
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of engineered nanomaterial interactions with organ barriers for enhanced drug transport
Vincent
Lenders†
 a,
Xanthippi
Koutsoumpou†
a,
Philana
Phan†
b,
Stefaan J.
Soenen
ac,
Karel
Allegaert
defg,
Steven
de Vleeschouwer
hij,
Jaan
Toelen
fgk,
Zongmin
Zhao
b and
Bella B.
Manshian
*a
a,
Xanthippi
Koutsoumpou†
a,
Philana
Phan†
b,
Stefaan J.
Soenen
ac,
Karel
Allegaert
defg,
Steven
de Vleeschouwer
hij,
Jaan
Toelen
fgk,
Zongmin
Zhao
b and
Bella B.
Manshian
*a
aTranslational Cell and Tissue Research Unit, Department of Imaging and Pathology, KU Leuven, Herestraat 49, B3000 Leuven, Belgium. E-mail: bella.manshian@kuleuven.be
bDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60612, USA
cNanoHealth and Optical Imaging Group, Department of Imaging and Pathology, KU Leuven, Herestraat 49, B3000 Leuven, Belgium
dDepartment of Hospital Pharmacy, Erasmus MC University Medical Center, CN Rotterdam, 3015, The Netherlands
eClinical Pharmacology and Pharmacotherapy, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, B3000 Leuven, Belgium
fLeuven Child and Youth Institute, KU Leuven, 3000 Leuven, Belgium
gWoman and Child, Department of Development and Regeneration, KU Leuven, 3000 Leuven, Belgium
hDepartment of Neurosurgery, University Hospitals Leuven, Leuven, Belgium
iLaboratory of Experimental Neurosurgery and Neuroanatomy, Department of Neurosciences, KU Leuven, Leuven, Belgium
jLeuven Brain Institute (LBI), KU Leuven, Leuven, Belgium
kDepartment of Pediatrics, University Hospitals Leuven, 3000 Leuven, Belgium
First published on 20th June 2023
Abstract
The biomedical use of nanoparticles (NPs) has been the focus of intense research for over a decade. As most NPs are explored as carriers to alter the biodistribution, pharmacokinetics and bioavailability of associated drugs, the delivery of these NPs to the tissues of interest remains an important topic. To date, the majority of NP delivery studies have used tumor models as their tool of interest, and the limitations concerning tumor targeting of systemically administered NPs have been well studied. In recent years, the focus has also shifted to other organs, each presenting their own unique delivery challenges to overcome. In this review, we discuss the recent advances in leveraging NPs to overcome four major biological barriers including the lung mucus, the gastrointestinal mucus, the placental barrier, and the blood–brain barrier. We define the specific properties of these biological barriers, discuss the challenges related to NP transport across them, and provide an overview of recent advances in the field. We discuss the strengths and shortcomings of different strategies to facilitate NP transport across the barriers and highlight some key findings that can stimulate further advances in this field.
 Karel Allegaert | Karel Allegaert (MD, PhD) is a pediatrician (1999)-neonatologist (2000) and clinical pharmacologist (2003). He is full professor at KU Leuven, departments of Development and Regeneration, and Pharmaceutical and Pharmacological Sciences, and senior consultant (part-time) at Erasmus University, Rotterdam, Department of Clinical Pharmacy (https://www.researchgate.net/profile/Karel-Allegaert; ORCID: 0000-0001-9921-5105). His research is mainly focused on perinatal and pediatric clinical pharmacology and its co-variates (maturational, non-maturational, pharmacogenetics, disease-related), with the subsequent use of in vivo datasets to develop prediction models and translation to support drug development in these special populations (neonates, children and pregnant women, or during lactation). |
 Jaan Toelen | Jaan Toelen (MD, PhD) is a staff member in Pediatrics at the University Hospitals Leuven and an associate-professor at KU Leuven, Department of Development and Regeneration (ORCID: 0000-0001-9339-5408). His research is mainly focused on pre- and postnatal lung development and neonatal lung disorders such as bronchopulmonary dysplasia and congenital diaphragmatic hernia with both a basic science and a translational-clinical perspective. His research group uses several animal models to study these conditions and to test experimental strategies before further clinical translation. |
 Zongmin Zhao (left) and Philana Phan (right) | Zongmin Zhao is an Assistant Professor in the College of Pharmacy at the University of Illinois Chicago. He received a PhD degree from Virginia Tech and was a postdoctoral fellow at Harvard University. The research interests of his group are focused on developing bioinspired and biomimetic strategies to improve the diagnosis and treatment of diseases including cancer, autoimmune diseases, acute injuries, and drug addictions. Dr Zhao has published >50 articles in journals including Cell, Nature Biomedical Engineering, PNAS, Science Advances, Advanced Materials, etc. He is currently an Associate Editor of Bioengineering & Translational Medicine. Philana Phan is currently a second-year PhD student in the laboratory of Dr Zongmin Zhao. She completed her BA in biochemistry from New College of Florida in 2020. Her research interests at UIC are focused on the development of cell-based immunotherapies and understanding the immunomodulatory mechanisms behind these therapeutics in the treatment of cancer. |
 From left to right: Bella B. Manshian, Vincent Lenders, Xanthippi Koutsoumpou and Stefaan J. Soenen | The group of Bella Manshian (PhD), part of the Translational Cell and Tissue Research Unit, works on translational nanomedicine using advanced 3D precision cut tissue models and engineered nanoformulations for drug delivery. Vincent Lenders and Xanthippi Koutsoumpou (4th year PhD students) have been working in the group on cell based drug delivery systems across biological barriers. They also collaborate closely with the NanoHealth and Optical Imaging Group of Stefaan J. Soenen (PhD), focusing on non-invasive monitoring of nanoparticle distribution in preclinical model systems. |
1. Introduction
Since its first introduction, nanomedicine has aimed to manifest itself as a major solution to problems in the drug delivery field. Indeed, nanomaterials have been able to mitigate major therapeutic limitations, including protection from rapid degradation, improved drug absorption, improved targeted delivery and – related to this – reduced off-target side effects. These improved therapeutic traits have led to promising preclinical and clinical therapeutic applications.3 Application areas include nanovaccines,4 hemostasis,5 targeted cancer therapy,6–8 with well-known commercial examples Abraxane® and Doxil®, and inflammation.9 Recently, the clinical translation of nanomaterials has been boosted by the introduction of the COVID-19 lipid nanoparticle mRNA vaccines, illustrated by the start of over 55 clinical trials using new nanoparticle technologies since 2019.10However, despite these achievements, major challenges remain in the field of nanomedicine. For example, industrial manufacturing of nanomaterials on a large scale remains, although evolving, a grey area, with limited knowledge of key parameters and process conditions during synthesis.11 However, also regarding the selective delivery of therapeutic agents to specific targeted tissues, nanomedicine has not been able to live up to its original hype. Ever since the sobering meta-analysis by Wilhelm and colleagues, revealing that only a median nanomaterial delivery efficiency of 0.7% to solid tumors has been achieved, increased attention has been focused on investigating the reasons behind this low target efficiency.12 After intravenous injection of nanoparticles (NPs), adsorption of opsonin results in the formation of a protein layer around the NPs, referred to as the protein corona. Subsequently, NPs are recognized by the mononuclear phagocyte system (MPS). Uptake of the NPs by macrophages then clears the NPs from the bloodstream, usually within minutes.13 In a similar fashion, inhalation and subsequent alveolar deposition of NPs is subjected to clearance by alveolar macrophages, significantly reducing the lung residence time of these NPs.14 Rapid opsonization and subsequent clearance of nanoparticles are therefore major contributors to low targeting efficiencies and underwhelming therapeutic effects of NP-based strategies. A well-accepted solution to avoid interaction of nanomaterials with immune cells is stealth coating, for example surface PEGylation, which prevents adsorption of proteins.15 Also, circulatory cell mimicking or hitchhiking particles have been shown to decrease the clearance rates, by leveraging the biological features of, for example, red blood cells or circulatory immune cells, and thereby avoiding MPS phagocytosis.16–18 Alternatively, partial blocking of the MPS may enhance the performance of NPs, for example through pretreatment with liposomes or by inducing a partial depletion of erythrocytes by injection of allogeneic anti-erythrocyte antibodies.19,20
The second reason for the only modest nanomedicine success has been the failure of several active and passive targeting strategies to, effectively, increase NP accumulation in target organs. For example, many NP delivery strategies have relied (solely) on the enhanced permeability and retention (EPR) effect for passive tumor targeting, where interstitial accumulation of NPs could be achieved due to the leakiness of the tumor vasculature. However, reliance on the EPR effect has not shown promising clinical results and the significance of the EPR role as the main driver of tumor targeting has been challenged.21–23
Finally, the third challenge is the presence of physical biological barriers, effectively blocking the passage of cargo-loaded nanoparticles to the organs or regions of interest. The endothelial barrier is the most predominant barrier, impeding translocation over the vascular vessel, when NPs are injected intravenously, or limiting uptake by target endothelial organ cells (barriers of organs). The exact mechanisms underlying vascular crossing or organ uptake are not yet elucidated, but some mechanisms have been suggested. Possible pathways for endothelial uptake include phagocytosis, micropinocytosis, and clathrin- and caveolin-dependent or receptor-mediated endocytosis.24,25 The NP uptake efficiency by endothelial cells of distinct organs, including liver, lungs, brain and kidney, has been shown to be significantly different, likely due to differences in, among others, surface receptors.26 Overcoming the vascular endothelial barrier, referred to as extravasation, has been suggested to be possible through dysfunction of the tight junction, for example by nanoparticle induced endothelial leakiness (NanoEL). Some NPs can induce micrometer sized gaps in the vascular endothelial barrier, by disrupting the VE-cadherin–VE-cadherin interactions, which eventually leads to the induction of actin remodeling.27 While this offers a significant opportunity for nanomedicines, a recent study highlights the potential effects it may have on facilitating cancer metastasis.28
While many barriers of organs can be considered as an endothelial barrier, there are some special cases where the barrier is complexified. For example, the endothelial blood–brain barrier is generally considered as a stronger barrier compared to the liver or kidney barrier, as brain endothelial cells are non-fenestrated and more tightly packed allowing for more controlled brain protection.29 Similarly, the placental barrier consists of, next to an endothelial layer, 3 additional barrier layers, which is necessitated by the crucial protection of the developing fetus. Other special barriers include the lung mucus and gastrointestinal barrier, where a superficial mucus layer strengthens control on intake or inhalation of unwanted particles.
As a thorough understanding of the interaction mechanisms of nanomaterials with these physical barriers is critical for better design of nanomedicine-based therapies, we herein offer an overview of the research that has been performed on nano-barrier interactions (Fig. 1). In this review, we have specifically focused on the 4 special barriers of organs discussed earlier. Administration of NPs through inhalation is challenged by the presence of the lung mucus barrier, while the gastro-intestinal mucus barrier is the main physical barrier to be crossed for oral delivery strategies. Within systemic administration, we have focused on the blood–brain barrier, being a major research focus for brain-related diseases and given its non-fenestrated endothelial barrier, and the placental barrier, given its unique role in both maternal and fetal therapeutic strategies and its unique 4-layer barrier composition. Other special systemic barriers, such as the blood–testis barrier and blood–milk barrier, are not discussed given their very limited nanomedical research focus. For each biological barrier, the NP characteristics that can be tuned to enhance or hinder the transport through these barriers are analyzed and advanced technologies to overcome these barriers are discussed. Finally, suggestions are given for improved translatability of barrier-crossing nanomaterials.
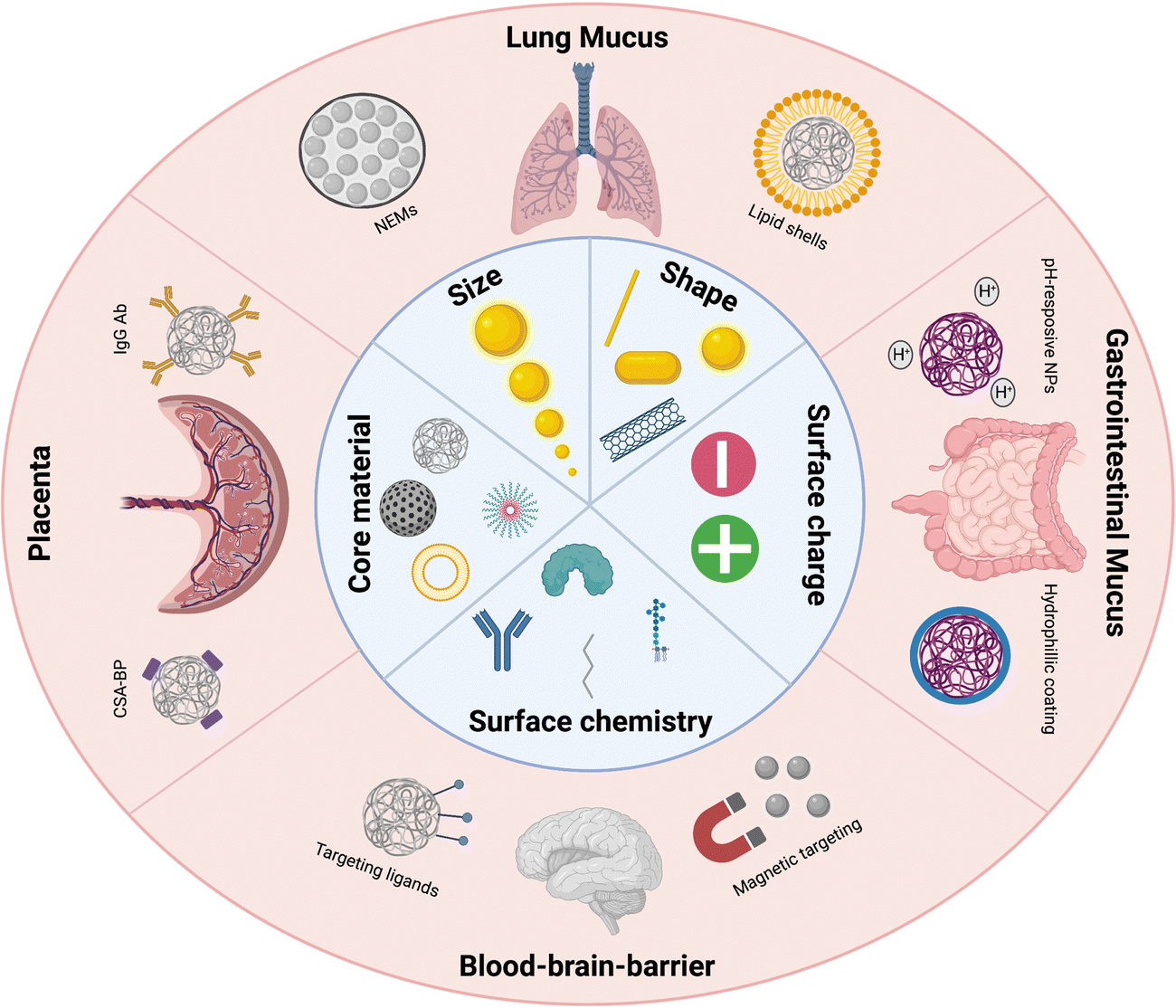 | ||
| Fig. 1 A representative image of the various biological barriers discussed in this review and some of the successful strategies to overcome these barriers. Illustration was made using https://BioRender.com. | ||
2. Lung mucus barrier
The mucus forms a very effective protection layer against injury at multiple sites in (in)direct contact with environmental exposure, such as the intestine, nose and lungs. However, in addition to protection against environmental toxins and microbes,30 the lung mucus also complexifies administration routes for drug delivery, both for localized lung delivery and for systemic delivery through inhalation. For localized lung delivery, the airway route is one of the most straightforward administration routes, as lungs are easily accessible via inhalation.31 However, major hurdles remain for effective airway drug delivery, mainly due to natural safeguard barriers of the lung, protecting against deep inhalation of large particles or microbe entry. Biological barriers include the typical branched structure of the respiratory tract, the mucus layer, the periciliary layer and alveolar macrophages.32 These natural protection barriers complicate airway drug delivery by filtration of inhaled agents, restricted permeation and mucociliary clearance, resulting in poor therapeutic efficiencies. In this section, we will describe more in depth the lung mucus as a biological barrier for airway drug delivery and analyze how NP formulation strategies are used to overcome the lung mucus barrier, improving current therapeutic strategies for asthma, cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), bronchopulmonary dysplasia (BPD) and cancer.33–352.1. Mucus barrier characteristics
The typical biological features of the lung mucus (Box 1) lead to the formation of a steric filter through a size-exclusion gradient towards the epithelial surface. The molecular mesh tightens towards the cellular surface, so that the particles with a diameter (d) larger than the local correlation length (ξ) are impeded from reaching the cell surface.36 The mesh size ranges from 100 to 1000 nm, depending on the airway site. The protective mucus layer progressively reduces in thickness as the alveolar region is approached, decreasing from a thickness of 10–30 μm at the tracheal level to 2–5 μm in the smaller bronchi. At the alveolar level, type II pneumocytes excrete a surfactant, a mixture of phospholipids and proteins, which lines the alveoli with the main function of reducing surface tension.37 This site-dependent mucus volume, combined with the size-exclusion gradient, is essential in balancing the successful entrapment and removal of particulates, while allowing the passage of small molecules for gas exchange at the lung alveoli.38Mucins, containing negatively charged side chains, and mucin-associated compounds such as lipids and DNA, can interact with particulates through electrostatic interactions, hydrophobic interactions and H-bonding. Therefore, the mucus also forms an interaction filter, capable of entrapping small, interacting particles.39
Box 1: Lung mucus characteristicsAlthough the basic properties are shared, mucus secretions are adapted to suit their specific mucosal location. In the conducting airways, the main structural trait of bronchial mucus is its 2-layer system: the actual mucus and the underlying periciliary liquid layer (PCL) (Fig. 2). The mucus is a hydrogel consisting of water (90–95%), mucins, lipids, electrolytes, DNA, enzymes and cellular debris, with mucins, secreted by goblet cells, as the main functional components.40 Mucins are glycoproteins that, through cysteine rich regions, undergo dimerization and subsequent polymerization of the monomers via disulfide bonds.41 This aggregation behavior, further stabilized by weaker hydrophobic and electrostatic interactions, leads to the formation of a gel-like structure.The PCL has been shown to consist of membrane-spanning mucins and large mucopolysaccharides tethered to the cilia, microvilli and the epithelial cells, providing an effective ‘gel-on-brush’ system.36 This structural feature allows for an added dimension to the mucus as an effective biological barrier. Through continuous secretion of mucins by the goblet cells, removal of excessive mucus is facilitated, creating a clearance mechanism. The PCL is less viscous and allows for beating of the cilia as well as lubrication of the cellular layer, allowing for the upwards movement of the mucus in the airways. Clearance of foreign material can be achieved within 15 to 20 minutes after capturing in the mucus. Mucociliary clearance can be further assisted by reflexive coughing if the airways are irritated by foreign matter.42 |
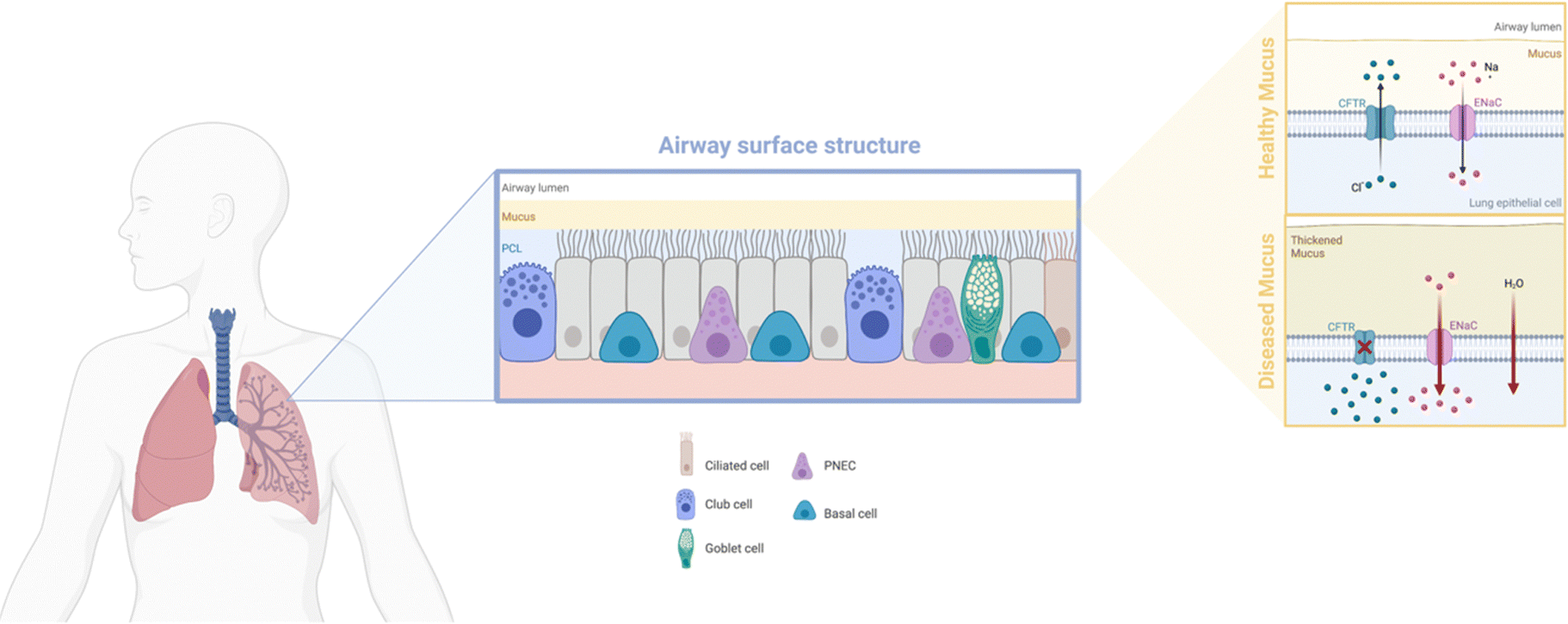 | ||
| Fig. 2 Schematic depicting the makeup of the airway surface structure, with bronchial mucus in the healthy or diseased state. Illustration was made using https://BioRender.com. | ||
Of note, mucus characteristics can change significantly in disease states, depending on the disease type and stage. The thickness of healthy mucus is approximately 30 μm and can easily be transported through ciliary beating. However, a decrease in elasticity or an increase in viscosity and thickness can impede mucus transport.42 Lung disorders, such as cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD) or primary ciliary dyskinesia, often show defects in ciliary transport.38 Clearance defects in lung disorders are found to be beneficial for therapy purposes as, due to the slowed or abnormal ciliary beating, retention times of drug-loaded nanoparticles are increased at the mucus site.43 Although the mechanisms underlying the mucociliary clearing defects are not yet elucidated, the contributing factors are as follows: (i) mucus dehydration: water is crucial in governing the gel-like state of the mucus. For example, exposure to cigarette smoke has been linked to mucus dehydration, leading to an increased mucus concentration. This, in turn, generates a partial osmotic pressure exceeding basal PCL values and eventually reduced mucociliary clearance, aiding in the pathophysiological development of chronic bronchitis.44,45 Similarly, absence of ion channels, and consequently disruption of the ion streams, after mutation of the cystic fibrosis transmembrane conductance regulator gene, leads to dehydration and acidification of CF airways.46 (ii) Mucus hypersecretion: upregulation of mucin expression has been associated with chronic airway diseases, with MUC5B becoming dominant in mucus in disease states. Hypersecretion leads to increased mucus concentration, changing the rheological properties of the mucus and hampering mucociliary clearance.47,48
2.2. NM engineering for lung mucus penetration
With mucus acting as a multiparametric barrier, airway drug delivery strategies are required to overcome the size exclusion gradient, interaction filter and clearance mechanism of the mucus. Multiple NP formulations and designs have been researched over the years and are commonly referred to as mucus penetrating particles (MPPs); a general overview of these is given in Fig. 3. The details of the most recent strategies are tabulated in Table 1. Strategies not focused on overcoming the lung biological barrier, for example alveolar macrophage targeting therapies, are not considered in the scope of this review.49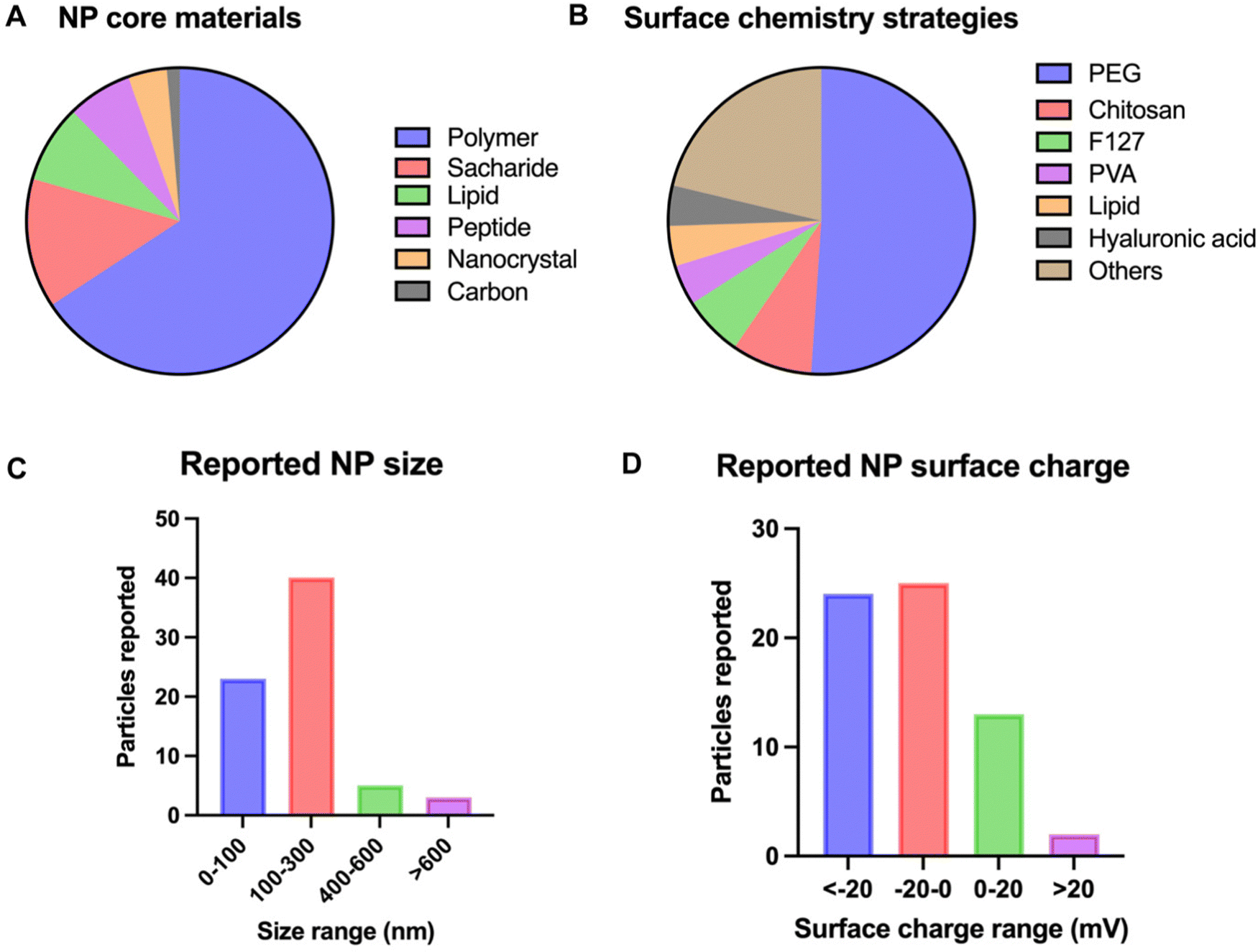 | ||
| Fig. 3 Overview of NP characteristics used for airway drug delivery. The characteristics of interest are (A) NP core material, (B) NP size, (C) surface chemistry and (D) NP surface charge. Graphs are based on data extracted from the PubMed database of the last 10 years using the search terms ‘lung’, ‘mucus’, ‘nanoparticle’, and ‘delivery’, identifying 97 manuscripts. 62 articles were used for data insights. Only original research articles and articles within the scope were included. | ||
| NM engineering strategy | Surface modification | Core material | Size (nm) | Zeta potential (mV) | Model system | Diseased/healthy state | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Core material | / | Stearic acid/F127/Tween 20 | 90 ± 6 | −9.5 ± 1.4 | Artificial mucus | Diseased | Zeta potential and size effect were evaluated, favoring a near-zero charge and small size | 50 |
| DPPC | 102.6 ± 0.3 | −34 ± 10 | LPS-induced mouse model | Diseased | 51 | |||
| DSPC/cholesterol/DSPE-PEG | 161 ± 1 | −7.9 ± 0.6 | Artificial mucus | Healthy | 52 | |||
| Hyaluronic acid | 280 | −61 ± 4 | Artificial mucus | Healthy | 53 | |||
| Dextran | 200 to 300 | −30 to −50 | Artificial mucus | Diseased | 54 | |||
| Sizing | / | PLGA | 100 to 2500 | Around -20 | Artificial mucus | Diseased | Smaller NPs (100 nm) have better lung retention and adsorption properties than larger NPs (300, 800 and 2500 nm) | 55 |
| Curcumin (nanocrystal) | 246 to 1089 | / | Rat model | Healthy | Small NCs show higher dissolution rates. Crossing of the mucus occurs mainly by the free drug form | 56 | ||
| Surface engineering: polymeric coatings | PEG | PBAE | 53 ± 2 | 0.7 ± 0.3 | Scnn1b-Tg mouse model | Diseased | 57 | |
| C190 (nanocrystal) | 823 ± 123 | -21.2 ± 6.07 | Artificial mucus | Diseased | Rod shaped NPs | 58 | ||
| FLR (peptide) | Around 100 | Around 5 | Mouse model | Healthy | Effect of the PEGylation rate was evaluated, with 40% the most optimal | 59 | ||
| PBAE | 55 ± 1 | 1.6 ± 0.3 | Orthotopic lung cancer model | Diseased | 60 | |||
| PLGA | Around 3000 | / | Rat model | Healthy | Effect of PEG molecular weight was evaluated, with 2 kDa the most optimal | 61 | ||
| PHEA-PCL | 51.1 | −14.4 ± 4.6 | Artificial mucus | Healthy | NEM (see nano-embedded microparticles) | 62 | ||
| TPFE | 73.4 ± 0.7 | 1.3 ± 0.2 | Scnn1b-Tg mouse model | Diseased | 63 | |||
| PS | 104.6 ± 1.2 | −4.9 ± 0.3 | Artificial mucus | Healthy + diseased | 64 | |||
| PLGA | 130.9 ± 2.17 | 1.97 ± 0.1 | Bleomycin sulfate-induced mouse model | Diseased | 65 | |||
| oxi-aCD | 254.2 ± 9.5 | −32.4 to −37.4 | P. aeruginosa mouse model | Diseased | Folic acid was added for better cellular uptake | 66 | ||
| Hyaluronic acid | Poly(b-amino ester) | 150 | 10 | LPS-induced mouse model | Diseased | 67 | ||
| PLGA | 228 | Around -50 | P. aeruginosa mouse model | Diseased | 68 | |||
| Pluronic F127 | PLGA | 307.5 ± 9.54 | −11.3 ± 0.4 | / | / | No mucus interaction experiments performed | 69 | |
| PVA | PLGA | 261 to 282 | −0.67 to −0.84 | P. aeruginosa mouse model | Diseased | 70 | ||
| CS-A | pDNA/siRNA (CRHC-2/M9) | 200 to 400 | 20 to 25 | Mouse model | Healthy | 71 | ||
| PMeOzi | Co-polymer: grafted PHEA and PLA | 95 ± 5 | −7.2 ± 4.7 | Artificial mucus | Healthy | NEM (see nano-embedded microparticles) | 72 | |
| PMeOx | 78 ± 3 | −5.8 ± 4.5 | ||||||
| Surface engineering: lipid shells | DPPC | PLGA | 177.6 ± 9.2 | −28.7 ± 1.6 | In vitro model with mucus-covered Calu-3 cells | Healthy + diseased | Bare lipid shell NPs showed better epithelial internalization compared to PEGylated lipid shell NPs | 73 |
| 174 ± 2.03 | −29.2 ± 1.58 | Artificial mucus | Diseased | 74 | ||||
| 238 ± 9 | −25 ± 1 | Mouse model | Healthy | More neutrally charged lipids DPPC and DPPE led to macrophage uptake inhibition, while negatively charged lipids DPPG and DPPS led to increased macrophage uptake | 75 | |||
| DPPE | 230 ± 10 | −26 ± 1 | ||||||
| Surface engineering: peptide coating | Peptide CPSSSREKC | PS | 180 ± 3.8 | −21.4 ± 1.6 | Ex vivo human CF sputum + mouse model | Diseased (ex vivo) and healthy (in vivo) | 76 | |
| Nano-embedded microparticles | PMeOzi | Co-polymer: grafted PHEA and PLA | 95 ± 5 | −7.2 ± 4.7 | Artificial mucus | Healthy | NEM size: around 4 μm | 72 |
| PMeOx | 78 ± 3 | −5.8 ± 4.5 | ||||||
| PEG | PHEA-PCL | 51.1 | −14.4 ± 4.6 | Artificial mucus | Healthy | NEM size: 2 μm | 62 | |
| Others: redox-responsive NPs | PEG | PLGA | 120 | −30 | Artificial mucus | Healthy | 77 | |
| Others: enzyme-modified NPs | Papain | Dextran | 200 | −50 | Artificial mucus | Diseased | 78 | |
| Others: size-shifting NPs | Phosphate ester and octadecylamine | Lipid | 126.4 ± 3.5 | −27.9 ± 1.3 | In vitro model with mucus-covered Caco-2 cells | Healthy | 79 |
The choice of the core material appears to be mainly driven by the specific application and the drug types to be delivered. Limited research has been performed on the effect of the core material on mucus crossing, probably because most unmodified NPs, especially hydrophobic polymers, fail to penetrate the mucus in a satisfactory manner. The main exception to this is liposomes. Surfactant-mimicking liposomes can be designed, consisting of dipalmitoyl-phosphatidylcholine (DPPC), a component of the pulmonary surfactant. This biomimetic approach protects the cargo against degradation, improves NP diffusivity and increases lung retention.51 Furthermore, liposomes offer excellent epithelial cell uptake after mucus penetration, as was shown for pulmonary fibrosis treatment with PGE2.84 The potential of liposomal formulation is further illustrated by its translation to clinical trials and FDA approval of, for example, Arikayce, a liposomal formulation of amikacin.85,86
Although the size constraints for mucus penetration are widely accepted, mucus penetrating particles have been formulated with sizes up to 800 nm by aspect ratio engineering. For example, Costabile et al. made benzothiadiazole nanocrystals of 823 nm that showed diffusion in artificial CF mucus due to their elongated nanorod shape.58
Consequently, overlooking the reported NP formulations in the past decade, this study shifted design strategies for airway NP delivery towards creating negatively or near-neutrally charged NPs (Fig. 3D), in most cases combined with a hydrophilic coating to limit attraction with mucin glycoproteins through electrostatic and hydrophobic attraction. This effect was also seen in coarse-grained molecular dynamics (CGMD) simulations after modeling the surfactant monolayer translocation behavior of PEG-grafted gold NPs (Fig. 4A).89 All neutral NPs could penetrate the surfactant, regardless of grafting density or monomer number per chain. Charged NPs with a low monomer number per chain and a high grafting density were impeded from undergoing translocation. Lowering the grafting density or the length of the grafting polymer reallows penetration, likely due to the decrease in surface density. Furthermore, through analyzing the interaction energies between differently charged NPs and the lipid heads, it was observed that positively charged particles take longer to penetrate and adhere to the film after penetration. This phenomenon can be attributed to stronger electrostatic interactions with the lipid heads, as well as stronger van der Waals interactions (Fig. 4B). Reducing the hydrophobicity of the particle surface has also been shown to improve pulmonary biocompatibility in vivo.90
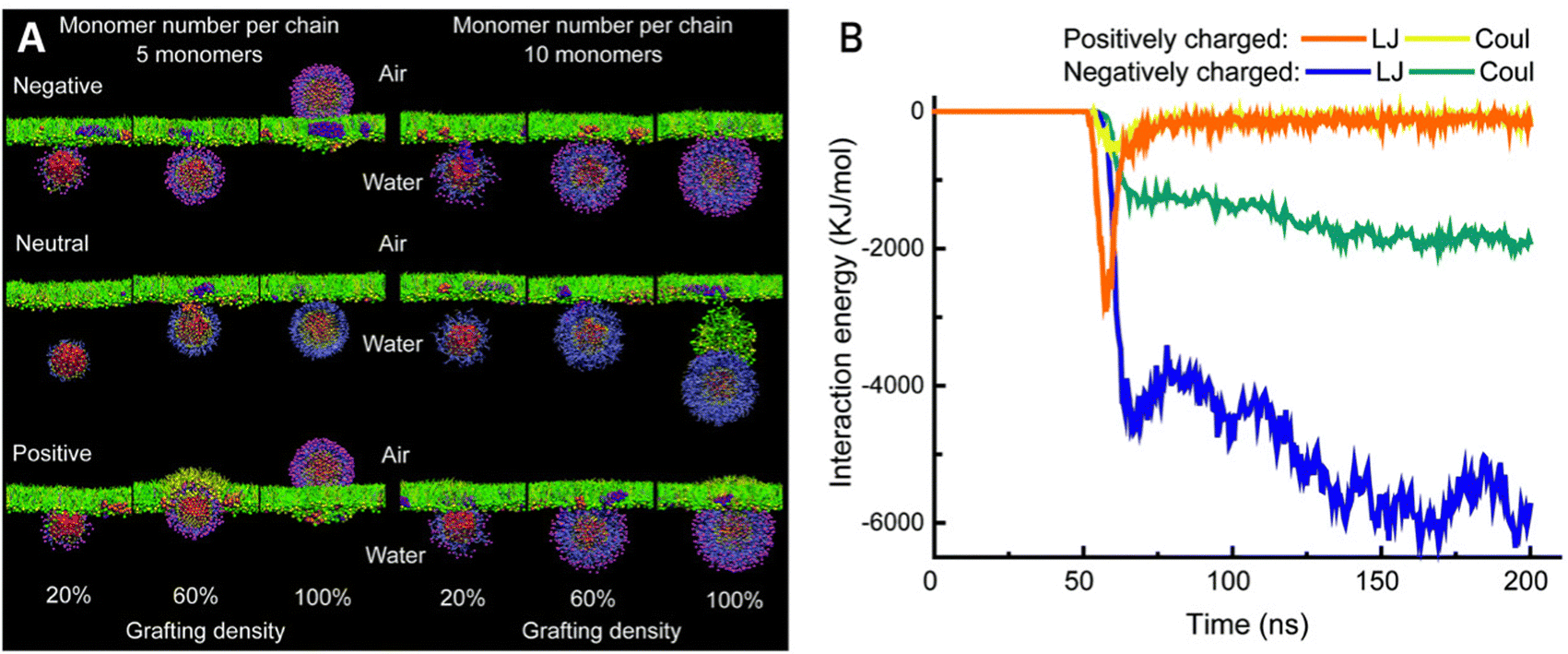 | ||
| Fig. 4 (A) Snapshots of NPs interacting with a lung surfactant monolayer in a CGMD model. NP variations include monomer number per chain, grafting density and terminal charge. (B) Interaction energy diagram of differently charged NPs with the lipid heads of the surfactant monolayer. Coul stands for electrostatic interactions, and LJ stands for van der Waals interactions. Adapted from ref. 89 with permission from The Royal Society of Chemistry. | ||
2.2.3.1. Polymeric coatings. PEG is by far the most used surface modification for this purpose (Fig. 3), and has been shown to improve mucus penetration and therapy effectiveness for cystic fibrosis,57 inflammation62 and cancer.91 Recently, siRNA against IL11 was co-loaded into PLGA-PEG diblock polymeric NPs with a cationic lipid-like molecule G0-C14, which facilitates transmucosal delivery. Inhalation of these RNAi NPs was shown to effectively inhibit fibrosis in a post-bleomycin challenged mouse model.65 Surface coating with PEG, however, should be carefully optimized for its surface density and molecular weight. A 5 wt% PEG content is believed to be needed for effectively shielding the nanoparticle core from mucus interactions.92 Although some studies have reported on effective bronchial epithelial cell uptake of PEGylated NPs,93 it is important to note that an increased PEG content or PEG molecular weight may limit cellular uptake and can therefore hamper delivery effectiveness.94
Multicomponent coatings have been proposed as a strategy to leverage the transmucosal traits of PEG, while still ensuring uptake by target cells. Wang et al. formulated oxy-α-cyclodextrin particles coated with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG and DSPE-PEG-folic acid. While the PEG layer was shown to improve mucus penetration, the use of folic acid improved the uptake by the targeted macrophages, mediated by membrane folate receptors.66 Another example of multicomponent coatings is related to DNA delivery applications. Dense PEG coating may interfere with DNA compaction, entailing larger NPs with poorer mucus penetrating and cellular characteristics. However, Suk et al. reported that using polyethylenimine (PEI)/PEG-PEI or poly-L-lysine (PLL)/PEG-PLL mixtures in optimal ratios can reduce the hydrodynamic size by ∼15% compared to particles using PEG-PEI or PEG-PLL coatings only. This approach reduced the mean square displacement ratio MSDw/〈MSD〉 by ∼16 fold and ∼136 fold for PEI and PLL NPs, respectively, indicating a significant improvement of diffusivity in CF mucus.95 Finally, non-covalent modification of PEG-NPs with Pluronic F127 has been reported to improve drug activity duration, likely due to an increased colloidal stability of the NP.96 However, due to its protective nature and possible shielding of mucus interactions, Pluronic F127 alone, without additional PEG shielding, has been reported as a potential, alternative surface engineering strategy for mucus penetration.69
Various other hydrophilic coatings have been reported as an alternative to PEG, especially since research indicated possible immune response after repeated administration of PEGylated therapeutics.97 Casciaro et al. have developed PLGA NPs coated with poly(vinyl alcohol) (PVA), which not only reduces NP aggregation, but also provides a neutral, hydrophilic surface. On loading the NPs with an antimicrobial peptide (Esc), an improved efficacy in inhibiting Pseudomonas aeruginosa was achieved, proven by a 3-log reduction of pulmonary bacterial burden.70 In a similar fashion, d’Angelo et al. developed colistin loaded PVA-coated PLGA NPs for Pseudomonas aeruginosa treatment. Moreover, they tested coating with another hydrophilic polymer, namely chitosan (CS). Although it would be expected that the more positively charged CS-NPs have a higher tendency to interact with mucins, this did not hamper mucus penetration. Instead, CS facilitated mucus penetration to a greater extent than PVA, probably due to the induced collapse of mucus fibers, creating larger mesh pores.98 The use of cationic chitosan-coated nanoparticles is especially of interest, given their good cell uptake characteristics, as shown for example in asthma treatment, although more mucus interaction studies should be performed to elucidate the dynamics of CS-particles in the mucus.99
Alternatively, hyaluronic acid (HA) has recently received attention for its successful creation of a hydrophilic shell and, consequently, improved mucus penetration.68 For example, Zhu and colleagues designed HA-coated poly(β-amino ester) (BP) NPs successfully penetrating the mucus. Furthermore, once inside the interstitium, uptake by the target interstitial M1 macrophages was achieved.67 Of note, the radical scavenging property of HA gives, together with any loaded cargo, a synergetic anti-inflammatory benefit, in this case down-regulating TNF-α siRNA.
Improved delivery of siRNA with NPs has also been achieved by using chondroitin sulfate A (CS-A) as hydrophilic coating. Kumari and colleagues recently did indeed show improved mucus penetration for CS-A-coated nanocomplexes, which was further improved by surface conjugation of mannitol, acting as a mucolytic agent. The presence of mannitol reduces mucus viscosity, likely by increasing water influx.71
Instead of coating NPs, grafting hydrophilic chains to hydrophobic core materials has been suggested as a design strategy for mucus penetration. Drago and colleagues reported the grafting of the hydrophobic polymer PLA and the hydrophilic chains PMeOx or PMeOzi on the poly(2-hydroxyethyl acrylate) (PHEA) backbone. The pseudo-polypeptide POx structures have similar mucus shielding properties as PEG but can offer the added advantage of faster excretion from the organism.72 Whether they show similar or better cellular uptake properties as PEG should be evaluated in future work.
2.2.3.2. Peptide coating. While most surface engineering relies on polymeric hydrophilic coatings, the use of peptides as surface coatings was suggested by Leal and colleagues.76 They developed a peptide-presenting phage library, which can be used for high-throughput screening to identify peptide coatings with the desired mucus inert functionalities. This screening allowed identification of neutral net-charge, hydrophilic sequences (CGGQDLKSC, CSNLTSP*C and CPSSSREKC), mainly composed of glycine, serine, glutamic acid and aspartic acid. CPSSSREKC was shown to be the most promising peptide, as CPSSSREKC-coated PS NPs were more abundantly taken up by cells in a transwell co-culture assay with CF sputum and showed 90% retainment in mouse lungs 24 hours after administration. Interestingly, the peptide-coated PS NPs significantly outperformed PEGylated PS particles (Fig. 5).
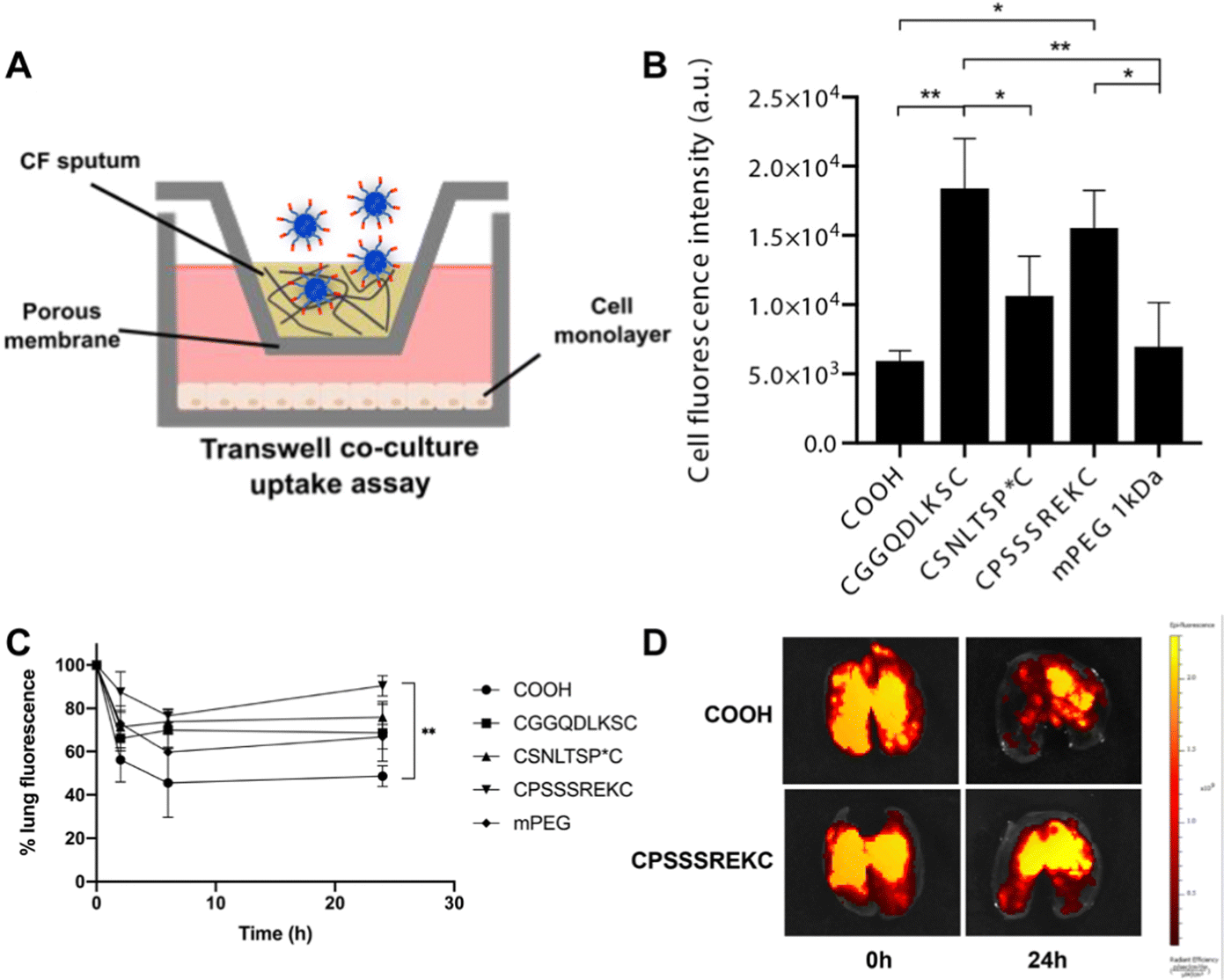 | ||
| Fig. 5 (A) Schematic of transwell co-culture. (B) Fluorescence intensity of cells after incubation with bare, peptide-coated and mPEG coated PS NPs. (C) NP retention in the lung, measured up to 24 h post-administration. (D) Representative ex vivo lung images at time point 0 and 24h. *p < 0.05, **p < 0.01. Adapted from ref. 76 with permission from Elsevier. | ||
2.2.3.3. Lipid shells. A more established surface engineering strategy is the use of lipid shell NPs. While liposomes tend to have several drawbacks in terms of stability and drug release properties, they do offer mucus penetrating traits, as discussed earlier. A lipid shell-enveloped polymeric NP formulation, as reported by Wan et al., combines mucus penetration with sustained drug release from the polymeric core.100 Conte et al. reported a hybrid lipid/polymer NP that effectively achieved gene silencing in an in vitro CF model. Moreover, it was demonstrated that bare polymer/lipid nanoparticles, following mucus penetration, are capable of internalization by epithelial cells, whereas epithelial internalization is hampered for PEGylated polymer/lipid NPs.73 The use of lipids for improved mucus penetration and epithelial cellular uptake was also reported by Liu et al., who developed mucus-inert NPs by biomimetic modification with endogenous surfactants, in particular DPPC.75
These formulations consist of mucus penetrating NPs, which are embedded in a microparticle carrier containing an excipient. Once the NEMs reach the mucus, the embedded NPs are released and can spread along and penetrate the mucus (Fig. 6). For example, Craparo et al. synthesized rapamycin loaded, PEGylated copolymer nanoparticles embedded in mannitol-based microparticles by spray drying. The NEMs released rapamycin in artificial lung fluid, indicating the successful disintegration of mannitol.62 Mannitol NEMs have also been successfully designed for gene editing purposes by co-loading siRNA with the cationic lipid dioleoyl-3-trimethylammonium propane (DOTAP) for improving gene silencing properties.102 Excipients other than mannitol have been researched as well. In an earlier study, lactose was successfully employed as an excipient. Within this study, a comparison was made between PVA and chitosan as a NP stabilizer for antibiotic loaded PLGA NPs. Although both yielded good NEMs, the aerodynamic properties of both differed as illustrated by the difference in disposition of the NPs. While PVA NPs reached alveoli, chitosan NPs were mainly found in high amounts in the upper airways.103
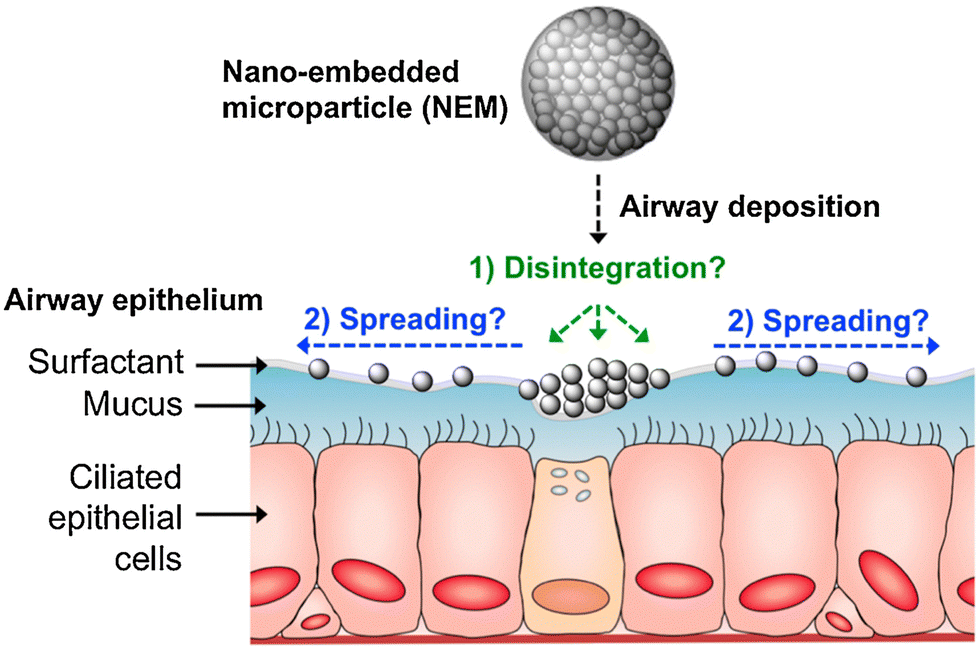 | ||
| Fig. 6 Schematic overview of the fate of NEMs after deposition on the pulmonary surface. (1) Possible disintegration of the excipient after contact with the mucus. (2) Possible epithelial spreading of the released NP due to ciliary beating movement. Reproduced from ref.104 with permission from Elsevier. | ||
In a study by Porsio et al. for the treatment of microbial infections in cystic fibrosis, NEMs were synthesized with either mannitol or PVA, both resulting in NEMs of desired aerodynamic properties. However, mannitol-based NEMs showed better antimicrobial activity and improved CF lung function. The latter was further improved by mixing mannitol with cysteamine.105 The supremacy of mannitol as an excipient is due to its potential to improve mucus penetration by increasing the fluidity, and thereby the mesh size, of the mucus.105,106
Key to the performance of NEMs is the disintegration of mannitol, releasing the embedded NPs. Meticulous analysis of excipient disintegration in an in vivo-like environment is thus needed. Torge et al. performed a disintegration study under lung-like conditions and showed that exposure to high air humidity is sufficient for mannitol disintegration. However, the disintegration time was significantly influenced by the mannitol content. In their study 20% mannitol content ensured fast release of NPs before clearance.107 In another study, performed by Ruge et al., the disintegration of NEMs was shown to be only successful when mechanical forces are exerted on the mucus, implicating possible limitations to the use of NEMs for efficient drug delivery.104 Better models, more closely mimicking the in vivo setup, are expected to bring more clarity on this in the future.
In an effort to overcome the hampered epithelial cellular uptake of some mucus inert NPs, Conte and colleagues reported a redox-responsive delivery system by creating PEG and PLGA block copolymer NPs, which were synthesized through disulfide bridges between the 2 polymers. This system allows for (i) mucus penetration due to the hydrophilic external PEG layer, (ii) reductive cleavage of the disulfide bond by reducing agents at the cancer cell surface, which reduces the outer PEG layer and improves cellular uptake, and (iii) complete removal of the PEG layer by intracellular GSH, leading to NP breakdown and intracellular drug release (Fig. 7).77 An alternative strategy for improved absorption has been proposed by Le-Vinh et al., using size-shifting nanocarriers.79 Solid lipid nanoparticles with a phosphate ester and octadecylamine surfactant provided negatively charged NPs that could penetrate the mucus. However, when in contact with epithelial cells, the membrane bound alkaline phosphatase cleaves and removes the phosphate ester outer layer, exposing the positively charged octadecylamine groups. The lack of negative charge leads to particle aggregation, thereby preventing back-diffusion of particles and thus extending exposure to the absorption membrane.
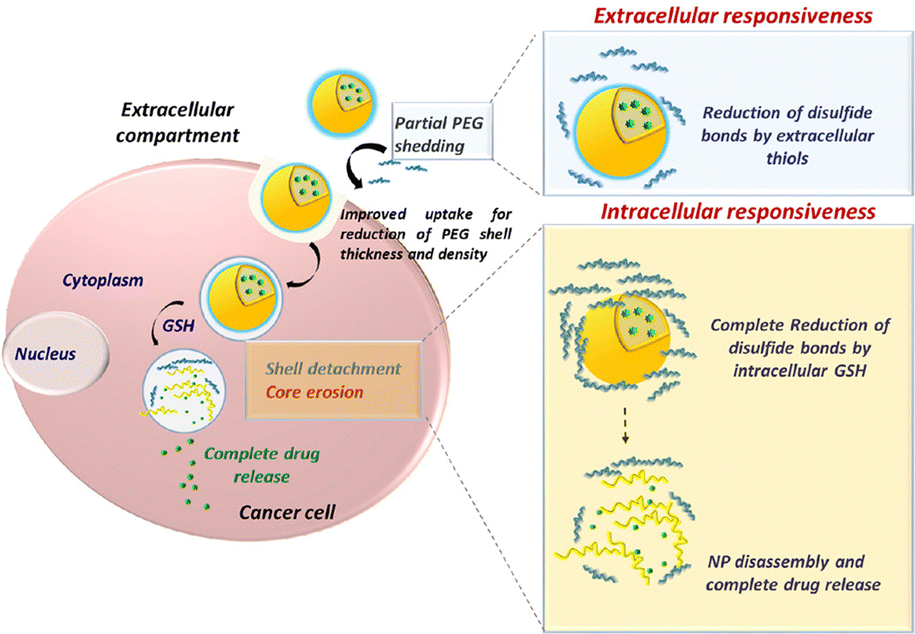 | ||
| Fig. 7 Improved cellular uptake mechanism using redox-responsive, mucus penetrating nanoparticles for lung cancer therapy. Adapted from ref. 77 with permission from Elsevier. | ||
2.3. Considerations for airway drug delivery
2.3.1.1. Inhalation devices. The type of nebulizer used has been shown to influence delivery efficiency and should be chosen carefully. Additionally, current nebulizers often fail to achieve deep lung disposition of active pharmaceutical ingredients (APIs).112 Recent efforts in further improving inhalation devices include mesh nebulizers113 and smart nebulizers (e.g. Akita®Jet).114 Additionally, computational fluid dynamics tools can be used to analyze and predict the transport and disposition behavior of various formulations in the airways.115
2.3.1.2. Formulation considerations. Depending on the nebulization process used, certain formulation constraints are imposed. For example, the aerosolization of liposomes has been a major issue. Tolerance against shear forces during the nebulization process has been shown to depend on surface characteristics of the liposomes, with positively charged liposomes tending to aggregate and lose the encapsulated cargo during the process.116 Alternatively, liposomes can be stabilized through membrane addition of cholesterol or phosphatidic acid. Using PEG as a stabilizer is also possible, albeit only at high concentrations.117 Additionally, to ensure stable shelf-life, lyophilization and subsequent rehydration before nebulization of the lysosomal formulation may be needed. In that case, addition of cryoprotectants can modulate the membrane properties and affect membrane integrity during nebulization.118 While liposomal NPs may need additional consideration, polymeric nanoparticles have shown to be nebulized without artefacts, for example by employing PVA as a surfactant, shielding the core NP from high shear forces.119
Although intravenous administration provides a sound alternative to airway delivery, (dis)advantages of both should be weighed in for every specific formulation or disease type (Table 2). For example, for certain pulmonary diseases, such as COPD or asthma, airway administration of the therapeutic compound is desired as it improves drug delivery to relevant cells.127 Also for lung carcinoma treatment, intratracheal liposomal administration has been shown to be more therapeutically effective than intravenous administration.128 However, for other lung disorders, such as bronchopulmonary dysplasia, a systemic approach might still be preferred, given the strong interlink between alveolarization and angiogenesis.129,130
| Airway administration | Systemic administration |
|---|---|
| Advantages | |
| • Noninvasive administration | • Avoidance of the mucus barrier |
| • Direct delivery | • Capable of reaching the capillary/alveoli interface |
| • Low systemic side effects | |
| Disadvantages | |
| • Mucus clearance | • Invasive administration |
| • Formulation restrictions | • Targeting strategy needed |
| • Specialized administration equipment needed | • Systemic side effects common |
| • Loss of API by sedimentation in the upper tract or through exhalation | • High clearance by the MPS |
3. Gastrointestinal mucus barrier
3.1. GI mucus characteristics
The gastrointestinal (GI) barrier is coated with mucus in a protective manner that maintains the integrity of the organs from foreign entities. The GI tract is composed of the mouth, pharynx, esophagus, stomach, small and large intestines, colon, and liver amongst others. Delivery to target organs surrounding the GI tract can be achieved via localized (direct injection)135 and systemic (oral) routes of administration.136 The most common delivery method involves oral administration; however the nanomaterials that follow this route rely on methods that strengthen the stability during transit against drastic pH changes through surface functionality or other modifications. Challenges with orally administered nanomaterials include low proportions absorbed within the gut lumen137,138 despite taking advantage of higher oral bioavailability.Certain disease states like inflammatory bowel diseases also influence the properties of mucus. Irritable bowel disease, Crohn's disease and ulcerative colitis all have similar properties such as inflammation of the GI mucosal tissue that can result in impaired mucus barrier operation.139 In particular, Crohn's disease can affect all portions of the GI tract in a non-uniform distribution, which can exaggerate immune responses throughout the barrier and compromise its integrity.140 Specific to the disease state, there are also changes in pH within the GI tract where some patients experiencing Crohn's disease have a colon pH between 5 and 7, whereas ulcerative colitis patients can have a pH of 2.7–5.5; for comparison a healthy patient generally has a colon pH of 6.2–7.4.141 Notably, the lower pH may present an additional barrier to orally administered drugs, and biologics in particular. Additional characteristics of the GI mucus can be further examined in Box 2. By examining the makeup and physical characteristics of the GI mucosa, it is evident that nanovehicles must be designed with robustness to withstand mucus cycling and acidic pH for effective delivery especially in disease states with more drastic pH shifts.
Within this section, we will examine the current formulations used in targeting the GI tract in regard to physical properties of the nanomaterial as well as potential surface modifications in order to best optimize its translatability.
Box 2: GI mucus characteristicsThe gut microbiota of the GI tract maintains the balance of the human flora. Within the secretions of the mucosa, bacteria closely associated to the immune system and tolerated by it must navigate secretory immunoglobulin A that modulates pathogenic access to the intestinal lumen.142 Goblet cells make up the cell layers that secrete mucin and mucin-associated proteins such as MUC2 and MUC3, which make up the majority of components within the mucus. These proteins have been previously characterized to prevent the adherence of foreign objects and microbes such as Salmonella enterica from infiltrating the inner layers of the mouse intestine.143,144 Bacteria that form the makeup within the mucus facilitate an antimicrobial layer when moving towards the inner layers of the intestinal lumen of the small intestine.145 With its constant supply of mucus that is secreted and recycled there, researchers have found that the innate protease meprin β plays a role in the detachment of mucus and establishes the adherence of the asymmetrical mucosal outer layer to its inner layer of the colon.146 Within the colon of the GI tract exists a bilayer of mucus that allows for the passage of endogenous bacterium to maintain the intestinal microbiome balance. Acting as the first line of defense, the mucosal layers inhibit the adherence of foreign pathogens and nanovehicles alike and promote their clearance through the cycling mucus. A brief overview of the human gastrointestinal tract and small intestine makeup is presented in Fig. 8.Fluid characteristics of the GI tract also play roles in buffering capacity as well as fluid volume that is available for drugs to be dissolved and metabolized. Luminal fluid volume in the GI tract of mice when administered atenolol and/or metoprolol with varying osmolarity indicated varying degrees of permeability.147 With regard to drugs with poor solubility loaded into polymers, the result is often a lipophilic nanoproduct that has poor adhesion to the mucosal layer and poor aqueous solubility within the GI tract due to the flux in fluid volume across the GI tract.148 |
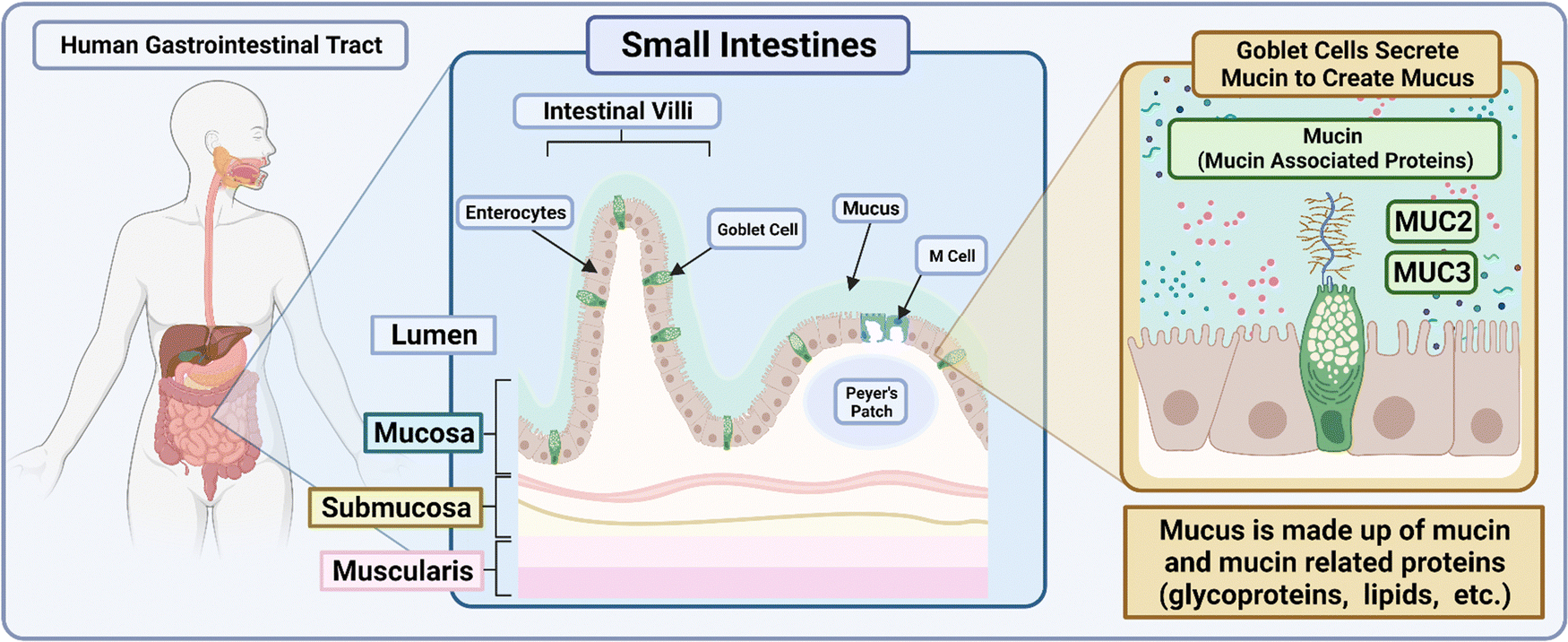 | ||
| Fig. 8 Schematic depicting the makeup of the small intestines within the human GI tract. The illustration was made using https://BioRender.com. | ||
3.2. NM engineering considerations influencing the interaction with GI mucus
Mucin, the major macromolecule responsible for the gel-like characteristic of mucus, has been found to inhibit the diffusion of various drugs when exposed to a phospholipid vesicle-based permeation assay with stimulated mucin concentrations.149 In particular, the study determined that drugs would have difficulties diffusing at thicker mucus layered tissues and that the characteristics of the drug–mucin interactions would play a significant role in their diffusibility; lipophilic drugs such as naproxen had reduced diffusion coefficients at higher mucin concentrations similarly to hydrophilic drugs such as atenolol.Nanomaterials in general have ideal dissolution properties as they have a large surface-to-volume ratio for loaded cargos and quick release of payloads can be achieved to reach saturation.150 While in theory the nanoscale dimension application seems simple, there exists issues with the biological form of the nanoparticle once taken orally; the outer coating of the nanoparticles once interacting with the mucosal layers can acquire proteins to its surface that alters lipophilicity, size and even different surface charges or moieties.151,152 This could be due to the protein rich regions of the GI tract as well as any harsh changes in the pH environment such as pH 1–2 in the stomach and up to pH 8 within the intestines and colon. Physical characteristics to keep in mind when formulating nanosystems are delineated in Fig. 9.
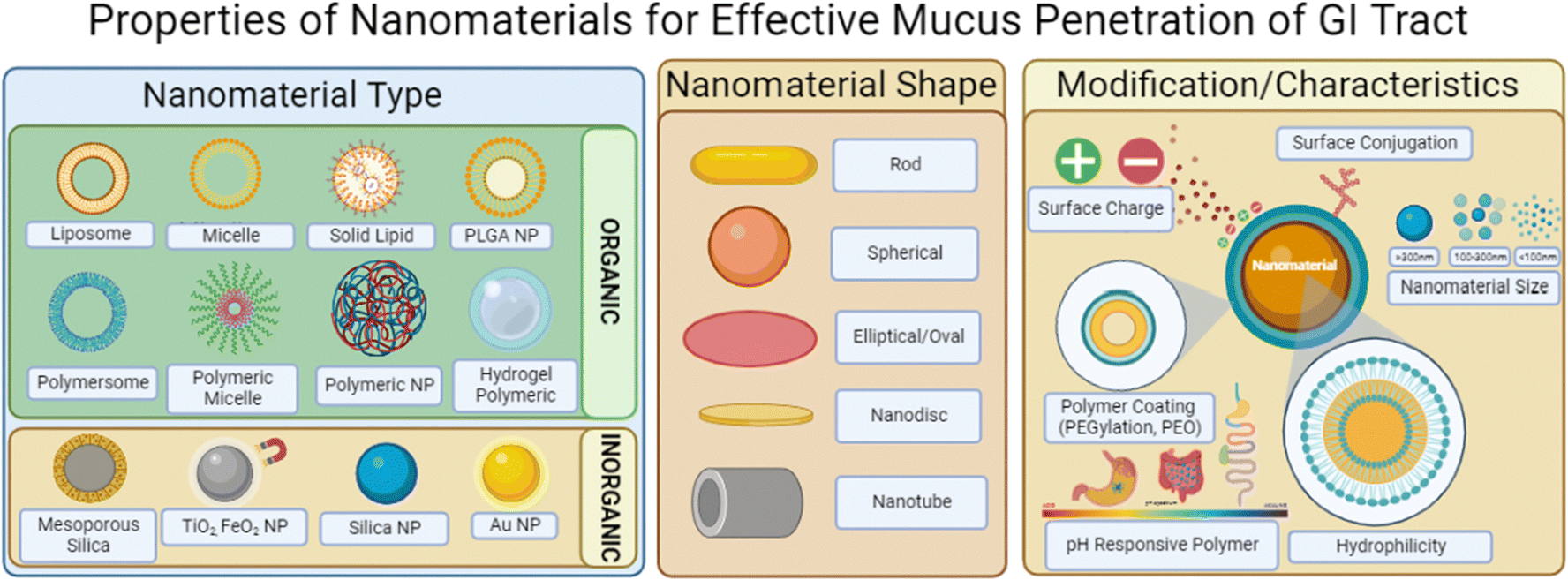 | ||
| Fig. 9 Image depicting nanomaterial characteristics that influence mucus penetration within the GI tract. Illustrations were made using BioRender. | ||
When performing a search on PubMed Central to identify physical properties of nano delivery systems that penetrate the GI mucus barrier, a total of 59 studies published within the last 10 years were identified. The search terms included “nanoparticle” AND “gastrointestinal tract” within the abstracts. By excluding studies that did not contain relevance to mammalian GI tracts and reviews, a total of 36 studies of interest were isolated. When examining common sizes of these nanosystems, it was found that over half of the studies had nanoparticles with sizes less than 200 nm (61.11%) and the majority of studies contained nanoparticle systems less than 350 nm (86.11%) as seen in Fig. 10 and detailed in Table 3. Similarly, when examining the zeta potential for reported systems, the majority of studies utilized a negatively charged particle (64%) with approximately half (52%) of the studies designing nanosystems with reported zeta potentials between −10 mV and +10 mV. Polymeric nanomaterials were the most common (70.3%) amongst all studies compared to metal- and lipid-based formulations; polymers observed included PLGA,153 Eudragit,154 chitosan,155 and polystyrene156 amongst others. Metal-based formulations varied from synthesized to commercial grade and included titanium dioxide,157 zinc oxide,158 iron oxide,159 silver,160 and gold.161 Studies using lipids were the least common among those isolated studies and included micelles.162 When analyzing these data, it can be observed that research has found success in developing nanosystems at smaller sizes and negatively/neutrally charged for optimized mucus permeability.
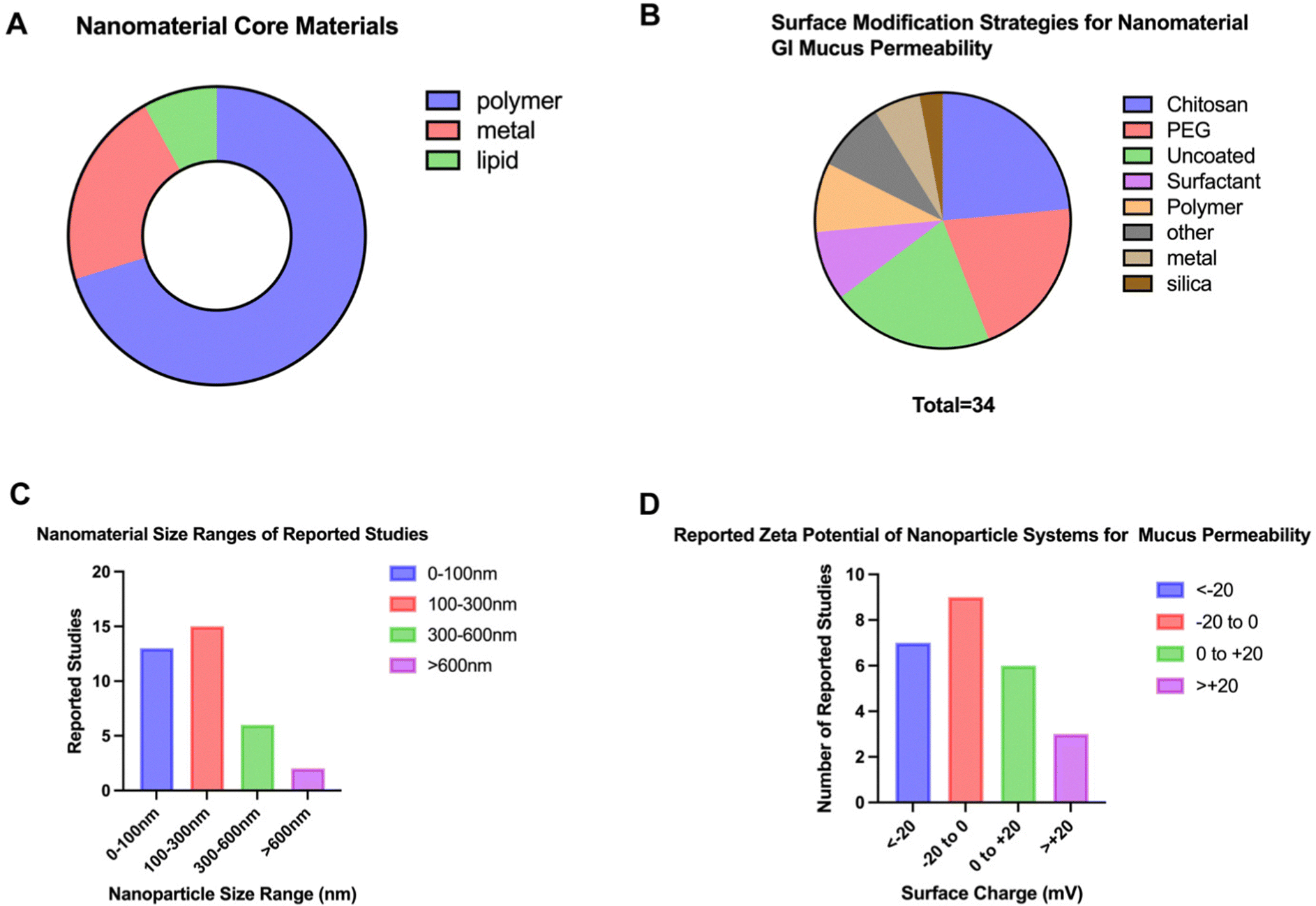 | ||
| Fig. 10 Characteristics of nanodelivery systems for GI mucus permeation. Among common characteristics of these platforms is (A) the nanomaterial core material, (B) the surface modification, (C) the size ranges and (D) reported surface charges. 37 Pubmed research articles were used to determine common sizes for nano delivery systems in penetrating the mucus barrier of the GI tract. The search terms included ‘GI tract’, ‘mucus’, and ‘nano’ for research articles limited to the last ten years. | ||
| NM engineering strategy | Surface modification | Core material | Size (nm) | Zeta potential (mV) | Model system | Disease/normal state | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Core material | — | PLGA | 200–300 | −34 to −33 | C57BL/6 intestinal stem cells organoid unit | IBD | PLGA nanoparticles with Matrigel and organoid suspension increase luminal absorption | 163 |
| PHOSPHOLIPON 90G (lipid) | 23 | — | Premalignant oral epithelial cells (SCC83) | Oral cancer | Lipid micelle delivery targeted drug effectively in a short amount of time (15 minutes) | 162 | ||
| Chitosan | 350 | — | Sprague Dawley rats (fasted) | Diabetes mellitus | Mucoadhesive particles adhere within the stomach for prolonged time periods | 164 | ||
| Iron | Zinc oxide (metal) | 100 | −40 | Balb/c mice | Normal state | Iron embedded zinc oxide nanoparticles improve iron absorption in the gastrointestinal tract in a supplemental manner | 158 | |
| Surface engineering: polymeric coatings | PEGylation | Chitosan | 47 | 8 | Caco-2, HT29-MTX | Normal state | Nanoparticles travel paracellularly through the intestinal epithelium | 165 |
| Coacervate/catechol | 150 | — | Sprague Dawley rats | DSS induced colitis | Improved controlled release was achieved as well as improved retention time within the intestines | 166 | ||
| Surface engineering: chitosan | Chitosan | PLGA | 350 | 9.7 | Balb/c acute colitis | Acute colitis | CS-PLGA-NP mucoadhesion increased colonic drug delivery in GI | 153 |
| Chitosan | Polylysine-bilirubin | 233.9 | 6.2 | Balb/c ethanol induced acute gastric ulcer | Ethanol induced gastric ulcer | Positively charged CS-bilirubin nanoparticles enhance the anti-inflammatory effect | 167 | |
| Chitosan, PEG | Hydroxypropyl ethylcellulose phthalate | 294 | −26.7 | Caco-2-cell monolayer, ICR mice | Normal state | HTCC modified chitosan increased mucoadhesion | 168 | |
| Chitosan | Chitosan/fucoidan | 300 | 30 | Dialysis bag for nanoparticle release | Normal state | Increased fucoidan composition improves gastrointestinal simulated environments | 155 | |
| Chitosan | Tripolyphosphate/chitosan | 700 | 33.06 | Kunming mice | E. coli challenged (cow mastitis) | Chitosan encapsulated OmpA protected from acidic pH | 169 | |
| Surface engineering: pH responsive polymer | Eudragit | PLGA | 397.1 | — | Gastrointestinal pH simulated conditions | Normal state | Eudragit NP protects the contents from gastric pH | 170 |
With considerations to nanosystem design, it is imperative to consider size, shape, hydrophilicity, surface modifications and polymer type in order to optimize mucus permeability and enhance bioavailability of loaded cargos. Studies mimicking the mucus permeability of viruses have modified the surface of the nanomaterials to have hydrophilic properties to reduce mucus adhesion and maintain a neutral surface charge. As research has mainly focused on one property of the nanomaterial for optimization, the best formulation for circumventing the issues that arise with delivery to the mucus GI barrier involves utilizing size, shape, and surface properties as mentioned above. In silico simulations have been performed in order to optimize the shape for the drug delivery system. With these considerations in mind, design for mucus penetration can be achieved.
Conversely, lipid nanoparticles (LNPs) have been found to have stable release profiles within a large range of pH values which would allow for the delivery through the GI tract. LNPs loaded with siRNA against the luciferase gene were found to effectively achieve gene silencing across a broad pH range.174 When exposed to pH values ranging from 1 to 8, LNP encapsulation efficiency of the siRNA was insignificantly affected with minimal disturbances to the polydispersity index and zeta potential. In the presence of low levels of mucin, the gene silencing capabilities of LNP siRNA delivery were significantly reduced. Formulating LNPs with increased percentages of PEG was found to improve gene silencing; however, the trade-offs for using high percentages of PEG include smaller diameters and lower encapsulation efficiencies of the siRNA. Incorporation of chitosan for siRNA delivery is also advantageous for mucosal delivery across the gastric mucosal layer and not the colonic mucus for selective delivery in the treatment of CDX2 gastric lesions;175 this selectivity resulted from the lowered mucosal adhesion from a lower charge density compared to the colonic mucus.
Within the same category, lipid based micelles have also been modified using a zwitterionic betaine polymer in order to permeate the mucus in a similar manner as viruses in a more efficient approach compared to PEG particles of similar magnitude.176 The micelles were manufactured using 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) with poly carboxybetaine (PCB) and were able to transport through cellular tight junctions without opening them and showed increased uptake by Caco-2 cells in order to deliver insulin. This formulation could also achieve a high encapsulation efficiency of 98% and maintain the integrity of the mucus barrier in porcine models.
Apart from lipid and polymer based nanomaterials, the use of metal inorganic systems has also been characterized for GI delivery. Nanoparticles with antimicrobial properties such as those made with titanium oxide have been found to influence the thickness of the mucus layer as well as its composition when exposed to bacteria.177 In particular, titanium oxide particle treatment for one week was found to result in rats developing oxidative damage to the glycolic proteins within the GI tract mucus in animals receiving nanoparticle treatment over fine particles.178 Similarly, studies have shown that silver nanoparticles and their interactions with human derived ileal explant tissues indicated different cytokine responses depending on treatment exposure time, the size of the nanoparticle and even depending on the differences in the sex of the derived tissues;179 tissues derived from human males indicated increased RANTES cytokines compared to those from females which may suggest that males have increased sensitivity to inflammation upon exposure. Within the same study, smaller nanoparticles (10 nm and 20 nm size) could invoke changes in mRNA expression of cell junction genes after nanoparticle treatment. Compared to other inorganic nanoparticle types, cerium oxide nanoparticles manufactured with the sol–gel process have biological modulative effects such as superoxide dismutase and catalase mimicking activity which may suggest a protective effect against ethanol-induced ulcers within the GI tract.180 Due to the oxidative state of cerium oxide, reactive oxygen species are able to be scavenged by the particles to reduce stress in the local mucosa caused by ethanol. As an intrinsic property of the nanomaterial, antioxidant properties can be dictated by the particle makeup.
Comparatively, the magnetic properties of iron oxide nanoparticles in chitosan-alginate core–shell beads effectively localized the loaded drug to the intestine of rats and increased bioavailability.181 By taking advantage of the magnetic properties of the iron oxide nanoparticles and incorporating them into the system formulation, there can be improved localization within the targeted areas of the intestines while surpassing first-pass metabolism. In parallel, these systems effect the charge of the NP formulation rendering them more positively charged and thus improving their adsorption properties. Dextran nanoparticles with iron oxide cores provided improved catalytic activity when exposed to the acidic pH to selectively treat oral biofilms.182
With regard to rigidity, the mechanism for cellular uptake is correlated to the formulation. Plant derived nanoparticle-based formulations have a similar rigidity to lipid-based nanoparticles.185 With PLGA-lipid nanoparticles formulated with varying rigidity, it was found that less rigid particles had lowered penetration into the trans-epithelial layer compared to the stiffer ones; it should be noted that functionalization of the Fc neonatal domain binding peptide on the softer nanoparticles increased their uptake in the liver fibrosis model.186 Physiochemical characteristics of the formulation can be observed as they relate to stability (Fig. 11). Overall, the researchers found that rigidity of the nanoparticles improved mucus penetration over ligand functionalization and/or PEGylation but the most rigid formulation with ligand modification had the best results for transcytosis.
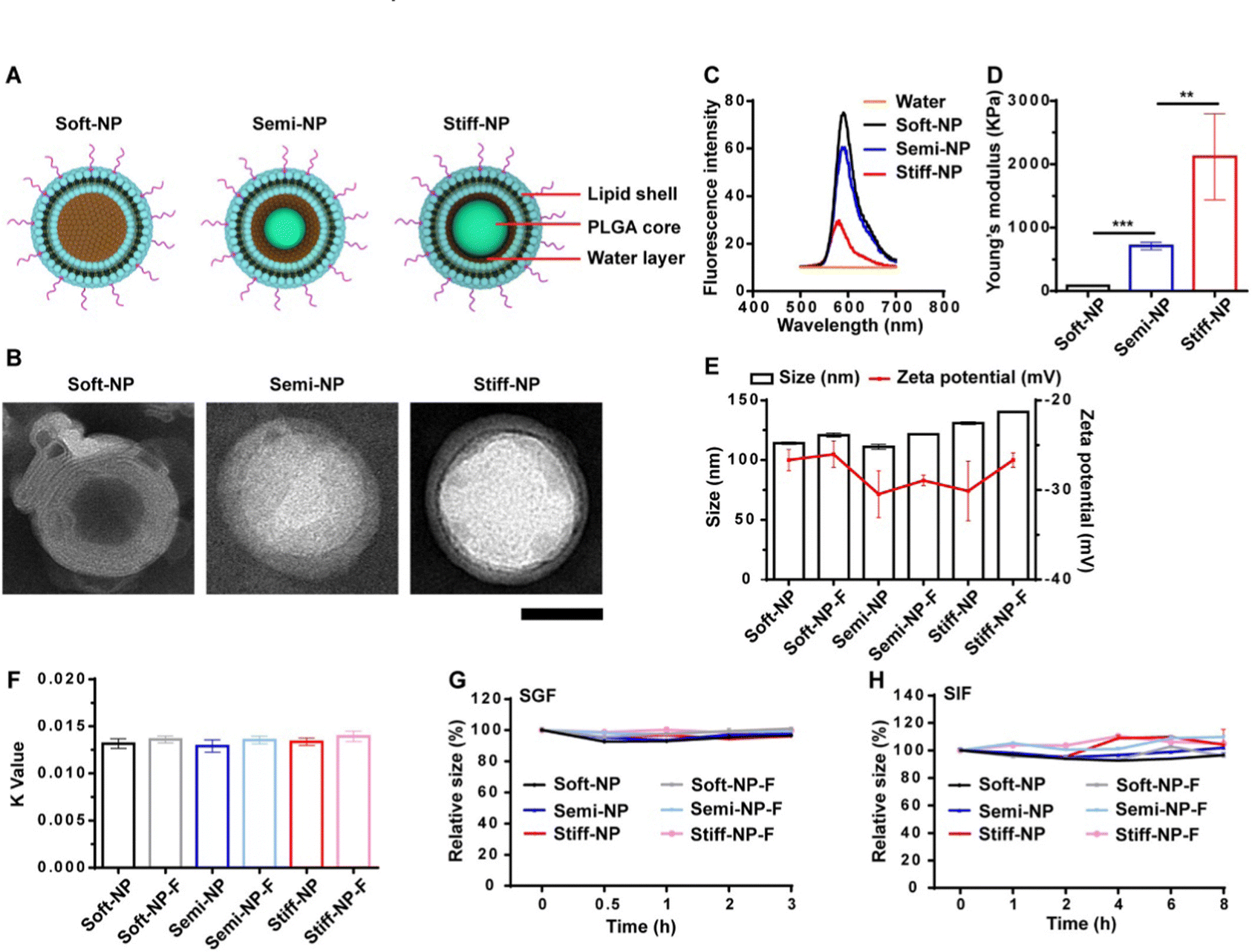 | ||
| Fig. 11 Characterization of the NP. (A) Schematic illustration of the PLGA-lipid NP. (B) TEM images of the NP. Scar bar: 50 nm. (C) The fluorescence emission spectrum of the rhodamine B-loaded PLGA-lipid NP. (D) The Young's modulus of the PLGA-lipid NP. **p < 0.01, ***p < 0.001. (E) Hydrodynamic diameter and zeta potential of the NP. (F) Hydrophobicity was measured by the rose Bengal method. Stability of the NP in simulated gastric fluid (SGF, G) and simulated intestinal fluid (SIF, H). Data represent mean ± SD (n = 3). Adapted from ref. 186 with permission from Elsevier. | ||
The formulations of nanotube systems have also been advantageous due to their modifiable rigidity and shape.193 The researchers utilized tubular peptosomes with various levels of rigidity according to the level of α-lactalbumin incorporated into their surface. As the protein assembled tubes had a net negative surface charge, through multiple particle tracking it was found that compared to their spherical counterparts the shorter assembled nanotubes had an advantage in mucosal penetration due to their ability to navigate the shear flow within the intestines and mucus mesh structure. With the highest bioavailability of loaded curcumin, the short nanotube structures indicated good tolerance with no inflammation observable through microscopy of the tissue samples and had the best therapeutic effect compared to longer and more rigid nanotube formulations.
In the comparison of spherical nanoparticles with short and long sized nanorods, spherical nanoparticles were found to have faster clearance in in vivo studies.194 With regard to in vivo studies, mesoporous silica nanorods were found to accumulate in a uniform manner along the intestinal villi of rats compared to spherical nanoparticles which had more difficulty in reaching epithelial cells.195 Longer nanorods were found to be difficult to eliminate from the mouse GI tract as they achieved a residence time of up to 7 days compared to the other groups with accumulation being observed mostly in the liver and kidney when orally dosed. Additionally with this formulation loaded with camptothecin, mesoporous silica nanorods had up to 2.6-fold higher transportation of the drug compared to the other shaped/control groups. Likewise, the structural rigidity of the nanoparticle system also seems to be instrumental in mucus penetration as well. PLGA nanoparticles that were developed to have a lipid shell in a ‘semi-elastic’ fashion were able to achieve increased bioavailability of delivered doxorubicin to Caco-2 cells. When the particles were made to be rigid, there were steric effects that hindered deformation; however, soft nanoparticles were found to interact with the hydrogel network and inhibit its permeability.196 When visualized using microscopy, the nanoparticles were more ellipsoid in shape. Overall, it seemed that rod-shaped and ellipsoid shaped nanoparticles are able to navigate more easily through the mucus compared to nanoparticles of other shapes. Needle shaped nanosystems driven by magnetic properties for gastric mucus penetration were also found to increase drug penetration of doxorubicin;197 the pepsin bridge structure allows for increased interaction between the nanocapsule needle platform and the mucus itself for improved mucosal adhesion. Utilizing physiochemical properties such as magnetism as well as nanomaterial shape was the key factor in this system's success in improved cellular retention and uptake of the drug.
3.3. NM design strategies and surface engineering
When designing nanomaterials to specifically target the GI tract, it is imperative to address the challenge of pH in the stability of the nanosystem as it travels to its targeted site.198,199 With this examination, the material to coat the outside of the nanomaterial must be considered. Nanoparticles in general are able to be formulated to encapsulate many pharmaceutically active molecules and are often a popular choice due to their good biodistribution. pH-responsive polymers should be able to change their properties according to their chemical environment such as being dissolvable at higher pH values and insoluble at lower ones.200Considerations to circumvent the issue of early dissolution include coating the particles with a pH-responsive polymer such as Eudragit® and its derivatives,201 which allows for nanoparticle transport through the GI tract until a specific pH is reached. Eudragit® FS30D was recently incorporated to form zinc oxide nanohybrids which could withstand a stimulated succus entericus environment and achieve an increased release rate from 9.5% to 74.7% with a pH shift from 1 to 6.8.202 This formulation was also found to remain stable and did not exhibit large physiological changes at 25 °C at 60% humidity for one month. Formulations of Eudragit®-S100 coated PLGA nanoparticles loaded with etoricoxib revealed colon targeting capabilities in healthy human volunteers as a way to treat irritable bowel syndrome.203
Strategies incorporating inspirations from H. pylori infections within the GI have also found success in biomimetic engineering of nanomaterials. Nucleic acid mimics incorporated using a bacterial cell envelope are also a technique to hybridize H. pylori in the treatment of stomach infections allowing for advantageous mucus permeability.204 With this unique method, antibacterial therapy is achievable through the delivery of antisense oligonucleotides in in vivo hybridization. Under this methodology, the cell envelope did not require permeabilization that is generally necessary for cell-membrane based therapeutics. Interestingly, the use of micro propellers to navigate the biological barrier of the GI tract has been implemented to integrate urease to the surface to avoid mucosal degradation.205 As the bacterium also utilizes the urease mechanism to navigate the mucus by altering the mucus viscosity, systems with magnetized properties with the enzyme on their surface can mobilize through their screw-like shape configuration. In addition, artificial zinc-based micromotors have shown effective travel through acidic gastric conditions for drug retention in the GI.206 When compared to simple passive diffusion of the free drug, the hydrogen bubble projection allowed for the micromotors to remain stable in low pH environments and retained on the stomach lining in a time-dependent manner for extended retention and drug delivery.
While optimization of nanomaterial delivery platforms is generally done in vitro, there are advantages that come with computational simulations; molecular docking technologies are able to provide more insight into the protein–drug interactions for delivery such as the potential effects of metabolism from enzymes and protein coronas that may form. Nanovectors designed for dual encapsulation of evodiamine and curcumin drugs for the treatment of gastric mucosal lesions allowed for improved insight into how the drug formulations would interact in the GI environment in the presence of efflux pumps; this technology allowed researchers to estimate the absorption of the drugs in the supramolecular nanocomplex according to the epithelial cellular uptake models.207 Similarly, to investigate the gastroprotective effect in a rat model, in silico response and contour plots have been used to optimize the globule size of self-nano emulsion particles, where the predicted sizes were similar to the actual value measured (67.7 nm calculated versus 64.8 nm actual).208 Adding automation for the high throughput screening of nanoparticle formulations is necessary to improve optimization speed and predict biological interactions such as solubility and potential penetration capabilities.209,210
In parallel studies, mesoporous silica nano-particles have been modified with poly(lactic acid)-methoxy poly(ethylene glycol) polymers and cysteine modified protamine. While the use of cyteine modified protamine did increase cellular uptake, PEG remained the main factor that contributed to the overall net neutrality of the nanosystem for avoiding mucus inhibition.218 While the use of cysteine modified protamine did increase cellular uptake, PEG remained the main factor that contributed to the overall net neutrality of the nanosystem for avoiding mucus inhibition. Similar copolymer mixtures of PEG and PLGA linked by a hydrazone bond were able to exhibit increased penetration with high stability in oral insulin delivery;219 with a surface that can switch from hydrophilic to hydrophobic the researchers found increased nanoparticle reach within the jejunum villi of diabetic rats (Fig. 12).
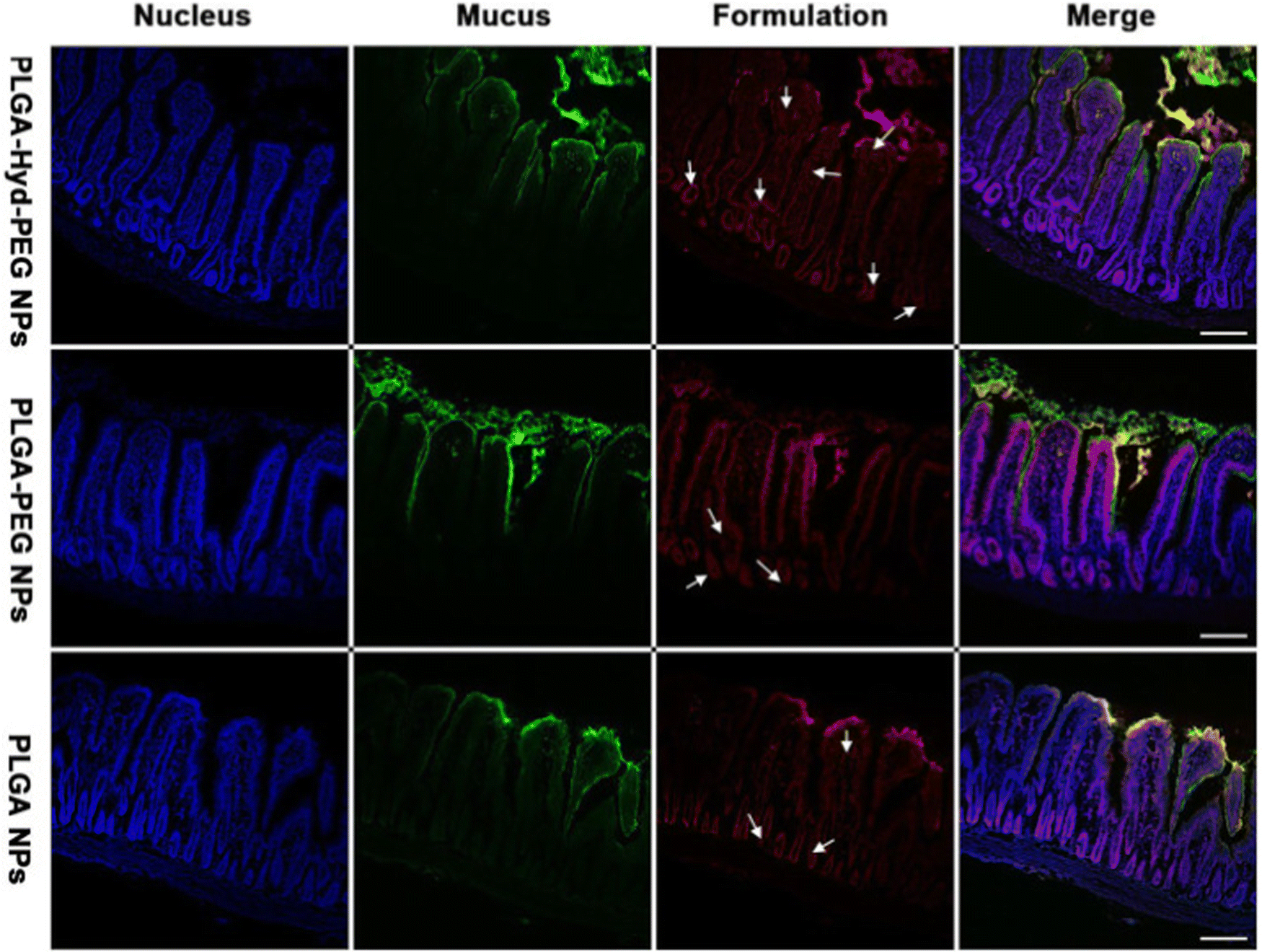 | ||
| Fig. 12 Confocal micrographs of frozen sections of jejunum villi after 1-h incubation with various formulations. The white arrows indicate representative NPs absorbed into the villi. Scale bars, 100 μm. Adapted from ref. 219 with permission from Elsevier. | ||
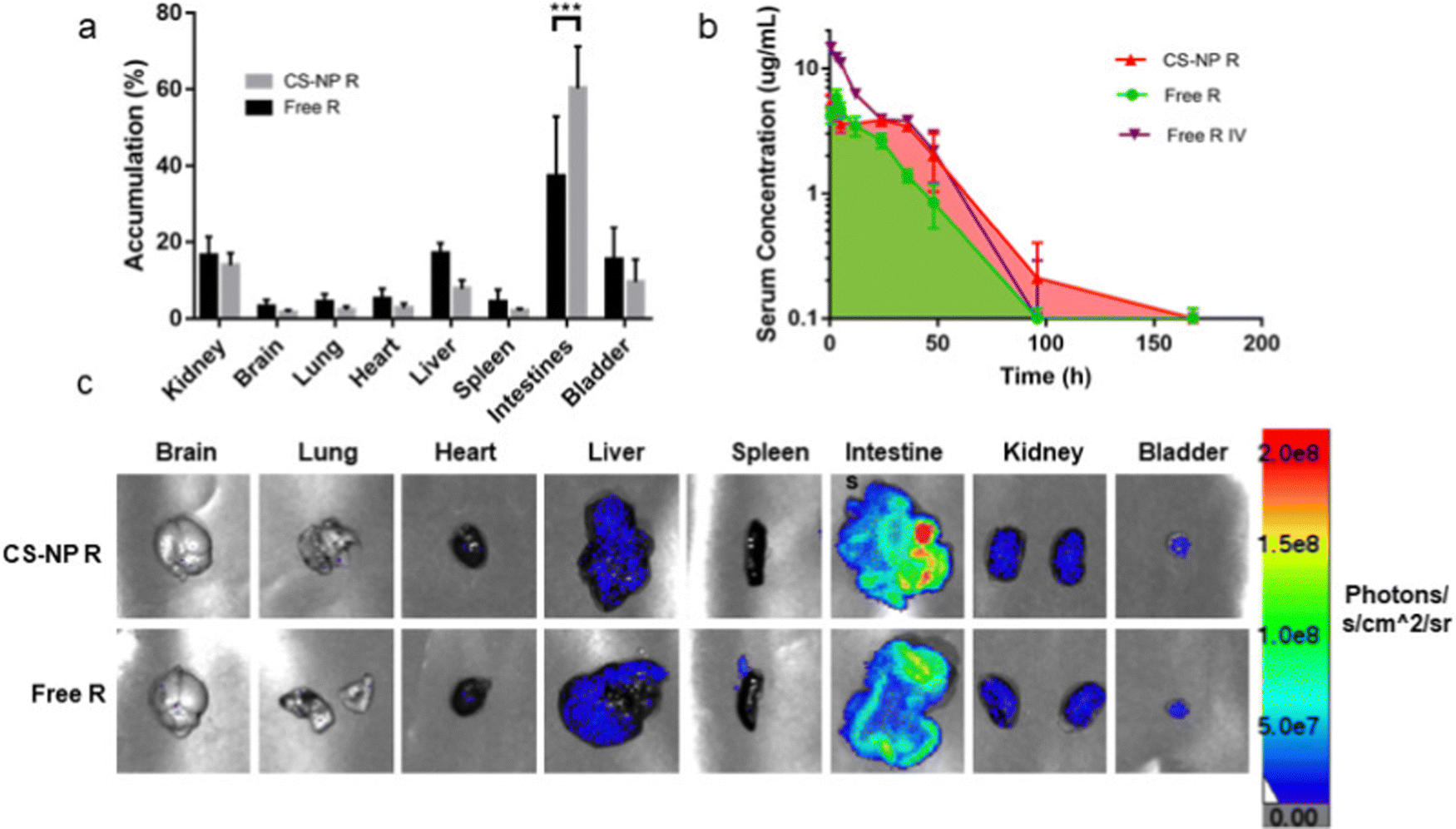 | ||
| Fig. 13 Semi-quantitative biodistribution of mice treated with 10 mg kg−1 rhodamine in 200 μL of CS-NP R and free R 24 h after oral gavage. (a) Comparison of ex vivo imaging between rhodamine fluorescence levels showed higher accumulation in the intestines for CS-NP R vs. free R 24 h post-oral gavage. (b) Serum fluorescence of CS-NP R shows a greater absolute bioavailability and an extended release profile for the CS-NP formulation for up to 7 days (76.2% for CS-NP R and 47.9% for free R; ***p ≤ 0.001, N ≥ 4). (c) Representative ex vivo images confirm the highest signal in the intestines in the CS-NP R condition 24 h after oral gavage. Adapted from ref. 223 with permission from Elsevier. | ||
In a different study, researchers attempting to circumvent the acidic environment that is present in the stomach have modified chitosan-alginate core–shell beads with magnetic nanoparticles and mucoadhesive properties.225 The beads were able to travel and accumulate in the targeted area through the center of a magnet positioned on the skin. The results delineated that the chitosan coating of the beads improved adhesion to the mucosal membrane layer of the rat jejunum and facilitated drug release.
Micelles formed from chitosan–vitamin E succinate copolymers have been previously reported as a self-assembling nanocomplex capable of encapsulation of the hydrophobic drug paclitaxel for intestinal penetration.230 Bioconjugation of N-acetyl-l-cysteine improved mucosal adhesion 2-fold compared to the unconjugated form for improved pharmacokinetic properties. Similar to the nanoparticle formulation, the surface charge, hydrophilicity and physicochemical characteristics of micelles will affect its stability in the mucus environment and affect its mucus penetrating capabilities.231
3.4. GI mucus delivery
With these points in mind, the properties of the nanosystem must consider the delivery method such as mucoadhesion or mucus permeability; mucus adhesive nanosystems are optimal for long term drug delivery due to their ability to bypass mucosal clearance when adhered to the mucus while mucus permeability has greater potential for increased protection of sensitive cargos such as proteins due to direct interactions with the epithelium.211,232 While mucoadhesive nanomaterials are more commonly found within the literature, mucus penetrating systems are also increasingly developed in recent years. As mucoadhesion relies on the exploitation of electrostatic interactions, there exist various molecular interactions that can be artificially engineered for improved delivery system efficiencies such as hydrophobic forces, van der Waals interactions and even non-specific recognition.233 In parallel, it is imperative to address the properties associated with mucoadhesion within the GI tract with reference to nanomaterials and their efficiency in uptake. Mucins are negatively charged due to their glycosidic moieties and, therefore, positively charged nanomaterials are mucosally adhesive and may prompt swift elimination by the dynamic top layer of the mucus.234 Mucosally adhesive particles were found to generally accumulate outside the range of the epithelium for absorption whereas non-mucoadhesive mucus particles were able to have a more uniform distribution.213 It is indicated that for best particle distribution within the GI tract one should formulate their nanosystems according to non-mucoadhesive properties.3.5. Translatability
Issues also persist with translatability between animal models to study biodistribution of various delivered nanomaterials: induction of inflammation in nonmammalian and mammalian models based on techniques used as well as the type of inflammation whether long or short term.237 Mice are commonly used as their intestinal system is similar to humans; however, they can be engineered to display acute or chronic inflammation which may not always fit the origins of the disease state. Similarly, lesions present in humans presenting irritable bowel syndrome differ from those of mice that have been induced to have lesions through chemical irritation/injury. Even structurally in pig models, there is a high concentration of B-cell follicles within Peyer's patches in the intestinal jejunum and a lack of T-cells, which differs from humans.238 This is despite the fact that pigs and their immune system show more similarities to humans compared to mice models of inflammation.
Mice subject to dextran sulfate sodium (DSS) in order to induce inflammatory colitis must be administered daily in order to maintain the studied disease state which is unlike the progressive conditions experienced by humans;239 considerations with the model also indicate variability in inflammatory severity according to gender and the living environment. In order to effectively translate biodistribution and inflammation studies to human models, the use of animal models must also be considered with varying degrees of mucus producing capabilities as well as inflammation marker variance. When examining these issues through in vitro models like the Caco-2 cell line capable of producing mucus, researchers often get variability between monolayers of different passages and clone variabilities.240 These issues in combination make studies with human inflammation more difficult in terms of reproducibility and consistency in designing drug delivery systems according to GI mucus characterization.
When determining the formulation for nanomaterial systems, specific cell populations can be considered for more targeted approaches beyond mucus-adhesion and mucus-penetration. In general, these techniques focus on additional surface modifications to target cell receptors within the targeted area.185 For example, targeting of M-cells within the intestines can be achieved through dapsone and mannosylated solid lipid nanoparticles to target M-cell mannose receptors.243 Similarly for Peyer's patch M-cell targeting, rifampicin lipid–polymer hybrid nanoparticles had increased adhesion when hydrophobicity was prioritized in the formulation; in vivo studies demonstrated increased uptake with Gantrez incorporated nanoparticles for gut-associated lymphoid tissues as M-cells have a thinner layer of mucus and may be easier to deliver.244
Immune cell targeting for gut immunity has also found success; in the cases of ulcerative colitis/IBS treatment, galactosylated trimethyl chitosan–cysteine nanoparticles could improve uptake in macrophages for TNF-α production within the colon.245 Leukocytes localized within the gut were also targeted for siRNA delivery owing to the decorated lipid nanoparticles specific to the α4β7 integrin through a PEG linked mucosal vascular addressin cell-adhesion molecule-1 to its surface.246
The necessity of surface modification of nanomaterials is advantageous for navigating the changing pH environment within the GI tract but there is an increased chance of off-target effects that challenge effective targeting.250 As the complexity of the nanosystem increases, the chance for variability also increases due to the intricate biological processes occurring within the GI. Simplicity in formulation and manufacturing allows for the greatest translatability across different organisms from murine cellular studies to human clinical trials.
4. Blood–brain barrier
The blood–brain barrier (BBB) is a critical component of the neurovascular unit, and its characteristics (Box 3) serve important physiological roles, which include (1) the protection of the brain from toxic substances, (2) the modulation of nutrients, (3) the response to different plasma proteins, (4) and the protection of the central nervous system.251–253 This strong protective barrier function makes the BBB a serious impediment for effective treatment of brain diseases, such as brain tumors or neurodegenerative disorders. Strategies that have been employed for brain drug delivery include among others local drug delivery with convection-enhanced delivery, or drug-loaded polymeric wafer implants.254,255 However, due to their invasiveness, these methods exhibit a high chance of infection. Other methods include BBB disruption with osmotic agents or ultrasound, but these methods can potentially lead to neurotoxicity.256Systemic administration of nanoparticles, engineered to feature BBB crossing characteristics, is an innovative way for brain targeted drug delivery.257 Extracellular vesicles, biomimetic NPs and cell-based drug delivery systems are yet other promising strategies employed for crossing the BBB. Therefore, in this section, we elaborate on the characteristics of NPs that influence BBB crossing and the strategies that are employed to penetrate the BBB (Fig. 15). The details of the most recent strategies (published in the last 5 years) are presented in Table 4.
Box 3: BBB characteristicsThe BBB separates the vascular space from the brain parenchyma, which consists of brain cells and extracellular (interstitial) space.251 It is formed by endothelial cells that line the brain microvessels, impeding the paracellular route of large or hydrophilic molecules across the BBB, due to tight junctions (TJ) between the adjacent endothelial cells.258,259 This barrier is supported by a closely associated basement membrane consisting of type IV collagen, fibronectin and laminin. Additionally, pericyte and astrocyte endfeet are located in the abluminal site and regulate and maintain BBB integrity.260,261 On the luminal side there is a glycocalyx layer covering the brain endothelial cells, which hinders the passage of anionic macromolecules and substances with sizes over 70 kDa. Molecules can cross the BBB through the paracellular or transcellular pathway.259,261–264 Paracellular transport, which is the passage between endothelial cells, is highly restricted to small molecules less than 180 Da and is concentration gradient dependent.263 Larger substances, including NPs, use the transcellular pathway to cross the BBB, travelling through endothelial cells. The transcellular mechanisms include (a) the passive diffusion of lipophilic molecules, (b) receptor-mediated transcytosis, which is used for hydrophilic molecules, (c) carrier-mediated transport, (d) adsorptive-mediated transcytosis and (e) cell-mediated transcytosis (Fig. 14). |
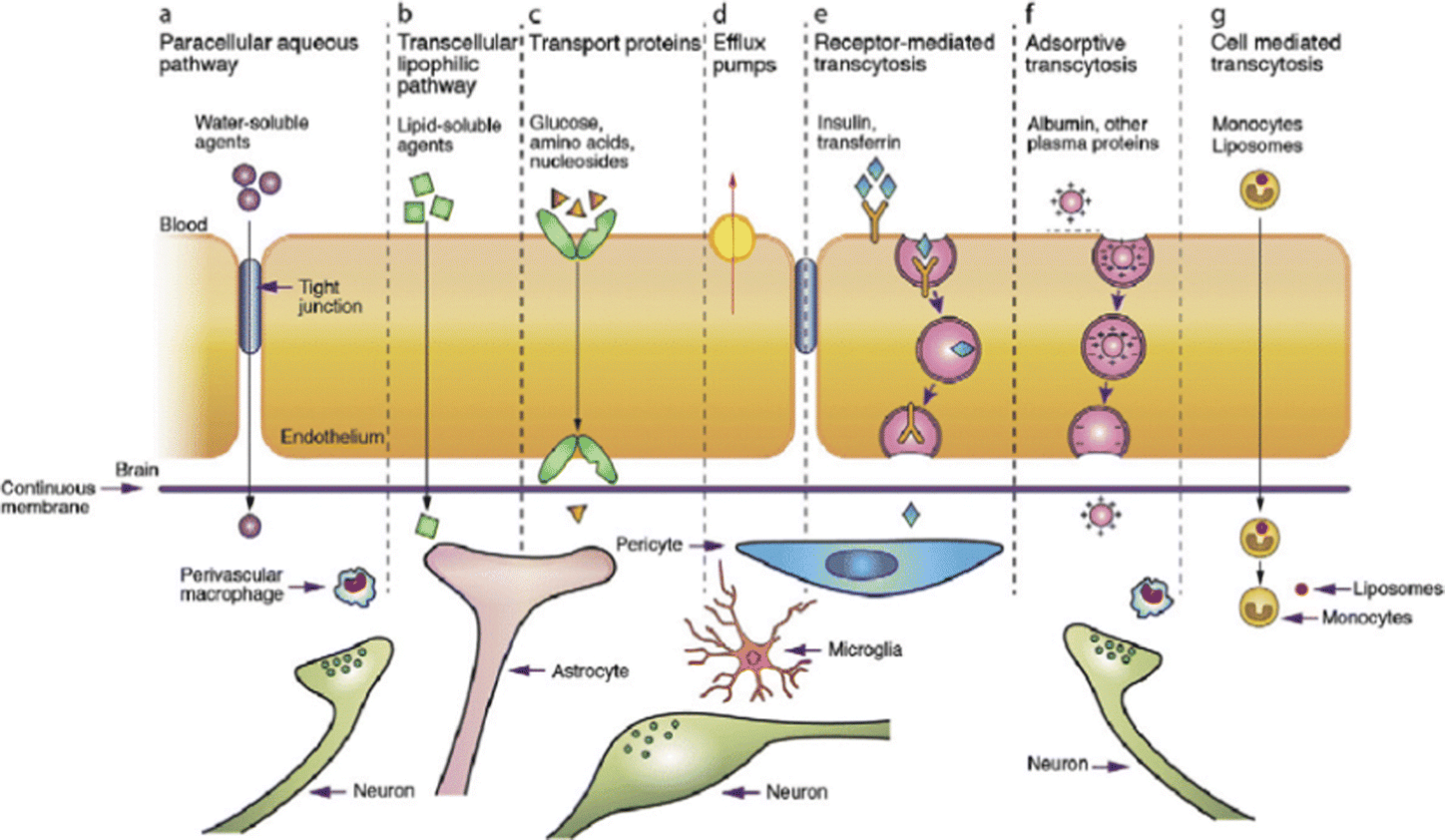 | ||
| Fig. 14 Transport pathways of substances across the BBB. Reproduced from Ref.1 with permission from Elsevier. | ||
| NM engineering strategy | Surface modification | Core material | Size (nm) | Zeta potential (mV) | Model system | Diseased/normal state | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Targeting ligand to low density lipoprotein receptors | Angiopep-2 | Ceria | 20 | +5 | In vitro (BCECs) and in vivo (rat) | Diseased (stroke) | 265 | |
| PMLA | 4.45 | −11.6 | In vivo (mouse) | Normal | 266 | |||
| PMLA | 9.4 | −8.2 | In vivo (mouse) | Diseased (glioblastoma) | Contrast-enhanced MRI agent | 267 | ||
| Solid lipid | 111.4 | −16.4 | In vivo (mouse) | Diseased (glioblastoma) | 268 | |||
| PEG-PLL | 18.8 | +2 | In vitro (BCECs) and in vivo (mouse) | Diseased (brain metastasis of breast cancer) | 269 | |||
| L-carnosine | PLGA | 50 | −7.2 | In vitro (BCECs) | Diseased/normal | 270 | ||
| Tween 80 | Lipid | 165 | / | In vivo (zebrafish) | Normal | 271 | ||
| Lactoferrin | Lipid | 170 | −15.9 | In vivo (mouse) | Normal | + Lactoferrin receptor binding | 272 | |
| Silica | 30 | −32.3 | In vitro (HBEC-5i) and 3D U87 tumor spheroids | Diseased/normal | 273 | |||
| Gold | 5 | / | In vivo (mouse) | Diseased (glioblastoma) | *Oral administration | 274 | ||
| Gold | 5 | −26.8 | In vitro (BMVECs) and in vivo (mouse) | Diseased (glioblastoma) | *Oral/i.v. administration | 275 | ||
| Apolipoprotein E peptide | Polymersome | 42–45 | +1.06 to +2.02 | In vivo (mouse) | Diseased (glioma) | *Intranasal administration | 276 | |
| Chimeric polymersome | 80–86 | +1.6 | In vivo (mouse) | Diseased (glioblastoma) | 277 | |||
| PBCA | 196.4 | −8.8 | In vitro (hCMEC/D3) and in vivo (mouse) | Diseased (cryptococcal meningitis) | 278 | |||
| Polymersome | 44 | −5.2 | In vivo (mouse) | Diseased (glioma) | *Intranasal/i.v. administration | 279 | ||
| Polysorbate 80 | PLGA | 220 | −32.9 | In vivo (rat) | Diseased (seizures)/normal | 280 | ||
| Phosphatic acid | HDL | 26.4 | −7.2 | In vitro (hCMEC/D3) and in vivo (mouse) | Diseased (Alzheimer's disease) | 281 | ||
| Targeting ligand to transferrin receptors | Anti-TfR antibody | PMLA | 28.0–28.5 | −9.9 to -11.0 | In vivo (mouse) | Diseased (glioblastoma) | 282 | |
| H-ferritin | Iron oxide | 6.6 | / | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma)/normal | 283 | ||
| Transferrin targeted peptide | Chitosan-c-PGA | 132 | −38 | In vivo (mouse) | Diseased (cerebral infarction) | 284 | ||
| Disease targeting ligand | RAP peptide decoration (ligand of RAGE) | PCL-PEG | 86.9 | −38.3 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (Alzheimer's disease) | In AD lesion sites, the receptor of advanced glycation end products (RAGE) is specifically and highly expressed on the diseased neurovascular unit, including cerebral vascular endothelia l cells, astrocytes and neurons | 285 |
| Diphtheria toxin receptor (DTR) ligand | Poly(ethylene oxide-co-propylene oxide) | 129–240 | −25.6 | In vitro (hCMEC/D3) | Diseased (glioblastoma) | DTR is up-regulated on the cerebral blood vessels in gliomas, ischemic stroke, and under hypoxic conditions | 286 | |
| Neuropilin-1 targeted peptide | Poly(levodopamine) | 313 | −37.9 | In vivo (mouse) | Diseased (Glioma) | Neuropilin-1 expressed on neo-vasculature/glioma cells | 287 | |
| Claudin-1 targeted peptide | PEG-Gd | 14.5 | +7.8 | In vivo (mouse) | Diseased (aging-induced altered brain) | Claudin-1 is upregulated in aging brain vasculature | 288 | |
| cRGD | PEG-b-poly-(L-glutamic acid) | 29 | −2.3 | In vivo (mouse) | Diseased (glioblastoma) | Affinity of cRGD to Rvβ3 and Rvβ5 integrins that are overexpressed on endothelial cells of tumor angiogenic vessels as well as GBM cells | 289 | |
| Tryptamine (Try) | Try- Cinnamaldehyde nanoprodrug | 200 | −0.2 | In vivo (mouse) | Diseased (glioblastoma) | Try is more easily internalized by gliomas cells via the overexpressed 5-HT receptors | 290 | |
| CXCR4 targeted anatagonist | PEG-PCL | 100 | / | In vivo (mouse) | Diseased (ischemia) | CXCR4 overexpession in the ischemic brain | 291 | |
| VCAM-1 targeted peptide | lanthanide | 80 | −4 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (ischemia) | VCAM-1 is overexpressed in the cerebral ischemia area | 292 | |
| IL13 | PEG-PLGA | 92 | −30.5 | In vitro (human brain microvascular endothelial cells with human astrocytes and human brain vascular pericytes) and in vivo (mouse and rat) | Diseased (glioma) | IL13 binds to the tumor-specific interleukin-13 alpha 2 receptor | 293 | |
| iRGD | Solid lipid | 300 | −8 | In vitro (hBMEC or mBMEC) and in vivo (mouse) | Diseased (glioblastoma) | iRGD: tumor-targeting transcytotic peptide | 294 | |
| Zinc phthalocyanine | 122 | −31.6 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 295 | |||
| p-Hydroxybenzoic acid (p-HA) | Liposome | 123 | 0.14 | In vivo (mouse) | Diseased (glioma) | p-HA has high affinity for dopamine and sigma receptors which are prominent in the BBB. And overexpressed in brain tumors | 296 | |
| Targeting ligand: Glucose transporter (GLUT-1) | Cholesterol-undecanoate-glucose conjugate | Liposome | 96.2 | / | In vivo (mouse) | Diseased (malaria) | Better brain targeting via i.n. administration | 297 |
| Glycose | PEG-PLL | 45 | / | In vivo (mouse) | Diseased (glycemic conditions, Alzheimer's) | 298 | ||
| Targeting ligand: large neutral amino acid transporter (LAT)-1 | L-DOPA | Gold | 90 | / | In vitro (hCMEC/D3 and primary rat brain microvascular endothelial cells) | Normal | 299 | |
| Targeting ligand: acetylcholine and choline tranbsporters | PMPC | PAMAM dendrimer | 8 | 1.48 | In vivo (mouse) | Diseased (glioblastoma) | 300 | |
| Choline analogue (MPC) | PEGMA | 74 | 0.12 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioma) | 301 | ||
| BBB targeting antibody | RG3 single-domain antibody | Liposome | 110 | / | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioma)/normal | 302 | |
| JAM-A targeting antibody | Gold | 70 | / | In vivo (mouse) | Normal | +Increased BBB permeability via laser pulse excitation | 303 | |
| Targeting ligand: folate receptor | Folic acid | Carbon | 359.24 | −24.7 | In vivo (mouse) | Diseased (glioblastoma) | 304 | |
| Targeting ligand: NGF receptor | NGF | MgO micelle | 120 | −10 | In vivo (mouse) | Diseased (Parkinson's) | 305 | |
| Targeting ligand: BBB targeting peptide | TGN | Glycolipid nanomicelle | 146.2 | +28.2 | In vivo (rat) | Normal | 306 | |
| BBB crossing ligand | LysoGM1 | PLGA | 246.8 | −33.2 | In vivo (zebrafish and mouse) | Diseased (Glioma)/Normal | 307 | |
| Extracellular vesicles (EVs) | MSC-derived exosomes | Gold | 129.2 | −39.9 | In vivo (mouse) | Diseased (stroke) | 308 | |
| Grapefruit EVs | Heparin-based | 192 | / | In vitro (hCMEC/D3) and in vivo (mouse) | Diseased (glioma) | 309 | ||
| / | Neuron-2a cell derived exosomes | 114 | In vivo (mouse) | Diseased (Alzheimer's) | 310 | |||
| MSC-derived exosomes | Gold | 100 | / | In vivo (mouse) | Diseased (stroke, Parkinson's, Alzheimer's, autism) | 311 | ||
| / | EVs from hiPSC-derived NSCs | 145.3 | / | In vivo (mouse and rat) | Diseased (status epilepticus) | i.n. administration | 312 | |
| RGD | EVs from ReN VM cells | 100–250 | / | In vivo (mouse) | Diseased (glioblastoma) | 313 | ||
| RAW264.7 exosome membrane decorated with cRGD | DSPE-PEG-PEI | 152 | −20 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (diffuse intrinsic pontine glioma) | 314 | ||
| CXCR4 | NSCs derived EVs decorated with CXCR4 | 84.3 | −24.4 | In vivo (mouse) | Diseased (glioblastoma) | CXCR4 is a chemokine receptor specific for CXCL12 that is overexpressed in cancer cells, including GBM | 315 | |
| Combination of targeting ligands | CGN BBB targeting ligand + Tet-1 neuronal targeting ligand | PEG-PDMAEM | 70–80 | +10 | In vivo (mouse) | Diseased (Alzheimer's) | 316 | |
| Folic acid + Transferrin + Polysorbate 80 | PLGA | 109.7 | −9.38 | In vivo (rat) | Diseased (Glioma) | 317 | ||
| Lactoferrin + RGD dimer | Fe3O4/Gd2O3 | 14.7 | / | In vitro (HBEC-5i) and in vivo (mouse) | Diseased (glioblastoma)/normal | 318 | ||
| Cell mediated delivery | Bone marrow derived macrophages | Iron oxide | 190 | / | In vitro (only transwell inserts) and in vivo (rat) | Diseased (glioma) | 319 | |
| Bacteria (Escherichia coli 25922) | GP-ICG-SiNPs | 4.1 | / | In vitro (human brain microvascular endothelial cells) and in vivo (mouse) | Diseased (glioblastoma) | 320 | ||
| (MOG35–55) T-cells | PLGA-PEG iron oxide | 44.1 | −40 | In vivo (mouse) | Diseased (autoimmune encephalomyelitis)/normal | 321 | ||
| Cell membrane coated | Apolipoprotein E peptide-decorated red blood cell membrane | dextran | 180 | −5 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 322 | |
| Angiopep-2-decorated red blood cell membrane | PEI | 150–500 | −30 × changes with pH | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 323 | ||
| Macrophage membrane expressing PD-1 | PLGA | 124.1 | −18.9 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 324 | ||
| / | Hybrid membrane nanocomposites using DC and C6 glioma cell membranes | 154 | −20 to −33 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioma) | 325 | ||
| Natural killer cell membrane | AIEdots | 80 | −33.6 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 326 | ||
| Macrophage membrane | MnO2@PVCL | 270 | −8.2 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | 327 | ||
| Magnetic targeting | BaTiO3 (BTO) @CoFe2O4 | 20–30 | +30 | In vitro (primary human brain microvascular endothelial cells with human astrocytes and human brain vascular pericytes) | Diseased (HIV) | 328 | ||
| PLGA-MgO/SPIONs | 68.4 | −33.2 | In vivo (rat) | Diseased (glioma) | 329 | |||
| Anti-TfR antibody | Lipid-SPIONs | 101.3 | / | In vitro (C8D1A with bEnd.3) | Diseased (glioblastoma) | 330 | ||
| FUS + micro/nano bubbles | Fluorinated surfactant | Perfluorooctyl bromide (PFOB) | 60 | / | In vivo (mouse) | Normal | 331 | |
| Sonosensitive liposome + microbubbles | 123.9 | / | In vivo (mouse) | Diseased (glioblastoma)/normal | 332 | |||
| PVG peptide | PEG-liposomes + nanobubbles | 115.4 | +13.6 | In vivo (mouse) | Normal | 333 | ||
| Others: virus mimicking NPs | Viral envelope (E) protein | Pbs QDs wrapped in TPE-BPA@8CTAB | 50 | +25 | In vivo (mouse) | Diseased (inflammation) | 334 | |
| Others: surface functionalization with cell penetrating peptide | CB5005 peptide | liposome | 110 | −5 | In vivo (mouse) | Diseased (glioblastoma) | 335 | |
| Others: BBB targeting core material | / | Albumin | 150.4 | −35 | In vitro (bEnd.3) and in vivo (mouse) | Diseased (glioblastoma) | Albumin binding proteins (SPARC) present in glioma | 336 |
| Albumin/PLGA | 353–497 | −37 | In vivo (mouse) | Diseased (Parkinson's) | Albumin has the ability to permeate the BBB via receptor-mediated pathways | 337 |
4.1. NM characteristics influencing interaction with the BBB
As the BBB is a strong barrier, BBB crossing requires effective strategies to deliver therapy for brain diseases. Multiple NP formulations and designs have been researched over the years, and a general overview of these is given in Fig. 15. Here, we report an overview of different nanoparticle properties such as size, surface charge, surface chemistry and BBB targeting strategies that showed effective transport through an in vitro or in vivo BBB model. Please note that while BBB transport has been widely studied using both organic and inorganic NPs, the majority of studies focused on organic NPs (Fig. 15A). Given the large variety of NP types, it is furthermore rather difficult to properly evaluate the influence of the NP core material on BBB crossing ability. We therefore do not address the topic of NP composition directly, but rather indirectly, where specific aspects such as magnetism or NP rigidity will be discussed in view of the specific NP types. | ||
| Fig. 15 Overview of NP characteristics and BBB targeting strategies used for brain delivery. Characteristics of interest are (A) core material, (B) size, (C) surface charge, (D) BBB targeting strategies and (E) surface chemistry strategy. Graphs are based on data extracted from the PubMed database of the last 10 years and the search terms ‘nano’ and ‘blood brain barrier’, identifying 287 research articles from which 141 articles were selected and used for data insights, with the criteria that show evidence of BBB crossing in vivo (animal model) or in vitro (Boyden chamber). Only original research articles and articles within the scope were included. | ||
Furthermore, there is evidence that size is not a determining factor regarding BBB crossing, if the particles are in an acceptable size range.263,264 To shed light on the effect of NP physicochemical characteristics on BBB passage, Voigt et al. evaluated the influence of size, zeta potential and surfactant composition on BBB transport.264 For this purpose, they designed fluorescently labeled polybutylcyanoacrylate (PBCA) NPs with a size range from 64 nm to 464 nm and imaged the NP transport through the retina barrier, a model for the BBB barrier, using in vivo confocal neuroimaging. Their results indicated that NP size does not influence BBB transport, but the most important parameter proved to be surface chemistry. However, it should be noted that for comparing the effects of NPs of different sizes, it is not easy to decide how the equivalent injected NP dose will be determined (total mass, NPs mL−1, total surface area) and specifically for fluorescence imaging, bigger NPs will be brighter and hence more easily detected, which may result in biased results.
Additionally, the employed BBB model should be taken into consideration when NP size is investigated. A lot of research on the influence of NP size on BBB permeation has been performed using in vitro BBB models, which cannot fully recapitulate the in vivo biodistribution clearance mechanisms. For example, in the study of Ohta et al., the size-dependence of Au NP brain delivery, assisted by FUS-induced BBB opening, was investigated.340 They compared the permeability efficiency of 3 nm, 15 nm and 120 nm Au NPs across an in vitro and in vivo BBB mouse model. Interestingly, it was shown that the permeability of smaller NPs was higher in vitro, while the highest permeability in vivo was with the medium sized NPs.
This size-dependence was attributed to competition for permeation through gaps in the BBB and excretion of particles from blood circulation. Of note, the authors determined the equivalent dose of the different NP sized formulation based on the total mass of gold. This means that in their setups, the concentration of NPs mL−1 or total surface area for a particular NP size would vary. This is important to take into consideration, in particular upon considering active targeting strategies where at equal ligand densities, the NPs with the highest total surface area will have a higher absolute number of targeting ligands, which may affect receptor binding efficacy and associated transcytosis across the BBB.
The work of Lockman et al. demonstrated that high concentrations of negatively charged NPs and both high and low concentration of positively charged NPs affected BBB integrity, while neutral NPs had no acute effect.345 In addition, the researchers showed that neutral NPs and low concentrations of anionic NPs can be delivered safely across the BBB. Therefore, these results indicate that not only the cationic NPs have the potential to cross the BBB, but the neutral and negatively charged NPs do so too.
On the other hand, there are studies showing that spherical NPs exhibit higher permeation across the brain endothelium than their rod- or star-shaped counterparts. Liu et al. investigated the permeabilization of TiO2 NPs across an in vitro as well as an in vivo BBB model, and their findings suggest that spherical TiO2 NPs have higher BBB permeabilization efficiency than rod-shaped particles with similar size.349 In addition, Enea et al. showed that spherical Au NPs internalized more efficiently in BBB endothelial model cells than their counterpart star-shaped NPs.350
The discrepancies between the above-mentioned studies regarding the shape effect on BBB permeation can be attributed to variation of other parameters influencing BBB crossing, such as the NP material, BBB model and experimental method used for the validation of BBB permeation. For example, other parameters play a role in vivo, such as the sequestration of NPs by macrophages, on the biodistribution and BBB permeation efficiency of NPs. Several studies have shown that NPs with a higher aspect ratio are phagocytosed less efficiently by macrophages, which should be taken into consideration when engineering NPs for BBB targeting through systemic delivery.351,352
4.2. Targeted delivery strategies
In the last decade, a plethora of research has been conducted on surface functionalization of NPs with moieties that target components present on the BBB or on chemotaxis exploitation of biological vehicles for enhancing NP passage across the BBB. In this section we give an overview of the most studied strategies for targeted delivery of NPs (Fig. 16). In most of the studies, the BBB targeting strategy followed is the surface modification of NPs using a targeting ligand (Fig. 16D). In this section we are discussing the main receptors/transporters on the BBB that have been exploited as targets of ligand-functionalized NPs. | ||
| Fig. 16 Schematic representation of the BBB targeted NP delivery strategies. Created with https://BioRender.com. | ||
4.2.1.1. Low-density lipoprotein receptor (LDLR). LDLR is a transmembrane receptor involved in lipid metabolism and is found in different organs such as the liver, the adrenal cortex, the ovarian corpus luteum and cell structures like neurons and glial cells. The main ligands for LDLR are apolipoproteins in the blood such as apolipoprotein A-I and/or E (ApoE). LDLR has often been used for targeting BBB crossing by NPs.353–361 It has been shown that the use of surfactants, such as polysorbate 80 and poloxamer 188, enables the adsorption of apolipoproteins from the blood on the NP surface.339,340 After the adsorption of apolipoproteins on the NP surface, LDL receptor-mediated transcytosis through the BBB becomes possible. Similarly, in a study by Voigt et al., efficient BBB passage was achieved when PBCA-NPs were fabricated with non-ionic surfactants (Tween80, LutensolAT80, Tween20, Brij35) or cationic stabilizers (such as DEAEdextran), but not when anionic compounds were added (SDS).337 Thus, non-anionic surfactants allow the adsorption of Apo-E from blood, which leads to an efficient BBB passage through the LDL receptor-mediated transcytosis. Additionally, the successful BBB passage of the cationic DEAEdextran NPs suggests that adsorption mediated transcytosis may also play a role. A study by Piazzini et al. further confirms the advantages of surfactant usage for BBB crossing. Sterically stabilized liposomes with Tween 80 alone or in combination with didecyldimethylammonium bromide (DDAB) increased the permeability of andrographolide (AG) across hCMEC/D3 monolayer cells. Their results suggest that caveolae-mediated endocytosis is the main mechanism involved in NP uptake.362 Exploiting the beneficial BBB crossing effect of surfactants, Dal Margo et al. proposed a translational NP targeting strategy by using lipid NPs with polysorbate 80 as a surfactant and incubating with recombinant human ApoE4.363 The ApoE4 decorated NPs showed a 3-fold increased brain accumulation in BALB/c mice, compared to the non-decorated NPs (Fig. 17).
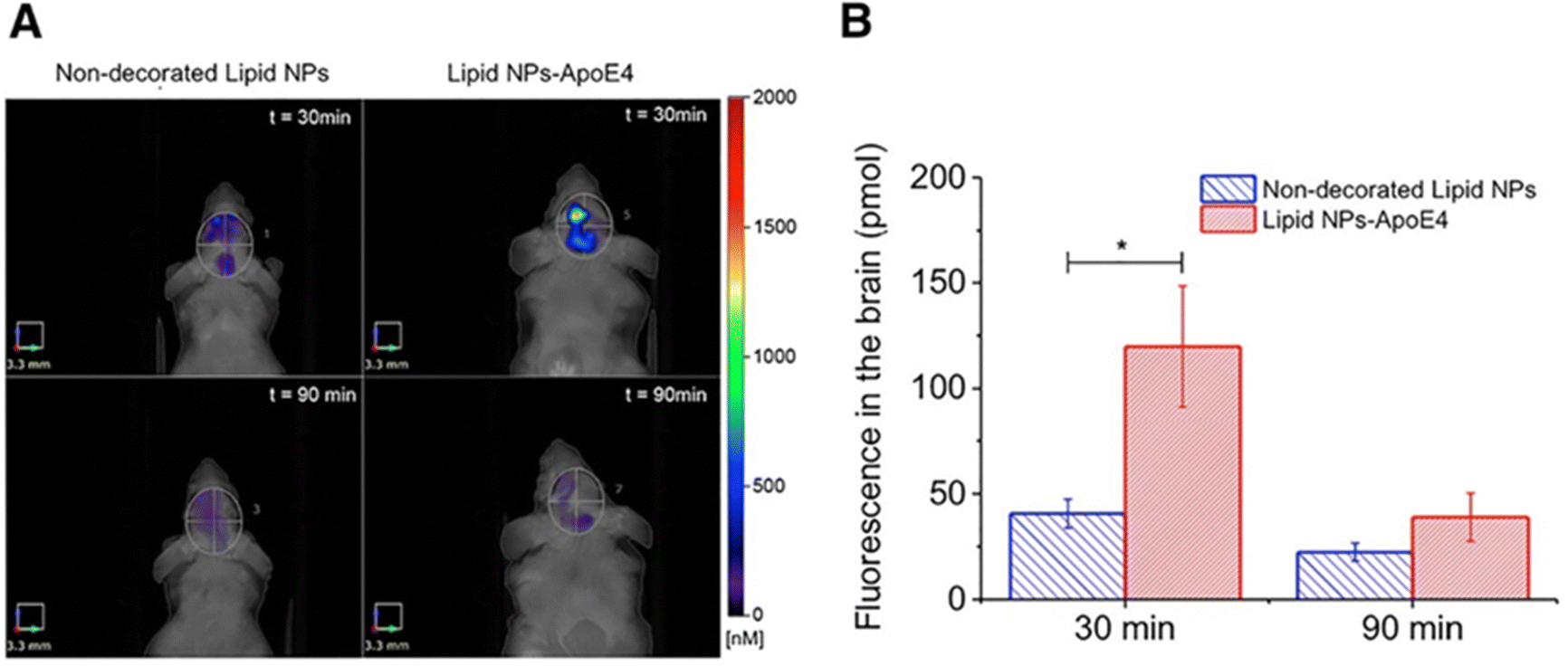 | ||
| Fig. 17 Comparison of fluorescence of non-decorated and ApoE4-decorated lipid NPs in the brain. (A) Mice were treated with 100 μL of non-decorated lipid NPs or lipid NPs-ApoE4 (ApoE4: 5 μg mL−1) by IV injection and each mouse was analysed 30 min (upper panels) and 90 min (lower panels) after the administration. (B) Quantification of fluorescence (pmol) in the selected region of interest. *p < 0.01 by Student's t-test. Reproduced from ref. 363 with permission from Elsevier. | ||
In addition to (indirect) absorption of apolipoproteins on the NP surface, ApoE can be directly coated on the NP surface. For example, Wei et al. used a short ApoE peptide, which is less immunogenic, easy to synthesize and amenable to chemical modifications than the large apolipoprotein E, to decorate polymersomes for glioma immunotherapy.364 They used a combination treatment of ApoE-decorated polymersomes loaded with granzyme B, a serine protease granzyme mostly found in natural killer cells and cytotoxic T-cells, or loaded with immunoadjuvants like CpG oligonucleotide (CpG). This combination treatment induced significantly more immunogenic cell death compared with free GrB and non-decorated polymersomes. Additionally, Neves et al. functionalized solid lipid nanoparticles with ApoE to assess the advantage of functionalization.365 Their results showed an increased transcytosis of ApoE modified NPs through an in vitro BBB model compared to non-modified NPs, which could be attributed to the interaction between the ApoE ligand and overexpressed LDL receptors in the BBB.
4.2.1.2. Transferrin receptor (TfR). The iron-transport protein transferrin (Tf) is a glycoprotein abundant in plasma, which, upon binding to the transferrin receptor, shuttles iron between cells. Iron binding to transferrin serves three main purposes: (i) it maintains Fe3+ in a soluble form under physiologic conditions, (ii) it facilitates iron transport and cellular uptake, and (iii) it maintains Fe3+ in a redox-inert state, preventing the generation of toxic free radicals.366 Two different TfRs have been recognized, TfR1 and TfR2. In the brain, iron deficiency leads to increased translocation of TfR1 to the membrane of microvascular endothelial cells. As TfR is overexpressed at the BBB in diseases such as Alzheimer's disease (AD) and Parkinson's disease (PS) and in brain tumors such as glioblastoma (GBM), TfR-targeted brain drug delivery systems have been extensively explored the recent years.367–372
The advantage of using Tf-decorated NPs to target GBM was exploited by Lam et al., who showed that Tf-decorated liposomes co-loaded with temozolomide and bromodomain inhibitor JQ1 resulted in a significantly prolonged survival of U87MG and GL261 glioma mouse models.373 Similarly, Sun et al. showed that the decoration of PTX loaded PEG-PLA micelles with the TfR-T12 peptide presented a prolonged median survival of nude mice bearing glioma.374
An important parameter that should be taken into consideration for successful translocation of Tf-NPs from the endothelium into the brain parenchyma is the avidity of Tf-decorated NPs for TfR. In their study, Wiley et al. investigated how NP avidity affects their permeation across the BBB into the brain parenchyma.375 As the avidity of NPs for TfR increases with respect to NP size and Tf content, their study showed that NPs with high binding avidity to TfR resulted in an increased association of the NPs with the brain endothelial cells and thus lower accumulation in the brain parenchyma. NPs with less avidity showed an increased accumulation in the brain parenchyma, which could be attributed to the transcytosis pathway. Continuing this work, a solution for boosting the transcytosis of Tf-NPs across the BBB was proposed by synthesizing 80 nm Au NPs with an acid-cleavable linkage between Tf and NP core.376 This system exploits the drop in pH during NP transcytosis, so that after the NPs reach the brain, they are free from TfR and can be released into the brain parenchyma.
A second parameter that should be optimized is the density of the TfR antibody on the surface of NPs. A higher density of the TfR antibody has been proven to be more efficient for transmigration of NPs across the BBB, which indicates that there is a lower density limit for successful targeting. However, a higher density comes with the consequence of off-targeted accumulation in the lungs and spleen.377
4.2.1.3. Insulin receptor. Insulin receptors (IRs) are highly expressed on the BBB endothelial cells and transport insulin to the brain from the blood through receptor-mediated transcytosis.378 These receptors can be exploited for the active targeting of the brain using surface functionalized NPs with anti-insulin receptor antibodies. In their study, Ulbrich et al. surface functionalized human serum albumin (HSA) NPs with insulin or an anti-insulin receptor antibody (29B4), with the goal to transfer the drug loperamide into the brain.379 They tested the antinociceptive effect of the drug through a tail flick test in an ICR (CD-1) mouse model and showed that both insulin and 29B4 surface coated NPs resulted in higher antinociceptive effects compared with the drug alone, while a pre-injection with 29B4 antibody before NP administration inhibited the antinociceptive effects, attributed to saturation of the IRs from the plain antibodies. Furthermore, Kuo et al. developed carmustine (BCNU) loaded SLNs grafted with an 83–14 MAb for targeting BBB IRs, which resulted in an improved permeability across an in vitro BBB model.380 Similarly, the study of Shilo et al. showed a 5 times greater accumulation of Au NPs in the brain of BALB/c mice when NPs are surface functionalized with insulin compared to non-functionalized NPs.381
4.2.1.4. Nicotinic acetylcholine receptor (nAchR). Nicotinic acetylcholine receptors (nAchRs) are receptors for the acetylcholine neurotransmitter and can be found in the peripheral and central nervous system. nAchRs are also expressed on the BBB and BBB permeability can be increased by nicotine treatment.382 With this in mind, Huey et al. conjugated doxorubicin (DOX) loaded PLGA NPs with a 39 amino acid rabies-virus derived peptide (RDP), possessing nAchR binding capability.383 Their study showed doubled cytotoxicity of RDP-DOX NPs in vitro, on SH-SY5Y neural cells, compared to non-decorated DOX-NPs, while nAChR was proven to be essential for RDP uptake. In another study by Hua et al., RVG was used as well, with a 29-amino-acid sequence for an active brain-targeting NP delivery.384 They developed docetaxel (DTX) loaded RVG29-PEG-PLGA NPs as an anti-glioma targeted therapy and investigated NP biodistribution as well as the anti-glioma effect on a rat glioma model. The results showed that the RVG29 decorated NPs reached the brain with a 2.1 higher concentration than the non-decorated NPs, while the group treated with the RVG decorated NPs had the highest medium survival time.
In a later study, Park et al. exploited the RVG brain targeting system for delivery of siRNA for the treatment of Alzheimer's disease.385 siRNA was loaded in poly(mannitol-co-PEI) NP complexes (R-PEG-PMT), showing an increased brain uptake for R-PEG-PMT in vivo, while a significant decrease in the expression level of GAPDH mRNA in the brain was noted. Furthermore, R-PEG-PMT/siBACE1 complexes showed significant downregulation of BACE1 in both cortex and hippocampus, whereas PEI, R-PEG-PEI and PEG-PMT/siBACE1 only showed significant downregulation in the cortex.
Another promising group of peptides with high binding affinity to the nAChR are snake neurotoxins. CDX is a peptide sequence isolated from candoxin, a snake venom, and was investigated by Sepasi et al. for its brain delivery potential.386 CDX-modified chitosan NPs for gene delivery into the brain indeed showed efficient entering of the brain parenchyma. Furthermore, by using DCDX surface decoration, which is resistant to proteolytic degradation, the integrity of NPs can be maintained. This was shown for CDX-decorated nanoliposomes, which were able to not only target the brain, but also reach the brain in an intact form (Fig. 18).
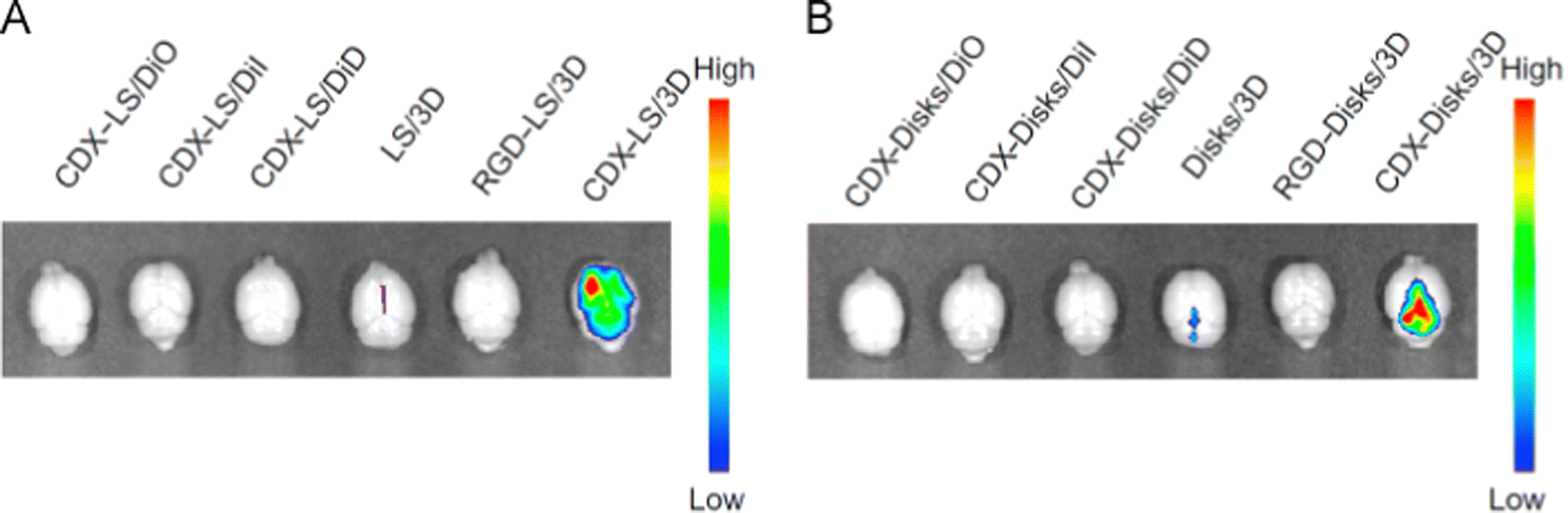 | ||
| Fig. 18 The ex vivo imaging of liposomes (A) and disks (B) in the brains of nude mice bearing an intracranial glioma 15 days post-implantation. The excitation wavelength was 488 nm and the emission filter was 680 nm. C + R, CDX + RGD. Reproduced from ref. 386 (CC BY 4.0). | ||
4.2.2.1. Glucose transporter (GLUT1). The GLUT1 transporter is a sodium-independent transporter which facilitates the transportation of glucose across the BBB and is highly expressed on the microvascular endothelial cells of the BBB, facilitating BBB transport of glucose-modified NPs.388–390 Using this as a BBB target, Anraku et al. developed 30 nm polymeric micelles decorated with multiple glucose molecules with a density of 10%, 25% and 50% glucose.391 The glucose molecules were linked onto the micelles via an ether linkage at the C3 or C6 position of glucose. 25% of glycosylated micelles with the glucose linked at the C6 position accumulated significantly in the brain of mice. However, these results were only obtained for mice that had been fasting for 1 day and after administration of a 20% glucose solution 30 minutes post NP administration, in order to have an elevated blood glucose concentration.
GLUT1 was also exploited by Zhou et al., to deliver BACE1 siRNA polymeric nanocomplexes into the brain, for the treatment of Alzheimer's disease.392D-Galactose modified Cy5-siRNA nanocomplexes were synthesized, which demonstrated a 5.8-fold higher accumulation in the brain of mice and led to restoration of the cognitive performance of AD mice. Again, advantage was taken of GLUT1 recycling by treating the mice after 1 day of fasting and 30 min after 20 wt% injection of glucose solution.
4.2.2.2. Choline transporter (ChT). Similar to GLUT1 exploiting strategies, Wang et al. developed anti-PDL1 polymeric nanocapsules (PEGMA) with a choline analogue, 2-methacryloyloxyethyl phosphorylcholine (MPC), for an effective immunotherapy of glioma. Glioma bearing mice treated with the MPC-anti-PDL1-PEGMA nanocapsules presented a prolonged survival compared to the ones treated with anti-PDL1 alone or anti-PDL1-PEGMA nanocapsules without MPC.393 In another study by Wu et al., MPC was also used for surface decoration of polymeric nanocapsules, successfully delivering protein therapeutics to the brain of mice and non-human primates.394
4.2.2.3. Cell-penetrating peptides (CPPs). Cell-penetrating peptides (CPPs) are a group of naturally derived peptides that have been identified with the capability of efficiently crossing cell membranes. They can be classified into three groups: cationic, amphipathic and hydrophobic CPPs.395 Well-studied CPPs include polyarginine, HIV-1 trans-activator of transcription peptide (TAT), penetratin and mastoparan.396 The BBB crossing capability of CPPs has not been studied in depth yet, but was explored by Kanazawa et al., who developed MPEG-PCL nanocarriers decorated with TAT. Using intranasal delivery, TAT decorated nanocarriers showed increased brain accumulation compared to naked ones.397 However, as CPPs can cross various cell membranes, off-target uptake by other organs occurs as well.396
4.2.2.4. Considerations on the targeted ligand strategy. It is rather difficult to make solid conclusions about the most efficient targeting ligand for brain NP delivery, as the methods that have been used in the different studies to prove NP targeting efficiency are not comparable. There are very few studies that mention the dose of the encapsulated cargo that reached the brain related to the initial administered dose, while a lot of studies prove their point with qualitative methods such as optical imaging, microscopy or other indirect methods, such as intensity of an encapsulated dye or therapeutic outcome. In the majority of the surface engineering strategy though, NPs are functionalized with a ligand that targets LDL receptors, while in a lot of cases ligands that are related to a specific disease are aimed to be targeted (Table 4). It is worth mentioning that it would be extremely valuable to the field that scientists investigating NP targeting would give quantitative results of the dose that reached the targeted tissue related to the initial administered dose.
4.2.3.1. Extracellular vesicles (EVs). Extracellular vesicles (EVs) are small membrane particles produced by cells, loaded with genetic material, lipids and proteins for intercellular communication.398 EVs are divided into two main groups: (i) EVs formed from the cell's plasma membrane with average sizes of 100–1000 nm, known as microvesicles, ectosomes, or microparticles, and (ii) exosomes, which are smaller than 150 nm and are generated inside multivesicular endosomes or multivesicular bodies.399 Given their natural property to deliver messages between cells and cross the BBB, they have been exploited as delivery vehicles to deliver therapeutics to the brain.398 For example, Tsivion-Visbord et al. used EVs derived from mesenchymal stem cells (MSCs-EVs), which can home towards sites of injury, in order to improve schizophrenia-like behaviors in a schizophrenia mouse model.400 Alternatively, Tang et al. used neuron-2a cell isolated exosomes loaded with neferine, a compound known for mitigating anxiety, and reported improved motor function and a reduced level of β-amyloid (Aβ) in the brain of an Alzheimer's disease mouse model.401 Additionally, they found that only exosomes encapsulated with compounds of molecular weight lower than 1109 Da are able to cross the BBB, which is an important finding for future use of exosomes as a brain delivery vehicle.
4.2.3.2. Cell membrane coating. Biomimetic nanomaterials, including cell membranes, have gained considerable attention as delivery vehicles due to their excellent capability of escaping immune recognition. Functionalization of cell membranes with BBB targeting moieties can facilitate passage across the BBB. For example, Duan et al. exploited the homotypic targeting capabilities of brain cancer cell membranes for targeting brain tumors for imaging purposes, and used them as a shell for polymeric NPs, loaded with superparamagnetic iron oxide NPs (SPIOs).402 Additionally, they engineered these cell membranes with cRGD, an endothelial integrin receptor targeting peptide, which is overexpressed in the tumor vasculature, to enhance their targeting efficiency towards glioma. Similarly, Liu et al. engineered red blood cell membrane (RBCm) nanovehicles loaded with anti-glioma therapeutic siRNA.403 The nanovehicles were further decorated with Angiopep-2, resulting in superior anti-tumor efficacy in GBM bearing mice.
Apart from membrane functionalization, fusion of cell membranes to obtain a delivery vehicle with desired properties is a promising alternative strategy. For example, Hao et al. prepared hybrid membrane-coated nanosuspensions using the membranes of glioma cancer cells and dendritic cells and loaded them with docetaxel.404 The exploitation of antigen presentation on the membrane of dendritic cells and the homotypic-targeting mechanism of glioma cells resulted in a delivery system exhibiting the synergistic effects of chemotherapy and immunotherapy, resulting in an enhanced anticancer effect.
4.2.3.3. Cell-mediated delivery. Cells as a whole have also been used for delivery purposes, with immune cells, such as monocytes, macrophages, dendritic cells, lymphocytes, neutrophils and stem cells, exhibiting both homing properties to sites of injury, inflammation or cancer and BBB crossing capabilities. Immune cells have the capability of crossing the BBB in response to cytokines produced in neuroinflammation or cancer, via changing their shape through a process called diapedesis.405 NPs can be attached on the cell membrane with covalent or ligand–receptor coupling, the so-called cell–NP hitchhiking system, or be internalized inside the cell cytoplasm, the so called ‘Trojan Horse method’.406 Ayer M. et al. exploited the migration capability of activated CD4+ T-cells across the BBB and used them for delivering NPs conjugated to the membranes of these cells for enhancing their brain targeting.407 Alternatively, Tong H. et al. used SPIO internalized bone marrow derived monocytes to target the inflamed brain of mice.408 Transiently disrupting the BBB using mannitol, bradykinin and serotonin further increased the transmigration efficiency of monocytes into the inflamed brain tissues (1.3–1.8-fold increase). In addition, Wu M. et al. exploited the chemotactic ability of neutrophils to inflamed tissues and used them as Trojan horses of doxorubicin loaded core–shell magnetic mesoporous silica nanoparticles to increase NP delivery into an incomplete resected U87 glioma model that mimics postsurgical glioma. This strategy enabled MRI diagnosis of the residual tumor as well as an improved survival rate and delayed glioma relapse.409
Besides its potential, engineering of live cells ex vivo also has limitations, such as the risk of mitigating cell transmigration abilities, reduced cell viability and fast degradation of the cargos before reaching their target, high cost and insufficient quantities of harvested cells.410,411 An emerging strategy is the in vivo hitchhiking of immune cells with NPs which give them an ‘eat me’ signal in order to exploit their migratory abilities to the diseased tissue. For example, Li M. et al. cloaked cisplatin loaded NPs with bacteria-secreted outer membrane vesicles so as to be recognized by neutrophils upon intravenous administration and internalized by them. Following NP internalization, neutrophils targeted residual s.c. microtumors after photothermal therapy, released their cargo and eradicated the residual tumors.411 In another example, Gao C. et al. intravenously injected bacteria-mimetic gold NPs in a melanoma mouse model, which were internalized by immune cells in circulation and converted to gold aggregates with a photothermal effect inside the cell cytoplasm. Next, the intracellular gold nano-aggregates were transferred to melanoma tissue via inflammatory tropism and showed highly effective antitumor PTT/immunotherapy.412 These strategies show great potential for NP targeted delivery and still need to be explored for their potential to cross the BBB.
4.3. Combination strategies
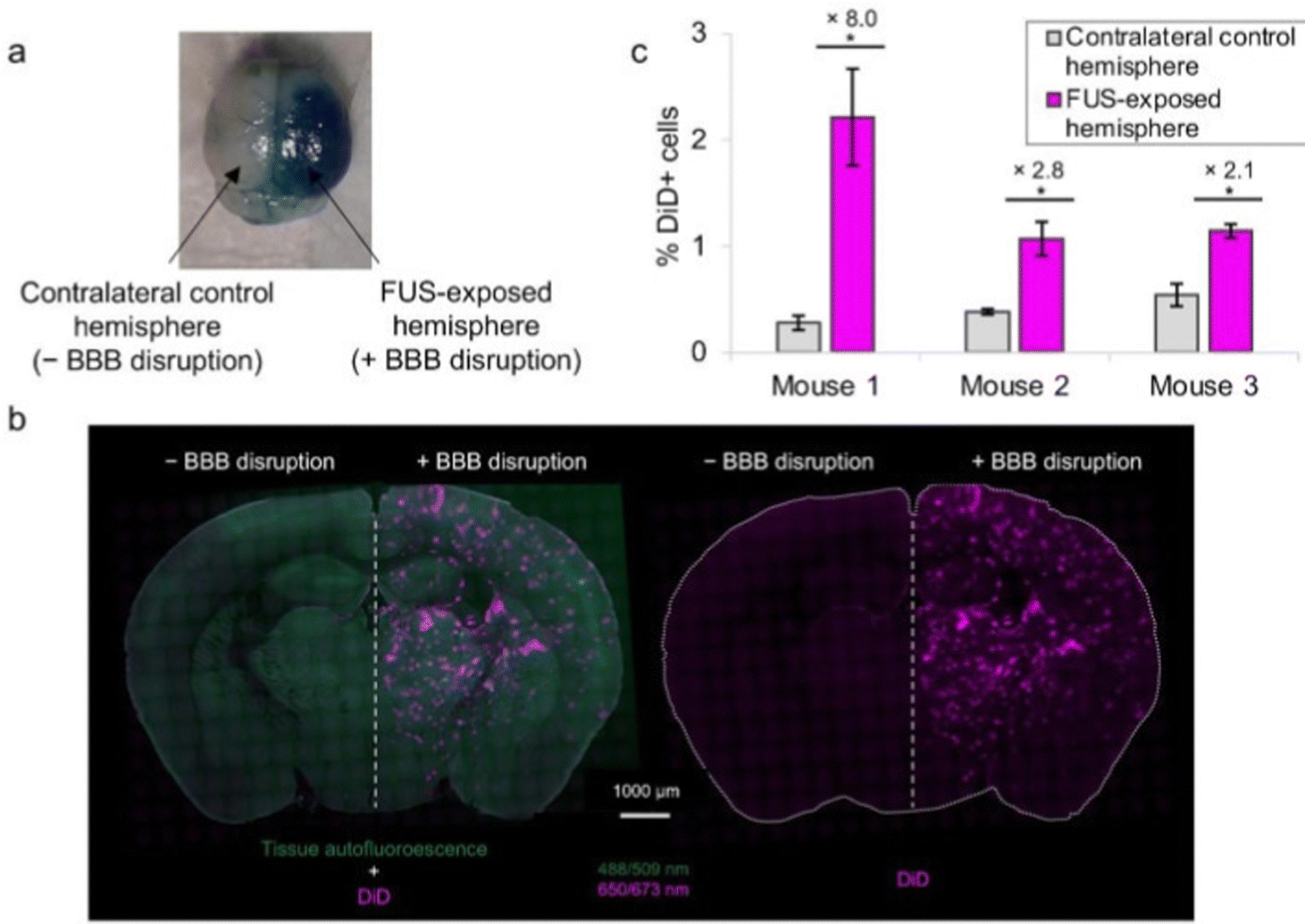 | ||
| Fig. 19 In vivo intracerebral accumulation of nanodroplets after BBB disruption. (a) Evans blue coloration in the sonicated hemisphere after FUS exposure to 0.33 MPa acoustic pressure. (b) Fluorescence microscopy imaging of coronal brain slices after retro-orbital i.v. administration with STE (pale blue-purple) and hemisphere FUS procedure application. (c) Fluorescence-activated cell-sorting analysis of DiD positive cells in sonicated and non-sonicated (control) brain hemispheres after retro-orbital i.v. administration with STE and hemisphere FUS procedure application (n = 3 areas for each hemisphere, *p < 0.05, student's t-test). Adapted from ref. 414 (CC BY 4.0). | ||
Apart from co-administration of NPs with microbubbles, several studies have shown more sufficient BBB crossing when NPs are linked to the microbubbles. For example, Burke et al. demonstrated that 5-fluorouracil (5FU) loaded PLGA NPs, covalently linked to microbubbles with FUS activation, significantly increased NP delivery to C6 glioma subcutaneous tumors compared to PLGA NPs alone or PLGA NPs co-administrated with microbubbles.416 Similarly, Aslund et al. showed that poly(butyl cyanoacrylate) (PBCA) NPs used together with casein could be self-assembled into microbubbles (NPMBs).417 Application of FUS after intravenous administration of the NPMBs resulted in a reversible BBB disruption allowing NP delivery across the BBB.
4.4. Considerations for BBB delivery
Apart from the commonly chosen intravenous administration route for brain delivery of NPs, other routes of administration have been investigated. Nose-to-brain delivery is considered a non-invasive way to bypass the BBB, by delivering therapeutics directly from the nasal cavity, via the olfactory and trigeminal neural pathways, to the central nervous system. Comparing the delivery of dendrimers to the brain after intranasal or intravenous administration, studies have showed an increased brain accumulation after intranasal administration (Fig. 20).421 Furthermore, Wei et al. compared the immunotherapy effects of (ApoE)-directed polymersomes CpG NPs in LCPN glioma bearing mice after intravenous or intranasal administration.247 Both administration routes showed an effective glioma immunotherapy, with the intranasal administered group showing around 45 days overall survival versus 55 days overall survival for the intravenous administered group. While promising, intranasal delivery has some intrinsic drawbacks, as there is a limited volume and surface area of the olfactory mucosa and mucociliary clearance resulting in low drug concentrations reaching the brain. In their extensive review, D. Lee and T. Minko give the details of the pharmacokinetics and physicochemical properties of NPs that play an important role for an effective nose-to-brain delivery. We advise the readers to refer to their review for more details on nose-to-brain NP delivery.422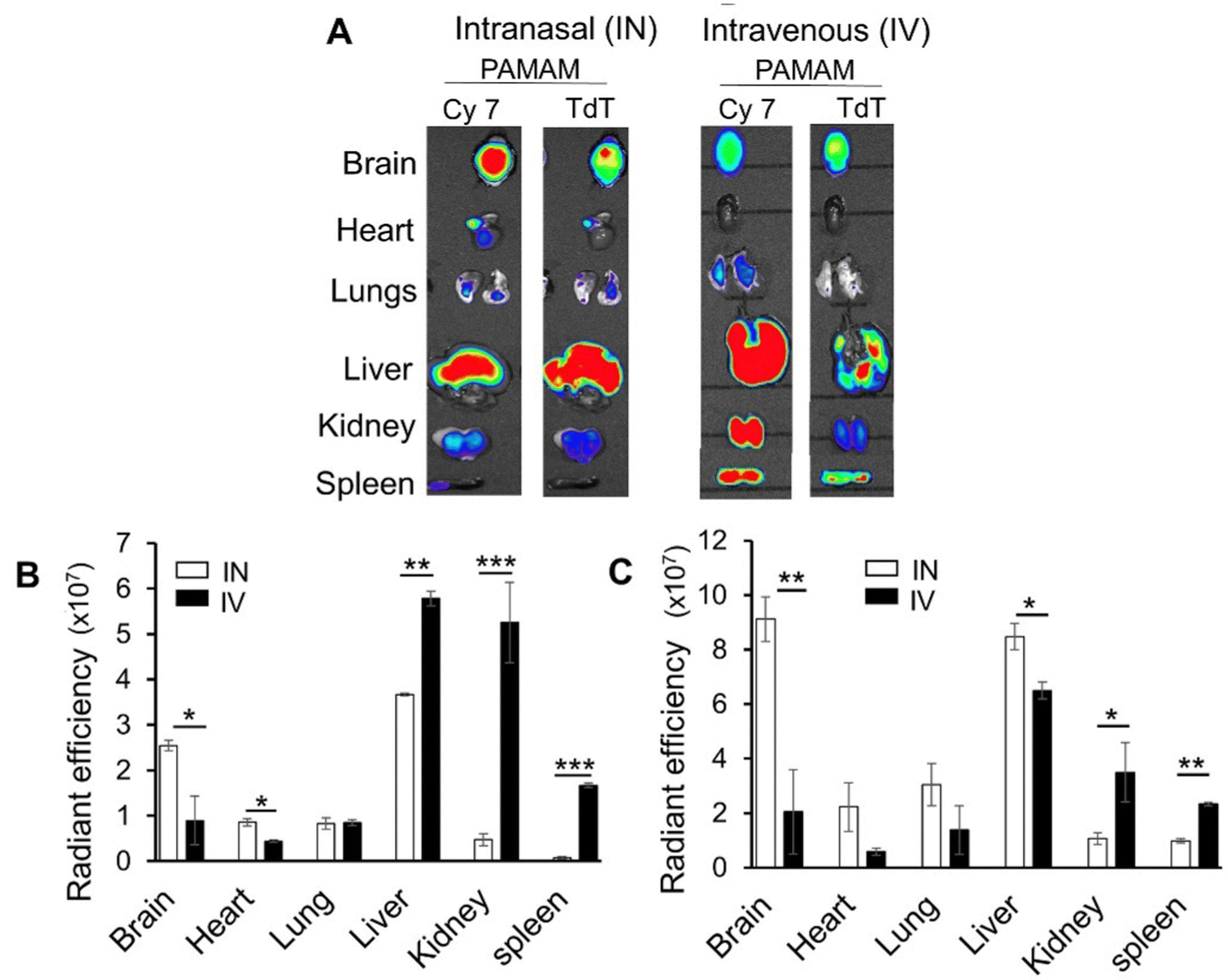 | ||
| Fig. 20 PAMAM NPs with labeled with cy7 and complexed with the Td tomato plasmid were delivered IN or IV. The mice were sacrificed 72 h post administration. (A) Ex vivo images of the explanted organs. (B and C) Histograms representing the level of Cy7 fluorescence (B) and RFP fluorescence (C). Reproduced from Ref. 421 with permission from Elsevier. | ||
For NM to cross the BBB, a clear distinction must be made in the disease state that will be targeted. In neurodegenerative diseases, the BBB may be affected, but its integrity overall will not change too much. In invasive tumors such as glioblastoma, the effects on BBB integrity can be far more outspoken. However, it is difficult to assess the extent of it in all conditions and how to relate this to NM delivery efficacy. Preclinical imaging setups such as dynamic contrast-enhanced MRI or PET imaging are indispensable tools in this, but these are not available to all researchers. While various strategies are employed to enhance NM delivery across the BBB, as for other ‘tissue barriers’, one unique concept of the BBB is to purposefully break down the BBB to allow for transfer of blood-borne components to enter the brain more easily. These transient pores, generated through ablation or focused ultrasound, serve as a special tool to allow the researcher or clinician to open up small delivery portals through the barrier. As such, the excellent filtration capacity of the BBB becomes less of a direct issue, but more information is needed on the safety and efficacy of generating these pores.
Other administration routes for brain delivery include delivery by the lymphatic system, which has been shown to be more effective than iv administration,278 and oral delivery, which is often considered the most convenient administration route, because of the ease of administration and patient compliance. However oral delivery requires formulations to cross both the intestinal barrier and the BBB to reach the brain.279 Nevertheless, Kim et al. have reported the accumulation of gold (Au) NPs in the brain of glioma bearing mice after oral administration, by surface functionalizing the NPs with lactoferrin, which is expressed in the GI tract, BBB and GBM.280
4.5. Translatability
Despite a wealth of promising research in nanomedicine for targeting the brain and in the use of nanoparticle-mediated delivery across the BBB, only a limited number of these discoveries have made it to clinical trials. Most studies focused on tumor models such as GBM, where, due to the invasive nature of the tumor, the integrity of the BBB itself may have been compromised facilitating the delivery of the investigated nano-therapeutics across the BBB. An initial study (phase I; NCT00734682) examined the pharmacokinetics of a liposomal irinotecan derivative in patients with recurrent, high-grade gliomas. While the trial stopped in 2014, no follow-up trial was set up, likely indicating the poor brain delivery. A second trial (phase II; NCT02340156) studied the efficacy of temozolomide together with p53 cDNA encapsulated in cationic liposomes. This study was, however, terminated due to the low number of participants that could be enrolled. A first-in-human trial was concluded in 2022 (NCT03020017) where patients were given NU-0129, a gold nanoparticle coated with nuclear acids that target Bcl2L12, where the primary objective was to study safety and delivery into the brain. The study indicated that most patients had not suffered any side effects, but a low percentage did indicate changes in lymphocytes, platelets and vascular disorders. It remains unclear whether these studies will be continued and whether the delivery was sufficiently high to reach therapeutic efficacy. Another ongoing trial was set up (phase I/II) in 2022, where polysiloxane Gd-chelates based nanoparticles (AGuIX) are tested together with radiotherapy and concomitant temozolomide in the treatment of newly diagnosed glioblastoma (NCT04881032). The study will be a combined dose escalation (phase I) and randomized (phase II) trial to study the recommended dose and therapeutic efficacy, next to pharmacokinetic properties. While the number of trials is low, it is important to notice as well that none of the formulations mentioned make any modifications to enhance the delivery of their nanoparticles across the BBB. They rely on the intrinsic ability of some nanoparticles to cross the BBB, albeit at low efficacy, while simultaneously expecting some BBB damage that would help the nanoparticles to cross more efficiently. It is, therefore, important to note that most strategies described here are preclinical and would need to be tested in human settings. If they could efficiently promote BBB transfer, this would have a strong impact on future clinical trials. Other settings, such as the transient opening of the BBB using external stimuli, have been more widely researched (NCT03712293; NCT02474966; NCT03744026; NCT04440358; NCT04417088; NCT04614493; NCT04417088… a total of 58 studies were found on ClinicalTrials.gov), but have thus far not been combined with nanotherapies; instead they have been used in combination with classical chemotherapies. Therefore, more preclinical studies are needed which are based on patient derived tissues and cells for precision medicine and to facilitate the translation of discoveries in the nanomedicine and nano targeting field into the clinic.5. Placental barrier
During intrauterine life, the fetus is protected from environmental factors by the mother and the placenta. The latter functions both as a barrier and a central metabolic organ where the two main pathways (maternal–fetal and fetal–maternal transport) enable fetal protection, detoxification, growth and development. Optimizing treatment and minimizing side effects through targeted drug delivery are therefore of critical importance for maternal diseases that co-occur in pregnant women, for example cancer, pregnancy-related complications, for example preeclampsia, or specific fetal diseases. Although major therapeutic advancements have been implemented, such as antenatal corticosteroid use for improved fetal outcome and flecainide administration for fetal supraventricular tachycardia,423,424 true custom-made therapies for mothers and fetuses are rare. This can, at least in part, be attributed to the abovementioned presence of the placenta as a biological barrier. The placental barrier plays a dual role, as it allows and facilitates transport of nutrients/waste to and from the fetus, and also protects the fetus by impeding exposure to damaging substances, making tuned antenatal drug delivery more complex (Box 4).In this section, we analyze how nanomaterials have been employed for crossing the placental barrier and improving prenatal medicine. The latter can be subdivided into fetal, maternal or placental disorders and each category poses specific challenges for targeted therapeutics. Fetal therapeutics are needed for diseases such as fetal tachycardia and genetic diseases, but also for lung maturation acceleration in case of expected preterm delivery.425 The aim of these fetal therapeutics should be to target the fetus by enabling the crossing of the placental barrier, while avoiding maternal side effects, for example in the case of pregnancy-related diabetes after antenatal corticosteroid therapy.426 For maternal therapeutics, for example in the case of cancer, the therapeutic agent should be targeted towards the tumor while the crossing of the placenta, with potential fetal exposure, should be avoided. The third category are the placental therapeutics, for example for pre-eclampsia management, for which only the placenta should be targeted and side effects in both the fetus and the mother should be minimized.
Box 4: Placental barrier characteristicsThe placenta is composed of both maternal, derived from the endometrium, and fetal tissue, derived from the chorionic sac. In between the two regions, the intervillous space is characterized by the villous structures containing fetal blood vessels. These vessels transport oxygenated blood from the microvilli to the umbilical vein, while deoxygenated fetal blood is returned from the fetus to the placental microvilli via the umbilical arteries. Nutrients, waste and oxygen are then interchanged with maternal blood, which flows through the intervillous space.427 Blood perfusion of the villi and maternal–fetal exchange are not established until the end of the first trimester and abnormal vascular development has been linked to preeclampsia onset.428 In humans, fetal and maternal blood are separated by 3 layers: the syncytiotrophoblast, connective tissue and the vascular endothelium of fetal blood vessels. These 3 layers form the placental membrane or barrier. Molecules larger than ∼600 Da do not cross the human placenta, unless through active energy-dependent pathways, as is the case for IgG antibodies.429,430 However, this size exclusion can vary over time as the placenta is a dynamic structure, developing right after blastocyst implantation up to expulsion right after birth. Multiple changes in the placenta can be noted during pregnancy: (i) the syncytiotrophoblast (STB) develops from a primitive to a layer with apical–basal polarity (Fig. 21A); (ii) continued regeneration of the STB by differentiation and fusion of cytotrophoblasts (Fig. 21B); (iii) thinning of the placental barrier, as the cytotrophoblast layer is depleted, from 50–100 μm to 2–4 μm at term; and (iv) changes in placental transporters at the placental surface.2,427 Of note, important differences in the placental structure exist between species, so interspecies extrapolation has its limits. |
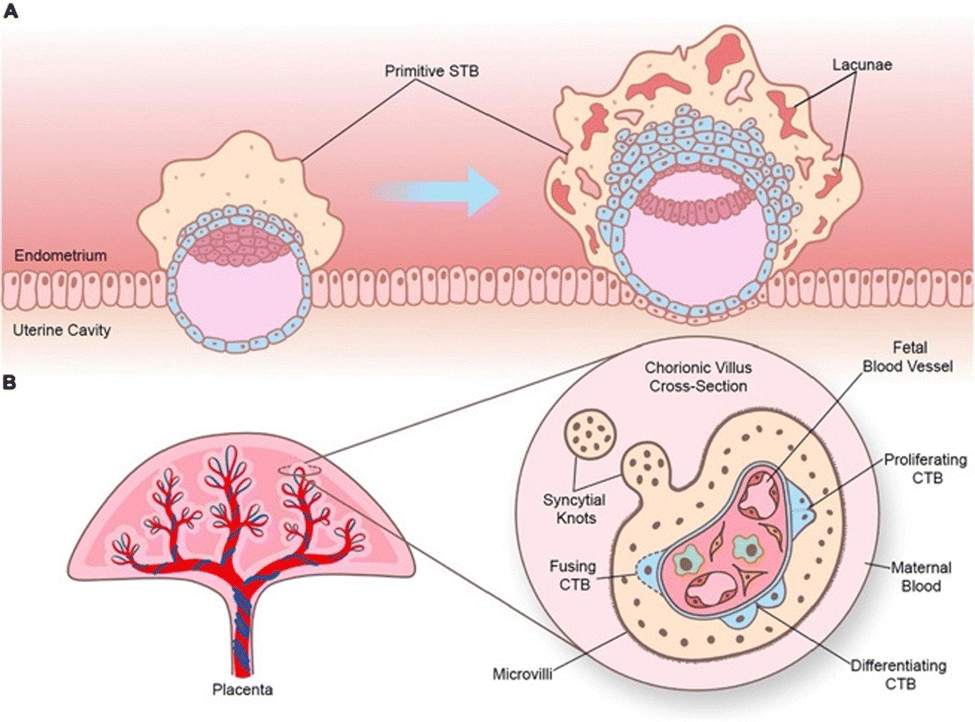 | ||
| Fig. 21 Placental structure during pregnancy. (A) Formation of the primitive syncytiotrophoblast (STB) from GD 7–8 to 8–9. First formation of lacunae. (B) Well-developed chorionic villus in late pregnancy. Reproduced from ref. 2 (CC BY 4.0). | ||
5.1. NM design strategies for placental interaction
Evidently, simply encapsulating drugs in NPs does not suffice for (avoiding) placental crossing. As with any barrier, multiple parameters can determine whether nanoparticles can cross the placental barrier. While sizing of NPs is considered to play a role, reports vary significantly. For example, small inorganic nanoparticles, such as titanium dioxide and silver NPs, show high (toxic) fetal concentrations after maternal exposure.435,436 On the other hand, polystyrene nanoparticles up to 500 nm have been reported to cross the placenta, albeit at significantly lower concentrations.437 Furthermore, a study by Huang and colleagues in an in vivo mouse model showed that 20 nm PS NPs showed significantly less placental uptake than 40 nm PS NPs, suggesting a non-linear size exclusion mechanism for placental uptake.438 Small NPs may even be impeded from undergoing translocation, as was shown for Cu NPs with a size of 15 nm.439 Likely, the varying effect can, at least partly, be attributed to differing material compositions. Of note, the lack of placental translocation does not exclude possible fetal side effects. Maternal exposure to Cu NPs did lead to strong immunomodulatory effects in the offspring.
The non-linear size exclusion implies the involvement of active transportation of particles. In a study by Grafmueller and colleagues, it was shown that passive diffusion, through concentration equilibration, was indeed not the main driver of placental translocation and, therefore, active transport pathways are involved in NP transport.440 Various active transport pathways have later been described to play a role. Through selective inhibition of the caveolae-mediated endocytosis pathways, barrier crossing in vitro of pullulan acetate (PA) NPs was reduced by 55%, indicating that active caveolae-mediated transport has an important role in NP transport (Fig. 22A). Inhibition of macropinocytosis and clathrin-mediated endocytosis reduced transport as well, but by only. 24.2% and 23.9% respectively.441 With the role of active transporters in mind, Jiang and colleagues developed folate-conjugated pullulan acetate (FPA) NPs, which showed placental translocation in vitro, by leveraging folate receptor-mediated transport.442
 | ||
| Fig. 22 (A) Fluorescence intensity of BeWo after uptake of PA-FITC NPs in the presence of the endocytic inhibitor chlorpromazine (CPZ), colchicine (Col), nystatin (NY) or amiloride (AMR). **p < 0.01. Adapted from ref. 441 with permission from Dove Medical Press Limited. (B) Confocal microscopy images of BeWo cells and FITC-labelled CNPs or IgG-CNPs (green) with immunostaining for FcRn (red) and counterstaining with DAPI (blue). Co-localization (yellow; white arrows) of the IgG-CNPs with FcRn was observed. Scale bars = 20 μm. Adapted from ref. 456 with permission from the Royal Society of Chemistry. | ||
The role of surface charge is still unclear, but studies have showed its impact on placental transport, with negatively charged NPs tending to show better barrier penetration compared to positively charged NPs.443 However, highly negatively charged nanoparticles, for example through COOH-surface groups, show less placental translocation.338 Neutrally charged NPs, for example NPs coated with PEG, have been shown to pass in higher quantity compared to negatively charged particles in ex vivo human placenta models.444 However, the precise effect of PEGylation on placental crossing remains unclear, as other reports have suggested impeding of placental translocation by particle PEGylation.443,445
Finally, in a recent study by Tse and colleagues, inspiration was taken from the maternal–fetal transfer of passive immunity for their design of IgG-modified chitosan nanoparticles.446 IgG antibodies are known to be transported across the placental barrier through involvement of Fc receptors.447 Based on this, IgG-NPs were shown to be translocated across the placenta in vitro (Fig. 22B).
Besides active targeting strategies, tuning nanoparticle properties can improve placental targeting, although available studies are scarce. A study by Ho and colleagues reported positively charged PEI-PGMA with significant higher retention proportions than plain PGMA particles.429 Additionally, Swingle and colleagues recently reported on a systematic study in which they screened lipid NPs with differing ionizable lipid components for both their mRNA loading and their placental targeting.455 While lipid NPs have commonly been used for liver accumulation for, among others, vaccination purposes, oxygen-containing lipids were found to significantly increase extrahepatic delivery towards the placenta. In a similar fashion, systematically varying the molar ratios of C12–200, DOPE, cholesterol and PEG allowed optimal lipid NP design for maximizing placental accumulation.456
5.2. Considerations for antenatal drug delivery
Fetal targeting can be invasively enhanced, for example by intra-amniotic administration of PAMAM dendrimer-based therapeutics.459 Alternatively, administration via the umbilical vein was recently reported in clinical settings for enzyme-replacement therapy for infantile-onset Pompe's disease.460 However, it should be noted that more invasive procedures pose risks to the unborn child and mother; however recent studies indicate that risk mitigation might be feasible.461,462
Furthermore, clinical applications of therapeutics during pregnancy are subjected to significant constraints which make translationability cumbersome. Ethical considerations regarding fetal therapies need to balance harm and benefits for both mother and child and may change depending on the gestational age.466 These considerations further impact clinical trial designs, where exclusion of pregnant women has been common practice. Illustratively, only 5% of available medicines have been tested and labeled for use in pregnancy.467 Given the proven differences of changing drug pharmacokinetics during pregnancy, translationability and clinical use of new drugs and advanced drug delivery technologies for pregnancy-related complications remain a gray area.468
6. Conclusions and final remarks
The biological barriers described in this review portrait a unique challenge, where each individual barrier requires tailormade engineering of nanomaterials to modulate their translocation. While we provide an overview of key parameters, and their respective effect on barrier penetration, a general lack of systematic studies impedes in-depth analysis of the relative contribution of these different parameters to (un)successful barrier crossing and subsequent interactions with the target cells. We reckon that future studies should include homogeneous standardization, for example, in the case of mucus penetration by including multiple particle tracking analysis, offering interstudy comparable parameters for penetration probability. Such standardized systematic studies may not only allow better translational research, but they will also aid in our fundamental understanding of the bio-nano interface and the in vivo fate of these nanoformulations.469 While this review has mainly focused on 4 special, yet highly relevant, cases of organ barriers, the insights on nanoformulation engineering are transferable to other physical barriers, albeit with a significant required effort on specific barrier customization.Upcoming technologies that may aid in the execution of these standardized studies include precision-cut-tissue slices and organ-on-chip technologies, providing a tool wherein, similar to in vitro studies, barrier penetration studies are possible but in a more closely in vivo mimicking environment.470 Additionally, mathematical modelling tools can aid in screening relevant nanoparticles and calculating their barrier crossing probability.471–473
Despite the knowledge gap on fundamental bio-nano interactions, advanced strategies have been discussed in this review showing potential to efficiently transport the cargo across the barriers. For systemic barriers, such as the blood–brain barrier and placental barrier, initial reports on, for example, cell-based nanoformulations show a very promising outlook and are expected to drastically improve biodistribution control and barrier translocation efficiency.474,475 On the other hand, for mucus-based barriers, intelligent or switchable nanoparticles are expected to drive formulation efficiency, not only in terms of mucus penetration, but also for subsequent target cell interactions. These intelligent nanoparticles can be redox-responsive, pH-responsive or ROS-triggered and, subsequently, may change their size, charge or shape accordingly.77,476,477 Other NP types, such as Janus NPs, may serve as hyper-engineered platforms, leveraging the strengths of different crossing strategies or allow dual-drug treatment with simultaneous delivery in multifunctional NPs.478,479 While simplicity in formulation has often been regarded as having the greatest translatability from preclinical studies to human clinical trials, these recently suggested engineered formulations may hold the greatest promise for effectively penetrating the barriers of organs.
In conclusion, the current review has demonstrated that ample efforts have been made to improve the transport of engineered NPs across biological barriers. While the efficacy of nanomedicine strategies has remained, generally, rather low, the introduction of more advanced engineered nanomaterials is expected to be the foundation for future clinical success.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Commission under the Horizon 2020 framework for ERC (ERC StG 750973), KU Leuven Internal Funds C2 (C24/18/101) and Fonds Wetenschappelijk Onderzoek (G0B2919N).References
- Y. Chen and L. Liu, Adv. Drug Deliv. Rev., 2012, 64, 640–665, DOI:10.1016/j.addr.2011.11.010.
- A. Jaremek, M. J. Jeyarajah, G. Jaju Bhattad and S. J. Renaud, Front. Cell Dev. Biol., 2021, 9, 674162, DOI:10.3389/fcell.2021.674162.
- V. Lenders, X. Koutsoumpou, A. Sargsian and B. B. Manshian, Nanoscale Adv., 2020, 2, 5046–5089, 10.1039/d0na00478b.
- Z. Zhao, K. Powers, Y. Hu, M. Raleigh, P. Pentel and C. Zhang, Biomaterials, 2017, 123, 107–117, DOI:10.1016/j.biomaterials.2017.01.038.
- Y. Gao, A. Sarode, N. Kokoroskos, A. Ukidve, Z. Zhao, S. Guo, R. Flaumenhaft, A. S. Gupta, N. Saillant and S. Mitragotri, Sci. Adv., 2020, 6, eaba0588 CrossRef CAS PubMed.
- H. Naatz, B. B. Manshian, C. Rios Luci, V. Tsikourkitoudi, Y. Deligiannakis, J. Birkenstock, S. Pokhrel, L. Madler and S. J. Soenen, Angew. Chem., Int. Ed., 2020, 59, 1828–1836, DOI:10.1002/anie.201912312.
- M. Van Woensel, N. Wauthoz, R. Rosière, V. Mathieu, R. Kiss, F. Lefranc, B. Steelant, E. Dilissen, S. W. Van Gool, T. Mathivet, H. Gerhardt, K. Amighi and S. De Vleeschouwer, J. Controlled Release, 2016, 227, 71–81, DOI:10.1016/j.jconrel.2016.02.032.
- M. Van Woensel, T. Mathivet, N. Wauthoz, R. Rosière, A. D. Garg, P. Agostinis, V. Mathieu, R. Kiss, F. Lefranc, L. Boon, J. Belmans, S. W. Van Gool, H. Gerhardt, K. Amighi and S. De Vleeschouwer, Sci. Rep., 2017, 7, 1217, DOI:10.1038/s41598-017-01279-1.
- A. Gauthier, A. Fisch, K. Seuwen, B. Baumgarten, H. Ruffner, A. Aebi, M. Rausch, F. Kiessling, M. Bartneck, R. Weiskirchen, F. Tacke, G. Storm, T. Lammers and M. G. Ludwig, Biomaterials, 2018, 178, 481–495, DOI:10.1016/j.biomaterials.2018.04.006.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2021, 6, e10246, DOI:10.1002/btm2.10246.
- S. M. Stavis, J. A. Fagan, M. Stopa and J. A. Liddle, ACS Appl. Nano Mater., 2018, 1, 4358–4385, DOI:10.1021/acsanm.8b01239.
- S. Wilhelm, A. Tavares, Q. Dai, S. Ohta, J. Audet, H. Dvorak and W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- Y. Hayashi, M. Takamiya, P. B. Jensen, I. Ojea-Jimenez, H. Claude, C. Antony, K. Kjaer-Sorensen, C. Grabher, T. Boesen, D. Gilliland, C. Oxvig, U. Strahle and C. Weiss, ACS Nano, 2020, 14, 1665–1681, DOI:10.1021/acsnano.9b07233.
- C. A. Ruge, U. F. Schaefer, J. Herrmann, J. Kirch, O. Canadas, M. Echaide, J. Perez-Gil, C. Casals, R. Muller and C. M. Lehr, PLoS One, 2012, 7, e40775, DOI:10.1371/journal.pone.0040775.
- J. Yoo, K. Kim, S. Kim, H. H. Park, H. Shin and J. Joo, Nanoscale, 2022, 14, 14482–14490, 10.1039/d2nr02995b.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Deliv. Rev., 2016, 99, 28–51, DOI:10.1016/j.addr.2015.09.012.
- Z. Zhao, A. Ukidve, V. Krishnan, A. Fehnel, D. C. Pan, Y. Gao, J. Kim, M. A. Evans, A. Mandal, J. Guo, V. R. Muzykantov and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 441–454, DOI:10.1038/s41551-020-00644-2.
- C. Gao, Q. Wang, J. Li, C. H. T. Kwong, J. Wei, B. Xie, S. Lu, S. M. Y. Lee and R. Wang, Sci. Adv., 2022, 8, eabn1805 CrossRef CAS PubMed.
- L. Liu, T. K. Hitchens, Q. Ye, Y. Wu, B. Barbe, D. E. Prior, W. F. Li, F. C. Yeh, L. M. Foley, D. J. Bain and C. Ho, Biochim. Biophys. Acta, 2013, 1830, 3447–3453, DOI:10.1016/j.bbagen.2013.01.021.
- M. P. Nikitin, I. V. Zelepukin, V. O. Shipunova, I. L. Sokolov, S. M. Deyev and P. I. Nikitin, Nat. Biomed. Eng., 2020, 4, 717–731, DOI:10.1038/s41551-020-0581-2.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575, DOI:10.1038/s41563-019-0566-2.
- F. Danhier, J. Controlled Release, 2016, 244, 108–121, DOI:10.1016/j.jconrel.2016.11.015.
- M. Izci, C. Maksoudian, F. Goncalves, L. Aversa, R. Salembier, A. Sargsian, I. Perez Gilabert, T. Chu, C. Rios Luci, E. Bolea-Fernandez, D. Nittner, F. Vanhaecke, B. B. Manshian and S. J. Soenen, J. Nanobiotechnol., 2022, 20, 518, DOI:10.1186/s12951-022-01727-9.
- Y. X. Li, Y. Wei, R. Zhong, L. Li and H. B. Pang, Pharmaceutics, 2021, 13, 552, DOI:10.3390/pharmaceutics13040552.
- T. Kang, X. Gao, Q. Hu, D. Jiang, X. Feng, X. Zhang, Q. Song, L. Yao, M. Huang, X. Jiang, Z. Pang, H. Chen and J. Chen, Biomaterials, 2014, 35, 4319–4332, DOI:10.1016/j.biomaterials.2014.01.082.
- A. Aliyandi, S. Satchell, R. E. Unger, B. Bartosch, R. Parent, I. S. Zuhorn and A. Salvati, Int. J. Pharm., 2020, 587, 119699, DOI:10.1016/j.ijpharm.2020.119699.
- M. I. Setyawati, C. Y. Tay, B. H. Bay and D. T. Leong, ACS Nano, 2017, 11, 5020–5030, DOI:10.1021/acsnano.7b01744.
- F. Peng, M. I. Setyawati, J. K. Tee, X. Ding, J. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nat. Nanotechnol., 2019, 14, 279–286, DOI:10.1038/s41565-018-0356-z.
- G. W. Liu, E. B. Guzman, N. Menon and R. S. Langer, Pharm. Res., 2023, 40, 3–25, DOI:10.1007/s11095-023-03471-7.
- C. R. Bakshani, A. L. Morales-Garcia, M. Althaus, M. D. Wilcox, J. P. Pearson, J. C. Bythell and J. G. Burgess, npj Biofilms Microbiomes, 2018, 4, 14, DOI:10.1038/s41522-018-0057-2.
- A. M. Shen and T. Minko, J. Controlled Release, 2020, 326, 222–244, DOI:10.1016/j.jconrel.2020.07.011.
- K. S. LeMessurier, M. Tiwary, N. P. Morin and A. E. Samarasinghe, Front. Immunol., 2020, 11, 3, DOI:10.3389/fimmu.2020.00003.
- A. Gie, Y. Regin, A. Mersanne and J. Toelen, JoVE, 2021, e61982, DOI:10.3791/61982.
- C. C. Campa, R. L. Silva, J. P. Margaria, T. Pirali, M. S. Mattos, L. R. Kraemer, D. C. Reis, G. Grosa, F. Copperi, E. M. Dalmarco, R. C. P. Lima-Junior, S. Aprile, V. Sala, F. Dal Bello, D. S. Prado, J. C. Alves-Filho, C. Medana, G. D. Cassali, G. C. Tron, M. M. Teixeira, E. Ciraolo, R. C. Russo and E. Hirsch, Nat. Commun., 2018, 9, 5232, DOI:10.1038/s41467-018-07698-6.
- P. J. Kuehl, C. S. Tellez, M. J. Grimes, T. H. March, M. Tessema, D. A. Revelli, L. M. Mallis, W. W. Dye, T. Sniegowski, A. Badenoch, M. Burke, D. Dubose, D. T. Vodak, M. A. Picchi and S. A. Belinsky, Br. J. Cancer, 2020, 122, 1194–1204, DOI:10.1038/s41416-020-0765-2.
- B. Button, L.-H. Cai, C. Ehre, M. Kesimer, D. B. Hill, J. K. Sheehan, R. C. Boucher and M. Rubinstein, Science, 2012, 337, 937–941, DOI:10.1126/science.1223012.
- E. Hermans, M. S. Bhamla, P. Kao, G. G. Fuller and J. Vermant, Soft Matter, 2015, 11, 8048–8057, 10.1039/c5sm01603g.
- C. Karamaoun, B. Sobac, B. Mauroy, A. Van Muylem and B. Haut, PLoS One, 2018, 13, e0199319, DOI:10.1371/journal.pone.0199319.
- J. Witten, T. Samad and K. Ribbeck, Curr. Opin. Biotechnol, 2018, 52, 124–133, DOI:10.1016/j.copbio.2018.03.010.
- F. Taherali, F. Varum and A. W. Basit, Adv. Drug Deliv. Rev., 2018, 124, 16–33, DOI:10.1016/j.addr.2017.10.014.
- R. Bansil and B. S. Turner, Curr. Opin. Colloid Interface Sci., 2006, 11, 164–170, DOI:10.1016/j.cocis.2005.11.001.
- S. Ren, W. Li, L. Wang, Y. Shi, M. Cai, L. Hao, Z. Luo, J. Niu, W. Xu and Z. Luo, Sci. Rep., 2020, 10, 2030, DOI:10.1038/s41598-020-58922-7.
- S. M. Vanaki, D. Holmes, K. Suara, P. G. Jayathilake and R. Brown, Comput. Biol. Med., 2020, 117, 103595, DOI:10.1016/j.compbiomed.2019.103595.
- W. H. Anderson, R. D. Coakley, B. Button, A. G. Henderson, K. L. Zeman, N. E. Alexis, D. B. Peden, E. R. Lazarowski, C. W. Davis, S. Bailey, F. Fuller, M. Almond, B. Qaqish, E. Bordonali, M. Rubinstein, W. D. Bennett, M. Kesimer and R. C. Boucher, Am. J. Respir. Crit. Care Med., 2015, 192, 182–190, DOI:10.1164/rccm.201412-2230OC.
- V. Y. Lin, N. Kaza, S. E. Birket, H. Kim, L. J. Edwards, J. LaFontaine, L. Liu, M. Mazur, S. A. Byzek, J. Hanes, G. J. Tearney, S. V. Raju and S. M. Rowe, Eur. Respir. J., 2020, 55, 1900419, DOI:10.1183/13993003.00419-2019.
- A. L. Garland, W. G. Walton, R. D. Coakley, C. D. Tan, R. C. Gilmore, C. A. Hobbs, A. Tripathy, L. A. Clunes, S. Bencharit, M. J. Stutts, L. Betts, M. R. Redinbo and R. Tarran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15973–15978, DOI:10.1073/pnas.1311999110.
- S. Yuan, Q. Liu, Z. Hu, Z. Zhou, G. Wang, C. Li, W. Xie, G. Meng, Y. Xiang, N. Wu, L. Wu, Z. Yu, L. Bai and Y. Li, Cell Death Dis., 2018, 9, 450, DOI:10.1038/s41419-018-0472-6.
- B. A. Helling, A. N. Gerber, V. Kadiyala, S. K. Sasse, B. S. Pedersen, L. Sparks, Y. Nakano, T. Okamoto, C. M. Evans, I. V. Yang and D. A. Schwartz, Am. J. Respir. Cell Mol. Biol., 2017, 57, 91–99, DOI:10.1165/rcmb.2017-0046OC.
- I. R. Scolari, X. Volpini, M. L. Fanani, B. La Cruz-Thea, L. Natali, M. M. Musri and G. E. Granero, Mol. Pharm., 2021, 18, 807–821, DOI:10.1021/acs.molpharmaceut.0c00692.
- G. Alp and N. Aydogan, Eur. J. Pharm. Biopharm., 2020, 149, 45–57, DOI:10.1016/j.ejpb.2020.01.017.
- S. Arber Raviv, M. Alyan, E. Egorov, A. Zano, M. Y. Harush, C. Pieters, H. Korach-Rechtman, A. Saadya, G. Kaneti, I. Nudelman, S. Farkash, O. D. Flikshtain, L. N. Mekies, L. Koren, Y. Gal, E. Dor, J. Shainsky, J. Shklover, Y. Adir and A. Schroeder, J. Controlled Release, 2022, 346, 421–433, DOI:10.1016/j.jconrel.2022.03.028.
- R. M. Derbali, V. Aoun, G. Moussa, G. Frei, S. F. Tehrani, J. C. Del'Orto, P. Hildgen, V. G. Roullin and J. L. Chain, Mol. Pharm., 2019, 16, 1906–1916, DOI:10.1021/acs.molpharmaceut.8b01256.
- C. I. Camara, L. Bertocchi, C. Ricci, R. Bassi, A. Bianchera, L. Cantu, R. Bettini and E. Del Favero, Int. J. Mol. Sci., 2021, 22, 10480, DOI:10.3390/ijms221910480.
- T. T. Tran, C. Vidaillac, H. Yu, V. F. L. Yong, D. Roizman, R. Chandrasekaran, A. Y. H. Lim, T. B. Low, G. L. Tan, J. A. Abisheganaden, M. S. Koh, J. Teo, S. H. Chotirmall and K. Hadinoto, Int. J. Pharm., 2018, 547, 368–376, DOI:10.1016/j.ijpharm.2018.06.017.
- Q. Liu, X. Zhang, J. Xue, J. Chai, L. Qin, J. Guan, X. Zhang and S. Mao, J. Controlled Release, 2022, 347, 435–448, DOI:10.1016/j.jconrel.2022.05.006.
- Y. He, Y. Liang, J. C. W. Mak, Y. Liao, T. Li, R. Yan, H.-F. Li and Y. Zheng, Colloids Surf., B, 2020, 186, 110703, DOI:10.1016/j.colsurfb.2019.110703.
- N. Kim, G. Kwak, J. Rodriguez, A. Livraghi-Butrico, X. Zuo, V. Simon, E. Han, S. K. Shenoy, N. Pandey, M. Mazur, S. E. Birket, A. Kim, S. M. Rowe, R. Boucher, J. Hanes and J. S. Suk, Thorax, 2022, 77, 812–820, DOI:10.1136/thoraxjnl-2020-215185.
- G. Costabile, R. Provenzano, A. Azzalin, V. C. Scoffone, L. R. Chiarelli, V. Rondelli, I. Grillo, T. Zinn, A. Lepioshkin, S. Savina, A. Miro, F. Quaglia, V. Makarov, T. Coenye, P. Brocca, G. Riccardi, S. Buroni and F. Ungaro, Nanomedicine, 2020, 23, 102113, DOI:10.1016/j.nano.2019.102113.
- G. Osman, J. Rodriguez, S. Y. Chan, J. Chisholm, G. Duncan, N. Kim, A. L. Tatler, K. M. Shakesheff, J. Hanes, J. S. Suk and J. E. Dixon, J. Controlled Release, 2018, 285, 35–45, DOI:10.1016/j.jconrel.2018.07.001.
- Y. C. Kim, H. T. Hsueh, N. Kim, J. Rodriguez, K. T. Leo, D. Rao, N. E. West, J. Hanes and J. S. Suk, Adv. Ther., 2020, 3, 2000013, DOI:10.1002/adtp.202000013.
- J. Li, H. Zheng, E. Y. Xu, M. Moehwald, L. Chen, X. Zhang and S. Mao, Acta Biomater., 2021, 123, 325–334, DOI:10.1016/j.actbio.2020.12.061.
- E. F. Craparo, S. E. Drago, F. Quaglia, F. Ungaro and G. Cavallaro, Drug Deliv. Transl. Res., 2022, 12, 1859–1872, DOI:10.1007/s13346-021-01102-5.
- D. Chen, S. Liu, D. Chen, J. Liu, J. Wu, H. Wang, Y. Su, G. Kwak, X. Zuo, D. Rao, H. Cui, C. Shu and J. S. Suk, Angew. Chem., Int. Ed., 2021, 60, 15225–15229, DOI:10.1002/anie.202101732.
- G. Chai, A. Hassan, T. Meng, L. Lou, J. Ma, R. Simmers, L. Zhou, B. K. Rubin, Q. T. Zhou, P. W. Longest, M. Hindle and Q. Xu, Nanomedicine, 2020, 29, 102262, DOI:10.1016/j.nano.2020.102262.
- X. Bai, G. Zhao, Q. Chen, Z. Li, M. Gao, W. Ho, X. Xu and X.-Q. Zhang, Sci. Adv., 2022, 8, eabn7162 CrossRef CAS PubMed.
- Y. Wang, Q. Yuan, W. Feng, W. Pu, J. Ding, H. Zhang, X. Li, B. Yang, Q. Dai, L. Cheng, J. Wang, F. Sun and D. Zhang, J. Nanobiotechnol., 2019, 17, 103, DOI:10.1186/s12951-019-0537-4.
- J. Zhu, M. Guo, Y. Cui, Y. Meng, J. Ding, W. Zeng and W. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 5090–5100, DOI:10.1021/acsami.1c23069.
- J. Wu, T. Zhai, J. Sun, Q. Yu, Y. Feng, R. Li, H. Wang, Q. Ouyang, T. Yang, Q. Zhan, L. Deng, M. Qin and F. Wang, J. Colloid Interface Sci., 2022, 624, 307–319, DOI:10.1016/j.jcis.2022.05.121.
- V. Puri, K. R. Chaudhary, A. Singh and C. Singh, Curr. Res. Pharmacol. Drug Discov., 2022, 3, 100084, DOI:10.1016/j.crphar.2022.100084.
- B. Casciaro, I. d'Angelo, X. Zhang, M. R. Loffredo, G. Conte, F. Cappiello, F. Quaglia, Y. P. Di, F. Ungaro and M. L. Mangoni, Biomacromolecules, 2019, 20, 1876–1888, DOI:10.1021/acs.biomac.8b01829.
- A. Kumari, S. Pal, B. R. G, F. P. Mohny, N. Gupta, C. Miglani, B. Pattnaik, A. Pal and M. Ganguli, Mol. Pharm., 2022, 19, 1309–1324, DOI:10.1021/acs.molpharmaceut.1c00770.
- S. E. Drago, E. F. Craparo, R. Luxenhofer and G. Cavallaro, Nanomedicine, 2021, 37, 102451, DOI:10.1016/j.nano.2021.102451.
- G. Conte, G. Costabile, D. Baldassi, V. Rondelli, R. Bassi, D. Colombo, G. Linardos, E. V. Fiscarelli, R. Sorrentino, A. Miro, F. Quaglia, P. Brocca, I. d'Angelo, O. M. Merkel and F. Ungaro, ACS Appl. Mater. Interfaces, 2022, 14, 7565–7578, DOI:10.1021/acsami.1c14975.
- M. Comegna, G. Conte, A. P. Falanga, M. Marzano, G. Cernera, A. M. Di Lullo, F. Amato, N. Borbone, S. D'Errico, F. Ungaro, I. d'Angelo, G. Oliviero and G. Castaldo, Sci. Rep., 2021, 11, 6393, DOI:10.1038/s41598-021-85549-z.
- Q. Liu, J. Xue, X. Zhang, J. Chai, L. Qin, J. Guan, X. Zhang and S. Mao, Acta Biomater., 2022, 147, 391–402, DOI:10.1016/j.actbio.2022.05.038.
- J. Leal, X. Peng, X. Liu, D. Arasappan, D. C. Wylie, S. H. Schwartz, J. J. Fullmer, B. C. McWilliams, H. D. C. Smyth and D. Ghosh, J. Controlled Release, 2020, 322, 457–469, DOI:10.1016/j.jconrel.2020.03.032.
- C. Conte, F. Mastrotto, V. Taresco, A. Tchoryk, F. Quaglia, S. Stolnik and C. Alexander, J. Controlled Release, 2018, 277, 126–141, DOI:10.1016/j.jconrel.2018.03.011.
- T. T. Tran and K. Hadinoto, Colloids Surf., B, 2020, 193, 111095, DOI:10.1016/j.colsurfb.2020.111095.
- B. Le-Vinh, C. Steinbring, R. Wibel, J. D. Friedl and A. Bernkop-Schnurch, Eur. J. Pharm. Biopharm., 2021, 163, 109–119, DOI:10.1016/j.ejpb.2021.03.012.
- L. Gomes Dos Reis, W. H. Lee, M. Svolos, L. M. Moir, R. Jaber, N. Windhab, P. M. Young and D. Traini, Pharmaceutics, 2019, 11, 12, DOI:10.3390/pharmaceutics11010012.
- N. Varga, V. Hornok, L. Janovak, I. Dekany and E. Csapo, Colloids Surf., B, 2019, 176, 212–218, DOI:10.1016/j.colsurfb.2019.01.012.
- N. Nafee, A. Husari, C. K. Maurer, C. Lu, C. de Rossi, A. Steinbach, R. W. Hartmann, C. M. Lehr and M. Schneider, J. Controlled Release, 2014, 192, 131–140, DOI:10.1016/j.jconrel.2014.06.055.
- C. P. Hollis, H. L. Weiss, M. Leggas, B. M. Evers, R. A. Gemeinhart and T. Li, J. Controlled Release, 2013, 172, 12–21, DOI:10.1016/j.jconrel.2013.06.039.
- V. Ivanova, O. B. Garbuzenko, K. R. Reuhl, D. C. Reimer, V. P. Pozharov and T. Minko, Eur. J. Pharm. Biopharm., 2013, 84, 335–344, DOI:10.1016/j.ejpb.2012.11.023.
- B. P. H. Wittgen, P. W. A. Kunst, K. van der Born, A. W. van Wijk, W. Perkins, F. G. Pilkiewicz, R. Perez-Soler, S. Nicholson, G. J. Peters and P. E. Postmus, Clin. Cancer Res., 2007, 13, 2414–2421, DOI:10.1158/1078-0432.Ccr-06-1480.
- X. Jin, J. Oh, J.-Y. Cho, S. Lee and S.-J. Rhee, Antibiotics, 2020, 9, 784 CrossRef CAS PubMed.
- X. Murgia, P. Pawelzyk, U. F. Schaefer, C. Wagner, N. Willenbacher and C. M. Lehr, Biomacromolecules, 2016, 17, 1536–1542, DOI:10.1021/acs.biomac.6b00164.
- C. S. Schneider, Q. Xu, N. J. Boylan, J. Chisholm, B. C. Tang, B. S. Schuster, A. Henning, L. M. Ensign, E. Lee, P. Adstamongkonkul, B. W. Simons, S.-Y. S. Wang, X. Gong, T. Yu, M. P. Boyle, J. S. Suk and J. Hanes, Sci. Adv., 2017, 3, e1601556 CrossRef PubMed.
- X. Bai, M. Li and G. Hu, Nanoscale, 2020, 12, 3931–3940, 10.1039/c9nr09251j.
- M.-C. Jones, S. A. Jones, Y. Riffo-Vasquez, D. Spina, E. Hoffman, A. Morgan, A. Patel, C. Page, B. Forbes and L. A. Dailey, J. Controlled Release, 2014, 183, 94–104, DOI:10.1016/j.jconrel.2014.03.022.
- Q. Zhong, B. V. Humia, A. R. Punjabi, F. F. Padilha and S. R. P. da Rocha, Eur. J. Pharm. Sci., 2017, 109, 86–95, DOI:10.1016/j.ejps.2017.07.030.
- Q. Xu, L. M. Ensign, N. J. Boylan, A. Schon, X. Gong, J.-C. Yang, N. W. Lamb, S. Cai, T. Yu, E. Freire and J. Hanes, ACS Nano, 2015, 9, 9217–9227 CrossRef CAS PubMed.
- O. B. Garbuzenko, N. Kbah, A. Kuzmov, N. Pogrebnyak, V. Pozharov and T. Minko, J. Controlled Release, 2019, 296, 225–231, DOI:10.1016/j.jconrel.2019.01.025.
- X. Huang, J. Chisholm, J. Zhuang, Y. Xiao, G. Duncan, X. Chen, J. S. Suk and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6595–E6602, DOI:10.1073/pnas.1705407114.
- J. S. Suk, A. J. Kim, K. Trehan, C. S. Schneider, L. Cebotaru, O. M. Woodward, N. J. Boylan, M. P. Boyle, S. K. Lai, W. B. Guggino and J. Hanes, J. Controlled Release, 2014, 178, 8–17, DOI:10.1016/j.jconrel.2014.01.007.
- A. Popov, L. Schopf, J. Bourassa and H. Chen, Int. J. Pharm., 2016, 502, 188–197, DOI:10.1016/j.ijpharm.2016.02.031.
- J. J. Verhoef, J. F. Carpenter, T. J. Anchordoquy and H. Schellekens, Drug Discov. Today, 2014, 19, 1945–1952, DOI:10.1016/j.drudis.2014.08.015.
- I. d'Angelo, B. Casciaro, A. Miro, F. Quaglia, M. L. Mangoni and F. Ungaro, Colloids Surf., B, 2015, 135, 717–725, DOI:10.1016/j.colsurfb.2015.08.027.
- A. S. Morris, S. C. Sebag, J. D. Paschke, A. Wongrakpanich, K. Ebeid, M. E. Anderson, I. M. Grumbach and A. K. Salem, Mol. Pharm., 2017, 14, 2166–2175, DOI:10.1021/acs.molpharmaceut.7b00114.
- F. Wan, T. Nylander, S. N. Klodzinska, C. Foged, M. Yang, S. G. Baldursdottir and H. M. Nielsen, ACS Appl. Mater. Interfaces, 2018, 10, 10678–10687, DOI:10.1021/acsami.7b19762.
- J. Heyder, Proc. Am. Thorac. Soc., 2004, 1, 315–320, DOI:10.1513/pats.200409-046TA.
- D. K. Jensen, L. B. Jensen, S. Koocheki, L. Bengtson, D. Cun, H. M. Nielsen and C. Foged, J. Controlled Release, 2012, 157, 141–148, DOI:10.1016/j.jconrel.2011.08.011.
- F. Ungaro, I. d'Angelo, C. Coletta, R. d'Emmanuele di Villa Bianca, R. Sorrentino, B. Perfetto, M. A. Tufano, A. Miro, M. I. La Rotonda and F. Quaglia, J. Controlled Release, 2012, 157, 149–159, DOI:10.1016/j.jconrel.2011.08.010.
- C. A. Ruge, A. Bohr, M. Beck-Broichsitter, V. Nicolas, N. Tsapis and E. Fattal, Colloids Surf., B, 2016, 139, 219–227, DOI:10.1016/j.colsurfb.2015.12.017.
- B. Porsio, M. G. Cusimano, D. Schillaci, E. F. Craparo, G. Giammona and G. Cavallaro, Biomacromolecules, 2017, 18, 3924–3935, DOI:10.1021/acs.biomac.7b00945.
- Z. Wang, J. L. Cuddigan, S. K. Gupta and S. A. Meenach, Int. J. Pharm., 2016, 512, 305–313, DOI:10.1016/j.ijpharm.2016.08.047.
- A. Torge, P. Grutzmacher, F. Mucklich and M. Schneider, Eur. J. Pharm. Sci., 2017, 104, 171–179, DOI:10.1016/j.ejps.2017.04.003.
- F. Wan, S. S. Bohr, S. N. Klodzinska, H. Jumaa, Z. Huang, T. Nylander, M. B. Thygesen, K. K. Sorensen, K. J. Jensen, C. Sternberg, N. Hatzakis and H. Morck Nielsen, ACS Appl. Mater. Interfaces, 2020, 12, 380–389, DOI:10.1021/acsami.9b19644.
- M. Y. T. Chow, R. Y. K. Chang and H. K. Chan, Adv. Drug Deliv. Rev., 2021, 168, 217–228, DOI:10.1016/j.addr.2020.06.001.
- A. Yildiz-Pekoz and C. Ehrhardt, Pharmaceutics, 2020, 12, DOI:10.3390/pharmaceutics12100911.
- J. G. Chan, J. Wong, Q. T. Zhou, S. S. Leung and H. K. Chan, AAPS PharmSciTech, 2014, 15, 882–897, DOI:10.1208/s12249-014-0114-y.
- A. Ari, Pediatr. Pulmonol., 2019, 54, 1735–1741, DOI:10.1002/ppul.24449.
- K. H. Chang, S. H. Moon, S. K. Yoo, B. J. Park and K. C. Nam, Pharmaceutics, 2020, 12, DOI:10.3390/pharmaceutics12080721.
- W. B. van den Bosch, S. F. Kloosterman, E. R. Andrinopoulou, R. Greidanus, M. W. H. Pijnenburg, H. Tiddens and H. M. Janssens, J. Asthma, 2022, 59, 2223–2233, DOI:10.1080/02770903.2021.1996597.
- L. Shachar-Berman, S. Bhardwaj, Y. Ostrovski, P. Das, P. Koullapis, S. Kassinos and J. Sznitman, Pharmaceutics, 2020, 12, DOI:10.3390/pharmaceutics12030230.
- B. Lehofer, F. Bloder, P. P. Jain, L. M. Marsh, G. Leitinger, H. Olschewski, R. Leber, A. Olschewski and R. Prassl, Eur. J. Pharm. Biopharm., 2014, 88, 1076–1085, DOI:10.1016/j.ejpb.2014.10.009.
- J. Szabova, O. Misik, M. Havlikova, F. Lizal and F. Mravec, Colloids Surf., B, 2021, 204, 111793, DOI:10.1016/j.colsurfb.2021.111793.
- M. Zaru, S. Mourtas, P. Klepetsanis, A. M. Fadda and S. G. Antimisiaris, Eur. J. Pharm. Biopharm., 2007, 67, 655–666, DOI:10.1016/j.ejpb.2007.04.005.
- M. Beck-Broichsitter, P. Kleimann, T. Gessler, W. Seeger, T. Kissel and T. Schmehl, Int. J. Pharm., 2012, 422, 398–408, DOI:10.1016/j.ijpharm.2011.10.012.
- Y. Yang, Y. Ding, B. Fan, Y. Wang, Z. Mao, W. Wang and J. Wu, J. Controlled Release, 2020, 321, 463–474, DOI:10.1016/j.jconrel.2020.02.030.
- Y. Gao, J. Wang, M. Chai, X. Li, Y. Deng, Q. Jin and J. Ji, ACS Nano, 2020, 14, 5686–5699, DOI:10.1021/acsnano.0c00269.
- W. Zhang, W. Xu, Y. Lan, X. He, K. Liu and Y. Liang, Int. J. Nanomed., 2019, 14, 5287–5301, DOI:10.2147/IJN.S203113.
- F. Guo, J. Wu, W. Wu, D. Huang, Q. Yan, Q. Yang, Y. Gao and G. Yang, J. Nanobiotechnol., 2018, 16, DOI:10.1186/s12951-018-0384-8.
- V. Lenders, R. Escudero, X. Koutsoumpou, L. Armengol Álvarez, J. Rozenski, S. J. Soenen, Z. Zhao, S. Mitragotri, P. Baatsen, K. Allegaert, J. Toelen and B. B. Manshian, J. Nanobiotechnol., 2022, 20, 0, DOI:10.1186/s12951-022-01544-.
- X. Wang, H. Chen, X. Zeng, W. Guo, Y. Jin, S. Wang, R. Tian, Y. Han, L. Guo, J. Han, Y. Wu and L. Mei, Acta Pharm. Sin. B, 2019, 9, 167–176, DOI:10.1016/j.apsb.2018.08.006.
- Z. Zhao, A. Ukidve, Y. Gao, J. Kim and S. Mitragotri, Sci. Adv., 2019, 5, eaax9250 CrossRef CAS PubMed.
- A. Kuzmov and T. Minko, J. Controlled Release, 2015, 219, 500–518, DOI:10.1016/j.jconrel.2015.07.024.
- O. B. Garbuzenko, M. Saad, S. Betigeri, M. Zhang, A. A. Vetcher, V. A. Soldatenkov, D. C. Reimer, V. P. Pozharov and T. Minko, Pharm. Res., 2009, 26, 382–394, DOI:10.1007/s11095-008-9755-4.
- L. Bonadies, P. Zaramella, A. Porzionato, G. Perilongo, M. Muraca and E. Baraldi, J. Clin. Med., 2020, 9, DOI:10.3390/jcm9051539.
- S. H. Abman, Am. J. Respir. Crit. Care Med., 2001, 164, 1755–1756, DOI:10.1164/rccm2109108.
- A. De Soyza, T. Aksamit, T.-J. Bandel, M. Criollo, J. S. Elborn, E. Operschall, E. Polverino, K. Roth, K. L. Winthrop and R. Wilson, Eur. Respir. J., 2018, 51, 1702052, DOI:10.1183/13993003.02052-2017.
- B. S. Schuster, J. S. Suk, G. F. Woodworth and J. Hanes, Biomaterials, 2013, 34, 3439–3446, DOI:10.1016/j.biomaterials.2013.01.064.
- M. A. U. Khalid, Y. S. Kim, M. Ali, B. G. Lee, Y.-J. Cho and K. H. Choi, Biochem. Eng. J., 2020, 155, DOI:10.1016/j.bej.2019.107469.
- D. Baptista, L. Moreira Teixeira, D. Barata, Z. Tahmasebi Birgani, J. King, S. van Riet, T. Pasman, A. A. Poot, D. Stamatialis, R. J. Rottier, P. S. Hiemstra, A. Carlier, C. van Blitterswijk, P. Habibovic, S. Giselbrecht and R. Truckenmuller, ACS Biomater. Sci. Eng., 2022, 8, 2684–2699, DOI:10.1021/acsbiomaterials.1c01463.
- G. Cafri, J. J. Gartner, T. Zaks, K. Hopson, N. Levin, B. C. Paria, M. R. Parkhurst, R. Yossef, F. J. Lowery, M. S. Jafferji, T. D. Prickett, S. L. Goff, C. T. McGowan, S. Seitter, M. L. Shindorf, A. Parikh, P. D. Chatani, P. F. Robbins and S. A. Rosenberg, J. Clin. Invest., 2020, 130, 5976–5988, DOI:10.1172/JCI134915.
- J. Byrne, H.-W. Huang, J. C. McRae, S. Babaee, A. Soltani, S. L. Becker and G. Traverso, Adv. Drug Delivery Rev., 2021, 177, 113926, DOI:10.1016/j.addr.2021.113926.
- A. Pietroiusti, E. Bergamaschi, M. Campagna, L. Campagnolo, G. De Palma, S. Iavicoli, V. Leso, A. Magrini, M. Miragoli, P. Pedata, L. Palombi and I. Iavicoli, Part. Fibre Toxicol., 2017, 14, 47, DOI:10.1186/s12989-017-0226-0.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71, DOI:10.1186/s12951-018-0392-8.
- J. Sun, X. Shen, Y. Li, Z. Guo, W. Zhu, L. Zuo, J. Zhao, L. Gu, J. Gong and J. Li, Nutrients, 2016, 8, 44, DOI:10.3390/nu8010044.
- O. Hartwig, M. A. Shetab Boushehri, K. S. Shalaby, B. Loretz, A. Lamprecht and C.-M. Lehr, Adv. Drug Delivery Rev., 2021, 175, 113828, DOI:10.1016/j.addr.2021.113828.
- E.-M. Collnot, H. Ali and C.-M. Lehr, J. Controlled Release, 2012, 161, 235–246, DOI:10.1016/j.jconrel.2012.01.028.
- G. P. Donaldson, S. M. Lee and S. K. Mazmanian, Nat. Rev. Microbiol., 2016, 14, 20–32, DOI:10.1038/nrmicro3552.
- M. Zarepour, K. Bhullar, M. Montero, C. Ma, T. Huang, A. Velcich, L. Xia and A. Vallance Bruce, Infect. Immun., 2013, 81, 3672–3683, DOI:10.1128/IAI.00854-13.
- J.-F. Sicard, G. Le Bihan, P. Vogeleer, M. Jacques and J. Harel, Front. Cell. Infect. Microbiol., 2017, 7, 387 CrossRef PubMed.
- M. Herath, S. Hosie, J. C. Bornstein, A. E. Franks and E. L. Hill-Yardin, Front. Cell. Infect. Microbiol., 2020, 10, 248 CrossRef CAS PubMed.
- R. Wichert, A. Ermund, S. Schmidt, M. Schweinlin, M. Ksiazek, P. Arnold, K. Knittler, F. Wilkens, B. Potempa, B. Rabe, M. Stirnberg, R. Lucius, J. W. Bartsch, S. Nikolaus, M. Falk-Paulsen, P. Rosenstiel, M. Metzger, S. Rose-John, J. Potempa, G. C. Hansson, P. J. Dempsey and C. Becker-Pauly, Cell Rep., 2017, 21, 2090–2103, DOI:10.1016/j.celrep.2017.10.087.
- Y. Tanaka, T. Goto, M. Kataoka, S. Sakuma and S. Yamashita, J. Pharm. Sci., 2015, 104, 3120–3127, DOI:10.1002/jps.24433.
- L. M. Ensign, R. Cone and J. Hanes, Adv. Drug Delivery Rev., 2012, 64, 557–570, DOI:10.1016/j.addr.2011.12.009.
- M. Falavigna, P. C. Stein, G. E. Flaten and M. P. di Cagno, Pharmaceutics, 2020, 12, 168, DOI:10.3390/pharmaceutics12020168.
- V. Muraleetharan, J. Mantaj, M. Swedrowska and D. Vllasaliu, RSC Adv., 2019, 9, 40487–40497, 10.1039/C9RA08403G.
- A. Berardi and F. Baldelli Bombelli, Expert Opin. Drug Delivery, 2019, 16, 563–566, DOI:10.1080/17425247.2019.1610384.
- Y. Wang, M. Li, X. Xu, W. Tang, L. Xiong and Q. Sun, J. Agric. Food Chem., 2019, 67, 2296–2306, DOI:10.1021/acs.jafc.8b05702.
- J. Zhang, A. Ou, X. Tang, R. Wang, Y. Fan, Y. Fang, Y. Zhao, P. Zhao, D. Chen, B. Wang and Y. Huang, J. Nanobiotechnol., 2022, 20, 389, DOI:10.1186/s12951-022-01598-0.
- B. J. Müller, A. V. Zhdanov, S. M. Borisov, T. Foley, I. A. Okkelman, V. Tsytsarev, Q. Tang, R. S. Erzurumlu, Y. Chen, H. Zhang, C. Toncelli, I. Klimant, D. B. Papkovsky and R. I. Dmitriev, Adv. Funct. Mater., 2018, 28, 1704598, DOI:10.1002/adfm.201704598.
- A. I. Barbosa, S. A. Costa Lima and S. Reis, Molecules, 2019, 24, 346, DOI:10.3390/molecules24020346.
- K. S. Kim, K. Suzuki, H. Cho, Y. S. Youn and Y. H. Bae, ACS Nano, 2018, 12, 8893–8900, DOI:10.1021/acsnano.8b04315.
- E. Baranowska-Wójcik, K. Gustaw, D. Szwajgier, P. Oleszczuk, B. Pawlikowska-Pawlęga, J. Pawelec and J. Kapral-Piotrowska, Foods, 2021, 10, 939, DOI:10.3390/foods10050939.
- P. Kiełbik, A. Jończy, J. Kaszewski, M. Gralak, J. Rosowska, R. Sapierzyński, B. Witkowski, Ł. Wachnicki, K. Lawniczak-Jablonska, P. Kuzmiuk, P. Lipiński, M. Godlewski and M. M. Godlewski, Pharmaceuticals, 2021, 14, 859, DOI:10.3390/ph14090859.
- G. M. DeLoid, Y. Wang, K. Kapronezai, L. R. Lorente, R. Zhang, G. Pyrgiotakis, N. V. Konduru, M. Ericsson, J. C. White, R. De La Torre-Roche, H. Xiao, D. J. McClements and P. Demokritou, Part. Fibre Toxicol., 2017, 14, 40, DOI:10.1186/s12989-017-0221-5.
- S. Nallanthighal, C. Chan, D. J. Bharali, S. A. Mousa, E. Vásquez and R. Reliene, NanoImpact, 2017, 5, 92–100, DOI:10.1016/j.impact.2017.01.003.
- G. K. Hinkley, P. Carpinone, J. W. Munson, K. W. Powers and S. M. Roberts, Part. Fibre Toxicol., 2015, 12, 9, DOI:10.1186/s12989-015-0085-5.
- L. Cosby, K. H. Lee, T. Knobloch, C. Weghorst and J. Winter, Int. J. Nanomed., 2020, 15, 8217–8230, DOI:10.2147/IJN.S259202.
- Z. Davoudi, N. Peroutka-Bigus, B. Bellaire, M. Wannemuehler, T. A. Barrett, B. Narasimhan and Q. Wang, J. Biomed. Mater. Res., Part A, 2018, 106, 876–886, DOI:10.1002/jbm.a.36305.
- N. SreeHarsha, C. Ramnarayanan, B. E. Al-Dhubiab, A. B. Nair, J. G. Hiremath, K. N. Venugopala, R. T. Satish, M. Attimarad and A. Shariff, BioMed Res. Int., 2019, 2019, 3950942, DOI:10.1155/2019/3950942.
- J. C. Imperiale, I. Schlachet, M. Lewicki, A. Sosnik and M. M. Biglione, Polymers, 2019, 11, 1862, DOI:10.3390/polym11111862.
- P. Zhao, X. Xia, X. Xu, K. K. C. Leung, A. Rai, Y. Deng, B. Yang, H. Lai, X. Peng, P. Shi, H. Zhang, P. W. Y. Chiu and L. Bian, Nat. Commun., 2021, 12, 7162, DOI:10.1038/s41467-021-27463-6.
- Z. Huang, Y. Shi, H. Wang, C. Chun, L. Chen, K. Wang, Z. Lu, Y. Zhao and X. Li, Int. J. Nanomed., 2021, 16, 8235–8250, DOI:10.2147/IJN.S344805.
- Z. Zhang, H. Li, G. Xu and P. Yao, Drug Delivery, 2018, 25, 1224–1233, DOI:10.1080/10717544.2018.1469685.
- X. Liu, W. Sun, N. Wu, N. Rong, C. Kang, S. Jian, C. Chen, C. Chen and X. Zhang, Vaccines, 2021, 9, 304, DOI:10.3390/vaccines9030304.
- A. Larrea, M. Arruebo, C. A. Serra and V. Sebastián, Micromachines, 2022, 13, 878, DOI:10.3390/mi13060878.
- C. L. Okolie, B. Mason, A. Mohan, N. Pitts and C. C. Udenigwe, Food Biosci., 2020, 37, 100672, DOI:10.1016/j.fbio.2020.100672.
- M. A. Oshi, J. Lee, J. Kim, N. Hasan, E. Im, Y. Jung and J.-W. Yoo, Pharmaceutics, 2021, 13, 1412, DOI:10.3390/pharmaceutics13091412.
- L. G. Gómez-Mascaraque, M. Martínez-Sanz, S. A. Hogan, A. López-Rubio and A. Brodkorb, Carbohydr. Polym., 2019, 223, 115121, DOI:10.1016/j.carbpol.2019.115121.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Sci. Rep., 2018, 8, 2178, DOI:10.1038/s41598-018-20632-6.
- A. Sadio, J. K. Gustafsson, B. Pereira, C. P. Gomes, G. C. Hansson, L. David, A. P. Pêgo and R. Almeida, PLoS One, 2014, 9, e99449, DOI:10.1371/journal.pone.0099449.
- X. Han, Y. Lu, J. Xie, E. Zhang, H. Zhu, H. Du, K. Wang, B. Song, C. Yang, Y. Shi and Z. Cao, Nat. Nanotechnol., 2020, 15, 605–614, DOI:10.1038/s41565-020-0693-6.
- R. Limage, E. Tako, N. Kolba, Z. Guo, A. García-Rodríguez, C. N. H. Marques and G. J. Mahler, Small, 2020, 16, 2000601, DOI:10.1002/smll.202000601.
- N. Krivova, O. Zaeva, T. Misina and N. Kuvshinov, Key Eng. Mater., 2016, 683, 447–453, DOI:10.4028/www.scientific.net/KEM.683.447.
- K. Gokulan, K. Williams, S. Orr and S. Khare, Int. J. Mol. Sci., 2021, 22, 9, DOI:10.3390/ijms22010009.
- V. Prasad, R. Davan, S. Jothi, P. Ayalasomayajula and D. B. Raju, Nano Biomed. Eng., 2013, 5, DOI:10.5101/nbe.v5i1.p46-49.
- A. Seth, D. Lafargue, C. Poirier, T. Badier, N. Delory, A. Laporte, J.-M. Delbos, V. Jeannin, J.-M. Péan and C. Ménager, Eur. J. Pharm. Sci., 2017, 100, 25–35, DOI:10.1016/j.ejps.2016.12.022.
- P. C. Naha, Y. Liu, G. Hwang, Y. Huang, S. Gubara, V. Jonnakuti, A. Simon-Soro, D. Kim, L. Gao, H. Koo and D. P. Cormode, ACS Nano, 2019, 13, 4960–4971, DOI:10.1021/acsnano.8b08702.
- L. Liu and F. Kong, J. Food Eng., 2021, 292, 110346, DOI:10.1016/j.jfoodeng.2020.110346.
- M. P. Sarparanta, L. M. Bimbo, E. M. Mäkilä, J. J. Salonen, P. H. Laaksonen, A. M. K. Helariutta, M. B. Linder, J. T. Hirvonen, T. J. Laaksonen, H. A. Santos and A. J. Airaksinen, Biomaterials, 2012, 33, 3353–3362, DOI:10.1016/j.biomaterials.2012.01.029.
- C. Yang and D. Merlin, Int. J. Nanomed., 2019, 14, 8875–8889, DOI:10.2147/IJN.S210315.
- Y. Yu, L. Xing, L. Li, J. Wu, J. He and Y. Huang, J. Controlled Release, 2022, 341, 215–226, DOI:10.1016/j.jconrel.2021.11.026.
- B. H. Bajka, N. M. Rigby, K. L. Cross, A. Macierzanka and A. R. Mackie, Colloids Surf., B, 2015, 135, 73–80, DOI:10.1016/j.colsurfb.2015.07.038.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692, DOI:10.2217/nnm.16.5.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124, DOI:10.1038/s41573-020-0090-8.
- A. Banerjee, J. Qi, R. Gogoi, J. Wong and S. Mitragotri, J. Controlled Release, 2016, 238, 176–185, DOI:10.1016/j.jconrel.2016.07.051.
- N. Zheng, J. Li, C. Xu, L. Xu, S. Li and L. Xu, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1132–1140, DOI:10.1080/21691401.2017.1362414.
- S.-j Cao, S. Xu, H.-m Wang, Y. Ling, J. Dong, R.-d Xia and X.-h Sun, AAPS PharmSciTech, 2019, 20, 190, DOI:10.1208/s12249-019-1325-z.
- C. Bao, B. Liu, B. Li, J. Chai, L. Zhang, L. Jiao, D. Li, Z. Yu, F. Ren, X. Shi and Y. Li, Nano Lett., 2020, 20, 1352–1361, DOI:10.1021/acs.nanolett.9b04841.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 4131, DOI:10.1038/s41598-017-03834-2.
- M. Yu, J. Wang, Y. Yang, C. Zhu, Q. Su, S. Guo, J. Sun, Y. Gan, X. Shi and H. Gao, Nano Lett., 2016, 16, 7176–7182, DOI:10.1021/acs.nanolett.6b03515.
- M. Yu, L. Xu, F. Tian, Q. Su, N. Zheng, Y. Yang, J. Wang, A. Wang, C. Zhu, S. Guo, X. Zhang, Y. Gan, X. Shi and H. Gao, Nat. Commun., 2018, 9, 2607, DOI:10.1038/s41467-018-05061-3.
- X. Cai, Y. Xu, L. Zhao, J. Xu, S. Li, C. Wen, X. Xia, Q. Dong, X. Hu, X. Wang, L. Chen, Z. Chen and W. Tan, Nano Today, 2021, 36, 101032, DOI:10.1016/j.nantod.2020.101032.
- A. A. Date, J. Hanes and L. M. Ensign, J. Controlled Release, 2016, 240, 504–526, DOI:10.1016/j.jconrel.2016.06.016.
- Y. Mu, L. Gong, T. Peng, J. Yao and Z. Lin, OpenNano, 2021, 5, 100031, DOI:10.1016/j.onano.2021.100031.
- L. Liu, W. Yao, Y. Rao, X. Lu and J. Gao, Drug Delivery, 2017, 24, 569–581, DOI:10.1080/10717544.2017.1279238.
- A. Sood, A. Dev, S. J. Mohanbhai, N. Shrimali, M. Kapasiya, A. C. Kushwaha, S. Roy Choudhury, P. Guchhait and S. Karmakar, ACS Appl. Nano Mater., 2019, 2, 6409–6417, DOI:10.1021/acsanm.9b01377.
- F. Luo, M. Wang, L. Huang, Z. Wu, W. Wang, A. Zafar, Y. Tian, M. Hasan and X. Shu, ACS Omega, 2020, 5, 11799–11808, DOI:10.1021/acsomega.0c01216.
- E. El-Maghawry, M. Tadros, S. Elkheshen and A. Abd-Elbary, Int. J. Nanomed., 2020, 15, 3965–3980, DOI:10.2147/IJN.S244124.
- R. S. Santos, G. R. Dakwar, R. Xiong, K. Forier, K. Remaut, S. Stremersch, N. Guimarães, S. Fontenete, J. Wengel, M. Leite, C. Figueiredo, S. C. De Smedt, K. Braeckmans and N. F. Azevedo, Mol. Ther. – Nucleic Acids, 2015, 4, DOI:10.1038/mtna.2015.46.
- D. Walker, B. T. Käsdorf, H.-H. Jeong, O. Lieleg and P. Fischer, Sci. Adv., 2015, 1, e1500501, DOI:10.1126/sciadv.1500501.
- W. Gao, R. Dong, S. Thamphiwatana, J. Li, W. Gao, L. Zhang and J. Wang, ACS Nano, 2015, 9, 117–123, DOI:10.1021/nn507097k.
- Y. Chen, M. Zhang, H. Zhao, Y. Liu, T. Wang, T. Lei, X. Xiang, L. Lu, Z. Yuan, J. Xu and J. Zhang, Nanoscale, 2022, 14, 8967–8977, 10.1039/D2NR01469F.
- M. F. Radwan, M. A. El-Moselhy, W. M. Alarif, M. Orif, N. K. Alruwaili and N. A. Alhakamy, Dose-Response, 2021, 19, 15593258211013655, DOI:10.1177/15593258211013655.
- J. N. Chu and G. Traverso, Nat. Rev. Gastroenterol. Hepatol., 2022, 19, 219–238, DOI:10.1038/s41575-021-00539-w.
- B.-H. Mao, Y.-K. Luo, B. Wang, Jr., C.-W. Chen, F.-Y. Cheng, Y.-H. Lee, S.-J. Yan and Y.-J. Wang, Part. Fibre Toxicol., 2022, 19, 6, DOI:10.1186/s12989-022-00447-0.
- D. A. Subramanian, R. Langer and G. Traverso, J. Nanobiotechnol., 2022, 20, 362, DOI:10.1186/s12951-022-01539-x.
- M. Liu, J. Zhang, W. Shan and Y. Huang, Asian J. Pharm. Sci., 2015, 10, 275–282, DOI:10.1016/j.ajps.2014.12.007.
- K. Maisel, L. Ensign, M. Reddy, R. Cone and J. Hanes, J. Controlled Release, 2015, 197, 48–57, DOI:10.1016/j.jconrel.2014.10.026.
- L. Mahlert, J. Anderski, D. Mulac and K. Langer, Eur. J. Pharm. Sci., 2019, 133, 28–39, DOI:10.1016/j.ejps.2019.03.010.
- V. Bourganis, T. Karamanidou, E. Samaridou, K. Karidi, O. Kammona and C. Kiparissides, Eur. J. Pharm. Biopharm., 2015, 97, 239–249, DOI:10.1016/j.ejpb.2015.01.021.
- G. Deng, Y. Wu, Z. Song, S. Li, M. Du, J. Deng, Q. Xu, L. Deng, H. S. Bahlol and H. Han, ACS Appl. Mater. Interfaces, 2022, 14, 13001–13012, DOI:10.1021/acsami.1c23342.
- M. Dawson, E. Krauland, D. Wirtz and J. Hanes, Biotechnol. Prog., 2004, 20, 851–857, DOI:10.1021/bp0342553.
- X. Tan, N. Yin, Z. Liu, R. Sun, J. Gou, T. Yin, Y. Zhang, H. He and X. Tang, Mol. Pharmaceutics, 2020, 17, 3177–3191, DOI:10.1021/acs.molpharmaceut.0c00223.
- J. Li, H. Qiang, W. Yang, Y. Xu, T. Feng, H. Cai, S. Wang, Z. Liu, Z. Zhang and J. Zhang, J. Controlled Release, 2022, 341, 31–43, DOI:10.1016/j.jconrel.2021.11.020.
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133–1174, DOI:10.3390/md13031133.
- J. Xie, A. Li and J. Li, Macromol. Rapid Commun., 2017, 38, 1700413, DOI:10.1002/marc.201700413.
- A. Murthy, P. R. Ravi, H. Kathuria and R. Vats, Int. J. Pharm., 2020, 588, 119731, DOI:10.1016/j.ijpharm.2020.119731.
- J. Wang, D. Chin, C. Poon, V. Mancino, J. Pham, H. Li, P.-Y. Ho, K. R. Hallows and E. J. Chung, J. Controlled Release, 2021, 329, 1198–1209, DOI:10.1016/j.jconrel.2020.10.047.
- H. Anter, I. Abu Hashim, W. Awadin and M. Meshali, Int. J. Nanomed., 2019, 14, 4911–4929, DOI:10.2147/IJN.S209987.
- A. Seth, D. Lafargue, C. Poirier, J.-M. Péan and C. Ménager, Eur. J. Pharm. Biopharm., 2014, 88, 374–381, DOI:10.1016/j.ejpb.2014.05.017.
- A. Parodi, P. Buzaeva, D. Nigovora, A. Baldin, D. Kostyushev, V. Chulanov, L. V. Savvateeva and A. A. Zamyatnin, J. Nanobiotechnol., 2021, 19(354), 2, DOI:10.1186/s12951-021-01100-.
- T. Ren, Q. Wang, Y. Xu, L. Cong, J. Gou, X. Tao, Y. Zhang, H. He, T. Yin, H. Zhang, Y. Zhang and X. Tang, J. Controlled Release, 2018, 269, 423–438, DOI:10.1016/j.jconrel.2017.11.015.
- Y. Zhang, M. Xiong, X. Ni, J. Wang, H. Rong, Y. Su, S. Yu, I. S. Mohammad, S. S. Y. Leung and H. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 18077–18088, DOI:10.1021/acsami.1c00580.
- R. Rao, X. Liu, Y. Li, X. Tan, H. Zhou, X. Bai, X. Yang and W. Liu, Biomater. Sci., 2021, 9, 685–699, 10.1039/D0BM01772H.
- H. Lian, T. Zhang, J. Sun, X. Liu, G. Ren, L. Kou, Y. Zhang, X. Han, W. Ding, X. Ai, C. Wu, L. Li, Y. Wang, Y. Sun, S. Wang and Z. He, Mol. Pharmaceutics, 2013, 10, 3447–3458, DOI:10.1021/mp400282r.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. Del Favero, L. Cantù and S. Nicoli, J. Controlled Release, 2021, 332, 312–336, DOI:10.1016/j.jconrel.2021.02.031.
- H. Cheng, Z. Cui, S. Guo, X. Zhang, Y. Huo and S. Mao, Acta Biomater., 2021, 135, 506–519, DOI:10.1016/j.actbio.2021.08.046.
- S. K. Lai, Y.-Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171, DOI:10.1016/j.addr.2008.11.002.
- M. Vitulo, E. Gnodi, R. Meneveri and D. Barisani, Int. J. Mol. Sci., 2022, 23, 4339, DOI:10.3390/ijms23084339.
- M. Boegh and H. M. Nielsen, Basic Clin. Pharmacol. Toxicol., 2015, 116, 179–186, DOI:10.1111/bcpt.12342.
- A.-C. Groo and F. Lagarce, Drug Discovery Today, 2014, 19, 1097–1108, DOI:10.1016/j.drudis.2014.01.011.
- J. A. Jiminez, T. C. Uwiera, G. Douglas Inglis and R. R. E. Uwiera, Gut Pathogens, 2015, 7, 29, DOI:10.1186/s13099-015-0076-y.
- M. Bailey, Z. Christoforidou and M. C. Lewis, Vet. Immunol. Immunopathol., 2013, 152, 13–19, DOI:10.1016/j.vetimm.2012.09.022.
- A. Melero, C. Draheim, S. Hansen, E. Giner, J. J. Carreras, R. Talens-Visconti, T. M. Garrigues, J. E. Peris, M. C. Recio, R. Giner and C.-M. Lehr, Eur. J. Pharm. Biopharm., 2017, 119, 361–371, DOI:10.1016/j.ejpb.2017.07.004.
- I. Hubatsch, E. G. E. Ragnarsson and P. Artursson, Nat. Protoc., 2007, 2, 2111–2119, DOI:10.1038/nprot.2007.303.
- H. Takedatsu, K. Mitsuyama and T. Torimura, World J. Gastroenterol., 2015, 21, 11343–11352, DOI:10.3748/wjg.v21.i40.11343.
- S. Hua, E. Marks, J. J. Schneider and S. Keely, Nanomedicine, 2015, 11, 1117–1132, DOI:10.1016/j.nano.2015.02.018.
- A. Vieira, L. Chaves, M. Pinheiro, D. Ferreira, B. Sarmento and S. Reis, Design and statistical modeling of mannose-decorated dapsone-containing nanoparticles as a strategy of targeting intestinal M-cells, 2016.
- S. S. Bachhav, V. D. Dighe, D. Kotak and P. V. Devarajan, Int. J. Pharm., 2017, 532, 612–622, DOI:10.1016/j.ijpharm.2017.09.040.
- J. Zhang, C. Tang and C. Yin, Biomaterials, 2013, 34, 3667–3677, DOI:10.1016/j.biomaterials.2013.01.079.
- N. Dammes, M. Goldsmith, S. Ramishetti, J. L. J. Dearling, N. Veiga, A. B. Packard and D. Peer, Nat. Nanotechnol., 2021, 16, 1030–1038, DOI:10.1038/s41565-021-00928-x.
- H. S. Kim and C. H. Jung, Int. J. Mol. Sci., 2021, 22, 9936, DOI:10.3390/ijms22189936.
- M. M. Smits and D. H. Van Raalte, Front. Endocrinol., 2021, 12, 645563, DOI:10.3389/fendo.2021.645563.
- R. Ismail, A. Bocsik, G. Katona, I. Gróf, M. A. Deli and I. Csóka, Pharmaceutics, 2019, 11, 599, DOI:10.3390/pharmaceutics11110599.
- J. W. Shreffler, J. E. Pullan, K. M. Dailey, S. Mallik and A. E. Brooks, Int. J. Mol. Sci., 2019, 20, 6056, DOI:10.3390/ijms20236056.
- R. A. Yokel, Nanomedicine, 2020, 15, 409–432 CrossRef CAS PubMed.
- E. M. Rhea and W. A. Banks, Front. Neurosci., 2019, 13, 521, DOI:10.3389/fnins.2019.00521.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924, DOI:10.7150/thno.49577.
- C. D. Nwagwu, A. V. Immidisetti, M. Y. Jiang, O. Adeagbo, D. C. Adamson and A. M. Carbonell, Pharmaceutics, 2021, 13, DOI:10.3390/pharmaceutics13040561.
- S. A. Chowdhary, T. Ryken and H. B. Newton, J. Neurooncol., 2015, 122, 367–382, DOI:10.1007/s11060-015-1724-2.
- S. M. Lombardo, M. Schneider, A. E. Tureli and N. Gunday Tureli, Beilstein J. Nanotechnol., 2020, 11, 866–883, DOI:10.3762/bjnano.11.72.
- W. Tang, W. Fan, J. Lau, L. Deng, Z. Shen and X. Chen, Chem. Soc. Rev., 2019, 48, 2967–3014, 10.1039/c8cs00805a.
- N. J. Abbott, L. Ronnback and E. Hansson, Nat. Rev. Neurosci., 2006, 7, 41–53, DOI:10.1038/nrn1824.
- G. Li, W. Yuan and B. M. Fu, J. Biomech., 2010, 43, 2133–2140, DOI:10.1016/j.jbiomech.2010.03.047.
- S. Ruan, Y. Zhou, X. Jiang and H. Gao, Adv. Sci., 2021, 8, 2004025, DOI:10.1002/advs.202004025.
- J. Keaney and M. Campbell, FEBS J., 2015, 282, 4067–4079, DOI:10.1111/febs.13412.
- A. E. Caprifico, P. J. S. Foot, E. Polycarpou and G. Calabrese, Pharmaceutics, 2020, 12, DOI:10.3390/pharmaceutics12111013.
- L. Ribovski, N. M. Hamelmann and J. M. J. Paulusse, Pharmaceutics, 2021, 13, DOI:10.3390/pharmaceutics13122045.
- N. Voigt, P. Henrich-Noack, S. Kockentiedt, W. Hintz, J. Tomas and B. A. Sabel, Eur. J. Pharm. Biopharm., 2014, 87, 19–29, DOI:10.1016/j.ejpb.2014.02.013.
- Q. Bao, P. Hu, Y. Xu, T. Cheng, C. Wei, L. Pan and J. Shi, ACS Nano, 2018, 12, 6794–6805, DOI:10.1021/acsnano.8b01994.
- L. L. Israel, O. Braubach, A. Galstyan, A. Chiechi, E. S. Shatalova, Z. Grodzinski, H. Ding, K. L. Black, J. Y. Ljubimova and E. Holler, ACS Nano, 2019, 13, 1253–1271, DOI:10.1021/acsnano.8b06437.
- R. Patil, A. Galstyan, Z. B. Grodzinski, E. S. Shatalova, S. Wagner, L. L. Israel, H. Ding, K. L. Black, J. Y. Ljubimova and E. Holler, Int. J. Nanomed., 2020, 15, 3057–3070, DOI:10.2147/IJN.S238265.
- A. Kadari, D. Pooja, R. H. Gora, S. Gudem, V. R. M. Kolapalli, H. Kulhari and R. Sistla, Eur. J. Pharm. Biopharm., 2018, 132, 168–179, DOI:10.1016/j.ejpb.2018.09.012.
- M. Meenu, K. H. Reeta, A. K. Dinda, S. K. Kottarath and Y. K. Gupta, Epilepsy Res., 2019, 158, DOI:10.1016/j.eplepsyres.2019.106219.
- X. Lu, Y. Zhang, L. Wang, G. Li, J. Gao and Y. Wang, Drug Deliv., 2021, 28, 380–389, DOI:10.1080/10717544.2021.1883158.
- H. Azhari, M. Younus, S. M. Hook, B. J. Boyd and S. B. Rizwan, Int. J. Pharm., 2021, 600, DOI:10.1016/j.ijpharm.2021.120411.
- C. Zhao, J. Zhang, H. Hu, M. Qiao, D. Chen, X. Zhao and C. Yang, Mater. Sci. Eng., C, 2018, 92, 1031–1040, DOI:10.1016/j.msec.2018.02.004.
- T. I. Janjua, A. Ahmed-Cox, A. K. Meka, F. M. Mansfeld, H. Forgham, R. M. C. Ignacio, Y. Cao, J. A. McCarroll, R. Mazzieri, M. Kavallaris and A. F. Popat, Nanoscale, 2021, 13, 16909–16922, 10.1039/d1nr03553c.
- H. S. Kim, S. J. Lee and D. Y. Lee, Bioact. Mater., 2022, 8, 35–48, DOI:10.1016/j.bioactmat.2021.06.026.
- H. S. Kim, M. Seo, T. E. Park and D. Y. Lee, J. Nanobiotechnol., 2022, 20, 14, DOI:10.1186/s12951-021-01220-9.
- J. Wei, D. Wu, Y. Shao, B. Guo, J. Jiang, J. Chen, J. Zhang, F. Meng and Z. Zhong, J. Controlled Release, 2022, 347, 68–77, DOI:10.1016/j.jconrel.2022.04.048.
- Y. Jiang, J. Zhang, F. Meng and Z. Zhong, ACS Nano, 2018, 12, 11070–11079, DOI:10.1021/acsnano.8b05265279.
- P. Thammasit, C. S. Tharinjaroen, Y. Tragoolpua, V. Rickerts, R. Georgieva, H. Bäumler and K. Tragoolpua, Front. Pharmacol., 2021, 12, 723727, DOI:10.3389/fphar.2021.723727.
- J. Wei, D. Wu, S. Zhao, Y. Shao, Y. Xia, D. Ni, X. Qiu, J. Zhang, J. Chen, F. Meng and Z. Zhong, Adv. Sci., 2022, 9, e2103689, DOI:10.1002/advs.202103689.
- M. Meenu, K. H. Reeta, A. K. Dinda, S. K. Kottarath and Y. K. Gupta, Epilepsy Res., 2019, 158, DOI:10.1016/j.eplepsyres.2019.106219.
- H. Zhang, W. Jiang, Y. Zhao, T. Song, Y. Xi, G. Han, Y. Jin, M. Song, K. Bai, J. Zhou and Y. Ding, Nano Lett., 2022, 22, 2450–2460, DOI:10.1021/acs.nanolett.2c00191.
- A. Galstyan, J. L. Markman, E. S. Shatalova, A. Chiechi, A. J. Korman, R. Patil, D. Klymyshyn, W. G. Tourtellotte, L. L. Israel, O. Braubach, V. A. Ljubimov, L. A. Mashouf, A. Ramesh, Z. B. Grodzinski, M. L. Penichet, K. L. Black, E. Holler, T. Sun, H. Ding, A. V. Ljubimov and J. Y. Ljubimova, Nat. Commun., 2019, 10, 3850, DOI:10.1038/s41467-019-11719-3.
- K. Fan, X. Jia, M. Zhou, K. Wang, J. Conde, J. He, J. Tian and X. Yan, ACS Nano, 2018, 12, 4105–4115, DOI:10.1021/acsnano.7b06969.
- J. Shen, Z. Zhao, W. Shang, C. Liu, B. Zhang, Z. Xu and H. Cai, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 192–200, DOI:10.1080/21691401.2018.1548471.
- X. He, X. Wang, L. Yang, Z. Yang, W. Yu, Y. Wang, R. Liu, M. Chen and H. Gao, Acta Pharm. Sin. B, 2022, 12, 1987–1999, DOI:10.1016/j.apsb.2022.02.001.
- S. Singh, N. Drude, L. Blank, P. B. Desai, H. Königs, S. Rütten, K. J. Langen, M. Möller, F. M. Mottaghy and A. Morgenroth, Adv. Healthcare Mater., 2021, 10, 2100812, DOI:10.1002/adhm.202100812.
- T. Dube, N. Kumar, M. Bishnoi and J. J. Panda, Bioconjug. Chem., 2021, 32, 2014–2031, DOI:10.1021/acs.bioconjchem.1c00321.
- B. A. Bony, A. W. Tarudji, H. A. Miller, S. Gowrikumar, S. Roy, E. T. Curtis, C. C. Gee, A. Vecchio, P. Dhawan and F. M. Kievit, ACS Nano, 2021, 15, 18520–18531, DOI:10.1021/acsnano.1c0843.
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and K. Kataoka, ACS Nano, 2013, 7, 8583–8592, DOI:10.1021/nn402662d.
- Z. Wang, J. Yao, Z. Guan, H. Wu, H. Cheng, G. Yan and R. Tang, Colloids Surf., B, 2021, 207, DOI:10.1016/j.colsurfb.2021.112052.
- X. Guo, G. Deng, J. Liu, P. Zou, F. Du, F. Liu, A. T. Chen, R. Hu, M. Li, S. Zhang, Z. Tang, L. Han, J. Liu, K. N. Sheth, Q. Chen, X. Gou and J. Zhou, ACS Nano, 2018, 12, 8723–8732, DOI:10.1021/acsnano.8b04787.
- M. Zhang, Z. Wang, C. Wang, Y. Wu, Z. Li and Z. Liu, ACS Nano, 2021, 15, 11940–11952, DOI:10.1021/acsnano.1c03117.
- Y. J. Kang, C. K. Holley, M. R. Abidian, A. B. Madhankumar, J. Connor and S. Majd, Adv. Healthcare Mater., 2021, 10, DOI:10.1002/adhm.202001261.
- M. S. Alghamri, K. Banerjee, A. A. Mujeeb, A. Mauser, A. Taher, R. Thalla, B. L. McClellan, M. L. Varela, S. M. Stamatovic, G. Martinez-Revollar, A. V. Andjelkovic, J. V. Gregory, P. Kadiyala, A. Calinescu, J. A. Jiménez, A. A. Apfelbaum, E. R. Lawlor, S. Carney, A. Comba, S. M. Faisal, M. Barissi, M. B. Edwards, H. Appelman, Y. Sun, J. Gan, R. Ackermann, A. Schwendeman, M. Candolfi, M. R. Olin, J. Lahann, P. R. Lowenstein and M. G. Castro, ACS Nano, 2022, 16, 8729–8750, DOI:10.1021/acsnano.1c07492.
- H. Zhang, C. Shi, F. Han, M. Li, H. Ma, R. Sui, S. Long, W. Sun, J. Du, J. Fan, H. Piao and X. Peng, Biomaterials, 2022, 289, DOI:10.1016/j.biomaterials.2022.121770.
- J. Lu, R. Li, B. Mu, Y. Peng, Y. Zhao, Y. Shi, L. Guo, L. Hai and Y. Wu, Eur J. Med. Chem., 2022, 230, DOI:10.1016/j.ejmech.2021.114093.
- Y. Tian, Z. Zheng, X. Wang, S. Liu, L. Gu, J. Mu, X. Zheng, Y. Li and S. Shen, J. Nanobiotechnol., 2022, 20, 318, DOI:10.1186/s12951-022-01493-8.
- J. Xie, D. Gonzalez-Carter, T. A. Tockary, N. Nakamura, Y. Xue, M. Nakakido, H. Akiba, A. Dirisala, X. Liu, K. Toh, T. Yang, Z. Wang, S. Fukushima, J. Li, S. Quader, K. Tsumoto, T. Yokota, Y. Anraku and K. Kataoka, ACS Nano, 2020, 14, 6729–6742, DOI:10.1021/acsnano.9b09991.
- D. A. Gonzalez-Carter, Z. Y. Ong, C. M. McGilvery, I. E. Dunlop, D. T. Dexter and A. E. Porter, Nanomedicine, 2019, 15, 1–11, DOI:10.1016/j.nano.2018.08.011.
- J. Ban, S. Li, Q. Zhan, X. Li, H. Xing, N. Chen, L. Long, X. Hou, J. Zhao and X. Yuan, Macromol. Biosci., 2021, 21, DOI:10.1002/mabi.202000392.
- H. Wang, Y. Chao, H. Zhao, X. Zhou, F. Zhang, Z. Zhang, Z. Li, J. Pan, J. Wang, Q. Chen and Z. Liu, ACS Nano, 2022, 16, 664–674, DOI:10.1021/acsnano.1c08120.
- S. I. Aguiar, J. N. R. Dias, A. S. André, M. L. Silva, D. Martins, B. Carrapiço, M. Castanho, J. Carriço, M. Cavaco, M. M. Gaspar, R. J. Nobre, L. P. de Almeida, S. Oliveira, L. Gano, J. D. G. Correia, C. Barbas, J. Gonçalves, V. Neves and F. Aires-Da-silva, Pharmaceutics, 2021, 13, DOI:10.3390/pharmaceutics13101598.
- X. Li, V. Vemireddy, Q. Cai, H. Xiong, P. Kang, X. Li, M. Giannotta, H. N. Hayenga, E. Pan, S. R. Sirsi, C. Mateo, D. Kleinfeld, C. Greene, M. Campbell, E. Dejana, R. Bachoo and Z. Qin, Nano Lett., 2021, 21, 9805–9815, DOI:10.1021/acs.nanolett.1c02996.
- C. K. Elechalawar, D. Bhattacharya, M. T. Ahmed, H. Gora, K. Sridharan, P. Chaturbedy, S. H. Sinha, M. M. Chandra Sekhar Jaggarapu, K. P. Narayan, S. Chakravarty, M. Eswaramoorthy, T. K. Kundu and R. Banerjee, Nanoscale Adv., 2019, 1, 3555–3567, 10.1039/c9na00056a.
- M. Li, K. Hu, D. Lin, Z. Wang, M. Xu, J. Huang, Z. Chen, Y. Zhang, L. Yin, R. You, C. H. Li and Y. Q. Guan, ACS Biomater. Sci. Eng., 2021, 7, 1216–1229, DOI:10.1021/acsbiomaterials.0c01474.
- S. Meenu Vasudevan, N. Ashwanikumar and G. S. Vinod Kumar, Biomater. Sci., 2019, 7, 4017–4021, 10.1039/c9bm00955h.
- Y. Yin, J. Wang, M. Yang, R. Du, G. Pontrelli, S. McGinty, G. Wang, T. Yin and Y. Wang, Nanoscale, 2020, 12, 2946–2960, 10.1039/c9nr08741a.
- O. Betzer, N. Perets, A. Angel, M. Motiei, T. Sadan, G. Yadid, D. Offen and R. Popovtzer, ACS Nano, 2017, 11, 10883–10893, DOI:10.1021/acsnano.7b04495.
- W. Niu, Q. Xiao, X. Wang, J. Zhu, J. Li, X. Liang, Y. Peng, C. Wu, R. Lu, Y. Pan, J. Luo, X. Zhong, H. He, Z. Rong, J. B. Fan and Y. A. Wang, Nano Lett., 2021, 21, 1484–1492, DOI:10.1021/acs.nanolett.0c04753.
- B. Tang, W. Zeng, L. L. Song, H. M. Wang, L. Q. Qu, H. H. Lo, L. Yu, A. G. Wu, V. K. W. Wong and B. Y. K. Law, Pharmaceuticals, 2022, 15, DOI:10.3390/ph15010083.
- N. Perets, O. Betzer, R. Shapira, S. Brenstein, A. Angel, T. Sadan, U. Ashery, R. Popovtzer and D. Offen, Nano Lett., 2019, 19, 3422–3431, DOI:10.1021/acs.nanolett.8b04148.
- R. Upadhya, L. N. Madhu, S. Attaluri, D. L. G. Gitaí, M. R. Pinson, M. Kodali, G. Shetty, G. Zanirati, S. Kumar, B. Shuai, S. T. Weintraub and A. K. Shetty, J. Extracell Vesicles, 2020, 9, DOI:10.1080/20013078.2020.1809064.
- T. Tian, R. Liang, G. Erel-Akbaba, L. Saad, P. J. Obeid, J. Gao, E. A. Chiocca, R. Weissleder and B. A. Tannous, ACS Nano, 2022, 16, 1940–1953, DOI:10.1021/acsnano.1c05505.
- S. Shan, J. Chen, Y. Sun, Y. Wang, B. Xia, H. Tan, C. Pan, G. Gu, J. Zhong, G. Qing, Y. Zhang, J. Wang, Y. Wang, Y. Wang, P. Zuo, C. Xu, F. Li, W. Guo, L. Xu, M. Chen, Y. Fan, L. Zhang and X. J. Liang, Adv. Sci., 2022, 9, DOI:10.1002/advs.202200353.
- K. Wang, U. S. Kumar, N. Sadeghipour, T. F. Massoud and R. A. Paulmurugan, ACS Nano, 2021, 15, 18327–18346, DOI:10.1021/acsnano.1c07587.
- P. Wang, X. Zheng, Q. Guo, P. Yang, X. Pang, K. Qian, W. Lu, Q. Zhang and X. Jiang, J. Controlled Release, 2018, 279, 220–233, DOI:10.1016/j.jconrel.2018.04.034.
- A. Parmar, A. Jain, S. Uppal, S. K. Mehta, K. Kaur, B. Singh, R. Sandhir and S. Sharma, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 704–719, DOI:10.1080/21691401.2018.1468768.
- Z. Shen, T. Liu, Y. Li, J. Lau, Z. Yang, W. Fan, Z. Zhou, C. Shi, C. Ke, V. I. Bregadze, S. K. Mandal, Y. Liu, Z. Li, T. Xue, G. Zhu, J. Munasinghe, G. Niu, A. Wu and X. Chen, ACS Nano, 2018, 12, 11355–11365, DOI:10.1021/acsnano.8b06201.
- S. Wang, H. Shen, Q. Mao, Q. Tao, G. Yuan, L. Zeng, Z. Chen, Y. Zhang, L. Cheng, J. Zhang, H. Dai, C. Hu, Y. Pan and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 56825–56837, DOI:10.1021/acsami.1c12406.
- R. Sun, M. Liu, J. Lu, B. Chu, Y. Yang, B. Song, H. Wang and Y. He, Nat. Commun., 2022, 13, DOI:10.1038/s41467-022-32837-5.
- M. M. D’Elios, A. Aldinucci, R. Amoriello, M. Benagiano, E. Bonechi, P. Maggi, A. Flori, C. Ravagli, D. Saer, L. Cappiello, L. Conti, B. Valtancoli, A. Bencini, L. Menichetti, G. Baldi and C. Ballerini, RSC Adv., 2018, 8, 904–913, 10.1039/c7ra11290d.
- W. He, X. Li, M. Morsch, M. Ismail, Y. Liu, F. U. Rehman, D. Zhang, Y. Wang, M. Zheng, R. Chung, Y. Zou and B. Shi, ACS Nano, 2022, 16(4), 6293–6308, DOI:10.1021/acsnano.2c00320.
- Y. Liu, Y. Zou, C. Feng, A. Lee, J. Yin, R. Chung, J. B. Park, H. Rizos, W. Tao, M. Zheng, O. C. Farokhzad and B. Shi, Nano Lett., 2020, 20, 1637–1646, DOI:10.1021/acs.nanolett.9b04683.
- T. Yin, Q. Fan, F. Hu, X. Ma, Y. Yin, B. Wang, L. Kuang, X. Hu, B. Xu and Y. Wang, Nano Lett., 2022, 22, 6606–6614, DOI:10.1021/acs.nanolett.2c01863.
- W. Hao, Y. Cui, Y. Fan, M. Chen, G. Yang, Y. Wang, M. Yang, Z. Li, W. Gong, Y. Yang and C. Gao, J. Nanobiotechnol., 2021, 19, 378, DOI:10.1186/s12951-021-01110-0.
- G. Deng, X. Peng, Z. Sun, W. Zheng, J. Yu, L. Du, H. Chen, P. Gong, P. Zhang, L. Cai and B. Z. Tang, ACS Nano, 2020, 14, 11452–11462, DOI:10.1021/acsnano.0c03824.
- T. Xiao, M. He, F. Xu, Y. Fan, B. Jia, M. Shen, H. Wang and X. Shi, ACS Nano, 2021, 15, 20377–20390, DOI:10.1021/acsnano.1c08689.
- A. Kaushik, A. Yndart, V. Atluri, S. Tiwari, A. Tomitaka, P. Gupta, R. D. Jayant, D. Alvarez-Carbonell, K. Khalili and M. Nair, Sci. Rep., 2019, 9, DOI:10.1038/s41598-019-40222-4.
- S. Shirvalilou, S. Khoei, S. Khoee, S. R. Mahdavi, N. J. Raoufi, M. Motevalian and M. Y. Karimi, J. Photochem. Photobiol., B, 2020, 205, DOI:10.1016/j.jphotobiol.2020.111827.
- A. Marino, A. Camponovo, A. Degl’Innocenti, M. Bartolucci, C. Tapeinos, C. Martinelli, D. de Pasquale, F. Santoro, V. Mollo, S. Arai, M. Suzuki, Y. Harada, A. Petretto and G. Ciofani, Nanoscale, 2019, 11, 21227–21248, 10.1039/c9nr07976a.
- C. Berard, S. Desgranges, N. Dumas, A. Novell, B. Larrat, M. Hamimed, N. Taulier, M. A. Esteve, F. Correard and C. Contino-Pepin, Pharmaceutics, 2022, 14, 1498, DOI:10.3390/pharmaceutics14071498.
- H. Moon, K. Hwang, K. M. Nam, Y. S. Kim, M. J. Ko, H. R. Kim, H. J. Lee, M. J. Kim, T. H. Kim, K. S. Kang, N. G. Kim, S. W. Choi and C. Y. Kim, Biomater. Adv., 2022, 141, 213102, DOI:10.1016/j.bioadv.2022.213102.
- Y. Endo-Takahashi, R. Kurokawa, K. Sato, N. Takizawa, F. Katagiri, N. Hamano, R. Suzuki, K. Maruyama, M. Nomizu, N. Takagi and Y. Negishi, Pharmaceutics, 2021, 13, 1003, DOI:10.3390/pharmaceutics13071003.
- H. J. Chen, Y. Qin, Z. G. Wang, L. Wang, D. W. Pang, D. Zhao and S. L. Liu, Angew. Chem., Int. Ed., 2022, 61, DOI:10.1002/anie.202210285.
- A. K. Singh, S. K. Mishra, G. Mishra, A. Maurya, R. Awasthi, M. K. Yadav, N. Atri, P. K. Pandey and S. K. Singh, Drug Dev. Ind. Pharm., 2020, 46, 8–19, DOI:10.1080/03639045.2019.1698594.
- Z. Yang, Y. Du, Q. Sun, Y. Peng, R. Wang, Y. Zhou, Y. Wang, C. Zhang and X. Qi, ACS Nano, 2020, 14, 6191–6212, DOI:10.1021/acsnano.0c02249.
- V. Monge-Fuentes, A. Biolchi Mayer, M. R. Lima, L. R. Geraldes, L. N. Zanotto, K. G. Moreira, O. P. Martins, H. L. Piva, M. S. S. Felipe, A. C. Amaral, A. L. Bocca, A. C. Tedesco and M. R. Mortari, Sci. Rep., 2021, 11, 15185, DOI:10.1038/s41598-021-94175-8.
- M. Shilo, A. Sharon, K. Baranes, M. Motiei, J. P. Lellouche and R. Popovtzer, J. Nanobiotechnol., 2015, 13, 19, DOI:10.1186/s12951-015-0075-7.
- C. H. Li, M. K. Shyu, C. Jhan, Y. W. Cheng, C. H. Tsai, C. W. Liu, C. C. Lee, R. M. Chen and J. J. Kang, Toxicol. Sci, 2015, 148, 192–203, DOI:10.1093/toxsci/kfv176.
- S. Ohta, E. Kikuchi, A. Ishijima, T. Azuma, I. Sakuma and T. Ito, Sci. Rep., 2020, 10, 18220, DOI:10.1038/s41598-020-75253-9.
- F. R. Walter, A. R. Santa-Maria, M. Meszaros, S. Veszelka, A. Der and M. A. Deli, Tissue Barriers, 2021, 9, 1904773, DOI:10.1080/21688370.2021.1904773.
- L. Zhang, J. Fan, G. Li, Z. Yin and B. M. Fu, Cardiovasc. Eng. Technol., 2020, 11, 607–620, DOI:10.1007/s13239-020-00496-6.
- C. Saraiva, C. Praca, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47, DOI:10.1016/j.jconrel.2016.05.044.
- Y. H. Tsou, X. Q. Zhang, H. Zhu, S. Syed and X. Xu, Small, 2017, 13, 1701921, DOI:10.1002/smll.201701921.
- P. R. Lockman, J. M. Koziara, R. J. Mumper and D. D. Allen, J. Drug Target, 2004, 12, 635–641, DOI:10.1080/10611860400015936.
- S. Ding, A. I. Khan, X. Cai, Y. Song, Z. Lyu, D. Du, P. Dutta and Y. Lin, Mater. Today, 2020, 37, 112–125, DOI:10.1016/j.mattod.2020.02.001.
- M. Cooley, A. Sarode, M. Hoore, D. A. Fedosov, S. Mitragotri and A. Sen Gupta, Nanoscale, 2018, 10, 15350–15364, 10.1039/c8nr04042g.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753–10758, DOI:10.1073/pnas.1308345110.
- M. Nowak, T. D. Brown, A. Graham, M. E. Helgeson and S. Mitragotri, Bioeng. Transl. Med., 2020, 5, e10153, DOI:10.1002/btm2.10153.
- X. Liu, B. Sui and J. Sun, J. Mater. Chem. B, 2017, 5, 9558–9570, 10.1039/c7tb01314k.
- M. Enea, M. Peixoto de Almeida, P. Eaton, D. Dias da Silva, E. Pereira, M. E. Soares, M. L. Bastos and H. Carmo, Nanotoxicology, 2019, 13, 990–1004, DOI:10.1080/17435390.2019.1621398.
- G. Sharma, D. T. Valenta, Y. Altman, S. Harvey, H. Xie, S. Mitragotri and J. W. Smith, J. Controlled Release, 2010, 147, 408–412, DOI:10.1016/j.jconrel.2010.07.116.
- J. A. Champion and S. Mitragotri, Pharm. Res., 2009, 26, 244–249, DOI:10.1007/s11095-008-9626-z.
- X. Lu, Y. Zhang, L. Wang, G. Li, J. Gao and Y. Wang, Drug Deliv., 2021, 28, 380–389, DOI:10.1080/10717544.2021.1883158.
- Q. Bao, P. Hu, Y. Xu, T. Cheng, C. Wei, L. Pan and J. Shi, ACS Nano, 2018, 12, 6794–6805, DOI:10.1021/acsnano.8b01994.
- D. Ni, J. Zhang, W. Bu, H. Xing, F. Han, Q. Xiao, Z. Yao, F. Chen, Q. He, J. Liu, S. Zhang, W. Fan, L. Zhou, W. Peng and J. Shi, ACS Nano, 2014, 8, 1231–1242, DOI:10.1021/nn406197c.
- Q. Song, M. Huang, L. Yao, X. Wang, X. Gu, J. Chen, J. Chen, J. Huang, Q. Hu, T. Kang, Z. Rong, H. Qi, G. Zheng, H. Chen and X. Gao, ACS Nano, 2014, 8, 2345–2359, DOI:10.1021/nn4058215.
- J. Wei, D. Wu, S. Zhao, Y. Shao, Y. Xia, D. Ni, X. Qiu, J. Zhang, J. Chen, F. Meng and Z. Zhong, Adv. Sci., 2022, 9, e2103689, DOI:10.1002/advs.202103689.
- Y. Jiang, J. Zhang, F. Meng and Z. Zhong, ACS Nano, 2018, 12, 11070–11079, DOI:10.1021/acsnano.8b05265.
- A. Joseph, C. Contini, D. Cecchin, S. Nyberg, L. Ruiz-Perez, J. Gaitzsch, G. Fullstone, X. Tian, J. Azizi, J. Preston, G. Volpe and G. Battaglia, Sci. Adv., 2017, 3, e1700362, DOI:10.1126/sciadv.1700362.
- L. L. Israel, O. Braubach, A. Galstyan, A. Chiechi, E. S. Shatalova, Z. Grodzinski, H. Ding, K. L. Black, J. Y. Ljubimova and E. Holler, ACS Nano, 2019, 13, 1253–1271, DOI:10.1021/acsnano.8b06437.
- V. Piazzini, E. Landucci, G. Graverini, D. E. Pellegrini-Giampietro, A. R. Bilia and M. C. Bergonzi, Pharmaceutics, 2018, 10, 128, DOI:10.3390/pharmaceutics10030128.
- R. Dal Magro, B. Albertini, S. Beretta, R. Rigolio, E. Donzelli, A. Chiorazzi, M. Ricci, P. Blasi and G. Sancini, Nanomedicine, 2018, 14, 429–438, DOI:10.1016/j.nano.2017.11.008.
- J. Wei, D. Wu, Y. Shao, B. Guo, J. Jiang, J. Chen, J. Zhang, F. Meng and Z. Zhong, J. Controlled Release, 2022, 347, 68–77, DOI:10.1016/j.jconrel.2022.04.048.
- A. R. Neves, J. F. Queiroz, S. A. C. Lima and S. Reis, Bioconjug. Chem., 2017, 28, 995–1004, DOI:10.1021/acs.bioconjchem.6b00705.
- K. Gkouvatsos, G. Papanikolaou and K. Pantopoulos, Biochim. Biophys. Acta, 2012, 1820, 188–202, DOI:10.1016/j.bbagen.2011.10.013.
- K. Fan, X. Jia, M. Zhou, K. Wang, J. Conde, J. He, J. Tian and X. Yan, ACS Nano, 2018, 12, 4105–4115, DOI:10.1021/acsnano.7b06969.
- C. Cao, X. Wang, Y. Cai, L. Sun, L. Tian, H. Wu, X. He, H. Lei, W. Liu, G. Chen, R. Zhu and Y. Pan, Adv. Mater., 2014, 26, 2566–2571, DOI:10.1002/adma.201304544.
- J. Yang, L. Fan, F. Wang, Y. Luo, X. Sui, W. Li, X. Zhang and Y. Wang, Nanoscale, 2016, 8, 9537–9547, 10.1039/c5nr06658a.
- A. Galstyan, J. L. Markman, E. S. Shatalova, A. Chiechi, A. J. Korman, R. Patil, D. Klymyshyn, W. G. Tourtellotte, L. L. Israel, O. Braubach, V. A. Ljubimov, L. A. Mashouf, A. Ramesh, Z. B. Grodzinski, M. L. Penichet, K. L. Black, E. Holler, T. Sun, H. Ding, A. V. Ljubimov and J. Y. Ljubimova, Nat. Commun., 2019, 10, 3850, DOI:10.1038/s41467-019-11719-3.
- J. Shen, Z. Zhao, W. Shang, C. Liu, B. Zhang, Z. Xu and H. Cai, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 192–200, DOI:10.1080/21691401.2018.1548471.
- S. S. Kim, A. Rait, E. Kim, K. F. Pirollo, M. Nishida, N. Farkas, J. A. Dagata and E. H. Chang, ACS Nano, 2014, 8, 5494–5514, DOI:10.1021/nn5014484.
- F. C. Lam, S. W. Morton, J. Wyckoff, T. L. Vu Han, M. K. Hwang, A. Maffa, E. Balkanska-Sinclair, M. B. Yaffe, S. R. Floyd and P. T. Hammond, Nat. Commun., 2018, 9, 1991, DOI:10.1038/s41467-018-04315-4.
- P. Sun, Y. Xiao, Q. Di, W. Ma, X. Ma, Q. Wang and W. Chen, Int. J. Nanomed., 2020, 15, 6673–6688, DOI:10.2147/IJN.S257459.
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667, DOI:10.1073/pnas.1307152110.
- A. J. Clark and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12486–12491, DOI:10.1073/pnas.1517048112.
- K. B. Johnsen, M. Bak, F. Melander, M. S. Thomsen, A. Burkhart, P. J. Kempen, T. L. Andresen and T. Moos, J. Controlled Release, 2019, 295, 237–249, DOI:10.1016/j.jconrel.2019.01.005.
- K. H. Wong, M. K. Riaz, Y. Xie, X. Zhang, Q. Liu, H. Chen, Z. Bian, X. Chen, A. Lu and Z. Yang, Int. J. Mol. Sci., 2019, 20, 381, DOI:10.3390/ijms20020381.
- K. Ulbrich, T. Knobloch and J. Kreuter, J. Drug Target, 2011, 19, 125–132, DOI:10.3109/10611861003734001.
- Y. C. Kuo and C. Y. Shih-Huang, J. Drug Target, 2013, 21, 730–738, DOI:10.3109/1061186X.2013.812094.
- M. Shilo, M. Motiei, P. Hana and R. Popovtzer, Nanoscale, 2014, 6, 2146–2152, 10.1039/c3nr04878k.
- B. T. Hawkins, R. D. Egleton and T. P. Davis, Am. J. Physiol. Heart Circ. Physiol., 2005, 289, H212–219, DOI:10.1152/ajpheart.01210.2004.
- R. Huey, B. O'Hagan, P. McCarron and S. Hawthorne, Int. J. Pharm., 2017, 525, 12–20, DOI:10.1016/j.ijpharm.2017.04.023.
- H. Hua, X. Zhang, H. Mu, Q. Meng, Y. Jiang, Y. Wang, X. Lu, A. Wang, S. Liu, Y. Zhang, Z. Wan and K. Sun, Int. J. Pharm., 2018, 543, 179–189, DOI:10.1016/j.ijpharm.2018.03.028.
- T. E. Park, B. Singh, H. Li, J. Y. Lee, S. K. Kang, Y. J. Choi and C. S. Cho, Biomaterials, 2015, 38, 61–71, DOI:10.1016/j.biomaterials.2014.10.068.
- T. Sepasi, F. Bani, R. Rahbarghazi, A. Ebrahimi-Kalan, M.-R. Sadeghi, S. Z. Alamolhoda, A. Zarebkohan, T. Ghadiri and H. Gao, BioImpacts, 2022, 13, 133–144, DOI:10.34172/bi.2022.23876.
- N. U. Khan, T. Miao, X. Ju, Q. Guo and L. Han, Brain Targeted Drug Delivery System, 2019, pp. 129–158 DOI:10.1016/b978-0-12-814001-7.00006-8.
- Y. Tian, Z. Zheng, X. Wang, S. Liu, L. Gu, J. Mu, X. Zheng, Y. Li and S. Shen, J. Nanobiotechnol., 2022, 20, 318, DOI:10.1186/s12951-022-01493-8.
- J. Xie, D. Gonzalez-Carter, T. A. Tockary, N. Nakamura, Y. Xue, M. Nakakido, H. Akiba, A. Dirisala, X. Liu, K. Toh, T. Yang, Z. Wang, S. Fukushima, J. Li, S. Quader, K. Tsumoto, T. Yokota, Y. Anraku and K. Kataoka, ACS Nano, 2020, 14, 6729–6742, DOI:10.1021/acsnano.9b09991.
- X. Jiang, H. Xin, Q. Ren, J. Gu, L. Zhu, F. Du, C. Feng, Y. Xie, X. Sha and X. Fang, Biomaterials, 2014, 35, 518–529, DOI:10.1016/j.biomaterials.2013.09.094.
- Y. Anraku, H. Kuwahara, Y. Fukusato, A. Mizoguchi, T. Ishii, K. Nitta, Y. Matsumoto, K. Toh, K. Miyata, S. Uchida, K. Nishina, K. Osada, K. Itaka, N. Nishiyama, H. Mizusawa, T. Yamasoba, T. Yokota and K. Kataoka, Nat. Commun., 2017, 8, 1001, DOI:10.1038/s41467-017-00952-3.
- Y. Zhou, F. Zhu, Y. Liu, M. Zheng, Y. Wang, D. Zhang, Y. Anraku, Y. Zou, J. Li, H. Wu, X. Pang, W. Tao, O. Shimoni, A. I. Bush, X. Xue and B. Shi, Sci. Adv., 2020, 6, eabc7031 CrossRef CAS PubMed.
- H. Wang, Y. Chao, H. Zhao, X. Zhou, F. Zhang, Z. Zhang, Z. Li, J. Pan, J. Wang, Q. Chen and Z. Liu, ACS Nano, 2022, 16, 664–674, DOI:10.1021/acsnano.1c08120.
- D. Wu, M. Qin, D. Xu, L. Wang, C. Liu, J. Ren, G. Zhou, C. Chen, F. Yang, Y. Li, Y. Zhao, R. Huang, S. Pourtaheri, C. Kang, M. Kamata, I. S. Y. Chen, Z. He, J. Wen, W. Chen and Y. Lu, Adv. Mater., 2019, 31, e1807557, DOI:10.1002/adma.201807557.
- S. Stalmans, N. Bracke, E. Wynendaele, B. Gevaert, K. Peremans, C. Burvenich, I. Polis and B. De Spiegeleer, PLoS One, 2015, 10, e0139652, DOI:10.1371/journal.pone.0139652.
- G. Sharma, S. Lakkadwala, A. Modgil and J. Singh, Int. J. Mol. Sci., 2016, 17, 806, DOI:10.3390/ijms17060806.
- T. Kanazawa, F. Akiyama, S. Kakizaki, Y. Takashima and Y. Seta, Biomaterials, 2013, 34, 9220–9226, DOI:10.1016/j.biomaterials.2013.08.036.
- J. Saint-Pol, F. Gosselet, S. Duban-Deweer, G. Pottiez and Y. Karamanos, Cells, 2020, 9, 851, DOI:10.3390/cells9040851.
- M. Tkach and C. Thery, Cell, 2016, 164, 1226–1232, DOI:10.1016/j.cell.2016.01.043.
- H. Tsivion-Visbord, N. Perets, T. Sofer, L. Bikovski, Y. Goldshmit, A. Ruban and D. Offen, Transl. Psychiatry, 2020, 10, 305, DOI:10.1038/s41398-020-00988-y.
- B. Tang, W. Zeng, L. L. Song, H. M. Wang, L. Q. Qu, H. H. Lo, L. Yu, A. G. Wu, V. K. W. Wong and B. Y. K. Law, Pharmaceuticals, 2022, 15, DOI:10.3390/ph15010083.
- Y. Duan, M. Wu, D. Hu, Y. Pan, F. Hu, X. Liu, N. Thakor, W. H. Ng, X. Liu, Z. Sheng, H. Zheng and B. Liu, Adv. Funct. Mater., 2020, 30, DOI:10.1002/adfm.202004346.
- Y. Liu, Y. Zou, C. Feng, A. Lee, J. Yin, R. Chung, J. B. Park, H. Rizos, W. Tao, M. Zheng, O. C. Farokhzad and B. Shi, Nano Lett., 2020, 20, 1637–1646, DOI:10.1021/acs.nanolett.9b04683.
- W. Hao, Y. Cui, Y. Fan, M. Chen, G. Yang, Y. Wang, M. Yang, Z. Li, W. Gong, Y. Yang and C. Gao, J. Nanobiotechnol., 2021, 19, 378, DOI:10.1186/s12951-021-01110-0.
- G. C. Terstappen, A. H. Meyer, R. D. Bell and W. Zhang, Nat. Rev. Drug Discov., 2021, 20, 362–383, DOI:10.1038/s41573-021-00139-y.
- A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2014, 190, 531–541, DOI:10.1016/j.jconrel.2014.03.050.
- M. Ayer, M. Schuster, I. Gruber, C. Blatti, E. Kaba, G. Enzmann, O. Burri, R. Guiet, A. Seitz, B. Engelhardt and H. A. Klok, Adv. Healthcare Mater., 2021, 10, e2001375, DOI:10.1002/adhm.202001375.
- H. I. Tong, W. Kang, P. M. Davy, Y. Shi, S. Sun, R. C. Allsopp and Y. Lu, PLoS One, 2016, 11, e0154022, DOI:10.1371/journal.pone.0154022.
- M. Wu, H. Zhang, C. Tie, C. Yan, Z. Deng, Q. Wan, X. Liu, F. Yan and H. Zheng, Nat. Commun., 2018, 9, 4777, DOI:10.1038/s41467-018-07250-6.
- J. Lu, X. Gao, S. Wang, Y. He, X. Ma, T. Zhang and X. Liu, Exploration, 2023, 3, 20220045, DOI:10.1002/EXP.20220045.
- M. Li, S. Li, H. Zhou, X. Tang, Y. Wu, W. Jiang, Z. Tian, X. Zhou, X. Yang and Y. Wang, Nat. Commun., 2020, 11, 1126, DOI:10.1038/s41467-020-14963-0.
- C. Gao, Q. Wang, J. Li, C. Kwong, J. Wei, B. Xie, S. Lu, S. Lee and R. Wang, Sci Adv., 2022, 8, eabn1805, DOI:10.1126/sciadv.abn1805.
- C. Bing, M. Ladouceur-Wodzak, C. R. Wanner, J. M. Shelton, J. A. Richardson and R. Chopra, J. Ther. Ultrasound, 2014, 2, 1–11 CrossRef PubMed.
- C. Berard, S. Desgranges, N. Dumas, A. Novell, B. Larrat, M. Hamimed, N. Taulier, M. A. Esteve, F. Correard and C. Contino-Pepin, Pharmaceutics, 2022, 14, 1498, DOI:10.3390/pharmaceutics14071498.
- H. Moon, K. Hwang, K. M. Nam, Y. S. Kim, M. J. Ko, H. R. Kim, H. J. Lee, M. J. Kim, T. H. Kim, K. S. Kang, N. G. Kim, S. W. Choi and C. Y. Kim, Biomater. Adv., 2022, 141, 213102, DOI:10.1016/j.bioadv.2022.213102.
- C. W. Burke, E. t Alexander, K. Timbie, A. L. Kilbanov and R. J. Price, Mol. Ther., 2014, 22, 321–328, DOI:10.1038/mt.2013.259.
- A. K. O. Aslund, S. Berg, S. Hak, Y. Morch, S. H. Torp, A. Sandvig, M. Wideroe, R. Hansen and C. de Lange Davies, J. Controlled Release, 2015, 220, 287–294, DOI:10.1016/j.jconrel.2015.10.047.
- L. L. Israel, A. Galstyan, E. Holler and J. Y. Ljubimova, J. Controlled Release, 2020, 320, 45–62, DOI:10.1016/j.jconrel.2020.01.009.
- Y. Huang, B. Zhang, S. Xie, B. Yang, Q. Xu and J. Tan, ACS Appl. Mater. Interfaces, 2016, 8, 11336–11341, DOI:10.1021/acsami.6b02838.
- S. D. Kong, J. Lee, S. Ramachandran, B. P. Eliceiri, V. I. Shubayev, R. Lal and S. Jin, J. Controlled Release, 2012, 164, 49–57, DOI:10.1016/j.jconrel.2012.09.021.
- K. Mayilsamy, E. Markoutsa, M. Das, P. Chopade, D. Puro, A. Kumar, D. Gulick, A. E. Willing, S. S. Mohapatra and S. Mohapatra, Nanomedicine, 2020, 29, 102247, DOI:10.1016/j.nano.2020.102247.
- D. Lee and T. Minko, Pharmaceutics, 2021, 13, 2049, DOI:10.3390/pharmaceutics13122049.
- M. W. Kemp, J. P. Newnham, J. G. Challis, A. H. Jobe and S. J. Stock, Hum. Reprod. Update, 2016, 22, 240–259, DOI:10.1093/humupd/dmv047.
- S. Sridharan, I. Sullivan, V. Tomek, J. Wolfenden, J. Skovranek, R. Yates, J. Janousek, T. E. Dominguez and J. Marek, Heart Rhythm, 2016, 13, 1913–1919, DOI:10.1016/j.hrthm.2016.03.023.
- B. F. Cuneo and J. F. Strasburger, Obstetrics Gynecology, 2000, 96, 575–581 CAS.
- D. Vejrazkova, J. Vcelak, M. Vankova, P. Lukasova, O. Bradnova, T. Halkova, R. Kancheva and B. Bendlova, J. Steroid Biochem. Mol. Biol., 2014, 139, 122–129, DOI:10.1016/j.jsbmb.2012.11.007.
- N. M. Gude, C. T. Roberts, B. Kalionis and R. G. King, Thromb. Res., 2004, 114, 397–407, DOI:10.1016/j.thromres.2004.06.038.
- E. Kurtoglu, B. Z. Altunkaynak, I. Aydin, A. Z. Ozdemir, G. Altun, A. Kokcu and S. Kaplan, J. Obstet. Gynaecol. Res., 2015, 41, 1533–1540, DOI:10.1111/jog.12751.
- D. Ho, J. W. Leong, R. C. Crew, M. Norret, M. J. House, P. J. Mark, B. J. Waddell, K. S. Iyer and J. A. Keelan, Sci. Rep., 2017, 7, 2866, DOI:10.1038/s41598-017-03128-7.
- T. Clements, T. F. Rice, G. Vamvakas, S. Barnett, M. Barnes, B. Donaldson, C. E. Jones, B. Kampmann and B. Holder, Front. Immunol., 2020, 11, 1920, DOI:10.3389/fimmu.2020.01920.
- E. Bongaerts, L. L. Lecante, H. Bove, M. B. J. Roeffaers, M. Ameloot, P. A. Fowler and T. S. Nawrot, Lancet Planet Health, 2022, 6, e804–e811, DOI:10.1016/S2542-5196(22)00200-5.
- X. Huang, F. Zhang, X. Sun, K. Y. Choi, G. Niu, G. Zhang, J. Guo, S. Lee and X. Chen, Biomaterials, 2014, 35, 856–865, DOI:10.1016/j.biomaterials.2013.10.027.
- F. R. De Bie, D. Sharma, D. Lannoy, K. Allegaert, L. Storme, J. Deprest and F. M. Russo, Fetal Diagn. Ther., 2021, 48, 411–420, DOI:10.1159/000515435.
- M. M. Barzago, A. Bortolotti, F. F. Stellari, L. Diomede, M. Algeri, S. Efrati, M. Salmona and M. Bonati, J. Pharmacol. Exp. Ther., 1996, 277, 79–86 CAS.
- P. Pourali, M. Nouri, F. Ameri, T. Heidari, N. Kheirkhahan, S. Arabzadeh and B. Yahyaei, Naunyn Schmiedebergs Arch Pharmacol, 2020, 393, 867–878, DOI:10.1007/s00210-019-01796-y.
- E. Paul, M. L. Franco-Montoya, E. Paineau, B. Angeletti, S. Vibhushan, A. Ridoux, A. Tiendrebeogo, M. Salome, B. Hesse, D. Vantelon, J. Rose, F. Canoui-Poitrine, J. Boczkowski, S. Lanone, C. Delacourt and J. C. Pairon, Nanotoxicology, 2017, 11, 484–495, DOI:10.1080/17435390.2017.1311381.
- S. Grafmuller, P. Manser, H. F. Krug, P. Wick and U. von Mandach, J. Vis. Exp., 2013, 50401, DOI:10.3791/50401.
- J. P. Huang, P. C. Hsieh, C. Y. Chen, T. Y. Wang, P. C. Chen, C. C. Liu, C. C. Chen and C. P. Chen, Placenta, 2015, 36, 1433–1441, DOI:10.1016/j.placenta.2015.10.007.
- A. Adamcakova-Dodd, M. M. Monick, L. S. Powers, K. N. Gibson-Corley and P. S. Thorne, Part. Fibre Toxicol., 2015, 12, 30, DOI:10.1186/s12989-015-0105-5.
- S. Grafmueller, P. Manser, L. Diener, P. A. Diener, X. Maeder-Althaus, L. Maurizi, W. Jochum, H. F. Krug, T. Buerki-Thurnherr, U. von Mandach and P. Wick, Environ. Health Perspect., 2015, 123, 1280–1286, DOI:10.1289/ehp.1409271.
- H. Tang, Z. Jiang, H. He, X. Li, H. Hu, N. Zhang, Y. Dai and Z. Zhou, Int. J. Nanomed., 2018, 13, 4073–4082, DOI:10.2147/IJN.S161319.
- Z. Jiang, H. Tang, Q. Xiong, M. Li, Y. Dai and Z. Zhou, Colloids Surf., B, 2022, 216, 112553, DOI:10.1016/j.colsurfb.2022.112553.
- Z. Sezgin-Bayindir, A. E. Elcin, M. Parmaksiz, Y. M. Elcin and N. Yuksel, J. Microencapsul, 2018, 35, 149–164, DOI:10.1080/02652048.2018.1447615.
- J. Vidmar, K. Loeschner, M. Correia, E. H. Larsen, P. Manser, A. Wichser, K. Boodhia, Z. S. Al-Ahmady, J. Ruiz, D. Astruc and T. Buerki-Thurnherr, Nanoscale, 2018, 10, 11980–11991, 10.1039/c8nr02096e.
- P. K. Myllynen, M. J. Loughran, C. V. Howard, R. Sormunen, A. A. Walsh and K. H. Vahakangas, Reprod. Toxicol., 2008, 26, 130–137, DOI:10.1016/j.reprotox.2008.06.008.
- W. H. Tse, S. Higgins, D. Patel, M. Xing, A. R. West, H. I. Labouta and R. Keijzer, Biomater. Sci., 2022, 10, 5243–5253, 10.1039/d2bm00293k.
- S. Borghi, S. Bournazos, N. K. Thulin, C. Li, A. Gajewski, R. W. Sherwood, S. Zhang, E. Harris, P. Jagannathan, L. X. Wang, J. V. Ravetch and T. T. Wang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12943–12951, DOI:10.1073/pnas.2004325117.
- M. Resende, M. A. Nielsen, M. Dahlback, S. B. Ditlev, P. Andersen, A. F. Sander, N. T. Ndam, T. G. Theander and A. Salanti, Malar. J., 2008, 7, 104, DOI:10.1186/1475-2875-7-104.
- B. Zhang, L. Tan, Y. Yu, B. Wang, Z. Chen, J. Han, M. Li, J. Chen, T. Xiao, B. K. Ambati, L. Cai, Q. Yang, N. R. Nayak, J. Zhang and X. Fan, Theranostics, 2018, 8, 2765–2781, DOI:10.7150/thno.22904.
- K. Zhao, D. Li, G. Cheng, B. Zhang, J. Han, J. Chen, B. Wang, M. Li, T. Xiao, J. Zhang, D. Zhou, Z. Jin and X. Fan, Int. J. Mol. Sci., 2019, 20, 5458, DOI:10.3390/ijms20215458.
- L. Li, H. Yang, P. Chen, T. Xin, Q. Zhou, D. Wei, Y. Zhang and S. Wang, Front. Bioeng. Biotechnol., 2020, 8, 64, DOI:10.3389/fbioe.2020.00064.
- A. King, C. Ndifon, S. Lui, K. Widdows, V. R. Kotamraju, L. Agemy, T. Teesalu, J. D. Glazier, F. Cellesi, N. Tirelli, J. D. Aplin, E. Ruoslahti and L. K. Harris, Sci. Adv., 2016, 2, e1600349 CrossRef PubMed.
- Q. Yu, Y. Qiu, X. Wang, J. Tang, Y. Liu, L. Mei, M. Li, M. Yang, L. Tang, H. Gao, Z. Zhang, W. Xu and Q. He, Int. J. Pharm., 2018, 546, 115–124, DOI:10.1016/j.ijpharm.2018.05.001.
- T. J. Kaitu'u-Lino, S. Pattison, L. Ye, L. Tuohey, P. Sluka, J. MacDiarmid, H. Brahmbhatt, T. Johns, A. W. Horne, J. Brown and S. Tong, Endocrinology, 2013, 154, 911–919, DOI:10.1210/en.2012-1832.
- K. L. Swingle, H. C. Safford, H. C. Geisler, A. G. Hamilton, A. S. Thatte, M. M. Billingsley, R. A. Joseph, K. Mrksich, M. S. Padilla, A. A. Ghalsasi, M.-G. Alameh, D. Weissman and M. J. Mitchell, J. Am. Chem. Soc., 2023, 145, 4691–4706, DOI:10.1021/jacs.2c12893.
- R. E. Young, K. M. Nelson, S. I. Hofbauer, T. Vijayakumar, M.-G. Alameh, D. Weissman, C. Papachristou, J. P. Gleghorn and R. S. Riley, bioRxiv, 2022 DOI:10.1101/2022.12.22.521490.
- A. Azagury, E. Amar-Lewis, E. Mann, R. Goldbart, T. Traitel, R. Jelinek, M. Hallak and J. Kost, J. Controlled Release, 2014, 183, 105–113, DOI:10.1016/j.jconrel.2014.03.040.
- N. Abd Ellah, L. Taylor, W. Troja, K. Owens, N. Ayres, G. Pauletti and H. Jones, PLoS One, 2015, 10, e0140879, DOI:10.1371/journal.pone.0140879.
- I. Burd, F. Zhang, T. Dada, M. K. Mishra, T. Borbiev, W. G. Lesniak, H. Baghlaf, S. Kannan and R. M. Kannan, Nanomedicine, 2014, 10, 1343–1351, DOI:10.1016/j.nano.2014.03.008.
- J. L. Cohen, P. Chakraborty, K. Fung-Kee-Fung, M. E. Schwab, D. Bali, S. P. Young, M. H. Gelb, H. Khaledi, A. DiBattista, S. Smallshaw, F. Moretti, D. Wong, C. Lacroix, D. El Demellawy, K. C. Strickland, J. Lougheed, A. Moon-Grady, B. R. Lianoglou, P. Harmatz, P. S. Kishnani and T. C. MacKenzie, N. Engl. J. Med., 2022, 387, 2150–2158, DOI:10.1056/NEJMoa2200587.
- T. Elger, R. Akolekar, A. Syngelaki, C. De Paco Matallana, F. S. Molina, M. Gallardo Arozena, P. Chaveeva, N. Persico, V. Accurti, K. O. Kagan, N. Prodan, J. Cruz and K. H. Nicolaides, Ultrasound Obstet. Gynecol., 2021, 58, 48–55, DOI:10.1002/uog.23694.
- R. Akolekar, J. Beta, G. Picciarelli, C. Ogilvie and F. D'Antonio, Ultrasound Obstet. Gynecol., 2015, 45, 16–26, DOI:10.1002/uog.14636.
- A. Abostait, J. Tyrrell, M. Abdelkarim, S. Shojaei, W. H. Tse, I. M. El-Sherbiny, R. Keijzer and H. I. Labouta, Mol. Pharm., 2022, 19, 3757–3769, DOI:10.1021/acs.molpharmaceut.2c00216.
- N. S. Irvin-Choy, K. M. Nelson, M. N. Dang, J. P. Gleghorn and E. S. Day, Nanomedicine, 2021, 36, 102412, DOI:10.1016/j.nano.2021.102412.
- A. Schmidt, D. M. Morales-Prieto, J. Pastuschek, K. Frohlich and U. R. Markert, J. Reprod. Immunol., 2015, 108, 65–71, DOI:10.1016/j.jri.2015.03.001.
- F. R. De Bie, S. D. Kim, S. K. Bose, P. Nathanson, E. A. Partridge, A. W. Flake and C. Feudtner, Am. J. Bioeth., 2022, 23, 67–78, DOI:10.1080/15265161.2022.2048738.
- K. Allegaert, Int. J. Environ. Res. Public Health, 2022, 19, DOI:10.3390/ijerph191811336.
- F. M. Russo, P. Mian, E. H. Krekels, K. Van Calsteren, D. Tibboel, J. Deprest and K. Allegaert, Xenobiotica, 2019, 49, 98–105, DOI:10.1080/00498254.2017.1422217.
- X. Tian, Y. Chong and C. Ge, Front. Chem., 2020, 8, 446, DOI:10.3389/fchem.2020.00446.
- R. Bartucci, C. Åberg, B. N. Melgert, Y. L. Boersma, P. Olinga and A. Salvati, Small, 2020, 16, 1906523, DOI:10.1002/smll.201906523.
- L. T. Curtis, M. Wu, J. Lowengrub, P. Decuzzi and H. B. Frieboes, PLoS One, 2015, 10, e0144888, DOI:10.1371/journal.pone.0144888.
- T. A. Brocato, E. N. Coker, P. N. Durfee, Y. S. Lin, J. Townson, E. F. Wyckoff, V. Cristini, C. J. Brinker and Z. Wang, Sci. Rep., 2018, 8(7538), 8, DOI:10.1038/s41598-018-25878-.
- W. M. Pardridge and T. Chou, Pharmaceuticals, 2021, 14, 535 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, S. Li, Y. Cui, X. Liang, J. Shan, W. Gu, J. Qiu, Y. Li and G. Wang, J. Nanobiotechnol., 2021, 19(331), 0, DOI:10.1186/s12951-021-01067-.
- J. S. Brenner, D. C. Pan, J. W. Myerson, O. A. Marcos-Contreras, C. H. Villa, P. Patel, H. Hekierski, S. Chatterjee, J. Q. Tao, H. Parhiz, K. Bhamidipati, T. G. Uhler, E. D. Hood, R. Y. Kiseleva, V. S. Shuvaev, T. Shuvaeva, M. Khoshnejad, I. Johnston, J. V. Gregory, J. Lahann, T. Wang, E. Cantu, W. M. Armstead, S. Mitragotri and V. Muzykantov, Nat. Commun., 2018, 9(2684), 7, DOI:10.1038/s41467-018-05079-.
- X. Zhang, X. Zhao, Z. Hua, S. Xing, J. Li, S. Fei and M. Tan, Biomaterials, 2023, 292, 121937, DOI:10.1016/j.biomaterials.2022.121937.
- X. Li, E. C. Montague, A. Pollinzi, A. Lofts and T. Hoare, Small, 2022, 18, 2104632, DOI:10.1002/smll.202104632.
- O. B. Garbuzenko, J. Winkler, M. S. Tomassone and T. Minko, Langmuir, 2014, 30, 12941–12949, DOI:10.1021/la502144z.
- B. Shaghaghi, S. Khoee and S. Bonakdar, Int. J. Pharm., 2019, 559, 1–12, DOI:10.1016/j.ijpharm.2019.01.020.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |