
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRenaissance of elemental phosphorus materials: properties, synthesis, and applications in sustainable energy and environment
Haijiang
Tian
 ac,
Jiahong
Wang
*bhi,
Gengchang
Lai
bi,
Yanpeng
Dou
bh,
Jie
Gao
ad,
Zunbin
Duan
bh,
Xiaoxiao
Feng
b,
Qi
Wu
ad,
Xingchen
He
bh,
Linlin
Yao
a,
Li
Zeng
a,
Yanna
Liu
a,
Xiaoxi
Yang
a,
Jing
Zhao
ad,
Shulin
Zhuang
c,
Jianbo
Shi
adi,
Guangbo
Qu
*adi,
Xue-Feng
Yu
*bhi,
Paul K.
Chu
efg and
Guibin
Jiang
acdi
ac,
Jiahong
Wang
*bhi,
Gengchang
Lai
bi,
Yanpeng
Dou
bh,
Jie
Gao
ad,
Zunbin
Duan
bh,
Xiaoxiao
Feng
b,
Qi
Wu
ad,
Xingchen
He
bh,
Linlin
Yao
a,
Li
Zeng
a,
Yanna
Liu
a,
Xiaoxi
Yang
a,
Jing
Zhao
ad,
Shulin
Zhuang
c,
Jianbo
Shi
adi,
Guangbo
Qu
*adi,
Xue-Feng
Yu
*bhi,
Paul K.
Chu
efg and
Guibin
Jiang
acdi
aState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China. E-mail: gbqu@rcees.ac.cn
bShenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China. E-mail: jh.wang1@siat.ac.cn; xf.yu@siat.ac.cn
cKey Laboratory of Environment Remediation and Ecological Health, Ministry of Education, College of Environmental and Resource Sciences, Zhejiang University, Hangzhou 310058, P. R. China
dSchool of Environment, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, P. R. China
eDepartment of Physics, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
fDepartment of Materials Science and Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
gDepartment of Biomedical Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
hHubei Three Gorges Laboratory, Yichang, Hubei 443007, P. R. China
iUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 17th July 2023
Abstract
The polymorphism of phosphorus-based materials has garnered much research interest, and the variable chemical bonding structures give rise to a variety of micro and nanostructures. Among the different types of materials containing phosphorus, elemental phosphorus materials (EPMs) constitute the foundation for the synthesis of related compounds. EPMs are experiencing a renaissance in the post-graphene era, thanks to recent advancements in the scaling-down of black phosphorus, amorphous red phosphorus, violet phosphorus, and fibrous phosphorus and consequently, diverse classes of low-dimensional sheets, ribbons, and dots of EPMs with intriguing properties have been produced. The nanostructured EPMs featuring tunable bandgaps, moderate carrier mobility, and excellent optical absorption have shown great potential in energy conversion, energy storage, and environmental remediation. It is thus important to have a good understanding of the differences and interrelationships among diverse EPMs, their intrinsic physical and chemical properties, the synthesis of specific structures, and the selection of suitable nanostructures of EPMs for particular applications. In this comprehensive review, we aim to provide an in-depth analysis and discussion of the fundamental physicochemical properties, synthesis, and applications of EPMs in the areas of energy conversion, energy storage, and environmental remediation. Our evaluations are based on recent literature on well-established phosphorus allotropes and theoretical predictions of new EPMs. The objective of this review is to enhance our comprehension of the characteristics of EPMs, keep abreast of recent advances, and provide guidance for future research of EPMs in the fields of chemistry and materials science.
 Haijiang Tian | Haijiang Tian is currently a PhD student at the College of Environmental & Resource Sciences, Zhejiang University, and a joint student at the Research Center for Eco-Environmental Sciences (RCEES), Chinese Academy of Sciences (CAS). He received his bachelor's degree in chemistry at School of Chemistry and Chemical Engineering, Shandong University in 2021. His research interests are the synthesis of low-dimensional phosphorus-based materials and the evaluation of their biological effects. |
 Jiahong Wang | Jiahong Wang is an associate professor in the Shenzhen Institute of Advanced Technology (SIAT), CAS. He obtained his PhD in Optics from Wuhan University in 2016. His major research interests are crystal growth and electronic or energy applications. Currently, Dr. Wang focuses on the growth and anodic performance of phosphorus-based materials, such as black phosphorus. |
 Guangbo Qu | Guangbo Qu is a professor in RCEES, CAS. He obtained his PhD in environmental science at RCEES, CAS in 2011. His major research interests are the evaluation of the biological effects of emerging nanomaterials and halogenated organic pollutants, as well as their molecular mechanisms. Prof. Qu is working on the chemico–physical properties of phosphorus materials as well as biological effects induced by phosphorus materials. |
 Xue-Feng Yu | Xue-Feng Yu is a professor and director of Materials and Interfaces Center at SIAT, CAS. He received his PhD degree in physics from Wuhan University in 2008, and then worked at Wuhan University and City University of Hong Kong. He leads an interdisciplinary research group focused on the synthesis and applications of nanomaterials. He has coauthored more than 100 publications in SCI-indexed journals with over 16 |
 Paul K. Chu | Paul K. Chu is Chair Professor of Materials Engineering in the Department of Physics, Department of Materials Science and Engineering, and Department of Biomedical Engineering at City University of Hong Kong. He obtained his PhD in chemistry from Cornell University in 1982. He is fellow of the APS, AVS, IEEE, MRS, Hong Kong Institution of Engineers, and the Hong Kong Academy of Engineering Sciences. His research interests are quite diverse spanning plasma surface engineering, materials science and engineering, surface science, and functional materials. He is a highly cited researcher and his papers have been cited more than 100 |
 Guibin Jiang | Guibin Jiang is a full professor at RCEES, CAS. He obtained his PhD at RCEES, CAS in 1991. Prof. Jiang is an academician of the Chinese Academy of Sciences and the Academy of Sciences of Developing Countries. He won the 2021 Outstanding Achievements in Environmental Science & Technology Award for his basic and translational research, leadership, and service to understand and manage POPs and other emerging contaminants. His research focuses on environmental analysis and toxicology, including environmental occurrence, processes, toxicology and health effects of environmental toxic substances. |
1. Introduction
Phosphorus (P) is one of the elements serving as the foundation for life on Earth. The Earth's crust contains around 1200 mg P kg−1, ranking it as the eleventh most plentiful element.1 Phosphorus plays a significant part in global biogeochemical cycles in conjunction with other bioactive elements such as carbon (C), nitrogen (N), and trace metals.2,3 It has direct and indirect impacts on the rock cycle,4 marine biogeochemistry,5,6 biomass accumulation,7 and nutrient availability.8,9 Phosphorus is also crucial to numerous technological applications, including optical engineering and communication,10,11 electronics,4,12 biomedicine and drug delivery,13–15 agriculture and food,16 cosmetics,17 and textiles,18 and the development of cost-effective phosphorus-based materials for environmental and energy applications has been rapid recently.19–21 This is because of the ability of phosphorus to self-bond and form bonds with many metal and non-metal elements with different orbital hybridization, thus resulting in a myriad of physical and chemical properties as well as structural diversity of phosphorus compounds and molecules.Among the diverse materials containing phosphorus, elemental phosphorus materials (EPMs) are the fundamental materials and origin of many complex products.22–24 The polymorphism of EPMs arises from different types of phosphorus clusters serving as the building blocks. Phosphorus allotropes such as tetrahedral white phosphorus (white-P), puckered honeycomb-like black phosphorus (black-P), and polygonal chain-like red phosphorus (red-P) have distinctive micro and nanostructures resulting from the diverse covalent bonding nature and arrangement of phosphorus atoms. Different crystal structures lead to different physicochemical properties including the bandgap, optical absorption, electrical conductivity, redox activity, and material stability. For example, white-P consisting of tetrahedral P4 molecules is the most active allotrope.25 Black-P is composed of irregular honeycomb interlayers and is the most thermodynamically stable allotrope.26 Amorphous red-P is the most versatile phosphorus allotrope. Because of its low cost, environmental friendliness, availability, and stability, it finds applications in various fields such as flame retardants,27 energy harvesting,23,28 ion storage,29 and pollution sensing.30,31 During the synthesis of red-P, Hittorf and Thurn et al. observed the formation of plate-like and fiber bundles of phosphorus crystals, namely, violet phosphorus (violet-P) and fibrous phosphorus (fibrous-P), respectively.32,33 In earlier studies, violet-P and fibrous-P were often mentioned as two of the red-P modifications (type V and type IV).34,35 However, violet-P and fibrous-P have been shown to have several unique properties by unambiguous structural characterization, and a growing number of researchers prefer to use the names violet phosphorus and fibrous phosphorus instead of type IV or type V red-P.36,37 It is recommended to use the term “red-P” only to describe type I amorphous red-P, since it is often produced under mild crystallization conditions. In this review, unless otherwise noted, red-P refers to amorphous red phosphorus.
EPMs are unusual in the elemental material family because their allotropes do not exist in nature and must be produced artificially.38 Each EPM is “made” rather than “found”, and the target allotrope needs to artfully match the structural motif assembly with the achievable reaction conditions. They are witnesses to the development of modern synthetic chemistry. Before 2010, studies in this field seemed to be dominated by inorganic chemists or physicists, probably due to the complicated and risky manufacturing procedures of EPMs. The majority of research has focused on the improvement of synthetic approaches and crystal quality of EPMs. For example, Percy Williams Bridgman, who received the Nobel Prize in Physics in 1946, sought to prepare black-P crystals using high-temperature or high-pressure approaches,39,40 and subsequently Akimoto et al. obtained black-P single crystals with diameters up to 4 × 2 × 0.2 mm3.41 Nilges' group developed Sn-based mineralizers to alleviate the harsh conditions for synthesizing high-quality black-P crystals.42–44 Phosphorene, one of the structural analogs of graphene, was regarded as a purely theoretical material for a very long time. In 2014, however, black-P was successfully scaled down to phosphorene by mechanical exfoliation, spurring the advance of EPM synthetic chemistry to the nanoscale.45,46 Synthetic low-dimensional EPMs are a developing class of intriguing atomic structures with amazing materials properties. On the heels of a comprehensive understanding of the physicochemical properties of black-P based materials, researchers have extended their curiosity to rediscover other EPMs. Amorphous red-P, violet-P, and fibrous-P can also be scaled down to form low-dimensional sheets, ribbons, and dots with exciting properties.28,47,48 Concurrently, researchers have predicted the existence of new fancy EPMs such as blue phosphorus (blue-P, also known as gray phosphorus) and green phosphorus (green-P) based on ab initio density functional theory (DFT) calculations.49–51 In 2016, Zhang et al. fabricated blue-P on Au(111),52 and Wang et al. synthesized greenish phosphorus on carbon substrates in 2021.53 A developing trend is the use of computational simulation to assist the synthesis of EPMs. Since 2018, several research groups have employed machine learning-based atomic simulations to predict new EPMs or attempt to answer fundamental structural questions (for instance, the precise structure of amorphous red-P).54–58 It is expected that more computational simulation systems will be developed with artificial intelligence for structural analysis and prediction.
The development of new materials has the potential to address the challenges faced by humanity today.59 Among the pressing challenges of the 21st century, energy crisis and environmental pollution are most critical. As the global population continues to grow, the current energy consumption of 12.8 TW is projected to more than double to 28–35 TW by 2050 and triple by 2100.60–62 This increase in energy demand is limited by the availability of fossil fuels, which currently account for 77% of energy consumption and are associated with a 16% rise in anthropogenic carbon dioxide (CO2) emissions by 2040.28,63 The International Panel on Climate Change (IPCC) estimates that by 2100, the atmosphere will contain up to 570 ppm of CO2, resulting in an average global temperature rise of approximately 1.9 °C.64,65 The consequences of global warming will severely impact agriculture, labor productivity, human health, and economic development.66,67 To meet the growing global energy demand, sustainable and inexpensive energy sources must be developed as alternatives to fossil fuels. Moreover, due to the intermittent nature of renewable energy, cost-effective and high-performance energy storage devices are urgently needed for electric vehicles and scalable energy storage.68 Environmental stewardship is also important in aiming for sensitive sensing and complete removal of pollutants, or selectively converting pollutants into useable chemicals. Synthetic EPMs have the potential to address both sustainable energy and environmental remediation challenges. Although traditional EPMs (white-P, black-P, red-P, violet-P, and fibrous-P) are not novel materials, the recent progress in synthesizing high-quality, large-size bulk crystals and various low-dimensional structures has enabled the rediscovery of many previously unknown physicochemical properties, including chemical sensing, tunable bandgaps, and anisotropic mobility.36,48,69–72 At the same time, theoretical studies have identified several unique photoelectric properties of new EPMs (blue-P and green-P).73–75 The implementation of these EPMs will offer innovative opportunities and new approaches for energy conversion, energy storage, and environmental remediation.
EPMs are experiencing a renaissance in the post-graphene era. Researchers have explored various phosphorus allotropes and synthesized corresponding nanostructures to date. This rich variety of materials allows for the construction of a library of elemental phosphorus materials to facilitate the selection of specific phosphorus allotropes and corresponding micro/nanostructures and to address practical needs for particular applications. Although black-P and red-P have been reviewed in recent years, there is still a lack of comprehensive understanding regarding the differences and relationships between numerous EPMs, their intrinsic physical and chemical properties, synthesis of specific structures, and selection of suitable EPMs for specific applications. To fill this gap, we herein provide a comprehensive review of the physicochemical properties, synthetic methods, and sustainable applications of EPMs (white-P, red-P, black-P, fibrous-P, violet-P, blue-P, and green-P), as illustrated in Fig. 1. The aim of this review is to not only describe the latest research advancements in black-P, red-P, and other emerging EPMs, but also summarize the intrinsic properties of elemental phosphorus by examining the broader research context of this field, which will guide future materials design and applications. It is important to note that EPMs are a big family of phosphorus materials that also include theoretically predicted materials such as phosphorus fullerenes,76 crimson phosphorus,77 phosphorus nanotubes,78 and others. However, owing to the challenges associated with chemical synthesis and their limited practical applications, these materials are not covered in this review.
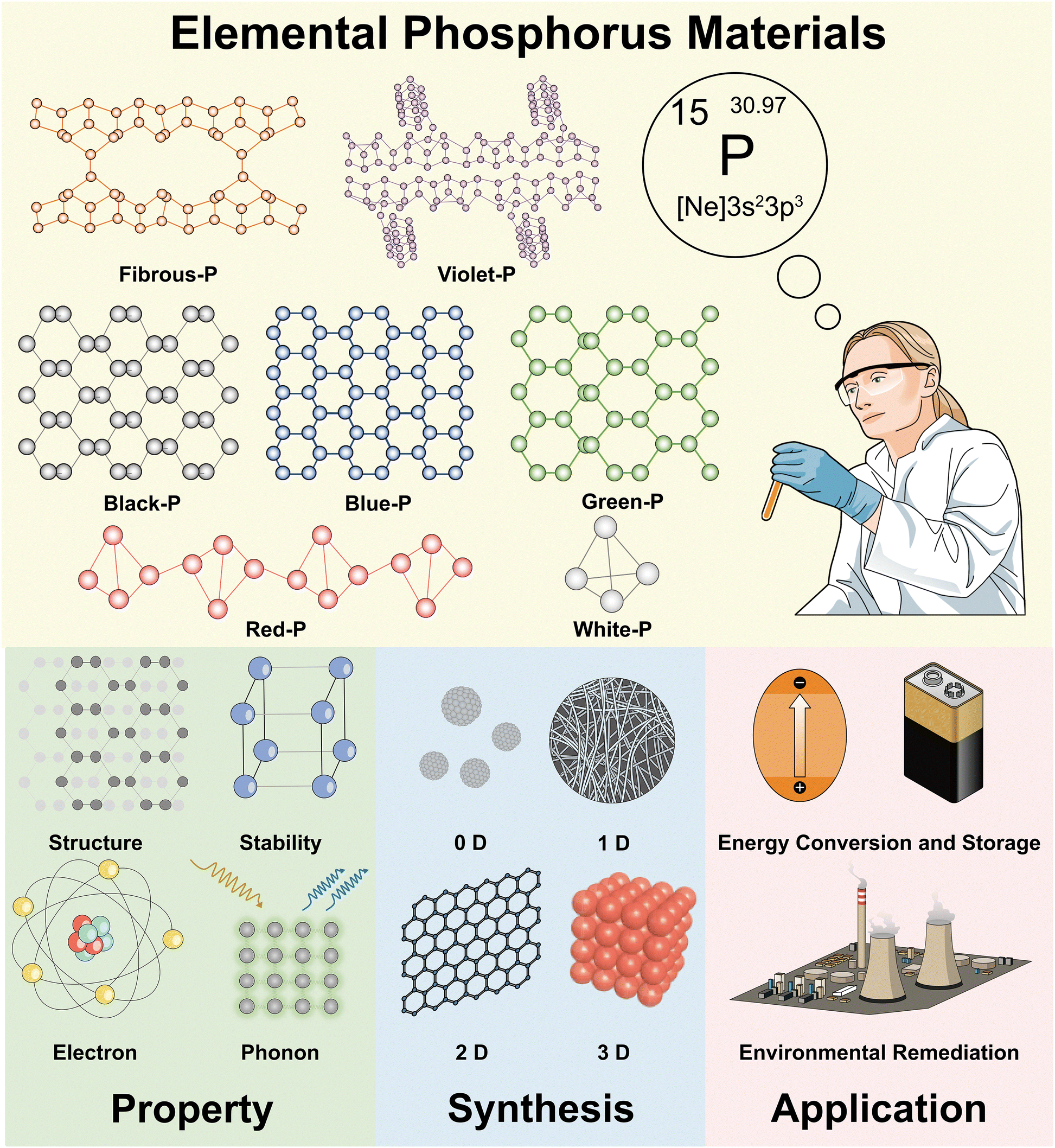 | ||
| Fig. 1 Overview of the properties, synthesis, and sustainable applications of elemental phosphorus materials. | ||
This review is organized as follows. We start with a concise overview of the discovery of common phosphorus bulk crystals and phosphorene. The latest studies on machine learning-driven stimulations that aid the description and prediction of EPM structures are described in this part (Section 2). The structural, electron, and phonon properties of EPMs which are theoretically and/or experimentally studied are described in Section 3. The comparisons of building blocks, material stability, bandgaps, and charge carrier mobility among different EPMs are highlighted. Section 4 critically discusses the synthesis of EPMs and their nanostructures with different dimensions (0D, 1D, 2D, or 3D). The advantages and disadvantages of different synthetic routes are discussed and characterization of the corresponding products is described. In Section 5, recent advancement in EPMs and EPM-derived systems for energy conversion (photocatalytic/electrocatalytic HER, OER, CO2RR, and NRR), energy storage (hydrogen storage, LIBs, SIBs, PIBs, and SCs), and environmental remediation (sensing and gas/liquid/soil treatment) is summarized. In each area, the mechanisms, advantages of EPMs, performance comparison among EPMs, and modification strategies are emphasized. The conclusions and outlook are finally presented to provide guidance for future research in this burgeoning field.
2. Development of elemental phosphorus materials
As the fifteenth element in the periodic table, phosphorus derives its name from the Greek words ‘phôs’ and ‘phoros’, translating literally to “light-bearer”.79 The first isolated form of the element glows and emits visible light. The origin of phosphorus is unknown. Some investigations demonstrate that phosphorous might have been used in the Roman era, but this record was lost over time.80 However, elemental phosphorus in its molecular form was only isolated in the 17th century and afterwards, scientists inaugurated the age of synthetic EPMs.Between the 17th and 20th centuries, researchers synthesized several elemental phosphorus bulk crystals. Self-styled doctor Hennig Brand discovered a strange substance (white-P) from human urine in 1669 and Anton Schrötter von Kristelli presented red-P before the Vienna Academy of Sciences in 1847. The first black-P crystals were fabricated at a high pressure by the Nobel prize winner Percy Williams Bridgman in 1914.81 At the turn of the 21st century, the rapid development of nano-synthesis and characterization technology enabled researchers to expand their interest into low-dimensional materials, and phosphorene, corresponding to the elemental phosphorus bulk crystals, was isolated one after another. Concurrently, breakthroughs in computational chemistry have enabled chemists to discover metastable phosphorus allotropes that can be a potent supplement to common EPMs in technological applications.82,83Fig. 2 demonstrates the development of phosphorus crystals and phosphorene, highlighting key milestones such as the discovery of allotropes and low-dimensional materials. In addition, artificial intelligence has gained traction in materials science over the last few years, and atomistic simulations of phosphorus can now be achieved using machine learning-based algorithms.54,55 This section reviews the discovery of bulk crystals and phosphorene, renowned scientists, and recent progress in machine learning-driven stimulations for structural description and prediction of EPMs.
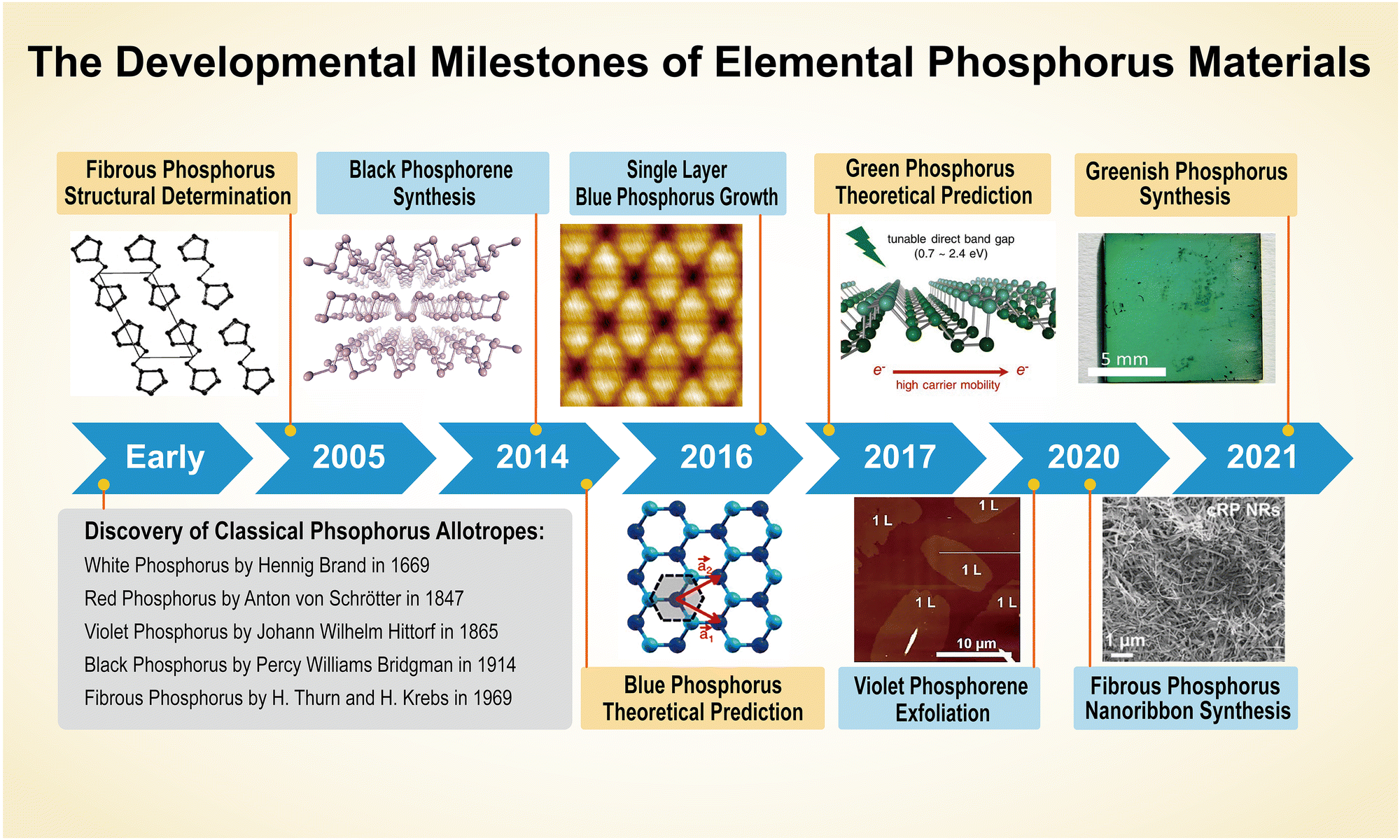 | ||
| Fig. 2 Timeline of the developmental milestones of elemental phosphorus materials (bulk crystals and phosphorene). Inset images are reproduced from the following sources: fibrous phosphorus structural determination, reproduced with permission from ref. 88. Copyright 2005 John Wiley and Sons. Black phosphorene synthesis, reproduced with permission from ref. 45. Copyright 2014 Nature Publishing Group. Blue phosphorus prediction, reproduced with permission from ref. 49. Copyright 2014 American Physical Society. Blue phosphorus synthesis, reproduced with permission from ref. 52. Copyright 2016 American Chemical Society. Green phosphorus prediction, reproduced with permission from ref. 50. Copyright 2017 American Chemical Society. Violet phosphorene synthesis, reproduced with permission from ref. 48. Copyright 2020 John Wiley and Sons. Fibrous phosphorus nanoribbon synthesis, reproduced with permission from ref. 47. Copyright 2020 John Wiley and Sons. Greenish phosphorus synthesis, reproduced with permission from ref. 53. Copyright 2021 Royal Society of Chemistry. | ||
2.1 History of phosphorus allotropes
EPMs cannot exist in nature alone because of the high chemical reactivity and must be synthesized chemically. Their synthesis dates back to the end of the renaissance and physicists and chemists made great contributions to the discovery of phosphorus allotropes (Fig. 3). | ||
| Fig. 3 Timeline of the discovery of white phosphorus, red phosphorus, violet phosphorus, black phosphorus, and fibrous phosphorus bulk crystals as well as renowned scientists. Painting of Hennig Brand taken from “The Alchymist, In Search of the Philosopher's Stone” by Joseph Wright of Derby. Portrait of Anton Schrötter von Kristelli taken from Smithsonian Archives by Adolf Dauthage. Portrait of Johann Wilhelm Hittorf taken from the special issue in honour of the 80th anniversary of Hittorf's birth, Leipzig: Barth 1904. Portrait of Percy Williams Bridgman taken from ref. 81. Copyright 1946 The Nobel Foundation. For H. Thurn and H. Krebs, the discoverers of fibrous phosphorus, in absence of their photographs, we have chosen to represent them with the first page of their original research paper published in 1969 documenting the discovery. The research paper taken from ref. 33. Copyright 1969 International Union of Crystallography. | ||
White-P is the first known phosphorus allotrope, and its discovery is intrinsically related to medieval alchemy. In 1669, Hennig Brand, a German alchemist and a self-styled doctor, was in search of the mythical Philosopher's Stone, which was believed to have the power to turn ordinary metals into gold.80,84 Brand condensed 50 barrels of urine by heating and distillation. Surprisingly, he did not get the magical stone with mutant powers, but did find the pure form of flammable phosphorous, which gave out a faint green glow in the dark. After a period of keeping his discovery a secret, Brand revealed the discovery of white-P in 1675, and his colleague alchemist Daniel Kraft gained fame and fortune by exhibiting this enigmatic new light source to European nobles. Later in the 19th century, chemists developed white-P matches,85 but these matches were extremely reactive to fire and generated noxious fumes. Due to its pyrophoric properties, white-P was subsequently used as a bombing agent during World War II, causing severe harm to human health, and its usage became obsolete after the war. White-P is now mostly used as an industrial intermediate for the production of phosphorus-containing compounds such as herbicides, fertilizers, and food additives.86
Red-P is the second known phosphorus allotrope, and Anton Schrötter von Kristelli is the acknowledged discoverer of this allotrope, even before other scientists had this substance in their hands.87 In 1844, chemist Berzelius reported that when white-P is exposed to sunshine, it could be transformed into a red modification that would not spontaneously oxidize or ignite. However, many chemists recognized this red modification as phosphorus oxide. This controversy was settled by Schrötter's work. He presented his findings to the Vienna Academy of Sciences on December 9, 1847. He discovered that white-P could be converted to red-P by exposure to sunlight, even in the absence of moisture and atmospheric oxygen. The red substance was isolated by treating the residual white-P with carbon disulfide. It could also be produced from white-P by heating it to ∼250 °C in an inert atmosphere. Heating to a higher temperature would reconvert red-P back to white-P. Schrötter emphasized the potential of this allotrope in the match industry. This was confirmed by the creation of safe Swedish matches made of red-P, which only ignited upon friction with a rough surface and did not generate harmful fumes like white-P matches.85,87 Red-P (amorphous form) is now a widely used industrial allotrope along with white-P. Its commercial chemical, with a purity of around 90%, sells for USD 2–10 per kilogram in the Chinese market.22 In addition, research on the atomic structure of amorphous red-P has advanced over the decades, and this part will be discussed later.
Violet-P, also known as Hittorf's phosphorus (or α-metallic phosphorus), is the third known allotrope of phosphorus that can be produced by annealing white-P/red-P above 500 °C. This phosphorus form was first observed in 1865 by Johann Wilhelm Hittorf, who noted that recrystallizing phosphorus from molten lead resulted in red-to-violet colored platelets.32 Furthering this discovery, Thurn and Krebs synthesized violet-P and identified its structure in 1969.33
In the same year, Thurn and Krebs also made an intriguing observation of formations resembling capillary fibers beside plate-like violet-P crystals in the reaction kettle.33 These fibrous formations, later referred to as fibrous phosphorus (fibrous-P), appeared to possess a structure that was similar yet distinct from violet-P. Based on X-ray rotation images of the fibers, they proposed the layout of tubes of the same sort as in violet-P but bound to double tubes in parallel. Ruck and colleagues validated the presumed structure of fibrous-P using single-crystal XRD and TEM in 2005.88
The discovery of black-P can be traced back to 1914, when American physicist Percy Williams Bridgeman attempted to convert commercial white-P into red-P by applying high hydrostatic pressure at a temperature below the transition point that occurs with an appreciable velocity at atmospheric pressure.39 The density of the obtained black-P was 2.691 kg m−3, which was greater than those of white-P (1.83 kg m−3) and red-P (2.05–2.34 kg m−3) at ambient temperature. Percy Williams Bridgman won the 1946 Nobel Prize in Physics for his pioneering work on the physics of high pressures.89 He subsequently made several improvements to the reaction conditions for the synthesis of black-P. For example, he developed a room-temperature method for converting red-P into black-P crystals, although extremely high pressure (8.0 GPa) was required in the process.40 Today, black-P has become a promising elemental material and plays an important role in materials science, chemistry, condensation physics, engineering, and biomedical applications.
2.2 Emergence of phosphorene
In the 1960s, the interest in superconductivity brought attention to phosphorus-rich phase diagrams.90 However, because of the narrow bandgaps and harsh synthetic conditions of crystalline phosphorus, the research on EPMs remained lukewarm for a long time. Black phosphorus, for example, was mentioned in only about 150 research publications before 2014. Since the successful exfoliation of graphene layers using Scotch tape, scientists have turned to other elemental crystals that can be layered through van der Waals (vdW) forces, and the fabrication of atomic-layered elemental nanomaterials has gained popularity.91 For this reason, research interests in EPMs, particularly black-P, have been reignited.In 2014, atomic-layered black-P was synthesized for the first time, and this breakthrough launched a new section in research on what came to be known as phosphorene.45,46 Liu and colleagues referred to single- or few-layered black-P as “phosphorene” with the suffix “-ene,” emphasizing its structural similarity to graphene.46 However, the designation of single- or few-layered black-P is still a subject of debate. As Favron and co-workers have pointed out,92 the term phosphorene does not correlate with the IUPAC nomenclature since the suffix “-ene” is allotted to systems having sp2 hybridized orbitals. They suggested renaming a single layer of black-P as 2D-phosphane to ensure consistency with other sp3-hybridized monolayers such as graphane and germanane. Gusmão et al.,93 on the other hand, argued that the term phosphane should be reserved for saturated phosphorus hydrides with the general formula PnHn+2, and is inappropriate for designating the black-P monolayer. They preferred the term “black-P single sheet” to directly designate single-layered black-P. However, the term phosphorene has surfaced in an increasing number of publications in the past few years, and the majority of academics have embraced this designation. Nowadays, the term phosphorene encompasses not only black phosphorene, but also blue phosphorene, violet phosphorene, fibrous phosphorene, and other atomic-layer allotropes of the phosphorus family.94
The discovery of phosphorene in silico was encouraged by the development of structural prediction algorithms. Zhu et al. first proposed an in-plane hexagonal structure termed blue phosphorus (blue-P) in 2014,49 and this new phase is almost as stable as black phosphorene. In the side view, blue phosphorene displays less puckered zigzag ridges, in contrast to the armchair ridges of the black counterpart. The structural shift from the black to blue counterpart can be viewed as flipping every fourth row of phosphorus atoms, transforming all armchair ridges into zigzag ridges.49,50 In 2016, Zhang et al. reported the growth of atomic-layered blue-P on Au(111),52 and its fabrication encouraged other metastable phosphorene discoveries. One year later, Han et al. predicted the presence of green phosphorene (λ-P) combining both armchair and zigzag ridges, indicating that its structure is a hybrid of black and blue phosphorene.50 The structural shift from the black to green counterpart can be viewed as flipping every twelfth row of phosphorus atoms following the dislocation of every fourth row.50 Other materials with honeycomb-like structures such as γ-P and δ-P, have also been predicted based on ab initio DFT calculations, and both of them are semiconductors.76 While these theoretical transformations provide valuable insights into the exploration of potential phosphorus allotropes, it is important to note that the actual synthesis process is often more complex. The transformation from one phosphorus allotrope to another in practice requires precise control over reaction conditions, highlighting the necessity to understand the phosphorus phase diagram in detail.
Apart from the honeycomb lattice structures, Wu et al. presented four distinct structural phosphorenes, named ε-P, ζ-P, η-P, and θ-P, and these allotropes are dynamically stable based on phonon-spectrum calculations.95 Each building block unit in ε-P and ζ-P comprises two P4 squares which, when viewed from the side, resembles the armchair and zigzag ridges found in black and blue phosphorene, respectively. For η-P and θ-P, each unit is composed of two P5 units oriented in opposing directions, and pentagonal blocks are arranged in a chain along a certain direction, forming a monolayer. Each pentagonal block contains four intrachain bonds and one interchain bond.95 Among the four phosphorene allotropes mentioned, θ-P has been proven to be energetically close to black phosphorene with a relative energy disparity of less than 0.01 eV per atom, suggesting its theoretically stable nature. In principle, new kinds of phosphorene can be created by reorganizing different unit blocks, which can considerably expand the family of atomic-layer EPMs. It is envisaged that other types of phosphorene with hybridized building blocks will be predicted and eventually synthesized.
2.3 Machine learning for prediction of phosphorus materials
Classical phosphorus allotropes such as black, red, and white phosphorus were discovered directly by chemical synthesis, normally relying on chemists' extraordinary intuition and wealth of experience. However, direct synthesis can be difficult to accomplish when we attempt to predict crystal structures that go beyond chemical intuition. At this point, DFT-based computational chemistry techniques come in handy, enabling us to effectively predict complex structures (e.g., ε-P, ζ-P, η-P, and θ-P).95 Of note, quantum-mechanical DFT computations entail solving the Schrödinger equations for the electronic structures of molecules and periodic systems, limiting them to systems with small unit cells and high symmetry.54,96,97 However, for sophisticated systems with numerous atoms per cell and low symmetry (e.g., violet-P and fibrous-P), DFT-based calculations become prohibitively costly and time-consuming.To circumvent this issue, application of machine learning (ML) in atomic-scale simulations and predictions is emerging. By “learning” small structures commonly generated from DFT results, ML-based force field method achieves high accuracy but are orders of magnitude faster.97–99 ML modeling differs from empirically fitted force fields in that it does not make any assumptions about the interatomic potential and instead gathers all information from a large reference database. Building an appropriate reference database is vital for ML-based force field training, which should include enough atomic configurations while having a manageable number of entries. Following that, an appropriate mathematical representation of the atomic structure is required as input for the ML algorithm and ultimately, ML methodology involves regression or “learning” for materials simulations.97 In 2020, Deringer et al. achieved notable success in building a reference database for phosphorus and using it to develop an ML-based Gaussian approximation potential (GAP) model that accurately depicts various bulk crystalline allotropes, liquid structures, and nanostructured phases of phosphorus (Fig. 4).55 This GAP can even be utilized to explore the local structure of amorphous phosphorus's and how it changes under pressure.58 In 2022, Koneru et al. presented a multi-reward reinforcement learning algorithm for training flexible and high-fidelity bond-order potential models for 2D phosphorene.57 The resulting ML-based model accurately portrays various properties of diverse phosphorene polymorphs such as the structure, energetics, transformation barriers, equation of state, elastic constants, and phonon dispersions.57 This work paves a way for the dynamic simulation of low-dimensional EPMs on a mesoscale, leading to faster design and discovery.
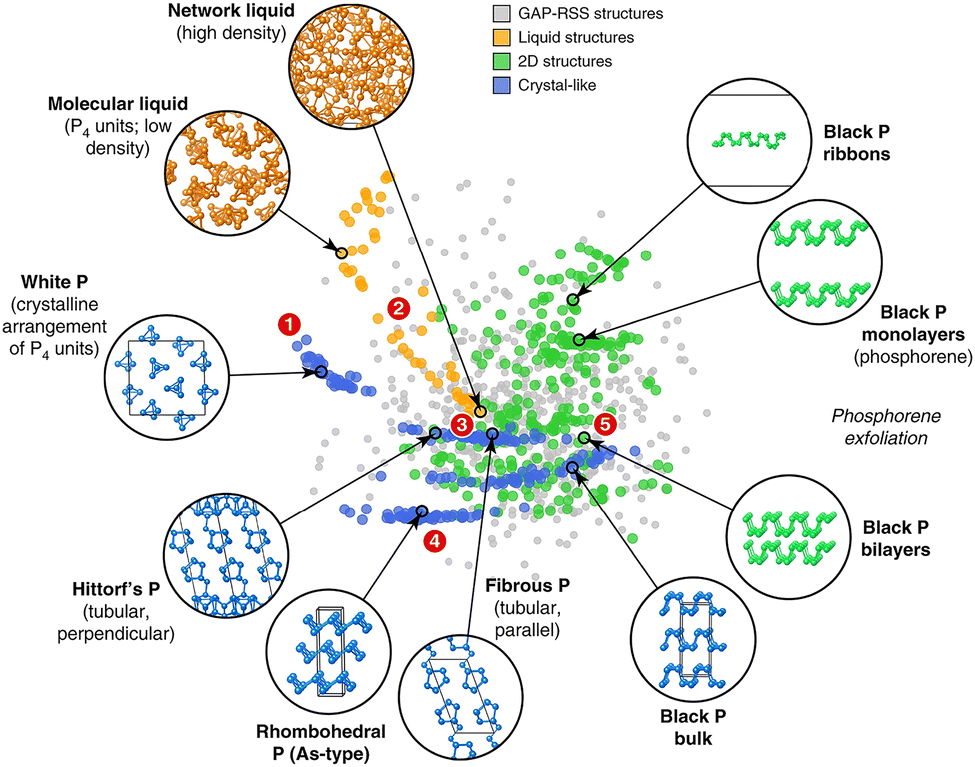 | ||
| Fig. 4 GAP (Gaussian approximation potential) fitting database designed for elemental phosphorus. Reproduced with permission from ref. 55. Copyright 2020 Nature Publishing Group. | ||
Atomic-level simulations offer significant advantages in studying materials with complex structures. ML-based force field method is rapidly emerging as a powerful tool in materials modeling, capable of solving a wide range of materials science challenges. However, ML modeling for phosphorus-based materials is still in its infancy, and the academic community must solve several algorithmic and adaptability issues. For instance, ML-based force fields that are trained to be very accurate for a certain set of configurations will not be highly accurate elsewhere.97 Thus, a reference database with representative configurations is needed to balance simulation accuracy and versatility. In addition, balancing precision and speed in describing the diverse and complex structures of phosphorus-based materials is crucial. We expect that in the future, ML-based force field method will tackle key issues in phosphorus materials science, and data-driven structure searching will reveal more structural allotropes of phosphorus.
In summary, this section has delved into the development of EPMs, tracing the discovery of classic allotropes to the emergence of phosphorene and the use of machine learning in this field. As we forge ahead, future work should leverage a multidisciplinary approach to discover new phosphorus allotropes, further investigate the properties of newly predicted phosphorene allotropes, and advance the use of machine learning models for predicting and simulating these materials. This integrated approach could open new frontiers in EPM research. Importantly, it is crucial to note that every claim of a newly discovered phosphorus allotrope must be substantiated by solid crystallographic data from free-standing samples, such as single-crystal X-ray diffraction, single-crystal neutron diffraction, and electron diffraction.
3. Fundamental physicochemical properties of elemental phosphorus materials
The characteristics of materials such as electronic, optical, and mechanical properties are not solely determined by the chemical composition but are also significantly influenced by allotropes, which refer to the arrangement of atoms within the lattice structure. EPMs that possess different crystal lattices have been investigated and demonstrated to exhibit fascinating properties, such as structural polymorphism, moderate carrier mobility, adjustable bandgaps, as well as in-plane anisotropic electronic and phonon properties. These properties are both structure-dependent and versatile, favoring potential applications of EPMs. In this section, we analyze these properties which have been theoretically and/or experimentally studied (Fig. 5). In particular, there are commonalities and differences among various EPMs and the comparisons of building blocks, materials stability, bandgaps, and charge carrier mobility of EPMs are highlighted. In addition, we briefly introduce the passivation techniques for improving the materials stability by, for example, doping, encapsulation, and surface functionalization.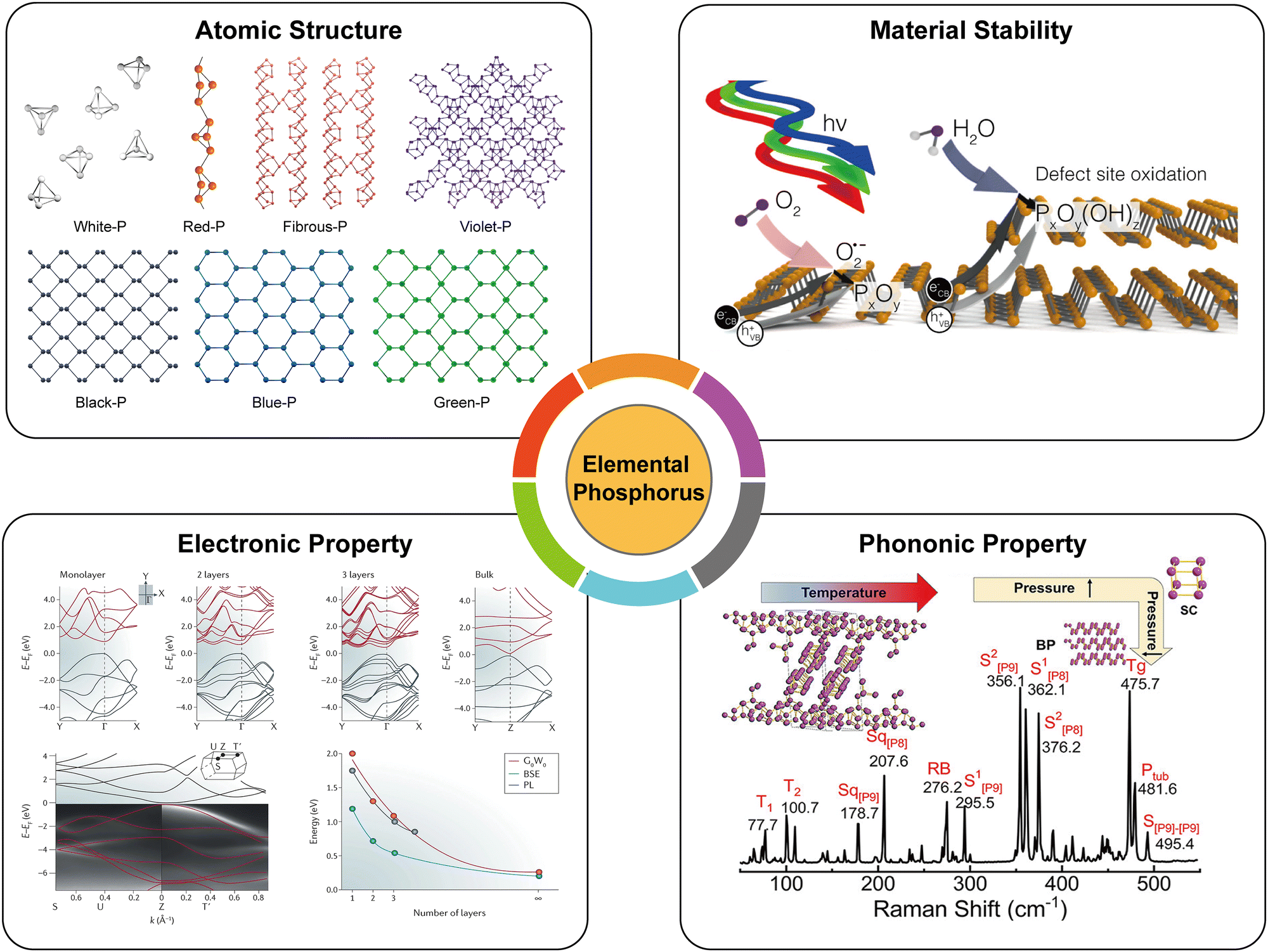 | ||
| Fig. 5 Fundamental physicochemical properties of elemental phosphorus materials. Atomic structure, originally drawn by our group. Material stability, reproduced with permission from ref. 72. Copyright 2017 American Chemical Society. Electronic property, reproduced with permission from ref. 112. Copyright 2016 Nature Publishing Group. Phonon property, reproduced with permission from ref. 173. Copyright 2021 American Chemical Society. | ||
3.1 Atomic structure
Many intriguing properties of EPMs are intimately related to the geometric configuration of phosphorus atoms. As shown in Fig. 6, EPMs possess different building blocks that can be divided into three categories: (1) white-P containing tetrahedral P4 cages; (2) black-P and its derivatives with a puckered honeycomb structure; and (3) red-P allotropic modifications composed of chain-like and polygonal connected rings. In the following parts, we will discuss these categories in greater detail, providing insights into their unique geometries and the consequent impacts on the properties of EPMs.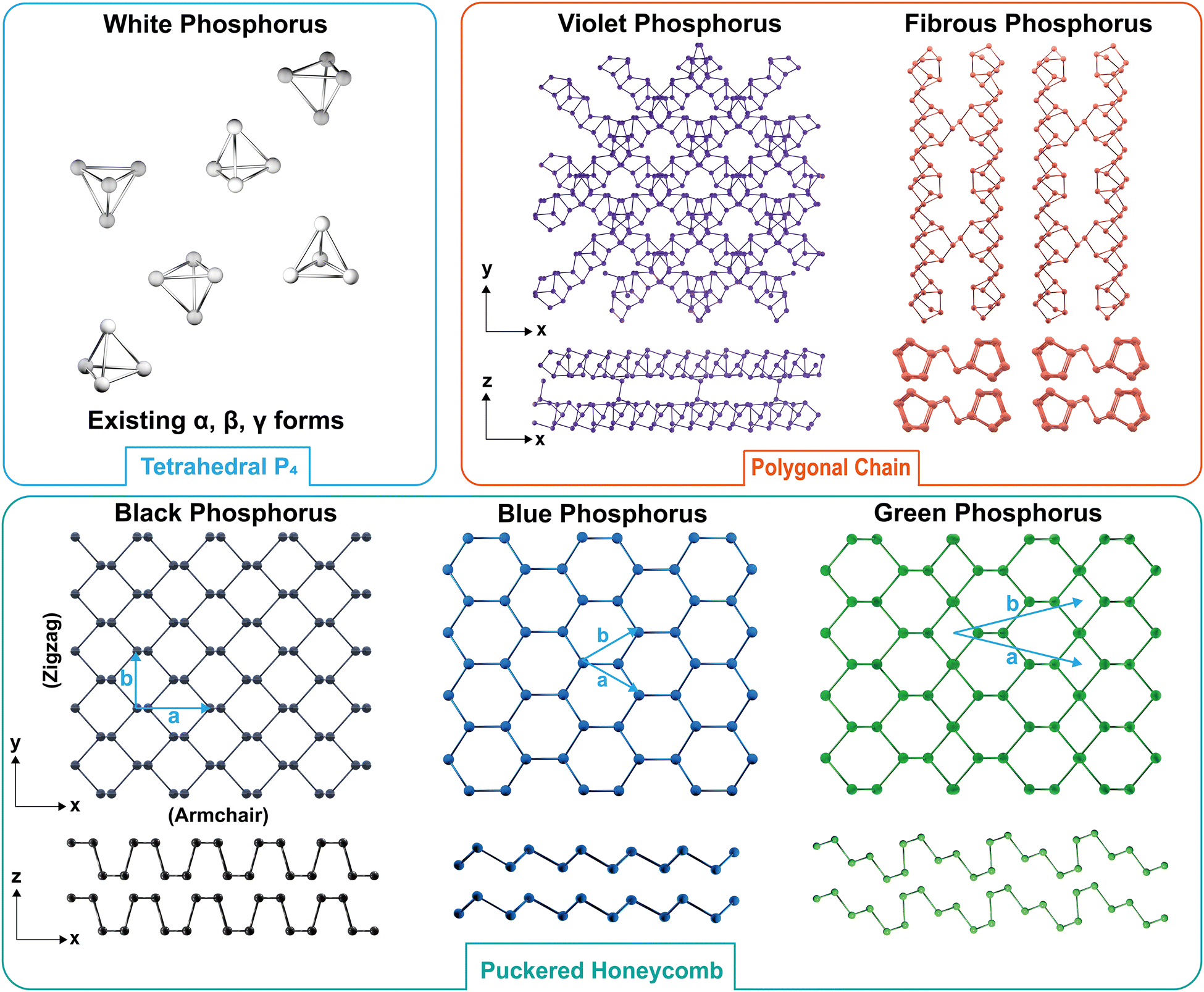 | ||
| Fig. 6 Atomic structures of white, violet, fibrous, black, blue, and green phosphorus. Three categories are divided by the building blocks: tetrahedral P4 (white phosphorus), polygonal chain (violet and fibrous phosphorus), and puckered honeycomb (black, blue, and green phosphorus). | ||
Bulk black-P exists in three crystalline phases, orthorhombic, simple cubic, and rhombohedral, and the most intriguing is the semiconducting orthorhombic phase belonging to the space group Cmca(64) with eight phosphorus atoms per cell.109 Orthorhombic black-P can change its structure reversibly at a high pressure morphing into the denser rhombohedral or simple cubic forms.110 However, these two forms are unstable under normal conditions and are not suitable for use in practice. In this review, unless specified, black-P refers to the orthorhombic phase which is the focus of most research. The lattice constants a, b, and c of black-P crystals, determined by time-of-flight neutron powder diffractometry by Cartz et al., are 4.374(2), 3.3133(9), and 10.473(5) Å, respectively.111 Owing to orbital hybridization, black-P has a nonplanar bilayer structure with two different bond angles (α = 96.34° and β = 103.09°). From the side view, there are two types of P–P bonds, a short bond (2.224 Å) and a long bond (2.244 Å) connecting two close atoms in different sublayers.112 This results in anisotropy between the zigzag (y) and armchair (x) directions and the crystalline symmetry of single-layered black-P is two-fold rotational (D2h), which is far less than the six-fold rotational (D6h) symmetry observed in single-layered graphene, which has a flat hexagonal lattice.71 The reduction in crystalline symmetry is fundamental to the various chemical and physical properties. For instance, the band structure of black-P exhibits anisotropy irrespective of the thickness, giving rise to in-plane anisotropic properties in charge transport, thermal conductivity, and optics.113,114 The interlayers are held together by vdW forces with a layer spacing of 3.214–3.729 Å,115 depending on different stacking orders. The relatively weak vdW forces make black-P suitable for exfoliation to prepare phosphorene and allow for property turnability.
The atomic structure of the blue-P monolayer, as discussed in Section 2, can be viewed as the flipping of specific atoms, transforming the armchair ridges into zigzag ridges in the black-P monolayer without altering the local bond angles. This leads to a non-planar but isotropic structure for single-layered blue-P, a significant difference from the anisotropic structure of its black counterpart. Zhu et al. noted the possibility of a new phosphorus phase resulting from a certain dislocation of black-P and conducted ab initio calculations to investigate the basic properties of the allotrope.49 They dubbed this new allotrope blue phosphorus (blue-P) with the color defined by its fundamental bandgap. Blue-P atoms are covalently bonded in the layer with a bond length of 2.27 Å, and a weak interlayer interaction of 6 meV per atom keeps the layered structure intact with an interlayer distance of 5.63 Å.49 The optimized blue-P monolayer has a hexagonal unit cell with lattice vectors of |![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) | = |
| = |![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) | = 3.33 Å. The interlayer interaction shows little effects on the in-layer structure, causing only a slight shift in the lattice vector from 3.324 Å in bulk to 3.326 Å in the monolayer. The free-standing bulk crystals of blue-P do not exist, although single-layered blue-P has been reported to grow on several noble metals.52
| = 3.33 Å. The interlayer interaction shows little effects on the in-layer structure, causing only a slight shift in the lattice vector from 3.324 Å in bulk to 3.326 Å in the monolayer. The free-standing bulk crystals of blue-P do not exist, although single-layered blue-P has been reported to grow on several noble metals.52
Green-P is a metastable EPM that combines features of armchair-ridged black-P and zigzag-ridged blue-P. Han et al. were the first to report the structural and electronic properties in silico.50 Bulk green-P has a monoclinic C2/m structure with six atoms per unit cell and in-plane lattice constants of a = b = 7.13 Å. Green-P exhibits the greatest layer thickness (2.91 Å) among the three allotropes due to the presence of three slightly curved sublayers in each layer, compared to 2.11 Å for black phosphorene and 1.24 Å for blue phosphorene. All the atoms in green-P are three-fold bonded with in-plane lengths of 2.23 Å and out-of-plane bond lengths of 2.26 Å. The AB stacking order is energetically preferred and the interlayer distance is 2.16 Å. Although green-P has a shorter interlayer distance than black-P and blue-P, the interaction between green-P layers at 74 meV per atom is similar to that of Black-P (79 meV per atom), indicating its potential for mechanical exfoliation. Moreover, the buckled structure of green phosphorene aligns well with corrugated Au and Ag substrates, suggesting that epitaxial growth is favored with this allotrope.50
The structure of type I amorphous red-P has been the subject of debate and two main models are currently being considered.28 Model 1 postulates that it consists of chain-like pentagonal nanotubes resembling monoclinic violet-P,118,119 while model 2 proposes a layered structure similar to that of orthorhombic black-P.120–122 Model 2, which explains the alterations in the intermediate-range order in phosphorus layers based on growth factors,120 has been widely adopted and accepted by many studies. However, several structural studies using Raman spectroscopy and XPS have found evidence of the presence of pentagonal tubes in the microstructure of amorphous red-P,100,118 suggesting that model 1 may also play a role.
In recent years, innovative methodologies have been employed to determine the microstructure of amorphous red-P. Zhang et al.123 discussed the four possible linear molecular structures of amorphous red-P that were previously proposed by chemists (Fig. 7a). These included the double triangle P4 (1) proposed by Pauling and Simonetta in 1952,108 the zigzag ladder P4 (2) proposed by Salzmann et al.,124 as well as [P8]P2[ (3) and [P10]P2[ (4) suggested by Häser and colleagues.125,126 Through their single-molecule studies, they discovered that amorphous red-P is a straight-chain inorganic polymer with a zigzag ladder structure, which aligns more closely with structure (2). However, Deringer et al. adopted ML-based simulations to visualize the local structure and pressure-induced changes in amorphous red-P at the atomic scale.58 They inferred that amorphous red-P is an open network of covalently connected phosphorus clusters. They analyzed the types and counts of cluster fragments, finding that P5 rings, P2[P3]P2 and P3]P2[P3 clusters, and P10 cages are the dominant building units, whereas P3 rings and P4 tetrahedra are much less abundant (Fig. 7b). The precise molecular structure of amorphous red-P still awaits verification through further computational and experimental investigation.
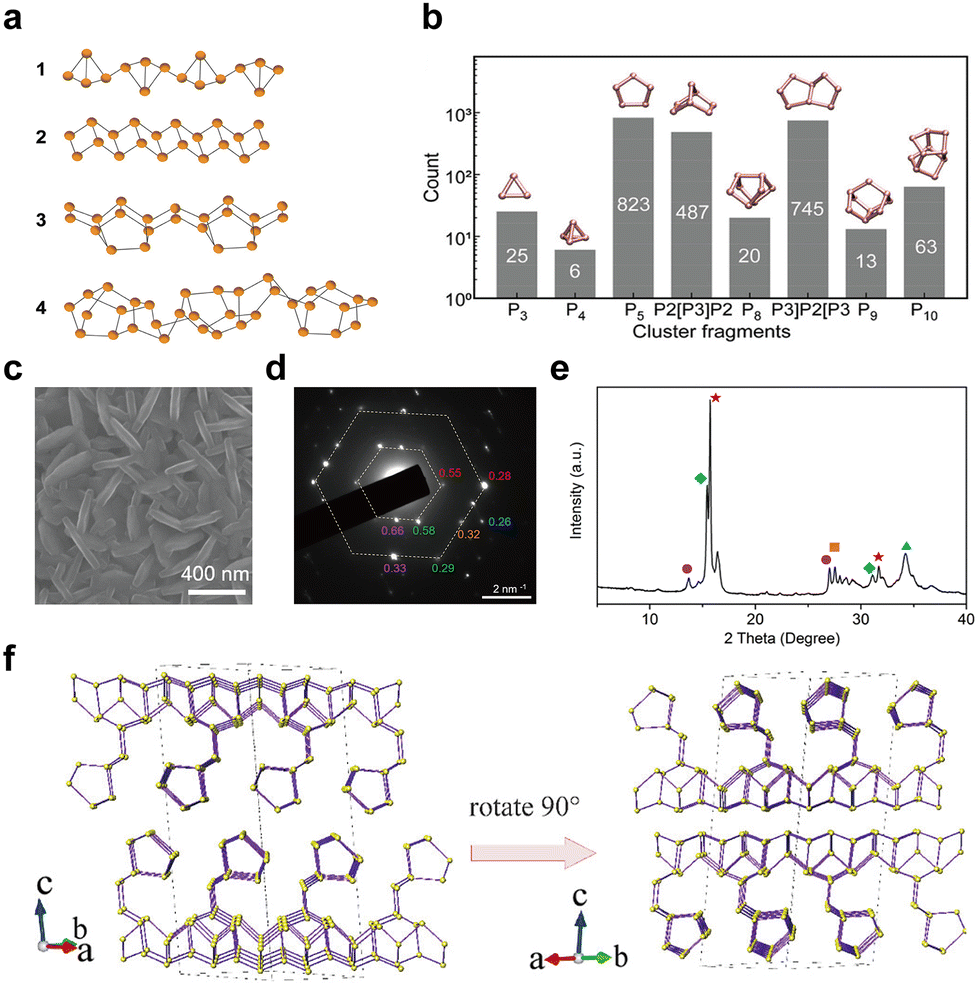 | ||
| Fig. 7 Structure and characterization of type I amorphous red-P, type II crystalline red-P, and violet-P11. (a) Possible linear molecular structures of amorphous red-P. Reproduced with permission from ref. 123. Copyright 2019 John Wiley and Sons. (b) Counts of relevant cluster fragments in the simulated model. Reproduced with permission from ref. 58. Copyright 2021 John Wiley and Sons. (c) SEM image of type II crystalline red-P. (d) SAED pattern of type II crystalline red-P with interplanar spacings. (e) XRD peaks marked with the same colour as spots in (d) represent a set of corresponding lattice planes with primary and secondary diffraction peaks. Reproduced with permission from ref. 129. Copyright 2021 Royal Society of Chemistry. (f) Crystal structure of violet-P11 viewed along two directions. Reproduced with permission from ref. 136. Copyright 2023 American Chemical Society. | ||
Roth et al.34 were the first to observe type II and III crystalline red-P using differential thermal analysis. In 1966, Rubenstein and Ryan127 verified the presence of type II crystalline red-P by annealing in an inert atmosphere and matching the powder XRD patterns with Roth's findings. Nechaeva et al.128 later produced type II crystalline red-P by chemical vapor deposition and identified the monoclinic structure with specific lattice parameters. Recently, Yan and colleagues have produced type II red-P crystals with a platelet morphology using a solution phase synthesis method (Fig. 7c).129 The high crystallinity of the product is evident from the XRD results, with the most intense peak at 15.66° and a plane spacing of 5.66 Å, which is in close agreement with the results obtained by Roth and colleagues in 1947.34,129 The nearly hexagonal symmetrical SAED pattern of the crystal suggests a monoclinic crystal system, and the plane spacing is consistent with the XRD pattern (Fig. 7d and e). However, so far, limited structural information is available for type III crystalline red-P. It is imperative to develop reliable methods for synthesizing type III crystalline red-P to facilitate the analysis of its crystal structure.
The structures of two other types of crystalline red-P have been studied.23,28 Type IV fibrous-P and type V violet-P have been confirmed to consist of repeating polymeric tubes with a pentagonal segment arranged in a regular pattern of [P9]P2 [P8]P2[. The key difference between fibrous-P and violet-P lies in the manner the double tubes are connected. Violet-P consists of two interpenetrating tube systems that run perpendicular to each other, forming double stacks held together by vdW forces in the c direction. The double tubes in fibrous-P, on the other hand, are parallel and held together by vdW forces in both the a and c directions, making it a member of the unique 1D vdW crystal family with distinct bonding behavior and anisotropic physical properties.36 The structural parameters of violet-P and fibrous-P are available. By using single-crystal XRD, Zhang et al.48 have determined the lattice constants of violet-P crystals to be 9.210, 9.128, and 21.893 Å for the parameters a, b, and c, respectively. Meanwhile, Du et al.36 have used XRD to show that the lattice constants of fibrous-P crystals are 12.198, 12.986, and 7.075 Å for a, b, and c, respectively, which are consistent with theoretical simulation results. Although both fibrous-P and violet-P have similar plate-like shape and color, they can be differentiated through mechanical stress tests, in which fibrous-P crystals break down into thin filaments, while violet-P crystals shatter into smaller platelets.88 Furthermore, high-resolution TEM shows distinct morphological differences between the two crystalline phases.
Bulk crystals of violet-P can be peeled into few- or single-layered violet phosphorene by applying external forces. DFT calculation has been used to compute the binding energy per layer needed for complete exfoliation of the bulk crystal into monolayers. The binding energy of violet phosphorene is 0.35 J m−2, which is comparable to that of graphene (0.34 J m−2) but lower than that of black phosphorene (0.40 J m−2) or blue phosphorene (0.42 J m−2).130 This indicates that exfoliating violet phosphorene is easier than other types of phosphorene. In 2020, violet phosphorene was synthesized and characterized as a 2D nanobelt composed of tubular strands with an interlayer spacing of 11 Å in AB stacking.48 Regarding fibrous phosphorene, Hu and Guo have confirmed that fibrous-P bulk crystals have a layered structure along the (001) direction and can be exfoliated experimentally.131 Each layer consists of four [P9]P2 [P8]P2[ polytubes with 84 phosphorus atoms held together by vdW interactions. The average interlayer distances along the (001) and (200) facets are 5.83 Å and 5.57 Å, respectively. The binding energy for fibrous phosphorene, calculated to be 0.88 J m−2,132 is at least two times greater than that of violet phosphorene, making exfoliation of fibrous-P more challenging. However, fibrous phosphorene is held together by vdW interactions in two directions (a and c) similar to selenene and tellurene133–135 and can be fabricated by epitaxy or other methods.
Very recently, Cicirello and colleagues synthesized a new two-dimensional allotrope of violet phosphorus, dubbed violet-P11, using a salt flux method in the presence of bismuth.136 The crystal structure of this allotrope from different views is shown in Fig. 7f. The structure of violet-P11 is similar to that of violet-P. However, violet-P11 possesses a higher structural symmetry compared to that of violet-P. The asymmetric unit of violet-P11 is entirely occupied by 11 distinct phosphorus atoms, while that of violet-P comprises 21 different phosphorus atoms. Single-crystal XRD has revealed that violet-P11 crystallizes in the monoclinic space group C2/c(15), with unit cell parameters of 9.166(6), 9.121(6), and 21.803(14) Å for a, b, and c, respectively.136 Violet-P11 exhibits substantial stability in ambient air even after being exfoliated for at least an hour.
3.2 Electronic properties
Understanding the electronic properties of a material is pivotal for unraveling the mechanisms underlying its applications in catalysis, ion storage, sensing, and environmental remediation. In the following parts, we will discuss the differences and interrelationships in the band structure and charge mobility among different EPMs.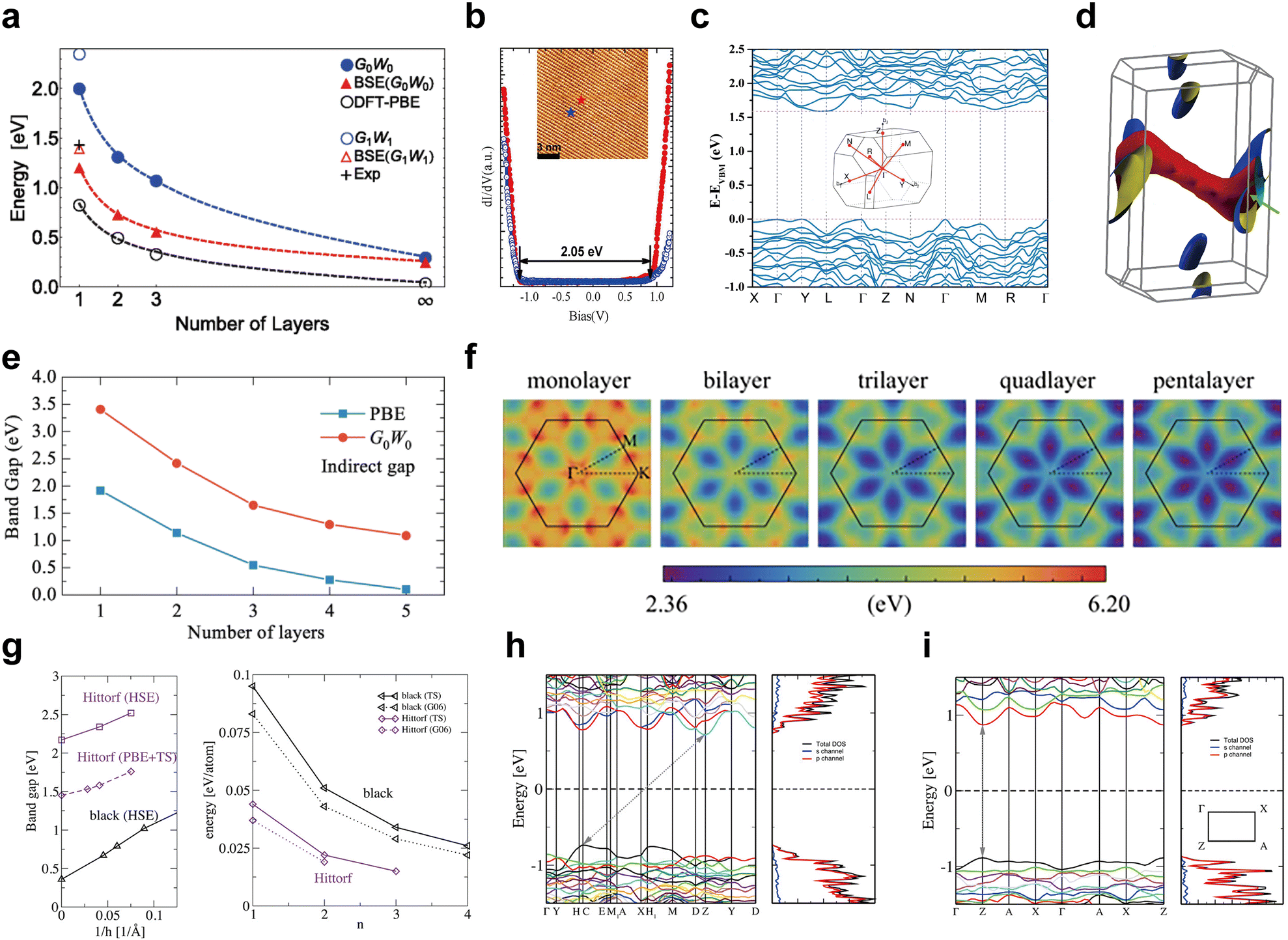 | ||
| Fig. 8 Electronic band structures of black, violet, fibrous, blue, and green phosphorus. (a) Layer dependence of bandgaps for black-P as calculated by various methods. Reproduced with permission from ref. 139. Copyright 2014 American Physical Society. (b) Spectroscopic measurement of the bandgap for black-P. Reproduced with permission from ref. 140. Copyright 2014 American Chemical Society. (c) Theoretical band structure and (d) the mapping of the Fermi surface of bulk fibrous-P. Reproduced with permission from ref. 36. Copyright 2021 Nature Publishing Group. (e and f) Layer dependence of quasiparticle bandgaps for few-layered blue-P. Reproduced with permission from ref. 142. Copyright 2021 American Physical Society. (g) Layer-dependent bandgaps and relative energy of violet-P and black-P. The electronic band structure of the violet-P monolayer (h) and bulk form (i). Reproduced with permission from ref. 130. Copyright 2016 American Chemical Society. | ||
Fibrous-P and atomically layered blue-P have indirect bandgaps. Du et al. have plotted the electronic band structures of bulk fibrous-P and shown an indirect bandgap of approximately 1.57 eV (Fig. 8c).36 The conduction band minimum (CBM) and valence band maximum (VBM) can be found around the L and Γ/Y points of the Brillouin zone, respectively. To correct for the underestimated value, they applied the modified Becke–Johnson functional, resulting in an indirect bandgap range of ∼1.80 to 2.11 eV. The 3D mapping of the Fermi surface reveals weak dispersion in the valence band (Fig. 8d), which is caused by electron confinement in one dimension. Lu et al. used the HSE06 hybrid functional and determined the indirect bandgap of fibrous-P bulk crystals and monolayers to be 2.25 and 2.46 eV, respectively.132 The bandgap of a fibrous-P bilayer is 2.34 eV, which is only slightly narrower than that of the monolayer, indicating that the number of stacked layers has minimal effects on the bandgap of fibrous-P. This makes fibrous-P based materials ideal for catalysis because bulk crystals can be directly used without the need for extra exfoliation or precise control over layer numbers. Zhou et al.142 have assessed the evolution of quasiparticle bandgaps in few-layered blue-P (Fig. 8e and f). The indirect bandgap diminishes as the layer count rises starting at 3.41 eV for the monolayer, reaching 1.09 eV for the penta-layer, and finally changing into a metallic bandgap in the bulk form. The bandgap calculated using PBE is significantly smaller than that determined using quasiparticle approximation, yet the degree of band dispersion remains largely unchanged. This suggests that the bandgap width of blue-P is larger than that of black-P and green-P, and bulk blue-P even exhibits metallic characteristics. However, whether bulk blue-P is semiconducting or metallic is controversial,49,142,143 and further theoretical studies on the electrical structure of blue-P crystals are required. Experimentally, the bandgap of 4 × 4 blue phosphorene on Au(111) is measured to be approximately 1.10 eV by angle-resolved photoemission spectroscopy,144 which is significantly lower than the theoretical bandgap of free-standing blue phosphorene. This results from the strong in-layer strain caused by the interaction between phosphorene and the Au(111) substrate,52,145 which disturbs the lattice symmetry of free-standing blue-P, hence impacting the electrical characteristics.
Violet-P differs from the other four allotropes as it undergoes an indirect-to-direct bandgap cross-over when downscaling from the bulk to monolayers. This cross-over is also found in many semiconducting transition metal dichalcogenides (TMDs),146–148 making it a useful way to tune the electronic properties of layered semiconductors. Schusteritsch et al.130 have developed several calculation methods for determining the electronic band structure of Violet-P (Fig. 8g). The PBE method with dispersion corrections shows the bandgap of bulk violet-P to be 1.45 eV, while monolayer violet-P exhibits a direct bandgap of 1.76 eV at the Z point in the Brillouin zone (Fig. 8h–i).130 A more accurate HSE(TS) hybrid calculation gives wider bandgaps of 2.17 and 2.52 eV for the violet-P bulk form and monolayer, respectively. UV/vis diffuse reflectance spectroscopy reveals an optical bandgap of 1.7 eV for violet-P crystal powder, similar to 1.77 eV obtained in photoluminescence of single-crystal violet-P.48 The optical bandgap is narrower than the calculated value because it is measured on a substrate-supported sample rather than in vacuum.112 Both fibrous-P and violet-P have similar bandgaps that are not greatly affected by the number of layers due to similarities in the atomic structures.88 Further research is needed to fully explain why violet-P has an indirect-to-direct cross-over instead of an indirect bandgap like fibrous-P.
The bandgaps of the aforementioned EPMs are influenced by the number of layers, and the bandgap becomes smaller with increasing number of layers due to the influence of vdW interactions between the layers. As the layer number increases, the interlayer interaction becomes stronger to increase band dispersion and produce a narrower band gap.11,149,150 Single- and few-layered phosphorene displays pronounced quantum confinement, and the valence and conduction bands mimic those of a quantum-well structure. However, in the bulk counterpart, energy dispersion becomes densely packed, causing the discrete energy bands to turn into quasi-continuous sub-bands.11 In addition, the bandgaps of phosphorus allotropes are sensitive to electric fields,151 molecule adsorption,152 and tensile strain.153 These external factors offer significant control over the bandgap, particularly in the single-and few-layered structures. As shown in Fig. 9, EPMs with tunable and layer-dependent bandgaps cover a wide range of energy and exhibit strong interaction with electromagnetic waves across the mid-infrared, near-infrared, visible, and near-ultraviolet frequencies, making them suitable for various applications such as thermal imaging (0.1–1.0 eV), photovoltaics/photocatalysis (1.2–2.2 eV), and blue LED techniques (2.5–2.9 eV).
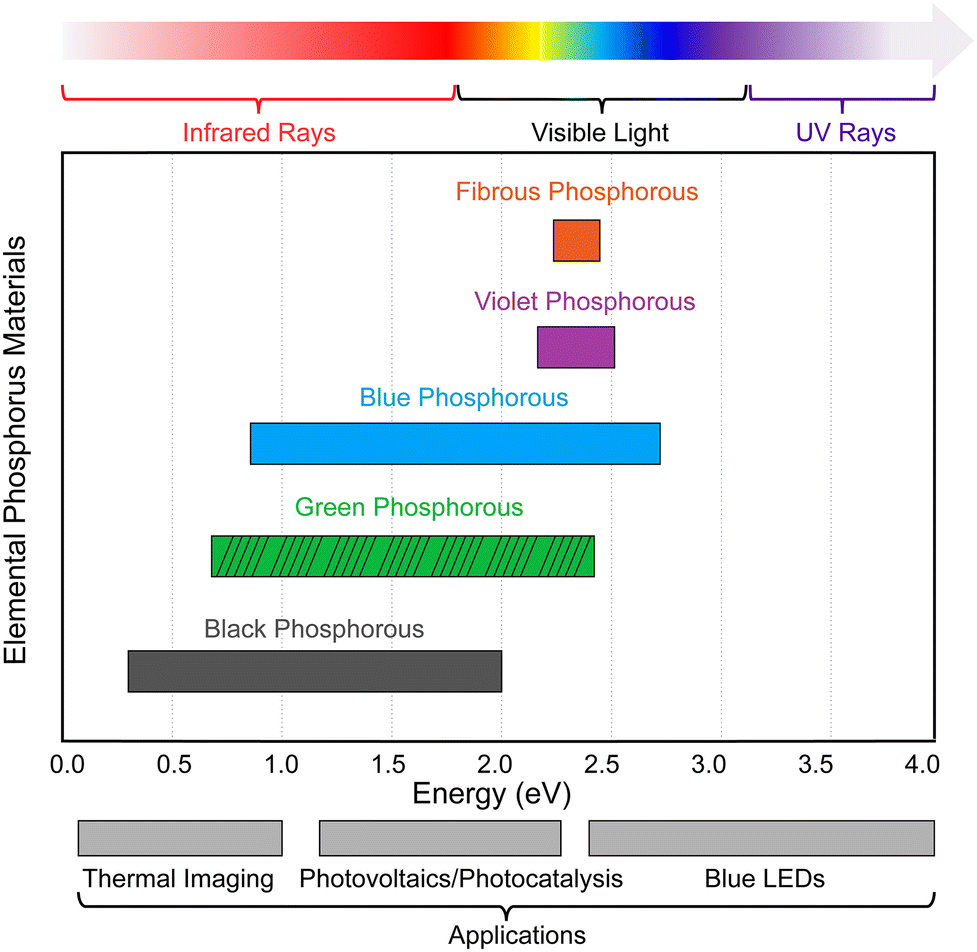 | ||
| Fig. 9 Comparison of the bandgaps of different EPMs and potential applications. The horizontal bars displaying a range of bandgap values indicate that the bandgap can be adjusted by changing the number of layers. The bandgap of green-P is currently based on theoretical calculations. Bandgap values are taken from ref. 50, 130, 132, 139, and 143. | ||
| System |
|
|
E 1x | E 1y | C x_2D | C y_2D | μ x_2D | μ y_2D |
|---|---|---|---|---|---|---|---|---|
| e, electrons; h, holes; Blue-P*, 4 × 4 phosphorene on Au(111). Data extracted from ref. 69, 75, 130, 132, and 144. | ||||||||
| Black-P(e) | 0.17 | 1.12 | 2.72 ± 0.02 | 7.11 ± 0.02 | 28.94 | 101.60 | 1.10–1.14 | ∼0.08 |
| Black-P(h) | 0.15 | 6.35 | 2.50 ± 0.06 | 0.15 ± 0.03 | 28.94 | 101.60 | 0.64–0.70 | 10–26 |
| Violet-P(e) | 0.69 ± 0.06 | 3.58 ± 0.14 | 1.40 ± 0.03 | 0.66 ± 0.01 | 49.70 ± 0.2 | 49.92 ± 0.4 | 0.50 ± 0.02 | 0.43 ± 0.03 |
| Violet-P(h) | 1.24 ± 0.03 | 2.45 ± 0.06 | 1.26 ± 0.06 | 0.18 ± 0.05 | 49.70 ± 0.2 | 49.92 ± 0.4 | 0.31 ± 0.02 | 7.68 ± 4.3 |
| Fibrous-P(e) | 0.41 | 0.56 | 3.98 | 0.64 | 9.96 | 91.65 | 0.07 | 17.7 |
| Fibrous-P(h) | 2.95 | 0.44 | 0.56 | 1.83 | 9.96 | 91.65 | 0.19 | 1.17 |
| Green-P(e) | 0.27 | 0.20 | −5.02 | 2.35 | 26.5 | 84.9 | 0.355 | 7.02 |
| Green-P(h) | 0.21 | 1.71 | −9.93 | 7.71 | 26.5 | 84.9 | 0.048 | 0.024 |
| Blue-P*(e) | 0.130 | 0.132 | −2.289 | −2.261 | 42.826 | 47.248 | 10.263 | 11.429 |
| Blue-P*(h) | 2.231 | 1.587 | −1.007 | −1.005 | 42.826 | 47.248 | 0.215 | 0.335 |
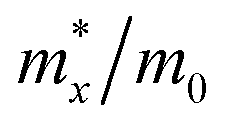
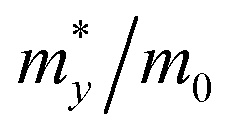
Black-P and violet-P exhibit intrinsic p-type conductivity with a positive Hall coefficient, indicating that conductivity is primarily driven by holes.37,69 Based on Qiao's phonon-limited scattering model, the hole and electron mobility of black-P can be extended to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–26000 cm2 V−1 s−1 and 1100–1140 cm2 V−1 s−1, respectively, which are comparable to those of graphene and greater than those of 2D TMDs.69 Black-P shows in-plane anisotropic carrier mobility with electron mobility along the x-axis being greater than that along the y-axis. Regarding the hole mobility, the preference for migration along the x or y-axis depends on the number of layers. In the few-layered black-P, the hole mobility along the y-axis is about half that along the x-axis. In a monolayer, the hole mobility along the y-axis is surprisingly 16–38 times greater than along the x-axis, that is, 10000–26000 cm2 V−1 s−1versus 640–700 cm2 V−1 s−1, indicating that the y-axis is the direction of enhanced hole conductivity.69 Despite the carriers being relatively heavy (6.35m0), the high mobility in a monolayer is due to the low deformation potential (0.15 ± 0.03 eV).69 Several people have designed black-P based field-effect transistors (FETs) and examined the charge carrier mobility.45,46,109,157 Violet-P has been predicted to have hole mobilities between 3000–7000 cm2 V−1 s−1 based on theoretical calculations.130 Applying PBE without dispersion corrections, the effective masses of electrons and holes are found to be greater along the y-axis and the effective mass of electrons along the x-axis (0.69 ± 0.01m0) is the smallest. The hole deformation potential along the y-axis is nearly 7 times smaller than the value along the x-axis, that is, 0.18 ± 0.05 eV versus 1.26 ± 0.06 eV, resulting in a significant hole mobility of ∼7000 cm2 V−1 s−1 along the y-axis. However, when PBE with dispersion corrections (PBE + TS) is employed, the hole mobility is reduced to ∼3000 cm2 V−1 s−1 as PBE + TS calculates a magnitude of the deformation potential that is twice that of PBE. Recently, Ricciardulli et al. have created a FET using exfoliated violet-P thin films and found that the hole mobility of the film is 2.25 cm2 V−1 s−1 at room temperature.37 Although charge transport in films may be impeded by inter-flake resistances and structural defects caused by the exfoliation process,158 this work experimentally demonstrates the potential of violet-P as a p-type semiconductor.
000–26000 cm2 V−1 s−1 and 1100–1140 cm2 V−1 s−1, respectively, which are comparable to those of graphene and greater than those of 2D TMDs.69 Black-P shows in-plane anisotropic carrier mobility with electron mobility along the x-axis being greater than that along the y-axis. Regarding the hole mobility, the preference for migration along the x or y-axis depends on the number of layers. In the few-layered black-P, the hole mobility along the y-axis is about half that along the x-axis. In a monolayer, the hole mobility along the y-axis is surprisingly 16–38 times greater than along the x-axis, that is, 10000–26000 cm2 V−1 s−1versus 640–700 cm2 V−1 s−1, indicating that the y-axis is the direction of enhanced hole conductivity.69 Despite the carriers being relatively heavy (6.35m0), the high mobility in a monolayer is due to the low deformation potential (0.15 ± 0.03 eV).69 Several people have designed black-P based field-effect transistors (FETs) and examined the charge carrier mobility.45,46,109,157 Violet-P has been predicted to have hole mobilities between 3000–7000 cm2 V−1 s−1 based on theoretical calculations.130 Applying PBE without dispersion corrections, the effective masses of electrons and holes are found to be greater along the y-axis and the effective mass of electrons along the x-axis (0.69 ± 0.01m0) is the smallest. The hole deformation potential along the y-axis is nearly 7 times smaller than the value along the x-axis, that is, 0.18 ± 0.05 eV versus 1.26 ± 0.06 eV, resulting in a significant hole mobility of ∼7000 cm2 V−1 s−1 along the y-axis. However, when PBE with dispersion corrections (PBE + TS) is employed, the hole mobility is reduced to ∼3000 cm2 V−1 s−1 as PBE + TS calculates a magnitude of the deformation potential that is twice that of PBE. Recently, Ricciardulli et al. have created a FET using exfoliated violet-P thin films and found that the hole mobility of the film is 2.25 cm2 V−1 s−1 at room temperature.37 Although charge transport in films may be impeded by inter-flake resistances and structural defects caused by the exfoliation process,158 this work experimentally demonstrates the potential of violet-P as a p-type semiconductor.
Dominated by high electron mobilities, fibrous-P and green-P are potential materials for n-type devices.50,132 In the fibrous-P monolayer, the effective masses of electrons (holes) along the x- and y-axis are calculated to be 0.41 (2.95) and 0.56 (0.44)m0, respectively.132 The deformation potential (E1) and elastic modulus (C2D) are highly anisotropic in the plane. The calculated deformation potentials of electrons along the x- and y-axis are approximately 3.98 and 0.64 eV, respectively, with an anisotropy ratio greater than 6. C2D_y is almost nine times larger than C2D_x. This results in a significant electron mobility of ∼17700 cm2 V−1 s−1 along the y-axis, which is comparable to that of other layered semiconductors like black phosphorene,69 violet phosphorene,130 and Al2C monolayers.159 However, the FET made from fibrous-P nanoribbons has a lower hole mobility (236.7 cm2 V−1 s−1),160 which is far less than expected. This may be due to the high contact resistance between the channel materials and electrodes. High mobility can be improved by modifying the contact electrodes or using high-k materials as the gate layer.156,160 In green phosphorene, the effective masses of electrons (holes) along the x- and y-axis are predicted to be 0.274 (0.166) and 0.162 (1.333)m0, respectively.50 This suggests that the hole mobility is highly anisotropic, while the electron mobility is nearly isotropic. For a carrier concentration of 5 × 1012 cm−2, the electron mobility is determined to be around 200 cm2 V−1 s−1 at 300 K, which is higher than the hole mobility (∼60 cm2 V−1 s−1). This relatively high electron mobility is a result of lower scattering rates near the CBM.
Zhuang et al. have investigated the carrier mobility characteristics of 4 × 4 blue phosphorene on Au(111) theoretically.144 The effective mass of holes is slightly greater along the x-axis than along the y-axis (2.231m0versus 1.587m0), whereas the effective mass of electrons along both axes is very close and small (∼0.130m0). This small effective mass leads to high electron mobility, which can reach up to 11429 cm2 V−1 s−1. The limited anisotropy in carrier mobilities of 4 × 4 blue phosphorene on Au(111) is related to the more symmetrical atomic structure, but even minor changes in the structure of layered materials can produce significant variations in the electronic properties. The strong in-layer strain caused by the blue-P–Au(111) interaction greatly influences the electron mobility.144,161 Therefore, they have also calculated the carrier mobilities of 4 × 4 blue phosphorene in the absence of tensile strain.144 The effective mass of electrons increases significantly in free-standing blue phosphorene, which greatly reduces the electron mobility by one or two orders of magnitude.
So far, studies on the carrier mobility of elemental phosphorus semiconductors, excluding black-P, have mainly been theoretical. In general, theoretical calculations for the room-temperature carrier mobilities (μ2D) of layered materials use an acoustic phonon limited method and the following expression:69,162
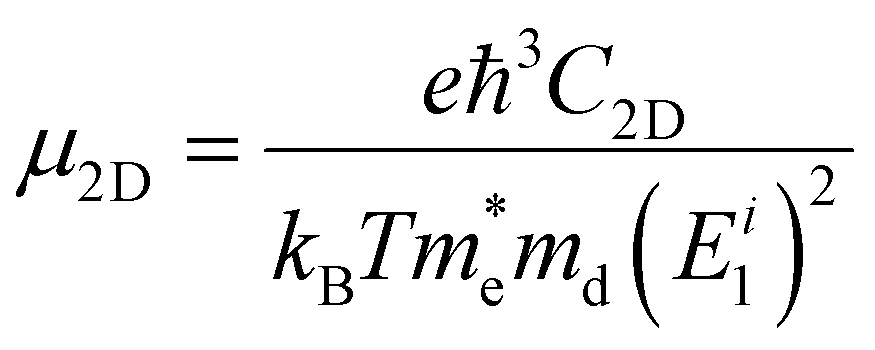 000 cm2 V−1 s−1. The unmanageable layer number and structural defects in preparation as well as the interface resistance in detection should be ascribed to the discrepancy.
000 cm2 V−1 s−1. The unmanageable layer number and structural defects in preparation as well as the interface resistance in detection should be ascribed to the discrepancy.
3.3 Phonon properties
Phonons are the quantized vibrational modes of the material's lattice structure and strongly influence its thermal and mechanical properties as well as electronic behavior.164–166 To obtain a comprehensive understanding of the performance of low-dimensional EPMs in various applications including nanoelectronics, optoelectronics, and spintronic devices, it is necessary to conduct systematic investigations of the phonon-related properties. Raman spectroscopy serves as an important characterization tool for acquiring information about the phonon band structure and properties of materials. To comprehend the Raman scattering and Raman selection rules in EPM crystals, it is essential to understand the crystal symmetry, lattice vibration, and phonon dispersion.167Generally, group theory can be employed to predict the normal modes in crystals and classify lattice vibrations based on the irreducible representations of the corresponding crystal symmetry groups.168 Recent theoretical and experimental investigations have primarily focused on characterizing the vibrational properties of three phosphorus allotropes: black-P, blue-P, and violet-P. Considering that there is an extensive literature discussing the vibrational modes of bulk black-P,169–171 here we primarily focus on the vibrational modes of black-P monolayers. Black-P monolayers belong to the D2h7 space group and the primitive cell consists of four atoms. This space group exhibits 12 phonon branches including 3 acoustic and 9 optical modes.172 The irreducible representations for the black-P monolayer are as follows: Γ = 2Γ1+ + Γ2+ + 2Γ3+ + Γ4+ + Γ1− + 2Γ2− + Γ3− + 2Γ4−, where the acoustic modes are Γ2−, Γ3−, and Γ4−. The remaining optical modes in the one-dimensional representations Γ1+, Γ2+, Γ3+, and Γ4+ are Raman active, while Γ2− and Γ4−, are infrared active. The associated non-normalized eigenvectors for black-P monolayer lattice vibration modes are shown in Fig. 10a. Different numbers of layers and different stacking arrangements can also give rise to variations in the symmetry and lattice vibration modes. When multiple layers of black-P are stacked in the AA manner as shown in Fig. 10b, the space group is always D2h7 (Pbmn, #53), irrespective of the odd or even number of stacked layers. The corresponding irreducible representations are as follows: Γ = 2N(Γ1+ + Γ3+ + Γ2− + Γ4−) + N(Γ2+ + Γ4+ + Γ1− + Γ3−), where Γ2− + Γ3− + Γ4− are acoustic modes, 2N(Γ1+ + Γ3+) + N(Γ2+ + Γ4+) are Raman active, (2N − 1)(Γ2− + Γ4−)(N − 1)Γ3− are infrared active, and NΓ1− are silent. In contrast, when multiple layers of black-P are stacked in the AB manner, odd and even layers exhibit distinct vibrational modes. For odd-numbered layers, the space group and irreducible representations remain consistent with those stacked in the AA manner. However, for even-numbered layers, the lattice vibration belongs to the D2h11 (Pbmn, #57) space group, and the irreducible representations are as follows: Γ = 2N(Γ1+ + Γ3+ + Γ2− + Γ4−) + N(Γ2+ + Γ4+ + Γ1− + Γ3−), where the acoustic and optical modes are the same as in the space group of D2h7 (Pbmn, #53).
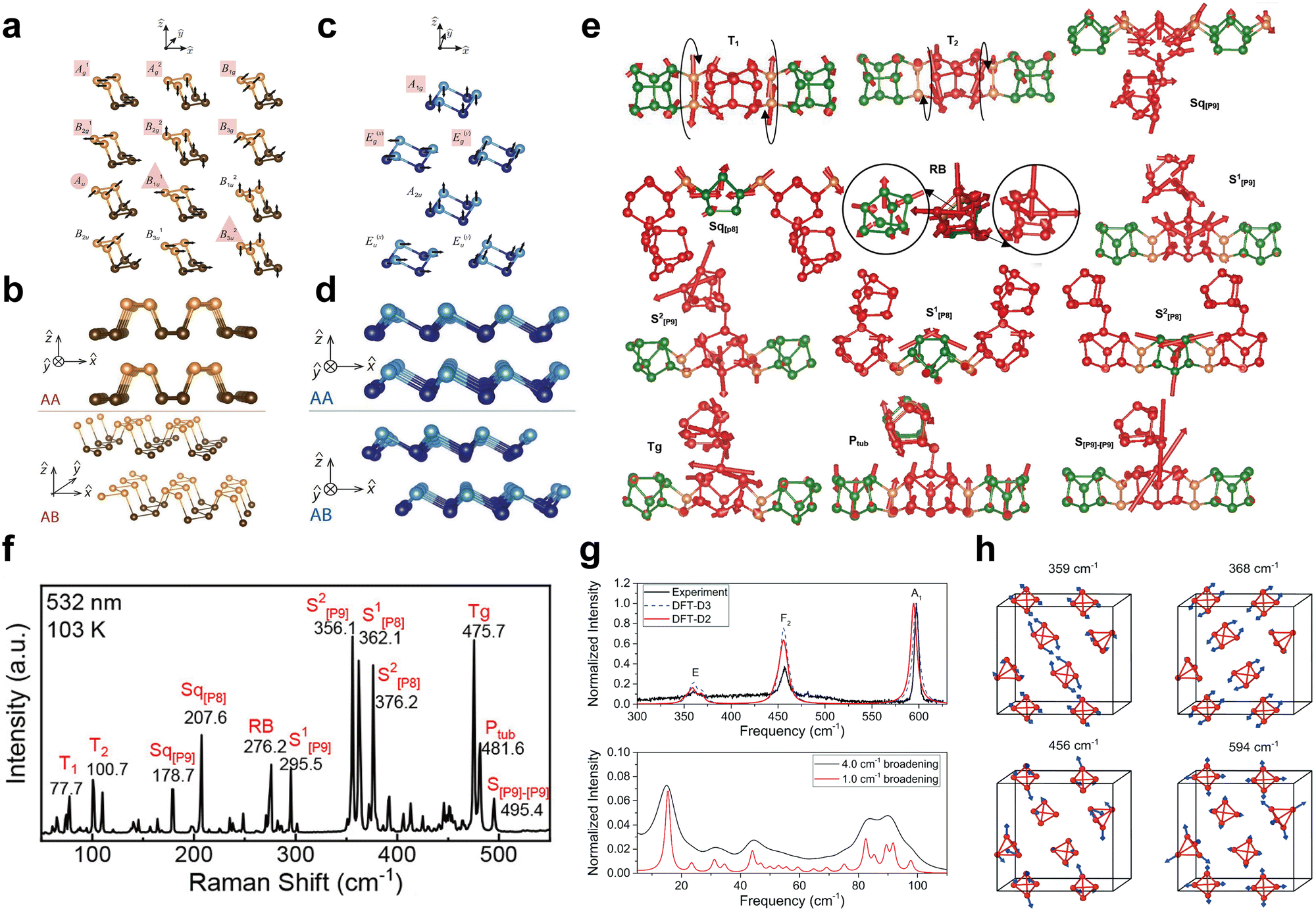 | ||
| Fig. 10 Phonon-related properties of EPMs. (a) Non-normalized eigenvector representations for Black-P monolayer vibrational modes. (b) Side view of the AA and AB stacking arrangements for the Black-P bilayer. (c) Non-normalized eigenvector representations for blue-P monolayer vibrational modes. (d) Side view of the AA and AB stacking arrangements for the blue-P bilayer. Reproduced with permission from ref. 172. Copyright 2015 American Physical Society. (e) Vibrations of the typical Raman modes of bulk violet-P. (f) Raman spectra of violet-P single crystals at 103 K. Reproduced with permission from ref. 173. Copyright 2021 American Chemical Society. (g) DFT calculated the Raman spectrum of bulk white-P together with the recorded experimental spectrum and (h) calculated eigenvectors of key Raman active modes. Reproduced with permission from ref. 174. Copyright 2021 Royal Society of Chemistry. | ||
Blue-P has a lattice structure similar to that of Black-P. Especially, the blue-P monolayer primitive unit cell comprises two atoms and belongs to the D3d3 subgroup, which has the triple rotational symmetry. The irreducible representations of the blue-P monolayer are as follows: Γ = Γ1+ + Γ3+ + Γ2− + Γ3−, where Γ2− + Γ3− are acoustic modes. The remaining optical modes in the one-dimensional representations Γ1+ and Γ3+ are Raman active with no infrared activity. The corresponding non-normalized eigenvectors for blue-P monolayer lattice vibration modes are shown in Fig. 10c. However, when dealing with multiple layers of blue-P, the lattice vibration modes differ. For layers stacked in the AA manner, as shown in Fig. 10d, the lattice vibration belongs to the D3d3 (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1, #164) group, and irreducible representations are as follows: Γ = N(Γ1+ + Γ3+ + Γ2− + Γ3−), where Γ2− + Γ3− are acoustic modes, N(Γ1+ + Γ3+) are Raman active, and (N − 1)(Γ2− + Γ3−) are infrared active. In contrast, when multiple layers of blue-P are stacked in the AB manner, odd and even layers exhibit distinct vibrational modes. For odd-numbered layers, the lattice vibration belongs to the C3v1 (P3m1, #156) group and the irreducible representations are as follows: Γ = 2N(Γ1 + Γ3), where Γ1 + Γ3 are acoustic modes and (2N − 1)(Γ1 + Γ3) are both Raman active and infrared active. For even-numbered layers, the space group and irreducible representations are consistent with the AA stacking pattern.172
m1, #164) group, and irreducible representations are as follows: Γ = N(Γ1+ + Γ3+ + Γ2− + Γ3−), where Γ2− + Γ3− are acoustic modes, N(Γ1+ + Γ3+) are Raman active, and (N − 1)(Γ2− + Γ3−) are infrared active. In contrast, when multiple layers of blue-P are stacked in the AB manner, odd and even layers exhibit distinct vibrational modes. For odd-numbered layers, the lattice vibration belongs to the C3v1 (P3m1, #156) group and the irreducible representations are as follows: Γ = 2N(Γ1 + Γ3), where Γ1 + Γ3 are acoustic modes and (2N − 1)(Γ1 + Γ3) are both Raman active and infrared active. For even-numbered layers, the space group and irreducible representations are consistent with the AA stacking pattern.172
Violet-P in its bulk form has the monoclinic lattice structure with a space group of P2/n (13, C2h(2/m)).173 The lattice vibrations of bulk violet-P at the center of the Brillouin zone involve 252 phonon modes, consisting of 3 acoustic modes and 249 optical modes. These modes can be represented as follows: Γ = (Au + 2Bu) + 63(Ag + Bg) + (62Au + 61Bu), where Au + 2Bu are the acoustic modes, 63(Ag + Bg) are both Raman and infrared active, and 62Au + 61Bu are inactive in terms of both Raman and infrared. Based on this information, 12 primary vibration modes can be identified (Fig. 10e) and the corresponding Raman features of violet-P will be discussed further below.
Raman scattering is a straightforward, nondestructive, and efficient technique to examine crystal lattice vibrations. During Raman scattering in a crystal, the incident photons can either absorb or emit crystal phonons and subsequently scatter in various directions at different angles.165 By detecting and analyzing the scattered light that has gained or lost phonon energy, one can determine the relevant phonon properties of the materials. Additionally, since the Raman intensity is proportional to the derivative of the dielectric tensor concerning vibrational coordinates, DFT calculations can be combined with Raman scattering experiments to conduct in-depth studies. Impellizzeri et al.174 applied the Placzek approximation to DFT calculations and combined it with experiments to simulate Raman spectra for various phosphorus allotropes including bulk and low-dimensional structures. The simulated Raman spectrum of white-P (Fig. 10g and h) reveals three different modes: a bending mode E at 371.5 cm−1, and symmetrical stretching modes F2 and A1 at 459.5 cm−1 and 602.1 cm−1, respectively. The simulated Raman spectra for bulk and monolayer black-P display three unique peaks at 357.3 cm−1, 425.2 cm−1, and 453.5 cm−1, corresponding to the Ag1, B2g, and Ag2 modes associated with out-of-plane, zigzag, and armchair vibrations, respectively. Violet-P with its more intricate crystal structure produces a more complex Raman spectrum than white-P and Black-P. The monolayer blue-P simulated Raman spectrum exhibits only two prominent Raman peaks, reflecting its higher symmetry relative to black-P. The Eg mode at 422 cm−1 corresponds to the longitudinal displacement in the plane direction, while the A1g mode at 534 cm−1 arises from out-of-plane vibrations. Although DFT calculations are effective in simulating Raman spectra to discern the vibrational modes of phosphorus allotropes, refinements are necessary to achieve a closer match with experimental results. Raman spectroscopic studies of bulk violet-P have been reported by Zhang's group.173 As shown in Fig. 10e and f, the 12 peaks correspond to the 12 main vibrational modes, ranging from strong to moderate intensity.
The thermal properties of phosphorus allotropes are heavily influenced by the crystal orientation due to structural anisotropy. For instance, the thermal conductivity of black-P demonstrates a significant dependence on the thickness and high degree of anisotropy.166 Theoretical calculations indicate that the thermal conductivity of monolayer phosphorene is lower than that of bulk black-P,175 showing that the thermal conductivity tends to increase as the black-P thickness increases. However, defects can result in thermal conductivity values lower than the intrinsic ones. Single-crystal black-P exhibits strong in-plane anisotropy in the thermal conductivity across the zigzag and armchair directions, with the armchair direction having lower thermal conductivity values—up to half—compared of those in the zigzag direction.176 This anisotropy primarily originates from the anisotropic dispersion of phonons and, to a lesser extent, the phonon–phonon scattering rate. Ge et al. have investigated the lattice vibrational properties and thermal expansion of blue phosphorene through DFT calculations.177 Slight buckling of the blue phosphorene atoms in the Z-direction disrupts the mirror symmetry, resulting in hybridization of ZA and ZO modes with other modes. Consequently, a phonon band gap develops and the phonon group velocity decreases, effectively reducing the thermal conductivity.
3.4 Materials stability
The stability of EPMs under ambient conditions is crucial to applications because the presence of small molecules like water and oxygen can be problematic.178 Prolonged exposure to the environment can cause significant changes in the physicochemical properties of phosphorene, including a rougher surface, higher ohmic resistance, and reduced charge mobility due to the formation of a surface oxide layer.179,180 The detrimental modifications negatively impact the efficiency of catalysts and electrodes. Therefore, comprehending the vulnerabilities of EPMs and potential factors that may interfere with their performance is important for designing more durable EPM-based materials that can be used under industrial conditions.The stability of materials is influenced by the intrinsic atomic and electronic structures. First, layered EPMs generally have an elevated surface area-to-volume ratio, which accelerates the rate of surface chemical reactions and makes them susceptible to deterioration. Second, the valence electron configuration of phosphorus atoms (3s23p3) results in the formation of covalent bonds with three neighboring atoms, leaving a lone electron pair that is highly reactive to ambient components such as oxygen and water.181 Recently, Lyu and colleagues have conducted a systematic evaluation of the relative stability of 10 layered EPMs with a focus on structure and bonding and found that the gauche effect is essential in determining the stability of these materials.182 Based on DFT calculations, violet-P has the lowest energy relative to other structures and is deemed the most stable (Fig. 11a). Analysis of the torsion-bonding strength relationship reveals that the majority of the P–P bonds in violet-P fall into the gauche region (Fig. 11b), indicating that these bonds predominantly exhibit the gauche conformation. The phosphorus atom in the layered structure is sp3 hybridized with a lone electron pair, which contributes to the stability of the gauche conformation. This is because the orbitals of two adjacent lone pairs are positioned near each other's nodal planes (Fig. 11c), thus avoiding strong interaction.183–185 The energetically favorable structure of EPMs has a greater proportion of gauche bonds, and an increase in the proportion of eclipse or trans conformations will result in a corresponding rise in the energy of the ground state.182 Other structural factors that can impact the stability include bond length, bond angle, and vdW interactions, and some studies have also evaluated the stability of EPMs from these perspectives.75,186,187
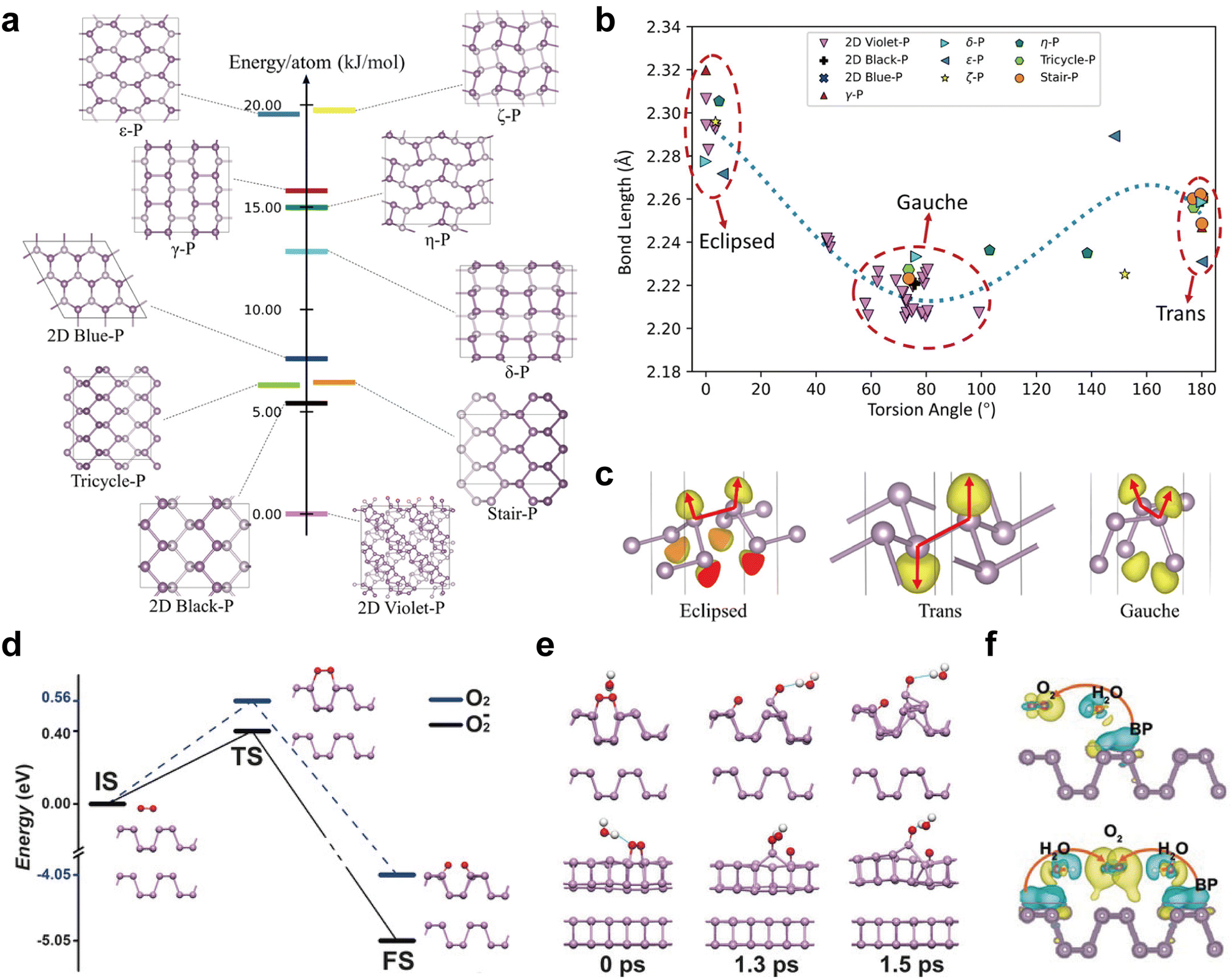 | ||
| Fig. 11 Theoretical calculations of the stability of EPMs. (a) The relative energy of 10 layered EPMs. (b) Torsion-bonding strength relationship of all P–P bonds in 10 layered EPMs. (c) Eclipsed, trans, and gauche conformations of P–P bond in layered EPMs. Reproduced with permission from ref. 182. Copyright 2022 American Chemical Society. (d and e) The oxidization and disintegration process of bilayer black-P. Reproduced with permission from ref. 192. Copyright 2016 John Wiley and Sons. (f) The formation of water–oxygen clusters on the black-P surface. Reproduced with permission from ref. 195. Copyright 2017 John Wiley and Sons. | ||
Under environmental conditions, the stability of EPMs is also influenced by the thickness and form of crystals. Single-layered phosphorene, composed entirely of surface phosphorus atoms with fully exposed lone-pair electrons, show more surface defects than thicker crystals.188 Thinner phosphorene tends to degrade from the edges inward due to edge effects, while bulk crystals primarily degrade on the surface, and surface oxidation can extend the overall stability of the materials.189–191 Additionally, as the thickness decreases, the phosphorene bandgap moves to higher energies due to quantum confinement, bringing the valence and conduction bands closer to O2 acceptor states.192 As a result, thinner phosphorene has lower stability and is more susceptible to oxidative degradation than thicker ones. EPMs with different crystal forms also show variations in their degradation under environmental conditions. Fali et al. have studied chemical degradation of violet-P flakes and compared surface oxidation with that of black-P under ambient conditions with time.193 Initially, both violet-P and black-P show similar degradation patterns with random nanoparticles appearing on the surface shortly after exfoliation. However, the sustained growth kinetics of the nanoparticles differs between the two allotropes. In black-P, the degradation area expands rapidly along an S-shaped curve due to merging of neighboring nanoparticles, but no noticeable merging is observed in violet-P, and the deteriorated surface elements have minimal impact on the non-degraded surface elements. As a result, the degradation rate of violet-P is nearly 9 times slower than that of black-P under the same ambient conditions. This work provides experimental proof of the superior stability of violet-P.
External factors that impact the stability of EPMs include light, oxygen, and water. However, the exact degradation mechanism remains a subject of debate. The degradation mechanism of black-P has been extensively investigated under various environmental conditions. Martel's group92 and Wang's group192 have put forth a light-induced oxidation mechanism based on in situ Raman spectroscopy and theoretical calculations, in which the superoxide anions on the surface, triggered by light exposure, are critical in the degradation of black-P. This light-induced degradation process can be divided into three stages: (1) ambient light promotes the generation of superoxide anions through a charge transfer reaction; (2) the superoxide anions dissociate on the surface and form two P–O bonds with black-P, resulting in the formation of a native surface oxide; and (3) water molecules interact with the bonded O and remove P atoms from the surface via hydrogen bonding, leading to the deterioration of the top layer and further oxidation of the inner layer. Throughout the process, superoxide anions stride over a small potential barrier through a triplet–singlet transition, generating dangling O atoms on the surface (Fig. 11d). As a result, the surface becomes hydrophilic, causing the O adsorbed phosphorene to dissolve in water (Fig. 11e).192 Huang et al. have performed isotopic labeling and wetting experiments to confirm the effects of O2 on the surface hydrophilicity.194 The pristine Black-P is hydrophobic, but with dissociative chemisorption of O2, the surface becomes progressively more hydrophilic. Interestingly, a distinctive degradation mechanism for the water-catalyzed oxidation of a few-layered black-P in the absence of light has been proposed.195 This theory suggests that water and oxygen molecules co-adsorb on the surface of black-P to generate water–oxygen clusters (Fig. 11f), and then electron transfer from black-P to oxygen initiates oxidation even in the absence of light. This can be attributed to the strong polarization effect of water molecules, which significantly enhances the electron affinity of oxygen to improve the capacity to accept electrons. Yu et al. recently found that persistent generation of reactive oxygen species on the surface of few-layered black-P also plays a significant role in the dark degradation.196 Zhao et al. have studied the degradation kinetics of fibrous-P nanoribbons under four different exposure conditions.197 They found that fibrous-P nanoribbons deteriorate rapidly in both oxygenated and deoxygenated water, while deterioration is slower in the absence of light, suggesting that the degradation behavior of fibrous-P differs from that of black-P. For black-P, the influence of light on the degradation kinetics is insignificant and oxygen plays a substantial role.198 It is also necessary to study the degradation kinetics of other EPMs as the degradation mechanisms may be different from those of black-P.
A variety of physical and chemical protection techniques such as capping layers,199 liquid protection,200 surface functionalization,201 and doping with other elements,202 have been developed to passivate EPMs. However, in practical applications, striking a balance between material stability and performance is crucial, as blindly pursuing stability at the expense of performance is not a viable option. Our group has proposed a metal-ion modification strategy that can improve both the stability and performance of black-P sheets.203 The strategy involves the cation–π interaction between metal ions (Ag+) and the conjugated π bond formed by the lone pair electrons of phosphorus atoms in each layer (Fig. 12a). The high-resolution Raman spectra confirm that the surface of black-P is modified with Ag+ and no noticeable PxOy characteristic peaks are observed (Fig. 12b–d). The intensity of the typical black-P peak at 360.9 cm−1 progressively increases until 60 minutes and then stabilizes as a result of saturated adsorption (Fig. 12e),203 indicating that modifying black-P with Ag+via cation–π interaction is a manageable and straightforward process. As shown in Fig. 12f–i, the modification process improves the stability of black-P sheets, allowing them to remain unchanged in the air with a relative humidity of up to 95% for several days and without any major alterations in the surface morphology or roughness, despite only a slight increase in thickness (∼9.5 nm) compared to the pristine black-P sheets (∼8.2 nm). Furthermore, the modified black-P FET exhibits improved hole mobility and ON/OFF ratios. The hole mobility reaches 1666 cm2 V−1 s−1, which is more than double the value for pristine black-P. The ON/OFF ratio improves to 2.6 × 106, which is 44 times greater than that of pristine black-P. This strategy has the added advantage of allowing control over the ion concentration and modification time, and it can be extended to other metal ions also. In the future, it is expected that more innovative methods will be developed, which will not only secure the stability of EPMs in practical applications but also enhance their performance.
 | ||
| Fig. 12 Metal-ion modified black-P with enhanced stability. (a) Schematic illustration of the interaction of Ag ions with black-P. (b) High-resolution XPS spectra of P 2p and (c) Ag 3d. (d) Raman spectra of a black-P sheet before and after Ag+ modification. (e) Series of Raman spectra of a black-P sheet after Ag+ modification for 0 to 120 min. (f) AFM images of pristine (top) and modified (bottom) black-P sheets exposed to air for several days. (g) Corresponding height profile of pristine and (h) modified black-P sheet. (i) Surface roughness of the modified and pristine black-P sheet with exposure time. Reproduced with permission from ref. 203. Copyright 2017 John Wiley and Sons. | ||
In summary, this section have offered an in-depth analysis of the fundamental physicochemical properties of EPMs, elucidating the differences and interrelationships among their atomic structures, electronic and phonon properties, and material stability. The section underscored the unique features of these materials and ongoing debates in the field. A specific point of focus, moving forward, should be the structural intricacies of EPMs, particularly surface and edge structures. Despite the advances made in the research of bulk crystals, our comprehension of surface or edge reconstruction and amorphization remains limited. The arrangement change of the surface or edge atoms would bring about significant modifications in physicochemical properties. Furthermore, there is a noticeable lack of emphasis on the structural phase transitions occurring at the sub-nanometer scale, and associated structure–property relationship studies are still insufficient. Further research should also focus on the exotic properties of phosphorene, potentially revealing novel phenomena and applications. In addition, several fundamental debates remain unresolved, including the precise atomic structure of amorphous red-P and type II and III crystalline red-P, the disparity between theoretical predictions and actual measurements of carrier mobility in diverse “violet-P”, and the different degradation behavior observed in various EPMs. Addressing these issues will provide a more solid foundation for the field and guide the design and application of these intriguing materials.
4. Synthetic strategies of elemental phosphorus materials
Phosphorus is a versatile element in the periodic table that exists in a variety of allotrope forms and sizes, ranging from bulk crystals to layered sheets, nanoribbons, and quantum dots. Synthetic EPMs provide a broad range of properties that can be tailored through the choice of constituent allotropes and reaction conditions. To fully realize the potential of EPMs and their “on-demand” properties, it is necessary to develop and optimize the synthetic methods. High-quality, uniform, and mass-producible synthesis of EPMs is crucial to applications. Currently, the synthesis of white-P still relies on pyrolysis of phosphate ores using the J. B. Readman electrothermal approach reported in 1888.104 However, new and improved synthesis methods have been developed for other EPMs such as black-P, red-P, fibrous-P, and violet-P. These methods have enabled the synthesis of high-quality 3D bulk crystals as well as low-dimensional (0D, 1D, and 2D) nanostructures. Bulk crystals of blue-P have not yet been synthesized, but 2D blue phosphorene has been prepared. Additionally, recent reports have described the synthesis of new nanostructured EPMs. The varying dimensional material structures that have been demonstrated for each of the phosphorus allotropes are summarized in Fig. 13. This section critically discusses the synthesis of EPMs and their nanostructures. The advantages and disadvantages of different synthetic routes are discussed and characterization of the corresponding products is described.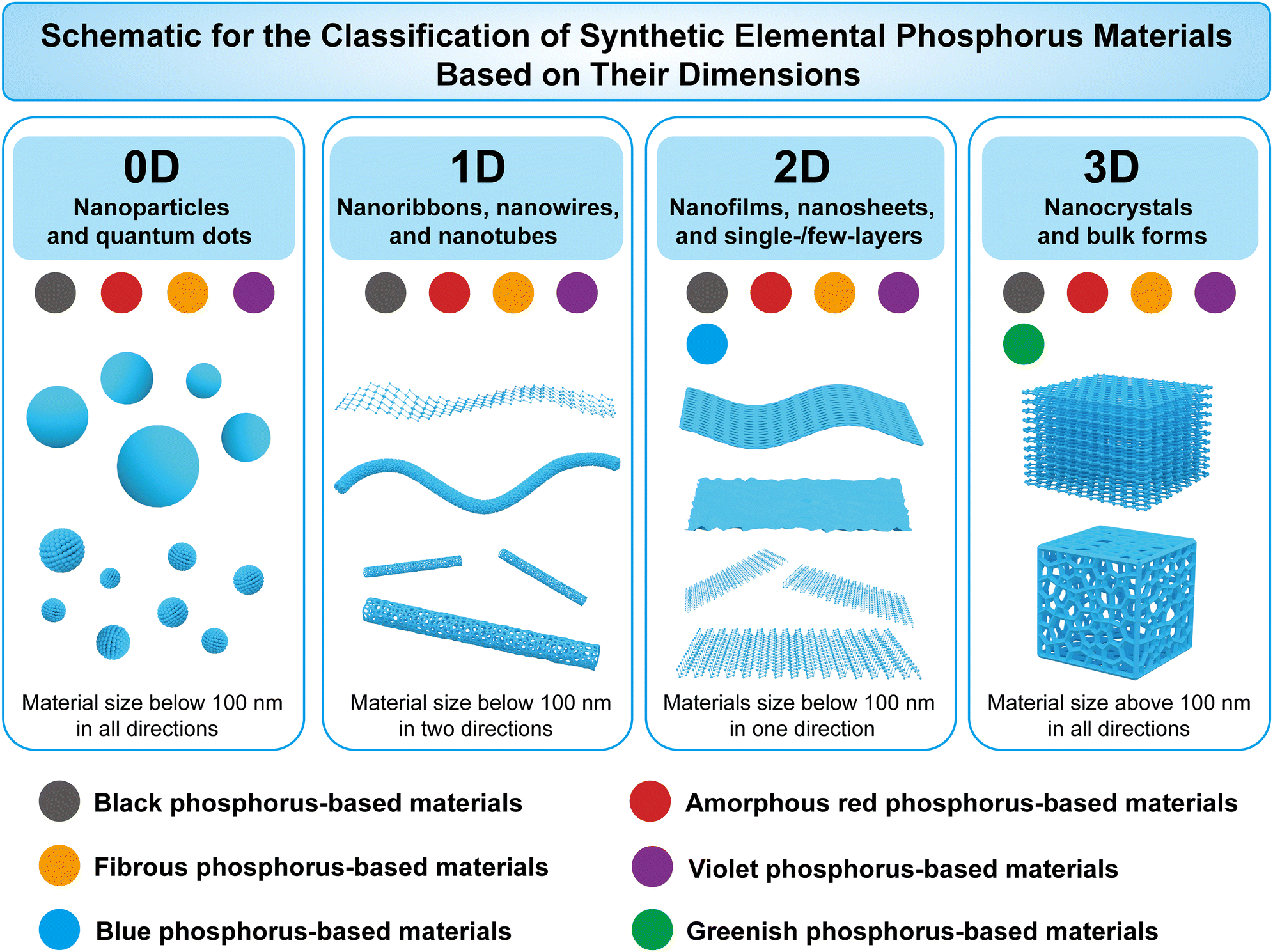 | ||
| Fig. 13 Schematic diagram showing classification of synthetic elemental phosphorus materials based on the dimensions and structural examples for each dimension. Elemental phosphorus materials (EPMs) are classified into three categories: zero-dimensional (0D) EPMs (nanoparticles and quantum dots), one-dimensional (1D) EPMs (nanoribbons, nanowires, and nanotubes), two-dimensional (2D) EPMs (nanofilms, nanosheets, and single-/few-layers), and three-dimensional (3D) EPMs (nanocrystals and bulk forms). Each coloured circle within the EPM structural box is indicative of different phosphorus allotropes reported with corresponding dimensional structures in the literature. It should be noted that the EPM nanotubes have not been experimentally synthesized so far, but the synthesis of black or blue phosphorus nanotubes is theoretically feasible. | ||
4.1 Synthesis of black phosphorus
To address the requirement for high-pressure conditions in black-P crystal production, mercury flux or bismuth flux was introduced as supplementary catalysts.205–207 In 1955, Krebs and his colleagues reported that under high-temperature conditions (370–410 °C), addition of 30 to 40 atomic percent of mercury would initiate the transition of white-P into black-P.205 The reaction was found to take a few days to complete, yielding Black-P crystals as the final product. However, the use of mercury or bismuth as catalysts is associated with several drawbacks, including mercury contamination, bismuth toxicity, prolonged reaction times, and limited yield, making the mercury or bismuth flux method far from ideal. Despite the limitations, this method of introducing additional catalysts seems to provide insights into the later-developed chemical vapor transport (CVT) method, which efficiently produces black-P crystals at moderate temperature and pressure through the addition of transfer agents. CVT refers to a chemical reaction that transforms a condensed phase into a gaseous phase in the presence of a transfer agent, followed by deposition elsewhere and eventual formation of a crystalline structure.208 In 2007, Nilges and colleagues introduced a pioneering CVT-based approach for the preparation of black-P crystals at low pressure in evacuated silica ampules.42 By heating red-P with a mixture of Au, Sn, and SnI4 as transport agents, they were able to produce black-P crystals, where a sample batch was grown on the surface of the Au3SnP7/AuSn materials (Fig. 14a). One year later, this research group improved the previous reaction system by using the AuSn alloy instead of Au and Sn metals.43 This change, along with the optimal temperature program, reduced the crystal growth time to 32.5 hours and increased the crystal size to 1.5 cm in diameter. However, the cost of synthesis remained high due to the use of noble metals. In 2014, they further refined the synthetic procedure by eliminating expensive Au and substituting it with Sn/SnI4 as transfer agents, which significantly reduced costs and the number of impurities.44 Building on these advances, Nilges et al. recently utilized DFT calculations to probe the formation mechanism of black phosphorus and phosphorene.209 They discovered that the gaseous species P4 and SnI2 play a crucial role in the gas-phase growth of black phosphorus and phosphorene, with SnI2 directly inserting into the P–P bonds of P4. This work not only contributes to the optimization of the gas-phase growth system for black-P crystals but also aids in understanding the principles of epitaxial growth of phosphorene on substrates, which will be discussed further below.
 | ||
| Fig. 14 CVT-based methods for black-P bulk crystal preparation. (a) Photographs of the obtained black-P crystals. Reproduced with permission from ref. 42. Copyright 2007 American Chemical Society. (b) A comparison of the yield and purity of black-P synthesized by different methods, including high-pressure high temperature (HPHT) methods, sonochemistry, flux methods, and CVT methods. (c) Schemes of the LDT method and (d) SDT method to grow black-P crystals and their corresponding photographs of the resulting products. Reproduced with permission from ref. 210. Copyright 2020 Elsevier. | ||
In contrast to Nilges' CVT growth approach,42,43 which requires a temperature gradient for long-distance transport (LDT), Liu's group have devised an efficient short-distance transport (SDT) growth method that utilizes a uniform temperature.210 As shown in Fig. 14c and d, a substantial number of black-P crystals formed during the SDT process, with 98% of red-P being converted to black-P, while only a limited number of black-P crystals formed during the LDT process. The comparison of the yield and purity of black-P synthesized by different methods including high-pressure high temperature (HPHT) methods, sonochemistry, flux methods, and CVT methods is shown in Fig. 14b, and the results indicate that this work has achieved the highest yield and purity of black-P crystals among the compared methods. Two key factors for obtaining high black-P production yields are the short-distance transport and uniform temperature.210 As the temperature in the quartz ampoule rises, the reactants (red-P, Sn, and SnI4) start vaporizing and fill the ampoule. This results in a reaction between gaseous phosphorus and Sn and SnI4 gases, forming P–Sn–I complexes that accumulate at the bottom of the ampoule. These complexes act as nucleation sites for further crystal growth. The transport agent aids in the crystallization and growth of black-P during the gradual cooling process until all the gaseous phosphorus is transformed into black-P crystals. However, the Sn/SnI4/red-P reaction system still faces several challenges, such as the time-consuming synthesis of SnI4 and the requirement for precise temperature control. The amount of red-P loading, ampoule capacity, and Sn/SnI4 proportion all play crucial roles in the synthesis of black-P crystals. Scaling up the production of black-P necessitates appropriate modification of the experimental conditions, as excessive red-P loading can cause ampoule breakage, while inadequate loading results in the production of only white-P or violet-P.93 In addition, this method can introduce iodine- or tin-doped impurities during the crystallization process of black-P. However, the ability to dope black-P allows for the alteration of properties, thereby offering potential to broaden the range of applications.
Top–down methods. Inspired by the successful mechanical exfoliation of graphene, few- and single-layered black phosphorene was first prepared by a micro-mechanical cleavage method in 2014.45,46 When the Scotch tape is repeatedly detached from bulk crystals, single-layered phosphorene with a thickness of ∼0.85 nm can be obtained and transferred onto a Si/SiO2 substrate.46 Although this method can produce controllable layers of black phosphorene by recycling the detaching process at different times, adhesive contamination and low yields must to be addressed. A modified mechanical exfoliation method has been developed to improve the yield of atomically thin black-P flakes.211 In this method, the commercially available black-P bulk crystals are repeatedly exfoliated using blue Nitto tape and the thinnest region of the flake has a thickness of 1.6 nm equivalent to three layers of black phosphorene. The use of sticky tape coupled with a viscoelastic polydimethylsiloxane substrate increases the yield of the products and reduces contamination.
Dry and wet ball milling methods are commonly used mechanical techniques to grind bulk black-P powders into few-layered phosphorene.212 Zhu et al. have developed a solid-state ball milling method to prepare ultrathin black-P nanosheets (Fig. 15a).213 They mixed 300 mg of black-P powder with 700 mg of anhydrous LiOH powder in a vessel brimming with an Ar atmosphere and ball milled for 24 hours,213 resulting in the formation of black-P nanosheets with lateral sizes ranging from 30–60 nm and a thickness of ∼0.7–6 nm. Addition of LiOH plays a crucial role in the ball milling process by functionalizing the nanosheet edges with hydroxyl groups, which improves the stability of nanosheets. Without LiOH, the exfoliated black-P nanosheets exhibit instability, and some of them are converted into red-P. Liu et al. have used wet ball milling to prepare a few-layered black-P and evaluated the impact of different organic solvents.214 Dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), and dimethyl sulfoxide (DMSO) are more effective solvents than absolute ethanol (AE) and ethylene glycol (EG), with DMSO being the optimal solvent for 3–5 layers of black-P. However, ball milling requires extended processing to achieve high yield, and the irregular movement of hard balls induces high stress that leads to the fragmentation of bulk crystals into nanosheets with reduced lateral dimensions. To tackle these challenges, Liu's group introduced glue-assisted grinding for exfoliating large-size 2D materials and obtained hexagonal boron nitride nanosheets with an average lateral size of 2.18 μm.215 Similar methods are expected to be developed for the production of large-size black phosphorene.
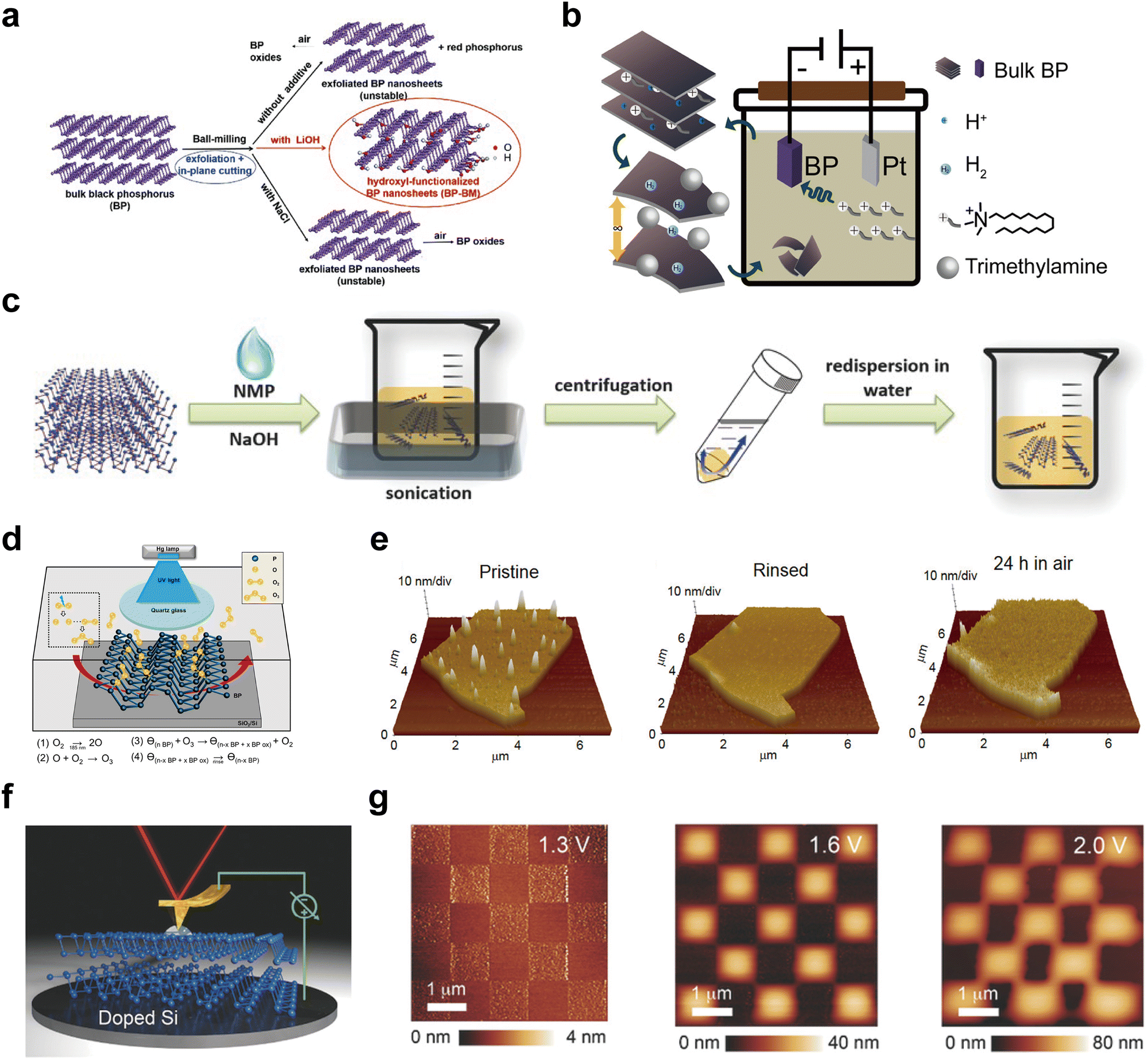 | ||
| Fig. 15 Typical top–down methods for the black phosphorene preparation. (a) Scheme of solid-state ball milling process of black-P with or without additives. Reproduced with permission from ref. 213. Copyright 2017 John Wiley and Sons. (b) Scheme of cathode exfoliation of few-layered black-P. Reproduced with permission from ref. 234. Copyright 2019 American Chemical Society. (c) Scheme of basic NMP exfoliation of black phosphorene. Reproduced with permission from ref. 218. Copyright 2015 John Wiley and Sons. (d) Scheme of the ozone etching method for black phosphorene preparation. (e) 3D AFM height images of black-P flakes before and after rinsing with water. Reproduced with permission from ref. 240. Copyright 2016 American Chemical Society. (f) Scheme of CAFM patterning setup for black-P. (g) Tapping-mode AFM images of checkerboard patterns created under various sample biases. Reproduced with permission from ref. 241. Copyright 2016 John Wiley and Sons. | ||
Liquid-phase exfoliation is another widely used method to produce black phosphorene, with NMP being the first solvent used for this purpose.200 The process involves immersing bulk black-P into NMP (5 mg mL−1) and exfoliating it in an ultrasonic bath (820 W, 37 kHz) for 24 hours. The resulting phosphorene nanosheets had dimensions of ca. 200 × 200 nm and a thickness of 3.5–5 nm. To date, NMP has been the most widely utilized solvent for black-P exfoliation, with the size of the resulting black phosphorene nanosheets ranging from 0.6 to 60 nm depending on the time (from 1.5 to 24 hours) and power (from 200 to 820 W) of ultrasonication.216 In NMP exfoliation, the functional groups work as “scissors”, cleaving bulk black-P into phosphorene through sharing of electron pairs from the exposed phosphorus atoms and oxygen from NMP.217 The surface tension of NMP is comparable to that of black phosphorene, which effectively disperses the exfoliated nanosheets and prevents restacking and aggregation. However, pure NMP exfoliation leads to surface contamination with NMP and instability of phosphorene in water. To address this, our group has developed a basic-NMP solvent process, in which bulk black-P is added to a NaOH-saturated NMP solution to perform liquid exfoliation (Fig. 15c). The resulting phosphorene exhibits improved water stability and controllable size and number of layers.218 Addition of OH− to the solution adsorbed on the surface of phosphorene makes it negatively charged (−30.9 mV) and further increases the stability in water. Other polarized solvents have been also used to synthesize black phosphorene, including DMF, DMSO, isopropyl alcohol (IPA), N-cyclohexyl-2-pyrrolidone (CHP), bis(2-methoxyethyl) ether (DIGLYM), acetonitrile (AN), distilled water, tetrahydrofuran, ethanol, benzonitrile, acetone, chloroform, and hexane.219–230 Liquid-phase exfoliation is more scalable than mechanical exfoliation for the preparation of black phosphorene. However, Young's modulus of phosphorene is smaller compared to other 2D materials, and strong ultrasonic treatment results in nanosheets that are smaller than a few hundred nanometers.231 Long-term sonication can also lead to fragile crystallinity and irreversible surface damage. Therefore, it is important to develop a large-scale method for the liquid synthesis of high-quality black phosphorene.
Electrochemical exfoliation is considered to a more efficient and controllable method that allows for the production of black phosphorene with precise control over the size and thickness by applying different voltages for different time. This method can be divided into anode and cathode exfoliation, depending on the location of the exfoliating materials. In 2015, Erande and colleagues were the first to use anode exfoliation to produce black phosphorene.232 After applying a positive bias voltage of +7 V and a current of ∼0.2 A to the working electrode for 50 minutes,232 black phosphorene displayed a nanosheet-like characteristic with lateral dimensions ranging from 5 to 10 μm and a thickness of around 1.4 nm. Different from anode exfoliation, cathode exfoliation does not involve the generation of oxygen-containing free radicals, leading to the formation of phosphorene products with no visible surface defects or oxidative damage.233 Cationic surfactant hexadecyltrimethylammonium chloride (CTAC) can be used as an intercalating agent to exfoliate bulk black-P in water at a cathodic potential (Fig. 15b), resulting in the formation of few-layered black-P flakes within 20 seconds.234 Delamination of bulk black-P is primarily driven by intercalation of large-sized hexadecyltrimethylammonium cations (CTA+) into the interlayer of black-P, which then decompose into gaseous bubbles at a negative potential, further improving the cathodic exfoliation of black-P.235,236 The interface-assisted strategy is useful for the synthesis of 2D materials.237 Our group has developed a cathodic electrochemical exfoliation method which uses the plasma–liquid interface to facilitate the transfer of active species and initiate chemical reactions in solution.238 This method allows for mass production of few-layered black phosphorene in pure N,N-dimethylformamide (DMF) without any intercalating agents. The entire exfoliation process can be completed in merely five minutes with a yield of ∼63%. During exfoliation, the plasma and DMF molecules interact strongly to form a series of cationic species. These species are drawn towards the cathode black-P crystals under the action of electric field and intercalated into the interlayer. Afterwards, these cationic species decompose into gaseous bubbles, causing the black-P crystals to expand and the phosphorene nanosheets to separate. The plasma–liquid exfoliation system has the advantage of being easily scalable and cost-effective, making it a viable option for commercial production of black phosphorene.
Plasma etching is an alternative strategy for preparing large-size black phosphorene. Lu et al. have reported a combination approach of mechanical cleavage and Ar+ plasma treatment to produce stable monolayer phosphorene.239 Subsequently, ozone etching has been used to prepare black phosphorene for efficient and precise control over the thickness of Black-P flakes (Fig. 15d).240 By adjusting the flow velocity of O2 in the reactor vessel, an average etching rate of 12 ± 2 nm h−1 or 0.40 ± 0.07 layer min−1 can be achieved, allowing for near-monolayer control.240 However, ozone molecules generated by O2 photolysis partially oxidize black-P, necessitating an additional step of oxide removal to obtain pristine black phosphorene. This can be accomplished by rinsing with deionized water and then blow-drying with N2. This clean-up procedure also results in phosphorene with good resistance to ambient oxidation (Fig. 15e). However, plasma etching operates on a large scale, and laser oxidation is spatially constrained by optical diffraction, resulting in either the unavailability or significant difficulty in achieving nanoscale thinning and patterning.241 Scanning probe nanolithography is well known for its high spatial resolution, which makes it a potential solution to overcome the limitations of plasma etching and laser oxidation.242 Liu et al. have used conductive force microscopy (CAFM) to accomplish lateral nanopatterning and layer-by-layer thinning of black-P (Fig. 15f), and the checkerboard pattern is produced on a newly exfoliated black-P crystal.241 The height of the pattern protrusions increases in proportion to the applied bias voltage during contact-mode CAFM scanning (Fig. 15g). However, inevitable oxidation of black-P during the process as well as the high cost of CAFM probe consumption limit the widespread use of this technology.
Bottom–up methods. Chemical vapor deposition (CVD) is a commonly used bottom–up method to synthesize high-quality thin films of 2D materials. This requires volatile precursors in controlled reactions or decomposition onto the substrate surface under specific atmospheres, temperatures, and pressures.243 Smith et al. have demonstrated an in situ CVD method for growing black-P films on a silicon substrate.244 Initially, an amorphous red-P film is vapor-deposited onto the substrate, which is then heated in the presence of an Sn/SnI4 (10 mg) mineralizer under an Ar atmosphere and high pressure (27.2 atm).244 This process results in the direct growth of black-P films with varying thicknesses on the silicon substrate. Nevertheless, this method may encounter uncontrolled nucleation and incomplete conversion issues throughout the growth process. To achieve effective nucleation and spatial control, Zhang's group have utilized polyphosphide Au3SnP7 as nucleation seeds to grow large-area black-P thin films on silicon substrates (Fig. 16a).245 Au3SnP7 is a suitable nucleation seed for black-P epitaxial growth because its lattice plane (010) matches the (100) plane of black-P. The process involves three stages: precursor evaporation, Au3SnP7 formation, and black-P nucleation and growth. The process begins by depositing a gold film on the silicon substrate and heating it with precursors (Red-P, Sn, and SnI4) to 750 °C for 1 hour in an evacuated quartz tube, where red-P evaporates and transforms into P4 molecules, and SnI4 decomposes into Sn and I2. The temperature is then lowered to 500 °C, leading to the formation of dispersed Au3SnP7 (Fig. 16b) on the silicon substrate through a series of chemical reactions between excessive red-P, Sn, and gold films. Due to lattice plane matching, vaporous P4 molecules are continuously deposited on the surface of Au3SnP7, completing the nucleation of black-P (Fig. 16c). As the temperature decreases, P4 molecules fully transform into black-P, and small black-P nanosheets spatially grow over the nucleation center (Fig. 16d), expanding and fusing to ultimately form a large-area black-P thin film with a lateral size of over 200 μm and a thickness of ∼150 nm (Fig. 16e). The resulting black-P films have a flawless orthogonally symmetrical structure with high crystallinity and purity, as well as an exceptional field effect and Hall mobility of over 1200 cm2 V−1 s−1 and 1400 cm2 V−1 s−1 at room temperature, respectively.245 Researchers have addressed the nucleation challenge of black-P films on substrates. However, controlling the lateral growth of these films is difficult due to the complexities in managing the vapor pressure of P4 molecules, leading to limited domain sizes of only a few hundred nanometers. To resolve this issue, the group has recently implemented a sustained feedstock release strategy for synthesizing single-crystal black-P thin films, focusing on promoting the lateral growth mode at low nucleation rates.246
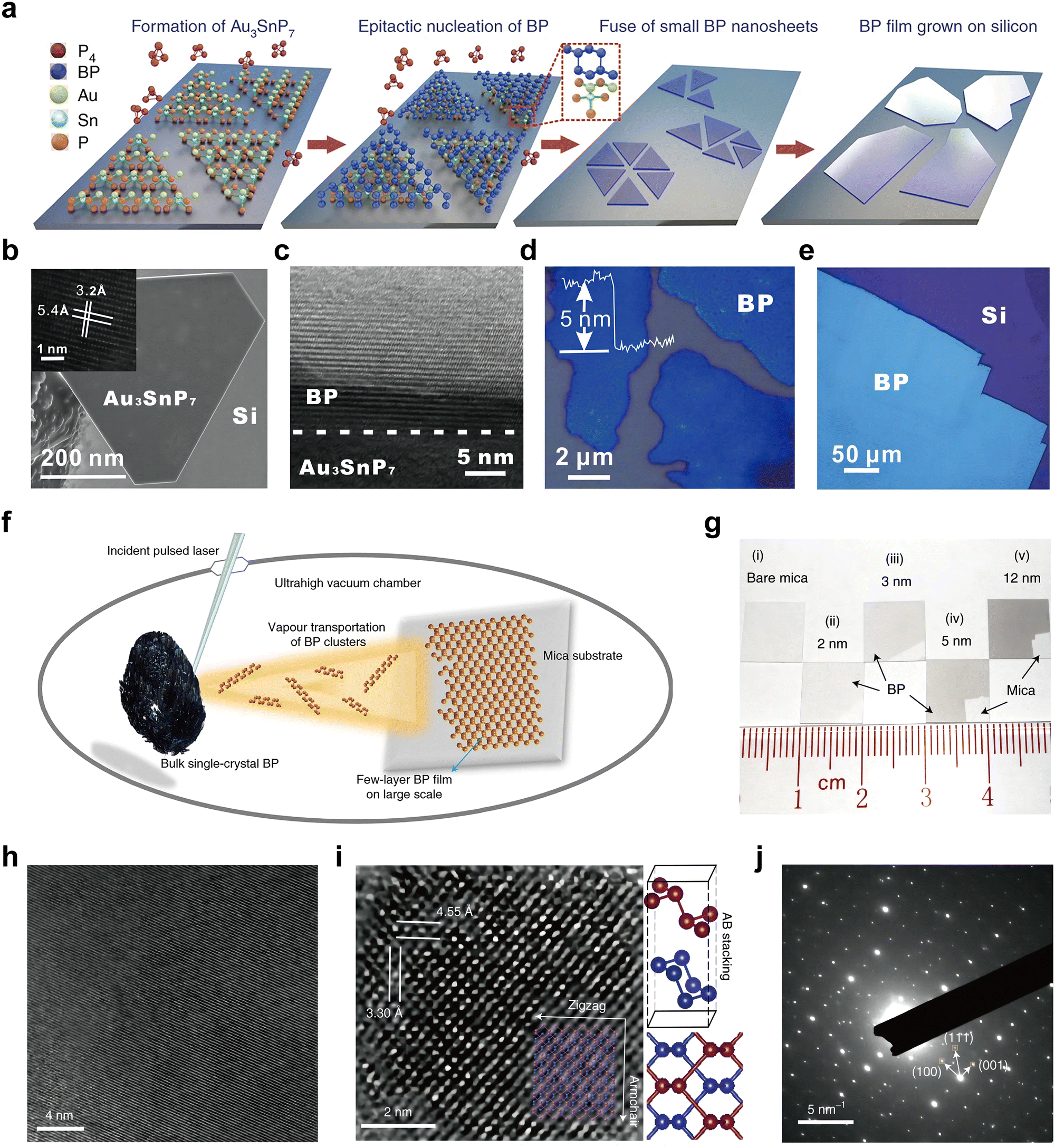 | ||
| Fig. 16 Typical bottom–up methods for black-P film preparation. (a) Scheme of epitaxial nucleation and lateral growth process of black-P films. (b) SEM image of Au3SnP7 grown on a silicon substrate. The inset shows the lattice structure of Au3SnP7. (c) Cross-sectional HRTEM image of a layered black-P nanosheet grown on Au3SnP7. (d) Optical image of the fusion of small black-P nanosheets to form a large-area film. (e) Optical image of a black-P film grown on a silicon substrate with a lateral size over 200 μm. Reproduced with permission from ref. 245. Copyright 2020 Nature Publishing Group. (f) Scheme of a controlled PLD method for few-layered black-P film growth. (g) Photographs of bare mica and as-synthesized centimetre-scale black-P films of different thicknesses. (h) Plan-view HRTEM image of a few-layered black-P film. (i) Detailed HRTEM image of the lattice structure of black-P films. (j) The corresponding SAED pattern of black-P films. Reproduced with permission from ref. 249. Copyright 2021 Nature Publishing Group. | ||
Under quasi-static equilibrium conditions and a continuous, stable P4 supply, individual domains progressively expand into millimeter-sized single-crystal black-P films. Subsequently, these films merge to form centimeter-sized, continuous black-P films, covering nearly the entire Si/SiO2 substrate. The as-grown black-P single-crystal film exhibits outstanding single crystallinity, as evidenced by the full-width at half-maximum of the (040) reflection rocking curve, measured at ∼0.08° using high-resolution XRD.246 Furthermore, the film has a remarkable carrier mobility of around 6500 cm2 V−1 s−1 at low temperature. This study marks the first observation of Shubnikov-de Haas oscillations in directly grown black-P films.
Pulsed laser deposition (PLD) is an approach that involves a laser to irradiate the target to deposit materials onto a substrate, resulting in the formation of a thin film or multilayer structure.247 By employing a pulsed laser, large black-P clusters can be formed within the transported physical vapor, which reduces the formation energy and allows for the growth of centimeter-scale black-P thin films.248,249 Yang et al. have used the PLD approach to fabricate wafer-scale black-P films with controllable thicknesses ranging from 2 to 10 nm.250 During the synthesis process, the substrate temperature is held at 150 °C. A KrF pulsed laser operating at a repetition rate of 5 Hz is used to irradiate the black-P bulk crystals. To achieve uniform film growth, both the black-P crystals and substrate are continuously rotated. However, structural characterization reveals that the as-synthesized black-P films are highly disordered and have an amorphous nature. By utilizing a black-P single crystal as the target source, Wu et al. have developed a controlled PLD method to grow centimeter-scale black-P ultrathin films on mica substrates, as demonstrated in Fig. 16f.249 The resulting black-P films are firmly deposited onto the 1 cm2 mica surfaces and the thickness can be accurately modulated by varying the number of laser pulses (Fig. 16g). Throughout the growth process, the vaporous black-P clusters generated by laser irradiation are uniformly dispersed over the homogenous mica surface, facilitating the thermodynamically-driven unidirectional growth and intermingling of small black-P nanosheets to form a large-area film. As observed by high-resolution TEM, the as-grown black-P film exhibits high ordering of phosphorus atoms with no apparent crystal defects (Fig. 16h). The lattice constants in the zigzag and armchair directions were measured to be 3.30 and 4.55 Å, respectively, indicating an orthorhombic phase for the black-P film (Fig. 16i). The selected-area electron diffraction (SAED) pattern confirms the presence of a typical orthorhombic lattice of black-P with the homologous fourfold symmetry (Fig. 16j). This work addresses three critical challenges that have impeded the production of high-quality black-P thin films by deposition-based methods. First, traditional deposition methods often yield black-P films that are relatively thick (>10 nm), suggesting that the resulting black-P has bulk crystal properties rather than the desired two-dimensional features. This work, however, successfully produces few-layered black-P films with a thickness of ∼1.1 nm for the bilayer. Second, earlier attempts in growing black-P films resulted in films with limited crystallinity, whereas the lattice structure of the black-P films grown in this work is highly ordered and devoid of significant crystal defects. Third, this work extends the lateral area of high-quality black-P films to the centimeter scale, facilitating the manufacture of wafer-scale optoelectronic and electrical devices based on black-P.
Wet chemical synthesis is an effective bottom–up approach known for its ease of use, versatility, high yield, and remarkable reproducibility.251 Some studies have claimed successful synthesis of black-P nanosheets by this method.252–254 However, the wet chemical method for synthesizing black-P nanosheets often encounters challenges of partial oxidation and poly-phosphorous phase complexes. XRD analysis of the resulting structures commonly reveals broad peaks with amorphous features, indicating the presence of impurities. Consequently, the synthesized nanosheets cannot be considered strictly crystalline black-P. To the best of our knowledge, currently there is a lack of definitive XRD and TEM characterization results demonstrating the efficacy of wet chemical synthesis in producing crystalline black-P nanosheets.
Nano-etching of black-P nanoribbons is achieved by electron beam lithography, reactive ion etching, and scanning TEM nano-sculpting techniques. In 2015, Lee et al. reported the successful fabrication of black-P nanoribbons with desired width and length through the use of electron beam lithography and reactive ion etching.176 The process involved placing an exfoliated black-P flake on a SiO2 substrate, followed by spin-coating PMMA on the flake for electron beam lithography in either the armchair or zigzag direction. Reactive ion etching (90% SF6 and 10% O2) was then used to eliminate the exposed black-P stripes, and PMMA protective stripes were removed with acetone to obtain black-P nanoribbons (Fig. 17a). However, the produced nanoribbons had a thickness of ∼50 nm and width within the range of 500 nm, resulting in their electronic properties being similar to those of black-P in its bulk form. To produce atomic-thick black-P nanoribbons, a scanning TEM nano-sculpting technique has been reported to synthesize sub-10 nm wide black-P nanoribbons composed of a few layers.256 However, this method has low throughput, making it challenging to produce black-P nanoribbons in large quantities. Feng et al. have utilized reactive ion etching to prepare anisotropic black-P nanoribbon field-effect transistors (Fig. 17b).257 This process allows for precise positioning of the nanoribbon and control of its width (∼60 nm) and height (∼3 nm) (Fig. 17c). However, the reliance on exfoliated black-P flakes and need for crystallographic alignment still limit the potential of this etching technique in practice.
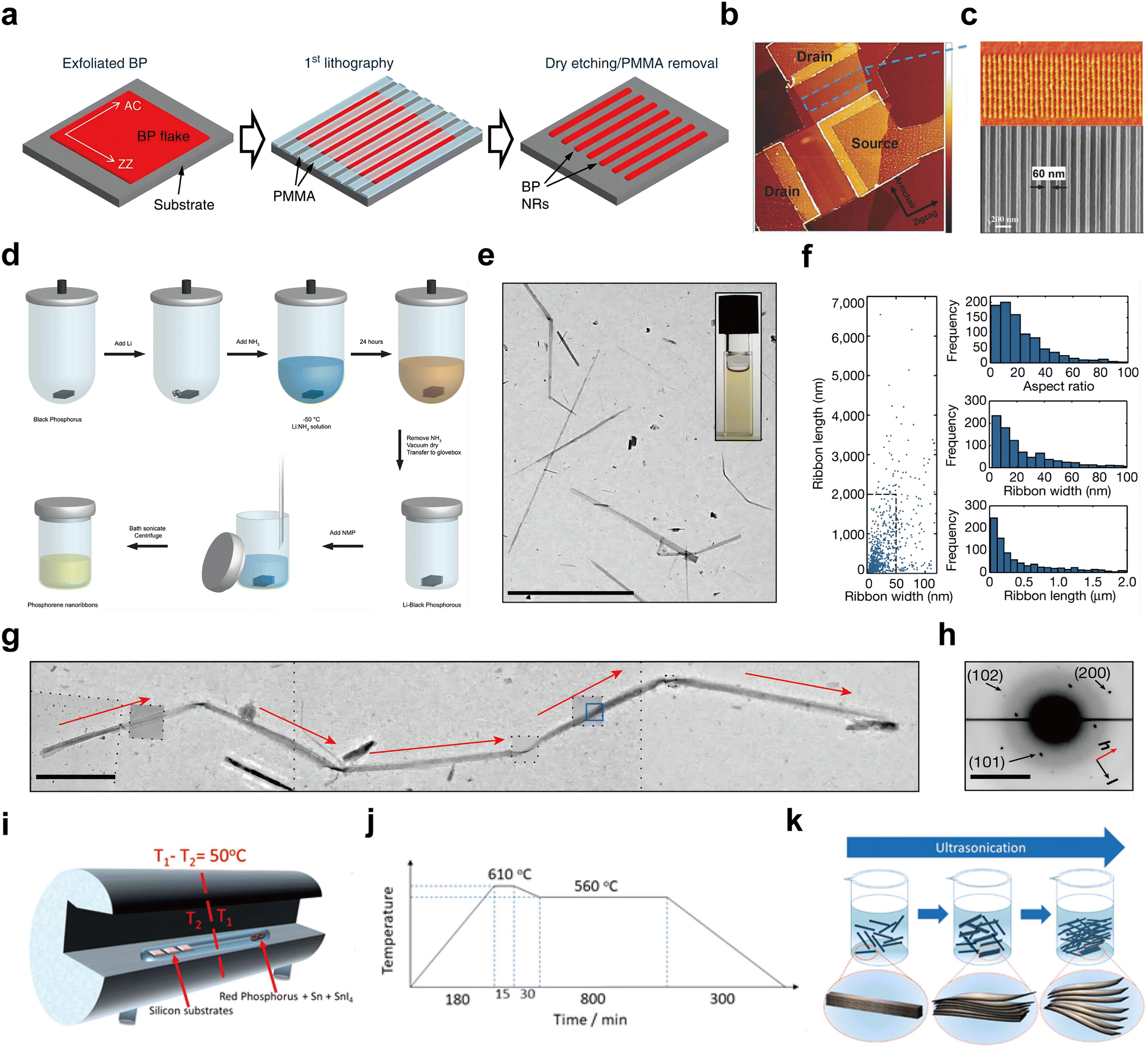 | ||
| Fig. 17 Synthesis and characterization of black-P nanoribbons. (a) Scheme of electron beam lithography and reactive ion etching process of black-P nanoribbons. Reproduced with permission from ref. 176. Copyright 2015 Nature Publishing Group. (b) AFM image of the black-P nanoribbon field-effect transistor along the armchair and zigzag directions. (c) An enlarged AFM image of the nanoribbons (top) and SEM image showing the nanoribbon width of 60 nm (bottom). Reproduced with permission from ref. 257. Copyright 2018 John Wiley and Sons. (d) Schematic diagram of phosphorene nanoribbon production by lithium intercalation and liquid exfoliation. (e) TEM image of phosphorene nanoribbons drop-cast from the liquid dispersion shown in the inset. Scale bar is 10 μm. (f) Scatter diagram depicting the relationship between the widths and lengths of 940 phosphorene nanoribbons (left); aspect ratios (top right), widths (middle right), and lengths (bottom right) of only those nanoribbons falling within a dashed rectangle located in the scatter diagram. (g) A combined TEM image of a phosphorene nanoribbon with an approximate length of 11 μm. The crystallographic direction in the zigzag direction, identified by SAED, is indicated by the use of red arrows. The scale bar is 1 μm. (h) SAED pattern corresponding to the blue box shown in (g). The scale bar is 5 nm−1. Reproduced with permission from ref. 258. Copyright 2019 Nature Publishing Group. (i) Scheme of CVT-growth of black-P columns. (j) Temperature profile of the CVT heating process. (k) Scheme of nanoribbon formation through ultrasonic treatment. Reproduced with permission from ref. 265. Copyright 2021 American Chemical Society. | ||
In 2019, Watts and colleagues introduced a novel approach for synthesizing high-quality black-P nanoribbons on a large scale.258 Their method relied on the combination of lithium intercalation and liquid exfoliation and involved two steps.
Black-P bulk crystals are first immersed in an aprotic lithium-ion solution at −50 °C for 24 hours, resulting in the intercalation of lithium into black-P. After ammonia is removed, the intercalated black-P is transferred to a glovebox and added to a vial containing NMP. The resulting mixture is sonicated for 1 hour and then centrifuged at a low speed to obtain a black-P nanoribbon solution (Fig. 17d). These nanoribbons exhibit a predominantly monolayer thickness, widths ranging from 4–50 nm, lengths up to 75 μm, and aspect ratios of up to 1000 (Fig. 17e and f). The nanoribbons are highly ordered and align well along the zigzag crystallographic orientation (Fig. 17g and h). The responsible mechanism for the formation of black-P nanoribbons involves the difference in diffusion kinetics between the armchair and zigzag directions.259,260 Specifically, the preferred diffusion of alkaline metal ions in the zigzag direction creates a strain between intercalated and non-intercalated regions. This strain eventually leads to the breakage of the longer armchair P–P bonds, resulting in the formation of stripes oriented in the zigzag direction. Using this method, McDonald et al. synthesized black-P nanoribbons and utilized the nanoribbons in photovoltaic devices.261 A recent report demonstrated an alternative method for the synthesis of black-P nanoribbons, utilizing an electrochemical-based sodium intercalation process. The resulting nanoribbons have a constrained width of 10.3 ± 3.8 nm and a length of 250 ± 156 nm. The width of the nanoribbons obtained by this method is narrower compared to that of the nanoribbons prepared by Watts et al.258 Furthermore, this electrochemical-based method eliminates the requirement for cryogenic conditions (−50 °C), making it more cost-effective and readily scalable for the synthesis of black-P nanoribbons. Other electrochemical-based exfoliation approaches have also been developed to synthesize black-P nanoribbons. For example, Liu et al.262 have synthesized zigzag-phosphorene nanobelts by an electrochemical method with oxygen-driven BF4− intercalations. Yu et al.263 have optimized the electrochemical exfoliation conditions and employed two different types of quaternary ammonium salt cations (TPA and THA) as intercalating agents in the preparation of zigzag black-P nanoribbons with a yield of over 80%.
The bottom–up CVT method has been recently reported as an alternative to the top–down synthesis methods to produce black-P nanoribbons. Wang's group developed a one-step CVT method for the synthesis of black-P nanoribbons under mild conditions.264 The process involves heating commercial red-P with iodine precursors at a rate of 2 K min−1 in a sealed quartz ampoule to 873 K. The temperature is maintained for 2 hours, followed by cooling to 738 K after 490 minutes and keeping for another 2 hours before natural cooling to room temperature. Ultimately, the black-P nanoribbons are grown at the bottom of the ampoule. The nanoribbons are several tens of microns in length, ∼400 nm in width, and ∼20 nm in thickness. Macewicz et al. optimized the CVT synthesis method of producing black-P nanoribbons and introduced a two-step process involving CVT growth of black-P columns, followed by ultrasonic treatment and centrifugation.265 The process is initiated by placing red-P, Sn, and SnI4 precursors at one end of a vacuum quartz ampoule with the silicon substrate being positioned on the other end (Fig. 17i). To maintain the CVT process, a temperature gradient of 50 °C is maintained between the precursor and silicon substrate during heating (Fig. 17j). After the reaction, a uniform layer of columnar black-P is formed on the substrate surface. The columnar black-P is then transferred to a beaker containing DMF and sonicated for 10 minutes (Fig. 17k), after which black-P nanoribbons are collected by centrifugation. The black-P nanoribbons have an aspect ratio in the range of a few hundred with the majority of the columns having a width of up to 1.5 μm and length of more than 500 μm.
Overall, nano-etching is capable of producing black-P nanoribbons with different crystallographic orientations, but achieving precise control over the thickness and width at the atomic level remains challenging. Liquid-based exfoliation methods, including ion intercalation and electrochemical exfoliation, can lead to the fabrication of high-quality black-P nanoribbons on a large scale, but the nanoribbons obtained are predominantly oriented along the zigzag direction. CVT synthesis offers a relatively straightforward experimental procedure, but effectively separating the nanoribbons from impurities during crystal growth is an obstacle. It is anticipated that further improvement of existing methods or emergence of new ones will hasten the incorporation of black-P nanoribbons into real-world applications.
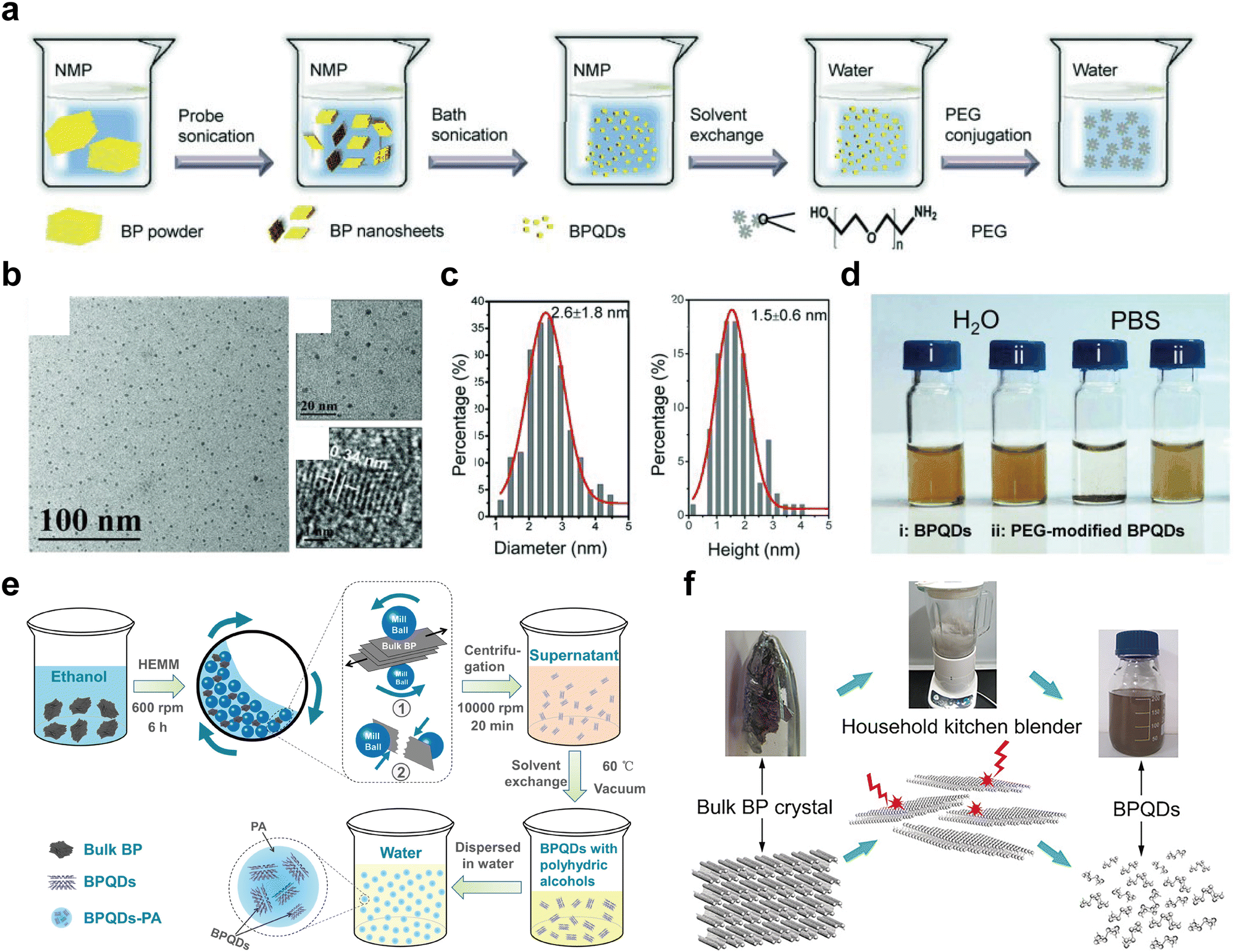 | ||
| Fig. 18 Synthesis and characterization of black-P quantum dots. (a) Scheme of the two-step sonication method for BPQD preparation and modification. (b) TEM images of BPQDs. (c) Lateral sizes (right) and heights (left) of BPQDs. (d) BPQDs and PEG-modified BPQDs in H2O and PBS. Reproduced with permission from ref. 269. Copyright 2015 John Wiley and Sons. (e) Scheme of the HEBM process of BPQDs and modification. Reproduced with permission from ref. 278. Copyright 2020 American Chemical Society. (f) Scheme of the synthesis of BPQDs by using a household kitchen blender. Reproduced with permission from ref. 279. Copyright 2016 John Wiley and Sons. | ||
Electrochemical exfoliation with bipolar electrodes for the synthesis of BPQDs has been developed. In 2016, Mayorga-Martinez and co-workers devised a simple electrochemical exfoliation-based approach to prepare black-P nanoparticles, yielding nanoparticles with a size ranging from 40 to 200 nm.276 Two years later, Tang et al. succeeded in preparing fluorinated BPQDs (F-BPQDs) using an electrochemical exfoliation and synchronous fluorination technique.277 This process involves using black-P bulk crystals as the working electrode in a sealed three-electrode electrochemical system, where 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4])/acetonitrile (MeCN) serves as the electrolyte.277 Application of a +8 V voltage against a Ag wire anode initiates a series of chemical reactions in solution, leading to gradual exfoliation of black-P and electrolysis of BF4− to generate BF3 molecules and F− anions. The resulting BF3 molecules form donor–acceptor bonds with MeCN, while the F− anions synchronously exfoliate BPQDs. The as-synthesized BPQDs exhibit robust environmental stability, allowing them to withstand persistent ambient exposure for up to 7 days due to the hindrance of charge transfer from the phosphorus atoms to external oxygen, induced by fluorine adatoms.277
Ball milling is a viable choice for the synthesis of BPQDs. Ren et al. have developed a liquid-phase high energy ball milling (HEBM) and centrifugal separation process to synthesize brown transparent BPQDs ethanol suspensions, as shown in Fig. 18e.278 First, the HEBM approach is used to prepare black-P powders from amorphous red-P which are then placed in a sealed stainless ball milling jar containing ethanol and milling balls. The milling procedure is carried out at 600 revolutions per minute for 6 hours. After centrifugation and dispersion, the BPQD solution is mixed with various polyhydric alcohols to enhance the stability in aqueous environments. The as-prepared BPQDs show stable dispersion in ethyl alcohol and ethylene glycol, with an average lateral size of 6.5 ± 3 nm and a thickness of 3.4 ± 2.6 nm. Interestingly, BPQDs can also be fabricated by utilizing a household kitchen blender to generate a high turbulent shear rate for layer-by-layer disassembly (Fig. 18f).279 Using DMSO as a stabilized solvent, black-P bulk crystals are processed for 40 minutes in a household kitchen blender set to 17000–21000 rpm. The high-shear turbulence gradually peels off and disintegrates the black-P, ultimately resulting in the formation of the BPQDs suspension.
Pulsed laser ablation with high peak power densities (usually >1013 W cm−2) and ultra-short periods (femto- and nanosecond) has been applied for the exfoliation of 2D materials and production of their low-dimensional structures.268 Some studies have demonstrated that this approach can be utilized to prepare BPQDs. Ge et al. have synthesized phosphorene quantum dots (PQDs) by pulsed laser ablation of a bulk black-P target in diethyl ether.280 Diethyl ether molecules are cleaved by laser ablation to passivate the surface and edges of the phosphorene domains. The PQDs exhibit strong photoluminescence in the blue–violet range, with an average lateral size of ∼7 nm. Ren and co-workers have fabricated a few-layered BPQDs using pulsed laser ablation of black-P bulk crystal in isopropyl ether.281 A neodymium-doped yttrium aluminum garnet (Nd:YAG) pulsed laser with a repetition rate of 10 Hz and wavelength of 1064 nm is used in the experiment.281 The ablation process lasts for about 30 minutes and the BPQDs exhibit a higher photoluminescence quantum yield (∼20.7%) than PQD (11.92%),280 showing potential in cell bioimaging.
4.2 Synthesis of red phosphorus
Using white-P as starting materials, early synthesis of red-P often involves the pyrochemical method, which requires temperatures over 250 °C in the absence of air.87 Vvedenskii and Frost studied the reaction kinetics of thermal conversion and claimed that the conversion of melted white-P to red-P followed a first-order reaction in the temperature range of 176–373 °C.282 However, the increasing trend of constants with a rise in the percentage conversion at temperature below 263 °C suggests an autocatalytic conversion. To validate this phenomenon, DeWitt and co-workers conducted more experiments on the reaction kinetics in 1946.282 The reaction mixture initially comprises red-P particles suspended in white-P. As the reaction progresses and the percentage conversion reaches approximately 50%, the particles grow in number and size forming a semi-fluid before forming a solid product. The reaction in the temperature range of 250–350 °C strictly follows a first-order rate law, with an activation energy of 37800 calories per mole. In addition, it is noted that addition of iodine or sulfur has a significant accelerating effect on the thermal conversion process. However, this effect is only noticeable in the initial stages and rapidly diminishes, resulting in the conversion proceeding at a rate similar to that without accelerators.
In contrast to the pyrochemical method of red-P synthesis, electromagnetic radiation utilizes a gentler synthetic approach to convert white-P to red-P (Fig. 19a, route a).283 Linus Pauling had proposed that red-P can be formed from white-P by cleaving a single bond in each P4 molecule, resulting in an expansion of the unit into a figure comprising two equilateral triangles with a shared base.108 The two apical phosphorus atoms in each P4 unit can then bond with similar phosphorus atoms in neighboring P4 complexes, leading to the formation of long chains of bonded atoms and ultimately the formation of red-P. The formation of red-P from white-P is easier than that of black-P because breaking a single bond is sufficient to convert each P4 unit into red-P, whereas the formation of black-P requires breaking of three out of the six bonds in each P4 unit.108 A. Pedler and G. Rathenau have published two seminal papers demonstrating how light triggers the conversion of white-P to red-P.284,285 These studies reveal two mechanisms that drive the transformation through light activation. One mechanism involves the symmetry cleavage of the P4 tetrahedron and subsequent rearrangement of P2 into the final polymeric red-P.285 The other mechanism, which occurs in non-polar media, involves a radical process.286 The dispersed phase also influences the concentrations of P4 and P2. When white-P is heated to 1000–1500 °C in an inert atmosphere, the resulting gas is a mixture of P4 and P2 molecules, with their vapor pressure ultimately reaching equilibrium (Fig. 19a, route f),283,287 but when the dispersed phase is a solution at room temperature, P4 molecules are photolyzed to P2.288 M. Kraft and V. Parini have described a photochemical reaction between white-P and alkyl iodides at 20 °C, yielding a small quantity of red-P containing organic molecules.289 D. Perner and A. Henglein have carried out a similar reaction of white-P with alkyl (or aryl) iodides in CCl4 under γ-60Co at temperatures ranging from 20 to 140 °C.290 The primary product is red-P (79%) at 25 °C, but the primary product is alkyl-phosphorylated derivatives (80%) at 130 °C. However, when white-P is subjected to visible light up to 100 °C, red-P becomes the primary product. There seems to be a more sophisticated mechanism behind the transformation of white-P into red-P through electromagnetic radiation, and a further study is necessary.
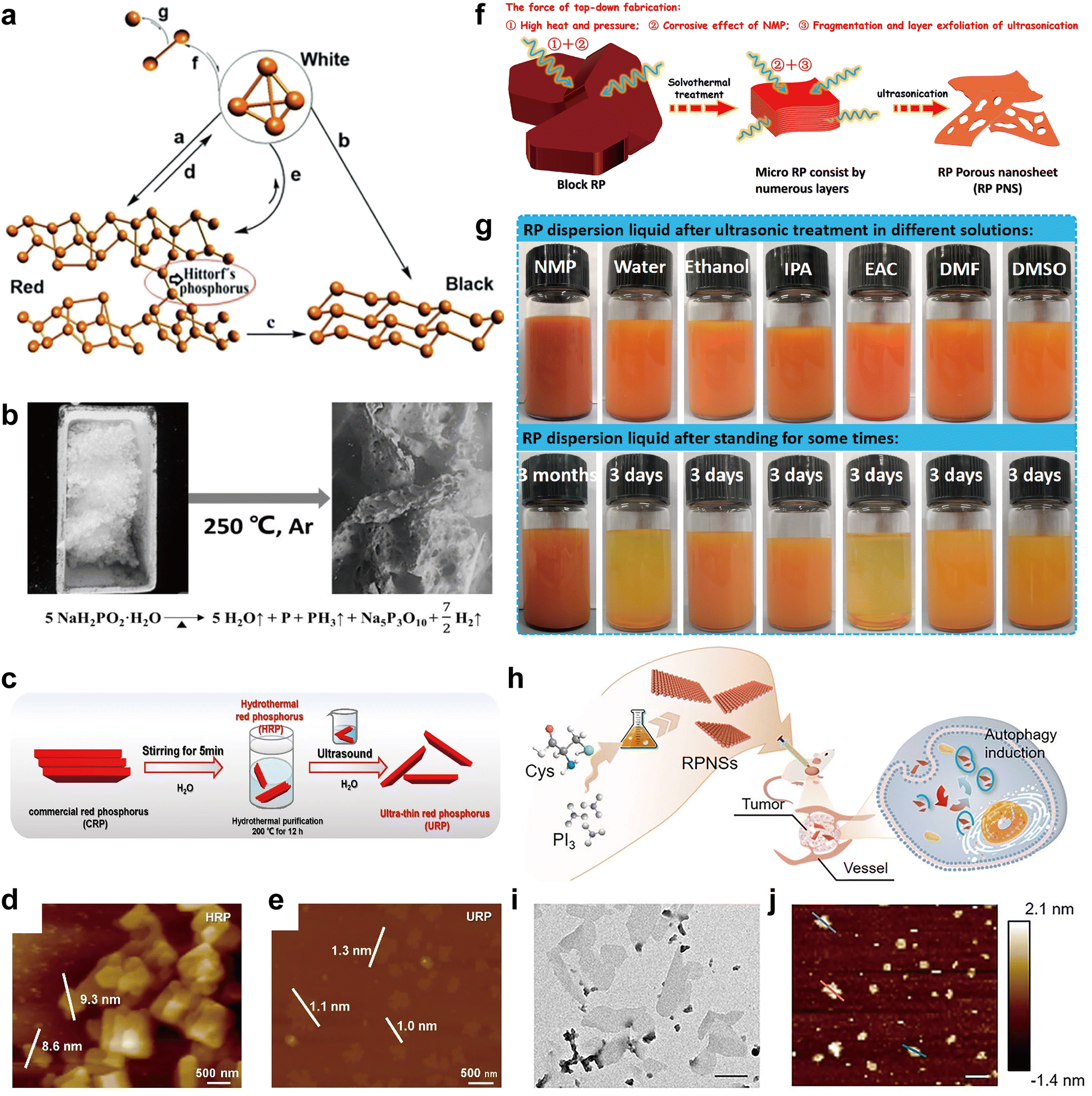 | ||
| Fig. 19 Synthesis and characterization of the red-P bulk form and nanosheets. (a) Scheme of the conversion of the main significant phosphorus allotropes through different routes. Reproduced with permission from ref. 283. Copyright 2014 John Wiley and Sons. (b) Images of samples before and after thermal decomposition. Reproduced with permission from ref. 292. Copyright 2018 Elsevier. (c) Scheme of the fabrication procedure of ultra-thin red-P. (d) and (e) AFM images of samples before and after sonication. Reproduced with permission from ref. 293. Copyright 2021 Springer. (f) Scheme of the fabrication procedure of red-P porous nanosheets. (g) Photographs of the red-P dispersion after ultrasonic treatment in different solvents and after standing for some days. Reproduced with permission from ref. 294. Copyright 2020 Elsevier. (h) Scheme of the synthesis of red-P nanosheets and their potential anticancer applications. (i) TEM image of the obtained red-P nanosheets. The scale bar is 200 nm. (j) AFM image of the as-synthesized red-P nanosheets. The scale bar is 500 nm. Reproduced with permission from ref. 298. Copyright 2021 John Wiley and Sons. | ||
Ultrasound-assisted liquid exfoliation can be utilized to prepare highly dispersed red-P nanosheets. Water exfoliation is carried out to produce ultra-thin red-P from commercial red-P (Fig. 19c), with the resulting nanosheets being highly dispersed and having an average thickness of ∼1 nm (Fig. 19d–e).293 When a similar liquid exfoliation process is conducted using probe ultrasonication in the NMP solvent, red-P porous nanosheets are produced (Fig. 19f).294 This is because of the strong synergistic effects of NMP and ultrasonication facilitating layered exfoliation and partial surface fragmentation of bulk red-P, leading to the formation of nanosheets with porous topologies. In contrast, bulk red-P exfoliated in a water solution displays insufficient fragmentation and fails to form porous structures. To further assess the suitability of red-P nanosheets with dispersants and the role of NMP in ultrasonication, other solvents are used in exfoliating nanosheets (Fig. 19g).294 However, the nanosheets are found to be unstable in these solvents and visible deposition occurs to varying degrees after three days. On the other hand, the nanosheets can remain stable in the NMP solvent for over three months, thus confirming the superior stability of NMP in the red-P exfoliation systems. However, the specific interaction between bulk red-P and solvents of varying polarity requires further elucidation.
While red-P nanosheets prepared by liquid exfoliation have shown potential for a variety of applications, they suffer from issues such as sheet-edge corrosion and uncontrollable size. Fortunately, an alternative method using amine-induced wet chemical synthesis has made significant progress in the production of red-P nanosheets.295–297 This method involves a nucleophilic attack on phosphorus sources, which triggers the breakage of the P–P bond and the formation of polyphosphoric ions (Pn−). Subsequent treatment with water and acid replaces external ions with electrophiles, resulting in the decomposition of Pn− to form red-P nanosheets. Recently, Mo and colleagues have described a unique wet-chemical approach for the synthesis of red-P nanosheets through a cysteine-mediated redox reaction and demonstrated their potential for anticancer applications (Fig. 19h).298 In this method, phosphorus triiodide (PI3) serves as the phosphorus source, while L-cysteine (L-Cys) acts as the reductant. The confinement material CATB and stabilizer PVP are also employed. Through a mild redox reaction between PI3 and L-Cys, phosphorus atoms are gradually released and then assembled into nanosheets. The optimal concentration of CATB is critical in the process, since it suppresses red-P natural 3D growth, allowing for 2D growth instead. The weakened reduction, combined with the appropriate confinement material, enables a mild reaction equilibrium, resulting in the formation of high-quality red-P nanosheets. TEM and AFM reveal that the RPNSs are atomically flat with a thickness of around 1.7 nm (Fig. 19i and j). Moreover, this method enables doping of metal atoms in red-P nanosheets to regulate the basic properties and expand the applications.
Song et al. have developed a drug delivery platform by constructing a nanocomposite of bismuthene and RPQDs for synergistic photothermal/photodynamic/chemotherapy.299 The nanocomposite is synthesized through an electrostatic assembly of exfoliated bismuth nanosheets and RPQDs, followed by PEGylation and loading of the organic drug molecule DOX (Fig. 20a). RPQDs are prepared by dispersing 0.5g of red-P powder and 0.2g of triphosphate in water and subjecting them to sonication in an ice bath for 10 hours, followed by centrifugation and filtration. The resulting RPQDs are then PEGylated to make them more suitable for physiological conditions and the average diameters of RPQDs before and after PEGylation are around 2.5 and 55 nm, respectively. The combination of bismuthene and RPQDs exhibits synergistic effects achieving photothermal conversion efficiencies of 54.2% and 38.5% upon 1064 nm and 808 nm (1.5 W cm−2) laser irradiation, respectively.
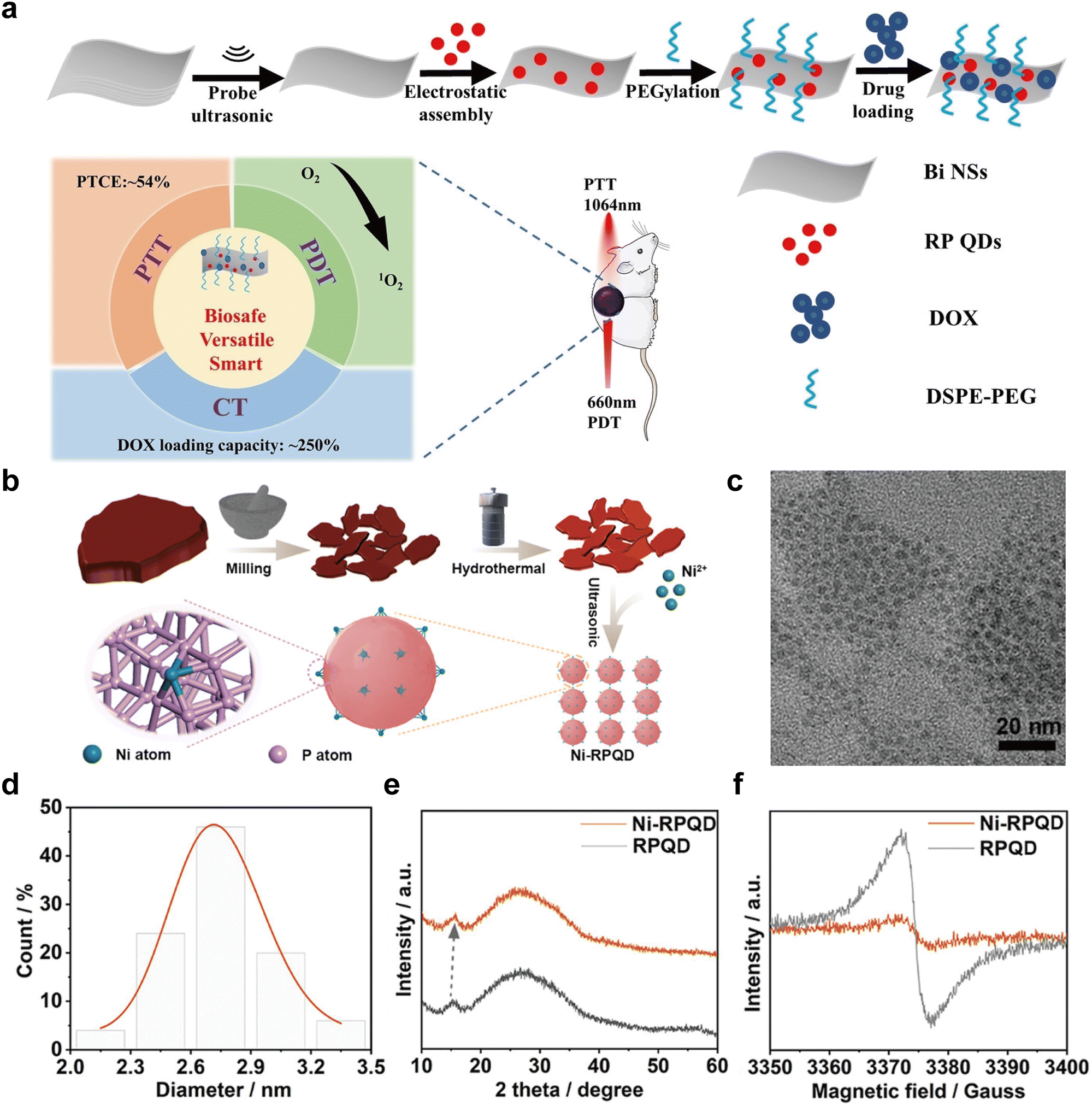 | ||
| Fig. 20 Synthesis and characterization of RPQD-based materials. (a) Scheme of the fabrication procedure of Bi@RP-PEG-DOX. Reproduced with permission from ref. 299. Copyright 2022 John Wiley and Sons. (b) Scheme of the synthesis of Ni-RPQD. (c) XRD pattern of Ni-RPQD and RPQD. (d) TEM image of Ni-RPQDs. (e) Statistical analysis of Ni-RPQD diameters. (f) EPR spectra of Ni-RPQD and RPQD. Reproduced with permission from ref. 300. Copyright 2022 John Wiley and Sons. | ||
Jia and colleagues have reported in situ growth of single-atom nickel on the surface of RPQDs using the ultrasonic method.300 In the process, commercial bulk red-P is first ground into fine particles, followed by hydrothermal treatment to remove the oxide layer from the surface. The purified red-P and NiCl2·6H2O are introduced to 100 mL of NMP and sonicated in an ice bath for 48 h to form the nanocomposite Ni-RPQD (Fig. 20b). TEM demonstrates successful synthesis of a homogeneous nanocomposite with a particle size of around 2.7 nm (Fig. 20c and d). The uniform distribution of single-atom Ni is attributed to the unique coordination of single atoms docked with P vacancies on the RPQD surface, which is supported by significant reduction of the electron paramagnetic resonance signal and high-angle shift in the XRD patterns (Fig. 20e and f). The Ni–P bond potentially serves as an electronic antenna to promote facile extraction and transfer of charges. Furthermore, incorporation of Ni single atoms reduces the bandgap and enhances light absorption to improve photocatalytic activity under visible light irradiation.300
4.3 Synthesis of fibrous phosphorus
Ruck's detailed experimental synthesis procedure for fibrous-P bulk crystals using the CVT approach has become a widely adopted practice. Researchers have utilized various iodine-containing transport agents in experiments to improve the short-distance transport reaction and obtain high-purity fibrous-P crystals. Smith et al. developed a two-step method for producing ultra-long and uniform fibrous-P nanowires.302 They first prepared thin films of red-P on a SiO2/Si wafer, and then heated a mixture of purified red-P (110 mg), SnI4 (15 mg), and Sn powder (30 mg) in a sealed ampoule with the red-P-coated silicon wafer plate to 630 °C for 1 hour, followed by programmed cooling. The resulting fibrous-P nanowires have a diameter of 300–800 nm and a length of over 1 mm. However, the nanowires are found to be sensitive to air due to their larger specific surface area and the presence of other crystalline forms of red-P. Other iodine-containing transporters such as CuI and SbI3 are also found to produce other phosphorus allotropes besides fibrous-P. To obtain single-phase fibrous-P crystals, Nilges’ group substituted the iodine-containing transport agents with chloride compounds and used CuCl2 as an assisting agent.303 They designed a temperature-control program that involves heating the precursors to 550 °C within 8 hours, followed by holding the temperature for 15 hours, cooling to 275 °C within 75 hours, maintaining this temperature for 1 hour, and finally cooling to room temperature within 75 hours.303 This method leads to the production of fibrous-P crystals in high yield with reasonable quality. However, not all products synthesized by the CVT method have well-defined fibrous structures and some crystals instead show different morphologies such as bunches-like, nanowire, and urchin-shaped structures.302–305 These diverse morphologies may be attributed to the complex phase transformation that occurs during the gas phase process and the dispersion interaction between each tubular unit.
Aside from the transport agent, the vapor transport process is highly influenced by the reaction conditions. Our group has developed a low-temperature CVT method that effectively produces high-quality fibrous-P crystals on a large scale (Fig. 21a).47 In this process, amorphous red-P is transported in the presence of iodine from a high-temperature region (480 °C) to a low-temperature region (450 °C), where crystallization occurs within a day. Compared to previous reports in which the typical temperature is over 550 °C and annealing time lasts several days to weeks, our method greatly reduces the overall reaction temperature and annealing time. In a single preparation, we are able to obtain a total amount of 9.937 g crystals with a yield exceeding 99% (Fig. 21b and c). Mass production of fibrous-P bulk crystals allows for exfoliation of fibrous-P nanoribbons, which will be discussed in the next part. Yan's group has investigated the impact of growth kinetics on the crystallinity of fibrous-P in the CVT reaction and found that the kinetics of CVT is regulated by the rate-determining steps of gas convection and diffusion. To obtain high-quality crystalline fibrous-P, it is important to slow down gas convection and diffusion. To achieve this, there are several strategies including reducing the amount of red-P during the reaction, increasing the length of the ampoule, lowering the average reaction temperature, and designing a “neck” on the ampoule to reduce the cross-sectional area and slowing down the reaction rate (Fig. 22a).160 These measures are effective in modifying the growth kinetics of the CVT reaction, ultimately leading to the production of high-quality and well-crystallized fibrous-P crystals. Recently, Zhang et al. have developed a low-pressure CVT approach for the production of centimeter-scale fibrous-P micropillar arrays without the use of transport agents.306 The approach utilizes directional phase transformation of amorphous red-P at a specific temperature and pressure to generate fibrous-P micropillar arrays by self-assembly. In contrast to traditional transport agent-assisted CVT methods, this approach avoids nucleation aggregation induced by transport agents, paving the way for the bottom–up synthesis of fibrous-P microstructures and nanostructures.
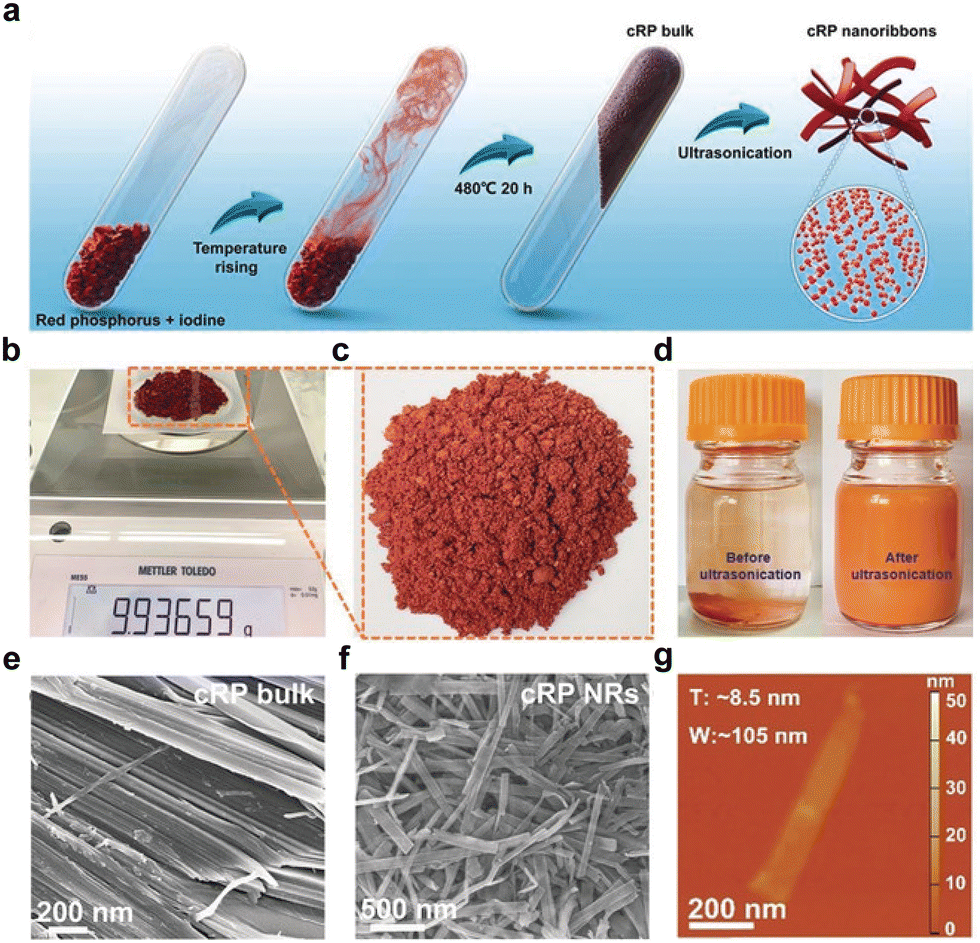 | ||
| Fig. 21 Low-temperature CVT method for the preparation of fibrous-P bulk crystals and nanoribbons. (a) Scheme of the low-temperature CVT method. (b) Photograph and weight of the fibrous-P bulk powder. (c) Photograph of the bulk powder under high magnification. (d) Photograph of the fibrous-P dispersion before and after exfoliation. (e) TEM image of fibrous-P bulk crystals and (f) nanoribbons. (g) AFM image of a representative fibrous-P nanoribbon. For the sake of consistency, it should be noted that the term “fibrous-P” is used throughout this review to refer to the “triclinic crystalline red phosphorus (cRP)” mentioned in the original text. Reproduced with permission from ref. 47. Copyright 2020 John Wiley and Sons. | ||
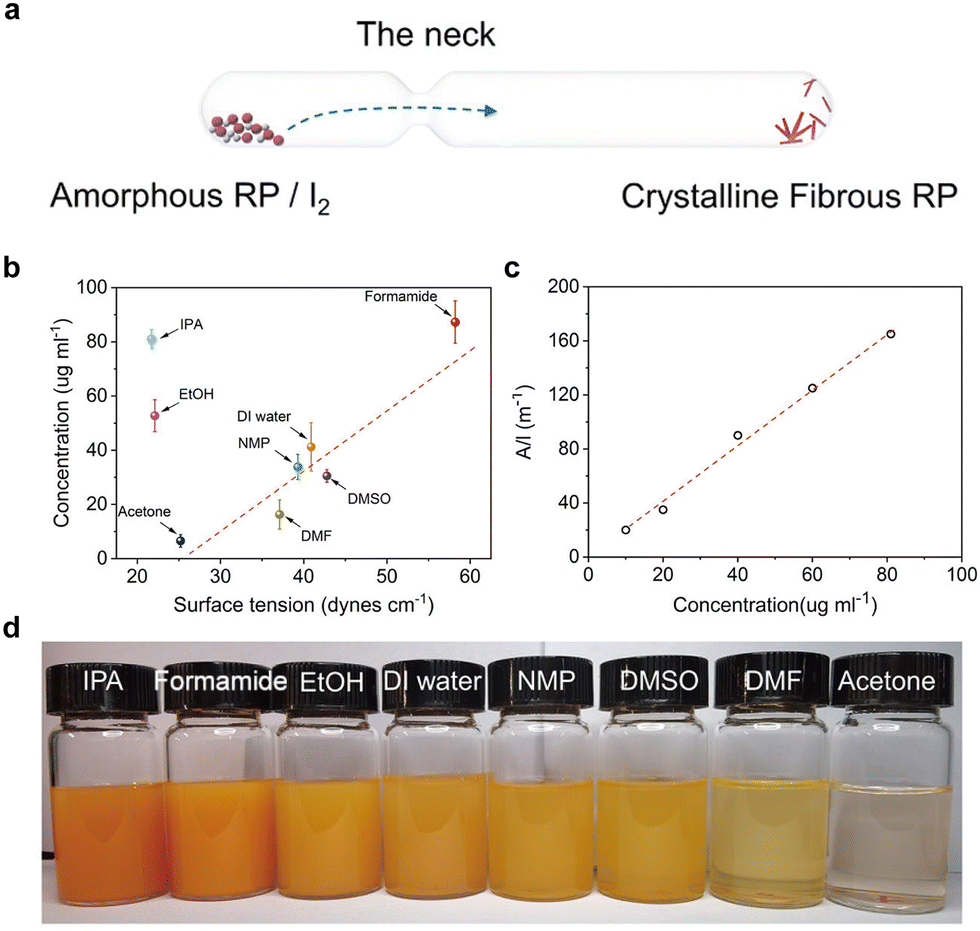 | ||
| Fig. 22 Modification of the growth kinetics in the CVT reaction for the preparation of fibrous-P bulk crystals and nanoribbons. (a) Scheme of a “neck” on the ampoule in the CVT reaction. (b) Dispersion concentration of fibrous-P nanoribbons versus solvent surface tensions. (c) Lambert–Beer's plot at λ = 650 nm of fibrous-P nanoribbons in IPA. (d) Photograph of fibrous-P nanoribbon dispersions in different solvents. Reproduced with permission from ref. 160. Copyright 2021 American Chemical Society. | ||
Solution-based deposition of elemental phosphorus is an alternative method to prepare low-cost and scalable fibrous-P thin films. Ban et al. have demonstrated a wet chemical process that utilizes soluble polyphosphides as precursors to generate fibrous-P through moderate heating without the need for external agents.307 Amorphous red-P is the source in the polyphosphide synthesis and reacts with potassium ethoxide to produce a potassium mixture of polyphosphide powder. The resulting polyphosphide powder is then refined by ethanol dispersion and insoluble precipitate removal to eliminate unreacted precursors and by-products. To fabricate fibrous-P thin films, the polyphosphide solution is spin-coated or drop-cast onto glass or Si/SiO2 substrates followed by annealing for 30 minutes at 250 °C. The Raman signals of fibrous-P films formed on glass and Si/SiO2 substrates are almost identical, indicating that fibrous-P formation is mainly due to the precursor itself rather than the precursor–substrate interaction. However, similar to the issues encountered during the preparation of black-P crystals by wet chemical methods,252 fibrous-P produced by this method lacks obvious lattice structures and may contain mixed phosphorus phases. Hence, investigation of the interaction between phosphorus clusters and solvents is necessary to better understand the dynamic growth process of crystalline phosphorus in solutions.
Liquid-phase exfoliation is currently the primary method to fabricate fibrous-P nanoribbons due to its scalability and high-yield production. As aforementioned in the preceding section, our group has developed a low-temperature CVT method for mass production of high-quality fibrous-P bulk crystals.47 After dispersing these crystal powders in absolute ethanol and performing probe sonication, the resulting supernatant changes from colorless to a uniform orange color (Fig. 21d), indicating the formation of a stable fibrous-P colloid solution. The fibrous-P nanoribbons obtained from this solution are compact, micro-pillar-like lumps, which are quite distinct from the micrometer-scale fiber structures of the bulk materials (Fig. 21e and f). The width of the nanoribbons is approximately 120 nm and lengths range from hundreds of nanometers to several micrometers. The majority of the fibrous-P nanoribbons have an aspect ratio greater than 10, with some having an aspect ratio close to 100. AFM reveals that the thickness of a representative fibrous-P nanoribbon is ∼8.5 nm (Fig. 21g), indicating that liquid-phase exfoliation can lead to the production of few-layered fibrous-P nanoribbons. Hu et al. have proposed the concept of fibrous phosphorene based on the feature that fibrous-P can be exfoliated.131 Using N-cyclohexyl-2-pyrrolidone (CHP) as the liquid-sonication solvent, they were able to prepare fibrous phosphorene along the [001] direction with a thickness of roughly 1.67 nm. Given that the [001] facet has an average spacing of 0.583 nm,131 the fibrous phosphorene should consist of three layers. NMP is also used to exfoliate bulk fibrous-P. In the process, single-layered phosphorene is observed, indicating that NMP may have a greater exfoliation capability than CHP. This study demonstrates that the choice of solvents plays a critical role in the exfoliation of Fibrous-P.
To investigate the impact of solvents on the efficiency of fibrous-P exfoliation, Yan's group selected eight solvents with different surface tensions, including isopropanol (IPA), DMSO, NMP, DMF, formamide, ethanol, deionized water, and acetone (Fig. 22d).160 Each solvent shows a different exfoliation efficiency and there is a broad linear relationship between the dispersed concentration of nanoribbons and solvent surface tension (Fig. 22b). Solvents with higher surface tension tend to have stronger phosphorus–solvent adhesion, which stabilizes the nanoribbon surface for a higher exfoliation efficiency.308 However, IPA and ethanol, which have relatively low surface tension, also show efficient exfoliation due to the appropriate molecular interaction with fibrous-P interlayers.309 Although formamide has the highest exfoliation concentration, IPA is recommend as the primary solvent for fibrous-P exfoliation due to its high efficiency, low boiling point, ease of handling, and cost-effective features. The yield of exfoliated nanoribbons in IPA is 16%, and the dispersion extinction coefficient is measured to be ∼2087 L g−1 m−1 under 650 nm incident light (Fig. 22c).160 Liu et al. have used the Hansen solubility parameters (HSPs) to quantify the effects of the solvents on the exfoliation efficiency of fibrous-P nanoribbons.310 The HSP approach considers three key parameters: δD (intermolecular dispersion force), δH (intermolecular hydrogen bond), and δP (intermolecular polar force).311 The desired exfoliating solvent should have HSPs that closely match those of fibrous-P. After comparing 19 common solvents, acetone and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) were found to be more suitable for fibrous-P exfoliation. Moreover, the HSPs of a solvent can be modified by varying the solvent ratios. The water/acetone mixture is chosen as the solvent because it covers the HSPs of Fibrous-P, and both water and acetone are easy to remove without leaving a residual solution. The optimal solvent mixture for fibrous-P exfoliation is determined to be 10% water and 90% acetone, which yields 40% nanoribbons with a thickness of less than 10 nm and 70% with a thickness of less than 20 nm. These results demonstrate the potential of HSP modeling in identifying suitable solvents for exfoliating 2D materials and highlight the importance of solvent selection in the exfoliation process.
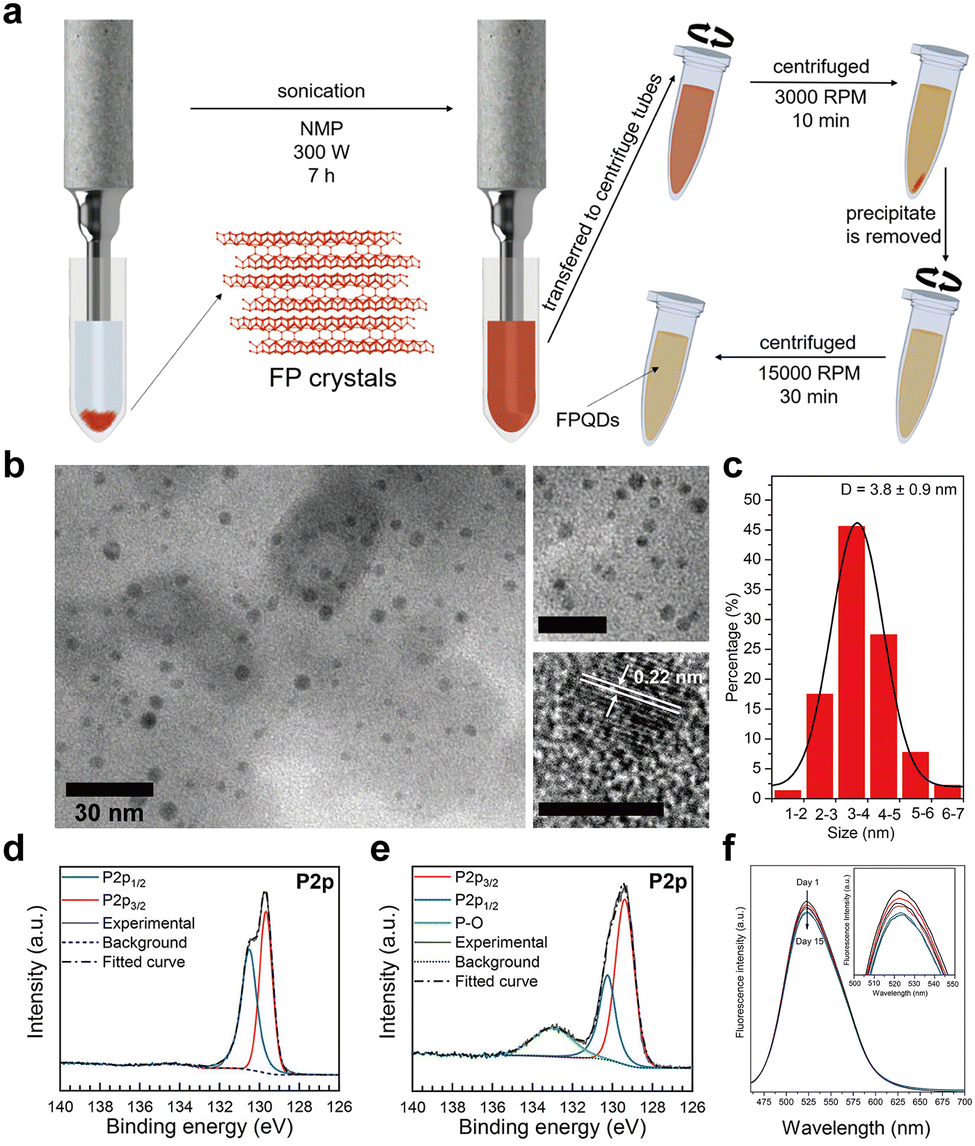 | ||
| Fig. 23 Synthesis and characterization of FPQDs. (a) Scheme of the fabrication procedure of FPQDs. (b) TEM, high-resolution TEM images, and (c) statistical analysis of the size of FPQDs. The middle–upside and middle–downside images have scale bars of 20 and 5 nm, respectively. (d) XPS spectra of bulk fibrous-P and (e) FPQDs. (f) Photoluminescence intensity of FPQDs was measured over 15 days, with excitation at λ = 370 nm. The inset is a magnified view of the photoluminescence peak. Reproduced with permission from ref. 312. Copyright 2020 American Chemical Society. | ||
4.4 Synthesis of violet phosphorus
In 2019, Yu's group developed a thermal evaporation method that utilized liquid bismuth nanodroplets to produce large quantities of single-crystal violet-P microbelts measuring up to 100 × 10 × 0.3 μm3 (Fig. 24a).314 The growth mechanism of microbelts is shown in Fig. 24c. The process involves the sealing of appropriate amounts of red-P and Bi powders in vacuum quartz capsules and heating to 550 °C, inducing the sublimation of the red-P precursor while Bi melts. At this temperature, the molecular motion of the phosphorus vapor becomes highly turbulent producing quick disturbances within the capsule to impinge into the Bi droplets and stretch then into nanodroplets. As the temperature drops to 300 °C, the sublimed phosphorus progressively adsorbs onto the surface of the Bi nanodroplets and nucleates, forming seed crystals that eventually grow into nanorods.315 Due to the high surface energy of the nanodroplets, they tend to consolidate, causing the nanorods to cohere and form a thicker crystal that gradually expands into a microbelt. The Bi nanodroplets may depart during growth and after cooling to ambient temperature, most of them aggregate, with only a few droplets condensing singly on the microbelts.316,317 The XRD pattern of violet-P microbelts reveals a high-intensity peak at 15.66 degrees from the (013) facet. This peak is markedly distinct from the corresponding peak in the standard violet-P bulk crystal with low intensity. The difference in the peak intensity suggests the specific orientation of facets within the crystal.314 Additionally, high-resolution TEM shows a well-resolved interference fringe spacing of 5.67 Å along the crystal's edge (Fig. 24b), which is in good agreement with the interplane distance between the (013) planes of violet-P.314 These findings provide evidence of successful synthesis of high-quality violet-P microbelts with the exposed active (013) facets.
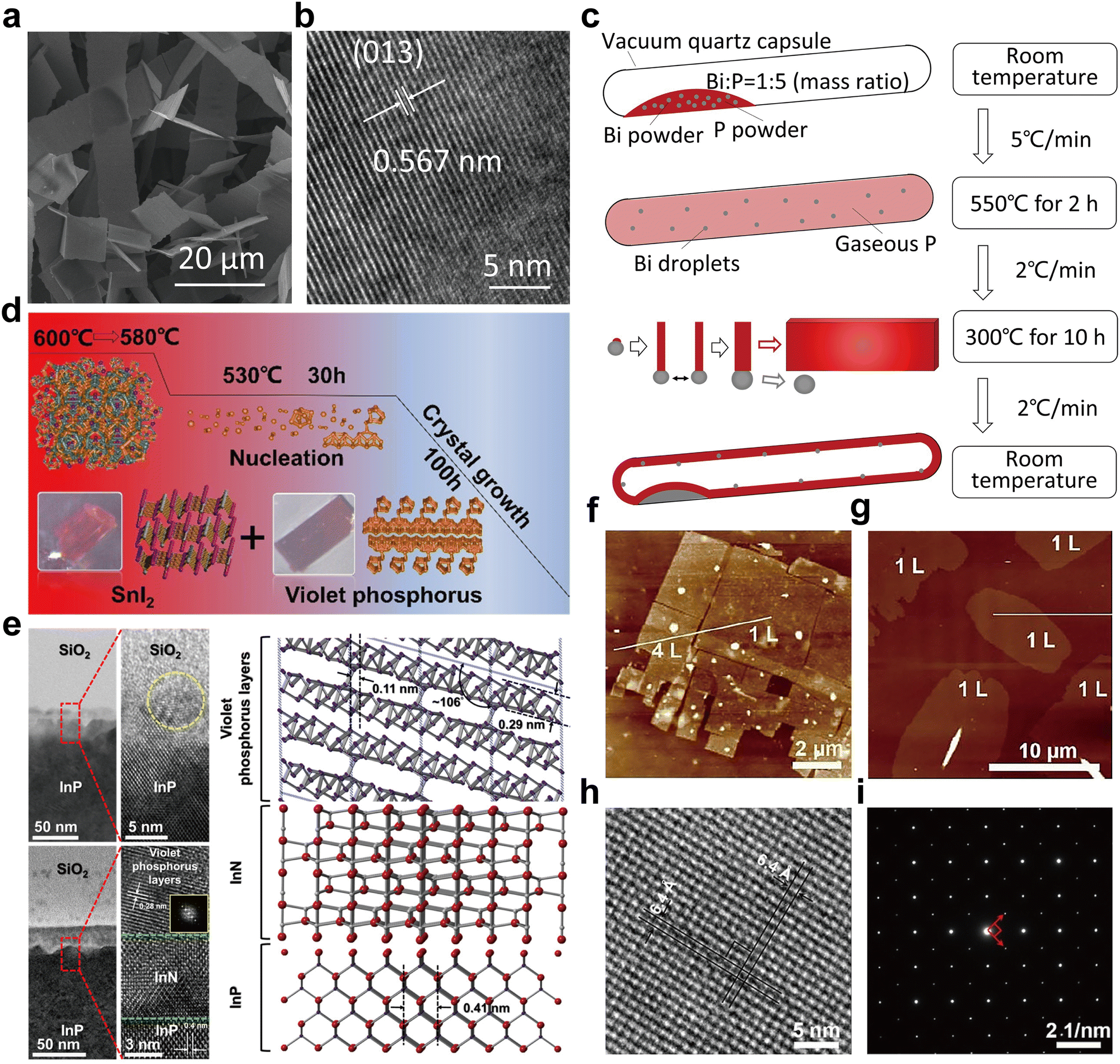 | ||
| Fig. 24 Synthesis and characterization of the violet-P bulk form and phosphorene. (a) SEM image and (b) high-resolution TEM patterns of violet-P microbelts. (c) Scheme of the growth mechanism of microbelts initiated by liquid Bi through thermal vaporization. Reproduced with permission from ref. 314. Copyright 2019 Elsevier. (d) Scheme of the fabrication procedure of violet-P crystals through an improved CVT method. Reproduced with permission from ref. 319. Copyright 2020 American Chemical Society. (e) TEM images and atomic model of the violet-P/InN/InP layered structure. Reproduced with permission from ref. 320. Copyright 2015 American Chemical Society. (f) AFM images of violet phosphorene from mechanical and (g) liquid exfoliation. (h) High-resolution TEM image of a violet-P nanobelt. (i) SAED pattern corresponding to (h) with the zone axis of [001]. Reproduced with permission from ref. 48. Copyright 2020 John Wiley and Sons. | ||
Different from direct thermal evaporation, CVT typically involves separate heating zones, which allow for the movement of gaseous intermediates in defined directions.318 Zhang's group has made contributions to the synthesis of high-quality violet-P bulk crystals using the CVT method. In 2020, they obtained single-crystal violet-P and conducted a detailed analysis of its structure.48 This achievement is of great significance, as it represents the first experimental determination of the lattice structure of violet-P and provides valuable information about the properties and potential applications of violet-P. However, the violet-P crystals obtained in this study contain a mixture of black-P with a yield of only about 36% after separation. To address this issue and improve the production yield, the CVT process has been optimized by using 10 mg of Sn and 18 mg of SnI4 as the transport agents and 470 mg of amorphous red-P as the source materials in a vacuum quartz tube.319 As shown in Fig. 24d, the tube is heated slowly in a three-zone muffle furnace for 8 hours to reach 600 °C at the sample end (source zone) and 580 °C at the other end (reaction zone).319 After 5 hours, the tube is cooled for 10 hours to 550 °C in the source zone and 530 °C in the reaction zone. The tube is kept at these temperatures for 30 hours before slowly cooling to room temperature for about 100 hours. The violet-P crystals are produced with a high yield of ∼80%. The holding time of 550–530 °C and cooling time to room temperature are critical for violet-P crystal nucleation and growth.319 A long holding time at 550–530 °C increases the nucleation of violet-P, with nucleation peaking at approximately 30 hours. Longer cooling time results in larger crystal formation with crystal size reaching its maximum at around 100 hours.319 The ability to produce high-quality and high-yield violet-P bulk crystals serves as the foundation for the subsequent synthesis of nanostructured violet-P.
To date, violet phosphorene has been fabricated by both mechanical and liquid exfoliation of violet-P single crystals.48,321–323 Zhang's group has demonstrated that violet phosphorene can be produced by immersing single-crystal violet-P (∼0.30 × 0.23 × 0.13 mm3) in 30 mL of ethanol and treating it by tip sonication at room temperature.48 They also utilized Scotch tape to peel violet-P crystals into thin flakes several times and then transferred them to SiO2/Si substrates. Fig. 24f and g show the AFM images of violet phosphorene prepared by both mechanical and liquid exfoliation methods. Mechanical exfoliation produces single-layered violet-P with a thickness of ∼1.9 nm, but most of the flakes are multi-layered. On the other hand, liquid phase exfoliation can more easily produce thin violet-P flakes (∼2.2 nm), but the edges of the flakes are irregular due to defects generated during liquid exfoliation. However, the internal lattice in the liquid-exfoliated flakes remains intact and high-resolution TEM shows that most flakes have two sets of crystal planes intersecting at an angle of 90° (Fig. 24h) consistent with the (110) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) lattice planes determined by single-crystal XRD.48 The SAED pattern obtained from the flake with the zone axis [001] in Fig. 24i clearly demonstrates the high degree of crystallinity in the as-prepared phosphorene. Furthermore, EDS confirms the high stability of violet phosphorene by showing no detectable oxygen signal. These findings make a significant contribution to the development of high-quality violet phosphorene materials and potential applications in various fields.
0) lattice planes determined by single-crystal XRD.48 The SAED pattern obtained from the flake with the zone axis [001] in Fig. 24i clearly demonstrates the high degree of crystallinity in the as-prepared phosphorene. Furthermore, EDS confirms the high stability of violet phosphorene by showing no detectable oxygen signal. These findings make a significant contribution to the development of high-quality violet phosphorene materials and potential applications in various fields.
The VPQDs synthesized in NMP emit a high fluorescence intensity. To rule out the possibility that the fluorescence originates from the NMP solvent, the excitation wavelength is set at 430 nm, as fluorescence from NMP disappears completely at excitation wavelengths longer than 430 nm. Upon 430 nm excitation, a robust fluorescence band at 522 nm is observed from the VPQDs.324 The VPQDs emit strong green light compared to conventional QDs such as BPQDs (450 nm) and MoS2 QDs (414 nm),326,327 which emit light in the blue–violet range. Conventional QDs emitting blue–violet light penetrate biological tissues poorly due to the predominantly blue luminescence of biological tissues thus limiting in vivo optical imaging of deep tissues.328 However, the green fluorescence emitted by VPQDs may greatly mitigate this issue. The fluorescence quantum yield of VPQDs is determined to be 7.02% and the fluorescence properties have been demonstrated to be adjustable by varying the solvothermal temperature and time conditions, making them potential candidates for nanosensors in biology and medicine.324,329
In addition to the solvothermal temperature and reaction time, Zhang's group demonstrated that the solvent used in the VPQD synthesis influences the fluorescence properties.330 They used six different solvents, including DMF, NMP, H2O, 1,3-dimethyl-2-imidazolidinone (DMI), isopropanol (IPA), and ethanol (EA), to prepare VPQDs based on the solvothermal method (Fig. 25a).330 The quantum dots prepared in the six solvents had the characteristic lattice structure of violet-P with similar lateral dimensions (2.5–4.0 nm) and thickness (1.5–2.0 nm). However, FTIR reveals that the VPQDs have different oxygen-containing functional groups in different solvents (Fig. 25b), which can stabilize the edge-dangling phosphorus bonds of VPQDs during synthesis. This modification affects the bandgap size of VPQDs, and thus their fluorescence properties. By functionalizing the edge-dangling phosphorus bonds with different reaction solvents, fluorescence from VPQDs can be adjusted from 400 nm (3.10 eV) to 536 nm (2.31 eV) to span the violet to green spectrum.330 The VPQDs prepared in protic solvents (IPA, H2O, and EA) exhibit shorter emission wavelengths (<430 nm) than those prepared in aprotic solvents (DMF, NMP, and DMI), suggesting larger bandgaps for VPQDs prepared in protic solvents (Fig. 25c and d).330 Theoretical calculations show that different functional groups can alter the HOMO and LUMO positions of VPQDs. In protic solvents, hydroxyl groups are ionized, leading to the formation of P–O bonds between the O atoms of solvent molecules and the edge-dangling phosphorus bonds. This results in significant changes in the HOMO–LUMO bandgaps. Functionalizing the edge-dangling phosphorus bonds with different reaction solvents to adjust the bandgap properties is a simpler and more feasible method than traditional doping and particle size control methods, thereby expanding the potential optoelectronic applications of the VPQDs.
 | ||
| Fig. 25 Synthesis and characterization of VPQDs in various solvents. (a) Scheme of the synthesis of VPQDs with tunable bandgaps by using different solvents. (b) Fourier transform infrared spectroscopy analysis of VPQDs in different solvents. (c) Fluorescence emission spectra of VPQDs in protic solvents and (d) in aprotic solvents. The excitation spectrum is represented by the dotted line, and the emission spectrum is represented by the solid line. Reproduced with permission from ref. 330 Copyright 2022 American Chemical Society. | ||
4.5 Synthesis of blue phosphorus
Since Zhu et al. predicted the existence of blue-P using DFT calculations,49in silico research on the structure and properties of this allotrope has been intense. However, to confirm theoretical proposals of blue-P, it is necessary to explore the practical properties through experiments. One method to synthesize 2D inorganic materials with precise control over the deposition rate, composition, and thickness is molecular-beam epitaxy (MBE).331,332 This technique involves depositing a directed beam of atoms or molecules onto the substrate in an ultrahigh vacuum environment. The beam of atoms or molecules is generated by heating the source in a chamber and the substrate is typically heated to promote the growth of the thin film. MBE has been the primary method for the synthesis of atomically layered blue-P. However, synthesis of blue-P bulk crystals has not been achieved so far.In 2016, Chen's group reported the growth of single-layered blue-P on Au(111) using MBE under ultrahigh-vacuum conditions.52 They deposited the black-P precursor on a heated Au(111) surface (230 °C) to produce a highly organized hexagonal superstructure of blue phosphorene on Au(111). Low-temperature scanning tunneling microscopy (STM) shows that this structure consists of a dark center surrounded by six equilateral triangles (Fig. 26a) with an average distance of 14.7 Å between the dark centers. DFT calculations show that the hexagonal surface structure is derived from the 4 × 4 blue-P supercell, which highly matches with the 5 × 5 Au(111) supercell. When the precursor is annealed at a lower temperature of 150 °C for 60 minutes, phosphorus clusters with a regular geometric structure are formed on the substrate surface instead of the blue-P layer. The edges of the phosphorus clusters are found to exhibit a preferential orientation towards the [110] direction of the Au(111) surface.52 STM cross-line scanning reveals that the height of the monolayer phosphorus cluster was 2.3 Å, which is consistent with the height of monolayer blue-P. Therefore, the growth of blue phosphorene entails the initial formation of triangular phosphorus clusters at a lower temperature followed by gradual merging to form a blue-P monolayer at a higher temperature.
 | ||
| Fig. 26 Synthesis and characterization of blue phosphorene. (a) High-resolution STM image of single-layered blue-P on Au(111). Reproduced with permission from ref. 52. Copyright 2016 American Chemical Society. (b-c) STM images of single-layered blue-P on the tellurium layer. Reproduced with permission from ref. 145. Copyright 2017 American Chemical Society. (d) STM images of blue-P grown on Au(111) before and after Si intercalation. Reproduced with permission from ref. 334. Copyright 2020 American Chemical Society. (e) STM image of the two different phosphorus phases (purple and red domains). Reproduced with permission from ref. 336. Copyright 2021 IOP Publishing. | ||
The substrate is critical in the MBE growth of blue phosphorene and regulating the interactions between the substrate and phosphorus source is necessary to form the desirable structures.333 Chen's group has developed a two-step deposition strategy to grow quasi-free-standing single-layered blue-P on tellurium (Te) functionalized Au(111).145 In this approach, a well-defined tellurium layer is deposited on Au(111) as a buffer layer, followed by the deposition of phosphorus onto the Te/Au(111) surface at 250 °C by evaporating the black-P precursor at 290 °C.145 The single-layered blue-P film has a low degree of contact with the underlying tellurium layer (Fig. 26b and c), indicating that inserting Te atoms between the phosphorus and Au surface can reduce their interaction. Alternatively, intercalation is a powerful technique to modify interfacial interactions by in situ introduction of an atomic layer between the surface and substrate. The same group has synthesized a pure blue-P monolayer by silicon intercalation (Fig. 26d).334 In contrast to the tellurium-layer intercalation described above, a blue-P–Au alloy is first formed by directly depositing the black-P precursor on pure Au(111). Si is then evaporated and intercalated beneath the blue-P–Au alloy to form a gold silicide buffer layer. This results in P–Au bond breakage and re-establishment of P–P bonds between nearby blue-P nanoclusters. These nanoclusters eventually fuse into a solid 1 × 1 blue-P monolayer on the buffer layer. The band structure related to the blue-P–Au alloy disappears and a new feature corresponding to 1 × 1 blue-P appears. Angle-resolved photoemission spectroscopy shows that the band maximum at the Γ point is about −1.2 eV with a parabolic shape, consistent with the theoretical band characteristics of the free-standing blue-P monolayer at the Γ point.334
The choice of the substrate is also crucial to blue phosphorene growth, and several experiments have explored different substrates. The Ag(111) surface is a fundamental substrate for the synthesis of a variety of elemental materials such as silicene, germanene, and borophene. Yang et al. used the Ag(111) substrate and high-quality bulk red-P as the precursor to produce phosphorus films consisting of regular 2D blue phosphorene clusters with a buckled 1 × 1 lattice.335 The average lattice constant of this film is 3.08 Å, which is smaller than the theoretical lattice constant of free-standing blue phosphorene (3.28 Å) but contradicts the lattice constant of Ag(111) (2.88 Å).335 This stems from compression of the phosphorus domains and formation of twisted blue phosphorene clusters due to the lattice mismatch with the Ag(111) substrate. Schaal et al. found that growing phosphorus on the Au(100) surface forms two phases depending on the phosphorus coverage (Fig. 26e).336 A low-density 2 × 2 phosphorous superstructure is observed in the low coverage regime. By increasing the phosphorus amount, the 2 × 2 superstructure vanishes and a roughly hexagonal structure emerges. The lattice constant of the latter phase (3.4(2) Å) resembles that of free-standing blue-P (3.33 Å).49 However, the unit cell angle of this structure is different from 120°, indicating that it is not completely hexagonal. The domains in this structure are extended, likely as a consequence of on-line epitaxy and strain occurring in diverse directions.337 Some theoretical studies on the growth of blue phosphorene on different substrates have also contributed to our understanding of the growth process.338–341
The most promising method to prepare atomically layered blue-P is MBE-based evaporative thermal decomposition of black-P and deposition on selected and highly ordered single-crystal metal surfaces.342 This approach produces a complete structure of blue phosphorene with relatively low total energy required for the atomic arrangement. However, the interfacial behavior between phosphorus and substrate requires further experimental investigation. Moreover, the MBE technique involves a sophisticated experimental procedure requiring precise temperature control and ultrahigh vacuum conditions,343 thereby hindering the cost-effective and large-scale production of blue-P. Currently, the synthesized blue phosphorene remains on the nanoscale level in terms of lateral size. Top–down approaches, such as mechanical or liquid exfoliation, can be utilized to scale up the manufacture of blue phosphorus/phosphorene, but producing blue-P bulk crystals remains a challenge.
4.6 Synthesis of other phosphorus allotropes
The discovery of new phosphorus allotropes is promising but challenging, requiring a combination of experimental synthesis and theoretical calculations. Researchers have gained a better understanding of phosphorus clusters that constitute EPMs, and theoretical studies have predicted a variety of energetically supported allotropes such as phosphorus fullerenes and nanotubes,76 crimson phosphorus,77 Haeckelite phosphorus,344 and phosphorene derivatives.345–347 However, despite these predictions, most of these allotropes are still in the theoretical stage lacking effective experimental synthetic methods. This section focuses on the experimental synthesis of three uncommon EPMs, namely, phosphorus nanorods, greenish phosphorus, and one-dimensional phosphorus nanostructures confined within carbon nanotubes.At the dawn of the 21st century, several inorganic chemists uncovered the potential of copper halides as a unique synthetic tool for the creation of compounds featuring novel phosphorus-based structures.348–350 This was achieved by reacting a stoichiometric mixture of copper halides and elemental phosphorus, which led to the synthesis of polymeric phosphorus chains that were complexed with copper halide adducts. Leveraging these studies, Pfitzner et al., in 2004, successfully extracted pure phosphorus nanorods that displayed deep red/brown colors from two distinctive adducts, (CuI)8P12 and (CuI)3P12.351 These allotropes were identified by their unique repeating units, [P8]P4(4)[ and [P10]P2[, respectively.
After the prediction of blue-P in 2014,49 a new phosphorus allotrope called green-P was predicted in 2017.50 However, due to its complicated structure, no experimental synthesis of green-P has been reported to date. Nevertheless, it is noteworthy that “greenish phosphorus” on carbon substrates was prepared by the CVD method in 2021 with red-P being the source which was encapsulated in a quartz tube with a glassy carbon substrate under a vacuum of 0.1 MPa.53 The mixture is heated to 480 °C in a tube furnace to sublimate phosphorus and then cooled to 260 °C to generate greenish phosphorus. The quartz tube holding red-P and the glassy carbon substrate before and after deposition is shown in Fig. 27a. Many nanoparticles are deposited on the substrate, and most of the phosphorus particles show a diameter of ∼100 nm, while a few are bigger with a diameter of about 700 nm (Fig. 27b–d). By sonication or scraping, the greenish phosphorus is hardly removed from the substrate, demonstrating strong interactions between phosphorus and the substrate. Other substrates such as carbon paper and silicon wafer have been investigated for the synthesis of greenish phosphorus. Greenish phosphorus can be deposited on carbon paper but not the silicon wafer. The partial pressure and edge structure of carbon materials plays essential roles in the production of this allotrope. The Raman spectra, XRD patterns, and lattice fringes observed by TEM confirm that greenish phosphorus has an atomic structure distinct from those of existing phosphorus allotropes including amorphous red-P, fibrous-P, black-P, and blue-P. Although the synthesis of greenish phosphorus on carbon substrates is promising, some questions remain unanswered. The structural analysis of greenish phosphorus is incomplete, and there is a lack of detailed analysis of the grazing incidence XRD data. Moreover, the name “greenish phosphorus” is given based on the observed color of the materials on the substrate, but this color may be a structural color caused by thin film interference.352 Therefore, additional structural characterization is necessary to establish that this is indeed a new phosphorus allotrope.
 | ||
| Fig. 27 Synthesis and characterization of greenish phosphorus. (a) Photograph of the sealed quartz tube before and after the deposition of greenish phosphorus onto glassy carbon. (b) Photograph, (c) optical microscopy image, and (d) SEM image of greenish phosphorus on glassy carbon. Reproduced with permission from ref. 53. Copyright 2021 Royal Society of Chemistry. | ||
Carbon nanotubes (CNTs) have been shown to be efficient nanoreactors to synthesize nanostructured materials for confined and specific orientation material growth. The type of phosphorus precursor and the wall layer and inner diameter of the CNTs significantly influence the nanostructured phosphorus. Using white-P as a precursor, Hart et al. have polymerized melted P4 molecules inside single-walled CNTs at 50 °C to form a single-stranded phosphorus zigzag chain (Fig. 28a–e).124,353 This polymerized chain may be an intermediate between white-P and red-P referred to as “pink phosphorus”,354 although its true color remains unknown. Zhang et al.355 prepared sp3 hybridized ring-shaped phosphorus nanostructures inside multi-walled CNTs using red-P as a precursor by the CVT method at 500 °C. They observed a ring with a diameter of 5.30 nm in a multi-walled CNT with an inner diameter of 5.90 nm, comprising 23 P8 and 23 P2 units with 230 phosphorus atoms, and a distance of 6.4 Å between adjacent rings (Fig. 28f and g).355 Under similar reaction conditions, they obtained a square columnar phosphorus structure with an intersectional size of less than 1.4 nm inside single-walled CNTs with an inner diameter of ∼1 nm (Fig. 28h and i), while planar zigzag black-P nanoribbons were formed inside multi-walled CNTs with an inner diameter of ∼4.1 nm (Fig. 28j and k),356 indicating that the host size of the CNTs plays a more significant role than the synthesis conditions in determining the structure of the encapsulated phosphorus.
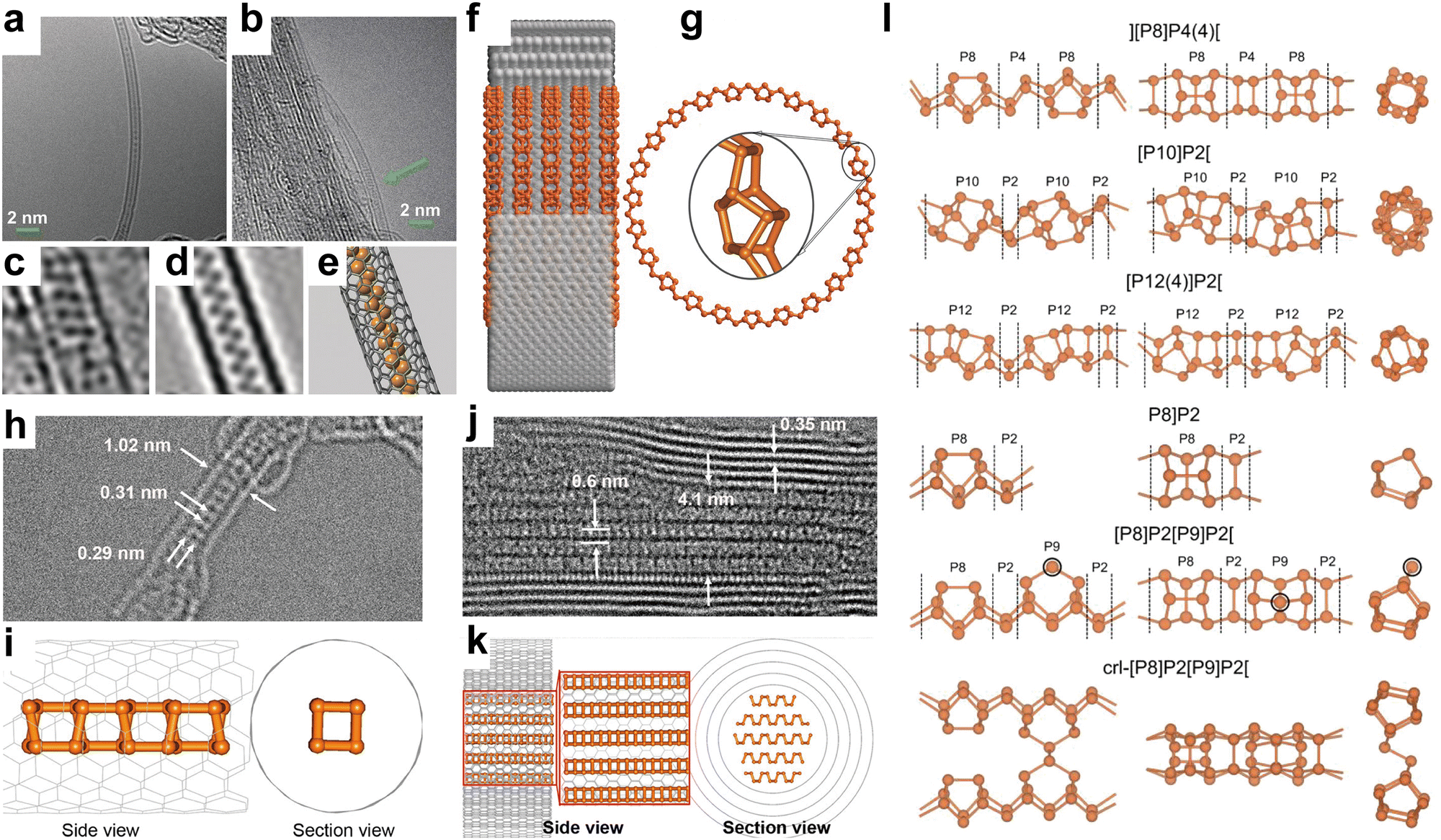 | ||
| Fig. 28 Synthesis and characterization of 1D phosphorus nanostructures confined within CNTs. (a and b) High-resolution TEM images of polymerized P4 chain within single-walled CNTs. (c) Noise-filtered high-resolution TEM image of the region highlighted in (b). (d) Simulated high-resolution TEM image and (e) the corresponding atomic structure of a polymerized P4 chain within a single-walled CNT. Reproduced with permission from ref. 124. Copyright 2017 John Wiley and Sons. (f) Structural model of ring-shaped phosphorus inside a multi-walled CNT. (g) Side view of ring-shaped phosphorus. The inset shows the repeating units. Reproduced with permission from ref. 355. Copyright 2017 John Wiley and Sons. (h) Negative spherical aberration imaging and (i) structural model of square columnar phosphorus inside a single-walled CNT. (j) Negative spherical aberration imaging and (k) structural model of planar zigzag Black-P nanoribbons inside a multi-walled CNT. Reproduced with permission from ref. 356. Copyright 2020 American Chemical Society. (l) Side, top, and end views of six potential phosphorus chain types. Reproduced with permission from ref. 357. Copyright 2022 American Chemical Society. | ||
Identification of motif fragments of one-dimensional red-P chains confined in CNTs is crucial to understand the relationship between the host size of CNTs and phosphorus nanostructures. Recently, Rybkovskiy et al.357 have obtained one-dimensional red-P-based chains in intermediate-diameter (1.6–2.9 nm) single-walled CNTs, and identified the main phosphorus clusters comprising the nanochains by combining experimental characterization and theoretical calculations. By screening six potential phosphorus chain types based on previously reported fibrous-P, (CuI)nPm crystals and free-standing vdW bonded chain crystals (Fig. 28l), they have simulated the associated phosphorus using DFT calculations to match the product morphology obtained from high-resolution TEM observations. The obtained one-dimensional red-P-based chains in CNTs are derived from a mixture of fibrous-P building blocks P8]P2 and crl-[P8]P2[P9]P2[. This chain can be the most common nanostructure for encapsulating red-P within CNTs with inner diameters of at least ∼1 nm and represents a new phosphorus phase. Additionally, based on previous reports and their findings, a phosphorus phase diagram is constructed to illustrate the behavior in 1D channels with varying diameters. In narrow channels with diameters less than 0.8 nm, phosphorus prefers to form zigzag chains. In the diameter range of 0.8–1 nm, tetrahedral P4 cages and their polymeric derivatives are formed.124,353 As the diameter increases beyond 1 nm, a thermodynamically stable single P8]P2 chain is confined within the larger cavity, which then forms bundles of P8]P2 chains and paired cross-linked counterparts as the cavity size increases.357 In larger cavities, [P8]P2 nanorings are formed possibly due to different synthesis conditions and the natural curvature of the P8]P2 chains.355 This study is of great significance for the predictable growth of phosphorus materials confined within tube-related nanostructures.
The future trend in synthesizing new phosphorus allotropes involves integrating nanotechnology with artificial intelligence-based theoretical calculations. Specifically, machine learning-driven theoretical calculations can accelerate the exploration of phosphorus motif fragments and enable the assembly of diverse architecture types based on identified phosphorus cluster configurations obtained from experiments.56 After the desired nanostructures are designed, specific cavity nanoreactors such as CNTs, metal-organic frameworks (MOFs), and covalent organic frameworks (COFs) with various dimensions are selected for the confined growth of nanostructures. This synthetic route is expected to facilitate the experimental synthesis of more phosphorus allotropes and requires exploring diverse phosphorus phase diagrams for different confined materials.
In summary, this section has discussed the synthesis of each EPMs across various dimensions. Looking ahead, efforts should be directed towards optimizing reaction conditions for the production of high-quality single crystals and large-scale thin films, and establishing reliable synthesis methods for low-dimensional structures such as phosphorene. Despite the multitude of synthesized allotropes, only a few, notably white-P and amorphous red-P, have reached industrial-scale production of thousands of tons. This underscores the urgency and importance of enhancing growth techniques for black-P, with the goal of achieving ton-level production in a single batch, which would significantly propel the evolution of the future phosphorus chemical industry.
5. Applications of elemental phosphorus materials to sustainable energy and environment
Growing concerns about energy sustainability and security arise from the rapid depletion of fossil fuels and their adverse environmental effects,358 and developing novel, stable, and efficient materials for green energy exploitation, storage, and environmental remediation is crucial.68,359,360 EPMs with diverse microstructures and nanostructures have emerged as versatile toolkits offering a range of application possibilities. The electron-rich nature of EPMs facilitates interactions with a variety of metal and non-metal materials, promoting the development of advanced composite systems. These elemental phosphorus-based materials exhibit exciting and tunable physicochemical properties including bandgaps, carrier mobility, and ion absorption. Consequently, EPMs demonstrate significant potential in applications such as chemical sensing, ion storage, photo(electro)catalysis, and energy conversion (Fig. 29). In this section, we systemically discuss the functional performance of EPMs and their derivatives in applications of energy conversion, energy storage, and environmental remediation. For each type of application discipline, the analysis of reaction mechanisms, the unique advantages of EPMs in specific scenarios, and strategies for enhancing material performance are emphasized. | ||
| Fig. 29 Diverse applications of elemental phosphorus-based materials in the fields of energy conversion, energy storage, and environmental remediation. | ||
5.1 Energy conversion applications
Photocatalysis is a process in which the photoreaction is accelerated in the presence of a semiconductor photocatalyst.369 The semiconductor absorbs solar photons and converts the reactant into the final product without depletion.370 The overall mechanism of photocatalysis involves three steps. First, solar photons excite electrons in the semiconductor, allowing them to move from the valence band to the conduction band, generating charge-negative electron (e−) and charge-positive hole (h+) pairs. Second, the photoexcited electrons and holes move to the semiconductor surface, where they act as redox agents. Third, the transfer of electrons leads to the formation of superoxide radicals (˙O2−) or initiates reduction events with a corresponding reduction potential. Hole transfer, on the other hand, generates hydroxyl radicals (˙OH) or initiates oxidation events with an appropriate oxidation potential. The two critical steps in a photocatalytic process are light absorption and charge carrier excitation, and the application of this process in energy conversion is largely dependent on the light radiation intensity and the semiconductor's bandgap. To achieve photocatalytic hydrogen generation, the semiconductor bandgap should be greater than 1.23 eV with a suitable conduction band position for the H+/H2 potential. Several common semiconductors such as TiO2 and MoS2371,372 have been reported for photocatalytic water splitting to generate hydrogen. However, as these metal oxides consist of rare elements with relatively high manufacturing costs, their widespread industrial use is restricted. Moreover, these semiconductors have wider bandgaps (>3.2 eV), resulting in the primary absorption of ultraviolet light (<400 nm), which corresponds to only ∼4% of the solar spectrum, leaving a significant portion of the solar spectrum unused. Consequently, these disadvantages significantly limit the practical applications of conventional semiconductors and necessitate the development of innovative and cost-effective photocatalysts for hydrogen production.
EPMs are promising materials in the mono-elemental family, with the potential to serve as metal-free photocatalysts for hydrogen generation. First, phosphorus is one of the most earth-abundant elements on the earth,373,374 and so the production cost of phosphorus-based catalysts is relatively low. Second, EPMs have narrow bandgaps (<2.7 eV), making them suitable for absorbing solar photons primarily in the visible light region and even down to the near-infrared region, with their conduction band minimum position being more negative than the water reduction potential. Third, EPMs have several layered allotropes with tunable bandgaps, allowing the selection of different allotropes in bulk or few-layered nanosheet forms for specific photocatalytic situations. Apart from black-P, amorphous red-P, fibrous-P, violet-P, and blue-P have all demonstrated their viability as candidates for photocatalytic hydrogen generation, both theoretically and experimentally.21,23,375–377Table 2 summarizes the progress made in EPMs and EPM-based systems for photocatalytic hydrogen generation.
| Allotropes | Materials | Methodology | Light source | H2 generation rate | Ref. |
|---|---|---|---|---|---|
| Black phosphorus | BP-BM | Ball milling | 300 W Xe lamp with a 420 nm cut-off filter | 512 μmol h−1 g−1 | 213 |
| BP nanosheets and nanoparticles | Liquid phase exfoliation | 300 W Xe lamp with a 420 nm cut-off filter | 64 μmol from the two cycles fornanosheets/45 μmol h−1 g−1 for nanoparticles | 381 | |
| BPQDs/g-C3N4 | Sonication approach | 300 W Xenon lamp with a 420 nm cut-off filter | 271 μmol h−1 g−1 | 382 | |
| Ni2P/BP | Solvothermal process | 150 W Xe arc lamp l | 406.08 μmol h−1 g−1 | 383 | |
| BP-10000/MoS2 | Solvothermal method | 300 W Xenon lamp with a UV cutoff filter (λ > 420 nm) | 1286 μmol h−1 g−1 | 384 | |
| BP/CN | High-energy ball milling | Blue LED lamp (λ = 440–445 nm) | 786 μmol h−1 g−1 | 385 | |
| FPS/CS | Mechanical mixing | 300 W Xenon arc lamp with a UV-cutoff filter (λ ≥ 420 nm) | 11192 μmol h−1 g−1 |
386 | |
| BP/PCN-HKM | Ultrasonication and wet-chemical self-assembly | 300 W Xenon lamp with an optical filter (λ > 400 nm) | 7380 μmol h−1 g−1 | 387 | |
| BP/MBWO | Sonication | 300 W Xenon lamp | 21042 μmol g−1 |
388 | |
| Amorphous red phosphorus | URP | Liquid exfoliation | 300 W Xe lamp with a UV-cut 420 nm filter | 180 μmol h−1 g−1 | 293 |
| RP PNS | Solvothermal treatment-coupled ultrasonic exfoliation | 300 W Xe lamp | 1169.4 μmol h−1 g−1 | 294 | |
| Red-P/g-C3N4 | Solid state annealing | 300 W Xenon lamp with a UV cut-off filter (λ > 420 nm) | 310 μmol h−1 g−1 | 389 | |
| Hierarchical P/YPO4 hollow microspheres | Hydrothermal method | 300 W Xenon lamp with a 400 nm cut-off filter | 65 μmol h−1 g−1 | 390 | |
| g-C3N4/Red-P hybrid nanosheets | Sono-chemical treatment | 300 W Xenon lamp with a 420 nm cut-off filter | 1691 μmol h−1 g−1 | 391 | |
| Co2P/RP | Hydrothermal method | 300 W Xe lamp irradiation | 5992.5 μmol h−1 g−1 | 392 | |
| Fibrous phosphorus | Micro-fibrous P/SiO2 and smashed-fibrous P | CVD and ultrasonication | 300 W Xenon lamp | 633 and 684 μmol h−1 g−1, respectively | 393 |
| Red P/g-C3N4 | One-step CVD | 300 W Xenon lamp equipped with L40 cutoff filter | 2565 μmol h−1 g−1 | 394 | |
| Fibrous phosphorene | Ultrasonication | 300 W Xenon lamp | 1021 μmol h−1 g−1 | 131 | |
| Violet phosphorus | Violet phosphorus | CVT | 300 W Xenon lamp | 553 μmol h−1 g−1 | 395 |
| CRPMR | CVT | Solar simulator-300 W Xenon lamp | 324.2 μmol h−1 g−1 | 396 | |
| PCN@HP | CVD | 300 W Xe lamp with an AM 1.5G filter as simulated sunlight (λ > 300 nm) or a 420 nm cutoff filter as visible light (λ > 420 nm) | 33.2 and 17.5 μmol h−1 g−1 for simulated sunlight and visible light, respectively | 397 | |
Although theoretical studies suggest that pristine bulk EPMs may serve as photocatalysts for water splitting,75,378,379 their inadequate catalytic efficiency limits the competitiveness compared to conventional photocatalysts. This is due to their narrow bandgaps, limited catalytic sites, and elevated recombination of photogenerated electron–hole pairs. As discussed in the electronic property section, exfoliation of EPM bulk crystals into few-layered nanosheets or phosphorene leads to a positive shift in the valence band and a negative shift in the conduction band, resulting in a significant increase in the bandgap. This promotes the efficient generation of strongly oxidative holes, preventing undesirable photoinduced charge pair recombination. In addition, the exfoliation process provides a large surface area and increases the molecule absorption space and active sites. As a result, low-dimensional EPMs are becoming increasingly popular for photocatalytic hydrogen generation. Zhu et al. conducted a systematic study of the visible-light photocatalytic efficiency of black-P bulk powder and nanosheets and found that black-P nanosheets exhibit an 18-fold greater hydrogen generation activity compared to the bulk form ref. 205. It is noteworthy that anhydrous LiOH powder is added during the synthetic process, and the photocatalytic hydrogen generation rate of black-P nanosheets in the presence of LiOH is 512 μmol h−1 g−1, which is greater than that of bulk black-P (28 μmol h−1 g−1) and well-known metal-free photocatalyst g-C3N4 (107 μmol h−1 g−1).380 After illumination for 9 hours with 420 nm homochromatic light, the apparent quantum yield of hydrogen generation is calculated to be approximately 0.47%. However, another study also using black-P nanosheets for photocatalytic hydrogen generation showed a lower generation rate.213,381 This discrepancy may arise from the different fabrication methods (liquid exfoliation and ball milling, respectively) and addition of LiOH, which terminates the reactive black-P nanosheet edges and provides a stable structure for photocatalytic reaction.
Amorphous red-P boasting a lower manufacturing cost and well-established production technique compared to black-P has attracted a considerable interest due to its great photocatalytic characteristics. Chen and co-workers investigated the photocatalytic hydrogen generation efficiency of ultra-thin amorphous red-P nanosheets produced by liquid exfoliation.293 The nanosheets exhibited a good hydrogen generation activity of 180 μmol h−1 g−1, which is 2.88 times higher than that of bulk red-P. The apparent quantum efficiency of amorphous red-P nanosheets is approximately 0.28%. Cycling stability measurements show that the amorphous red-P nanosheets retain 89.1% of their original catalytic activity after three irradiation cycles, indicative of great chemical stability. The primary advantage is the low cost of the raw materials and environmentally friendly preparation process, boding well for commercializing. Zhang et al.294 used a probe ultrasonication technique to synthesize porous red-P nanosheets with a significantly higher photocatalytic hydrogen generation rate of 1169.4 μmol h−1 g−1. The BET-specific surface area of the porous nanosheets reach 84.37 m2 g−1 and this vast surface area significantly increases the number of reactive sites and molecule adsorption areas, leading to higher hydrogen production efficiency. Furthermore, other red-P derivatives such as fibrous phosphorene (1021 μmol h−1 g−1),131 and violet-P microbelts (553.4 μmol h−1 g−1)396 have also delivered excellent performance in photocatalytic hydrogen generation.
However, low-dimensional EPMs are not invincible, and they also face some challenges in photocatalytic hydrogen generation. Experimental findings show that the quantum efficiency of two-dimensional EPMs is relatively low due to the poor stability of pristine phosphorene. The electron-rich nature of the phosphorene surface and the presence of multiple unpaired lone pairs of electrons leads to the recombination of layers, preventing the materials from maintaining a long-term stability in the photocatalytic process. To overcome these issues, three potential strategies can be applied to improve the photocatalytic efficiency. First of all, chemical modification of phosphorene with hydroxyl (–OH) or pseudo-halogen (–CN and –OCN) functional groups can passivate the phosphorene surface and improve the durability.213,398 Second, phosphorene can be utilized as a matrix to load photocatalytically active electron mediators (Ag/Au nanoparticles)399,400 to produce co-catalytic effects in photocatalysis. Third, metal/nonmetal doping and heterostructure construction are powerful methods for regulating the electronic structure and mechanical strength of phosphorene.401,402
Heterostructures play an important role in promoting the spatial separation of electrons and holes and the photocatalytic activity of materials like black-P. Zhu et al. have designed a binary nanohybrid composed of black-P nanoflakes and graphitic carbon nitride nanosheets to investigate the photocatalytic hydrogen performance in the presence of methanol as a hole quencher.403 At the interface between black-P and graphitic carbon nitride, a type-I heterojunction is formed. Under light irradiation above 420 nm, graphitic carbon nitride is the main excited materials generating electrons and holes in the conduction band and valence band, respectively. Black-P serves as an electron acceptor and accepts electrons from the conduction band of adjacent graphitic carbon nitride, while the transferred holes in black-P are quenched by methanol (Fig. 30a). This process inhibits recombination of electron–hole pairs in graphitic carbon nitride. Electrons and holes in black-P recombine rapidly under irradiation (lifetime of less than 1 ps) to prevent black-P from functioning as a photocatalyst. However, this type-I heterojunction can produce hydrogen under light irradiation above 780 nm. Time-resolved diffuse reflectance spectroscopy indicates that the P–N coordination bond at the interface between Black-P and graphitic carbon nitride can act as a trap site for electrons and captures electrons in the conduction band of black-P to generate hydrogen. This strong interfacial interaction and effective charge transfer between black-P and graphitic carbon nitride improves the photocatalytic efficiency by inhibiting the recombination of photogenerated charges in black-P. This demonstrates that the interfacial P–N coordination bond plays a critical role in photocatalytic hydrogen generation in both visible and near-infrared regions. Zhu et al. constructed 2D black-P/BiVO4 heterostructures as a novel Z-scheme photocatalyst for overall water splitting.404 Under >420 nm light irradiation, the photocatalyst generates H2 and O2 yields of approximately 160 and 102 μmol h−1 g−1, respectively, without the need for any sacrificial agents or external bias. The staggered band positions of black-P and BiVO4 facilitate separation of charges, leading to the reduction and oxidation of water on black-P and BiVO4, respectively. In addition to binary heterostructures, researchers have constructed ternary nanostructures for photocatalytic hydrogen generation. Black-P–Au/La2Ti2O7 shows high efficiency as a photocatalyst over a broad spectrum of wavelengths spanning the ultraviolet to near-infrared regime.405 The optimal hydrogen generation rates of black-P–Au/La2Ti2O7 are 0.74 and 0.30 mmol h−1 g−1 at wavelengths longer than 420 nm and 780 nm, respectively. This work highlights that the strong surface plasmon resonance absorption of gold nanoparticles in the hybrid nanostructure can transform incident photon energy into plasma energy and transmit electrons to the semiconductor to induce charge separation (Fig. 30b), thus improving the photocatalytic performance. Furthermore, the strong optical absorption of black-P and plasma gold nanoparticles in the region from ultraviolet to near-infrared, along with the interfacial interaction of black-P and Au/La2Ti2O7, enables effective electron transfer to La2Ti2O7 by excited black-P and Au.
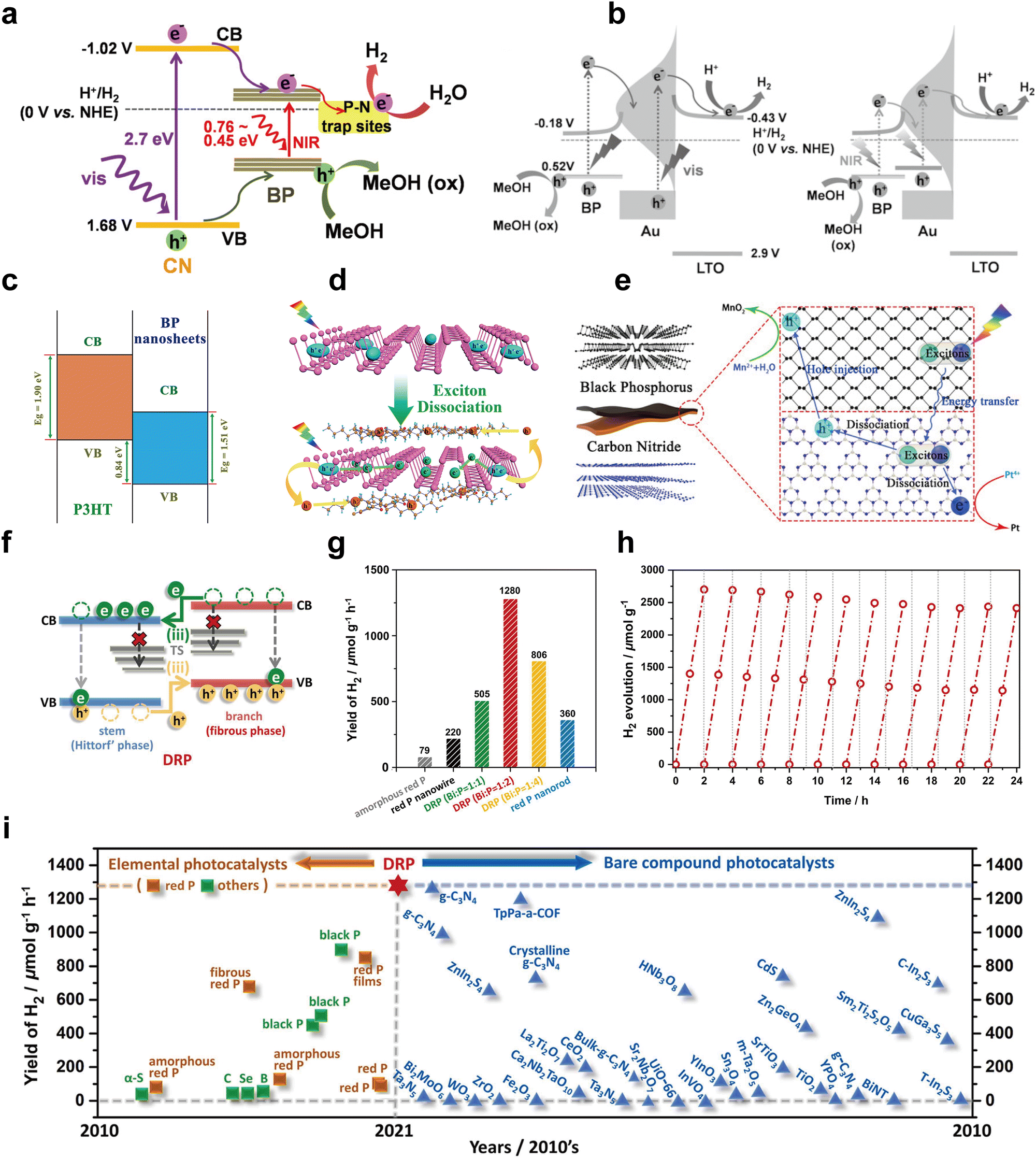 | ||
| Fig. 30 Applications of elemental phosphorus materials to photocatalytic hydrogen generation. (a) Proposed mechanism of photocatalytic hydrogen generation for the black-P/graphitic carbon nitride (CN) heterostructure under visible and near-infrared irradiation in the presence of methanol. Reproduced with permission from ref. 403. Copyright 2017 American Chemical Society. (b) Proposed mechanism of photocatalytic hydrogen generation for the black-P–Au/La2Ti2O7 (LTO) composite under visible (left) and near-infrared (right) irradiation in the presence of methanol. Reproduced with permission from ref. 405. Copyright 2017 John Wiley and Sons. (c) Band alignment of black-P/P3HT type-II heterojunctions. (d) Charge transfer at the heterojunction interface. Reproduced with permission from ref. 408. Copyright 2019 American Chemical Society. (e) Photoexcitation processes in black-P/carbon nitride heterojunctions. Reproduced with permission from ref. 410. Copyright 2021 John Wiley and Sons. (f) Proposed photocatalytic process of dendritic red-P (DRP). (g) Photocatalytic hydrogen evolution yield of DRP and red-P-based materials under visible light irradiation. (h) Photocatalytic stability of DRP. (i) Comparison of the visible-light photocatalytic hydrogen generation rate between DRP and other bare elemental/compound photocatalysts. Reproduced with permission from ref. 412. Copyright 2022 Elsevier. | ||
The low-dimensional black-P materials exhibit powerful multibody effects resulting in strong exciton that dominates the photoexcitation process.219,406,407 To enhance the photocatalytic effects of black-P, it is crucial to make reasonable use of these excitons. Xie's group has proposed a strategy to modulate exciton dissociation by heterojunction engineering,408 which combines black-P nanosheets with poly(3-hexylthiophene) (P3HT) leading to the construction of type-II heterojunctions (Fig. 30c). This strategy weakens the stability of excitons in black-P and promotes the dissociation and conversion to free carriers. The internal electric field generated at the heterojunction interface due to the difference in the band structure further promotes directional transfer of holes to P3HT and electrons to black-P (Fig. 30d), consequently increasing the output of the loaded carriers. Benefiting from this, the black-P/P3HT heterojunction shows efficient exciton dissociation and greatly improved photoresponsive properties of black-P such as a photocurrent switching ratio of approximately 18.3. Excitons can undergo resonant transfer between systems possessing compatible spin states and energy levels.409 In 2021, Xie's group proposed an exciton-mediated energy transfer strategy in heterojunction engineering for efficient infrared-light-driven photocatalysis.410 They constructed a black-P/polymeric carbon nitride heterojunction in which black-P acted as an infrared light absorber and transferred its energy to activate polymeric carbon nitride through exciton interaction (Fig. 30e). The excitons in carbon nitride separated into free charge carriers at the heterojunction interface, where holes were further transferred into black-P and electrons were transferred into carbon nitride to achieve great photocatalytic efficiency under infrared light irradiation.
The strategy has also been widely used for other EPMs, and Yu's group has made contributions in the preparation of crystalline red-P composites to promote sustainable application of low-cost and effective materials in photocatalysis. In 2016, they used different synthetic methods to prepare two kinds of fibrous-P-based materials, namely, micro-fibrous-P/SiO2 composites and smashed-fibrous-P crystals.411 These materials show stable and high hydrogen release rates of 633 μmol h−1 g−1 and 684 μmol h−1 g−1, respectively. The high photocatalytic efficiency is due to the high conduction band minimum of fibrous-P (−0.9 eV vs. NHE) and the favorable full contact of the small-size microstructure with water. One year later, they developed a crystalline red-P/g-C3N4 composite with a record-high hydrogen generation rate of 2565 μmol h−1 g−1.394 The apparent quantum yield of the red-P/g-C3N4 composite is 13.8% at 420 nm, which is about 3.5 times that of pristine g-C3N4 (3.9%).394 The great catalytic performance of the composite is attributed to the formation of chemical bonds between red-P (fibrous phase phosphorus) nanoparticles and g-C3N4, which reduces the number of structural defects in g-C3N4. This, in turn, inhibits the inactive charge capture and prolongs the lifetime of the active charge in g-C3N4, resulting in improved photocatalytic activity. Recently, Yu's group412 has developed a bismuth-catalyzed CVD method to prepare violet-fibrous hetero-phase dendritic structures from amorphous red-P for photocatalytic hydrogen generation (Fig. 30f). The hetero-phase dendritic catalyst shows stable and efficient photocatalytic activity such as a hydrogen generation rate of 1280 μmol h−1 g−1, which is about 16 times greater than that of amorphous red-P and exceeds those of many elemental and bare compound photocatalysts (Fig. 30g–i). The alignment of band structures between two crystalline phases and intimate heterojunction binding at the stem-branch intersection contributes to the construction of a well-organized structure that reduces the interfacial resistance, promotes charge transfer, and inhibits charge trapping and recombination. Other phosphorus-based composites have also been explored for photocatalytic hydrogen generation, for example, red-P/YPO4,390 fibrous-P/TiO2,413 violet-P/polymeric C3N4,414 blue-P/Mg(OH)2,415 blue-P/g-C3N4,416 and green-P/MoSe2.75
Photocatalytic CO2 conversion is more challenging than photocatalytic H2 generation. CO2 is a non-polar linear molecule with a symmetric structure, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy is 804.4 kJ mol−1 at 298 K, indicating that a large amount of energy is required to break the bond.423,424Eqn (1)–(5) show the electrode equations and potentials (V vs. NHE) for CO2 reduction to free radicals and typically produced compounds.425,426 Although CO2 can form a free radical by accepting one electron directly as shown in eqn (1), the reduction potential of −1.90 V is substantially negative, indicating a higher positive Gibbs free energy required, which is thermodynamically unfavorable. As a result, photocatalytic CO2 reduction generally occurs via a proton-coupled multielectron transfer process.425 The typical reduction products are methane, methanol, carbon monoxide, and formic acid (eqn (2)–(5)). In addition to producing photoexcited electrons and delivering them to adsorbed carbon dioxide molecules for activation and reduction, the photocatalyst also plays a crucial role in stabilizing the corresponding intermediates in their respective reduction pathways, thus increasing the reaction selectivity.427
O bond energy is 804.4 kJ mol−1 at 298 K, indicating that a large amount of energy is required to break the bond.423,424Eqn (1)–(5) show the electrode equations and potentials (V vs. NHE) for CO2 reduction to free radicals and typically produced compounds.425,426 Although CO2 can form a free radical by accepting one electron directly as shown in eqn (1), the reduction potential of −1.90 V is substantially negative, indicating a higher positive Gibbs free energy required, which is thermodynamically unfavorable. As a result, photocatalytic CO2 reduction generally occurs via a proton-coupled multielectron transfer process.425 The typical reduction products are methane, methanol, carbon monoxide, and formic acid (eqn (2)–(5)). In addition to producing photoexcited electrons and delivering them to adsorbed carbon dioxide molecules for activation and reduction, the photocatalyst also plays a crucial role in stabilizing the corresponding intermediates in their respective reduction pathways, thus increasing the reaction selectivity.427
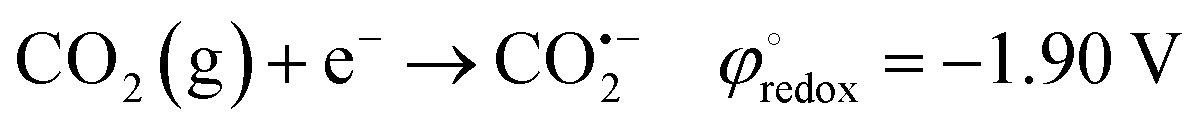 | (1) |
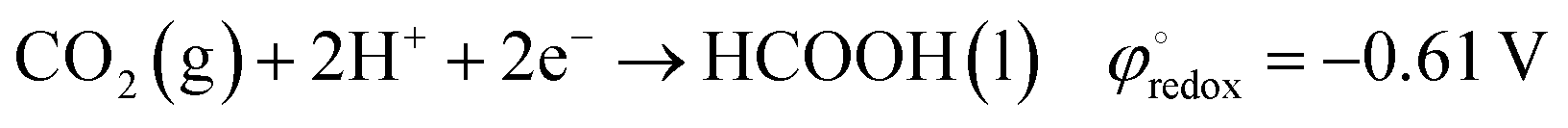 | (2) |
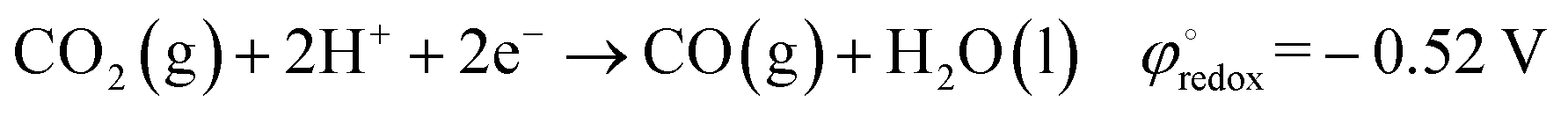 | (3) |
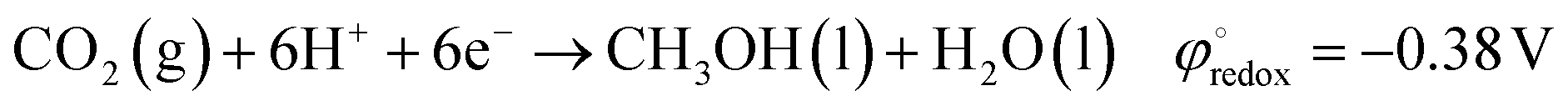 | (4) |
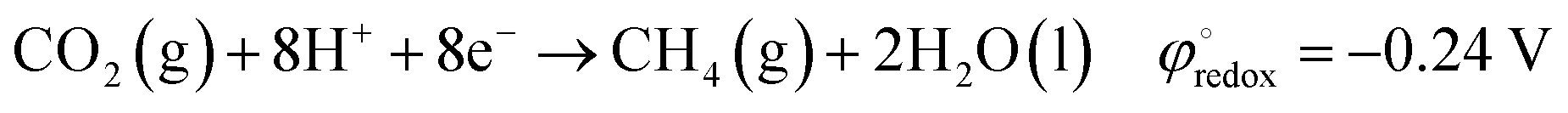 | (5) |
In 2020, Zhu et al. reported a hydroxyl-modified black-P monolayer showing outstanding photocatalytic efficiency, and it was capable of converting CO2 into CO with a generation rate of 112.6 μmol h−1 g−1.428 This value is four times that of bulk black-P, and comparable to that of 2D g-C3N4. Competing reactions leading to the production of by-products such as H2 are constantly in progress. However, hydroxyl-modified black-P exhibits a better reaction selectivity to CO (90.8%) than pristine black-P. This can be attributed to the electron-donating character of a hydroxyl group (Fig. 31a), resulting in the formation of modified black-P with a higher bandgap (∼2.2 eV) than the unmodified one (2.0 eV). This elevated bandgap makes black-P more accessible to the CO2/CO redox potential. In addition, hydroxyl-modified black-P has outstanding stability due to surface passivation, and its degradation rate is 8.8 times slower than the pristine black-P monolayer during light cycling measurements. This shows that an appropriate choice of electron-acquiring/electron-donating functional groups influences the electron distribution of the materials. Reasonable control over the reaction conditions can lead to the formation of elemental homojunction comprising mixed crystalline phases of red-P and black-P. Band alignment of red-P and black-P allows for the formation of a Z-scheme structure and introduction of another suitable semiconductor into the system results in the formation of a ternary dual Z-scheme system. Chai's group designed a dual Z-scheme homo-heterojunction of red-P/black-P@WO3,429 with great photocatalytic efficiency under visible light irradiation by converting CO2 into CH4 with a yield of 6.21 μmol g−1 within 6 hours, whereas the original components, red-P, WO3, and red-P/black-P, do not produce any CH4. In the dual Z-scheme system, red-P acts as a key bridge connecting black-P and WO3, forming a unified and synergistic ternary homo-heterojunction composite with the appropriate bandgap energy for CO2 reduction (Fig. 31b). Specifically, red-P has a higher conduction band position than black-P and WO3,430 indicating its more negative reduction potential. Under visible light irradiation, black-P, red-P, and WO3 generate charges in their respective conduction bands and valence bands. The photoinduced electrons in the conduction bands of black-P and WO3 follow the Z-scheme vectorial transfer pathway to reach the valence band of red-P, where they recombine with holes in red-P. This effective separation and directional transport of the photoinduced electrons and holes in the ternary system contribute to the continuous progress of the photocatalytic reduction reaction.
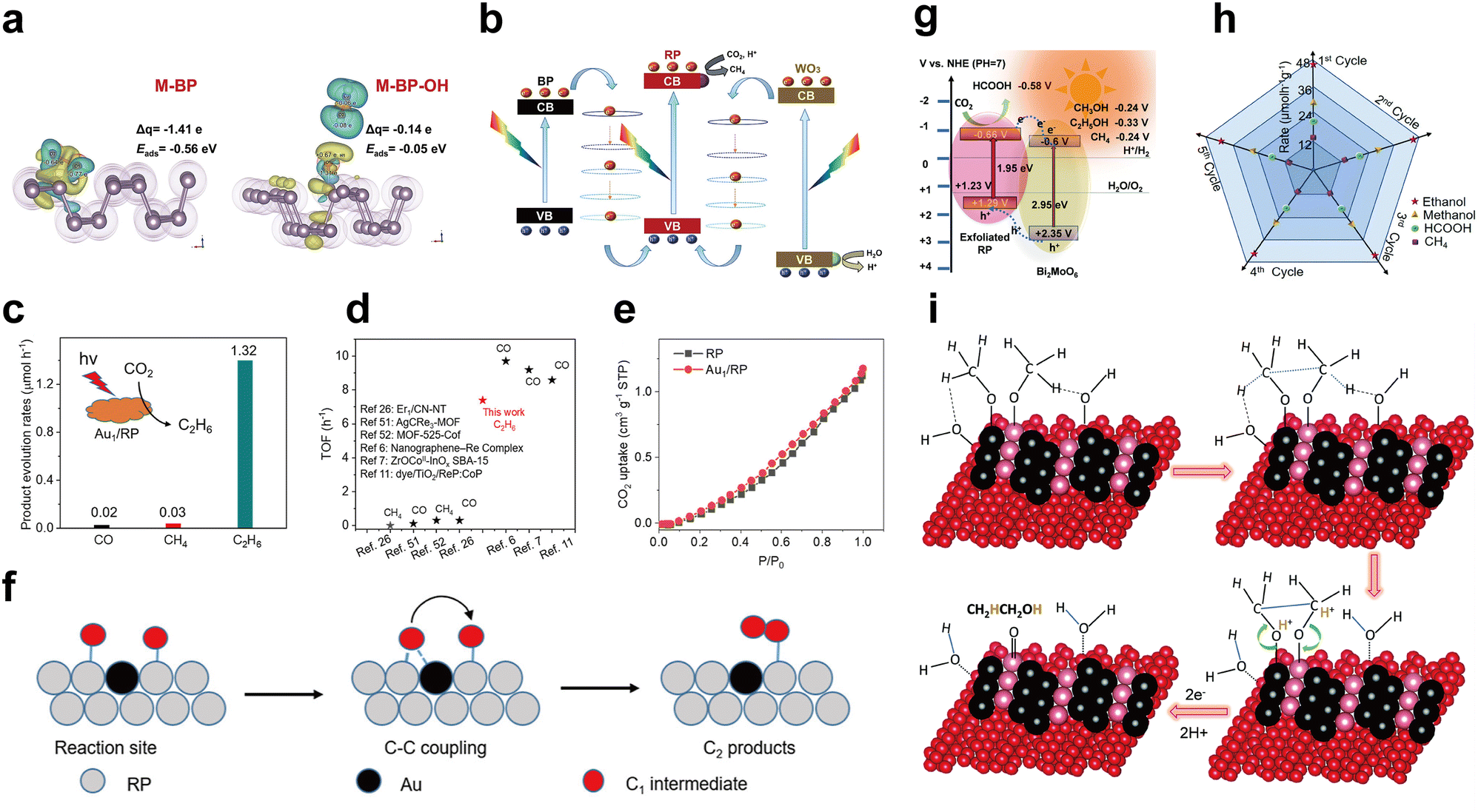 | ||
| Fig. 31 Applications of elemental phosphorus materials to photocatalytic carbon dioxide conversion. (a) Charge difference of O2 adsorbed on the black-P monolayer (M-BP) (left) and the hydroxyl-modified black-P monolayer (M-BP-OH). Reproduced with permission from ref. 428. Copyright 2020 Elsevier. (b) Scheme of the dual Z-scheme homo-heterojunction of red-P/black-P@WO3 for photocatalytic CO2 reduction. Reproduced with permission from ref. 429. Copyright 2022 American Chemical Society. (c) Photocatalytic CO2 reduction rates of single-atom Au/red-P (Au1/RP). (d) Comparison of TOF for photocatalytic CO2 reduction of single-atom Au/red-P and other photocatalysts. (e) CO2 adsorption curves of red-P and single-atom Au/red-P. (f) Proposed mechanism of single-atom Au/red-P for C–C coupling. Reproduced with permission from ref. 432. Copyright 2022 American Chemical Society. (g) Band structure alignment of the red-P/Bi2MoO6 heterostructure following the type-II mechanism. (h) The product formation rate of red-P/Bi2MoO6 in different cycles. (i) Proposed mechanism of photocatalytic ethanol production for red-P/Bi2MoO6. Reproduced with permission from ref. 434. Copyright 2022 Royal Society of Chemistry. | ||
Generating C1+ compounds in the photocatalytic reduction of CO2 is typically more challenging than producing C1 compounds (CO, CH4, and CH3OH). This process involves multiple electron and proton transfer reaction pathways, resulting in unsatisfactory catalytic efficiency and selectivity. Nonetheless, several C2 compounds such as ethanol are crucial industrial chemicals and potential energy storage materials, and it is imperative to produce them in a cost-effective and environmental-friendly manner. Photocatalytic reduction of CO2 to C2 compounds involves a C–C coupling process, which requires overcoming a larger energy barrier during the reaction.431 However, EPMs exhibit great potential for photocatalytic C2 compound production based on the above discussion. From the physical perspective, EPMs are semiconducting materials with relatively narrow bandgaps, making them efficient for light harvesting in the infrared-visible light range, and they possess suitable conduction band edges for CO2 photoreduction. From the chemical point of view, electron-rich EPMs can adsorb CO2 on the surface through Lewis acid–base interactions, interact with O in the molecule, promote cleavage of C–O bonds, and generate large C1 intermediates.432,433 These physical and chemical properties have recently been exploited by some research groups.
In 2022, Li's group designed a single-atom Au loaded red-P photocatalyst for efficient ethane production.432 The optimized single-atom Au/red-P catalyst exhibits a high ethane yield of 1.32 μmol g−1 h−1 without the need of any sacrificial agents. In contrast, the yields of CO and methane are only 0.02 and 0.03 g−1 h−1, respectively, indicating excellent selectivity (96%) for ethane production (Fig. 31c). The turnover frequency (TOF) of the catalyst is also noteworthy, reaching as high as 7.39 h−1, which exceeds those of many C1 compound conversion photocatalysts (Fig. 31d). Various characterization techniques have been employed to demonstrate that CO2 preferentially adsorbs on the surface of red-P (Fig. 31e) and provides active sites near each single-atom Au. This proximity effectively reduces the transfer distance of photoinduced electrons and C1 intermediates. The presence of single-atom Au aids the desorption and movement of the C1 intermediate at the phosphorus reactive site, followed by C–C coupling at another phosphorus site (Fig. 31f).432 Das et al. have developed a type-II red-P/Bi2MoO6 heterostructure photocatalyst capable of converting CO2 into ethanol under visible light irradiation and water conditions (Fig. 31g).434 The composite shows an excellent ethanol conversion rate of 51.8 mmol g−1 h−1, whereas pristine red-P shows no detectable ethanol production as it mainly produces methane and formic acid after light irradiation for 10 hours. To expand the applicability of the catalyst, researchers have investigated the sunlight-directed photoreduction performance of the optimized red-P/Bi2MoO6 composite. The results reveal an ethanol yield of 34.1 mmol g−1 h−1, which is 90.91% of that under Xe lamp irradiation. To evaluate the catalytic stability, the optimized composite undergoes five consecutive photocatalytic cycles and a continuous 30 hour catalytic test. The catalyst shows great recyclability and stability in CO2 photoreduction and no noticeable change in the photoactivity (Fig. 31h). In the catalytic process, transformation of the C1 intermediate is crucial for ethanol production. It has been found that using methane as a reactant does not lead to ethanol formation. However, a 300 mM methanol solution produces a high yield of 996.3 mmol h−1 g−1 of ethanol, indicating that methanol plays a crucial role as an intermediate in ethanol formation. A new ethanol formation mechanism has been proposed for the red-P/Bi2MoO6 heterostructure based on the kinetic isotope effects and in situ diffuse reflectance infrared Fourier transform spectroscopy, showing that one OCH3* on the Bi atom and another OCH3* on the Mo atom interact with the adjacent OH* groups on red-P and promote C–C coupling and C–H bond cleavage in OCH3.434 This leads to the formation of a C2H4O2* intermediate with protons adsorbed on O centers (Bi) and C centers (Mo–O), ultimately resulting in ethanol production (Fig. 31i). The red-P in the composite provides strong visible light harvesting capability and abundant active sites, forms a heterojunction interface with Bi2MoO6 nanoparticles, prevents recombination of photoinduced charges, and improves the charge transfer efficiency of the system.
The electrocatalytic HER mechanism is a multi-step reaction that typically takes place on the surface of the catalytic electrode layer.438 It involves three main steps (adsorption, reduction, and desorption) and requires either hydronium ions in acidic electrolytes or water molecules in basic media (Fig. 32a).439 The initial step in this process is the Volmer reaction (eqn (6) and (7)), in which a proton adsorbs at the active sites of the electrocatalyst and accepts an electron from the external circuit to form an adsorbed hydrogen atom (H*). Subsequently, a hydrogen molecule is generated through the Heyrovsky (eqn (8) and (9)) or Tafel (eqn (10)) routes, or both. In the Heyrovsky route, another proton migrates to the electrode surface to capture a second electron, which then combines with the adjacent H* to produce a hydrogen molecule. In contrast, in the Tafel route, two neighbouring H* interact on the electrode surface to generate H2. Finally, hydrogen molecules are released from the surface of the catalytic electrode. The electrocatalytic HER can be written as follows:435,440
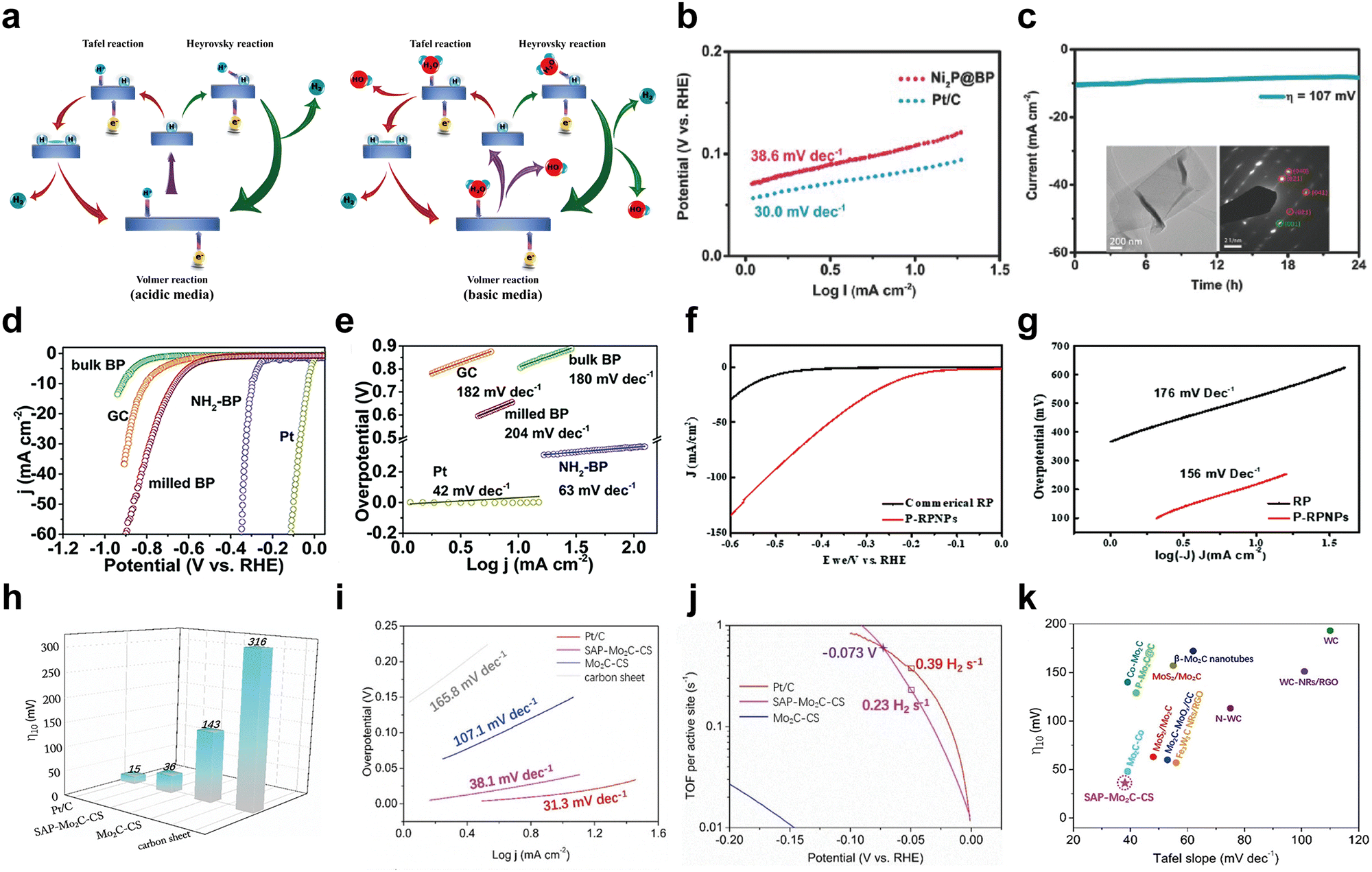 | ||
| Fig. 32 Applications of elemental phosphorus materials to electrocatalytic hydrogen evolution. (a) Scheme of the hydrogen evolution mechanism on the surface of a catalyst in acidic (left) and basic (right) media. Reproduced with permission from ref. 439. Copyright 2019 Elsevier. (b) Corresponding Tafel plots for Ni2P@black-P (BP) and Pt/C. (c) Time-dependent current density curves of Ni2P@black-P under an overpotential of 107 mV. The inset shows the TEM image and SAED pattern after continuous sweeps for 24 hours. Reproduced with permission from ref. 444. Copyright 2016 John Wiley and Sons. (d) Voltammetry of different materials for the electrocatalytic HER at a scan rate of 5 mV s−1 in 1 M KOH. (e) Tafel plots derived from (d). Reproduced with permission from ref. 446. Copyright 2018 Royal Society of Chemistry. (f) Voltammetry of porous red-P nanoparticles (P-RPNPs) and commercial red-P at a scan rate of 5 mV s−1. (g) Corresponding Tafel plots for porous red-P nanoparticles and red-P. Reproduced with permission from ref. 448. Copyright 2020 Royal Society of Chemistry. (h) Corresponding overpotentials at a current density of 10 mA cm−2 (η10), (i) Tafel plots, and (j) TOF plots of four electrocatalysts. (k) The η10vs. Tafel slope of SAP-Mo2C-CS compared to the reported TMC-based catalysts. Reproduced with permission from ref. 454. Copyright 2020 John Wiley and Sons. | ||
(1) Electrochemical hydrogen adsorption (Volmer reaction)
| H3O+ + M + e− → M – H* + H2O (acidic electrolyte) | (6) |
| H2O + M + e− → M – H* + OH− (alkaline electrolyte) | (7) |
(2) Electrochemical desorption (Heyrovsky reaction)
| H+ + e− + M – H* → H2 + M (acidic electrolyte) | (8) |
| H2O + e− + M – H* → H2 + OH− + M (alkaline electrolyte) | (9) |
(3) Chemical desorption (Tafel reaction)
| 2M – H* → H2 + 2M (acidic and alkaline electrolytes) | (10) |
Based on the analysis above, EPMs exhibit electron-rich properties, abundant active sites, and reasonable stability, boding well for the HER. Black-P has been extensively studied as an electrocatalyst or as a conductive scaffold with a large surface area. Luo et al.444 utilized a simple solvothermal approach for preparing a 0D-2D Ni2P@black-P heterostructure and demonstrated its excellent electrocatalytic activity for the HER. In the synthesis process, black-P nanosheets are used as precursors to react with nickel chloride, resulting in the formation of Ni2P nanocrystals that are monodispersed and incorporated in the black-P nanosheets to create a co-catalytic system. The electrocatalytic performance of Ni2P@black-P was evaluated using a conventional three-electrode setup with a 0.5 M sulfuric acid solution. The overpotential of Ni2P@black-P is only ∼107 mV at 10 mA cm−2, which is substantially lower than that of commercial Ni2P particles (∼311 mV) and pristine black-P nanosheets (600 mV). As shown in Fig. 32b, Ni2P@black-P shows a small Tafel slope (38.6 mV dec−1), which is comparable to that of a commercial Pt electrocatalyst (20 wt% Pt/Vulcan XC-72, 30 mV dec−1), and the entire electrocatalytic process follows the Volmer–Tafel pathway.444,445 Furthermore, the Ni2P@black-P electrode maintains a stable current density at an overpotential of 107 mV and a well-defined crystalline morphology even after continuous scanning for 24 hours (Fig. 32c). The high performance of Ni2P@black-P is related to the well-designed heterostructure which efficiently prevents Ni2P nanoparticles from agglomeration and black-P nanosheets from restacking, thereby increasing the surface area and abundance of active sites. Additionally, the presence of Ni2P tethered to the black-P surface modulates the charge carrier concentration and optimizes the HER kinetic rate. The electrocatalytic HER performance of NH2-functionalized black-P nanosheets in alkaline electrolytes is also outstanding as exemplified by an overpotential of 290 mV at 10 mA cm−2 (Fig. 32d).446 The Tafel slope of NH2-black-P is 63 mV dec−1 (Fig. 32e) indicative of the Volmer–Heyrovsky mechanism. Moreover, NH2-black-P exhibits good stability in the 12 hour chronoamperometric test, and the polarization curve remains nearly unchanged after 1000 consecutive cycles. Li et al.447 have recently employed a modified CVT approach to synthesize black-P bulk crystals cost effectively, which are peeled off into nanostructured sheets for use as an HER electrocatalyst. The improved electrocatalytic activity of black-P nanosheets derived from low-cost bulk crystals can be attributed to the presence of more edge defects than in high-cost black-P, abundant exposed active sites, and promoted charge transport. Compared to the traditionally high price of black-P bulk crystals (about $600 per gram) made from ultrapure red-P,93 there is potential to reduce the manufacturing costs of black-P and in fact, cost reduction by hundreds of times will promote widespread use of black-P-based electrocatalysts in industrial production.
Porous red-P nanoparticles have been reported as a potential metal-free electrocatalyst for the HER.448 Uniform porous red-P nanoparticles have been prepared by a simple one-pot injection method involving decomposition of the precursor PxIy and adding oleic acid for shape control. To evaluate the electrocatalytic performance, the nanoparticles are dissolved in toluene as ink and applied to enable red-P to stick to the surface of the working electrode. As shown in Fig. 32f, the porous red-P nanoparticles show an overpotential of 218 mV at a current density of 10 mA cm−2, which is substantially lower than that of commercial red-P (523 mV), indicating that the porous morphology improves the HER electrocatalytic efficiency. At this current density, the overpotentials of most metal-free electrocatalytic materials such as CNTs449 and N-S co-doped graphene450 are in the range of 200–425 mV,451 and porous red-P nanoparticles have excellent properties in this range, which are even comparable to metal catalysts such as MoS2 nanosheets with an overpotential of 205 mV.452 The Tafel slope is 156 mV dec−1 (Fig. 32g), suggesting that the overall reaction rate is controlled by diffusion.441,448 The long-term stability of porous red-P nanoparticles is confirmed in an acidic medium after 45 hours at a static potential of −0.5 V (vs. RHE) and the cathode current density remains constant even after 6000 cyclic voltammetry cycles from −0.6 to 0.2 V.
Single-atom catalysts have garnered significant attention in electrocatalysis due to their exceptional atom utilization efficiency and notable reaction selectivity.453 However, the majority of single-atom catalysts employ metal elements and the synthesis of non-metal single-atom catalysts is uncommon. Nevertheless, the electron-rich active sites of non-metal catalysts exhibit reasonable interactions with hydronium ions in acidic media, indicating their great potential for the electrocatalytic process. To maximize the utilization efficiency of phosphorus in the electrocatalytic process, Fu et al. prepared a highly dispersed single-atom phosphorus embedded on a single-crystal Mo2C nanosheet array supported by a carbon sheet (SAP-Mo2C-CS) and demonstrated the robust performance as an electrocatalyst for the HER.454 As shown in Fig. 32h, SAP-Mo2C-CS shows an overpotential of 36 mV at a current density of 10 mA cm−2, which is comparable to that of the commercial Pt/C electrode (15 mV), and significantly lower than that of Mo2C on carbon sheets (143 mV) and pristine carbon sheets (316 mV). The Tafel slope of SAP-Mo2C-CS (38.1 mV dec−1) indicates that the HER process mainly follows the Volmer–Tafel mechanism (Fig. 32i) and hence, recombination of the adsorbed hydrogen species determines the overall reaction rate. To elucidate the intrinsic catalytic activity of SAP-Mo2C-CS, researchers have calculated the turnover frequency (TOF) of each active site (Fig. 32j). At an overpotential of 0.05 V, the TOF of SAP-Mo2C-CS is 0.23 H2s−1, which is similar to the value of Pt/C (0.39 H2s−1) and considerably higher than that of Mo2C on carbon sheets, confirming that the phosphorus single atoms are the primary active sites in SAP-Mo2C-CS. Notably, SAP-Mo2C-CS exhibits higher catalytic activity in acidic electrolytes than many transition metal carbides (Fig. 32k) and also maintains the stable HER activity during the chronopotentiometry test for 11 hours. To probe the mechanism for the high-performance activity of SAP-Mo2C-CS, researchers have conducted DFT calculations to simulate the entire catalytic process. The results indicate that the presence of anchored phosphorus single atoms on Mo2C results in chemical coupling and accompanying electron redistribution for the unique HER kinetics. This leads to a free energy of hydrogen adsorption (ΔGH*) that is almost zero and hydrogen binding strength that is sufficient to cover the surface, but weak enough to facilitate desorption of H2 molecules for the exceptional HER activity.
Regarding the mechanism, owing to the complexity of the basic reaction involving multi-electron transfer, a comprehensive and unified mechanism for OER electrocatalysis is yet to be established. Several studies have proposed alternative processes for the OER under different conditions. Most of the reported studies on OER electrocatalysis have been conducted under alkaline conditions, and the mechanism of OER electrocatalysis in the alkaline medium can be described as follows:457,458
| M + OH− → MOH + e− | (11) |
| MOH + OH− → MO + H2O + e− | (12) |
| 2MO → 2M + O2 | (13) |
| MO + OH− → MOOH + e− | (14) |
| MOOH + OH− → M + O2 + H2O | (15) |
Significant efforts have been dedicated to investigating the OER electrocatalytic activity of EPMs. In 2016, Jiang et al.460 evaluated the OER performance of black-P materials and observed that even black-P bulk forms supported on Ti foil or carbon nanotubes required only 1.6 V (vs. RHE) to achieve a current density of 10 mA cm−2, which is comparable to that of well-known commercial RuO2 (1.59 V)461 and IrO2 (1.57 V)462 electrocatalysts. This work demonstrates the potential of EPMs for OER electrocatalysis and encourages further research into the electrocatalytic efficiency of low-dimensional black-P and other phosphorus allotropes. Zhang's group systematically investigated the performance of few-layered black-P nanosheets in the electrocatalytic OER process.463 The 2D structure of nanosheets has more active sites for higher electrocatalytic efficiency than the bulk counterpart, even allowing it to compete with some heterostructure materials.460,464,465 To examine the electrocatalytic performance of black-P nanosheets, a three-electrode setup with a gradient concentration of potassium hydroxide (KOH) electrolyte is used and surprisingly, when the electrolyte concentration goes up, the electrocatalytic capability of black-P nanosheets improves approximately linearly. Under optimal conditions, i.e., in 1 M KOH solution, black-P nanosheets exhibit an enhanced current density of 2.7 mA cm−2 at 1.6 V potential and a reduced onset potential (∼1.45 V). The Tafel slopes of black-P nanosheets in 1, 0.2, 0.1, and 0.05 M KOH are 88, 138, 218, and 307 mV dec−1,463 respectively, demonstrating that the OH− concentration influences the electrocatalytic performance (Fig. 33a and b). The high-concentration hydroxide solution promotes electron transport between the electrolyte and electrode and also makes the nanosheets more stable. Fig. 33c shows the current density fluctuation of black-P nanosheets around 6.2 mA cm−2 in 1 M KOH. It is higher than the current densities for lower concentrations of KOH over time. After 10000 s, the black-P nanosheets in 1 M, 0.2 M, and 0.1 M KOH show no decay in the current density, while a slight drop in the current density is observed from 0.05 M KOH. Furthermore, the polarization curves of black-P nanosheets before and after the reaction remain almost unchanged, except in 0.05 M KOH (Fig. 33d), indicating that few-layered Black-P electrocatalysts exhibit great long-term stability within a certain range of alkaline electrolyte concentrations. DFT-based theoretical calculations have shown that the oxidation level of pristine EPMs influences the electrocatalytic activity of the OER.466 Due to the lone electron pair feature of phosphorus atoms, the catalytic site shows excessive adsorption to the reaction intermediates (MOH, MO and MOOH), blocking the subsequent reactions or impeding O2 desorption. Changes in the local oxidation degree 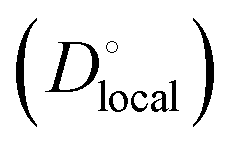 of phosphorene can affect the adsorption energy of O*. When
of phosphorene can affect the adsorption energy of O*. When 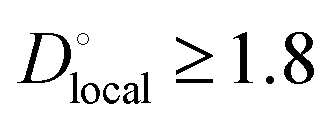 , the O* adsorption strength decreases rapidly with increasing
, the O* adsorption strength decreases rapidly with increasing  . Therefore, partially oxidizing the phosphorus surfaces may alleviate the adsorption strength and increase the catalytic efficiency.
. Therefore, partially oxidizing the phosphorus surfaces may alleviate the adsorption strength and increase the catalytic efficiency.
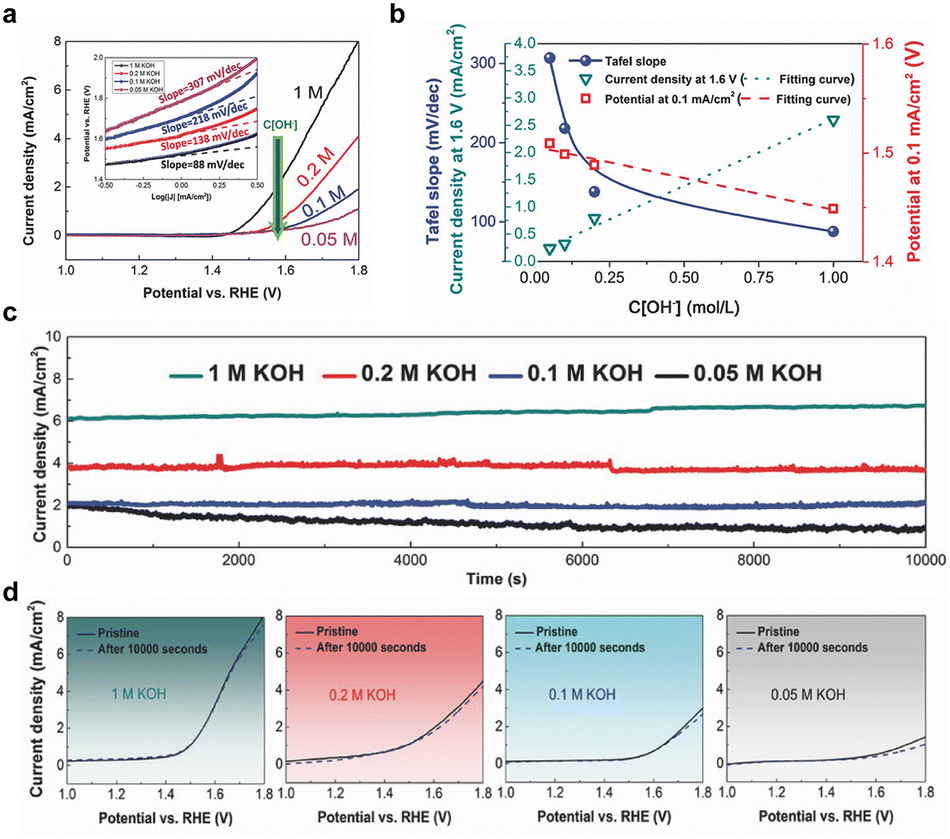 | ||
| Fig. 33 Application of few-layered black-P nanosheets to electrocatalytic oxygen evolution. (a) Polarization curves of black-P nanosheets in KOH electrolyte with varied concentrations. The inset shows their corresponding Tafel plots. (b) Calculated Tafel slope, the current density at 1.6 V, and potential at 10 mV cm−2 in relation to the concentration of OH−. (c) Electrocatalytic OER stability of black-P nanosheets in KOH electrolyte with varied concentrations. (d) Polarization curves of black-P nanosheets in corresponding KOH electrolytes before and after 10000 seconds. Reproduced with permission from ref. 463. Copyright 2017 John Wiley and Sons. | ||
Similar to HER electrocatalysis, element doping,467 combination with other materials,468 and designing catalyst scaffolding469 are strategies to enhance the OER electrocatalytic activity of EPMs. Inspired by a novel photocatalyst of red-P–black-P heterostructure created by Shen and colleagues,470 Xu's group synthesized a series of composites of red-P–black-P chemically bonded with expanded graphite (RP-BP/EG) or carbon nanotubes (RP-BP/CNTs) using a high-energy ball milling technique and compared the OER electrocatalytic activities.471,472 The primary factors influencing the catalytic performance of the composites are the ball-milling time and relative mass ratio of the carbon-based materials. The P–C bond cannot be formed if the ball-milling time is too short but if it is too long, the heterogeneous structure collapses, and the catalyst surface is damaged, hindering O* adsorption. Excess or insufficient EG or CNTs can make it difficult for the active sites to form or be buried. Under optimum circumstances, the overpotential of RP-BP/EG reaches 328 mV vs. RHE at 10 mA cm−2. Although this value is similar to that of some nitrogen-doped carbon materials and surface-oxidized multiwall carbon nanotubes,471,473,474 the overpotential of RP-BP/CNTs is only 263 mV at the same current density.472 The Tafel slope of RP-BP/EG is smaller than that of RP-BP/CNTs, indicating that the former has a faster electron transfer rate or the two systems have different rate-determining steps. However, the strength of the P–C bond varies due to the diversity of carbon nanostructures thus altering the stability of heterostructures, and the nanostructures also impact the catalytic area and density of active sites. Further investigations are required to determine the influence of composite nanostructures on OER electrocatalytic efficiency.
EPMs have shown promise in both the HER and OER, and researchers have attempted to develop these materials as bifunctional electrocatalysts for overall water splitting. In 2018, our group synthesized monodispersed in-plane black-P/Co2P heterostructures by a simple solvothermal method in which Co2P nanocrystals were selectively grown on black-P edge defects.475 Co2P in the heterostructure not only occupies the black-P defects to improve the stability, but also provides more effective electrocatalytic active sites. Moreover, the bulging morphology alleviates agglomeration of black-P nanosheets and increases the exposure of active sites and gas effusion. The electrocatalytic activities and stability for the HER and OER are improved compared to pristine black-P and they are even comparable to those of commercial electrocatalysts (Fig. 34a–d). Given the outstanding performance of black-P/Co2P in the HER and OER, a dual-electrode system has been constructed with black-P/Co2P as both the cathode and anode to evaluate the overall water-splitting activity in 1.0 M KOH electrolyte (Fig. 34e).475 The system achieves a current density of 10 mA cm−2 at a voltage of 1.92 V (Fig. 34f), demonstrating the great potential of this electrocatalyst for overall water splitting. Similarly, Li et al. prepared a bifunctional HER/OER catalyst (CoFeO@Black-P) by growing amorphous multi-transition metal (cobalt and iron) oxides on 2D black-P nanosheets.476 The water electrolysis cell with CoFeO@Black-P electrodes reaches a current density of 10 mA cm−2 at a voltage of 1.58 V in 1 M KOH, and maintains 93% of the initial current density after the 24 hour of operation (Fig. 34g and h). As shown in Fig. 34i, metastable CoFe oxides can react with the phosphorus source provided by black-P to in situ form CoFe phosphide as the active sites in HERs. On the other hand, the rich oxygen vacancies in CoFe oxides catalyze the OER through lattice oxygen oxidation mechanism. Other effective bifunctional electrocatalysts including BPQDs/MXene nanohybrids,477 black-P@N-doped graphene,478 and black-P@red-P-g-C3N4,479 have also been reported for overall water splitting.
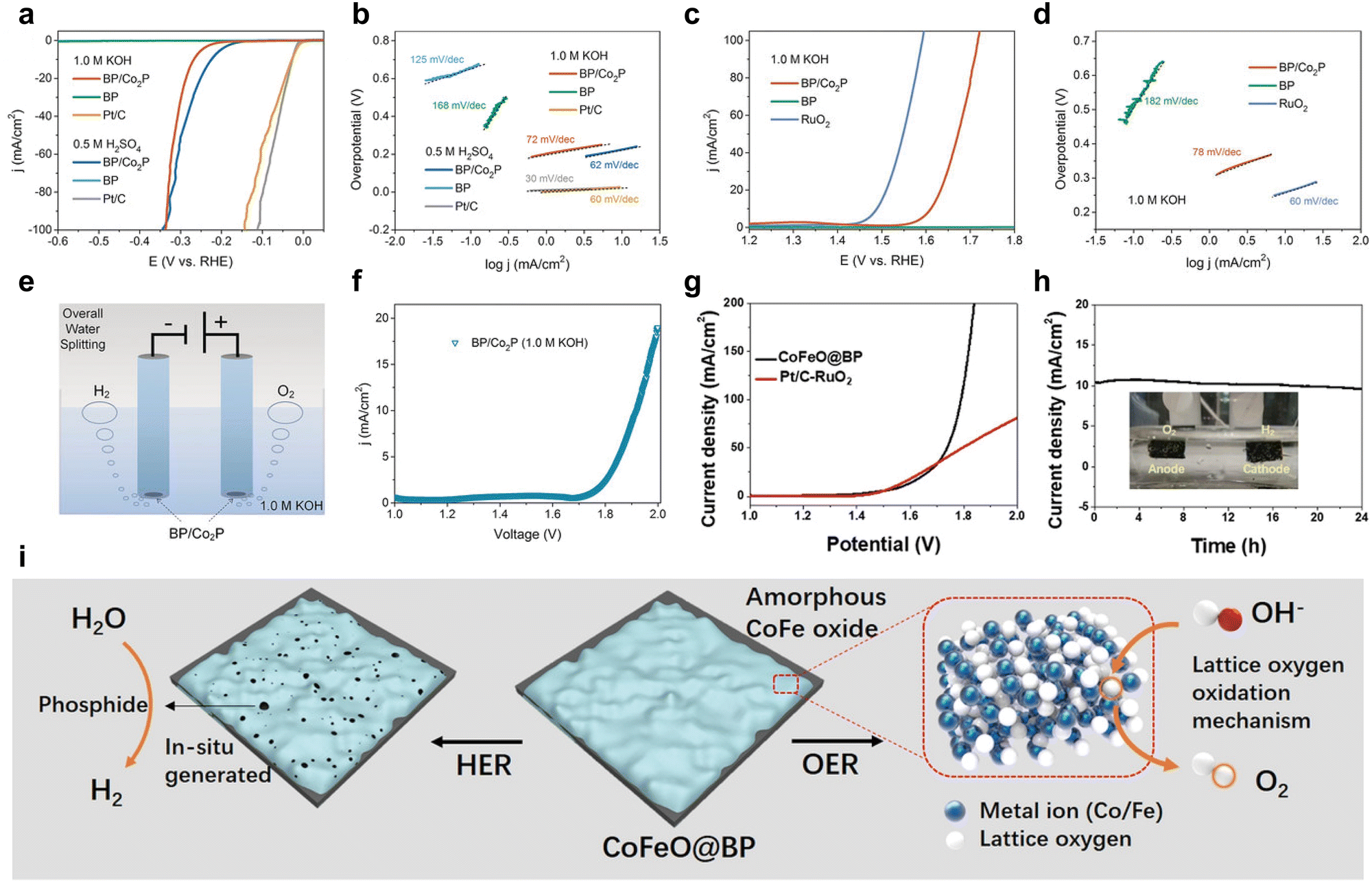 | ||
| Fig. 34 Applications of elemental phosphorus materials to electrocatalytic overall water splitting. (a) HER polarization curves of pristine black-P (BP), BP/Co2P, and Pt/C in 0.5 M H2SO4 and 1.0 M KOH. (b) Corresponding Tafel plots for BP, BP/Co2P, and Pt/C in (a). (c) OER polarization curves of BP, BP/Co2P, and RuO2 in 1.0 M KOH. (d) Corresponding Tafel plots for BP, BP/Co2P, and RuO2 in (c). (e) Scheme of electrocatalytic overall water splitting for BP/Co2P. (f) Polarization curve of BP/Co2P for overall water splitting in 1.0 M KOH. Reproduced with permission from ref. 475. Copyright 2018 John Wiley and Sons. (g) Polarization curves of CoFeO@black-P and Pt/C-RuO2 for overall water splitting. (h) Stability test of CoFeO@black-P. The inset shows bubble formation from both electrodes during electrolysis. (i) Proposed mechanism of CoFeO@black-P for electrocatalytic overall water splitting. Reproduced with permission from ref. 476. Copyright 2020 John Wiley and Sons. | ||
Understanding the mechanism of the NRR process is critical to designing an ideal NRR catalyst. Generally, nitrogen reduction on a heterogeneous catalyst surface follows either an associative or dissociative pathway, where the primary difference between the two is the time required to break the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond (Fig. 35a).484,489 In the associative process, a nitrogen molecule initially adsorbs on the catalyst surface and is then hydrogenated. Following hydrogenation, the NN triple bond breaks, leading to the formation of ammonia molecules. Since the nitrogen molecule contains two nitrogen atoms, hydrogenation occurs through two distinct routes: distal and alternating hydrogenation. In the distal route, hydrogenation commences on the N atom farthest from the catalyst surface, and one NH3 molecule is formed from this N atom. The hydrogenation process then proceeds to the N atom closest to the catalyst surface, generating the second NH3 molecule. In contrast, in the alternating route, hydrogenation occurs alternately between the two N atoms, resulting in a continuous hydrogenation cycle where the first NH3 molecule is released immediately followed by the second NH3 molecule. On the other hand, in the dissociative process, the NN triple bond breaks before hydrogenation, leaving two adsorbed N atoms on the catalyst surface. These two atoms are then hydrogenated individually before forming NH3 molecules. It is worth mentioning that the specific NRR mechanism (associative or dissociative) followed by different NRR catalysts is mostly driven by their atomic and electronic structures,490 which must be verified by theoretical calculations and experiments.
N triple bond (Fig. 35a).484,489 In the associative process, a nitrogen molecule initially adsorbs on the catalyst surface and is then hydrogenated. Following hydrogenation, the NN triple bond breaks, leading to the formation of ammonia molecules. Since the nitrogen molecule contains two nitrogen atoms, hydrogenation occurs through two distinct routes: distal and alternating hydrogenation. In the distal route, hydrogenation commences on the N atom farthest from the catalyst surface, and one NH3 molecule is formed from this N atom. The hydrogenation process then proceeds to the N atom closest to the catalyst surface, generating the second NH3 molecule. In contrast, in the alternating route, hydrogenation occurs alternately between the two N atoms, resulting in a continuous hydrogenation cycle where the first NH3 molecule is released immediately followed by the second NH3 molecule. On the other hand, in the dissociative process, the NN triple bond breaks before hydrogenation, leaving two adsorbed N atoms on the catalyst surface. These two atoms are then hydrogenated individually before forming NH3 molecules. It is worth mentioning that the specific NRR mechanism (associative or dissociative) followed by different NRR catalysts is mostly driven by their atomic and electronic structures,490 which must be verified by theoretical calculations and experiments.
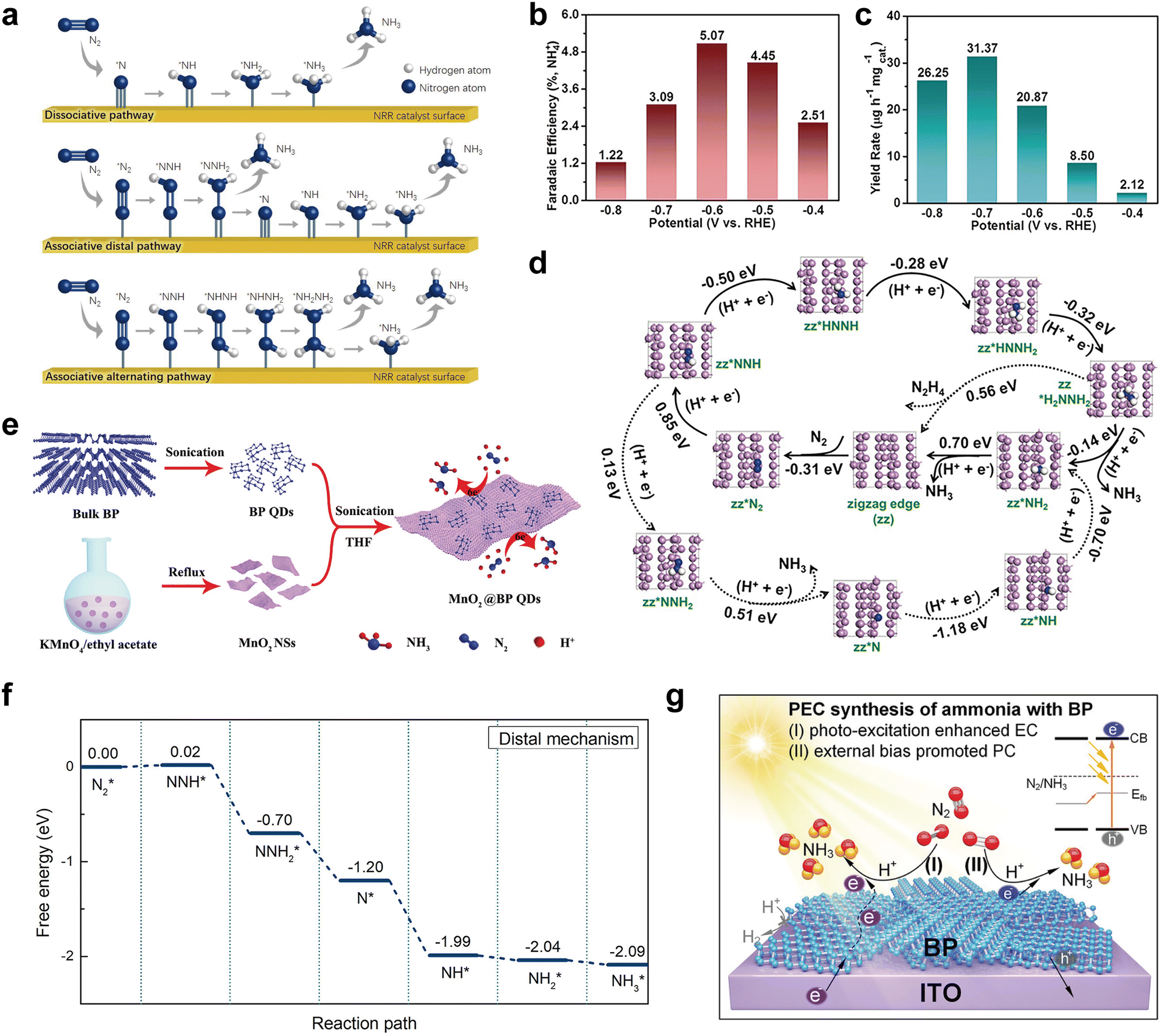 | ||
| Fig. 35 Applications of elemental phosphorus materials to electrocatalytic and photocatalytic nitrogen reduction. (a) Scheme of possible mechanisms of the NRR process on the catalyst surface. Reproduced with permission from ref. 489. Copyright 2019 Elsevier. (b) Faradaic efficiency and (c) NH3 yield rate of few-layered black-P nanosheets at different potentials. (d) Scheme of the possible reaction mechanisms (solid line: low-energy pathway and dotted line: unfavourable pathway) for the electrocatalytic NRR at the zigzag edge of few-layered black-P nanosheets under a N2 atmosphere. Reproduced with permission from ref. 494. Copyright 2019 John Wiley and Sons. (e) Scheme of the preparation of MnO2 nanosheets, BPQDs, and the vdW assembly of BPQDs onto MnO2 nanosheets for NRR electrocatalysis. Reproduced with permission from ref. 497. Copyright 2020 John Wiley and Sons. (f) Free energy diagrams of the NRR process on W-N3@blue-P along the distal mechanism. Reproduced with permission from ref. 498. Copyright 2020 Elsevier. (g) Photoelectrochemical NRR mechanisms of black-P under light irradiation and external electric field. Reproduced with permission from ref. 502. Copyright 2020 John Wiley and Sons. | ||
Developing high-performance NRR catalysts is a challenging task. The cleavage of the NN triple bond requires a significant amount of energy (941 kJ mol−1),491 and throughout the catalytic process, the HER always competes with the NRR at the catalytically active sites,487,488,492 leading to a catalytic selectivity issue. Therefore, a state-of-the-art NRR catalyst must not only fulfil the conventional requirements for a high density of active sites and material stability, but also exhibit exceptional catalytic selectivity. This means that the catalyst surface binding strength of nitrogen must be greater than that of hydrogen to favour electron transfer along the NRR pathway. Due to the similar valence electronic structures of phosphorus and nitrogen (3s23p3 and 2s22p3, respectively),21,493 nitrogen has a stronger affinity for the surface of phosphorus-based materials, giving EPMs a unique advantage as NRR catalysts.
In 2019, Wang's group demonstrated that few-layered black-P nanosheets perform well as NRR electrocatalysts under ambient conditions.494 To determine the electrocatalytic activity, they selected 0.01 M HCl as the acidic electrolyte in a standard three-electrode setup, and a few-layered black-P suspension was dropped onto carbon fibers as the working electrode. As shown in Fig. 35b and c, the catalytic electrode achieved the highest Faradaic efficiency of 5.07% at −0.6 V, with a maximum NH3 production rate of 31.37 μg h−1 mgcat.−1 at −0.7 V. The few-layered black-P electrocatalyst outperforms its bulk counterpart (0.13% Faradaic efficiency, 0.036 μg h−1 mgcat.−1), as well as various metal-free catalysts (e.g., N-doped porous carbon with a Faradaic efficiency of 1.42% and 23.8 μg h−1 mgcat.−1) and metal-based catalysts (e.g., Au–TiO2 sub-nanoclusters with a Faradaic efficiency of 8.11%and 21.4 μg h−1 mgcat.−1).495,496 Moreover, the UV/vis absorption spectra and chromogenic reactions indicate that no obvious side-product (N2H4) is generated throughout the electrocatalytic process, confirming the high catalytic selectivity of few-layered black-P. Based on DFT calculations for the molecular orbitals of black-P, the NRR catalytic process follows an alternating hydrogenation pathway in an association mechanism (Fig. 35d).494 This study not only demonstrates the feasibility of using black-P materials for the electrocatalytic NRR under ambient conditions, but also serves as a model for investigating the NRR mechanisms of EPMs. Zhu's group has reported a straightforward vdW assembly approach for loading BPQDs onto wrinkled MnO2 nanosheets (Fig. 35e), which leads to enhanced NRR electrocatalytic activity compared to pristine materials.497 The maximum NH3 generation rate of BPQDs/MnO2 is 25.3 μg h−1 mgcat.−1 and its Faradaic efficiency is 6.7% at an applied potential of −0.5 V (vs. RHE). The outstanding electrocatalytic performance can be ascribed to the strong synergistic effects of BPQDs/MnO2.497 The wrinkled MnO2 nanosheets act as a flexible substrate to prevent severe BPQD aggregation during the catalytic process. The edges and defect sites of MnO2 nanosheets exhibit reasonable electrocatalytic activity. Moreover, BPQDs with an abundance of active sites serve as the primary NRR catalyst and physical spacer to prevent restacking of MnO2 nanosheets and maintain the structural stability of the composite.
Other EPMs have also shown potential for NRR electrocatalysis. Our group has conducted a series of DFT calculations and found that crystalline red-P (fibrous phase) nanoribbons have potential electrochemical nitrogen fixation activity. On the surface of crystalline red-P nanoribbons, the NRR process can be carried out through the proton-coupled electron transfer pathway, with the associative mechanism being preferable, followed by distal hydrogenation. The initial step of adsorbed N2 hydrogenation to NNH* is proposed to be the rate-determining step due to the largest energy input required. Experimentally, we have carried out the electrocatalytic test in a gastight H-type electrolytic cell with 0.1 M Na2SO4 electrolyte and placed crystalline red-P nanoribbons onto a nickel foam as the catalytic electrode. The catalytic electrode delivers efficient and durable NRR performance, at an optimal NH3 production rate of 15.4 μg h−1 mgcat.−1 at −0.4 V and a faradaic efficiency of 9.4% at −0.2 V. Compared to the few-layered black-P electrocatalyst developed by Wang's group, crystalline red-P nanoribbons exhibit a lower optimal voltage but a higher Faradaic efficiency due to the increased charge transfer and exposed-edge active sites of the quasi-1D structure.47 However, the ammonia production rate of crystalline red-P nanoribbons is still limited and the development of red-P-based materials for NRR electrocatalysis still has a long way to go.
Compared to the deeper armchair ridges of black-P, the hexagonal lattice structure of blue-P with zigzag ridges is relatively isotropic, making it an ideal candidate for multi-electron transfer. Recently, Zhang et al. have demonstrated that a kinetically stable W-N3 center supported by blue-P exhibited great NRR catalytic activity with a limit potential of −0.02 V based on DFT calculations.498 The W atom plays a crucial role in weakening the NN triple bond by accepting lone electron pairs from N2 and donating electrons to the anti-bonding orbitals of N2,499 enabling N2 molecules to readily adsorb on the composite surface. The Gibbs free energy variations of the intermediates suggest that the associative distal mechanism is the most favourable in this system (Fig. 35f). Additionally, the high selectivity of the NRR can be achieved due to the high free energy barriers for the HER (0.83 eV) and the ultralow free energy barriers for the NRR (0.02 eV). This work provides theoretical guidance for the development of blue-P-based NRR electrocatalysts.
The solar-powered photocatalytic NRR is a promising low-cost, green, and sustainable strategy for ammonia production. However, the high energy barrier of the six-electron reaction that activates and breaks the strong NN bond by photocatalysts, hinders the reaction kinetics, resulting in relatively low photocatalytic NRR efficiency. Thus, developing highly active photocatalysts is crucial for achieving an efficient photocatalytic NRR. Our group has synthesized edge-rich black-P nanoflakes using a simple chemical etching exfoliation method, which achieves an efficient photocatalytic NRR without using any cocatalysts.500 The abundant edges of the resulting nanoflakes offer numerous active sites for N2 adsorption and complete activation of the NN bond to enhance the reduction of N2. Moreover, the black-P nanoflakes exhibit good water dispersibility, enabling full contact with the reactants. As a result, edge-rich black-P shows a high catalytic nitrogen fixation rate of 2.37 mmol h−1 g−1 under visible light irradiation, which is higher than 0.9 mmol h−1 g−1 shown by conventional black-P nanoflakes. This work highlights the impact of black-P morphology on catalytic efficiency, and the development of black-P materials with nanostructured design for NRR electrocatalysis is a promising research direction. Alternatively, incorporating co-catalysts has demonstrated acceleration of charge transfer and improvement in photocatalytic nitrogen conversion to some extent. However, controlling the interface state and studying the charge transfer at the interface remain a challenge. To address this, Shen et al. have selectively deposited a Ni2P layer at the edge of black-P nanosheets, forming a 2D–2D heterojunction with plentiful Ni–P bonds.501 The resulting Ni2P–BP heterojunction has improved photocatalytic NRR properties such as an NH3 generation rate of 6.14 μmol h−1 g−1 under visible light irradiation, which is 1.56 times higher than that of the Ni–P bond-free Ni2P nanoparticle loaded black-P photocatalyst. This indicates that the interfacial Ni–P bonds plays a critical role in the catalytic process by facilitating interfacial charge transfer and enabling photoexcited electrons on the conduction band of black-P to efficiently transfer to Ni2P and enhance the separation efficiency of electron–hole pairs. Moreover, the Ni2P layer chemically passivates the edges of the black-P nanosheets and improves the stability of the system during the electrocatalytic process.
Compared to the electrocatalytic process with continuous and stable external bias potentials, the photocatalytic NRR faces challenges of low conversion efficiency and unstable conversion processes. However, by combining the advantages of both photocatalysis and electrocatalysis, the N2-to-NH3 conversion efficiency can be further improved through photoelectrochemical reactions. With these principles in mind, our group has developed a black-P photoelectrochemical electrode and investigated its catalytic nitrogen conversion activity under normal environmental conditions.502 The electrode shows remarkable catalytic activity with an ammonia yield rate of 102.4 μg h−1 mgcat.−1 and a Faradaic efficiency of 23.3% at −0.4 V, outperforming most metal-free photocatalysts or electrocatalysts for the NRR.502 Moreover, the black-P electrode exhibits great stability after 6 consecutive cycles. The exceptional NRR activity of the black-P electrode can be ascribed to the synergistic effect generated by photoexcitation and external bias (Fig. 35g), in which the photoexcitation boosts the average electron energy and charge exchange efficiency and facilitates the electrocatalytic process, while the external bias potentials improve the separation of photoexcited electron–hole pairs, promoting the photocatalytic process. Considering the excellent charge mobility of black-P and the strong visible-light absorption and chemical stability of red-P based materials such as amorphous red-P, violet-P, and fibrous-P, the future development of black-P/red-P homojunctions for photoelectrochemical ammonia production holds great promise.
This section has discussed the mechanism and performance of EPMs in photocatalytic/electrocatalytic water splitting, carbon dioxide reduction, and nitrogen fixation. It has underscored the need for a deeper understanding of reaction pathways and intermediate products in the catalytic processes involving different phosphorus materials. Attention should be given to the role of excitons in photocatalysis, the selectivity in carbon dioxide reduction, and how certain material structures lead to different products. For electrocatalysis, the establishment of standardized criteria to assess the catalytic activity of EPMs and evaluating their contributions in composite systems are areas warranting further exploration.
5.2 Energy storage applications
Solid-state storage technologies such as metal hydride techniques, physisorption, and chemical adsorption have garnered considerable attention as a potential alternative to conventional hydrogen storage technologies.507 The search for suitable solid-state storage materials is ongoing and promises to yield exciting possibilities.508–510 According to the US Department of Energy (USDOE) targets for 2020,511–513 high-efficiency hydrogen storage materials with a gravimetric density of 5.5 wt% should be employed with hydrogen reversibility occurring at temperatures ranging from −40 to 60 °C at a filling/releasing rate of 1.5 kg H2 min−1. This criterion requires that the energy associated with adsorbing each hydrogen molecule must be within an appropriate range to regulate the adsorption/desorption kinetics. To date, many publications have demonstrated that carbonaceous materials can be used for hydrogen storage.514–516 However, pure carbon atoms have a low affinity for hydrogen, and therefore, metal doping is a viable strategy for enhancing hydrogen adsorption. Nonetheless, metal atoms are prone to clustering on the carbon matrix, resulting in low reversibility and an inability to meet the long-term hydrogen storage requirement.
In comparison to carbonaceous materials, metal atoms on phosphorene exhibit significantly lower tendencies for cluster formation. Although phosphorene is heavier than graphene, it is lighter than many other prominent 2D materials, including MXenes and TMDs, resulting in a high gravimetric density. In 2014, Li et al.517 conducted the first study on Li-decorated monolayer black-P using theoretical calculations. Li-decorated black phosphorene exhibits a hydrogen storage capacity of 8.11 wt% and average adsorption energy of 0.18 eV per H2, exceeding the hydrogen storage targets established by USDOE. Luo et al.513 have demonstrated better hydrogen storage performance for Li-decorated single-layered blue-P. At a Li/P ratio of 1:1, the interaction between Li atoms and blue-P is sufficiently strong, maintaining the Li atom dispersion. Due to the space constraint surrounding Li atoms, double-layer adsorption occurred, and each atom can adsorb two molecules of H2, leading to a hydrogen storage capacity of 9.52 wt%. This can be attributed to blue phosphorene having a flatter in-plane structure than black phosphorene, and the zigzag ridges aiding in the dispersion of Li atoms. Recently, Xu et al.518 have used a solvothermal approach to prepare Co-doped black-P nanosheets and evaluated their efficacy in electrochemical hydrogen storage. The optimal hydrogen storage capacity of Co-doped black-P reaches 6200 mA h g−1 (i.e., 18 wt% of composite weight), and the average capacity is ∼10.3 times that of pristine black-P nanosheets. The active catalytic sites of Co atoms that facilitate hydrogen transfer are primarily responsible for the electrochemical hydrogen storage ability of the composite. Additionally, Co doping reduces the bandgap and increases the conductivity to provide favourable conditions for electrochemical hydrogen storage. However, most of the research on hydrogen storage in phosphorene is still in the theoretical stage, and several studies based on DFT calculations have been conducted to investigate the hydrogen capture capability of phosphorene.519–523
In LIBs, the specific capacities of anode and cathode materials primarily determine the energy capacity and charge/discharge rate. Commercially available LIBs often employ graphite anodes and lithium metal oxide cathodes, resulting in a theoretical energy capacity of approximately 400 W h kg−1 and a practical capacity of around 200 W h kg−1.530 However, this performance falls short of the increasing requirements for electrochemical energy storage devices, thus necessitating enhancements in the specific capacity of LIBs. Beyond graphite anodes, considerable efforts have been devoted to layered materials, which can be divided into four categories: 2D monoelemental materials (Xenes),531 metal nitrides/carbides (MXenes),532 transition metal dichalcogenides (TMDs),533 and transition metal oxides (TMOs).534 Low-dimensional EPMs have garnered significant interest as potential LIB anodes among the Xenes. For example, black-P possesses a low diffusion energy barrier for lithium ions (0.08 eV),535 enabling it to exhibit 102 and 104 times faster lithium ion diffusion rates than MoS2 and graphene, respectively, thereby suggesting a rapid charge/discharge rate. Moreover, the theoretical specific capacities of EPMs reach 2596 mA h g−1 surpass those of most TMOs and TMDs.536,537 These characteristics render EPMs a promising anode material for the next-generation LIBs and other electrochemical energy storage devices (Table 3).
| Allotropes | Materials | Applications | Methodology | Capacity | Capacity retention rate (%) (cycle number, rate or current density) | Ref. |
|---|---|---|---|---|---|---|
| Black phosphorus | BP-Graphite | LIB | High energy mechanical milling | Initial discharge capacity of 2786 mA h g−1 at 0.2C | 80 (100, 0.2C) | 549 |
| Sandwich-structure G-BPGO | LIB | Vacuum-filtration | Initial discharge specific capacity of 2587 mA h g−1 at 0.1 A g−1 | 1401 mA h g−1 (200, 0.1 A g−1) | 550 | |
| Bi–P/C | LIB | Ball-milling method | High-rate discharge capacities of 1788.2 mA h g−1 at 13 A g−1 | 86.3 (300, 7.8 A g−1) | 551 | |
| BP/NiCo MOF | LIB | Solution reaction route | Reversible capacity of 853 mA h g−1 at 0.5 A g−1 | 91.3 (1000, 5 A g−1) | 552 | |
| BP/G/CNTs | LIB | Ball-milling method | Initial reversible capacity of 1375 mA h g−1 at 0.15 A g−1 | 79 (1000, 1 A g−1) | 553 | |
| (BP-G)/PANI | LIB | Sonication and ball milling | Reversible discharge capacity of 1520 mA h g−1 at 0.26 A g−1 | 440 mA h g−1 (2000, 13 A g−1) | 554 | |
| PCNF/S/BPQD | LSB | Sonication and wet chemistry | Rate capacity of 784 mA h g−1 at 4C | ∼90 (200, 0.1C) | 555 | |
| BP-coated separator | LSB | Vacuum-filtration | Specific capacity of 725 mA h g−1 at 1.8 A g−1 | 86 (100, 0.4 A g−1) | 556 | |
| BPQD/δ-MnO2 | LAB | Hydrothermal route | Discharge capacity of 8463 mA h g−1 at 0.1 A g−1 | 1000 mA h g−1 (182, 0.4 A g−1) | 557 | |
| Phosphorene-coated Li metal | LAB | Ultrasonication and spin coating | Constant discharge capacity 1000 mA h g−1 at 0.25 A g−1 | 100 (50, 0.25 A g−1) | 558 | |
| BP–CNT | LIB and SIB | Surface oxidation-assisted chemical bonding procedure | LIB: Initial discharge capacity of ∼1922 mA h g−1 at 0.2C | LIB: 87.5 (400, 0.2C) | 559 | |
| SIB: Initial discharge capacity of ∼2073 mA h g−1 at 0.2C | SIB: 75.3 (200, 0.2C) | |||||
| BPQD/TNS | LIB and SIB | Interfacial assembly strategy | LIB: Reversible capacity of 828 mA h g−1 at 0.1 A g−1 | LIB: over 100% (2400, 1 A g−1) | 560 | |
| SIB: Initial discharge capacity of ≈723 mA h g−1 at 0.2 A g−1 | SIB: nearly 100% (1000, 1 A g−1) | |||||
| 4-RBP | SIB | 4-NBD modification and solvothermal reaction | Initial discharge specific capacity of 2500 mA h g−1 at 0.1 A g−1 | 1472 mA h g−1 (50, 0.1 A g−1) | 561 | |
| Layered BP/graphene | SIB | Flash-heat-treatment and straightforward pressure synthesis | Specific charge capacity of 1377.6 and 720.8 mA h g−1 at 1 and 40 A g−1, respectively | 1250 mA h g−1 (500, 1 A g−1) | 562 | |
| 640 mA h g−1 (500, 40 A g−1) | ||||||
| RP@BP/3DNG | SIB | Solvothermal strategy | Reversible capacity of 1440.2 and 521.3 mA h g−1 at 0.05 and 10 A g−1, respectively | 89.3 (1200, 10.0 A g−1) | 563 | |
| Few-layer black phosphorene | SIB | Electrochemical exfoliation | Activated capacity of 1968 mA h g−1 at 0.1 A g−1 | 60.5 (50, 0.1 A g−1) | 564 | |
| Phosphorene–graphene | SIB | Self-assembly | Specific capacity of 2440 mA h g−1 at 0.05 A g−1 | 84 (100, 8 A g−1) | 565 | |
| Phosphorene/MXene | SIB | Self-assembly | Reversible capacity of 535 mA h g−1 at 0.1 A g−1 | 87 (1000, 1 A g−1) | 566 | |
| MoS2/BP | SIB | Hydrothermal method | Reversible capacity of 435.5 mA h g−1 at 1.0 A g−1 after 150 cycles | 263.2 mA h g−1 (1000, 10 A g−1) | 567 | |
| BP–C | PIB | Ball-milling method | Initial specific capacity of 617 mA h g−1 at 0.05 A g−1 | ∼30 mA h g−1 (50, 0.05 A g−1) | 568 | |
| (P-CNTs)/PANI | PIB | Ball-milling and in situ solution polymerization | Reversible capacity of 642.7 mA h g−1 at 50 mA g−1 | 94 (500, 0.5 A g−1) | 569 | |
| Restacked BP nanoflakes | SC | Liquid exfoliation | Specific capacitance of 13.75 F cm−3 at 0.01 V s−1 | 71.8 (30000, 0.5 V s−1) |
570 | |
| BP sponges | SC | Electrochemical technique | Specific capacitance of 80 F g−1 at 0.01 V s−1 | 80 (15000, 0.1 V s−1) |
571 | |
| BP/CNTs | SC | High-heat treatment and microfluidic-spinning technique | Specific volumetric capacitance of 308.7 F cm−3 at 0.1 A cm−3 | 90.2 (10000, 0.4 A cm−3) |
572 | |
| E-BP/ZIF-67 | SC | Droplet microfluidic | Specific capacitance of 506 F cm−3 at 500 mA cm−3 | 89.3 (12000, 2 A cm−3) |
573 | |
| Amorphous red phosphorus | RP/A-TiO2 | LIB | In situ hydrolyzation | Rate capacity of 202 mA h g−1 at 1 A g−1 | 89.3 (100, 0.1 A g−1) | 574 |
| P/C | LIB | Vaporization-adsorption method | Specific capacity of 2173 mA h g−1 at 1C | 90 (500, 0.2C) | 29 | |
| RPN/rGF | LIB | Evaporation–condensation method | Rate capacity of 1073 mA h g−1 at 3C | 78.4 (300, 0.3C) | 575 | |
| RP/TiN/CNT | LIB | High-energy ball milling method | Reversible capacity of 868.7 mA h g−1 at 2.3 A g−1 | 96.9 (3rd ∼ 850th, 2.3 A g−1) | 576 | |
| HPNs | LIB and SIB | Wet solvothermal method | Specific capacity of 1285.7 and 1364.7 mA h g−1 at 0.2C for LIBs and SIBs, respectively | LIB: 1048.4 mA h g−1 (600, 1C) | 577 | |
| SIB: 969.8 mA h g−1 (600, 1C) | ||||||
| P@CMK-3 | LIB and SIB | Vaporization–condensation–conversion process | LIB: Specific capacity of 2185 mA h g−1 at 1.2C | LIB: ∼80 (1000, 1.2C) | 578 | |
| SIB: Specific capacity of 2331 mA h g−1 at 0.6C | SIB: 1600 mA h g−1 (140, 1C) | |||||
| BP/RP | LIB and SC | Sono-chemical process | LIB: Initial discharge capacity of 2449 mA h g−1 at 0.05 A g−1 | LIB: 491 mA h g−1 (100, 0.05 A g−1) | 579 | |
| SC: Specific capacitance of ≈60.1 F g−1 at 0.5 A g−1 | SC: 83.3 (2000, 1 A g−1) | |||||
| RPEN@CF | LSB | Laser-assisted exfoliation | Specific capacity of 782.3 mA h g−1 at 3C | 769.5 mA h g−1 (500, 1C) | 580 | |
| P@C | SIB | Shear emulsifying and electrospinning | Reversible capacity of 1308 mA h g−1 at 0.2 A g−1 | 81 (1000, 2 A g−1) | 581 | |
| P-CNT | SIB | Ball-milling method | Specific capacity of 2134 mA h g−1 at 0.26 A g−1 | ∼91 (100, 0.52 A g−1) | 582 | |
| P@RGO | SIB | Physical vapor deposition | Specific capacity of 1165.4 mA h g−1 at 159.4 mA g−1 | 87.9 (300, 1.5939 A g−1) | 583 | |
| Pred@CNF | SIB | Vaporization–condensation method | Specific capacities of ∼1850 mA h g−1 over 500 cycles at 0.1 A g−1 | >1000 mA h g−1 (5000, 1 A g−1) | 584 | |
| NPRP@RGO | SIB | Redox reaction and boiling process | Specific capacity of 1848.67 mA h g−1 at 0.2563 A g−1 after 150 cycles | 775.3 mA h g−1 (1500, 5.12 A g−1) | 585 | |
| RP@Ni–P | SIB | Electroless deposition and chemical dealloying | Reversible specific capacity of 1256.2 mA h g−1 at 0.26 A g−1 | 409.1 mA h g−1 (2000, 5 A g−1) | 586 | |
| P@N-MPC | SIB | Vaporization-condensation method | Specific capacity of ≈600 mA h g−1 at 0.15 A g−1 | ∼450 mA h g−1 (1000, 1 A g−1) | 587 | |
| P2@N-SGCNT | SIB and PIB | High-temperature infiltration | SIB: Reversible capacity of 2480 mA h g−1 at 0.1 A g−1 | SIB: 1936 mA h g−1 (2000, 1 A g−1) | 588 | |
| PIB: Reversible capacity of 762 mA h g−1 at 0.05 A g−1 | PIB: 319 mA h g−1 (1000, 2 A g−1) | |||||
| P50@ZRCod-0.025 | PIB | Solvothermal synthesis | Reversible capacity of 595.8 mA h g−1 at 50 m A g−1 | 150.7 mA h g−1 (400, 2.5 A g−1) | 589 | |
| Red P@N-PHCNFs | PIB | Vaporization–condensation | Reversible capacity of 650 mA h g−1 at 0.1 A g−1 | 84 (200, 1 A g−1) | 590 | |
| H-P@NCNS/NCNT | PIB | Magnetic field assisted methodology | Reversible charge capacity of ≈527 mA h g−1 at 2.0 A g−1 | ≈85% (500, 2 A g−1) | 591 | |
| R-BP/SPC | SC | Sonochemical process | Specific capacitance of 364.5 F g−1 at 500 mA g−1 | 89 (10000, 2 A g−1) |
592 | |
| P-rGO | SC | Potentiodyna-mic deposition | Specific capacitance of 334.2 F g−1 at 1 A g−1 | 99.2 (10000, 1 A g−1) |
593 | |
| Fibrous phosphorus | Fibrous phosphorus | LIB | Iodine-assisted CVD | Initial discharge capacity of 1783 mA h g−1 at 0.1 A g−1 | 75 (10, 0.1 A g−1) | 305 |
| FP-C | LIB | CVT | Reversible specific capacity of 1621 mA h g−1 at 0.2 A g−1 | 742.4 mA h g−1 (700, 2 A g−1) | 594 | |
| MWCNT@f-RP@BP hybrid | LIB | CVD | Initial discharge capacity of ∼1333 mA h g−1 at 0.5 A g−1 | 560 mA h g−1 (250, 0.5 A g−1) | 595 | |
| Violet phosphorus | Hittorf's phosphorus | LIB | Iodine-assisted CVD | Initial discharge capacity of 2113.8 mA h g−1 at 0.1 A g−1 | 558 mA h g−1 (40, 0.1 A g−1) | 305 |
| Hittorf's violet phosphorene | LIB and SIB | DFT calculations | Adsorption energies of 1.65 eV for Li and 1.32 eV for Na (theoretical) | — | 596 | |
| Blue phosphorus | Single-layer blue phosphorus | LIB | DFT calculations | Charge capacity of 865 mA h g−1 (theoretical) | — | 597 |
| Double-layer blue phosphorus | LIB | DFT calculations | Charge capacity of 649 mA h g−1 (theoretical) | — | 597 | |
| BlueP/graphene heterostructure | LIB | DFT calculations | Specific capacity of 626 mA h g−1 (theoretical) | — | 598 | |
| BlueP/MS2 heterostructures (M = Nb, Ta) | LIB | DFT calculations | Specific capacities of 528.257 mA h g−1 for BlueP/NbS2 and 392.154 mA h g−1 for BlueP/TaS2 (theoretical) | — | 599 | |
| Blue phosphorene | LSB | DFT calculations | Adsorption energies in the range of −0.86 to 2.45 eV for lithium polysulfides | — | 600 | |
| 2D-N-doped blue phosphorus | LAB | DFT calculations | Storage capacity of >314.22 mA h g−1 (theoretical) | — | 601 | |
| h-BN/blue-P heterostructure | LIB and SIB | DFT calculations | Specific capacities of 801 mA h g−1 for Li and 541 mA h g−1 for Na (theoretical) | — | 602 | |
| C3N/blue-P heterostructure | LIB and SIB | DFT calculations | Specific capacities of 333 mA h g−1 for Li and 658 mA h g−1 for Na (theoretical) | — | 603 | |
| Monolayer blue phosphorene | SIB and PIB | DFT calculations | Specific capacities of 865 mA h g−1 in both cases (theoretical) | — | 540 | |
| Green phosphorus | Single-layer GP | LIB | DFT calculations | Specific capacity of 432.7 mA h g−1 (theoretical) | — | 74 |
| 2D green phosphorene | SC | DFT calculations | Specific capacitance of ≈90 F g−1 (theoretical) | — | 604 | |
The working mechanism of LIBs is characterized by two primary phases: lithiation and delithiation.538,539 During the charging process, energy is stored as chemical energy and an external current drives lithium ions to the anode, where they are intercalated into the phosphorus interlayer, forming a new phase of Li3P. During the charging process, the thermodynamically favourable delithiation continues. Li3P progressively decomposes, and the intercalated Li atoms contribute their valence electrons to the external circuit, resulting in the formation of Li+. Eventually, the contributed electrons congregate at the cathode, dragging Li+ across the electrolyte and accumulating in the cathode materials. Given the significant volume changes that occur during each lithiation/delithiation process, maintaining the anode structure is critical to achieving long-cycle performance in LIBs.
Experimental studies have been conducted on black-P, amorphous red-P, violet-P, and fibrous-P as anode materials for LIBs, with blue-P and green-P LIBs being theoretically proven to be viable.74,540 In the past few years, black-P has garnered significant interest, and the mechanism of the lithiation/delithiation process in black-P has been more explicitly defined. The structure of black-P is highly anisotropic, and lithium ions prefer diffusing along the shallower zigzag ridges rather than the armchair ridges with a relatively higher energy barrier of 0.68 eV.535 Moreover, a semiconductor-to-metallic transition occurs during the Li+ intercalation process,541 leading to increased electronic conductivity of the black-P anode. Parker et al.542 were the first to report the black-P-based anodes for LIBs and found that bulk black-P exhibited a reversible rated charge capacity of 1279 mA h g−1. While this value exceeds that of commercial graphite (372 mA h g−1), it only reaches half of the theoretical capacity, and the initial cycle efficiency remains relatively low (57%). In 2019, Chen et al.305 synthesized fibrous-P and violet-P bulk crystals through a CVD method and demonstrated that these two EPMs have the potential to be LIB anodes as well. The first discharge capacity of the fibrous-P anode is 1783 mA h g−1 and that of the violet-P anode is 2113.8 mA h g−1, with Coulombic efficiencies of 81.04% and 75.76%, respectively. However, fibrous-P and violet-P anodes display irreversible capacities after several dozen cycles, limiting their long-term capacity retention. Generally, pristine bulk materials lack rational structural designs,543 and abrupt pulverization of the anode materials during the delithiation process leads to undulation of the lattice structure, resulting in a substantial volume expansion of up to 300%.544 This can lead to the loss of electrical contact within the active material, resulting in a rapid decay of the battery capacity.
Nanostructured design can enhance electrochemical reactions and improve active material utilization.545,546 Among various nanostructured strategies, exfoliating the bulk phase into 2D nanosheets is a widely adopted method that can decrease electrode thickness, reduce electron and ion diffusion distances within the electrode layer, and ultimately enhance the volumetric energy density of LIBs. However, 2D phosphorene nanosheets with large specific surface areas suffer from restacking and detrimental parasitic reactions with the electrolyte. These reactions often occur at the edge of the phosphorene zigzag diffusion channel, resulting in the distortion of the surrounding phosphorus atoms.547 Alternatively, the development of 3D electrode architectures is another design option for enhancing performance. Zhu et al. have developed a green and template-free hydrothermal approach to synthesize honeycomb-like micron-sized red-P with a tunable porous architecture.548 The optimal porous red-P anode delivers exceptional lithium storage performance, achieving a substantial reversible capacity of 2587.4 mA h g−1 at 50 mA g−1 and a capacity retention of ∼81.9% at 500 mA g−1 after 500 cycles. The results from in situ TEM and ex situ SEM measurements indicate that the porous structure buffers the volume variations throughout the lithiation/delithiation process, improving the long-cycle stability of the anode materials.
Currently, commercial LIBs offer limited power densities typically ranging from 100 to 300 W kg−1 and require long charging time to ensure safe operation.605,606 To enhance battery utilization efficiency in portable electronics and electric vehicles, it is crucial to develop high-energy-density LIBs that possess superior fast-charging capabilities. EPMs are promising candidates as anode materials due to their high theoretical specific capacity (2596 mA h g−1) and moderate lithiation potential (0.7 V vs. Li+/Li). However, challenges such as limited conductivity, sluggish reaction kinetics, and unstable solid-electrolyte interphase (SEI) impede the high-rate capability of the phosphorus anode during the lithiation/delithiation process. Despite self-structure redesign efforts, there are still limitations, and new strategies are required to optimize the performance of EPMs as anode materials in fast-charging LIBs.526 Carbonaceous materials possess good electrical conductivity and material stability and their incorporation into the anode can effectively improve the fast-charging capabilities of EPMs. In carbon–phosphorus (P/C) composites, the presence of high-conductive carbon-rich materials amplifies lithium ion diffusion in the anode by reducing the polarization effect,607 and carbon-rich materials also act as an external buffer layer to control volume changes.608 Cui's group designed a red-P/C nanocomposite for fast-charging LIBs by an adsorption and surface cleaning approach.29 The average thickness of red-P/C electrodes is 21.5 μm, featuring an industrial-grade areal capacity of around 3.5 mA h cm−2 at 0.5 mA cm−2. This thickness is considerably less than that of the commercial graphite (76.3 μm) and Li4Ti5O12 (124.5 μm) electrodes at the same areal capacity (Fig. 36a). Consequently, the total volumetric capacity of the red-P/C electrode is 1628 mA h cm−3, far surpassing that of graphite (459 mA h cm−3) and Li4Ti5O12 (281 mA h cm−3) electrodes (Fig. 36b). The red-P/C nanocomposite also exhibits an extended cycle life. After 500 discharge–charge cycles, the red-P/C electrode shows a specific capacity of 1625 mA h g−1 and retention rate of around 90% at 0.86 mA cm−2 (0.2C). Following long-cycling, the red-P/C electrode retains its morphological integrity and electrical connectivity without fractures or contact losses after long-cycling tests. A uniform and thin solid-electrolyte interphase layer (<50 nm) is formed on the surface of a red-P/C nanoparticle, while the interior nanoscale void spaces are well preserved to buffer the volume changes after 100 cycles.29 These findings demonstrate that the red-P/C nanocomposite is structurally robust at both the electrode and particle levels, enabling stable battery cycling and high specific capacity at relatively high current densities.
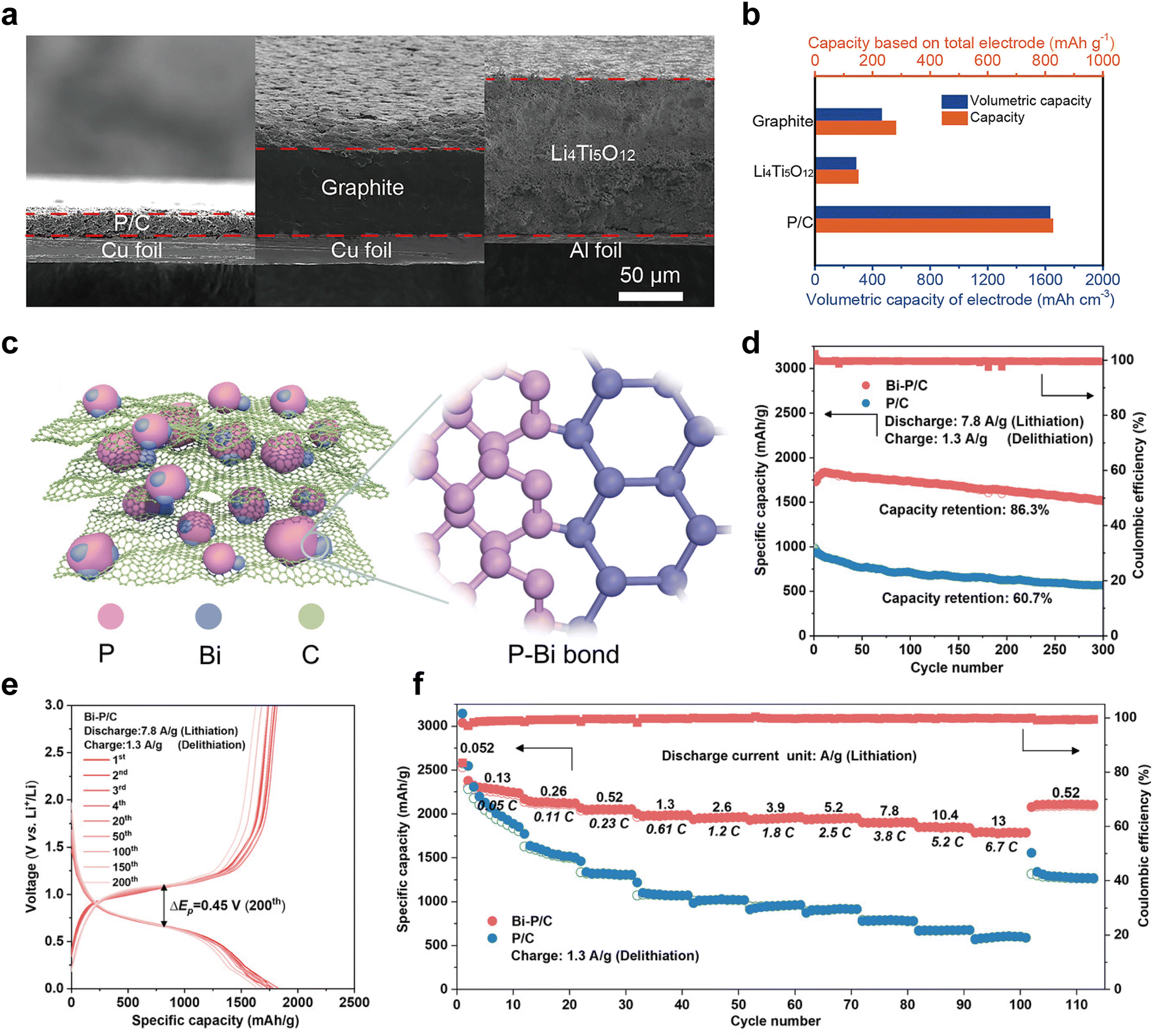 | ||
| Fig. 36 Applications of elemental phosphorus materials to LIBs. (a) Cross-sectional SEM images of P/C, graphite, and Li4Ti5O12 anodes with the same areal capacity of 3.5 mA h cm−2 at 0.5 mA cm−2. (b) Comparison of capacities and volumetric capacities of P/C, graphite, and Li4Ti5O12 anodes with the same areal capacity of 3.5 mA h cm−2 at 0.5 mA cm−2. Reproduced with permission from ref. 29. Copyright 2019 Elsevier. (c) Scheme of the synthesis of Bi–P/C composites. (d) Long-cycling stability of Bi–P/C and P/C at 7.8 A g−1. (e) Corresponding voltage profiles of Bi–P/C at 7.8 A g−1. (f) Rate performance comparison between Bi–P/C and P/C anodes across a range of rates from 0.052 to 13 A g−1. Reproduced with permission from ref. 551. Copyright 2022 John Wiley and Sons. | ||
Sun et al. have proposed that the formation of robust P–C bonds in P/C composites can minimize electrical contact loss between the active materials and the carbon conductor and improve the cyclic stability of P/C composites.549 They evaluated the impact of the carbon structure on stable P–C bond formation by comparing four common carbon materials: graphite, graphite oxide, carbon black, and fullerene. Their findings demonstrate that the black-P–graphite composite has the greatest proportion of P–C bonds, making it an optimal choice as a LIB anode material with extended cycle life and high-rate performance. At a constant current of 0.2C, the black-P–graphite composite displays an impressive initial discharge capacity of 2786 mA h g−1 and excellent stability. Specifically, it maintains a remarkable capacity of 1849 mA h g−1 after 100 cycles, whereas the capacity of black-P/graphite without robust P–C bonds declines rapidly after only 40 cycles. However, the fast-charging capabilities of P/C composites are limited by the high lithium diffusion barrier (0.34 eV) of carbon carriers and the heterogeneous interface.609 To address these issues, Sun et al. have recently incorporated electrochemically active bismuth nanoparticles into P/C composites by ball milling (Fig. 36c) to improve fast-charging and cycling.551 In principle, Bi possesses a slightly higher initial lithiation potential than P, enabling it to act as a modest Li reservoir that traps Li during lithiation. Meanwhile, initial delithiation potential of Bi is slightly lower than that of P, allowing it to emit Li before P during delithiation. Moreover, the Bi anode exhibits a low Li+ diffusion barrier (0.08 eV) and excellent electron transfer ability. Its strong interaction with P promotes Li+ transport while reducing large transient stress and capturing dissolvable polyphosphide at elevated current densities. The optimized Bi–P/C anode delivers excellent electrochemical performance, with a substantial fast-charging capacity of 1755.7 mA h g−1 at 7.8 A g−1 (5.2C) and it maintains a capacity retention rate of 86.3% after 300 cycles, which is better than the 60.7% retention rate of the pristine P/C anode after 300 cycles (Fig. 36d).551 Morphological studies reveal that the Bi–P/C anode maintains a smooth and dense surface with a volume expansion of ∼164.7% after 300 cycles, while the P/C anode has huge surface cracks with a larger volume expansion of ∼268.2%. Moreover, the discharge platform of the Bi–P/C anode is around 0.66 V at the 200th cycle (Fig. 36e), effectively inhibiting Li dendrite formation. The ΔEp of the Bi–P/C anode (0.45 V) at the 200th cycle is considerably lower than that of P/C (0.91 V) at 7.8 A g−1, suggesting that the Bi–P/C electrode provides ultrafast lithiation/delithiation reaction kinetics. As depicted in Fig. 36f, the Bi–P/C anode exhibits remarkably high-rate discharge capacities at rates ranging from 0.052 to 13 A g−1, while the P/C anode shows inferior discharge capacities. Additionally, both Bi and LiBi are calculated to have high adsorption energies for LiP7 and LiP5, indicating their ability to capture and facilitate the conversion of soluble lithium polyphosphides, resulting in increased reversible capacity and improved cycle stability.
Introduction of a conductive polymer coating layer into P/C composites has been shown to be effective in stabilizing the solid–electrolyte interphase and preventing the formation of low-conductivity by-products during fast charging. Ji's group has developed a covalently bonded black-P/graphite anode coated with a thin polyaniline gel ((BP-G)/PANI), which has a high rate, high capacity, and robust cycling stability.554 The covalent bonding to graphite inhibits edge reconstruction of layered black-P and maintains the structure of phosphorus atoms surrounding the zigzag diffusion channel, while the conductive PANI facilitates charge transfer at the electrode–electrolyte interface (Fig. 37a). After 2000 cycles at current densities of 2.6, 5.2 and 13 A g−1, the reversible capacities reach 910, 790 and 440 mA h g−1, respectively (Fig. 37b). The high-rate performance of (BP-G)/PANI exceeds that of conventional carbon anodes and advanced silicon anodes and is comparable to that of several well-known high-rate materials (Fig. 37c). To probe the role of different components in (BP-G)/PANI composites, researchers have prepared several composite materials for comparison (Fig. 37d). The (BP-G)/PANI composite exhibits a reversible capacity of 1520 mA h g−1 at 0.26 A g−1, which is 10 times higher than that of BP-G without the PANI coating at the same rate. Furthermore, the discharge capacity of (BP-G)/PANI is ∼3 times that of (RP-G)/PANI, indicating the significant contribution of black-P to the capacity of (BP-G)/PANI. The synergistic effects of the composite enable excellent Li+ transport kinetics and high-rate and high-capacity lithium storage.
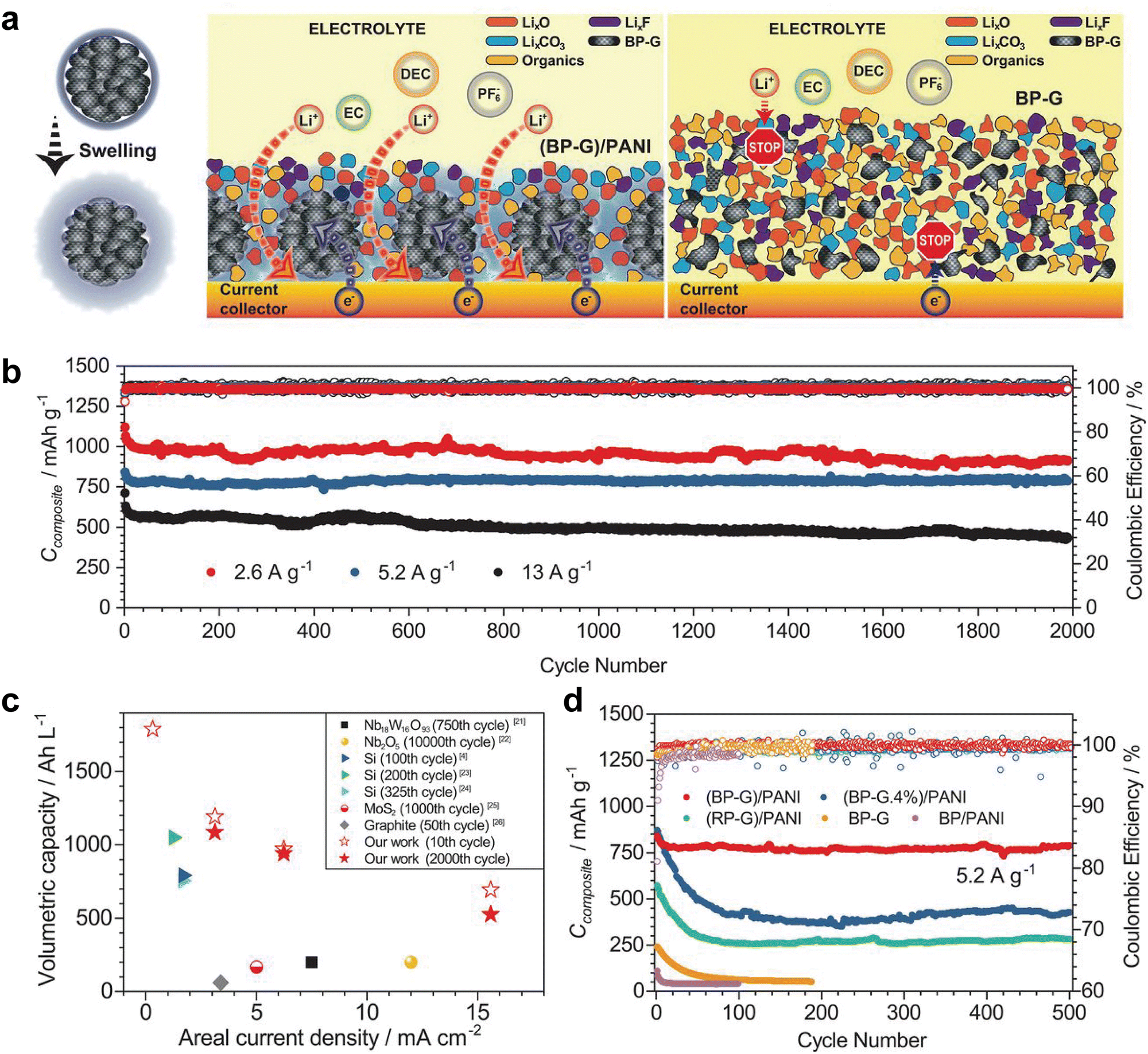 | ||
| Fig. 37 Engineered interface black-P composites designed for high-rate and high-capacity lithium storage. (a) Scheme of electrolyte-swollen (BP-G)/PANI that retains the solid–electrolyte interphase and pristine BP-G that suffers from the formation of less-conductive by-products during lithiation/delithiation. (b) Long-cycling tests of (BP-G)/PANI at current densities of 2.6, 5.2 and 13 A g−1. (c) Volumetric performance metrics of the (BP-G)/PANI composite in comparison to a range of high-rate materials. (d) Cycling performance comparison between the (BP-G)/PANI composite and various control samples. Reproduced with permission from ref. 554. Copyright 2020 Science. | ||
Graphene is typically arranged in a layered honeycomb-like pattern, which enables ion insertion and extraction. However, the limited interlayer spacing in graphene is insufficient for sodium ions, significantly constraining the gravimetric capacity of graphene-based anode materials. In contrast, black-P exhibits a considerably larger interlayer spacing than graphene (3.08 Å vs. 1.86 Å), indicating its potential to accommodate more sodium ions per gram, resulting in a higher theoretical specific capacity (2596 mA h g−1). The sodiation mechanism in black-P involves two processes: intercalation and alloying (Fig. 38a).565In situ TEM and ex situ XRD studies have revealed that intercalation occurs within interlayer spaces of black-P, where Na+ preferentially diffuses along the one-dimensional pore channels (zigzag ridges) due to the wide channel size. The channel along the zigzag direction has a sufficient width of 3.08 Å to permit Na+ (2.04 Å) diffusion, whereas the channel width along the armchair direction is only 1.16 Å and cannot accommodate Na+ diffusion. During the intercalation process, the host structure of black-P remains largely unaltered, resulting in a highly reversible cycle in the 0.54–1.5 V range. However, the specific capacity is limited to 150 mA h g−1 at this point, with a spatial conformation of Na0.17P. Further sodiation below 0.54 V proceeds to the alloying reaction stage, where the P–P bond is broken, leading to the formation of amorphous NaxP. Eventually, as the potential reaches 0.1 V, the Na3P phase is formed, and the alloying reaction is almost complete. However, this is accompanied by a significant volume expansion of over 500%. First-principles calculations also support this proposed mechanism. The diffusion energy barriers for sodium atoms along the zigzag, armchair, and interlayer directions are 0.18, 0.76, and 4.2 eV, respectively.619 These values indicate that sodium atoms primarily follow one-dimensional pathways along the zigzag ridges in black-P. This diffusion behaviour is similar to that observed in nanotube materials along their pores, suggesting that black-P could potentially exhibit exceptional rate capacity when employed as an anode material for SIBs.619,620
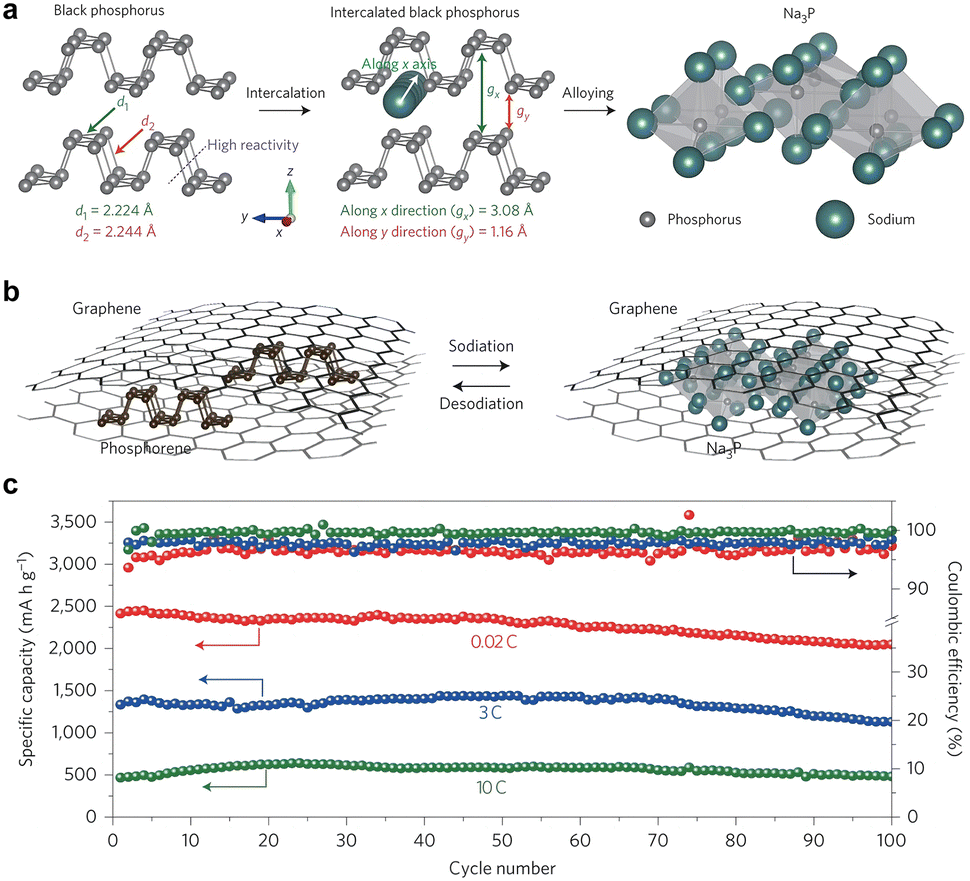 | ||
| Fig. 38 Sodiation process and electrochemical performance in black-P-based anodes. (a) Scheme of the mechanism of sodiation in black-P. (b) Scheme of sandwich-like phosphorene–graphene nanohybrids during sodiation/desodiation. (c) Cycling performance of phosphorene–graphene nanohybrids evaluated at different current densities. Reproduced with permission from ref. 565. Copyright 2015 Nature Publishing Group. | ||
Huang et al. have electrochemically exfoliated black-P bulk crystals into few-layered phosphorene and directly utilized the obtained phosphorene as an anode material for SIBs.564 Electrochemical tests reveal that few-layered phosphorene exhibited an activation capacity of up to 1968 mA h g−1 at 100 mA g−1 and a capacity retention of 60.5% after 50 cycles. At a higher current density of 1500 mA g−1, the retained capacity is 603.3 mA h g−1 after 100 cycles, suggesting the great potential of phosphorene as an SIB anode material. Given that phosphorene is directly used in this work, the electrochemical performance can be further enhanced by doping and compositing strategies. As a representative example, Cui's group has prepared sandwich-like phosphorene–graphene nanohybrids using a self-assembly method (Fig. 38b).565 In this hybrid material, phosphorene nanosheets reduce the diffusion distance for both sodium ions and electrons to improve the rate performance. Simultaneously, the graphene nanosheets facilitate electron transport from phosphorene to the current collector and provide a buffer space for the anisotropic expansion of phosphorene during Na3P alloy formation. These lead to exceptional electrochemical performance with a high specific capacity, rate capability, and capacity retention. Specifically, the phosphorene–graphene composite shows a specific capacity of 2440 mA h g−1 at 0.02C (50 mA g−1) and maintains a reversible capacity of 2080 mA h g−1 after 100 cycles (with a decay of only 0.16% per cycle). Moreover, even at the significantly higher rate of 10C (10 A g−1), a specific capacity of 645 mA h g−1 can be achieved (Fig. 38c).
As one of the most prevalent phosphorus allotropes, cost-effective red-P possesses a high theoretical sodium capacity and a relatively low safe working potential (0.4 V for Na+/Na),621 which promotes fast charging without the risk of dendrite formation. However, the fundamental mechanism behind its long-cycle capacity decay remains a subject of debate. It has been assumed that electrochemical fading of red-P-based anodes is similar to the decay process of silicon anodes in LIBs, wherein the anode undergoes significant volume expansion during long-term cycling, leading to severe particle pulverization and active material losses.583,622,623 Nevertheless, Liu et al. have discovered the “liquefication” phenomenon of red-P in SIBs by in situ TEM and chemo-mechanical simulations.584 This finding suggests that red-P should be less susceptible to volume fluctuation and fracture compared to the silicon anodes in LIBs. Instead, the primary cause of capacity decay in red-P-based anodes stems from side reactions that occur during the formation of reactive sodium phosphides. To mitigate capacity decay, researchers have developed an effective encapsulation technique, that is, synthesis of red-P-impregnated carbon nanofiber composites (Pred@CNF), which has been confirmed to prevent undesirable side reactions in red-P anodes. They employed a dry-format electrochemical cell setup (Fig. 39a) for real-time observation of the sodiation process.584Fig. 39b illustrates the “liquid-like” material behaviour of red-P during sodiation, where red-P may produce low-yielding stress and exhibit low stiffness (i.e., softening) as sodiation continues, thus allowing red-P to flow plastically inside the CNF like a fluid.584 Model stimulations in the various states of sodiation (Fig. 39c) align well with the longitudinal flow of red-P observed by in situ TEM. The average longitudinal expansion of red-P along the CNF is ∼330% during the complete sodiation process (Fig. 39d and e). Another series of cropped time-lapse STEM images display two distorted and blended red-P segments during the sodiation expansion (Fig. 39f), resembling two droplets and directly confirming the sodiation induced red-P softening effect. In contrast to a free-standing red-P segment, the CNF-encapsulated red-P exhibits reduced tensile stresses near the segment surface and is mechanically constrained by the CNF shell (Fig. 39g and h) to prevent fracture formation in the red-P segments. The designed Pred@CNF anode demonstrates a specific capacity of 1850 mA h g−1 at 0.1 A g−1 after 500 cycles, and over 1000 mA h g−1 after 5000 cycles at 1 A g−1.
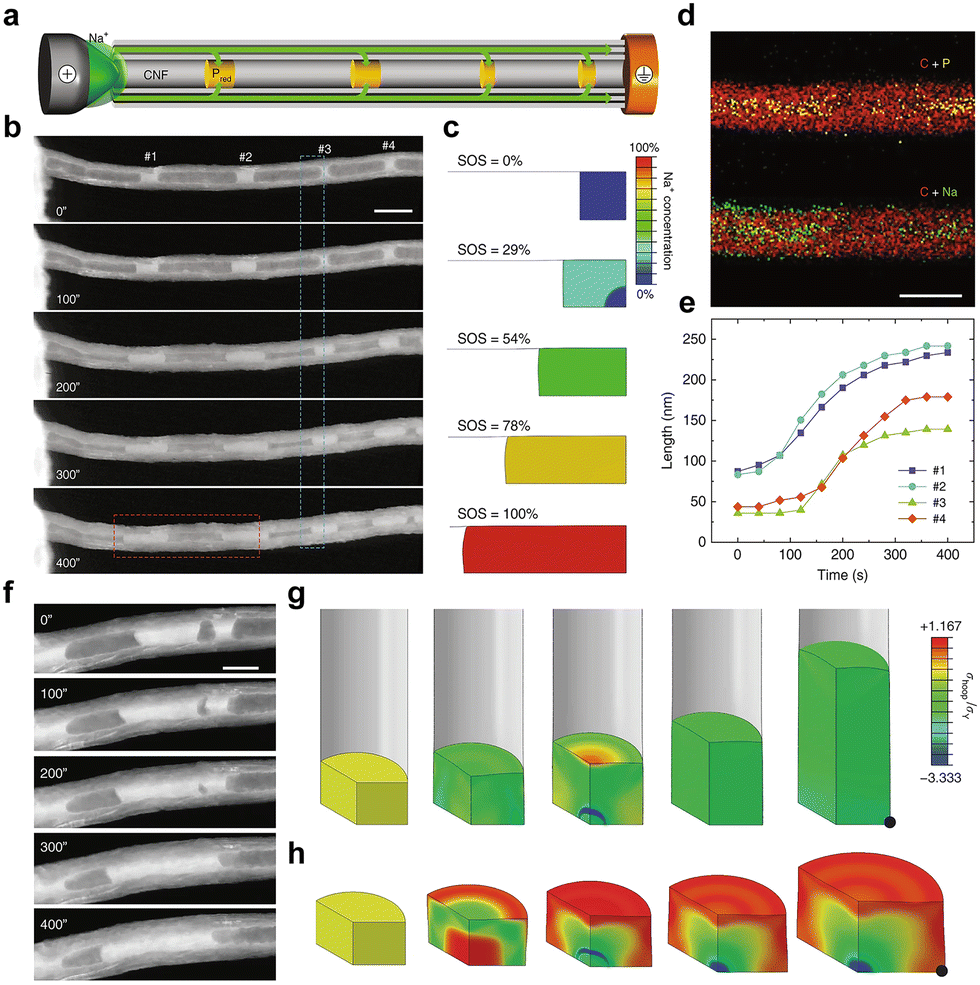 | ||
| Fig. 39 In situ TEM analysis and corresponding chemo-mechanical simulations of Pred@CNF composites. (a) Scheme of in situ TEM experimental setup of a dry-format electrochemical cell. (b) STEM image series captured in real-time showing red-P volume expansion during sodiation. The scale bar is 200 nm. (c) Simulated state of sodiation (SOS) corresponding to the morphological changes in red-P in the region labelled with a blue box in (b). (d) Energy dispersive spectroscopy mapping of phosphorus (green), sodium (yellow), and carbon (red) elements of the region labelled with a red box in (b). The scale bar is 200 nm. (e) The length change of four red-P segments marked in (b) over time during sodiation. (f) STEM image series captured in real-time showing two red-P segments merging during sodiation. The scale bar is 100 nm. (g) Simulated hoop stress distribution of Pred@CNF and (h) a free-standing red-P particle. Reproduced with permission from ref. 584. Copyright 2020 Nature Publishing Group. | ||
Heterojunction construction can enhance the electrochemical stability and conductivity of phosphorus-based electrodes.578,624,625 However, the incorporation of another component in these heterojunctions often results in a lower theoretical capacity (e.g., carbon materials with a theoretical capacity of 530 mA h g−1),626 sacrificing a part of the total electrode capacity. To optimize the stability of phosphorus-based electrodes while preserving the theoretical capacity as much as possible, it is beneficial to develop monoelemental phosphorus heterojunctions. Recently, Ma et al. have fabricated RP@BP core–shell heterostructures on 3D nitrogen-doped graphene using a straightforward one-step solvothermal method.563 The resulting RP@BP core–shell heterogeneous structure features a crystalline black-P shell tightly enclosing an amorphous red-P core. The internal electric field at the heterojunction not only provides exceptional electron conductivity and an extremely low Na+ diffusion barrier, but also induces electron cloud transfer from black-P to red-P. This suppresses the reactivity of lone-pair electrons in black-P atoms, resulting in excellent air stability of the composite. Electrochemical tests reveal that the composite anode exhibits an impressive capacity of 1440.2 mA h g−1 at 0.05 A g−1, superior to that of both pristine red-P (1137.9 mA h g−1) and black-P (713.3 mA h g−1) supported on 3D nitrogen-doped graphene.563 The composite anode also delivers an exceptional rate performance with a reversible capacity of 521.3 mA h g−1 achievable at 10.0 A g−1. Moreover, the electrode displays great electrochemical and air stability with a retention rate of 89.3% after 1200 cycles at 10.0 A g−1 and stable cycling over 500 cycles at 10.0 A g−1 after exposure to air.
The synthesis of crystalline red-P phases, such as fibrous-P and Violet-P, remains a challenging endeavour with limited reports on their utilization for SIBs. Nevertheless, the crystalline phase offers several distinct advantages over its amorphous counterpart. First, the crystalline lattice structure enhances charge mobility and electrode conductivity.33 Second, crystalline red-P generates polymeric chains,351 which mitigate the occurrence of side reactions during the charge/discharge process. Third, the crystalline structural phase enables a more comprehensive understanding of the sodiation mechanism in red-P anodes. Yan et al.627 have synthesized a 3D structured nanocomposite by incorporating highly dispersed crystalline red-P (violet-P phase) nanorods within a reduced graphite oxide aerogel matrix. This composite anode has a remarkable initial specific capacity of 2427 mA h g−1 with an initial Coulombic efficiency of 82% at 0.1 A g−1 and capacity retention even at higher current densities. This investigation highlights the potential of crystalline red-P as promising anode materials for SIBs.
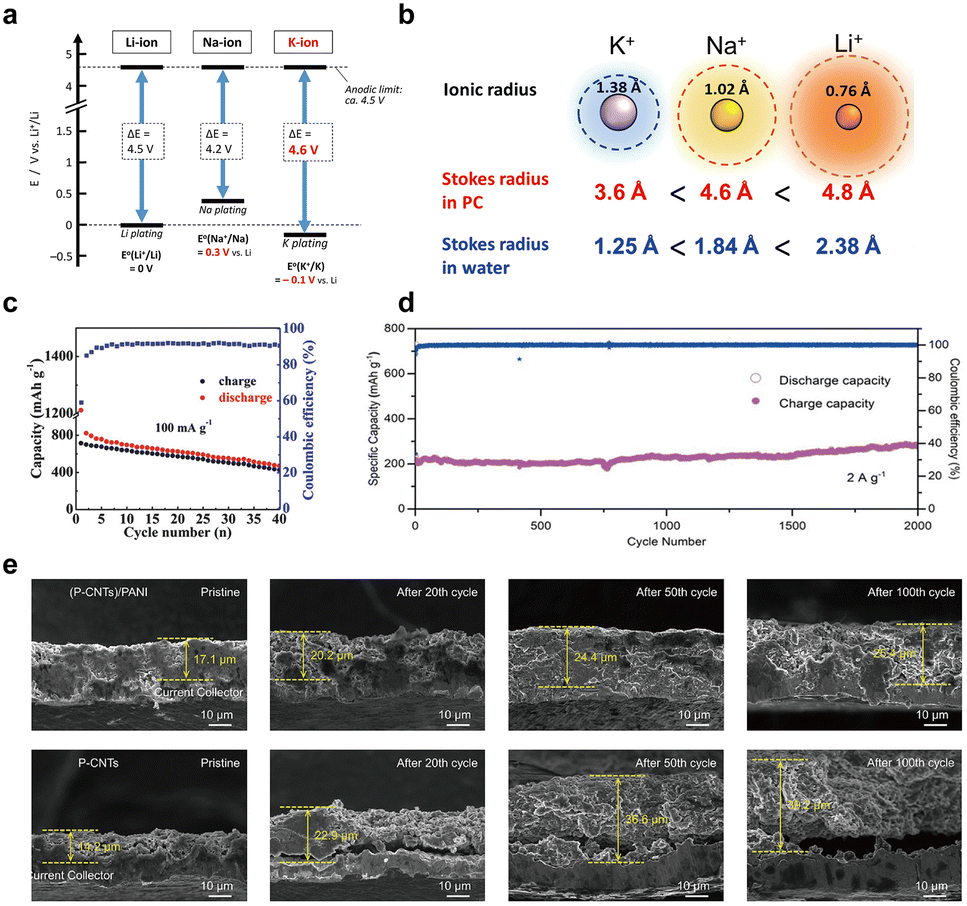 | ||
| Fig. 40 Applications of elemental phosphorus materials to PIBs. (a) Electrochemical voltage windows of LIBs, SIBs, and PIBs in carbonate electrolyte solutions. (b) Comparison of ionic radii and Stokes radii in propylene carbonate and water among Li+, Na+, and K+. Reproduced with permission from ref. 629. Copyright 2020 American Chemical Society. (c) Cycling performance of the red-P/3D carbon nanosheet framework nanocomposite at 100 mA g−1. Reproduced with permission from ref. 634. Copyright 2018 John Wiley and Sons. (d) Long-term cycling performance of red-P/3D sulphur and nitrogen co-doped carbon nanofiber electrodes at 2 A g−1. Reproduced with permission from ref. 637. Copyright 2022 John Wiley and Sons. (e) SEM images of the thickness of (P-CNTs)/PANI (top) and P-CNTs without PANI coating (bottom) in the initial and depotassiation states after 20, 50, and 100 cycles at 1000 mA g−1. Reproduced with permission from ref. 569. Copyright 2022 John Wiley and Sons. | ||
However, the large ion radius of K+ leads to sluggish potassiation kinetics,631 which poses a challenge in selecting suitable electrode materials. For example, silicon-based materials that exhibit a high capacity for LIBs are inactive in PIBs.632 The commercial graphite anode for KIBs only achieves a capacity of 279 mA h g−1.633 Despite various modification strategies being developed for graphite, the capacity improvement has remained limited due to the intrinsic properties of carbonous materials. In contrast, the theoretical capacities of EPMs upon K4P3 and KP formation are 1154 and 843 mA h g−1, respectively.590,634 To the best of our knowledge, these are the highest specific capacities among all elements for PIB anodes, indicating that phosphorus-based anodes have a higher upper limit for PIB development. However, the existence of potassium phosphide-rich polymorphisms (such as K3P, KP, K4P3, and K3P11) makes it difficult to investigate the potassium storage mechanism of phosphorus.635 Currently, research on PIBs for phosphorus-based anodes mainly focuses on well-established EPMs including amorphous red-P and black-P, as well as their heterostructures with carbon or metal species.
Amorphous red-P has been investigated for PIBs due to its non-toxicity and cost-effectiveness for mass production. However, its intrinsic low conductivity (∼10−14 S cm−1) and significant volume expansion during the potassiation/depotassiation make pure red-P unsuitable as electrodes. To address this issue, researchers have explored the incorporation of highly conductive carbon materials into the phosphorus anode to enhance its storage performance. In 2017, Zhang et al. measured the K+ storage capacity of red-P-based composites.636 They utilized red-P and carbon black as precursors to prepare the P/C anode via ball milling and electrochemical tests showed that this P/C anode exhibited an initial discharge capacity of 2171.7 mA h g−1 at 50 mA g−1, significantly surpassing the performance of the Sn4P3/C anode (588.7 mA h g−1). However, the P/C anode displayed rapid capacity fading, experiencing a 91% reversible capacity decay by the 20th cycle and reaching near-zero capacity by the 50th cycle. This capacity fading arose from the disordered microstructure of the composite, which impeded interfacial charge transport. To address the significant structural changes during the potassiation/depotassiation process, Xiong et al. anchored red-P nanoparticles to a 3D porous carbon nanosheet framework.634 This approach yielded an initial charge capacity of 715.2 mA h g−1 at 100 mA g−1 through KP alloy formation, with a capacity retention of 427.4 mA h g−1 after 40 cycles (Fig. 40c). Although this method demonstrates improved cycle stability compared to Zhang's work,636 there are limitations. Yu's group has further addressed the aggregation and pulverization issues of red-P particles by incorporating them into free-standing nitrogen-doped porous hollow carbon nanofibers, achieving a remarkable reversible capacity and exceptionally long cycle life (465 mA h g−1 at 2 A g−1 after 800 cycles).590 Compared to nitrogen, lower electronegativity of sulphur leads to stronger chemical interactions with phosphorus atoms, increasing the adsorption energy of phosphorus atoms on sulphur-doped carbon matrices. Feng and colleagues have demonstrated that encapsulating strain-relaxed red-P within 3D interconnected multi-channel sulphur and nitrogen co-doped carbon nanofibers can enhance potassium ion capture and promote electrochemical reaction kinetics.637 This composite structure not only buffers the stress and strain caused by significant volume expansion, but also utilizes the defect sites and high electronic conductivity generated by the synergistic effects of dual heteroatom doping. Impressively, the composite sustains a reversible capacity of 282 mA h g−1 after 2000 continuous cycles even at a high current rate of 2 A g−1 (Fig. 40d). In addition, employing metallic Bi as an electrochemically active coating has recently been shown to boost the electrical conductivity and cycle stability of red-P-based composites.638
The unique thermodynamically stable structure and high electronic conductivity (∼102 S cm−1) of black-P make it a potential anode material for PIBs. However, pristine black-P is not practical for PIBs, and engineering is necessary to achieve reasonable electrochemical stability. Sultana et al.568 have synthesized a black-P-carbon nanocomposite by ball milling and compacted the nanocomposites into dense aggregates to improve packaging of the black-P active surface. The nanocomposite shows a first cycle capacity of 617 mA h g−1 at 50 mA g−1 in 0.75 M KPF6 in ethylene carbonate/diethyl carbonate (1:1 by vol.), which is more than twice that of the graphite anode (270 mA h g−1). However, the cycle stability of the nanocomposite is relatively limited. After 50 cycles at the same current rate, the anode capacity decays to around 30 mA h g−1. The gradual degradation of black-P during the cycle is responsible for the destruction of the solid electrolyte interphase layer and subsequent effects on the heterogeneous charge transfer kinetics. To improve the cycle stability of black-P-based materials, Lu's group has recently proposed the use of a PANI coating layer to alleviate black phosphorene degradation and replacement of graphite with regular nanostructured carbon materials to improve electrode conductivity.569 They have assembled carbon nanotubes and black-P flakes by ball milling and performed in situ polymerization of PANI to obtain a phosphorene–carbon nanotubes/polyaniline ((P-CNTs)/PANI) composite. In 3 M KFSI/dimethoxyethane electrolyte, (P-CNTs)/PANI shows a maximum charge capacity of 417.6 mA h g−1 at 500 mA g−1 and maintains a reversible capacity of 392.5 mA h g−1 and an impressive retention rate of 94% after 500 cycles. Morphological studies show that the (P-CNTs)/PANI electrode has good structural stability during the potassiation/depotassiation process (Fig. 40e). After 20, 50, and 100 cycles at 1000 mA g−1, the thickness of the (P-CNTs)/PANI electrode shows a slight increase with the number of cycles, but the overall electrode structure remains largely unchanged. In contrast, the P-CNT electrode without the PANI coating undergoes significant alterations in thickness and there are noticeable cracks in the electrode material. These observations suggest that the PANI coating mitigates volume expansion of the composite and prevents contact loss with the current collector during the charge/discharge process. Zhang et al.639 have demonstrated that the incorporation of a localized high-concentration electrolyte enhances the ionic conductivity of the electrolyte and facilitates the formation of a robust solid electrolyte interphase for improved long-cycling stability of black-P/C electrodes. This finding emphasizes the role of the electrolyte in determining the electrochemical performance of KIBs and motivates further exploration to develop electrolytes that complement the characteristics of phosphorus-based electrodes.
The electrode is a critical component of SCs and the material selection and structural design impact the electrochemical and cycling properties.642 EPMs comprise various phosphorus allotropes and corresponding nanostructures boding well for electrodes. EPMs have the following advantages as SC electrodes: (i) both black-P derivatives and red-P modifications feature nanoscale channels within their structures, allowing for shorter charge transfer paths and facilitating electrochemical reaction kinetics. (ii) Low-dimensional EPMs exhibit great mechanical flexibility and packing density,643 suggesting their potential use in fabricating sophisticated and flexible SCs. (iii) Low-dimensional EPMs possess large lateral dimensions and exposed active-site surfaces. In comparison with chemically inert carbon materials, EPMs are more likely to form composites with metal/non-metal species, enhancing electrochemical performance.
In 2016, Hao et al.570 investigated the electrochemical performance of liquid-exfoliated black-P nanoflakes in all-solid-state SCs. The fabricated flexible electrodes achieved a high stack volumetric capacitance of 13.75 F cm−3 (48.5 F g−1) at a scan rate of 0.01 V s−1 and a great capacitance retention of 71.8% at 0.5 V s−1 over 30000 cycles (Fig. 41a and b). Although this charge storage performance is better than that of several graphene-based and multi-walled carbon nanotube electrodes,646–648 the as-fabricated SCs still have some limitations, including a rapid volumetric capacitance reduction with increased charge/discharge rates, which is due to morphological degradation caused by long-term sonication. Yang et al.649 have described a modified electrochemical exfoliation approach for the production of high-quality few-layered black-P nanoflakes. Vacuum filtering of the black-P nanoflake dispersion yields free-standing thin films for flexible quasi-solid-state micro-SCs. The as-prepared micro-SCs show excellent steady electrochemical performance at a high scanning rate of even up to 100 V s−1 (Fig. 41c) comparable to that of MXene-based quasi-solid-state micro-SCs at the same scanning rate.649 The micro-SCs have a remarkable energy density of 3.63 mW h cm−3 at a power density of 0.247 W cm−3, surpassing that of many 2D metal/non-metal materials (Fig. 41d). The device also exhibits an ultralong cycling lifetime of over 50000 cycles at 0.2 A cm−3 and remarkable flexibility by retaining more than 91.3% of capacity after 500 bending cycles (Fig. 41e). Using 2D nanosheets as the building blocks in the 3D architecture can enhance the electrochemical performance of electrode materials, particularly for LIBs. To expand this strategy to SC electrodes, our group has reported the synthesis of a semi-connected 3D black-P sponge in three minutes under ambient conditions using a facile and simple electrochemical approach.571 As an electrode material for all-solid-state SCs, black-P sponge shows a specific capacitance of 80 F g−1 at 10 mV s−1, which is better than those of bulk black-P and black-P nanosheets. On the one hand, the high-quality ultra-thin nanosheets as a basic unit of the black-P sponge offer expanded active sites and lateral dimensions and remove the high energy barrier of individual nanosheets to promote the charge transfer kinetics. On the other hand, the pores and channels inside the sponge allow contact with the electrolyte for better ion diffusion.
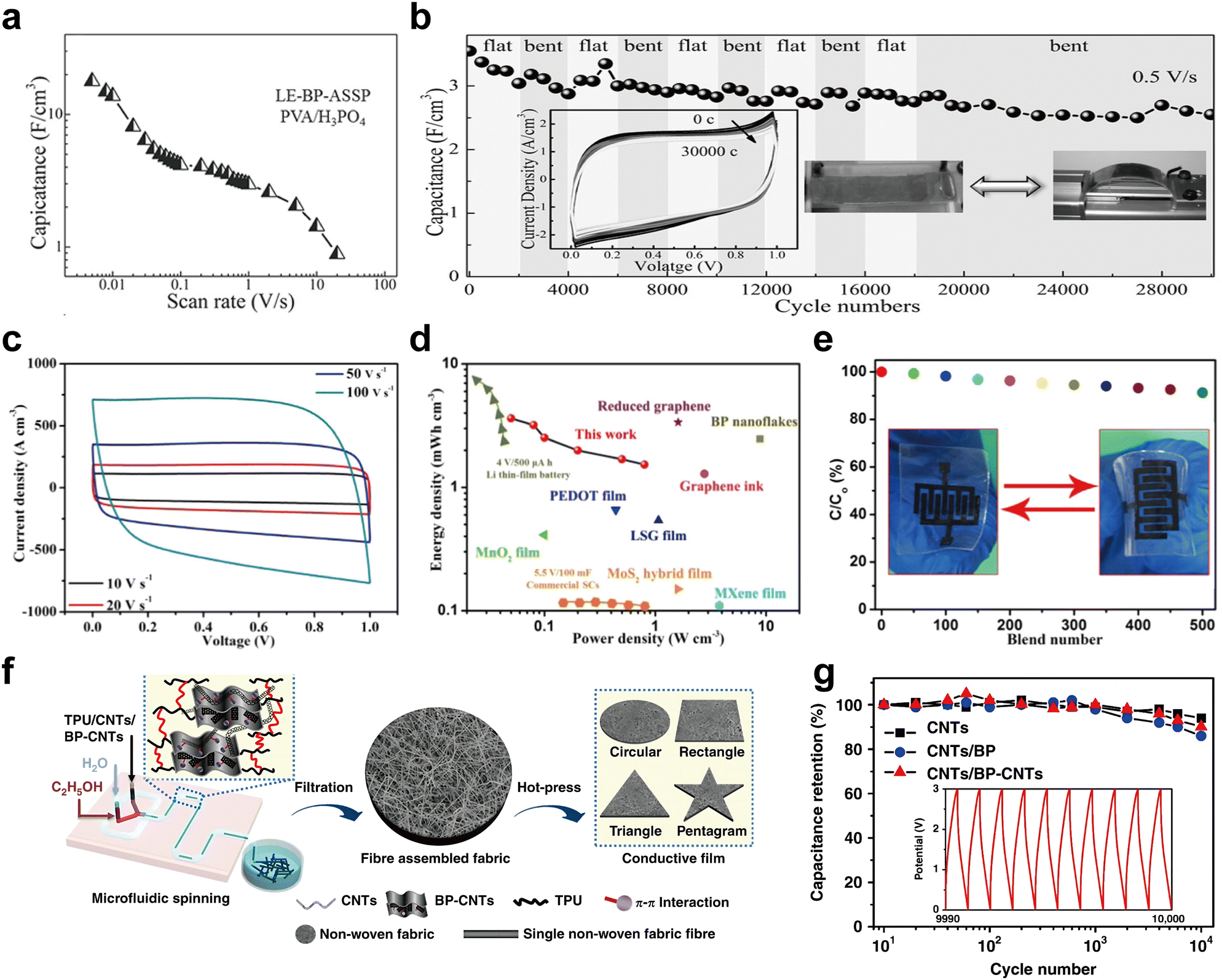 | ||
| Fig. 41 Applications of elemental phosphorus materials to SCs. (a) Stack capacitances of the liquid-exfoliated black-P based all-solid-state SC (LE-BP-ASSP) with PVA/H3PO4 gel electrolyte at different scan rates. (b) Cycle stability of LE-BP-ASSP at 0.5 V s−1 in alternating flat and bent configurations. The inset shows representative cyclic voltammetry curves and photographs of the device in both flat and bent configurations. Reproduced with permission from ref. 570. Copyright 2016 John Wiley and Sons. (c) Cyclic voltammetry curves of the as-prepared micro-SCs at various scan rates. (d) Energy densities and power densities of the micro-SCs compared to those of various previously reported SC devices. (e) The capacitance retention rate of different bending cycles for the micro-SCs. Reproduced with permission from ref. 649. Copyright 2019 American Chemical Society. (f) Microfluidic spinning technique for the assembly of non-woven fabrics, which can be tailored into various shapes. (g) Cycle stability of black-P-carbon nanotube based SCs at 0.4 A cm−3. The inset shows galvanostatic charge/discharge curves after 10000 cycles. CNTs refer to pristine carbon nanotubes. CNTs/BP refers to a physical mixture of carbon nanotubes and black-P. CNTs/BP–CNTs refer to the physical mixture of carbon nanotubes and carbon nanotubes chemical-bridged black-P, which achieved the best electrochemical performance among the three materials. Reproduced with permission from ref. 572. Copyright 2018 Nature Publishing Group. | ||
Nanostructures can enhance the specific capacitances of pure black-P electrodes, but achieving capacitance values in the hundreds or thousands remains highly challenging. To fabricate flexible SCs with a high energy density, the development of black-P-based multicomponent materials is necessary. Chen's group has chemically engineered black-P with carbon nanotubes to produce heterostructure composites.572 With the aid of the microfluidic spinning technique, the composites are assembled into non-woven fabrics (Fig. 41f) for high-performance SC electrodes. The optimal flexible SC shows an outstanding volumetric capacitance of 308.7 F cm−3 at 0.1 A cm−3, an energy density of 96.5 mW h cm−3, cycle stability with a 90.2% retention rate after 10000 cycles (Fig. 41g), and sustained bending durability. This enhancement stems from the open network structure of the composites featuring abundant well-defined micro/mesopores for enhanced conduction, rapid ion transport, and mechanical stability. Recently, Wu et al. have employed a droplet microfluidics method to fabricate an anisotropic black-P/ZIF-67 heterostructure composite and utilized 3D printing to construct fully integrated solid-state flexible SCs.573 The SC has a high volumetric energy density of 109.8 mW h cm−3 at a power density of 0.675 W cm−3, a capacitance of 506 F cm−3, and long-term stability for 12000 cycles. These research findings demonstrate the significant potential of SCs in next-generation wearable devices.
Researchers have also shown interests in red-P as an electrode material for SCs due to its environmental-friendless, cost-effectiveness, chemical stability, and high theoretical capacitance similar to that of black-P.650 However, as previously stated, the poor electrical conductivity of pristine red-P limits the charge storage effectiveness. To enhance the conductivity, red-P is often coupled with carbon materials such as graphene and carbon nanotubes through chemical bonding or vdW interactions.593 However, the main drawback of carbon materials is the low specific capacitance that impacts the energy storage efficiency. In this regard, black-P owns high conductivity and theoretical capacitance as well as compatibility with red-P. In 2017, Chen et al. reported the in situ synthesis of red-P/black-P composites using a facile sonication method and their application as SC electrodes.579 The cyclic voltammetry curves of the resulting electrode exhibited a rectangular-like shape, indicating the electric double-layer charge mechanism at the electrolyte/electrode interface.651 At a current density of 0.5 A g−1, the hybrid composite showed a specific capacitance of around 60.1 F g−1 and 83.3% capacity retention for 2000 cycles. This work not only establishes the excellent energy storage properties of red-P/black-P hybrids, but also provides a straightforward and cost-effective preparation method. Inspired by Chen's work, Gopalakrishnan and colleagues incorporated sulfonated porous carbon materials into red-P/black-P hybrids to boost the electrochemical performance of elemental phosphorus hybrids.592 As an SC electrode, the ternary composite has a high specific capacitance of 364.5 F g−1 and maintains long-cycling stability retaining 89% of capacity after 10000 charge/discharge cycles. The improved capacitive properties are associated with the synergistic effects between elemental phosphorus hybrids and porous carbon materials. The highly stable carbon frameworks featuring phosphorus bonding and heteroatom doping provide numerous active sites for ionic interactions and improve the surface wettability for seamless electrode/electrolyte interactions. Other EPMs such as blue-P and green-P have recently also shown promising theoretical potential as electrodes in SCs.604
Few-layered black-P nanosheets offer several advantages such as large interlayer spacing (∼3.08 Å), high sulfophilicity, and high lithium-ion mobility, rendering them ideal encapsulating materials for sulphur cathodes. Zhou et al. have constructed a 2D heterostructure of black-P/graphene for encapsulating sulphur species to provide catalytic and chemisorptive benefits for electrolyte-lean Li–S batteries.658 The 2D heterostructure not only enhances the electrochemical stability of black-P nanosheets, but also facilitates electronic regulation between black-P and graphene. The electric field generated at the heterostructure interface increases the intrinsic conductivity of the materials and accelerates electron transfer in the redox conversion of polysulfides. Concurrently, it promotes chemisorption of polysulfides and reduces the migration barrier for lithium ions to catalyze the conversion of polysulfides. These features contribute to low capacity fading with an average decline as low as 0.021% per cycle after 600 cycles at 2C. Xu et al.555 have demonstrated that BPQDs have a higher lithium polysulfide adsorption capacity and more substantial Li2S precipitation capacity than large black-P flakes (Fig. 42a and b). BPQDs show strong interactions with lithium polysulphide through P–S and P–Li bonds and the abundant active sites at the edges of BPQDs catalyze the conversion of lithium polysulphide. As a result, the porous carbon nanofibers/sulphur cathodes containing a small amount (2 wt% of the cathode weight) of BPQDs exhibit fast reaction kinetics, no polysulphide shuttling, and excellent cycling stability. The porous carbon nanofibers/sulphur cathode with BPQDs has an initial capacity of 1234 mA h g−1 at 0.1C and after 200 cycles, it retains a capacity of 1072 mA h g−1 (Fig. 42c) and shows a small capacity decay rate of 0.06% per cycle. In comparison, the porous carbon nanofibers/sulphur cathode without BPQDs has an initial capacity of only 1036 mA h g−1 and a decay rate of 2% per cycle. The BPQD containing cathode also delivers greater rate performance than the BPQD-free cathode (Fig. 42d).
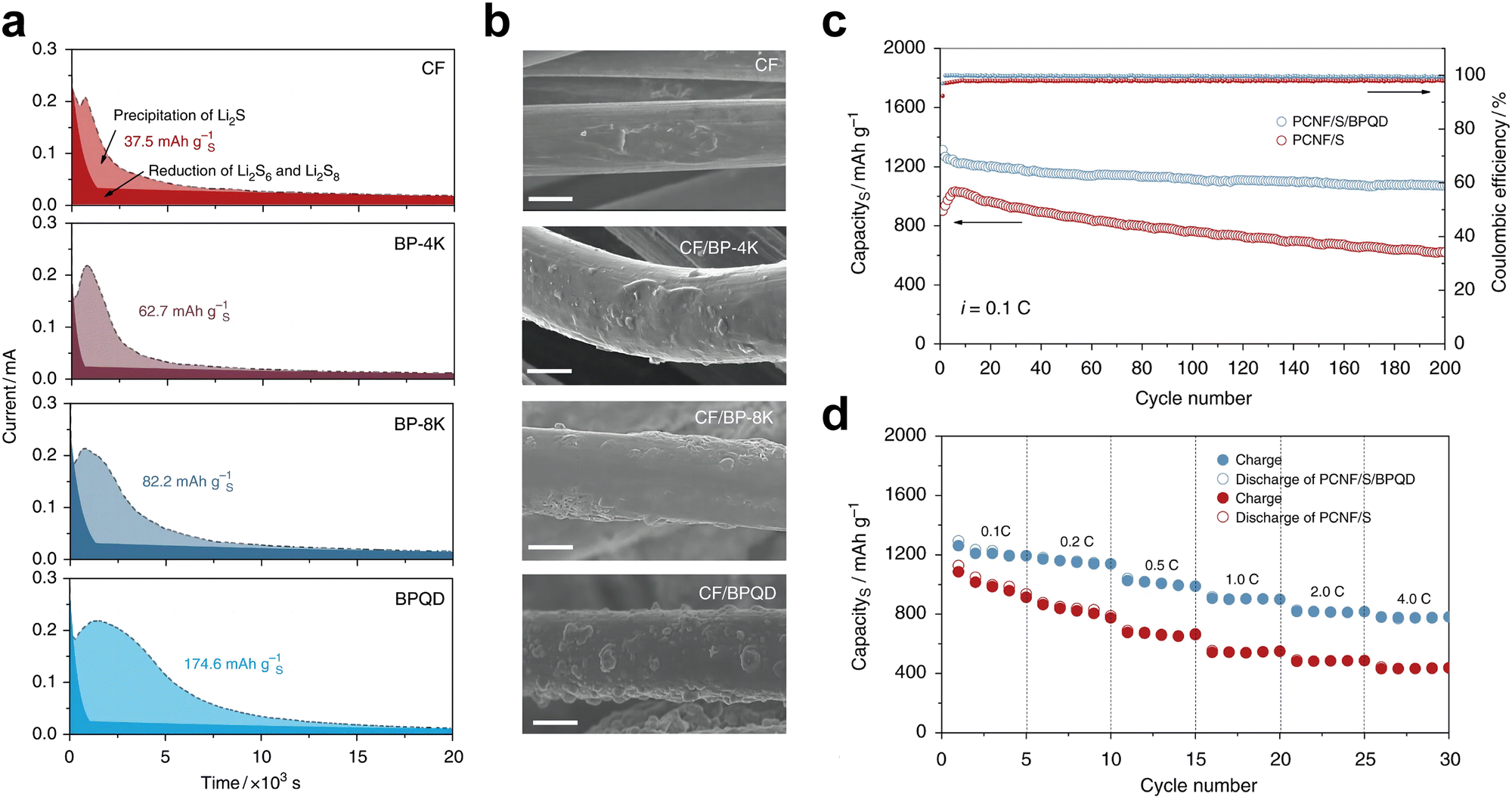 | ||
| Fig. 42 Applications of BPQDs to LSBs. (a) Potentiostatic discharge curves of Li2S8 tetraglyme solution on various substrates at 2.05 V. The dark/light colours denote the reduction of Li2S8/Li2S6 and the precipitation of Li2S, respectively. CF refers to carbon fibers. BP-4K and BP-8K refer to 4000 rpm and 8000 rpm centrifugated black-P products, respectively. (b) SEM images of the precipitation of Li2S on various substrates as indicated in (a). The scale bar is 5 μm. (c) Cyclic capacities and Coulombic efficiencies of porous carbon nanofibers/sulphur cathode containing BPQDs (PCNF/S/BPQD) and pristine PCNF/S at 0.1C for 200 cycles. (d) Cyclic capacities of PCNF/S/BPQD and PCNF/S at various rates. Reproduced with permission from ref. 555. Copyright 2018 Nature Publishing Group. | ||
While both the component and structural design of the cathode materials discussed above inhibit polysulfide shuttling and enhance the cycle stability, they do not eliminate shuttling, particularly during extended cycling. Consequently, researchers have explored separator modification by introducing active materials to physically restrict polysulfides or chemically anchoring polysulfides.656,659,660 The modified separators also decrease the impedance between the cathode and separator, enhance the wettability between the separator and electrolyte, and improve the electrochemical performance. Cui's group has prepared a uniform, dense black-P nanoflake coating on a Celgard commercial polypropylene separator by vacuum filtration deposition.556 The black-P coating intercepts and combines the polysulfides to prevent diffusion through the separator to the anode. The binding energy of the black-P coating to lithium polysulfide ranges from 1.32 to 2.82 eV (without vdW interaction) and 1.86 to 3.05 eV (with vdW interaction) and increases as the Li2Sx molecular chain shortens (x decreasing from 8 to 1). This energy is higher than that of the graphene coating (0.21–0.79 eV). The capacity of the cells with a conventional separator decays to below 100 mA h g−1 at 0.4 A g−1 after 100 cycles, while the capacity retention of separators modified with classical graphene is around 66%. In contrast, the black-P modified separator displays an initial discharge capacity of 930 mA h g−1 and retains a capacity of 800 mA h g−1 after 100 cycles. In further high-rate cycling studies, the specific capacity of the black-P modified separator remains at 623 mA h g−1 even at a high rate of 3.5 A g−1.
In addition to black-P, red-P is used in separator modification. Red-P is a weak Lewis base agent that can undergo reasonable Lewis acid–base interactions with weak Lewis acids of lithium polysulphide, allowing it to chemically anchor the polysulphide. Concurrently, red-P can anchor polysulphides through sulphur chain interactions to inhibit shuttling and the interaction between red-P and polysulfide leads to the production of a high-conductivity by-product Li3PO4 which aids in Li+ transport in the separator and promotes the sulphur reaction kinetics. Wang et al. were the first to utilize red-P nanoparticles to modify conventional polypropylene separators, capturing and reusing lithium polysulphide through strong chemical interactions between red-P and polysulfides.661 The LSBs based on red-P modified separators have excellent cycling properties. The unmodified polypropylene separators experienced substantial pore blockage due to polysulfide penetration with polysulphide decomposing, precipitating, and obstructing the pores. In contrast, the red-P modified separator maintained a highly porous surface to inhibit polysulfide penetration. The surface of lithium after cycling with the red-P modified separator was the smoothest, while the surface of lithium with the pristine polypropylene separators exhibited a large number of cracks and dendrites. This result suggests that red-P-based separators inhibit polysulphides and safeguard the lithium anode from corrosion. Consequently, the red-P modified separators achieved a high capacity retention of 82% during a long cycle test of 500 cycles at a current density of 1C, better than that of super P-coated separators (65%) and pristine polypropylene separators (43%). The red-P-based separators also had an excellent rate capability of 809 mA h g−1 at a high rate of 2C. Moreover, incorporating an independent functional interlayer between the cathode and separator of LSBs can physically block and chemically absorb lithium polysulfide. An interlayer with exceptional conductivity can function as a secondary current collector to improve the cathode conductivity. Lee et al.662 prepared a red-P nanoparticle-coated carbon nanotube film as an interlayer by pulsed laser ablation and direct spinning. The interlayer provides efficient Li+ and electron transport as well as strong chemical anchoring of lithium polysulfide. Consequently, the interlayer has a superior specific capacity of 782.3 mA h g−1 at 3C, long-term cycle stability, and high Coulombic efficiency. At a rate of 1C, the specific capacity initially reaches 1050.2 mA h g−1 and remains at 769.5 mA h g−1 after 500 cycles, with a Coulomb efficiency exceeding 99%.
Lithium-air batteries (LABs), which operate through the reversible reaction of Li + O2 ↔ Li2O2, have the potential to offer an impressive theoretical energy density of around 3500 W h kg−1. However, the sluggish kinetics of the cathode leads to high overpotentials, substantial polarization, and poor reversibility. As discussed in Section 5.1.4, EPMs inherently offer several benefits for electrocatalytic oxygen evolution, rendering them promising candidates to enhance the reaction kinetics of LABs. Cheng et al.557 have designed a BPQD-modified δ-MnO2 catalyst and prepared it on carbon cloth using a hydrothermal method as a binder-free cathode for LABs. The Li–O2 cell with this BPQD modification shows a high capacity of 8463 mA h g−1 at 100 mA g−1 (Fig. 43a) which is greater than the capacity of 2965 mA h g−1 without BPQD modification (Fig. 43b), demonstrating the high catalytic activity of BPQD/δ-MnO2 in the oxygen reduction reaction (ORR). In the cyclic voltammetry test (Fig. 43c), the BPQD/δ-MnO2 electrode exhibits a higher onset potential for the ORR (2.64 V) and a lower onset potential for the OER (3.04 V) than the δ-MnO2 electrode without BPQD modification.557 Additionally, the peak OER current of the BPQD/δ-MnO2 electrode increases significantly, suggesting that the introduction of BPQDs enhances the electrochemical reaction kinetics. Cycling stability tests (Fig. 43d–f) reveal that BPQD/δ-MnO2 polarization gradually increases with the cycle number, whereas δ-MnO2 polarization increases rapidly. These findings indicate that BPQDs enhance the ORR/OER catalytic activity, leading to superior discharge capacity and cycling stability for LABs. Moreover, researchers have identified black phosphorene and nitrogen-doped blue-P as highly effective catalysts for cathodic reactions through theoretical calculations.601,663
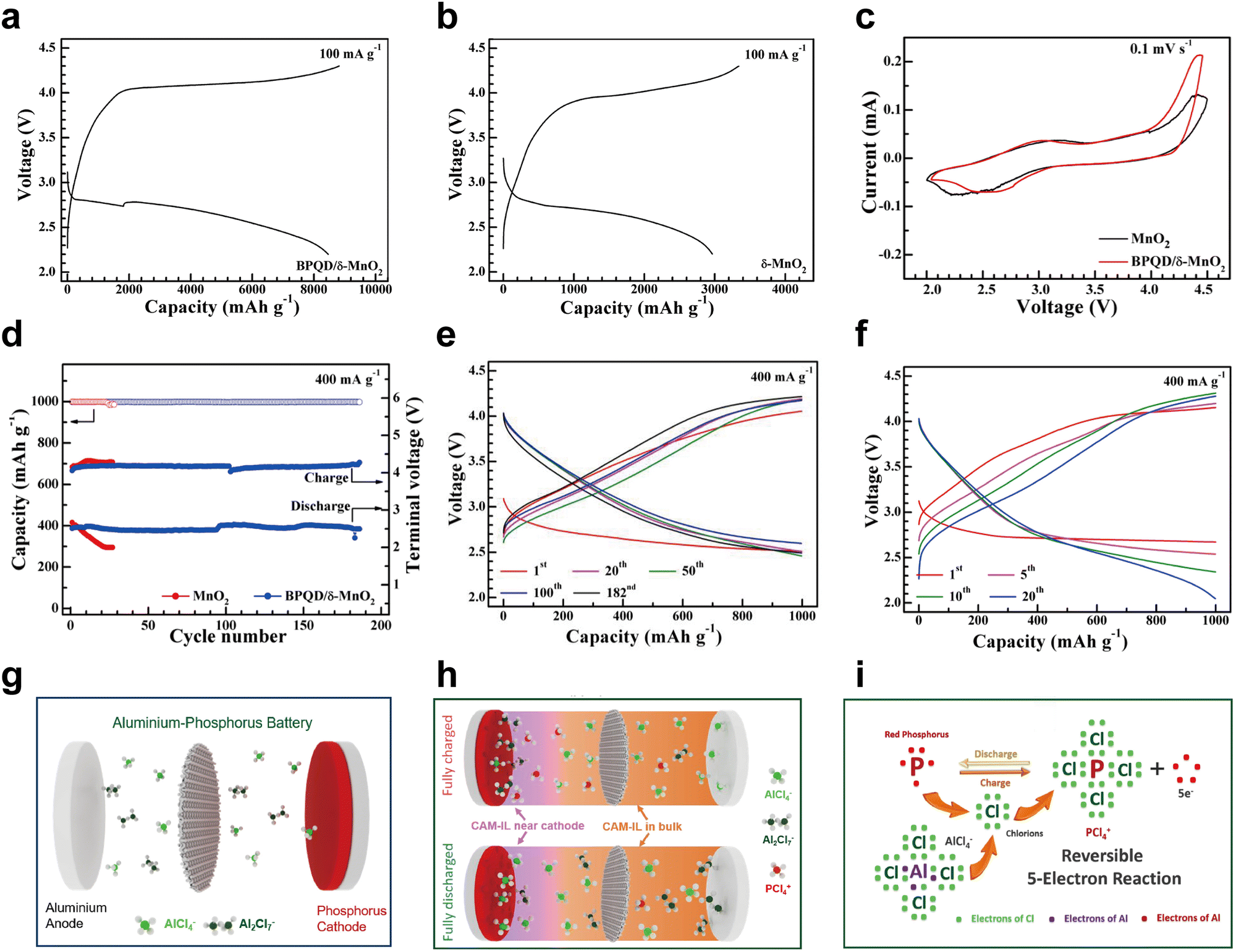 | ||
| Fig. 43 Applications of elemental phosphorus materials to other energy storage applications. (a) Voltage curves of BPQD/δ-MnO2 and (b) δ-MnO2-catalyzed Li–O2 cells at 100 mA g−1. (c) Cyclic voltammetry curves of BPQD/δ-MnO2 and δ-MnO2-catalyzed Li–O2 cells at 0.1 mV s−1. (d) Cycling performance of Li–O2 cells with BPQD/δ-MnO2 and δ-MnO2 cathodes at 400 mA g−1 with a capacity limit of 1000 mA h g−1, and (e) voltage curves of BPQD/δ-MnO2 and (f) δ-MnO2-catalyzed Li–O2 cells. Reproduced with permission from ref. 557. Copyright 2019 Elsevier. (g) Scheme of an aluminium–phosphorus battery comprising a phosphorus cathode, aluminium anode, and CAM-IL electrolyte. (h) Scheme of the distribution of AlCl, Al2Cl, and PCl in an Al–P battery under a fully charged/discharged state. (i) Scheme of the reversible phosphorus-based five-electron transfer processes. Reproduced with permission from ref. 664. Copyright 2022 Elsevier. | ||
Recent advances have shown that EPMs are being developed for new types of energy storage devices. Cai et al. have reported an innovative aluminium–phosphorus battery which incorporates red-P as the cathode, aluminium as the anode, and CAM-IL as the electrolyte (Fig. 43g).664 This battery boasts a high theoretical capacity of 4325 mA h g−1 due to the reversible, phosphorus-based five-electron transfer reaction as shown in Fig. 43h and i. The researchers have assembled a prototype aluminium–phosphorus battery showing a capacity of 1512 mA h g−1 and an energy density of 1176 W h kg−1, which surpass those of many alkaline metal ion batteries. By employing modification strategies, the cutting-edge aluminium–phosphorus battery is anticipated to have large potential in low-cost, high-performance energy storage applications due to the multiple electron transfer reactions and abundant availability of both phosphorus and aluminium.
In summary, EPMs possess many intrinsic advantages including high theoretical capacity, resource abundance, and environmental compatibility and are promising candidates for various energy storage devices such as hydrogen storage systems, alkali metal batteries, and supercapacitors. As researchers delve deeper into various modification strategies including structural design and nanosizing, practical application challenges such as structural and electrochemical stability degradation stemming from the volume expansion of phosphorus allotrope materials must be addressed. The development of innovative energy storage devices with various phosphorus allotropes like aluminium–phosphorus batteries will offer more viable alternatives for energy storage systems. Arguably, the most prominent potential application of EPMs currently lies in the realm of energy storage. However, all these applications are still in their infancy and lack industrial-grade standardized testing. Therefore, further efforts are necessary to transition from research to commercial application.
5.3. Environmental remediation applications
Previous theoretical studies have demonstrated that EPMs exhibit ultra-high sensitivity and selectivity for NO2 detection at room temperature. The strong interaction between the frontier orbitals of NO2 molecules and pz orbitals of phosphorus atoms leads to charge transfer, enabling NO2 to act as an oxidant and remove electrons from EPMs.671–673 Among the various allotropes of phosphorus, black phosphorene is currently the predominantly used sensing material. In 2015, Abbas et al.674 developed a multilayer black-P field-effect transistor for gas sensing (Fig. 44a) showing an NO2 detection limit of 5 ppb (Fig. 44b). Cho et al.675 have investigated the sensing characteristics of black-P, graphene, and MoS2 and found that black-P has an intrinsic sensing capacity that is 20 times greater than that of graphene and MoS2 (Fig. 44c). Furthermore, only black-P shows selective response to NO2 while being unresponsive to oxygen-functionalized molecules.675 A plausible explanation for the superior NO2 detecting capability of black-P has been postulated. First, since NO2 molecules are paramagnetic, their adsorption on black-P can provoke spin polarization, leading to the production of a spin-polarized current in the channel. Second, black-P has larger molecular adsorption energy than the other two materials rendering it more sensitive to NO2 (Fig. 44d).
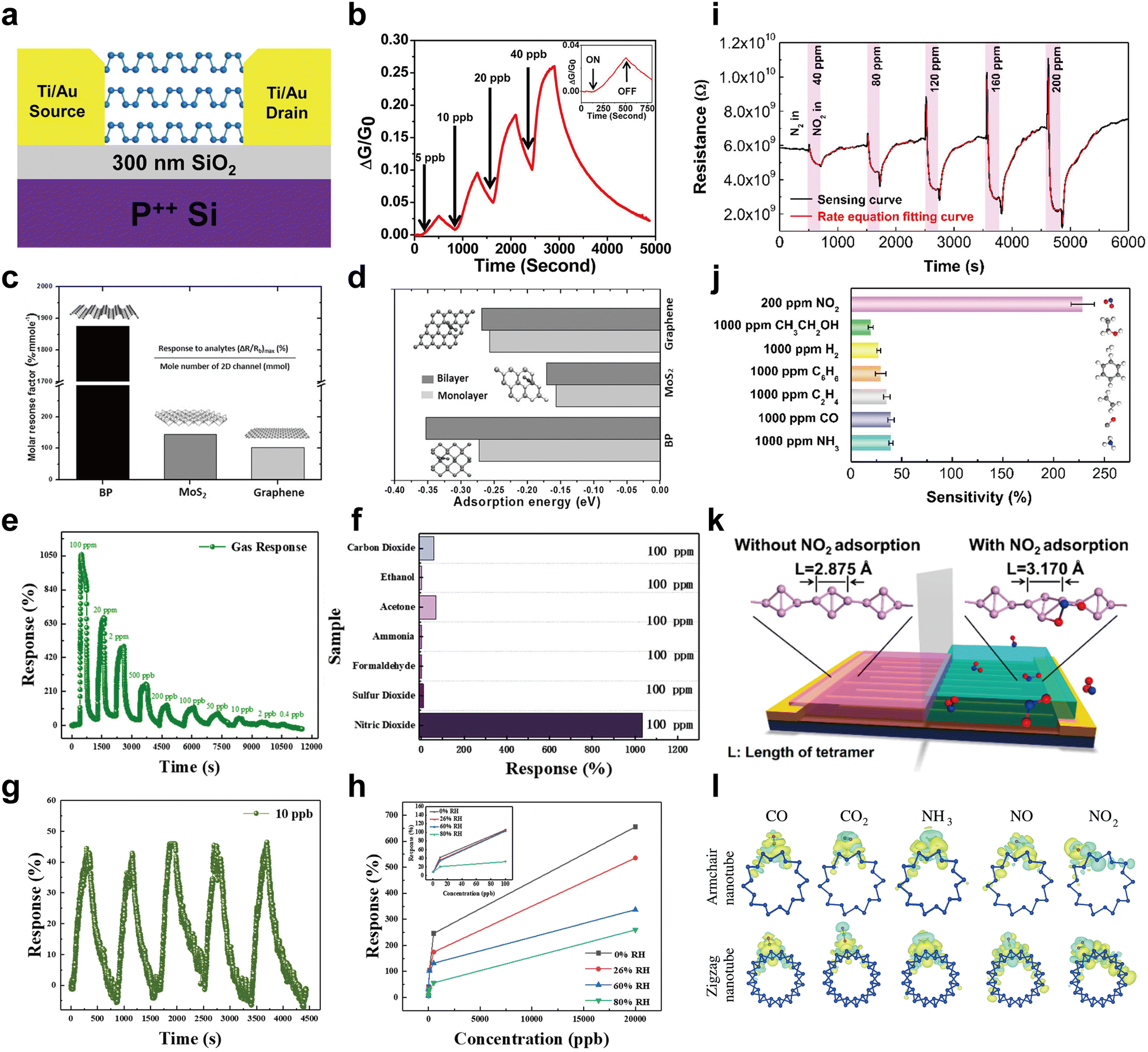 | ||
| Fig. 44 Applications of elemental phosphorus materials to pollutant sensing. (a) Scheme of a multilayer black-P field-effect transistor. (b) A plot of the time-dependent relative conductance change (ΔG/G0) is presented for a multilayer black-P sensor, which exhibits sensitivity to NO2 concentrations ranging from 5 ppb to 40 ppb. The inset displays an enlarged image of the response to a 5 ppb NO2 exposure, with marked time points denoting the activation and deactivation of the NO2 gas. Reproduced with permission from ref. 674. Copyright 2015 American Chemical Society. (c) Calculated molar response factor of the black-P sensor and comparison with that of MoS2 and graphene. (d) Minimum NO2 adsorption energy for monolayer and bilayer for black-P, MoS2, and graphene. Reproduced with permission from ref. 675. Copyright 2016 John Wiley and Sons. (e) A plot of the time-dependent relative conductance change (ΔG/G0) is presented for a black-P microribbon sensor under zero relative humidity, which exhibits sensitivity to NO2 concentrations ranging from 0.4 ppb to 100 ppm. (f) Black-P microribbon sensor response to NO2 and other common indoor air pollutants under dry room temperature conditions. (g) Five cycles of the on–off switching mode through the black-P microribbon sensor to 10 ppb NO2 under dry room temperature conditions. (h) Plots of the black-P microribbon sensor response to NO2 concentrations are presented under various relative humidity conditions of 0%, 26%, 60%, and 80% in air. The inset displays the response to concentrations of NO2 in the range of 0.4 ppb to 100 ppb under the same relative humidity conditions. Reproduced with permission from ref. 676. Copyright 2022 American Chemical Society. (i) Dynamic sensing response-recovery curves (black) measured for different NO2 concentrations ranging from 40 to 200 ppm. A fitting function based on an adsorption rate is used to fit the data, and the results are shown in red. (j) Red-P based sensor response to NO2 (200 ppm) and other gas molecules (1000 ppm). (k) Expansion of the amorphous red-P tetramer induced by NO2 adsorption. Reproduced with permission from ref. 30. Copyright 2018 American Chemical Society. (l) The adsorption of different molecules on the armchair and zigzag blue-P nanotubes with variances in electron density. Reproduced with permission from ref. 678. Copyright 2017 Royal Society of Chemistry. | ||
To further improve the detection ability of NO2, Sang's group has recently prepared a few-layered black-P microribbon-based gas sensor that achieves sub-ppb level detection under nitrogen and ambient conditions.676 As shown in Fig. 44e, the gas sensor shows response (ΔG/G0) to different concentrations of NO2 over time at room temperature and zero relative humidity. After exposing the sensor to 0.4 ppb of NO2, the response is about 8.09%. The specific adsorption capacity of the black-P microribbon for NO2 is assessed by comparing the response to 100 ppb NO2 and other common indoor air pollutants under dry ambient conditions (Fig. 44f). When the sensor is exposed to 10 ppm NO2 for five cycles (on–off mode) under dry conditions at room temperature (Fig. 44g), good repeatability is demonstrated and the selective detection limit is 0.4 ppb for NO2 at room temperature and zero relative humidity. However, in practical applications, ambient humidity often complicates the calibration process and reduces the sensor accuracy. If the relative humidity goes up from 0 to 80%, the response of the black-P microribbon sensor to NO2 is no longer evident in the range of 100 ppb to 20 ppm (Fig. 44h) because adsorbed water molecules capture electrons and degrade the sensitivity.677 Therefore, gas sensors made of black-P need to be surface passivated.
An alternative strategy is to select a relatively chemically stable allotrope of phosphorus. Zhu et al.30 deposited a red-P thin film on an Au electrode-patterned SiO2/Si substrate. The film is composed of micrometer-scale particles assembled from amorphous red-P nanoclusters with different sizes (10 or 100 nm). As shown in Fig. 44i, the sensor shows dynamic response-recovery curves for different concentrations of NO2 (40 to 200 ppm) at room temperature in nitrogen. The sensor has high sensitivity (229%) to 200 ppm NO2, but due to the poor conductivity of amorphous red-P, the bias stress and high noise levels (0.107%) reduce the detection limit.30 The conductivity of red-P can be improved by modulating the internal structure. The selectivity of the sensor is evaluated in the presence of potentially interfering gases (Fig. 44j) and the response to NO2 is significantly stronger, even though the concentration of the other gases is higher (1000 ppm) compared to NO2 (200 ppm). This selectivity originates from the intrinsic physicochemical properties of elemental phosphorus. Amorphous red-P has several unique sensing features, including amphoteric sensing that allows it to respond negatively to both oxidizing and reducing gas species. In addition, unlike the adsorption mechanism of black-P,674 the adsorption process on amorphous red-P is dominated by synergistic Langmuir isotherm adsorption and P–P bond expansion (Fig. 44k).
DFT calculations show that other EPMs are also promising gas sensors. Montes et al. investigated the adsorption of inorganic gas molecules (CO, CO2, NH3, NO, and NO2) on blue-P nanotubes (Fig. 44l).678 In comparison with black phosphorene, blue-P nanotubes exhibit better selectivity and sensitivity for N-containing gases. The current change before and after adsorption of NO2 is 294% at a voltage of 0.2 V, which is more than 2.4 times higher than those of other materials such as phosphorene (122%),679 MoS2 (57%),680 and PtSe2 (12%).681 Singsen et al. have observed that silicon-doped green phosphorene exhibits high selectivity towards carbonyl-containing volatile organic compounds such as acetone, propane, and formaldehyde because the pi and lone pairs of electrons in the carbonyl group interact with electron-deficient Si atoms in the doped green phosphorene to produce strong adsorption.682 Dharani et al.683 and Bhuvaneswari et al.684 have studied the adsorption characteristics of hexanal, benzyl chloride, and chlorobenzene on violet phosphorene. Zhang et al.685 have conducted spin-polarized first-principles computations with vdW correction to investigate the properties of fibrous phosphorene in air pollution monitoring. Fibrous phosphorene with a lone pair of electrons in the p orbital shows strong attraction to electron-deficient substances, giving rise to high detection sensitivity for NO, NO2, O3, and SO2. However, gas sensors made of blue, green, violet, and fibrous phosphorus are still in the theoretical stage.
Yellow phosphorus, also known as white phosphorus, is typically unsuitable for energy storage and catalysis due to its high reactivity. However, its cost-effective and efficient reducing properties make it a popular in flue gas treatment. In the 1990s, Chang and Liu utilized a yellow phosphorus/alkaline emulsion to remove NOx and SO2 from the flue gas.690 The emulsified yellow phosphorus droplets come in contact with O2 in the flue gas, releasing O3 and O, which oxidize NO into water-soluble NO2, and convert SO2 into (NH4)2SO4 and CaSO4.691 The alkaline buffer in the yellow phosphorus emulsion plays a crucial role in maintaining an appropriate pH range for NOx absorption to enhance the adsorption efficiency. Ning's group has reported that red mud could replace CaCO3 as the alkaline buffer in the yellow phosphorus emulsion and suggested economic benefits in removing insoluble NO and reducing the ecological risk of red mud accumulation.692 The primary reaction products between NO and yellow phosphorus are high-priced NO3−, NO2−, and PO43−, which have potential applications in the production of nitrogen and phosphorus fertilizers. Subsequently, an improved oxidation process has been developed to simultaneously remove NOx and SO2 using yellow phosphorus and red mud and the removal capabilities are 97.9% and 100%, respectively.693 This method is more cost-effective than traditional oxidative denitrification and desulfurization techniques while achieving the purpose of “waste control by waste”.
Photo-oxidation is an innovative and promising technique for the degradation of gaseous pollutants, and Lu's group has developed photocatalytic black-P composites for NO removal. The 2D heterojunction of black-P/monolayer Bi2WO6 is prepared by integrating Bi2WO6 into the surface of black-P nanosheets (Fig. 45a).388 Active radicals are identified by DMPO-ESR spin-trap measurements and a charge transfer mechanism for photodegradation is postulated based on the Z-scheme. In the process, the electron in the valence band of black-P reduces O2 to ˙O2−, which then oxidizes NO to NO3− (Fig. 45b). Owing to the excellent electron transfer ability and broader absorption range (from UV to NIR) of black-P, the NO removal efficiency goes up to 67% after irradiation for 30 minutes, which is 2.6 times greater than that of the pristine monolayer Bi2WO6 (26%). A type I heterojunction of black-P/porous g-C3N4-HKUST-1 has also been designed (Fig. 45c). HKUST-1 is a copper benzene tricarboxylate metal–organic framework (MOF) with good NO adsorption properties and visible light response.694 The ternary composite is employed to photocatalyze the oxidation of NO at the ppb level under visible-light irradiation (Fig. 45d).387 The optimal composite membrane achieves an NO removal ratio of 74%, which is better than those of previously reported g-C3N4-based heterojunctions.695–697 This high photo-oxidation performance can be ascribed to the synergistic effects of the heterojunction as well as superior NO adsorption capability of the MOF.
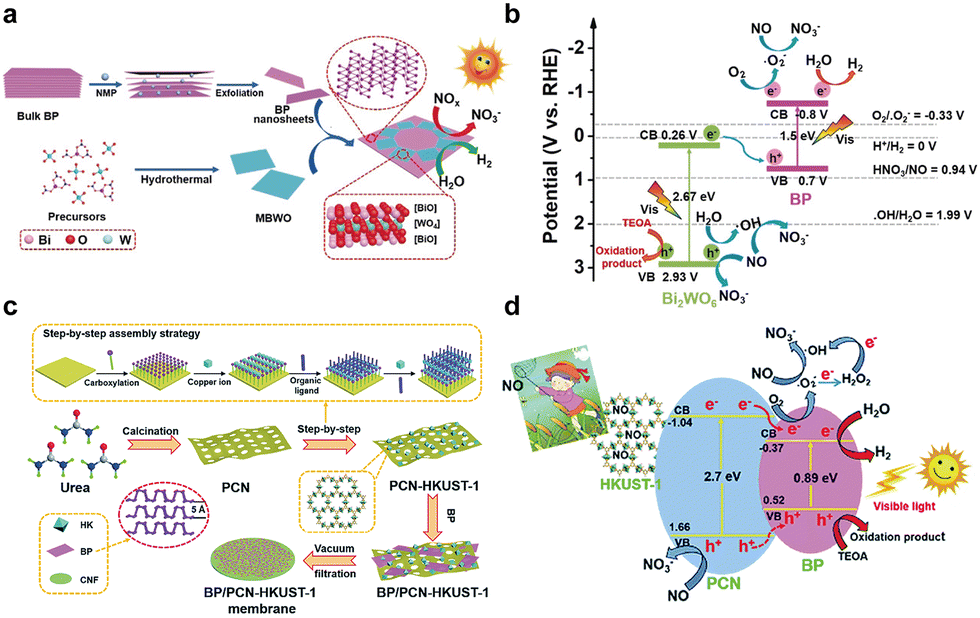 | ||
| Fig. 45 Applications of elemental phosphorus materials to gaseous pollutant degradation. (a) Scheme of the fabrication of a 2D heterojunction black-P/monolayer Bi2WO6. (b) Photocatalytic mechanism of the black-P/monolayer Bi2WO6 heterojunction under visible-light irradiation. Reproduced with permission from ref. 388. Copyright 2019 John Wiley and Sons. (c) Scheme of fabrication of the black-P/porous g-C3N4-HKUST-1 membrane. (d) Photocatalytic mechanism of the black-P/porous g-C3N4-HKUST-1 membrane under visible-light irradiation. Reproduced with permission from ref. 387. Copyright 2019 Royal Society of Chemistry. | ||
Synthetic organic dyes which are widely used industrially have become a growing concern due to their impact on the ecosystem. Azo dyes containing the azo-functional group R–NN–R′ (R and R′ can be aryl or alkyl) constitute more than 70% of synthetic organic dyes.28,704 Methyl orange is a representative azo dye that can be used to evaluate the effectiveness of degradation for other azo dyes. Zhang et al. have investigated the photo-degradation efficiency of 50 mL of methyl orange (100 ppm) using amorphous red-P, crystalline red-P ([P12(4)]P2[ phase) microbelts and violet-P microrods.396 Both crystalline red-P and violet-P exhibit higher efficiency under visible light irradiation compared to the amorphous red-P sample. Crystalline red-P degrades 90% of the methyl orange within 40 minutes and the rate is twice that of violet-P. The transient photocurrents of crystalline red-P microbelts, which have a bigger surface area, are higher than those of the violet-P microrods, indicating that the former has superior photo-induced charge separation and transfer efficiency.
Ag2CO3 has attracted the attention of many researchers due to the unique photocatalytic mechanism. Ag+ in Ag2CO3 can react with photo-induced electrons to form the Ag/Ag2CO3 system,705 in which an appropriate amount of Ag can lead to the production of surface plasmon resonance for enhanced light absorption and photocatalytic efficiency,706 but excessive Ag may cause photo-corrosion and suppress the photocatalytic activity.707 Therefore, precise control of the Ag concentration on the surface is critical. By taking advantage of the conduction band and valence band positions of Ag2CO3 and black-P, a Z-scheme black-P/Ag2CO3 heterojunction can be constructed to avoid excessive formation of Ag nanoparticles and improve the photocatalytic activity to degrade organic dyes. Wang et al. have synthesized a direct Z-scheme heterojunction photocatalyst composed of PDDA-functionalized black-P nanosheets and Ag2CO3 by co-precipitation.708 The black-P/Ag2CO3 composite has a high degradation efficiency as exemplified by the mineralization of 94.1% of methyl orange into CO2 and H2O within 30 minutes, which is better than that of Ag2CO3 (43.3%). After four irradiation cycles, the activity of the black-P/Ag2CO3 composite photocatalytic activity is 87.1%, whereas the pristine Ag2CO3 shows a 19.2% degradation rate (Fig. 46a). Ag2CO3 has a lower conduction band potential than the ˙O2− redox potential and the conduction band of Ag2CO3 is incapable of reducing O2 to ˙O2− based on the type-II mechanism. However, capture experiments show that both ˙O2− and ˙OH play important roles in the photocatalytic degradation of methyl orange (Fig. 46b and c), confirming that it is not a type-II heterojunction but rather a direct Z-scheme photocatalytic mechanism. The amount of black-P affects the photocatalytic activity, because excess black-P obstructs the photoactive sites on the Ag2CO3 surface but insufficient black-P cannot mitigate photo-corrosion. Therefore, the Ag concentration must be controlled to optimize the transfer and separation of photo-induced charges to function as an “electron sink” to enhance the stability of the black-P/Ag2CO3 heterojunction.
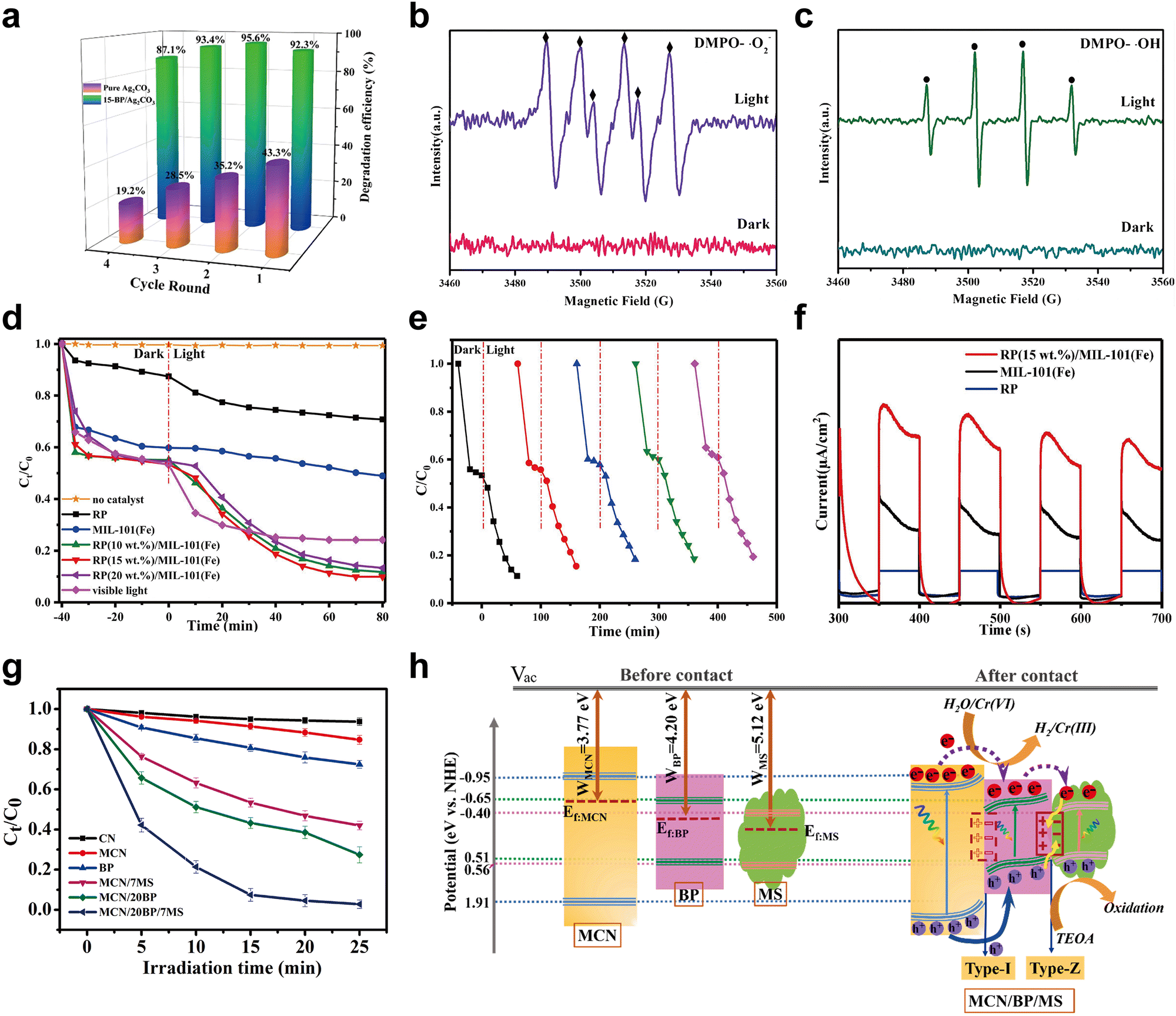 | ||
| Fig. 46 Applications of elemental phosphorus materials to liquid phase pollutant degradation. (a) Cycling performance of Ag2CO3 and optimal black-P/Ag2CO3 composite for methyl orange degradation. (b) Electron spin resonance spectra of the black-P/Ag2CO3 composite for DMPO-˙O2− and (c) DMPO-˙OH. Reproduced with permission from ref. 708. Copyright 2021 Springer. (d) The degradation of tetracycline using various photocatalysts. (e) The cycling stability of red-P/MIL-101(Fe) for tetracycline degradation. (f) Photocurrent responses of red-P/MIL-101(Fe), pristine MIL-101(Fe), and red-P. Reproduced with permission from ref. 718. Copyright 2020 Elsevier. (g) Photocatalytic Cr(VI) reduction rate curves in the presence of g-C3N4 (CN), ball-milled g-C3N4 (MCN), black phosphorus (BP), and binary and ternary composites. (h) The band energy alignment scheme and photocatalytic reaction mechanism of the g-C3N4/black-P/MoS2 composite (MCN/BP/MS). Reproduced with permission from ref. 721. Copyright 2021 Elsevier. | ||
Rhodamine B is a well-known hydrophilic industrial colorant used in food processing, textile dyeing, and fluorescent labelling. Rhodamine B is classified as a group 3 carcinogen by The International Agency for Research on Cancer in 1987,709 and it is considered a carcinogenic, genotoxic, and neurotoxic inducer or toxin in animals.710 Jeong et al. synthesized amorphous partial oxidized violet-P through an open-air ball milling approach with controlled mechanical energy and the materials have excellent catalytic activity for the removal of rhodamine B in a light-free environment.711 In contrast to crystalline violet-P prepared by conventional CVD, violet-P is amorphous. When 10 mg of partially oxidized violet-P is added to 40 mL of 30 mg L−1 rhodamine B solution, complete reduction occurs within 15 minutes and the fastest rate occurs in the first 5 minutes. Violet-P with strong electron affinity and surface PO as the redox-active species are responsible for the outstanding activity.711 Removal of the oxidized phosphorus moiety normally decreases the catalytic reduction rate, but amorphous violet-P retains the reducing ability due to the substantial negative charge on the surface which persists in a highly unstable state.
Inadvertent discharge of antibiotics is detrimental to the environment and organisms with antibiotic resistance and superbugs can develop, which pose threats to human beings.712–714 Fe-based MOFs are a unique class of MOFs exhibiting excellent stability, large porosity, large surface area, ease of production, and environmental friendliness.715–717 Among them, MIL-101(Fe) has found many applications in photocatalysis, although the fundamental limitation is fast electron–hole recombination that decreases the photocatalytic efficiency. In this respect, red-P is an excellent dopant to improve the photocatalytic activity of MOF-based photocatalysts. Lei et al. have prepared tunable red-P/MIL-101(Fe) composites by a low-temperature solvothermal technique and investigated the efficacy in photo-degradation of tetracycline.718 Morphological analysis reveals that the purified red-P is evenly anchored on MIL-101(Fe) in the form of tiny particles thus producing a rough surface. At a pH of 5, tetracycline concentration of 50 mg L−1, catalyst dosage of 0.50 g L−1, and full-range wavelength exposure, the photocatalytic efficiency of the Red-P (15 wt%)/MIL-101(Fe) composite towards tetracycline is 90.1% in 80 minutes (Fig. 46d), which is higher than those of pristine MIL-101(Fe) (51.1%), red-P (29.2%), and red-P (1.5 wt%)/TiO2 (75.9%) under the same conditions.718 Furthermore, the composite has remarkable stability (Fig. 46e) and the photocurrent of red-P (15 wt%)/MIL-101(Fe) is also higher than that of the pristine MIL-101(Fe) (Fig. 46f).
Besides organic pollutants, inorganic pollutants such as lead, mercury, arsenic, and chromium contribute to water pollution. Cr(VI), a by-product of electroplating and leather manufacturing, can cause liver and renal disorders in humans.719 The metal-free photocatalyst g-C3N4 with a bandgap of ∼2.7 eV shows good chemical stability but its poor electroconductivity limits the catalytic activity.720 Introduction of black-P with a smaller bandgap can improve the conductivity of g-C3N4 and the stability of black-P can be enhanced by covering the noncovalent surface with unreactive g-C3N4. Meng et al. have synthesized a ternary heterojunction photocatalyst of g-C3N4/Black-P/MoS2 by the solvent-free high-energy ball milling method.721 The grinding balls deliver high-temperature pulses to g-C3N4, black-P, and MoS2 causing chemical bond breakage and formation and creating good interfacial contact to reduce the interfacial charge transfer impedance and facilitate carrier migration and separation, making g-C3N4/Black-P/MoS2 an efficient photocatalyst. Under light illumination, the optimal g-C3N4/Black-P/MoS2 composite eliminates nearly 100% Cr(VI) after 25 minutes (Fig. 46g). However, the reduction efficiency of Cr(VI) by black-P is inadequate with only 27.5% of Cr(VI) reduced in the same time. Hence, light absorption by the ternary composite increases considerably throughout the UV–vis range and charge transfer in the ternary composite shows the mixed type-I/Z scheme (Fig. 46h). This uncommon scheme allows integration of the electric field at the interface, making it difficult for photogenerated electrons to migrate from the high conduction band to the low conduction band or to recombine with valence band holes. As a result, more photogenerated electrons with the strong reducing capacity remain in the conduction band of g-C3N4 and black-P to boost the photocatalytic Cr(VI) reduction efficacy.722
Amorphous red-P has been extensively studied in photocatalysis as an inexpensive and ambient-stable semiconductor.28 More importantly, it has low toxicity and can be used to disinfect drinking water.730 In 2015, Xia et al.731 reported inactivation of Escherichia coli by amorphous red-P under visible light irradiation. The purified amorphous red-P has superior bacterial inactivation efficiency due to the water stability and moderate bandgap (1.42 eV). At a concentration of 100 mg L−1, the cell loss is 2 × 107 cfu mL−1 within 90 minutes during visible light irradiation (Fig. 47a), outperforming C3N4 and even metal-based B-Ni-co-doped TiO2.732,733 The disinfection mechanism of amorphous red-P has been studied by monitoring the leakage of membrane-functional enzymes and biomolecules. ROS attacks the cell membrane as the primary target, increases the cell membrane permeability, and ultimately kills the bacteria via intracellular protein carboxylation and DNA destruction (Fig. 47b). Athira et al.734 have investigated the photocatalytic efficiency of commercial red-P in inactivating Escherichia coli using direct sunlight. The photo-induced ROS, mostly O2˙− and HO2˙, sterilizes the bacteria within 50 minutes with a red-P concentration of 150 mg L−1. The conduction band potential is −0.497 V, which is more negative than the potential required for the generation of O2˙− by single-electron transfer reduction (−0.16 V vs. NHE) and it is also adequate for the production of HO2˙. These findings suggest that amorphous red-P has promising prospects in photocatalytic bacterial disinfection and is suitable for outdoor sterilization of water under natural light.
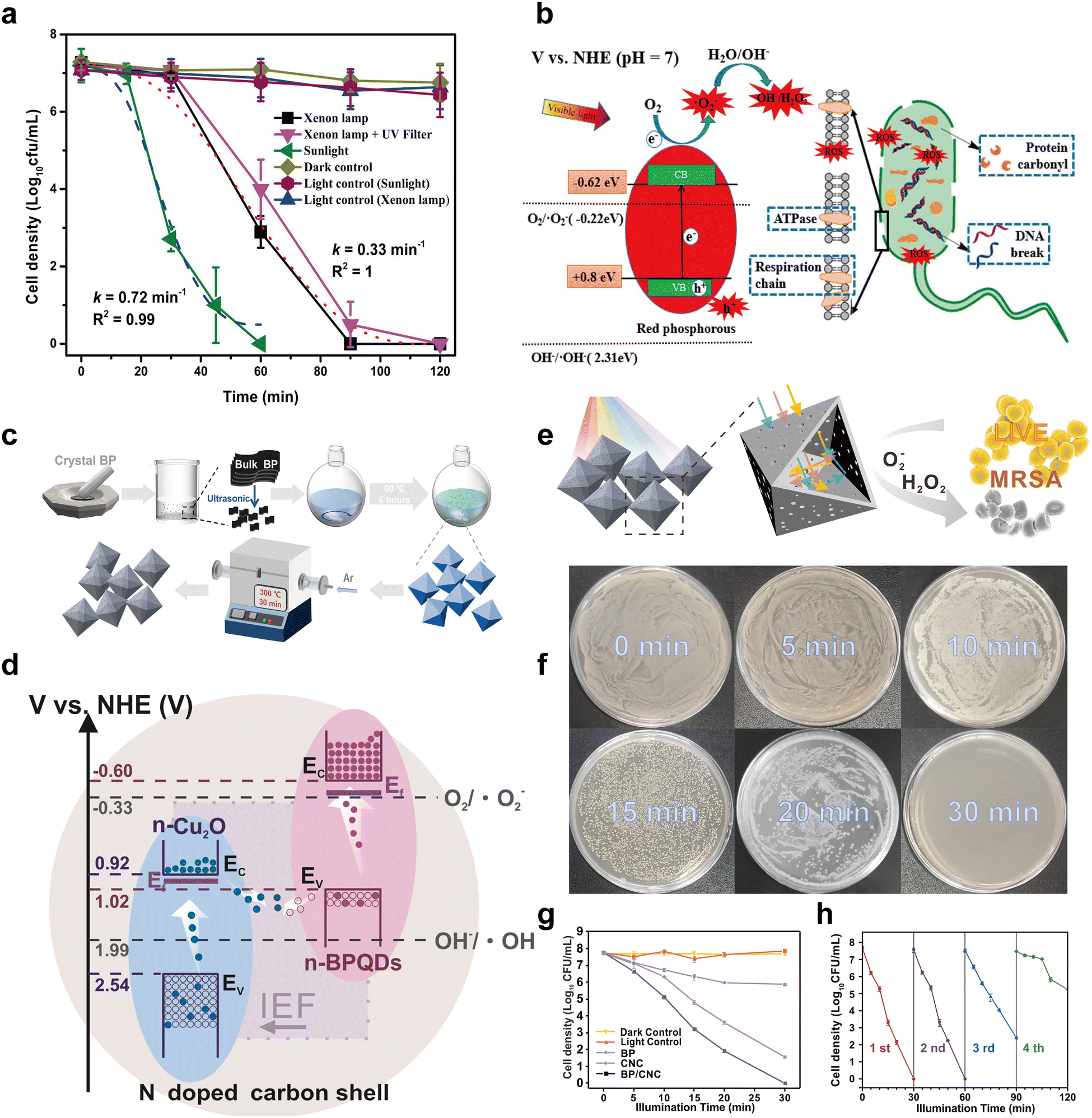 | ||
| Fig. 47 Applications of elemental phosphorus materials to bacterial disinfection. (a) Photocatalytic inactivation kinetics under different irradiation conditions in the presence of amorphous red-P. (b) The proposed mechanism for bacterial disinfection by amorphous red-P under visible-light irradiation. Reproduced with permission from ref. 731. Copyright 2015 American Chemical Society. (c) The synthetic process for preparing BPQDs/Cu2O/N-doped hollow porous carbon composite. (d) Z-scheme band structure and electron transfer pathway of the composite. (e) Scheme of the photo-responsive multidrug-resistant bacterial disinfection of BPQDs/Cu2O/N-doped hollow porous carbon composites. (f) Photographs of the agar plates with the composite (10 mg L−1) at different illumination times. (g) Disinfection efficiency of materials under different conditions. (h) The cycling stability of BPQDs/Cu2O/N-doped hollow porous carbon composite for photocatalytic disinfection. Reproduced with permission from ref. 741. Copyright 2021 Elsevier. | ||
On the heels of the development of CVD for fibrous-P, several studies have focused on the photocatalytic performance pertaining to bacterial disinfection. Roshith and colleagues have prepared urchin-like fibrous-P with a bandgap of 1.9 eV and an absorption edge extending to 650 nm for more efficient sunlight absorption.304 Under direct sunlight irradiation, a high capacity to inactivate Escherichia coli is at 108 cfu mL−1 in 30 minutes is demonstrated by urchin-like fibrous-P, which is more effective than most red-P-based systems. An optofluidic reactor capable of continuous flow photocatalytic disinfection of Escherichia coli has been developed by immobilizing fibrous-P to the inner wall of a quartz capillary.735 After sunlight exposure for 14 minutes, the immobilized photocatalyst reduces the bacterial concentration by 6.7log10 (>99.9999%). This continuous photocatalytic disinfection technique is promising for purposes as it can be scaled up. Zhang et al. have reported a two-step approach to prepare a crystalline red-P/graphene oxide layer on Ti for rapid microbial inactivation.736 An irregular pyramidal crystalline red-P layer is first prepared on a Ti plate by CVD and then a graphene oxide layer is deposited on the red-P layer via the shellac heating transition. After exposure to simulated sunlight for 20 minutes, the composite film shows an inactivation rate of 99.9% against Staphylococcus aureus and Escherichia coli and also has good antibacterial properties upon exposure to visible (>410 nm), infrared (808 nm), and LED light.
The sharp edges of 2D nanosheets can physically damage bacterial membranes producing leakage of intracellular contents and bacterial death, the so-called “nanoknife” effect.737–739 Hence, black-P nanosheets used as photocatalysts also produce physical antibacterial effects. He et al.740 have constructed a metal-free 2D/2D black-P and graphitic carbon nitride heterojunction for efficient photocatalytic water disinfection under visible light. The disinfection activity against Escherichia coli is about 7 times higher than that of pristine graphitic carbon nitride, and bacterial cells (107 cfu mL−1) can be completely inactivated within 60 minutes. Both black-P and graphitic carbon nitride nanosheets are 2D materials with synergistic physical antibacterial properties, and graphitic carbon nitride can further mitigate the recombination of photoinduced electrons/holes during the photocatalytic process to give electrons sufficient time to form ˙OH and H2O2 to attack bacteria.
A stable and well-ordered hierarchical structure can improve the sunlight usage and lifespan of photo-induced charge carriers. In particular, porous hollow structures have excellent light-harvesting properties because of the longer light path. Luo et al. have reported an MOF-derived BPQDs/Cu2O/N-doped hollow porous carbon structure for inactivation of methicillin-resistant Staphylococcus aureus.741 The synthesis involves cross-linking copper ions with trimesic acid through coordination assembly to form Cu-BTC MOFs. Benzimidazole is added to the solution to control the MOF shape and introduce the N source.742 Cu-BTC MOFs are then used as the self-sacrificial templates in the annealing process to generate Cu2O/N-doped hollow porous carbon. The sonicated BPQDs interact with Cu2O/N-doped hollow porous carbon to produce a regular octahedral-like ternary composite (Fig. 47c). Cu2O nanoparticles and BPQDs are evenly distributed throughout the hollow sphere to form an indirect Z-type heterojunction (Fig. 47d). This heterojunction constitutes a broad spatial barrier to prevent charge congestion and recombination thus allowing unimpeded transmission of photo-induced carriers and preserving the high redox potential. Upon visible light irradiation, the ternary composite shows photo-responsive multidrug-resistant bacterial disinfection (Fig. 47e) and completely inactivates 108 cfu mL−1Staphylococcus aureus within 30 minutes with a dosage of only 10 mg L−1 (Fig. 47f). Control experiments show that visible light irradiation or the photocatalyst alone has no disinfection effects on drug-resistant bacteria and the disinfection efficiency of single BPQD or Cu2O/N-doped hollow porous carbon is thus not significant (Fig. 47g).741 The photocatalytic stability of BPQDs/Cu2O/N-doped hollow porous carbon is very good in the first three cycles, but the efficiency decreases in the 4th cycle (Fig. 47h) on account of photo-corrosion of Cu2O.743 Future research should aim at developing more stable and efficient hierarchical photocatalysts.
Introducing phosphorus-containing substances (phosphate, phosphorus oxide, and phosphorus allotrope) into biochar has been proven to be effective modification technology to improve remediation of saline-alkali soil. Phosphorus-containing substances undergo biodegradation in the soil to generate phosphate groups which can neutralize the soluble alkaline ions in biochar.754 Moreover, phosphorus acts as a nutrient element for soil microorganisms and plant growth and enhances plant phosphorus absorption, soil microbial activity, and metal-ion immobilization.755 In 2019, Li's group reported that Ca(H2PO4)2 engineered swine manure biochar could immobilize toxic metal ions in smelting contaminated soil by forming insoluble phosphate precipitates.756 The application of this modified biochar not only reduced the accumulation of toxic metals in the plants, but also increased the plant biomass to simultaneously promote remediation and crop production. Red-P is a possible fertilizer that slowly converts to phosphate in the presence of H2O and O2.757 Recently, Sun's group has developed red-P-modified biochar through a ball milling approach for saline-alkali soil remediation.750 The ball milling process converts red-P powder into nanoparticles that are evenly distributed on the biochar surface with a portion being oxidized into phosphorus oxide and phosphate. The biochar is further ground into submicron particles to increase the specific surface area and enhance soil remediation. Through a series of experiments in amended soil, researchers have proposed a remediation mechanism for red-P-modified biochar (Fig. 48). First, biochar has a strong salt adsorption capacity to allay saline-alkali stress or stress responses in plants. Both biochar-based materials and ball-milled red-P reduce the soil electrical conductivity. Second, biochar improves the physical and chemical properties of saline-alkali soil by increasing the organic carbon content, cation exchange capacity, and water storage while reducing the soil pH. Red-P-modified biochar can reduce the soil pH to 7.36 and maintain it at a near-neutral pH because phosphoric acid in red-P neutralizes hydroxyl ions in soil.758 Third, biochar is nutrient-rich and improves soil fertility for plant growth. For the same level of phosphorus addition, red-P-modified biochar contains more available phosphorus than ball-milled red-P, suggesting its high potential as a fertilizer with the ability to accumulate and release soil nutrients beneficial for microorganisms and plant reproduction. Overall, red-P-modified biochar is a promising soil amendment to enhance the soil productivity and biological activity and in turn to promote the growth of plants in saline-alkali soils.
 | ||
| Fig. 48 Mechanism of red-P-modified biochar for saline-alkali soil remediation. Reproduced with permission from ref. 750. Copyright 2021 Elsevier. | ||
The use of EPMs in soil remediation is still in its early stages and only red-P has been directly applied for this purpose. Zhang et al. have found that black-P nanosheets have no significant effects on the enzyme activity in black soil during 60 day exposure, demonstrating that black-P nanosheets are eco-friendly compared with many other nanomaterials.759 This study highlights the potential of black-P nanomaterials in soil remediation. Black-P nanomaterials possess a larger specific surface area and higher chemical activity than nanoscale red-P, so that black-P can quickly deposit oxidized metal ions in contaminated soil and release high-valence phosphate in a shorter time. Therefore, it is anticipated that modified black-P nanomaterials have superior soil remediation characteristics compared to red-P. Moreover, the current production cost of black-P-based materials has been reduced to 0.235 Euros per gram, boding well for more widespread use in soil remediation.385
In summary, EPMs have developed distinctive applications in the field of sensing and environmental remediation. Among these, black-P exhibits superior conductivity in comparison to other EPMs, rendering it an optimal choice for the development of highly sensitive sensors. For pollutant treatment and bacterial disinfection, amorphous red-P, crystalline fibrous-P, and violet-P have demonstrated notable performance. This is largely due to their appropriate bandgaps and environmental stability, making them highly promising for sustained roles in environmental catalysis. Furthermore, given that phosphorus serves as a suitable fertilizer, exploring the application of EPMs in soil remediation could prove to be a strategy with dual benefits, both repairing the soil and enhancing its fertility. However, research in this particular domain is still relatively sparse at the present.
6. Conclusions and outlook
This review presents a comprehensive and up-to-date overview of the progress pertaining to the development, fundamental properties, synthetic methodologies, and applications of EPMs to energy conversion, storage, and environmental remediation. Significant progress has been made in understanding the structure–property–function relationships of various phosphorus allotropes to provide insights into the optimal constituent materials and systems for specific applications. Although considerable achievements have been made in the design and engineering of EPMs, there are still ample opportunities for further advancements. To facilitate sustained development of EPMs in the post-graphene era, this review provides critical information about the four important aspects: (1) new allotrope discovery, (2) fundamental property exploration, (3) synthetic methods and optimization, and (4) applications and performance improvement.The synergistic integration of artificial intelligence-based theoretical calculations and chemical synthesis presents a promising avenue for the discovery of new phosphorus allotropes. Theoretically, phosphorus allotropes can be obtained by the mixed assembly of distinct phosphorus cluster motifs. However, as the range of EPMs continues to expand, the challenge of identifying new allotropes becomes increasingly complex. Traditional reliance on experience and intuition may no longer be sufficient to uncover intricate phosphorus allotrope structures. This is where the evolving realm of artificial intelligence steps in. With its potential applications in structural prediction, functional forecasting, performance analysis, and structural optimization, artificial intelligence offers a plethora of opportunities for improving both efficiency and accuracy in these areas. Implementation of machine learning-driven theoretical predictions can screen different phosphorus cluster motifs with high throughput in order to reduce the need for substantial human labour and computing resources.56 We anticipate that more advanced and sophisticated artificial intelligence algorithms will be developed, especially structural prediction of new EPMs, in the future to expedite discovery and enhance our understanding of these materials.
The primary aim of exploring fundamental properties is to elucidate the precise structures of existing phosphorus allotropes and the microscopic and nanoscopic interactions between EPMs and exotic materials. Currently, the research on properties focuses on black-P based materials but there is an urgent need to characterize the fundamental properties of other EPMs. It is imperative to conduct a comprehensive examination of the structures of all five red-P modifications and evaluate the associated structure–property relationships. Furthermore, it is essential to investigate the alloying and dealloying mechanisms of different crystalline phosphorus for alkaline metal ion batteries. To observe these interactions, appropriate in situ characterization techniques should be employed in conjunction with ex situ methods.
Optimization of the synthetic methods and conditions is crucial to both bulk crystals and low-dimensional nanomaterials of EPMs. Developing high-quality, cost-effective synthesis for bulk crystal production is important. Although several techniques have been reported for the preparation of gram-level black-P single crystals, the high production cost, elevated temperature, and high pressure have impeded widespread adoption in many laboratories. Successful synthesis of blue and green phosphorus bulk crystals remains elusive but presents a potential avenue for future inorganic synthesis. Although crystalline red-P bulk crystals can be prepared by chemical vapor transport and other methods, contamination from other crystalline forms is prevalent necessitating improvements in quality control. The synthesis of nanostructured EPMs grapples with challenges such as low yield and inadequate quality control. The liquid-phase exfoliation method shows promise for large-scale EPMs nanosheet production, but often encounters issues like edge defects. In the synthesis of blue phosphorene, molecular beam epitaxy is the only reported technique so far. It typically produces domain sizes ranging from a few nanometers to several tens of nanometers, which is significantly smaller than that required for practical device fabrication. Additionally, there is a need to optimize the synthesis of 1D nanoribbons and 0D quantum dots for different phosphorus allotropes.
The current state of EPM application is primarily in the proof-of-concept stage. The majority of applications are confined to laboratory settings with insufficient attention being paid to the cost, processability, and scalability of the synthesized EPMs. Among the various EPMs, black-P and amorphous red-P have greater potential for industrialization due to more advanced development. Energy storage is one of the most promising applications of EPMs and the construction of 3D hierarchical structures represents a promising approach to enhance the EPM energy storage capability by promoting ion diffusion and accommodating volume changes during charging-discharging cycles. Furthermore, a deeper understanding of the intercalation processes involving multivalent ions and organic ions will be useful for the development of EPM-based electrodes in next-generation batteries and supercapacitors.
In conclusion, substantial advancements have been made in the field of EPMs in the past decade. Cutting-edge analytical methodologies, computational chemistry, and nano-techniques have fueled this progress and deepened our understanding of the intricate structure–property–function relationships of EPMs. At the same time, it is important to keep in mind that phosphorus is a critical resource. Application of phosphorus beyond its dominant use for fertilizers should be critically evaluated for sustainability, including in the field of EPMs. Future progress is anticipated from extensive characterization of phosphorus allotropes and development of a diverse range of micro and nanostructures, with an increasing focus on sustainable usage and recycling of phosphorus. This balanced approach will open new opportunities for the practical application of EPMs.
Author contributions
J. Wang, G. Qu, and X.-F. Yu proposed the topic of the review. H. Tian surveyed the literature and compiled the manuscript. G. Lai supplemented the tables and figures. P. K. Chu, G. Jiang, S. Zhuang, and J. Shi refined the manuscript and provided constructive insight. J. Gao, Q. Wu, L. Yao, L. Zeng, Y. Liu, and X. Yang discussed and revised the tables and figures. Y. Dou, Z. Duan, X. Feng, X. He, and J. Zhao discussed and revised the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (92143301, 92043302, and 21975280), National Key Research and Development Program of China (2020YFA0907500), Shenzhen Science and Technology Program Grant (JCYJ20220818100806014), and Youth Innovation Promotion Association of the Chinese Academy of Sciences (2020354).References
- H. Tiessen, P. White and J. Hammond, The Ecophysiology of Plant-Phosphorus Interactions, Springer, 2008, https://www.springer.com/gp/book/9781402084348 Search PubMed.
- C. T. Reinhard, N. J. Planavsky, B. C. Gill, K. Ozaki, L. J. Robbins, T. W. Lyons, W. W. Fischer, C. Wang, D. B. Cole and K. O. Konhauser, Nature, 2017, 541, 386–389 CrossRef CAS PubMed.
- A. R. Jupp, S. Beijer, G. C. Narain, W. Schipper and J. C. Slootweg, Chem. Soc. Rev., 2021, 50, 87–101 RSC.
- M. Pasek and K. Block, Nat. Geosci., 2009, 2, 553–556 CrossRef CAS.
- S. Duhamel, J. M. Diaz, J. C. Adams, K. Djaoudi, V. Steck and E. M. Waggoner, Nat. Geosci., 2021, 14, 359–368 CrossRef CAS.
- A. C. Martiny, C. T. A. Pham, F. W. Primeau, J. A. Vrugt, J. K. Moore, S. A. Levin and M. W. Lomas, Nat. Geosci., 2013, 6, 279–283 CrossRef CAS.
- X. Wei, Y. Hu, B. S. Razavi, J. Zhou, J. Shen, P. Nannipieri, J. Wu and T. Ge, Soil Biol. Biochem., 2019, 131, 62–70 CrossRef CAS.
- C. M. Moore, M. M. Mills, K. R. Arrigo, I. Berman-Frank, L. Bopp, P. W. Boyd, E. D. Galbraith, R. J. Geider, C. Guieu, S. L. Jaccard, T. D. Jickells, J. La Roche, T. M. Lenton, N. M. Mahowald, E. Maranon, I. Marinov, J. K. Moore, T. Nakatsuka, A. Oschlies, M. A. Saito, T. F. Thingstad, A. Tsuda and O. Ulloa, Nat. Geosci., 2013, 6, 701–710 CrossRef CAS.
- M. Delgado-Baquerizo, F. T. Maestre, A. Gallardo, M. A. Bowker, M. D. Wallenstein, J. L. Quero, V. Ochoa, B. Gozalo, M. García-Gómez, S. Soliveres, P. García-Palacios, M. Berdugo, E. Valencia, C. Escolar, T. Arredondo, C. Barraza-Zepeda, D. Bran, J. A. Carreira, M. Chaieb, A. A. Conceição, M. Derak, D. J. Eldridge, A. Escudero, C. I. Espinosa, J. Gaitán, M. G. Gatica, S. Gómez-González, E. Guzman, J. R. Gutiérrez, A. Florentino, E. Hepper, R. M. Hernández, E. Huber-Sannwald, M. Jankju, J. Liu, R. L. Mau, M. Miriti, J. Monerris, K. Naseri, Z. Noumi, V. Polo, A. Prina, E. Pucheta, E. Ramírez, D. A. Ramírez-Collantes, R. Romão, M. Tighe, D. Torres, C. Torres-Díaz, E. D. Ungar, J. Val, W. Wamiti, D. Wang and E. Zaady, Nature, 2013, 502, 672–676 CrossRef CAS PubMed.
- S. Huang and X. Ling, Small, 2017, 13, 1700823 CrossRef PubMed.
- Y. Yi, Z. Sun, J. Li, P. K. Chu and X.-F. Yu, Small Methods, 2019, 3, 1900165 CrossRef CAS.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- G. Qu, T. Xia, W. Zhou, X. Zhang, H. Zhang, L. Hu, J. Shi, X. F. Yu and G. Jiang, Chem. Rev., 2020, 120, 2288–2346 CrossRef CAS PubMed.
- X. Shao, Z. Ding, W. Zhou, Y. Li, Z. Li, H. Cui, X. Lin, G. Cao, B. Cheng, H. Sun, M. Li, K. Liu, D. Lu, S. Geng, W. Shi, G. Zhang, Q. Song, L. Chen, G. Wang, W. Su, L. Cai, L. Fang, D. T. Leong, Y. Li, X. F. Yu and H. Li, Nat. Nanotechnol., 2021, 16, 1150–1160 CrossRef CAS PubMed.
- J. Shao, H. Xie, H. Huang, Z. Li, Z. Sun, Y. Xu, Q. Xiao, X. F. Yu, Y. Zhao, H. Zhang, H. Wang and P. K. Chu, Nat. Commun., 2016, 7, 12967 CrossRef CAS PubMed.
- E. T. Alori, B. R. Glick and O. O. Babalola, Front. Microbiol., 2017, 8, 971 CrossRef PubMed.
- A. Seweryn and T. Bujak, ACS Sustainable Chem. Eng., 2018, 6, 17294–17301 CrossRef CAS.
- J. Xiong, P. Cui, X. Chen, J. Wang, K. Parida, M.-F. Lin and P. S. Lee, Nat. Commun., 2018, 9, 4280 CrossRef PubMed.
- H. Liu, K. Hu, D. Yan, R. Chen, Y. Zou, H. Liu and S. Wang, Adv. Mater., 2018, 30, 1800295 CrossRef PubMed.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef.
- W. Gao, Y. Zhou, X. Wu, Q. Shen, J. Ye and Z. Zou, Adv. Funct. Mater., 2021, 31, 2005197 CrossRef CAS.
- Y. Duan, R. Li, Q. Liu and Z. Shen, Green Chem., 2022, 24, 3475–3501 RSC.
- Y. Zhu, J. Ren, X. Zhang and D. Yang, Nanoscale, 2020, 12, 13297–13310 RSC.
- L. Zeng, L. Huang, J. Zhu, P. Li, P. K. Chu, J. Wang and X. F. Yu, Small, 2022, 18, 2201808 CrossRef CAS PubMed.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- L. Zeng, X. Zhang, Y. Liu, X. Yang, J. Wang, Q. Liu, Q. Luo, C. Jing, X.-F. Yu, G. Qu, P. K. Chu and G. Jiang, Chem, 2021, 8, 632–662 Search PubMed.
- K. Song, Q. Li, Y. Yuan, S. Hu, J. Liu, Y. Zhang, Y.-T. Pan, W. Zhao and J. He, ACS Appl. Nano Mater., 2022, 5, 18080–18092 CrossRef CAS.
- C. M. Fung, C. C. Er, L. L. Tan, A. R. Mohamed and S. P. Chai, Chem. Rev., 2021, 122, 3879–3965 CrossRef PubMed.
- Y. Sun, L. Wang, Y. Li, Y. Li, H. R. Lee, A. Pei, X. He and Y. Cui, Joule, 2019, 3, 1080–1093 CrossRef CAS.
- Q. Zhu, H. Wang, J. Yang, C. Xie, D. Zeng and N. Zhao, ACS Sens., 2018, 3, 2629–2636 CrossRef CAS PubMed.
- J. Zhang, X. Liu, G. Neri and N. Pinna, Adv. Mater., 2016, 28, 795–831 CrossRef CAS PubMed.
- W. Hittorf, Ann. Phys., 1865, 202, 193–228 CrossRef.
- H. Thurn and H. Krebs, Acta Crystallogr., Sect. B: Struct. Sci., 1969, 25, 125–135 CrossRef CAS.
- W. L. Roth, T. W. DeWitt and A. J. Smith, J. Am. Chem. Soc., 1947, 69, 2881–2885 CrossRef CAS PubMed.
- C. C. Stephenson, R. L. Potter, T. G. Maple and J. C. Morrow, J. Chem. Thermodyn., 1969, 1, 59–76 CrossRef CAS.
- L. Du, Y. Zhao, L. Wu, X. Hu, L. Yao, Y. Wang, X. Bai, Y. Dai, J. Qiao, M. G. Uddin, X. Li, J. Lahtinen, X. Bai, G. Zhang, W. Ji and Z. Sun, Nat. Commun., 2021, 12, 4822 CrossRef CAS PubMed.
- A. G. Ricciardulli, Y. Wang, S. Yang and P. Samori, J. Am. Chem. Soc., 2022, 144, 3660–3666 CrossRef CAS PubMed.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef CAS.
- P. W. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS.
- P. W. Bridgman, Proc. Am. Acad. Arts Sci., 1937, 71, 387–460 CrossRef CAS.
- I. Shirotani, R. Maniwa, H. Sato, A. Fukizawa, N. Sato, Y. Maruyama, T. Kajiwara, H. Inokuchi and S.-I. Akimoto, Nippon Kagaku Kaishi, 1981, 10, 1604–1609 CrossRef.
- S. Lange, P. Schmidt and T. Nilges, Inorg. Chem., 2007, 46, 4028–4035 CrossRef CAS PubMed.
- T. Nilges, M. Kersting and T. Pfeifer, J. Solid State Chem., 2008, 181, 1707–1711 CrossRef CAS.
- M. Köpf, N. Eckstein, D. Pfister, C. Grotz, I. Krüger, M. Greiwe, T. Hansen, H. Kohlmann and T. Nilges, J. Cryst. Growth, 2014, 405, 6–10 CrossRef.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- Q. Liu, X. Zhang, J. Wang, Y. Zhang, S. Bian, Z. Cheng, N. Kang, H. Huang, S. Gu, Y. Wang, D. Liu, P. K. Chu and X.-F. Yu, Angew. Chem., Int. Ed., 2020, 59, 14383–14387 CrossRef CAS PubMed.
- L. Zhang, H. Huang, B. Zhang, M. Gu, D. Zhao, X. Zhao, L. Li, J. Zhou, K. Wu, Y. Cheng and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 1074–1080 CrossRef CAS PubMed.
- Z. Zhu and D. Tománek, Phys. Rev. Lett., 2014, 112, 176802 CrossRef PubMed.
- W. H. Han, S. Kim, I.-H. Lee and K. J. Chang, J. Phys. Chem. Lett., 2017, 8, 4627–4632 CrossRef CAS PubMed.
- L. Craco, T. A. d S. Pereira, S. R. Ferreira, S. S. Carara and S. Leoni, Phys. Rev. B, 2018, 98, 035114 CrossRef CAS.
- J. L. Zhang, S. Zhao, C. Han, Z. Wang, S. Zhong, S. Sun, R. Guo, X. Zhou, C. D. Gu, K. D. Yuan, Z. Li and W. Chen, Nano Lett., 2016, 16, 4903–4908 CrossRef CAS PubMed.
- H. Wang, C. Liu, H. Wang, X. Han, S. Zhang, J. Sun, Y. Zhang, Y. Cao, Y. Yao and J. Sun, Chem. Commun., 2021, 57, 3975–3978 RSC.
- V. L. Deringer, D. M. Proserpio, G. Csanyi and C. J. Pickard, Faraday Discuss., 2018, 211, 45–59 RSC.
- V. L. Deringer, M. A. Caro and G. Csanyi, Nat. Commun., 2020, 11, 5461 CrossRef CAS PubMed.
- V. L. Deringer, C. J. Pickard and D. M. Proserpio, Angew. Chem., Int. Ed., 2020, 59, 15880–15885 CrossRef CAS PubMed.
- A. Koneru, R. Batra, S. Manna, T. D. Loeffler, H. Chan, M. Sternberg, A. Avarca, H. Singh, M. J. Cherukara and S. Sankaranarayanan, J. Phys. Chem. Lett., 2022, 13, 1886–1893 CrossRef CAS PubMed.
- Y. Zhou, W. Kirkpatrick and V. L. Deringer, Adv. Mater., 2022, 34, 2107515 CrossRef CAS PubMed.
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- S. Lewis Nathan and G. Nocera Daniel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- D. G. Nocera, 2007 Renewable Energy: Solar Fuels Gordon Research Conference-January 21-26, Gordon Research Conferences, 2008 Search PubMed.
- S. L. Suib, New and Future Developments in Catalysis: Solar Photocatalysis, Elsevier Science, 2013 Search PubMed.
- R. Showstack, World's Heavy Dependence on Fossil Fuels Projected to Continue, EOS, 2017 DOI:10.1029/2017EO082549.
- C. Stewart and M.-A. Hessami, Energy Convers Manage., 2005, 46, 403–420 CrossRef CAS.
- H. Zhou, T. Fan and D. Zhang, ChemSusChem, 2011, 4, 1344–1387 CrossRef CAS PubMed.
- N. S. Diffenbaugh and M. Burke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9808–9813 CrossRef CAS PubMed.
- A. Carleton Tamma and M. Hsiang Solomon, Science, 2016, 353, aad9837 CrossRef PubMed.
- F. Zhang, W. Zhang, D. Wexler and Z. Guo, Adv. Mater., 2022, 34, 2107965 CrossRef CAS PubMed.
- J. Qiao, X. Kong, Z. X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed.
- P. E. M. Amaral, G. P. Nieman, G. R. Schwenk, H. Jing, R. Zhang, E. B. Cerkez, D. Strongin and H. F. Ji, Angew. Chem., Int. Ed., 2019, 58, 6766–6771 CrossRef CAS PubMed.
- F. Xia, H. Wang, J. C. M. Hwang, A. H. C. Neto and L. Yang, Nat. Rev. Phys., 2019, 1, 306–317 CrossRef CAS.
- K. L. Kuntz, R. A. Wells, J. Hu, T. Yang, B. Dong, H. Guo, A. H. Woomer, D. L. Druffel, A. Alabanza, D. Tománek and S. C. Warren, ACS Appl. Mater. Interfaces, 2017, 9, 9126–9135 CrossRef CAS PubMed.
- B. Ghosh, S. Nahas, S. Bhowmick and A. Agarwal, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115433 CrossRef.
- H. Wang, C. Liu, Y. Cao, S. Liu, B. Zhang, Z. Hu and J. Sun, ACS Appl. Energy Mater., 2022, 5, 2184–2191 CrossRef CAS.
- S. Kaur, A. Kumar, S. Srivastava, K. Tankeshwar and R. Pandey, J. Phys. Chem. C, 2018, 122, 26032–26038 CrossRef CAS.
- J. Guan, Z. Zhu and D. Tománek, Phys. Rev. Lett., 2014, 113, 226801 CrossRef PubMed.
- H. Huang, L. Zhang, B. Xiao, Y. Cheng and J. Zhang, Appl. Phys. Lett., 2019, 115, 163101 CrossRef.
- H. Guo, N. Lu, J. Dai, X. Wu and X. C. Zeng, J. Phys. Chem. C, 2014, 118, 14051–14059 CrossRef CAS.
- F. Krafft, Angew. Chem., Int. Ed. Engl., 1969, 8, 660–671 CrossRef CAS PubMed.
- J. Emsley, 13th element, John Wiley & Sons, Inc., 2000 Search PubMed.
- The Nobel Foundation, Percy W. Bridgman – Biographical, 1946.
- J. Xiao, M. Long, C.-S. Deng, J. He, L.-L. Cui and H. Xu, J. Phys. Chem. C, 2016, 120, 4638–4646 CrossRef CAS.
- A. Singh, H. Bae, S. Lee, K. Shabbiri, T. Hussain and H. Lee, Appl. Surf. Sci., 2020, 512, 145641 CrossRef CAS.
- K. Ashley, D. Cordell and D. Mavinic, Chemosphere, 2011, 84, 737–746 CrossRef CAS PubMed.
- M. F. Crass, J. Chem. Educ., 1941, 18, 316 CrossRef.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383–1385 CrossRef CAS PubMed.
- M. Kohn, J. Chem. Educ., 1944, 21, 522 CrossRef CAS.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 44, 7616–7619 CrossRef CAS PubMed.
- P. F. McMillan, Nat. Mater., 2005, 4, 715–718 CrossRef CAS PubMed.
- J. Wittig and B. T. Matthias, Science, 1968, 160, 994–995 CrossRef CAS PubMed.
- W. Tao, N. Kong, X. Ji, Y. Zhang, A. Sharma, J. Ouyang, B. Qi, J. Wang, N. Xie, C. Kang, H. Zhang, O. C. Farokhzad and J. S. Kim, Chem. Soc. Rev., 2019, 48, 2891–2912 RSC.
- A. Favron, E. Gaufres, F. Fossard, A. L. Phaneuf-L'Heureux, N. Y. Tang, P. L. Levesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- R. Gusmão, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 8052–8072 CrossRef PubMed.
- T. Gorkan, E. Aktürk and S. Ciraci, Phys. Rev. B, 2019, 100, 125306 CrossRef CAS.
- M. Wu, H. Fu, L. Zhou, K. Yao and X. C. Zeng, Nano Lett., 2015, 15, 3557–3562 CrossRef CAS PubMed.
- K. Burke, J. Chem. Phys., 2012, 136, 150901 CrossRef PubMed.
- V. L. Deringer, M. A. Caro and G. Csanyi, Adv. Mater., 2019, 31, 1902765 CrossRef CAS PubMed.
- J. Behler, Angew. Chem., Int. Ed., 2017, 56, 12828–12840 CrossRef CAS PubMed.
- Z. Wang, Z. Sun, H. Yin, X. Liu, J. Wang, H. Zhao, C. H. Pang, T. Wu, S. Li, Z. Yin and X.-F. Yu, Adv. Mater., 2022, 34, 2104113 CrossRef CAS PubMed.
- N. B. Goodman, L. Ley and D. W. Bullett, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 7440–7450 CrossRef CAS.
- M. Häser and O. Treutler, J. Chem. Phys., 1995, 102, 3703–3711 CrossRef.
- N. J. Brassington, H. G. M. Edwards and D. A. Long, J. Raman Spectrosc., 1981, 11, 346–348 CrossRef CAS.
- L. R. Maxwell, S. B. Hendricks and V. M. Mosley, J. Chem. Phys., 1935, 3, 699–709 CrossRef CAS.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- A. Simon, H. Borrmann and J. Horakh, Chem. Ber., 2006, 130, 1235–1240 CrossRef.
- H. Okudera, R. E. Dinnebier and A. Simon, Z. Kirchenges., 2005, 220, 259–264 CAS.
- R. R. Hart, M. B. Robin and N. A. Kuebler, J. Chem. Phys., 1965, 42, 3631–3638 CrossRef CAS.
- L. Pauling and M. Simonetta, J. Chem. Phys., 1952, 20, 29–34 CrossRef CAS.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 CrossRef CAS PubMed.
- J. C. Jamieson, Science, 1963, 139, 1291–1292 CrossRef CAS PubMed.
- L. Cartz, S. R. Srinivasa, R. J. Riedner, J. D. Jorgensen and T. G. Worlton, J. Chem. Phys., 1979, 71, 1718–1721 CrossRef CAS.
- A. Carvalho, M. Wang, X. Zhu, A. S. Rodin, H. Su and A. H. Castro Neto, Nat. Rev. Mater., 2016, 1, 16061 CrossRef CAS.
- S. Zhou, C. Bao, B. Fan, H. Zhou, Q. Gao, H. Zhong, T. Lin, H. Liu, P. Yu, P. Tang, S. Meng, W. Duan and S. Zhou, Nature, 2023, 614, 75–80 CrossRef CAS PubMed.
- X. Ling, H. Wang, S. Huang, F. Xia and M. S. Dresselhaus, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4523 CrossRef CAS PubMed.
- J. Dai and X. C. Zeng, J. Phys. Chem. Lett., 2014, 5, 1289–1293 CrossRef CAS PubMed.
- Y. Cai, G. Zhang and Y.-W. Zhang, Phosphorene: Physical Properties, Synthesis, and Fabrication, Jenny Standford Publishing, 2019 Search PubMed.
- J. M. Zaug, A. K. Soper and S. M. Clark, Nat. Mater., 2008, 7, 890–899 CrossRef CAS PubMed.
- D. J. Olego, J. A. Baumann, M. A. Kuck, R. Schachter, C. G. Michel and P. M. Raccah, Solid State Commun., 1984, 52, 311–314 CrossRef CAS.
- R. O. Jones and D. Hohl, J. Chem. Phys., 1990, 92, 6710–6721 CrossRef CAS.
- B. V. Shanabrook and J. S. Lannin, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 4771–4780 CrossRef CAS.
- G. E. Jellison, Solid State Commun., 1979, 30, 481–485 CrossRef CAS.
- H. Farman, J. C. Dore and S. R. Elliott, Phys. Lett. A, 1994, 186, 410–414 CrossRef CAS.
- S. Zhang, H. J. Qian, Z. Liu, H. Ju, Z. Y. Lu, H. Zhang, L. Chi and S. Cui, Angew. Chem., Int. Ed., 2019, 58, 1659–1663 CrossRef CAS PubMed.
- M. Hart, E. R. White, J. Chen, C. M. McGilvery, C. J. Pickard, A. Michaelides, A. Sella, M. S. P. Shaffer and C. G. Salzmann, Angew. Chem., Int. Ed., 2017, 56, 8144–8148 CrossRef CAS PubMed.
- S. Böcker and M. Häser, Z. Anorg. Allg. Chem., 1995, 621, 258–286 CrossRef.
- M. Häser, U. Schneider and R. Ahlrichs, J. Am. Chem. Soc., 1992, 114, 9551–9559 CrossRef.
- M. Rubenstein and F. M. Ryan, J. Electrochem. Soc., 1966, 113, 1063 CAS.
- V. V. Nechaeva, N. D. Talanov and A. I. Soklakov, Zh. Neorg. Khim., 1979, 24, 1979–1981 CAS.
- Z. Sun, B. Zhang and Q. Yan, Inorg. Chem. Front., 2022, 9, 4385–4393 RSC.
- G. Schusteritsch, M. Uhrin and C. J. Pickard, Nano Lett., 2016, 16, 2975–2980 CrossRef CAS PubMed.
- Z. Hu and W. Guo, Small, 2021, 17, 2008004 CrossRef CAS PubMed.
- Y. L. Lu, S. Dong, J. Li, Y. Wu, L. Wang and H. Zhao, Phys. Chem. Chem. Phys., 2020, 22, 13713–13720 RSC.
- Z. Zhu, X. Cai, S. Yi, J. Chen, Y. Dai, C. Niu, Z. Guo, M. Xie, F. Liu, J.-H. Cho, Y. Jia and Z. Zhang, Phys. Rev. Lett., 2017, 119, 106101 CrossRef PubMed.
- X. Huang, J. Guan, Z. Lin, B. Liu, S. Xing, W. Wang and J. Guo, Nano Lett., 2017, 17, 4619–4623 CrossRef CAS PubMed.
- L. Xian, A. Pérez Paz, E. Bianco, P. M. Ajayan and A. Rubio, 2D Mater., 2017, 4, 041003 CrossRef.
- G. Cicirello, M. Wang, Q. P. Sam, J. L. Hart, N. L. Williams, H. Yin, J. J. Cha and J. Wang, J. Am. Chem. Soc., 2023, 145, 8218–8230 CrossRef CAS PubMed.
- T. Nilges, P. Schmidt and R. Weihrich, Encycl. Inorg. Bioinorg. Chem., 2018 DOI:10.1002/9781119951438.eibc2643.
- M. Hermes and F. Scholz, Electrochem. Commun., 2000, 2, 845–850 CrossRef CAS.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- L. Liang, J. Wang, W. Lin, B. G. Sumpter, V. Meunier and M. Pan, Nano Lett., 2014, 14, 6400–6406 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Zhou, T.-Y. Cai and S. Ju, Phys. Rev. B, 2021, 104, 245401 CrossRef CAS.
- R. B. Pontes, R. H. Miwa, A. J. R. da Silva, A. Fazzio and J. E. Padilha, Phys. Rev. B, 2018, 97, 235419 CrossRef CAS.
- J. Zhuang, C. Liu, Q. Gao, Y. Liu, H. Feng, X. Xu, J. Wang, J. Zhao, S. X. Dou, Z. Hu and Y. Du, ACS Nano, 2018, 12, 5059–5065 CrossRef CAS PubMed.
- C. Gu, S. Zhao, J. L. Zhang, S. Sun, K. Yuan, Z. Hu, C. Han, Z. Ma, L. Wang, F. Huo, W. Huang, Z. Li and W. Chen, ACS Nano, 2017, 11, 4943–4949 CrossRef CAS PubMed.
- J. K. Ellis, M. J. Lucero and G. E. Scuseria, Appl. Phys. Lett., 2011, 99, 261908 CrossRef.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- I. G. Lezama, A. Arora, A. Ubaldini, C. Barreteau, E. Giannini, M. Potemski and A. F. Morpurgo, Nano Lett., 2015, 15, 2336–2342 CrossRef CAS PubMed.
- T. Low, A. S. Rodin, A. Carvalho, Y. Jiang, H. Wang, F. Xia and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 075434 CrossRef CAS.
- G. Zhang, S. Huang, A. Chaves, C. Song, V. O. Özçelik, T. Low and H. Yan, Nat. Commun., 2017, 8, 14071 CrossRef CAS PubMed.
- Q. Liu, X. Zhang, L. B. Abdalla, A. Fazzio and A. Zunger, Nano Lett., 2015, 15, 1222–1228 CrossRef CAS PubMed.
- T. Kaewmaraya, L. Ngamwongwan, P. Moontragoon, W. Jarernboon, D. Singh, R. Ahuja, A. Karton and T. Hussain, J. Hazard. Mater., 2021, 401, 123340 CrossRef CAS PubMed.
- X. Peng, Q. Wei and A. Copple, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 085402 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- L. Liao, Y.-C. Lin, M. Bao, R. Cheng, J. Bai, Y. Liu, Y. Qu, K. L. Wang, Y. Huang and X. Duan, Nature, 2010, 467, 305–308 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- M. Buscema, D. J. Groenendijk, S. I. Blanter, G. A. Steele, H. S. van der Zant and A. Castellanos-Gomez, Nano Lett., 2014, 14, 3347–3352 CrossRef CAS PubMed.
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. van Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Nat. Commun., 2019, 10, 522 CrossRef CAS PubMed.
- Y. Xu, J. Dai and X. C. Zeng, J. Phys. Chem. Lett., 2016, 7, 302–307 CrossRef CAS PubMed.
- Z. Sun, B. Zhang, Y. Zhao, M. Khurram and Q. Yan, Chem. Mater., 2021, 33, 6240–6248 CrossRef CAS.
- E. Golias, M. Krivenkov, A. Varykhalov, J. Sanchez-Barriga and O. Rader, Nano Lett., 2018, 18, 6672–6678 CrossRef CAS PubMed.
- R. Fei, A. Faghaninia, R. Soklaski, J.-A. Yan, C. Lo and L. Yang, Nano Lett., 2014, 14, 6393–6399 CrossRef CAS PubMed.
- T. Yin, L. Long, X. Tang, M. Qiu, W. Liang, R. Cao, Q. Zhang, D. Wang and H. Zhang, Adv. Sci., 2020, 7, 2001431 CrossRef CAS PubMed.
- X. Qian, J. Zhou and G. Chen, Nat. Mater., 2021, 20, 1188–1202 CrossRef CAS PubMed.
- X. Zhang, X. F. Qiao, W. Shi, J. B. Wu, D. S. Jiang and P. H. Tan, Chem. Soc. Rev., 2015, 44, 2757–2785 RSC.
- Y. Zhang, J. Wang, Q. Liu, S. Gu, Z. Sun, P. K. Chu and X. Yu, APL Mater., 2020, 8, 120903 CrossRef CAS.
- N. Mao, S. Zhang, J. Wu, J. Zhang and L. Tong, Small Methods, 2018, 2, 1700409 CrossRef.
- J. Hlinka, T. Ostapchuk, E. Buixaderas, C. Kadlec, P. Kuzel, I. Gregora, J. Kroupa, M. Savinov, A. Klic, J. Drahokoupil, I. Etxebarria and J. Dec, Phys. Rev. Lett., 2014, 112, 197601 CrossRef CAS PubMed.
- S. Sugai and I. Shirotani, Solid State Commun., 1985, 53, 753–755 CrossRef CAS.
- C. Kaneta, H. Katayama-Yoshida and A. Morita, J. Phys. Soc. Jpn., 1986, 55, 1213–1223 CrossRef CAS.
- N. Suzuki and M. Aoki, Solid State Commun., 1987, 61, 595–600 CrossRef CAS.
- J. Ribeiro-Soares, R. M. Almeida, L. G. Cançado, M. S. Dresselhaus and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 205421 CrossRef.
- L. Zhang, H. Huang, Z. Lv, L. Li, M. Gu, X. Zhao, B. Zhang, Y. Cheng and J. Zhang, ACS Appl. Electron. Mater., 2021, 3, 1043–1049 CrossRef CAS.
- A. Impellizzeri, A. A. Vorfolomeeva, N. V. Surovtsev, A. V. Okotrub, C. P. Ewels and D. V. Rybkovskiy, Phys. Chem. Chem. Phys., 2021, 23, 16611–16622 RSC.
- B. Smith, B. Vermeersch, J. Carrete, E. Ou, J. Kim, N. Mingo, D. Akinwande and L. Shi, Adv. Mater., 2017, 29, 1603756 CrossRef PubMed.
- S. Lee, F. Yang, J. Suh, S. Yang, Y. Lee, G. Li, H. Sung Choe, A. Suslu, Y. Chen, C. Ko, J. Park, K. Liu, J. Li, K. Hippalgaonkar, J. J. Urban, S. Tongay and J. Wu, Nat. Commun., 2015, 6, 8573 CrossRef CAS PubMed.
- X.-J. Ge, K.-L. Yao and J.-T. Lü, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 165433 CrossRef.
- Y. Zhao, H. Wang, H. Huang, Q. Xiao, Y. Xu, Z. Guo, H. Xie, J. Shao, Z. Sun, W. Han, X. F. Yu, P. Li and P. K. Chu, Angew. Chem., Int. Ed., 2016, 55, 5003–5007 CrossRef CAS PubMed.
- D. K. Sang, H. Wang, Z. Guo, N. Xie and H. Zhang, Adv. Funct. Mater., 2019, 29, 1903419 CrossRef CAS.
- L. Zhao, X. Chao, N. Xu, G. Ling and P. Zhang, Adv. Eng. Mater., 2021, 23, 2100450 CrossRef CAS.
- X. Liu, K. Chen, X. Li, Q. Xu, J. Weng and J. Xu, Adv. Mater., 2021, 33, 2005924 CrossRef CAS PubMed.
- H. Lyu, W. Wilwin, Z. Lin and H. Su, J. Phys. Chem. C, 2022, 126, 8883–8888 CrossRef.
- J. K. Burdett, P. Haaland and T. J. McLarnan, J. Chem. Phys., 1981, 75, 5774–5781 CrossRef CAS.
- L. Radom, W. J. Hehre and J. A. Pople, J. Am. Chem. Soc., 1972, 94, 2371–2381 CrossRef CAS.
- S. F. Nelsen and J. M. Buschek, J. Am. Chem. Soc., 1973, 95, 2011–2013 CrossRef CAS.
- F. Bachhuber, J. von Appen, R. Dronskowski, P. Schmidt, T. Nilges, A. Pfitzner and R. Weihrich, Angew. Chem., Int. Ed., 2014, 53, 11629–11633 CrossRef CAS PubMed.
- K. Ding, L. Wen, S. Huang, Y. Li, Y. Zhang and Y. Lu, RSC Adv., 2016, 6, 80872–80884 RSC.
- S. Kuriakose, T. Ahmed, S. Balendhran, V. Bansal, S. Sriram, M. Bhaskaran and S. Walia, 2D Mater., 2018, 5, 032001 CrossRef.
- J.-S. Kim, Y. Liu, W. Zhu, S. Kim, D. Wu, L. Tao, A. Dodabalapur, K. Lai and D. Akinwande, Sci. Rep., 2015, 5, 8989 CrossRef CAS PubMed.
- F. Alsaffar, S. Alodan, A. Alrasheed, A. Alhussain, N. Alrubaiq, A. Abbas and M. R. Amer, Sci. Rep., 2017, 7, 44540 CrossRef CAS PubMed.
- M. T. Edmonds, A. Tadich, A. Carvalho, A. Ziletti, K. M. O’Donnell, S. P. Koenig, D. F. Coker, B. Özyilmaz, A. H. C. Neto and M. S. Fuhrer, ACS Appl. Mater. Interfaces, 2015, 7, 14557–14562 CrossRef CAS PubMed.
- Q. Zhou, Q. Chen, Y. Tong and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 11437–11441 CrossRef CAS PubMed.
- A. Fali, M. Snure and Y. Abate, Appl. Phys. Lett., 2021, 118, 163105 CrossRef CAS.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- Z. Hu, Q. Li, B. Lei, Q. Zhou, D. Xiang, Z. Lyu, F. Hu, J. Wang, Y. Ren, R. Guo, E. Goki, L. Wang, C. Han, J. Wang and W. Chen, Angew. Chem., Int. Ed., 2017, 56, 9131–9135 CrossRef CAS PubMed.
- W. Yu, J. Xu, F. Chen, Y. Wang, S. Tang, F. Geng, J. Lv, G. Qu, L. Zhao and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213595 CAS.
- Y. Zhao, Z. Sun, B. Zhang and Q. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 9925–9932 CrossRef CAS PubMed.
- T. Zhang, Y. Wan, H. Xie, Y. Mu, P. Du, D. Wang, X. Wu, H. Ji and L. Wan, J. Am. Chem. Soc., 2018, 140, 7561–7567 CrossRef CAS PubMed.
- L. Li, M. Engel, D. B. Farmer, S.-J. Han and H. S. P. Wong, ACS Nano, 2016, 10, 4672–4677 CrossRef CAS PubMed.
- J. R. Brent, N. Savjani, E. A. Lewis, S. J. Haigh, D. J. Lewis and P. O'Brien, Chem. Commun., 2014, 50, 13338–13341 RSC.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef CAS PubMed.
- W. Lv, B. Yang, B. Wang, W. Wan, Y. Ge, R. Yang, C. Hao, J. Xiang, B. Zhang, Z. Zeng and Z. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 9663–9668 CrossRef CAS PubMed.
- Z. Guo, S. Chen, Z. Wang, Z. Yang, F. Liu, Y. Xu, J. Wang, Y. Yi, H. Zhang, L. Liao, P. K. Chu and X. F. Yu, Adv. Mater., 2017, 29, 1703811 CrossRef PubMed.
- R. W. Keyes, Phys. Rev., 1953, 92, 580–584 CrossRef CAS.
- H. Krebs, H. Weitz and K. H. Worms, Z. Anorg. Allg. Chem., 1955, 280, 119–133 CrossRef CAS.
- A. Brown and S. Rundqvist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS.
- Y. Maruyama, S. Suzuki, K. Kobayashi and S. Tanuma, Phys. B, 1981, 105, 99–102 CrossRef CAS.
- M. Binnewies, M. Schmidt and P. Schmidt, Z. Anorg. Allg. Chem., 2017, 643, 1295–1311 CrossRef CAS.
- M. R. P. Pielmeier and T. Nilges, Angew. Chem., Int. Ed., 2021, 60, 6816–6823 CrossRef CAS PubMed.
- M. Liu, S. Feng, Y. Hou, S. Zhao, L. Tang, J. Liu, F. Wang and B. Liu, Mater. Today, 2020, 36, 91–101 CrossRef CAS.
- A. Castellanos-Gomez, L. Vicarelli, E. Prada, J. O. Island, K. L. Narasimha-Acharya, S. I. Blanter, D. J. Groenendijk, M. Buscema, G. A. Steele, J. V. Alvarez, H. W. Zandbergen, J. J. Palacios and H. S. J. van der Zant, 2D Mater., 2014, 1, 025001 CrossRef CAS.
- Z. Duan, Y. Wang, S. Bian, D. Liu, Y. Zhang, X. Zhang, R. He, J. Wang, G. Qu, P. K. Chu and X. F. Yu, Nanoscale, 2022, 14, 2599–2604 RSC.
- X. Zhu, T. Zhang, Z. Sun, H. Chen, J. Guan, X. Chen, H. Ji, P. Du and S. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed.
- W. Liu, Y. Zhu, X. Xu, S. Wang and X. Zhang, J. Mater. Sci.: Mater. Electron., 2020, 31, 9543–9549 CrossRef CAS.
- L. Yang, D. Wang, M. Liu, H. Liu, J. Tan, Z. Wang, H. Zhou, Q. Yu, J. Wang, J. Lin, X. Zou, L. Qiu, H.-M. Cheng and B. Liu, Mater. Today, 2021, 51, 145–154 CrossRef CAS.
- X. Liu, B. Gaihre, M. N. George, Y. Li, M. Tilton, M. J. Yaszemski and L. Lu, Biomater. Sci., 2021, 9, 2768–2803 RSC.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- Z. Guo, H. Zhang, S. Lu, Z. Wang, S. Tang, J. Shao, Z. Sun, H. Xie, H. Wang, X.-F. Yu and P. K. Chu, Adv. Funct. Mater., 2015, 25, 6996–7002 CrossRef CAS.
- H. Wang, S. Jiang, W. Shao, X. Zhang, S. Chen, X. Sun, Q. Zhang, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2018, 140, 3474–3480 CrossRef CAS PubMed.
- G. Abellan, V. Lloret, U. Mundloch, M. Marcia, C. Neiss, A. Gorling, M. Varela, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 14557–14562 CrossRef CAS PubMed.
- D. Hanlon, C. Backes, E. Doherty, C. S. Cucinotta, N. C. Berner, C. Boland, K. Lee, A. Harvey, P. Lynch, Z. Gholamvand, S. Zhang, K. Wang, G. Moynihan, A. Pokle, Q. M. Ramasse, N. McEvoy, W. J. Blau, J. Wang, G. Abellan, F. Hauke, A. Hirsch, S. Sanvito, D. D. O'Regan, G. S. Duesberg, V. Nicolosi and J. N. Coleman, Nat. Commun., 2015, 6, 8563 CrossRef CAS PubMed.
- J. Kang, J. D. Wood, S. A. Wells, J.-H. Lee, X. Liu, K.-S. Chen and M. C. Hersam, ACS Nano, 2015, 9, 3596–3604 CrossRef CAS PubMed.
- S. Lin, S. Liu, Z. Yang, Y. Li, T. W. Ng, Z. Xu, Q. Bao, J. Hao, C.-S. Lee, C. Surya, F. Yan and S. P. Lau, Adv. Funct. Mater., 2016, 26, 864–871 CrossRef CAS.
- H. Mu, S. Lin, Z. Wang, S. Xiao, P. Li, Y. Chen, H. Zhang, H. Bao, S. P. Lau, C. Pan, D. Fan and Q. Bao, Adv. Opt. Mater., 2015, 3, 1447–1453 CrossRef CAS.
- M. Qiu, D. Wang, W. Liang, L. Liu, Y. Zhang, X. Chen, D. K. Sang, C. Xing, Z. Li, B. Dong, F. Xing, D. Fan, S. Bao, H. Zhang and Y. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 501–506 CrossRef CAS PubMed.
- Z. Sofer, D. Bousa, J. Luxa, V. Mazanek and M. Pumera, Chem. Commun., 2016, 52, 1563–1566 RSC.
- A. H. Woomer, T. W. Farnsworth, J. Hu, R. A. Wells, C. L. Donley and S. C. Warren, ACS Nano, 2015, 9, 8869–8884 CrossRef CAS PubMed.
- J. Y. Xu, L. F. Gao, C. X. Hu, Z. Y. Zhu, M. Zhao, Q. Wang and H. L. Zhang, Chem. Commun., 2016, 52, 8107–8110 RSC.
- P. Yasaei, B. Kumar, T. Foroozan, C. Wang, M. Asadi, D. Tuschel, J. E. Indacochea, R. F. Klie and A. Salehi-Khojin, Adv. Mater., 2015, 27, 1887–1892 CrossRef CAS PubMed.
- B. Zhang, F. Lou, R. Zhao, J. He, J. Li, X. Su, J. Ning and K. Yang, Opt. Lett., 2015, 40, 3691–3694 CrossRef CAS PubMed.
- J.-W. Jiang and H. S. Park, J. Phys. D: Appl. Phys., 2014, 47, 385304 CrossRef.
- M. B. Erande, S. R. Suryawanshi, M. A. More and D. J. Late, Eur. J. Inorg. Chem., 2015, 3102–3107 CrossRef CAS.
- L. Li, D. Zhang, J. Deng, Y. Gou and J. Fang, J. Energy Chem., 2020, 49, 365–374 CrossRef.
- F. Luo, D. Wang, J. Zhang, X. Li, D. Liu, H. Li, M. Lu, X. Xie, L. Huang and W. Huang, ACS Appl. Nano Mater., 2019, 2, 3793–3801 CrossRef CAS.
- J. S. Mayell and A. J. Bard, J. Am. Chem. Soc., 1963, 85, 421–425 CrossRef CAS.
- C. Wang, Q. He, U. Halim, Y. Liu, E. Zhu, Z. Lin, H. Xiao, X. Duan, Z. Feng, R. Cheng, N. O. Weiss, G. Ye, Y.-C. Huang, H. Wu, H.-C. Cheng, I. Shakir, L. Liao, X. Chen, W. A. Goddard Iii, Y. Huang and X. Duan, Nature, 2018, 555, 231–236 CrossRef CAS PubMed.
- X. Jiang, Z. e Lin, X. Zeng, J. He, F. Xu, P. Deng, J. Jia, X. Jiang, X. Hou and Z. Long, Chem. Commun., 2019, 55, 12192–12195 RSC.
- H. Huang, M. Gao, Y. Kang, J. Li, J. Wang, L. Wu, P. K. Chu, Y. Huang, M. R. Ibarra and X. F. Yu, Chem. Commun., 2019, 56, 221–224 RSC.
- W. Lu, H. Nan, J. Hong, Y. Chen, C. Zhu, Z. Liang, X. Ma, Z. Ni, C. Jin and Z. Zhang, Nano Res., 2014, 7, 853–859 CrossRef CAS.
- H. Kwon, S. W. Seo, T. G. Kim, E. S. Lee, P. T. Lanh, S. Yang, S. Ryu and J. W. Kim, ACS Nano, 2016, 10, 8723–8731 CrossRef CAS PubMed.
- X. Liu, K. S. Chen, S. A. Wells, I. Balla, J. Zhu, J. D. Wood and M. C. Hersam, Adv. Mater., 2017, 29, 1604121 CrossRef PubMed.
- R. Garcia, A. W. Knoll and E. Riedo, Nat. Nanotechnol., 2014, 9, 577–587 CrossRef CAS PubMed.
- Z. Cai, B. Liu, X. Zou and H.-M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS PubMed.
- J. B. Smith, D. Hagaman and H. F. Ji, Nanotechnology, 2016, 27, 215602 CrossRef PubMed.
- Y. Xu, X. Shi, Y. Zhang, H. Zhang, Q. Zhang, Z. Huang, X. Xu, J. Guo, H. Zhang, L. Sun, Z. Zeng, A. Pan and K. Zhang, Nat. Commun., 2020, 11, 1330 CrossRef CAS PubMed.
- C. Chen, Y. Yin, R. Zhang, Q. Yuan, Y. Xu, Y. Zhang, J. Chen, Y. Zhang, C. Li, J. Wang, J. Li, L. Fei, Q. Yu, Z. Zhou, H. Zhang, R. Cheng, Z. Dong, X. Xu, A. Pan, K. Zhang and J. He, Nat. Mater., 2023, 22, 717–724 CrossRef CAS PubMed.
- H. Lowndes Douglas, D. B. Geohegan, A. A. Puretzky, D. P. Norton and C. M. Rouleau, Science, 1996, 273, 898–903 CrossRef PubMed.
- P. Shriber, A. Samanta, G. D. Nessim and I. Grinberg, J. Phys. Chem. Lett., 2018, 9, 1759–1764 CrossRef CAS PubMed.
- Z. Wu, Y. Lyu, Y. Zhang, R. Ding, B. Zheng, Z. Yang, S. P. Lau, X. H. Chen and J. Hao, Nat. Mater., 2021, 20, 1203–1209 CrossRef CAS PubMed.
- Z. Yang, J. Hao, S. Yuan, S. Lin, H. M. Yau, J. Dai and S. P. Lau, Adv. Mater., 2015, 27, 3748–3754 CrossRef CAS PubMed.
- C. Tan and H. Zhang, Nat. Commun., 2015, 6, 7873 CrossRef CAS PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, Q. Lei, R. Hua, Y. Tian and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4345–4350 CrossRef CAS PubMed.
- Y. Zhang, X. Rui, Y. Tang, Y. Liu, J. Wei, S. Chen, W. R. Leow, W. Li, Y. Liu, J. Deng, B. Ma, Q. Yan and X. Chen, Adv. Energy Mater., 2016, 6, 1502409 CrossRef.
- G. Zhao, T. Wang, Y. Shao, Y. Wu, B. Huang and X. Hao, Small, 2017, 13, 1602243 CrossRef PubMed.
- N. Wei, Y. Chen, Y. Zhang, C. Zhou, X. Hao, K. Xu, K. Cai and J. Chen, Nanoscale, 2018, 10, 4385–4390 RSC.
- P. Masih Das, G. Danda, A. Cupo, W. M. Parkin, L. Liang, N. Kharche, X. Ling, S. Huang, M. S. Dresselhaus, V. Meunier and M. Drndic, ACS Nano, 2016, 10, 5687–5695 CrossRef CAS PubMed.
- X. W. Feng, X. Huang, L. Chen, W. C. Tan, L. Wang and K. W. Ang, Adv. Funct. Mater., 2018, 28, 1801524 CrossRef.
- M. C. Watts, L. Picco, F. S. Russell-Pavier, P. L. Cullen, T. S. Miller, S. P. Bartus, O. D. Payton, N. T. Skipper, V. Tileli and C. A. Howard, Nature, 2019, 568, 216–220 CrossRef CAS PubMed.
- Y. C. Cheng, Y. H. Zhu, Y. Han, Z. Y. Liu, B. C. Yang, A. M. Nie, W. Huang, R. Shahbazian-Yassar and F. Mashayek, Chem. Mater., 2017, 29, 1350–1356 CrossRef CAS.
- G. Abellan, C. Neiss, V. Lloret, S. Wild, J. C. Chacon-Torres, K. Werbach, F. Fedi, H. Shiozawa, A. Gorling, H. Peterlik, T. Pichler, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2017, 56, 15267–15273 CrossRef CAS PubMed.
- T. J. Macdonald, A. J. Clancy, W. Xu, Z. Jiang, C. T. Lin, L. Mohan, T. Du, D. D. Tune, L. Lanzetta, G. Min, T. Webb, A. Ashoka, R. Pandya, V. Tileli, M. A. McLachlan, J. R. Durrant, S. A. Haque and C. A. Howard, J. Am. Chem. Soc., 2021, 143, 21549–21559 CrossRef CAS PubMed.
- Z. Liu, Y. Sun, H. Cao, D. Xie, W. Li, J. Wang and A. K. Cheetham, Nat. Commun., 2020, 11, 3917 CrossRef CAS PubMed.
- W. Yu, J. Yang, J. Li, K. Zhang, H. Xu, X. Zhou, W. Chen and K. P. Loh, Adv. Mater., 2021, 33, 2102083 CrossRef CAS PubMed.
- Y. Yu, J. Yao, X. Niu, B. Xing, Y. Liu, X. Wu, M. Li, X. Yan, J. Sha and Y. Wang, CrystEngComm, 2020, 22, 3824–3830 RSC.
- L. Macewicz, K. Pyrchla, R. Bogdanowicz, G. Sumanasekera and J. B. Jasinski, J. Phys. Chem. Lett., 2021, 12, 8347–8354 CrossRef CAS PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS PubMed.
- X. Zhang, H. Xie, Z. Liu, C. Tan, Z. Luo, H. Li, J. Lin, L. Sun, W. Chen, Z. Xu, L. Xie, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 3653–3657 CrossRef CAS PubMed.
- M. Ozhukil Valappil, S. Alwarappan and V. K. Pillai, Nanoscale, 2022, 14, 1037–1053 RSC.
- Z. Sun, H. Xie, S. Tang, X. F. Yu, Z. Guo, J. Shao, H. Zhang, H. Huang, H. Wang and P. K. Chu, Angew. Chem., Int. Ed., 2015, 54, 11526–11530 CrossRef CAS PubMed.
- Z. Sun, Y. Zhao, Z. Li, H. Cui, Y. Zhou, W. Li, W. Tao, H. Zhang, H. Wang, P. K. Chu and X. F. Yu, Small, 2017, 13, 1602896 CrossRef PubMed.
- L. Chan, P. Gao, W. Zhou, C. Mei, Y. Huang, X. F. Yu, P. K. Chu and T. Chen, ACS Nano, 2018, 12, 12401–12415 CrossRef CAS PubMed.
- S. Geng, L. Wu, H. Cui, W. Tan, T. Chen, P. K. Chu and X. F. Yu, Chem. Commun., 2018, 54, 6060–6063 RSC.
- M. Meng, Z. Gan, J. Zhang, K. Liu, L. Wang, S. Li, Y. Yao, Y. Zhu and J. Li, Phys. Status Solidi B, 2017, 254, 1700011 CrossRef.
- W. Chen, K. Li, Y. Wang, X. Feng, Z. Liao, Q. Su, X. Lin and Z. He, J. Phys. Chem. Lett., 2017, 8, 591–598 CrossRef CAS PubMed.
- W. Wang, X. Niu, H. Qian, L. Guan, M. Zhao, X. Ding, S. Zhang, Y. Wang and J. Sha, Nanotechnology, 2016, 27, 505204 CrossRef PubMed.
- C. C. Mayorga-Martinez, N. Mohamad Latiff, A. Y. S. Eng, Z. Sofer and M. Pumera, Anal. Chem., 2016, 88, 10074–10079 CrossRef CAS PubMed.
- X. Tang, H. Chen, J. S. Ponraj, S. C. Dhanabalan, Q. Xiao, D. Fan and H. Zhang, Adv. Sci., 2018, 5, 1800420 CrossRef PubMed.
- X. Ren, X. Yang, G. Xie and J. Luo, ACS Appl. Nano Mater., 2020, 3, 4799–4809 CrossRef CAS.
- C. Zhu, F. Xu, L. Zhang, M. Li, J. Chen, S. Xu, G. Huang, W. Chen and L. Sun, Chem. – Eur. J., 2016, 22, 7357–7362 CrossRef CAS PubMed.
- S. Ge, L. Zhang, P. Wang and Y. Fang, Sci. Rep., 2016, 6, 27307 CrossRef CAS PubMed.
- X. Ren, F. Zhang and X. Zhang, Chem. – Asian J., 2018, 13, 1842–1846 CrossRef CAS PubMed.
- T. W. DeWitt and S. Skolnik, J. Am. Chem. Soc., 1946, 68, 2305–2309 CrossRef CAS.
- M. Serrano-Ruiz, A. Romerosa and P. Lorenzo-Luis, Eur. J. Inorg. Chem., 2014, 1587–1598 CrossRef CAS.
- A. Pedler, J. Chem. Soc., Trans., 1890, 57, 599–613 RSC.
- G. Rathenau, Physica, 1937, 4, 503–514 CrossRef CAS.
- N. P. Tarasova, Y. V. Smetannikov, I. M. Artemkina, I. A. Lavrov, M. A. Sinaiskii and V. I. Ermakov, Dokl. Chem., 2006, 410, 189–191 CrossRef CAS.
- A. Piro Nicholas, S. Figueroa Joshua, T. McKellar Jessica and C. Cummins Christopher, Science, 2006, 313, 1276–1279 CrossRef CAS PubMed.
- M. E. Barr, B. R. Adams, R. R. Weller and L. F. Dahl, J. Am. Chem. Soc., 1991, 113, 3052–3060 CrossRef CAS.
- M. Grayson, Pure Appl. Chem., 1964, 9, 193–204 CrossRef CAS.
- D. Perner and A. Henglein, Z. Naturforsch. B, 1962, 17, 703–711 CrossRef.
- R. Saravanan, E. Thirumal, V. K. Gupta, V. Narayanan and A. Stephen, J. Mol. Liq., 2013, 177, 394–401 CrossRef CAS.
- Y. Sun, Z. Ren, Y. Liu and R. Fu, Mater. Lett., 2019, 236, 542–546 CrossRef CAS.
- F. Chen, K. Mu, D. Zhang, X. Mi, Y. Li, Z. Shen and S. Zhan, Top. Catal., 2021, 64, 559–566 CrossRef CAS.
- M. Zhang, J. Liu, L. Liu, K. Sun, X. Liang, J. Wan and F. Fu, Ceram. Int., 2020, 46, 23165–23172 CrossRef CAS.
- L. Liu, J. Shen, K. Wu and N. Yang, Small Methods, 2021, 5, 2100720 CrossRef CAS PubMed.
- Y. Li, S. Jiang, Y. Qian, Y. Han, J. Zhou, T. Li, L. Xi, N. Lin and Y. Qian, Chem. Commun., 2019, 55, 6751–6754 RSC.
- W. Liu, S. Ju and X. Yu, ACS Nano, 2020, 14, 974–984 CrossRef CAS PubMed.
- J. Mo, Y. Xu, L. Zhu, W. Wei and J. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12524–12531 CrossRef CAS PubMed.
- H. Song, J. Wang, B. Xiong, J. Hu, P. Zeng, X. Liu and H. Liang, Angew. Chem., Int. Ed., 2022, 134, e202117679 Search PubMed.
- G. Jia, M. Sun, Y. Wang, X. Cui, B. Huang and J. C. Yu, Adv. Funct. Mater., 2022, 33, 2212051 CrossRef.
- G. H. Han, D. L. Duong, D. H. Keum, S. J. Yun and Y. H. Lee, Chem. Rev., 2018, 118, 6297–6336 CrossRef CAS PubMed.
- J. B. Smith, D. Hagaman, D. DiGuiseppi, R. Schweitzer-Stenner and H. F. Ji, Angew. Chem., Int. Ed., 2016, 55, 11829–11833 CrossRef CAS PubMed.
- N. Eckstein, A. Hohmann, R. Weihrich, T. Nilges and P. Schmidt, Z. Anorg. Allg. Chem., 2013, 639, 2741–2743 CrossRef CAS.
- M. Roshith, M. S. Kumar, A. K. Nanda Kumar, S. Ramasubramanian, J. Stanley, T. G. Satheesh Babu and D. V. Ravi Kumar, J. Photochem. Photobiol., A, 2019, 384, 112034 CrossRef CAS.
- Z. Chen, Y. Zhu, Q. Wang, W. Liu, Y. Cui, X. Tao and D. Zhang, Electrochim. Acta, 2019, 295, 230–236 CrossRef CAS.
- S. Zhang, S. Ma, B. Cao, Q. Zhuang, Y. Xu, J. Wang, X. Zhang, X. Nan, X. Hao and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202217127 CAS.
- H. W. Ban, J. G. Oh, S. Jo, H. Jeong, D. H. Gu, S. Baek, S. Y. Lee, Y. I. Park, J. Jang and J. S. Son, Chem. Mater., 2019, 31, 5909–5918 CrossRef CAS.
- G. Cunningham, M. Lotya, C. S. Cucinotta, S. Sanvito, S. D. Bergin, R. Menzel, M. S. P. Shaffer and J. N. Coleman, ACS Nano, 2012, 6, 3468–3480 CrossRef CAS PubMed.
- V. Sresht, A. A. H. Pádua and D. Blankschtein, ACS Nano, 2015, 9, 8255–8268 CrossRef CAS PubMed.
- G. Zhang, D. Liu, N. Tian, X. Wang, W. Yan, Z. Huang and Y. Zhang, Inorg. Chem., 2021, 60, 4883–4890 CrossRef CAS PubMed.
- D. Nakamura and H. Nakano, Chem. Mater., 2018, 30, 5333–5338 CrossRef CAS.
- P. E. M. Amaral, D. C. Hall, Jr., R. Pai, J. E. Krol, V. Kalra, G. D. Ehrlich and H.-F. Ji, ACS Appl. Nano Mater., 2020, 3, 752–759 CrossRef CAS.
- D. Geng, B. Wu, Y. Guo, L. Huang, Y. Xue, J. Chen, G. Yu, L. Jiang, W. Hu and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7992 CrossRef CAS PubMed.
- Y. Liu, Z. Hu and J. C. Yu, Appl. Catal., B, 2019, 247, 100–106 CrossRef CAS.
- F. Wang and W. E. Buhro, ACS Nano, 2017, 11, 12526–12535 CrossRef CAS PubMed.
- J. Li, Z. Wang and F. L. Deepak, ACS Nano, 2017, 11, 5590–5597 CrossRef CAS PubMed.
- Y. Li, B. R. Bunes, L. Zang, J. Zhao, Y. Li, Y. Zhu and C. Wang, ACS Nano, 2016, 10, 2386–2391 CrossRef CAS PubMed.
- D. Wang, F. Luo, M. Lu, X. Xie, L. Huang and W. Huang, Small, 2019, 15, 1804404 CrossRef PubMed.
- L. Zhang, M. Gu, L. Li, X. Zhao, C. Fu, T. Liu, X. Xu, Y. Cheng and J. Zhang, Chem. Mater., 2020, 32, 7363–7369 CrossRef CAS.
- H.-S. Tsai, C.-C. Lai, C.-H. Hsiao, H. Medina, T.-Y. Su, H. Ouyang, T.-H. Chen, J.-H. Liang and Y.-L. Chueh, ACS Appl. Mater. Interfaces, 2015, 7, 13723–13727 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, Y. Wang, Q. Liu, Q. Li and M. Dong, ACS Appl. Nano Mater., 2021, 4, 9932–9937 CrossRef CAS.
- B. Zhang, L. Zhang, Z. Wang, Y. Li, Y. Cheng, L. Ma and J. Zhang, J. Mater. Chem. A, 2021, 9, 13855–13860 RSC.
- F. Baumer, Y. Ma, C. Shen, A. Zhang, L. Chen, Y. Liu, D. Pfister, T. Nilges and C. Zhou, ACS Nano, 2017, 11, 4105–4113 CrossRef CAS PubMed.
- R. Zhao, S. Liu, X. Zhao, M. Gu, Y. Zhang, M. Jin, Y. Wang, Y. Cheng and J. Zhang, J. Mater. Chem. A, 2022, 10, 245–250 RSC.
- W. M. Girma, M. Z. Fahmi, A. Permadi, M. A. Abate and J.-Y. Chang, J. Mater. Chem. B, 2017, 5, 6193–6216 RSC.
- L. Long, X. Niu, K. Yan, G. Zhou, J. Wang, X. Wu and P. K. Chu, Small, 2018, 14, 1803132 CrossRef PubMed.
- W. Dai, H. Dong, B. Fugetsu, Y. Cao, H. Lu, X. Ma and X. Zhang, Small, 2015, 11, 4158–4164 CrossRef CAS PubMed.
- S. H. C. Askes, G. U. Reddy, R. Wyrwa, S. Bonnet and A. Schiller, J. Am. Chem. Soc., 2017, 139, 15292–15295 CrossRef CAS PubMed.
- R. Zhao, S. Liu, X. Zhao, Y. Cheng and J. Zhang, Adv. Mater. Interfaces, 2022, 9, 2200705 CrossRef CAS.
- R. Zhao, X. Zhao, X. Xu, Y. Zhang, Y. Wang, M. Jin, Z. Liu, Y. Cheng, H. Zheng and J. Zhang, J. Phys. Chem. Lett., 2022, 13, 8236–8244 CrossRef CAS PubMed.
- H. Tian, J.-Q. Zhang, W. Ho, J.-P. Xu, B. Xia, Y. Xia, J. Fan, H. Xu, M. Xie and S. Y. Tong, Matter, 2020, 2, 111–118 CrossRef.
- F.-F. Zhu, W.-J. Chen, Y. Xu, C.-L. Gao, D.-D. Guan, C.-H. Liu, D. Qian, S.-C. Zhang and J.-F. Jia, Nat. Mater., 2015, 14, 1020–1025 CrossRef CAS PubMed.
- J. L. Zhang, C. Han, Z. Hu, L. Wang, L. Liu, A. T. S. Wee and W. Chen, Adv. Mater., 2018, 30, 1802207 CrossRef PubMed.
- J. L. Zhang, S. Zhao, S. Sun, H. Ding, J. Hu, Y. Li, Q. Xu, X. Yu, M. Telychko, J. Su, C. Gu, Y. Zheng, X. Lian, Z. Ma, R. Guo, J. Lu, Z. Sun, J. Zhu, Z. Li and W. Chen, ACS Nano, 2020, 14, 3687–3695 CrossRef CAS PubMed.
- S. Yang, Z. Hu, W. Wang, P. Cheng, L. Chen and K. Wu, Chin. Phys. Lett., 2020, 37, 096803 CrossRef CAS.
- M. Schaal, J. Picker, F. Otto, M. Gruenewald, R. Forker and T. Fritz, J. Phys.: Condens. Matter, 2021, 33, 485002 CrossRef CAS PubMed.
- R. Forker, M. Meissner and T. Fritz, Soft Matter, 2017, 13, 1748–1758 RSC.
- Y. Yin, V. Gladkikh, Q. Yuan and F. Ding, Chem. Mater., 2022, 34, 8230–8236 CrossRef CAS.
- S. Zhao and Z. Li, J. Phys. Chem. C, 2020, 125, 675–679 CrossRef.
- J. Zeng, P. Cui and Z. Zhang, Phys. Rev. Lett., 2017, 118, 046101 CrossRef PubMed.
- Y. Yin, V. Gladkikh, P. Li, L. Zhang, Q. Yuan and F. Ding, Chem. Mater., 2021, 33, 9447–9453 CrossRef CAS.
- A. Sala, M. Caporali, M. Serrano-Ruiz, F. Armillotta, E. Vesselli, F. Genuzio, T. O. Mentes, A. Locatelli, G. Comelli, C. Africh and A. Verdini, Nanoscale, 2022, 14, 16256–16261 RSC.
- W. Nunn, T. K. Truttmann and B. Jalan, J. Mater. Res., 2021, 36, 4846–4864 CrossRef CAS.
- G. Barik and S. Pal, Phys. Chem. Chem. Phys., 2021, 23, 26547–26560 RSC.
- P. G. Demingos and A. R. Muniz, J. Phys. Chem. C, 2020, 124, 21207–21214 CrossRef CAS.
- C. Dai, X. Cai, Y. Ni, Y. Chen and H. Wang, Phys. Chem. Chem. Phys., 2022, 24, 22572–22579 RSC.
- L. Qiu, J. Dong and F. Ding, Nanoscale, 2019, 11, 7135–7139 RSC.
- A. Pfitzner, S. Reiser and T. Nilges, Angew. Chem., Int. Ed., 2000, 39, 4160–4162 CrossRef CAS PubMed.
- S. Nilges, T. Nilges, H. Haeuseler and A. Pfitzner, J. Mol. Struct., 2004, 706, 89–94 CrossRef CAS.
- A. Pfitzner, Chem. – Eur. J., 2000, 6, 1891–1898 CrossRef CAS PubMed.
- A. Pfitzner, M. F. Bräu, J. Zweck, G. Brunklaus and H. Eckert, Angew. Chem., Int. Ed., 2004, 43, 4228–4231 CrossRef CAS PubMed.
- Y. Zhao, Z. Xie, H. Gu, C. Zhu and Z. Gu, Chem. Soc. Rev., 2012, 41, 3297–3317 RSC.
- M. Hart, J. Chen, A. Michaelides, A. Sella, M. S. P. Shaffer and C. G. Salzmann, Inorg. Chem., 2019, 58, 15216–15224 CrossRef CAS PubMed.
- J. Urquhart, Chem. World, 2017, 31 Search PubMed.
- J. Zhang, D. Zhao, D. Xiao, C. Ma, H. Du, X. Li, L. Zhang, J. Huang, H. Huang, C. L. Jia, D. Tománek and C. Niu, Angew. Chem., Int. Ed., 2017, 56, 1850–1854 CrossRef CAS PubMed.
- J. Zhang, C. Fu, S. Song, H. Du, D. Zhao, H. Huang, L. Zhang, J. Guan, Y. Zhang, X. Zhao, C. Ma, C. L. Jia and D. Tománek, Nano Lett., 2020, 20, 1280–1285 CrossRef CAS PubMed.
- D. V. Rybkovskiy, V. O. Koroteev, A. Impellizzeri, A. A. Vorfolomeeva, E. Y. Gerasimov, A. V. Okotrub, A. Chuvilin, L. G. Bulusheva and C. P. Ewels, ACS Nano, 2022, 16, 6002–6012 CrossRef CAS PubMed.
- N. Kittner, F. Lill and D. M. Kammen, Nat. Energy, 2017, 2, 17125 CrossRef.
- K. Liu, P. Cao, W. Chen, C. I. Ezeh, Z. Chen, Y. Luo, Q. Liu, H. Zhao, Z. Rui, S. Gao, Z. Yin, X. Sun and X. Yu, Mater. Adv., 2022, 3, 1359–1400 RSC.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 RSC.
- E. Shayan, V. Zare and I. Mirzaee, Energy Convers Manage., 2018, 159, 30–41 CrossRef CAS.
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC.
- J. M. Ogden, Annu. Rev. Energy Environ., 1999, 24, 227–279 CrossRef.
- A. Turner John, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- C. Wu and R. Liu, Int. J. Hydrogen Energy, 2011, 36, 2860–2868 CrossRef CAS.
- S. Kumar, N. L. Reddy, H. S. Kushwaha, A. Kumar, M. V. Shankar, K. Bhattacharyya, A. Halder and V. Krishnan, ChemSusChem, 2017, 10, 3588–3603 CrossRef CAS PubMed.
- N. Zhu, X. Dong, Z. Liu, G. Zhang, W. Jin and N. Xu, Chem. Commun., 2012, 48, 7137–7139 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- C. Xu, P. Ravi Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868–3902 RSC.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- Z. Li, X. Meng and Z. Zhang, J. Photochem. Photobiol., C, 2018, 35, 39–55 CrossRef CAS.
- S. A. Ansari, M. S. Ansari and M. H. Cho, Phys. Chem. Chem. Phys., 2016, 18, 3921–3928 RSC.
- X. J. Zhu, T. M. Zhang, Z. J. Sun, H. L. Chen, J. Guan, X. Chen, H. X. Ji, P. W. Du and S. F. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed.
- Y. Zhu, C. Lv, Z. Yin, J. Ren, X. Yang, C. L. Dong, H. Liu, R. Cai, Y. C. Huang, W. Theis, S. Shen and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 868–873 CrossRef CAS PubMed.
- A. Maibam, S. K. Das, P. P. Samal and S. Krishnamurty, RSC Adv., 2021, 11, 13348–13358 RSC.
- Z. Shen, Z. Hu, W. Wang, S.-F. Lee, D. K. L. Chan, Y. Li, T. Gu and J. C. Yu, Nanoscale, 2014, 6, 14163–14167 RSC.
- J. Xiao, M. Q. Long, X. J. Zhang, D. Zhang, H. Xu and K. S. Chan, J. Phys. Chem. Lett., 2015, 6, 4141–4147 CrossRef CAS PubMed.
- B. Li, C. C. Ren, S. F. Zhang, W. X. Ji, C. W. Zhang, P. Li and P. J. Wang, J. Nanomater., 2019, 2019, 4020762 Search PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. K. Muduli, E. Varrla, Y. Xu, S. A. Kulkarni, A. Katre, S. Chakraborty, S. Chen, T. C. Sum, R. Xu and N. Mathews, J. Mater. Chem. A, 2017, 5, 24874–24879 RSC.
- W. Lei, Y. Mi, R. Feng, P. Liu, S. Hu, J. Yu, X. Liu, J. A. Rodriguez, J.-O. Wang, L. Zheng, K. Tang, S. Zhu, G. Liu and M. Liu, Nano Energy, 2018, 50, 552–561 CrossRef CAS.
- A. Chouat, D. T. Nguyen, S. Mohan and T.-O. Do, ACS Appl. Nano Mater., 2022, 5, 13078–13089 CrossRef CAS.
- Y.-J. Yuan, P. Wang, Z. Li, Y. Wu, W. Bai, Y. Su, J. Guan, S. Wu, J. Zhong, Z.-T. Yu and Z. Zou, Appl. Catal., B, 2019, 242, 1–8 CrossRef CAS.
- M. Wen, J. Wang, R. Tong, D. Liu, H. Huang, Y. Yu, Z. K. Zhou, P. K. Chu and X. F. Yu, Adv. Sci., 2019, 6, 1801321 CrossRef PubMed.
- J. Ran, B. Zhu and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- J. Hu, Y. Ji, Z. Mo, N. Li, Q. Xu, Y. Li, H. Xu, D. Chen and J. Lu, J. Mater. Chem. A, 2019, 7, 4408–4414 RSC.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- Y.-P. Yuan, S.-W. Cao, Y.-S. Liao, L.-S. Yin and C. Xue, Appl. Catal., B, 2013, 140–141, 164–168 CrossRef CAS.
- F. Wang, C. Li, Y. Li and J. C. Yu, Appl. Catal., B, 2012, 119–120, 267–272 CrossRef CAS.
- W. Wang, G. Li, T. An, D. K. L. Chan, J. C. Yu and P. K. Wong, Appl. Catal., B, 2018, 238, 126–135 CrossRef CAS.
- C. Li, M. Fu, Y. Wang, E. Liu, J. Fan and X. Hu, Catal. Sci. Technol., 2020, 10, 2221–2230 RSC.
- Z. Hu, L. Yuan, Z. Liu, Z. Shen and J. C. Yu, Angew. Chem., Int. Ed., 2016, 55, 9580–9585 CrossRef CAS PubMed.
- L. Jing, R. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27, 1703484 CrossRef.
- M. Gu, L. Zhang, S. Mao, Y. Zou, D. Ma, J. Shi, N. Yang, C. Fu, X. Zhao, X. Xu, Y. Cheng and J. Zhang, Chem. Commun., 2022, 58, 12811–12814 RSC.
- S. Zhang, S. Ma, X. Hao, Y. Wang, B. Cao, B. Han, H. Zhang, X. Kong and B. Xu, Nanoscale, 2021, 13, 18955–18960 RSC.
- Y. Zhu, C. Lv, Z. Yin, J. Ren, X. Yang, C. L. Dong, H. Liu, R. Cai, Y. C. Huang, W. Theis, S. Shen and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 868–873 CrossRef CAS PubMed.
- S. Thurakkal and X. Y. Zhang, Adv. Sci., 2020, 7, 1902359 CrossRef CAS PubMed.
- H. Huang, Q. L. Xiao, J. H. Wang, X. F. Yu, H. Y. Wang, H. Zhang and P. K. Chu, npj 2D Mater. Appl., 2017, 1, 20 CrossRef.
- G. B. Tang, F. H. Hsu, X. Xu and P. K. Chu, Chem. Eng. J., 2020, 392, 123631 CrossRef CAS.
- A. Marjani, M. Ghambarian and M. Ghashghaee, Sci. Rep., 2021, 11, 842 CrossRef CAS PubMed.
- W. Y. Lei, G. Liu, J. Zhang and M. H. Liu, Chem. Soc. Rev., 2017, 46, 3492–3509 RSC.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 57, 2160–2164 CrossRef CAS PubMed.
- M. Zhu, X. Cai, M. Fujitsuka, J. Zhang and T. Majima, Angew. Chem., Int. Ed., 2017, 56, 2064–2068 CrossRef CAS PubMed.
- H. Wang, W. Liu, X. He, P. Zhang, X. Zhang and Y. Xie, J. Am. Chem. Soc., 2020, 142, 14007–14022 CrossRef CAS PubMed.
- H. Wang, X. Zhang and Y. Xie, ACS Nano, 2018, 12, 9648–9653 CrossRef CAS PubMed.
- W. Shao, L. Wang, H. Wang, Z. Zhao, X. Zhang, S. Jiang, S. Chen, X. Sun, Q. Zhang and Y. Xie, J. Phys. Chem. Lett., 2019, 10, 2904–2910 CrossRef CAS PubMed.
- H. Wang, S. Jin, X. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 22828–22839 CrossRef CAS PubMed.
- Y. Li, H. Wang, X. Zhang, S. Wang, S. Jin, X. Xu, W. Liu, Z. Zhao and Y. Xie, Angew. Chem., Int. Ed., 2021, 60, 12891–12896 CrossRef CAS PubMed.
- Z. Hu, L. Yuan, Z. Liu, Z. Shen and J. C. Yu, Angew. Chem., Int. Ed., 2016, 55, 9580–9585 CrossRef CAS PubMed.
- C. Wu, R. Zhu, W. Y. Teoh, Y. Liu, J. Deng, H. Dai, L. Jing, Y. H. Ng and J. C. Yu, Appl. Catal., B, 2022, 312, 121428 CrossRef CAS.
- S. Li, Y. H. Ng, R. Zhu, S. Lv, C. Wu, Y. Liu, L. Jing, J. Deng and H. Dai, Appl. Catal., B, 2021, 297, 120412 CrossRef CAS.
- Y. Zhu, C. Lv, Z. Yin, J. Ren, X. Yang, C.-L. Dong, H. Liu, R. Cai, Y.-C. Huang, W. Theis, S. Shen and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 868–873 CrossRef CAS PubMed.
- B. J. Wang, X. H. Li, X. L. Cai, W. Y. Yu, L. W. Zhang, R. Q. Zhao and S. H. Ke, J. Phys. Chem. C, 2018, 122, 7075–7080 CrossRef CAS.
- X. Zhan, Z. Deng, J. Nie, Y. Du, L. Li and X. Zu, J. Phys.: Condens. Matter, 2021, 33, 485703 CrossRef CAS PubMed.
- M. Tahir and N. S. Amin, Renewable Sustainable Energy Rev., 2013, 25, 560–579 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- J. Anjali, V. K. Jose and J.-M. Lee, J. Mater. Chem. A, 2019, 7, 15491–15518 RSC.
- G. Fu, Y. Liu, Z. Wu and J.-M. Lee, ACS Appl. Nano Mater., 2018, 1, 1904–1911 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- P. Prabhu, V. Jose and J. M. Lee, Adv. Funct. Mater., 2020, 30, 1910768 CrossRef CAS.
- Y. Jiang, X. Li, Z. Wu, C. Cao, Y. Mei and P. Lian, Inorg. Chem. Ind., 2021, 53, 59–71 Search PubMed.
- C. Wang, Z. Sun, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2019, 7, 865–887 RSC.
- X. Zhu, S. Huang, Q. Yu, Y. She, J. Yang, G. Zhou, Q. Li, X. She, J. Deng, H. Li and H. Xu, Appl. Catal., B, 2020, 269, 118760 CrossRef CAS.
- C.-M. Fung, B.-J. Ng, C.-C. Er, X. Y. Kong, L.-L. Tan, A. R. Mohamed and S.-P. Chai, ACS Appl. Energy Mater., 2022, 5, 15257–15268 CrossRef CAS.
- B. Wan, B. Yang, Y. Wang, J. Zhang, Z. Zeng, Z. Liu and W. Wang, Nanotechnology, 2015, 26, 435702 CrossRef PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS PubMed.
- M. Wang, S. Xu, Z. Zhou, C.-L. Dong, X. Guo, J.-L. Chen, Y.-C. Huang, S. Shen, Y. Chen, L. Guo and C. Burda, Angew. Chem., Int. Ed., 2022, 61, e202204711 CAS.
- R. Das, K. Das, B. Ray, C. P. Vinod and S. C. Peter, Energy Environ. Sci., 2022, 15, 1967–1976 RSC.
- J. Zhu, L. S. Hu, P. X. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- J. Li, M. N. Banis, Z. Ren, K. R. Adair, K. Doyle-Davis, D. M. Meira, Y. Z. Finfrock, L. Zhang, F. Kong, T. K. Sham, R. Li, J. Luo and X. Sun, Small, 2021, 17, 2007245 CrossRef CAS PubMed.
- W. Qian, S. Xu, X. Zhang, C. Li, W. Yang, C. R. Bowen and Y. Yang, Nano-Micro Lett., 2021, 13, 156 CrossRef CAS PubMed.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 21, 749–770 CrossRef CAS.
- P. Yu, F. Wang, T. A. Shifa, X. Zhan, X. Lou, F. Xia and J. He, Nano Energy, 2019, 58, 244–276 CrossRef CAS.
- J. Janata, Angew. Chem., Int. Ed., 2011, 50, 9538 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- Z.-Z. Luo, Y. Zhang, C. Zhang, H. T. Tan, Z. Li, A. Abutaha, X.-L. Wu, Q. Xiong, K. A. Khor, K. Hippalgaonkar, J. Xu, H. H. Hng and Q. Yan, Adv. Energy Mater., 2017, 7, 1601285 CrossRef.
- C. Xu, S. Peng, C. Tan, H. Ang, H. Tan, H. Zhang and Q. Yan, J. Mater. Chem. A, 2014, 2, 5597–5601 RSC.
- L. Shao, H. Sun, L. Miao, X. Chen, M. Han, J. Sun, S. Liu, L. Li, F. Cheng and J. Chen, J. Mater. Chem. A, 2018, 6, 2494–2499 RSC.
- W. Li, M. Li, J. Li, J. Liang, K. R. Adair, Y. Hu, Q. Xiao, X. Cui, R. Li, F. Brandys, R. Divigalpitiya, T.-K. Sham and X. Sun, ACS Appl. Nano Mater., 2020, 3, 7508–7515 CrossRef CAS.
- C. Y. Chan, C. H. Chang and H. Y. Tuan, Chem. Commun., 2020, 56, 2937–2940 RSC.
- W. Cui, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Chem. Commun., 2014, 50, 9340–9342 RSC.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131–2136 CrossRef CAS PubMed.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 RSC.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W. Fu, Y. Wang, W. Tian, H. Zhang, J. Li, S. Wang and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 23791–23799 CrossRef CAS PubMed.
- H. F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- M. Tahir, L. Pan, F. Idrees, X. W. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- Y. C. Liu, J. A. Koza and J. A. Switzer, Electrochim. Acta, 2014, 140, 359–365 CrossRef CAS.
- Q. Jiang, L. Xu, N. Chen, H. Zhang, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2016, 55, 13849–13853 CrossRef CAS PubMed.
- T. T. Sun, L. B. Xu, Y. S. Yan, A. A. Zakhidov, R. H. Baughman and J. F. Chen, ACS Catal., 2016, 6, 1446–1450 CrossRef CAS.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- X. Ren, J. Zhou, X. Qi, Y. Liu, Z. Huang, Z. Li, Y. Ge, S. C. Dhanabalan, J. S. Ponraj, S. Wang, J. Zhong and H. Zhang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- X. X. Xue, S. Shen, X. Jiang, P. Sengdala, K. Chen and Y. Feng, J. Phys. Chem. Lett., 2019, 10, 3440–3446 CrossRef CAS PubMed.
- R. Li, Z. D. Wei and X. L. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- T. Wu, S. N. Zhang, K. J. Bu, W. Zhao, Q. Y. Bi, T. Q. Lin, J. Huang, Y. S. Li and F. Q. Huang, J. Mater. Chem. A, 2019, 7, 22063–22069 RSC.
- H. Qiao, H. Liu, Z. Huang, Q. Ma, S. Luo, J. Li, Y. Liu, J. Zhong and X. Qi, Adv. Energy Mater., 2020, 10, 2002424 CrossRef CAS.
- Z. Shen, S. Sun, W. Wang, J. Liu, Z. Liu and J. C. Yu, J. Mater. Chem. A, 2015, 3, 3285–3288 RSC.
- K. Bai, J.-C. Fan, P.-H. Shi, Y.-L. Min and Q.-J. Xu, J. Power Sources, 2020, 456, 228003 CrossRef CAS.
- C. Gao, Z. Zhao, X. Qin, J. Teng, J. Fan, P. Shi, Q. Xu and Y. Min, Compos. Commun., 2021, 24, 100624 CrossRef.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- X. Lu, W.-L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS PubMed.
- J. Wang, D. Liu, H. Huang, N. Yang, B. Yu, M. Wen, X. Wang, P. K. Chu and X. F. Yu, Angew. Chem., Int. Ed., 2018, 57, 2600–2604 CrossRef CAS PubMed.
- X. Li, L. Xiao, L. Zhou, Q. Xu, J. Weng, J. Xu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 21106–21113 CrossRef CAS PubMed.
- X.-D. Zhu, Y. Xie and Y.-T. Liu, J. Mater. Chem. A, 2018, 6, 21255–21260 RSC.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS PubMed.
- F. Shiravani, J. Tashkhourian and B. Haghighi, Sustain. Energy Fuels, 2021, 5, 3229–3239 RSC.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- K. H. R. Rouwenhorst, A. G. J. Van der Ham, G. Mul and S. R. A. Kersten, Renewable Sustainable Energy Rev., 2019, 114, 109339 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. W. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- S. Mukherjee, D. A. Cullen, S. Karakalos, K. Liu, H. Zhang, S. Zhao, H. Xu, K. L. More, G. Wang and G. Wu, Nano Energy, 2018, 48, 217–226 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- X. Zhao, G. Hu, G. F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CrossRef CAS PubMed.
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- D. W. Boukhvalov, Phys. Chem. Chem. Phys., 2015, 17, 27210–27216 RSC.
- L. Zhang, L. X. Ding, G. F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- M.-M. Shi, D. Bao, B.-R. Wulan, Y.-H. Li, Y.-F. Zhang, J.-M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- C. Wang, J. Gao, J. G. Zhao, D. J. Yan and X. D. Zhu, Small, 2020, 16, 1907091 CrossRef CAS PubMed.
- Z. M. Zhang, X. Yao, X. Y. Lang, Y. F. Zhu, W. Gao and Q. Jiang, Appl. Surf. Sci., 2021, 536, 147706 CrossRef CAS.
- M.-A. Légaré, G. Bélanger-Chabot, D. Dewhurst Rian, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- S. Bian, M. Wen, J. Wang, N. Yang, P. K. Chu and X. F. Yu, J. Phys. Chem. Lett., 2020, 11, 1052–1058 CrossRef CAS PubMed.
- Z.-K. Shen, M. Cheng, Y.-J. Yuan, L. Pei, J. Zhong, J. Guan, X. Li, Z.-J. Li, L. Bao, X. Zhang, Z.-T. Yu and Z. Zou, Appl. Catal., B, 2021, 295, 120274 CrossRef CAS.
- D. Liu, J. Wang, S. Bian, Q. Liu, Y. Gao, X. Wang, P. K. Chu and X. F. Yu, Adv. Funct. Mater., 2020, 30, 2002731 CrossRef CAS.
- D. J. Durbin and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef CAS.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- E. Rivard, M. Trudeau and K. Zaghib, Materials, 2019, 12, 1973 CrossRef CAS PubMed.
- P. Kumar, S. Singh, S. A. R. Hashmi and K.-H. Kim, Nano Energy, 2021, 85, 105989 CrossRef CAS.
- R. Lotfi and Y. Saboohi, Phys. E, 2014, 60, 104–111 CrossRef CAS.
- R. Mohtadi and S.-i Orimo, Nat. Rev. Mater., 2016, 2, 16091 CrossRef.
- M. Darvish Ganji, S. M. Hosseini-khah and Z. Amini-tabar, Phys. Chem. Chem. Phys., 2015, 17, 2504–2511 RSC.
- K.-F. Aguey-Zinsou and J.-R. Ares-Fernández, Energy Environ. Sci., 2010, 3, 526–543 RSC.
- Y. F. Zhang and X. L. Cheng, Chem. Phys., 2018, 505, 26–33 CrossRef CAS.
- R. Y. Sathe and T. J. D. Kumar, Int. J. Hydrogen Energy, 2020, 45, 12940–12948 CrossRef CAS.
- D. Luo, J. Y. Li, Y. Zhang, Y. Song and H. S. Chen, Int. J. Hydrogen Energy, 2018, 43, 8415–8425 CrossRef CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- R. Nagar, B. P. Vinayan, S. S. Samantaray and S. Ramaprabhu, J. Mater. Chem. A, 2017, 5, 22897–22912 RSC.
- S. A. Shevlin and Z. X. Guo, Chem. Soc. Rev., 2009, 38, 211–225 RSC.
- Q.-F. Li, X. G. Wan, C.-G. Duan and J.-L. Kuo, J. Phys. D: Appl. Phys., 2014, 47, 465302 CrossRef.
- J. Xu, H. Li, Y. Li, M. Wang, Z. Li, F. Mo, C. Zhang, L. Zhu, R. Nan and P. Wang, Mater. Lett., 2021, 303, 130422 CrossRef CAS.
- M. Ghambarian, Z. Azizi and M. Ghashghaee, Int. J. Hydrogen Energy, 2020, 45, 16298–16309 CrossRef CAS.
- M. Garara, H. Benzidi, M. Lakhal, M. Louilidi, H. Ez-Zahraouy, A. El Kenz, M. Hamedoun, A. Benyoussef, A. Kara and O. Mounkachi, Int. J. Hydrogen Energy, 2019, 44, 24829–24838 CrossRef CAS.
- H.-P. Zhang, W. Hu, A. Du, X. Lu, Y.-P. Zhang, J. Zhou, X. Lin and Y. Tang, Appl. Surf. Sci., 2018, 433, 249–255 CrossRef CAS.
- M. Nagpal and R. Kakkar, Int. J. Hydrogen Energy, 2018, 43, 12168–12188 CrossRef CAS.
- S. Haldar, S. Mukherjee, F. Ahmed and C. V. Singh, Int. J. Hydrogen Energy, 2017, 42, 23018–23027 CrossRef CAS.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 RSC.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- E. Pomerantseva, F. Bonaccorso, X. L. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, 969 CrossRef PubMed.
- H. Jin, H. Wang, Z. Qi, D.-S. Bin, T. Zhang, Y. Wan, J. Chen, C. Chuang, Y.-R. Lu, T.-S. Chan, H. Ju, A.-M. Cao, W. Yan, X. Wu, H. Ji and L.-J. Wan, Angew. Chem., Int. Ed., 2020, 59, 2318–2322 CrossRef CAS PubMed.
- M. Qiu, Z. T. Sun, D. K. Sang, X. G. Han, H. Zhang and C. M. Niu, Nanoscale, 2017, 9, 13384–13403 RSC.
- T. T. Wang, H. D. Wang, Z. K. Kou, W. Y. Liang, X. L. Luo, F. Verpoort, Y. J. Zeng and H. Zhang, Adv. Funct. Mater., 2020, 30, 2002885 CrossRef CAS.
- J. B. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. F. Liu and M. H. Rummeli, Chem. Soc. Rev., 2019, 48, 72–133 RSC.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- M. Zheng, H. Tang, L. Li, Q. Hu, L. Zhang, H. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592 CrossRef PubMed.
- W. Li, Y. Yang, G. Zhang and Y.-W. Zhang, Nano Lett., 2015, 15, 1691–1697 CrossRef CAS PubMed.
- Y. Q. Fu, Q. L. Wei, G. X. Zhang and S. H. Sun, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- S. A. Ansari, Z. Khan, M. O. Ansari and M. H. Cho, RSC Adv., 2016, 6, 44616–44629 RSC.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- S. Mukherjee, L. Kavalsky and C. V. Singh, ACS Appl. Mater. Interfaces, 2018, 10, 8630–8639 CrossRef CAS PubMed.
- S. Zhao, W. Kang and J. Xue, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS.
- Y. Sun, F. Zeng, Y. Zhu, P. Lu and D. Yang, J. Energy Chem., 2021, 61, 531–552 CrossRef CAS.
- W. W. Xia, Q. B. Zhang, F. Xu, H. Y. Ma, J. Chen, K. Qasim, B. H. Ge, C. Y. Zhu and L. T. Sun, J. Phys. Chem. C, 2016, 120, 5861–5868 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Y. Lee, J.-Y. Yoon, D. Scullion, J. Jang, E. J. G. Santos, H. Y. Jeong and K. Kim, J. Phys. D: Appl. Phys., 2017, 50, 084003 CrossRef.
- J. Zhu, Z. Liu, W. Wang, L. Yue, W. Li, H. Zhang, L. Zhao, H. Zheng, J. Wang and Y. Li, ACS Nano, 2021, 15, 1880–1892 CrossRef CAS PubMed.
- J. Sun, G. Zheng, H. W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- H. Liu, Y. Zou, L. Tao, Z. Ma, D. Liu, P. Zhou, H. Liu and S. Wang, Small, 2017, 13, 1700758 CrossRef PubMed.
- S. Zhang, Y. Zhang, Z. Zhang, H. Wang, Y. Cao, B. Zhang, X. Liu, C. Mao, X. Han, H. Gong, Z. Yang and J. Sun, Adv. Energy Mater., 2022, 12, 2103888 CrossRef CAS.
- J. Jin, Y. Zheng, S.-Z. Huang, P.-P. Sun, N. Srikanth, L. B. Kong, Q. Yan and K. Zhou, J. Mater. Chem. A, 2019, 7, 783–790 RSC.
- M. Li, W. Li, Y. Hu, A. A. Yakovenko, Y. Ren, J. Luo, W. M. Holden, M. Shakouri, Q. Xiao, X. Gao, F. Zhao, J. Liang, R. Feng, R. Li, G. T. Seidler, F. Brandys, R. Divigalpitiya, T. K. Sham and X. Sun, Adv. Mater., 2021, 33, 2101259 CrossRef CAS PubMed.
- H. Jin, S. Xin, C. Chuang, W. Li, H. Wang, J. Zhu, H. Xie, T. Zhang, Y. Wan, Z. Qi, W. Yan, Y.-R. Lu, T.-S. Chan, X. Wu, B. Goodenough John, H. Ji and X. Duan, Science, 2020, 370, 192–197 CrossRef CAS PubMed.
- Z. L. Xu, S. Lin, N. Onofrio, L. Zhou, F. Shi, W. Lu, K. Kang, Q. Zhang and S. P. Lau, Nat. Commun., 2018, 9, 4164 CrossRef PubMed.
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797–9803 CrossRef CAS PubMed.
- H. Cheng, J. Xie, G. Cao, Y. Lu, D. Zheng, Y. Jin, K. Wang and X. Zhao, Energy Storage Mater., 2019, 23, 684–692 CrossRef.
- Y. Kim, D. Koo, S. Ha, S. C. Jung, T. Yim, H. Kim, S. K. Oh, D. M. Kim, A. Choi, Y. Kang, K. H. Ryu, M. Jang, Y. K. Han, S. M. Oh and K. T. Lee, ACS Nano, 2018, 12, 4419–4430 CrossRef CAS PubMed.
- S. Haghighat-Shishavan, M. Nazarian-Samani, M. Nazarian-Samani, H.-K. Roh, K.-Y. Chung, B.-W. Cho, S. F. Kashani-Bozorg and K.-B. Kim, J. Mater. Chem. A, 2018, 6, 10121–10134 RSC.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu, B. Chen, Y. Mi and J. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- H. Liu, L. Tao, Y. Zhang, C. Xie, P. Zhou, H. Liu, R. Chen and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36849–36856 CrossRef CAS PubMed.
- Y. Liu, Q. Liu, A. Zhang, J. Cai, X. Cao, Z. Li, P. D. Asimow and C. Zhou, ACS Nano, 2018, 12, 8323–8329 CrossRef CAS PubMed.
- X. Ma, C. Ji, X. Li, Y. Liu and X. Xiong, Mater. Today, 2022, 59, 36–45 CrossRef CAS.
- Z. Huang, H. Hou, Y. Zhang, C. Wang, X. Qiu and X. Ji, Adv. Mater., 2017, 29, 1702372 CrossRef PubMed.
- J. Sun, H. W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed.
- X. Guo, W. Zhang, J. Zhang, D. Zhou, X. Tang, X. Xu, B. Li, H. Liu and G. Wang, ACS Nano, 2020, 14, 3651–3659 CrossRef CAS PubMed.
- C. Zhang, T. Liang, H. Dong, J. Li, J. Shen, W. Yang, X. Wang, H. Geng and Z. Zhang, Mater. Chem. Front., 2021, 5, 6639–6647 RSC.
- I. Sultana, M. M. Rahman, T. Ramireddy, Y. Chen and A. M. Glushenkov, J. Mater. Chem. A, 2017, 5, 23506–23512 RSC.
- J. Guan, A. M. Rao, J. Zhou, X. Yu and B. Lu, Adv. Funct. Mater., 2022, 32, 2203522 CrossRef CAS.
- C. Hao, B. Yang, F. Wen, J. Xiang, L. Li, W. Wang, Z. Zeng, B. Xu, Z. Zhao, Z. Liu and Y. Tian, Adv. Mater., 2016, 28, 3194–3201 CrossRef CAS PubMed.
- M. Wen, D. Liu, Y. Kang, J. Wang, H. Huang, J. Li, P. K. Chu and X.-F. Yu, Mater. Horiz., 2019, 6, 176–181 RSC.
- X. Wu, Y. Xu, Y. Hu, G. Wu, H. Cheng, Q. Yu, K. Zhang, W. Chen and S. Chen, Nat. Commun., 2018, 9, 4573 CrossRef PubMed.
- T. Wu, Z. Ma, Y. He, X. Wu, B. Tang, Z. Yu, G. Wu, S. Chen and N. Bao, Angew. Chem., Int. Ed., 2021, 60, 10366–10374 CrossRef CAS PubMed.
- H. Xiao, Y. Xia, Y. Gan, H. Huang, C. Liang, X. Tao, L. Xu and W. Zhang, RSC Adv., 2014, 4, 60914–60919 RSC.
- T. Wang, F. Cheng, N. Zhang, W. Tian, J. Zhou, R. Zhang, J. Cao, M. Luo, N. Li, L. Jiang, D. Li, Y. Li, K. Liang, H. Liu, P. Chen and B. Kong, Adv. Eng. Mater., 2021, 23, 2001507 CrossRef CAS.
- X. Han, Z. Zhang, M. Han, Y. Cui and J. Sun, Energy Storage Mater., 2020, 26, 147–156 CrossRef.
- J. Zhou, X. Liu, W. Cai, Y. Zhu, J. Liang, K. Zhang, Y. Lan, Z. Jiang, G. Wang and Y. Qian, Adv. Mater., 2017, 29, 1700214 CrossRef PubMed.
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed.
- X. Chen, G. Xu, X. Ren, Z. Li, X. Qi, K. Huang, H. Zhang, Z. Huang and J. Zhong, J. Mater. Chem. A, 2017, 5, 6581–6588 RSC.
- J. Lee, H. Song, K. A. Min, Q. Guo, D. Kim, Z. Zheng, B. Han, Y. Jung and L. Y. S. Lee, Small Methods, 2021, 5, 2100215 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, X. Liu, C. Chen, L.-Z. Fan and L. Jiao, Energy Storage Mater., 2017, 9, 170–178 CrossRef.
- J. Song, Z. Yu, M. L. Gordin, X. Li, H. Peng and D. Wang, ACS Nano, 2015, 9, 11933–11941 CrossRef CAS PubMed.
- Y. Liu, A. Zhang, C. Shen, Q. Liu, X. Cao, Y. Ma, L. Chen, C. Lau, T.-C. Chen, F. Wei and C. Zhou, ACS Nano, 2017, 11, 5530–5537 CrossRef CAS PubMed.
- Y. Liu, Q. Liu, C. Jian, D. Cui, M. Chen, Z. Li, T. Li, T. Nilges, K. He, Z. Jia and C. Zhou, Nat. Commun., 2020, 11, 2520 CrossRef CAS PubMed.
- S. Liu, H. Xu, X. Bian, J. Feng, J. Liu, Y. Yang, C. Yuan, Y. An, R. Fan and L. Ci, ACS Nano, 2018, 12, 7380–7387 CrossRef CAS PubMed.
- S. Liu, J. Feng, X. Bian, J. Liu, H. Xu and Y. An, Energy Environ. Sci., 2017, 10, 1222–1233 RSC.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820 CrossRef PubMed.
- J. Ruan, F. Mo, Z. Long, Y. Song, F. Fang, D. Sun and S. Zheng, ACS Nano, 2020, 14, 12222–12233 CrossRef CAS PubMed.
- X. Sui, X. Huang, H. Pu, Y. Wang and J. Chen, Nano Energy, 2021, 83, 105797 CrossRef CAS.
- Y. Wu, S. Hu, R. Xu, J. Wang, Z. Peng, Q. Zhang and Y. Yu, Nano Lett., 2019, 19, 1351–1358 CrossRef CAS PubMed.
- G. Qin, Y. Liu, F. Liu, X. Sun, L. Hou, B. Liu and C. Yuan, Adv. Energy Mater., 2020, 11, 2003429 CrossRef.
- A. Gopalakrishnan and S. Badhulika, Chem. Commun., 2020, 56, 7096–7099 RSC.
- M. Khalid and H. Varela, J. Mater. Chem. A, 2018, 6, 3141–3150 RSC.
- L. Liu, X. Gao, X. Cui, B. Wang, F. Hu, T. Yuan, J. Li, L. Zu, H. Lian and X. Cui, Nanomaterials, 2023, 13, 1060 CrossRef CAS PubMed.
- D. Zhao, L. Zhang, C. Fu, J. Huang, H. Huang, Z. Li, J. Zhang and C. Niu, Carbon, 2018, 139, 1057–1062 CrossRef CAS.
- B. Cai, S. Dong, Z. Mao, Y.-L. Lu, Z. Pan, Y. Ou and J. Li, Solid State Commun., 2021, 330, 114276 CrossRef CAS.
- Q.-F. Li, C.-G. Duan, X. G. Wan and J.-L. Kuo, J. Phys. Chem. C, 2015, 119, 8662–8670 CrossRef CAS.
- Y. Li, W. Wu and F. Ma, J. Mater. Chem. A, 2019, 7, 611–620 RSC.
- Q. Peng, Z. Wang, B. Sa, B. Wu and Z. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 13449–13457 CrossRef CAS PubMed.
- S. Mukherjee, L. Kavalsky, K. Chattopadhyay and C. V. Singh, Nanoscale, 2018, 10, 21335–21352 RSC.
- Y. Xiao, J. Wang, Y. Wang and W. Zhang, Appl. Surf. Sci., 2019, 488, 620–628 CrossRef CAS.
- J. Bao, L. Zhu, H. Wang, S. Han, Y. Jin, G. Zhao, Y. Zhu, X. Guo, J. Hou, H. Yin and J. Tian, J. Phys. Chem. C, 2018, 122, 23329–23335 CrossRef CAS.
- J. Bao, H. Li, Q. Duan, D. Jiang, W. Liu, X. Guo, J. Hou and J. Tian, Solid State Ion., 2020, 345, 115160 CrossRef CAS.
- S. Hajibaba and Y. Abdi, J. Energy Storage, 2021, 33, 102062 CrossRef.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- G.-C. Guo, D. Wang, X.-L. Wei, Q. Zhang, H. Liu, W.-M. Lau and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 5002–5008 CrossRef CAS PubMed.
- J. Cheng, L. Gao, T. Li, S. Mei, C. Wang, B. Wen, W. Huang, C. Li, G. Zheng, H. Wang and H. Zhang, Nano-Micro Lett., 2020, 12, 179 CrossRef CAS PubMed.
- E. Olsson, G. Chai, M. Dove and Q. Cai, Nanoscale, 2019, 11, 5274–5284 RSC.
- S. Y. Chu, S. H. Guo and H. S. Zhou, Chem. Soc. Rev., 2021, 50, 13189–13235 RSC.
- A. K. Thakur, M. S. Ahmed, G. Oh, H. Kang, Y. Jeong, R. Prabakaran, M. P. Vikram, S. W. Sharshir, J. Kim and J. Y. Hwang, J. Mater. Chem. A, 2021, 9, 2628–2661 RSC.
- M. M. Lao, Y. Zhang, W. B. Luo, Q. Y. Yan, W. P. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef PubMed.
- W. Zhang, J. Lu and Z. Guo, Mater. Today, 2021, 50, 400–417 CrossRef CAS.
- T. Jin, Q. Q. Han and L. F. Jiao, Adv. Mater., 2020, 32, 1806304 CrossRef CAS PubMed.
- M. S. Balogun, Y. Luo, W. T. Qiu, P. Liu and Y. X. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- Y. M. Li, Y. X. Lu, C. L. Zhao, Y. S. Hu, M. M. Titirici, H. Li, X. J. Huang and L. Q. Chen, Energy Storage Mater., 2017, 7, 130–151 CrossRef.
- K. Kuratani, N. Uemura, H. Senoh, H. T. Takeshita and T. Kiyobayashi, J. Power Sources, 2013, 223, 175–182 CrossRef CAS.
- X. D. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS PubMed.
- K. P. S. S. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, J. Phys. Chem. C, 2015, 119, 15041–15046 CrossRef CAS.
- X. Liu, Y. Wen, Z. Chen, B. Shan and R. Chen, Phys. Chem. Chem. Phys., 2015, 17, 16398–16404 RSC.
- C. Liu, X. Han, Y. Cao, S. Zhang, Y. Zhang and J. Sun, Energy Storage Mater., 2019, 20, 343–372 CrossRef.
- W.-J. Li, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820 CrossRef PubMed.
- Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- C. Zhang, X. Wang, Q. Liang, X. Liu, Q. Weng, J. Liu, Y. Yang, Z. Dai, K. Ding, Y. Bando, J. Tang and D. Golberg, Nano Lett., 2016, 16, 2054–2060 CrossRef CAS PubMed.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef PubMed.
- Y. Yan, S. Xia, H. Sun, Y. Pang, J. Yang and S. Zheng, Chem. Eng. J., 2020, 393, 124788 CrossRef CAS.
- X. L. Huang, F. Zhao, Y. Qi, Y.-A. Qiu, J. S. Chen, H. K. Liu, S. X. Dou and Z. M. Wang, Energy Storage Mater., 2021, 42, 193–208 CrossRef.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- M. Okoshi, Y. Yamada, S. Komaba, A. Yamada and H. Nakai, J. Electrochem. Soc., 2017, 164, A54 CrossRef CAS.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- D. Yang, C. Liu, X. Rui and Q. Yan, Nanoscale, 2019, 11, 15402–15417 RSC.
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef CAS.
- P. Xiong, P. Bai, S. Tu, M. Cheng, J. Zhang, J. Sun and Y. Xu, Small, 2018, 14, 1802140 CrossRef PubMed.
- Y. Wu, H. B. Huang, Y. Feng, Z. S. Wu and Y. Yu, Adv. Mater., 2019, 31, 1901414 CrossRef CAS PubMed.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316–3319 CrossRef CAS PubMed.
- W. Feng, H. Wang, Y. Jiang, H. Zhang, W. Luo, W. Chen, C. Shen, C. Wang, J. Wu and L. Mai, Adv. Energy Mater., 2022, 12, 2103343 CrossRef CAS.
- X. Liu, J. Zhu, X. Wang, L. Yue, W. Wang, B. Wang, D. Shen and Y. Li, Adv. Funct. Mater., 2022, 33, 2209388 CrossRef.
- X. Du and B. Zhang, ACS Nano, 2021, 15, 16851–16860 CrossRef CAS PubMed.
- M. Yu and X. Feng, Joule, 2019, 3, 338–360 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- M. Hu, H. Zhang, T. Hu, B. Fan, X. Wang and Z. Li, Chem. Soc. Rev., 2020, 49, 6666–6693 RSC.
- Y. Wu, W. Yuan, M. Xu, S. Bai, Y. Chen, Z. Tang, C. Wang, Y. Yang, X. Zhang, Y. Yuan, M. Chen, X. Zhang, B. Liu and L. Jiang, Chem. Eng. J., 2021, 412, 128744 CrossRef CAS.
- D. P. Dubal, N. R. Chodankar, D.-H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- X. Tian, 2D Mater., 2021, 9, 012001 CrossRef.
- S.-K. Kim, H.-J. Koo, A. Lee and P. V. Braun, Adv. Mater., 2014, 26, 5108–5112 CrossRef CAS PubMed.
- Z. S. Wu, K. Parvez, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2487 CrossRef PubMed.
- F. El-Kady Maher, V. Strong, S. Dubin and B. Kaner Richard, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- J. Yang, Z. Pan, Q. Yu, Q. Zhang, X. Ding, X. Shi, Y. Qiu, K. Zhang, J. Wang and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 5938–5946 CrossRef CAS PubMed.
- J. S. Shaikh, N. S. Shaikh, S. Sabale, N. Parveen, S. P. Patil, Y. K. Mishra, P. Kanjanaboos, S. Praserthdam and C. D. Lokhande, Mater. Today Chem., 2021, 21, 100480 CrossRef CAS.
- R. Yi, S. Chen, J. Song, M. L. Gordin, A. Manivannan and D. Wang, Adv. Funct. Mater., 2014, 24, 7433–7439 CrossRef CAS.
- J. Chang, J. Shang, Y. Sun, L. K. Ono, D. Wang, Z. Ma, Q. Huang, D. Chen, G. Liu, Y. Cui, Y. Qi and Z. Zheng, Nat. Commun., 2018, 9, 4480 CrossRef PubMed.
- T. Tao, S. Lu, Y. Fan, W. Lei, S. Huang and Y. Chen, Adv. Mater., 2017, 29, 1700542 CrossRef PubMed.
- T. Wang, J. He, X.-B. Cheng, J. Zhu, B. Lu and Y. Wu, ACS Energy Lett., 2022, 8, 116–150 CrossRef.
- Z. Pan, D. J. L. Brett, G. He and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2103483 CrossRef CAS.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- H. J. Peng, J. Q. Huang and Q. Zhang, Chem. Soc. Rev., 2017, 46, 5237–5288 RSC.
- J. Zhou, T. Wu, Y. Pan, J. Zhu, X. Chen, C. Peng, C. Shu, L. Kong, W. Tang and S. L. Chou, Adv. Funct. Mater., 2021, 32, 2106966 CrossRef.
- H. Hao, T. Hutter, B. L. Boyce, J. Watt, P. Liu and D. Mitlin, Chem. Rev., 2022, 122, 8053–8125 CrossRef CAS PubMed.
- C. Li, R. Liu, Y. Xiao, F. Cao and H. Zhang, Energy Storage Mater., 2021, 40, 439–460 CrossRef.
- Z. Wang, M. Feng, H. Sun, G. Li, Q. Fu, H. Li, J. Liu, L. Sun, A. Mauger, C. M. Julien, H. Xie and Z. Chen, Nano Energy, 2019, 59, 390–398 CrossRef CAS.
- J. Lee, H. Song, K. A. Min, Q. Guo, D. Kim, Z. Zheng, B. Han, Y. Jung and L. Y. S. Lee, Small Methods, 2021, 5, 2100215 CrossRef CAS PubMed.
- L. Kavalsky, S. Mukherjee and C. V. Singh, ACS Appl. Mater. Interfaces, 2019, 11, 499–510 CrossRef CAS PubMed.
- T. Cai, T. Li, B. Li, Y. Hu, X. Li, T. Lin, H. Hu, B. Luo, Y. Zhang, X. Zhu, Y. Cui, L. Zhao, W. Xing, Z. Yan and L. Wang, Energy Storage Mater., 2022, 53, 415–423 CrossRef.
- H. S. Tsai, Y. Wang, C. Liu, T. Wang and M. Huo, J. Hazard. Mater., 2022, 423, 127148 CrossRef CAS PubMed.
- H.-J. Kim and J.-H. Lee, Sens. Actuators, B, 2014, 192, 607–627 CrossRef CAS.
- J. Zhang, X. H. Liu, G. Neri and N. Pinna, Adv. Mater., 2016, 28, 795–831 CrossRef CAS PubMed.
- S. Deng, V. Tjoa, H. M. Fan, H. R. Tan, D. C. Sayle, M. Olivo, S. Mhaisalkar, J. Wei and C. H. Sow, J. Am. Chem. Soc., 2012, 134, 4905–4917 CrossRef CAS PubMed.
- K. Belanger, T. R. Holford, J. F. Gent, M. E. Hill, J. M. Kezik and B. P. Leaderer, Epidemiol., 2013, 24, 320–330 CrossRef PubMed.
- Y.-H. Choi, D.-H. Kim and S.-H. Hong, ACS Appl. Mater. Interfaces, 2018, 10, 14901–14913 CrossRef CAS PubMed.
- Y. Cai, Q. Ke, G. Zhang and Y.-W. Zhang, J. Phys. Chem. C, 2015, 119, 3102–3110 CrossRef CAS.
- E. Montes and U. Schwingenschlögl, J. Mater. Chem. C, 2017, 5, 5365–5371 RSC.
- D. An, X. Zhang, Z. Bi, W. Shan, H. Zhang, S. Xia and M. Qiu, Adv. Funct. Mater., 2021, 31, 2106484 CrossRef CAS.
- A. N. Abbas, B. L. Liu, L. Chen, Y. Q. Ma, S. Cong, N. Aroonyadet, M. Köpf, T. Nilges and C. W. Zhou, ACS Nano, 2015, 9, 5618–5624 CrossRef CAS PubMed.
- S. Y. Cho, Y. Lee, H. J. Koh, H. Jung, J. S. Kim, H. W. Yoo, J. Kim and H. T. Jung, Adv. Mater., 2016, 28, 7020–7028 CrossRef CAS PubMed.
- D. Han, X. Han, L. Liu, D. Li, Y. Liu, Z. Liu, D. Liu, Y. Chen, K. Zhuo and S. Sang, ACS Appl. Mater. Interfaces, 2022, 14, 13942–13951 CrossRef CAS PubMed.
- H. Chen, Y. Chen, H. Zhang, D. W. Zhang, P. Zhou and J. Huang, Adv. Funct. Mater., 2018, 28, 1801035 CrossRef.
- E. Montes and U. Schwingenschlogl, J. Mater. Chem. C, 2017, 5, 5365–5371 RSC.
- L. Kou, T. Frauenheim and C. Chen, J. Phys. Chem. Lett., 2014, 5, 2675–2681 CrossRef CAS PubMed.
- J. Sun, N. Lin, H. Ren, C. Tang, L. Yang and X. Zhao, RSC Adv., 2016, 6, 17494–17503 RSC.
- M. Sajjad, E. Montes, N. Singh and U. Schwingenschlögl, Adv. Mater. Interfaces, 2017, 4, 1600911 CrossRef.
- S. Singsen, A. Watwiangkham, L. Ngamwongwan, I. Fongkaew, S. Jungthawan and S. Suthirakun, ACS Appl. Nano Mater., 2023, 6, 1496–1506 CrossRef CAS.
- S. Dharani, V. Nagarajan and R. Chandiramouli, J. Mol. Graphics Modell., 2019, 91, 22–29 CrossRef CAS PubMed.
- R. Bhuvaneswari, V. Nagarajan and R. Chandiramouli, Comput. Theor. Chem., 2019, 1165, 112563 CrossRef CAS.
- B. Zhang, Z. Mao and P. Wu, Appl. Surf. Sci., 2021, 565, 150546 CrossRef CAS.
- Y. X. Liu, J. Zhang, C. D. Sheng, Y. C. Zhang and L. A. Zhao, Chem. Eng. J., 2010, 162, 1006–1011 CrossRef CAS.
- R. Chen, T. S. Zhang, Y. Q. Guo, J. W. Wang, J. X. Wei and Q. J. Yu, Chem. Eng. J., 2021, 420, 127588 CrossRef CAS.
- F. Rezaei, A. A. Rownaghi, S. Monjezi, R. P. Lively and C. W. Jones, Energy Fuels, 2015, 29, 5467–5486 CrossRef CAS.
- Y. Sun, E. Zwolińska and A. G. Chmielewski, Crit. Rev. Environ. Sci. Technol., 2015, 46, 119–142 CrossRef.
- S. G. Chang and D. K. Liu, Nature, 1990, 343, 151–153 CrossRef CAS.
- D. K. Liu, D.-X. Shen and S.-G. Chang, Environ. Sci. Technol., 1991, 25, 55–60 CrossRef CAS.
- B. Li, Y. Liu, X. Zhao, P. Ning, X. Liu and T. Zhu, J. Hazard. Mater., 2021, 403, 123971 CrossRef CAS PubMed.
- Y. Liu, B. Li, X. Lei, S. Liu, H. Zhu, E. Ding and P. Ning, Chem. Eng. J., 2022, 428, 131991 CrossRef CAS.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- Z. Wang, Y. Huang, W. Ho, J. Cao, Z. Shen and S. C. Lee, Appl. Catal., B, 2016, 199, 123–133 CrossRef CAS.
- W. Wan, S. Yu, F. Dong, Q. Zhang and Y. Zhou, J. Mater. Chem. A, 2016, 4, 7823–7829 RSC.
- W. Cui, J. Li, F. Dong, Y. Sun, G. Jiang, W. Cen, S. C. Lee and Z. Wu, Environ. Sci. Technol., 2017, 51, 10682–10690 CrossRef CAS PubMed.
- X. Wu, S. J. Cobbina, G. Mao, H. Xu, Z. Zhang and L. Yang, Environ. Sci. Pollut. Res., 2016, 23, 8244–8259 CrossRef CAS PubMed.
- E. Nilsen, K. L. Smalling, L. Ahrens, M. Gros, K. S. B. Miglioranza, Y. Picó and H. L. Schoenfuss, Environ. Toxicol. Chem., 2019, 38, 46–60 CrossRef CAS PubMed.
- P. Morcillo, M. Á. Esteban and A. Cuesta, Chemosphere, 2016, 144, 225–233 CrossRef CAS PubMed.
- M. Feng, D. D. Dionysiou and V. K. Sharma, Chem. Eng. J., 2020, 389, 123460 CrossRef CAS.
- C. Wu, L. Jing, J. Deng, Y. Liu, S. Li, S. Lv, Y. Sun, Q. Zhang and H. Dai, Chemosphere, 2021, 274, 129793 CrossRef CAS PubMed.
- Y. Zheng, Y. Chen, B. Gao, B. Lin and X. Wang, Engineering, 2021, 7, 991–1001 CrossRef CAS.
- M. S. Tsuboy, J. P. F. Angeli, M. S. Mantovani, S. Knasmüller, G. A. Umbuzeiro and L. R. Ribeiro, Toxicol. Vitro, 2007, 21, 1650–1655 CrossRef CAS PubMed.
- Z. Zhang, W. Wang, E. Gao, S. Sun and L. Zhang, J. Phys. Chem. C, 2012, 116, 25898–25903 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- Y. Liu, J. Kong, J. Yuan, W. Zhao, X. Zhu, C. Sun and J. Xie, Chem. Eng. J., 2018, 331, 242–254 CrossRef CAS.
- P. Wang, H. Zhao, S. Li, R. Jing, Y. Liu, N. Jiang, F. Bian, Y. Liu, H. Liu and Q. Zhang, J. Mater. Sci., 2021, 56, 8060–8078 CrossRef CAS.
- Q. Lu, W. Gao, J. Du, L. Zhou and Y. Lian, J. Agric. Food Chem., 2012, 60, 4773–4778 CrossRef CAS PubMed.
- Y. Y. Cheng and T. H. Tsai, J. Agric. Food Chem., 2017, 65, 1078–1085 CrossRef CAS PubMed.
- R. H. Jeong, D. I. Kim, J. W. Lee, J.-H. Yu, B. G. Jeong, H. M. Oh, M. S. Jeong and J.-H. Boo, 2D Mater., 2019, 6, 045039 CrossRef CAS.
- F. Yu, Y. Li, S. Han and J. Ma, Chemosphere, 2016, 153, 365–385 CrossRef CAS PubMed.
- Y. Xiang, Z. Xu, Y. Wei, Y. Zhou, X. Yang, Y. Yang, J. Yang, J. Zhang, L. Luo and Z. Zhou, J. Environ. Manage., 2019, 237, 128–138 CrossRef CAS PubMed.
- M.-F. Li, Y.-G. Liu, G.-M. Zeng, N. Liu and S.-B. Liu, Chemosphere, 2019, 226, 360–380 CrossRef CAS PubMed.
- Q. Wu, H. Yang, L. Kang, Z. Gao and F. Ren, Appl. Catal., B, 2020, 263, 118282 CrossRef CAS.
- D. Wang and Z. Li, Res. Chem. Intermed., 2017, 43, 5169–5186 CrossRef CAS.
- K. G. M. Laurier, F. Vermoortele, R. Ameloot, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, J. Am. Chem. Soc., 2013, 135, 14488–14491 CrossRef CAS PubMed.
- X. Lei, J. Wang, Y. Shi, W. Yao, Q. Wu, Q. Wu and R. Zou, Appl. Surf. Sci., 2020, 528, 146963 CrossRef CAS.
- Z. Zhao, H. An, J. Lin, M. Feng, V. Murugadoss, T. Ding, H. Liu, Q. Shao, X. Mai, N. Wang, H. Gu, S. Angaiah and Z. Guo, Chem. Rec., 2019, 19, 873–882 CrossRef CAS PubMed.
- N. Tian, H. Huang, X. Du, F. Dong and Y. Zhang, J. Mater. Chem. A, 2019, 7, 11584–11612 RSC.
- A. Meng, W. Tian, H. Yang, X. Wang, X. Wang and Z. Li, J. Hazard. Mater., 2021, 413, 125400 CrossRef CAS PubMed.
- D. Wang, L. Huang, Z. Guo, S. Jin, C. Liu, W. Wang and W. Yuan, ACS Appl. Nano Mater., 2018, 1, 4594–4601 CrossRef CAS.
- S. Sharma and A. Bhattacharya, Appl. Water Sci., 2017, 7, 1043–1067 CrossRef CAS.
- Z. Teng, N. Yang, H. Lv, S. Wang, M. Hu, C. Wang, D. Wang and G. Wang, Chem, 2019, 5, 664–680 CAS.
- W. Wang, C. Zhou, Y. Yang, G. Zeng, C. Zhang, Y. Zhou, J. Yang, D. Huang, H. Wang, W. Xiong, X. Li, Y. Fu, Z. Wang, Q. He, M. Jia and H. Luo, Chem. Eng. J., 2021, 404, 126540 CrossRef CAS.
- E. M. Anastasi, T. D. Wohlsen, H. M. Stratton and M. Katouli, Water Res., 2013, 47, 6670–6679 CrossRef CAS PubMed.
- Y. Li, X. Liu, L. Tan, Z. Cui, D. Jing, X. Yang, Y. Liang, Z. Li, S. Zhu, Y. Zheng, K. W. K. Yeung, D. Zheng, X. Wang and S. Wu, Adv. Funct. Mater., 2019, 29, 1900946 CrossRef.
- X. Zhang, J. Zhang, J. Yu, Y. Zhang, Z. Cui, Y. Sun and B. Hou, Appl. Catal., B, 2018, 220, 57–66 CrossRef CAS.
- B. Zhang, S. Zou, R. Cai, M. Li and Z. He, Appl. Catal., B, 2018, 224, 383–393 CrossRef CAS.
- J. A. Young, J. Chem. Educ., 2004, 81, 945 CrossRef CAS.
- D. Xia, Z. Shen, G. Huang, W. Wang, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2015, 49, 6264–6273 CrossRef CAS PubMed.
- W. Wang, Y. Yu, T. An, G. Li, H. Y. Yip, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2012, 46, 4599–4606 CrossRef CAS PubMed.
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed.
- T. K. Athira, M. Roshith, R. Kadrekar, A. Arya, M. S. Kumar, G. Anantharaj, L. Gurrala, V. Saranyan, T. G. S. Babu and V. R. K. Darbha, Mater. Res. Express, 2020, 7, 104002 CrossRef CAS.
- M. Roshith, A. Pathak, A. K. Nanda Kumar, G. Anantharaj, V. Saranyan, S. Ramasubramanian, T. G. Satheesh Babu and D. V. Ravi Kumar, Appl. Surf. Sci., 2021, 540, 148398 CrossRef CAS.
- Q. Zhang, X. Liu, L. Tan, Z. Cui, Z. Li, Y. Liang, S. Zhu, K. W. K. Yeung, Y. Zheng and S. Wu, Chem. Eng. J., 2020, 383, 123088 CrossRef CAS.
- T. Guo, S. Zhuang, H. Qiu, Y. Guo, L. Wang, G. Jin, W. Lin, G. Huang and H. Yang, Part. Part. Syst. Charact., 2020, 37, 2000169 CrossRef CAS.
- H. Ji, H. Sun and X. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef CAS PubMed.
- M. D. Rojas-Andrade, G. Chata, D. Rouholiman, J. Liu, C. Saltikov and S. Chen, Nanoscale, 2017, 9, 994–1006 RSC.
- D. He, Z. Zhang, Y. Xing, Y. Zhou, H. Yang, H. Liu, J. Qu, X. Yuan, J. Guan and Y.-N. Zhang, Chem. Eng. J., 2020, 384, 123258 CrossRef CAS.
- S. Luo, R. Liu, X. Zhang, R. Chen, M. Yan, K. Huang, J. Sun, R. Wang and J. Wang, J. Hazard. Mater., 2022, 424, 127281 CrossRef CAS PubMed.
- X. Han, X. He, L. Sun, X. Han, W. Zhan, J. Xu, X. Wang and J. Chen, ACS Catal., 2018, 8, 3348–3356 CrossRef CAS.
- Y. Kwon, A. Soon, H. Han and H. Lee, J. Mater. Chem. A, 2015, 3, 156–162 RSC.
- Z. Bao, C. Shi, W. Tu, L. Li and Q. Li, Environ. Pollut., 2022, 313, 120184 CrossRef CAS PubMed.
- X. Pan, M. Shi, X. Chen, S. Kuang, H. Ullah, H. Lu and L. Riaz, Front. Environ. Sci., 2022, 10, 1057384 CrossRef.
- X. Zheng, W. Xu, J. Dong, T. Yang, Z. Shangguan, J. Qu, X. Li and X. Tan, J. Hazard. Mater., 2022, 438, 129557 CrossRef CAS PubMed.
- X. Li, A. Wang, W. Wan, X. Luo, L. Zheng, G. He, D. Huang, W. Chen and Q. Huang, Appl. Environ. Microbiol., 2021, 87, e01366 CAS.
- L. Stille, E. Smeets, B. Wicke, R. Singh and G. Singh, Energy Sustain. Dev., 2011, 15, 388–397 CrossRef CAS.
- S. Ullah, S. Dahlawi, A. Naeem, Z. Rengel and R. Naidu, Sci. Total Environ., 2018, 625, 320–335 CrossRef PubMed.
- P. Zhang, X. Bing, L. Jiao, H. Xiao, B. Li and H. Sun, Chem. Eng. J., 2022, 431, 133904 CrossRef CAS.
- H. Tang, M. Chen, P. Wu, M. Faheem, Q. Feng, X. Lee, S. Wang and B. Wang, Chemosphere, 2022, 311, 137025 CrossRef PubMed.
- S. K. Das, G. K. Ghosh and R. Avasthe, Biomass Conv. Bioref., 2023, 13, 1359–1369 CrossRef.
- J.-H. Yuan, R.-K. Xu and H. Zhang, Bioresour. Technol., 2011, 102, 3488–3497 CrossRef CAS PubMed.
- A. M. Puziy, O. I. Poddubnaya, B. Gawdzik and J. M. D. Tascon, Carbon, 2020, 157, 796–846 CrossRef CAS.
- H. Zhang, J. G. Shao, S. H. Zhang, X. Zhang and H. P. Chen, J. Hazard. Mater., 2020, 390, 121349 CrossRef CAS PubMed.
- J. Ren, Z. Zhao, A. Ali, W. Guan, R. Xiao, J. J. Wang, S. Ma, D. Guo, B. Zhou, Z. Zhang and R. Li, J. Soils Sediments, 2020, 20, 3041–3052 CrossRef CAS.
- H. P. Rothbaum, Outlook Agric., 1966, 5, 123–128 CrossRef CAS.
- H. Lyu, B. Gao, F. He, C. Ding, J. Tang and J. C. Crittenden, ACS Sustainable Chem. Eng., 2017, 5, 9568–9585 CrossRef CAS.
- X. Zhang, Z. Xiong, S. Zhang, Y. Ge, W. Ma, L. Yan, D. Li, D. Wang, S. Deng, Q. Zhao, W. Wang and B. Xing, Environ. Sci.: Nano, 2020, 7, 404–413 RSC.
| This journal is © The Royal Society of Chemistry 2023 |