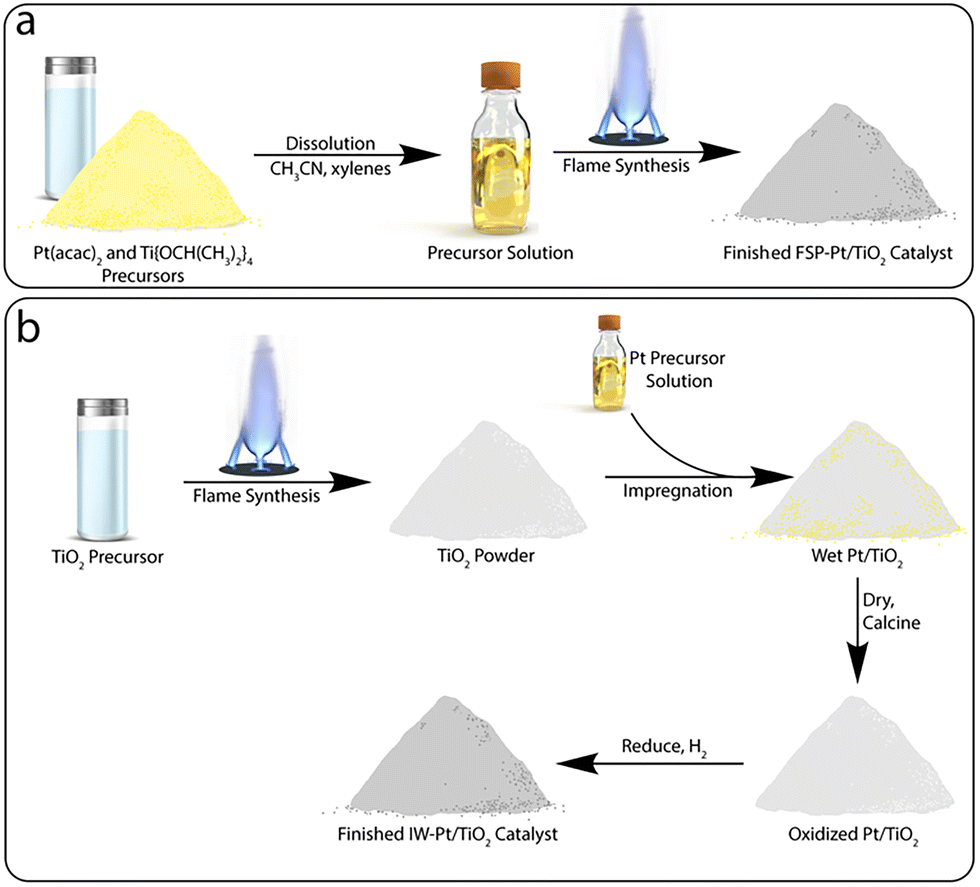 | ||
| Scheme 1 Simplified comparison of the synthetic steps involved in the preparation of a Pt/TiO2 catalyst by (a) flame spray pyrolysis and (b) a traditional incipient wetness impregnation approach. | ||
Technoeconomic evaluation of new synthesis pathways is an effective approach to assess catalyst viability at industrial scales and identify barriers to commercialization.3,29 To proliferate a new method offering reduced environmental impact compared to traditional wet chemistries,30,31 it is crucial to demonstrate that the method can achieve cost parity with incumbent methods via a preliminary techno-economic analysis. Moreover, cost assessment allows the quantification of benefits and cost reduction opportunities for novel synthesis approaches, such as the FSP route described herein. We employed the CatCost catalyst cost estimation tool32,33 to better understand the holistic impact of the FSP synthesis method.
Herein, we report the continuous synthesis of a family of bifunctional Pt/TiO2 catalysts in a single step via FSP and evaluation of their performance for the upgrading of biomass pyrolysis vapours in ex situ CFP. Catalysts synthesized by this method were found to exhibit higher selectivity towards aromatics, partially deoxygenated compounds, and alkenes compared to traditionally prepared IW-synthesized materials, which primarily yielded alkanes. Moreover, FSP-synthesized catalysts resulted in similar or higher total yields of carbon products when compared to IW-synthesized materials at comparable Pt loadings, indicating better utilization of the expensive noble metal. Detailed microscopy and spectroscopic characterization of the FSP-synthesized catalysts revealed notable differences in Pt speciation and dispersion compared to IW-based catalysts which we hypothesize to be responsible for the differences in observed selectivity and deactivation behaviour. Facile control over the relative speciation of single Pt atoms, clusters, and NPs as a function of FSP reaction conditions, and the impact of these features on the catalytic performance, highlight the ability of this synthetic method to tune for a desirable product slate. Finally, we performed a cost assessment to compare the FSP synthesis process with a conventional IW method, providing an economic outlook on the FSP synthesis method and areas in which further research is needed to lower cost and improve the commercial viability of this process.
Experimental section
Materials
Titanium tetraisopropoxide (TTiP, 98%, ACROS Organics), and platinum(II) acetylacetonate (Pt(acac)2, 97%, Sigma-Aldrich) were used as received without further purification. Titania support (Aeroxide P25) used in the IW impregnation synthesis was supplied by Evonik and used as received. All gases used in the synthesis and catalytic evaluation were ultra-high purity (UHP) unless otherwise noted. Loblolly pine was supplied by Idaho National Laboratory and elemental analyses showed that the feed contained 49.3 wt% carbon, 6.1 wt% hydrogen, 0.1 wt% nitrogen, and 44.5 wt% oxygen (by difference) on a dry biomass basis.Catalyst synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28:52. Platinum acetylacetonate (Pt(acac)2) was dissolved in 300 mL batches of this mixture at concentrations of 0.30, 0.75, and 1.51 mM targeting nominal Pt loadings of 0.10, 0.25, and 0.50 wt%. Actual Pt loadings determined by inductively coupled-optical emission spectroscopy (ICP-OES) were found to be 0.07, 0.14, 0.33 wt%, respectively, as shown in Table 1 and denoted as FSP-Pt(x)/TiO2 (x = 0.07, 0.14, and 0.33). A neat mixture of the TTiP, acetonitrile, and xylene was prepared to give the control FSP titania (FSP–TiO2). The prepared solutions were combusted in an enclosed FSP system using an array of six premixed methane–oxygen pilot flames arranged circumferentially around the spray nozzle and oriented to converge on the flame axis 35 mm from the tip of the spray nozzle, as shown in Fig. S1.† The pilot flames were operated continuously to ignite and support the combustion. Oxygen, used as a carrier gas flowing at 10 LPM, was used to atomize the liquid feed and provide a portion of the combustion oxidizer. An additional 25 LPM of oxygen was provided by a sheath flow located circumferential to the atomizing nozzle (Fig. S1†). The pilot methane and oxygen flows were 2 and 4 LPM respectively and the liquid feed was 8 mL min−1. Combustion was quenched with N2 gas that was fed circumferentially to the overall burner at 50 LPM. Finally, an additional 50 LPM of N2 quench gas was added to the combustion exhaust flow prior to collecting the produced Pt/TiO2 powder on a glass paper filter pad.
:28:52. Platinum acetylacetonate (Pt(acac)2) was dissolved in 300 mL batches of this mixture at concentrations of 0.30, 0.75, and 1.51 mM targeting nominal Pt loadings of 0.10, 0.25, and 0.50 wt%. Actual Pt loadings determined by inductively coupled-optical emission spectroscopy (ICP-OES) were found to be 0.07, 0.14, 0.33 wt%, respectively, as shown in Table 1 and denoted as FSP-Pt(x)/TiO2 (x = 0.07, 0.14, and 0.33). A neat mixture of the TTiP, acetonitrile, and xylene was prepared to give the control FSP titania (FSP–TiO2). The prepared solutions were combusted in an enclosed FSP system using an array of six premixed methane–oxygen pilot flames arranged circumferentially around the spray nozzle and oriented to converge on the flame axis 35 mm from the tip of the spray nozzle, as shown in Fig. S1.† The pilot flames were operated continuously to ignite and support the combustion. Oxygen, used as a carrier gas flowing at 10 LPM, was used to atomize the liquid feed and provide a portion of the combustion oxidizer. An additional 25 LPM of oxygen was provided by a sheath flow located circumferential to the atomizing nozzle (Fig. S1†). The pilot methane and oxygen flows were 2 and 4 LPM respectively and the liquid feed was 8 mL min−1. Combustion was quenched with N2 gas that was fed circumferentially to the overall burner at 50 LPM. Finally, an additional 50 LPM of N2 quench gas was added to the combustion exhaust flow prior to collecting the produced Pt/TiO2 powder on a glass paper filter pad.
| Catalyst | Pt loading (wt%) | Surface area (m2 g−1) | Acid-site density (μmol g−1) | Acid-site density (μmol m−2) |
|---|---|---|---|---|
| FSP–TiO2 | — | 49 | 377 | 7.7 |
| P25–TiO2 | — | 56 | 457 | 8.2 |
| FSP-Pt(0.07)/TiO2 | 0.07 | 42 | 297 | 7.1 |
| FSP-Pt(0.14)/TiO2 | 0.14 | 40 | 278 | 7.0 |
| FSP-Pt(0.33/TiO2 | 0.33 | 41 | 266 | 6.5 |
| IW-Pt(0.07)/TiO2 | 0.07 | 53 | 377 | 7.1 |
| IW-Pt(0.14)/TiO2 | 0.14 | 52 | 369 | 7.1 |
| IW-Pt(0.27)/TiO2 | 0.27 | 56 | 387 | 6.9 |
Catalyst characterization
Platinum metal content of the prepared materials was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) performed by Galbraith Laboratories (Knoxville, TN).562.76 eV) was collected simultaneously with each measurement for energy calibration. Spectra of the fresh catalysts were collected at room temperature in air. Reductive pretreatments (450 °C in 50 sccm 100% H2 for 0.5 h) were performed using a custom built in situ reactor.34 Following reduction, the samples were cooled to room temperature and spectra were collected under flowing H2.
XAS data processing and analysis were performed using the Demeter software suite.35 Edge energies were determined from the position of maximum intensity of the derivative of X-ray absorption near edge structure (XANES) spectra. X-ray absorption fine structure (EXAFS) coordination parameters were determined from simultaneous fits in R-space of the k1, k2, and k3 weighted Fourier transformed EXAFS using theoretical phase shift and backscattering amplitudes calculated from FEFF.36 Pt–O and Pt–Pt scattering were modeled from the structures of PtO2 and FCC Pt, respectively. The amplitude reduction factor for Pt was determined to be 0.86 from a fit of the metal foil and this value was held fixed in fits of the samples to determine coordination numbers.
Catalytic conversion of biomass pyrolysis vapours
Catalytic evaluation of the prepared Pt/TiO2 catalysts was performed in a tandem micro-furnace pyrolyzer (PY-2020iS, Frontier Laboratories, Japan) coupled with a gas chromatograph-mass spectrometer/flame ionization detector (GC-MS/FID). Pyrolysis of pine particles (at 500 °C and 1 atm under inert flow) and subsequent catalytic upgrading of pyrolysis vapours (at 400 °C and 1 atm in 50% H2/50% He) over the Pt/TiO2 catalysts was performed using a tandem micropyrolyzer-GC-MS system.37,38 A detailed description of its operation has been previously reported.39–41 The micropyrolyzer apparatus consists of a pyrolysis zone for generating biomass vapours coupled to a downstream zone for catalytic upgrading. Condensable products were collected in a liquid nitrogen trap downstream of the reactor where they were subsequently vapourised into the inlet of the GC-MS/FID during GC oven heat up. Samples of biomass (ca. 500 μg each) in deactivated stainless-steel cups were dropped automatically in intervals of 30 min into the preheated pyrolysis zone operating at 500 °C. The products from the pyrolysis zone were entrained in 54 mL min−1 of He carrier gas and passed over a fixed bed of 10 mg catalyst. For experiments conducted in reducing gas atmospheres, 54 mL min−1 of H2 was added prior to the fixed-bed reactor for a total flow of 108 mL min−1. The total pressure was 115 kPa. For each experiment, three successive cups of biomass were passed over the same catalyst bed for a total mass ratio of biomass fed to catalyst of 0.15 g g−1. For deactivation experiments, a biomass:catalyst weight ratio (B:C) of 1.0 was reached by successive injection of biomass cups. The catalysts were pretreated for 60 min in a mixture of 50% He/50% H2. The temperature of the catalysis zone was maintained at 400 °C. The condensable vapours produced during pyrolysis and upgrading were adsorbed on the liquid nitrogen trap, but light gases such as CH4 and CO passed through the trap and were separated in a GS-GasPro column and measured by a TCD. The condensable vapours were desorbed from the liquid nitrogen trap and separated in an Ultra-Alloy-5 capillary column, which utilized a stationary phase consisting of 5% phenyl and 95% dimethyl polysiloxane. The GC oven was programmed to hold at 40 °C for 4.5 min, then ramp to 300 °C at a rate of 20 °C min−1. The separated upgraded species were identified using the NIST GC-MS library and quantified based on calibration of twenty compounds representative of upgraded pyrolysis vapours. The CFP product carbon yields were calculated using eqn (1).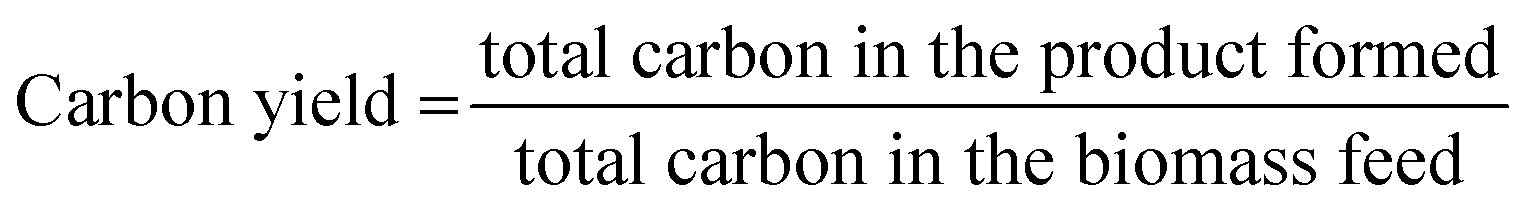 | (1) |
Economic assessment
Cost estimates were prepared in Microsoft Excel v16 using the spreadsheet version of CatCost v1.0.4. Further discussion of the CatCost methods and assumptions has been previously published.32 All prices were adjusted to 2016 USD using the U.S. Bureau of Labor Statistics Chemical Producer Price Index or, for equipment costs, the Chemical Engineering Plant Cost Index.Raw materials prices at ≥1000 kg scale were estimated through a combination of vendor quotations, freely available and proprietary price databases, and estimates from industry experts. Generally, several sources were consulted to develop an average and/or verify each assumed price. A factor of 3% was added to the raw materials costs to account for waste and spoilage.
Processing costs were estimated using the CapEx & OpEx Factors method (catalyst manufacture at a dedicated new-build plant).32 A production scale of 100 kg h−1 (788000 kg per year accounting for downtime) was used for all scenarios. Equipment was scaled from a base design automatically to the specified production scale by increasing the number of equipment items when the upper bound of the validity of that cost correlation was exceeded; similarly, when the equipment was required at less than the correlation's minimum size, the cost of the minimum size was used. Installation costs were estimated using the equipment-specific installation factors of Garrett,42 and labor factors (number of operators required per shift for that equipment item) were taken from Ulrich and Vasudevan.43 In both cases, the average value was used, or the midpoint if only a range was provided. The remainder of the capital costs—including direct capital costs like installation, piping, instrumentation, and buildings; indirect costs like engineering, legal, and contingencies; and working capital—were estimated as fixed factors (multipliers) of the total purchased equipment cost using the modified Lang factors44,45 of Peters and Timmerhaus.46 A similar calculation approach and factors were taken from the same source46 to determine operating costs such as supervisory labor and maintenance supplies, fixed/indirect costs such as insurance and overhead, and general expenses such as distribution and marketing. Direct labor was calculated by summing the labor factors of all the equipment items after scaling to the specified production rate, and then rounding up to the nearest whole number to determine the number of operators. Year-round operation (8760 hours) with full staffing during maintenance downtime (10%) was assumed. A labor rate, including benefits, of $48 per hour for US Gulf Coast production was used.
Results and discussion
Catalyst characterization
The FSP approach investigated here involves combustion of an aerosolized continuous feed of a solvent mixture comprised of liquid precursors (of the active metal phase and oxide support). The metals (Ti and Pt) are atomized into the gas phase followed by condensation (nucleation and agglomeration) into nano-sized catalyst particles. The relative volatility, or conversely, the temperature at which the individual metals condense into solid particles, establishes an order of condensation which results in Pt tending to be located on the TiO2 surface due to the higher volatility of Pt metal vapour compared to Ti. In this work, we synthesized a series of Pt/TiO2 catalysts via the FSP method modified from previous reports.28 Briefly, a solution of Pt(acac)2 was prepared in a mixture of TTiP, acetonitrile, and xylene and combusted in a custom enclosed FSP system schematically presented in Fig. S1.†Catalysts prepared by FSP with weight loadings of 0.07, 0.14, and 0.33 wt% Pt were attained by controlling the concentration of the Pt precursor solution. Actual loadings are presented in Table 1, with catalysts denoted as FSP-Pt(x)/TiO2 (x = 0.07, 0.14, and 0.33) based on these loadings. A lower feed-rate was employed to prepare the 0.33 wt% Pt catalyst to prevent further Pt particle aggregation at the higher required concentration. To assess the impact of catalyst synthesis method on the observed catalyst properties and performance, catalysts with similar nominal loadings were prepared via traditional IW impregnation and are presented in Table 1, denoted as IW-Pt(x)/TiO2 (x = 0.07, 0.14, and 0.27). The physico-chemical properties of all the catalysts are summarized in Table 1. The BET surface area measurements via N2 physisorption studies reveal that all catalysts synthesized by FSP, including the Pt-free TiO2 (FSP–TiO2), exhibit a consistent surface area between 41–49 m2 g−1, which is comparable to that of the commercially available Aeroxide P25 TiO2 support (56 m2 g−1). Notably, the TiO2 morphologies are similar between P25 and FSP–TiO2 (as observed by TEM and described below) despite their syntheses utilizing different titania precursors (TiCl4 and TTiP, respectively). Therefore, this commercial material was chosen as the support to prepare the IW-Pt/TiO2 catalysts. Inclusion of the Pt precursor in the FSP synthesis resulted in a minor reduction in the measured surface area from 49 m2 g−1 (FSP–TiO2) to 40–42 m2 g−1 (FSP-Pt/TiO2), although no relationship was observed between Pt weight loading and surface area. In addition, all FSP-Pt/TiO2 catalysts were found to have pore sizes and pore volumes consistent with the parent FSP–TiO2 (Table S1 and Fig. S2†) suggesting that the addition of Pt to the FSP synthesis does not disrupt the formation of the FSP–TiO2 pore structure.
The acid site density, as measured by ammonia TPD (NH3-TPD) (profiles shown in Fig. S3†), of the FSP–TiO2 (377 μmol g−1) was found to be lower than that of the commercial IW–TiO2 (457 μmol g−1) on a per-gram-catalyst basis, which is consistent with the lower measured surface area of the FSP–TiO2. The acid site density normalized by the surface area for FSP–TiO2 (7.7 μmol m−2) was within the experimental measurement error when compared to P25–TiO2 (8.2 μmol m−2). Pt incorporation at different metal loadings reduced the acid site density for both FSP–TiO2 and IW–TiO2 by 20–30% as compared to the parent TiO2.
XRD analysis of the as-synthesized FSP-Pt/TiO2 catalysts revealed the presence of titania with a predominantly anatase structure that corresponds well with the ICDD PDF 01-070-7348, with a minor fraction adopting the rutile phase (ICDD PDF 01-070-7347) in all cases, as shown in Fig. 1. The average composition of anatase and rutile phases, as determined by single-peak reference intensity ratio analysis, of all materials prepared by FSP (83.8 ± 1.5% anatase) were slightly below that of the P25-derived IW materials (91.9 ± 0.5% anatase), as shown in Table S2.† Unsurprisingly, the addition of Pt to the TiO2 support via IW had no impact on the resulting distribution of phases as determined by XRD (Fig. S4†). However, at all weight loadings studied, the introduction of Pt to the FSP synthesis increased the amount of rutile present (83.7–85.2%) compared to the bare FSP–TiO2 (81.7%). This observation is consistent with previous literature reports,28,47 and has been attributed to structural similarity between PtO2 and rutile TiO2, whereby PtO2 seeds promote the formation of the rutile phase.48–51 In addition, the presence of surface Pt has been found to have a dehydroxylating effect on TiO2 at the high temperatures of the FSP process, which has also been shown to promote rutile formation.28 Scherrer analysis of the (101) diffraction peak at 25.3° 2θ for the anatase titania revealed crystallites with diameters (dXRD) in the range of 26–34 nm, with a trend toward increasing size observed with higher Pt loadings. This trend is in good agreement with previous reports that reveal an increasing dXRD over a range of low Pt loadings (0–1 atom%) and is consistent with the materials synthesized herein (0.03–0.14 atom% Pt).28 This effect of increased crystallite size as a function of Pt loading is mirrored by the rutile phase as determined by Scherrer analysis of the (110) diffraction peak at 27.4° 2θ where the crystallite size was observed to increase from 33 to 43 nm over the interrogated weight loadings. All crystallite sizes determined by XRD are presented in Table S2.† Unsurprisingly, no diffraction peaks corresponding to Pt metal were observed in any of the Pt/TiO2 catalysts because of the low Pt loadings and small particle size, as well as the high crystallinity of the support.
 | ||
| Fig. 1 XRD patterns of FSP-derived Pt/TiO2 and TiO2 with reference patterns provided below. | ||
The as-synthesized FSP-Pt/TiO2 and IW-Pt/TiO2 catalysts were imaged with high resolution TEM and HAADF-STEM to assess any morphological changes induced by either Pt weight loading or synthetic method. All FSP-Pt/TiO2 materials were comprised of aggregates of titania particles approximately 35 ± 15 nm in diameter which is consistent with the titania particle size determined by XRD. TEM images revealed that the titania aggregates of FSP-Pt/TiO2 (Fig. S5a–c†) have similar particle size and size distributions for both FSP–TiO2 (Fig. S5d†) and P25–TiO2 (Fig. S5e†). HAADF-STEM was employed to elucidate the size distribution of the Pt species present on the FSP–TiO2 catalysts. As expected, the Pt speciation and size distribution was found to be dependent on concentration of the Pt(acac)2 solution used during synthesis (i.e., Pt weight loading).52 Analysis of the HAADF-STEM images of the as-synthesized FSP-Pt(0.07)/TiO2 revealed only the presence of highly dispersed single atoms of Pt (Fig. 2a and g), with no sub-nm clusters or NPs of Pt observed. Exemplary single atoms are highlighted with yellow circles in Fig. 2a. Conversely, both FSP-Pt(0.14)/TiO2 and FSP-Pt(0.33)/TiO2 (Fig. 2b and c) were found to possess a mixture of single-atoms, sub-nm clusters, and NPs with size distributions presented in the histograms in Fig. 2h and i, respectively. To characterize the morphology of the Pt-species present under conditions more relevant to CFP, the as-synthesized FSP-Pt/TiO2 catalysts were subjected to a reductive thermal treatment (450 °C, 100% H2, 6 h) and imaged with HAADF-STEM. For all FSP-Pt/TiO2 catalysts there was an increase in the average particle sizes and shift in the particle size distributions (Fig. 2g–i). After reduction of the FSP-Pt(0.07)/TiO2 material, both small NPs and Pt clusters were observed with an average size of 1.1 ± 0.2 nm, as shown in Fig. 2d, whereas only atomically dispersed Pt could be identified in the as-synthesized material. Despite the appearance of larger particles, the predominant species present in the FSP-Pt(0.07)/TiO2 catalyst remained atomically dispersed Pt. The observed particle growth was more severe in the cases of FSP-Pt(0.14)/TiO2 and FSP-Pt(0.33)/TiO2 (Fig. 2e and f), where, after reduction the ratio of sub-nm clusters-to-NPs of Pt decreased from 2.5 to 0.2 and from 0.6 to 0.1, respectively. HAADF-STEM of the analogous samples prepared by IW revealed Pt particles with sizes of 0.9 ± 0.2 nm and 1.1 ± 0.2 nm for IW-Pt(0.07)/TiO2 and IW-Pt(0.14)/TiO2, respectively (Fig. S6†). The average Pt particle size of IW-Pt(0.27)/TiO2 was found to be slightly larger at 1.3 ± 0.5 nm due to a population of particles approximately 2 nm in size. All Pt and titania particles sizes are presented in Table S3.†
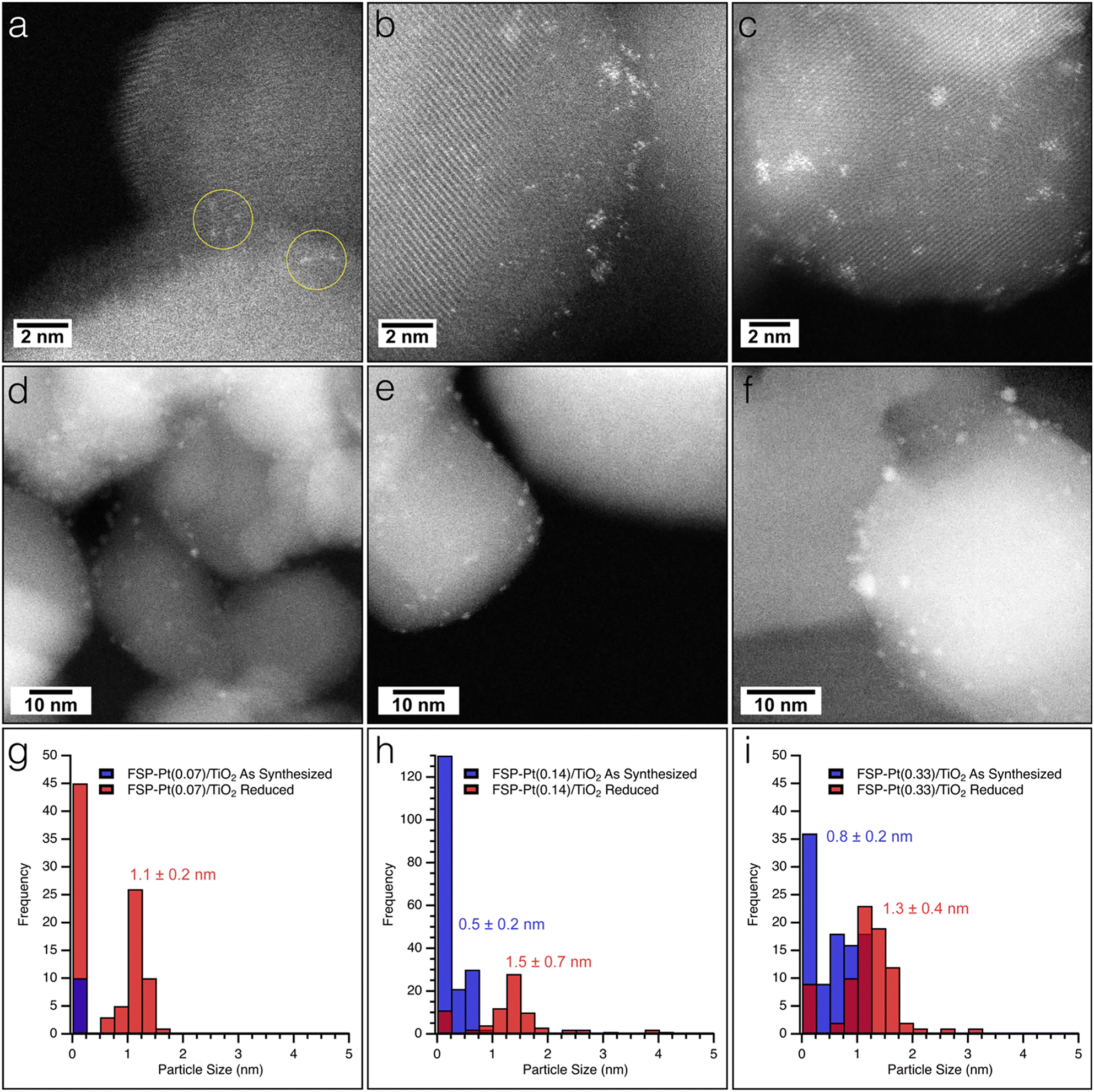 | ||
| Fig. 2 HAADF-STEM images of as-synthesized (a) FSP-Pt(0.07)/TiO2, (b) FSP-Pt(0.14)/TiO2, and (c) FSP-Pt(0.33)/TiO2 and after reduction in hydrogen (d) FSP-Pt(0.07)/TiO2, (e) FSP-Pt(0.14)/TiO2, and (f) FSP-Pt(0.33)/TiO2. Particle size histograms for as-synthesized and reduced (g) FSP-Pt(0.07)/TiO2, (h) FSP-Pt(0.14)/TiO2, and (i) FSP-Pt(0.33)/TiO2. Exemplary regions of single atoms are highlighted with yellow circles. | ||
XAS experiments were performed to complement the microscopy data to better understand changes in Pt oxidation state in the as-synthesized FSP-Pt/TiO2 catalyst and reduced FSP-Pt/TiO2 under reaction relevant pretreatment conditions. Notably, the as-synthesized FSP-Pt/TiO2 catalysts have an intermediate white line intensity compared to PtO2 and Pt foil (Fig. 3a). Because the white line intensity in the Pt LIII-edge XANES spectrum depends on the density of unoccupied 5d states, it has been frequently used as a metric to estimate the oxidation state of Pt.53,54 The white line intensity of the normalized XANES of all the as-synthesized samples (1.9–2.0) was found to be closer to that of the bulk PtO2 (2.3) than measured for the Pt foil (1.3). It is important to note that due to the nature of XAS as a bulk technique, the observed intermediate white line intensity cannot prove the presence of a mixed population of Pt4+ (i.e., PtO2) and Pt0, as such a mixed population could be indistinguishable from a homogenous population of Pt2+. However, this result does indicate the presence of a significant population of oxidized Pt present in the as-synthesized samples. This result is in contrast with the reduced samples, as upon treatment with H2 at 450 °C the white line intensities (1.3 norm. μeV) for all weight loadings are consistent with that of Pt0 suggesting complete reduction of the oxidized Pt species (Fig. 3b). The EXAFS of the as-synthesized FSP-Pt/TiO2 catalysts shown in Fig. 3c, reveals a Pt–O scattering peak at ca. 1.7 Å. However, despite a similar position to that of the first-shell peak in PtO2, the magnitude is lower, suggesting a lower coordination number than the bulk oxide. Likewise, the second-shell peaks (ca. 2.2–3.4 Å) of the three samples are similar in shape, but the intensities are also significantly different than PtO2, indicating the average local structure of Pt in the as-synthesized catalysts is not the same as the bulk oxide, which is consistent with the XANES analysis. Aside from intensity differences, the shapes of the real components of the Fourier transformed EXAFS of the second shell are essentially identical (Fig. S7†), suggesting that the as-synthesized materials contain the same species. Furthermore, similarities in the real components of the as-synthesized catalysts and Pt foil from 4.4–6.0 Å suggest a fraction of the Pt is present in metallic particles. After reduction, however, the EXAFS spectra (Fig. 3d) are consistent with metallic NPs (i.e., similar shape as foil, but lower intensity). In agreement with the XANES, there is no evidence of Pt–O scattering indicating all the Pt is reduced. Quantitative EXAFS fitting parameters for the FSP-Pt/TiO2 catalysts are summarized in Table S4.†
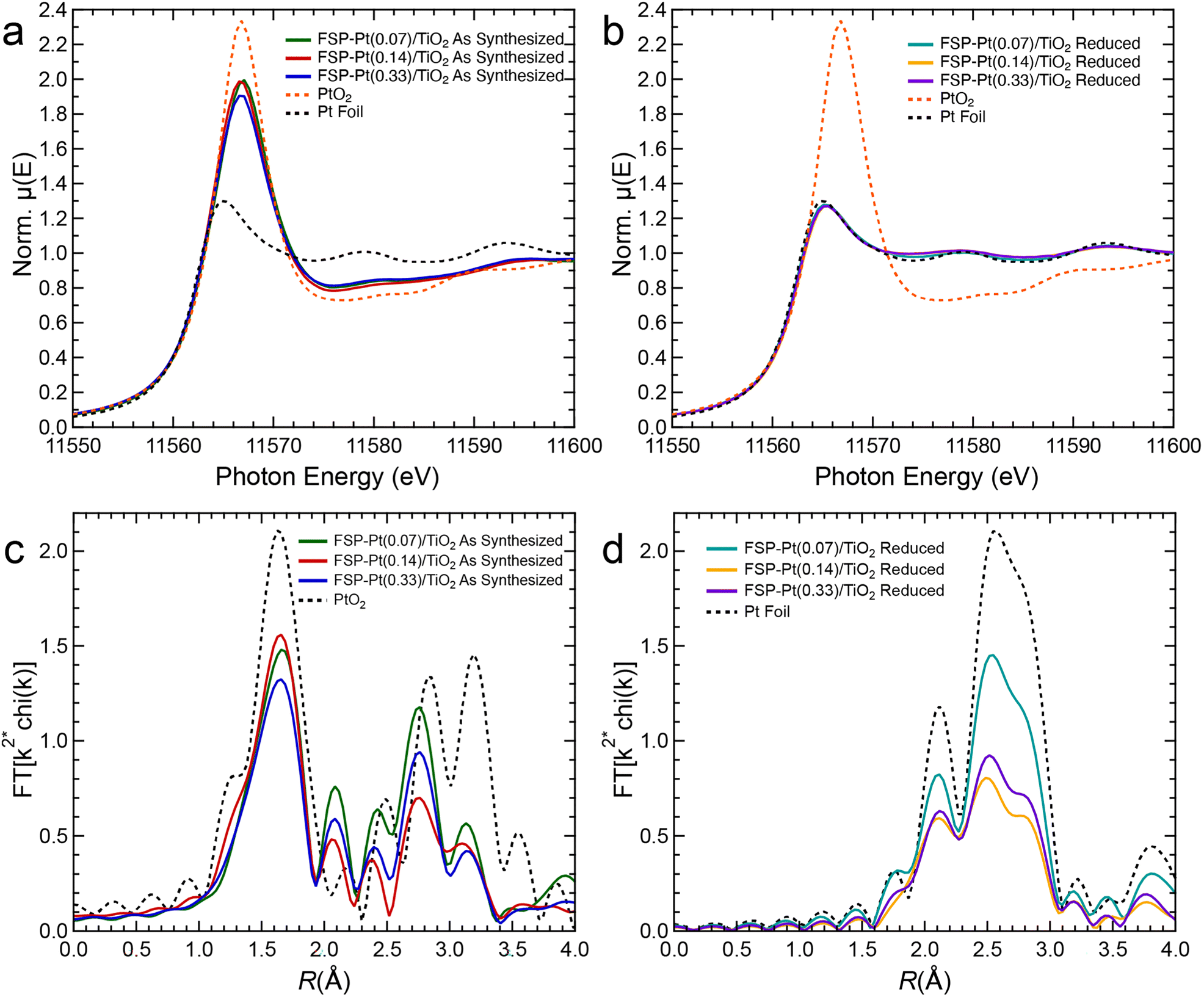 | ||
| Fig. 3 XANES spectra of as (a)-synthesized and (b) reduced FSP-Pt/TiO2 catalysts compared to Pt foil and PtO2 standards; and magnitude of the Fourier transformed k2-weighted Pt LIII edge EXAFS of (c) as prepared and (d) FSP-Pt/TiO2, Pt LIII-edge EXAFS. | ||
DRIFTS was performed to investigate the adsorption of CO on the FSP-Pt/TiO2 catalysts compared to their IW analogues (Fig. S8a and b†). Spectra were collected at 50 °C after a 450 °C pretreatment in 5% H2 to better emulate the speciation of the catalyst prior to reaction. All catalysts analyzed exhibited two distinct spectral features, a minor band at 2109–2115 cm−1 and a larger peak at 2071 cm−1. The band at 2071 cm−1 can be assigned to CO linearly adsorbed to Pt0 on NP surfaces.54,55 The relative intensity of this peak increased as a function of Pt loading in all cases, which is consistent with the increased population of metallic NPs present in the higher weight-loading materials as observed by STEM and TEM. The minor peak/shoulder observed at 2109–2115 cm−1 may be attributed to more oxidized Pt2+, Ptδ+, single Pt atoms, or very small clusters.54,56,57 Notably, the relative intensity of this feature is significantly higher for both FSP and IW catalysts at a Pt loading of 0.07 wt%. In the case of the FSP catalysts, this feature at 2109 cm−1 is diminished almost entirely at Pt loadings of 0.14 and 0.3 wt%, consistent with the sintering and loss of dispersed Pt atoms observed after pretreatment. In contrast, the IW catalysts retain this feature at all loadings.
Taken as a whole, the results of the spectroscopic characterization and imaging data reveal several distinct physical properties that differentiate the FSP-synthesized materials from their IW analogues. This ability to tune the distribution of Pt species (i.e., the population of Pt clusters, single atoms, and metallic NPs) as a function of FSP conditions highlights the potential of this synthetic method to afford control over the physico-chemical properties of the synthesized catalysts. In particular, differences in Pt speciation that were introduced by the FSP synthesis process may significantly alter their catalytic behaviour. To assess the impact of the physico-chemical changes afforded by the FSP method, we evaluated the performance of the FSP- and IW-synthesized materials for the upgrading of whole biomass pyrolysis vapours.
Whole biomass vapour upgrading
The catalytic performance of the FSP-Pt/TiO2 materials was evaluated with whole biomass vapours of pine particles using a micropyrolyzer-GC-MS system that has previously been described in detail.39–41 The biomass was pyrolyzed at 500 °C in He flow, while the catalytic upgrading reaction was operated at 400 °C in H2 flow at 115 kPa total pressure. Experiments were continued until a B:C of ca. 0.9–1.0 was reached based on literature precedent for evaluating the utility of catalysts for biomass conversion to fuels and chemicals, as well as to study the stability of these catalysts for continuous catalytic operation.40,58–60
The hydrocarbon products observed were grouped into 6 categories: alkanes, alkenes, aromatics, partially upgraded (intermediate) oxygenates, primary (unreacted) vapours, carbon monoxide, carbon dioxide, and a small fraction of unidentified species (unknowns, ≤1.5%). Representative species, that account for >90% of each category, are shown in Table S5.† Partially upgraded oxygenates are incompletely deoxygenated products formed from C–O cleavage via deoxygenation or hydrodeoxygenation of complex oxygenates present in the pyrolysis vapours (i.e., primary vapours) such as furanones, acids, methoxyphenols, acetaldehyde, hydroxybutanones. Aromatics are formed by aromatic C–O bond cleavage (via direct deoxygenation or hydrodeoxygenation) of the oxygenates without subsequent hydrogenation to form cycloalkenes (partial hydrogenation) and cycloalkanes (complete hydrogenation). Alkenes are a result of deoxygenation of linear oxygenates present in pyrolysis vapours or C–C bond breaking (cracking) of alkyl chains in branched oxygenates. Complete hydrogenation of the alkenes, due to high hydrogen activation and utilization, leads to the formation of alkanes. A generalized reaction scheme is presented in Fig. 4.
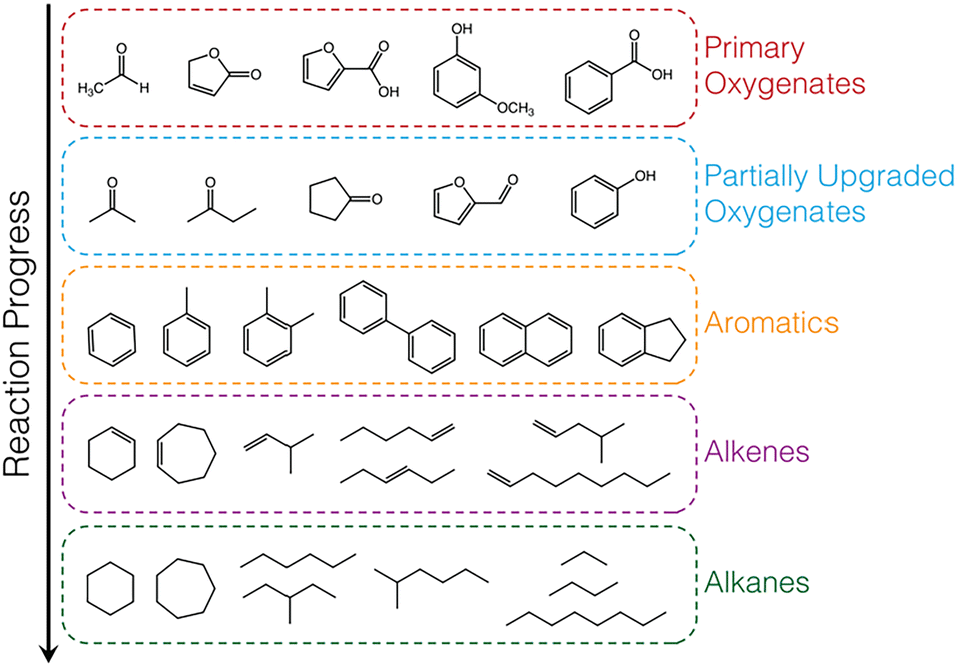 | ||
| Fig. 4 Generalized reaction scheme for biomass vapour upgrading with exemplary species shown for each category of product observed. | ||
As shown in Fig. 5, the total carbon yield for FSP–TiO2 and P25–TiO2 was low (<25%), with the product consisting mainly of primary vapours (not included in the yield) and CO2, and alkanes and aromatics as minor products. Unsurprisingly, the absence of hydrogen-activating Pt metal sites resulted in limited hydrodeoxygenation leading to the formation of intermediate oxygenates through direct C–O and C–C bond cleavage on the acid sites of TiO2 and/or thermolytic reaction. Incorporation of Pt resulted in an increased total carbon yield for all catalysts (Fig. 5a) with a concomitant enhancement of the yield of aromatics, alkanes, alkenes, and intermediate oxygenates (Fig. 5b and c) due to hydrogen activation on the metal sites and subsequent hydrodeoxygenation, hydrogenation, and hydrogenolysis reactions. Considering the hydrocarbon products observed, excluding the primary oxygenates, FSP-Pt(0.07)/TiO2 resulted in a carbon yield significantly higher than IW-Pt(0.07)/TiO2 (34.3 ± 0.9% vs. 22.3 ± 0.7%) at the same Pt loading (0.07 wt%). This is suggestive of enhanced metal utilization resulting from the FSP-based synthesis method as evidenced by the increased population of Pt clusters observed for this material. The carbon yield for the intermediate Pt loading catalysts, FSP-Pt(0.14)/TiO2 (39.4 ± 1.7%) and IW-Pt(0.14)/TiO2 (39.2 ± 1.0%), are within the error of the micropyrolyzer experiments, which is consistent with the predominance of Pt NPs as the form of Pt for both of these catalysts as observed by TEM after reduction. At the highest Pt loading of 0.3 wt%, both FSP-Pt(0.33)/TiO2 and IW-Pt(0.27)/TiO2 produced similar carbon yields of 41.4 ± 0.9% and 39.6 ± 1.5%, respectively, suggesting the enhanced dispersion afforded by FSP synthesis is not preserved at higher loadings after reduction. This conclusion is in agreement with the imaging and spectroscopic results detailed above that revealed a loss of atomic and clustered Pt species upon reduction for the FSP-Pt(0.33)/TiO2 catalyst. These results indicate that the FSP-synthesized Pt/TiO2 catalysts exhibit comparable activity to conventional IW-synthesized Pt/TiO2 catalysts at weight loadings ≥0.14%, with significantly greater activity at lower loadings. We hypothesize that the enhanced metal utilization at lower loadings observed for the FSP-synthesized materials results from the preservation of sub-nm Pt-clusters, as less sintering is observed in the case of the FSP-Pt(0.07)/TiO2 catalyst as revealed by the microscopy study above.
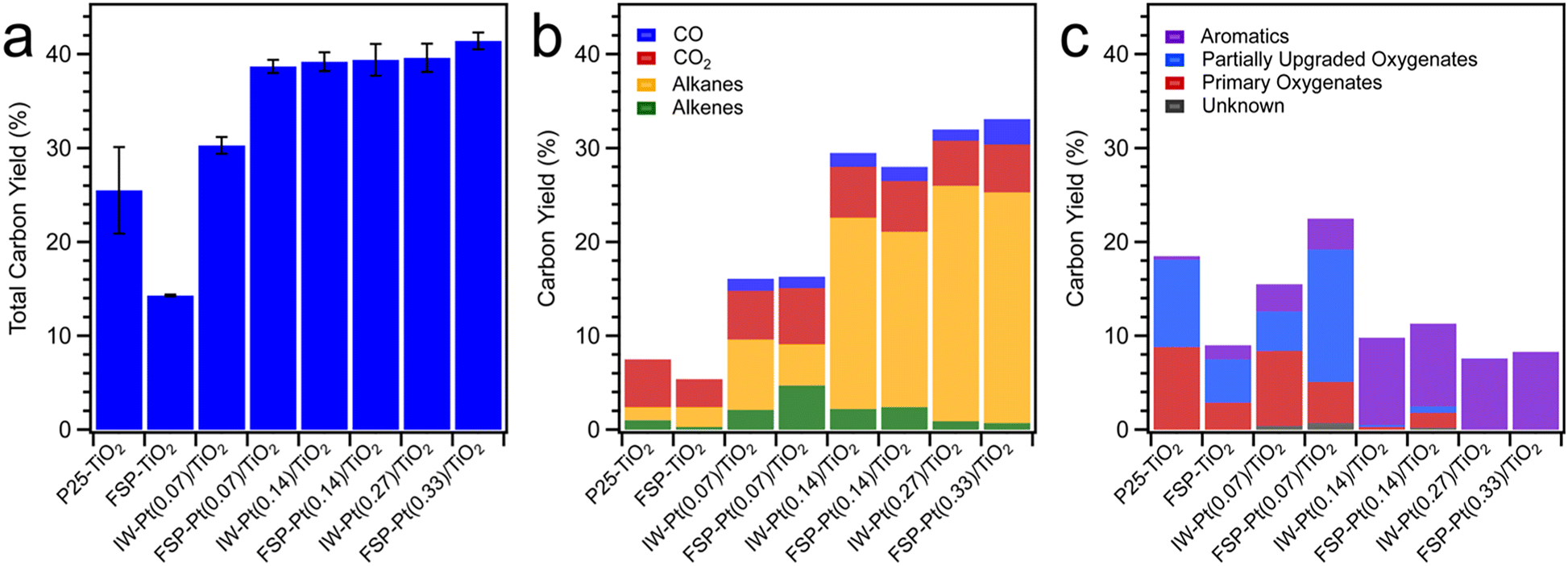 | ||
| Fig. 5 Category plots of (a) total carbon yield, (b) carbon yield of CO, CO2, alkanes, and alkenes, and (c) aromatics, partially upgraded oxygenates, and primary oxygenates for all catalysts. | ||
Analysis of the CFP product distributions reveals that the FSP-Pt/TiO2 catalysts generate a distinct product slate compared to the IW-Pt/TiO2 catalysts. At a Pt loading of 0.07 wt% and cumulative B:C of ca. 0.3, CFP over FSP-Pt(0.07)/TiO2 lead to greater formation of alkenes (4.7 ± 1.2 C%) and intermediate oxygenates (14.1 ± 2.1 C%) as compared to IW-Pt(0.07)/TiO2 (alkenes (2.1 ± 0.1 C%); intermediate oxygenates (4.2 ± 0.3 C%)) as shown in Fig. 5b and c. Furthermore, FSP-Pt(0.07)/TiO2 resulted in lower alkane production (4.4 ± 1.8 C%) and lower breakthrough of primary vapours (4.4 ± 1.9 C%) as compared to IW-Pt(0.07)/TiO2 (alkane (7.5 ± 0.6 C%) and primary vapours (8.0 ± 1.6 C%)). At higher Pt loadings (0.14 and 0.3 wt%) and similar B:Cs of ca. 0.3, the product distribution for FSP-synthesized and IW-synthesized Pt/TiO2 was similar as shown in Fig. 5b and c. The differences in product distribution at lower Pt loadings suggest that the single-atom site and Pt clusters present on the FSP catalysts (at low Pt loadings) reduce the hydrogenation activity of the FSP-Pt/TiO2 catalyst formulations and minimize the production of alkanes, thereby, leading to higher formation of partially upgraded oxygenates. This enables a facile synthetic handle for tuning the product slate in ex situ CFP towards ketones and alkenes, without adversely affecting C yield compared to IW synthesized catalysts. At higher Pt loadings of 0.14 wt% and 0.3 wt% Pt, similar product distributions for FSP and IW Pt/TiO2 catalysts are in line with the similar catalyst characterization results (acid site density, Pt speciation, and particle size), as noted above.
We investigated the evolution of catalyst performance with increasing B:C (representative of reaction time-on-stream) for FSP- and IW-synthesized Pt/TiO2. The results of these experiments are presented in Fig. 6 and S9–S11.† As shown in Fig. 6, FSP-Pt(0.07)/TiO2 resulted in increasing yields of intermediate oxygenates (11.5–18.9 C%) and primary vapours (2.5–10.0% C%) over the course of reaction (Fig. 6b and d), whereas at all B:Cs, IW-Pt(0.07)/TiO2 catalysts allowed breakthrough of more primary vapours (6.3–17.1 C%) and lower production of intermediate oxygenates (4.3–5.7 C%; Fig. 6a). These results indicate that FSP-Pt(0.07)/TiO2 enables enhanced catalytic deoxygenation of primary oxygenates to partially deoxygenated intermediates via C–O bond cleavage as compared to IW-Pt(0.07)/TiO2. Further, as shown in Fig. S9,† FSP-Pt(0.07)/TiO2 showed a consistently higher yield (0–3.5 C%) of phenolic molecules (alkyl phenols and phenol) and a lower yield (0–0.4 C%) of methoxyphenols for B:C ca. 0.1–1.0 whereas IW-Pt(0.07)/TiO2 produced higher yields (4.8–5.4 C%) of methoxyphenols and a lower yield (0–1.1 C%) of phenols from B:C ca. 0.1–0.9, further evidence of the enhanced deoxygenation functionality of FSP catalyst. Both the FSP- and IW-synthesized Pt/TiO2 catalysts exhibited low yields of 1-ring aromatics (Fig. S9a and b†). As shown in Fig. 6c and d, FSP-Pt(0.07)/TiO2 and IW-Pt(0.07)/Pt/TiO2 demonstrated low yields of alkanes and alkenes, however, FSP-Pt(0.07)/TiO2 maintained significantly higher yields of alkenes over the course of the reaction compared to the IW analogue (2.5–6.1 C% vs. 0.2–2.2 C%, respectively). This comparison further indicates the lower hydrogenation functionality of FSP Pt/TiO2 catalyst in comparison to IW catalyst at low Pt loadings.
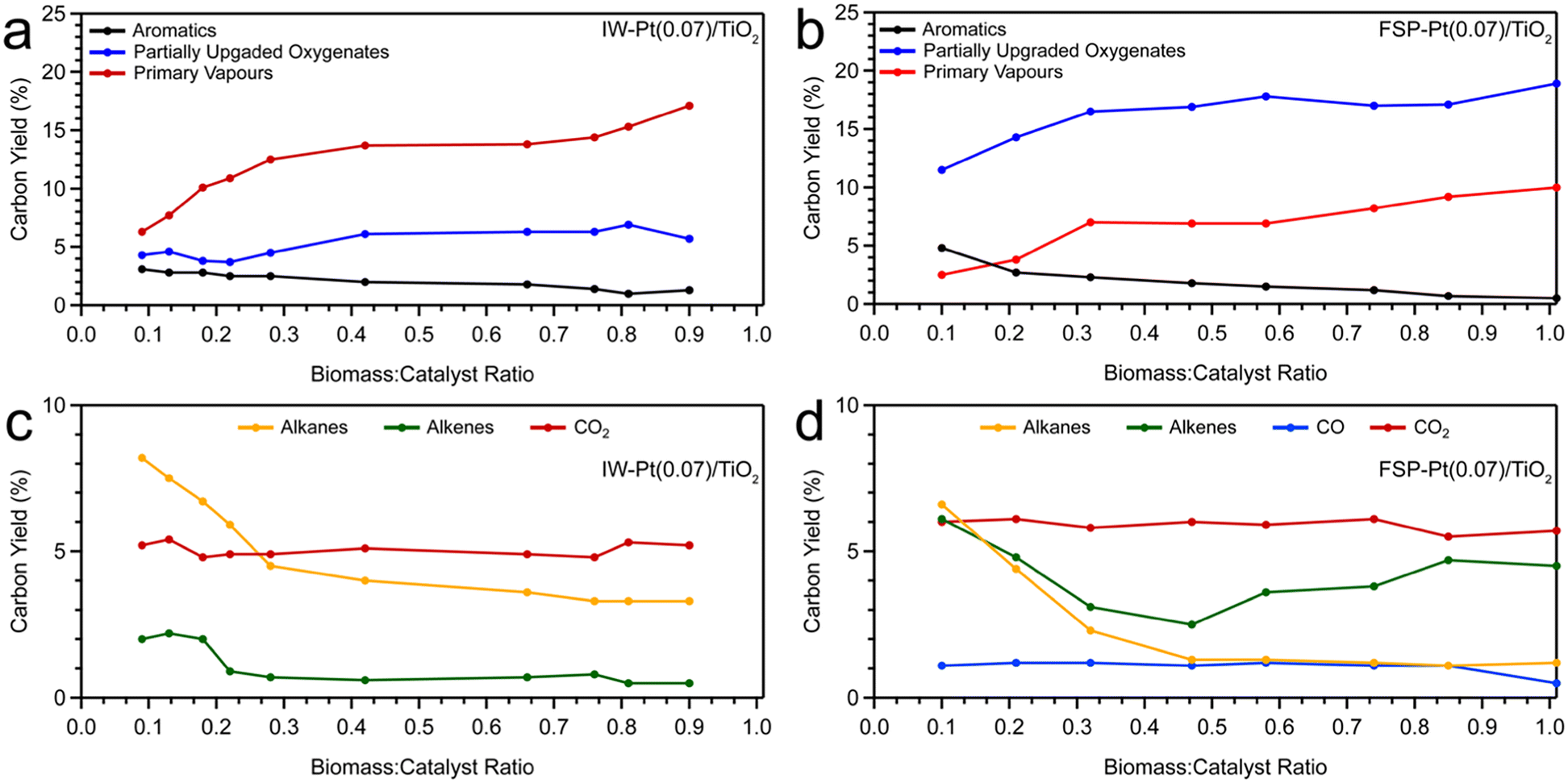 | ||
| Fig. 6 Carbon yield to aromatics, partially upgraded oxygenates, and primary vapours as a function of biomass-to-catalyst ratio for (a) IW-Pt(0.07)/TiO2 and (b) FSP-Pt(0.07)/TiO2 catalysts and carbon yield to alkenes, alkanes, CO, and CO2, as a function of biomass-to-catalyst ratio for (c) IW-Pt(0.07)/TiO2 and (d) FSP-Pt(0.07)/TiO2 catalysts. | ||
Fig. S10a–d† shows the results of deactivation studies for FSP-Pt(0.14)/TiO2 and IW-Pt(0.14)/TiO2. At B:C < 0.39, both catalysts demonstrate similar activity and product distribution. However, at B:C > 0.39, FSP-Pt(0.14)/TiO2 exhibited increasing yields of intermediate oxygenates (increases from 2.1% to 16.5% over the course of reaction) with a concurrent decrease in aromatics yield (from 9.7 to 4.0 C%). The yield of intermediate oxygenates produced over FSP-Pt(0.14)/TiO2 was observed to decrease starting at a B:C ca. 0.77 with a concurrent increase in the yield of primary vapours, indicative of catalyst deactivation. In contrast, CFP over the IW-Pt(0.14)/TiO2 resulted in a consistent yield of aromatics (8.6 ± 0.8 C%) until a B:C ca. 0.96 with a low intermediate oxygenates yield (2.0 ± 1.8%) and minimal breakthrough of primary vapours (<0.7%). Moreover, as shown in Fig. S10d,† the alkane yields decreased rapidly for FSP-Pt(0.14)/TiO2 after B:C ca. 0.17, while IW-Pt(0.14)/TiO2 showed consistent alkane yields until a B:C ca. 0.78. These results suggest a more rapid loss of hydrogenation activity for FSP(0.14)/TiO2 compared to the IW-Pt(0.14)/TiO2 analogue. FSP-Pt(0.33)/TiO2 and IW-Pt(0.33)/Pt/TiO2 catalysts result in similar product distribution and carbon yields with increasing B:C from ca. 0.08–0.98 (Fig. S11a–d†). Both catalysts demonstrated increased hydrogenation activity compared to their lower Pt loading analogues with high C yields to aromatics and alkanes and low yields of intermediate oxygenates, primary vapours, and alkenes. Both catalyst systems exhibited minimal deactivation over the B:Cs evaluated, with sustained high hydrocarbon yields (carbon yields > 28.5%) until a B:C ca. 0.83.
These performance data coupled with the detailed structural characterization suggest that (1) the effect of highly dispersed single-atom/sub-nm Pt clusters is more pronounced at low Pt loadings on FSP-synthesized catalysts leading to higher hydrocarbon yields and catalyst stability, (2) at higher Pt loadings, FSP catalysts are as active and stable as the conventional IW catalysts, and (3) FSP-synthesized catalysts show lower hydrogenation activity than IW-synthesized catalysts. We hypothesize that the changes in the product slate at increasing B:C for the FSP-Pt/TiO2 catalysts result from continued structural evolution under reaction conditions that is distinct from the deactivation observed for the traditionally prepared IW catalysts. Further study will seek to elucidate the basis of this structural evolution and its catalytic consequences.
Cost analysis of FSP synthesis
A cost estimate for the FSP-synthesized Pt/TiO2 material at a 0.07% weight loading was developed in CatCost32 on the basis of the lab-scale procedures. The lab-scale FSP equipment has a throughput of 200 g h−1 and approximate cost of $200000 and requires a dedicated operator. Unsurprisingly, this small-scale, labour-intensive equipment results in a very high estimated catalyst purchase cost of $1150 per kg, driven principally by operating expenditures (OpEx, Table 2, scenario A). However, we estimate that system throughput could be increased to 10 kg h−1 for only twice the purchase cost while maintaining the single operator. This increase in scale results in a dramatic decrease in both capital (CapEx) and OpEx, for a total cost of $142 per kg (scenario C). As part of the scale-up, the product collection equipment used in the lab-scale system is replaced with a more industrially relevant baghouse (bag filters), which produces a cost savings on its own (scenario B vs. A). Because Pt metal is routinely recovered from spent catalysts, we used the spent catalyst value (SCV) module of CatCost to estimate the recoverable value of Pt at the end of the catalyst's useful life. The catalysts were estimated to have a spent catalyst value of $10.23 per kg across all scenarios, which is not included in Table 2.
| Inputs | Costs, 2016 $ per kg catalyst | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Scenario | Details | Pt | TiO2/precursor | Solvent | Process gases | CapEx | OpEx | Margin | Total |
| A | FSP, 200 g h−1 | 26 | 14 | 44 | 16 | 65 | 822 | 164 | 1150 |
| B | FSP, 200 g h−1, baghouse | 26 | 14 | 44 | 16 | 56 | 810 | 140 | 1105 |
| C | FSP, 10 kg h−1, baghouse | 26 | 14 | 44 | 16 | 3 | 33 | 8 | 142 |
| D | IW, $3 per kg TiO2 | 18 | 3 | 0.01 | 3 | 0.16 | 3 | 0.40 | 27 |
| E | IW, $50 per kg P25 | 18 | 51 | 0.01 | 3 | 0.16 | 10 | 0.40 | 83 |
| F | IW, $75 per kg P25 | 18 | 77 | 0.01 | 3 | 0.16 | 14 | 0.40 | 113 |
| G | IW, $100 per kg P25 | 18 | 103 | 0.01 | 3 | 0.16 | 18 | 0.40 | 142 |
The cost to produce Pt/TiO2via FSP was compared to that of a conventional IW synthesis. The IW-Pt(0.07)/TiO2 cost estimates ranged from $27–142 per kg, depending on the type and cost of the titania support used for IW. The most relevant comparison is to a flame-synthesized titania powder such as the P25 titania support we have used throughout this work. Through a survey of quotes and publicly available prices, we determined a price range for P25 of ca. $50–100 per kg, depending on scale, purity, and other specifications. At the upper end of this range, the resulting catalyst cost of $142 per kg is identical to that of the FSP-derived material (Table 2, scenarios C and G). We also included a scenario using a low-cost ($3 per kg), non-flame-synthesized titania. The lower Pt cost for IW-Pt(0.07)/TiO2 relative to FSP-Pt(0.07)/TiO2 results from differences in Pt utilization (yield), which was assumed to be quantitative for IW but was observed to be 70% for FSP (targeted loading: 0.10%, actual loading: 0.07%).
In order to assess major cost drivers and identify R&D opportunities for the FSP material, a sensitivity analysis was performed based on scenario C, and the sensitivity results for the top contributors to cost uncertainty are shown in Fig. 7. The figure uses net catalyst cost, after spent catalyst value is subtracted. The costs of IW materials based on P25 prices of $75 and $100 per kg are included on the plot for reference. Consistent with the large contribution of solvent costs in Table 2, the largest opportunity for cost reduction is in solvent usage. A proportional reduction of solvent usage by approximately 3-fold (33 to 10 L kg−1) would yield cost savings of over $35 per kg, resulting in a purchase cost lower than the $75 per kg P25 IW scenario. Further opportunities for cost reduction through R&D include process gas usage and Pt utilization; on the latter point, it should be noted that industrial syntheses are expected to be substantially (i.e., 90+%) more materials efficient. Combining the modelled savings from these three areas would reduce purchase cost by $55 per kg total, to a cost of $87 per kg, approaching the level of the $50 per kg P25 IW scenario ($83 per kg). Other factors driving significant cost variation include TTiP price, distribution and marketing costs, production scale, and plant capacity factor (the ratio of actual to expected plant outputs). It is noteworthy that the TTiP precursor price was only the third most significant factor contributing to variation in catalyst cost, revealing that commercial applications of a TTiP/FSP route to titania may be limited more by solvent cost than TTiP cost.
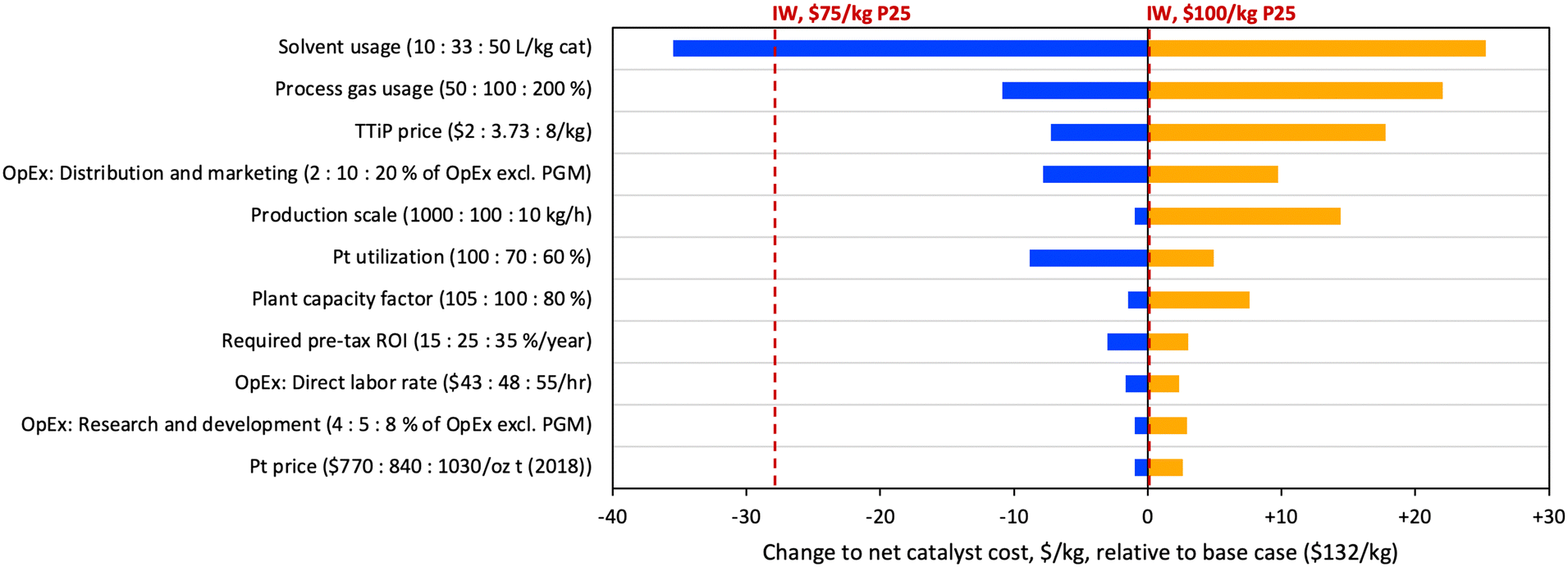 | ||
| Fig. 7 Tornado plot depicting the results of a sensitivity analysis on the FSP-Pt(0.07)/TiO2 material. | ||
Conclusions
The continuous FSP synthesis presented here highlights the utility and tunability of this approach for the scalable production of high-performance Pt/TiO2 catalysts for biomass upgrading applications. The ex situ CFP reaction studies demonstrate the (1) high deoxygenation activity of FSP-Pt/TiO2 catalysts revealed by formation of intermediate oxygenates, aromatics, (cyclo)alkanes, and (cyclo)alkenes as the dominant hydrocarbon products, and (2) decreased hydrogenation as revealed by the high alkene and low alkane product yield when compared to IW-Pt/TiO2. We hypothesize that the observed shift towards unsaturated hydrocarbons (alkenes, intermediate oxygenates, and aromatics) and improved deoxygenation performance at the lowest Pt loading compared to the IW materials results from the observed differences in Pt speciation and particle size. A preliminary cost analysis revealed several opportunities to improve the scalable FSP synthesis to make it more cost-competitive with traditionally synthesized IW catalysts including (1) reductions to solvent usage, (2) improving process gas usage, and (3) increasing Pt utilization. In conclusion, the FSP synthesis of Pt catalysts demonstrated process intensification of the synthesis procedure while offering changes to catalyst morphology that may be beneficial for tuning activity.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The usage of Electron Microscopy Service at University of Illinois at Chicago with help of Dr. Fengyuan Shi is gratefully acknowledged. This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, and in part by Argonne National Laboratory, operated by The University of Chicago for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308 and DE-AC02-06CH11357, respectively. This research used resources of the Advanced Photon Source; a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. MRCAT operations were supported by the DOE and the MRCAT member institutions. Funding provided by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. This research was conducted in collaboration with the Chemical Catalysis for Bioenergy (ChemCatBio) Consortium, a member of the Energy Materials Network (EMN). The views expressed in this article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell Iii, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC
.
- J.-M. Ha, K.-R. Hwang, Y.-M. Kim, J. Jae, K. H. Kim, H. W. Lee, J.-Y. Kim and Y.-K. Park, Renewable Sustainable Energy Rev., 2019, 111, 422–441 CrossRef CAS
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. D. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Energy Environ. Sci., 2018, 11, 2904–2918 RSC
- S. Boonyasuwat, T. Omotoso, D. E. Resasco and S. P. Crossley, Catal. Lett., 2013, 143, 783–791 CrossRef CAS
- M. B. Griffin, G. A. Ferguson, D. A. Ruddy, M. J. Biddy, G. T. Beckham and J. A. Schaidle, ACS Catal., 2016, 6, 2715–2727 CrossRef CAS
- M. Lu, H. Du, B. Wei, J. Zhu, M. Li, Y. Shan and C. Song, Energy Fuels, 2017, 31, 10858–10865 CrossRef CAS
- C. A. Teles, R. C. Rabelo-Neto, G. Jacobs, B. H. Davis, D. E. Resasco and F. B. Noronha, ChemCatChem, 2017, 9, 2850–2863 CrossRef CAS
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS
-
B. A. T. Mehrabadi, S. Eskandari, U. Khan, R. D. White and J. R. Regalbuto, in Advances in Catalysis, ed. C. Song, Academic Press, 2017, vol. 61, pp. 1–35 Search PubMed
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed
- F. G. Baddour, E. J. Roberts, A. T. To, L. Wang, S. E. Habas, D. A. Ruddy, N. M. Bedford, J. Wright, C. P. Nash, J. A. Schaidle, R. L. Brutchey and N. Malmstadt, J. Am. Chem. Soc., 2020, 142, 1010–1019 CrossRef CAS PubMed
- S. E. Habas, F. G. Baddour, D. A. Ruddy, C. P. Nash, J. Wang, M. Pan, J. E. Hensley and J. A. Schaidle, Chem. Mater., 2015, 27, 7580–7592 CrossRef CAS
- M. Schreier and J. R. Regalbuto, J. Catal., 2004, 225, 190–202 CrossRef CAS
- J. T. Miller, M. Schreier, A. J. Kropf and J. R. Regalbuto, J. Catal., 2004, 225, 203–212 CrossRef CAS
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed
- F. G. Baddour, C. P. Nash, J. A. Schaidle and D. A. Ruddy, Angew. Chem., Int. Ed., 2016, 55, 9026–9029 CrossRef CAS PubMed
- A. Dutta, J. A. Schaidle, D. Humbird, F. G. Baddour and A. Sahir, Top. Catal., 2016, 59, 2–18 CrossRef CAS
- W. Y. Teoh, R. Amal and L. Mädler, Nanoscale, 2010, 2, 1324–1347 RSC
- T. Johannessen, J. R. Jensen, M. Mosleh, J. Johansen, U. Quaade and H. Livbjerg, Chem. Eng. Res. Des., 2004, 82, 1444–1452 CrossRef CAS
- S. Li, Y. Ren, P. Biswas and S. D. Tse, Prog. Energy Combust. Sci., 2016, 55, 1–59 CrossRef
- K. Wegner and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 4581–4589 CrossRef CAS
- R. Mueller, L. Mädler and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 1969–1976 CrossRef CAS
- G. D. Ulrich, Chem. Eng. News, 1984, 62, 22–29 CrossRef CAS
- R. Koirala, S. E. Pratsinis and A. Baiker, Chem. Soc. Rev., 2016, 45, 3053–3068 RSC
- R. Strobel, A. Baiker and S. E. Pratsinis, Adv. Powder Technol., 2006, 17, 457–480 CrossRef CAS
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed
- S. Ding, H.-A. Chen, O. Mekasuwandumrong, M. J. Hülsey, X. Fu, Q. He, J. Panpranot, C.-M. Yang and N. Yan, Appl. Catal., B, 2021, 281, 119471 CrossRef CAS
- W. Y. Teoh, L. Mädler, D. Beydoun, S. E. Pratsinis and R. Amal, Chem. Eng. Sci., 2005, 60, 5852–5861 CrossRef CAS
- D. A. Ruddy, J. E. Hensley, C. P. Nash, E. C. D. Tan, E. Christensen, C. A. Farberow, F. G. Baddour, K. M. Van Allsburg and J. A. Schaidle, Nat. Catal., 2019, 2, 632–640 CrossRef CAS
- S. E. Pratsinis, AIChE J., 2010, 56, 3028–3035 CrossRef CAS
- P. Weyell, H. D. Kurland, T. Hülser, J. Grabow, F. A. Müller and D. Kralisch, Green Chem., 2020, 22, 814–827 RSC
- K. M. Van Allsburg, E. C. D. Tan, J. D. Super, J. A. Schaidle and F. G. Baddour, Nat. Catal., 2022, 5, 342–353 CrossRef CAS
- F. G. Baddour, L. Snowden-Swan, J. D. Super and K. M. Van Allsburg, Org. Process Res. Dev., 2018, 22, 1599–1605 CrossRef CAS
- T. B. Bolin, T. Wu, N. Schweitzer, R. Lobo-Lapidus, A. J. Kropf, H. Wang, Y. Hu, J. T. Miller and S. M. Heald, Operando IV 4th Int. Congr. Operando Spectrosc., 2013, vol. 205, pp. 141–147 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- J. J. Rehr, J. Mustre de Leon, S. I. Zabinsky and R. C. Albers, J. Am. Chem. Soc., 1991, 113, 5135–5140 CrossRef CAS
- C. Mukarakate, M. J. Watson, J. ten Dam, X. Baucherel, S. Budhi, M. M. Yung, H. Ben, K. Iisa, R. M. Baldwin and M. R. Nimlos, Green Chem., 2014, 16, 4891–4905 RSC
- C. Mukarakate, X. Zhang, A. R. Stanton, D. J. Robichaud, P. N. Ciesielski, K. Malhotra, B. S. Donohoe, E. Gjersing, R. J. Evans, D. S. Heroux, R. Richards, K. Iisa and M. R. Nimlos, Green Chem., 2014, 16, 1444–1461 RSC
- C. Mukarakate, K. Orton, Y. Kim, S. Dell'Orco, C. A. Farberow, S. Kim, M. J. Watson, R. M. Baldwin and K. A. Magrini, ACS Sustainable Chem. Eng., 2020, 8, 2652–2664 CrossRef CAS
- A. R. Stanton, K. Iisa, C. Mukarakate and M. R. Nimlos, ACS Sustainable Chem. Eng., 2018, 6, 10030–10038 CrossRef CAS
- K. Iisa, Y. Kim, K. A. Orton, D. J. Robichaud, R. Katahira, M. J. Watson, E. C. Wegener, M. R. Nimlos, J. A. Schaidle, C. Mukarakate and S. Kim, Green Chem., 2020, 22, 2403–2418 RSC
-
D. E. Garrett, Chemical Engineering Economics, Van Nostrand-Reinhold, New York, 1989 Search PubMed
-
G. D. Ulrich and P. T. Vasudevan, Chemical Engineering Process Design and Economics, Durham, New Hampshire, 2nd edn, 2004 Search PubMed
- H. J. Lang, Chem. Eng., 1947, 117–121 Search PubMed
- H. J. Lang, Chem. Eng., 1948, 112–113 CAS
-
M. S. Peters and K. D. Timmerhaus, Plant Design and Economics for Chemical Engineers, McGraw-Hill, New York, 5th edn, 2003 Search PubMed
- E. Sánchez, T. López, R. Gómez, Bokhimi, A. Morales and O. Novaro, J. Solid State Chem., 1996, 122, 309–314 CrossRef
- K. J. Range, F. Rau, U. Klement and A. M. Heyns, Mater. Res. Bull., 1987, 22, 1541–1547 CrossRef CAS
- K. Sugiyama and Y. Takéuchi, Z. Kristallogr. Cryst. Mater., 1991, 194, 305–313 CAS
- T. Sasaki, N. Koshizaki, S. Terauchi, H. Umehara, Y. Matsumoto and M. Koinuma, Nanostruct. Mater., 1997, 8, 1077–1083 CrossRef CAS
- T. Sasaki, N. Koshizaki, M. Koinuma and Y. Matsumoto, Nanostruct. Mater., 1999, 12, 511–514 CrossRef
- X. Sun, J. Lin, Y. Chen, Y. Wang, L. Li, S. Miao, X. Pan and X. Wang, Commun. Chem., 2019, 2, 27 CrossRef
- T. I. Hyde, P. W. Ash, D. A. Boyd, G. Randlshofer, K. Rothenbacher and G. Sankar, Platinum Met. Rev., 2011, 55, 233–245 CrossRef CAS
- Y. Zhou, D. E. Doronkin, M. Chen, S. Wei and J.-D. Grunwaldt, ACS Catal., 2016, 6, 7799–7809 CrossRef CAS
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed
- P. Bazin, O. Saur, J. C. Lavalley, M. Daturi and G. Blanchard, Phys. Chem. Chem. Phys., 2005, 7, 187–194 RSC
- C. A. Teles, P. M. de Souza, R. C. Rabelo-Neto, M. B. Griffin, C. Mukarakate, K. A. Orton, D. E. Resasco and F. B. Noronha, Appl. Catal., B, 2018, 238, 38–50 CrossRef CAS
- M. Xu, C. Mukarakate, K. Iisa, S. Budhi, M. Menart, M. Davidson, D. J. Robichaud, M. R. Nimlos, B. G. Trewyn and R. M. Richards, ACS Sustainable Chem. Eng., 2017, 5, 5477–5484 CrossRef CAS
- K. Murugappan, C. Mukarakate, S. Budhi, M. Shetty, M. R. Nimlos and Y. Román-Leshkov, Green Chem., 2016, 18, 5548–5557 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00550j |
| This journal is © The Royal Society of Chemistry 2023 |