
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of visible light-induced C–C cross-coupling by Csp3–H bond activation
Bholanath
Maity
 *,
Sayan
Dutta
and
Luigi
Cavallo
*
*,
Sayan
Dutta
and
Luigi
Cavallo
*
KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: bholanath.maity@kaust.edu.sa; Luigi.cavallo@kaust.edu.sa
First published on 19th July 2023
Abstract
Csp3–C cross-coupling by activating Csp3–H bonds is a dream reaction for the chemical community, and visible light-induced transition metal-catalysis under mild reaction conditions is considered a powerful tool to achieve it. Advancement of this research area is still in its infancy because of the chemical and technical complexity of this catalysis. Mechanistic studies illuminating the operative reaction pathways can rationalize the increasing amount of experimental catalysis data and provide the knowledge allowing faster and rational advances in the field. This goal requires complementary experimental and theoretical mechanistic studies, as each of them is unfit to clarify the operative mechanisms alone. In this tutorial review we summarize representative experimental and computational mechanistic studies, highlighting weaknesses, strengths, and synergies between the two approaches.
 Bholanath Maity | Bholanath Maity received his MSc in chemistry from Vidyasagar University, India in 2011. His PhD studies at the Indian Institute of Science Education and Research (IISER) Kolkata, India, focused on computational mechanistic studies of homogeneous catalysis and main group reactions. After completing his PhD in 2017, he joined Prof. Luigi Cavallo's group at King Abdullah University of Science and Technology (KAUST), Saudi Arabia, as a postdoctoral fellow. Since 2019, he has been a research scientist in the same group. His current research interest includes computational studies on photoredox catalysis. |
 Sayan Dutta | Sayan Dutta obtained his MSc in Chemistry from Visva-Bharati University, India in 2013. His PhD studies at the Indian Institute of Science Education and Research (IISER) Kolkata, India, focused on the mechanistic investigation of main group-mediated chemical transformations and exploring the bonding scenario in various donor–acceptor complexes using computational methods. After earning his PhD, he joined Prof. Luigi Cavallo's group in 2022 at King Abdullah University of Science and Technology (KAUST), Saudi Arabia, as a postdoctoral fellow. Theoretical exploration of mechanistic avenues in visible light-induced transition metal catalysis is his current research interest. |
 Luigi Cavallo | Luigi Cavallo earned his PhD in 1991 at the University of Naples (Italy) and completed his education on DFT working with Prof. Tom Ziegler at the University of Calgary, where he contributed to the development of combined quantum mechanics/molecular mechanics techniques. He joined the University of Naples, Italy, in 1994 as an assistant professor. He subsequently moved to the University of Salerno, Italy, in 2002 as an associate professor. He joined the Catalysis Center at KAUST in 2011, where he currently serves as a professor of chemistry. His research interests focus on understanding and solving problems in catalysis using the armory of tools known as computational chemistry. |
Key learning points(1) The different protocols available for visible light-induced Csp3–C cross-coupling involving Csp3–H activation.(2) The complexity and the challenges of visible light-induced Csp3–C cross-coupling involving Csp3–H activation. (3) Mechanistic aspects of visible light-induced Csp3–C cross-coupling catalysis involving Csp3–H activation. (4) The methodological limitations and the knowledge provided by quantum-mechanics investigations of this reaction. (5) Potential future directions and challenges. |
1. Introduction
Transition metal (TM) catalysed C–C cross-coupling is a powerful strategy that allows linking together two chemical fragments belonging to different classes (Scheme 1a).1,2 This tool has become an essential component of the methodological arsenal in synthetic chemistry, from the academic scale to industrial production, due to the variety of products that can be formed under mild conditions.3,4 The mechanism of the TM catalysed C–C cross-coupling reaction is well understood,5 and it is based on three sequential elementary steps, see Scheme 1b: (i) oxidative addition of an electrophilic substrate to a metal complex; (ii) transmetallation to load a nucleophilic substrate on the metal; and (iii) reductive elimination of the product from the metal complex. In classic TM catalysed C–C cross-coupling reactions, all elementary steps occur with intermediates in the electronic ground state and involve 2e exchange with the TM. As with any other reactivity paradigm, this scheme also has some limitations. Among them is the inability to activate inert bonds, such as the ubiquitous Csp3–H bonds, which would pave the way to effective cross-coupling enabling formation of Csp3–C bonds.6,7 For reference, the Csp3–H bond dissociation energies (BDEs) in various organic molecules are depicted in Scheme 2. It is worth mentioning that the BDE is usually referred to as the enthalpy change in homolytic dissociation of the concerned bond (C–H → C˙ + H˙) in the gas phase, while its free energy analogue, the bond dissociation free energy (BDFE), is usually determined in solution.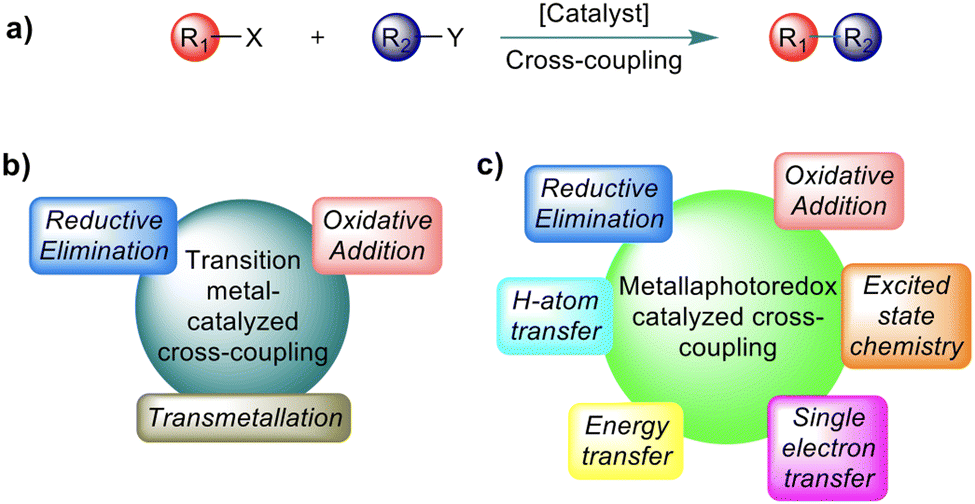 | ||
| Scheme 1 (a) General scope of C–C cross-coupling reactions. (b) General scheme outlying the elementary steps of ground state transition metal-catalysed C–C cross-coupling. (c) General scheme outlying the elementary steps available for metallaphotoredox-catalysed C–C cross-coupling. | ||
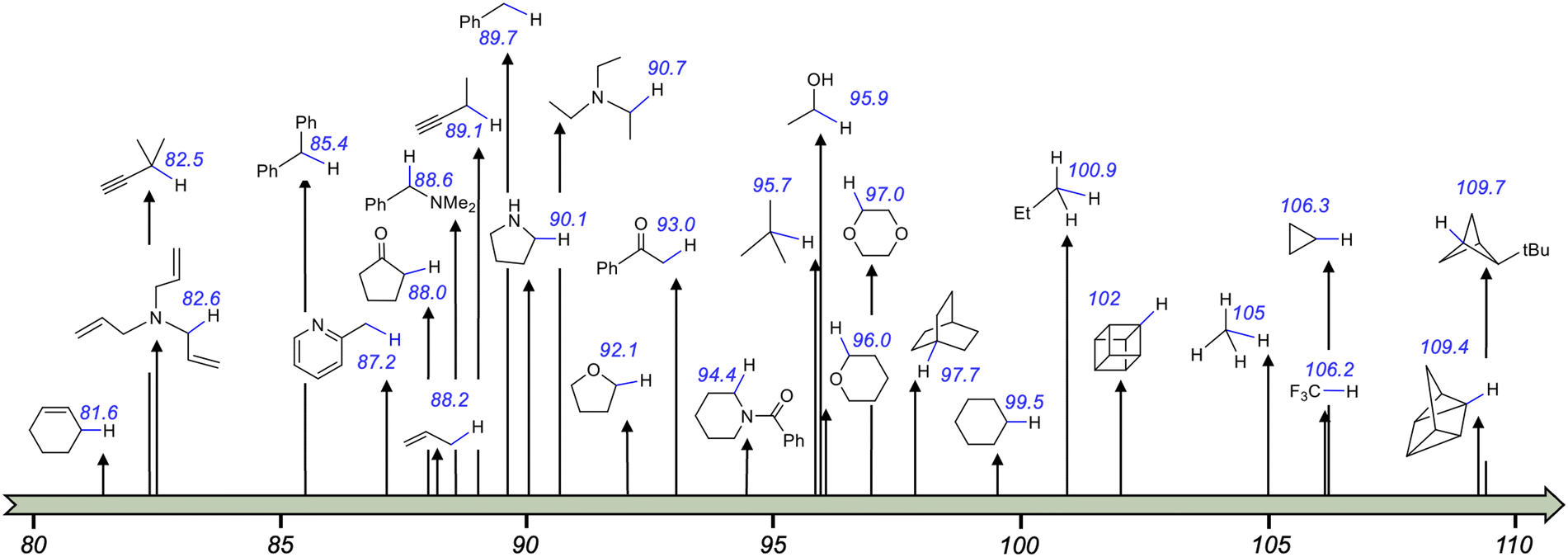 | ||
| Scheme 2 Homolytic bond dissociation energies (BDEs in kcal mol−1) of various Csp3–H bonds.8,9 | ||
Over the last several decades, remarkable progress in Csp2–H activation has been witnessed.9–11 In comparison, functionalization of Csp3–H bonds was underdeveloped, with the high bond energy and low acidity accounting for their poor reactivity.7,12 Moreover, controlling site-selectivity in a complex molecular system, containing several Csp3–H bonds with nearly similar strengths, represents another formidable challenge.13 This scenario has been changed in the last decade, through the merger of photoredox catalysis with TM catalysis, termed metallaphotoredox catalysis.14,15 This branch of catalysis allows unique reaction pathways that are not feasible in conventional ground-state reactivity, such as the classic TM catalysed C–C cross-coupling scheme described above. Consequently, this protocol has the potential to promote effective cross-coupling of inert bonds, including Csp3–C coupling under mild conditions. These spectacular advances are possible because of the large variety of mechanistic handles in metallaphotoredox catalysis. In addition to the elementary steps available in conventional TM catalysed cross-coupling, metallaphotoredox catalysed cross-coupling can involve intermediates in excited electronic states, single electron transfers (SET), energy transfer (EnT) between different species, and can benefit from an assisting hydrogen atom transfer (HAT) agent, see Scheme 1c. Furthermore, different from ground state TM catalysed cross-coupling, the exact sequence of the elementary steps in metallaphotoredox catalysed cross-coupling can be, to some extent, varied. This gives more flexibility for designing a reactivity profile transforming given substrates into the desired product and justifies the relevance of having a clear mechanistic understanding of the protocols developed to date. While numerous reviews and perspective articles on the experimental development of metallaphotoredox catalysis are available,16–21 the review presented herein will be a comprehensive assessment on mechanistic studies on Csp3–C cross-coupling reactions that involve Csp3–H functionalization.11,12,22–26 We believe that having a unified report of the available mechanistic knowledge, currently dispersed in different articles, can stimulate creative thinking.
The body of the review is composed of Section 2, providing a short description of the insights provided by computational modelling of metallaphotoredox catalysis and of the theoretical methods needed to tackle them, and of Section 3, illustrating the main four mechanistic paradigms currently available in visible light-induced TM catalysis to promote Csp3–C cross-coupling reactions of Csp3–H bonds. Section 4, providing an outlook on the field, will conclude the review.
Why computational tools are necessaryTime-resolved spectroscopic tools like Stern–Volmer analysis and laser flash photolysis techniques are frequently employed to study excited-state kinetics in metallaphotoredox cross-coupling reactions. These methods enable the assessment of the kinetics of both singlet and triplet excited states of the photocatalyst i.e., dynamic and static quenching processes. However, the short lifetime of key reactive intermediates, the competing side routes for the evolution of excited-state species, and the challenges of determining the kinetic parameters of individual steps make it extremely difficult to ascertain complex mechanistic scenarios in these reactions utilizing merely experimental techniques. This explains the necessity of computational tools to untangle the complete mechanistic picture for this fascinating chemical transformation. |
2. Computational approaches
Transition metal catalysed conventional cross-coupling occurs via TM complexes that have two relatively stable oxidation states separated by two electrons, such as Ni0/NiIIetc. Moreover, all intermediates and transition states along the reaction profile are in the electronic ground state. This allows experimental characterization of several intermediates that, coupled with ground state density functional theory (DFT) calculations, results in a comprehensive mechanistic understanding of this catalysis. Differently, visible light-induced TM cross-coupling catalysis can occur via a multiplicity of oxidation states, including less stable ones, such as NiI and NiIII, in addition to the more stable, and commonly used in catalysis, Ni0/NiII oxidation states. Additionally, intermediates can participate in reactivity in electronically excited states, which further complicates experimental mechanistic investigations. This explains why computational methods have become indispensable to elucidating the mechanisms of visible light-induced TM cross-coupling catalysis. Most of the metallaphotoredox-catalysed reactions involving a SET event can still be explained by combining ground state DFT and time-dependent DFT (TDDFT) methods.27–36 However, these methods are not the most appropriate ones to model some of the elementary steps depicted in Scheme 1c, such as energy transfer, as well as for the accurate characterization of excited states. These problems are successfully handled by less frequently used, and computationally much more expensive, multireference calculations.In this section we briefly introduce these state-of-the-art computational methods for non-specialists as we believe this will increase the readability of the following sections and literature works. We also clarify the area in which cutting-edge computational methods are applicable, their computational cost and handling challenges. Considering that ground state DFT has become an ordinary tool in molecular catalysis it will not be covered here. We only remark that the DFT functional and the basis set used can be chosen based on the extensive literature available. Differently, exploring the electronic, structural, thermodynamic, and kinetic aspects of open-shell transition metal complexes is less standard, and requires more careful selection of the computational method.37 In these cases it is usually advised to benchmark and calibrate the protocol on the systems in hand with the accessible experimental data. Furthermore, confirming the spin state by analysis of the expectation values of the total spin operator, 〈Ŝ2〉, to exclude contamination of higher excited states, is recommended. More reliable energetics can be achieved by using the wave-function based coupled-cluster method with iterative inclusion of single and double excitations, and perturbative inclusion of triple excitations, CCSD(T), which is considered as the golden standard in quantum mechanics calculations.38 However, due to its heavy computational cost this method is impracticable for large organometallic complexes. A promising alternative is the domain-based local pair natural orbital (DLPNO) version of CCSD(T), DLPNO-CCSD(T), which allows the calculation of molecular systems of interest to metallaphotoredox catalysis.39 In this case, to achieve accurate results it is recommended to enforce tight setup of the input parameters, at the expense of increased computational costs.40 As open-shell 3d transition metal complexes can exhibit different spin states, and often these spin states have very small energy gaps, the single-reference nature of DFT may produce inaccurate results. In these cases multireference or multiconfiguration methods, such as configuration interaction (CI) and multiconfiguration self-consistent field (MCSCF) methods, should be employed to validate the computational approach.41 For instance, to analyse the electronic structure and excited-state properties at nonequilibrium geometries of mono- and polynuclear transition metal complexes, the complete active space self-consistent field (CASSCF) method is proven to be efficient.42–48 Moreover, to examine conical intersections between various electronic states, multireference techniques like CASPT2, NEVPT2, and MRCI can also be used. However, the considerably high computational cost and the dependence of the results on the definition and size of active space of multireference methods limits their scope. The lack of efficient and fast multireference methods to explore excited-state processes with approachable computational costs explains the extensive application of TDDFT in photochemistry involving large transition metal complexes. However, one should be cautious about a few limitations of TDDFT methods while using them. The drawbacks include the dependency of the computed results on the choice of the exchange–correlation functional employed, the lack of double and higher excitations, and the limited precision in the calculation of higher spin states. Therefore, extensive electronic excited state calculations on larger catalytic systems necessitates the development of more precise and effective electronic structure calculation approaches. Of note, one advanced technique, multiconfiguration pair-DFT (MC-PDFT),49 combining multiconfigurational wave functions with generalized DFT, was recently developed to tackle multi electron excitations and can be applied to transition metal systems.50
Another challenge in the field is the accurate modelling of the energy barrier for the single electron transfer (SET) steps. Applying the Marcus–Hush theory,51–54 the free energy barrier (ΔG‡MH) of a single electron transfer process can be estimated according to eqn (1):
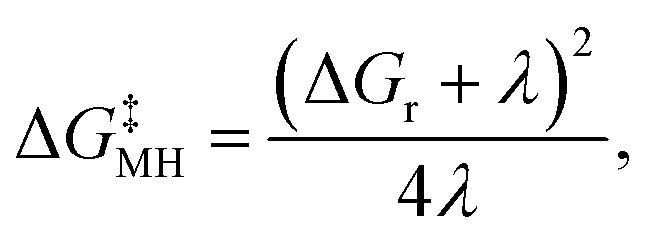 | (1) |
3. Protocols for visible light-induced transition metal catalysed Csp3–C cross-coupling of Csp3–H bonds
From a mechanistic perspective, visible light-induced transition metal catalysed methodologies for Csp3–C cross-coupling via Csp3–H bond activation can be classified into the four paradigms shown in Scheme 3, which suggest the possible interplay between the transition metal (TM) catalyst, an assisting photocatalyst (PC), with involvement of a H-atom transfer (HAT) catalyst. The four paradigms are usually defined as: (a) excited state TM catalysis; (b) PC/TM dual catalysis; (c) PC-HAT/TM dual catalysis; and (d) PC/HAT/TM triple catalysis. Since this review is not intended to cover experimental results comprehensively, for each reactivity paradigm we will discuss only a selection of experimental papers, giving priority to the publication date. Comprehensive survey of the experimental literature can be found in ref. 22.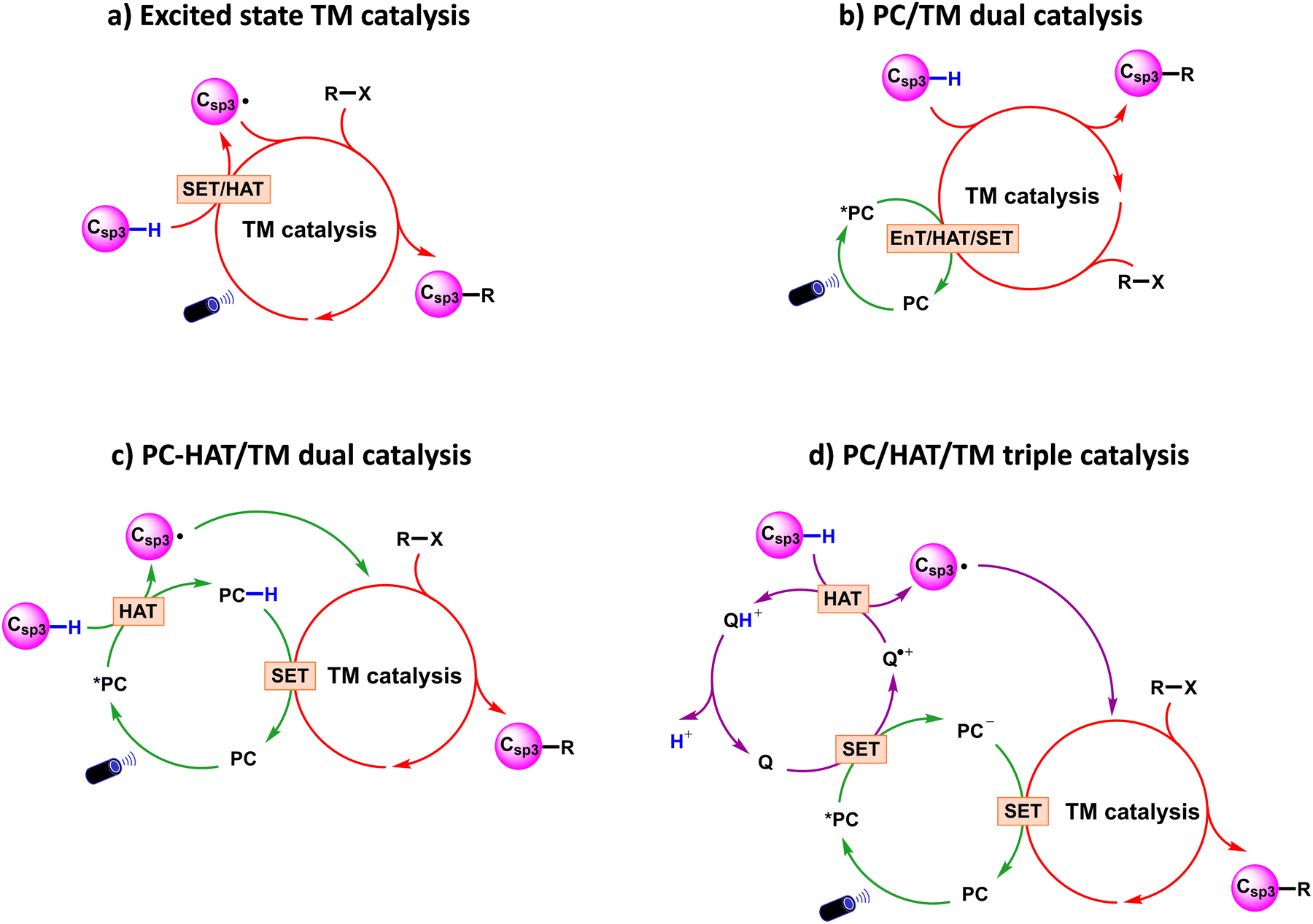 | ||
| Scheme 3 Representative reaction models in visible light-promoted transition metal catalysed Csp3–C cross-couplings by Csp3–H activation. PC: photocatalyst; TM: transition metal; Q: HAT catalyst. | ||
3.1. Excited state TM catalysed reactions
The outline of this protocol is shown in Scheme 3a, and representative reactions are given in Scheme 4. Along this reaction channel the visible light-induced excited state TM performs the reaction in the absence of an additional photocatalyst. Unlike the metallaphotoredox PC/TM dual catalysis that employs a conventional photocatalyst and a TM synergistically, in visible light-induced TM catalysis, the TM complex acts as both the PC and the cross-coupling catalyst. This protocol is highly desirable due to its conceptual simplicity, but it is chemically challenging and thus underdeveloped compared to other protocols. Very few reports are available in the literature and are discussed in this section. The first one reporting visible light-driven palladium-catalysed α-amino-Csp3–H alkylation with alkyl bromide (Scheme 4a) was published by Yu and co-workers in 2017.55 Similarly, more recently, Cao and Zhang have developed an example of alkynylation reaction of Csp3–H bonds via cross-dehydrogenative-coupling using CuI as the catalyst (Scheme 4b).56 Additionally, photo-induced cross-dehydrogenative coupling between Csp3–H bonds in alkanes and heteroarenes using catalytic chloride and cobalt catalyst was developed by Li and co-workers.57 A remarkable development, the alkynylation of a α-oxy-Csp3–H bond using photoexcited uranyl catalyst was reported by Wang and co-workers (Scheme 4c).58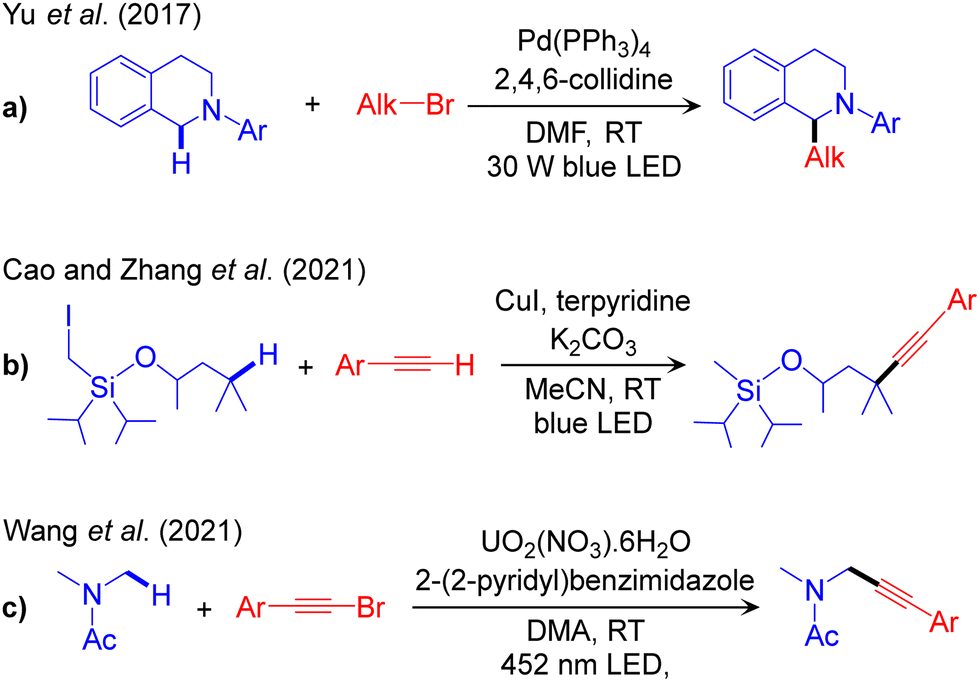 | ||
| Scheme 4 Representative methodologies for visible light-induced excited TM catalysed Csp3–C cross-couplings of Csp3–H bonds. | ||
No detailed mechanistic study of the above catalytic schemes is available in the literature. Based on radical trapping experiments, radical clock reactions and quenching studies, preliminary mechanisms of these methodologies have been proposed. These control experiments confirm one or more of the operative elementary steps but fail to define the complete mechanism. For example, Yu et al. conducted radical trapping experiments and radical clock reactions to characterize possible radical intermediates formed during the reaction (Scheme 4a). Based on these studies they concluded that the alkyl radical (Alk˙) is formed by interaction of Alk–Br with the photoexcited Pd-complex. This hypothesis was further supported by UV-Vis absorption and quenching experiments. Within the mechanism they proposed (Fig. 1), the light excited [LnPd0]* species reacts with Alk–Br in a SET step to generate the alkyl radical via debromination. In line with this hypothesis, a combined experimental and computational study by our group identified the visible light-active Pd(0) complex, (PPh3)3Pd0.59 Theoretical calculations suggested a barrier-less 1e oxidative addition of Alk-Br to the excited state of (PPh3)3Pd0 with liberation of the alkyl radical. Single electron reduction of LnPdIBr by 1 was suggested to generate the radical cation 6, which is deprotonated by the base B, resulting in the activation of the Csp3–H bond (Fig. 1). Finally, it was suggested that outer sphere radical–radical coupling of the resulting species 7 with the previously formed Alk˙ species yields the coupling product.
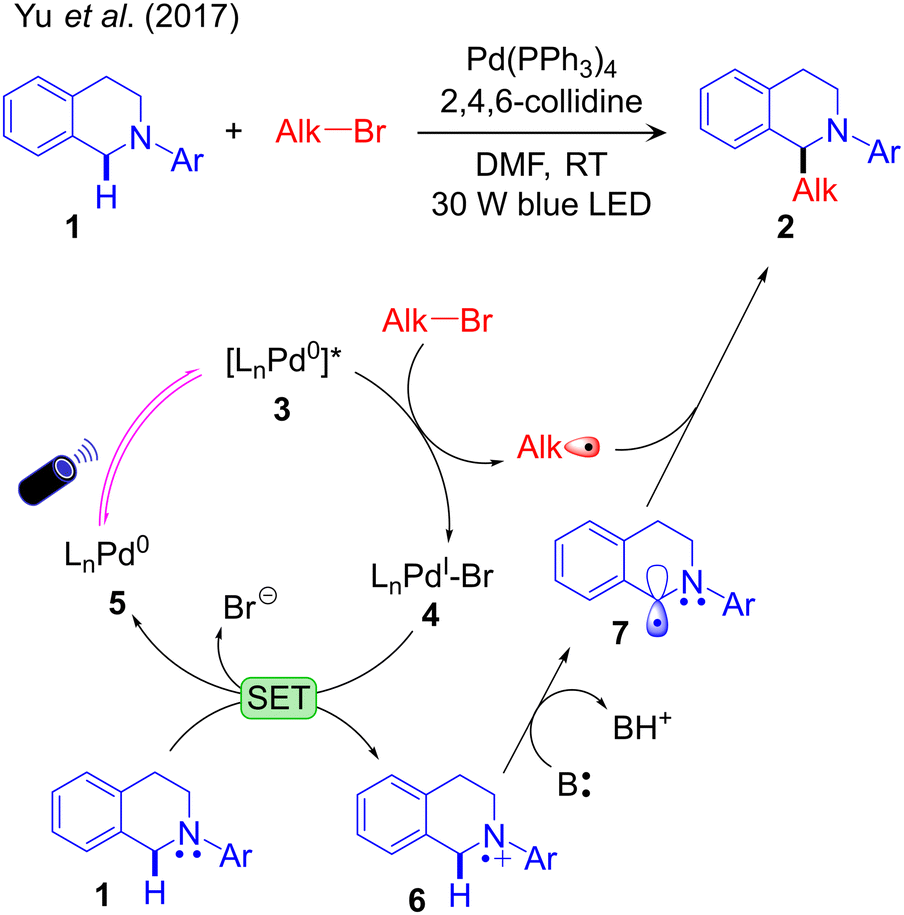 | ||
| Fig. 1 Visible light-driven palladium-catalysed Csp3–H alkylation. | ||
Despite its simplicity this protocol is underdeveloped compared to other strategies. In the proposed reaction profile easy 1e oxidation of the nucleophilic partner by the oxidized TM complex, e.g. oxidation of 1 by 4, is a step specific to this protocol and crucial for Csp3–H activation of the substrate by the base. This limits the applicability to a narrow range of substrates. A further challenge is extending this protocol to first row TMs, such as Ni.
3.2. PC/TM dual catalysed reactions
The outline of this protocol is shown in Scheme 3b, and representative reactions are provided in Scheme 5, with reactions classified according to the nature of the activated Csp3–H bond. We start with functionalization of rather weak α-amino-Csp3–H bonds, Scheme 2. The first report of this scheme, a combined Ru/Cu dual catalysed dehydrogenative alkynylation of an α-amino-Csp3–H bond, was reported by Rueping in 2012 (Scheme 5a).60 In the same year Fu reported a similar reaction replacing the Ru-based photocatalyst with an organic dye to extend the scope to the trifluoromethylation of the Csp3–H bond in diverse tetrahydroisoquinolines.61 A chiral ligand was incorporated by Li in 2015 to control enantioselectivity in the Csp3–H alkynylation step.62 More recently, Kapdi and co-workers utilized a diazo salt as a coupling partner to perform both arylation and alkylation.63 Another elegant example of α-amino-Csp3–H bond functionalization was provided by Doyle in 2016 using aryl iodides as the coupling partner and Ir/Ni dual catalysis. This scheme provides direct access to benzylic amines from inexpensive and readily available starting materials.64 As a recent addition, Huo achieved enantioselectivity in the cross-coupling step by incorporating chiral Ni-ligands.65 This dual catalytic platform has also been used for the arylation and alkylation of α-amido-Csp3–H bonds, as reported by Montgomery and Martin,66 and by König.67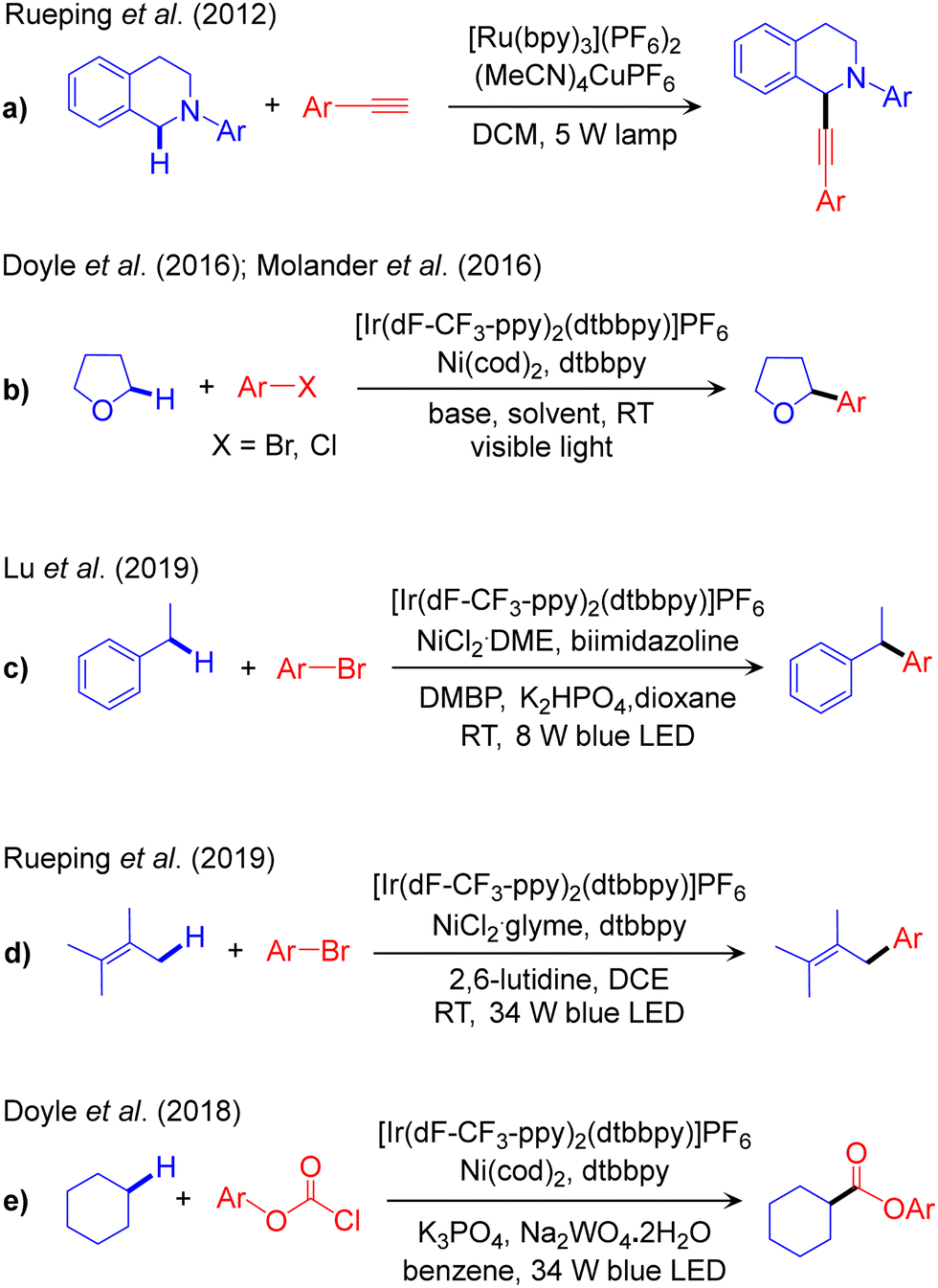 | ||
| Scheme 5 Representative reaction methodologies of visible light-induced PC/TM dual catalysed Csp3–C cross-couplings of Csp3–H bonds. | ||
Visible light-promoted Ir/Ni dual catalysis has been extended to the functionalization of stronger α-oxy-Csp3–H bonds (Scheme 2). Arylation was first reported by Molander68 and by Doyle (Scheme 5b).69,70 This protocol was further improved by Wu to the hydroalkylation of alkynes,71 by Hong to the thiocarbonylation of various cyclic/acyclic ether,72 and by Kumagai and Shibasaki to the α-benzoylation of ethers.73 Recently, König reported the alkylation of α-oxy-Csp3–H bonds in the cross-coupling of alkyl halides with ethers using the organic 4-CZIPN photocatalyst together with a Ni catalyst.74
The PC/Ni dual catalysis strategy is also widely used for the functionalization of benzylic-Csp3–H bonds. Arylation of these bonds catalysed by Ir/Ni was reported by Lu in 2019 (Scheme 5c)75 and, recently, by Deng.76 Benzylic-Csp3–H arylation of indole using the same catalyst composition has been reported by Lee,77 while alkenylation of the benzylic-Csp3–H bond was reported by Lu78 and Huo.79 The same methodology has been utilized for the acylation reaction in the presence of aldehyde as a coupling partner, reported by Ishida and Murakami.80 The same reaction has been performed in flow by Ley,81 while Huo modified this methodology by replacing the coupling partner from aldehydes to carboxylic acids.82
The first report of allylic-Csp3–H bond arylation using the Ir/Ni dual catalysis was achieved in 2018 by Rueping (Scheme 5d),83 with stereoselectivity controlled subsequently by ligand modulation.84 Recently, Cramer and Martin have used a similar catalyst platform for the Csp3–H alkylation of α-olefins.85 A different catalytic scheme, Ir/Cr dual catalysis, was used by Glorius for the Csp3–H functionalization of allyl(hetero-)arene in the presence of an aldehyde as a coupling partner.86 Dixon also reported Ru/Ni dual catalysed α-Csp3–H alkylation of ketimines.87
The Ir/Ni dual catalysis scheme is also used for the functionalization of stronger Csp3–H bonds, such as those of alkanes (Scheme 2). Doyle first reported the esterification of cyclohexane using chloroformate derivatives (Scheme 5e).88 A similar catalytic system has been explored by Hong for the cross-coupling of alkanes with amides89 and acid chlorides.90 Recently, Xu and Tambar developed direct allylation of unactivated Csp3–H bonds in unfunctionalized amides with simple allylic chlorides using a combined photoredox and nickel catalytic system and a [1,5]-hydrogen atom transfer (HAT) process.91 Due to their prevalence and relative similarity in most organic skeletons, unactivated Csp3–H bonds present a considerable obstacle for selective functionalization. Subsequently, Rovis reported the regioselective Csp3–Csp3 coupling of unactivated Csp3–H bonds in amides and alkyl bromides using a different Ir/Ni dual catalytic system.92 Their proposed mechanism suggests positional selectivity to be governed by a similar intramolecular [1,5]-HAT event in a pending amide.
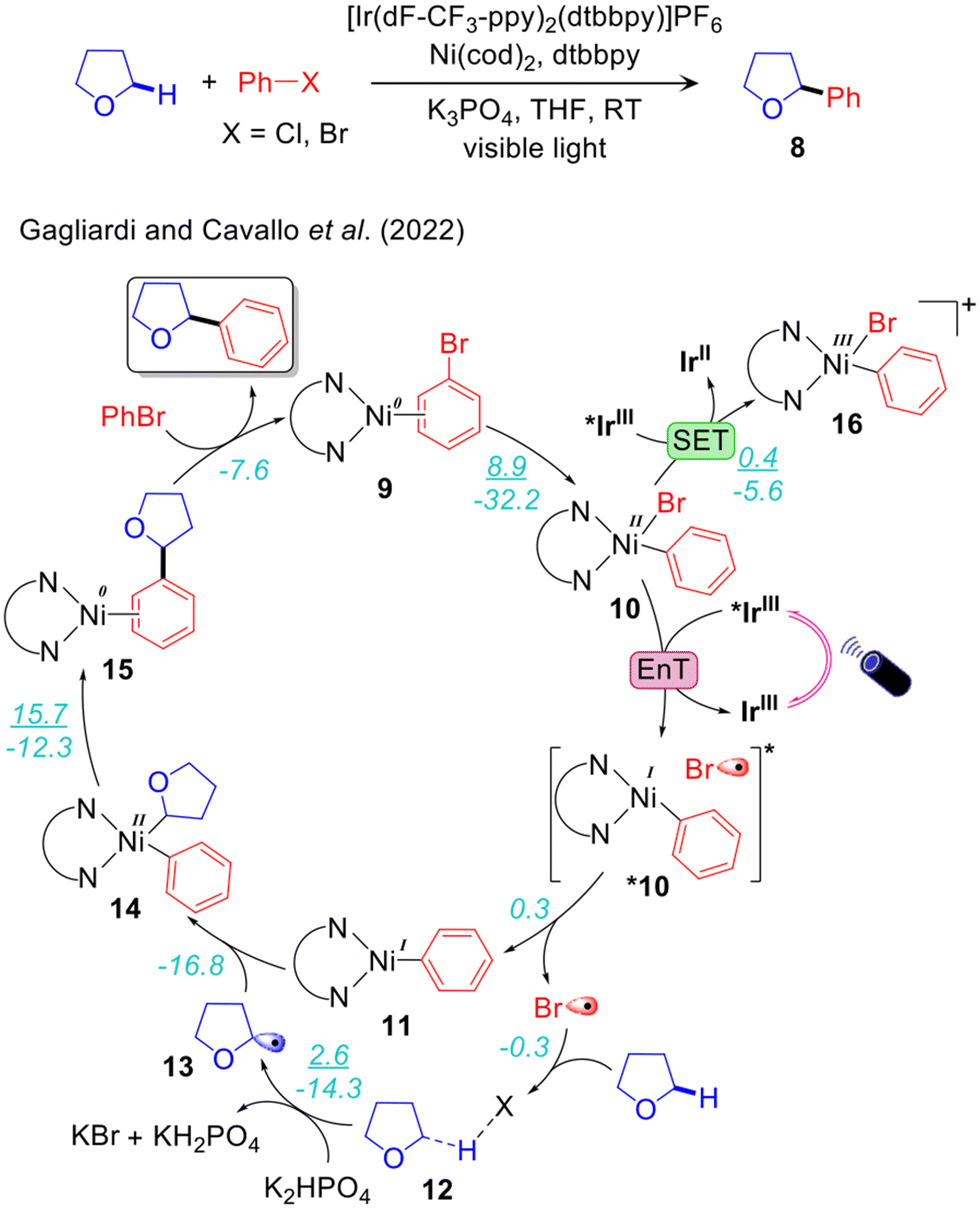
Despite these remarkable experimental advancements the mechanistic understanding still is incomplete. Based on control experiments, Molander68 and Doyle69 proposed two different mechanisms for the IrIII/Ni0 catalysed arylation of the α-oxy-Csp3–H bonds of THF. These two methodologies differ for the coupling partners used, ArCl by Doyle and ArBr in addition to 4,4′-dimetoxybenzophenone (DMBP) as HAT co-catalyst by Molander. Molander suggested that the LNiII(Ar)Br intermediate, resulting from the oxidative addition of ArBr to the initial catalyst LNi0, is promoted to the excited state via triplet EnT from the photoexcited *IrIII PC. The excited state *NiII-complex promotes activation of the α-oxy-Csp3–H bond of the THF coupling partner to generate an LNiII(Ar)Alk species. Subsequent reductive elimination liberates the desired product. The LNiII(Ar)Br complex was successfully isolated and was used as the starting complex in control experiments. The evidence that strongly oxidizing photocatalysts are unable to catalyse the reaction excluded the occurrence of a SET step with formation of a NiIII-complex.68 Differently, Doyle suggested that activation of the Csp3–H bond occurs via a SET step. Specifically, the photoexcited *IrIII PC promotes 1e oxidation of the LNiII(Ar)Cl complex.69 Photo-induced homolytic dissociation of the Ni–Cl bond of the resulting LNiIII(Ar)Cl+ complex would generate Cl˙, as previously suggested by Nocera.93 To probe this hypothesis the reaction was performed using LNiII(Ar)Cl in the presence of a non-photoredox oxidant. Formation of the desired product was considered as proof that the reaction proceeds via a SET step, which is 1e oxidation of NiII to NiIII.69
Our group compared these two mechanisms using DFT, TDDFT and HMC-PDFT tools (Fig. 2)48 on the reaction system developed by Molander.68 Since significant yields (85%) were obtained also in the absence of DMBP, we did not consider it in our calculations. Along the EnT pathway LNiII(Ph)Br (10) is excited to *10 by energy transfer from the photoexcited *IrIII PC. Conversely, along the SET pathway 10 is oxidized by *IrIII to the LNiIII(Ph)Br+ species 16, whose excitation by visible light triggers halogen radical elimination. TDDFT and HMC-PDFT calculations were performed on 10 and 16, and the corresponding chlorides analogues, to investigate the electronic structures of the excited states. These calculations suggested that the excited LNiII(Ph)Br is liable to Ni–Br bond homolysis to generate the halogen radical Br˙ promoting Csp3–H activation of the THF coupling partner. Interaction of the ground state LNiII(Ph)Br with the photoexcited IrIII PC could result in double electron exchange via the non-radiative Dexter mechanism, resulting in a transfer of energy from the *PC to the Ni complex as shown in Fig. 3.48 On the other hand, calculations of the excited states of the cationic NiIII complex LNiIII(Ph)Br+, upon the alternative 1e oxidation of LNiII(Ph)Br by the photoexcited IrIII PC, indicated that these states are not able to produce Br˙ in the energy range of visible light. Calculations also suggested that Ni–Ph bond dissociation from the photoexcited LNiIII(Ph)Br+ is preferred over Ni–Br bond homolysis, resulting in the liberation of Ph˙ rather than Br˙. This is consistent with Doyle's control experiment carried out in the presence of a photo innocent external oxidant replacing the Ir-photocatalyst and irradiated light.69 Under this condition Ph–H was observed as the major product (53%) over the desired α-oxy-Csp3–Ar product (28%). Therefore, our calculations suggest that the EnT mechanism is likely more viable than the SET one, particularly in the case of the ArBr coupling partner.48,68
 | ||
| Fig. 2 Computed mechanism of visible light-driven IrIII/Ni0 catalysed α-oxy-Csp3–H arylation. (ΔG‡/ΔG) represents the energy activation barrier and the thermodynamics of the given elementary step, in kcal mol−1. | ||
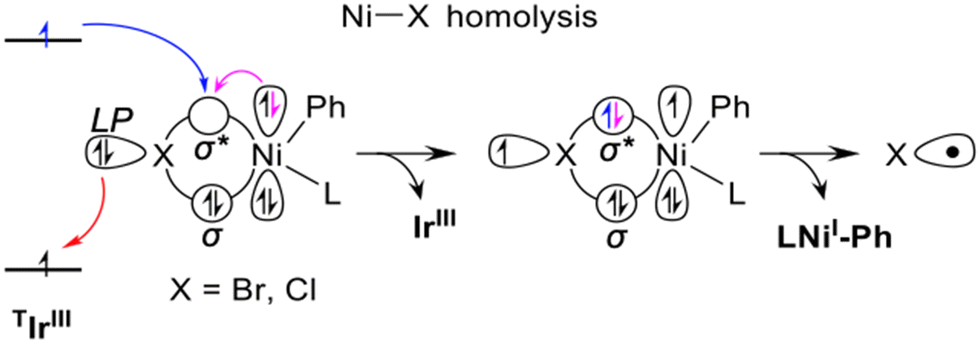 | ||
| Fig. 3 Schematic representation of the Dexter EnT step from the photoexcited PC catalysts in the triplet electronic state T1, TIrIII, to the ground state complex (bpy)NiII(Ph)X, leading to the (bpy)NiII(Ph)X excited states relevant for Ni–X homolytic dissociation. Adapted with permission from ref. 48 Copyright 2022 American Chemical Society. | ||
The overall reaction profile emerging from our work is thus consistent with the EnT mechanism (Fig. 2 and 3).48 The first step is the oxidative addition of PhBr, leading to 10 with an activation barrier of 8.9 kcal mol−1. The EnT step is from *IrIII to 10, affording the excited state *10, which serves as a source of Br˙. The next step is outer-sphere α-oxy-Csp3–H activation of THF by Br˙ via the THF-Br˙ intermediate 12, generating the THF˙ 13 while liberating HBr that is trapped by K2HPO4. The resulting THF˙ binds to doublet 11 to give intermediate 14 in a highly exergonic and barrierless step. Finally, the last step, reductive elimination from 14, liberates the Csp3–Csp3 coupling product with regeneration of the nickel catalyst via an energy barrier of 16.0 kcal mol−1. In line with this hypothesis, we have recently proposed a very similar mechanism, involving an EnT step, through a combined spectroscopic and computational analysis of the Ir/Ni dual catalysed α-oxy-Csp3–H alkylation.94
An alternative mechanism has been recently proposed by Chen and Shaik to explain Molander's experimental results (Fig. 4).47 Using CASSFT/CASPT2 and single-reference couple-cluster calculations they excluded both the mechanisms proposed by Molander and Doyle. They concluded that the energy required for homolysis of the Ni–Br bond of 10 is higher than the energy range provided by the excited *IrIII. They also ruled out the mechanism via a four-membered cyclic transition state involving Ni–Br–H–C, previously proposed by Molander. They also stated that Br˙ is not able to activate the α-oxy Csp3–H bond of THF via H-atom abstraction, despite the large number of photocatalysed halogen-radical mediated HAT for C–H bond functionalizations reported in the literature.95 To explain the experimental results they introduced a new mechanistic scenario involving valence inverted reactivity upon excitation of 10 (Fig. 4.) According to this proposal, d–d excited states by double electron excitation of 10 lead to the tricoordinate complex *10a, presenting a pyridine moiety of the bpy ligand dissociated from Ni. A THF molecule occupies the resulting vacant site on NiII and undergoes facile α-oxy-Csp3–H activation by the dangling pyridine group. This step takes place via concerted metal to ligand electron transfer and metalation-deprotonation, termed as ET-CMD. The resulting NiIII intermediate 17 undergoes fast reductive elimination, leading to the NiII-complex 18 having the Csp3–Ar product. The catalytic cycle is closed by deprotonation of the bpy ligand by K2HPO4via intermediates 19 and 20 (Fig. 4). Noteworthy, this study reported a unique mechanism in contrast with all the other mechanisms proposed for the same reaction by Molander,68 Doyle,69 and our group.48 One weakness of this mechanism is that the energy transfer step is unclear. Since intermediate 10 is a visible light active species,96,97 its direct excitation can also be operative to achieve targeted excited states. This point was not discussed thoroughly in ref. 47.
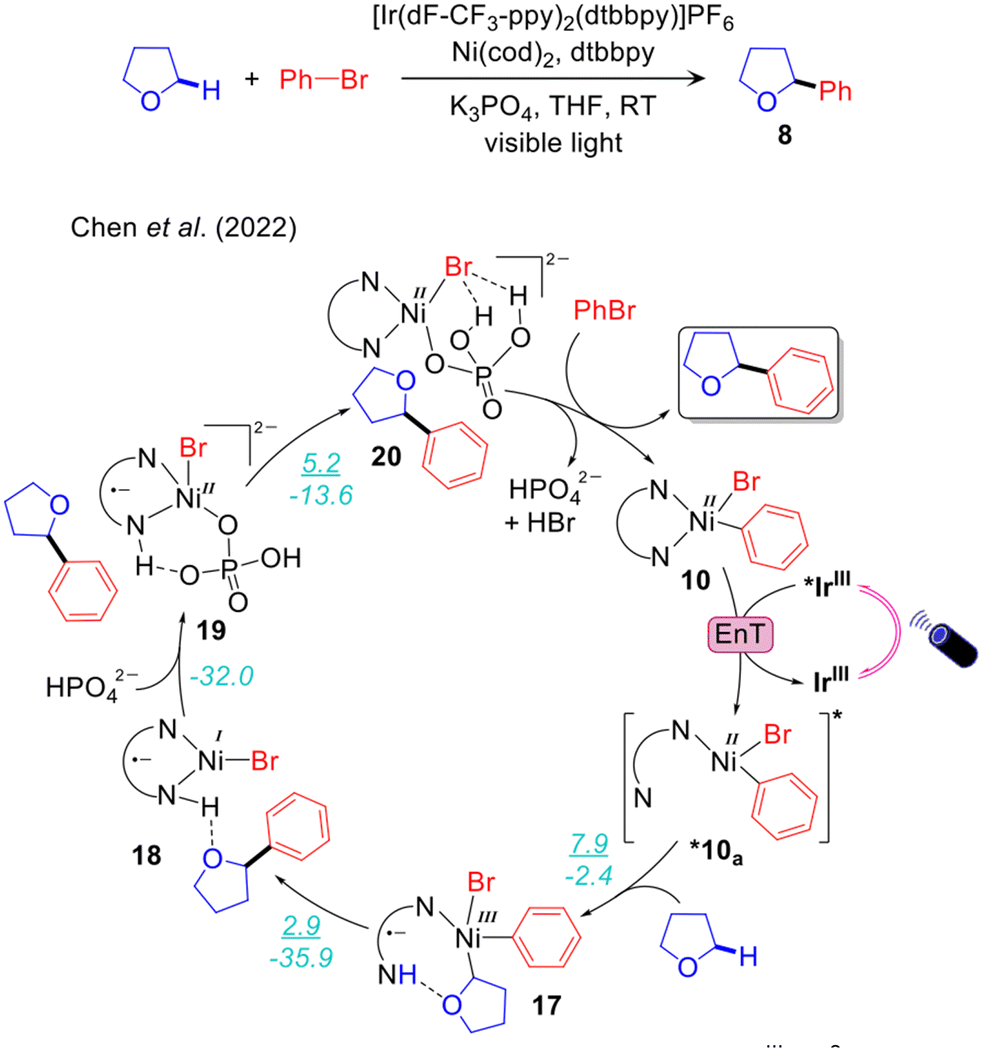 | ||
| Fig. 4 Computed mechanism of visible light-driven IrIII/Ni0 catalysed α-oxy-Csp3–H arylation via valence inverted reactivity. (ΔG‡/ΔG) represents the energy activation barrier and the thermodynamics of the given elementary step, in kcal mol−1. | ||
Furthermore, it is worth noting that different active spaces were selected in the two studies, as the focus was on the Ni–halogen bond homolysis in our work and Ni–N(dtbpy) dissociation in the work reported by Chen. This resulted in a clear difference in the energy of the Ni(I) complex and bromine radical relative to the ground state (59.9 kcal mol−1 in ref. 48 and 65.3 kcal mol−1 in ref. 47). It also underlines the importance of appropriately selecting the active space to match the chemistry of interest, and straightforward comparison of the results achieved with different active spaces should be avoided.
A combined experimental and computational mechanistic study on the IrIII/NiII-catalysed alkane-Csp3–H bond acylation has been performed by Baik and Hong.89 For the same acylation reaction they proposed entirely different mechanisms, depending on the specific electrophile used, N-acylsuccinimide or acyl chloride. In the case of N-acylsuccinimide alkane-Csp3–H activation occurs prior to oxidative addition, while the opposite sequence was proposed for acyl chloride. This result underlines again one of the additional complexities of visible light-induced cross-coupling. The sequence through which different elementary steps compose the overall reaction pathway is system dependent. The complete reaction pathway for the alkane-Csp3–H activation with N-acylsuccinimide is illustrated in Fig. 5. The catalytic cycle starts with the exergonic 1e oxidation of the LNiIICl2 complex 23 to the LNiIIICl3 complex 24. The resulting NiIII-complex is photoactive in the UV-Vis region, and thus can be promoted to the excited state *24. Based on Mulliken spin density and computed excitations using TDDFT calculation on 24, they predicted the ability of *24 in activating the alkane-Csp3–H bond. They also unsuccessfully tried to model both *24 and the subsequent excited state transition state.89 As anticipated in Section 2, modeling accurately excited state reactivity is one of challenges that have to be solved for more insightful computational mechanistic studies in photoredox catalysis. Despite these difficulties they were able to estimate a highly exergonic C–H bond activation step resulting in 25. Two successive 1e reduction of 25 by IrII leads to the NiII-alkyl complex 26 first, and subsequently to the NiI-alkyl complex 27 with elimination of Cl− and coordination of the electrophilic N-acylsuccinimide coupling partner. Complex 27 undergoes oxidative addition of N-acylsuccinimide to afford the NiIII-complex 28. Finally, reductive elimination from 28 liberates the desired product and generates the NiI-intermediate 29, which is further oxidized to complete the catalytic cycle.
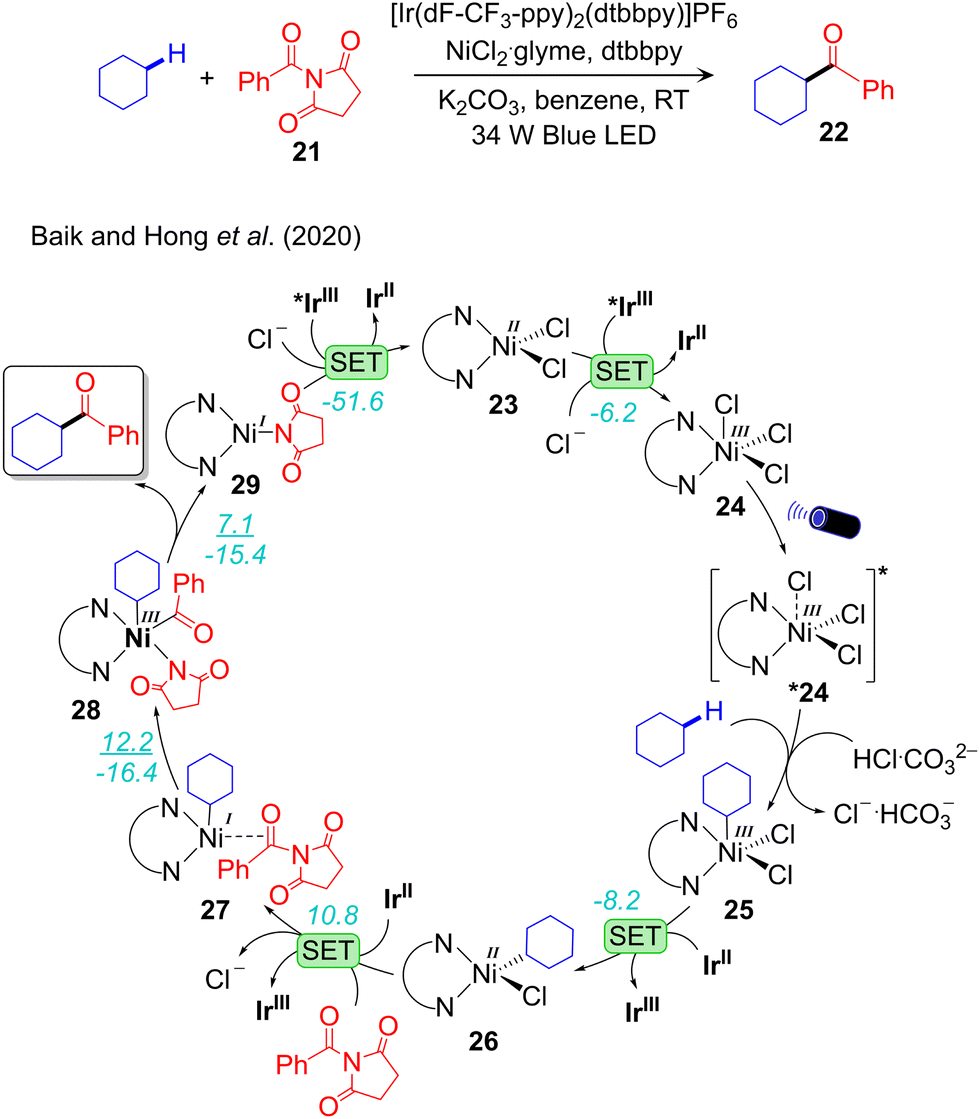 | ||
| Fig. 5 Computed mechanism of visible light-driven IrIII/NiII dual catalysed alkane-Csp3–H acylation. (ΔG‡/ΔG) represent the energy activation barrier and the thermodynamics of the given elementary step, in kcal mol−1. | ||
Additional mechanistic information on visible light-induced PC/TM dual catalysed Csp3–C cross-coupling reactivity was provided by Rand and Montgomery, who proposed a model to elucidate the mechanism and origin of regioselectivity in the metallaphotoredox-catalysed α-arylation of N-alkylbenzamides using aryl bromides. They performed NMR titration experiments and Stern–Volmer quenching studies for the identical catalytic system, IrIII/NiII, employed to arylate the amido-Csp3–H bond.98 They defined that the active complex in the Ni-catalytic cycle, LNiIBr, captures the radical species affording the LNiII(alkyl)Br complex 33 (Fig. 6). In the next step 33 is reduced to LNiI(alkyl) 34 by IrII with the generation of Br−. The resulting Br− is further oxidized by *IrIII to produce Br˙, which is then used to activate the Csp3–H bond in an outer-sphere mechanism. The oxidative addition of PhBr to 34 results in LNiIII(Alk)(Ph)Br 35, which readily undergoes reductive elimination to liberate the desired product and regenerate the LNiIBr. Recently, we provided a similar mechanistic model for the IrIII/NiII dual catalysed arylation of the allylic-Csp3–H bond.84 They also defined that the LNiII(allyl)Br is reduced to LNiI(allyl) by IrII with the generation of Br− which is further oxidized by *IrIII to produce Br˙ that activates the allylic-Csp3–H bond. The calculated mechanistic pathways indicate oxidative addition of the electrophile coupling partner to the NiI-complex as the rate-limiting step. This is consistent with the previously described mechanism outlined by Baik and Hong.89
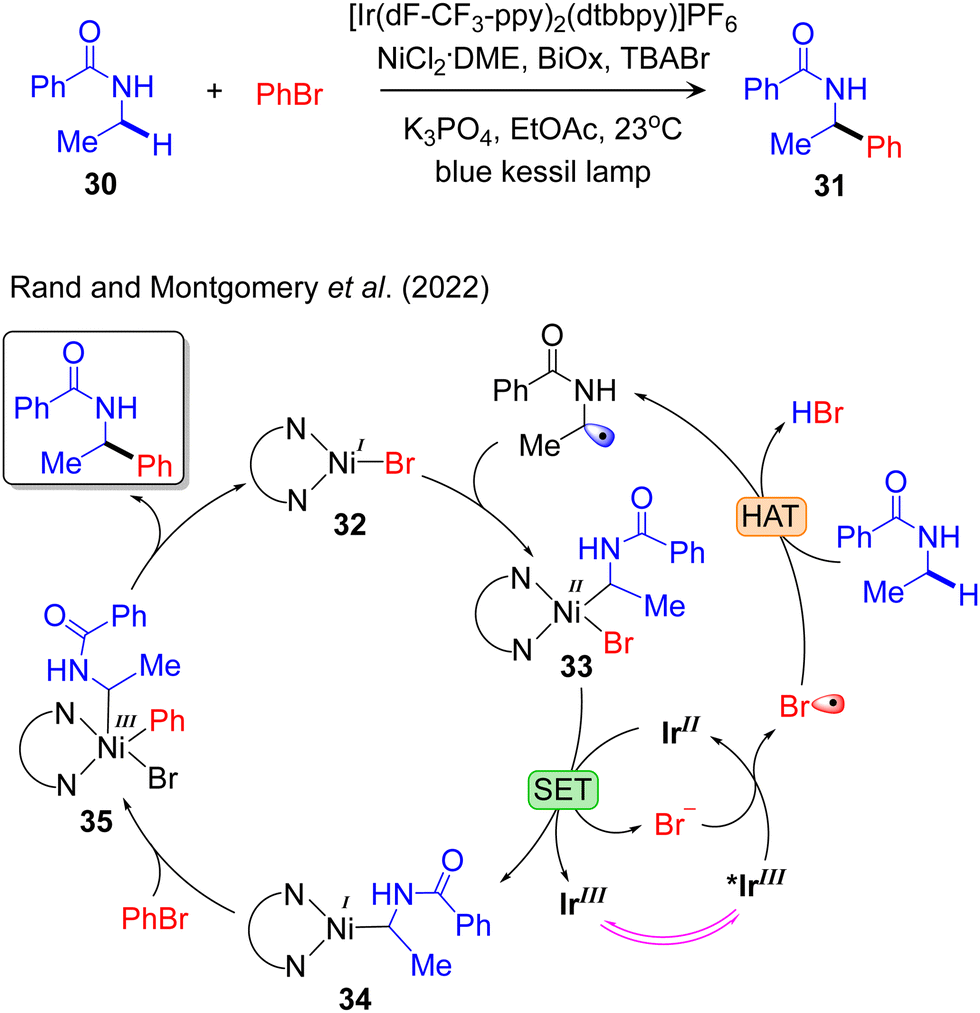 | ||
| Fig. 6 Proposed mechanism of visible light-driven IrIII/NiII dual catalysed amido-Csp3–H arylation. | ||
The above studies showcase the importance of synergic experimental-computational studies to achieve clear mechanistic understanding. Furthermore, converging and intersecting conclusions from different studies allow developing a comprehensive vision of the field.
3.3. PC-HAT/TM dual catalysed reactions
The outline of this protocol is shown in Scheme 3c, and representative catalysed reactions are given in Scheme 6. In this protocol the visible light-excited photocatalyst participates in a HAT reaction with the nucleophilic cross-coupling partner to activate the Csp3–H bond.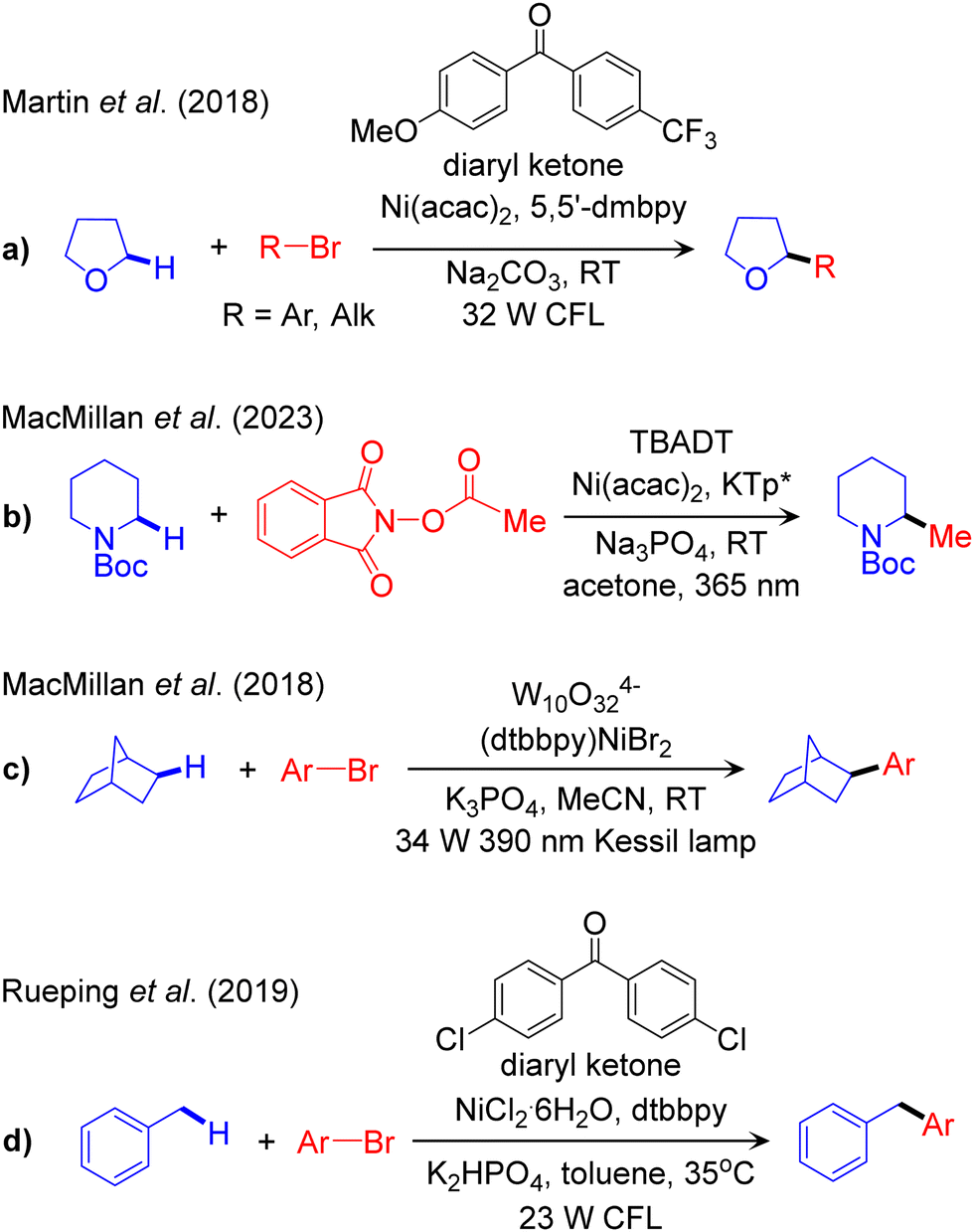 | ||
| Scheme 6 Representative reaction methodologies of visible light induced PC-HAT/TM dual catalysed Csp3–C cross-couplings of Csp3–H bonds. | ||
The first report of this scheme was published in 2018 by Martin, targeting both arylation and alkylation of α-oxy-Csp3–H bonds catalysed by a diaryl ketone as the PC and Ni as the TM catalyst (Scheme 6a).99 Recently, Wang and Kong expanded the scope of this protocol through incorporation of a chiral ligand to control the enantioselectivity of the arylation reaction.100 The first report of α-amino-Csp3–H cross coupling using the PC-HAT/TM dual catalysis strategy was recently published by MacMillan using decatungstate W10O324− (DT) as the photocatalyst.101 Using tetrabutylammonium decatungstate (TBADT) as the PC and nickel catalysis based on potassium-tris(3,5-dimethyl-1-pyrazolyl) borate (KTp*) as the source of the Ni ligand, he was able to achieve methylation of α-amino-Csp3–H bonds (Scheme 6b).101
As for the activation of unfunctionalized Csp3–H bonds, MacMillan reported the arylation of alkane-Csp3–H bonds in the presence of sodium decatungstate (NaDT) as the photocatalyst (Scheme 6c).102 Replacing the Ni catalyst by a Cu based catalyst allowed expanding the scope to the NaDT/Cu promoted trifluoro methylation of aliphatic-Csp3–H bonds.103 The NaDT/Ni platform was also used by Wang and Ackermann to promote the three-component carboacylation of various Csp3–H bonds in alkanes and aldehydes using acyl chlorides.104 They proposed that the photoexcited NaDT, resulting from visible light irradiation, is competent for abstracting a H-atom from a non-activated C–H bond of aldehydes or hydrocarbons. Similarly, Noël and co-workers used the PC-HAT/TM dual catalysis strategy based on TBADT/Ni for the rapid acylation and arylation of Csp3–H bonds in unfunctionalized alkyl derivatives in flow.105 They predicted the rate-determining step involves HAT, in accordance with kinetic isotope effect (KIE) measurements. Finally, arylation of benzylic-Csp3–H bonds was achieved by Rueping using the diaryl ketone/Ni strategy (Scheme 6d).106 They further extended the scope to the acylation of benzylic Csp3–H bonds using the same catalytic scheme.107
Aside of these advancements in scope, experimental mechanistic studies were performed to shed light on the underlying reaction pathway. Martin proposed a mechanism for the diaryl ketone/NiII(acac)2 catalysed arylation of the Csp3–H bond based on a series of control experiments.99 They imagined five different scenarios of Csp3–H activation steps via SET, ET, CT, σ-bond metathesis, and HAT, and concluded that HAT by the ketone in the excited state is viable. This hypothesis was supported by a productivity control experiment employing LNiII(aryl)Br and diaryl ketone under visible light irradiation. In a similar reaction, the arylation of a benzylic-Csp3–H bond catalysed by diaryl ketone/NiII, Rueping proposed that the photocatalyst plays a dual role, getting involved in both HAT and EnT.106
A synergic experimental/computational mechanistic study of the protocol developed by MacMillan for alkanes Csp3–H arylation, catalysed by W10O324−/NiII (Scheme 6c)102 was reported by our group (Fig. 7).36 The combined TDDFT and natural bond orbitals (NBO) analysis indicated that a ligand-to-metal charge transfer (LMCT) state of W10O324− provides active oxygen centres. Of note, the bridging oxygens were predicted to be more active than the terminal ones. Consequently, one of the bridging oxygen atoms activates the Csp3–H bond via HAT. This results in W10O325−H+ and liberates the alkyl radical 39, to be used in the nickel catalytic cycle. Disproportionation of W10O325−H+ results in W10O326−2H+, which reduces nickel from NiII in 41 to NiI in 42 inside the nickel cycle. Four different mechanisms were considered for the nickel cycle and two of them were identified to be operative in this reaction scheme. Along one pathway the reaction starts with 1e reduction of 40 followed by radical addition, while the opposite sequence was observed in the other possible pathway. Both pathways merged into the common NiI(alkyl) intermediate 42. In the progress of the reaction, the oxidative addition of ArBr to 42 leads to the NiIII-complex 44, which undergoes reductive elimination to liberate the desired product, and regenerates LNiIBr to initiate a new cycle. The conclusion that two reaction pathways are energetically feasible, one involving the NiI–Ni0–NiI–NiIII oxidation states sequence and the other the NiI–NiIII–NiI–NiIII one, highlights the importance of computational studies to achieve a clear understanding of this chemistry. Finally, oxidative addition of aryl bromide to a NiI-alkyl was calculated as the rate-limiting step (Fig. 7). Guan and Su defined a very similar mechanism in the same reaction system.108 In contrast to the conclusion made by us they claimed that the rate-limiting event is reductive elimination of the product instead of oxidative addition of the electrophilic coupling partner.
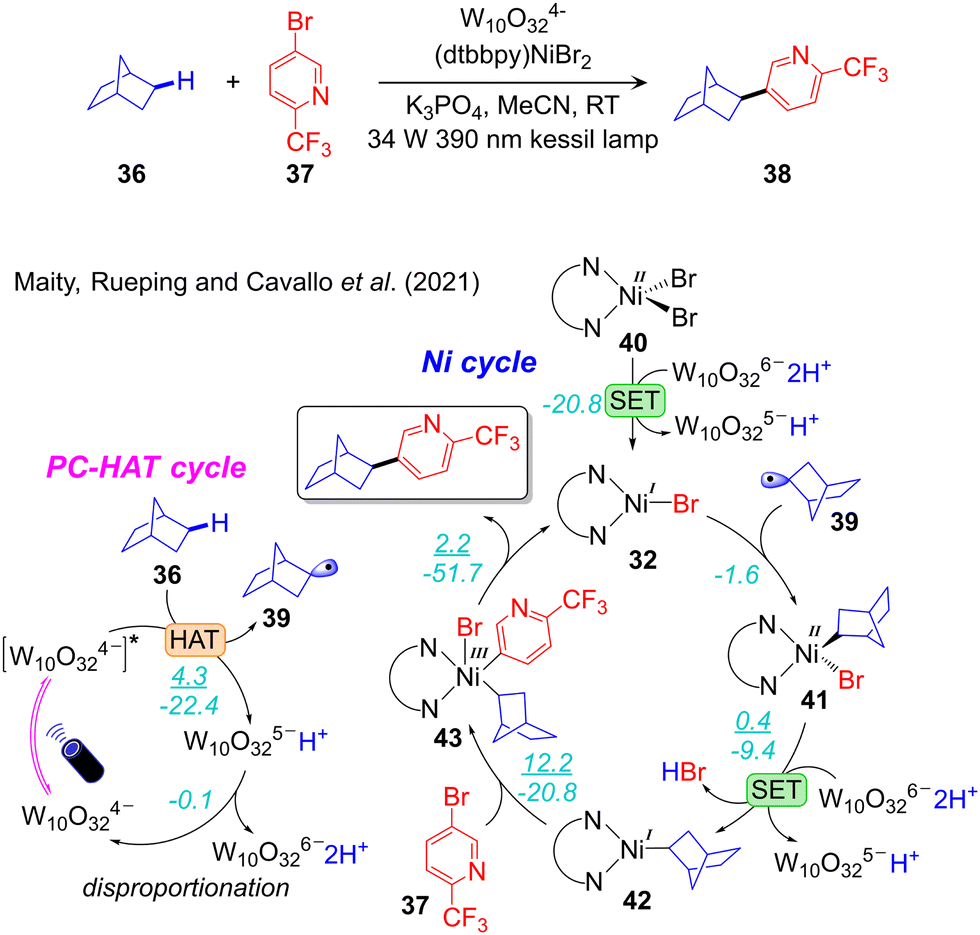 | ||
| Fig. 7 Computed mechanism of visible light-driven W10O324−/NiII catalysed alkane-Csp3–H arylation. (ΔG‡/ΔG) represent the energy activation barrier and the thermodynamics of the given elementary step, in kcal mol−1. | ||
Very recently, a similar Ni catalytic cycle has been developed by Wang and Kong to explain the enantioselective arylation of α-oxy-Csp3–H bond catalysed by diaryl ketone/NiII.100 They proposed that the reaction proceeds via SET-RA-OA-RE sequence, which is consistent with both reports by us36 and Su.108 Their calculated energy profile indicates that the oxidative addition is rate-limiting, which is in line with the mechanism proposed by our group. Based on steric effect arguments,109,110 they have explained that enantioselectivity is controlled at the step involving radical addition to LNiIBr.100
3.4. PC/HAT/TM triple catalysed reactions
The outline of this protocol is shown in Scheme 3d, and representative catalysed reactions are listed in Scheme 7. In this catalytic scheme an external HAT catalyst is used in addition to a photocatalyst and transition metal catalyst. Therefore, it is a triple catalytic system, with the corresponding three catalytic cycles, giving more flexibility in reactivity design and functionalization of Csp3–H bonds. Of note, ABCO (1-azabicyclo[2.2.2]octane), also known as quinuclidine, and DABCO (1,4-diazabicyclo[2.2.2]octane) and their derivatives are widely used as HAT catalysts in photo-induced reactions.111,112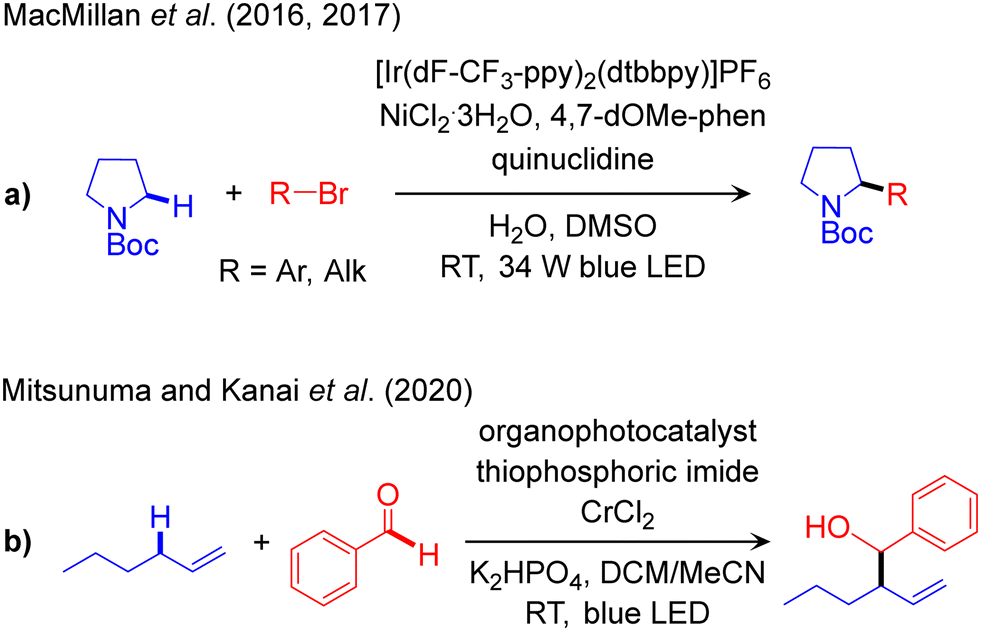 | ||
| Scheme 7 Representative reaction methodologies of visible light induced PC/HAT/TM triple catalysed Csp3–C cross-couplings of Csp3–H bonds. | ||
The first report using this methodology has been documented by MacMillan for the arylation of α-amino-Csp3–H bonds using the IrIII/Q/NiII (Q: quinuclidine derivatives as HAT catalyst) triple catalytic system (Scheme 7a).113 The same catalyst platform was later used to promote alkylation reactions.114 Mitsunuma and Kanai established another triple catalyst system incorporating an acridinium salt as the photoredox catalyst, thiophosphoric imide as the HAT catalyst, and CrCl2 to perform the Csp3–Csp2 cross-coupling of allylic-Csp3–H bonds with aldehydes as coupling partners (Scheme 7b).115 Recently, Houk and Glorius utilized a similar catalytic protocol to achieve arylation of allylic-Csp3–H bonds using a Ni-complex instead of the Cr salt.116
Computational work, and ad hoc control experiments, were performed by our group to elucidate the overall reactivity scenario operative in the α-amino-Csp3–H arylation and alkylation reactions catalysed by the IrIII/Q/NiII triple catalysts system (Fig. 8)29 developed by MacMillan (Scheme 7a).113,114 The three catalytic cycles and their connections were analysed, and the same mechanism was proposed to be operative both for arylation and alkylation reactions. Regarding the photoredox cycle, the IrIII-PC can be excited to the singlet metal-to-ligand charge-transfer 1MLCT state under visible light irradiation. Then, inter system crossing transforms the singlet 1MLCT state into the triplet 3MLCT state TIrIII, which is involved in the catalytic process. Depending on the reaction conditions two alternative pathways are possible: (i) oxidation of TIrIII to a IrIV species, followed by its reduction to IrIII, referred to as an oxidative quenching cycle; and (ii) the opposite sequence, with TIrIII reduction to IrII, followed by its oxidation to IrIII, referred to as the reductive quenching cycle. DFT calculations, supported by quenching experiments, indicated that both reductive and oxidative quenching cycles are possible under these reaction conditions. Along the reductive cycle quinuclidine (Q) acts as a quencher by 1e reduction of the photo excited IrIII PC to a IrII state, whereas the NiII-complex functions as a quencher in the oxidative cycle by 1e oxidation of the photo excited IrIII PC to a IrIV state. However, the reductive cycle was predicted to be operative, considering the much higher concentration of Q compared to that of the NiII-catalyst in the developed protocol. In the HAT cycle they proposed, the visible light excited *IrIII oxidizes the HAT catalyst 46 to generate the radical cation species 47 and closing the HAT cycle. Consistent with experimental observations, calculations indicated that the N-centered radical 47 is highly selective towards activation of α-Csp3–H bonds, compared to β-Csp3–H bonds, of the amino coupling partner 44. Within the PC cycle the reduced form of the photocatalyst, IrII, is oxidized by the LNiIIBr2 complex 40, affording the LNiIBr complex 32 that initiates the nickel catalytic cycle.
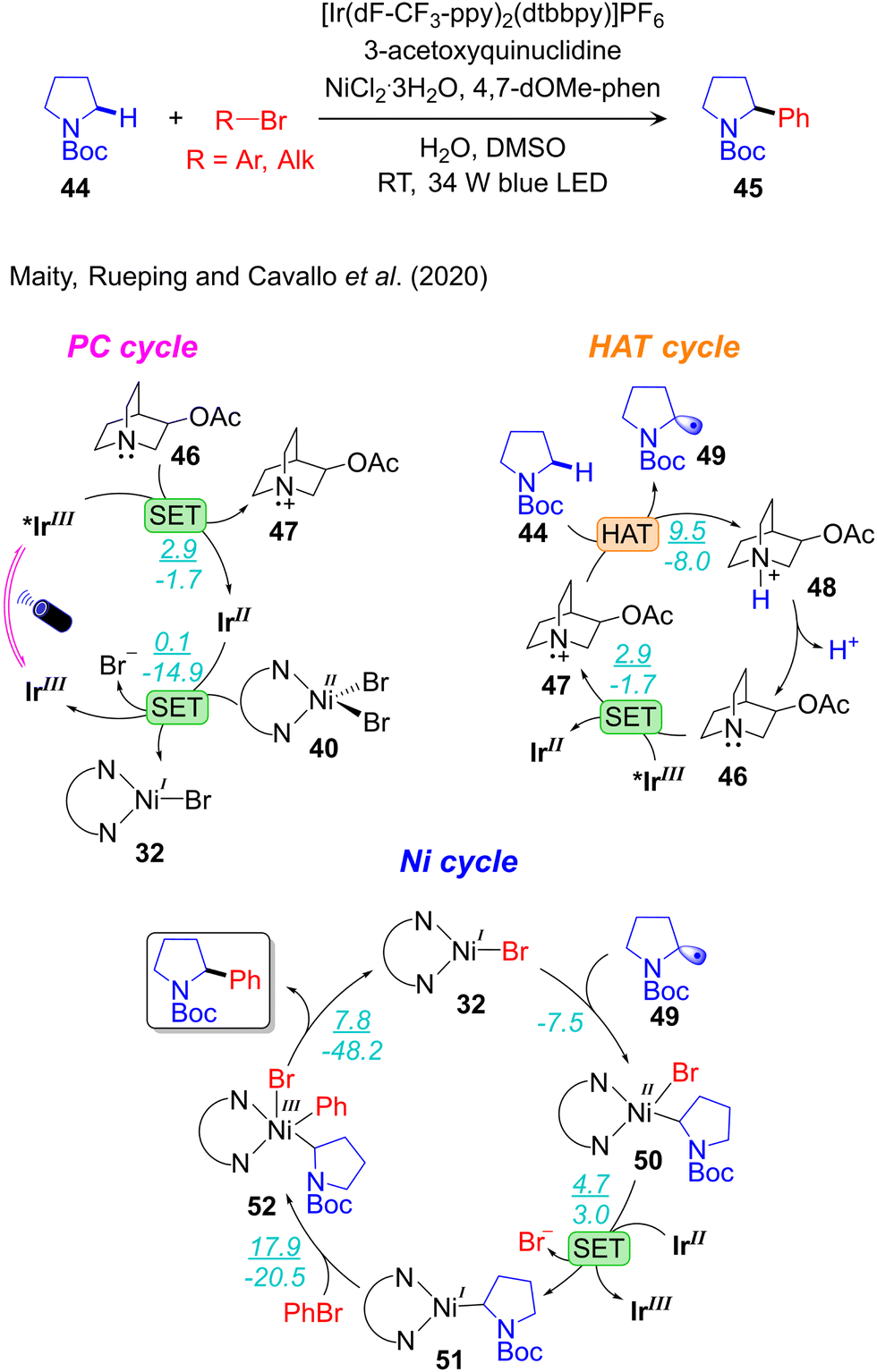 | ||
| Fig. 8 Computed mechanism of visible light-driven IrIII/Q/NiII triple catalysed α-amino-Csp3–H arylation. (ΔG‡/ΔG) represent the energy values in kcal mol−1. | ||
Four different pathways were composed for the Ni-cycle by varying the sequence of fundamental steps: oxidative addition (OA), radical addition (RA), single electron transfer (SET), and reductive elimination (RE). The conclusion was that two pathways are viable: one via the nickel oxidation states NiI–Ni0–NiI–NiIII with the sequence of elementary steps SET-RA-OA-RE, the other via the NiI–NiII–NiI–NiIII and RA-SET-OA-RE sequences. The latter was predicted to be energetically preferred. The two other reaction pathways via NiI–Ni0–NiII–NiIII proposed originally by MacMillan,113 and NiI–NiIII–NiII–NiIII can be excluded because of the higher energetics and supported by control experiments.29 Along the most feasible pathway the reaction starts with barrierless radical addition of 49 to the LNiIBr complex 32, resulting in the LNiII(Alk)Br intermediate 50 (Fig. 8). Slightly endergonic 1e reduction of 50 by IrII generates the NiI-complex 51 that undergoes oxidative addition of PhBr, leading to the LNiIII(Alk)(Ph)Br intermediate 52. Finally, highly facile reductive elimination from 52 liberates the product and regenerates the starting LNiIBr complex 32. The calculated results clearly indicated that oxidative addition of PhBr to the NiI(Alk) 51 is the rate-limiting step, which is consistent with other reports.29,36,89,100
Houk and Glorius performed a similar study to expound the reaction mechanism operative with the triple catalysts system they developed to arylate allylic-Csp3–H bonds.116 They also considered four different pathways for the Ni-catalytic cycle and identified the most feasible one involving the NiI–NiIII–NiII–NiIII oxidation states sequence, with the corresponding OA-SET-RA-RE sequence of elementary steps. Consistently with the work reports,29,36 they excluded participation of the Ni0 oxidation state, found a NiI-species serving as an active catalyst, and determined that oxidative addition of ArBr to the NiI-species is the rate-limiting step.29 The only discrepancy between the two studies is in the exact definition of the Ni-complex involved in the 1e reduction step in the PC cycle, which can be easily explained considering the different reduction potential of the used photocatalysts and the different stabilities of Ni-allyl and Ni-alkyl complexes.
Of note, nickel is the metal mostly used when designing a cross-coupling catalyst for visible light-induced catalysis, due to the existence of multiple oxidation states with small energy differences. Furthermore, oxidation states +1 and +2 of nickel are highly efficient radical capturing species.
4. Outlook
Visible light-induced transition metal catalysis has emerged as one the most powerful strategies to solve long-standing challenges in catalysis. The potential of this chemical technology is enormous, and the scope is broad. However, the advancements achieved so far have been more the results of brilliant intuitions of experimental chemists rather than the consequences of hypotheses cast on detailed mechanistic knowledge. Several reasons explain this limited understanding, among them: (i) the chemistry involved is extremely complex, with up to three catalytic cycles working together, as described in Section 3.4; (ii) reactions often proceed through unstable metal oxidation states, which limits the characterization of the intermediates involved in catalytic pathways; (iii) reactions involve systems in excited electronic states, whose photophysical characterization and photochemical behaviour are very hard to achieve experimentally; (iv) reactions often involve alkyl radical intermediates that, by definition, are highly reactive. The emerging scenario is a very complex reactivity pool with multiple mechanistic ways off, either leading to catalyst deactivation or to unwanted products, decreasing selectivity (Scheme 8). This explains the experimental and computational efforts dedicated to deciphering the undergoing reaction mechanisms. The field is large and to preserve conciseness we focused on visible light-induced transition metal catalysed Csp3–H functionalization to construct Csp3–C bonds. This chemical transformation is one of the nearly impossible tasks for ground state cross-coupling protocols.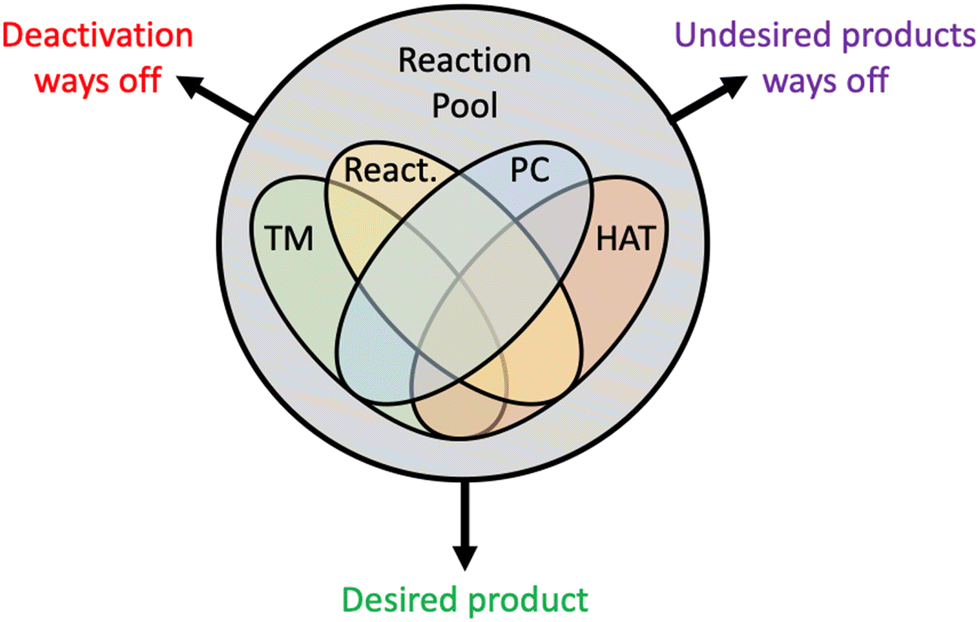 | ||
| Scheme 8 Cartoon representation showing the complexity of the reaction pool in visible light-induced PC/HAT/TM catalysed cross-couplings. Different zones include all possible intermediates involving TM, PC, HAT and reactants (React.) species and their dual, triple and quadruple combinations. | ||
The above chemical complexity, together with the experimental and computational challenges in investigating this reactivity, is certainly slowing the development of novel protocols. While we do not comment on the experimental challenges, we remark again that conventional DFT methods fail to model the reactivity of excited states. In principle, TDDFT could allow having an approximate description of excited state reactivity. However, attempts made by some groups currently failed. TDDFT also has intrinsic limitations in the description of multi-electron excited electronic states, which requires using far more expensive multireference methods if accurate description of the electronic structure is needed. This is in striking contrast with ground state cross-coupling reactivity, a perfectly suitable playground for conventional DFT, which has indeed been used to explain almost every fine details of this chemistry. The development of fast computational methods for accurate description of excited state reactivity would allow achieving a comprehension comparable to what we have for ground state cross-coupling.
Regarding the chemical challenges, the currently developed protocols have been largely used for the activation of relatively weaker Csp3–H bonds, approximately around 90 kcal mol−1, mostly using the PC/TM dual catalysis strategy merging the Ir-based photocatalyst and Ni-catalyst system. Development in this area could involve replacing the Ir photocatalyst with organic photocatalysts, or using lower energy visible light, considering that most current protocols use blue light. Another important direction is developing more protocols capable of activating stronger Csp3–H bonds, approximately in the 95–100 kcal mol−1 range characterizing alkanes. The goal being the strongest Csp3–H bonds, such as those of methane or cyclopropanes, beyond bond energies of 100 kcal mol−1. Having in hand effective protocols for the activation of unfunctionalized alkanes will open new challenges connected to the capability of activating selectively the desired Csp3–H bond. Most of the functionalized substrates used so far have one clearly weaker Csp3–H bond, which results in its selective activation controlled by the difference in strength with the other Csp3–H bonds in the molecule. Achieving similar selectivity with unfunctionalized alkanes will require accurate design of the photocatalyst and TM ligands, to access activation of specific Csp3–H bonds also based on steric differences between them. Solving all these challenges will remarkably benefit from a more comprehensive understanding of the reactivity scenario operative with the different protocols, which we tried to provide with this review. Ultimately, complementary experimental and computational studies will probably represent the main workhorse to shed light on this chemistry for the forthcoming years, as each of them is unfit to solve the problem alone.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia.References
- A. de Meijere and F. Diederich, Metal-catalyzed Cross-coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- Y. Nishihara, Applied Cross-coupling Reactions, Springer-Verlag, Germany, 1st edn, 2013 Search PubMed.
- J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed.
- L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- X. S. Xue, P. J. Ji, B. Y. Zhou and J. P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS.
- P. Bellotti, H. M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- D. L. Golden, S. E. Suh and S. S. Stahl, Nat. Rev. Chem., 2022, 6, 405–427 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- D. De Vos, K. Gadde and B. U. W. Maes, Synthesis, 2023, 193–231 CrossRef CAS.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- A. Shatskiy, E. V. Stepanova and M. D. Karkas, Nat. Rev. Chem., 2022, 6, 782–805 CrossRef CAS PubMed.
- S. L. Zheng, Y. Y. Hu and W. M. Yuan, Synthesis, 2021, 1719–1733 CAS.
- S. K. Kariofillis and A. G. Doyle, Acc. Chem. Res., 2021, 54, 988–1000 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- M. B. Yuan and O. Gutierrez, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1573 CAS.
- Z. H. Qi and J. Ma, ACS Catal., 2018, 8, 1456–1463 CrossRef CAS.
- B. Maity, C. Zhu, H. Yue, L. Huang, M. Harb, Y. Minenkov, M. Rueping and L. Cavallo, J. Am. Chem. Soc., 2020, 142, 16942–16952 CrossRef CAS PubMed.
- C. Zhu, H. F. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678–687 CrossRef CAS.
- M. V. Mane, S. Dutta, L. Cavallo and B. Maity, ACS Catal., 2023, 13, 6249–6260 CrossRef CAS.
- Y. J. Dong, Z. W. Zhao, Y. Geng, Z. M. Su, B. Zhu and W. Guan, Inorg. Chem., 2023, 62, 1156–1164 CrossRef CAS PubMed.
- Y. H. Liu, Y. Y. Yang, R. X. Zhu and D. J. Zhang, ACS Catal., 2020, 10, 5030–5041 CrossRef CAS.
- X. Zhao, Y. Liu, R. Zhu, C. Liu and D. Zhang, Inorg. Chem., 2019, 58, 12669–12677 CrossRef CAS PubMed.
- F. Calogero, S. Potenti, E. Bassan, A. Fermi, A. Gualandi, J. Monaldi, B. Dereli, B. Maity, L. Cavallo, P. Ceroni and P. G. Cozzi, Angew. Chem., Int. Ed., 2022, 61, e2021149 CrossRef.
- B. Maity, C. Zhu, M. Rueping and L. Cavallo, ACS Catal., 2021, 11, 13973–13982 CrossRef CAS.
- K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu and E. A. Pidko, Chem. Rev., 2019, 119, 2453–2523 CrossRef CAS PubMed.
- J. A. Pople, M. Headgordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- H. Lischka, D. Nachtigallova, A. J. A. Aquino, P. G. Szalay, F. Plasser, F. B. C. Machado and M. Barbatti, Chem. Rev., 2018, 118, 7293–7361 CrossRef CAS PubMed.
- P. G. Szalay, T. Müller, G. Gidofalvi, H. Lischka and R. Shepard, Chem. Rev., 2012, 112, 108–181 CrossRef CAS PubMed.
- P. Ma, S. Wang and H. Chen, ACS Catal., 2020, 10, 1–6 CrossRef CAS.
- L. S. Ma, W. H. Fang, L. Shen and X. B. Chen, ACS Catal., 2019, 9, 3672–3684 CrossRef CAS.
- D. A. Cagan, G. D. Stroscio, A. Q. Cusumano and R. G. Hadt, J. Phys. Chem. A, 2020, 124, 9915–9922 CrossRef CAS PubMed.
- D. A. Cagan, D. Bím, B. Silva, N. P. Kazmierczak, B. J. McNicholas and R. G. Hadt, J. Am. Chem. Soc., 2022, 144, 6516–6531 CrossRef CAS PubMed.
- S. H. Wang, P. C. Ma, S. Shaik and H. Chen, J. Am. Chem. Soc., 2022, 14607–14613 CrossRef CAS PubMed.
- B. Maity, T. R. Scott, G. D. Stroscio, L. Gagliardi and L. Cavallo, ACS Catal., 2022, 12, 13215–13224 CrossRef CAS.
- G. L. Manni, R. K. Carlson, S. Luo, D. Ma, J. Olsen, D. G. Truhlar and L. Gagliardi, J. Chem. Theory Comput., 2014, 10, 3669–3680 CrossRef PubMed.
- R. K. Carlson, G. L. Manni, A. L. Sonnenberger, L. Gagliardi and D. Truhlar, J. Chem. Theory Comput., 2015, 11, 82–90 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1957, 26, 872–877 CrossRef CAS.
- N. S. Hush, J. Chem. Phys., 1958, 28, 962–972 CrossRef CAS.
- N. S. Hush, Trans. Faraday Soc., 1961, 57, 557–580 RSC.
- W. J. Zhou, G. M. Cao, G. Shen, X. Y. Zhu, Y. Y. Gui, J. H. Ye, L. Sun, L. L. Liao, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed.
- Z. S. Cao, J. Y. Li, Y. W. Sun, H. W. Zhang, X. L. Mo, X. Cao and G. Z. Zhang, Chem. Sci., 2021, 12, 4836–4840 RSC.
- C. Y. Huang, J. B. Li and C. J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- Y. Mao, Y. Q. Liu, L. Yu, S. Y. Ni, Y. Wang and Y. Pan, Org. Chem. Front., 2021, 8, 5968–5974 RSC.
- R. Kancherla, K. Muralirajan, B. Maity, C. Zhu, P. E. Krach, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3412–3416 CrossRef CAS PubMed.
- M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem. – Eur. J., 2012, 18, 5170–5174 CrossRef CAS PubMed.
- W. J. Fu, W. B. Guo, G. L. Zou and C. Xu, J. Fluorine Chem., 2012, 140, 88–94 CrossRef CAS.
- I. Perepichka, S. Kundu, Z. Hearne and C. J. Li, Org. Biomol. Chem., 2015, 13, 447–451 RSC.
- M. R. Patil, J. Shah, A. V. Kumar and A. R. Kapdi, Chem. – Asian J., 2020, 15, 4302–4306 CrossRef CAS PubMed.
- D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC.
- X. M. Shu, D. Zhong, Y. M. Lin, X. Qin and H. H. Huo, J. Am. Chem. Soc., 2022, 144, 8797–8806 CrossRef CAS PubMed.
- A. W. Rand, H. F. Yin, L. Xu, J. Giacoboni, R. Martin-Montero, C. Romano, J. Montgomery and R. Martin, ACS Catal., 2020, 10, 4671–4676 CrossRef CAS.
- S. Das, K. Murugesan, G. J. V. Rodríguez, J. Kaur, J. P. Barham, A. Savateev, M. Antonietti and B. König, ACS Catal., 2021, 11, 1593–1603 CrossRef CAS.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- M. K. Nielsen, B. J. Shields, J. Y. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef CAS PubMed.
- H. P. Deng, X. Z. Fan, Z. H. Chen, Q. H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed.
- B. Kang and S. H. Hong, Chem. Sci., 2017, 8, 6613–6618 RSC.
- Z. D. Sun, N. Kumagai and M. Shibasaki, Org. Lett., 2017, 19, 3727–3730 CrossRef CAS PubMed.
- M. S. Santos, A. G. Corrêa, M. W. Paixão and B. König, Adv. Synth. Catal., 2020, 362, 2367–2372 CrossRef CAS.
- X. K. Cheng, H. Z. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed.
- Q. L. Wang, Z. Z. Sun, H. W. Huang, G. J. Mao and G. J. Deng, Green Chem., 2022, 24, 3293–3299 RSC.
- W. Kim, J. Koo and H. G. Lee, Chem. Sci., 2021, 12, 4119–4125 RSC.
- X. Cheng, T. Li, Y. Liu and Z. Lu, ACS Catal., 2021, 11, 11059–11065 CrossRef CAS.
- J. Xu, Z. Li, Y. Xu, X. Shu and H. Huo, ACS Catal., 2021, 11, 13567–13574 CrossRef CAS.
- T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed.
- O. M. Griffiths, H. A. Esteves, Y. D. Chen, K. Sowa, O. S. May, P. Morse, D. C. Blakemore and S. V. Ley, J. Org. Chem., 2021, 86, 13559–13571 CrossRef CAS PubMed.
- L. T. Huan, X. M. Shu, W. S. Zu, D. Zhong and H. H. Huo, Nat. Commun., 2021, 12, 3536 CrossRef CAS PubMed.
- L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed.
- L. Huang, M. Szewczyk, R. Kancherla, B. Maity, C. Zhu, L. Cavallo and M. Rueping, Nat. Commun., 2023, 14, 548 CrossRef CAS PubMed.
- M. B. Buendia, B. Higginson, S. Kegnaes, S. Kramer and R. Martin, ACS Catal., 2022, 12, 3815–3820 CrossRef CAS.
- J. L. Schwarz, F. Schafers, A. Tlahuext-Aca, L. Luckemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed.
- A. Franchino, A. Rinaldi and D. J. Dixon, RSC Adv., 2017, 7, 43655–43659 RSC.
- L. K. G. Ackerman, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- G. S. Lee, J. Won, S. Choi, M. H. Baik and S. H. Hong, Angew. Chem., Int. Ed., 2020, 59, 16933–16942 CrossRef CAS PubMed.
- G. S. Lee, B. Park and S. H. Hong, Nat. Commun., 2022, 13, 5200 CrossRef CAS PubMed.
- B. Xu and U. K. Tambar, ACS Catal., 2019, 9, 4627–4631 CrossRef CAS PubMed.
- S. M. Thullen, S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2019, 141, 14062–14067 CrossRef CAS PubMed.
- S. J. Hwang, B. L. Anderson, D. C. Powers, A. G. Maher, R. G. Hadt and D. G. Nocera, Organometallics, 2015, 34, 4766–4774 CrossRef CAS.
- R. Kancherla, K. Muralirajan, B. Maity, S. Karuthedath, G. S. Kumar, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2022, 13, 2737 CrossRef CAS PubMed.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CAS.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800–5810 CrossRef CAS PubMed.
- A. W. Rand, M. Chen and J. Montgomery, Chem. Sci., 2022, 13, 10566–10573 RSC.
- Y. Y. Shen, Y. T. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- S. Xu, Y. Ping, W. Li, H. Guo, Y. Su, Z. Li, M. Wang and W. Kong, J. Am. Chem. Soc., 2023, 145, 5231–5241 CrossRef CAS PubMed.
- E. Mao and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 2787–2793 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y. H. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- D. Y. Wang and L. Ackermann, Chem. Sci., 2022, 13, 7256–7263 RSC.
- D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noel, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS PubMed.
- A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed.
- P. E. Podsiadly, A. Dewanji, T. T. Yuan and M. Rueping, Chem. Commun., 2020, 56, 6082–6085 RSC.
- Y. J. Dong, B. Zhu, Y. J. Liang, W. Guan and Z. M. Su, Inorg. Chem., 2021, 60, 18706–18714 CrossRef CAS PubMed.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- Y. Kawamata, M. Yan, Z. Q. Liu, D. H. Bao, J. S. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- A. Matsumoto, M. Yamamoto and K. Maruoka, ACS Catal., 2022, 12, 2045–2051 CrossRef CAS.
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed.
- C. Le, Y. F. Liang, R. W. Evans, X. M. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- S. Tanabe, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 12374–12381 CrossRef CAS PubMed.
- H.-M. Huang, P. Bellotti, P.-P. Chen, K. N. Houk and F. Glorius, Nat. Synth., 2022, 1, 59–68 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |