
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypyridyl Ru(II) or cyclometalated Ir(III) functionalized architectures for photocatalysis
Yan-Lin
Li
a,
Ai-Juan
Li
a,
Sheng-Li
Huang
 *a,
Jagadese J.
Vittal
*b and
Guo-Yu
Yang
*a
*a,
Jagadese J.
Vittal
*b and
Guo-Yu
Yang
*a
aMOE Key Laboratory of Cluster Science, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: huangsl@bit.edu.cn; ygy@bit.edu.cn
bDepartment of Chemistry, National University of Singapore, 3, Science Drive 3, Singapore 117542, Singapore. E-mail: chmjjv@nus.edu.sg
First published on 29th June 2023
Abstract
The chemistry of polypyridyl Ru(II) and cyclometalated Ir(III) derivatives provides long-lasting interest to researchers due to the inherent advantage of their triplet states in a variety of photoactivities. The introduction of Ru(N^N)3 and Ir(C^N)2(X^N) modules into well-defined architectures extends the research areas of both photoactive metal complexes and network chemistry, generating a lot of new opportunities with interesting structural aesthetics and profound functional possibilities. The rapid development of research in integrating Ru(II) or Ir(III) metallotecons into the architectures has been apparent in recent years which makes this a fascinating subject for reviewing. This review focuses on the design and syntheses of Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures of metal–organic frameworks (MOFs), covalent–organic frameworks (COFs), metallasupramolecules, organic supramolecules and supramolecular organic frameworks (SOFs). Furthermore, the photocatalytic applications including the hydrogen evolution reaction (HER), carbon dioxide reduction reaction (CO2RR), photocatalytic oxidation and photoredox catalysis of organic transformation are also presented.
 Yan-Lin Li | Yan-Lin Li received her BS degree (2015) and MS degree (2018) from the China University of Petroleum. She is currently a PhD candidate under the supervision of Prof. Sheng-Li Huang at the Beijing Institute of Technology. Her current research interests are the preparation of polypyridyl Ru(ii) or cyclometalated Ir(iii) functionalized COFs for photocatalysis. |
 Ai-Juan Li | Ai-Juan Li is currently a PhD candidate under the supervision of Prof. Sheng-Li Huang at the Beijing Institute of Technology. Her current research interests are the preparation of polyoxometalate functionalized architectures, and polypyridyl Ru(ii) or cyclometalated Ir(iii) based supramolecular architectures for photocatalysis. |
 Sheng-Li Huang | Sheng-Li Huang received his PhD in chemistry in 2013 from Fudan University. Then he worked as a research scientist at the Institute of Materials Research & Engineering of A*STAR in Singapore. At the beginning of 2018, he moved to the Beijing Institute of Technology. His research interests contain well-organised architectures, functional polyoxometalates, Ru (Ir) chemistry and photosynthesis. |
 Jagadese J. Vittal | Jagadese J. Vittal obtained his PhD from the Indian Institute of Science, Bangalore (1982). He was a postdoctoral research associate and service crystallographer at the University of Western Ontario, Canada, before moving to NUS as a faculty member in 1997. He has been an Emeritus Professor since 2021. JJ is interested in solid-state science and structural chemistry including new materials, chemical reactivities, and structure–property relationships. |
 Guo-Yu Yang | Guo-Yu Yang received his MS and PhD degrees from Jilin University in 1991 and 1998, respectively. During the period 1998–2001, he worked as a Postdoctoral Fellow at the University of Notre Dame and Stockholm University. He became a full professor at FJIRSM-CAS in 2001. He was awarded the Cheung Kong Professorship in 2009. In 2014, he moved to the Beijing Institute of Technology. His research interests include oxo cluster chemistry and function development of oxo clusters. |
1. Introduction
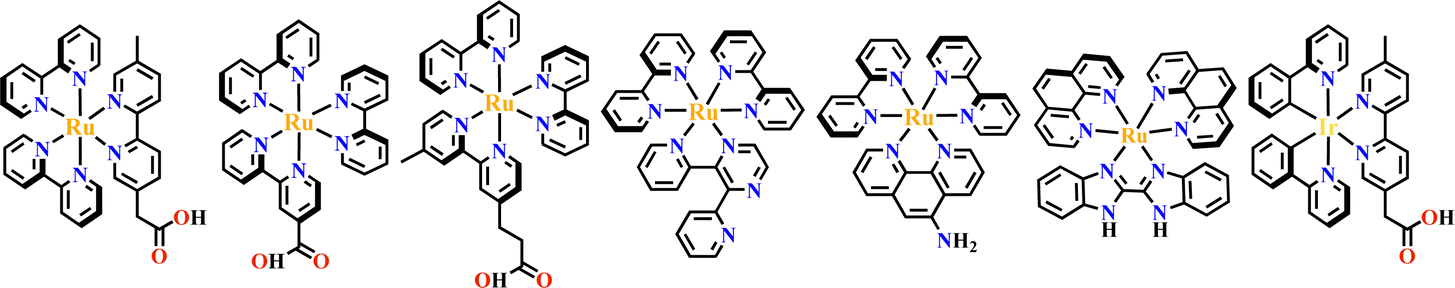
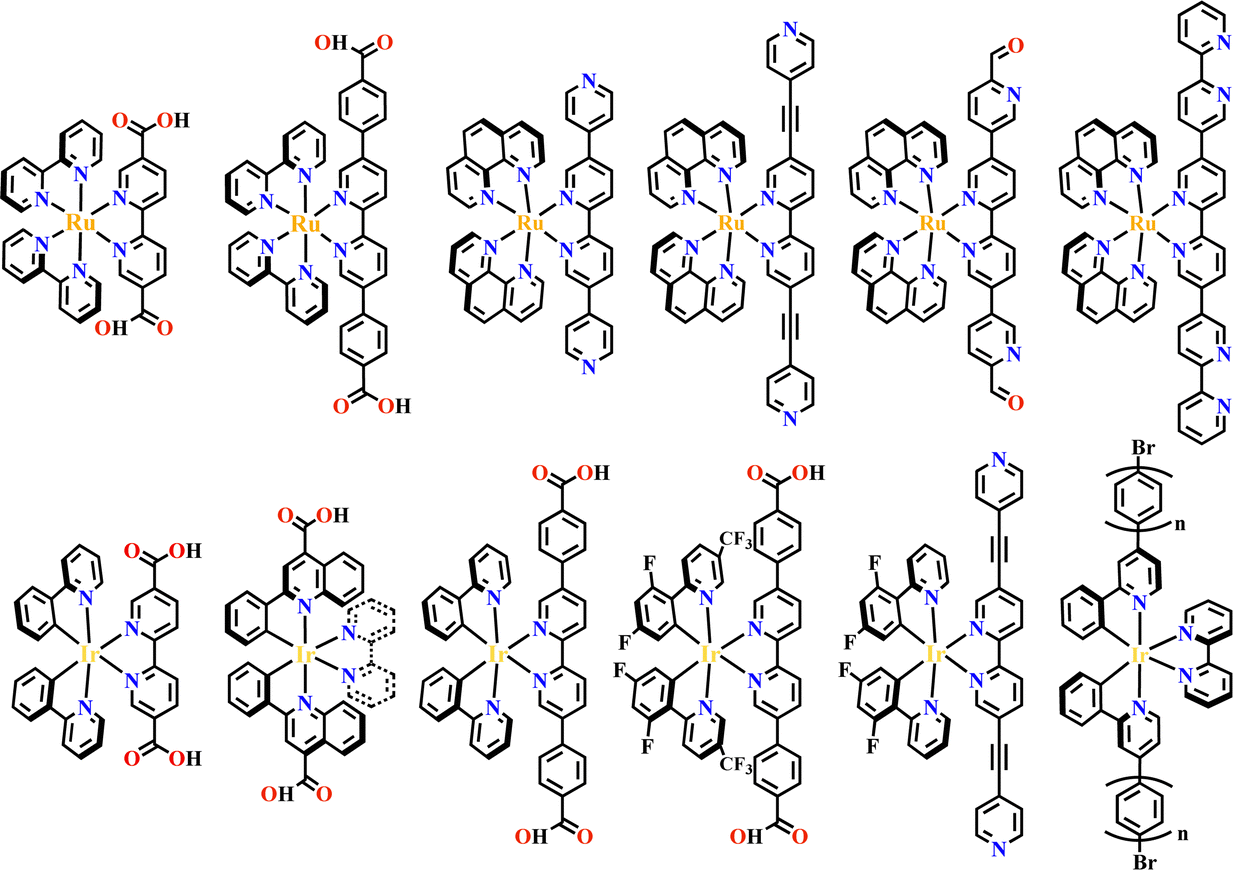
The octahedral d6-metal complexes, namely, polypyridyl–Ru(II) and cyclometalated–Ir(III) complexes have been exploited in photonic and optoelectronic applications due to their interesting electronic transitions and furthermore, charge-transfer of their triplet states is tunable over a wide range.1,2 Specifically, Ru(N^N)3 and Ir(C^N)2(X^N) (X = C or N), where (N^N) and (C^N) respectively represent the diimine ligand and cyclometalated ligand, have been mostly utilized for this purpose since the presence of Ru(II) and Ir(III) enhances the spin–orbit coupling and promotes the intersystem crossing of electrons from the excited singlet state to the triplet state, and their excited states are simultaneously influenced by the metal-to-ligand charge transfer (MLCT) and intraligand charge transfer (LLCT).3 The bonding of d6-metal ions and coordinating units provide enormous possibilities for assembling intriguing photoactive metal complexes.4,5 The highest occupied molecular orbital (HOMO) –lowest unoccupied molecular orbital (LUMO) gap, light absorption region, triplet lifetime, excitation potential, emission quantum yields (ΦPL) and other optoelectronic characteristics are readily modulated via substituent modification of the (C^N) and (N^N) units. The strong visible light absorption capacity and stable long-lived photoexcited states of Ru(N^N)3 and Ir(C^N)2(X^N) derivatives enabled their wide application in photophysics and photochemistry fields.6,7 For example, photoexcited Ir(ppy)3 (H-ppy = 2-phenylpyridine) exhibits special reducing features in photosynthesis owing to the strong electron-donating power of three ppy anions.8 Ru(N^N)3 and Ir(C^N)2(X^N) units have reliable synthesis and high chemical stability, and hence, their combinations with elaborate synthons yield desirable architectures with great significance in both skeletons and photo-functions (Fig. 1). | ||
| Fig. 1 Ru(N^N)3 or Ir(C^N)2(X^N) functionalized architectures and their photocatalytic applications. | ||
In the past two decades, well-defined architectures of MOFs,9–12 COFs,13–15 metallasupramolecules,16–18 organic supramolecules19–21 and SOFs22–25 have received greater attention due to their rich and designable structures, modifiable pore space and tunable functionality.26 These architectures provide the best models for the structure–property relationship study, and the well-organized skeletons commonly possess special functions for olefin separation in hierarchical porous MOFs27,28 for enzyme separation in mesoporous COFs,29,30 for unusual reactions in metallasupramolecules which act as molecular flasks,31 for photodynamic therapy efficacy in organic supramolecules32 and for accessible film production in SOFs.33 Like building skyscrapers, the molecular architectures can be precisely constructed by the judicious choice of divergent building blocks.34 The combination of the octahedral core of d6-metal ions and peripheral coordination components produces various three-dimensional (3D) building blocks,35,36 that may not be possible with pure organic components alone (Table 1). Furthermore, incorporation of Ru(N^N)3 and Ir(C^N)2(X^N) into well-defined architectures will not only yield unexpected and unusual properties of these materials but is also expected to have synergistic effects. For example, the structural confinement of the Ru(N^N)3 or Ir(C^N)2(X^N) moieties in MOFs, produced new photofunctional entities due to the weak conjugation of crystalline coordination frameworks.72 In photocatalysis, photogenerated charges are easily separated and transferred to Lewis acid sites for redox reactions.
















Efficient utilization of solar energy is an important way to solve the current global energy shortage and environmental crisis, and furthermore, photocatalysis can accomplish the transformation of the abundant solar energy into chemical energy.73 Molecular Ru(N^N)3 and Ir(C^N)2(X^N) complexes have been widely employed in various photosynthesis reactions, such as the HER,74,75 CO2RR,76–78 water oxidation,79,80 and particularly in organic transformations including electron transfer reactions, H atom transfer reactions and energy transfer reactions.81–83 Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures exhibit numerous applications from luminescence,7,84,85 sensing,86 solar fuels87,88 to bioimaging89–91 and furthermore, photocatalytic activity is always the most intriguing performance.92–98 They have great advantages in the photocatalysis system due to the following: (1) their light adsorption ranges are tunable from visible light to the near-infrared region.6,99 The integration of Ru(II)/Ir(III)–metallotecons within the architectures can broaden the spectral region, providing a platform for the efficient utilization of solar energy. (2) In these architectures, the varied potentials of building units linked to Ru(II)/Ir(III)-metallotecons lead to the efficient separation of photogenerated electrons and holes.100–102 (3) The conjugation of architectures is beneficial for highly efficient charge transfer, and the porous frameworks permitting more substrate to access the surface of the architectures and increase the utilization of charges.103 (4) Such architectures provide the required nano-space to govern the host–guest optoelectronic interactions, which accelerates photocatalysis arising from the photophysical interaction between bound guests and the emitting hosts. The nano-space can also accommodate other nanoparticles, producing interesting catalytic performance via the synergistic effect.70,104,105 (5) Most of these architectures are insoluble in common solvents, hence these noble metal catalysts can be easily recycled and reused.106 (6) These designed architectures are expected to provide a meaningful structure–function relationship which will help to design better catalysts and to improve our understanding of the catalytic process.54 Although some types of classical photocatalysts have been well explored, they have their disadvantages and limitations. Organic dyes have a tunable visible light absorption area, but they are readily decomposed during use.107,108 The cheap inorganic semiconductors of TiO2, Bi2WO6, α-Fe2O3, CdS and ZnO have better photostability, however the unavailable synthesis of nanoparticles without surfactant, narrow absorption spectrum of sunlight, high reduction potential, inefficient photogenerated charge separation and toxicity, in some cases restrict their wide application. The construction of heterojunctions and Schottky junctions led by doping of precious metals could improve some photochemical characteristics,109–111 but suffer from aggregation and deactivation of precious metals.112 In sharp contrast to other emerging photocatalysts, such as organic polymers, inorganic–organic hybrid materials, MOFs or COFs assembled from net organic building modules, the necessary conditions for photocatalysis including light absorption capacity, charge separation and utilization efficiency, interaction between substrate and catalyst of Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures could be greatly improved.
This review is focused on the design and synthesis of Ru(N^N)3 and Ir(C^N)2(X^N) containing architectures, which will be presented and discussed in separate sections based on MOFs, COFs, metallasupramolecules, organic supramolecules and SOFs. The Ru(N^N)3 and Ir(C^N)2(X^N) units can be obtained via reliable synthetic routes, the incorporation of these units into well-defined architectures commonly involves two methodologies (Fig. 2),113viz. deployment of the metalloligands70,71,114 and post-synthetic modification (PSM).115–118 High chemical stability of the Ru(N^N)3 and Ir(C^N)2(X^N) units makes the synthesis viable by the first method, even under harsh reaction conditions such as strong base or acid environments and high temperature. The post-synthetic incorporation of Ru(N^N)3 and Ir(C^N)2(X^N) units are achieved by the coordination linkage of (N^N) groups from the skeleton with Ru(N^N)2- or Ir(C^N)2- precursor. These architectures have many photocatalytic applications. The HER, CO2RR, photocatalytic oxidation and photoredox catalysis of organic transformation are concluded.
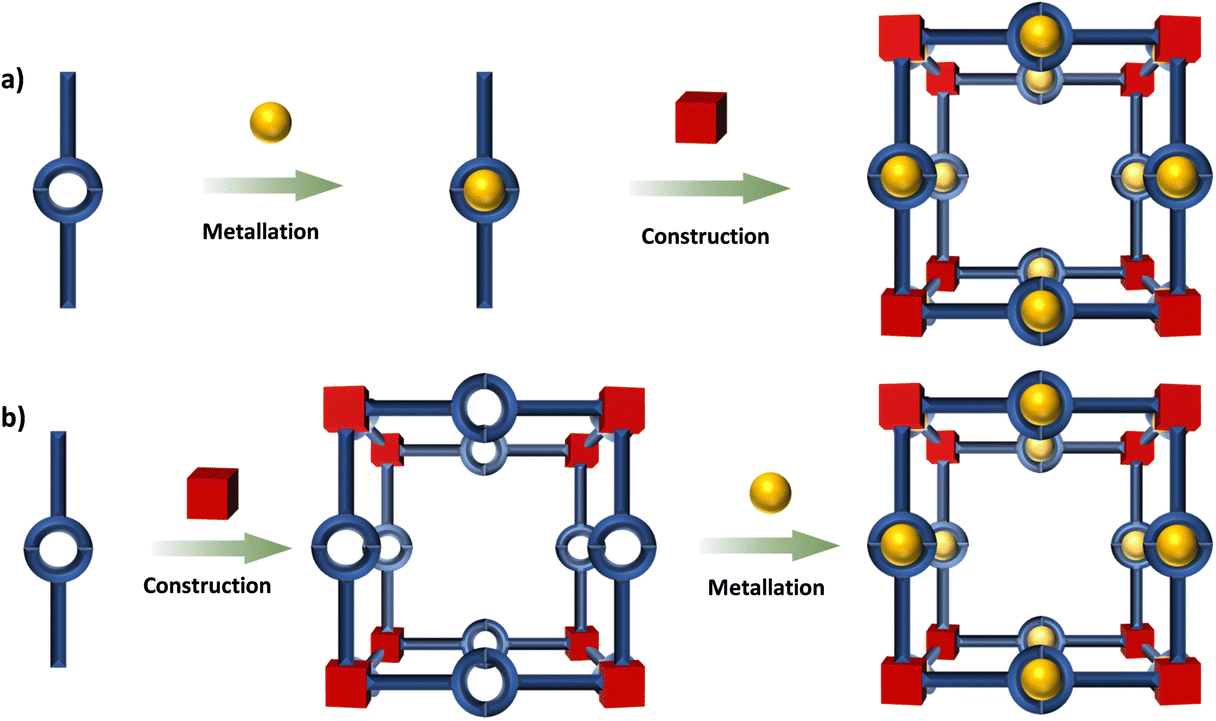 | ||
| Fig. 2 Two synthetic strategies for metalloarchitectures: (a) metalloligands; (b) PSM. | ||
2. Ru(N^N)3 or Ir(C^N)2(X^N) functionalized MOFs
MOF, also known as porous coordination polymer, is one of the fastest growing research fields in both chemistry and materials science in the last two decades.34,119–123 The structural diversity, ultrahigh surface area, crystalline skeleton and tunable functionality allow MOFs to be widely utilized in various disciplines.124–131 As featuring periodic networks, MOFs are formed by the self-assembly of organic linkers with metal ions or metal clusters (secondary building units (SBUs)).34 A large number of MOFs have been reported arising from different types of organic linkers, variable coordination numbers and flexible coordination modes of metal ions, as well as the in situ formed “OH−” and “O2−” species leading to the formation of wide-ranging MOFs.132,133 A judicious combination of organic linkers and SBUs control the topology of the resulting MOFs, whose photofunctions are commonly inherited from the photo-responsive ligands or post-introduced photosensitizers.34,134 In general, Ru(N^N)3 and Ir(C^N)2(X^N) functionalized MOFs are synthesized by two routes: (1) direct reaction of metalloligands with metal ions.35,49,135–140 (2) PSM: the bis-chelating sites of ligand skeleton in the MOFs, such as (N^N) groups, could be bonded to the Ru(II) or Ir(III) precursors, thus producing MOFs containing Ru(N^N)3 or Ir(C^N)2(X^N) units.50,141,142 In addition, the metalloligand could also be anchored at the unsaturated coordination sites of the MOFs via PSM.37,118 In the syntheses of Ru(N^N)3 and Ir(C^N)2(X^N) functionalized MOFs, the nature of metal ions has great influence on the SBUs, topology and function of the resulting MOFs. In the following section, the photoactive MOFs will be divided into three categories according to the metal ion species in the SBUs, viz. 3d metal ions, d0 metal ions and 4f metal ions.2.1 Photoactive MOFs constructed from the 3d metal ions
Transition metals of Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+, have been commonly used in the construction of various MOFs with the following advantages: (1) the variable coordination numbers and flexible coordination modes of these transition metal ions provide different SBUs for the construction of diverse MOFs; (2) the reaction conditions of solvents, acid or base, and temperature have great influence on the formation of metal nodes, providing infinite possibilities to synthesize different SBUs; (3) weak metal–ligand interactions facilitate the crystallization of the MOFs. But most of MOFs incorporating transition metal ions have low chemical stability, which has adverse effects on the catalytic applications. However, these reactions produce photoactive MOFs with diverse structures and interesting functions.A combination of Ru(II) and phenba (phenba = 4-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)benzoic acid) formed a C3 symmetric tridentate metalloligand [Ru(phenba)3]2+. The reaction of [Ru(phenba)3]2+ with Co(NO3)2 produced [Ru(phenba)3+Co]-MOF with two-dimensional (2D) hexagonal layers and large open channels. Mesoporous [Ru(phenba)3+Co]-MOF exhibited a selective gas sorption property, and its CO2 sorption capacities were far better than N2 (Fig. 3).61[Ru(ip)3+Co]-MOF (Hip = 1H-imidazo[4,5-f][1,10]phenanthroline) revealed a high symmetry ctn network consisting of 3-connected [Ru(ip)3]2+ and 4-connected Co2+ units. Under visible light, the overall conversion of CO2 reduction and H2O oxidation was achieved simultaneously by this multifunctional photocatalyst with a high conversion rate and CO selectivity.59 Ir(C^N)3-carboxy metalloligand was synthesized in a stepwise manner from the cyclometallation of (C^N)-ester derivatives and Ir(III) precursor, and then ester hydrolysis. Ir(C^N)3-cores have two stereochemical forms, namely, mer and fac. The fac isomer has better stability and a longer emission lifetime than mer, and in fac-isomer all nitrogen atoms are trans to carbon atoms. The introduction of a carboxy group into the para-position of the coordinated C or N atoms, resulting in the bowl shaped metalloligand, whose carboxy groups are nearly perpendicular to each other. Using the Ir(ppy-COOH)3-metalloligand, (H-ppy-COOH = 3-(pyridin-2-yl)benzoic acid) [Ir(ppy-COO)3+Co]-MOF and [Ir(ppy-COO)3+Zn4]-MOF were synthesized. [Ir(ppy-COO)3+Co]-MOF was applied as an efficient O2-evolution–reaction electrocatalyst.62 The highly phosphorescent [Ir(ppy-COO)3+Zn4]-MOF was used in O2 sensing63 as well as photocatalytic pollutant degradation.143 The position of the carboxyl groups at Ru(N^N)338,66,144 and Ir((C^N))2(N^N)-metalloligands can influence the topology of MOFs.57,145
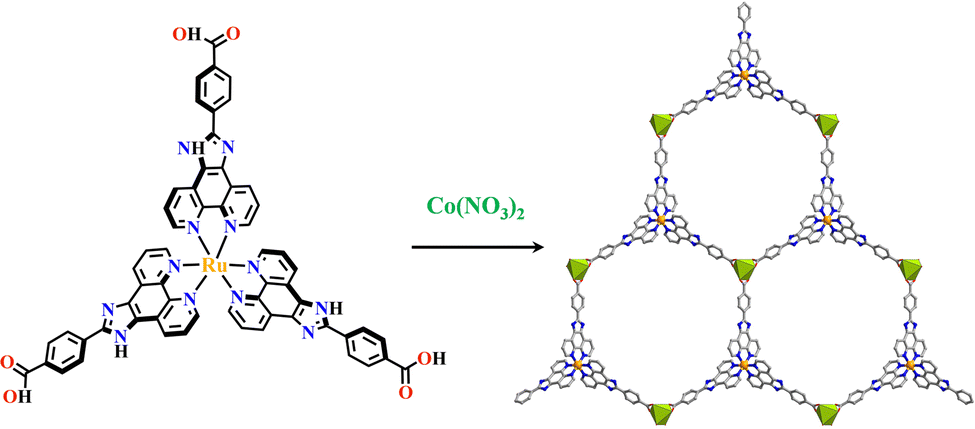 | ||
| Fig. 3 [Ru(phenba)3+Co]-MOF.61 | ||
In (C^N)-derivatives, the introduction of the carboxy groups into both the para positions of the coordinated C and N generated the hexadentate ligands Ir(H2dcppy)3 and [Ir(H2dcppy)2(H2dcbpy)]+ (H-H2dcppy = 2-(3-carboxyphenyl)pyridine-4-carboxylic acid, H2dcbpy = 2,2′-bipyridine-4,4′-dicarboxylic acid), and the carboxy groups of these two octahedral metalloligands linked with linear mono-node [Ni(cyclam)]2+ and formed the two isostructural complexes [Ir(dcppy)3+Ni]-MOF and [Ir(dcppy)2(dcbpy)+Ni]-MOF with pcu topology. They were 2-interpenetrating networks and exhibited a good CO2/N2 separation property (Fig. 4).35
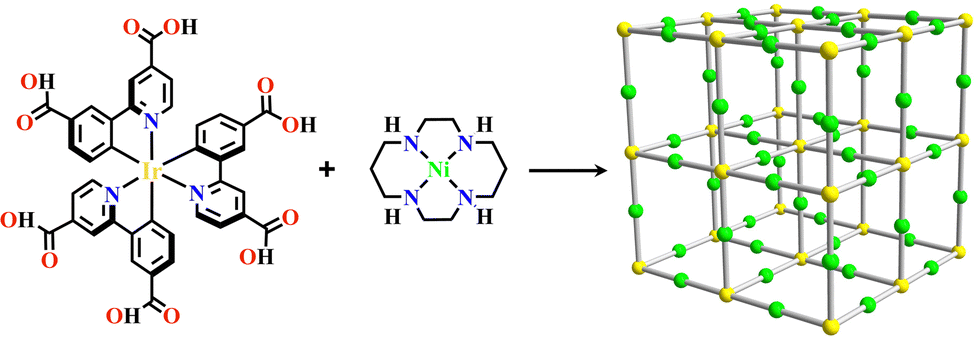 | ||
| Fig. 4 [Ir(dcppy)3+Ni]-MOF.35 | ||
2.2 Photoactive MOFs constructed from d0 metal ions
The d0 metal ions of Ti4+, Zr4+ and Hf4+ have high charge density and strong oxophilicity, and they have a particularly strong bonding ability with carboxylate groups. These d0 metal salts can easily form metal hydroxides or oxides when encountering water, further increasing the chemical stability of metal-carboxylate clusters. The MOFs synthesized from Ti4+, Zr4+ or Hf4+ have strong chemical stability and their skeletons are maintained even under a strong acid environment. They are commonly used as heterogeneous catalysts based on their stable and porous frameworks. The strong metal-carboxylate interactions result in a lower control over the crystallization process, and difficulty in growing the single crystals suitable for X-ray diffraction was encountered.146 Many d0 metal-based MOFs exhibited versatile catalytic reactions. The bonding energy between Zr4+ and carboxylate groups was weaker than Ti4+ but stronger than Hf4+, hence Zr6-MOFs have medium levels of chemical stability and crystallization ability, promoting the rapid development of functional Zr6-MOFs.146–148 The reaction of Zr4+ and COO− groups unexpectedly afford a Zr6(μ3-O)4(μ3-OH)4(CO2)12 cluster.Zhou and co-workers reported the synthesis of mesoporous MOFs, termed (TPTB+Zr6)-MOF (H4TPTB = 5′,5′′′-bis(4-carboxylatophenyl)-4′′′,6′-dimethoxy-[1,1′:3′,1′′:4′′,1′′′:3′′′,1′′′′-quinquephenyl]-4,4′′′′-dicarboxylate), via assembling the Zr6 clusters with the tetratopic carboxylate linkers H4TPTB. The carboxylate group of [Ru(bpy)2(H2bpydc)]2+-metalloligand (H2bpydc = 2,2′-bipyridine-5,5′-dicarboxylic acid) linked to the uncoordinated sites of the Zr6 cluster, leading to photoactive Ru(bpy)2(bpydc)@(TPTB+Zr6)-MOF with the expected porous network. It showed high catalytic efficiency in the aza-Henry reaction (Fig. 5).118
 | ||
| Fig. 5 Ru(bpy)2(H2bpydc)-metalloligand-functionalized (TPTB+Zr6)-MOF.118 | ||
In the construction of Ru(N^N)3 and Ir(C^N)2(X^N) functionalized MOFs, the most popular synthetic method was the direct reaction of metalloligand and metal source. In this way, the structural advantages of Ru(N^N)3 or Ir(C^N)2(X^N) units could be passed to the target frameworks and the photoactive units could be well organized in the pore. Using metalloligand [Ru(bpy)2(H2bpydc)] and 4,4′-biphenyldicarboxylic acid (H2bpdc), Lin and co-workers synthesized several heteroleptic MOFs that were isostructural with UIO-67 and exhibited high photocatalytic activity in aza-Henry reactions.43 The introduction of Cu(HxPO4)y into Cu@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF produced the dual-functional catalyst Cu@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF, and it promoted the conversion of CO2 to C2H5OH with light irradiation and CH3OH in the absence of light. TEM analysis showed that the catalysts formed Cu0 nanoparticles in the dark, which were the active species for CO2 transformation to CH3OH. With light illumination, {[Ru(bpy)2(bpydc)]2+}* transferred e− to Cu2+ and generated Cu+, simultaneously, {[Ru(bpy)2(bpydc)]2+}* was quenched by electron injection from Cu0 and generated Cu+, which were responsible for C2H5OH production.138 The encapsulation of [Ni(bpet)(H2O)2]2+ (bpet = 1,2-bis[(pyridin-2-ylmethyl)thio]ethane) into [Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF produced Ni⊂[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF, and the photogenerated electron reduced the Ni center with enhanced CO2 affinity and formed a Ni–CO2 adduct, achieving highly efficient CO2 transformation with good CO selectivity.139 Zhang and co-workers synthesized Co@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF by the insertion of Co2+ as a single-site catalyst in the open (N^N)-sites of [Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF, which supplied a molecular platform capable of rapid injection of multiple electrons from the photosensitizer to Co-catalyst, resulting in a MOF-based composite photocatalyst for efficient syngas production. The photogenerated electrons transferred from the photosensitizer to Co-catalyst, then reduced CO2 and H2O to syngas. In the photocatalytic process, the H2/CO ratios could be well controlled by adjusting the water content or the ratio of PS/Cat in the MOFs.149
In the construction of Ru(N^N)3-(UIO-67), the Ru(N^N)3-metalloligand was simultaneously used with other linear dicarboxylate linkers because the small cavity in UIO-67 could not accommodate Ru(N^N)3-units due to its small pore size. This problem was overcome by increasing the length between two terminal carboxyl groups of the metalloligands, producing more space for the accommodation of Ru(N^N)3 or Ir(C^N)2(X^N) units inside the framework. The linkage of Ru(bpy)2(H2bpydc)-metalloligands and [Zr6(μ3-O)4(μ3-OH)4]-SBUs produced Ru(bpy)2(dbpydc)-Zr6-MOF (H2dbpydc, 4,4′-([2,2′-bipyridine]-5,5′-diyl)dibenzoic acid). In the synthetic procedure, the addition of Wells–Dawson {P2W18} polyoxometalates (POMs) resulted in the formation of {P2W18}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF (Fig. 6).114{Ni4P2}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF, and {Ni4P2}⊂[Ir(ppy)2(dbpydc)+Zr6]-MOF were obtained via similar synthetic methods. Both [Ru(bpy)2(dbpydc)+Zr6]-MOF and [Ir(ppy)2(dbpydc)+Zr6]-MOF had the same topological network with UIO-67. The incorporation of the photosensitizer and POMs {Ni4P2} into a single framework shortened their distance, promoting the photo-generated electron transfer from photosensitizer to POMs, and the accumulation of electrons at POMs enhanced its reduction ability, thus achieving photocatalytic hydrogen production. However, {Ni4P2}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF could not photocatalyze hydrogen production because the Ru(N^N)3 photosensitizer does not have a low enough reduction potential to permit electron accumulation at {Ni4P2}.150 In POMs⊂Ru(Ir)-MOF, the enhanced multi-electron transfer from Ru(N^N)3 or Ir(C^N)2(X^N) photosensitizer to POMs catalyst was responsible for the photocatalytic hydrogen production.114,150 Ye and co-workers synthesized a series of Ir(pqc)2(N^N)-metalloligands (H-pqc = H-2-phenylquinoline-4-carboxylic acid), in which the (C^N)-units were located around the Ir(III) center with controllable trans arrangement and the carboxyl group was located at the para position of the N atom, resulting in linear [Ir(pqc)2(N^N)]+ metalloligands. The reaction of these metalloligands with ZrCl4 yielded [Ir(pqc)2(N^N)+Zr6]-MOF (N^N derivatives = bpy, 4,4′-dimethoxy-2,2′-bipyridine, or 1,10-phenanthroline (phen)) which showed excellent photocatalytic activity in the selective sulfide oxidation under a O2 atmosphere (Fig. 7).48 The attachment of the (N^N^N)-unit with the carboxylate group produced similar metalloligands, such as [Ru(cptpy)2]2+ (cptpy = 4′-(4-carboxyphenyl)-terpyridine), and its combination with simple Zr-node produced 1D framework [Ru(cptpy)2+Zr]-MOF, which had high stability and selectively photocatalyzed CO2 to formate (HCOOH) under visible light irradiation.151
 | ||
| Fig. 6 {P2W18}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF.114 | ||
 | ||
| Fig. 7 [Ir(pqc)2(N^N)+Zr6]-MOF.48 | ||
2D materials have unique photoelectric properties because the delamination has a great influence on their functions. Metal–organic layers (MOLs) are a new type of 2D organic–inorganic material.152,153 They are highly dispersible and have enough exposed active sites.154,155 A series of MOLs were constructed by linking Ir(C^N)2(N^N)-49,50,135,136,156,157 and Ru(N^N)3-44,158 metalloligands with SBUs of Hf12 or Hf6, exhibiting numerous applications. Ir(C^N)2(N^N) or Ru(N^N)3 derivatives are two efficient photosensitizers for the generation of 1O2.159,160 Hf atoms in the SBUs efficiently absorb X-rays and transfer its energy to Ir(C^N)2(N^N) and Ru(N^N)3 moieties, thus generating reactive oxygen species (ROSs) for photodynamic therapy. Hf6-MOLs were synthesized from the reaction of HfCl4 and H3BPY (H3BPY, 4′,6′-dibenzoato-[2,2′-bipyridine]-4-carboxylic acid), and the open (N^N)-coordination site of Hf6-MOL was occupied by the reaction with [Ir(ppy)2Cl]2 or Ru(bpy)2Cl2. An X-ray excited Hf6 cluster transferred energy to photosensitizer moieties, generating 1O2.156 The incorporation of POMs enhanced the generation of various ROSs. Under X-ray irradiation, different functional modules of {P2W18}⊂[Ir(ppy)2(dbpydc)+Hf12]-MOL synergistically produced the corresponding ROSs (˙OH from Hf12 cluster, 1O2 from Ir-photosensitizer and O2− from {P2W18}).49 Similarly, [Ir(dFCFppy)2(dbpydc)+Hf12]-MOL (H-dFCFppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine) and Ir(dFCFppy)2(N^N)@(BPY+Hf6)-MOL could simultaneously generate 1O2 from the Ir(C^N)2(N^N) unit and ˙OH from Hf6 or Hf12 SBUs.50 The weakly coordinated trifluoroacetic acid of [Ru(bpy)2(dbpydc)+Hf12]-MOL was replaced by 2-(5′-methyl-[2,2′-bipyridin]-5-yl)acetic acid, which was further coordinated with M(CO)3X (M = Mn or Re, X = Cl or Br), leading to Re@[Ru(bpy)2(dbpydc)+Hf12]-MOL or Mn@[Ru(bpy)2(dbpydc)+Hf12]-MOL with efficient photocatalytic CO2RR activities. The photogenerated electrons transferred from the Ru(N^N)3 photosensitizer to Mn or Re catalyst, and the coordination interaction between CO2 and catalyst enhanced its electron grabbing capacity from the catalyst, enhancing the selective CO2 transformation to CO.155 Furthermore, OTf@[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL (OTf = triflate) could effectively catalyse dehydrogenative cross-couplings. In this system, dehydrogenation occurred on [Ir(dFCFppy)2(H2dbpydc)]+ bridging ligands, and the OTf on the Hf12 SBUs working as Lewis acid.136 When [Ir(dFCFppy)2(dbpydc)+Hf12]-MOL was decorated with Ni(2-(4′-methyl-[2,2′-bipyridin]-4-yl)acetate)Cl2 at Hf12 centers (Fig. 8), the corresponding Ni@[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL showed high photoactivities in C–S, C–O, and C–C cross-coupling reactions.135OTf/Co@[Ru(bpy)2(dbpydc)+Hf12]-MOL hierarchically integrated three active sites into one complex, OTf as the strong Lewis acid, [Ru(bpy)2(H2dbpydc)]2+ ligand as the photosensitizer, and [Co(dimethylglyoxime)2(4-pyridinepropionate)]Cl as a hydrogen-transfer catalyst. They worked synergistically for tandem catalysis.158
 | ||
| Fig. 8 Ni@[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL.135 | ||
In contrast to numerous Zr-MOFs and Hf-MOFs, only a few Ti-MOFs were reported because Ti4+ ions are easily hydrolyzed and strong Ti–carboxylate bonds make the crystallization of Ti-MOFs difficult.146 The reaction of [Ti6O6(isopropoxy)6(4-aminobenzoate)6] and Ru(Ir)-metalloligand or 4,4′-biphenyldicarboxylic acid yielded [Ru(bpy)2(bpydc)/bpdc+Ti3]-MOF or [Ir(ppy)2(bpydc)/bpdc+Ti3]-MOF, and the combination of photosensitizer and Ti3 cluster facilitated the photocatalytic HER under visible light.140
2.3 Photoactive MOFs constructed from 4f metal ions
Rare-earth elements possess rich optical, electrical, and magnetic properties due to their large atomic magnetic moment, unique 4f sublayer electronic structure, and strong spin–orbit coupling. Lanthanide (Ln) ions have flexible coordination modes, high coordination numbers, strong Lewis acid142 and strong oxophilicity based on the Hard–Soft–Acid–Base theory.A few Ru(N^N)3 or Ir(C^N)2(X^N) functionalized Ln-MOFs have been reported with interesting functions.99,161–164 The reaction of planar triangular-shaped metalloligand [Ru(phenba)3]2+ and Co(NO3)2 generated a 2D network, however its reaction with Eu(NO3)3 produced [Ru(phenba)3+Eu2]-MOF with a 3D porous network, which efficiently promoted the selective transformation of CO2 to HCOOH.137 Under visible-light irradiation, (Eu3+)2-SBU obtained two electrons from the excited {[Ru(phenba)3]2+}* units and was reduced to (Eu2+)2, enabling the selective reduction of CO2 to HCOOH via a two-electron process. In [Ru(bpy)2(dcbpy)+Ln]-MOFs and [Ir(ppy)2(dcbpy)+Ln]-MOFs (Ln = Yb, Er, Nd, Gd, Yb, Dy),54,99,164 the photosensitizer groups acted as a good light-harvesting antenna to effectively sensitize Ln ions by d → f energy transfer, and achieving near-infrared luminescence. In addition, [Ru(dcbpy)3+La1.75]-MOF was constructed by the reaction of [Ru(H2dcbpy)3]2+ with LaCl3 in a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7. Water adsorption/desorption of [Ru(dcbpy)3+La1.75]-MOF triggered a reversible structural transformation, accompanying great influence on its 3MLCT emission energy and ion conductivity. Vapochromic luminescence occurred via water vapor adsorption–desorption, in addition, the increased ion conductivity rate was proportional to humidity.163 In isostructural complexes of [Ru(dcbpy)3+Ce1.75]-MOF and [Ru(dcbpy)3+Nd1.75]-MOF, different Ln ions have a great influence on their properties. MOFs synthesized with larger Ln ions could recover the ion conduction activation energy and the original porous structure at lower relative humidities.162 Similarly, in [RuH5.5(dpbpy)3+La1.5]-MOF (H4dpbpy = 2,2′-bipyridine-4,4′-bis(phosphonic acid)) and [RuH5.5(dpbpy)3+Pr1.5]-MOF, the 3MLCT emission exhibited a blue-shift following the decreased humidity and red-shift with increased humidity, because water-adsorption triggered proton release and reconstruction of the porous structure. The proton conductivity of [RuH5.5(dpbpy)3+La1.5]-MOF was much higher than [Ru(dcbpy)3+La1.75]-MOF due to the higher acidity of the phosphonic acid groups.161
:7. Water adsorption/desorption of [Ru(dcbpy)3+La1.75]-MOF triggered a reversible structural transformation, accompanying great influence on its 3MLCT emission energy and ion conductivity. Vapochromic luminescence occurred via water vapor adsorption–desorption, in addition, the increased ion conductivity rate was proportional to humidity.163 In isostructural complexes of [Ru(dcbpy)3+Ce1.75]-MOF and [Ru(dcbpy)3+Nd1.75]-MOF, different Ln ions have a great influence on their properties. MOFs synthesized with larger Ln ions could recover the ion conduction activation energy and the original porous structure at lower relative humidities.162 Similarly, in [RuH5.5(dpbpy)3+La1.5]-MOF (H4dpbpy = 2,2′-bipyridine-4,4′-bis(phosphonic acid)) and [RuH5.5(dpbpy)3+Pr1.5]-MOF, the 3MLCT emission exhibited a blue-shift following the decreased humidity and red-shift with increased humidity, because water-adsorption triggered proton release and reconstruction of the porous structure. The proton conductivity of [RuH5.5(dpbpy)3+La1.5]-MOF was much higher than [Ru(dcbpy)3+La1.75]-MOF due to the higher acidity of the phosphonic acid groups.161
Ln(III) ions have a high coordination number, and the combination of multifunctional carboxylate linker and Ln(III) ions can also form Ln6-MOFs. Like [Ru(Ir)+Zr6]-MOFs, the construction of [Ru(Ir)+Ln6]-MOFs should also simultaneously use both metalloligands and its isometric dicarboxylate ligand when the metalloligands have no sufficient length. Cu@[Ru(bpy)2(bpydc)/bpydc+Eu6]-MOF was constructed by mixed use of [Ru(bpy)2(H2bpydc)]2+, H2bpydc, and [Cu(H2bpydc)]Cl2, which photocatalyzed the selective transformation of CO2 to HCOOH with a high conversion rate (Fig. 9).165
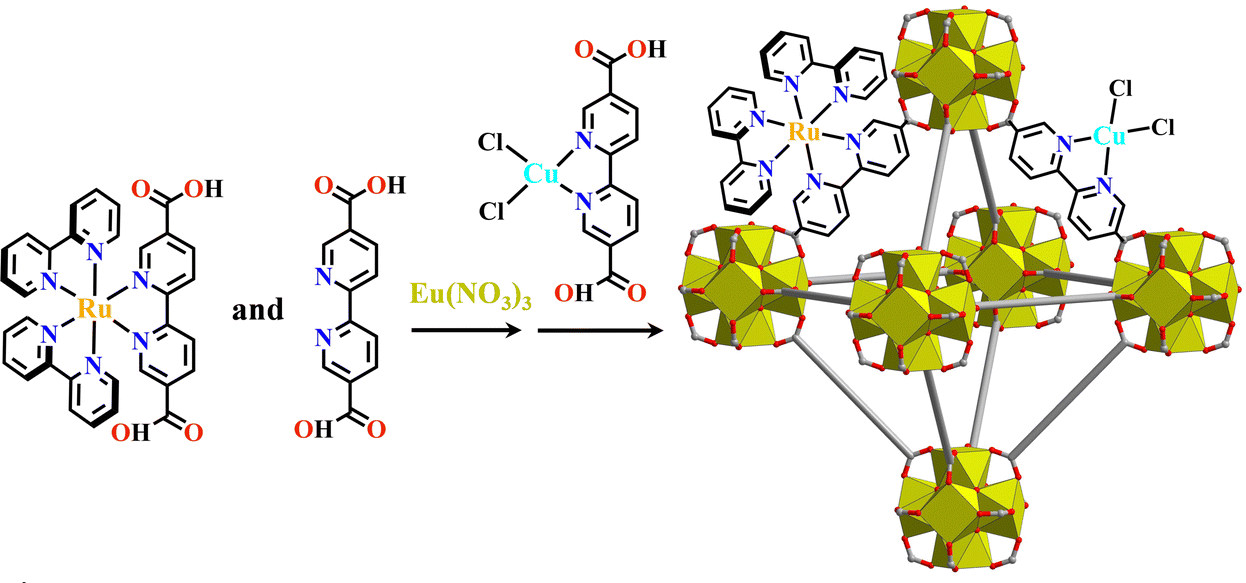 | ||
| Fig. 9 Cu@[Ru(bpy)2(bpydc)/bpydc+Eu6]-MOF.165 | ||
PSM is another effective method to fabricate multinuclear Ru(N^N)3 or Ir(C^N)2(X^N) functionalized Ln-MOFs. Ce6-MOLs. The combination of H3BTB (H3BTB = 1,3,5-benzenetribenzoate) and the photosensitive [Ir(ppy)2(HMBA)]+ (HMBA = 2-[5′-methyl-(2,2′-bipyridin)-5-yl]acetate) or [Ru(bpy)2(HMBA)]2+ group was introduced into the framework via the coordination with the Ce6 cluster. Ir(ppy)2(MBA)@(BTB+Ce6)-MOL and Ru(bpy)2(MBA)@(BTB+Ce6)-MOL showed good photocatalytic HER performance due to the synergistic effect of photosensitizer and Ce6 center (Fig. 10).37Ru(bpy)2(bpydc)@(bpydc+Ln6)-MOFs (Ln = Tb, Gd, Eu) were synthesized via the coordination linkage of Ru(N^N)2 and (N^N) groups of frameworks, exhibiting photocatalytic thioether oxidation with high sulfoxide selectivity under mild conditions.142
 | ||
| Fig. 10 Ru(bpy)2(MBA)@(BTB+Ce6)-MOL.37 | ||
The reaction of Ru(bpy)2Cl2 or Re(CO)3Cl with (N^N)-sites of Al-MOF produced [Re/Ru(bpy)2(bpydc)]@(bpydc+Al)-MOF, which showed high activity in photocatalytic CO2 reduction under visible light.166 The Al-OTf Lewis acid sites and Ir(ppy)2(bpydc) photocatalytic sites were installed on Al-MOF. The obtained [OTf/Ir(ppy)2(bpydc)]@(bpydc+Al)-MOF effectively catalysed the reductive cross-coupling to afford new aza arene derivatives. The close proximity of substrate binding centers and catalytic centers in the mesopore gave this heterogeneous catalyst better performance than the homogeneous counterpart. Furthermore, this MOF facilitated coupling reaction between the activated vinyl- or alkynyl-azaarenes and alkyl radicals.167
3. Ru(N^N)3 or Ir(C^N)2(X^N) functionalized COFs
A wide range of covalent organic frameworks (COFs) have been developed in the past two decades, because of their unique properties of good chemical and thermal stability, high specific surface area, and low skeletal density.168 They have been widely employed in photocatalytic systems due to their chromophore and long-range π–π conjugated structure providing a platform for light-harvesting and efficient charge transfer.108 Herein both crystalline COFs and amorphous COFs are described,26 which are classified by the type of covalent bonds, topology, porosity and functions.263.1 Ru(N^N)3 and Ir(C^N)2(X^N) functionalized crystalline COFs
Similar to MOFs, COFs are also crystalline materials which have well-defined skeletons formed by the orderly and infinitely extended molecular tecons.169 Different from MOFs, the covalent linkages give the whole framework with high conjugation and associated electronic properties.170,171 Crystalline COFs are also more advantageous than amorphous species because crystalline networks possess well-ordered building units and better framework conjugation that is beneficial for the charge transport.172 They exhibited numerous applications from drug delivery,105,173,174 gas adsorption175,176 and separation177,178 to optoelectronics.179 The introduction of the chelating coordination sites including N, O, and S elements into COFs makes them excellent metal catalyst supporters. The chelating sites could anchor the metal ions, permitting the uniform dispersion of metal catalysts into the supporters and preventing catalyst detachment. COFs can be easily functionalized to help the improvement of product selectivity. The high porosity and well-ordered pore channels of COFs accelerate the diffusion of substrates, and increase the approaching possibility between the substrates and catalytic center. These metal functionalized COFs have adjustable catalytic performances concerning high activity, product selectivity and reuse.The construction of metal functionalized COFs also includes two main methodologies of metalloligand and PSM (Fig. 2).113 The use of metalloligands is the most direct approach to fabricate metal-containing COFs with diverse topologies. For example, several interesting COFs were synthesized from metal–porphyrin180–183 and glyoximate184 precursors. PSM is another efficient approach to incorporate a metal catalyst into the frameworks via metal–ligand interactions, thus providing a reliable way to produce metal functionalized COFs for specific applications.185 Although porous, the crystallinity and structure of COFs are maintained186–190 by the PSM route, and an even distribution and precise location of the metal catalyst is difficult to achieve.
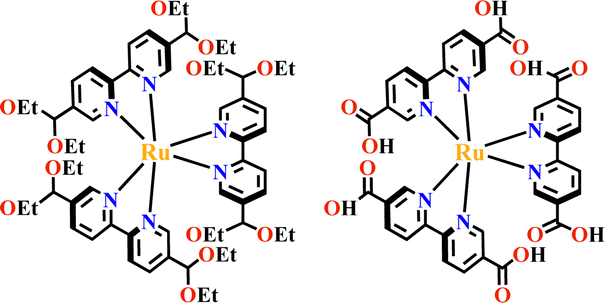
The condensation reaction of [Ru(bpy-OEt)3]2+ (bpy-OEt = 5,5′-bis(diethoxymethyl)-2,2′-bipyridine) metalloligands and multi-amine precursor including ETTA (ETTA = 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetraaniline), TPB (TPB = 1,2,4,5-tetrakis-(4-aminolphenyl)benzene) or ETTBA (ETTBA = 4′,4′′′,4′′′′′,4′′′′′′′-(ethene-1,1,2,2-tetrayl)tetrakis([1,1′-biphenyl]-4-amine)) produced three 3D stp topological networks [Ru(bpy-OEt)3+ETTA]-COF, [Ru(bpy-OEt)3+TPB]-COF and [Ru(bpy-OEt)3+ETTBA]-COF (Fig. 11). Based on the photosensitive Ru(N^N)3 unit, they possessed a strong light-harvesting ability and showed high photocatalytic activity in the HER process.71 Most of the Ru(N^N)3 or Ir(C^N)2(X^N) functionalized COFs were assembled via PSM, and the (N^N)-unit of COFs was a commonly used bis-chelating group to grab metal catalysts.187–189 For instance, Re(CO)3Cl(N^N)-functionalized COFs efficiently reduced CO2 to form CO under visible light illumination with high selectivity.186 The reaction of BPDCA (BPDCA = 2,2′-bipyridine-5,5′-dicarbaldehyde) and TAB (TAB = 1,3,5-tris-(4-aminophenyl) benzene) afforded an imine-linked 2D framework (BPDCA+TAB)-COF with an open (N^N)-site (Fig. 12), and its metalation with Ru(bpy)2Cl2 resulting in the formation of Ru(N^N)3@(BPDCA+TAB)-COF. The site-specific functionalization of sp3 C–H bonds of the C atom adjacent to a N atom is one less explored organic transformation. Ru(N^Ru(N^N)3@(BPDCA+TAB)-COF photocatalyzed cross-dehydrogenative coupling between secondary amines and indoles with excellent yield.116 Under visible light irradiation, the Ru(N^N)3 moiety was excited into [Ru(N^N)3]*, and it received one electron from the secondary amine offering a radical intermediate. The radical intermediate underwent deprotonation and formed a radical imine cation intermediate, and its quenching by indoles afforded the desired indolylglycine product. Another (N^N)-site-based 2D (phendda+TAB)-COF (phendda = 4,4′-(1,10-phenanthroline-3,8-diyl)dibenzaldehyde) was synthesized from TAB and phenanthroline-containing linear dialdehyde. The respective immobilization of [Ir(dFCFppy)2Cl]2 and Ni2+ into the porous framework led to [Ni/Ir(dFCFppy)2(N^N)]@(phendda+TAB)-COF, enabling heterogeneous photocatalysis of C–C cross-couplings.115 A similar complex, [Ni/Ir(C^N)2(N^N)]@(Tp+abpy)-COF (Tp = 1,3,5-triformylphloroglucinol; abpy = 5,5′-diamino-2,2′-bipyridine) exhibited good performance in photocatalyzing C–N cross-coupling reactions with broad substrate diversity and high recyclability (Fig. 13).117 The reaction of TAPM (TAPM = tetra(p-aminophenyl)methane) and BPDCA produced a 3D diamondoid network (BPDCA+TAPM)-COF with 9-fold interpenetration, and its coordination with Ru(bpy)2Cl2 enabled visible light mediated cross-dehydrogenative coupling of tertiary amines and indoles under mild conditions (Fig. 14).191
 | ||
| Fig. 11 3D [Ru(bpy-OEt)3+ETTA]-COF.71 | ||
 | ||
| Fig. 12 2D Ru(N^N)3@(BPDCA+TAB)-COF.116 | ||
 | ||
| Fig. 13 2D [Ni/Ir(C^N)2(N^N)]@(Tp+abpy)-COF.117 | ||
 | ||
| Fig. 14 3D Ru(N^N)3@(BPDCA+TAPM)-COF.191 | ||
Via host–guest interactions, the Ru(N^N)3 or Ir(C^N)2(X^N) derivatives could be incorporated into the pores of COFs,105 yielding PS⊂COFs with diverse photocatalytic properties. The combined use of hydrazone-based COF photosensitizer and [Ir(terpyridine)(ppy)Cl]+ catalyst exhibited efficient and durable photocatalytic CO2RR. The reactivity and durability were highly improved compared with the bare [Ir(terpyridine)(ppy)Cl]+, because the pore confinement effect from the COF facilitated the dispersion and weakened the aggregation of the Ir-catalyst.108
3.2 Ru(N^N)3 and Ir(C^N)2(X^N) functionalized amorphous COFs
The covalent bond strength determines the reversibility of the bond and crystallization of the COFs, thus imine-based COFs are readily formed with good crystallinity, while C–C bond-based COFs are extremely difficult to produce with crystalline networks. Amorphous COFs have disordered arrangement of building units and less porosity, but they commonly have strong chemical stability and are emerging as versatile platforms for exploring new functional materials.192–194 These cross-linked amorphous COFs are usually formed with inert covalent bonds through Suzuki reactions, alkyne trimerization reactions and oxidative coupling reactions, thus have high chemical stability. Amorphous COFs are not readily decomposed even under harsh reaction conditions,195–198 but with less skeleton conjugation and a low surface area.Heterogeneous photocatalysts can be formed by the incorporation of a photoredox catalyst into stable COF with excellent catalytic efficiency and easy separation from the reaction system.199 A series of amorphous [Ru(N^N)3+TPMB]-COF (TPMB = tetrakisphenylmethane borate) were synthesized from Pd-catalysed coupling reactions between one TPMB and two Br-Ru(N^N)3-derivatives, and they showed visible-light driven photocatalytic activity in enantioselective alkylation of aldehydes.199 The Suzuki coupling reaction of linear ditopic Ir(C^N)2(tbubpy)-linker (tbubpy = 4,4′-di(tert-butyl)-2,2′-bipyridine) and TPMB afforded diamond type [Ir(C^N)2(tbubpy)+TPMB]-COF, exhibiting good photocatalytic performance in a wide range of organic reactions, including aerobic oxidations of sulfides and oxidative hydroxylation of arylboronic acids, desulfurative conjugate addition to Michael acceptors, and Smiles-Truce rearrangement of alkyliodides (Fig. 15).52 Their photocatalytic efficiency is better than the homogeneous prototype Ir(III) complexes owing to the porous structure and conjugated backbone of COFs. The cross-coupling of [Ru(Ir)+TEPM]-COF (TEPM = tetra(4-ethynylphenyl)methane) was obtained via Co2(CO)8-catalysed alkyne trimerization reactions of TEPM and Ru(N^N)3 or Ir(C^N)2(N^N)-based linear alkyne-linker. [Ru(N^N)3+TEPM]-COF and [Ir(C^N)2(N^N) + TEPM]-COF were highly effective in a variety of important organic transformations, such as aza-Henry reactions, α-arylation of bromomalonate, and oxyamination of an aldehyde.200 FeCl3-promoted oxidative coupling reaction of carbazole derivatives was an efficient methodology for the construction of organic polymers, and the self-polymerization of Ir(C^N)3-carbazole derivatives produced [Ir(C^N)3+carbazole]-COF, showing intense phosphorescence and high activity in photocatalytic aza-Henry reactions.168 The Ir(C^N)2(N^N) analogue [Ir(C^N)2(N^N)+carbazole]-COF also showed good performance in aerobic photooxidation, such as sulfide oxidation, oxidative hydroxylation of arylboronic acids and cross-dehydrogenative coupling reactions.201 The self-polymerization of 4,4′-di(4-vinylphenyl)-2,2′-bipyridine led to 1D polymer with open (N^N)-sites, and its bondage with Ru(bpy)2Cl2 or [Ir(ppy)2Cl]2 produced Ru(N^N)3- or Ir(C^N)2(N^N)@polyethylene-COF with an electrochemiluminescence response to tri-n-propylamine.202
 | ||
| Fig. 15 Diamond type [Ir(C^N)2(tbubpy)+TPMB]-COF.52 | ||
4. Ru(N^N)3 or Ir(C^N)2(X^N) functionalized metallasupramolecules
Different from the polymeric networks of MOFs, COFs167 and SOFs, metallasupramolecules are discrete architectures with sufficient solubility in solvents, that exhibit unique properties in solution, such as host–guest and liquid luminescence.203,204 Like the synthesis of MOFs, the construction of metallasupramolecules is also based on the coordination-driven assembly of metal ions and ligands. However, the structures of target architectures are commonly precisely foreseen via the controlled combination of metal ions and organic linkers.203,205,206 The fruitful function and abundant structural configurations of these metallasupramolecules could be achieved by rational selection of well-designed ligands and metal precursors. The use of metalloligands with predesigned symmetry and configuration could contribute to the precise control of the architecture's geometry and their functionality could also be transferred to the metallasupramolecules.2074.1 Ir(C^N)2(N^N)-based metallasupramolecules
Similar to Pd(ethylenediamine) and cis-Pt(triethylphosphine)2208,209 groups, [Ir(ppy)2Cl]2 could also be utilized as a metal-corner with two available coordination sites and the ppy ligands are arranged in C,C-cis-N,N-trans orientation, which would sufficiently facilitate the self-assembly of Ir(C^N)2(N^N)-based metallasupramolecules.210 The combination of [Ir(ppy)2Cl]2-precursor and tritopic 1,3,5-tricyanobenzene produced octahedral metallasupramolecule [Ir6(1,3,5-tricyanobenzene)4], and compared to the mononuclear complex, the incorporation of the Ir(C^N)2(N^N) unit into the multimetallic array led to a significant luminescence enhancement (Fig. 16).211 Two homochiral [Ir3(tpmc)2] metallo-cryptophane cages (tpmc = tris(4-pyridyl-methyl)-cyclotriguaiacylene) were synthesized from the assembly of cyclotriveratrylene-based chiral tripodal ligand and [Ir(ppy)2Cl]2-precursor. Interestingly, homochiral self-sorting of both the metal-corner and ligand occurred simultaneously, which was accelerated by the addition of a chiral guest (Fig. 17).56 A similar [Ir3(ttpadtc)2] complex (ttpadtc = 2,7,12-trimethoxy-3,8,13-tris(4,4′-pyridyl-azophenylcarboxy)-10,15-dihydro-5H-tribenzo[a,d,g] cyclononane) was synthesized from the azobenzene-functionalized tripodal cyclotriguaiacylene derivative, showing reversible photo-isomerisation under blue light irradiation.212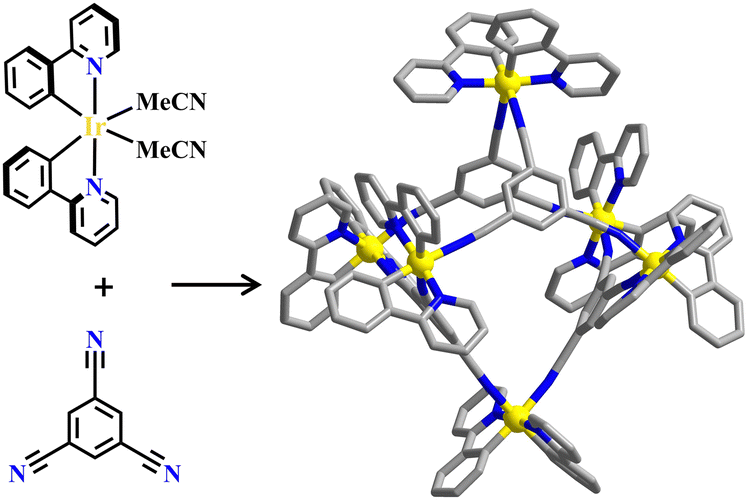 | ||
| Fig. 16 Ir(C^N)2(N^N)-based octahedral cage [Ir6(1,3,5-tricyanobenzene)4].211 | ||
 | ||
| Fig. 17 Homochiral Ir(C^N)2(N^N)-based cryptophane [Ir3(tpmc)2].56 | ||
The reaction of [Ir(ppy)2Cl]2-precursor and ligands with both bis-chelating (N^N)-unit and mono-pyridyl units produced [Ir(mesppy)2(qpy)]+ (H-mesppy = 2-phenyl-4-mesitylpyridinato; qpy = 4,4′:2′,2′′,4′′,4′′′-quaterpyridine) and [Ir(dFmesppy)2(qpy)]+ (H-dFmesppy = 2-(4,6-difluorophenyl)-4-mesitylpyridinato) metalloligands, and the assembly of them with Pd(MeCN)4(BF4)2 (MeCN = acetonitrile) afforded homochiral hetero-metallacages [Ir8Pd4]. The blue-emitting [Ir(dFppy)2(MeCN)2]+ ((H-dFppy = 2-(2,4-difluorophenyl)-4-phenyl)pyridine) guest was encapsulated into the cavity of [Ir8Pd4], and energy transfer between the red-emitting Δ-[Ir8Pd4] cage and the guest molecules was observed (Fig. 18).213 Another isostructural [Ir8Pd4] metallacage was constructed by [Ir(ppy)2(qpy)]+ and [Pd(MeCN)4](BF4)2. Under visible light irradiation, the cubic barrel-shaped [Ir8Pd4] showed large 1O2 quantum yields, which exhibited great potential in the photodynamic therapy and organelles-targeted cell imaging. The [Ir8Pd4] showed higher mitochondria-targeting efficiency and less dark toxicity compared with the [Ir(ppy)2(qpy)]+ metalloligand. The correlation coefficient of mitochondrial affected by the [Ir(ppy)2(qpy)]+ metalloligand was smaller than [Ir8Pd4], which might be attributed to the higher positive charge of the metallacage.58
 | ||
| Fig. 18 Ir(ppy)2(qpy)-based polyhedral cage [Ir8Pd4].213 | ||
4.2 Ir(C^N)3-based metallasupramolecules
Ir(C^N)3-derivatives are widely used in light-emitting diodes because of their strong ΦPL and large Stokes shift. The optical properties of Ir(C^N)3-based materials involving phosphorescence lifetime and emission colour can be regulated via modification of conjugation, heteroatom type, and push–pull electron groups of ligands.214,215 A triple helical organic cage was pre-organised with three (C–H^N)-units, and its cyclometallation with one Ir(III) ion formed a fac-Ir(C^N)3 metallacage with a wide range of light emission and suffering from photoluminescence quenching in the presence of O2 (Fig. 19).216,217 | ||
| Fig. 19 Fac-Ir(C^N)3-based triple helical architecture.217 | ||
Like organic linkers, the metallotecons containing organic active groups, such as amino and aldehyde groups could further react with organic precursors. The reaction of metallotecon fac-tris-Ir(appy)3 (H-appy = 4-(2-pyridinyl)phenylamine), 2-formylpyridine and Zn(BF4)2 afforded [Ir2Zn3] with the properties of photoluminescence, CO2 fixation and nitrite separation (Fig. 20).36 The isostructural complex [Ir2Co3] contained both a photosensitizer and Lewis catalytic center. Co2+ active sites were all in an open environment for favouring substrate bonding. [Ir2Co3] worked as a typical dual catalyst that synergistically combined photo-redox catalysis and metal coordination activation for a trichloromethylation of 2-acylpyridines with BrCCl3.65 In [Ir2Zn3] and [Ir2Co3] cages, the fac conformation of the Ir(C^N)3 moiety constrained the geometry of Zn2+ and Co2+ with four N donors positioned in a controllable fashion.
 | ||
| Fig. 20 Ir(appy)3-based heterometallic polyhedron [Ir2Zn3].36 | ||
4.3 Ru(N^N)3-based metallasupramolecules
Ru(N^N)3 is one type of renowned photosensitizer218 with intriguing photochemical applications in light-harvesting and photocatalysis.46 Ru(N^N)3 derivatives have a moderate redox state,6 and they have been widely employed in various types of photocatalytic reactions.219–222 As stable and functional building blocks, Ru(N^N)3 units are also used in the construction of interesting metallasupramolecules.223–227[Pd6Ru8] metallacages attached with a Ru(N^N)3-unit were synthesized from the reaction of bulky triangular [Ru(piphen)3]2+-metalloligand (piphen, 2-(pyridin-3-yl)-1H-imidazo[4,5-f][1,10]phenanthroline) and Pd(MeCN)4(BF4)2, and these metallacages featured a quite high ΦPL.39 The [Pd6Ru8] cage (Fig. 21) has a rhombododecahedral shape with a 5350 Å3 cavity and 12 open windows. The large hydrophobic size-selective cavity facilitated effective trapping of a nonpolar and water-immiscible aromatics guest in a water-containing hydrophilic solvent. The 1H NMR study of guest inclusion confirmed the formation of noticeable interactions between hosts and guests due to the encapsulation of aromatic guests, which was expected to form facile π–π interactions with the phenanthroline moieties, thus [Pd6Ru8] had potential applications in drug delivery. In addition, inclusion tests on photosensitive guest molecules against UV light radiation confirmed that the cage offered better photoprotection than the pure [Ru(piphen)3]2+-metalloligand, suggesting that the [Pd6Ru8] metallacage could well shield the guests to prevent undesired photolysis.228 Furthermore, the [Pd6Ru8] cage was incorporated with multiple photo- and Lewis acid centers,229 in which the Ru(N^N)3-unit acted as a photosensitizer and Pd2+ as the catalyst. Electron transfer from the [Ru(piphen)3]2+ photoactive center to the Pd2+ center resulted in intramolecular charge separation, and they worked synergistically for the efficient photocatalyzed HER. In the host–guest system, TTF (TTF = tetrathiafulvalene) guests acted as an electron relay mediator to improve the overall electron transfer efficiency by virtue of redox-guest modulation of the photo-induced electron transfer process. In this process, TTF strongly quenched [Pd6Ru8] emission due to the formation of a host–guest adduct. In contrast, without host–guest interactions, TTF-derivatives slightly influenced the emission quenching of [Pd6Ru8] and H2 evolution. This host–guest interaction between redox-active metallacages and a guest provided a model to understand and optimize redox events, such as photocatalytic activities in a confined nanosapce.230 The homochiral Δ/Λ-[Ru(piphen)3]2+-metalloligand led to the formation of homochiral [Pd6Ru8] (Δ-[Pd6Ru8] and Λ-[Pd6Ru8]) featured with large D4-symmetric chiral space, which was imposed by the predetermined [Ru(piphen)3]2+-octahedral stereo configuration. They had an enantio-separation ability for atropisomeric compounds with C2 symmetry, thus recognizing R- and S-BINOL (BINOL = 1,1′-bis(2-naphthol)) enantiomeric guests in solution.60 The homochiral metallasupramolecules possessed dual functionality of photoredox reactivity and stereoselectivity. Naphthol guests were encapsulated into the racemic or enantiopure cages, then underwent a regiospecific 1,4-coupling rather than the normal 1,1-coupling and formed 4-(2-hydroxy-1-naphthyl)-1,2-napthoquinones. This photoinduced regio- and enantioselective coupling was achieved in the confined chiral space of homochiral [Pd6Ru8]. Under aerobic conditions, the photo-excited [Ru(piphen)3]2+ centers were quenched by O2, affording hydrogen peroxide (H2O2) and hydroxyl radical (˙OH). The naphthol substrates were oxidized by [Pd6Ru8]+via single-electron-transfer to give radical species, and its further reaction with ˙OH produced intermediate naphthalene-1,2-dione. Through regioselective 1,4-coupling, 4-(2-hydroxy-1-naphthyl)-1,2-napthoquinone was exclusively obtained.231 By virtue of the molecular cage confinement effect and multi-functions coupling synergism, racemic [Pd6Ru8] accelerated [2+2] photodimerization of symmetric acenaphthylene with seteroselectivity and formed anti-products. Homochiral Δ- or Λ-[Pd6Ru8] photocatalyzed dimerization of 1-Br-acenaphthylene offered the corresponding (6bS,6cS,12bR,12cR)-6b,6c-dibromo-6b,6c,12b,12c-tetrahydrocyclobuta [1,2-a:3,4-a′]diacenaphthylene or (6bR,6cR,12bS,12cS)-6b,6c-dibromo-6b,6c,12b,12c-tetrahydrocyclobuta [1,2-a:3,4-a′]diacenaphthylene, respectively.232 Synergistic actions arising from imidazole-N coordination, imidazole-N protonation-deprotonation of [Pd6Ru8], cage hydrophobicity, and host–guest electrostatic interactions facilitated carbanionic intermediate stabilization of terminal alkynes and C–H activation to achieve Glaser-coupling and unusual H/D-exchange. The immiscible mixture of alkynes and [Pd6Ru8] in aqueous solution turned into a homogeneous phase rapidly, clearly owing to the inclusion of alkynes by the hydrophobicity of the cage via a phase transfer process. This provided a useful catalytic method combining advantages from heterogeneous, homogeneous, and phase transfer to enzymatic catalysis.233
 | ||
| Fig. 21 Ru(piphen)3-based [Pd6Ru8].228 | ||
A Ru(bpy)2Cl2 fragment was used as a 90° acceptor tecton, and the Cl− sites of cis-Ru(bpy)2Cl2 could be occupied by pyridine of the pyridyl linker, producing a [Ru4(4,4′-bipyridine)4] molecular square or [Ru6(TPT)4] (TPT = 2,4,6-tri(pyridin-4-yl)-1,3,5-triazine) truncated tetrahedral cage respectively. The [Ru6(TPT)4] cage possessed emergent properties attributed to its unique electronic structure, resulting in increased visible-light absorption and biexponential decay of an emission band.53 A heteronuclear metallacage [Pd4Ru8] was assembled by [Ru(tbubpy)2(qpy)]2+-metalloligand and Pd(MeCN)4(BF4)2 with high ΦPL.234
5. Ru(N^N)3 or Ir(C^N)2(X^N) functionalized organic supramolecules
Supramolecular architectures could be classified into metallasupramolecules and organic supramolecules. Metallasupramolecules are constructed by the metal–ligand interactions, and organic supramolecules are assembled via covalent linkages.19 The Ru(N^N)3 or Ir(C^N)2(X^N) functionalized organic supramolecules are described here. Luminescent tetrahedral molecular cages [Ru(phen)2(N^N)+tris(2-aminoethyl)amine] were formed via the reaction of linear precursor {Ru(phen)2[(3,3′:6′,2′′:5′′,3′′′-quaterpyridine)-6,6′′′-dicarbaldehyde]}2+ with tris(2-aminoethyl)amine and Zn{bis(trifluoromethanesulfonyl)imide}2. The stereoisomeric chirality of the Ru(N^N)3-center was passed to the synthesized cage (Fig. 22).46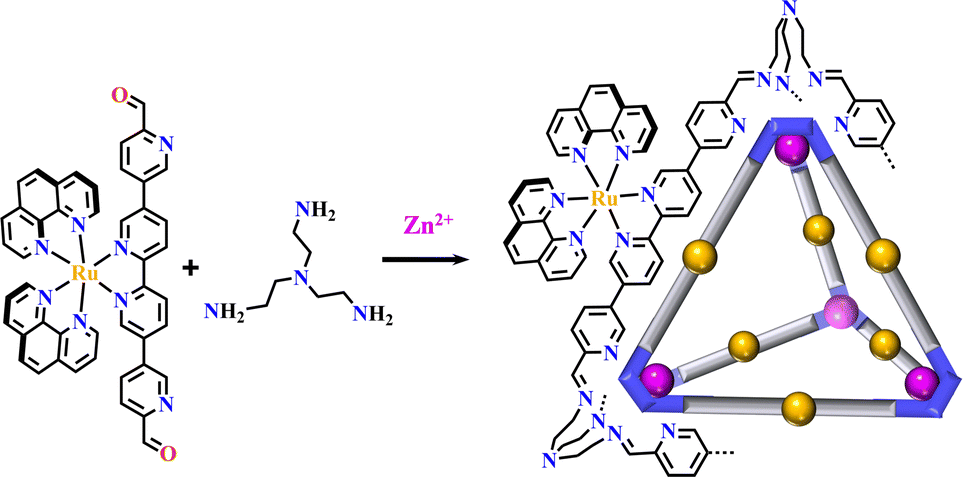 | ||
| Fig. 22 Luminescent tetrahedral cage [Ru(phen)2(N^N)+tris(2-aminoethyl)amine].46 | ||
Mitochondria DNA-targeted photodynamic therapy was used for cancer treatment with a series of [fac-Ir(ppy-CHO)3+diamine] metallohelices (H-ppy-CHO = 4-(2-Pyridyl)benzaldehyde). Aldehyde groups of fac-Ir(ppy-CHO)3 were helically arranged around the Ir(III) center with a facial propeller-like configuration. Connecting the preorganized Δ/Λ-fac-Ir(ppy-CHO)3 modules with different diamines by dynamic imine-coupling chemistry allowed for the formation of [fac-Ir(ppy-CHO)3+diamine] metallohelices with stereochemical information and suitable size. The odd-even character and length of the diamine alkyl linkers determined the conformations of two Ir(III) centers of one metallohelices with the same chirality or heterochirality (Fig. 23). The high degree of narcissistic chiral self-sorting occurred during the condensation, and a pair of enantiomers were obtained even when diamines with an even number of C atoms were introduced. For an odd number C-based diamine, only one achiral cage was obtained with two heterochiral Ir(III) centers. To improve their stabilities for biological applications, these imine-linked [fac-Ir(ppy-CHO)3+diamine] metallahelices were further reduced into amine-inked metallohelices by NaBH4. Notably, in sharp contrast to the corresponding imine complex, the ΦPL and 1O2 quantum yields of these amine-linked metallohelices were significantly enhanced. DNA-binding affinities influenced the photodynamic therapy treatment. 1,5-Pentanediamine had an appropriatelength between diamine spacers, thus showed stronger DNA-binding affinities in a minor groove manner with high photodynamic therapy efficacy.32 Employing Δ/Λ-fac-Ir(ppy-CHO)3 modules with chiral RR/SS-trans-1,2-diaminecyclohexane spacers, homochiral dinuclear ΔR-[fac-Ir(ppy-CHO)3+cyclohexylamine] and ΛS-[fac-Ir(ppy-CHO)3+cyclohexylamine] were obtained. Their chiral porous crystals could be used for the effective enantioseparation of atropisomers, yielding >99% ee for BINOL in a single separation cycle. These molecular crystals maintained consistent ee values even after 10 cycles of enantioseparation. Changing the helicands as achiral ethylenediamine, the resulting crystalline homochiral helicates Δ-[fac-Ir(ppy-CHO)3+cyclohexylamine] and Λ-[fac-Ir(ppy-CHO)3+cyclohexylamine] showed no enantioselectivity despite displaying a similar adsorption capacity. The strict chirality matching between the extrinsic 3D chiral channels and the enantiopure guests may dominate the enantioseparation process.64
 | ||
| Fig. 23 [fac-Ir(ppy-CHO)3+diamine]-helicates.32 | ||
A chiral Ir(C^N)2(N^N)-based building block was regionally coordinated to a bipyridyl-based strand, which subsequently extended into a chiral building block with six vacant pyridine-based chelating sites, and the building block was dimerized through coordination with four Zn(OTf)2 and formed a Ir(C^N)2(N^N)-based closed-loop helicate (Fig. 24). Helicate crossing was captured covalently by ring closing metathesis of pendant alkenes and a topological chiral star of David catenane was formed, where the Zn2+ could be removed with the maintenance of an interlocked structure. The kinetic stability of the coordinated Ir(III) center ensured that the circular helicate was formed as a single enantiomer. In the star of David [2] catenane, both complexes with and without coordinated Zn2+ ions retained the photophysical characteristic of the Ir(C^N)2(N^N) unit.67
 | ||
| Fig. 24 Ir(C^N)2(N^N)-based triply interlocked star.67 | ||
6. Ru(N^N)3 and Ir(C^N)2(X^N) functionalized SOFs
SOFs are crystalline, ordered supramolecular polymers with defined structures. Many reaction conditions including solvents, temperature, pH and reaction time could have a great influence on the synthesis of MOFs and COFs, however the construction of SOFs depends on the mild formation of intermolecular interactions,235 including hydrogen bonds, electrostatic, and π–π interactions, etc.235Using photosensitized building blocks, the homogeneity permitted SOFs with maximum visible light radiation.104Via host–guest interaction, the combination of six-armed Ru(N^N)3-based precursors with cucurbit[8]uril generated a {Ru(N^N)3+cucurbit[8]}-SOF at room temperature, which maintains its solid-state structure in solution, achieving the encapsulation of {P2W18} in a one-cage-one-guest manner. This material realized homogeneous catalytic H2 production under visible light irradiation with high efficiency (Fig. 25).70 Another similar cubic {Ru(N^N)3+cucurbit[8]}-SOF was obtained from the reaction of [Ru(bpy)3]2+-based acyl hydrazine with aldehyde in the presence of cucurbit[8]uril. The remarkable size could adsorb discrete anionic {P2W18} through electrostatic interaction, which was further stabilized in the cationic framework. The corresponding SOF remarkably facilitated visible light-induced electron transfer from Ru(N^N)3 units to POM guests, enabling highly efficient photocatalytic H2 production.69
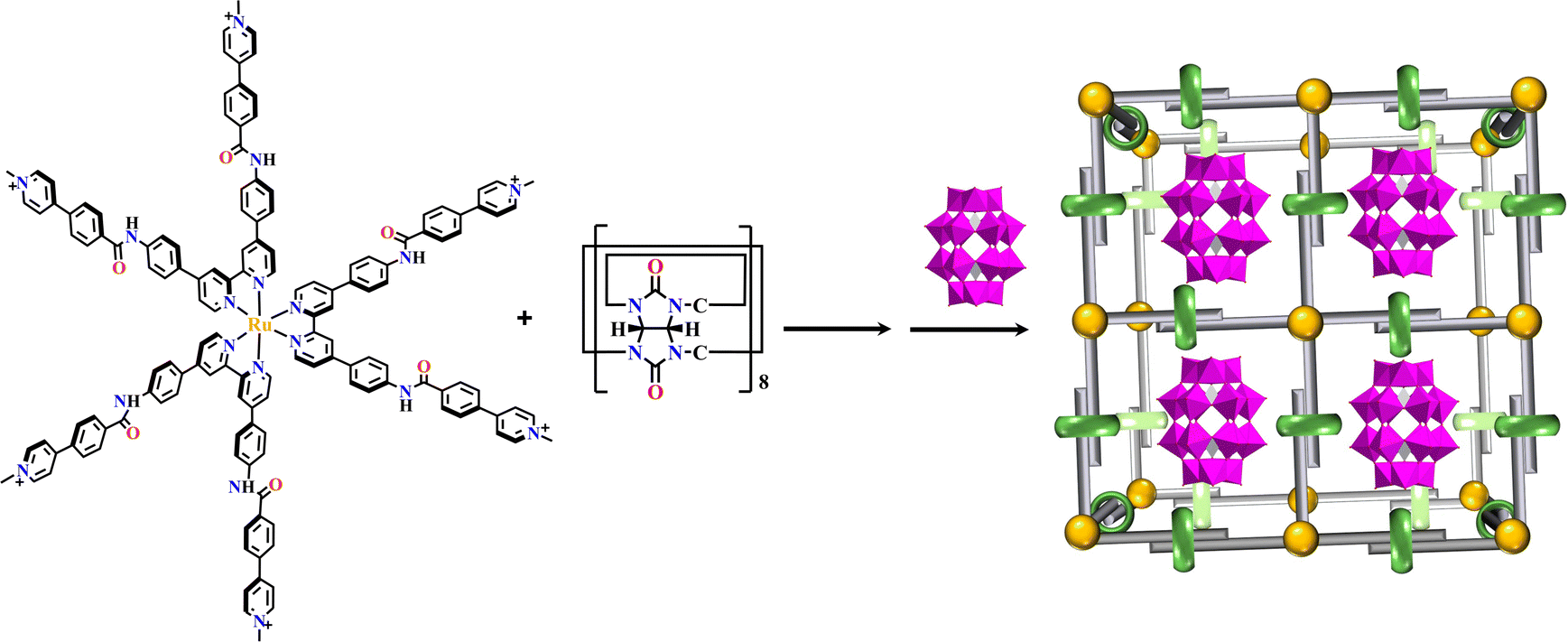 | ||
| Fig. 25 {P2W18}⊂{Ru(N^N)3+cucurbit[8]}-SOF.70 | ||
In addition, Ru(bpy)2(dcbpy) and [Ru(dcbpy)3]4− photosensitizers, {P2W18} and {PW12} POMs could also be encapsulated in the pores of SOFs as guests through host–guest interactions. The nanospace provided by SOFs simultaneously encapsulated Ru(N^N)3-photosensitizer and POMs. The improvement of electron transfer between the Ru(N^N)3-photosensitizer and POMs catalyst greatly improved the photocatalytic H2 production.104
7. Photocatalysis investigation of Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures
Ru(N^N)3 and Ir(C^N)2(X^N) derivatives are commonly employed as photocatalysts owing to their strong visible light absorption capability, long-lived photoexcited states, dually effective excited state oxidant and reductant. The light absorption range and redox potentials of three types of Ru(N^N)3, Ir(C^N)2(C^N) and Ir(C^N)3 samples are quite different. As shown in Table 2,81 photoexcited Ir(ppy)3 has a stronger reduction capability than [Ru(bpy)3]2+ and [Ir(ppy)2(bpy)]+ owing to its lower redox potentials. For the same type of PS, the redox potential of [Ir(ppy)2(bpy)]+ is higher than [Ir(ppy)2(tbubpy)]+, but lower than [Ir(dFCFppy)2(tbubpy)]+, indicating the influence of the electron donating or withdrawing group of (N^N) on redox potential. Their photophysical properties could be finely regulated via the judicious choice of (N^N) or (X^N) ligands, providing a designable and flexible photocatalyst platform for specific reactions.81
| Entry | Complex | Excitation λmax/(nm) | Emission λmax/(nm) | Excited-state lifetime t/(ns) | E 1/2(M/M−) | E 1/2(M+/M*) | E 1/2(M*/M−) | E 1/2(M+/M) |
|---|---|---|---|---|---|---|---|---|
| SCE = standard calomel electrode. M represents the Ru(N^N)3 or Ir(C^N)2(X^N) complex.a Measurements were performed in MeCN at room temperature unless otherwise noted.b Determined in 1:1 CH5OH/CH3OH at 77 K. | ||||||||
| 1 |

|
452 | 615 | 1100 | −1.33 | −0.81 | +0.77 | +1.29 |
| 2 |

|
375 | 494b | 1900 | −2.19 | −1.73 | +0.31 | +0.77 |
| 3 |

|
420 | 585 | 307 | −1.42 | −0.87 | +0.70 | +1.25 |
| 4 |

|
410 | 581 | 557 | −1.51 | −0.96 | +0.66 | +1.21 |
| 5 |

|
380 | 470 | 2300 | −1.37 | −0.89 | +1.21 | +1.69 |
Ru(N^N)3 and Ir(C^N)2(X^N) derivatives are good photosensitizers (PSs), and the combined use of the PS and Lewis acid catalyst (Cat) generated novel photocatalytic activities. For example, Ru(N^N)3 derivatives are good PS for water splitting because of their appropriate redox ability. According to thermodynamic criteria, Ru(N^N)3 catalysts can drive the photolysis of H2O, but this is rarely observed due to the high reaction barriers. Only when Ru(N^N)3 PS and other Cat are employed together, can the photocatalytic H2O splitting be achieved. Ru(N^N)3 or Ir(C^N)2(X^N) based PS and Cat photocatalytic systems commonly follow the three-component system of “PS-Cat-D/A” (D = electron donor, A = electron acceptor). As shown in Fig. 26, PS* acts as a mediator and transfers an electron from the electron donor D to Cat, which involves two mechanisms of oxidative quenching and reductive quenching.236 The oxidative quenching process involves single-electron-transfer from PS* to Cat, accompanied with the formation of high-oxidation-state PS+ and reduced Cat−. The PS+ gets one electron from D to achieve recovery. The reduction quenching process involves PS* grabbing the electrons from D associated with the formation of reduced PS− and single-electron-transfer occurs from PS− to Cat. In a word, reductive refers to the reduction of PS*, whereas the external D is oxidized in the same process. Oxidative means oxidation of the PS* concomitant with the reduction of Cat.237 The incorporation of both PS and catalytic sites into a confined nanospace not only increases the apparent concentration of both PS and Cat104 but also facilitates multielectron transfer to drive high photocatalytic performance, as well as stabilizing the PS and Cat via site isolation.47,114,238–240 Many types of photocatalytic reactions have been achieved using the Ru(N^N)3 or Ir(C^N)2(X^N) functionalized architectures.
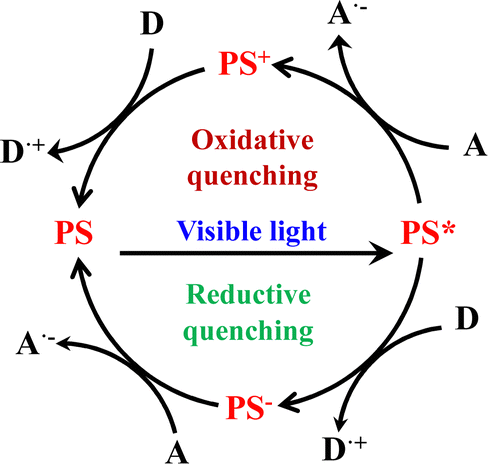 | ||
| Fig. 26 Mechanisms of oxidative quenching and reductive quenching.237 | ||
7.1 Photocatalytic hydrogen evolution reaction (HER)
Photocatalytic HER is an important approach to achieve solar-to-chemical energy conversion. The HER involves both oxidative quenching and reductive quenching mechanisms.140,150 Reductive quenching-based photoreduction can take place when both requirements of E(PS/PS−) < E(Cat/Cat−) < E(H+/H2) and E(PS*/PS−) > E(D+/D) are met. Oxidative quenching-based photoreduction takes place when both requirements of E(PS+/PS*) < E(Cat/Cat−) < E(H+/H2) and E(PS+/PS) > E(D+/D) are satisfied. Different Ru(N^N)3 and Ir(C^N)2(X^N) PSs should be matched with the corresponding Cat to achieve the HER. For instance, when [Ir(ppy)2(N^N)]+ and [Ru(bpy)2(N^N)]2+ were used as photosensitizers and {Ni4P2}-based POMs as catalysts. [Ir(ppy)2(N^N)]+ drove a highly efficient HER process, but [Ru(bpy)2(N^N)]2+ was proved useless, because −0.70 eV of E{[Ir(ppy)2(N^N)]2+/([Ir(ppy)2(N^N)]+)*} was negative enough to permit electron accumulation at {Ni4P2} POMs, enabling {Ni4P2} to have enough reduction activity for H2 production. In contrast, −0.62 eV of E{[Ru(bpy)2(N^N)]3+/([Ru(bpy)2(N^N)]2+)*} was higher than the −0.65 eV redox potential of {Ni4P2}, thus cannot give {Ni4P2} with a more negative reduction potential for driving H2 production (Fig. 27).150 | ||
| Fig. 27 HER process catalysed by {Ni4P2}⊂[Ir(ppy)2(dbpydc)+Zr6]-MOF.150 | ||
The incorporation of POMs into Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures produced interesting photocatalytic activities arising from the synergistic effect of the photosensitizer, POMs and well-defined nanospace. In {P2W18}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF114 and {Ni4P2}⊂[Ru(bpy)2(dbpydc)+Zr6]-MOF,150 the POMs were encapsulated into the porous cavity of the photosensitizer-functionalized frameworks via electrostatic force and host–guest interaction, and the proximity of POMs to the photosensitizer allowed a facile multi-electron transfer and enabled an efficient visible-light-driven HER with high turnover numbers. Photophysical and electrochemical studies proved that the oxidative quenching of PS* by POMs was the initiating step of the HER. The simultaneous encapsulation of Ru(N^N)3 derivatives and POMs into a crystalline framework, producing an excellent photocatalyst for the HER. In these networks, the electron transition distance was shortened because the photosensitizer unit and POM catalyst were confined together via electrostatic force and host–guest interactions, leading to sharply increased photocatalytic activity in the HER process.
Apart from POMs, the metal cluster in SBUs can also act as a catalyst. In [Ru(bpy)2(bpydc)/bpdc+Ti3]-MOF and [Ir(ppy)2(bpydc)/bpdc+Ti3]-MOF, photophysical and electrochemical studies proved that the photocatalytic HER proceeded reductive quenching of PS* and then an electron was transferred from the reduced PS to Ti3 SBUs. Density functional theory calculations revealed that key steps of the HER process included the Ti3+-OH was protonated to generate the Ti3+ species with a vacant coordination site, and proton-coupled electron transfer to provide the key Ti4+-H intermediate.140 In Ir(ppy)2(MBA)@(BTB+Ce6)-MOL and Ru(bpy)2(MBA)@(BTB+Ce6)-MOL, the proximity of photosensitizing ligands and Ce6-SBUs facilitated electron transfer to drive the photocatalytic HER under visible light. The PS* in the MOL was reductively quenched and then transferred electrons to Ce6-SBUs to generate reduced Ce3+ centers, and they were photoexcited to Ce3+* species for further HERs.37
In addition, the metal center anchored at the ligands can also be employed as a catalyst, and they were combined with a Ru(N^N)3 or Ir(C^N)2(X^N) photosensitizer for bifunctional catalysis. The incorporation of both photosensitizer and metal catalyst into one network, enabled the fast electron–hole separation and transfer to metal catalyst for excellent photocatalytic performance. The network of [Ir(ppy)2(bpydc)/bpydc+Zr6]-MOF had open (N^N)-sites that can capture K[PtCl3(C2H4)]. The catalyst-incorporated framework not only promoted photoelectron transfer from the Ir(C^N)2(X^N) photosensitizer to the Pt catalyst, but also increased the stability of the backbone.47 The stable Co@[Ru(bpy)2(bpydc)/bpydc+Zr6]-MOF exhibited extra high photocatalytic activity for H2 production with recycle and reuse performances. The rate-limiting step was the formation of Co+ intermediate species reduced by a photoelectron.241
7.2 Photocatalytic CO2 reduction reactions (CO2RR)
The ever-increasing atmospheric CO2 from fossil fuel consumption raises growing concerns about global warming.242,243 Photocatalytic CO2 transformation to other valued chemicals attracted great interest. However, selective CO2 transformation remains a challenge as the existing catalyst usually has low catalytic efficiency and poor product selectivity and often suffers from the competing HER.165,244In recent years, several Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures have been employed in the CO2RR process, in which Ru(N^N)3 and Ir(C^N)2(X^N) units are used as the photosensitizer, and metal clusters137,151 or metal complexes are used as coordination catalytic centers, such as Cu,138,165 Re, Mn,155 Ni,139 and Co.149 The CO2RR also obeys the three-component form of “PS-Cat-D” involving two mechanisms of oxidative quenching and reductive quenching. The electrons are transferred from PS* to Cat, and the photogenerated holes in PS* abstract the electrons from D.
CO2 can be reduced to various products of CO, HCOOH, HCHO, CH3OH, and CH4 by gaining different numbers of electrons and protons.244 During the CO2RR, the light-excitation attributes, band structure, and separation efficiency of photogenerated charge carriers are three important factors to influence photocatalytic CO2 transformation. Moreover, CO2 adsorption/activation, catalytic active sites, and intermediate adsorption/desorption are also critical in regulating the product selectivity.244 Redox potentials of photocatalytic CO2 reduction to different products (Fig. 28) can theoretically control the product selectivity of CO2 transformations. In addition, the C, H, and O affinity of the photocatalyst has a great influence on the adsorption and desorption of reactants/intermediates, the sequence of hydrogenation and deoxygenation reactions, reaction pathways and product selectivity.
 | ||
| Fig. 28 Reduction potentials of various products for the CO2RR (SHE = standard hydrogen electrode, CB = conduction band, VB = valence band). | ||
Herein, we elaborate on the materials based on the types of products formed. HCOOH is one potential compound for fuel cells and hydrogen storage.245,246[Ru(phenba)3+Eu2]-MOF has Eu2 clusters as connecting nodes and Ru(phenba)3-metalloligand as linkers, which selectively reduced CO2 to HCOOH in a two-electron process with an excellent rate. The efficient electron transfer from [Ru(phenba)3]2+-units to Eu2 clusters allowed high HCOOH production rates. The (Eu–H2O–Eu)-active sites not only accepted electrons but also captured CO2, thus could achieve highly efficient transformation of CO2 to HCOOH (Fig. 29).137[Ru(cptpy)2+Zr]-MOF was composed of [Ru(cptpy)2]2+-metalloligand and ZrO8 cluster and had both high chemical stability and photostability, and it efficiently photocatalyzed the reduction of CO2 to HCOOH under visible light irradiation.151 In addition, bimetallic catalysis can reduce CO2 to HCOOH. Highly selective photoreduction of CO2 to HCOOH was achieved with a high yield by hierarchical integration of [Ru(bpy)2(H2bpydc)]2+ photosensitizer and monometallic Cu(bpydc)Cl2 catalyst into a stable Cu@[Ru(bpy)2(bpydc)/bpydc+Eu6]-MOF.165 The multinuclear complexes of [Ru(N^N)3]x{Ru(N^N)2(CO)2}y combined PS and Cat together via covalent linkage, exhibited high photocatalytic efficiency and HCOOH selectivity in CO2 transformation. The ratio between PS and Cat strongly affected the photocatalytic activities, and the higher PS ratio gave a better yield of HCOOH. Weak conjugation of PS and Cat could be also beneficial for better photocatalytic performance.247
 | ||
| Fig. 29 Pathways of electron transfer from Ru(phenba)3 to the catalytic Eu2 oxo cluster in [Ru(phenba)3+Eu2]-MOF.137 | ||
Among all the possible technological routes, visible-light-driven two-electron reduction of CO2 to CO is a kinetically favourable option because it has a lower reaction barrier compared to one-electron and multi-electron reactions.248,249 Bimetallic synergistic catalysts are commonly explored for CO synthesis with enhanced performance over their monometallic counterparts because of multielectron accumulation at the catalytic centers. The carboxylate exchange at the Hf12 cluster of [Ru(bpy)2(dbpydc)+Hf12]-MOL with metal complexes yielded bimetallic catalysts M@[Ru(bpy)2(dbpydc)+Hf12]-MOL (M = Mn or Re, X = Cl or Br), that possessed both Ru(N^N)3 photosensitizer and M(CO)3X catalyst for efficient photocatalytic CO2 reduction (Fig. 30).155 The molecular catalyst [Ni(bpet)(H2O)2]2+ was encapsulated into a photo-responsive [Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF to fabricate a composite Ni⊂[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF for the photocatalytic transformation of CO2 to CO.139 Artificial photocatalytic CO2RR is a big challenge as the efficient cooperation of multiple functional units is difficult. Without use of an electron donor, the multifunctional photocatalyst [Ru(ip)3+Co]-MOF could convert CO2 to CO with a high CO production rate and nearly 100% selectivity accompanied by water oxidation to O2. Suitable photocatalytic redox potentials, efficient electron-hole separation and CO2 adsorption ability all contributed to this artificial photosynthesis.59
 | ||
| Fig. 30 Photocatalytic CO2RR to CO occurred in M@[Ru(bpy)2(dbpydc)+Hf12]-MOL.155 | ||
Syngas (a mixture of H2 and CO) is a versatile fuel precursor for producing chemical and synthetic liquid fuels. Photocatalytic CO2RR will open an avenue for consecutive production of syngas.250 Zhang and collaborators reported a Co@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF composite photocatalyst for efficient syngas production. Interestingly, the H2/CO ratios could be well regulated by precisely adjusting the water content of the reaction solution and the molar ratio of PS/Cat in the network.149
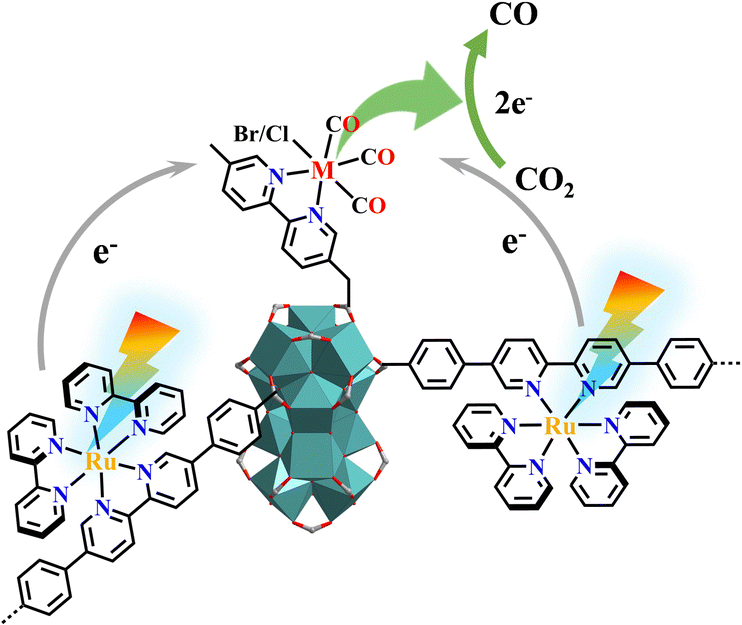
Photoirradiation CO2 hydrogenation to ethanol is of practical importance yet poses a significant challenge due to the formation of a C–C bond with an intact C–O bond. Lin and co-workers used low-intensity light to activate a Cu@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF catalyst for the selective hydrogenation of CO2 to C2H5OH. Under light illumination, [Ru(N^N)3]* underwent single-electron-transfer to Cu2+ to generate Cu+ which was active for C2H5OH production. In contrast, Cu0 nanoparticles were formed as the active species for CH3OH production under darkness (Fig. 31).138
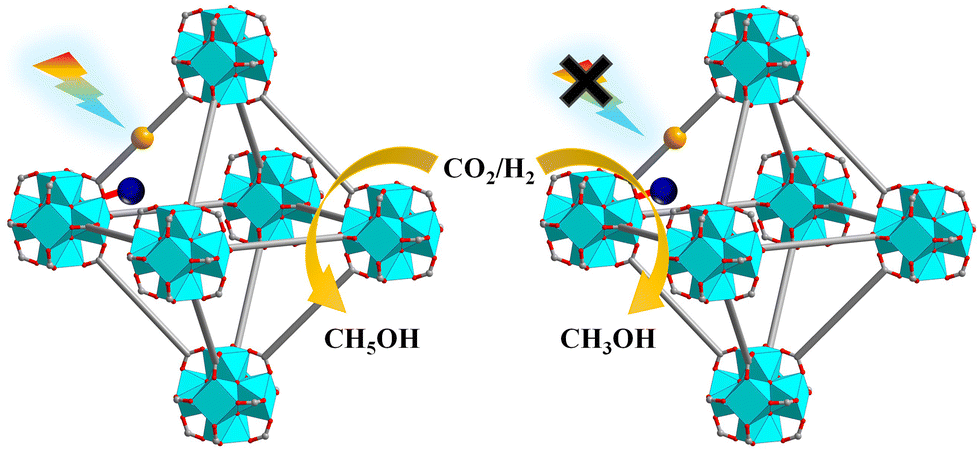 | ||
| Fig. 31 Selective photocatalytic CO2RR with or without light irradiation catalysed by Cu@[Ru(bpy)2(bpydc)/bpdc+Zr6]-MOF.138 | ||
7.3 Aerobic photooxidation
Aerobic photooxidation is very important in organic synthesis because photocatalysis provides a high-efficiency, safe and economic O2 utilization for developing diverse aerobic organic transformations and O2 could be transformed to other ROSs, such as H2O2, 1O2, ˙O2−, and ˙OH.251 ROSs of ˙OH, H2O2, ˙O2− and 1O2 could be generated in different ways. A stepwise reduction of O2 respectively generates ˙O2−, H2O2, and ˙OH.251 The quantum yield of ˙O2− is significantly higher, and its lifetime is only lower than H2O2. In the absence of reactants, ˙O2− is converted to the stable H2O2via a disproportionation process. The ˙OH radical has the highest oxidation activity, thus its oxidation performance is almost limited by the substrate diffusion rate. In addition, ˙OH is commonly pursued in photocatalytic organic decomposition. Although 1O2 has a lifetime of ten milliseconds in air, it was shortened to only 3 μs in H2O and thus it might be decayed by energy transfer with no chance of participating in any reaction.251By combining three intrinsically nontoxic components, namely, photosensitizer, light, and tissue O2 to generate cytotoxic ROS, photodynamic therapy provides an effective phototherapy against cancer.252 Ultrathin MOLs facilitate the diffusion of ROS to cell milieu to exert cytotoxic effects.50,156 Cationic [Ir(ppy)2(dbpydc)+Hf12]-MOL was built from Hf12-SBUs and [Ir(ppy)2(H2dbpydc)]+-metalloligand and then was loaded with POMs {P2W18} to afford {P2W18}⊂[Ir(ppy)2(dbpydc)+Hf12]-MOL. Upon X-ray irradiation, {P2W18}⊂[Ir(ppy)2(dbpydc)+Hf12]-MOL significantly enhanced ˙OH generation from Hf12-SBUs, 1O2 generation from [Ir(ppy)2(H2dbpydc)]+-metalloligand, and ˙O2− generation from {P2W18}, achieving synergistic cell killing by these ROSs (Fig. 32).49
 | ||
| Fig. 32 Three distinct ROS generated from {P2W18}⊂[Ir(ppy)2(dbpydc)+Hf12]-MOL upon X-ray irradiation.49 | ||
Aerobic photocatalytic sulfide oxidation to sulphoxide is a green pathway for sulphoxide production using O2 as an oxidant. Highly stable [Ir(pqc)2(phen)+Zr6]-MOF was constructed by [Ir(pqc)2(phen)]+-metalloigand linkers and Zr6-SBUs. [Ir(pqc)2(phen)+Zr6]-MOF exhibited high catalytic activity upon aerobic photooxidation of sulfide into sulfoxide in water at room temperature.48,142 Under visible light irradiation, the [Ir(pqc)2(phen)]+ photosensitizer was excited to a high-energy excited singlet state 1{[Ir(pqc)2(phen)]+}*, which quickly underwent intersystem crossing to an excited triplet due to the highly efficient spin–orbit coupling. The 3{[Ir(pqc)2(phen)]+}* transferred an electron to O2 to form ˙O2− and generated [Ir(pqc)2(phen)]2+. Sulfide was oxidized by [Ir(pqc)2(phen)]2+ to generate a sulfide radical cation and achieve the regeneration of neutral [Ir(pqc)2(phen)]+ photosensitizer in the photocatalytic cycle. The sulfide radical cation reacted with the ˙O2− to generate a persulfoxide intermediate, which further reacted with sulfide to afford the corresponding sulfoxides. In this system H2O not only accelerated the conversion of persulfoxide into sulfoxide but also prevented the over oxidation of sulfoxide into sulfone.48 Moreover, ˙O2− was the main ROS for efficient photocatalytic sulfide oxidation. Interestingly, by changing the auxiliary ligand of the Ir(pqc)2(N^N)-metalloligand, the excitation lifetime of the triplet state, the generation of ROS quantum yields, and the sulfide photooxidation efficiency could be tuned.253Ru(bpy)2(bpydc)@(bpydc+Ln6)-MOFs (Ln = Tb, Gd, Eu) showed excellent photocatalytic activity in sulfide oxidation with high conversion and sulfoxide selectivity under an O2 atmosphere, ˙O2− was the main ROS and h+ also took effect in oxidation. Ru(bpy)2(bpydc)@(bpydc+Ln6)-MOFs were excited by visible light and generated electrons and holes. Here, O2 accepted an electron and produced ˙O2−, which further attacked sulfide, with the assistance of h+, the active intermediate persulphoxide formed, and its reaction with another sulfide molecule led to the formation of sulfoxide products.142[Ru(cptpy)2+Ce]-MOF was synthesized from [Ru(cptpy)2]2+-metalloligands and Ce(NO3)3. [Ru(cptpy)2+Ce]-MOF exhibited high photocatalytic activities in sulfide oxidation, which is better than the isomorphic [Ru(cptpy)2+Zr]-MOF, revealing the important role of Ln(III) ions in the photocatalytic process. The doping of Zr(IV) enhanced the chemical stability of [Ru(cptpy)2+Ce/Zr]-MOF, and [Ru(cptpy)2+Ce/Zr]-MOF possessed both high chemical stability and catalytic efficiency in photocatalytic sulfide oxidation.254
Overall photocatalytic water splitting is a great challenge. Thermodynamically, the conduction band/valence band position of the semiconductor must match the reduction/oxidation potential of water, so that the excited electron–hole pair has a sufficient ability to carry out overall water splitting. Dynamically, the slow desorption rate of H2 and O2 generated by photocatalytic water splitting on the surface of the material also becomes the kinetic limiting factor, and the desorption rate is slower than the recombination of photo-generated electrons and holes.255–258HER-MOF and WOR-MOF nanosheets were integrated into liposomal structures for separation of the photogenerated charges. For example, the HER-MOF were constructed from hydrophobically modified Hf6 clusters and Zn–porphyrin or Pt–porphyrin linkers, while WOR-MOF were fabricated by Zr12 clusters and Ru(N^N)3 or Ir(C^N)2(N^N) metalloligands. The lipid membrane separated the oxidative and reductive components to prevent charges recombination, and this system was used in overall photocatalytic water splitting through the ‘Z-scheme’ electron-transfer.259
[Ru(bpy)2(bpydc)/BTB+Zr6]-MOF was assembled via the linkage of BTB-Zr6 layers and [Ru(bpy)2(H2bpydc)]2+-metalloligand, and it served as a highly efficient catalyst in the photooxidation reactions, including photooxidation of sulfide, coupling of amines and oxidative hydroxylation of arylboronic acids (Fig. 33). The main ROS of three types of reaction was ˙O2−, and the generated ROS was determined by the photocatalyst itself.260 Mesoporous Ru(bpy)2(bpydc)@(TPTB+Zr6)-MOF was composed of Zr6 clusters and Ru(bpy)2(H2bpydc)-metalloligand, and it exhibited exceptional high photocatalytic activity in the oxidation of dihydroartemisinic acid to artemisinin.118 The 3D diamondoid (BPDCA+TAPM)-COF had open (N^N)-sites, and the incorporation of the Ru(bpy)2Cl2 unit led to Ru(N^N)3@(BPDCA+TAPM)-COF, which performed as a highly efficient, visible-light-mediated photocatalyst for the oxidative cyanation reaction of tertiary amines with excellent yields.191
 | ||
| Fig. 33 Aerobic photooxidation catalysed by [Ru(bpy)2(bpydc)/BTB+Zr6]-MOF.260 | ||
7.4 Photocatalytic organic transformation
Organic transformations play a key role in the synthesis of pharmaceutical drugs and functional molecules. Photocatalytic reactions have appeared as a new tool to solve some critical problems met in the traditional reaction methods because photocatalysis could directly activate the substrates and bypass the reaction barrier. The last decade has witnessed the rapid development of photoredox catalysis.6,219,261 In particular, Ru(N^N)3 or Ir(C^N)2(X^N) derivatives have been widely used as the photocatalyst.262–264 A large number of Ru(N^N)3 or Ir(C^N)2(X^N) functionalized architectures have been explored for the photocatalytic organic transformation, including single-site catalysts,116,118 dual active sites115,135,136,141,265 and ternary active sites158 for synergistic catalysis.Ru(N^N)3 or Ir(C^N)2(X^N) derivatives acted as both photosensitizer and catalyst in the well-defined architectures. The Ru(N^N)3@(BPDCA+TAB)-COF displayed unprecedented photocatalytic activity of visible light and achieved cross-dehydrogenation coupling of secondary amine and indole with an excellent rate (Scheme 1).116 Mesoporous Ru(bpy)2(bpydc)@(TPTB+Zr6)-MOF photocatalyzed the aza-Henry reaction with high conversion yields (Scheme 2), showing a similar catalytic performance with [Ru(bpy)3]2+.118 Stereoselectivity in photoredox reactions was very difficult to control. One strategy to achieve enantioselectivity in photoredox reactions is to combine a photoredox catalyst and a stereochemically controlled cocatalyst into one catalytic system.266–268Δ/Λ-[Pd6Ru8] metallasupramolecules had dual functionality of photoredox reactivity and stereoselectivity. The photoinduced regio- and enantioselective coupling of naphthols and its derivatives thereof was achieved in the confined chiral space of [Pd6Ru8]. The enantiomer cages encased naphthol guests, and then underwent a regionally specific 1,4-coupling, instead of the normal 1,1-coupling, forming 4-(2-hydroxy-1-naphthyl)-1,2-napthoquinones.231Δ/Λ-[Pd6Ru8] was also an effective supramolecular reactor to achieve the enantioselective cycloaddition of 1-subsituted acenaphthylene derivatives.232
 | ||
| Scheme 1 Photocatalytic cross-dehydrogenative coupling of secondary amines with indoles catalysed by Ru(N^N)3@(BPDCA+TAB)-COF.116 | ||
 | ||
| Scheme 2 Aza-Henry reactions photocatalyzed by Ru(bpy)2(bpydc)@(TPTB+Zr6)-MOF.118 | ||
In the well-defined architectures, Ru(N^N)3 and Ir(C^N)2(X^N) units are synergistic with another catalyst in the catalytic reactions. The proximity and cooperativity between different catalytic sites are key issues in the design of dual-functional catalytic systems.269 The precise location of dual catalytic sites at porous architectures should be a feasible strategy.115,135,136,141,265 The proximity of two catalytic components in the cavities of the frameworks greatly facilitates charge transfer from photosensitizing centers to catalytic centers, reaching a higher turnover number than the free counterpart.135,136,265 Incorporation of Ni(N^N)Cl2 Cat into [Ir(dFCFppy)2(dbpydc)/dbpydc+Zr12]-MOL,265[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL,135Ir(dFCFppy)2(N^N)@(phendda+TAB)-COF and Ir(ppy)2((N^N))@(Tp+abpy)-COF115,117 efficiently catalysed four important cross-coupling reactions with broad substrate scopes, including C–S cross-coupling between thiols and aryl iodides,135,265 C–O cross-coupling between aryl bromides and alcohols,135 and C–C cross-coupling between aryl bromides and potassium benzyltrifluoroborates (Scheme 3)115,135 as well as C–N cross-coupling between iodides and amines.117 For example, upon light irradiation, [Ir(dFCFppy)2(H2dbpydc)]+ was excited to {[Ir(dFCFppy)2(H2dbpydc)]+}* and it was quenched by thiol and NiCl2 to generate the thiol radical and Ni+–Cl. The binding of thiol and Ni+–Cl formed Ni2+–Cl–sulphide species (Fig. 34). The concomitantly obtained Ir(dFCFppy)2(H2dbpydc) returned to [Ir(dFCFppy)2(H2dbpydc)]+ by transferring one electron to Ni2+–Cl–sulphide thus formed Ni+–sulphide. Oxidative addition of an aryl iodide to Ni+–sulphide delivered a Ni3+-complex, which underwent a facile reductive elimination process, leading to a C–S cross-coupled product and regeneration of Ni2+–Cl. The distance between the Ir(III) photosensitizer and Ni(II) catalytic center was shortened to only 0.6 nm, which facilitated both electron and thiophenol radical transfers and significantly enhanced its photocatalytic activity.265 Multifunctional OTf@[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL had a synergistic catalytic performance in dehydrogenative cross-couplings of heteroarenes with unactivated alkanes, amines, and ethers. Furthermore, it could also catalyse late-stage functionalization of bioactive and drug molecules such as caffeine, Fasudil, and Metyrapone.136 A mesoporous [OTf/Ir(ppy)2(bpydc)]@(bpydc+Al)-MOF had both strong Lewis acidic Al-OTf sites and photoredox sites, which effectively catalysed reductive cross-coupling to afford new azaarene derivatives.141 The employment of a homogeneous Au-catalyst typically required high catalyst loadings due to the relatively low reactivity of Au complexes and rapid catalyst deactivation. Deactivation of Au catalysts via a ligand redistribution formed [Au(phosphine)]+ and unstable Au+ complexes, then Au+ complexes disproportionated into catalytically inactive Au3+ species and Au0 nanoparticles.270 MOLs could hierarchically integrate photosensitizers and Au catalysts together, not only preventing Au catalyst deactivation but also enhancing photoredox catalytic activities with proximately placed photosensitizers and {Au[4-(diphenylphosphino)phenylacetate]}Cl. [Au/Ru(N^N)3]@(BPY+Hf6)-MOL was built from Ru(bpy)2Cl2, [Au(phosphine)]Cl, H3BPY ligands and Hf6 SBUs. Ru(bpy)2(N^N) photosensitizer and Au+ catalysts worked synergistically for the cross-coupling of allenoates, alkenes, or alkynes.271
 | ||
| Scheme 3 C–S, C–O, and C–C cross-coupling reactions photocatalyzed by Ni@[Ir(dFCFppy)2(dbpydc)+Hf12]-MOL.135 | ||
 | ||
| Fig. 34 Proposed mechanism for the C–S cross-coupling reaction catalysed by Ni@[Ir(dFCFppy)2(dbpydc)/dbpydc+Zr12]-MOL.265 | ||
The design of functional architectures with three or more catalytic centers remains a challenge.272 A multifunctional catalyst allows the combination of multiple transformations in a one-pot synthetic route, rather than the traditional stop-and-go approach.273 With three synergistic active sites, OTf/Co@[Ru(bpy)2(dbpydc)+Hf12]-MOL contained a Ru(bpy)2(N^N) photosensitizer, strong Lewis acid OTf-Hf12-SBUs and hydrogen transfer catalyst [Co(dimethylglyoxime)2(4-pyridinepropionate)]Cl, which efficiently catalysed dehydrogenative tandem transformations (Scheme 4). The hierarchical integration of three different catalytic centers into the MOL could prevent mutual inactivation and promote charge transfer to enhance the synergistic tandem catalytic activity. Under light irradiation, the excited {[Ru(bpy)2(N^N)]2+}* transferred one electron to the Co3+ unit and generated a Co2+ unit and [Ru(bpy)2(N^N)]3+, then indoline was oxidated to the radical cation intermediate by [Ru(bpy)2(N^N)]3+. Deprotonation of the radical cation intermediate formed the α-carbon radical, and its radical addition reaction with Co2+ formed Co3+–C bonded intermediate. Subsequently, the β-H elimination from the Co3+–C intermediate produced the indole and Co3+–H intermediate, and Co3+–H was protonated and enabled the H2 production and regeneration of Co3+ catalysts. Simultaneously, upon coordinating to the strongly Lewis acidic Hf12 SBUs, vinyl ketone became electron deficient and attacked the electron rich β-position of indole, and the resulting intermediate further reacted with vinyl ketone to form the corresponding product.158
 | ||
| Scheme 4 Dehydrogenative tandem transformations of indolines photocatalyzed by OTf/Co@[Ru(bpy)2(dbpydc)+Hf12]-MOL.158 | ||
8. Summary and future prospects
Here we reviewed the application of Ru(N^N)3 and Ir(C^N)2(X^N) based architectures for in-light-driven catalysis, which provide an interesting platform for photosynthesis by organizing photoactive components in well-organised architectures. The incorporation of Ru(N^N)3 and Ir(C^N)2(X^N) building blocks into well-defined skeletons can create interesting architectures with novel structures and the required photo-function, because the structural aesthetics and functional advantages of Ru(N^N)3 or Ir(C^N)2(X^N) are readily inherited in the target architectures, and the combination of functional tectons, nano-cavity, crystalline network and Lewis acid site produced new photofunctional entities. The absorption ability of light, absorption band, and the diversity of structures could be enhanced and broadened by the integration of Ru(N^N)3 or Ir(C^N)2(X^N) into distinct architectures. These Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures exhibited novel photocatalytic performances with a synergistic catalytic effect, high catalytic activity, product selectivity and recyclability. As highlighted in POMs⊂Ru(Ir)-MOF, the components of the photosensitizer and POMs were organised in close proximity, to promote the photogenerated electron transfer from the photosensitizer to POMs, and the accumulation of electrons at POMs achieved photocatalytic hydrogen production. The integration of Ir(C^N)2(N^N) and Ni2+ into the porous network of 2D (N^N)-COF enabled highly efficient photocatalysis of C–C or C–N cross-couplings. In the [Pd6Ru8] cage, the supramolecular interactions between host and guest can regulate the redox potentials of the photosensitizer and catalyst, enhance charger transfer, and stabilize the catalytic intermediates, resulting in special photocatalytic activity.The design and synthesis of Ru(N^N)3 and Ir(C^N)2(X^N) based architectures is ongoing research, while the use of these novel complexes for photosynthesis still remains underexplored. There are still some serious problems needed to be solved compared with plant photosynthesis. (1) Understanding of photo-generated charge and energy transfer processes, efficient charge separation, and stabilization of unstable intermediates should provide more insights into the fundamental mechanistic aspects for the development of efficient photocatalysts. (2) The simultaneous consumption of both electrons and holes is difficult, and the addition of sacrificial agents results in resource waste and pollution. (3) Ru(N^N)3 and Ir(C^N)2(X^N) functionalized architectures still face limited broadband light absorption and low ΦPL due to inefficient separation of photogenerated electrons and holes. (4) The design and construction of Ru(N^N)3 and Ir(C^N)2(X^N) photo-functionalized architectures remain sustainable developments because the architecture type, architecture porosity, linkage type and alignment mode of the photosensitizer and catalyst, and the introduction of a guest have a great influence on the functionality. These architectures showing interesting catalytic properties are also expected to show other photophysical properties like multi-photon absorption cross-sections, and second and third harmonic generations. Furthermore, the architectures may be able to include both the Ru(N^N)3 and Ir(C^N)2(X^N) moieties into the same architectures. Although this has not been accomplished successfully, the synergistic effect of the presence of these photofunctional metal complexes in the same materials will be of immense academic interest and may shed more light on understanding the design principles of new functional materials. The incorporation of Ru(N^N)3 and Ir(C^N)2(X^N) moieties into other well-defined architectures like a molecular sieve could produce stable mesoporous photocatalysts. Immobilizing these Ru(N^N)3 or Ir(C^N)2(X^N) based complexes on a conductive supporter will allow their implementation on photo-electrochemical devices. These are expected to promote their closer commercial applications.
Abbreviations
| MLCT | Metal-to-ligand charge transfer |
| ILCT | Intraligand charge transfer |
| MOFs | Metal–organic frameworks |
| COFs | Covalent organic frameworks |
| SOFs | Supramolecular organic framework |
| MOL | Metal–organic layer |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| Φ PL | Emission quantum yields |
| bpy | 2,2′-Bipyridine |
| H-ppy | 2-Phenylpyridine |
| SBUs | Secondary building units |
| PSM | Post-synthetic modification |
| H2bpydc | 2,2′-Bipyridine-5,5′-dicarboxylic acid |
| H4TPTB | 5′,5′′′-Bis(4-carboxylatophenyl)-4′′′,6′-dimethoxy-[1,1′:3′,1′′:4′′,1′′′:3′′′′,1′′′′-quinquephenyl]-4,4′′′′-dicarboxylate |
| H2bpdc | 4,4′-Biphenyldicarboxylic acid |
| H-pqc | H-2-Phenylquinoline-4-carboxylic acid |
| H3BPY | 4′,6′-Dibenzoato-[2,2′-bipyridine]-4-carboxylic acid |
| Hip | 1H-Imidazo[4,5-f][1,10]phenanthroline |
| H-H2dcppy | 2-(3-Carboxyphenyl)pyridine-4-carboxylic acid |
| H2dcbpy | 2,2′-Bipyridine-4,4′-dicarboxylic acid |
| H2dbpydc | 4,4′-([2,2′-Bipyridine]-5,5′-diyl)dibenzoic acid |
| H4dpbpy | 2,2′-Bipyridine-4,4′-bis(phosphonic acid) |
| phenba | 4-(1H-Imidazo[4,5-f][1,10]phenanthrolin-2-yl)benzoic acid |
| phen | 1,10-Phenanthroline |
| H-ppy-COOH | 3-(Pyridin-2-yl)benzoic acid |
| cptpy | 4′-(4-Carboxyphenyl)-terpyridine |
| Ln | Lanthanides |
| H3BTB | 1,3,5-Benzenetribenzoate |
| HMBA | 2-[5′-Methyl-(2,2′-bipyridin)-5-yl]acetate |
| bpy-OEt | 5,5′-Bis(diethoxymethyl)-2,2′-bipyridine |
| ETTA | 4,4′,4′′,4′′′-(Ethene-1,1,2,2-tetrayl)tetraaniline |
| TPB | 1,2,4,5-Tetrakis-(4-aminolphenyl)benzene |
| BPDCA | 2,2′-Bipyridine-5,5′-dicarbaldehyde |
| TAB | 1,3,5-Tris-(4-aminophenyl) benzene |
| ETTBA | 4′,4′′′,4′′′′′,4′′′′′′′-(Ethene-1,1,2,2-tetrayl)tetrakis([1,1′-biphenyl]-4-amine) |
| phendda | 4,4′-(1,10-Phenanthroline-3,8-diyl)dibenzaldehyde |
| abpy | 5,5′-Diamino-2,2′-bipyridine |
| Tp | 1,3,5-Triformylphloroglucinol |
| TAPM | Tetra(p-aminophenyl)methane |
| TPMB | Tetrakisphenylmethane borate |
| MeCN | Acetonitrile |
| TEPM | Tetra(4-ethynylphenyl)methane |
| piphen | 2-(Pyridin-3-yl)-1H-imidazo[4,5-f][1,10]phenanthroline |
| TTF | Tetrathiafulvalene |
| BINOL | 1,1′-Bis(2-naphthol) |
| H-dFCFppy | 2-(2,4-Difluorophenyl)-5-(trifluoromethyl)pyridine |
| H-ppy-CHO | 4-(2-Pyridyl)benzaldehyde |
| tbubpy | 4,4′-Di(tert-butyl)-2,2′-bipyridine |
| H-mesppy | 2-Phenyl-4-mesitylpyridinato |
| qpy | 4,4′:2′,2′′,4′′,4′′′-Quaterpyridine |
| H-dFmesppy | 2-(4,6-Difluorophenyl)-4-mesitylpyridinato |
| H-dFppy | (2-(2,4-Difluorophenyl)-4-phenyl)pyridine |
| H-appy | 4-(2-Pyridinyl)phenylamine |
| tpmc | Tris(4-pyridyl-methyl)-cyclotriguaiacylene |
| ttpadtc | 2,7,12-Trimethoxy-3,8,13-tris(4,4′-pyridyl-azophenylcarboxy)-10,15-dihydro-5H-tribenzo[a,d,g] cyclononene |
| bpet | 1,2-Bis[(pyridin-2-ylmethyl)thio]ethane |
| OTf | Triflate |
| TPT | 2,4,6-Tri(pyridin-4-yl)-1,3,5-triazine |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| PS | Photosensitizer |
| Cat | Catalyst |
| D | Electron donor |
| A | Electron acceptor |
| POMs | Polyoxometalates |
| {P2W18} | [P2W18O62]6− |
| {PW12} | [PW12O40]3− |
| {Ni4P2} | [Ni4(H2O)2(PW9O34)2]10− |
| CO2RR | Carbon dioxide reduction reactions |
| CO2 | Carbon dioxide |
| HCOOH | Formate |
| CO | Carbon monoxide |
| HCHO | Formaldehyde |
| CH3OH | Methanol |
| CH4 | Methane |
| O2 | Oxygen |
| ROS | Reactive oxygen species |
| H2O2 | Hydrogen peroxide |
| 1O2 | Singlet oxygen |
| ˙O2− | Superoxide radical |
| ˙OH | Hydroxyl radical |
| HER | Hydrogen evolution reaction. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (21971011 and 21831001).References
- S. Pete, N. Roy, B. Kar and P. Paira, Coord. Chem. Rev., 2022, 460, 214462–214516 CrossRef CAS.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem. – Eur. J., 2014, 20, 14004–14011 CrossRef CAS PubMed.
- A. J. Atkins and L. Gonzalez, J. Phys. Chem. Lett., 2017, 8, 3840–3845 CrossRef CAS PubMed.
- Y. Cho, S. Kim, M. Cho, K. Wee, H. Son, W. Han, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 15162–15169 RSC.
- S. Verma, P. Kar, A. Das and H. N. Ghosh, Dalton Trans., 2011, 40, 9765–9773 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 36–76 CrossRef PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- Z. Chang, D. Yang, J. Xu, T. Hu and X. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed.
- W. Cui, G. Zhang, T. Hu and X. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- X. Ge, R. Wong, A. Anisa and S. Ma, Biomaterials, 2022, 281, 121322–121339 CrossRef CAS PubMed.
- B. Gibbons, M. Cai and A. J. Morris, J. Am. Chem. Soc., 2022, 144, 17723–17736 CrossRef CAS PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410–1801443 CrossRef PubMed.
- H. Vardhan, A. Nafady, A. M. Al-Enizi and S. Ma, Nanoscale, 2019, 11, 21679–21708 RSC.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS PubMed.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- D. Chakraborty and P. S. Mukherjee, Chem. Commun., 2022, 58, 5558–5573 RSC.
- W. Liu, A. G. Oliver and B. D. Smith, J. Am. Chem. Soc., 2018, 140, 6810–6813 CrossRef CAS PubMed.
- P. K. Mandal, B. Kauffmann, H. Destecroix, Y. Ferrand, A. P. Davis and I. Huc, Chem. Commun., 2016, 52, 9355–9358 RSC.
- Y. Huang, R. Gao, M. Liu, L. Chen, X. Ni, X. Xiao, H. Cong, Q. Zhu, K. Chen and Z. Tao, Angew. Chem., Int. Ed., 2021, 60, 15166–15191 CrossRef CAS PubMed.
- M. Shen, X. Lin, J. Luo, W. Li, Y. Ye and X. Wang, Mol. Syst. Des. Eng., 2022, 7, 1570–1587 RSC.
- Y. Zhou, G. Zhang, B. Li and L. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 30761–30769 CrossRef CAS PubMed.
- L. Yue, S. Wang, D. Zhou, H. Zhang, B. Li and L. Wu, Nat. Commun., 2016, 7, 10742–10751 CrossRef CAS PubMed.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- K. Yang, Y. Ban and W. Yang, J. Membr. Sci., 2021, 639, 119771–119777 CrossRef CAS.
- A. H. Assen, T. Virdis, W. De Moor, A. Moussa, M. Eddaoudi, G. Baron, J. F. M. Denayer and Y. Belmabkhout, Chem. Eng. J., 2021, 413, 127388–127395 CrossRef CAS.
- S. Zhang, Y. Zheng, H. An, B. Aguila, C. Yang, Y. Dong, W. Xie, P. Cheng, Z. Zhang, Y. Chen and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 16754–16759 CrossRef CAS PubMed.
- H. Chen, C. Jin, X. Chen, J. Yu, D. Yan, P. Cheng, Z. Zhang, L. Zhao and Y. Chen, Chem. Eng. J., 2022, 444, 136624–136631 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- X. Li, J. Wu, L. Wang, C. He, L. Chen, Y. Jiao and C. Duan, Angew. Chem., Int. Ed., 2020, 59, 6420–6427 CrossRef CAS PubMed.
- G. Zhang, B. Li, Y. Zhou, X. Chen, B. Li, Z. Lu and L. Wu, Nat. Commun., 2020, 11, 425–433 CrossRef CAS PubMed.
- H. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- S. Huang, N. Liu, Y. Ling and H. Luo, Chem. – Asian J., 2017, 12, 3110–3113 CrossRef CAS PubMed.
- X. Li, J. Wu, C. He, R. Zhang and C. Duan, Chem. Commun., 2016, 52, 5104–5107 RSC.
- Y. Song, Y. Pi, X. Feng, K. Ni, Z. Xu, J. S. Chen, Z. Li and W. Lin, J. Am. Chem. Soc., 2020, 142, 6866–6871 CrossRef CAS PubMed.
- J. Lin, X. Hu, P. Zhang, A. Van Rynbach, D. N. Beratan, C. A. Kent, B. P. Mehl, J. M. Papanikolas, T. J. Meyer, W. Lin, S. S. Skourtis and M. Constantinou, J. Phys. Chem. C, 2013, 117, 22250–22259 CrossRef CAS.
- A. Schmidt, M. Hollering, J. Han, A. Casini and F. E. Kuehn, Dalton Trans., 2016, 45, 12297–12300 RSC.
- J. D. Knoll, S. M. Arachchige and K. J. Brewer, ChemSusChem, 2011, 4, 252–261 CAS.
- C. Hou, T. Li, S. Cao, Y. Chen and W. Fu, J. Mater. Chem. A, 2015, 3, 10386–10394 RSC.
- L. Zhang, S. Yin, M. Pan, W. Liao, J. Zhang, H. Wang and C. Su, J. Mater. Chem. A, 2017, 5, 9807–9814 RSC.
- C. Wang, Z. Xie, K. E. DeKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- G. Lan, K. Ni, E. You, M. Wang, A. Culbert, X. Jiang and W. Lin, J. Am. Chem. Soc., 2019, 141, 18964–18969 CrossRef CAS PubMed.
- H. Iranmanesh, K. S. A. Arachchige, M. Bhadbhade, W. A. Donald, J. Y. Liew, K. T. C. Liu, E. T. Luis, E. G. Moore, J. R. Price, H. Yan, J. Yang and J. E. Beves, Inorg. Chem., 2016, 55, 12737–12751 CrossRef CAS PubMed.
- E. T. Luis, H. Iranmanesh, K. S. A. Arachchige, W. A. Donald, G. Quach, E. G. Moore and J. E. Beves, Inorg. Chem., 2018, 57, 8476–8486 CrossRef CAS PubMed.
- D. Kim, D. R. Whang and S. Y. Park, J. Am. Chem. Soc., 2016, 138, 8698–8701 CrossRef CAS PubMed.
- L. Wei and B. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 41448–41457 CrossRef CAS PubMed.
- G. Lan, K. Ni, S. S. Veroneau, T. Luo, E. You and W. Lin, J. Am. Chem. Soc., 2019, 141, 6859–6863 CrossRef CAS PubMed.
- G. Lan, K. Ni, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2018, 140, 16971–16975 CrossRef CAS PubMed.
- C. Xu, A. Guenet, N. Kyritsakas, J. Planeix and M. W. Hosseini, Inorg. Chem., 2015, 54, 10429–10439 CrossRef CAS PubMed.
- Z. Xu, Y. Luo, D. Zhang, H. Wang, X. Sun and Z. Li, Green Chem., 2020, 22, 136–143 RSC.
- C. E. Hauke, A. N. Oldacre, C. R. P. Fulong, A. E. Friedman and T. R. Cook, Inorg. Chem., 2017, 57, 3587–3595 CrossRef PubMed.
- L. Wang, Z. Xie, S. Dang and Z. Sun, Chem. – Eur. J., 2017, 23, 2852–2857 CrossRef CAS PubMed.
- H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859–3861 RSC.
- V. E. Pritchard, D. R. Martir, S. Oldknow, S. Kai, S. Hiraoka, N. J. Cookson, E. Zysman-Colman and M. J. Hardie, Chem. – Eur. J., 2017, 23, 6290–6294 CrossRef CAS PubMed.
- L. Li, S. Zhang, L. Xu, L. Han, Z. Chen and J. Luo, Inorg. Chem., 2013, 52, 12323–12325 CrossRef CAS PubMed.
- C. Li, Y. Wang, Y. Lu, J. Guo, C. Zhu, H. He, X. Duan, M. Pan and C. Su, Chin. Chem. Lett., 2020, 31, 1183–1187 CrossRef CAS.
- N. Huang, J. Shen, X. Zhang, P. Liao, J. Zhang and X. Chen, J. Am. Chem. Soc., 2022, 144, 8676–8682 CrossRef CAS PubMed.
- K. Wu, K. Li, Y. Hou, M. Pan, L. Zhang, L. Chen and C. Su, Nat. Commun., 2016, 7, 10487–10496 CrossRef CAS PubMed.
- L. Martins, L. K. Macreadie, D. Sensharma, S. Vaesen, X. Zhang, J. J. Gough, M. O'Doherty, N. Zhu, M. Ruether, J. E. O'Brien, A. L. Bradley and W. Schmitt, Chem. Commun., 2019, 55, 5013–5016 RSC.
- C. Liao, K. Fan, S. Bao, H. Fan, X. Wang, Z. Hu, M. Kurmoo and L. Zheng, Chem. Commun., 2019, 55, 13920–13923 RSC.
- Z. Xie, L. Ma, K. E. DeKrafft, A. Jin and W. Lin, J. Am. Chem. Soc., 2010, 132, 922–923 CrossRef CAS PubMed.
- J. Wu, X. Li, P. Wu, Z. Shi, L. Chen, R. Zhang, C. He and C. Duan, Chem. Mater., 2022, 34, 4471–4478 CrossRef CAS.
- X. Li, J. Wu, L. Chen, X. Zhong, C. He, R. Zhang and C. Duan, Chem. Commun., 2016, 52, 9628–9631 RSC.
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS PubMed.
- D. P. August, J. Jaramillo-Garcia, D. A. Leigh, A. Valero and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2021, 143, 1154–1161 CrossRef CAS PubMed.
- A. Kobayashi, T. Ohba, E. Saitoh, Y. Suzuki, S. Noro, H. Chang and M. Kato, Inorg. Chem., 2014, 53, 2910–2921 CrossRef CAS PubMed.
- X. Li, S. Yu, B. Yang, J. Tian, H. Wang, D. Zhang, Y. Liu and Z. Li, Sci. China: Chem., 2018, 61, 830–835 CrossRef CAS.
- J. Tian, Z. Xu, D. Zhang, H. Wang, S. Xie, D. Xu, Y. Ren, H. Wang, Y. Liu and Z. Li, Nat. Commun., 2016, 7, 11580–11588 CrossRef CAS PubMed.
- W. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CAS.
- H. Yin, J. Yang and X. Yin, Anal. Chem., 2017, 89, 13434–13440 CrossRef CAS PubMed.
- J. S. Z. Qiao, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- H. Lv, W. Guo, K. Wu, Z. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 CrossRef CAS PubMed.
- X. Han, C. Qin, X. Wang, Y. Tan, X. Zhao and E. Wang, Appl. Catal., B, 2017, 211, 349–356 CrossRef CAS.
- X. Zhao, J. Zhou, C. Sun, S. You, X. Wang and Z. Su, Nanotechnology, 2020, 31, 255402–255407 CrossRef CAS PubMed.
- H. Xu, S. You, Z. Lang, Y. Sun, C. Sun, J. Zhou, X. Wang, Z. Kang and Z. Su, Chem. – Eur. J., 2020, 26, 2735–2740 CrossRef CAS PubMed.
- L. Qiao, M. Song, A. Geng and S. Yao, Chin. Chem. Lett., 2019, 30, 1273–1276 CrossRef CAS.
- J. Liu, S. Huang and G. Yang, Sci. Sin.: Chim., 2020, 50, 986–1000 Search PubMed.
- I. Bazzan, A. Volpe, A. Dolbecq, M. Natali, A. Sartorel, P. Mialane and M. Bonchio, Catal. Today, 2017, 290, 39–50 CrossRef CAS.
- A. Lemos, C. Lemaire and A. Luxen, Adv. Synth. Catal., 2019, 361, 1500–1537 CrossRef CAS PubMed.
- C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5491–5502 CrossRef PubMed.
- Q. Zhou, Y. Zou, L. Lu and W. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 761–784 RSC.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- W. Zhang, F. Zhang, Y. Wang, B. Song, R. Zhang and J. Yuan, Inorg. Chem., 2017, 56, 1309–1318 CrossRef CAS PubMed.
- G. B. Damas, D. A. Ivashchenko, I. Rivalta and C. M. Araujo, Sustainable Energy Fuels, 2021, 5, 6066–6076 RSC.
- I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef CAS PubMed.
- K. K. Lo, K. Y. Zhang, S. Leung and M. Tang, Angew. Chem., Int. Ed., 2008, 47, 2213–2216 CrossRef CAS PubMed.
- H. Wu, T. Yang, Q. Zhao, J. Zhou, C. Li and F. Li, Dalton Trans., 2011, 40, 1969–1976 RSC.
- M. Veerapandian, P. K. Avti and V. Ravichandiran, Colloids Surf., B, 2017, 154, 315–320 CrossRef CAS PubMed.
- S. K. Pagire, N. Kumagai and M. Shibasaki, Org. Lett., 2020, 22, 7853–7858 CrossRef CAS PubMed.
- V. Reddy Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915–10919 CrossRef PubMed.
- H. A. Sakai and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6185–6192 CrossRef CAS PubMed.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–93 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS PubMed.
- K. Fan, S. Bao, R. Huo, X. Huang, Y. Liu, Z. Yu, M. Kurmoo and L. Zheng, Inorg. Chem. Front., 2020, 7, 4580–4592 RSC.
- Z. He, C. Kim, L. Lin, T. H. Jeon, S. Lin, X. Wang and W. Choi, Nano Energy, 2017, 42, 58–68 CrossRef CAS.
- Q. Lin, X. Bu, C. Mao, X. Zhao, K. Sasan and P. Feng, J. Am. Chem. Soc., 2015, 137, 6184–6187 CrossRef CAS PubMed.
- W. Zhou, W. Li, J. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- J. Xiao, D. Li and H. Jiang, Sci. Sin.: Chim., 2018, 48, 1058–1075 Search PubMed.
- S. Yu, Q. Qi, B. Yang, H. Wang, D. Zhang, Y. Liu and Z. Li, Small, 2018, 14, 1801037–1801043 CrossRef PubMed.
- Z. Gao, Z. Wang, L. Wei, G. Yin, J. Tian, C. Liu, H. Wang, D. Zhang, Y. Zhang, X. Li, Y. Liu and Z. Li, ACS Appl. Mater. Interfaces, 2019, 12, 1404–1411 CrossRef PubMed.
- X. Yu and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 12320–12323 CrossRef CAS PubMed.
- C. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348–355 CrossRef CAS PubMed.
- S. You, J. Zhou, M. Chen, C. Sun, X. Qi, A. Yousaf, X. Wang and Z. Su, J. Catal., 2020, 392, 49–55 CrossRef CAS.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- S. Bai, J. Ge, L. Wang, M. Gong, M. Deng, Q. Kong, L. Song, J. Jiang, Q. Zhang, Y. Luo, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 5689 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. Jiang, Adv. Mater., 2018, 30, 1075112–1075118 Search PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- Z. Zhang, T. Zhang, C. Wang, Z. Lin, L. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed.
- A. López-Magano, B. Ortín-Rubio, I. Imaz, D. Maspoch, J. Alemán and R. Mas-Ballesté, ACS Catal., 2021, 11, 12344–12354 CrossRef PubMed.
- G. Kumar, R. S. Pillai, N. H. Khan and S. Neogi, Appl. Catal., B, 2021, 292, 120149 CrossRef CAS.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- J. Pang, Z. Di, J. Qin, S. Yuan, C. T. Lollar, J. Li, P. Zhang, M. Wu, D. Yuan, M. Hong and H. Zhou, J. Am. Chem. Soc., 2020, 142, 15020–15026 CrossRef CAS PubMed.
- H. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- X. Cui, Z. Niu, C. Shan, L. Yang, J. Hu, Q. Wang, P. C. Lan, Y. Li, L. Wojtas, S. Ma and H. Xing, Nat. Commun., 2020, 11, 5456–5463 CrossRef CAS PubMed.
- V. Van Speybroeck and G. Maurin, Nat. Mater., 2022, 22, 12–13 CrossRef PubMed.
- J. Wieme and V. Van Speybroeck, J. Mater. Chem. A, 2021, 9, 4898–4906 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- Y. Li, Z. Fu and G. Xu, Coord. Chem. Rev., 2019, 388, 79–106 CrossRef CAS.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- B. Li, M. Chrzanowski, Y. Zhang and S. Ma, Coord. Chem. Rev., 2016, 307, 106–129 CrossRef CAS.
- H. R. Moon, D. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC.
- D. Li, M. Kassymova, X. Cai, S. Zang and H. Jiang, Coord. Chem. Rev., 2020, 412, 213262–213277 CrossRef CAS.
- E. M. Johnson, S. Ilic and A. J. Morris, ACS Cent. Sci., 2021, 7, 445–453 CrossRef CAS PubMed.
- Q. Yang, Q. Xu and H. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- S. Cohen, Nature, 2019, 566, 464–465 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z. Gu, T. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. I. Gentle, M. Bosch and H. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- G. Lan, Y. Quan, M. Wang, G. T. Nash, E. You, Y. Song, S. S. Veroneau, X. Jiang and W. Lin, J. Am. Chem. Soc., 2019, 141, 15767–15772 CrossRef CAS PubMed.
- Y. Quan, G. Lan, Y. Fan, W. Shi, E. You and W. Lin, J. Am. Chem. Soc., 2020, 142, 1746–1751 CrossRef CAS PubMed.
- Z. Yan, M. Du, J. Liu, S. Jin, C. Wang, G. Zhuang, X. Kong, L. Long and L. Zheng, Nat. Commun., 2018, 9, 3353–3361 CrossRef PubMed.
- L. Zeng, Z. Wang, Y. Wang, J. Wang, Y. Guo, H. Hu, X. He, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 142, 75–79 CrossRef PubMed.
- Z. Yan, B. Ma, S. Li, J. Liu, R. Chen, M. Du, S. Jin, G. Zhuang, L. Long, X. Kong and L. Zheng, Sci. Bull., 2019, 64, 976–985 CrossRef CAS PubMed.
- Y. Song, Z. Li, Y. Zhu, X. Feng, J. S. Chen, M. Kaufmann, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 12219–12223 CrossRef CAS PubMed.
- Y. Quan, Y. Song, W. Shi, Z. Xu, J. S. Chen, X. Jiang, C. Wang and W. Lin, J. Am. Chem. Soc., 2020, 142, 8602–8607 CrossRef CAS PubMed.
- X. Zhang, X. Wei, S. Huang and G. Yang, Chem. – Asian J., 2021, 16, 2031–2034 CrossRef CAS PubMed.
- K. Fan, W. Nie, L. Wang, C. Liao, S. Bao and L. Zheng, Chem. – Eur. J., 2017, 23, 6615–6624 CrossRef CAS PubMed.
- C. A. Kent, D. Liu, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2011, 133, 12940–12943 CrossRef CAS PubMed.
- C. Wang and W. Lin, J. Am. Chem. Soc., 2011, 133, 4232–4235 CrossRef CAS PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2017, 7, 997–1014 CrossRef CAS.
- B. Wang, X. Lv, D. Feng, L. Xie, J. Zhang, M. Li, Y. Xie, J. Li and H. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- M. Liu, Y. Mu, S. Yao, S. Guo, X. Guo, Z. Zhang and T. Lu, Appl. Catal., B, 2019, 245, 496–501 CrossRef CAS.
- X. Kong, Z. Lin, Z. Zhang, T. Zhang and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 6411–6416 CrossRef CAS PubMed.
- M. Elcheikh Mahmoud, H. Audi, A. Assoud, T. H. Ghaddar and M. Hmadeh, J. Am. Chem. Soc., 2019, 141, 7115–7121 CrossRef CAS PubMed.
- C. Tan, X. Cao, X. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- J. I. Urgel, D. Ecija, W. Auwaerter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163–6167 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- G. Lan, Z. Li, S. S. Veroneau, Y. Zhu, Z. Xu, C. Wang and W. Lin, J. Am. Chem. Soc., 2018, 140, 12369–12373 CrossRef CAS PubMed.
- G. Lan, K. Ni, R. Xu, K. Lu, Z. Lin, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2017, 56, 12102–12106 CrossRef CAS PubMed.
- Y. Zhang, J. Phipps and S. Ma, Nat. Catal., 2022, 5, 973–974 CrossRef CAS.
- Y. Quan, G. Lan, W. Shi, Z. Xu, Y. Fan, E. You, X. Jiang, C. Wang and W. Lin, Angew. Chem., Int. Ed., 2021, 60, 3115–3120 CrossRef CAS PubMed.
- P. I. Djurovich, D. Murphy, M. E. Thompson, B. Hernandez, R. Gao, P. L. Hunt and M. Selke, Dalton Trans., 2007, 3763–3770 RSC.
- D. GarciaFresnadillo, Y. Georgiadou, G. Orellana, A. M. Braun and E. Oliveros, Helv. Chim. Acta, 1996, 79, 1222–1238 CrossRef CAS.
- A. Kobayashi, K. Shimizu, A. Watanabe, Y. Nagao, N. Yoshimura, M. Yoshida and M. Kato, Inorg. Chem., 2019, 58, 2413–2421 CrossRef CAS PubMed.
- A. Watanabe, A. Kobayashi, E. Saitoh, Y. Nagao, S. Omagari, T. Nakanishi, Y. Hasegawa, W. M. C. Sameera, M. Yoshida and M. Kato, Inorg. Chem., 2017, 56, 3005–3013 CrossRef CAS PubMed.
- A. Watanabe, A. Kobayashi, E. Saitoh, Y. Nagao, M. Yoshida and M. Kato, Inorg. Chem., 2015, 54, 11058–11060 CrossRef CAS PubMed.
- L. Li, S. Zhang, L. Xu, Z. Chen and J. Luo, J. Mater. Chem. C, 2014, 2, 1698–1703 RSC.
- T. Zhuo, Y. Song, G. Zhuang, L. Chang, S. Yao, W. Zhang, Y. Wang, P. Wang, W. Lin, T. Lu and Z. Zhang, J. Am. Chem. Soc., 2021, 143, 6114–6122 CrossRef CAS PubMed.
- X. Deng, J. Albero, L. Xu, H. Garcia and Z. Li, Inorg. Chem., 2018, 57, 8276–8286 CrossRef CAS PubMed.
- K. Wang, J. Zhang, Y. Hsu, H. Li, Z. Han, J. Pang, Z. Yang, R. Liang, W. Shi and H. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- L. Pan, M. Xu, L. Feng, Q. Chen, Y. He and B. Han, Polym. Chem., 2016, 7, 2299–2307 RSC.
- O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1295–1300 CrossRef CAS PubMed.
- M. Zhang, Y. Li, W. Yuan, X. Guo, C. Bai, Y. Zou, H. Long, Y. Qi, S. Li, G. Tao, C. Xia and L. Ma, Angew. Chem., Int. Ed., 2021, 60, 12396–12405 CrossRef CAS PubMed.
- Z. Mi, P. Yang, R. Wang, J. Unruangsri, W. Yang, C. Wang and J. Guo, J. Am. Chem. Soc., 2019, 141, 14433–14442 CrossRef CAS PubMed.
- H. Wang, Y. Yang, X. Yuan, W. Liang Teo, Y. Wu, L. Tang and Y. Zhao, Mater. Today, 2022, 53, 106–133 CrossRef CAS.
- L. Akyuz, Microporous Mesoporous Mater., 2020, 294, 109850 CrossRef CAS.
- L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC.
- J. Á. Martín Illán, D. Rodríguez San Miguel, O. Castillo, G. Beobide, J. Perez Carvajal, I. Imaz, D. Maspoch and F. Zamora, Angew. Chem., Int. Ed., 2021, 60, 13969–13977 CrossRef PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS PubMed.
- S. Xiong, L. Li, L. Dong, J. Tang, G. Yu and C. Pan, J. CO2 Util., 2020, 41, 101224 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed.
- Y. Meng, Y. Luo, J. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CAS.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- J. Chen, X. Tao, C. Li, Y. Ma, L. Tao, D. Zheng, J. Zhu, H. Li, R. Li and Q. Yang, Appl. Catal., B, 2020, 262, 118271–118278 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- R. Bu, L. Zhang, L. Gao, W. Sun, S. Yang and E. Gao, Mol. Catal., 2021, 499, 111319–111327 CrossRef CAS.
- Z. Mi, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642–9649 CrossRef CAS PubMed.
- G. Kumar, M. Singh, R. Goswami and S. Neogi, ACS Appl. Mater. Interfaces, 2020, 12, 48642–48653 CrossRef CAS PubMed.
- R. Liang and X. Zhao, Org. Chem. Front., 2018, 5, 3341–3356 RSC.
- K. Huang, J. Zhang, F. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS.
- Y. Zhao, Y. He and T. M. Swager, ACS Macro Lett., 2018, 7, 300–304 CrossRef CAS PubMed.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC.
- M. M. Unterlass, Angew. Chem., Int. Ed., 2018, 57, 2292–2294 CrossRef CAS PubMed.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998–3005 CrossRef PubMed.
- P. Wang, S. Ding, Z. Zhang, Z. Wang and W. Wang, J. Am. Chem. Soc., 2019, 141, 18004–18008 CrossRef CAS PubMed.
- Y. Luo, Z. Xu, H. Wang, X. Sun, Z. Li and D. Zhang, ACS Macro Lett., 2020, 9, 90–95 CrossRef CAS PubMed.
- Z. Xie, C. Wang, K. E. DeKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed.
- H. Liang, Q. Chen and B. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS.
- W. Guo, H. Ding and B. Su, J. Electroanal. Chem., 2018, 818, 176–180 CrossRef CAS.
- T. R. Cook, Y. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- Y. Liu, C. Hu, A. Comotti and M. D. Ward, Science, 2011, 333, 436–440 CrossRef CAS PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- O. Chepelin, J. Ujma, P. E. Barran and P. J. Lusby, Angew. Chem., Int. Ed., 2012, 51, 4194–4197 CrossRef CAS PubMed.
- O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334–19337 CrossRef CAS PubMed.
- S. Oldknow, D. R. Martir, V. E. Pritchard, M. A. Blitz, C. W. G. Fishwick, E. Zysman-Colman and M. J. Hardie, Chem. Sci., 2018, 9, 8150–8159 RSC.
- D. Rota Martir, D. Escudero, D. Jacquemin, D. B. Cordes, A. M. Z. Slawin, H. A. Fruchtl, S. L. Warriner and E. Zysman-Colman, Chem. – Eur. J., 2017, 23, 14358–14366 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- A. Ruggi, M. Berenguel Alonso, D. N. Reinhoudt and A. H. Velders, Chem. Commun., 2010, 46, 6726 RSC.
- H. Sato, M. A. Blemker, G. Hellinghausen, D. W. Armstrong, J. W. Nafie, S. T. Roberts and M. J. Krische, Chem. – Eur. J., 2019, 25, 8719–8724 CrossRef CAS PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176–1239184 CrossRef PubMed.
- B. Sahoo, J. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577–11580 CrossRef CAS PubMed.
- A. G. Amador, E. M. Sherbrook and T. P. Yoon, J. Am. Chem. Soc., 2016, 138, 4722–4725 CrossRef CAS PubMed.
- L. Zhang and X. Hu, Chem. Sci., 2017, 8, 7009–7013 RSC.
- A. Schultz, X. Li, B. Barkakaty, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2012, 134, 7672–7675 CrossRef CAS PubMed.
- B. Laramee-Milette, F. Nastasi, F. Puntoriero, S. Campagna and G. S. Hanan, Chem. – Eur. J., 2017, 23, 16497–16504 CrossRef CAS PubMed.
- Z. Zhang, H. Wang, X. Wang, Y. Li, B. Song, O. Bolarinwa, R. A. Reese, T. Zhang, X. Wang, J. Cai, B. Xu, M. Wang, C. Liu, H. Yang and X. Li, J. Am. Chem. Soc., 2017, 139, 8174–8185 CrossRef CAS PubMed.
- J. Y. Ryu, E. H. Wi, M. Pait, S. Lee, P. J. Stang and J. Lee, Inorg. Chem., 2017, 56, 5471–5477 CrossRef CAS PubMed.
- C. Shen, A. D. W. Kennedy, W. A. Donald, A. M. Torres, W. S. Price and J. E. Beves, Inorg. Chim. Acta, 2017, 458, 122–128 CrossRef CAS.
- K. Li, L. Zhang, C. Yan, S. Wei, M. Pan, L. Zhang and C. Su, J. Am. Chem. Soc., 2014, 136, 4456–4459 CrossRef CAS PubMed.
- S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y. Fan, J. Guo, J. Shi and C. Su, Nat. Commun., 2016, 7, 13169–13176 CrossRef CAS PubMed.
- K. Wu, K. Li, S. Chen, Y. Hou, Y. Lu, J. Wang, M. Wei, M. Pan and C. Su, Angew. Chem., Int. Ed., 2020, 59, 2639–2643 CrossRef CAS PubMed.
- J. Guo, Y. Xu, K. Li, L. Xiao, S. Chen, K. Wu, X. Chen, Y. Fan, J. Liu and C. Su, Angew. Chem., Int. Ed., 2017, 56, 3852–3856 CrossRef CAS PubMed.
- J. Guo, Y. Fan, Y. Lu, S. Zheng and C. Su, Angew. Chem., Int. Ed., 2020, 59, 8661–8669 CrossRef CAS PubMed.
- K. Li, K. Wu, Y. Lu, J. Guo, P. Hu and C. Su, Angew. Chem., Int. Ed., 2022, 61, e202114070 CAS.
- D. Rota Martir, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, Chem. Commun., 2018, 54, 6016–6019 RSC.
- J. Tian, T. Zhou, S. Zhang, S. Aloni, M. V. Altoe, S. Xie, H. Wang, D. Zhang, X. Zhao, Y. Liu and Z. Li, Nat. Commun., 2014, 5, 5574–5584 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Chem. Commun., 2014, 50, 10390–10394 RSC.
- G. Lan, Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- S. Yang, B. Pattengale, S. Lee and J. Huang, ACS Energy Lett., 2018, 3, 532–539 CrossRef CAS.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- Q. Schiermeier, Nature, 2011, 470, 316 CrossRef CAS PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W. Man, S. Yiu, K. Lau, T. Lau and M. Robert, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- W. Zhu, C. Zhang, Q. Li, L. Xiong, R. Chen, X. Wan, Z. Wang, W. Chen, Z. Deng and Y. Peng, Appl. Catal., B, 2018, 238, 339–345 CrossRef CAS.
- Y. Wang, N. Huang, J. Shen, P. Liao, X. Chen and J. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- S. Chu, S. Fan, Y. Wang, D. Rossouw, Y. Wang, G. A. Botton and Z. Mi, Angew. Chem., Int. Ed., 2016, 55, 14260–14264 Search PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- L. Li and B. Yeo, Inorg. Chem., 2019, 58, 7775–7784 CrossRef CAS PubMed.
- Y. Yu, S. Huang and G. Yang, CrystEngComm, 2022, 24, 3160–3164 RSC.
- A. P. Tuan, Y. Ping and G. Galli, Nat. Mater., 2017, 16, 401–408 CrossRef PubMed.
- Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 CrossRef CAS PubMed.
- Y. An, Y. Liu, P. An, J. Dong, B. Xu, Y. Dai, X. Qin, X. Zhang, M. Whangbo and B. Huang, Angew. Chem., Int. Ed., 2017, 56, 3036–3040 CrossRef CAS PubMed.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428–1702435 CrossRef PubMed.
- H. Hu, Z. Wang, L. Cao, L. Zeng, C. Zhang, W. Lin and C. Wang, Nat. Chem., 2021, 13, 358–366 CrossRef CAS PubMed.
- X. Wei, Y. Qi, S. Huang and G. Yang, Eur. J. Inorg. Chem., 2022, e202200204 CAS.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Xuan and W. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- S. Angerani and N. Winssinger, Chem. – Eur. J., 2019, 25, 6661–6672 CrossRef CAS PubMed.
- Y. Zhu, G. Lan, Y. Fan, S. S. Veroneau, Y. Song, D. Micheroni and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- Z. Zuo, H. Gong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Roese, L. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- H. Chen, W. Liu, A. Laemont, C. Krishnaraj, X. Feng, F. Rohman, M. Meledina, Q. Zhang, R. Van Deun, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2021, 60, 10820–10827 CrossRef CAS PubMed.
- Z. Lu, G. B. Hammond and B. Xu, Acc. Chem. Res., 2019, 52, 1275–1288 CrossRef CAS PubMed.
- H. Zheng, Y. Fan, Y. Song, J. S. Chen, E. You, S. Labalme and W. Lin, J. Am. Chem. Soc., 2022, 144, 10694–10699 CrossRef CAS PubMed.
- D. Ma, Y. Zhang, S. Jiao, J. Li, K. Liu and Z. Shi, Chem. Commun., 2019, 55, 14347–14350 RSC.
- S. P. Sancheti, Urvashi, M. P. Shah and N. T. Patil, ACS Catal., 2020, 10, 3462–3489 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |