
Olefin metathesis over supported MoOx catalysts: influence of the oxide support†
Bin
Zhang
 ,
Michael E.
Ford
,
Eli
Ream
and
Israel E.
Wachs
*
,
Michael E.
Ford
,
Eli
Ream
and
Israel E.
Wachs
*
Operando Molecular Spectroscopy and Catalysis Laboratory, Department of Chemical & Biomolecular Engineering, Lehigh University, Bethlehem, PA 18015, USA. E-mail: iew0@lehigh.edu
First published on 29th November 2022
Abstract
A series of supported MoOx catalysts on different oxide supports (Al2O3, TiO2, ZrO2, SiO2) were synthesized and investigated for propylene metathesis, characterized with in situ spectroscopies (DRIFTS, Raman, UV-vis) and chemically probed with propylene-TPSR-MS, propylene-TPSR-IR, and ethylene/2-butene titration. Under dehydrated conditions at monolayer coverage or maximum surface dispersion, the surface MoOx sites are present as a mixture of isolated di-oxo (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )2Mo(–O–Al)2 and oligomeric mono-oxo OMo(–O–Al)4/5 sites on Al2O3, primarily oligomeric mono-oxo OMo(–O–Ti)4/5 on TiO2, isolated di-oxo (O)2Mo(–O–Zr)2 and oligomeric mono-oxo OMo(–O–Zr)4/5 on ZrO2, and isolated di-oxo (O)2Mo(–O–Si)2 on SiO2. The bridged (S2-OH) and tri-coordinated (S3-OH) anchoring surface hydroxyls of the oxide supports with strong support cation electronegativity control the activation and number of active surface MoOx sites at low temperatures (<100 °C). The isolated anchoring surface hydroxyls (S-OH) of the oxide supports with strong support cation electronegativity control the activation and number of active surface MoOx sites at high temperatures (>350 °C). Olefin metathesis by the more redox active supported MoOx/TiO2 and MoOx/ZrO2 catalysts is retarded by the formation of stable surface acetone and acetate species that block olefin adsorption. The oxide supports are potent ligands that tune the activation and surface chemistry of the surface MoOx sites for olefin metathesis. This is the first time that the influence of oxide supports on the activation and surface chemistry of supported MoOx sites has been systematically examined.
)2Mo(–O–Al)2 and oligomeric mono-oxo OMo(–O–Al)4/5 sites on Al2O3, primarily oligomeric mono-oxo OMo(–O–Ti)4/5 on TiO2, isolated di-oxo (O)2Mo(–O–Zr)2 and oligomeric mono-oxo OMo(–O–Zr)4/5 on ZrO2, and isolated di-oxo (O)2Mo(–O–Si)2 on SiO2. The bridged (S2-OH) and tri-coordinated (S3-OH) anchoring surface hydroxyls of the oxide supports with strong support cation electronegativity control the activation and number of active surface MoOx sites at low temperatures (<100 °C). The isolated anchoring surface hydroxyls (S-OH) of the oxide supports with strong support cation electronegativity control the activation and number of active surface MoOx sites at high temperatures (>350 °C). Olefin metathesis by the more redox active supported MoOx/TiO2 and MoOx/ZrO2 catalysts is retarded by the formation of stable surface acetone and acetate species that block olefin adsorption. The oxide supports are potent ligands that tune the activation and surface chemistry of the surface MoOx sites for olefin metathesis. This is the first time that the influence of oxide supports on the activation and surface chemistry of supported MoOx sites has been systematically examined.
I. Introduction
Due to the feedstock shift in steam cracking and oil refineries, there is a growing gap between global propylene supply and demand. The olefin metathesis reaction is an on-purpose method to react ethylene and 2-butene to produce two propylene molecules.1–3 The supported transition metal oxide (Re, Mo, W) catalysts dispersed on high surface area oxide supports (Al2O3 or SiO2) are highly effective catalysts for the olefin metathesis reaction. The Shell Higher Olefin Process (SHOP) was developed to produce linear higher olefins with supported MoOx/Al2O3 catalysts.3 The activation of surface MoOx sites for olefin metathesis is reported to strongly depend on the specific oxide support. It's generally concluded that the supported MoOx/Al2O3 catalyst is ∼10× more active than the supported MoOx/SiO2 catalyst, while supported MoOx/ZrO2 and MoOx/TiO2 catalysts are inactive for olefin metathesis.4–8 Reductive pre-treatment of surface MoOx sites with H2 is claimed to activate surface MoOx sites on non-Al2O3 and non-SiO2 supports. Tanaka et al. showed that the H2 treated supported MoOx/TiO2 catalyst exhibits propylene metathesis activity at 25 °C and proposed that Mo(+4) and Mo(+5) are active sites with ex situ XPS.6 Indovina et al. studied the H2 reduced supported MoOx/ZrO2 catalyst with ex situ ESR and XPS and claimed that only H2 reduced Mo(+5) sites are active for propylene metathesis at 25 °C.7The molecular and electronic structure of surface MoOx sites on different oxide supports under oxidatively dehydrated conditions have been studied with in situ Raman,9–13 IR,14,15 UV-vis,10,12,13 XAS,16–18etc. It was found that the structure of the surface MoOx sites depends on the surface MoOx coverage and specific oxide support. For the Al2O3-, ZrO2- and TiO2-supported MoOx catalysts, the isolated surface MoO4 sites predominate at low surface coverage on the supports, and both isolated surface MoO4 and oligomeric surface MoO5/6 co-exist at high surface coverage. The surface MoOx sites, however, are only present as isolated MoO4 sites on SiO2 supports.9–12,16,17 The nature of the surface MoOx sites on the Al2O3 and SiO2 supports during propylene metathesis reaction conditions have been recently reported,10,13,15,19,20 but the nature of the surface MoOx sites on the ZrO2 and TiO2 supports during propylene metathesis reaction conditions have still not been reported.
Over the years, there have been studies trying to correlate the olefin metathesis catalytic activity to the nature of the surface MoOx sites,8–11,21,22 but correlations between the catalyst acidic or redox properties and olefin metathesis are still being debated. The reducibility of supported MoOx catalysts was determined from H2-TPR and followed the trend MoOx/TiO2 > MoOx/ZrO2 > MoOx/SiO2 > MoOx/Al2O3.21 This reducibility trend suggests that MoOx sites on ZrO2 and TiO2 supports may be over-reduced under propylene metathesis reaction conditions.23 Kim et al. reported a different reducibility trend from H2-TPR: MoOx/TiO2 > MoOx/ZrO2 > MoOx/Al2O3 > MoOx/SiO2.11 More recently, Otroshchenko et al. compared the metathesis activity of the surface MoOx sites on different oxides supports with the redox and Lewis/Brønsted acid properties of the surface MoOx sites, but could not reach any general structure–activity relationships.8 The simple correlations made between H2-TPR reducibility and propylene metathesis activity of surface MoOx sites23 has the following issues: (i) the reducibility determined from H2-TPR is not representative of the reducibility by olefins, and (ii) the nature of the surface MoOx sites during the propylene metathesis reaction conditions are still unknown.
The objective of this study is to determine the influence of the oxide support on the molecular structure and activity of the surface MoOx sites on multiple oxide supports (Al2O3, SiO2, TiO2 and ZrO2) under oxidatively dehydrated and propylene metathesis reaction conditions. The supported MoOx catalysts were physically characterized with in situ spectroscopy (DRIFTS, Raman, and UV-vis) and chemically probed with C3=-TPSR-MS, C3=-TPSR-IR, ethylene/2-butene titration, and steady state propylene self-metathesis. Comparison of these physical and chemical properties will allow building fundamental structure–activity relationships for propylene metathesis by the supported MoOx catalysts on different oxide supports.
II. Experimental details
Catalyst synthesis
The supported MoOx catalysts were prepared by incipient-wetness impregnation (IWI) of aqueous ammonium heptamolybdate ((NH4)6Mo7O24·4H2O, Matheson, Coleman & Bell, 99.9%) onto different oxide supports (Al2O3 (Puralox, SCCa 5/200, 197 m2 g−1), ZrO2 (Degussa, 60 m2 g−1), TiO2 (Evonik P25, 51 m2 g−1), SiO2 (Cabot, Cab-O-Sil, EH-5, 350 m2 g−1)). Before impregnation, the Al2O3 support was calcined at 500 °C for 2 hours under flowing air. The SiO2 support was treated with DI water and calcined at 500 °C for 4 h to condense the fluffy powders. The impregnated MoOx catalysts were dried at room temperature overnight, then dried at 120 °C for 2 h in flowing air (100 mL min−1), and finally calcined by ramping the temperature at 1 °C min−1 in flowing air (100 mL min−1) to 500 °C and hold 500 °C for 4 h. The loading (surface coverage) of Mo for Al2O3, TiO2, ZrO2 and SiO2 support MoOx catalysts is 18% (4.6 Mo/nm2), 5.8% (4.6 Mo/nm2), 5.8% (4.3 Mo/nm2), 7.5% (0.9 Mo/nm2) that are close to monolayer surface coverage, with the exception of SiO2 that is not able to form complete surface metal oxide monolayers because of the poor reactivity of the surface silanols.In situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS)
The in situ DRIFTS spectra of the supported MoOx catalysts were collected by a Thermo Scientific Nicolet 8700 FT-IR spectrometer equipped with a Harrick Praying Mantis attachment (DRA-2 with CaF2 window). A mercury–cadmium–telluride (MCT) detector was used to collect the spectra with a resolution of 4 cm−1 and an accumulation of 96 scans per min. For each experiment, ∼20 mg of catalysts powders were loaded into an in situ cell (Harrick, HVC-DR2 with a CaF2 window). The gas flow rates were controlled by Brooks 5850E mass flow controller. The procedure for collecting dehydrated in situ DRIFTS spectra was as follows: the catalyst was heated at 10 °C min−1 from room temperature to 500 °C under flowing 10% O2/Ar (Praxair, UHP, 30 mL min−1) and held for 1 h. Then the temperature was cooled at 10 °C min−1 to 120 °C under flowing 10% O2/Ar. The procedure for collecting in situ propylene-TPSR-IR was the same as the procedure below in TPSR with MS.In situ Raman spectroscopy
The in situ Raman spectra of supported MoOx catalysts were collected by a Horiba Labram HR Evolution spectrometer (532 nm). The laser was focused on the catalysts through a confocal microscope equipped with a X50 objective lens (Olympus BX-30). A 900 grooves per nm grating was selected to optimize the spectral resolution (∼1 cm−1). The calibration of Raman spectrometer was performed with a silicon standard possessing a reference peak of 520.7 cm−1. The gas flow rates were monitored with the same mass flow controllers as indicated above. Approximately 20 mg of catalysts powders were loaded into an in situ cell (Harrick Scientific HVC-MRA-5). The spectra were collected with an accumulation of 3 scans (20 s per scan) by a CCD camera detector (Horiba-Jobin Yvon CCD-3000 V). The dehydration procedure is the same as the DRIFTS above.In situ UV-vis Diffuse Reflectance Spectroscopy (DRS)
The in situ UV-vis spectra of the supported MoOx catalysts were obtained with an Agilent Cary 5000 UV-vis-NIR spectrophotometer. Approximately 20 mg of catalyst powders were loaded into an in situ cell described above. The UV-vis spectrum was collected in the 200–800 nm range. A MgO (Sigma-Aldrich, 99.999%) white standard was used as the standard for spectra baseline. The gas flow rates were monitored with the same mass flow controllers as indicated above. The edge energy (Eg) values were calculated from the intercept of the straight line for the low-energy rise of a plot of [F(R)hν]2versus the incident photon energy (hν).12 The dehydration procedure is the same as the DRIFTS above.Propylene-Temperature Programmed Surface Reaction (TPSR) with MS
The C3H6-TPSR spectroscopy was performed with an Altamira Instruments AMI-200. The catalysts powders (∼0.1 g) were loaded into a U-tube quartz reactor. The dehydration procedure was similar as indicated above in DRIFTS experiments. After dehydration, the reactor was flushed with Ar (Air Gas, UHP, 30 mL min−1) for 30 minutes at 30 °C, then 5% C3H6/Ar (Praxair, Purity 99%, 30 mL min−1) was flowed and held at 30 °C for several minutes to stabilize the MS signal. The reactor was heated at 10 °C min−1 to 600 °C. An online quadrupole mass spectrometer (Dycor ProLine Process) was equipped with AMI-200 to analyze the outgoing gases. The monitoring mass/charge channels were m/z = 18 (H2O), m/z = 28 (CO), m/z = 42 (C3H6), m/z = 44 (CO2) m/z = 56 (C4H8) and m/z = 58 (acetone).Ethylene/2-butene titration
The C2H4/C4H8-titration experiments were performed with the same Altamira Instruments AMI-200. The dehydration procedure was the same as the above DRIFTS experiments. After flushing with Ar at 30 °C for 30 min, 1% C4H8/Ar (Praxair, Purity 99%, 30 mL min−1) was flowing at 30 °C for 30 min to chemisorb 2-butene. The gas flow was then immediately switched to 1% C2H4/Ar (Praxair, Purity 99%, 30 mL min−1) for 30 min to titrate the surface intermediates achieved from 2-butene flowing. The same m/z channels were monitored as for the above C3H6-TPSR experiments. It was assumed that only one surface MoCHCH3 intermediate was present on the surface. The number of activated surface MoOx sites was calculated by the amount of C3H6 produced during C2H4–C4H8 titration accordingly.
Steady state propylene metathesis
The steady-state propylene self-metathesis activities were collected in a fix-bed reactor under differential conditions (propylene conversion <15%). The gas tubes were heated to 130 °C to avoid propylene reactant and products condensation. Approximately 0.1 g of catalyst was loaded in a tube reactor. A clam shell furnace was equipped around the reactor to control the reactor temperature. The dehydration procedure was similar as indicated above in DRIFTS experiments. After dehydration, the reactor was cooled to 100 °C with 10% O2/Ar and flushed with Ar (Air Gas, UHP, 30 mL min−1). 1% C3H6/Ar (Praxair, Purity 99%, 50 mL min−1) was flowing at 100 °C. The steady-state propylene self-metathesis conversion was collected after 60 min reaction. An online Agilent 6890 gas chromatograph (GC) equipped with a GS-Alumina (Agilent 1153552) column and an Agilent G1531 flame ionization detector (FID) was used to analyze the outgoing gases from the reactor. The catalytic activities (mmol g−1 h−1) were obtained by normalizing the conversion of propylene by the flow rate and catalyst weight. The turnover frequency (TOF) values were obtained by normalizing the steady-state activity by the number of activated MoOx sites derived from C2H4/C4H8 titration.III. Results
In situ DRIFTS of surface hydroxyl anchoring sites
The in situ DRIFTS difference spectra and original spectra reveal the surface hydroxyls of oxide supports are involved in anchoring MoOx species and are presented in Fig. 1 and S1.† The bare oxide supports have multiple surface hydroxyls (isolated S-OH, bridged S2-OH and tri-coordinated S3-OH, with S representing the support cation) that depend on the specific oxide support. The bare SiO2 support has isolated Si–OH (3737 cm−1) and unique geminal Si(–OH)2 (broad peak ∼3727–3755 cm−1) surface hydroxyls and the MoOx species primarily anchor at the isolated Si–OH surface hydroxyls. The slight perturbation of the vibrations of the germinal Si(–OH)2 surface hydroxyls occur because of band broadening from hydrogen bonding between the germinal surface hydroxyls and adjacent surface MoOx sites at maximum surface MoOx coverage. The bare TiO2 support contains isolated Ti–OH (3720 cm−1) and bridged Ti–(OH)–Ti (3670 cm−1) and the MoOx species anchors at both surface hydroxyls at monolayer coverage. The bare ZrO2 support exhibits isolated Zr–OH (3740 cm−1) and tri-coordinated Zr3–OH (3688 cm−1) and the MoOx species anchor at both surface hydroxyls at monolayer surface MoOx coverage. The bare Al2O3 support has multiple surface hydroxyls (isolated Al–OH (HO-μ1-AlIV at 3787 cm−1, HO-μ1-AlVI at 3765 cm−1, HO-μ1-AlV at 3741 and 3730 cm−1), bridged Al2–OH (HO-μ2-AlV at 3694 cm−1) and tri-coordinated Al3–OH (HO-μ3-AlVI at 3674 cm−1)) and the MoOx species anchor at all five surface hydroxyls at monolayer surface MoOx coverage.24–30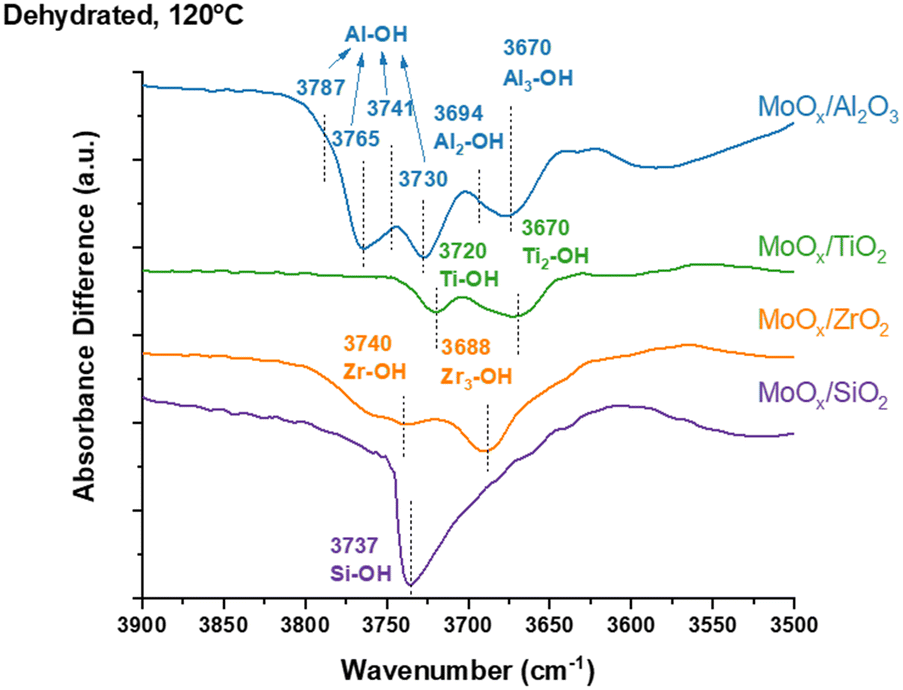 | ||
| Fig. 1 In situ DRIFTS difference spectra of the surface hydroxyl region of the supported MoOx catalysts under dehydrated conditions. The spectrum of the dehydrated oxide support was subtracted from the spectrum of each supported MoOx catalyst. | ||
In situ Raman under dehydrated conditions
The in situ Raman spectra of the supported MoOx catalysts under dehydrated conditions are presented in Fig. 2(A). The Al2O3, TiO2 and ZrO2 supports don't give rise to Raman bands in the 900–1200 cm−1 region. The TiO2 support has a very weak Raman band ∼800 cm−1 from anatase phase of TiO2. The SiO2 support has a weak band ∼970 cm−1 from Si–OH that is overshadowed by the stronger MoO band of the surface MoOx sites.31,32 Crystalline MoO3 nanoparticles (NPs) are not present on the Al2O3-, TiO2-, ZrO2-, and SiO2-supported MoOx catalysts because of the absence of sharp and strong Raman bands of crystalline MoO3 ∼820 and ∼997 cm−1. The surface MoOx sites on SiO2 give rise to both a strong vs(MoO) band at ∼986 cm−1 and a weaker vas(MoO) band at ∼965 cm−1 from dioxo (O)2MoO4 sites.33 Only the surface MoOx sites on Al2O3 and ZrO2 give rise to a broad band ∼862–865 cm−1 from the bridging Mo–O–Al and Mo–O–Zr vibrations. The bridging Mo–O–Ti and Mo–O–Si Raman bands are too weak to be observed. The Raman bands from the surface MoOx sites on the other supports give rise to broader bands in the ∼997–1006 cm−1 region associated with the vs(MoO) vibration of the surface mono-oxo OMoO4 sites.10,12 The Raman bands for the surface MoOx sites are broader on the non-SiO2 supports because of the presence of multiple anchoring surface hydroxyl sites and surface MoOx structures.
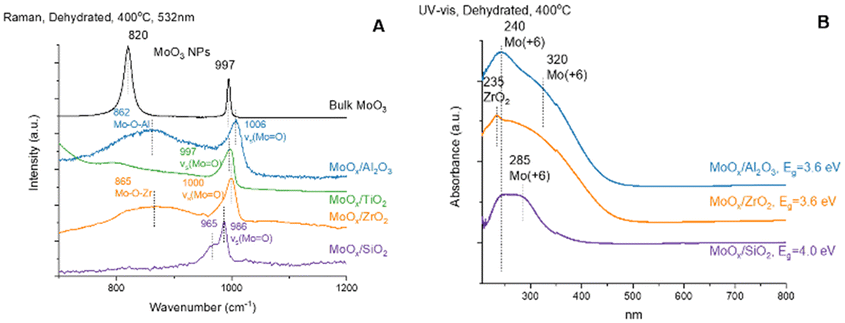 | ||
| Fig. 2 (A) In situ Raman spectra of the supported MoOx catalysts under dehydrated conditions at 400 °C. (B) In situ UV-vis DRS spectra of the supported MoOx catalysts under dehydrated conditions at 400 °C. | ||
In situ UV-vis spectroscopy under dehydrated conditions
The in situ UV-vis spectra of the supported MoOx catalysts under oxidatively dehydrated conditions are presented in Fig. 2(B). The dehydrated supported MoOx/SiO2 catalyst exhibits two LMCT bands at 240 and 285 nm (Eg ∼4.0 eV) corresponding to isolated surface MoOx sites. The TiO2 support possesses strong absorbance in the region below 350 nm and, thus, the bands of the surface MoOx sites are overshadowed by the much stronger TiO2 support bands and are not reported. The dehydrated supported MoOx/ZrO2 catalyst has one modest band at 235 nm from the ZrO2 support and one broad LMCT band at 250–350 nm corresponding to both isolated and oligomeric MoOx sites (Eg ∼3.6 eV), respectively. The dehydrated supported MoOx/Al2O3 catalyst exhibits two LMCT bands at ∼240 and 320 nm (Eg ∼3.6 eV) corresponding to isolated MoOx and oligomeric MoOx sites, respectively.10,12,31 The UV-vis LMCT bands at 240–320 nm and absence of UV-vis d–d bands at 350–800 nm indicate that the dehydrated surface MoOx sites on the Al2O3, ZrO2 and SiO2 supports are fully oxidized as Mo(+6). Although the LMCT bands for the dehydrated MoOx/TiO2 could not be determined because of the strong absorbance of the TiO2 support, the nature of the surface MoOx sites were determined from in situ XANES12,16 and found to primarily consist of oligomeric Mo(+6)Ox sites. The XANES study16 also revealed that MoOx sites on Al2O3 and ZrO2 supports are similar, and consist of both isolated Mo(+6)Ox and oligomeric Mo(+6)Ox sites. The surface MoOx sites on SiO2 support appear to possess only isolated Mo(+6)Ox sites.Propylene-TPSR-MS (C3=-TPSR-MS)
The surface chemistry of the supported MoOx catalysts were chemically probed with C3=-TPSR-MS. The C3=-TPSR-MS spectra of dehydrated supported MoOx catalysts and Mo-free oxide supports are presented in Fig. 3 and S2.† The Mo-free oxide supports don't produce any olefin or acetone reaction products between 30–600 °C. Only CO, CO2 and H2O formation from the combustion of propylene was observed above 500 °C. The supported MoOx/TiO2 and MoOx/ZrO2 catalysts essentially don't produce any 2-butenes over the 30–600 °C temperature range. The supported MoOx/SiO2 catalyst only produces 2-butenes at the high temperatures of ∼350–600 °C (peak temperature – Tp = 520 °C). The dehydrated supported MoOx/Al2O3 catalyst produces 2-butenes in two temperature ranges: ∼30–200 °C (Tp = 62 °C) and ∼400–600 °C (Tp = 530 °C). Oxygenated combustion products (CO, CO2, H2O) are also observed on all the supported MoOx catalysts during C3=-TPSR-MS, which reflects the partial reduction of the surface MoOx sites upon exposure to propylene at very high temperatures. Acetone is produced during the activation of the surface MoOx sites and the acetone desorption temperature varies slightly with the specific support: MoOx/Al2O3 (Tp = 221 °C), MoOx/TiO2 (Tp = 273 °C), MoOx/ZrO2 (Tp = 250 °C), and MoOx/SiO2 (Tp = 190, 520 °C). Comparison of the C3= signals during C3=-TPSR-MS in Fig. S3† indicates that propylene is only consumed and selectively converted to 2-butene by the supported MoOx/Al2O3 catalyst at low temperatures and both the supported MoOx/Al2O3 and MoOx/SiO2 catalysts at elevated temperatures. Consumption of propylene is very limited by the supported MoOx/TiO2 and MoOx/ZrO2 catalysts and CO2 is the dominant reaction product at elevated temperatures with essentially no 2-butene production.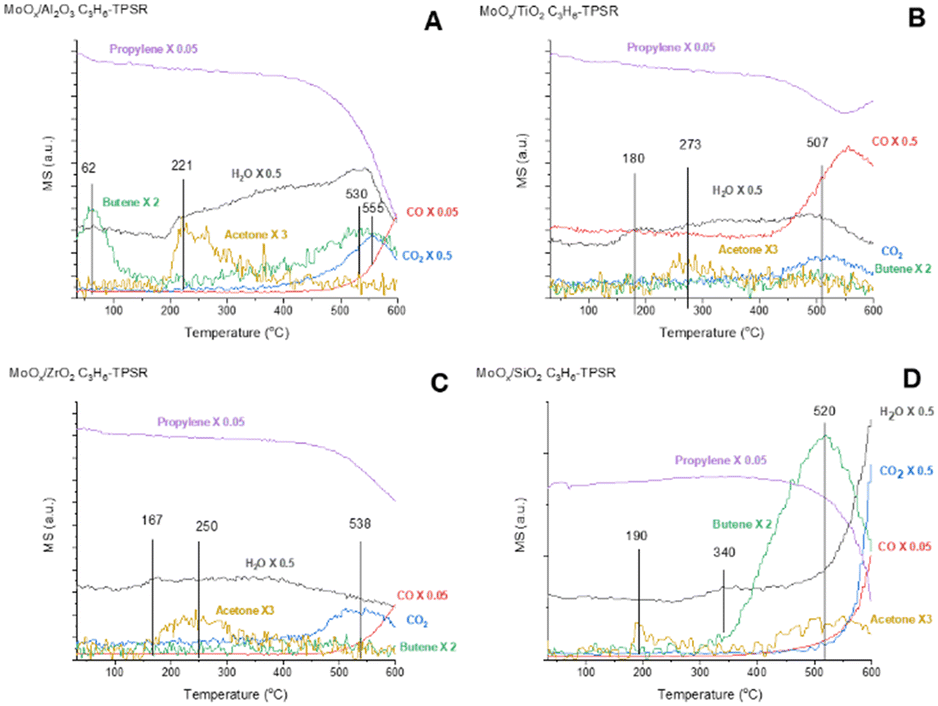 | ||
| Fig. 3 Propylene-TPSR (30–600 °C) with online MS for the supported MoOx catalysts: (A) MoOx/Al2O3, (B) MoOx/TiO2, (C) MoOx/ZrO2, (D) MoOx/SiO2. | ||
In situ propylene-TPSR-IR (C3=-TPSR-IR)
The in situ C3=-TPSR-IR spectra from the dehydrated supported MoOx catalysts are presented in Fig. 4. The gas phase propylene molecule gives rise to IR bands ∼1376, 1392, 1444, 1455, 1636, and 1664 cm−1.14,34 At 30 °C, the IR spectra are dominated by the vibrations of gas phase propylene as presented in Fig. S4.† Only the supported MoOx/Al2O3 shows the presence of a v(CO) vibration at ∼1686 cm−1 from surface acetone species.14 The gas phase propylene bands diminish above 120 °C from the supported MoOx/Al2O3 catalyst that reflect consumption of propylene. Strong bands from v(CC) ∼1664 cm−1 of adsorbed propylene on MoOx sites and v(C–C) ∼1250 cm−1, δs(CH3) ∼1375 cm−1 and v(CO) ∼1686 cm−1 of adsorbed acetone predominate between 120–220 °C. The surface acetone further reacts from 220–600 °C to form surface acetate intermediates with vs(COO−) at ∼1441 cm−1 and vas (COO−) at ∼1570 cm−1 vibrations.14 The supported MoOx/TiO2 and MoOx/ZrO2 catalysts behave similarly during C3=-TPSR-IR with the gas phase propylene bands diminishing above 120 °C and the IR bands from v(C–C) at ∼1250 cm−1, δs(CH3) at ∼1375 cm−1 and a much stronger v(CO) band at ∼1680–1665 cm−1 from vibration of adsorbed surface acetone predominates between 120–420 °C. The IR band from the v(CC) ∼1664 cm−1 vibration of adsorbed propylene on the surface MoOx sites are relatively weak and overshowed by the very strong IR v(CO) vibration at ∼1680–1665 cm−1. However, the strong IR bands for the surface acetate intermediates (vs(COO−) at ∼1441 cm−1 and vas(COO−) at ∼1570 cm−1 vibrations) reveals that the surface acetate intermediates dominate between 220–600 °C. The gas phase propylene bands only diminish above 320 °C for the supported MoOx/SiO2 catalyst reflecting the lower activity of this catalyst among the catalysts. Additionally, surface intermediates could not be detected during C3=-TPSR-IR, most probably due to the extremely low population of surface intermediates. The broad IR bands at ∼1279 and 1570 cm−1 from MoOx/SiO2 are induced by a thermal effect on the spectral baseline, and not from surface intermediates, as indicated in Fig. S5.† The C3=-TPSR-IR spectra reveal that while the surface of the supported MoOx/Al2O3 catalyst is dominated by adsorbed acetone species at 120–420 °C, the surfaces of the supported MoOx/TiO2 and MoOx/ZrO2 catalysts are dominated by adsorbed surface acetate intermediates in the 220–420 °C.
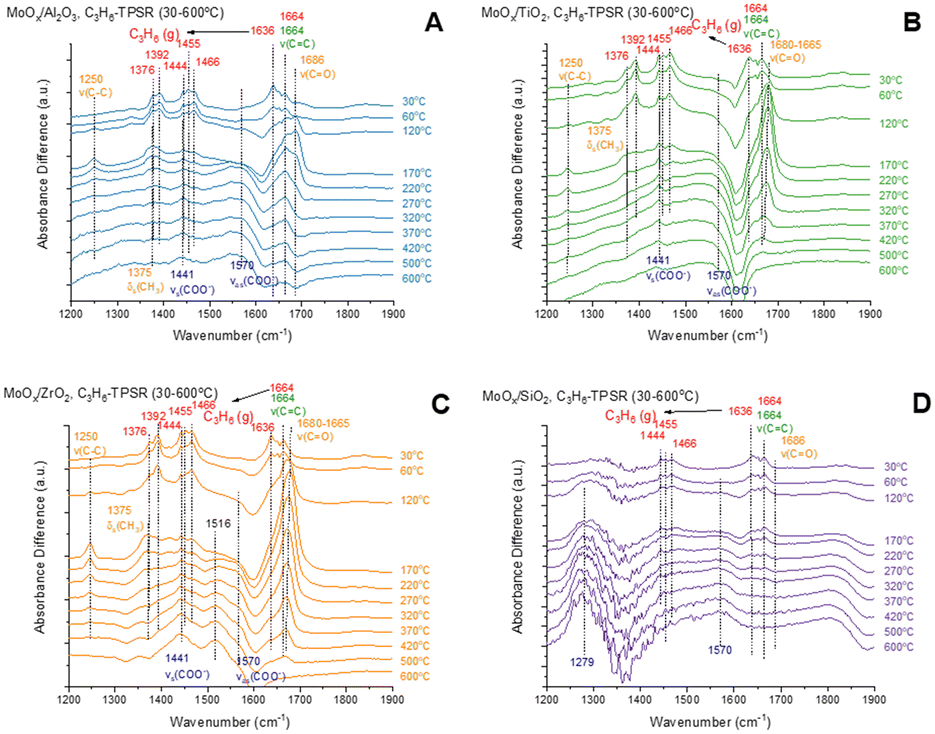 | ||
| Fig. 4 Propylene-TPSR (30–600 °C) with in situ DRIFTS difference spectra of supported MoOx catalysts. The spectra of dehydrated MoOx catalysts at 30 °C were subtracted from the spectra of MoOx catalysts during TPSR with propylene: (A) MoOx/Al2O3, (B) MoOx/TiO2, (C) MoOx/ZrO2, (D) MoOx/SiO2. | ||
Ethylene/2-butene titration and steady state propylene metathesis
Ethylene/2-butene titration measurements were undertaken to determine the number of activated sites for the supported MoOx catalysts and the values are presented in Table 1. Although the number of activated sites was relatively high for the supported MoOx/Al2O3 catalyst, the ZrO2, TiO2 and SiO2-supported catalysts barely contain any activated surface MoOx sites when exposed to 2-butene at 30 °C.| MoOx/Al2O3 | MoOx/TiO2 | MoOx/ZrO2 | MoOx/SiO2 | |
|---|---|---|---|---|
| Fraction of activated MoOx sites | 17% | 0.3% | 0.2% | 0.2% |
| Steady state activity (100 °C, mmol g−1 h−1) | 0.29 | 0 | 0 | 0 |
| TOF (s−1) | 3.8 × 10 −4 | 0 | 0 | 0 |
The steady state propylene self-metathesis activity of the supported MoOx catalysts at 100 °C are also shown in Table 1. The supported MoOx/TiO2, MoOx/ZrO2 and MoOx/SiO2 do not show any steady state activity for propylene metathesis at 100 °C. Only the supported MoOx/Al2O3 catalyst is active for propylene metathesis at 100 °C. The turnover frequency (TOF) values were calculated by normalizing the steady state activity values by the number of activated surface MoOx sites determined from the ethylene/2-butene titration at 30 °C. Given that the supported MoOx/TiO2, MoOx/ZrO2 and MoOx/SiO2 catalysts do not produce butene, their TOF values are zero within our detection limits. Therefore, the TOF can only be determined for the supported MoOx/Al2O3 catalyst. In contrast, the conversion and turnover frequency (TOF) value for the supported MoOx/Al2O3 catalyst could be determined and found to be 8.1% and 3.8 × 10−4 s−1.
IV. Discussion
Anchoring surface hydroxyls for MoOx species
Anchoring the maximum amount of surface MoOx sites on the oxide supports titrates all the accessible S-OH, S2-OH and S3-OH surface hydroxyls. Only the geminal Si(–OH)2 surface hydroxyls on the SiO2 support are unreactive or minimally reactive towards anchoring of the MoOx species (see Fig. 1).35,36 Besides the nature of the surface hydroxyl anchoring sites (isolated, bridged or tri-coordinated), the chemical properties of the surface hydroxyls may also be influenced by the local surface coordination (AlO5–6, TiO6, ZrO7, SiO4)24,25,37–39 and cation electronegativity (Si > Al > Ti > Zr)40 of the oxide support.Molecular and electronic structure of dehydrated surface MoOx sites
The dehydrated surface MoOx sites for all the supported MoOx catalysts are fully oxidized as Mo(+6) (only LMCT bands and absence of d–d bands from reduced surface MoOx sites). The supported MoOx/SiO2 catalyst only stabilizes the isolated dioxo surface (O)2MoO2 sites since only the Si–OH surface hydroxyl is available for anchoring.16,33,41,42 The supported MoOx/TiO2 catalyst mostly contains oligomeric mono-oxo surface MoO5/6 sites at monolayer surface coverage.16,41,43–45 The supported MoOx/ZrO2 catalyst consists of isolated dioxo surface MoO4 and oligomeric mono-oxo surface MoO5/6 sites at monolayer surface coverage.16,41,46,47 The supported MoOx/Al2O3 catalyst also contains a mixture of isolated dioxo surface MoO4 and oligomeric mono-oxo surface MoO5/6 sites at monolayer surface coverage.10,12,16,41 The presence of oligomeric surface MoO5/6 sites on the Al2O3, TiO2 and ZrO2 supports is related to the more acidic S2-OH and S3-OH surface hydroxyls available as anchoring sites on these oxide supports at high surface MoOx coverage. This is consistent with the observation that at low surface MoOx coverage, the MoOx species are preferentially anchored at the more basic S-OH sites that only stabilizes isolated surface MoOx sites.10,48,49
Surface chemistry of the supported MoOx catalysts
The surface chemistry of the supported MoOx catalysts was monitored with in situ IR spectroscopy. Only the supported MoOx/Al2O3 catalyst formed CO bonds from propylene adsorption at 30 °C (Fig. S4†) that is attributed to surface isopropoxide species formed by protonation of the surface propylene species. The surface isopropoxide species further form adsorbed acetone bonded to the surface MoOx sites.14 The supported MoOx/TiO2 and MoOx/ZrO2 catalysts are completely inactivated at 30 °C. At higher temperatures of 120–220 °C, the surface MoOx sites on Al2O3, TiO2 and ZrO2 produce surface acetone that desorbs from the surface above 220 °C or further oxidize to stable surface acetate intermediates (Fig. 4). Above 220 °C, the surface MoOx sites on TiO2 and ZrO2 produce more surface acetone (stronger v(CO) IR bands) than the surface MoOx sites on Al2O3. While the surface acetone intermediates persist until 420 °C on MoOx/TiO2 and MoOx/ZrO2, the surface acetone intermediates only persist up to 270 °C on MoOx/Al2O3 (Fig. 4). At the higher temperatures, the adsorbed acetone species further oxidize to stable surface acetate intermediates. The greater amount of surface acetate intermediates for the supported MoOx/TiO2 and MoOx/ZrO2 catalysts is related to the greater redox activity of these two catalysts probed by H2-TPR (MoOx/TiO2 > MoOx/ZrO2 > MoOx/Al2O3 > MoOx/SiO2).11,21 The greater population of more stable surface acetate species on the supported MoOx/TiO2 and MoOx/ZrO2 catalysts indicates that very few sites will be available for propylene adsorption and metathesis (site blocking).14 The supported MoOx/SiO2 catalyst might only have trace amounts of surface intermediates at both low and high temperatures because of the absence of IR peaks from surface intermediates. The formation of surface oxygenates are only related to surface MoOx sites, not related to the reduction of the oxide supports, even for the more reducible TiO2 and ZrO2 supports (see Fig. S2†).
Comparison of the TOF values of surface MoOx sites
The steady-state reaction was performed under differential reaction conditions with highly diluted (1%) propylene to avoid heat and mass transfer issues during the propylene metathesis reaction. The supported MoOx catalysts were extensively studied in the literature and the activities were measured with various oxide supports and reaction conditions: MoOx/SiO2 at 20 °C,13 MoOx/Al2O3–SiO2 at 150 °C,50,51 MoOx/SBA-15 at 50 °C,15 MoOx/Al2O3, MoOx/SiO2, and MoOx/Al2O3–SiO2 at 30 °C,52 MoOx/SBA-1,53 MoOx/Si-SBA-1 and MoOx/Al-SBA-154 at 50 °C. To make a quantitative comparison between TOF values reported in the literature, the TOF values need to be corrected for the following factors: (i) correction based on the number of activated sites Ns, which depends on the oxide support and surface MoOx coverage, (ii) correction based on the reaction temperatures and (iii) correction based on partial pressure of reactants (1st order reaction). The TOF values of our MoOx/Al2O3 catalyst and the TOF values reported in the literature can't be quantitatively compared since the number of activated sites Ns are not determined and reported in the literature. Earlier studies only reported olefin metathesis activities of H2 reduced MoOx/TiO2 and MoOx/ZrO2 catalysts. Non H2-reduced supported MoOx/TiO2 and MoOx/ZrO2 catalysts have been found to be inactive for olefin metathesis as reported in the present study.6,7 It is well established that maximum olefin metathesis activity corresponds to monolayer surface coverage,10,17 the loading of supported MoOx/TiO2 and MoOx/ZrOx catalysts were carefully controlled to maximize the coverage of surface MoOx sites without generating crystalline MoO3 nanoparticles, thus, the surface coverage of supported MoOx/TiO2 and MoOx/ZrOx catalysts in this study are properly selected to determine the activity of the catalysts. It's also well accepted that supported MoOx/SiO2 catalyst requires high temperature pre-activation under reducing environment for propylene metathesis.13,52–55 The activity of high temperature pre-activated MoOx/SiO2 catalysts, however, will be addressed in a forthcoming paper since the current study only focuses on the low temperature activity of supported MoOx catalysts without high temperature pre-activation.Influence of oxide support on the activation and activity of surface MoOx sites
The activation of the surface MoOx sites on the different oxide supports by propylene is reflected by the appearance of the propylene metathesis C4= products during C3=-TPSR-MS (Fig. 3). Only the surface MoOx sites on Al2O3 become activated below 100 °C and surface MoOx sites on Al2O3 and SiO2 also become activated above 500 °C, but the supported MoOx/TiO2 and MoOx/ZrO2 catalysts do not become activated for propylene metathesis. The facile formation of acetone and propylene metathesis products from the supported MoOx/Al2O3 catalysts below 100 °C indicates the ease of activation of the surface MoOx sites on Al2O3. The surface MoOx sites anchored at the isolated hydroxyls could only be activated at high temperatures, while the surface MoOx sites anchored at the bridged or tri-coordinated hydroxyls permit activation at low temperatures.10,56 Besides isolated surface hydroxyls, the Al2O3, TiO2 and ZrO2 contain either bridged (Al2O3, TiO2) or tri-coordinated surface hydroxyls (Al2O3, ZrO2) serving as the anchoring sites for MoOx. What is different between them is the different support cations with Al2O3 having a higher electronegativity than the TiO2 and ZrO2 cations.38,40 The SiO2 support, however, has a higher electronegativity than Al2O3 and SiO2 is not able to activate the surface MoOx sites at low temperatures. The difficulty of activating the surface MoOx sites on SiO2 at low temperature is related to the absence of S2-OH or S3-OH anchoring hydroxyls for surface MoOx sites. At high temperature, the surface MoOx sites anchored at the isolated Si–OH exhibit a better activation than the MoOx sites anchored at isolated Al–OH (Fig. S6†) due to the stronger electronegativity of SiO2. The stronger electronegative support cation will influence the electron deficiency of the anchoring surface hydroxyls, thus, making the anchored surface MoOx sites more electron deficient for interaction with electron rich olefins. Oxide supports with more electronegative cations will more readily activate the surface MoOx sites for olefin metathesis. It is concluded that surface MoOx sites anchored at isolated surface hydroxyls with strong support electronegativity (Al–OH, Si–OH) enables the activation of the surface MoOx sites at high temperatures, while surface MoOx sites anchored at bridged/tri-coordinated surface hydroxyls with strong support electronegativity (Al2–OH, Al3–OH) enables the activation of MoOx sites below 100 °C. Furthermore, the presence of stable surface acetone and acetate surface intermediates further retards olefin metathesis for the more redox MoOx/TiO2 and MoOx/ZrO2 catalysts. The influence of surface Lewis/Brønsted acidity on propylene metathesis has recently been reviewed in detail56 and a general conclusion about the influence of surface Lewis/Brønsted acidity on olefin metathesis could not be reached from the literature findings.V. Conclusions
The oxide support tunes the anchoring, molecular structure, activation and surface chemistry of the surface MoOx sites. Anchoring of molybdena species at the bridged and tri-coordinated surface hydroxyls (S2-OH and S3-OH) with higher support cation electronegativity (Al > Ti > Zr) permits the activation of surface MoOx sites at low temperatures (<100 °C). The isolated surface hydroxyls (S-OH) with strong support cation electronegativity (Si > Al > Ti > Zr) are responsible for the activation of surface MoOx sites at high temperatures (>350 °C). Oligomeric surface MoOx sites on Al2O3 activate at low temperatures and isolated surface MoOx sites on Al2O3 and SiO2 also activate at high temperatures. The significant accumulation of surface acetone and acetate species on the more redox active supported MoOx/TiO2 and MoOx/ZrO2 catalysts retards olefin metathesis. The above trends are responsible for the resulting propylene metathesis activity for supported MoOx catalysts: MoOx/Al2O3 > MoOx/SiO2 > MoOx/TiO2 ∼ MoOx/ZrO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy, Catalysis Science Program, under Award Number FG02-93ER14350.References
- S. Kim, S. Jeong and E. Heo, Effects of the shale boom on ethylene and propylene prices, Energy Sources, Part B, 2019, 14(3), 49–66 CrossRef.
- R. H. Grubbs, Handbook of Metathesis, Wiley-VCH, Weinheim, 2015 Search PubMed.
- J. C. Mol, Industrial applications of olefin metathesis, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- S. Lwin and I. E. Wachs, Olefin Metathesis by Supported Metal Oxide Catalysts, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, London, 1997 Search PubMed.
- K. Tanaka, K. Miyahara and K. Tanaka, The characterization of a novel metathesis catalysts, β-titanium oxide-supported molybdenum oxide, Bull. Chem. Soc. Jpn., 1981, 54, 3106–3109 CrossRef CAS.
- V. Indovina, A. Cimino, D. Cordischi, S. D. Bella, S. D. Rossi, D. Ferraris, D. Gazzoli, M. Occhiuzzi and M. Valigi, The Catalytic Activity of MoOx/ZrO2 in the Hydrogenation and Metathesis of Propene, Stud. Surf. Sci. Catal., 1993, 75, 875–887 CrossRef CAS.
- T. Otroshchenko, Q. Zhang and E. V. Kondratenko, Room-Temperature Metathesis of Ethylene with 2-Butene to Propene over MoOx-Based Catalysts: Mixed Oxides as Perspective Support Materials, Catal. Lett., 2022, 152, 2366–2374 CrossRef CAS.
- H. Hu and I. E. Wachs, Catalytic Properties of Supported Molybdenum Oxide Catalysts: In Situ Raman and Methanol Oxidation Studies, J. Phys. Chem., 1995, 99(27), 10911–10922 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, Molecular Structure–Reactivity Relationships for Olefin Metathesis by Al2O3-Supported Surface MoOx Sites, ACS Catal., 2018, 8(2), 949–959 CrossRef CAS.
- D. S. Kim, I. E. Wachs and K. Segawa, Molecular Structures and Reactivity of Supported Molybdenum Oxide Catalysts, J. Catal., 1994, 149(2), 268–277 CrossRef CAS.
- H. Tian, C. A. Roberts and I. E. Wachs, Molecular Structural Determination of Molybdena in Different Environments: Aqueous Solutions, Bulk Mixed Oxides, and Supported MoO3 Catalysts, J. Phys. Chem. C, 2010, 114, 14110–14120 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, T. L. Drake, W. Wu, Y. Lin, E. Weitz, L. D. Marks and P. C. Stair, Highly Efficient Activation, Regeneration, and Active Site Identification of Oxide-Based Olefin Metathesis Catalysts, ACS Catal., 2016, 6, 5740–5746 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, Activation Mechanism and Surface Intermediates during Olefin Metathesis by Supported MoOx/Al2O3 Catalysts, J. Phys. Chem. C, 2019, 123(19), 12367–12375 CrossRef CAS.
- K. Amakawa, S. Wrabetz, J. Krohnert, G. Tzolova-Muller, R. Schlogl and A. Trunschke, In Situ Generation of Active Sites in Olefin Metathesis, J. Am. Chem. Soc., 2012, 134, 11462–11473 CrossRef CAS.
- H. Hu, I. E. Wachs and S. R. Bare, Surface Structures of Supported Molybdenum Oxide Catalysts: Characterization by Raman and Mo L3-Edge XANES, J. Phys. Chem., 1995, 99, 10897–10910 CrossRef CAS.
- K. Amakawa, L. Sun, C. Guo, M. Havecker, P. Kube, I. E. Wachs, S. Lwin, I. E. Frenkel, A. Patlolla, K. Hermann, R. Schlogl and A. Trunschke, How Strain Affects the Reactivity of Surface Metal Oxide Catalysts, Angew. Chem., Int. Ed., 2013, 52, 13553–13557 CrossRef CAS PubMed.
- J. P. Thielemann, T. Ressler, A. Walter, G. Tzolova-Muller and C. Hess, Structure of molybdenum oxide supported on silica SBA-15 studied by Raman, UV–Vis and X-ray absorption spectroscopy, Appl. Catal., A, 2011, 399, 28–34 CrossRef.
- J. Handzlik, Properties and metathesis activity of monomeric and dimeric Mo centres variously located on γ-alumina − A DFT study, Surf. Sci., 2007, 601, 2054–2065 CrossRef.
- B. Zhang, N. Liu, Q. Lin and D. Jin, The effects of MO oxidation states on olefin metathesis, J. Mol. Catal., 1991, 65, 15–28 CrossRef.
- S. T. Oyama, R. Radhakrishnan, M. Seman, J. N. Kondo, K. Domen and K. Asakura, Control of Reactivity in C−H Bond Breaking Reactions on Oxide Catalysts: Methanol Oxidation on Supported Molybdenum Oxide, J. Phys. Chem. B, 2003, 107(8), 1845–1852 CrossRef.
- M. A. Bañares and S. J. Khatib, Structure–activity relationships in alumina-supported molybdena–vanadia catalysts for propane oxidative dehydrogenation, Catal. Today, 2004, 96(4), 251–257 CrossRef.
- M. A. Vuurman, D. J. Stufkens, A. Oskam and I. E. Wachs, Structural determination of surface rhenium oxide on various oxide supports (Al2O3, ZrO2, TiO2 and SiO2), J. Mol. Catal., 1992, 76, 263–285 CrossRef.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Hydroxyl Groups on yAlumina Surfaces: A DFT Study, J. Catal., 2002, 211, 1–5 CrossRef.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Use of DFT to achieve a rational understanding of acid-basic properties of γ-alumina surfaces, J. Catal., 2004, 226, 54–68 CrossRef.
- G. Busca, Catalytic materials based on silica and alumina: Structural features and generation of surface acidity, Prog. Mater. Sci., 2019, 104, 215–249 CrossRef.
- J. M. Kim, S. M. Chang, S. M. Kong, K. S. Kim, J. Kim and W. S. Kim, Control of hydroxyl group content in silica particle synthesized by the sol-precipitation process, Ceram. Int., 2009, 35, 1015–1019 CrossRef.
- A. A. Tsyganenko and V. N. Filimonov, Infrared spectra of surface hydroxyl groups and crystalline structure of oxides, J. Mol. Struct., 1973, 19, 579–589 CrossRef.
- N. Y. Topsoe, Characterization of the Nature of Surface Sites on Vanadia-Titania Catalysts by FTIR, J. Catal., 1991, 128, 499–511 CrossRef.
- A. M. Turek, I. E. Wachs and E. Decanio, Acidic Properties of Alumina-Supported Metal Oxide Catalysts: An Infrared Spectroscopy Study, J. Phys. Chem., 1992, 96, 5000–5007 CrossRef.
- E. L. Lee and I. E. Wachs, In Situ Spectroscopic Investigation of the Molecular and Electronic Structures of SiO2 Supported Surface Metal Oxides, J. Phys. Chem. C, 2007, 111, 14410–14425 CrossRef.
- B. Zhang, S. Lwin, S. Xiang, A. I. Frenkel and I. E. Wachs, Tuning the Number of Active Sites and Turnover Frequencies by Surface Modification of Supported ReO4/(SiO2−Al2O3) Catalysts for Olefin Metathesis, ACS Catal., 2021, 11, 2412–2421 CrossRef.
- E. L. Lee and I. E. Wachs, In Situ Raman Spectroscopy of SiO2-Supported Transition Metal Oxide Catalysts: An Isotopic 18O-16O Exchange Study, J. Phys. Chem. C, 2008, 112, 6487–6498 CrossRef.
- H. Y. Afeefy, J. F. Liebman and S. E. Stein, Neutral Thermochemical Data, in NIST Chemistry WebBook, ed. P. J. Linstrom and W. G. Mallard, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg, MD, 2005, p. 20899 Search PubMed.
- H. Knozinger and P. Ratnasamy, Catalytic Aluminas: Surface Models and Characterization of Surface Sites, Catal. Rev.: Sci. Eng., 1978, 17, 31–37 CrossRef.
- L. J. Burcham, J. Datka and I. E. Wachs, In Situ Vibrational Spectroscopy Studies of Supported Niobium Oxide Catalysts, J. Phys. Chem. B, 1999, 103, 6015–6024 CrossRef CAS.
- S. T. Korhonen, M. Calatayud and A. O. I. Krause, Stability of Hydroxylated (111) and (101) Surfaces of Monoclinic Zirconia: A Combined Study by DFT and Infrared Spectroscopy, J. Phys. Chem. C, 2008, 112, 6469–6476 CrossRef CAS.
- M. I. Zaki, M. A. Hasan, F. A. AI-Sagheer and L. Pasupulety, In situ FTIR spectra of pyridine adsorbed on SiO2−Al2O3, TiO2, ZrO2, and CeO2: general considerations for the identification of acid sites on surfaces of finely divided metal oxides, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS.
- K. Tanabe, Catalysis by Solid Bases and Related Subjects, in Studies in Surface Science and Catalysis, ed. B. Imelik, C. Naccache, G. Coudurier, Y. B. Taarit and J. C. Vedrine, Elsevier, 1985, vol. 20, pp. 1–14 Search PubMed.
- L. Pauling, The Nature of the Bond, J. Am. Chem. Soc., 1932, 54, 3570 CrossRef CAS.
- R. Radhakrishnan, C. Reed, S. T. Oyama, M. Seman, J. N. Kondo, K. Domen, Y. Ohminami and K. Asakura, Variability in the Structure of Supported MoO3 Catalysts: Studies Using Raman and X-ray Absorption Spectroscopy with ab Initio Calculations, J. Phys. Chem. B, 2001, 105, 8519–8530 CrossRef CAS.
- S. Chempath, Y. Zhang and A. T. Bell, DFT Studies of the Structure and Vibrational Spectra of Isolated Molybdena Species Supported on Silica, J. Phys. Chem. C, 2007, 111, 1291–1298 CrossRef CAS.
- C. Andriopoulou and S. Boghosian, Heterogeneity of deposited phases in supported transition metal oxide catalysts: reversible temperature-dependent evolution of molecular structures and configurations, Phys. Chem. Chem. Phys., 2018, 20, 1742–1751 RSC.
- G. Tsilomelekis and S. Boghosian, On the configuration, molecular structure and vibrational properties of MoOx sites on alumina, zirconia, titania and silica, Catal. Sci. Technol., 2013, 3, 1869–1888 RSC.
- G. Tsilomelekis and S. Boghosian, In Situ Raman and FTIR Spectroscopy of Molybdenum(VI) Oxide Supported on Titania Combined with 18O/16O Exchange: Molecular Structure, Vibrational Properties, and Vibrational Isotope Effects, J. Phys. Chem. C, 2011, 115, 2146–2154 CrossRef CAS.
- A. Christodoulakis and S. Boghosian, J. Catal., 2008, 260, 178–187 CrossRef CAS.
- S. Xie, K. Chen, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2000, 104, 10059–10068 CrossRef CAS.
- B. Zhang and I. E. Wachs, Identifying the Catalytic Active Site for Propylene Metathesis by Supported ReOx Catalysts, ACS Catal., 2021, 11, 1962–1976 CrossRef CAS.
- G. Ghiotti and F. Prinetto, FT-IR study of the nature and stability if NOx surface species on ZrO2, VOx/ZrOz and MoOx/ZrOz catalysts, Res. Chem. Intermed., 1999, 25(2), 131–156 CrossRef CAS.
- T. Hahn, U. Bentrup, M. Armbruster, E. V. Kondratenko and D. Linke, The Enhancing Effect of Brønsted Acidity of Supported MoOx Species on their Activity and Selectivity in Ethylene/trans-2-Butene Metathesis, ChemCatChem, 2014, 6, 1664–1672 CrossRef CAS.
- V. A. Kondratenko, T. Hahn, U. Bentrup, D. Linke and E. V. Kondratenko, Metathesis of ethylene and 2-butene over MoOx/Al2O3-SiO2: Effect of MoOx structure on formation of active sites and propene selectivity, J. Catal., 2018, 360, 135–144 CrossRef CAS.
- J. Handzlik, J. Ogonowski, J. Stoch, M. Mikolajczyk and P. Michorczyk, Properties and metathesis activity of molybdena-alumina, molybdena-silica-alumina and molybdena-silica catalysts-a comparative study, Appl. Catal., A, 2006, 312, 213–219 CrossRef CAS.
- P. Michorczyk, A. Wegrzyniak, A. Wegrzynowicz and J. Handzlik, Simple and Efficient Way of Molybdenum Oxide-Based Catalyst Activation for Olefins Metathesis by Methane Pretreatment, ACS Catal., 2019, 9(12), 11461–11467 CrossRef CAS.
- M. Myradova, A. Wegrzynowicz, A. Wegrzyniak, M. Gierada, P. Jodlowski, J. Lojewska, J. Handzlik and P. Michorczyk, Tuning metathesis performance of a molybdenum oxide-based catalyst by silica support acidity modulation and high temperature pretreatment, Catal. Sci. Technol., 2022, 12, 2134–2145 RSC.
- V. B. Kazansky, N. B. Shelimov and K. A. Vikulov, Designing of New Catalysts for Olefin Metathesis on the Base of Photoreduced Silica-Molybdena, Stud. Surf. Sci. Catal., 1993, 75, 515–527 CrossRef CAS.
- B. Zhang, S. Xiang, A. I. Frenkel and I. E. Wachs, Molecular Design of Supported MoOx Catalysts with Surface TaOx Promotion for Olefin Metathesis, ACS Catal., 2022, 12(5), 3226–3237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01612e |
| This journal is © The Royal Society of Chemistry 2023 |