
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbenes as privileged ligands for nickel-catalysed alkene functionalisation
Boon Chong
Lee
,
Chen-Fei
Liu
,
Leroy Qi Hao
Lin
,
Kang Zheng
Yap
,
NingXi
Song
,
Charyl Hui Min
Ko
,
Priscilla Hee
Chan
and
Ming Joo
Koh
 *
*
Department of Chemistry, National University of Singapore, 4 Science Drive 2, Singapore, 117544, Republic of Singapore. E-mail: chmkmj@nus.edu.sg
First published on 5th April 2023
Abstract
Alkene functionalisation is a powerful strategy that has enabled access to a wide array of compounds including valuable pharmaceuticals and agrochemicals. The reactivity of the alkene π-bond has allowed incorporation of a diverse range of atoms and functional groups through a wide variety of reaction pathways. N-Heterocyclic carbenes (NHCs) are a class of persistent carbenes that are widely employed as ancillary ligands due to their ability to act as strong σ-donors compared to widely-applied conventional phosphine-based ligands. NHCs are also unique as their molecular bulk provides steric influence for regio- and stereo-control in many alkene functionalisation reactions, illustrated by the examples covered in this review. A combination of the unique reactivity of NHC ligands and nickel's characteristics has facilitated the design of reaction pathways that show distinct selectivity and reactivity, including the activation of bonds previously considered “inert”, such as C–H bonds, the C–O bond of ethers and esters, and the C–N bonds of amides. This review summarises the advancements in Ni(NHC) catalysed alkene functionalisation up to 2022, covering the following major reaction classes: Heck-type reactions, hydrofunctionalisation and dicarbofunctionalisation.
 Boon Chong Lee | Boon Chong Lee was born in 1996 and raised in Singapore. In 2020, he obtained his bachelor's degree in chemistry from the National University of Singapore (NUS). In 2021, he started his PhD studies under the supervision of Prof. Ming Joo Koh at NUS. His research lies in synthetic organic chemistry and developing new methodologies for molecular functionalisation. |
 Chen-Fei Liu | Chen-Fei Liu earned his MS degree from Sichuan University under the supervision of Prof. Lin Dong. He is currently a PhD candidate at the National University of Singapore, working with his advisor Prof. Ming Joo Koh. His research focuses on base metal-catalysed alkene functionalization. |
 Leroy Qi Hao Lin | Leroy Qi Hao Lin was born in 1996 and raised in Singapore. In 2020, he obtained his bachelor's degree in chemistry from the National University of Singapore (NUS). In 20201, he started his PhD studies under the supervision of Prof. Koh Ming Joo. His research interest is focused on base metal catalysis and the novel synthesis of bioisosteres. |
 Ming Joo Koh | Ming Joo Koh is an Assistant Professor at the National University of Singapore (NUS). He received his PhD degree from Boston College in 2017 and carried out post-doctoral studies at the same institution. In 2018, he joined the Department of Chemistry at NUS as a President's Assistant Professor. His current research focuses on developing sustainable and practical catalytic solutions that address critical challenges in chemical synthesis through base metal catalysis and radical chemistry. |
1. Introduction
The field of N-heterocyclic carbene (NHC) complexes of 3d metals has grown significantly in recent years since the first synthesis of an isolable imidazol-2-ylidene by Arduengo in 1991.1 Consequently, NHCs have found numerous applications across fields such as catalysis, materials sciences and metallopharmaceuticals. Comprehensive and specialised reviews have described the synthesis, practical applications and recent developments (not limited to nickel) of NHCs including NHC-enabled organocatalysis.2–9The structure of NHCs can have a significant impact on their behaviour as ligands. Four key factors that influence this behaviour are the presence and location of heteroatoms (such as nitrogen), substituents at proximal positions, the backbone structure and the size of the ring.4 The adjacent electronegative nitrogen atoms can inductively withdraw electron density from the non-bonding lone pair at the carbenic carbon (HOMO). At the same time, nitrogen lone pairs can overlap with the empty p-orbital of the carbenic carbon as a π-electron donor. Steric bulk on the N-substituents serves to protect the reactive centre from kinetic dimerisation and decomposition whilst altering the electronic properties of the ring system. Unsaturation in the cyclic backbone provides enhanced thermodynamic stability due to the increased aromaticity of the ring.5 Different ring sizes in the backbone also have a marked influence on the overall stereoelectronic properties of the NHC ligand (Chart 1).
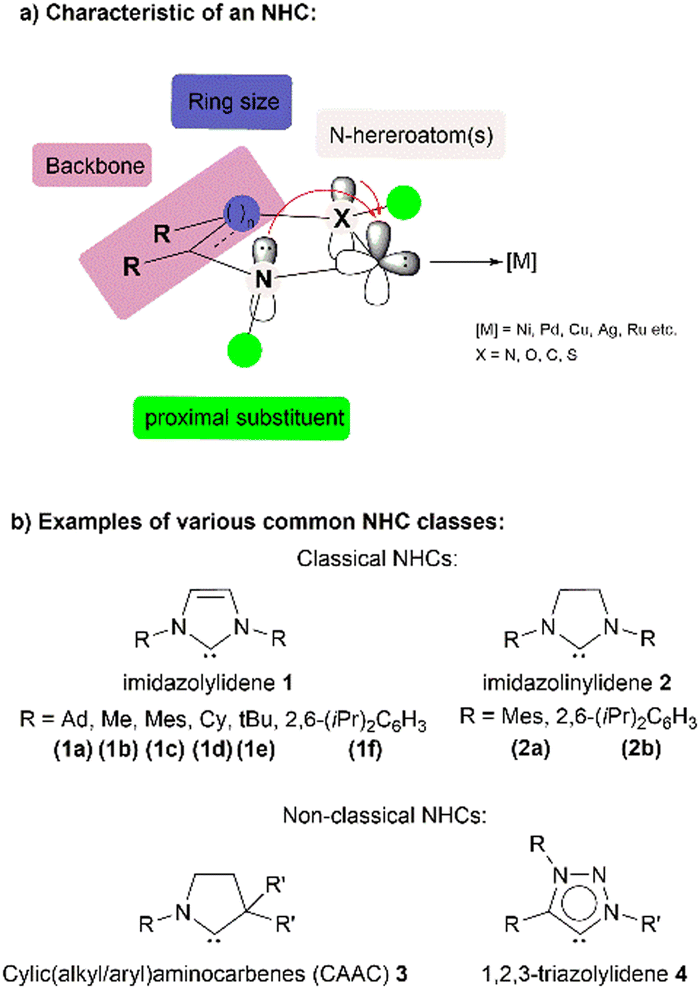 | ||
| Chart 1 Characteristic and classes of common NHCs. Ad: adamantyl, Me: methyl, Mes: mesityl, Cy: cyclohexyl, tBu: tert-butyl, and iPr: iso-propyl. | ||
Combining the abovementioned factors, NHCs are generally regarded to have a highly nucleophilic carbenic carbon atom, making them strong σ-donors that bind strongly to metal or non-metal centres. This characteristic has led to their widespread use as ligands in numerous catalytic applications. NHCs are also considered to be more electron-donating than phosphine ligands, as reflected in their lower calculated Tolman electronic parameter10 values. This results in the formation of thermodynamically stronger metal–ligand bonds in NHC–metal complexes, as reflected in the greater bond dissociation energies and shorter metal–ligand bond lengths observed compared to those of their phosphine-ligated complexes.5 This in turn confers greater thermal and oxidative stability to the Ni–NHC complex.11 In the domain of steric factors, the measurement of percentage buried volume (%Vbur) can be used to quantitatively compare the steric bulk of NHC ligands with that of phosphine ligands.12 Modelling studies on the %Vbur value of most NHC ligands have shown that they are as bulky or even more so than most regular phosphine ligands, particularly IAd (1a) and ItBu (1e). This ultimately has significant implications on the reactivity and selectivity of reactions, which we will cover in detail.
Research in this area has also expanded to include the use of non-classical NHCs (see Chart 1), most of which possess an even stronger electron-donating ability compared to their classical counterparts and have been proven to stabilise low coordination number metal (NHC) complexes, as well as reactive transient metal species. Numerous published reviews and book chapters13 have already covered the current state of research on well-defined NHC complexes of copper,14 nickel,15 cobalt,16 and iron.17 These articles extensively examine the development of these systems in terms of their structural, catalytic, and stoichiometric reactivities, as well as the aspects of their synthesis, bioinorganic chemistry, material chemistry, and photophysical properties.
Nickel, located just above palladium in the periodic table, is a group 10 metal and can perform many similar elementary reactions to palladium or platinum. Due to its smaller atomic radius, lower electronegativity, and lower redox potential, nickel exhibits faster rates of oxidative addition and slower rates of β-hydride elimination when compared to palladium in the same ligand framework.18 Thus, nickel is often used as a cost-effective alternative to its noble metal counterparts and can also offer a more facile access to multiple oxidation states (Ni0, NiI, NiII, NiIII and even NiIV), rendering radical pathways more accessible.
There are several reported methods for the synthesis of Ni–NHC complexes. These include the interaction of isolated or in situ generated NHCs with the common Ni0 and NiII precursors, Ag transmetalation protocols, and C–X (X = H, halide) bond activation methodologies.16 Rare oxidation states of nickel (i.e., NiI), proposed as reactive catalytic intermediates, can also be stabilised with various NHCs, and are usually synthesised from the disproportionation of Ni0 and NiIII precursors or single-electron reduction protocols of NiII, as described in a recent review.19 Synthesis and characterisation of various Ni(NHC) complexes of different nuclearity and oxidation states (Ni0, Nidi and NiII) are unfortunately outside the scope of this review. Nickel, being more electropositive than palladium, can under certain conditions activate bonds (C–O of ethers, C–N of amides) that are relatively inert to oxidative addition with palladium.20 The strong electron-donating properties of NHCs further facilitate this oxidative addition process, allowing Ni(NHC) catalysts to activate even more challenging C–H bonds.
Ni(NHC) complexes have a long history of use in homogenous catalysis and have been thoroughly reviewed for their use in C–C cross-couplings, C–H bond functionalisation, C–X (X = O, N, S, B) bond formation, oligo/polymerisation, and oxidation–reduction reactions.21,22 This review will not cover these previously mentioned classes of reactions. In this review, recent developments in Ni(NHC) catalysed alkene functionalisation will be discussed, mainly categorised by the three main reaction classes of alkene functionalisation: Heck-type reactions, hydrofunctionalisations and difunctionalisations (Scheme 1). The contents for this review will not include reactions with strongly activated enones, alkynes23–27 or cycloaddition-type reactions.28–31 The literature studies presented in this review are up to date until 2022. The origins of regioselectivity, diastereoselectivity and enantioselectivity in the discussed reactions will be thoroughly examined, including comprehensive mechanistic details, if deemed necessary.
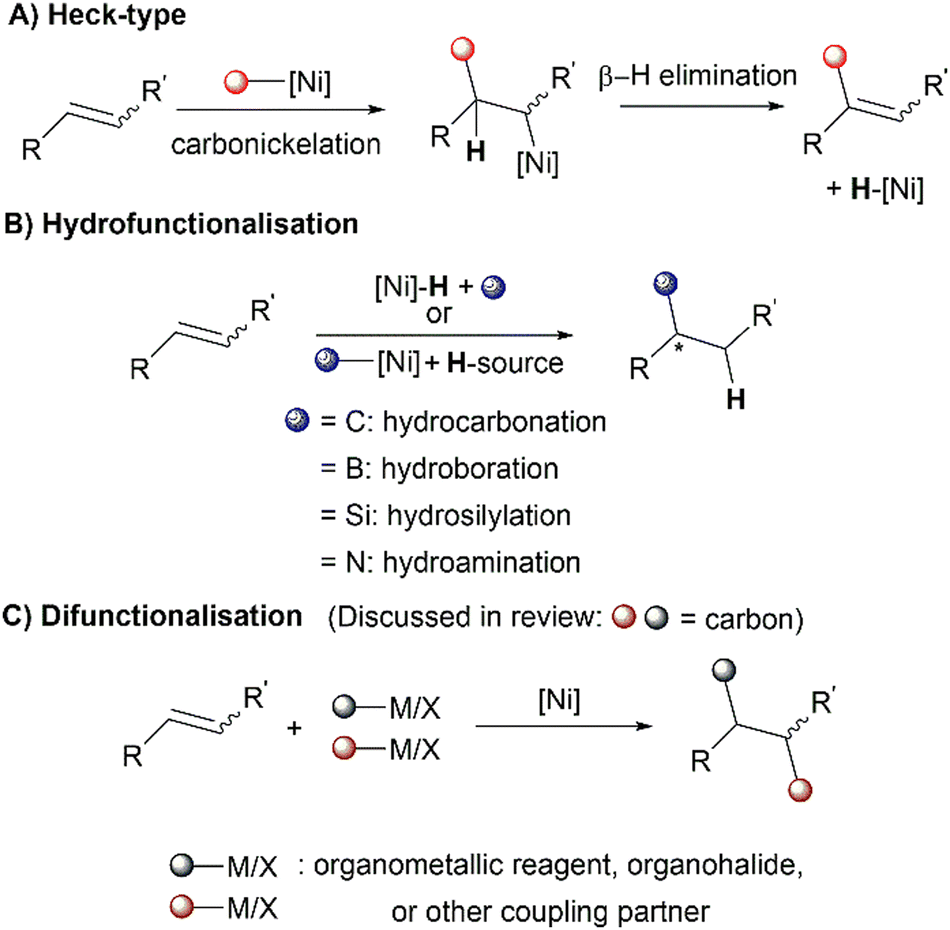 | ||
| Scheme 1 Different classes of alkene functionalisation covered in this review. | ||
Alkene functionalisation offers chemists a platform to rapidly assemble molecular complexity and stereogenicity from simple starting materials such as alkenes, due to their versatile reactivity and abundance as a feedstock chemical.32
There have been numerous developments in the use of homogenous nickel catalysis (not limited to NHC ligands) for the functionalisation of alkenes in recent years, which demonstrate its effectiveness in performing such valuable transformations. This has also been highlighted in several reviews.32,33 Nickel's high effectiveness as a catalyst for alkene functionalisation can be attributed to the following factors: nickel readily donates d-electrons to π-acceptors, resulting in strong olefin bonding. The less electronegative nickel undergoes more facile oxidative addition with electrophiles. β-Hydride elimination tends to be slower with nickel as compared to palladium, due to the significantly higher energy barrier for Ni–C bond rotation prior to β-hydride elimination in the organonickel species.34 As such, Ni(NHC) catalytic systems have also enabled the hydrofunctionalisation and difunctionalisation of alkenes beyond Heck-type reactions.
2. Heck-type reaction
The Mizoroki–Heck reaction has been established as one of the most important methods for the formation of valuable C–C bonds to afford substituted alkenes, and has been utilised significantly in organic chemistry, including the synthesis of various natural products.35 It falls under the class of “palladium-catalysed cross-coupling reactions” for which the 2010 Nobel Prize in Chemistry was awarded to Richard F. Heck, Ei-ichi Negishi, and Akira Suzuki for their pioneering efforts.36Nickel has also been found to be effective in catalysing Heck-type reactions,37 which follow a mechanism similar to that of the well-explored palladium-catalysed reaction.38 This is illustrated in a four-step catalytic cycle as shown in Scheme 2. First, the metal complex undergoes oxidative addition of the alkyl, aryl or alkenyl-(pseudo)halide bond, facilitated by the electron-donating ligand(s). Then, the olefin associates with the metal to form a π-complex, followed by the migratory insertion of the olefin into the carbon–metal bond. The regioselectivity of the Heck reaction, if any, is governed by this olefin insertion step, which can produce either 1,1-disubstituted or 1,2-substituted alkenes as the possible regioisomers. In the third step, β-hydride elimination occurs, furnishing the coupled product and a metal-hydride intermediate. Finally, the active metal catalyst is regenerated after the base-assisted removal of HX, completing the catalytic cycle. Variations in the Heck mechanism have also surfaced, suggesting the involvement of cationic Ni(II) intermediates.39
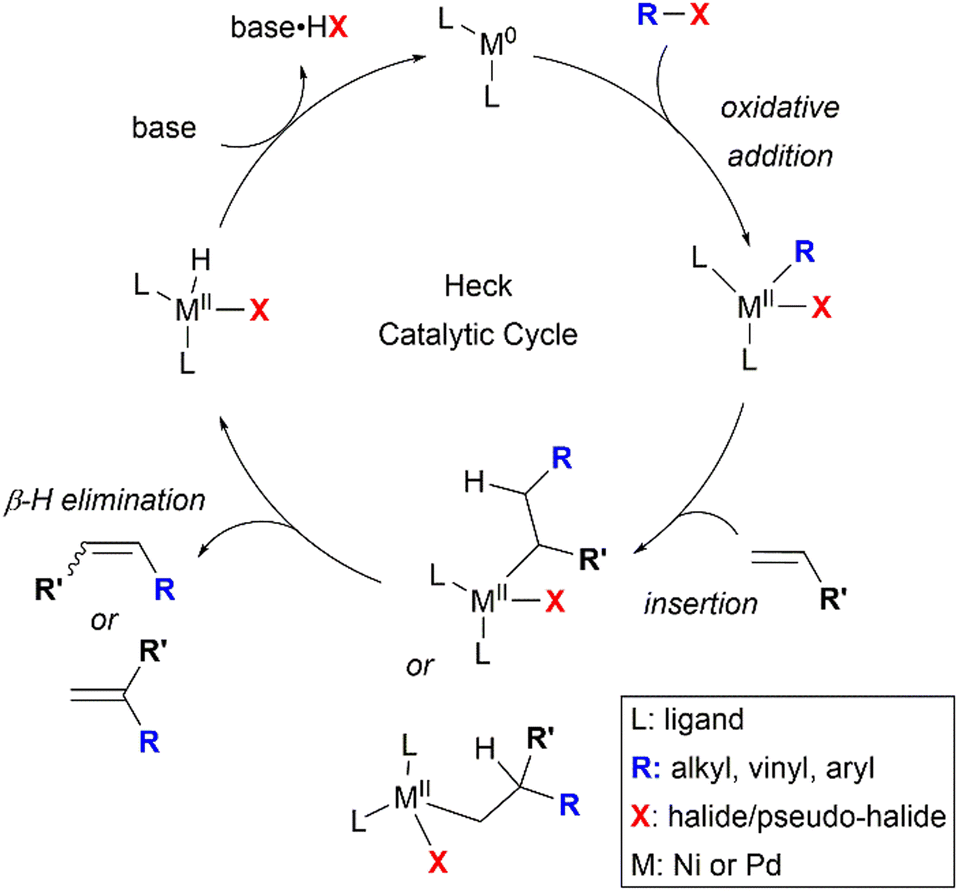 | ||
| Scheme 2 Typical Heck catalytic cycle (where M = nickel or palladium). | ||
Although numerous examples of the Heck reaction enabled with various phosphine-ligated Ni complexes have been reported,37 the use of NHC ligands to effect such transformations has been largely elusive and unexplored. The use of nickel–NHC complexes in the Heck reaction was first described by Sakamoto et al.40 in 2005. Their Ni(NHC) catalyst system was found to be highly efficient for the Heck reaction between aryl bromides/iodides with acrylates. The desired cinnamate-type products were obtained in high yields and with excellent trans-selectivity (Scheme 3). The reaction system was also evaluated for functional group tolerance and was found to work efficiently with nitriles (8), esters (10), aldehydes (9) and thiophenes (7).
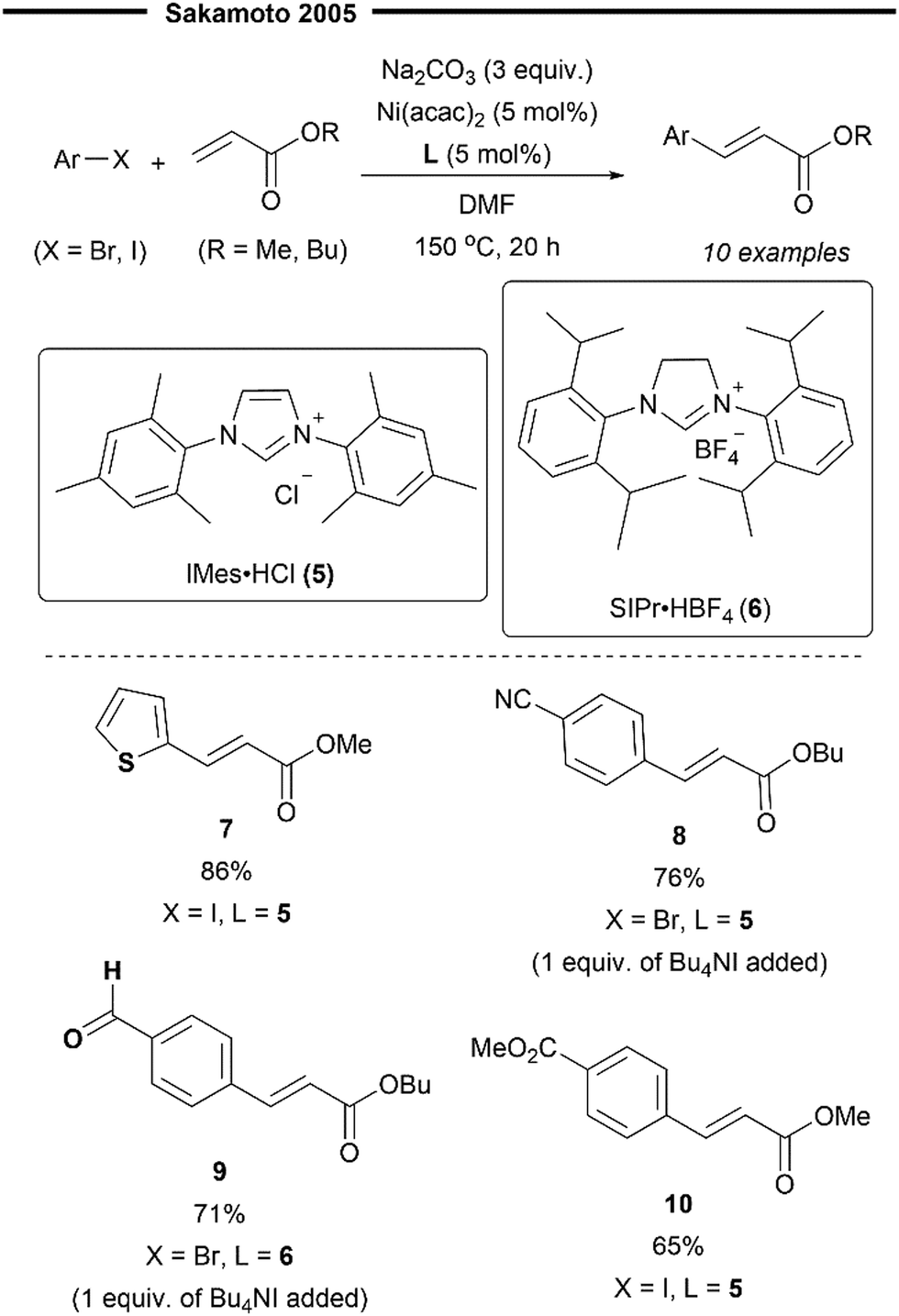 | ||
| Scheme 3 First Ni/NHC-catalysed Heck reaction of aryl halides with acrylates.40 | ||
In 2007, Schleicher and Jamison developed a Ni(0)/IPr catalysed formal intermolecular Heck coupling between α-olefins and isocyanates, affording N-alkylacrylamides in good yields and with excellent regioselectivity41 (Scheme 4). The observed regioselectivity for the C–C bond formation (prior to β-hydride elimination) was attributed to the sterically demanding NHC ligand, where the R1 substituent of the α-olefin prefers to be oriented away from the bulky isopropyl group of the IPr 1f ligand (Scheme 4), providing 1,1-disubstituted N-alkylated acrylamide as the major product. This is opposite in selectivity to previous reports under the Ni/PR3 catalytic system,42 where the major product was a trans-1,2-disubstituted α,β-unsaturated amide, instead. Aliphatic olefins with branching at the allylic position were highly effective substrates (11 and 13), furnishing the desired product in excellent yields and with regioselectivity. Compared to linear alkyl chains (n-hexyl in 14), the presence of coordinating groups (ketone in 12) in the olefins also resulted in enhanced selectivity of the products. However, the reaction appears to be only selective for monosubstituted terminal olefins and is also limited to bulky and electron-rich (Cy or tBu-substituted) alkyl isocyanates.
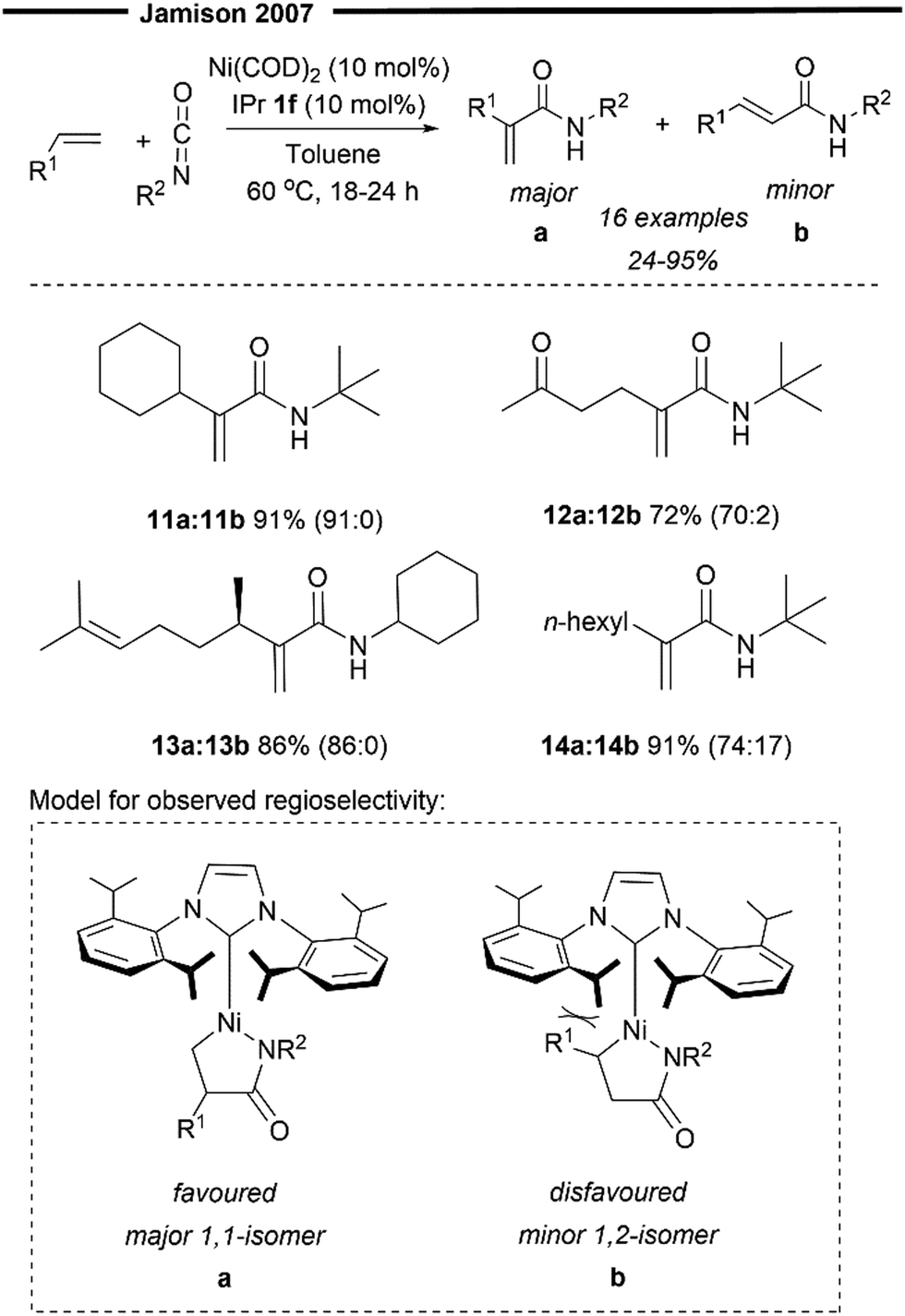 | ||
| Scheme 4 Regioselective Ni/NHC formal Heck reaction of α-olefins and isocyanates.41 | ||
In the same year, Ho and Jamison developed a Ni-catalysed three-component Heck-type coupling reaction between alkenes, aldehydes and silyl triflates to deliver the 1,1-disubstituted allylic alcohol product with excellent regioselectivity43 (Scheme 5) Their methodology utilises the synergistic relationship between a strong σ-donor ligand (NHC) and a strong π-acceptor phosphite ligand (P(OPh)3). Only when both are used in tandem will the reaction proceed catalytically with high selectivity and efficiency. From their preliminary optimisation and mechanistic studies, in the absence of the IPr 1f ligand, no product was observed. In the absence of P(OPh)3, there was no catalytic turnover, resulting in much lower conversion and yield, but without diminishing its selectivity. Their postulated mechanism (Scheme 5) involves the initial formation of the oxa-nickelacycle 20 from the Ni–NHC complex, alkene, and aldehyde, assisted by the Lewis Acid; silyl triflate. This step determines the regioselectivity of the oxanickelacycle assembly 20, which ultimately leads to the 1,1-disubstituted allylic alcohol product. The observed regioselectivity could be attributed to the bulky IPr 1f ligand which disfavours the other regioisomer (oxanickelacycle 20’). The role of the phosphite co-ligand was to accelerate the reductive elimination of the [Ni(IPr)H]OTf species 21 (via22 and 23), which in its absence, accumulates in the reaction mixture, leading to side reactions and hence, diminished yields. It was postulated that the formal reductive elimination of 21 to 19 to close the catalytic cycle was too slow (hence no catalytic behaviour in the absence of phosphite) due to the presence of the strong σ-donating IPr ligand.43 Anisaldehydes proved to be excellent substrates (15 and 17) while the presence of chlorine (16) and heteroaryl substituted aldehydes was less efficient (18).
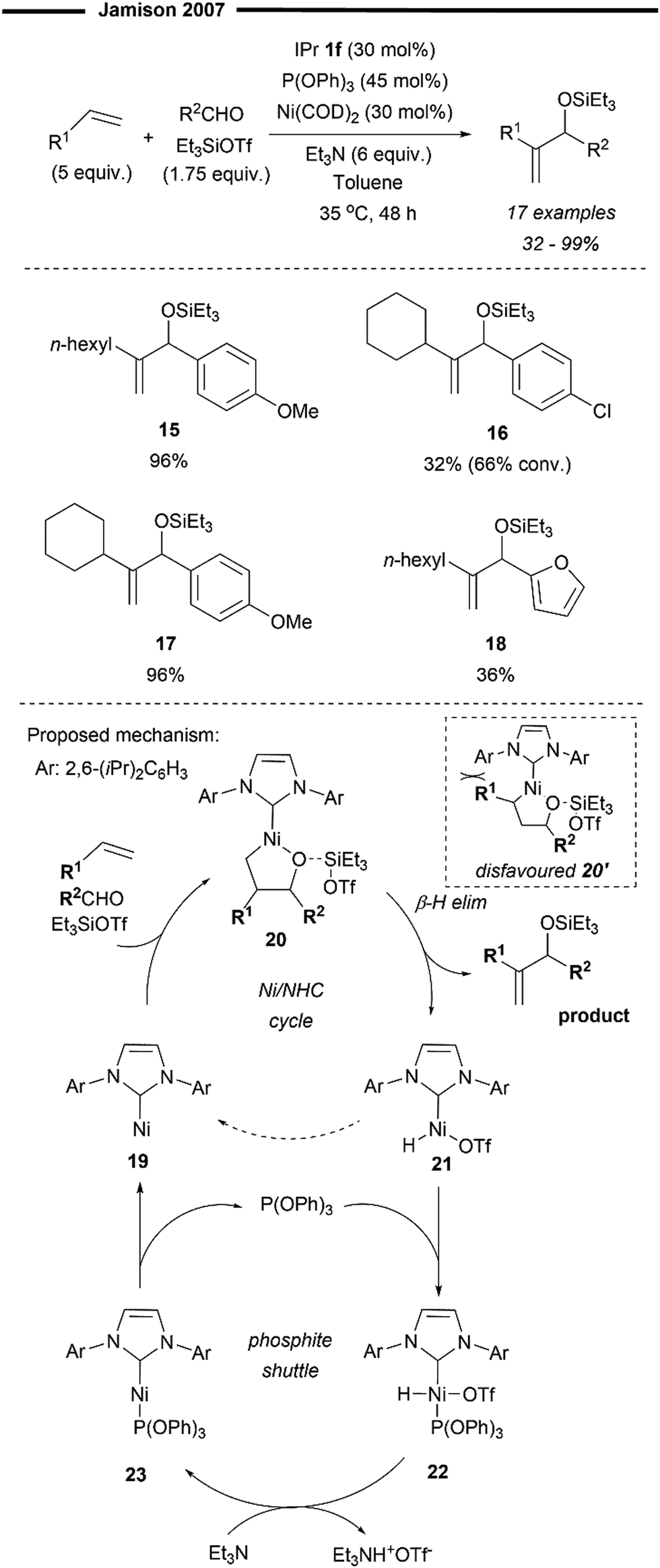 | ||
| Scheme 5 Regioselective formal Heck coupling of alkenes and aldehydes with [Ni(NHC)P(OPh3)].43 | ||
In addition to the commonly employed [Ni(COD)2]/IPr system, which can be used as a precursor for the in situ generation of [(NHC)NiH]OTf, Jamison and co-workers have developed an alternative, novel bench-stable nickelII-NHC precatalyst (24), which can be efficiently reduced to the NHC–Ni0 active species in the presence of silyl triflate (Scheme 6).44 The architecture of the nickel–NHC precatalyst was inspired by phosphine–nickel complexes, featuring a bidentate aryl ligand containing a piperidine moiety that occupies the coordination sites around the nickel, thereby stabilising the complex. Additionally, a pendant alkene linkage is present, which serves to reduce the NiII to Ni0 (in its active form) through an intramolecular Heck reaction.
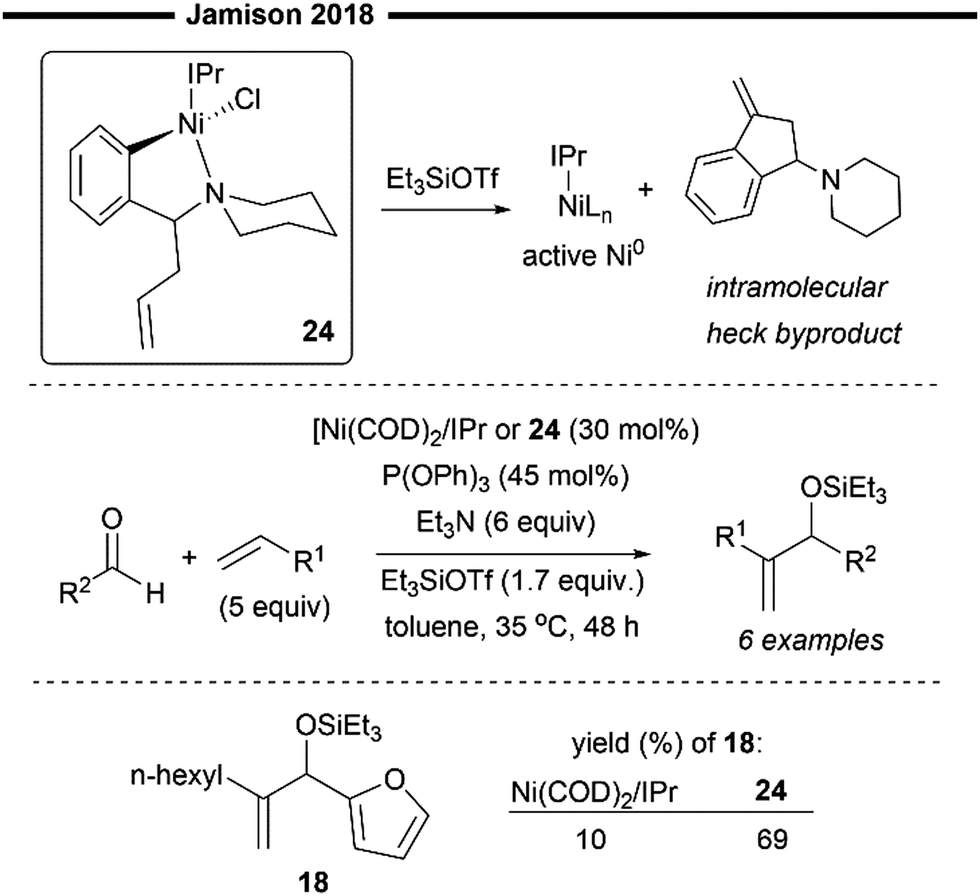 | ||
| Scheme 6 Bench-stable NiII/NHC precatalyst developed by Jamison and coworkers.44 | ||
This bright yellow nickelII–NHC precatalyst (24) also exhibits remarkable stability in air and can be easily isolated using column chromatography with neutral alumina. The precatalyst was found to be very effective in promoting the carbonyl-ene type coupling of alkenes and aldehyde, exhibiting even greater catalytic activity than the previously developed Ni(COD)2/IPr system (Scheme 5).
More recently, Koh and co-workers reported a novel Ni(I)/NHC-catalysed regio- and stereo-selective coupling reaction of monosubstituted alkenes with a variety of electrophilic reagents to access tri- and tetra-substituted alkenes in high yields (up to 92%) and selectivity (>98% E:Z).45 Mechanistic and computational studies support the idea of a novel tandem pathway involving a site-selective Heck-type reaction followed by a stereoselective C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond isomerisation, under a NiI/NiIII catalytic pathway (Scheme 7). Based on their proposed mechanism, initial dissociation of the dinuclear IPr-derived Ni(I) dimeric complex (25), and ligand exchange from sodium tert-pentoxide (NaOt-Am) forms the catalytically active Ni(I)-alkoxide 30. In the presence of an aryl triflate and a monosubstituted alkene, the Mizoroki–Heck reaction occurs, through an initial oxidative addition of 30 with aryl triflate, followed by a regioselective arylnickelation and β-hydride elimination. This produces an intermediate π-complex of NiIII-hydride and the 1,1-disubstituted alkene 33. Base-promoted reductive elimination of 33 then generates another olefin-coordinated NiI-alkoxide 34. Mechanistic studies and computational modelling of the transition states (of trans-TS-34 to 35versus cis-TS-34 to 35) suggest a probable stereoselective non-radical allyl isomerisation sequence of 34. 34 undergoes an uphill (hence requiring higher reaction temperatures) trans-selective allylic C–H insertion to give the η1-allylnickel(III) intermediate 35, followed by a subsequent allyl migration to form 36 and a final irreversible reductive elimination to furnish the desired trisubstituted alkene product, regenerating the catalytically active NiI-alkoxide 30. Despite the uphill olefin migration step, the overall process was computed to be thermodynamically favourable, with an energy change of −58.1 kcal mol−1.45
C bond isomerisation, under a NiI/NiIII catalytic pathway (Scheme 7). Based on their proposed mechanism, initial dissociation of the dinuclear IPr-derived Ni(I) dimeric complex (25), and ligand exchange from sodium tert-pentoxide (NaOt-Am) forms the catalytically active Ni(I)-alkoxide 30. In the presence of an aryl triflate and a monosubstituted alkene, the Mizoroki–Heck reaction occurs, through an initial oxidative addition of 30 with aryl triflate, followed by a regioselective arylnickelation and β-hydride elimination. This produces an intermediate π-complex of NiIII-hydride and the 1,1-disubstituted alkene 33. Base-promoted reductive elimination of 33 then generates another olefin-coordinated NiI-alkoxide 34. Mechanistic studies and computational modelling of the transition states (of trans-TS-34 to 35versus cis-TS-34 to 35) suggest a probable stereoselective non-radical allyl isomerisation sequence of 34. 34 undergoes an uphill (hence requiring higher reaction temperatures) trans-selective allylic C–H insertion to give the η1-allylnickel(III) intermediate 35, followed by a subsequent allyl migration to form 36 and a final irreversible reductive elimination to furnish the desired trisubstituted alkene product, regenerating the catalytically active NiI-alkoxide 30. Despite the uphill olefin migration step, the overall process was computed to be thermodynamically favourable, with an energy change of −58.1 kcal mol−1.45
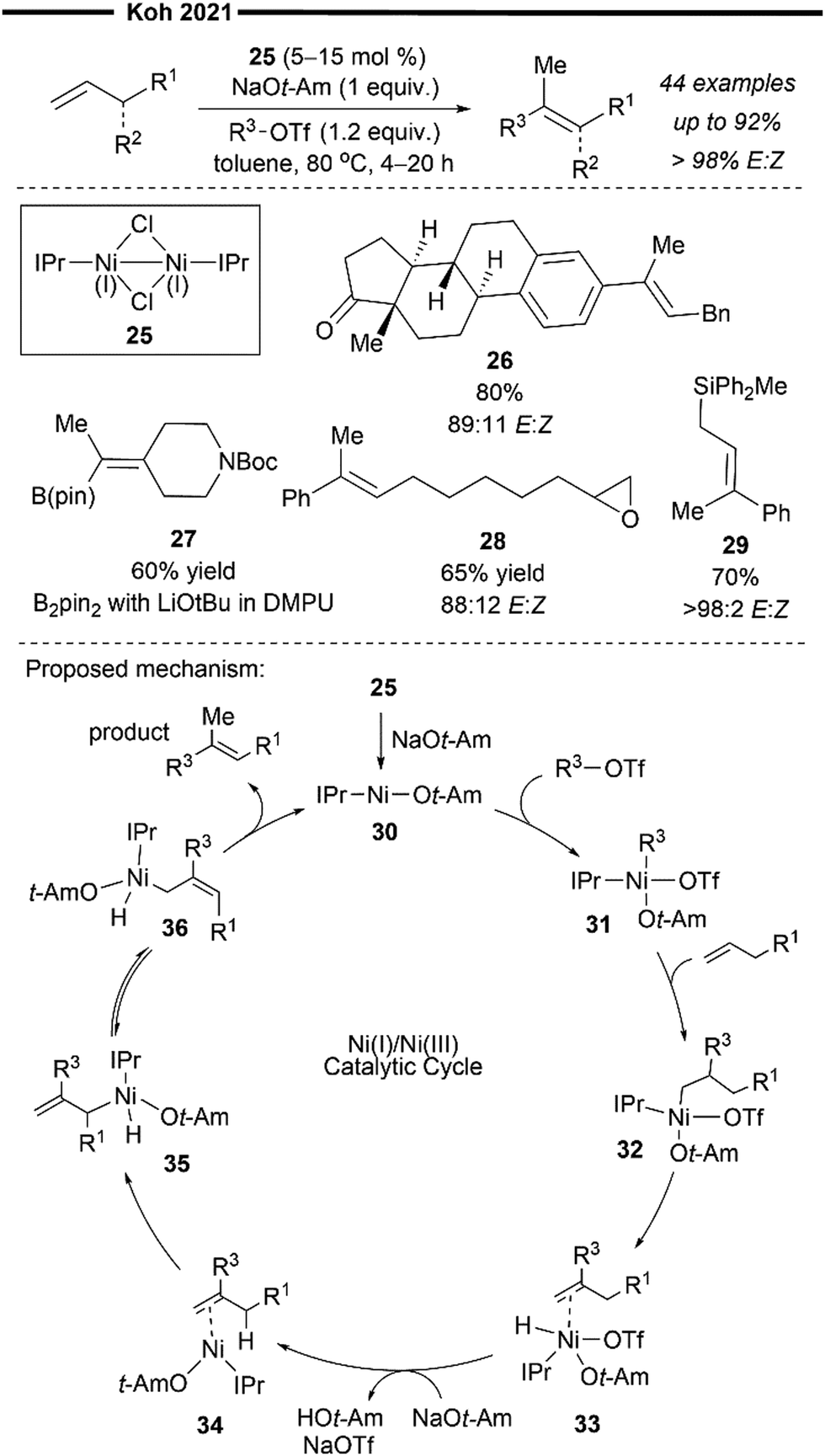 | ||
| Scheme 7 Ni(I)/IPr-catalysed stereoselective synthesis of alkenes via formal Heck reaction/alkene isomerisation.45 | ||
A plethora of functionalised monosubstituted alkenes and aryl triflates were found to be compatible under the optimised protocol. Electrophilic ketone-containing polycyclic triflates (26), epoxide (28) and silane (29) were effective substrates. With slight modifications, this reaction can also be extended to the synthesis of tri- or tetra-substituted alkenyl boronates (27), which are valuable intermediates in the preparation of many medicinally relevant pharmaceuticals.46,47 Analogous to the above proposed mechanism (Scheme 7), the alkenyl boronate product is likely formed from an initial boryl-Heck type reaction followed by an olefin isomerisation.
An extension to their previous work (Scheme 7), Koh and co-workers reported a Ni(I)/IPr catalysed branched-selective Heck-type benzylation-stereoselective olefin isomerisation methodology to access tri-substituted alkenes.48 The same dinuclear IPr-derived Ni(I) dimeric complex (25) was used as the precatalyst, which efficiently enabled the reaction of styrenes/vinylboronates with various functionalised benzyl chlorides under basic conditions, to afford trisubstituted alkenes with high trans-selectivity. The excellent branched selectivity (where the benzyl group is attached to the more substituted olefinic carbon) of the initial Heck-type benzylation is attributed to the steric control provided by the bulky NHC ligand, which effectively overrides the olefin's strong electronic bias for linear selectivity.
The proposed mechanism for the tandem Heck-type benzylation–isomerisation reaction (Scheme 8) is similar the non-radical NiI/NiIII pathway as illustrated previously (cf.Scheme 7). Reaction of the catalytically active monomeric NiI–X (X = Cl or Br) complex 40 with benzyl chloride forms the putative π-allyl nickel intermediate 41. Subsequent ligand exchange with TMSOTf forms a cationic NiIII intermediate 42 with the weakly bound non-nucleophilic triflate anion, accompanied with the TMS-halide by-product. The incoming styrene/vinylboronate substrate can form an initial π-complex with electrophilic nickel 42, followed by the regioselective migratory insertion of the benzyl group to yield a primary alkylnickel species 43. Regiochemical outcome for this branched-selective benzylation (instead of linear-selective insertion) can be rationalised by the favoured transition state of lowest energy with minimised steric repulsions between the bulky IPr NHC ligand and the R1 substituent of the olefin. β-H elimination then forms an olefin-associated NiI complex 44, which undergoes facile deprotonation with the weak Et3N base to give 45. Lastly, with motivation from the results of previous DFT calculations,45trans-selective insertion (from 45 to 46) into the benzylic C–H bond takes place (to minimise steric interactions between the aryl group Ar, halide X and IPr ligand) followed by a non-radical allyl migration to form the final allylic η1-Ni complex 47, which reductively eliminates to deliver the final trisubstituted alkene in high trans-selectivity.
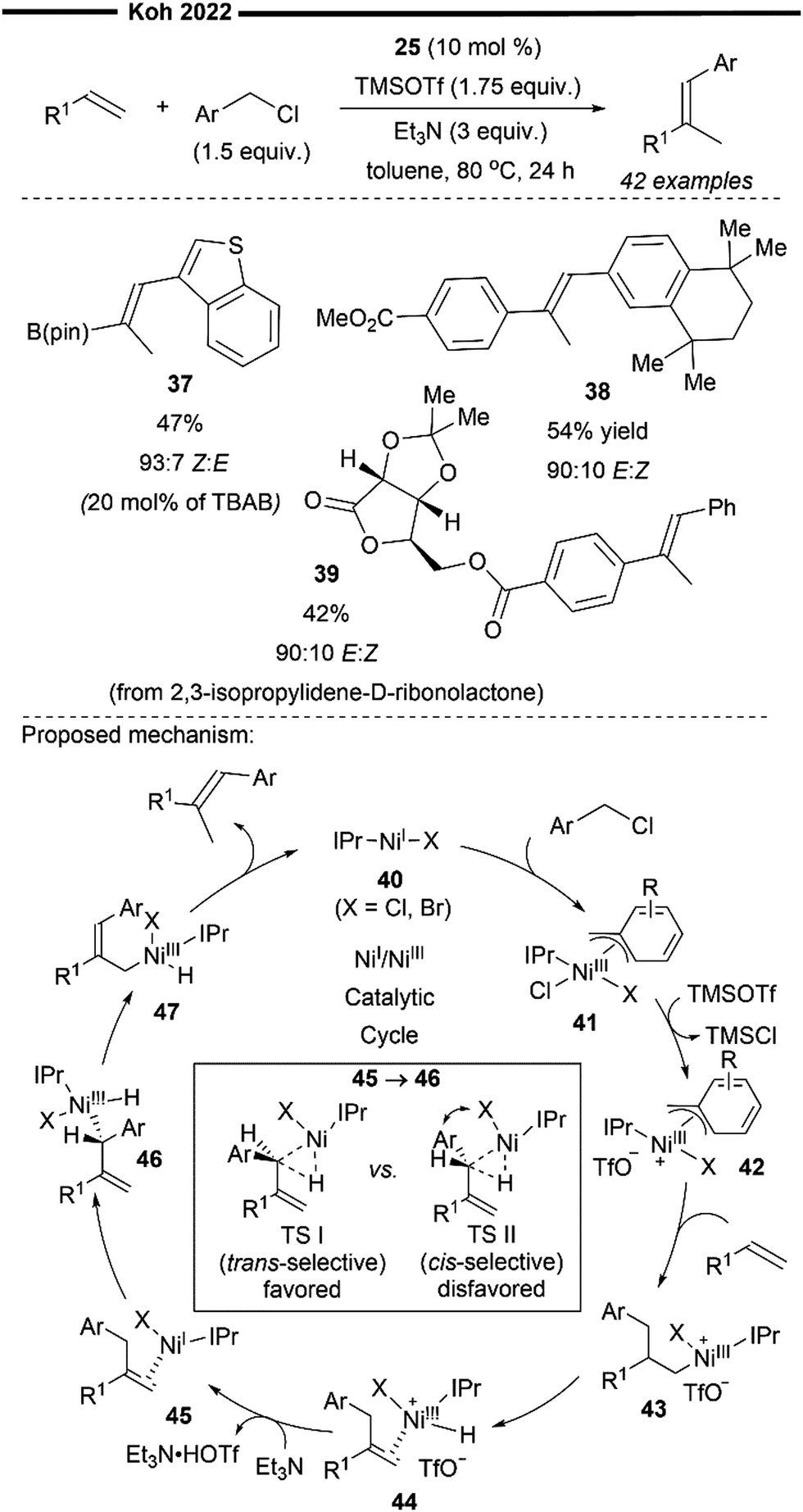 | ||
| Scheme 8 Ni(I)/IPr-catalysed Heck-type benzylation/isomerisation for stereoselective synthesis of tri-substituted alkenes.48 | ||
In the presence of TBAB additives, various benzyl chlorides bearing heteroaryl groups (37) were compatible with vinylboronate, to access highly substituted alkenylboronates in moderate yields. Highly biologically relevant carbohydrate-derived styrene substrates (39) were also effective. The utility of this regime was also demonstrated with the efficient synthesis of a retinoid precursor (38).
3. Hydrofunctionalisation of alkenes
In a broad sense, hydrofunctionalisation of alkenes refers to the installation of a hydrogen atom and a functional group across the CC π bond of the alkene, thereby generating a new C(sp3) centre. Numerous reports have surfaced in recent years,49–51 documenting the use of Ni catalysts for the hydrofunctionalisation of alkenes, including the development of asymmetric variants which enables the synthesis of valuable, optically active molecules using simple alkene feedstock. In this review, we will not restrict our analysis to C–C bond forming reactions (i.e., hydrocarbofunctionalisation), despite their importance. We will also be discussing various studies that demonstrate the versatility of hydrofunctionalisation using Ni/NHC catalysts in the hydrosilylation, hydroboration, and hydroamination of alkenes.
Not all hydrofunctionalisation reactions follow a universal reaction pathway. Herein, we illustrate certain subtle mechanistic nuances of hydrofunctionalisation reactions often disclosed in literature studies (Scheme 9). The C–H activation of the substrate as well as the migratory insertion into CC can proceed via a single transition state,52 known as metal-mediated ligand-to-ligand hydrogen transfer (LLHT), first termed by Perutz et al.53 in their DFT-supported mechanistic study of Nakao's Ni-catalysed hydrofluoroarylation reaction.54 This concerted pathway (Scheme 9a) proceeds without the involvement of nickel–hydride (Ni–H) species intermediates, which are commonly proposed in catalytic cycles of Ni-catalysed alkene transformation reactions.55
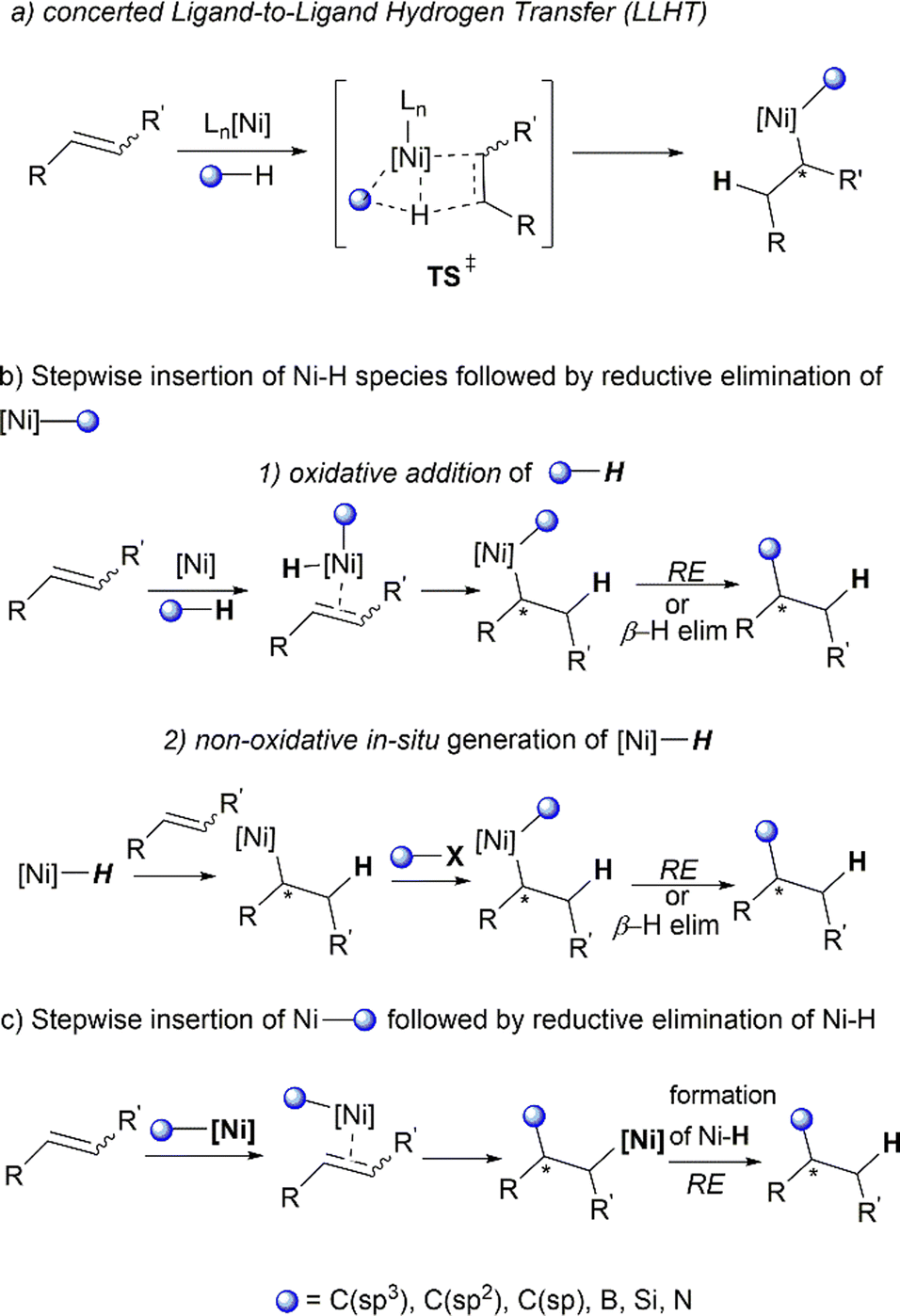 | ||
| Scheme 9 Different ionic mechanistic pathways for hydrofunctionalisation. | ||
A non-concerted stepwise mechanism of hydrofunctionalisation is also plausible, with Ni–H involved as intermediary species. An initial insertion of the alkene into the Ni–H bond generates an alkyl-Ni intermediate, which can react with the desired electrophile (via insertion or oxidative addition). Subsequent β-hydride elimination or reductive elimination affords the final product (Scheme 9b). The formation of the Ni–H intermediate can occur through an oxidative route which involves the oxidative addition of C–H or Si–H bonds by low valent Ni complexes55,56 (Scheme 9b1). The non-oxidative route usually involves the in situ formation of Ni–H via reaction of a main group hydride (e.g., LiEt3BH) and σ-bond metathesis with silanes (Si–H) or boranes (B–H).55 Other methods include the treatment of Ni0/IPr with silyltriflate, aldehyde and alkene, which effectively produces “[IPrNiH]OTf” species from an oxanickelacycle intermediate as discussed previously in Jamison's work43 (Scheme 9b2). An alternative feasible pathway for this transformation could be the initial carbonickelation followed by β-hydride elimination to produce an alkyl Ni–H intermediate,57,58 which undergoes reductive elimination to deliver the final hydrofunctionalised product (Scheme 9c).
In addition to the common ionic pathways involved in hydrofunctionalisation (illustrated in Scheme 9), there are also reports of radical-based pathways proceeding via metal–hydride hydrogen atom transfer (MHAT).59 These will be briefly discussed along with relevant literature studies. Intricate mechanistic details governing the regioselectivity (Markovnikov or anti-Markovnikov addition) and/or stereoselectivity of specific reactions will also be analysed in detail.
3.1 Hydroalkenylation of alkenes
Transition-metal catalysed alkene hydroalkenylation/arylation reactions offer an efficient way to construct highly valuable C(sp3)–C(sp2) bonds. However, as with most hydrofunctionalisation reactions, there are several key considerations beyond the reaction itself, including regioselectivity (Markovnikov vs. Anti-Markovnikov), stereoselectivity, and the catalyst's turnover frequency, all of which can greatly influence the outcome and efficiency of the reaction.Hydroalkenylation generally refers to the addition of a vinyl/alkenyl group and a hydrogen atom across an unsaturated CC bond. The products of a typical hydroalkenylation reaction are like those of a Heck reaction, but there are subtle variations in the mechanistic pathway. This process can also be viewed as a heterodimerisation or cross-coupling reaction between alkenes.
The exploration and utilisation of NHCs as ligands for hydroalkenylation reactions were pioneered and mainly carried out by Ho's group. In 2010, Ho and co-workers reported the Ni/NHC-catalysed intermolecular branched-selective (tail-to-tail) hetero-hydroalkenylation of styrenes with α-olefins.60 The tentative proposed catalytic species is an in situ generated [(NHC)NiH]OTf species, prepared from a modified procedure reported by Jamison and co-workers in their [(IPr)NiP(OPh)3]-mediated silyltriflate/alkene/aldehyde coupling reaction (Scheme 5). Building on this initial discovery, subsequent research by Ho's group primarily centered on the chemistry of an NHC–nickel hydride species generated in situ, which they identified as active catalysts in a variety of hydroalkenylation reactions (Schemes 11–13).
In their proposed mechanism (Scheme 10), addition of the Ni–H of the in situ generated (NHC)nickel–H species 48 to the vinylarene (chemoselective and regioselective hydronickelation) forms the resonance-stabilised benzylic nickel complex 49. The incoming α-olefin (with a less bulky substituent than the aryl group of the vinylarene) is then coordinated with 49 in an orientation that minimises steric repulsion between the bulky NHC ligand and the alkene substituent, forming the π-complex 50 (favoured over 50’), which is primed to undergo regioselective migratory insertion with the α-olefin to give the primary nickel intermediate 51. Finally, β-hydride elimination occurs, releasing the final hetero-coupled gem-disubstituted alkene product and regenerating the nickel hydride catalyst 48. It is noteworthy that, despite the thermodynamic driving force for the isomerisation of 1,1-disubstituted alkenes to the more stable internal or conjugated alkenes, only limited isomerisation was observed under those conditions. It is believed that the steric effect of the bulky IPr ligand explains this observation and governs the regioselectivity of the migratory insertion process as well as the chemoselectivity for the desired hetero-coupling reaction.
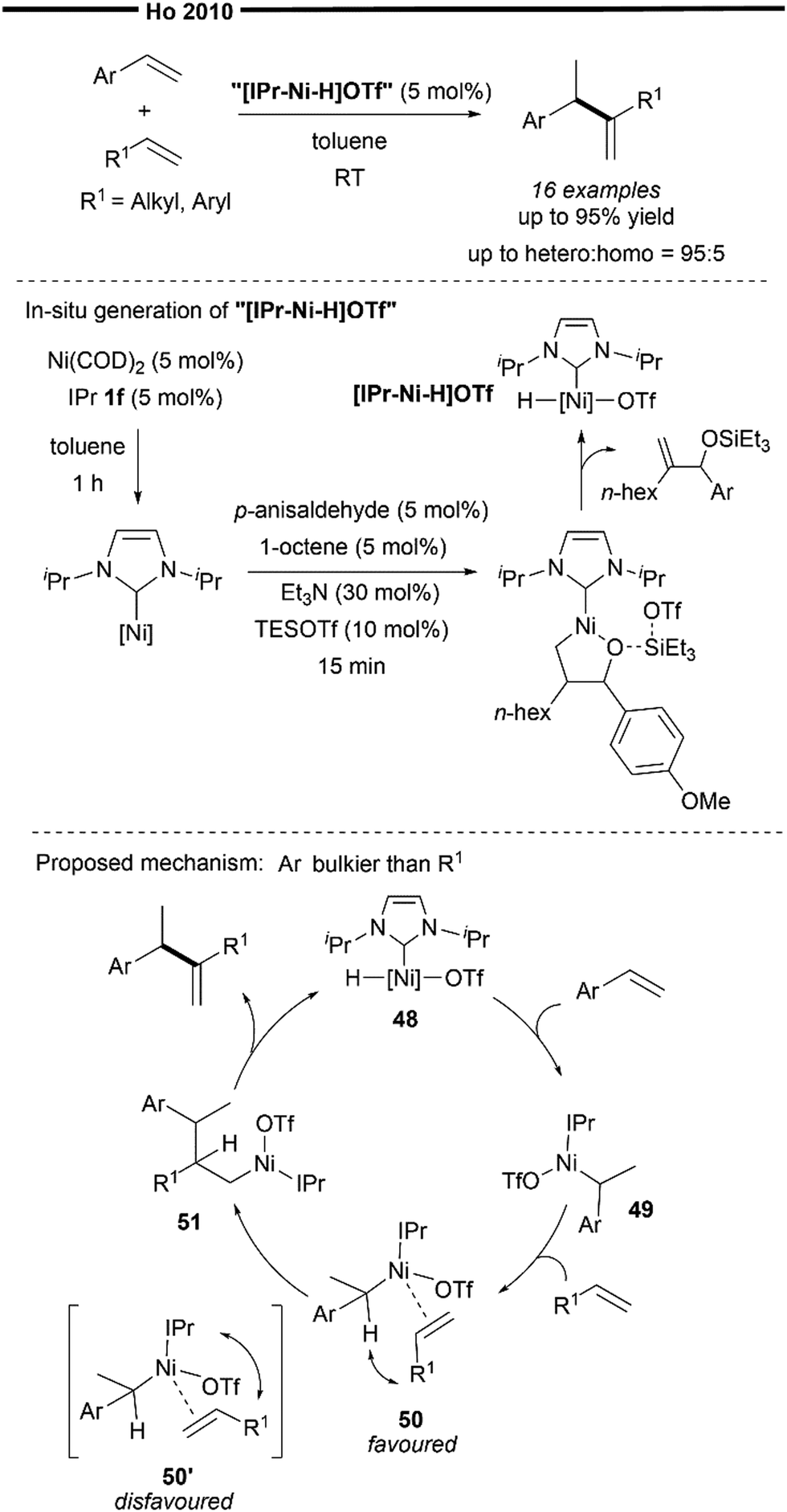 | ||
| Scheme 10 Ni/NHC catalysed branched-selective hetero-hydroalkenylation of styrenes and unactivated α-olefins.60 | ||
In 2015, Ho and co-workers developed the asymmetric variant of their previously reported hetero-hydroalkenylation of vinylarenes and unactivated α-olefins, with the use of an unsymmetrical chiral NHC ligand (52). This reaction also operates with an analogous mechanism to Scheme 10, where the active species is the in situ-generated [(52)NiH]OTf.43 Unsymmetrical gem-disubstituted olefins were obtained as the final major product in high yields and high ee (Scheme 11). Aryl-substituted vinylarenes like 3-methoxystyrene (56) and thioanisole (55) were well-tolerated, giving the corresponding product in good yields and ee. Regarding the scope of terminal olefins, the use of allylbenzenes (53) resulted in the highest obtained ee, while vinylcyclohexane (54) was also found to be an effective coupling partner.
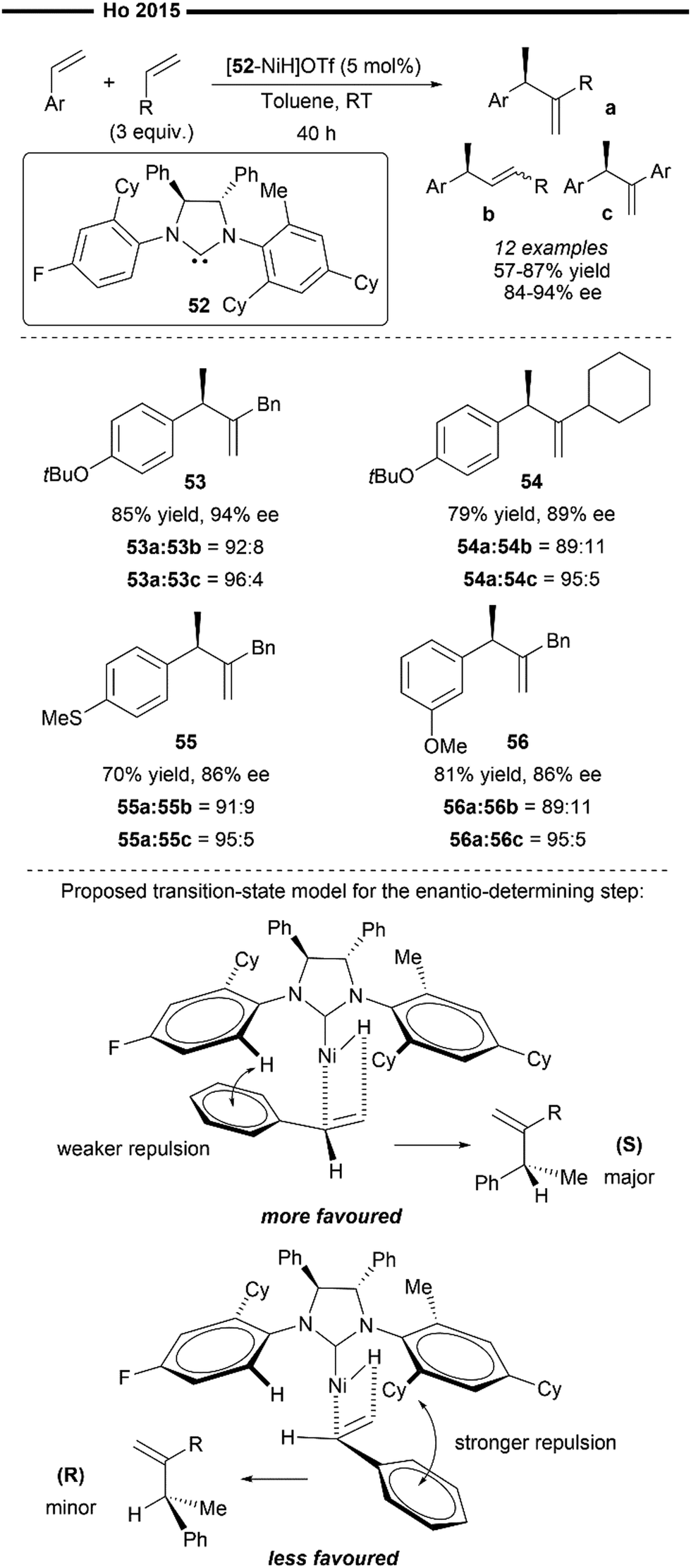 | ||
| Scheme 11 Asymmetric Ni/chiral NHC catalysed hetero-hydroalkenylation of styrenes and α-olefins.61 | ||
The author proposed a tentative stereochemical model to rationalise the enantio-determining vinylarene insertion step into Ni–H, purely based on steric factors, which minimises the unfavourable steric repulsion between the N-aryl substituent of the NHC and the aryl group of the vinylarene substrate (Scheme 11). This is exemplified with the increased ee values when more sterically bulky α-olefins were employed. The asymmetric induction by the Ni/chiral NHC complex can be influenced by the different N-aryl substituents on the NHC ligand as well as the external π-systems of the vinylarenes. However, their proposed stereochemical model has its limitations and does not fully explain how certain electronic factors can affect the enantioselectivity. For example, it is not fully understood why the presence of an ortho-methyl substituent on the N-aryl ring (of the NHC) is crucial for high enantioselectivity.
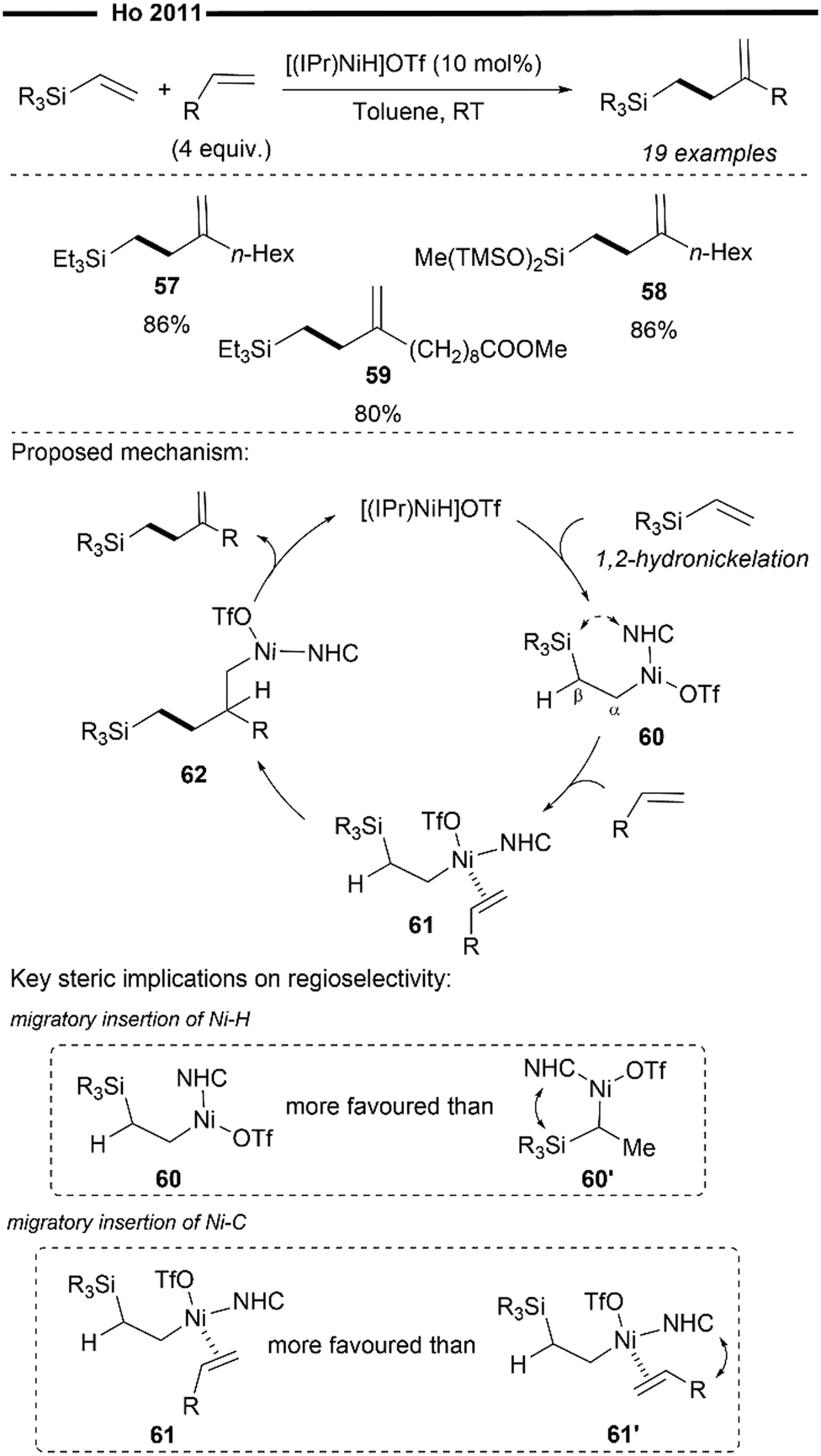
Not limited to only coupling with vinylarenes, Ho and co-workers further applied their chemistry on in situ generated [(NHC)NiH]OTf43 to design a novel method to access valuable organosilanes. In 2011, they developed the cross-hydroalkenylation reaction of vinylsilanes and α-olefins to afford homoallylsilanes in high chemoselectivity and regioselectivity (Scheme 12).62 Unlike their previous work on the cross-hydroalkenylation using vinylarenes,60,61 which affords the branched tail-to-tail product, the use of vinylsilanes results in a complete reversal of regioselectivity, governed exclusively by the steric properties of the NHC ligand.
 | ||
| Scheme 12 Ni/NHC catalysed cross-hydroalkenylation of vinylsilanes and α-olefins. | ||
The rationale for the observed chemoselectivity and regioselectivity in the reaction mechanism lies in the intricacies of the reaction mechanism. Specifically, the regioselectivity of Ni–H insertion is primarily directed by the steric influence of the bulky NHC ligand. In this reaction, 1,2-hydronickelation with the vinylsilane reagent occurs preferentially to 2,1-hydronickelation to give the primary alkylnickel intermediate 60, which is less sterically hindered. Unlike previous reports on reactions with vinylarenes,60,61 there is little thermodynamic incentive for 2,1-hydronickelation to occur with vinylsilanes due to the absence of adjacent aryl groups for π-stabilisation. Interestingly, β-silyl elimination of 60 is also strongly suppressed, presumably due to the attenuation of orbital overlap of Ni-β Si under the steric environment of the NHC. Coordination of α-olefin with 60 (in an orientation that minimises steric repulsion) forms 61, which subsequently undergoes another regioselective insertion to furnish 62. β-hydride elimination of 62 then regenerates the NHC nickel–hydride species and releases the homoallylsilane product in overall excellent regioselectivity.
Modifications to the steric and electronic properties of the Si centre do not appear to have a large influence on the reaction's efficiency (57,58). The reaction is also tolerant to a variety of substituents on the α-olefins, including enolisable esters (59).
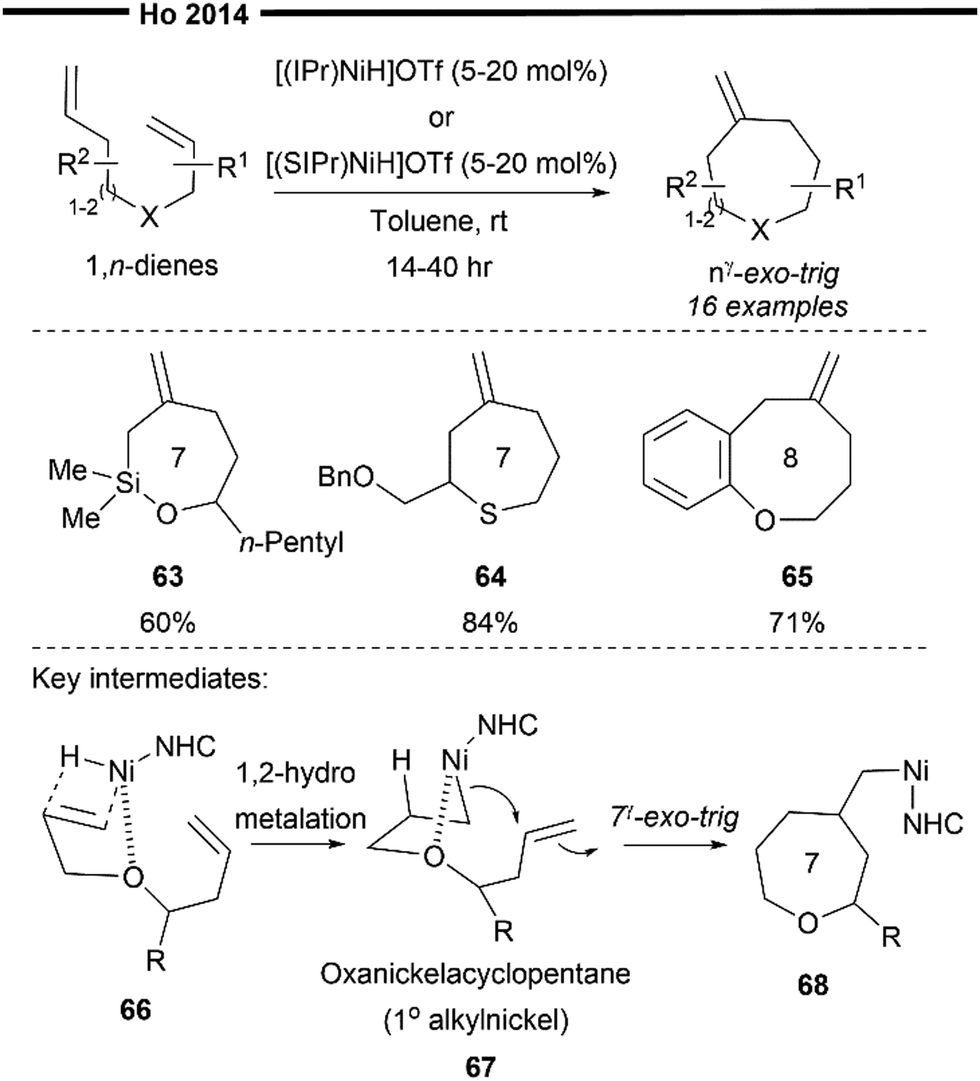
In 2014, Ho and co-workers developed a novel pathway to access seven-/eight-membered medium sized heterocycles via a Ni/NHC-catalysed nγ-exo-trig selective cycloisomerisation reaction of 1,n-heterodienes (n = 7,8).63 Their novel work utilises the synergistic effect between NHC's strong electron-donating and steric properties and γ-heteroatom chelation to selectively form medium-sized heterocycles despite the increased ring strain. Through their optimisation studies and competitive experiments, they found that the tethering length, γ-oxy chelation, and the steric effect of NHC all play important roles in the selective formation of the nγ-exo-trig product over the competitive (n − 1)γ-exo-trig product. Other than substituted oxepanes, siloxepane (63) and thiepane (64) can also be obtained through this reaction. 1,8-Oxadienes (65) also proved effective, successfully delivering the desired eight-membered oxecane product.
Their proposed mechanism (Scheme 13) postulates that the steric influence of the bulky NHC ligand promotes the initial 1,2-hydronickelation of the allyl ether arm of 66 (from an in situ generated [(NHC)NiH]OTf species), forming a less hindered primary carbocentre on nickel. Chelation assistance from the heteroatom (e.g., oxygen) present in the 1,n-diene also proves to be crucial for the reaction's efficiency and selectivity, possible stabilising the hydrometallated nickel species in the form of an oxanickelacyclopentane 67. The strongly electron-donating NHC ligand also led to improvement in functional group tolerance while mitigating the strength of γ-oxy chelation to the nickel centre. It must be balanced in a way that is strong enough as a stabilising group, yet not too strong that it deactivates the nickel catalyst. The stabilised oxanickelacyclopentane 67 then undergoes a facile intramolecular nγ-exo-trig ring closure reaction to form the cyclised intermediate 68, which releases the desired product after a syn β-H elimination step. While (n − 1)γ-exo-trig favours a product with a lower ring strain, the strong preference for nγ-exo-trig could be due to the less favourable 2,1-hydronickelation on the homoallyl ether terminus and the higher energy barrier for ring closure with a secondary alkylnickel-NHC species. However, it is noteworthy to point out that no in-depth mechanistic studies or calculations on the transition states were performed. It is therefore possible that other competitive insertion pathways may exist. However, due to the reversible nature of hydrometallation reactions, the steric influence of the NHC ligand may direct the mixed intermediates to converge into the lowest energy optimised geometry for ring closure, resulting in the observed selectivity for the nγ-exo-trig product.
 | ||
| Scheme 13 Ni/NHC catalysed cycloisomerisation of 1,n-dienes.63 | ||
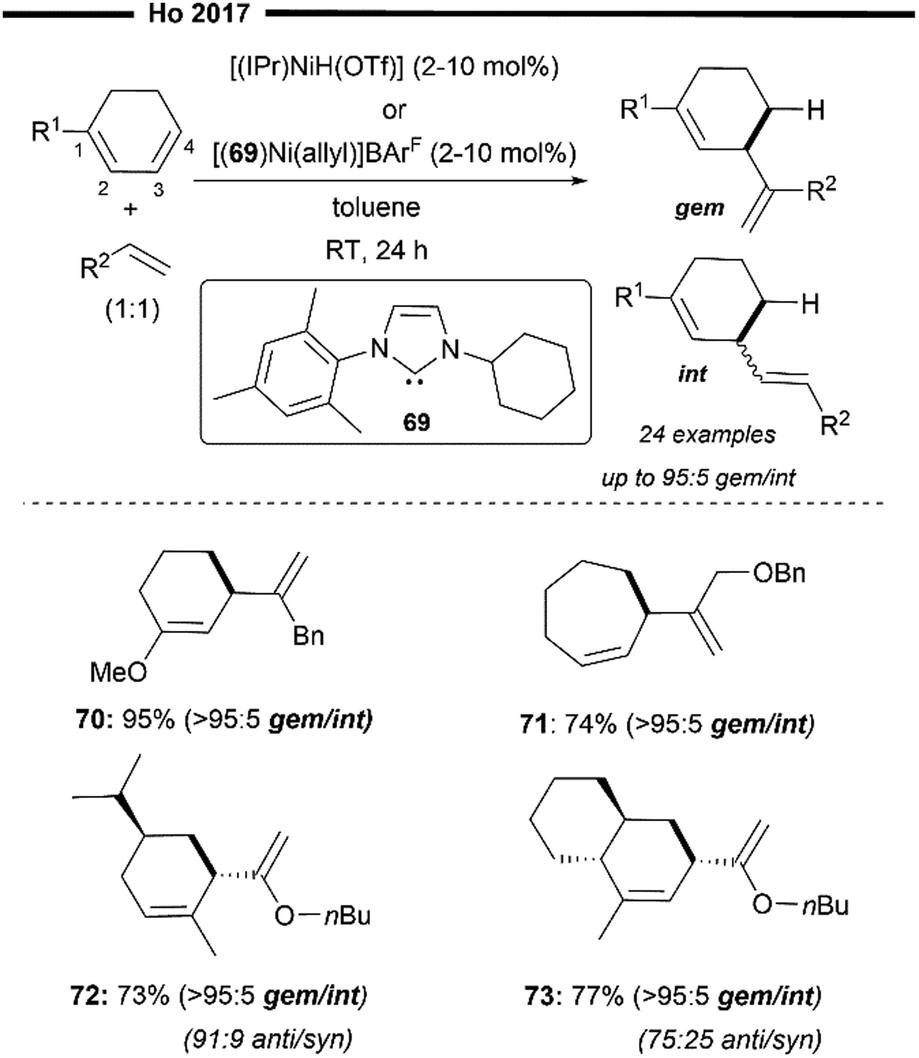
In 2014, Ho and co-workers described the first cross-hydroalkenylation of endocyclic 1,3-dienes with α-olefins enabled with neutral and cationic Ni/NHC precatalysts64 (Scheme 14). A rare product selectivity was observed under this protocol, where the 4,3-hydroalkenylation (the alkenyl group and H atom are introduced at the C3 and C4 position of the original diene substrate, respectively) skipped diene product was afforded with high chemo-, regio- and diastereo-selectivity. The reaction can be efficiently promoted with an in situ generated [(IPr)NiH(OTf)]43 or [(69)Ni(allyl)]+BarF− complex.65 Upon reaction with catalytic alkenes (1-octene was used), the precatalyst [(69)Ni(allyl)]+BArF− generates a similar [(NHC)NiH] hydride complex in an allylnickelation-β-hydride elimination sequence. Regioselective hydronickelation at the 4,3-position of the endocyclic 1,3-diene (at the least substituted CC) initially forms the allylic nickel intermediate (nickel at 3-position). The allylic shift of this nickel intermediate to a more substituted carbon of the diene (1- or 2-position) was not observed, possibly due to the sterically encumbered environment around the bulky NHC, which partly governs the observed regioselectivity for hydronickelation as well. The mechanism for this reaction could be thought of as analogous to that illustrated in Scheme 10.
 | ||
| Scheme 14 Ni/NHC catalysed cross-hydroalkenylation of endocyclic dienes with α-olefins.64 | ||
Another important criterion for desired reactivity is the presence of a coordinating heteroatom, either on the diene or the α-olefin substrate. It is postulated that the electronic effect of the heteroatom suppresses the undesired isomerisation of the alkene substrate. Cyclohepta-1,3-dienes were well tolerated, forming the corresponding skipped diene product in good yield (71). More sterically hindered fused bicyclic 1,3-dienes (73) were also feasible under their system, with the use of a less sterically hindered N-cyclohexyl-N′-mesityl NHC ligand (Scheme 14). Notably, high levels of diastereoselectivity of hydroalkenylation were also observed when chiral endocyclic dienes were employed (72). To minimise the steric strain between the diene substituents and the NHC ligand in the Ni-complex, the insertion of the α-olefin occurs on the face, with the newly formed stereogenic centre oriented anti to the existing chiral centre in the diene.
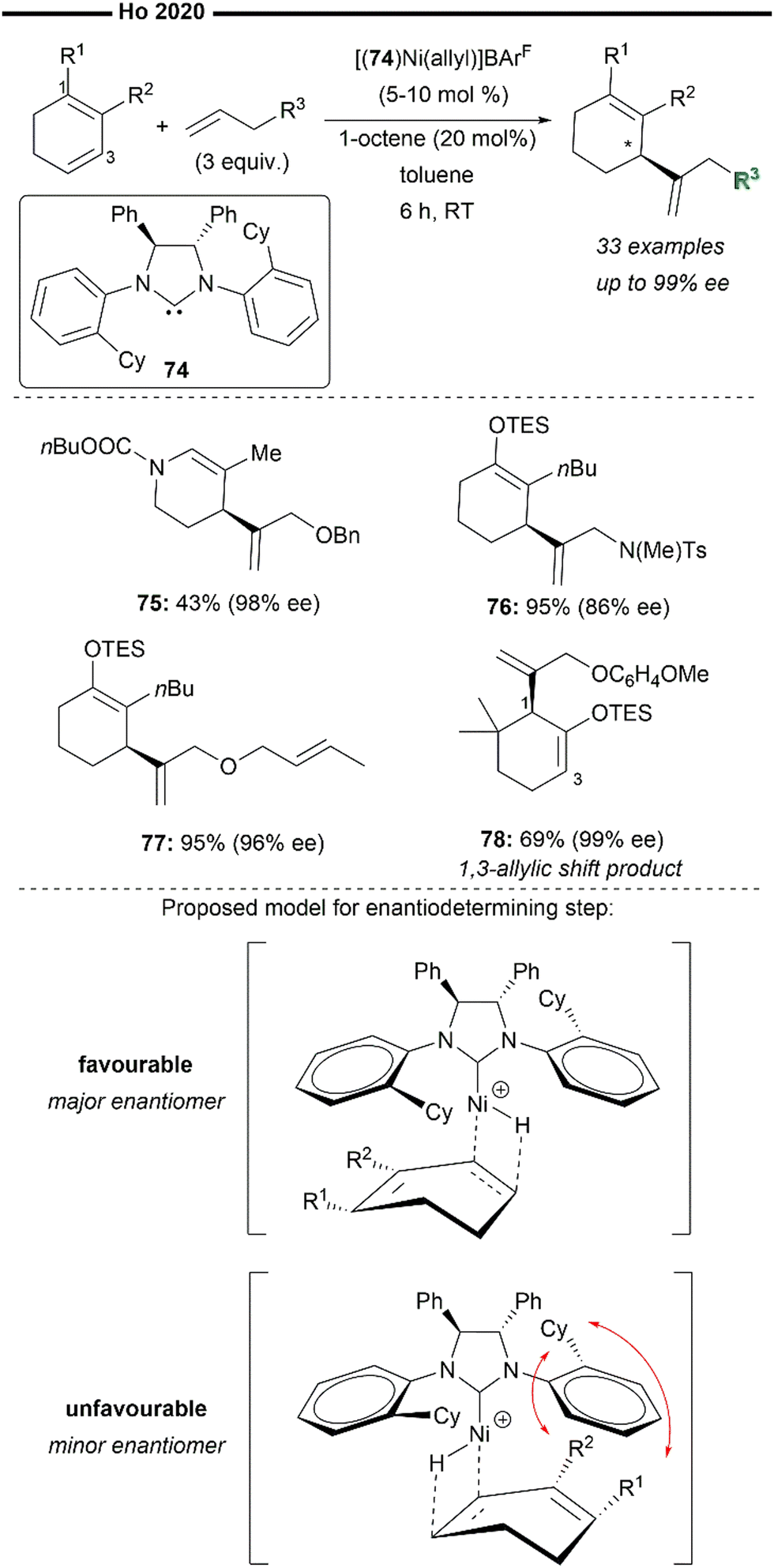
As an extension of their previous work (Scheme 14), Ho and co-workers developed an enantioselective cross-hydroalkenylation reaction of cyclic 1,3-dienes and hetero-substituted terminal alkenes/α-olefins to afford endocyclic 1,4 skipped dienes with a chiral allylic framework66 (Scheme 15). Similarly, the reaction employs the use of [(74)Ni(allyl)]+BArF− as the precatalyst, which forms the active nickel–hydride species upon reaction with catalytic amount of octene. A less bulky and structurally flexible C2-symmetric chiral NHC ligand was crucial for the balance of reactivity and selectivity of this reaction, considering the undesired steric interactions between the substituted dienes and the N-aryl substituents on the NHC. Hetero-substituted 1,3-dienes (75) without bulky R1 or R2 substituents proved to be compatible, delivering the product in moderate yield and excellent ee. N- or O-substituted terminal olefins were also efficient (76, 77). The reaction is also highly chemoselective for the insertion of the less hindered terminal alkenes over internal allylic ethers as exemplified with 77. Effective 1,3-allylic shift of the NHC–Ni intermediate can also be achieved without the erosion of enantioselectivity, which was found to be controlled by the steric properties of the substituents on the substrate. The 1,3-allylic shift equilibrium is also tunable with the electronic properties of the hetero-substituted terminal alkenes. Using a more electron-rich alkene (78) favours the 1,3-allylic shift diene product in excellent ee despite the 1-position being more hindered (adjacent to the gem-dimethyl and silyl ether moiety in 78). From the proposed stereochemical model of the transition state, the enantio-determining step is likely to be the hydronickelation of the diene, under the chiral environment of the NHC–Ni–H active species. Minimisation of the steric interactions between the substituents of the cyclic 1,3-diene and the cyclohexyl group of the C2-symmetric NHC ligand favours a face of nickel–hydride insertion in the conformation illustrated in Scheme 14. 1,3-Allylic shift of the allyl-nickel intermediate (if possible) then proceeds after nickel-hydride insertion. Subsequent regioselective insertion of the hetero-substituted terminal olefin (induced by the steric control of the NHC ligand) followed by β-H elimination affords the final enantioenriched allylic 1,4-diene product.
 | ||
| Scheme 15 Ni/chiral NHC catalysed enantioselective synthesis of 1,4-dienes by cross-hydroalkenylation of endocyclic 1,3-dienes and heterosubstituted terminal alkenes.66 | ||
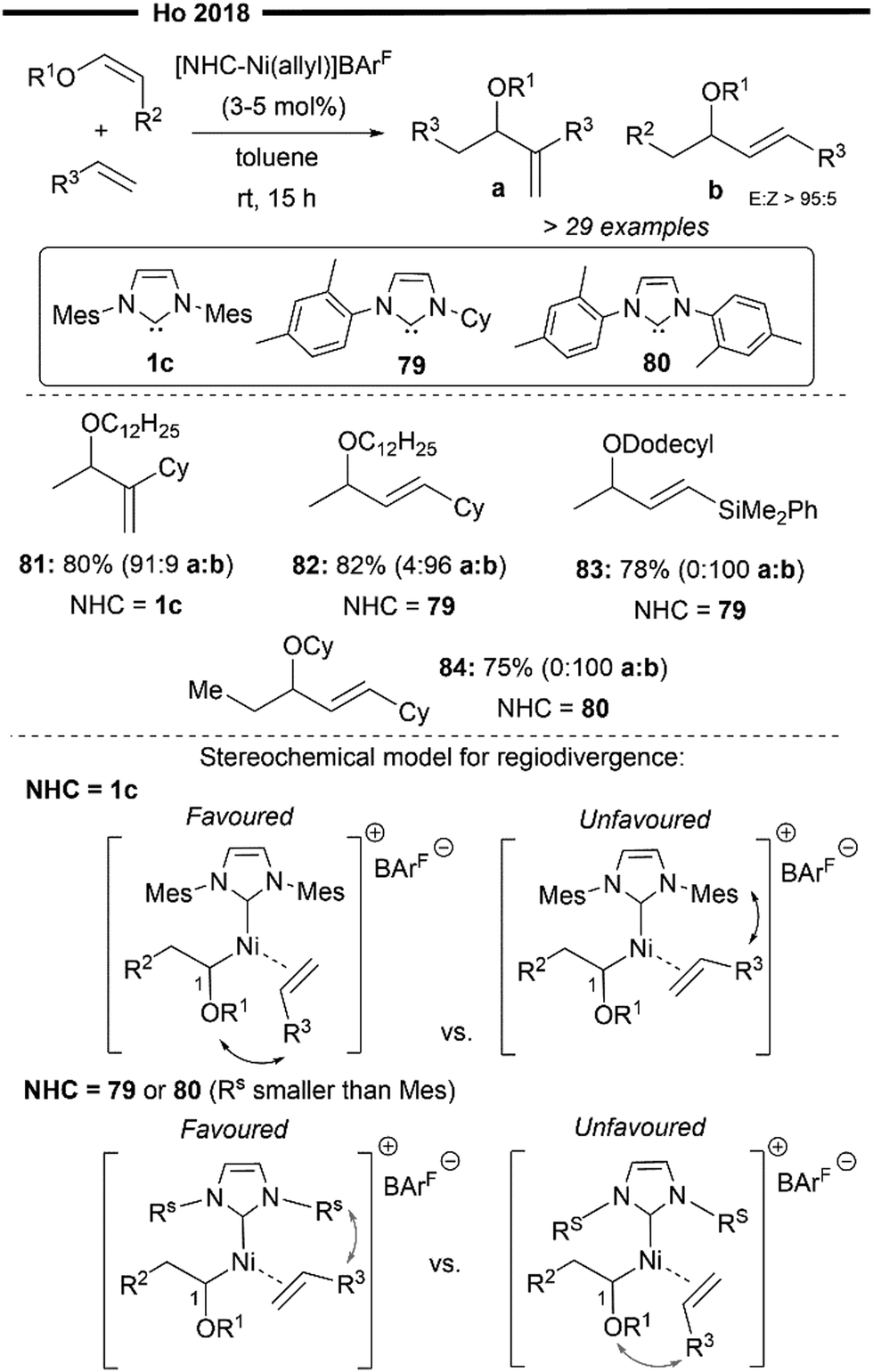
Leveraging the utility of [(NHC)Ni(allyl)]+BArF− as an efficient precatalyst for hydroalkenylation, Ho and co-workers successfully achieved a novel regiodivergent synthesis of 1,2- and 1,3-disubstituted allyl ethers in one step from vinyl ethers and terminal alkenes67 (Scheme 16). Under their mild protocol and by careful design of the NHC ligands (1c, 79 and 80 in Scheme 16), unwanted side reactions like cationic polymerisation, β-OR elimination and metal-catalyst deactivation (common for reactions with vinyl ethers)68 were effectively suppressed. This enabled electron-rich vinyl ethers to act as acceptors, leading to the formation of a new C(sp2)-C bond at the 1-position which delivers the allylic ether product, opening a new mode of reactivity for vinyl ethers.
 | ||
| Scheme 16 Ni/NHC catalysed regiodivergent cross-hydroalkenylation of vinyl ethers with α-olefins.67 | ||
Key elements of the reaction include the initial 2,1-hydronickelation (H at the terminal 2-position and Ni at the internal 1-position adjacent to the –OR group) of the vinyl ether substrate by the in situ generated [(NHC)Ni–H] active species. This suggests that the chemo- (coordination of vinyl ether over terminal alkene) and regio-selective (2,1-addition over 1,2-addition) hydronickelation is not controlled by the steric effects of the bulky NHC ligand, as the 2,1-addition of Ni–H leads to the formation of a more hindered 2° alkyl nickel intermediate. Rather, this step can be attributed to electronic control, favoured by the catalyst–substrate interaction. Subsequent regioselective migratory insertion of the terminal alkene was controlled by the steric interactions between the alkene substituent and the N-aryl substituent on the NHC (Rs being less bulky than mesityl), as illustrated in Scheme 16.
This allows for regiodivergence, where 1,2 or 1,3-disubstituted allyl ethers can be selectively obtained just by varying the nature of NHC ligands. The use of a bulky NHC ligand 1c allows for the selective formation of the 1,2-product (81, a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :b = 91:9), while the use of a sterically less demanding 79 and 80 favours the formation of 1,3-disubstituted allylic ether (82–84). 80 was designed to be less electron-rich than 79 while being less sterically bulky than 1c (absence of one methyl group of the N-aryl), which proved to be more efficient for the reaction of (Z)-configured internal vinyl ethers with terminal alkenes, delivering 84 with excellent chemo- and regio-selectivity. Even electron-deficient vinylsilanes (83) were compatible under their reaction protocol, despite being bulkier than the vinyl ether substrate. This further corroborates that the chemoselectivity for the reaction is largely governed by electronic effects, with cross-coupling being preferred over homo-coupling of the terminal alkenes/vinyl ethers.
:b = 91:9), while the use of a sterically less demanding 79 and 80 favours the formation of 1,3-disubstituted allylic ether (82–84). 80 was designed to be less electron-rich than 79 while being less sterically bulky than 1c (absence of one methyl group of the N-aryl), which proved to be more efficient for the reaction of (Z)-configured internal vinyl ethers with terminal alkenes, delivering 84 with excellent chemo- and regio-selectivity. Even electron-deficient vinylsilanes (83) were compatible under their reaction protocol, despite being bulkier than the vinyl ether substrate. This further corroborates that the chemoselectivity for the reaction is largely governed by electronic effects, with cross-coupling being preferred over homo-coupling of the terminal alkenes/vinyl ethers.
In 2020, Ho and co-workers devised a novel synthetic methodology for the synthesis of heterocycles, which involves the NHC/Ni-catalysed 1,3- and 1,4-diastereodivergent intramolecular hydroalkenylation of heteroenynes69 (Scheme 17). Although the authors did not explicitly detail the exact mechanism behind the origin of diastereoselectivity, preliminary mechanistic studies suggest the potential involvement of an NHC-nickelacyclopentene intermediate (illustrated in Scheme 17) instead of Ni–H insertion to the enyne. The different X heteroatom of the heteroenyne, in combination with the steric influence of the NHC, could favour a specific conformation leading to the diastereoselective formation of the nickelacyclopentene. Trapping of the NHC-nickelacyclopentene with an alcohol then results in a formal reductive hydroalkenylation process, leading to the expedient synthesis of various N- or O-heterocycles bearing an exocyclic olefin. The reductive hydroalkenylation reaction was shown to be effective with various substituted propargyl- or allyl–X (X = O or N) substrates, producing 1,3-syn/anti or 1,4-syn/anti products with high diastereoselectivity depending on the nature of X. Not limited to terminal alkenes, reductive hydroalkenylation with internal alkenes (85, 87) also proceeded efficiently. Notably, heterocycles with stereodefined quaternary centres can also be accessed using this strategy.
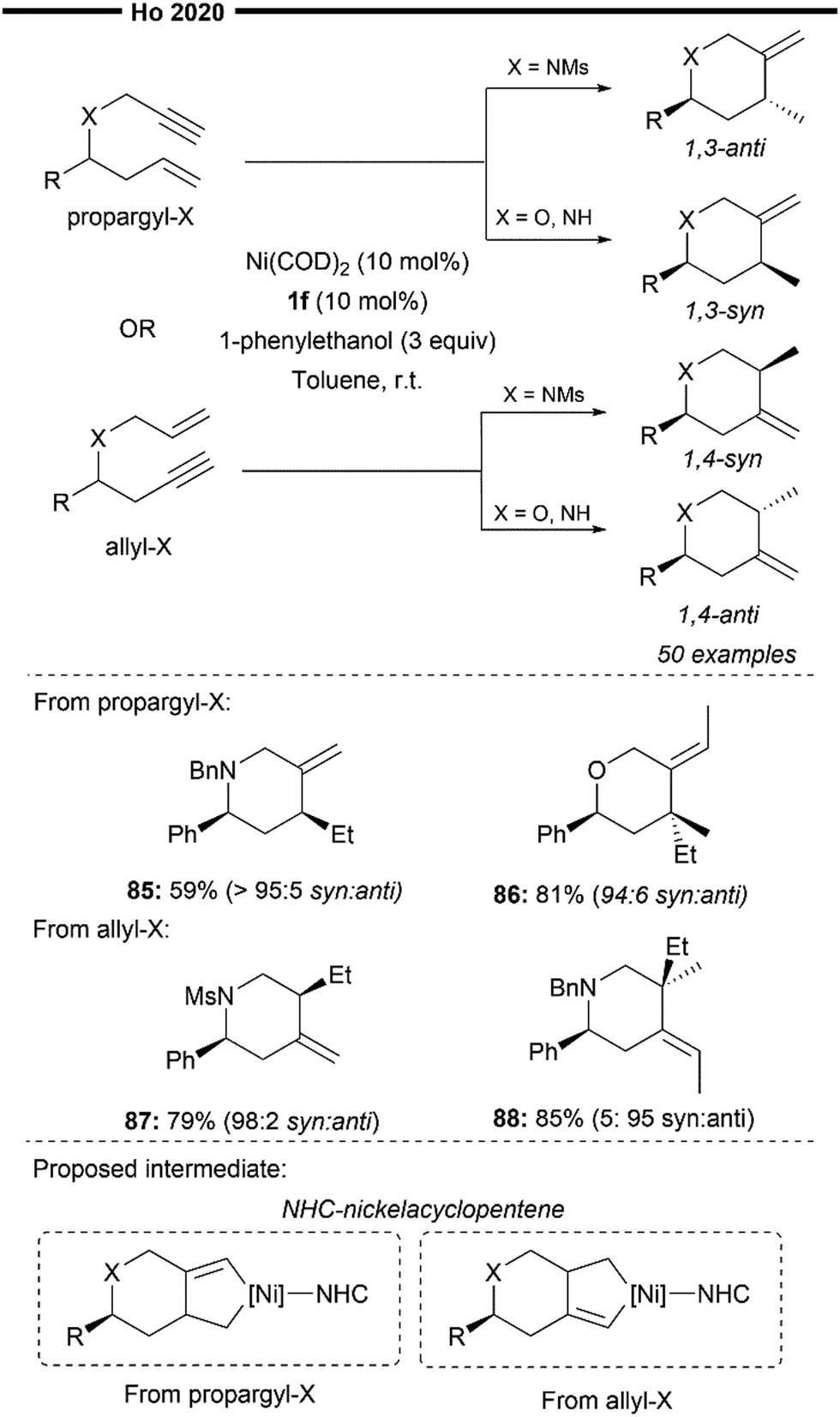 | ||
| Scheme 17 NHC/Ni-catalysed 1,3- and 1,4-diastereodivergent heterocycle synthesis from enynes.69 | ||
3.2 Hydroarylation of alkenes
Hydro(hetero)arylation, akin to hydroalkenylation, represents a straightforward method for the construction of C(sp3)–C(sp2) bonds and a means to access synthetically valuable alkyl(hetero)arenes from simple olefin feedstock. Traditional methods of hydroarylation involve the classical Friedel-Crafts type alkylation of electron-rich arenes,70 palladium-catalysed reductive Heck coupling of alkenes71 and transition metal-catalysed C–H oxidative addition – hydrometallation – reductive elimination sequence51 (Scheme 9b). The last of these methods includes examples of Ni/NHC catalysis which will be discussed in this section. The ligand-to-ligand hydrogen transfer (LLHT)-type mechanism and arylnickelation – nickel hydride reductive elimination are also possible pathways suggested for hydroarylation reactions, as supported with DFT calculations of the different transition state energies.Seminal work in this field was disclosed by Hiyama and co-workers in 2009 with the development of a C6-selective functionalisation reaction of pyridone derivatives – by insertion into unsaturated CC bonds – enabled with nickel/Lewis acid catalysis.72 This reaction also represents a formal hydroheteroarylation reaction of alkenes. While most of their optimisation studies and reaction scope mainly involves the use of a bulky, electron-rich triisopropylphosphine, two successful examples (89–90) using IMes 1c were also highlighted, demonstrating the effectiveness of NHC ligands in this process as well.
The N-heteroaryl substrate is first activated by the Lewis acid catalyst (AlMe3) upon coordination with the more basic carbonyl oxygen. η2-coordination of the activated pyridone ring with (NHC)Ni0 primed it for a subsequent oxidative addition of the C6–H bond, forming the heteroaryl-nickel hydride intermediate 92. Coordination of the 2-vinylnapthalene substrate to the Ni centre in 92, with the bulky IMes ligand oriented away from the naphthalene group (to minimise steric repulsion) forms 93, which undergoes a regioselective hydronickelation to furnish the π-allyl nickel intermediate 94. The stabilisation of the benzylic nickel 94 by the adjacent π-system of the naphthalene is presumably responsible for the observed regioselectivity in migratory insertion. Final reductive elimination of 94 affords the hydroarylated product.
In 2010, Hiyama and co-workers expanded upon their preliminary scope of Ni/Lewis acid co-catalysed hydroarylation of 2-vinylnaphthalene (Scheme 18)72 and reported a Ni/IMes-catalysed hydroheteroarylation reaction of vinylarenes, to furnish a diverse range of heteroaryl-bearing 1,1-diarylalkanes (Scheme 19).73 In contrast to their previous work, no Lewis acid co-catalyst is required for heteroaryl activation, possibly due to the use of relatively more electron-poor heteroarenes. A variety of heteroarenes were well-tolerated, including an enolisable ketone-containing indole derivative (97) and a benzothiazole (98). Electron-rich vinylarenes were also effective substrates (95). Notably, 1,2-disubstituted internal alkenes (96) also participated well in the reaction.
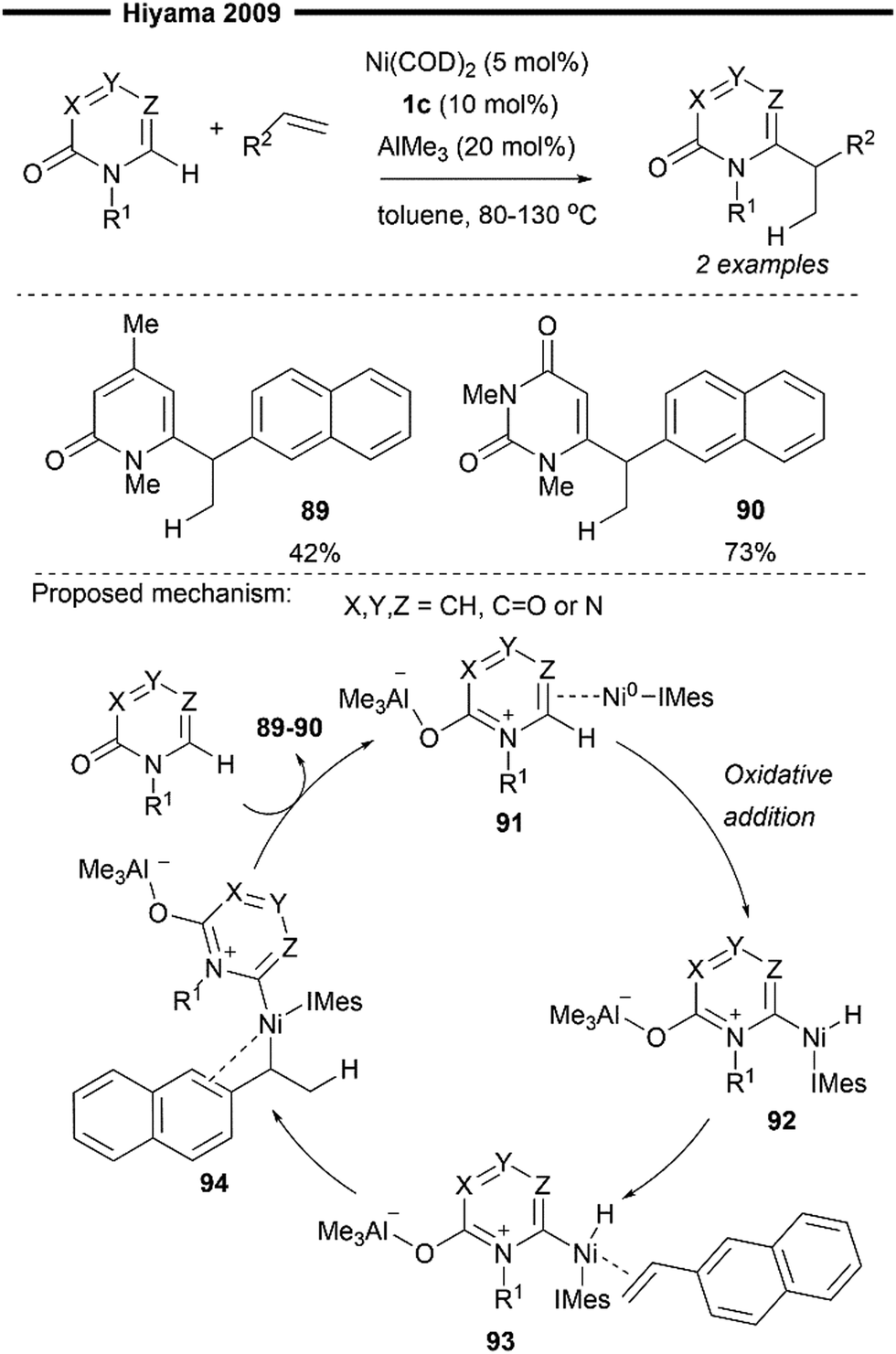 | ||
| Scheme 18 Hydroarylation of 2-vinylnapthalene with pyridone derivatives under (NHC)Ni/AlMe3 catalysis.72 | ||
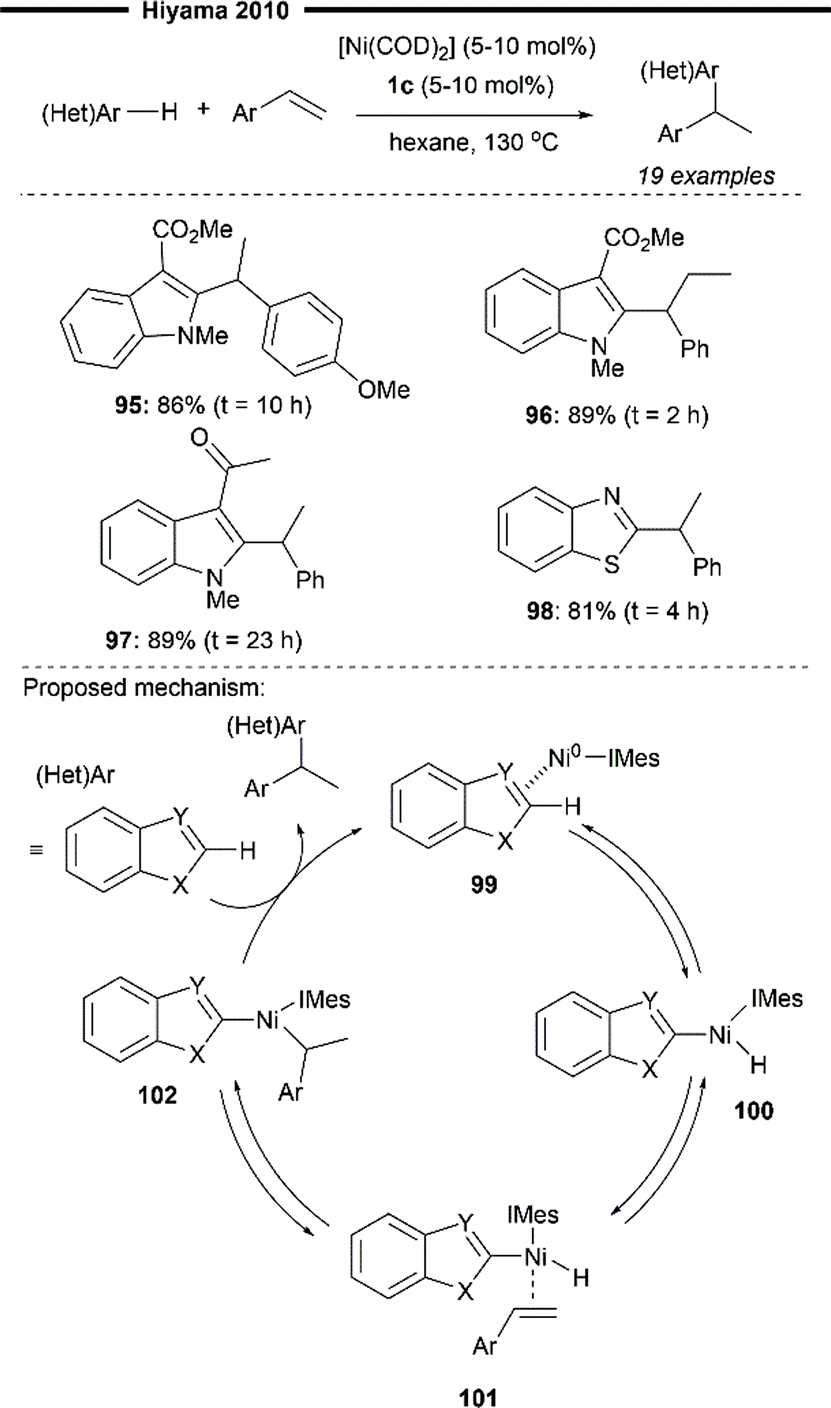 | ||
| Scheme 19 Ni/IMes-catalysed hydroheteroarylation of vinylarenes.73 | ||
Deuterium labelling experiments and previous mechanistic investigation into similar systems suggest a plausible catalytic pathway as illustrated (Scheme 19). Coordination of the heteroarene with Ni0/IMes forms the η2-arenenickel 99 which undergoes reversible oxidative addition of the C(sp2)–H bond of the heteroarene to give the nickel–hydride intermediate 100. A series of reversible coordination of the vinylarene substrate followed by a regioselective hydronickelation affords the 1-arylethylnickel 102, stabilised by the adjacent π-system. Lastly, irreversible reductive elimination of 102 regenerates the Ni/IMes species and releases the 1,1-diarylethane product.
In the same year, Hiyama and co-workers reported a (NHC)Ni/Lewis acid co-catalysed C-4-selective addition of pyridine across alkenes74 (Scheme 20). Direct alkylation of the C-4 position of pyridines has been more elusive and challenging to achieve compared to functionalisation at the C-2 position. Their protocol avoids the stoichiometric activation of the pyridine substrate and achieves direct C-4 alkylation under the steric control provided by both the highly bulky NHC ligand (IPr 1f) and the aluminium-based Lewis acid MAD (MAD = methylaluminium bis(2,6-di-tert-butyl-4-methylphenoxide)).
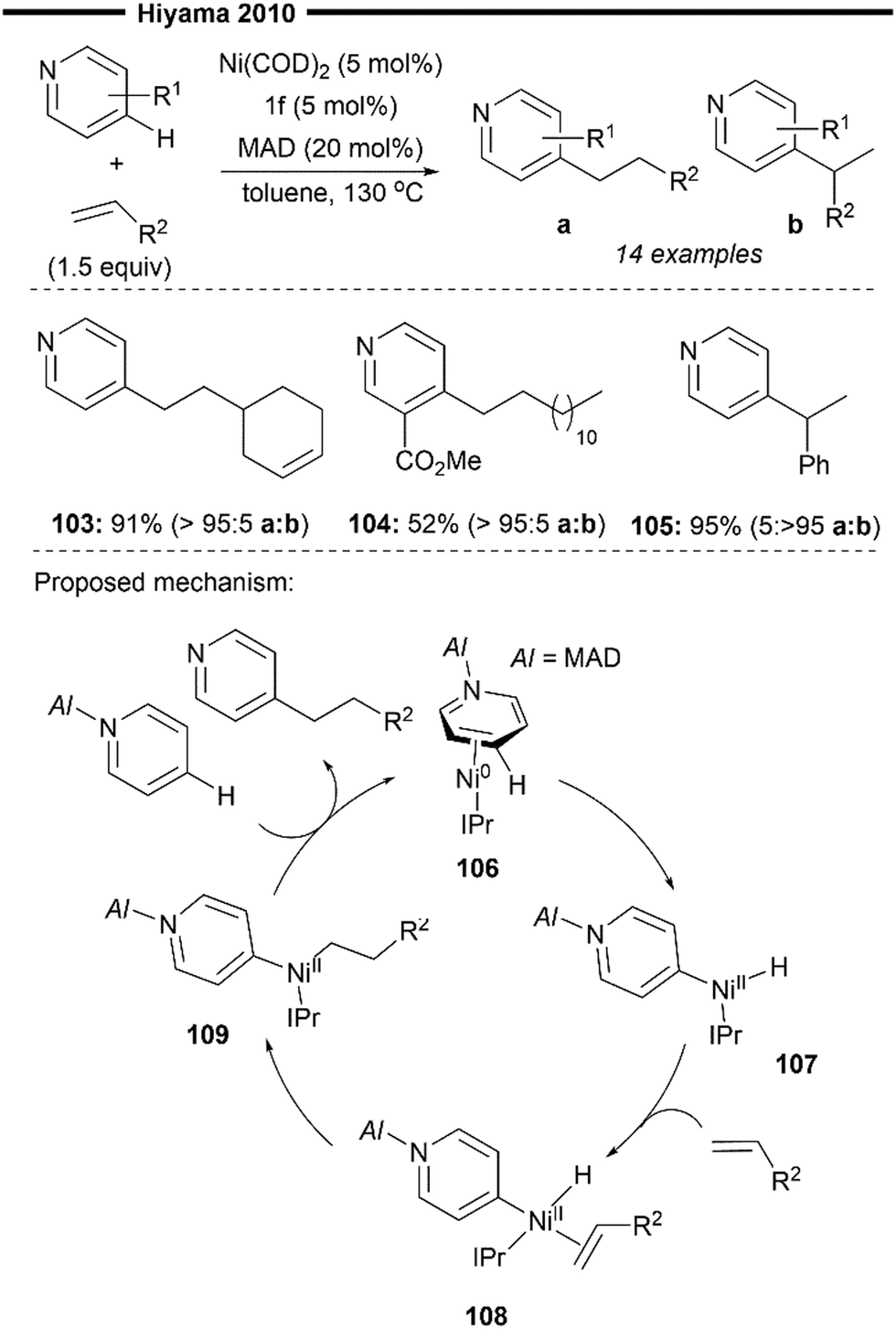 | ||
| Scheme 20 (NHC)Ni/Lewis acid catalysed selective C4-alkylation of pyridine/hydropyridylation of alkenes.74 | ||
Chemoselective addition to terminal alkenes over internal alkene was demonstrated (103), and no detectable second addition of pyridine was observed. Ester-containing pyridine derivatives like methyl nicotinate (104) also participated well in the reaction. Using styrene as a coupling partner leads to exclusive formation of the branched C-4 alkylated product, instead of the linear product obtained with aliphatic alkenes. This highlights the substrate-controlled regiodivergence in the insertion step, with the selective formation of a stabilised benzylic nickel intermediate (with vinylarene substrates) over the primary nickel intermediate (with an aliphatic alkene substrate).
A plausible reaction mechanism is illustrated in Scheme 20. Coordination of the Ni0/NHC with the activated pyridine substrate (after N-coordination to MAD) forms η2-arenenickel 106. Kinetically favoured oxidative addition of the C(4)–H bond of the activated pyridine substrate (preferred over the C(2)–H or C(3)–H bonds) forms the nickel–hydride intermediate 107. Regioselective hydronickelation into the aliphatic alkenes from the π-complex 108 forms the less sterically hindered primary alkylnickel intermediate 109 (if a vinylarene substrate was used, a π-stabilised benzylic nickel intermediate was preferentially formed instead73,74). The regioselectivity for insertion is solely governed by steric factors, due to the lack of electronic stabilisation of the branched 2° alkylnickel regioisomer (after insertion to aliphatic alkenes), in addition to the increased steric repulsion between the NHC and alkene substituent. Rate-determining, irreversible reductive elimination of 109 yields the final branched C4-alkylated pyridine product and regenerates 106.
More recently, building upon the pioneering efforts from Nakao and Hiyama,74 Shi and co-workers reported the enantioselective hydropyridylation of β-substituted styrene derivatives enabled by a cooperative chiral NHC/Ni–Al bimetallic catalysis.75 Similarly, the C(4)–H bond of the pyridine is selectively activated and added across an alkene, to deliver 1,1-diarylalkanes with high levels of regio- and enantio-control. Tunable bulky C2-symmetric chiral NHC – ANIPE-type ligand – as developed by their group76 was used, with the bulky 3,4-xylyl substituted ANIPE ligand 110 providing the best regioselectivity and enantioselectivity. The steric environment of bulky NHC is postulated to supress the undesired β-H elimination side reaction which gives the Heck product whilst facilitating reductive elimination.
The use of β-substituted styrenes as acceptors is crucial for efficient hydroarylation, possibly due to their weaker ability to coordinate to the nickel catalyst. Two molecules of unsubstituted styrenes were found to strongly coordinate to the nickel centre, deactivating the catalyst. cis-Substituted alkenes also result in poorer regioselectivity (with a greater proportion of a linear anti-Markovnikov product being formed) and enantioselectivity, likely due to the smaller steric repulsion between them and the NHC ligand. The authors performed detailed DFT first-principles calculations to elucidate the mechanism and the origin of the enantioselectivity. Supported with the results from previously done calculations,77 their calculations also suggest a reaction pathway involving a ligand-to-ligand hydrogen transfer (LLHT) mechanism, contrary to the stepwise oxidative addition-hydronickelation pathway as suggested by Hiyama and co-workers.74 LLHT involving the ortho or meta-C–H bonds was also less feasible due to a higher energy barrier, which supports the observed regioselectivity. Calculated energies of the transition state for the rate- and enantio-determining LLHT process (Scheme 21) also revealed the lower energy barrier incurred in the formation of the (S)-isomer over the (R)-isomer due to the stronger π–π stacking between the NHC aryl ring and the coordinated trans-styrene. The computed relative energies align well with the experimental enantiomeric ratio, where the (S)-isomer is the dominant product.
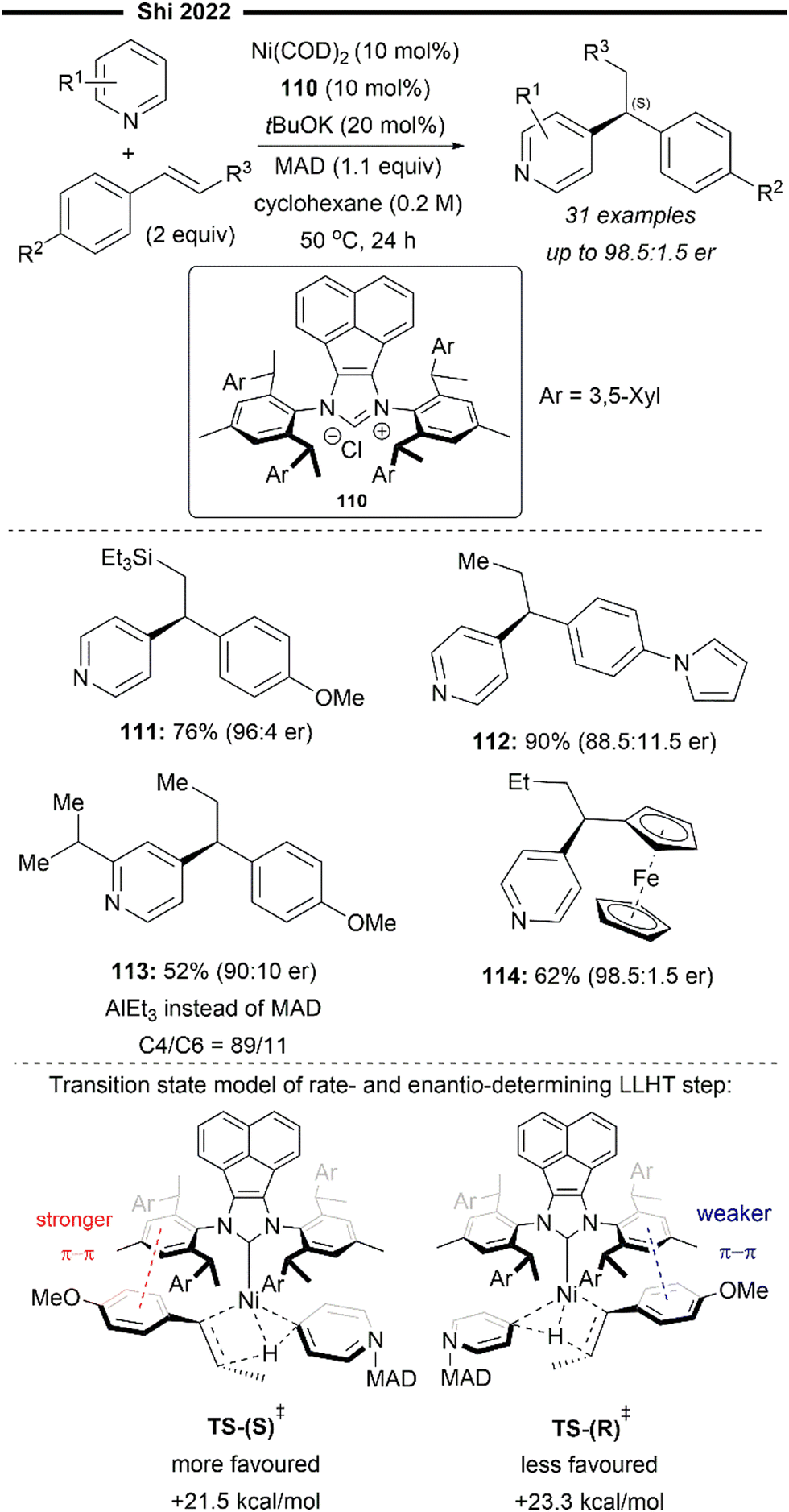 | ||
| Scheme 21 Enantioselective (NHC)Ni/Lewis acid-catalysed hydroarylation of styrene/C4-alkylation of pyridines.75 | ||
β-Silyl-substituted styrenes (111) and medicinally relevant N-heterocycles (112) were effective substrates, delivering the enantioenriched 1,1-diarylethane product in high yields and enantiomeric ratio. Ferrocene-containing alkene also participated well (114). Utilisation of an ortho-substituted pyridine substrate (113) which presents increased steric hindrance led to greater complexity, potentially attributable to the inhibition of coordination with the aluminium Lewis acid. Switching to a less bulky triethylaluminium Lewis acid was imperative in catalysing the required transformation, albeit with a lower yield due to formation of a competing C6-alkylated side product.
Hiyama and co-workers continued their efforts towards NHC–nickel/Lewis acid co-catalysed hydroarylation of alkenes, this time enabling the selective formation of linear alkylated pyridones78 (Scheme 22). This work explores the novel use of unactivated alkenes as viable coupling partners instead of activated vinylarene-type substrates. The optimal NHC ligand of choice is 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr 1f), in synergy with MAD as the Lewis acid co-catalyst, which efficiently delivers the linear hydroarylation product in good yields and selectivity. Pyridone derivatives like 1-methyl-quinazolone delivered the desired alkylated product (115) in good yields. Bulky vinylsilanes (116), gem-disubstituted alkenes (117), or cyclohexene (118) also participated well, and the linear products were obtained in high yields. The reaction mechanism is analogous to their previous report,74 where the linear selectivity originates from the regioselective insertion to form the less sterically hindered primary alkylnickel intermediate. With no electronic stabilisation possible (for unactivated aliphatic alkene substrates), this step is solely governed by steric control from the bulky IPr NHC.
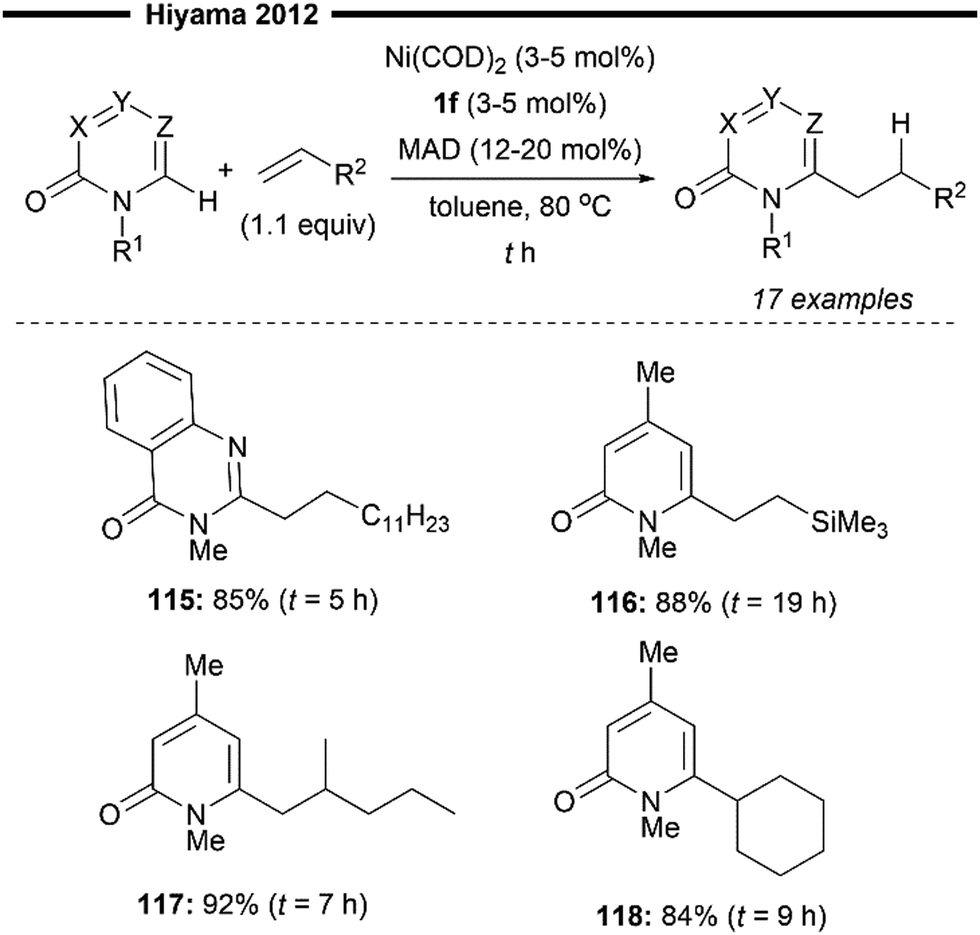 | ||
| Scheme 22 (NHC)Ni/Lewis acid catalysed formation of linear alkylated pyridones.78 | ||
In 2012, Ong and co-workers reported the heteroaromatic C–H activation of benzimidazole derivatives following a vinylarene insertion step, enabled by cooperative (NHC)Ni/Lewis acid catalysis66 (Scheme 23). This is, in essence, a hydroheteroarylation reaction of styrene. While the work of Hiyama et al.59,65 predominantly uses substrate control for the selective formation of branched (with vinylarene as a substrate) or linear (with aliphatic alkenes as a substrate) hydroarylated products, Ong and co-workers developed a chemical regioselective switch to selectively obtain either the linear or branched product from the same vinylarene substrate. Their system utilises an amino-NHC (with a pendant tert-butyl amino arm) ligand, with the chemical switch being the trimethylaluminum AlMe3 Lewis acid. Addition of the AlMe3 co-catalyst forms the (NHC)Ni–Al bimetallic complex which enforces a steric environment favouring the formation of a linear insertion product while the absence of AlMe3 switches the regioselectivity, favouring the branched-selective product probably due to the favourable electronic stabilisation of the benzylic nickel intermediate (120vs.121 and 122vs.123), which was established in previous reports, as well.59,60,62
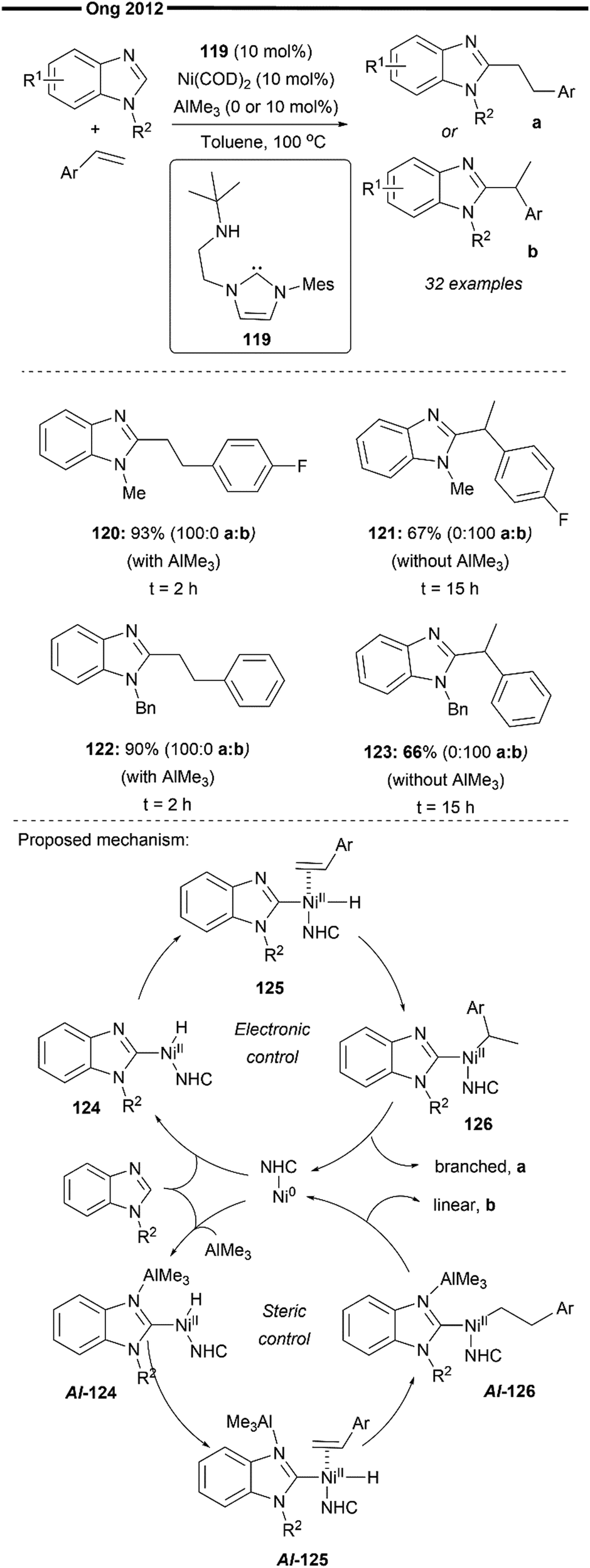 | ||
| Scheme 23 Regioselective switching for the linear/branched hydroarylation product via (amino-NHC)/Ni–Al synergistic catalysis.79 | ||
In-depth mechanistic studies to probe the nature of this regioselective switch enabled with AlMe3 were also conducted by the authors,67 with the proposed mechanism as illustrated in Scheme 23. There are two distinct pathways differing in the regioselectivity of insertion (in the presence or absence of AlMe3). Generally, a concerted oxidative addition of the C–H bond of the benzimidazole substrate generates the nickel–hydride intermediate Al-124. Facile coordination of the styrene substrate followed by regioselective hydronickelation yields the linear primary nickel species (Al-126), as controlled by the steric environment around the NHC–Ni/Al bimetallic catalyst or an electronically stabilised branched benzylic nickel species (126) in the absence of AlMe3. Reductive elimination of 126 under electronic control and Al-126 under steric control delivers the branched and linear products, respectively. Notably, the addition of AlMe3 was found to accelerate the hydroarylation reaction, as evident from the increased product yields and lower reaction times (120vs.121 and 122vs.123). The bidentate amino-NHC with a reversibly coordinating tert-butyl amino pendant group as well as the nature of the R2 substituent on the benzimidazole substrate could also play an important role in providing the additional steric pressure around the nickel complex, hence increasing the sterically-controlled linear selectivity.
In 2013, Ong and co-workers reported the first (NHC)Ni-catalysed hydroarylation reaction of allylarenes that operated under a tandem alkene isomerisation-heteroaryl C(sp2)–H bond activation process80 (Scheme 24), selectively obtaining the branched 1,1-di(hetero)arylethane product. A linear hydroarylated product can also be selectively achieved by using a chemical toggle (Lewis acidic AlMe3)79 as previously described, which effectively retards alkene isomerisation while promoting the hydroarylation process. Naturally occurring compounds like safrole participated well, delivering the branched (128) and linear (127) products with high selectivity. Other heteroarenes like imidazoles (129) or indoles (130) were also well-tolerated.
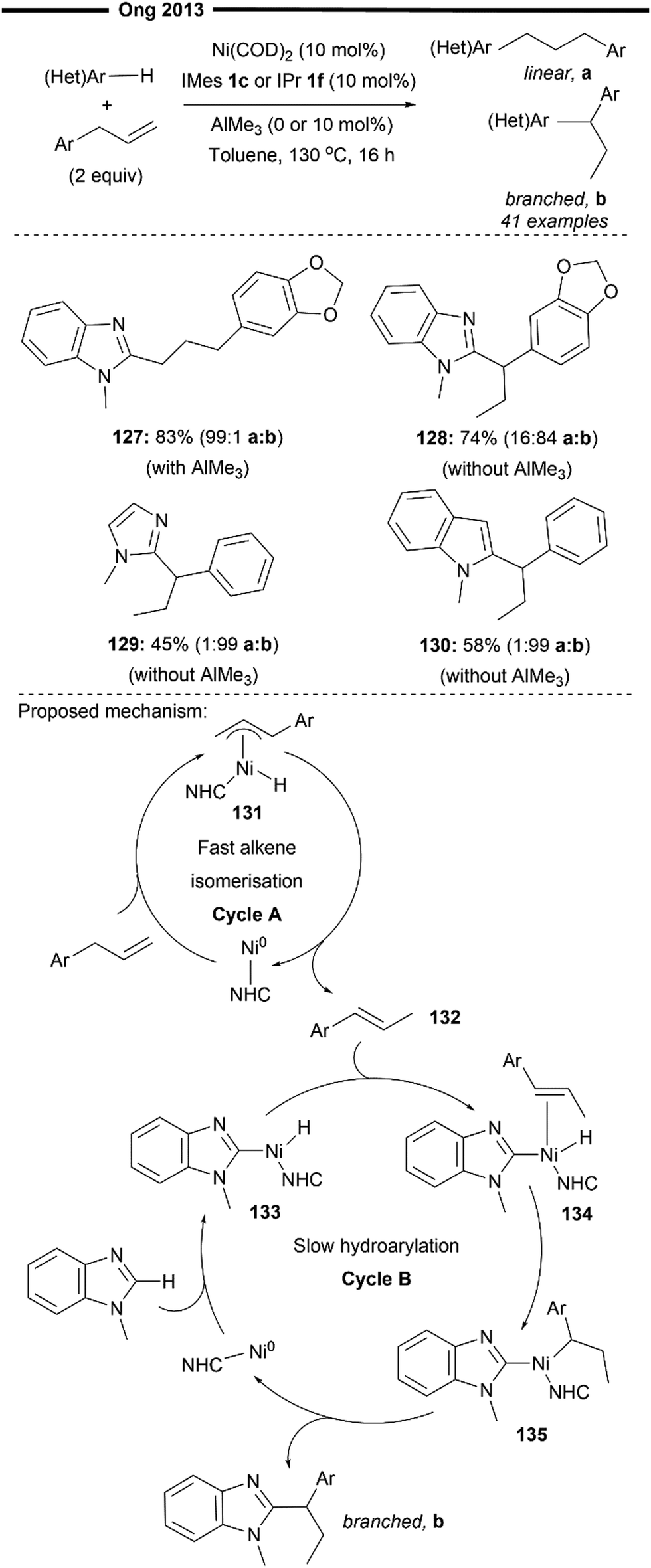 | ||
| Scheme 24 Regioselective hydroarylation of allylarenes via tandem alkene isomerisation and C–H bond activation.80 | ||
The criterion for an efficient branched-selective reaction hinges on the alkene isomerisation cycle being relatively faster than the C–H bond activation process, allowing for the two catalytic cycles to operate independently. Preliminary mechanistic studies by the authors and inspiration from previous similar literature studies81 suggest a mechanism with two operating catalytic cycles (Scheme 24, cycle A and B), involving a more facile alkene chain-walking mechanism (cycle A) and a hydroarylation mechanism (cycle B). In cycle A, oxidative addition of the allylic and benzylic C–H bond forms the η3-π-allylnickel hydride intermediate 131, which undergoes a formal 1,3-hydride shift to form the thermodynamic internal alkene 132. Cycle B is initiated with the oxidative addition of the C–H bond of the heteroarene to form the arylnickel hydride complex 133. Coordination of the isomerised alkene 132 to the nickel hydride intermediate 133 followed by the regioselective hydronickelation forms the electronically stabilised benzylic nickel intermediate 135. Facile reductive elimination of 135 affords the final branched hydroheteroarylation product, regenerating the active catalyst. The pathway responsible for the formation of the linear product follows an analogous mechanism outlined in Scheme 23 (under steric control). Addition of the chemical trigger AlMe3 increases the steric bulk around the (NHC)Ni–Al bimetallic active catalyst (NHC = IMes 1c or IPr 1f), which deactivates alkene isomerisation and further enhances the selectivity of migratory insertion to form the less hindered primary alkylnickel intermediate, leading to the formation of the linear product.
Ong and co-workers further expanded on their previous work on bimetallic (NHC)Ni/Al catalysed hydroarylation reaction of allylbenzenes80 and achieved convenient access to both linear or branched hydropyridylation products using their regiodivergent protocol82 (Scheme 25). The para-C–H bond of pyridines was selectively activated and inserted into allylbenzene substrates, following a similar mechanism to that described in Scheme 20 (for linear selective insertion) and Scheme 24 (tandem alkene isomerisation and branched selective insertion).
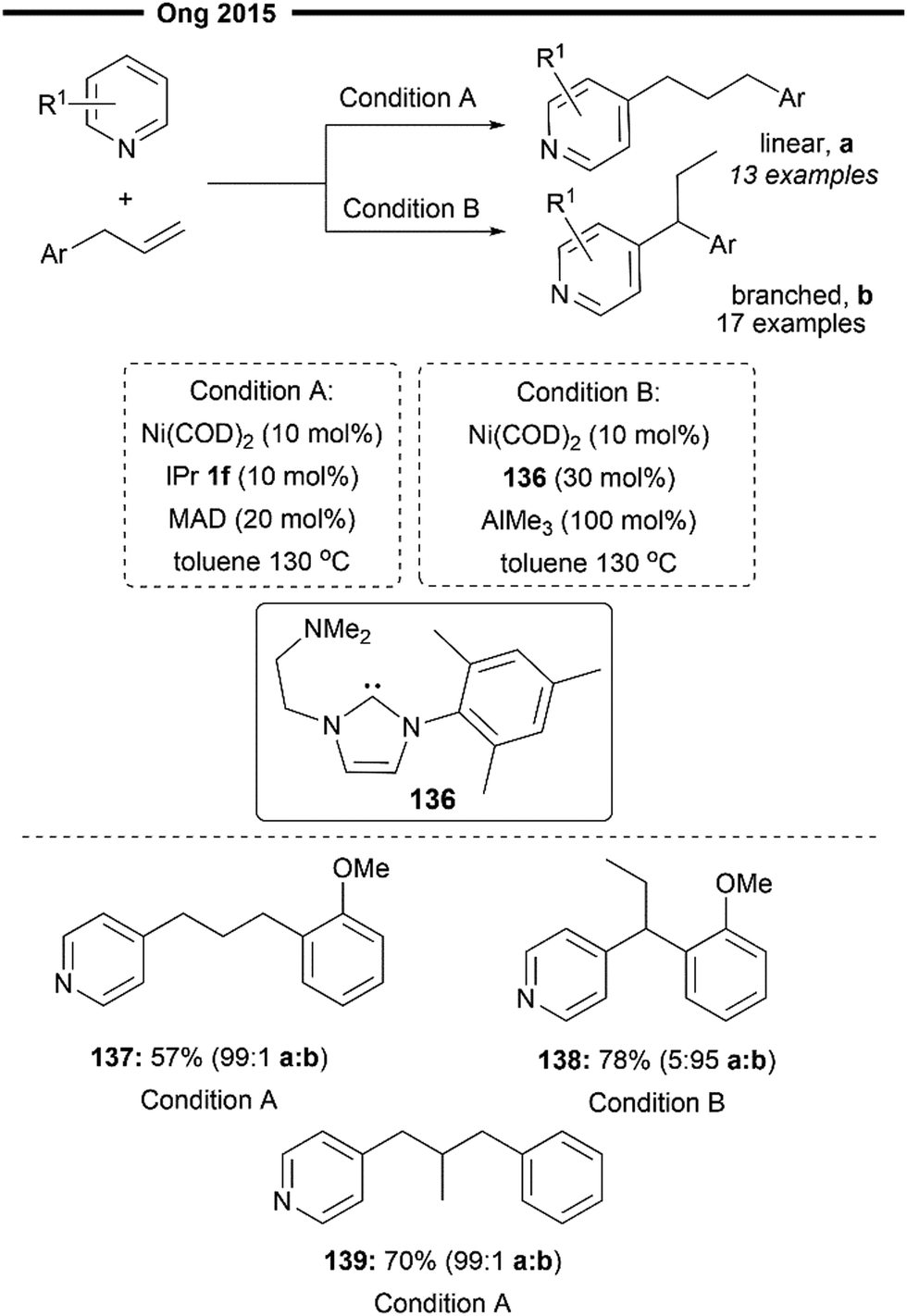 | ||
| Scheme 25 (NHC)Ni/Lewis acid-catalysed hydropyridylation of allylarenes with switchable regioselectivity.82 | ||
Unlike their previous work, this regiodivergence does not arise from the absence or presence of the aluminium Lewis acid as a chemical trigger,80 but rather from the size of the NHC ligand/Lewis acid pair. Using a very sterically demanding NHC ligand and a Lewis acid (i.e., condition A with IPr 1f and MAD) was found to favour the formation of a linear hydropyridylation isomer 137 via the less hindered migratory insertion process, with the suppression of alkene isomerisation as well. On the other end, employing a less bulky NHC/Lewis acid pair (i.e., condition B with amino-NHC-2 and AlMe3) toggles the isomerisation of allylbenzene to the internal alkene before hydroarylation takes place, delivering the final product with high branched selectivity 138. Subjecting gem-disubstituted allylbenzene to the reaction (condition A) also afforded the desired product 139 without diminishment in positional selectivity.
In 2017, Mandal and co-workers reported the use of an abnormal NHC (aNHC) ligand in combination with a nickel(0) precatalyst to promote the hydroheteroarylation of vinylarenes with benzoxazole derivatives, resulting in the exclusive formation of the branched 1,1-diarylalkane product (Scheme 26).83 This is a novel application of a non-Arduengo-type NHC ligand for alkene functionalisation (hydroarylation) despite its emerging utility in organometallic catalysis. Preliminary mechanistic studies led to the observation and isolation of a Ni(II) cyclooctenyl complex 145 (characterised with NMR and X-ray crystallography), formed by the reaction of the free aNHC ligand and Ni(COD)2. Reduction of this NiII–aNHC precatalyst forms the active Ni0–aNHC species, which initiates the reaction by the oxidative addition of the C–H bond (of benzoxazole). Subsequent migratory insertion of aNHC–nickel hydride into vinylarene forms an electronically stabilised benzylic nickel intermediate, which then undergoes reductive elimination to form the desired branched hydroheteroarylation product, overall similar to previously postulated hydroarylation mechanisms.82,84 While electronic perturbation of the benzoxazole and styrene substrates with the introduction of electron donating or withdrawing groups does not significantly affect the reaction yields (141–144), the scope of the heteroarene is largely limited to derivatives of benzoxazole only.
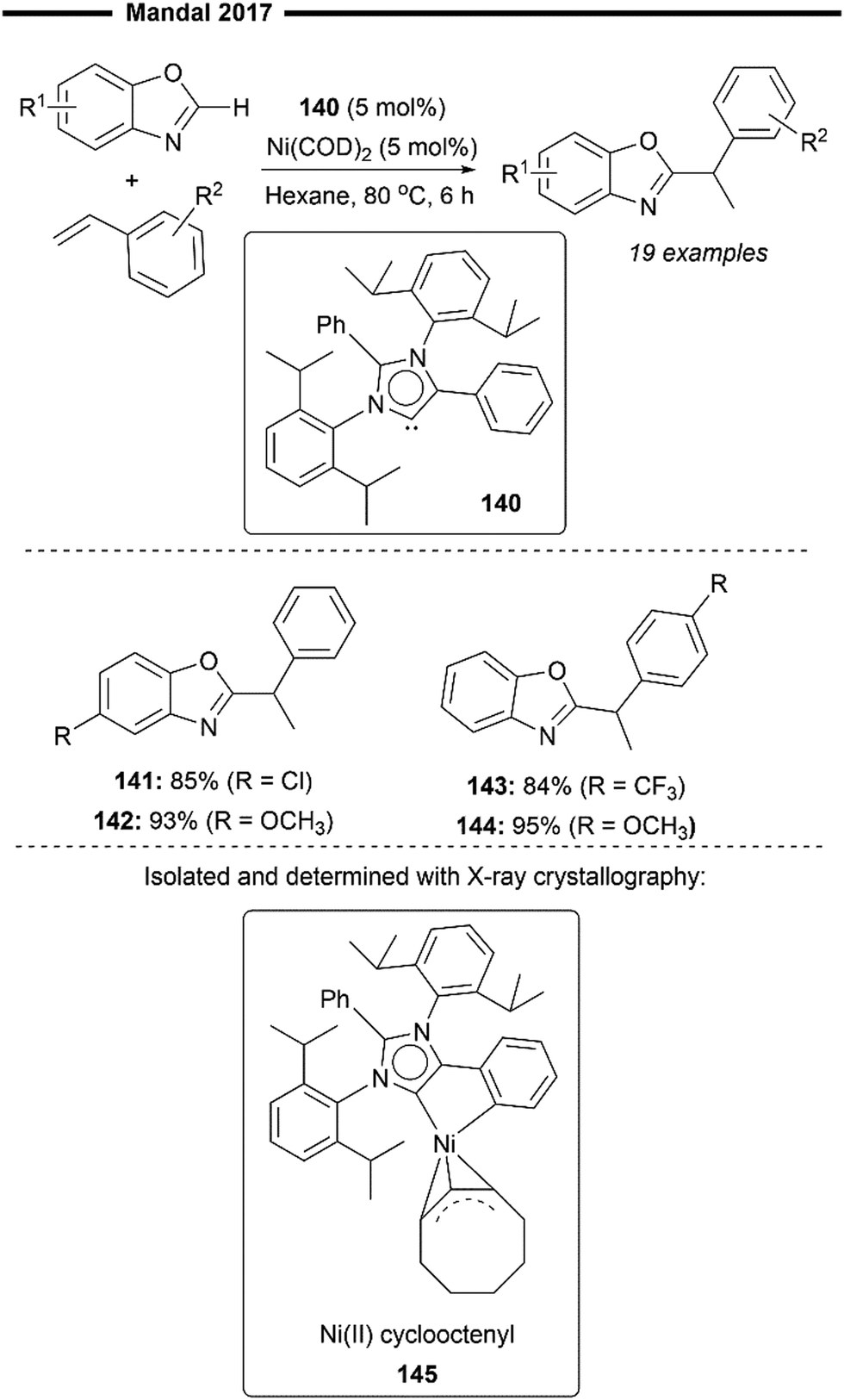 | ||
| Scheme 26 Abnormal NHC/Ni-catalysed hydroheteroarylation of vinylarenes.83 | ||
In 2012, Ananikov and co-workers presented a novel catalytic system for the hydroheteroarylation of alkenes. Their catalytic system utilises a commercially available bench stable Ni(Cp)2 (bis(cyclopentadienyl)nickel) precatalyst 147, an NHC salt as a pro-ligand, and sodium formate as a non-toxic reductant to generate the active Ni0 catalyst85 (Scheme 27, condition A). This approach presents an alternative methodology which obviates the necessity for the utilisation of highly air and moisture sensitive Ni(COD)2 or (NHC)Ni0 catalysts, which are commonly employed in many Ni/NHC-catalysed hydrofunctionalisation reactions. The pre-synthesised well-defined Ni0/NHC complex [(IMes)Ni(Cp)Cl] 146 was found to be equally efficacious in their hydroheteroarylation system.
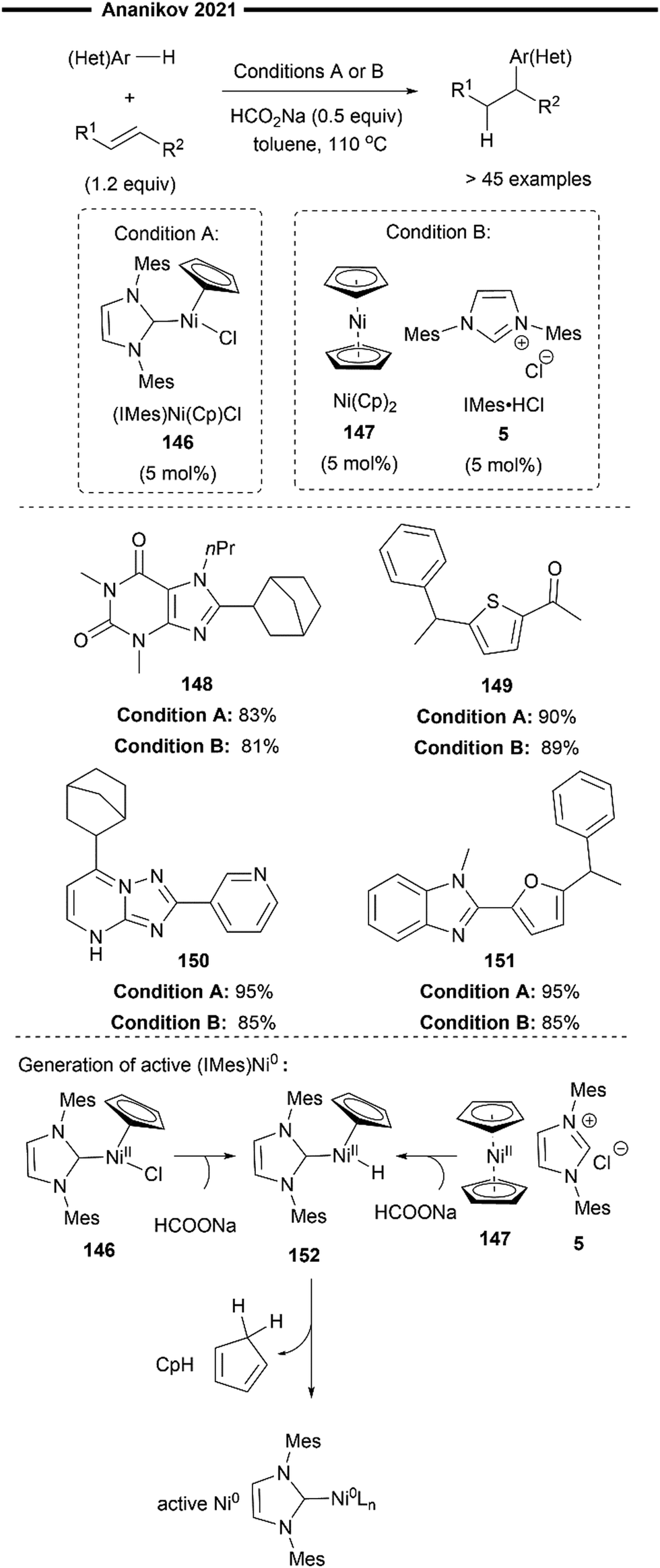 | ||
| Scheme 27 Hydroheteroarylation of alkenes with air-stable nickelocene and NHC salt as a precatalyst.85 | ||
The cyclopentadienyl (Cp) ring of the precatalyst was postulated to serve as an internal mild base for the deprotonation of the azolium salt (NHC proligand), thereby facilitating Ni–NHC bond formation and circumventing the need for the addition of strong external bases that may cause the decomposition of base-sensitive substrates. Sodium formate was utilised as a mild reducing agent in the in situ reduction of the Ni(II) precatalyst to the commonly proposed active species, Ni(0). The widely accepted mechanism for this catalytic activation process86 involves the initial ligand exchange of the formate anion with one Cp ligand. Facile decarboxylation yields a nickel–hydride intermediate 152. Reductive elimination of this nickel–hydride intermediate then forms the active NHC–Ni(0) species with concomitant release of cyclopentadiene. The proposed mechanism for the hydroarylation, as put forth by the authors, is in agreement with previous works,73,74 proceeding through a sequential oxidative addition of the C(sp2)–H bond, regioselective hydronickelation and reductive elimination.
Activated vinylarenes and norbornenes serve as representative substrates for the scope of alkenes. A wide variety of heterocycles including thiophene 149 and furans 151 were well-tolerated. Notably, pharmaceutically relevant heterocycles such as theophylline 148 (n-propyl derivative of caffeine) and 1,2,4-triazolo[1,5-a]pyrimidine derivatives (proposed surrogates of purines) 150 were demonstrated to proceed efficiently under either reaction conditions.
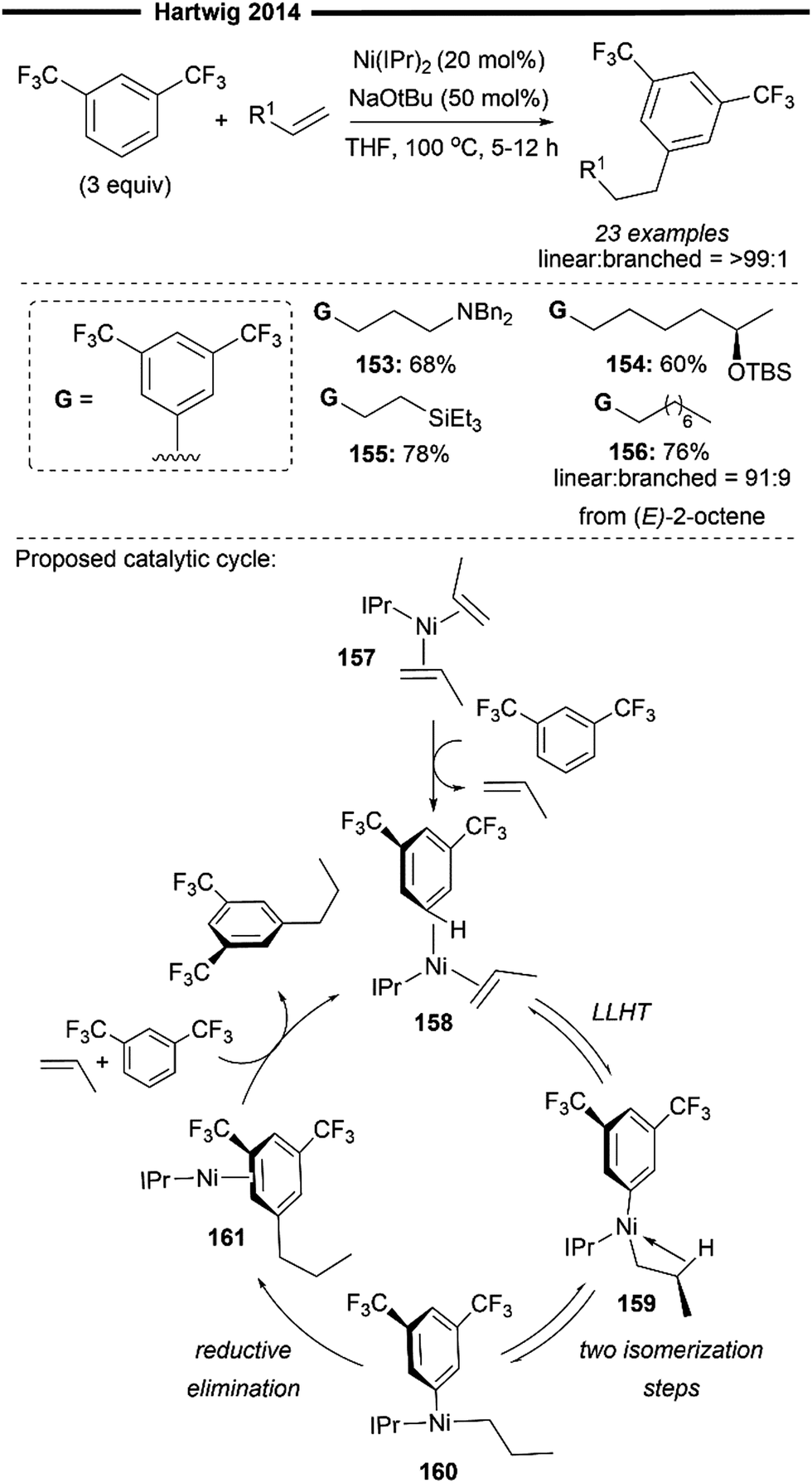
The Hartwig group has made significant contributions to the field of alkene activation via hydroarylation, with regard to the utilisation of non-activated aliphatic alkenes. In 2014, they achieved initial success through the reaction of unactivated alkenes with electron-poor trifluoromethyl-substituted arenes, obtaining anti-Markovnikov alkylarene products with remarkable regioselectivity (Scheme 28).87 Optimal results were obtained using the bulky IPr 1f ligand as the NHC ligand in the bis(NHC)Ni0 precatalyst employed for reaction. The regioselectivity of the terminal arylated product appears to be primarily driven by steric factors, with the aryl group being introduced at the least hindered position, untethered by any directing moieties.
 | ||
| Scheme 28 NHC/Ni-catalysed linear-selective hydroarylation of unactivated terminal and internal alkenes with trifluoromethyl arenes.87 | ||
The reaction proved to be efficacious with a diverse array of substituted olefins like vinylsilanes (155) and allylic amines (153). Optically pure secondary siloxyethers also reacted smoothly, yielding the product (154) without erosion of stereochemistry. Notably, internal alkenes (such as (E)-2-octene) also underwent hydroarylation to furnish terminal arylated products (156), implying that an alkene isomerisation occurs prior to hydroarylation. Deuterium labelling studies indicates that the isomerisation does not follow a chain-walking pathway involving proton-transfer steps with the arene. Although the precise mechanism for alkene isomerisation remains elusive, it is suggested that the nickel-catalyst selectively coordinates to the more reactive terminal alkene, leading to hydroarylation. As such, isomeric mixtures of internal alkenes could still be utilised in the reaction since they would eventually converge to the terminal alkenes from the equilibrium mixture.
Mechanistic studies and DFT calculations have shed light on a plausible reaction pathway (Scheme 28). Oxidative addition of the aryl C–H bond followed by hydronickelation of the alkene from an unstable, intermediary nickel–hydride species was computed to be much less favourable than a concerted ligand-to-ligand hydrogen transfer (LLHT) pathway (Scheme 9a). Coordination of the arene to bis(alkene)-coordinated resting state nickel 157 forms the active complex 158, which undergoes a reversible LLHT to afford the alkylnickel-aryl intermediate 159 that is stabilised by a β-C–H agnostic interaction. 159 subsequently undergoes a two-step isomerisation to form a T-shaped intermediate 160, in which the hydrocarbyl groups are now positioned mutually cis to each other. Turnover-limiting reductive elimination forms the desired C(sp2)–C(sp3) bond, yielding the product-coordinated nickel complex 161. Release of the linear hydroarylation product through ligand exchange with incoming substrates regenerates the active nickel species 158. It is noteworthy that the excellent linear selectivity observed results from the lower energy barrier for reductive elimination from the primary alkylnickel complex, rather than the absolute difference in computed free energy between the linear or branched alkylnickel complex.
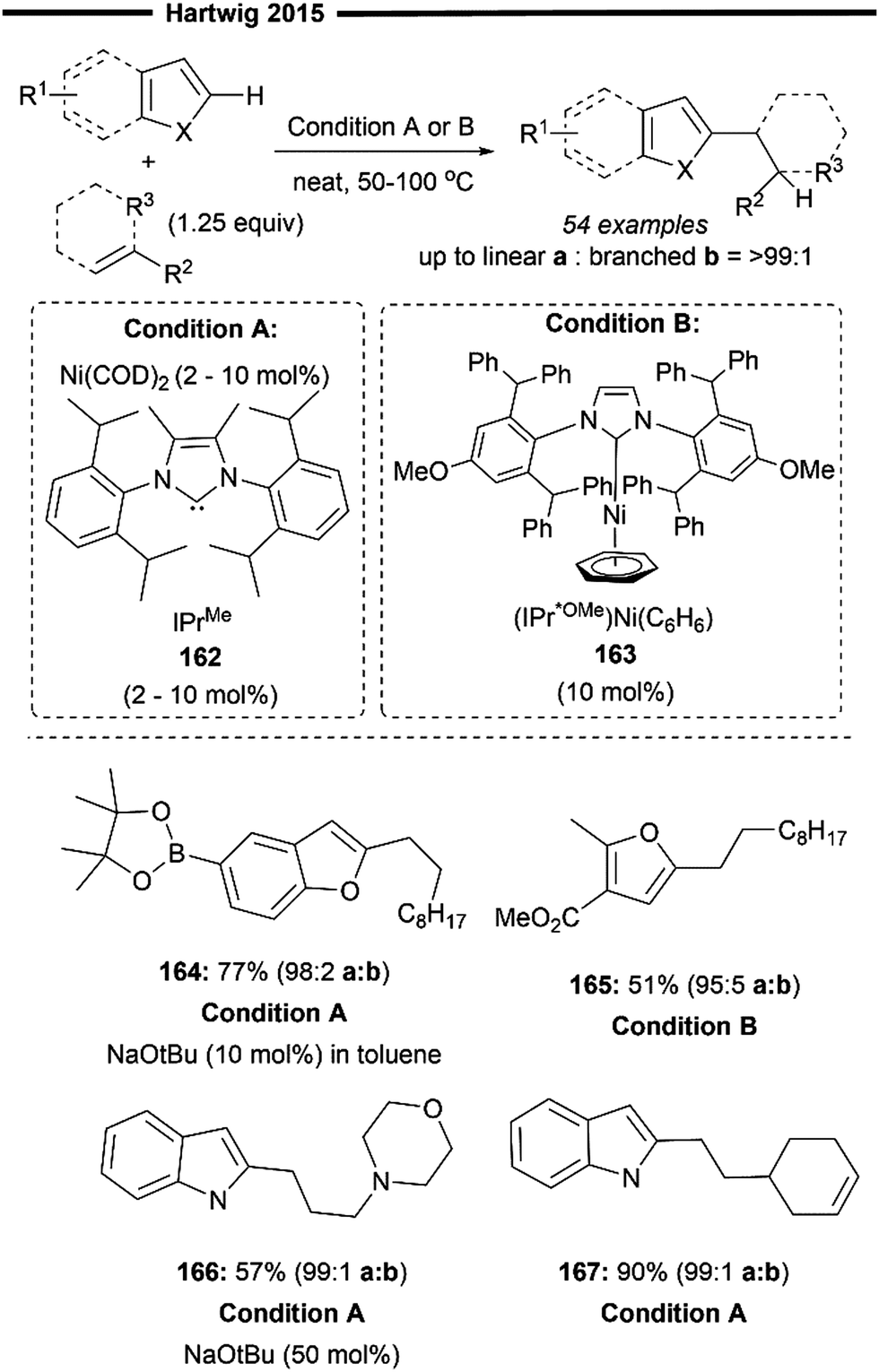
Later in 2015, Hartwig and co-workers expanded upon their previous findings,87 by broadening the substrate scope beyond just trifluoro-substituted arenes. In that work, utilising a more sterically bulky IPrMe162 or IPr* ligand, they achieved regioselective hydroheteroarylation of unactivated alkenes with a wide range of heterocycles (Scheme 29)88 under solvent-free conditions using the Ni(COD)2 precatalyst and the free carbene ligand. The linear alkylheteroarene product was generally obtained in high yields with high anti-Markovnikov selectivity. Alternatively, the use of a pre-synthesised IPr*OMe-ligated Ni-benzene complex 163 was also efficient, sometimes yielding better results. Effective hydroheteroarylation was observed with benzofurans, furans and indoles as heterocyclic coupling partners. Boronate esters (164) and potential coordinating esters (165) were all well-tolerated and led to the formation of the C2-selective linear hydroheteroarylation product. With respect to the scope of alkenes, a polar amine group at the allylic position gave moderate yields of the linear product (166). Non-conjugated 1,5-dienes 167 also underwent chemoselective hydroheteroarylation at the less substituted terminal olefin unit, leaving the internal cyclic olefin unreacted. The postulated reaction pathway involves an initial LLHT between the bound arene and alkene facilitated by the NHC/Ni catalyst, followed by isomerisation and cis-reductive elimination, as previously described.87
 | ||
| Scheme 29 Ni/NHC-catalysed anti-Markovnikov hydroheteroarylation of unactivated alkenes with N- and O-heterocycles.88 | ||
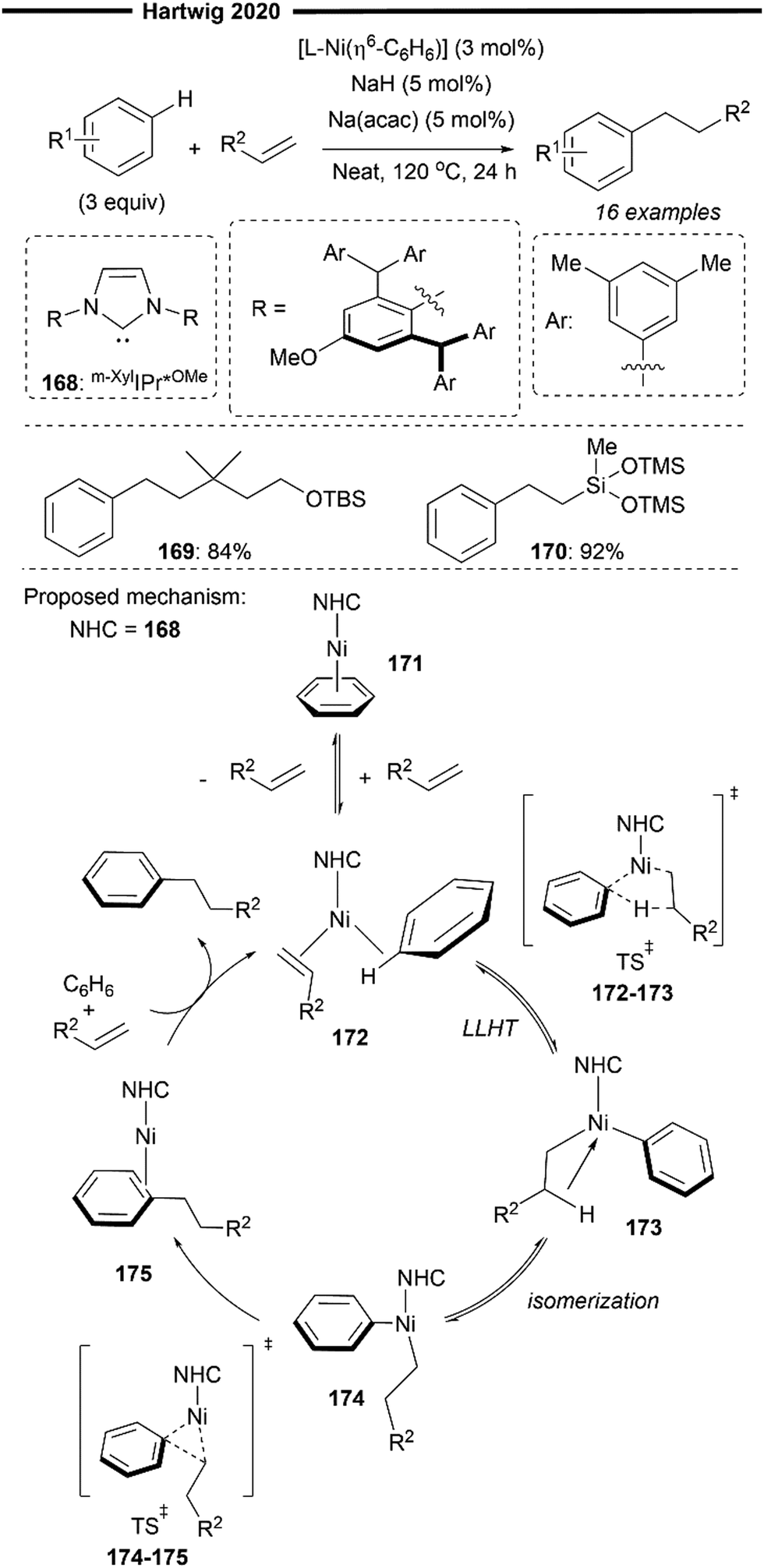
Going beyond selective hydroarylation of challenging unreactive aliphatic alkenes, Hartwig and co-workers extended their work by developing a Ni/NHC catalysed linear-selective hydroarylation of unactivated alkenes with unactivated arene substrates, facilitated by non-covalent interactions (Scheme 30).89 The use of the benzene-bound NHC-ligated nickel complex [NHC–Ni(η6-C6H6)], with NHC being m-XylIPr*OMe168, as a precatalyst led to excellent yields of the anti-Markovnikov hydroarylation product, with a linear/branched selectivity of >50:1 or no detectable branched isomer. Basic additives, such as NaH and Na(acac), was crucial for high turnover numbers especially at low catalyst loading, presumably removing traces of water which could significantly affect the reaction.
 | ||
| Scheme 30 Ni/NHC catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions.89 | ||
Electron-neutral, unactivated arenes such as benzene participated well in the hydroarylation reaction. Sterically hindered alkene bearing a quaternary carbon at the α-position (169) or vinlysiloxanes (170) were also tolerated. DFT calculations provided further insight into the mechanism, consistent with previous findings.87 Reversible, direct transfer of the C–H bond of the coordinated arene and ligand in 172 (in a concerted LLHT process) was computationally lower in energy than oxidative addition of the aryl C–H bond. The resulting linear, agostic Cβ–H stabilised alkylnickel-aryl complex 173 then isomerises to the cis-aryl-alkyl nickel complex 174. Rate-determining reductive elimination of 174 then forms a new carbon–carbon bond in the linear alkylarene product.
Unexpectedly, the rate control for reductive elimination under their reaction system goes beyond simple steric control within the transition state, as conventionally thought, despite the use of an extremely bulky NHC ligand. Energy decomposition analysis reveals a different reason for rate acceleration in reductive elimination: extensive non-covalent interactions within the coordination sphere of the catalyst. While Pauli's repulsion factor due to steric effects, cannot be ignored, the stabilising electrostatic interactions between the multiple aryl groups of the NHC and favourable London dispersion interactions resulting from the peripheral methyl groups in the m-XylIPr*OMe ligand (Scheme 30) outweigh the energy cost due to steric influence, thus explaining the high activity of the catalytic system. The significant stabilisation by non-covalent interactions was also corroborated by visualisation using a non-covalent interaction (NCI) plot.90
Recently, Montgomery and Chen contributed to an underdeveloped area of nickel catalysis, with their report on the Ni/NHC-catalysed intermolecular enantioselective hydroarylation of alkenes (Scheme 31).91 This novel approach is distinct from Shi's prior work that employed an SIPE/ANIPE-type ligand75 where the stereochemical induction was provided by the chiral 1-phenethyl substituents on the N-aryl ring. Instead, the authors used a sterically hindered NHC 176 bearing a 2,4,6-triisopropyl N-aryl ring, with a chiral backbone derived from chiral diamines. This chiral NHC ligand provided appreciable enantioinduction while displaying high reactivity with a variety of heterocycles. Paired with an alternative condition that employs a discrete C2-symmetric 1,5-hexadiene-supported chiral NHC–Ni0 complex 177 (instead of generating the active NHC–Ni in situ), this combination allowed for increased functional group tolerance and enhanced the catalytic activity with certain heterocyclic substrates.
 | ||
| Scheme 31 Ni/NHC-catalysed intermolecular enantioselective hydroheteroarylation of alkenes.91 | ||
Most coupling reactions with norbornene as the alkene partner gave the desired product in good yields, as a single exocyclic diastereomer in high enantiomeric ratios. Hydroarylation of norbornene with 1,2,4-triazole (178) surprisingly afforded the C5-alkylated product with high regio- and enantio-selectivity, despite the greater steric hindrance (closer to the benzyl substituent). Notable N-heterocycles like caffeine (179) were also highly efficient in the reaction. With condition B employing the presynthesised NHC–Ni(1,5-hexadiene) catalyst, the use of previously unreactive substrates under condition A could now be feasible, such as chloro-substituted benzoxazole (180) and indoles bearing electron-withdrawing substituents (i.e., esters) (181).
From the preliminary mechanistic studies and analogies with previous computations,87–89 the authors proposed a similar reaction pathway. Initiation of the catalytic cycle involves ligand exchange with the alkene and heteroarene substrate to form the active Ni species, which undergoes reversible, enantio-determining ligand-to-ligand hydrogen transfer (LLHT). Facile isomerisation to a cis-aryl-alkyl T-shaped intermediate preceding the rate-limiting reductive elimination delivers the final enantioenriched exo-selective hydroarylation product.
Significant advancements have been made in the field of intramolecular hydroarylation enabled by Ni/NHC catalysts. In 2015, Cramer and Donets reported NHC ligand-controlled regiodivergent 1,6-annulation of 2-pyridone derivatives, through a selective intramolecular olefin hydroarylation (Scheme 32). The simple cyclooctadiene (COD) ligand favours an exo-selective cyclisation while bulky IPr NHC (1f) favours the endo-selective mode via formation of the alkylnickel at the less hindered end of the alkene. This approach offers an alternative way for controlling the cyclisation mode apart from the ring size, which is highly dependent on substrates. Vinylfuran-containing pyridones (184) underwent endo-cyclisation smoothly (with exo-184’ formed preferentially in the absence of NHC). The desired cis-fused bicyclic ring (182) was also obtained from a cyclopentene-tethered pyridone in good yields. Medium-sized heterocycles (183) can also be efficiently accessed under their protocol. endo-Selectivity is purely ligand-controlled, presumably due to the favoured formation of the less hindered nickelacycle intermediate (Scheme 32). Initial enantioselective endo-cyclisation attempts using the chiral NHC ligand 185 based on the isoquinoline framework were successful, yielding the desired pyridone (−)-186 in a modest enantiomeric ratio.
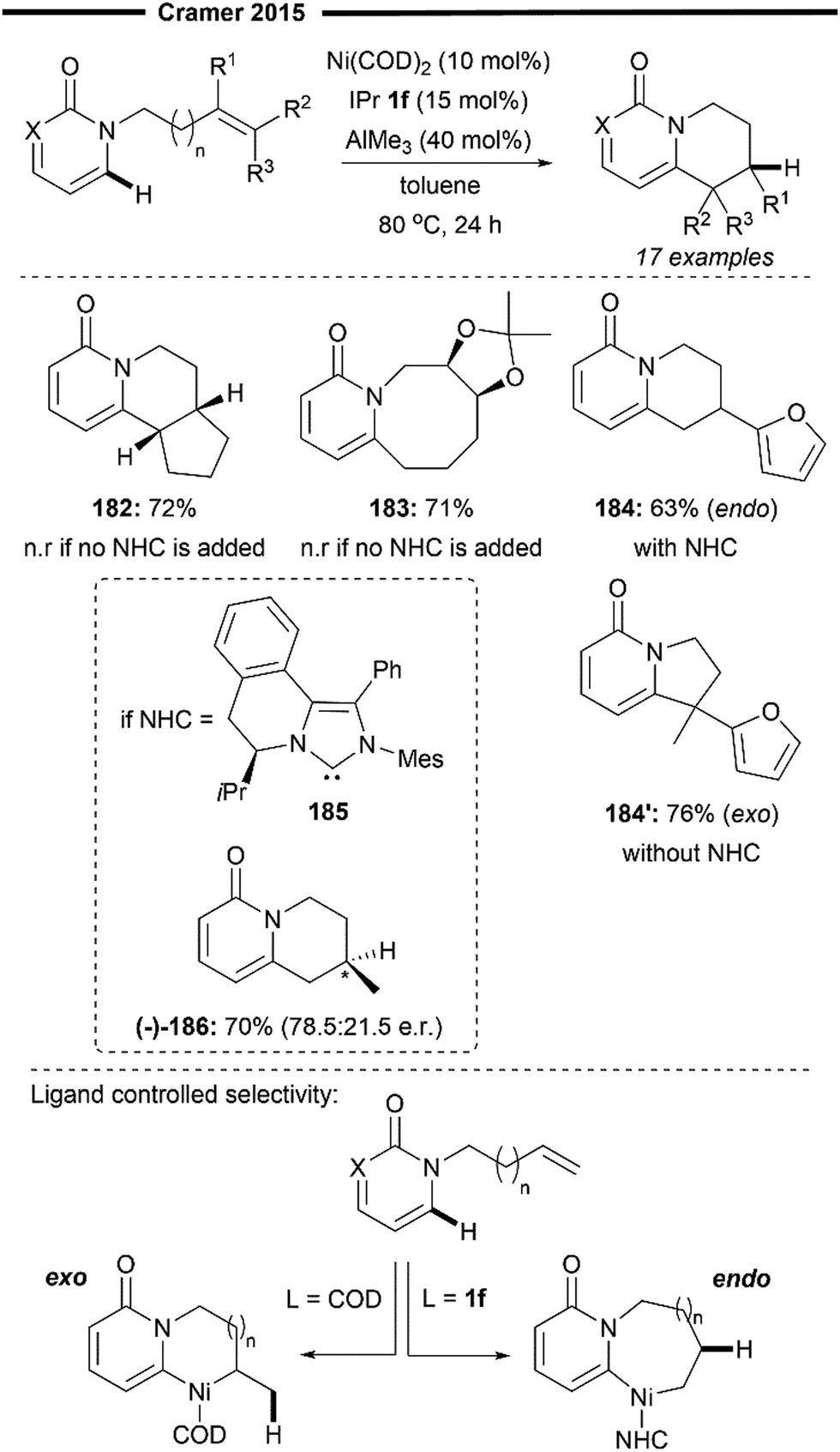 | ||
| Scheme 32 NHC–ligand controlled regiodivergent Ni-catalysed annulation of pyridones.92 | ||
With the demonstration of the Ni-catalysed regiodivergent annulation of pyridines by control of the ancillary NHC ligand,92 Cramer and co-workers recently improved on the control of enantioselectivity for this class of reactions (Scheme 33). A modular class of chiral NHC 187, which mimics the properties of the privileged achiral IPr 1f ligand, was utilised for the intramolecular enantioselective endo-cyclisation of 2-and 4-pyridones with their tethered olefin unit. The most effective chiral ligand was a modified C2-symmetric carbene first reported by Gawley,94 with a bis(imino)acenaphthene backbone.95 Enantiocontrol of the reaction was enhanced through the introduction of 3,5-xylyl groups to the N-aryl side arms of the NHC. This class of ligand was termed, in the same year, by Shi as an ANIPE-type76 ligand.
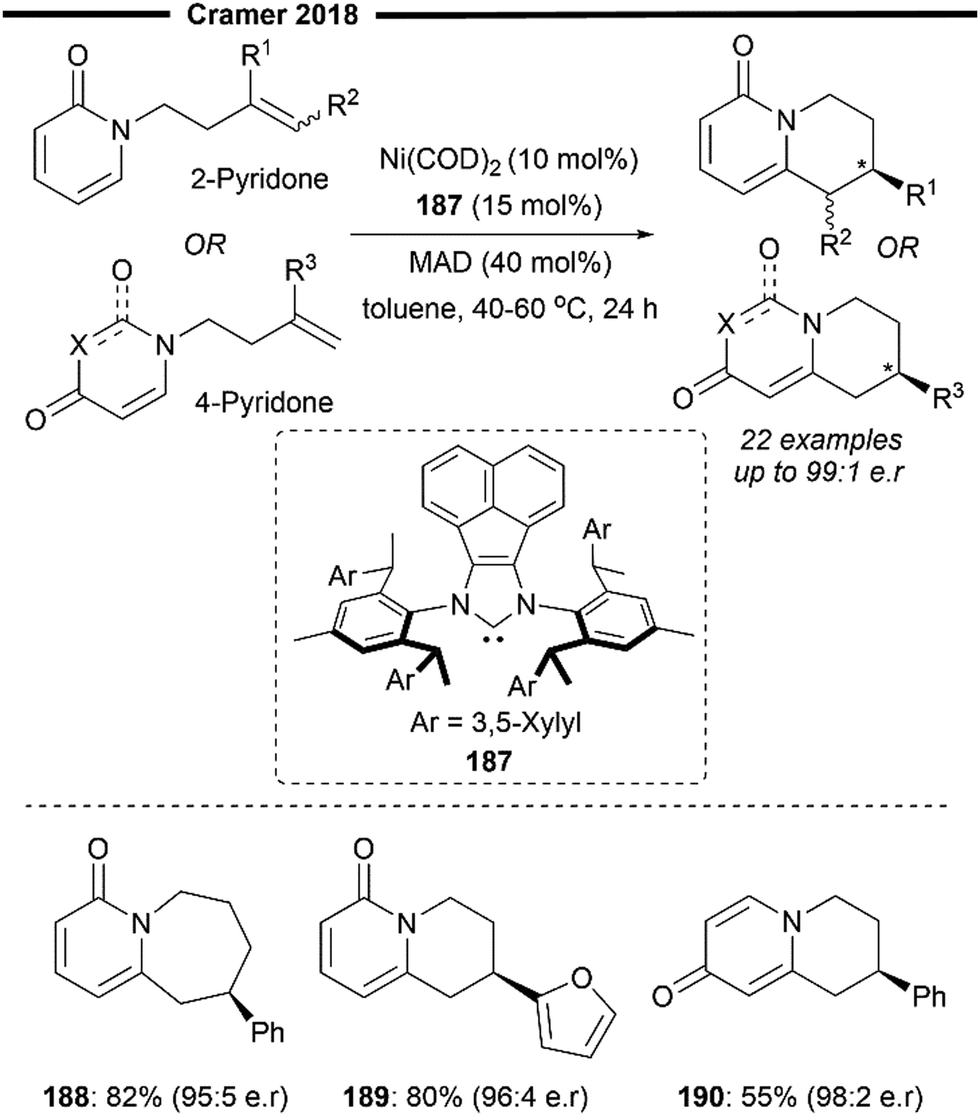 | ||
| Scheme 33 Ni/NHC-catalysed enantioselective annulation of 2- and 4-pyridones via intramolecular olefin hydroarylation.93 | ||
The generality of the reaction was assessed using various substituted 2- and 4-pyridones and was found to be tolerant to heterocyclic substituents such as furans (189). The reaction was also amenable to the synthesis of medium-sized 7-membered heterocycles (188). The chiral and steric influence of the backbone modifications and the N-aryl arms of the chiral NHC was studied in-depth, with the analysis of analogous NHC–Ni and Au complexes. Preliminary data suggest a degree of preorganisation of the flanking bulky N-aryl groups by π-stacking interactions with the acenaphthene backbone, reducing the overall buried volume %Vbur and enhancing the accessibility of the C2-symmetric binding pocket. This, in turn, improves the catalytic performance and enantiocontrol provided by the chiral ligand.
In 2019, Shi and co-workers reported an unprecedented Ni/NHC-catalysed enantioselective C–H alkylation reaction of alkene-tethered polyfluoroarenes through an endo-selective intramolecular hydroarylation process (Scheme 34).96 Chiral fluorotetralin derivatives–which are important bioisosteric analogues of tetralin for drug design97–were efficiently synthesised under this protocol, with chemoselective activation of the C–H bond over the C–F bond. Competitive side reactions such as hydrodefluorination and alkene chain-walking were also suppressed.
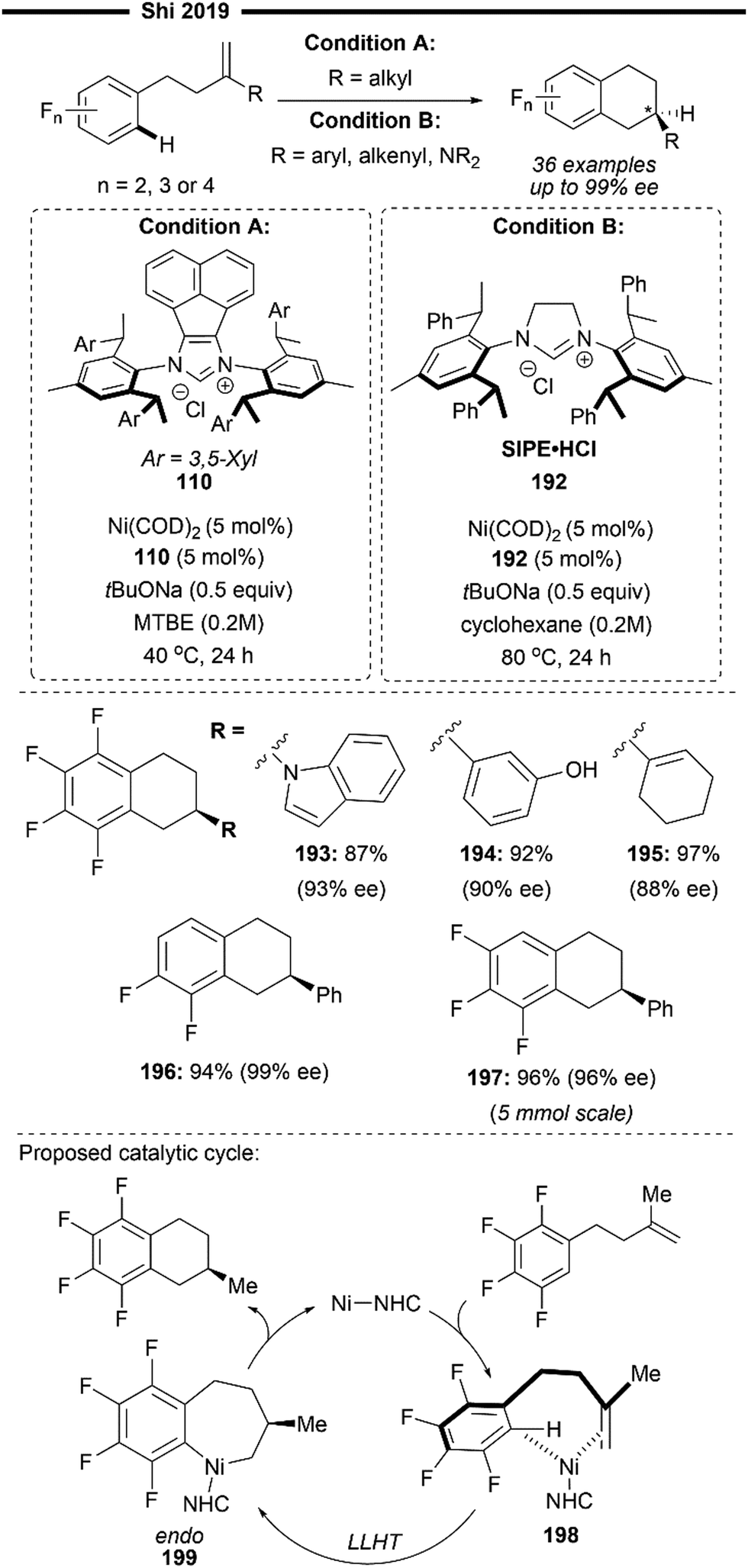 | ||
| Scheme 34 Ni/NHC-catalysed asymmetric intramolecular hydroarylation of olefin-tethered fluoroarenes.96 | ||
The most effective chiral ligands were found to be the bulky 3,5-Xyl ANIPE or SIPE-type NHC ligands, which led to high yields and enantioselectivity of the fluorotetralin product. The large steric bulk of the NHC was proposed to play a role in accelerating the reductive elimination of the nickelacycle 199 and favouring the endo-cyclisation pathway. Selective activation of the C–H bond over the C–F bond was also attributed to the monomeric Ni/NHC active catalyst as the bulky NHC disfavours aggregation.
The method was successfully applied to difluro-(196), trifluoro-(197) and tetrafluorobenzenes (193–195), even on a gram scale, showcasing the general practicality of their method. Notably, substituents bearing acidic protons, (phenol in 194), pharmaceutically relevant heterocycles like indole 193 and 1,3-dienes 195 were all well tolerated, furnishing the product with high levels of enantiocontrol. Preliminary mechanistic studies suggest a direct ligand-to-ligand hydrogen transfer (LLHT) from the fluoroarene to the alkene within the coordinated complex 198, under the influence of the chiral environment of the NHC–Ni complex. The endo-cyclised nickelacycle 199 that was formed underwent reductive elimination to yield the chiral fluorotetralin product.
By employing the same bulky chiral (R,R,R,R)-SIPE NHC ligand, Shi and co-workers, in successive years, developed the Ni/Lewis acid catalysed asymmetric C–H functionalisation of alkene-tethered pyridines98 and pyridones99 through an intramolecular hydroarylation process, affording the corresponding annulated chiral N-heterocycles from racemic building blocks (Scheme 35). From their preliminary mechanistic investigations involving deuterium labelling studies, a similar operating mechanism seems to be the case for both reactions. Dual coordination of the substrate with the aluminium-based Lewis acid additive and the Ni/NHC active catalyst activates the substrate, forming a Ni/Al bimetallic complex 206. Oxidative addition of Ni(0) into the C–H/D bond forms a tentative Ni–H species 207, which subsequently undergoes enantioselective anti-Markovnikov hydrometallation to form the seven-membered nickelacycle intermediate 208. The observed regioselectivity could be influenced by the combined steric effect from the bulky NHC ligand and the Lewis acid. Final reductive elimination yields the enantioenriched (R)-N-heterocycle and regenerates the active NHC/Ni0 species. The strongly electron-donating SIPE ligand is believed to facilitate the process of C–H oxidative addition, with enantiocontrol arising from the efficient enantio-discrimination of one face for alkene insertion, owing to the steric repulsion of the pyridone/pyridine moiety and the phenyl rings of the 1-phenethyl arm on SIPE.
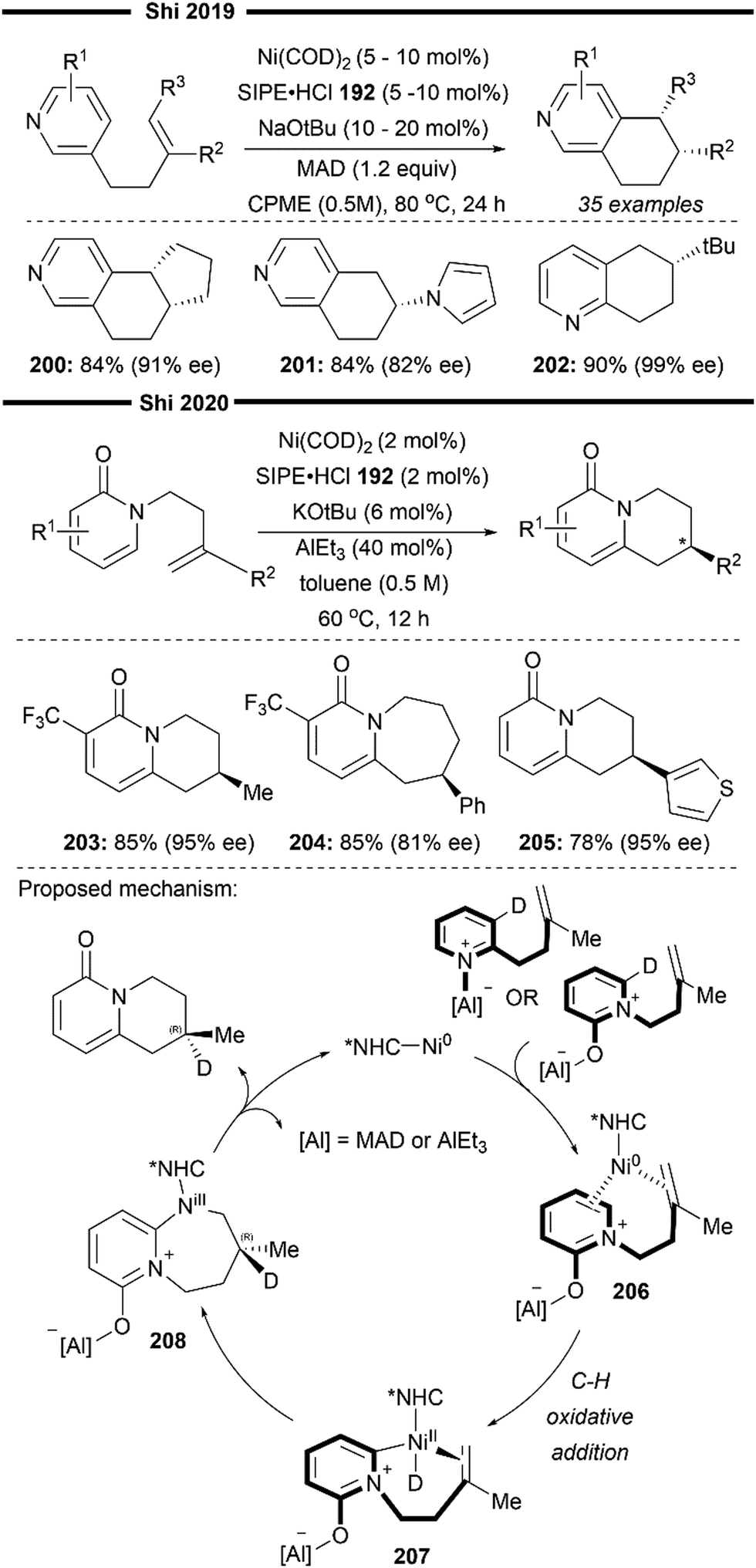 | ||
| Scheme 35 Ni/SIPE-catalysed intramolecular enantioselective hydroarylation of alkene-tethered pyridines98 and pyridones.99 | ||
Endo-Selective annulation of C3-tethered alkenes with trisubstituted cyclopentene and heterocyclic enamines successfully results in the formation of the corresponding tricyclic (200) and pyrrole-containing (201) products with high enantioselectivity. Reaction with pyridine C2-tethered alkenes was also viable, affording enantioenriched 5,6,7,8-tetrahydroquinolines in excellent yield. In the annulation of pyridones, trifluoromethyl substituted pyridone cores (203) and heterocyclic functional groups like thiophene (205) were well-tolerated. Medium-sized heterocycles (204) could also be accessed in good yields and with high enantioselectivity under this regime.
Cramer and co-workers took the concept of previously reported enantioselective C–H functionalisation of various heterocycles via hydroarylation,92,93,100 and applied it to biologically relevant electron-rich heterocycles like pyrroles and indoles. In 2019, they reported the Ni/NHC-catalysed intermolecular enantioselective hydroarylation of alkene-tethered pyrroles or indoles, with a high anti-Markovnikov selectivity despite the absence of a directing group100 (Scheme 36). Usage of a highly sterically demanding 3,5-di-tert-butylphenyl substituted SIPE-type ligand 209 proved to be superior in delivering the endo-cyclised tricyclic tetrahydropyridyl indole scaffolds in high yields and with high enantioselectivities.
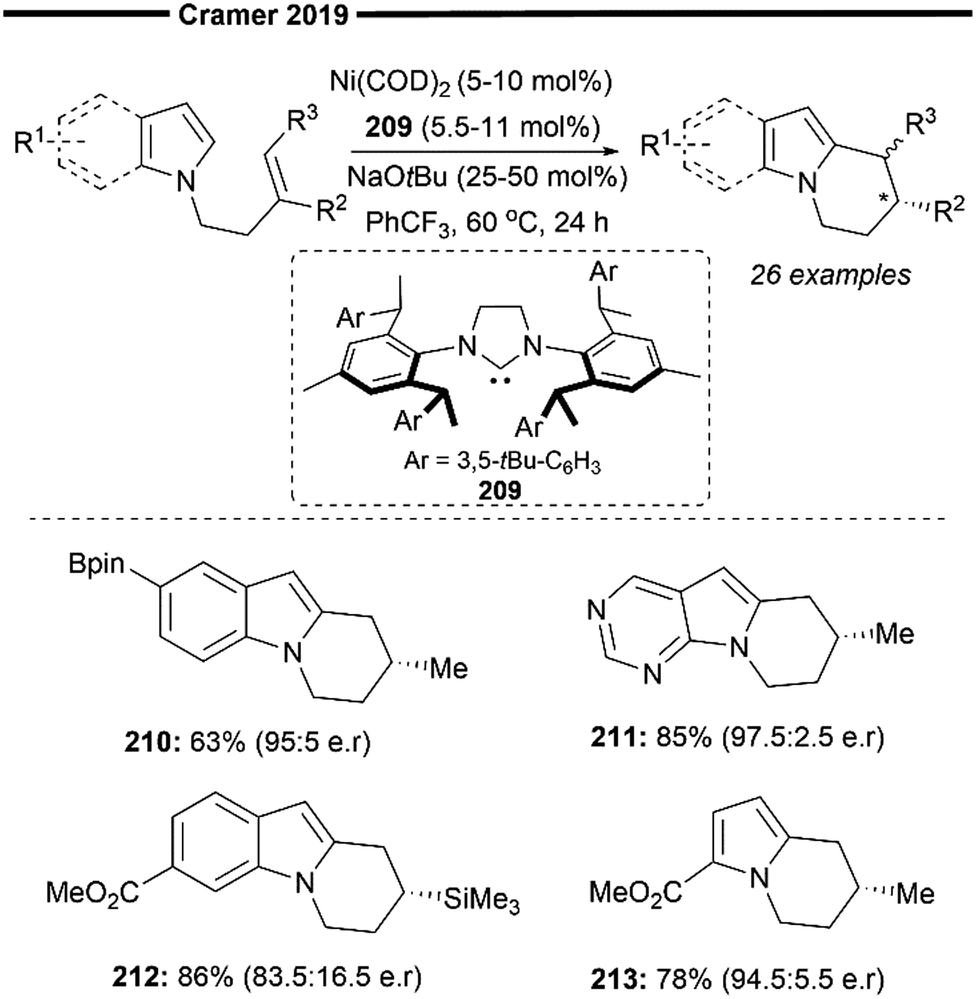 | ||
| Scheme 36 Ni/NHC-catalysed intermolecular enantioselective hydroarylation of alkene-tethered indoles and pyrroles.100 | ||
Base-sensitive boronate ester (210) functionality was tolerated and remained intact after the reaction. Other pharmaceutically relevant azaheterocycles like pyrrolopyrimidine (211) and pyrroles (213) also participated well in the reaction. Indole bearing tethered vinylsilanes (212) also reacted successfully, albeit with a lower enantioselectivity, presumably due to the steric influence of the bulky trimethylsilyl group at the newly formed stereogenic centre.
In 2021, Ye and co-workers reported the construction of medium sized 7-membered heterocycles via an NHC/Ni-Lewis catalysed C7–H bond cyclisation reaction of benzimidazoles with a tethered alkene moiety101 (Scheme 37). Formally an intramolecular hydroarylation process, enantioenriched tricyclic imiazdoles can be achieved, with exclusive endo-selectivity in the cyclisation mode, despite the challenging enantiocontrol due to the flexible large ring transition state. The synergistic (NHC)Ni–Al bimetallic system responsible for the success of this reaction comes from the bulky chiral ANIPE type ligand 214 and trimethylaluminum Lewis acid.
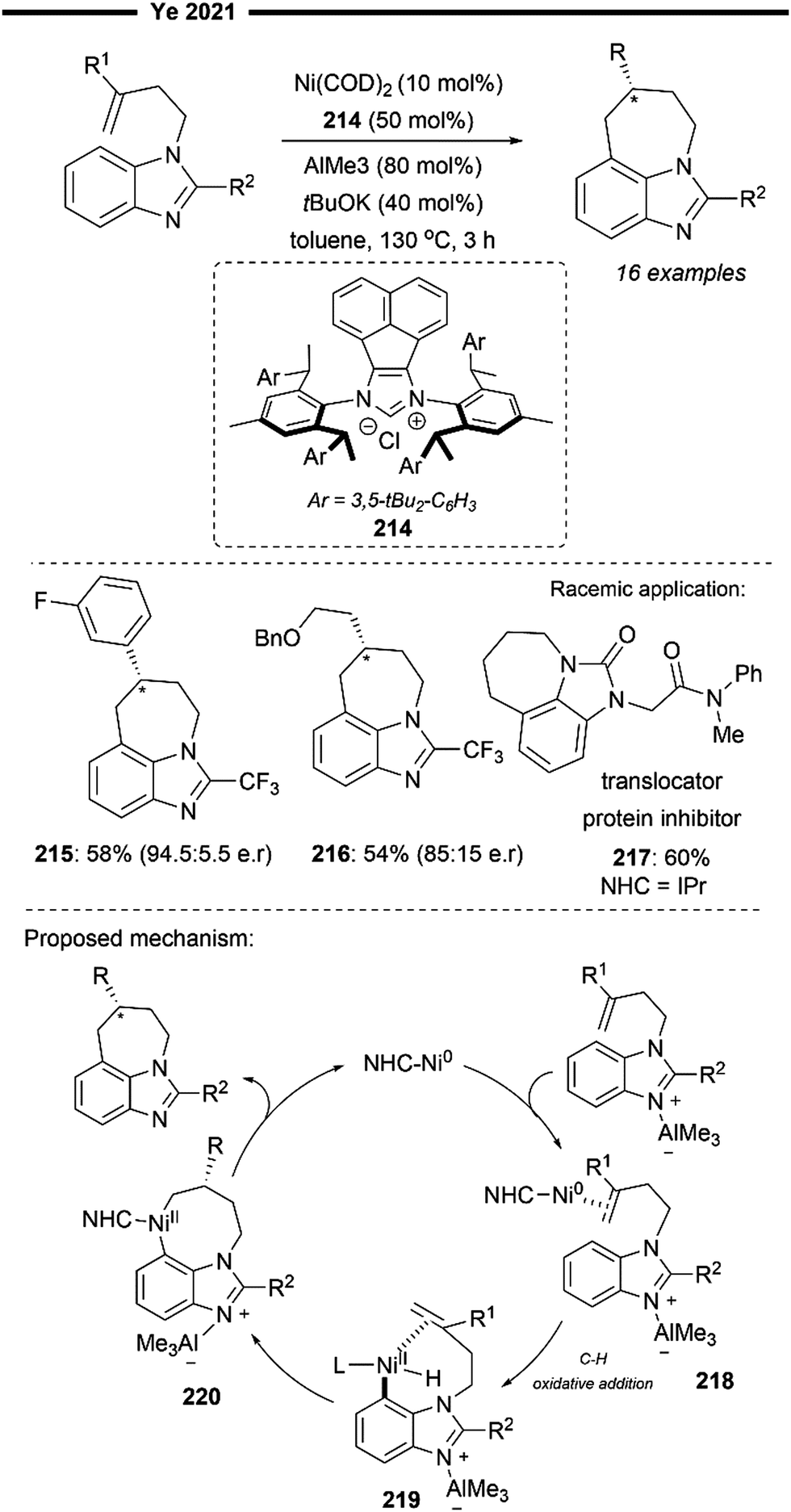 | ||
| Scheme 37 (NHC)Ni/Lewis acid catalysed synthesis of tricyclic imidazoles via intramolecular enantioselective hydroarylation of alkene-tethered benzimidazole.101 | ||
The reaction is quite general, where various aryl or alkyl alkenes were well compatible under the reaction conditions, with the latter resulting in a poorer ee of the seven-membered cyclised product (cf.215 and 216), presumably due to greater structural flexibility. Racemic version of the reaction could also be easily realised with the use of the achiral IPr 1f ligand, and the utility of the reaction was demonstrated to access bioactive molecules like a translocator protein inhibitor (217) from imdazol-2-one. Preliminary mechanistic studies provide some insight into a possible mechanism (Scheme 37). Activation of the benzimidazole substrate by AlMe3 and coordination of the activated substrate with NHC–Ni active species forms 218. This facilitates the subsequent rate-determining oxidative addition of the C7–H bond, forming the alkene-coordinated nickel–hydride intermediate 219. Governed by the steric and chiral environment of the ANIPE ligand, an irreversible endo-type alkene migratory insertion occurs, forming the 8-membered nickelacycle 220 with the stereochemistry being determined. Lastly, stereoretentive reductive elimination releases the final tricyclic product after dissociation with the aluminium Lewis acid, to initiate the next cycle.
In 2021, Koh and co-workers developed a new regime for NHC–Ni catalysed Markovnikov-selective hydroarylation(alkenylation) of olefins, which operates through a novel pathway involving sequential carbonickelation followed by hydrogenation57 (Scheme 38). This reversed order is distinct from the conventional hydroarylation pathway, with hydronickelation preceding carbofunctionalisation, predominantly delivering the anti-Markovnikov product especially in reactions with unactivated aliphatic alkenes due to the absence of the adjacent electronic π-stabilisation from the alkylnickel intermediate. The authors used a dimeric IPr-Ni(I) precatalyst and NaOiPr as the hydride-source, which furnished the branched-selective hydroarylation product in high yields. Excess alkoxide base was also found to be crucial for suppressing the undesired Heck-type reaction.
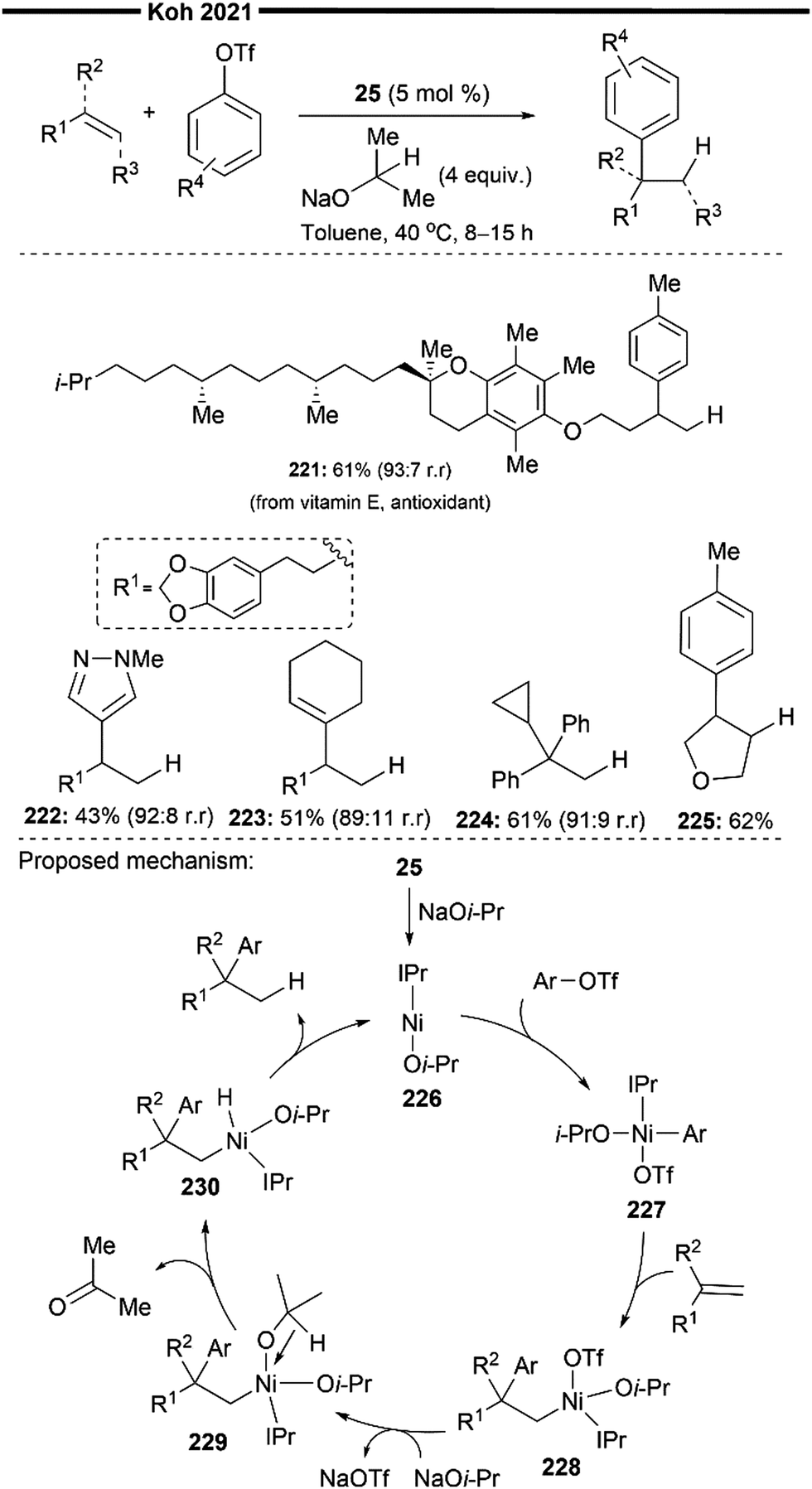 | ||
| Scheme 38 Ni/NHC-catalysed regioselective hydrocarbofunctionalisation of alkenes via sequential carbonickelation – hydrogenation.57 | ||
A series of investigations were carried out to gain a deeper understanding of the underlying mechanisms. In a series of radical clock experiments, no cyclopropyl ring-ruptured or radical annulation side-products were observed, supporting the proposition that no long-lived radical intermediate species were present, even if a Ni(I) complex was employed. A plausible non-radical pathway was proposed by the authors (Scheme 38). The active monomeric Ni(I)-isopropoxide intermediate 226 is formed through the alkoxide assisted dissociation of the dimeric Ni(I) precatalyst 25. Oxidative addition of the aryl/alkenyl triflate electrophile affords the aryl-Ni(III) species 227, which undergoes Markovnikov-selective carbonickelation across the incoming olefin substrate, forming the least hindered primary alkylnickel complex 228. The observed regioselectivity in the insertion is likely attributed to the steric influence of the sizeable IPr ligand, where steric repulsion between the IPr ligand and present alkene substituent is minimised. Ligand exchange with excess NaOiPr forms the Ni-bis(isopropoxide) 229. Dehydrogenation of the ligated isopropoxide ligand through β-H elimination as facilitated by the adjacent Cβ–H agostic interaction yields the nickel–hydride intermediate 230, with concomitant release of acetone as a by-product. Final reductive elimination releases the Markovnikov-selective hydroarylation/alkenylation product, regenerating 226. Although a tentative proposal of a Ni(I)/Ni(III) operating catalytic pathway was presented, the authors acknowledge that a Ni(0)/Ni(II) pathway cannot be entirely ruled out. The main emphasis of the mechanism and the observed regiocontrol lies in the preference for carbonickelation over the adventitious hydronickelation, contrary to many previous reports.102
The reaction has been shown to be versatile, accommodating a variety of functionalities, including heterocycles like pyrazoles (222). Challenging quaternary carbon-centres can also be efficiently accessed from 1,1-disubstituted alkenes (224). Notably, the cyclopropane moiety of 224 was not disrupted after the reaction with no traces of radical-induced ring ruptured products. Furthermore, the reaction is effective with unactivated alkenes like 2,5-dihydrofurans (225), and alkenyltriflates could also be utilised as complementary electrophiles to afford the corresponding hydroalkenylation product in good yields and with high selectivity. The utility of the reaction was further demonstrated by the efficient reaction with alkenes derived from bioactive molecules (221).
In continuation of their previous work,57 Koh and co-workers have developed a versatile enantioselective nickel-catalysed strategy for olefin hydrocarbofunctionalisation and dicarbofunctionalisation103 (Scheme 39). This three-component reaction involves the union of an activated olefin or diene, an organotriflate electrophile and a metal alkoxide as a hydride donor in the presence of a sterically encumbered chiral NHC–Ni(0) catalyst (where NHC = (R,R,R,R)-IPE 231). This work allows for practical access to the corresponding high value enantioenriched secondary, tertiary, or quaternary stereogenic centres. Substitution of the metal alkoxide with an organometallic reagent leads to the formation of useful dicarbofunctionalisation adducts through the installation of two different carbogenic motifs across the CC bond.
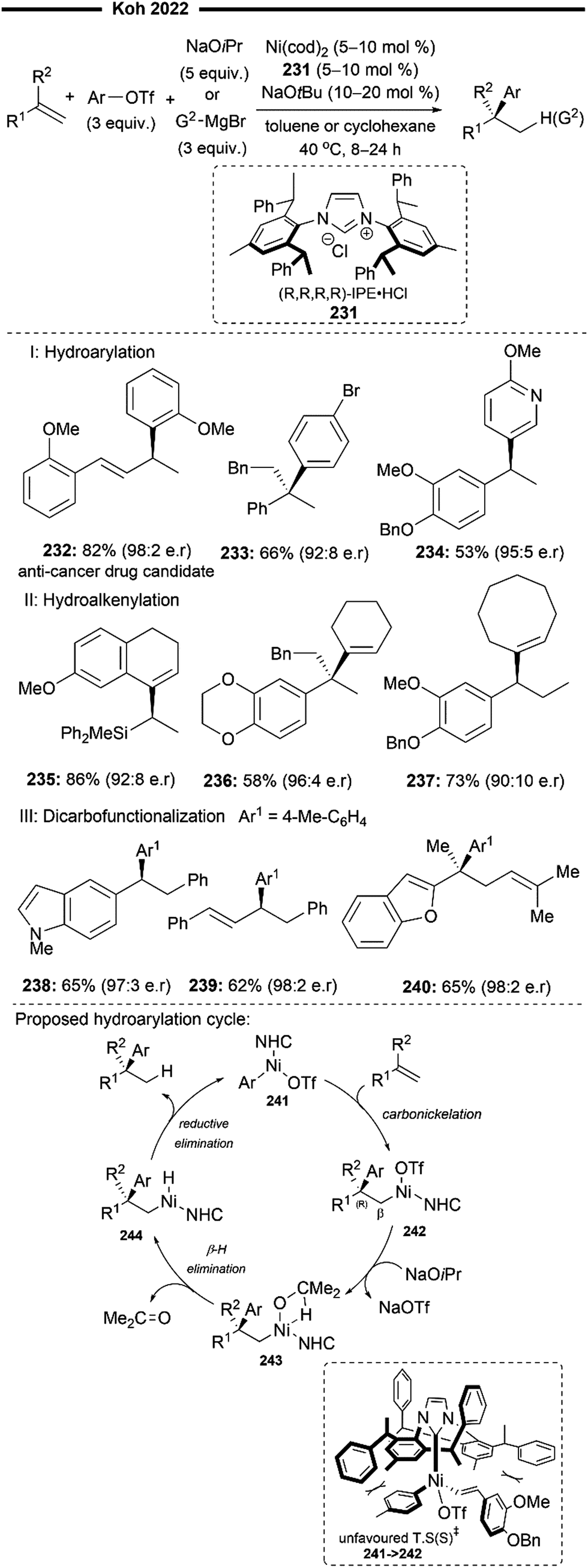 | ||
| Scheme 39 Generation of tri- and tetra-substituted stereocentres by NHC/Ni-catalysed enantioselective olefin cross-coupling.103 | ||
Electron paramagnetic resonance spectroscopy reveals the absence of paramagnetic radical species in the reaction system, suggesting that it follows a Ni(0)/Ni(II) pathway. Initial oxidative addition of the C(sp2) aryl or alkenyltriflate electrophile to NHC–Ni(0) forms the chiral arylnickel species 241. Regio- and enantio-selective carbonickelation across the π-complexed alkene substrate forms the less hindered primary alkylnickel intermediate 242. Minimisation of the unfavourable steric interactions between the olefinic substituents and the β arms of the NHC in the chiral environment governs the regio- and enantio-control for the insertion (an unfavoured transition state leading to the minor (S)-enantiomer). Ligand exchange with the excess alkoxide forms the Ni(alkoxide) species 243, which participates in β-H elimination to give the nickel–hydride intermediate 244, with the concomitant expulsion of acetone. The formation of the C–H bond occurs following a reductive elimination reaction. Since the enantio-determining C–C bond forming step occurs during carbonickelation, appropriate interception of intermediate 242 with a carbon-based nucleophile instead of an alkoxide led to the formation of dicarbofunctionalisation products.
The catalytic regime is highly general and compatible with even highly hindered substrates like 1,1-disubstituted alkenes, leading to the formation of quaternary stereocentres (233, 236 and 240) with excellent regio- and enantio-selectivity. Heterocyclic substituents, such as Lewis basic indoles (238), benzofurans (240) and pyridines (234) were well-tolerated. Chemoselective activation of triflates instead of bromides (233) was also highlighted, providing the potential for orthogonal functionalisation. Medium-sized alkenyltriflates (237) participate well in the hydroalkenylation, and dicarbofunctionalisation adducts can be efficiently converted from various styrene derivatives or dienes with high enantiocontrol, exemplified by various diarylation (238 and 239) products and a notable skipped 1,5-diene arylalkenylation product 240. Utility of the reaction was highlighted with the synthesis of an anti-cancer drug candidate product (232) without the observation of any undesired allylic rearrangement.
Having previously developed the para-C–H alkylation of pyridine derivatives using NHC–Ni/Al cooperative catalysis,74 Nakao and co-workers applied the concept to the direct arene C–H activation of benzamides and aromatic ketones, in a formal hydroarylation reaction with exogenous alkenes104 (Scheme 40). The use of a sterically encumbered C2-symmetric NHC ligand with 3,5-xylyl substituted N-aryl arms in conjunction with the bulky MAD cocatalyst resulted in exceptional para-C–H and anti-Markovnikov site-selectivity, aided by the combination of electronic activation and steric repulsive forces.
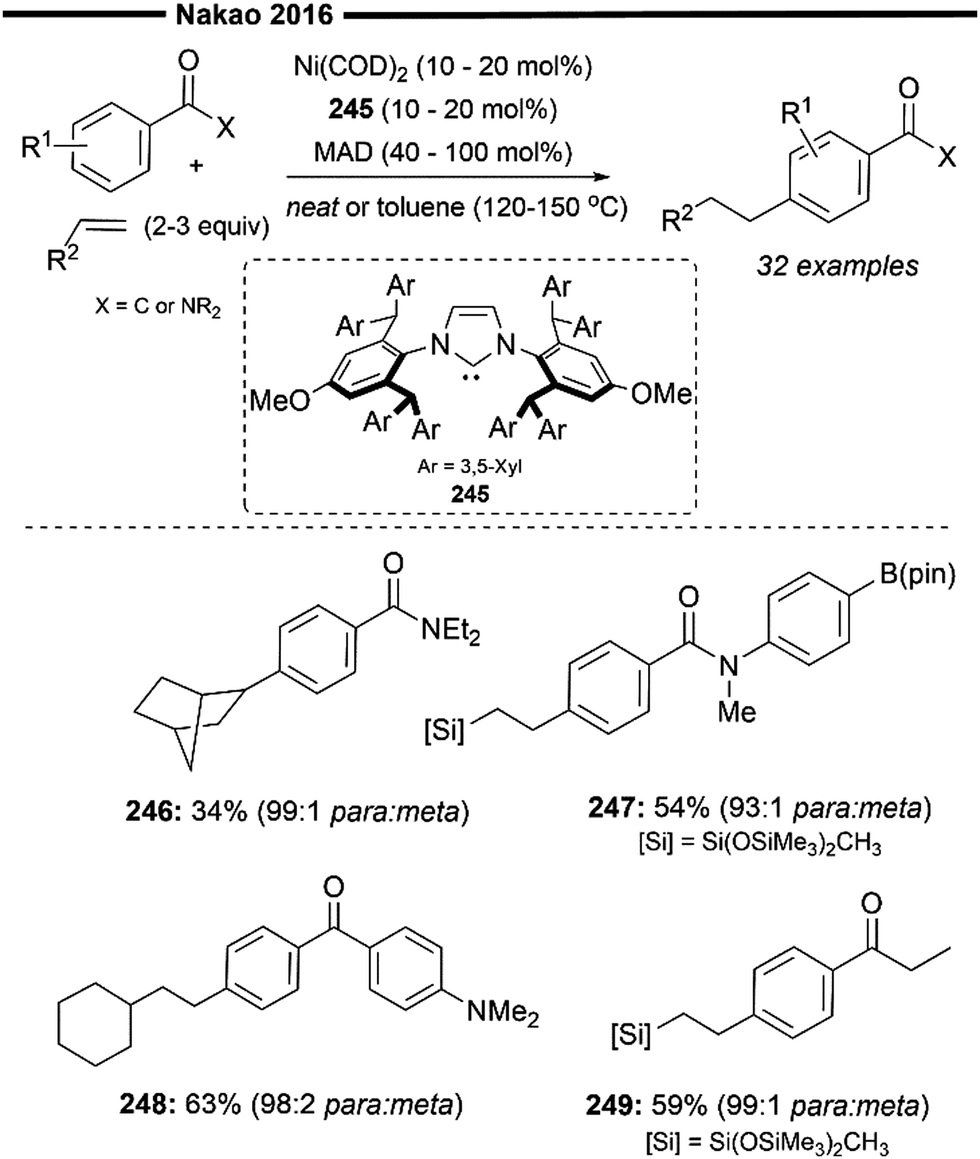 | ||
| Scheme 40 NHC–Ni/Al catalysed para-selective C–H alkylation of benzamides and aromatic ketones via hydroarylation with alkenes.104 | ||
Bulky olefin substrates such as vinylcyclohexanes (248) and methylbis(trimethylsilyloxy)vinylsilane (247, 249) were well-tolerated, yielding the corresponding para-alkylated benzamides. The 1,2-disubstituted CC bond of norbornene (246) was also reacted under the protocol even though it was less efficient. Enolisable alkylaryl ketones (249) also underwent reaction smoothly to deliver the C4-alkylated arene.
Theoretical calculations using DFT are in agreement with the previously divulged mechanism,87 involving a concerted LLHT as the key rate-determining C–H activation step. Calculations using the model AlMe3 reveal the mode of rate-acceleration by the Lewis acid. Benzamide coordinating to AlMe3 was calculated to have a lower LUMO (σ* of C–H and aromatic ring) energy than the uncoordinated substrate. AlMe3 also enhances the selectivity for the para-position due to the stronger repulsive interactions with N-aryl substituents of the NHC in the transition state leading to the formation of the meta-functionalised product.
With successful para-C–H alkylation of arenes with selectivity governed by both steric and electronic factors,104 Nakao and colleagues sought to achieve a site-selective linear C–H functionalisation protocol by variation in the nature of NHC ligands used. As previously established, electron-poor C–H bonds are more reactive due to the ease of activation under a concerted LLHT mechanism. Using anilides as a model substrate, one would expect the meta-C–H bond of anilides to be the most electron poor and electronically activated, assuming negligible steric effects. This presents the possibility for selective and switchable site-functionalisation, which they were able to achieve. The use of a smaller, less sterically demanding IPrMe ligand 162 (condition A, Scheme 41) enables meta-selective functionalisation, while the bulkier ligand 245 (condition B, Scheme 41) sterically favours para-selective functionalisation. Vinylsilanes (251, 253) or vinylcyclohexanes (250, 252) were identified as suitable alkene substrates for this formal hydroarylation reaction. Significant enhancement of para-selectivity was achieved with a bulkier N-alkyl substituent R2 on the anilide substrate (R2 = sec-isopentyl for 252 or isopropyl for 253). Formation of the meta-isomer could be disfavoured as a result of steric hindrance and the strong steric repulsion between R2, the sizeable MAD Lewis acid and the NHC ligand.
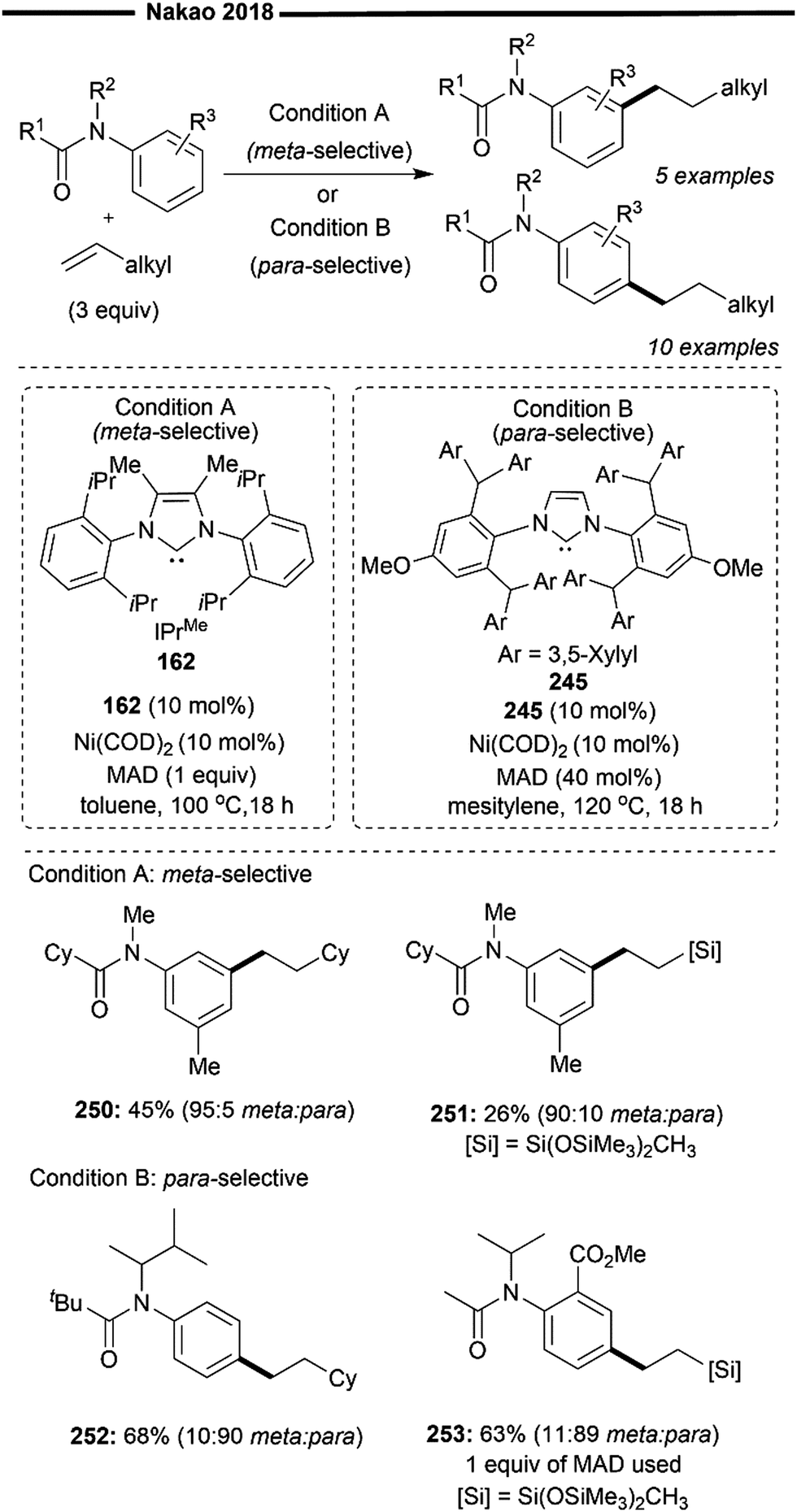 | ||
| Scheme 41 NHC–Ni/Al catalysed site-selective linear alkylation of anilides.105 | ||
Recently, Chen and co-workers developed a novel synthetic route to access biologically and pharmaceutically relevant monoterpenoids (Scheme 42).106 Their atom-economic regime involves an NHC/Ni-catalysed asymmetric heteroarylative cyclotelomerisation of isoprenes (a commercially available bulk chemical), to generate cyclic monoterpene derivatives bearing quaternary carbon stereogenic centres with high enantioselectivity. The best enantioselectivity was achieved with the use of a bulky C2-symmetric (R,R,R,R)-ANIPE NHC ligand (254) with a structurally rigid acenaphthene backbone.
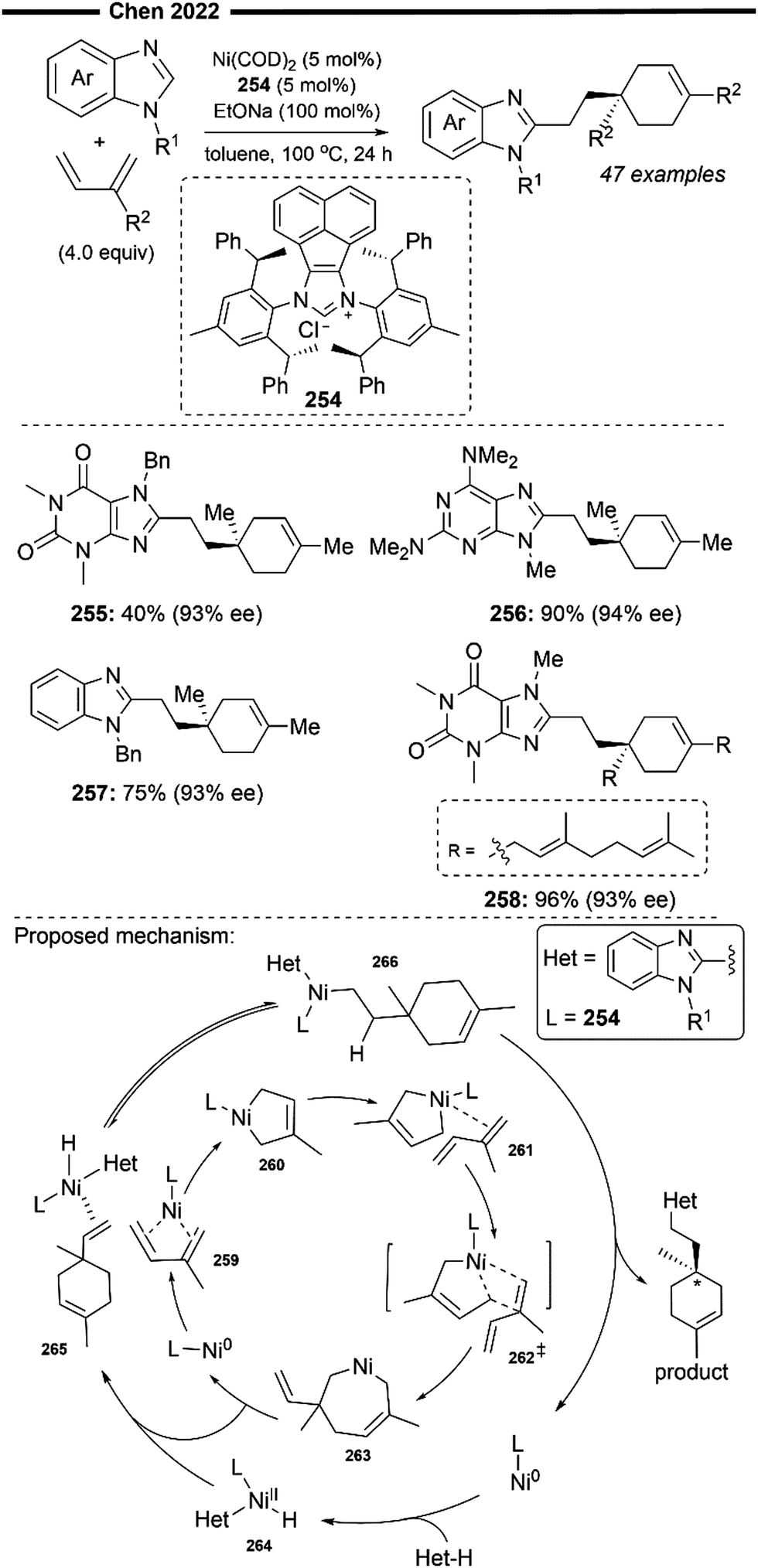 | ||
| Scheme 42 NHC/Ni-catalysed asymmetric heteroarylative cyclotelomerisation of isoprene.106 | ||
Various naturally occurring and medicinally important heterocycles were amendable to the procedure, delivering the telomer in excellent enantioselectivity. N7-benzyl theophylline (255), an analogue of caffeine, participated well in the reaction. Adenine, a nucleobase found in nucleic acids DNA and RNA, is also a viable substrate for this asymmetric telomerisation reaction. Dimethylamino-substituted adenine (256) was converted to the corresponding product in excellent yield and with high enantioselectivity. N-Heterocycles such as benzimidazole (257) which can be found in numerous clinical drugs are also tolerated under their regime. In addition to isoprene, other terpenes such as sesquiterpene farnesene with trisubstituted olefin moieties also reacted efficiently with caffeine to deliver the expected product (258).
The key components of their asymmetric heteroarylative cyclotelomerisation reaction include the cyclisation of isoprene and the C–H alkylation of the heterocycle (via a hydroheteroarylation process). Extensive mechanistic investigations were conducted to unveil the sequence of events, with the conclusion that the enantioselective dimerisation of isoprene precedes the C–H bond functionalisation of the heterocyclic coupling partner. Initial coordination of isoprene with the active NHC–Ni0 species forms the π-complex 259, which leads to the formation of the nickelacyclopentene intermediate 260 after oxidative cyclometallation. Subsequent coordination with a second molecule of isoprene forms the complex 261, which undergoes regio- and enantio-selective carbonickelation (with a depicted transition state) to furnish a seven-membered nickelacycle 263. The quaternary carbocentre formed is already stereodefined at this stage. The simultaneous control of the regio- and enantio-selectivity of the migratory insertion is due to the encumbered chiral environment provided by the ANIPE ligand. Reductive elimination of 263 then releases the isoprene-dimer which enters a separate catalytic cycle. In the second cycle, C–H bond activation of the heterocycle via oxidative addition forms the nickel(II)-hydride species 264, which coordinates with the isoprene-dimer to yield a π-complex 265. 265 then undergoes an anti-Markovnikov selective hydronickelation, under the steric control of the bulky NHC, resulting in the formation of a primary alkylnickel intermediate 266. Reductive elimination of 266 affords the desired cyclised terpenoid product and regenerates the active NHC–Ni0 catalyst.
3.3 Hydroboration of alkenes
Organoboronates are known for their stability, versatility, and ease of diversification, making them a popular alternative to sensitive organometallic reagents. These compounds can be transformed into a variety of synthetically useful motifs using well-established C–B to C–X (such as X = C, N, O or halide) transformations.107,108 A highly efficient method for synthesising aliphatic organoboron compounds involves employing catalytic hydroboration or protoboration of alkenes,109 which has been achieved with some success under nickel-catalysed systems.110 Although NHCs are widely utilised as ligands in many nickel-catalysed hydro(hetero)arylation and hydroalkenylation reactions, their application in the hydroboration of alkenes is more limited.In 2016, Schomaker and co-workers first reported the Markovnikov-selective hydroboration of alkenes with a heteroleptic NHC/phosphine nickel catalyst 267.111 Benzyl boronic esters were obtained with excellent branched Markovnikov selectivity and in moderate yields (Scheme 43). Both electron-rich (ortho-methoxy, 270) and electron-deficient (meta-methoxy, 269) styrenes were well-tolerated. Notably, internal 1,2-disubstituted alkenes (268) also performed well under the regime. However, 4-vinylpyridine (271) proved to be ineffective, likely due to the deactivation of the catalyst by the Lewis basic nitrogen atom.
 | ||
| Scheme 43 Heteroleptic Ni/IMes-catalysed Markovnikov selective hydroboration of styrenes111 | ||
The authors proposed a Ni0/NiII catalytic cycle for their hydroboration protocol. Initial reduction of the NiII precatalyst 267 by HBpin generates the 14 e− Ni0 active catalytic species 269. Dissociation of the existing tricyclohexylphosphine ligand (facilitated by the strong trans effect of the electron-donating NHC ligand) followed by the oxidative addition into the B–H bond of pinacolborane yields the boryl-Ni-hydride intermediate 270. Addition of external PCy3 proved to be highly detrimental to the reaction, supporting the notion for the dissociation of the phosphine ligand. The incoming styrene substrate then proceeds to form a π-complex with the coordinatively unsaturated boryl-Ni-hydride complex 270 to give 271. Markovnikov-selective migratory insertion of the alkene into the Ni–H bond forms the benzylic-Ni intermediate 272, which is presumably stabilised by conjugation with the π-aromatic system. By exploiting the steric properties of NHC ligands, the authors discovered that the use a bulkier IPr* NHC ligand can disfavour the formation of the π-benzylic-Ni intermediate 272, due to a greater steric repulsion between the aryl substrate and bulkier NHC. This results in the production of a higher proportion of linear boronic esters, despite the decreased overall reactivity. Final reductive elimination of 272 then delivers the final Markovnikov product, regenerating the active Ni0 catalyst to close the catalytic cycle.
Later in 2020, Ritleg and co-workers developed an anti-Markovnikov hydroboration reaction of alkenes with a half-sandwich type Ni/NHC complex [Ni(Cp*)Cl(IMes)] 273, using catecholborane (HBCat) as the boron source in the presence of catalytic potassium tert-butoxide (Scheme 44).
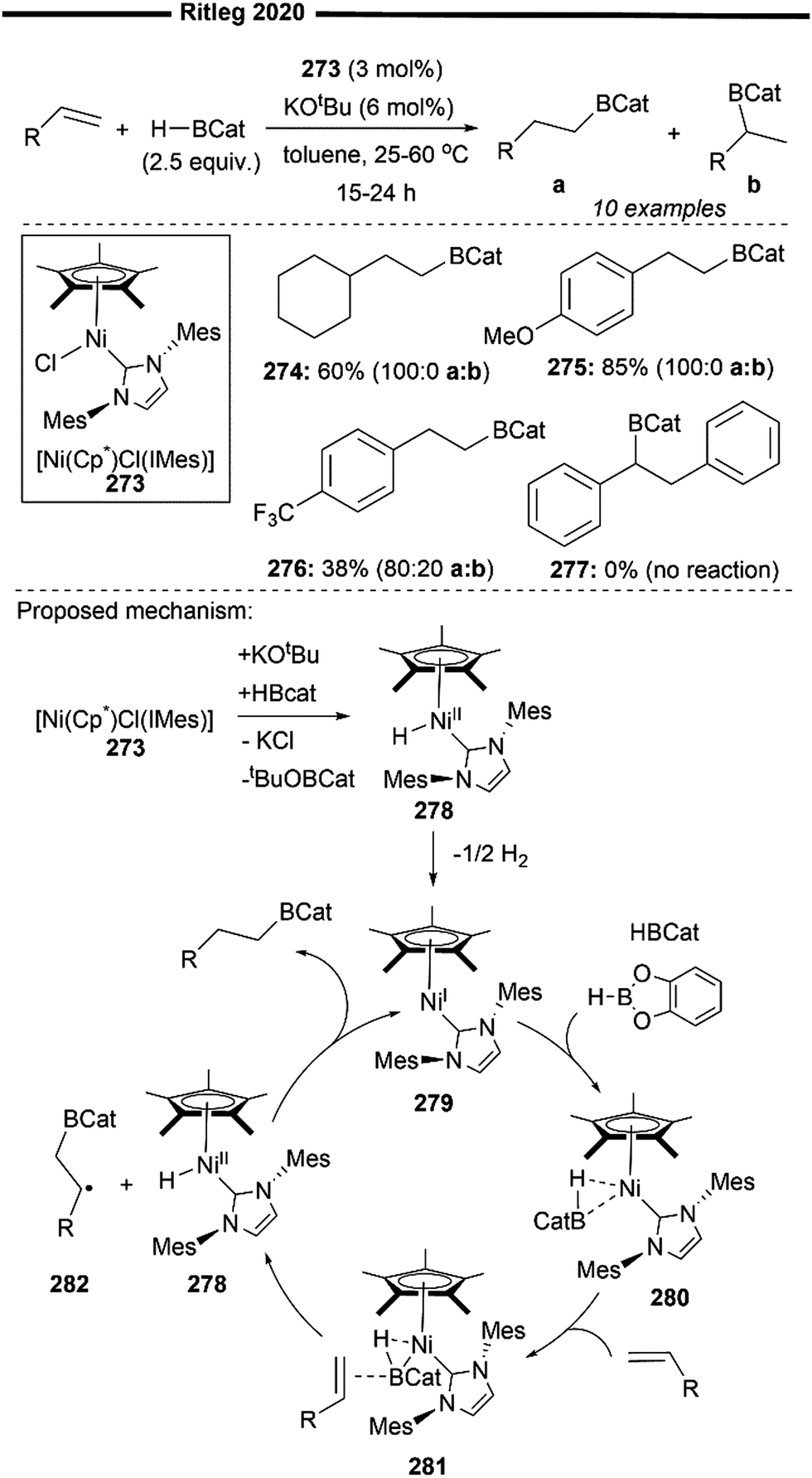 | ||
| Scheme 44 Anti-Markovnikov selective hydroboration of alkenes with the [Ni(Cp*)Cl(IMes)] complex.112 | ||
Electron-rich styrenes were all fully converted with high linear, anti-Markovnikov selectivity 275. In comparison, electron-poor styrenes underwent hydroboration with poorer yields and selectivity 276. Surprisingly, a non-aryl derivative like vinylcyclohexane was still effective (274). Limitations of the reported regime include 1,2-disubstituted internal alkenes which showed no conversion (277 was not observed after subjecting (E)-stilbene to their reaction protocol).
Detailed investigation into the mechanism revealed that the nickel–hydride complex [NiCp*H(IMes)] (278) is the key catalytic precursor which would be reduced with concomitant release of H2 gas to form the active NiI species, [NiCp*(IMes)] (279). It was proposed that the reaction of [Ni(Cp*)Cl(IMes)] with KOtBu and HBcat generates the initial nickel–hydride complex 278via a σ-bond metathesis mechanism. The active species 279 forms an initial agostic intermediate 280 with HBCat, which then associates with the styrene substrate to furnish 281. An energetically favourable homolytic fission of the B–H bond followed by a radical Giese addition to the styrene forms back the nickel–hydride intermediate 278 and the linear borylated benzylic radical 282. It is noteworthy to point out that the reaction was completely inhibited with the addition of one equivalent of a radical scavenger such as TEMPO. Final hydrogen atom abstraction from 278 releases the final hydroboration product, regenerating the active NiI species 279 to close the catalytic cycle.
In 2022, the Kennedy research group developed a class of mononuclear, anionic Ni0 complexes 283, stabilised by a unique bidentate NHC-pyridone ligand which exclusively coordinates to the Ni centre in a κ2-C,N coordination mode113 (Scheme 45). From single-crystal diffraction analysis, the [K(18-crown-6)]+ counterion of this anionic nickelate was observed to be in a contact ion pair with the pyridone oxygen, allowing for monomeric isolation of the complex instead of multimetallic aggregation and clustering induced by the pyridone.113 The low-valent Ni0 core is highly electron-rich, with an asymmetric electronic and steric coordination environment around the nickel centre. In their work, this class of complexes was demonstrated to exhibit exceptional catalytic activity towards the hydroboration of alkenes (with the evaluation of only styrenes 285 and β-methylstyrene 284), affording the desired hydroboration product in good yields and with excellent branched Markovnikov selectivity (Scheme 45). Related stoichiometric experiments reveal that the pyridone ligand plays a role in facilitating the transfer of the Lewis acidic HBpin (pinacolborane) reagent to the alkene through its involvement in the secondary coordination sphere.
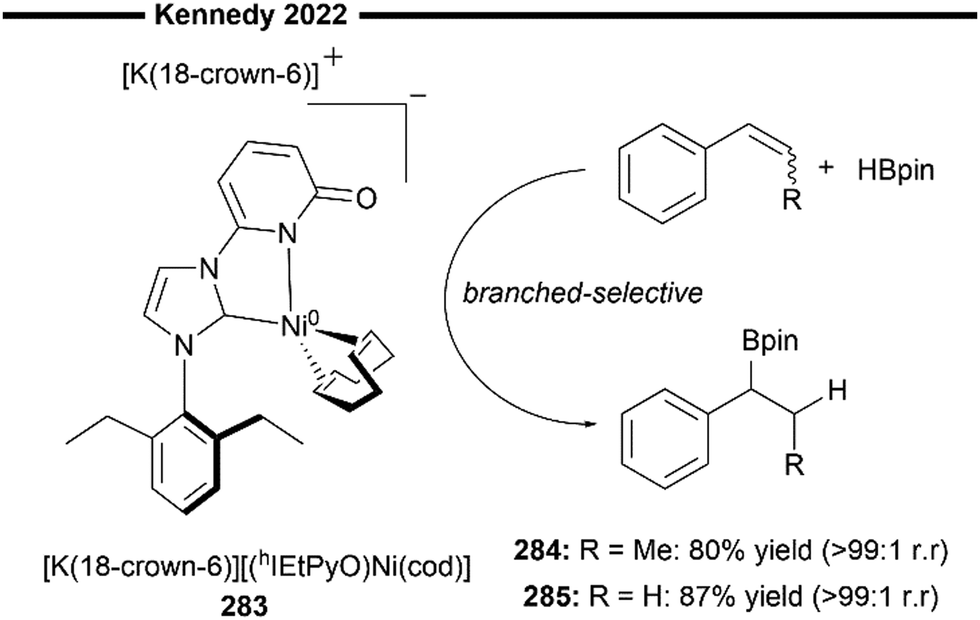 | ||
| Scheme 45 κ2-C,N anionic NHC/pyridone Ni0 complex for hydroboration of alkenes.113 | ||
3.4 Hydrosilylation of alkenes
Hydrosilylation of alkenes remains a powerful method for the formation of organosilicon compounds, with the addition of silicon hydrides across CC p-bonds.114 It is widely used in industry to produce silane coupling agents and silicone polymers such as oils, rubbers, and resins. Various phosphine-ligated nickel complexes have been reported as hydrosilylation catalysts, but face the issue of undesirable site-selectivity and activity due to the competing side reactions like dehydrogenative silylation.114 Recently, ligand-free Ni-catalysed electrophilic hydrosilylation of alkenes was also developed.115 However, regioselective nickel-catalysed hydrosilylation of CC bonds enabled by NHC ligands has been rather elusive and difficult to achieve, with only a few studies having been reported on this topic.
The first attempt of Ni/NHC-catalysed hydrosilylation of alkenes was demonstrated by Valerga and co-workers in 2012116 (Scheme 46). Various NiII(allyl)–NHC complex bearing bidentate 2-picolyl-imidazolylidene ligands (286 and 287) were found to show high activity for the hydrosilylation of simple styrene substrates. Different substituents on the NHC and allyl ligand have a marked impact on the efficiency and selectivity of the hydrosilylation reaction. Only complex 286 was found to display activity when α-methylstyrene was used, giving the hydrosilylated product with complete anti-Markovnikov selectivity albeit in diminished yield. With regular styrene as the substrate, complex 287 affords excellent Markovnikov selectivity whereas complex 286 proved to be much less selective. Preliminary mechanistic studies show the presence of Ni–H and Ni–silyl intermediates, suggesting that the mechanism could proceed as illustrated in Schemes 8b and c, even though a full proposal was not provided.
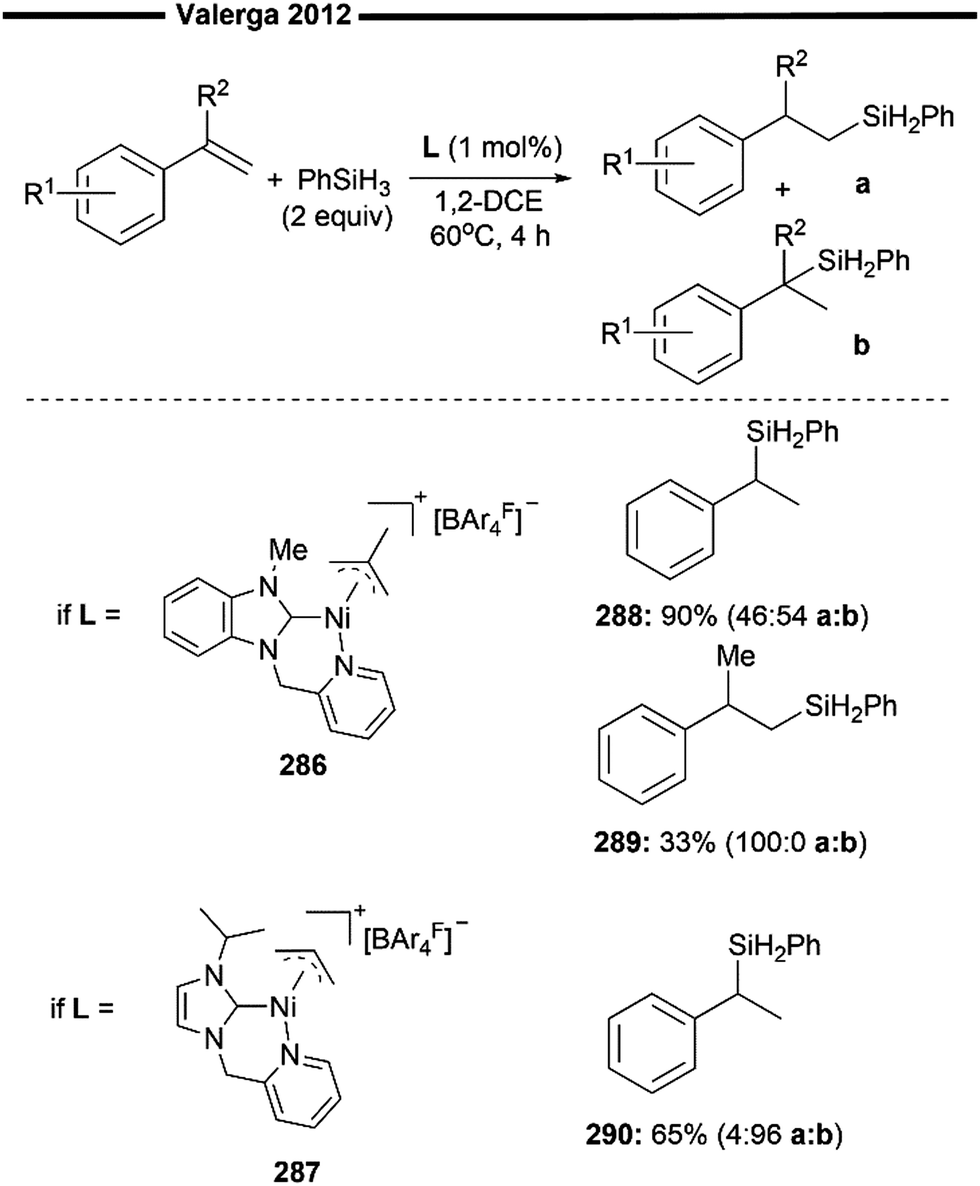 | ||
| Scheme 46 Allyl-NiII(NHC)-catalysed hydrosilylation of styrenes.116 | ||
In 2013, Montgomery and co-workers developed a method for the selective hydrosilylation of monosubstituted allenes using nickel–NHC catalysts.117 Building upon their initial discovery, in 2015 they expanded the scope of their research with the development of a regioselective and stereoselective hydrosilylation protocol for 1,3-disubstituted allenes118 (Scheme 47). The steric bulk of the NHC ligand played a key role in the successful synthesis of vinyl silane 292–294 and (Z)-alkenylsilanes 296–297 under the optimised regime, both of which were achieved with exceptional regio- and stereo-selectivity.
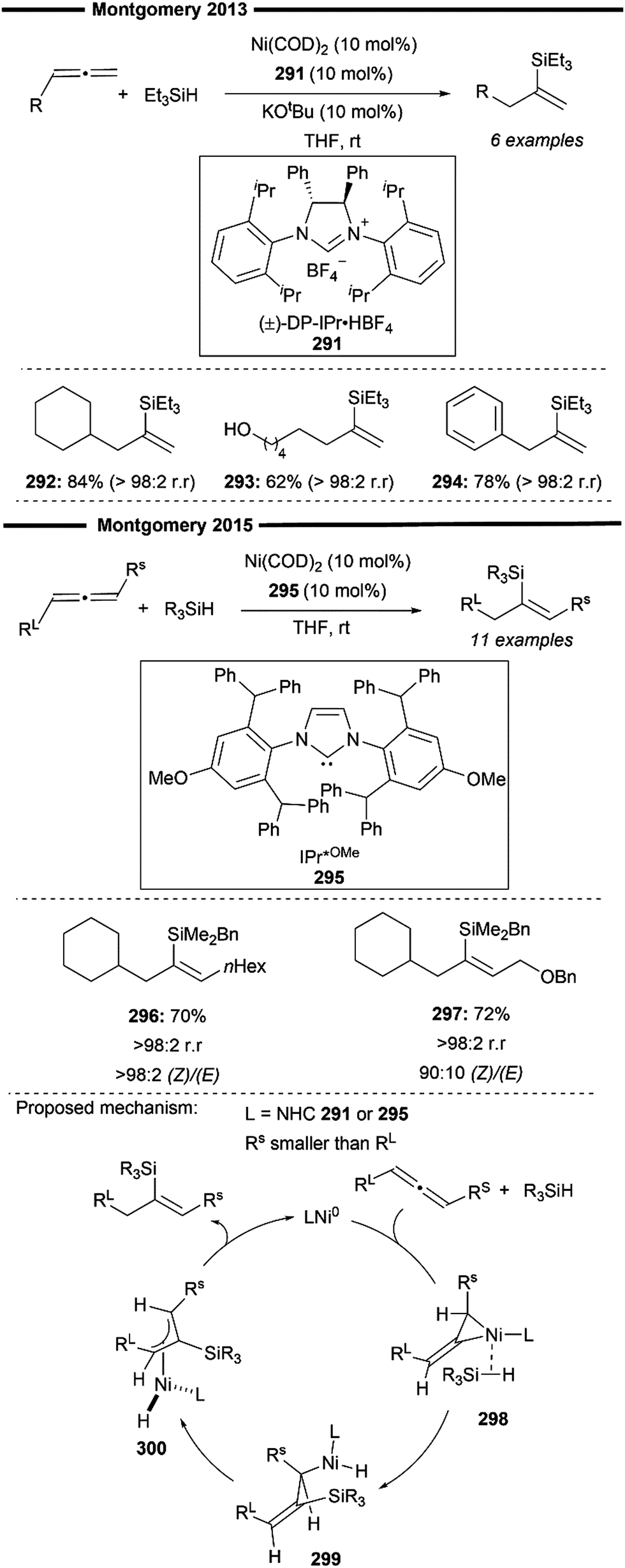 | ||
| Scheme 47 Ni/NHC-catalysed regioselective and stereoselective hydrosilylation of allenes.117,118 | ||
Deuterium-labelling studies were performed to further investigate the origin of regioselectivity and the reaction mechanism. The presence of only traces of cross-over products when using deuterated and regular silanes suggests that the mechanism does not involve the formation of nickel–hydride species that are not bonded to a silyl group, or vice versa. Extensive computational studies84 have provided insight into a plausible mechanism for the reaction (Scheme 47). Coordination of the silane and least sterically hindered face of the allene (thereby differentiating the different π-systems of the unsymmetrical allenes) to the nickel centre forms the putative complex 298. This is followed by the rate-determining step, in which a concerted Si–H bond oxidative addition – silyl insertion occurs to form the β-silyl nickel–hydride intermediate 299. This step also determines the regioselectivity of the reaction with silyl-insertion being more thermodynamically favourable than hydride-insertion. The sterically bulky NHC ligand further reinforces this regiocontrol. Facile formation of the η3-allyl intermediate (300) occurs, with minimisation of allylic 1,3-strain where the RS and RL substituents prefer to be trans in the π-allyl intermediate. A final C–H reductive elimination, avoiding the formation of a more congested olefin in the transition state,119 delivers the (Z)-alkenylsilane product with excellent regioselectivity and (Z)-selectivity.
In 2022, Cook et al. reported a Ni0/NHC-catalysed branched-selective hydrosilylation reaction of styrenes with 2°, 3° and chlorosilanes120 (Scheme 48). Industrially relevant silanes, particularly those substituted with chlorine or alkoxy groups, have not been extensively studied in the field of base-metal catalysis. Branched selective hydrosilylation of alkenes remains a challenging task, as it often faces the issue of lower selectivity and a narrower scope.121 (NHC)Ni0(alkene)2 complexes, which can be easily prepared, have been shown to be effective in catalysing this transformation.
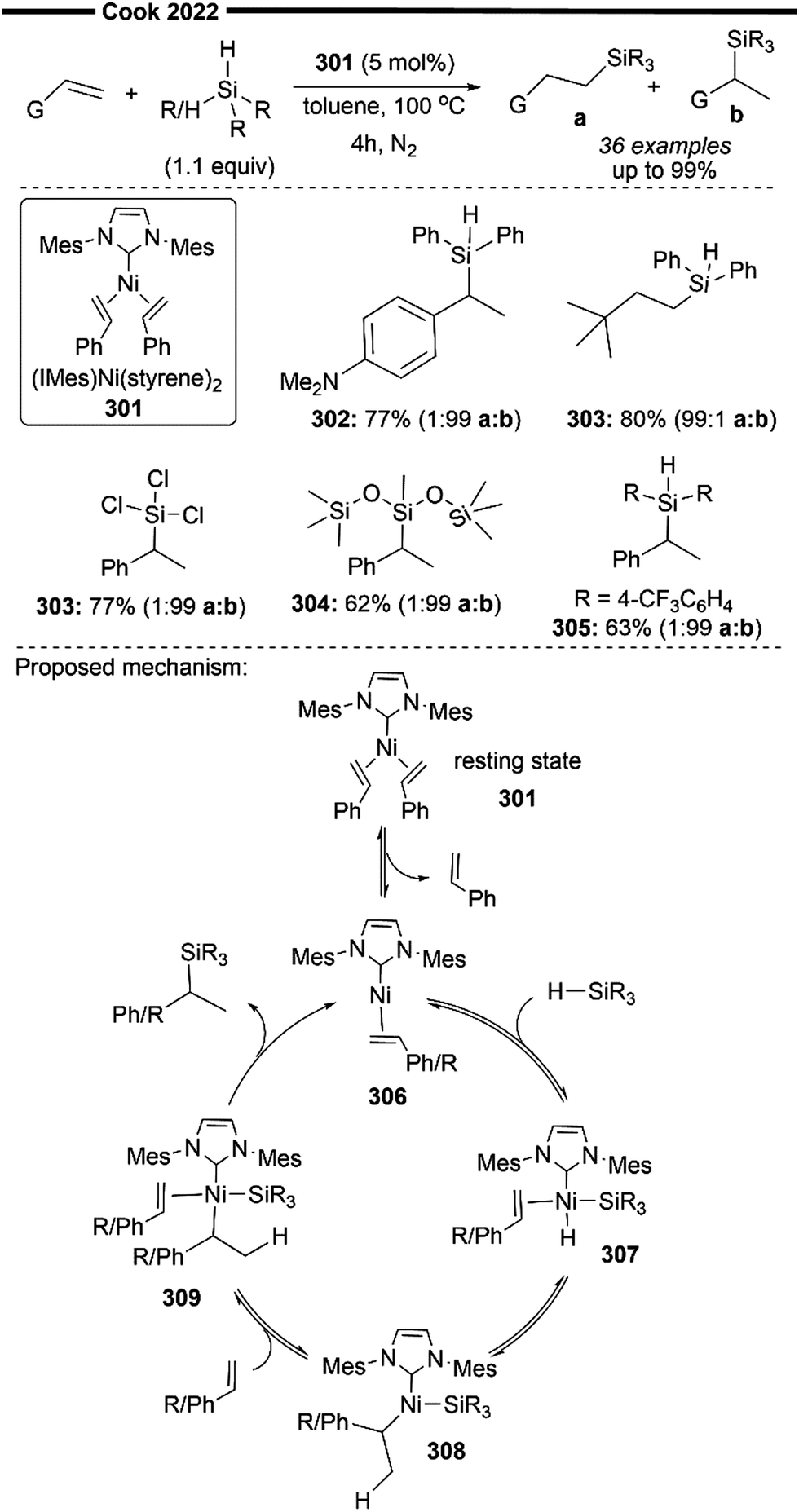 | ||
| Scheme 48 Ni/NHC-catalysed hydrosilylation of alkenes with 2°, 3° and chlorosilanes.120 | ||
Extensive mechanistic investigations to probe into the reaction mechanism were carried out by the authors. Deuterium-labelling experiments reveals minimal 2D incorporation at the benzylic position, hence supporting the Chalk–Harrod122 mechanism as the primary reaction pathway. In the Chalk–Harrod mechanism, insertion of the alkene into the nickel–hydride (Ni–H) bond occurs preferentially to the insertion into the nickel–silyl (Ni–Si) bond. Radical-trapping experiments validate an ionic pathway as expected by the authors, rather than a one-electron radical pathway. In summary, initial alkene dissociation from the resting state catalyst 301 forms the 14-electron active complex 306. Oxidative addition of silane into the complex forms the silyl–Ni–hydride intermediate 307. Subsequent migratory insertion of the coordinated styrene into the Ni–H bond affords a benzylic nickel species (if R = aryl) 308 with an open coordination site available for coordination with another alkene substrate. This results in the formation of the 16-electron complex 309 which undergoes facile C–Si reductive elimination, releasing the branched product while regenerating the active coordinatively unsaturated Ni species 306.
Electron-donating amine-containing styrenes (302) were well-tolerated, giving the product in moderate yield and with excellent branched selectivity. Terminal aliphatic alkenes are also viable, but with the selectivity being reversed to favour the linear-selective hydrosilylated product (303). This demonstrates the importance of an aryl substituent adjacent to the CC bond for exclusive formation of the branched product, validating the involvement of a benzylic nickel intermediate 308. Without adjacent π-stabilisation of the nickel intermediate following insertion, the regioselectivity was predominantly governed by the steric interactions between the NHC ligand and alkyl substituent, giving rise to a preference for the linear product resulting from the less hindered transition state. Various electronically altered silanes such as diarylsilanes (305), 3° bis(alkoxy)-substituted silanes (304) and trichlorosilane (303) also reacted well, delivering the product with remarkable branched selectivity and in good yields.
3.5 Hydroamination of alkenes
Nitrogen-containing compounds play a vital role in biological systems and are also widely used in various industries such as bulk chemicals, specialty chemicals, and pharmaceuticals.123 Among the plethora of reported methods for synthesising them, the direct construction of the desired C–N bond through the addition of an amine to an unsaturated CC bond (known as hydroamination) is still of elegance and significant importance in industries. This route provides a highly atom-economical synthetic route starting with inexpensive and readily accessible alkenes. In addition to the challenge of regioselectivity, hydroamination reactions present an additional hurdle as they are only slightly exergonic due to a large entropic cost, particularly for the intermolecular variant.124 To the best of our knowledge, the Ni/NHC-catalysed hydroamination reaction remains elusive, with only one reported instance of it.
The only work to date detailing the hydroamination of alkenes with Ni/NHC was reported by Ghosh and co-workers in 2010. Well-defined complexes of nickel with 1,2,4-triazole derived NHC as precatalysts (310 and 311) were effective in promoting the hydroamination of activated electron-deficient alkenes with secondary amines125 (Scheme 49).
 | ||
| Scheme 49 Ni/NHC-catalysed hydroamination of activated alkenes with 2° amines.125 | ||
Preliminary computational calculations to determine the nature of bonding of the (NHC)2NiBr2 complex were performed, which showed that the 1,2,4-triazole based NHC is a strong σ-donating ligand like the common imidazole based NHC ligands (Chart 1). An amine activation route which involves the oxidative addition of the N–H bond to the nickel centre was ruled out as the Ni–N (amido) bond energy is known to be very low. Extensive DFT calculations to probe into the mechanism for this reaction were published separately in 2017.126 The putative mechanism with the least energy penalty is illustrated in Scheme 47. Ag-promoted ligand exchange with the bound halide ion forms the triflate coordinated Ni/NHC complex. Initial alkene coordination with the active [(NHC)2Ni(OTf)]+ species forms 316. Anti-Markovnikov type attack of the secondary amine to the less substituted end of the alkene bound to Ni yields intermediate 317 which can be depicted in two resonance forms (α-C bound or N-bound if EWG = CN). Subsequent 1,3-proton shuttle of the N-bound complex 317 assisted by another equivalent of the basic amine (via intermediate 318), furnished the final hydroamination product upon dissociation from 319, closing the catalytic cycle.
Under their protocol, pyrrolidine (312), morpholine (314), piperidine (313) and diethylamine (315) are examples of 2° amine substrates that have been successfully employed. Only activated alkenes adjacent to strongly electron-withdrawing groups like nitriles and esters proved to be suitable, delivering the product in moderate yields and with excellent anti-Markovnikov selectivity (312–315).
3.6 Hydroacylation and hydrocarbamoylation of alkenes
The use of transition metal catalysts to formally add an aldehyde C–H bond across unsaturated CC bonds (termed alkene and alkyne hydroacylation) has become a popular and efficient method for synthesising ketones in recent years.127
In the seminal work reported by Ogoshi and co-workers in 2012, they developed a Ni0/ItBu catalysed intramolecular alkene hydroacylation reaction to access five/six-membered benzocyclic ketones128 (Scheme 50). Mechanistic studies involving stoichiometric experiments128 and DFT calculations129 provide valuable insights into the plausible reaction mechanism which involves a dioxanickelacycle intermediate in the lowest energy pathway.
 | ||
| Scheme 50 Ni/NHC-catalysed intramolecular hydroacylation of alkenes.128 | ||
Coordination of the substrate to the Ni/ItBu active catalyst forms the (η2:η2-enal)Ni0(ItBu) complex 326. Subsequent oxidative cyclisation of 326 forms the monomeric oxanickelacycle intermediate 327 which undergoes facile dimerisation to the dioxanickelacycle 328. The dimer 328 was isolated in 84% yield in a separate stoichiometric experiment and was characterised with 1H NMR and X-ray crystallography. A later DFT study129 delved deeper into the mechanism and revealed that the facile dimerisation of 327, which results in the formation of 328, significantly reduces the free energy required for the subsequent rate-determining β-hydride elimination step that leads to the formation of intermediate 329. This is followed by a formal reductive elimination step, which releases the final benzocyclic ketone product and regenerates the active Ni/ItBu species. Their protocol presents a significant advantage, as their efficient hydroacylation process takes place through an oxanickelacycle intermediate without the occurrence of any decarbonylation. Decarbonylation of acyl-metal intermediates is a common undesired side reaction that leads to a decrease in atom efficiency and deactivation of the active catalyst through the coordination with carbon monoxide.130
Fluorine and the silyl substituted o-allylbenzaldehyde substrate delivers the corresponding indanone product in high yields (320–321). 1,1-Dimethyldisubstituted alkenes (322 and 324) and 1,2-disubstituted alkenes (323) were also effective substrates. Notably, the reaction can also be extended to the synthesis of six-membered ring systems (324 and 325) with the desired 1-tetralone derivatives furnished in excellent yields.
Ogoshi and co-workers expanded on their previous development of Ni/NHC catalysed intramolecular hydroacylation of alkenes (Scheme 50) and reported the successful intramolecular coupling of alkenes and aldehydes in the presence of hydrosilanes under reducing conditions (Scheme 51). (η6-toluene)Ni(SIPr) 330 which can be conveniently synthesised in a single step on a gram-scale was used as the precatalyst. This modified approach provides access to a variety of silyl-protected 1-indanol derivatives with high syn-diastereoselectivity (between the silyl ether and methyl group), instead of benzocyclic ketones. Although a plausible mechanism has not been described in the literature, some insights into the mechanism can be inferred by drawing parallels to analogous mechanisms involving the reductive coupling of aldehydes and hydrosilanes, which proceed through oxanickelacycle intermediates.128,132 The diastereoselective formation of a similar oxanickelacycle (331) is believed to be facilitated by the Lewis acidic hydrosilane. Cleavage of the Ni–O bond of the oxanickelacycle by σ-bond metathesis with the Si–H bond of the hydrosilane forms a nickel–hydride intermediate (332) which undergoes reductive elimination to afford the desired product.
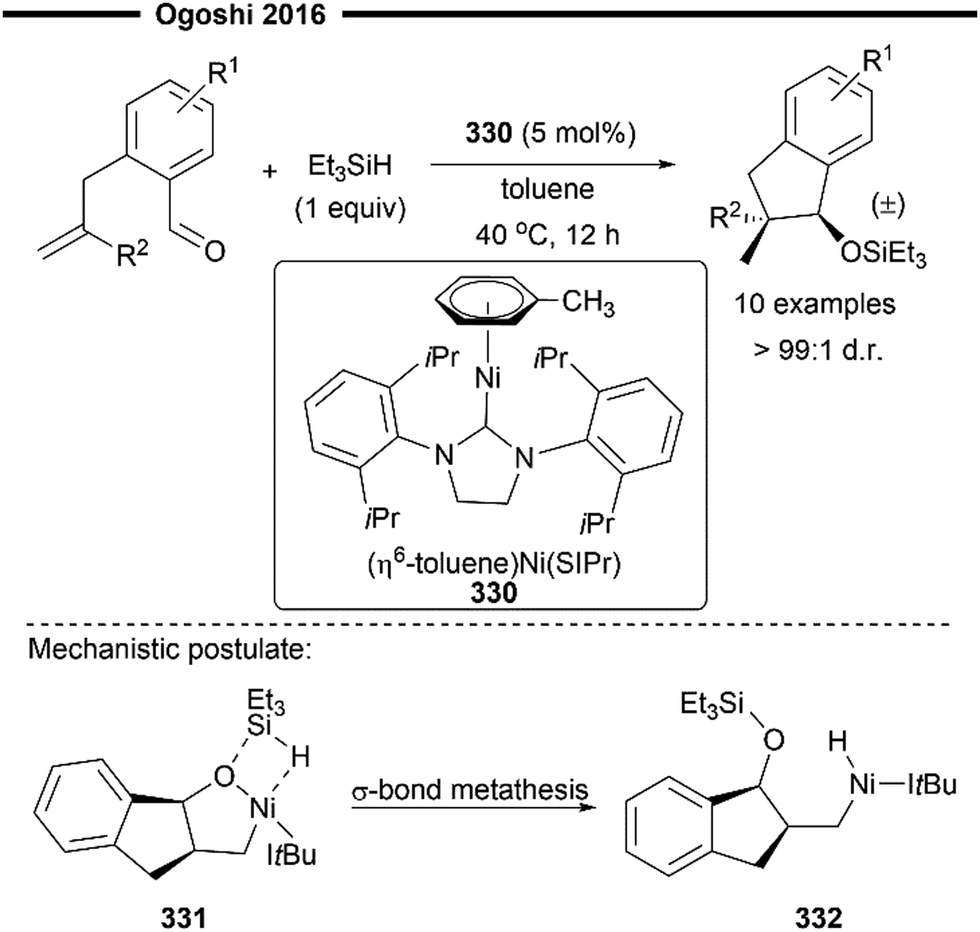 | ||
| Scheme 51 Ni/NHC reductive coupling of alkenes and carbonyls with hydrosilanes.131 | ||
In 2016, Stanley and co-workers reported the intramolecular, exo-selective hydroacylation of N-allylindole-2-carboxaldehyde enabled with Ni0/IAd (IAd = 1,3-bis(1-adamantyl)imidazol-2-ylidene)133 (Scheme 52). Their research aimed to expand upon the previous advancements in and scope of Ni/NHC-catalysed hydroacylation, as developed by Ogoshi,128 by enabling the formation of heterocyclic ketones (beyond just carbocyclic ketones), which is a key intermediate for a sequential enantioselective α-arylation reaction, thus allowing access to valuable N-containing heterocyclic ketones with α-chiral quaternary stereocentres.
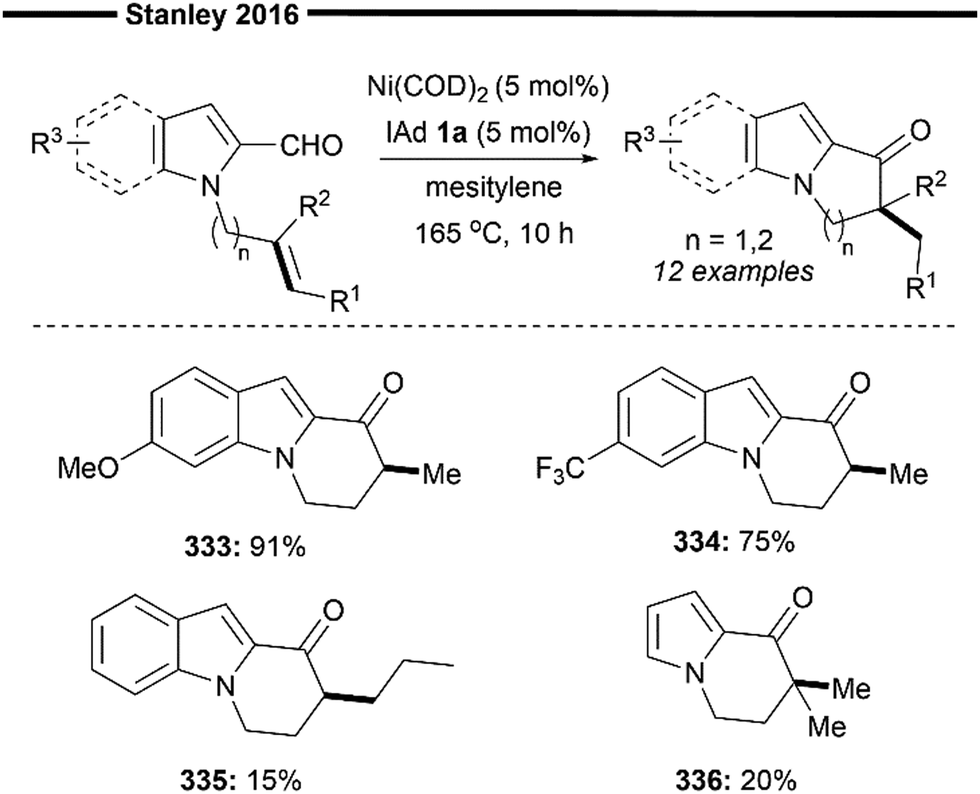 | ||
| Scheme 52 Ni0/IAd-catalysed intramolecular hydroacylation of alkenes for synthesis of heterocyclic ketones.133 | ||
Various indole moieties bearing electron-donating (333), or electron-withdrawing groups (334) were successfully tolerated under the protocol giving six-membered heterocyclic ketones in moderate yield. However, the scope of the substrates used is primarily limited to monosubstituted alkenes. The presence of additional β-substituents on the alkene was found to be detrimental to the hydroacylation reaction, resulting in poor yields (335). Likewise, 1,1-disubstituted alkenes also performed poorly (336) under their protocol.
Hydrocarbamoylation refers to the formal insertion of unsaturated CC bonds into the C–H bond of formamide. It provides yet another alternative to access alkanamides, circumventing the need for the introduction of toxic carbon monoxide, which is commonly used in the aminocarbonylation of electrophiles134 to access amides.
To date, the only study of Ni/NHC-catalysed hydrocarbamoylation of alkenes was reported by Hiyama and co-workers135 in 2012. Using [Ni(COD)2] and IAd together with triethylaluminium as a co-catalyst, they could obtain linear alkanamides in high yields with excellent anti-Markovnikov selectivity (Scheme 53).
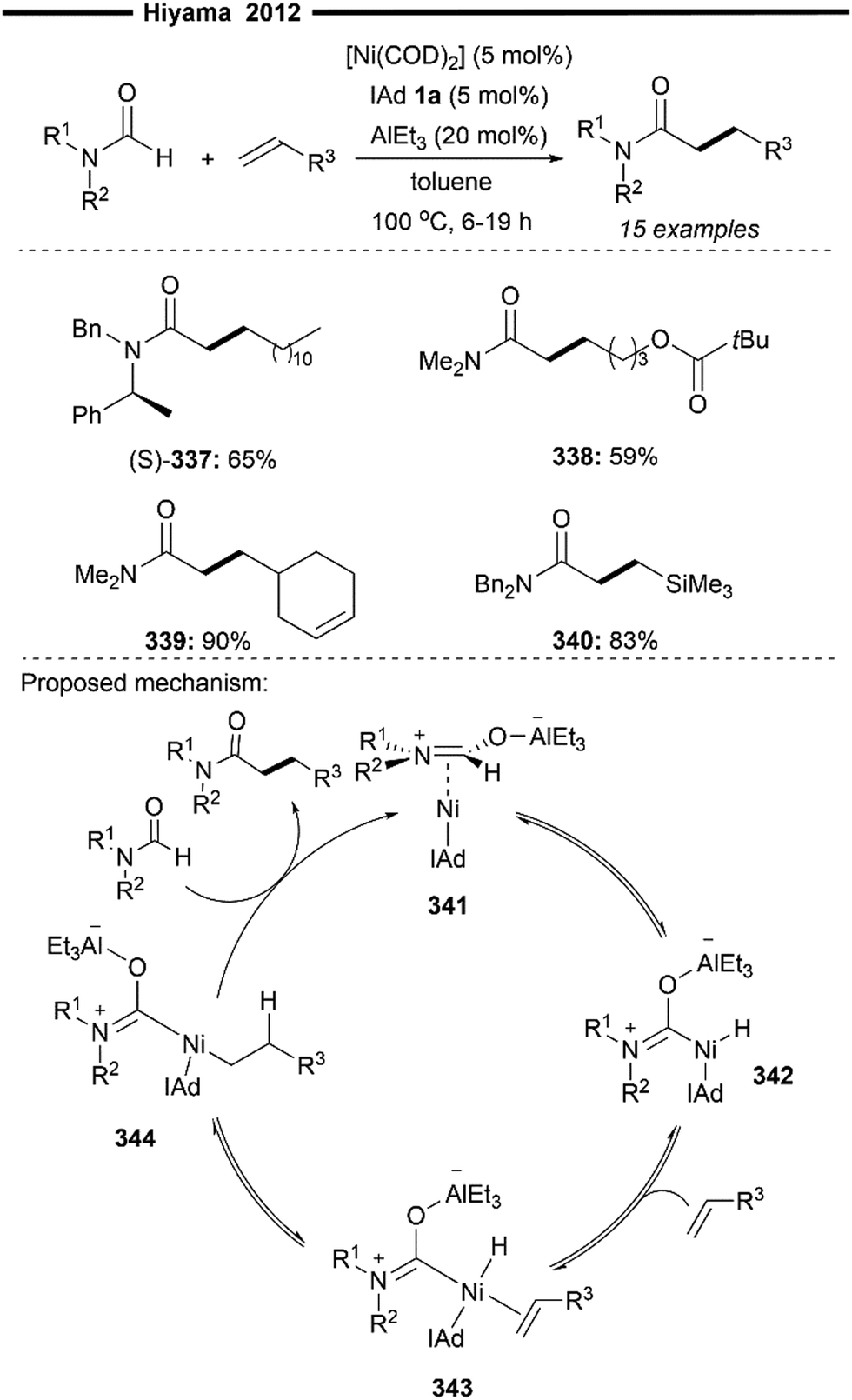 | ||
| Scheme 53 Linear-selective hydrocarbamoylation of 1-alkenes under Ni/NHC/Lewis acid cooperative catalysis.135 | ||
Various N-alkyl substituted formamides (substituted with Me or Bn) proved to be effective in their hydrocarbamoylation regime. When an enantioenriched formamide is used, the corresponding optically pure linear alkanamide is likewise formed without the erosion of chirality ((S)-337). However, formamides derived from primary amines (with an acidic N–H) were incompatible under the Lewis acidic conditions. Monosubstituted alkenes bearing internal CC linkages (339), silyl groups (340) and alkoxycarbonyl groups (338) were also compatible. Limitations in the scope include 1,1-disubstituted or 1,2-disubstituted internal alkenes, which were unsuccessful in delivering the desired product.
In a tentative mechanism provided by the authors (Scheme 53), the activated formamide substrate (via O-coordination with triethylaluminium Lewis acid) forms an η2-formamidenickel intermediate 341. Subsequent oxidative addition of the Ni0 centre into the C(sp2)–H bond forms the nickel–hydride intermediate 342. Coordination of the alkene substrate forms 343, which participates in regioselective migratory insertion to deliver a primary alkyl-nickel intermediate 344. The less hindered primary alkyl-nickel intermediate is formed preferentially over the secondary alkyl-nickel intermediate which results in excellent linear-selectivity. Lastly, reductive elimination of 344 forms the crucial C(sp3)–C(sp2) bond, with the release of the linear alkanamide product. The presence of a highly bulky NHC ligand is believed to govern the regioselectivity of insertion and to improve the efficiency of the reaction through the facilitation of reductive elimination of 344.
3.7 Dicarbofunctionalisation of alkenes
Dicarbofunctionalisation of alkenes, involving the incorporation of two carbogenic moieties across the carbon–carbon double bond, has emerged as an attractive method for elevating molecular complexity from coupling of simple alkene precursors.32 Complex sp3-carbon frameworks can be easily accessed in this manner. Nickel-catalysed dicarbofunctionalisation has gained significant traction over the years, and with appropriate adjustment of the catalyst's electronic and steric features, β-H elimination leading to the undesired Heck-type product can be suppressed. Two major pathways typically proceed, the two-electron ionic pathway and the radical pathway which involves a single electron transfer (SET) process. The former pathway is more frequently documented with the utilisation of a robust σ-donating NHC ligand. Another proposed mechanism involving oxidative cyclisation has also been put forth.136,137 Often cited in literature studies, redox-neutral dicarbofunctionalisation refers to the coupling of alkene with a nucleophilic (e.g., organometallics) and electrophilic (e.g., halides or pseudohalides) reagent, while reductive dicarbofunctionalisation refers to the coupling of alkene with two electrophilic partners in the presence of a reductant.32,138The reaction is initiated either through transmetalation with an organometallic compound or through the oxidative addition of an electrophile (e.g., halides and pseudohalides), resulting in the formation of a carbon–nickel species. This species undergoes a site-selective insertion with an alkene, forming a regiodefined alkyl-nickel intermediate. Regioselectivity of the insertion is primarily dictated by steric factors, minimising unfavourable steric repulsions between the olefinic substituent and the NHC. The second carbogenic component can then be trapped and incorporated through cross-coupling with another organometallic compound or electrophile. The challenges associated with dicarbofunctionalisation, such as the control of regio- and stereo-selectivities, competitive β-H elimination and undesired coupling of nucleophilic and electrophilic reagents, have been addressed in recent studies, through the utilisation of N-heterocyclic carbene ligands and their distinctive properties translated to the NHC–Ni complex (Scheme 54).
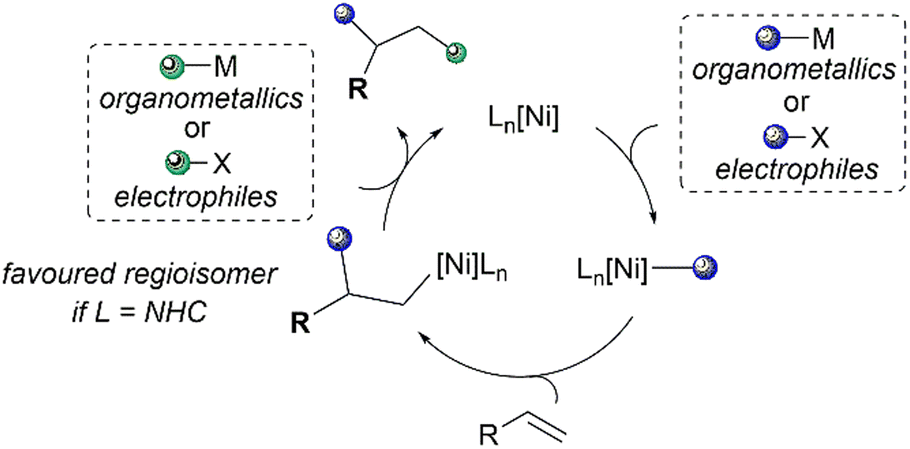 | ||
| Scheme 54 Generalised two-electron pathway of NHC/Ni-catalysed dicarbofunctionalisation. | ||
One of the earlier reports on NHC/Ni-catalysed dicarbofunctionalisation of alkenes was that of Ogoshi's work, who described a selective three-component coupling reaction of tetrafluoroethene, ethylene and aldehydes to furnish fluorine-containing ketone derivatives (Scheme 55).139 This reaction can be formally conceptualised as 1,2-alkylacylation of ethene or hydroalkylation of tetrafluoroethene. The optimal ligand found for this reaction was IPr 1f, which enabled the formation of the desired cross-trimerisation product while suppressing the formation of the side product originating from the dimerisation of aldehyde. A plausible reaction mechanism was described by the authors. Coordination of ethylene and tetrafluoroethene with the active IPr-Ni0 species forms the η2:η2-Ni(IPr) complex 349, which undergoes an oxidative cyclisation with the bound alkenes, to form a 2,2,3,3-tetrafluoronickelacyclopentane intermediate 350. Chemoselective nucleophilic addition from the more electron-rich alkylnickel (–CH2Ni) bond to the aldehyde produces the seven-membered oxanickelacycle species 351. β-Hydride elimination of 351 precedes the final reductive elimination of 352 to afford the tetrafluorinated ketone product, while regenerating the active IPr-Ni0 species.
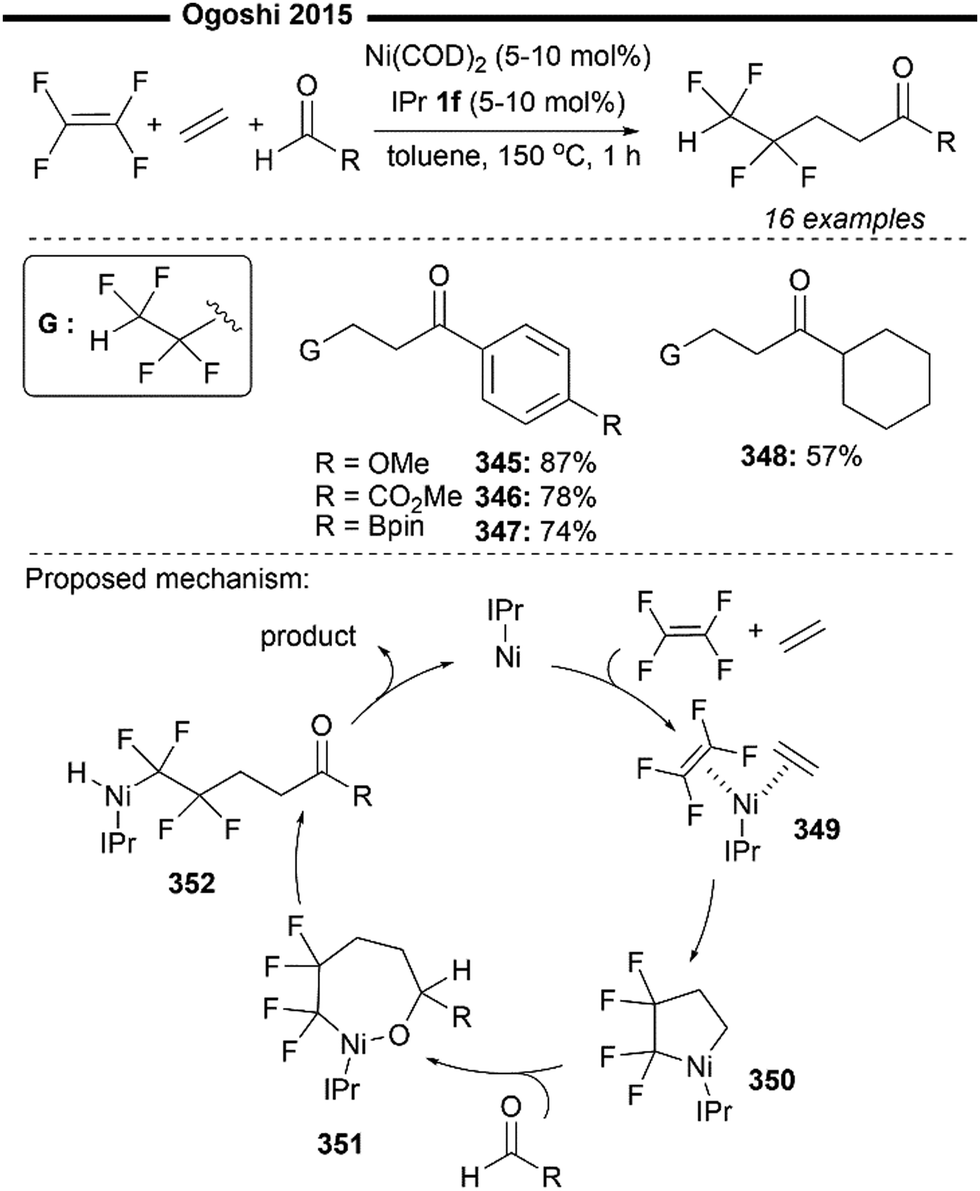 | ||
| Scheme 55 NHC/Ni-catalysed cross-trimerisation of tetrafluoroethene, ethene and aldehyde.139 | ||
While olefinic substituents were not varied, different electronic environments of the aldehydes were tested under their regime. Benzaldehyde derivatives such as p-anisaldehyde (345), 4-formylbenzoate (346) or boronic ester-substituted aldehydes (347) were well tolerated. Aliphatic aldehydes, such as, cyclohexyl carboxaldehyde (348), also gave the corresponding aliphatic ketone product in moderate yields.
In 2016, Cramer and co-workers reported an enantio- and diastereo-selective NHC/Ni-catalysed three-component coupling reaction of aldehydes, silanes and norbornenes, to deliver enantioenriched silyl-protected indanols140 (Scheme 56). Survey of a variety of chiral NHC ligands led to the most effective C2-symmetric NHC ligand 353 with a 1,2-di(napthalen-1-yl)ethylene diamine backbone scaffold and an ortho-isopropylphenyl group as the N-aryl arm.
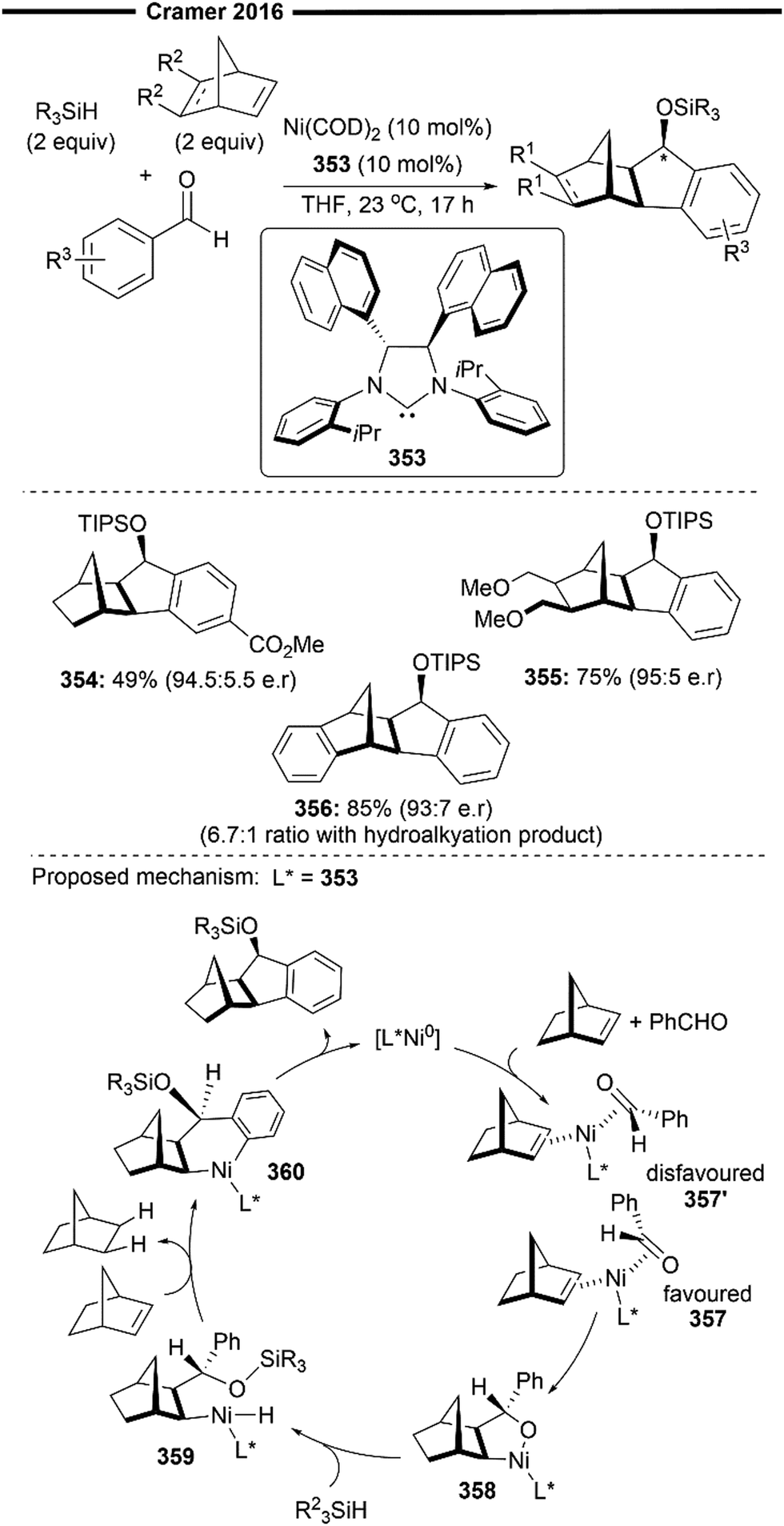 | ||
| Scheme 56 NHC/Ni-catalysed enantioselective three-component reaction to access silylated indanols.140 | ||
A plausible reaction mechanism commences with the η2-coordination of the norbornene and aldehyde to the chiral NHC–Ni0 complex. The favoured orientation mode of the aldehyde group is proposed to be 357, where the aryl substituent of the aldehyde group is positioned away from both the methylene bridge of norbornene and the flanking bulky N-aryl substituents of the NHC to minimise unfavourable steric interactions. Enantio-determining oxidative cyclisation of 357 leads to the formation of the fused oxanickelacycle intermediate 358. Reaction of the nickel-alkoxide with silane breaks the Ni–O bond of the oxanickelacycle, rupturing the ring to form intermediate 359, which consists of a silyl-protected alcohol and a nickel–hydride moiety. Another molecule of norbornene enters the cycle and reacts with 359 in a sequence, hydronickelation, C(sp2)–H activation (of the aldehyde substrate) and reductive elimination, to afford the cyclonickelated species 360, with concomitant release of the hydrogenated norbornane species. Final reductive elimination leads to the formation of the second C(sp3)–C(sp2) bond, releasing the silylated indanol product and regenerating the active NHC–Ni0 species.
Reaction with electron-deficient benzaldehyde 354 (with a para-substituted ester moiety) resulted in slightly reduced yields but enantioselectivity remained high. Norbornene derivatives, such as the exo-methylenealkoxy-substituted derivative 355 or benzonorbornadiene 356, were effective substrates, delivering the annulated products in good yields and enantioselectivity. Certain limitations of the catalytic regime were highlighted. A minor hydroalkylated side product was observed for some substrates, possibly formed from the competing reductive elimination of nickel–hydride 359. No appreciable reaction with unstrained alkenes like cyclopentenes was observed under the reaction conditions.
In 2017, Stanley and co-workers described an NHC/Ni-catalysed carboacylation reaction of o-allylbenzamides with aryl boronic pinacol esters to access a variety of 2-benzyl-2,3-dihydro-1H-inden-1-ones141 (Scheme 57). Their methodology was noteworthy as it involves the key NHC–Ni-mediated amide C–N bond activation step and obviates the requirement for strained cyclic ketones or directing groups, which had been conventionally used to facilitate carboacylation.
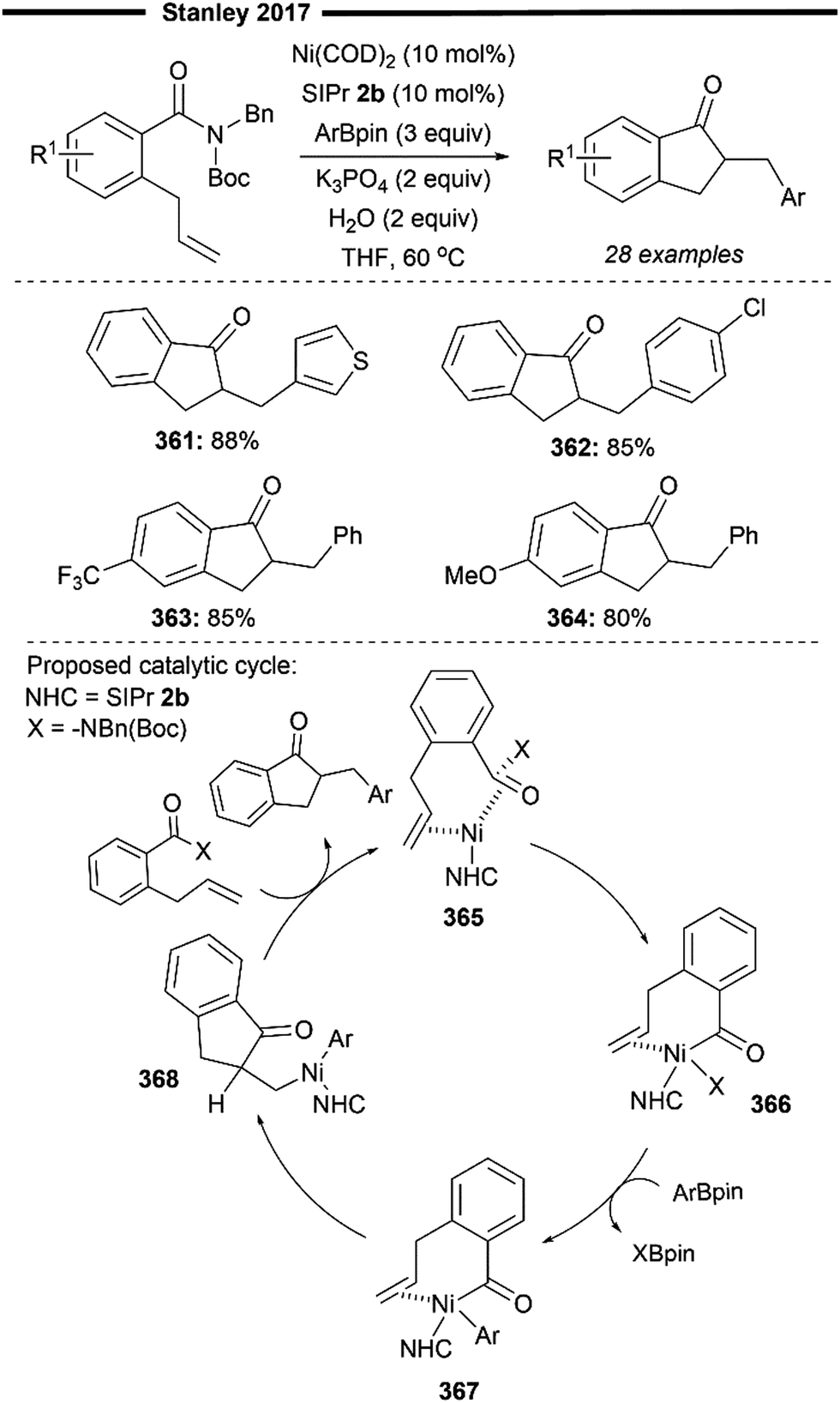 | ||
| Scheme 57 NHC/Ni-catalysed carboacylation of alkenes via amide C–N bond activation.141 | ||
The carboacylation methodology enabled with the strongly electron-donating SIPr 2b (1,3-bis(2,6- diisopropylphenyl)-4,5-dihydroimidazol-2-ylidine) ligand was evaluated using various substituted arylboronic pinacol esters (ArBpin) and o-allylbenzamides. The regime was compatible with heteroarylboronic acid pinacol esters featuring a thiophene 361 group. Electron-deficient para-chlorinated ArBpin 362 was well-tolerated, without competing activation of the C–Cl bond. Carboacylation of o-allylbenzamide with electron-withdrawing (363) or electron-donating (364) substituents afforded the corresponding cyclised product in excellent yields. However, the reaction had certain substrate limitations which include o-allylbenzamides with a 1,1-disubstituted alkene moiety, or acyclic 5-hexenamide derivatives.
The authors proposed a possible reaction pathway, initiated by the η2(alkene):η2(amide) coordination of the o-allylbenzamide substrate with the NHC–Ni0 catalyst. Subsequent oxidative addition of the C–N amide bond generates the acyl-Ni(II)-amido complex 366. Base-assisted transmetalation with the arylboronic pinacol ester produced the acyl-Ni(II)-aryl intermediate 367, followed by regioselective insertion of the acyl-nickel bond into the tethered alkene, to form the primary alkyl-Ni(II)-aryl complex 368. Reductive elimination releases the indanone product and regenerates the active species 365 through coordination with another o-allylbenzamide molecule. Although the proposed pathway involves transmetalation followed by migratory insertion, the reverse order of events with migratory insertion preceding transmetalation cannot be completely ruled out.
In 2019, Newman's group reported a NHC/Ni-catalysed intramolecular carboacylation and hydroacylation reaction of methyl ortho-allylbenzoate derivatives (Scheme 58), via C(acyl)–O bond activation, building upon Stanley's prior work.141 Despite the strong C(acyl)–O bond of methyl esters142 and the high energy cost incurred in the generation of acyl-Ni intermediates, this challenging transformation was rendered feasible through an intramolecular cyclisation step to trap the unstable acyl-Ni intermediate.141,143 The most effective proligand was determined to be the saturated SIPr salt, and a combination of CsF and KF in the reaction media was found to be crucial in enhancing the yield and ensuring the reproducibility of the reaction. Both electron-withdrawing (enolisable para-acetyl group) 370 and electron-donating substituents (para-methoxy) 371 on the arylboronic acid substrate were well tolerated. In their attempt to realise an alkylacylation reaction with the formation of challenging C(sp3)–C(sp3) bonds, alkylboronic acid was used but instead produced a significant amount of the reduced indanone product. With this serendipitous finding, their group proceeded to optimise this reductive hydroacylation reaction, with 1-phenylethanol being the most effective hydride source and reductant. A monodeuterated derivative of the alcohol also led to the formation of the corresponding indanone product 372 with high (85%) deuterium incorporation in the methyl group, opening opportunities for pharmaceutical applications.
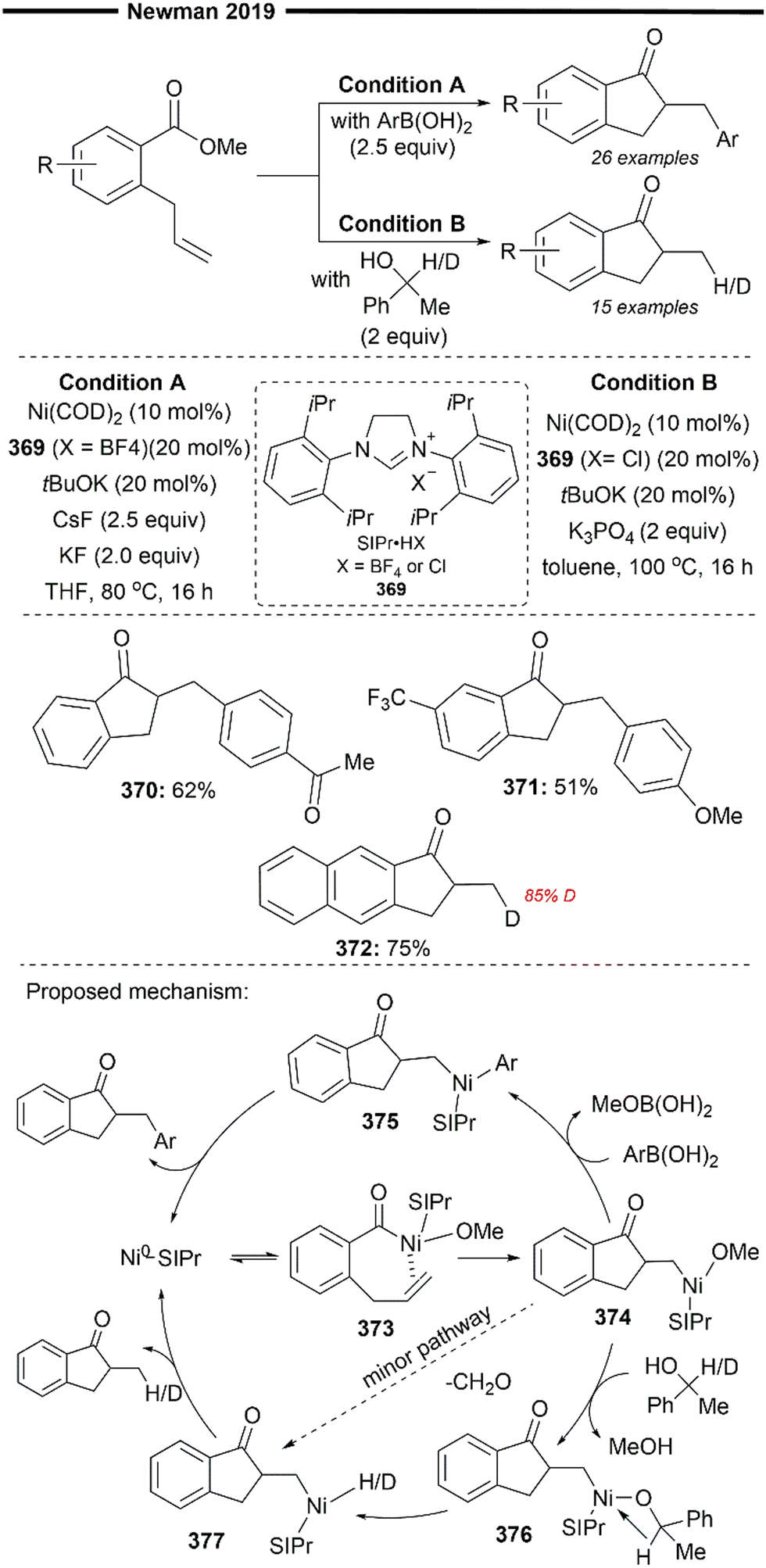 | ||
| Scheme 58 NHC/Ni-catalysed intramolecular carboacylation and hydroacylation of alkenes via ester C–O bond activation.58 | ||
A plausible mechanism for the two distinct transformations is depicted in Scheme 57. Reversible oxidative addition of the C(acyl)–O bond of the methyl ester forms the acyl-Ni intermediate 373. Regioselective migratory insertion into the tethered alkene yields the primary alkylnickel intermediate 374. The selectivity is likely governed by the steric influence of the bulky SIPr ligand. In the carboacylation pathway, fluoride-assisted transmetalation with the arylboronic acid results in the formation of the alkyl-Ni-aryl complex 375, which furnishes the desired product upon reductive elimination. In the reductive hydroacylation pathway, ligand exchange of the methoxy ligand with 1-phenylethanol (or the monodeuterated derivative) initially occurs, giving the alkoxy-nickel species 376. β-H-elimination from the alkoxide moiety results in the formation of the nickel–hydride (or deuteride) intermediate 377, accompanied with the release of the acetophenone by-product. A possible minor pathway involving the direct β-H-elimination from the methoxy group, albeit slow, could also produce the same 377 species, without the intervention of the exogenous alcohol. Importantly, no intramolecular Heck-type enone product was observed under their protocol.
In their ongoing pursuit of developing regio- and stereo-selective olefin functionalisation protocols enabled with Ni–NHC catalysts, Koh and co-workers reported a regioselective 1,2-diarylation reaction of the less electronically activated aliphatic 1,3-dienes with site-selectivity fully controlled by the bulky IPE ligand144 (Scheme 59). This redox-neutral reaction system facilitated the effective three-component combination of a diene, an aryltriflate electrophile and an aryl organometallic nucleophile, resulting in the formation of the desired diarylation products consisting of tertiary and quaternary carbon centres. Notably, minimal Heck-type or homo-/cross-coupling biaryl by-products were observed. Previous challenges of allylic rearrangement from the dissymmetrical η3-π-allyl-metallointermediate leading to mixtures of 1,2- and 1,4-addition regioisomers,145 and stereoisomeric E/Z mixtures were not an issue under this protocol.
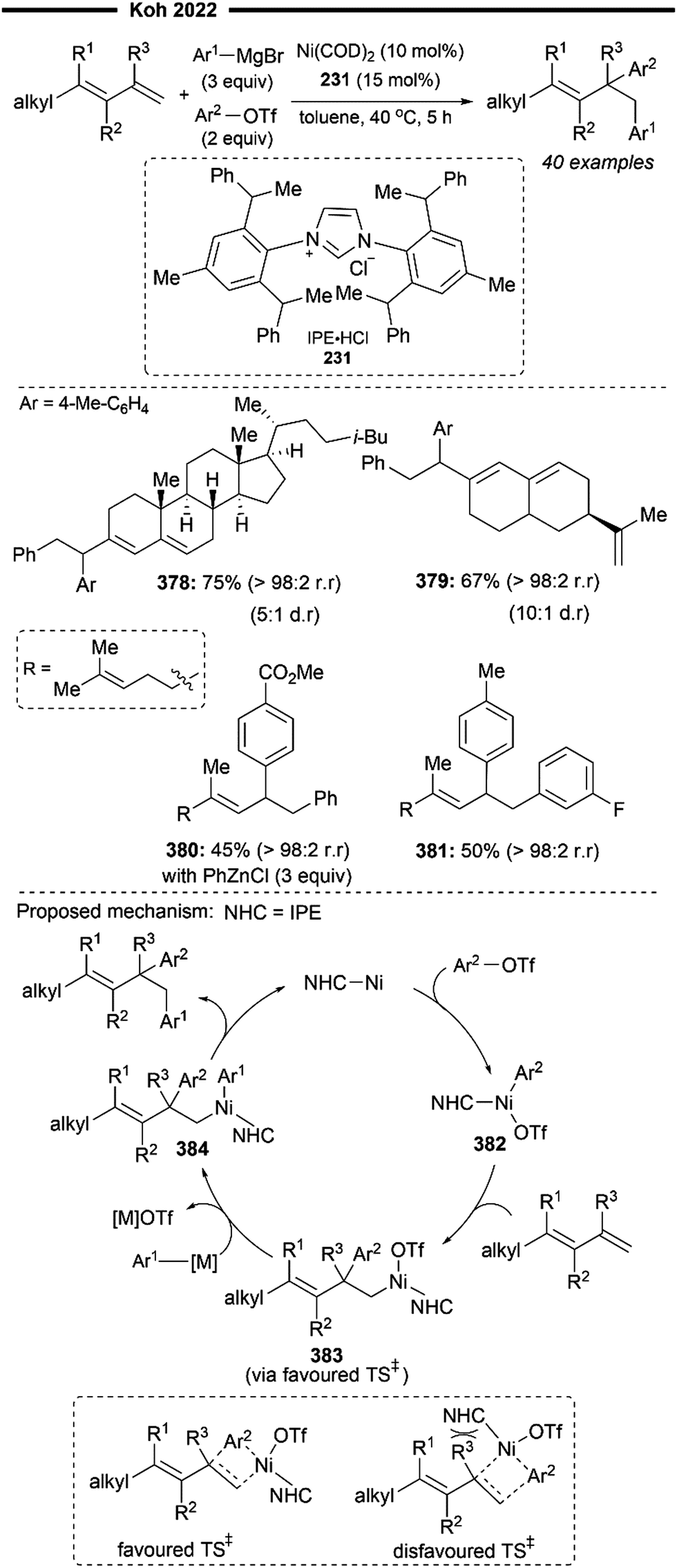 | ||
| Scheme 59 NHC/Ni-catalysed regioselective diarylation of aliphatic 1,3-dienes.144 | ||
Naturally occurring and bioactive metabolites such as (+)4-cholesten-3-one (378) and (+)-nootkatone (379), consisting of a 1,3,5-triene moiety, performed well under the reaction protocol, delivering the products in a single regioisomer in a high diastereomeric ratio (5:1–10:1). Besides using a Grignard reagent as the organometallic coupling partner, the milder arylzinc nucleophile could also be employed, allowing for the tolerance of more sensitive functionalities such as esters (380). Arylmagnesium bromide with an electron-withdrawing meta-fluoro substituent (381) also successfully delivered the adduct.
Preliminary mechanistic studies led to a postulated mechanism depicted in Scheme 59. Oxidative addition of the aryltriflate with the active NHC–Ni0 species forms the aryl-NiII intermediate 382, which selectively inserts into the less substituted CC bond of the 1,3-diene, producing the aryl-branched primary alkylnickel species 383. The generation of a rearrangement- and stereoerosion-prone allylnickel intermediate was precluded with this branched-selective carbonickelation. The observed regioselectivity is solely determined from steric effects, proceeding via the lower energy transition state that minimises steric repulsions between the sizeable NHC ligand and the diene substituent. Transmetalation with the aryl organometallic reagent led to the formation of an alkyl-Ni-aryl intermediate 384, which underwent reductive elimination to turn over the catalytic cycle and release the desired 1,2-diarylation product.
In 2022, Tobisu and co-workers developed a NHC/Ni-catalysed 1,2-carboaminocarbonylation reaction of norbornene via C–C bond cleavage of arylamide derivatives146 (Scheme 60). This reaction does not depend on the prerequisite of ring strain nor directing groups for C–C bond cleavage and resembles the net atom economical addition of a C–C bond of an arylamide across the carbon–carbon double bond.
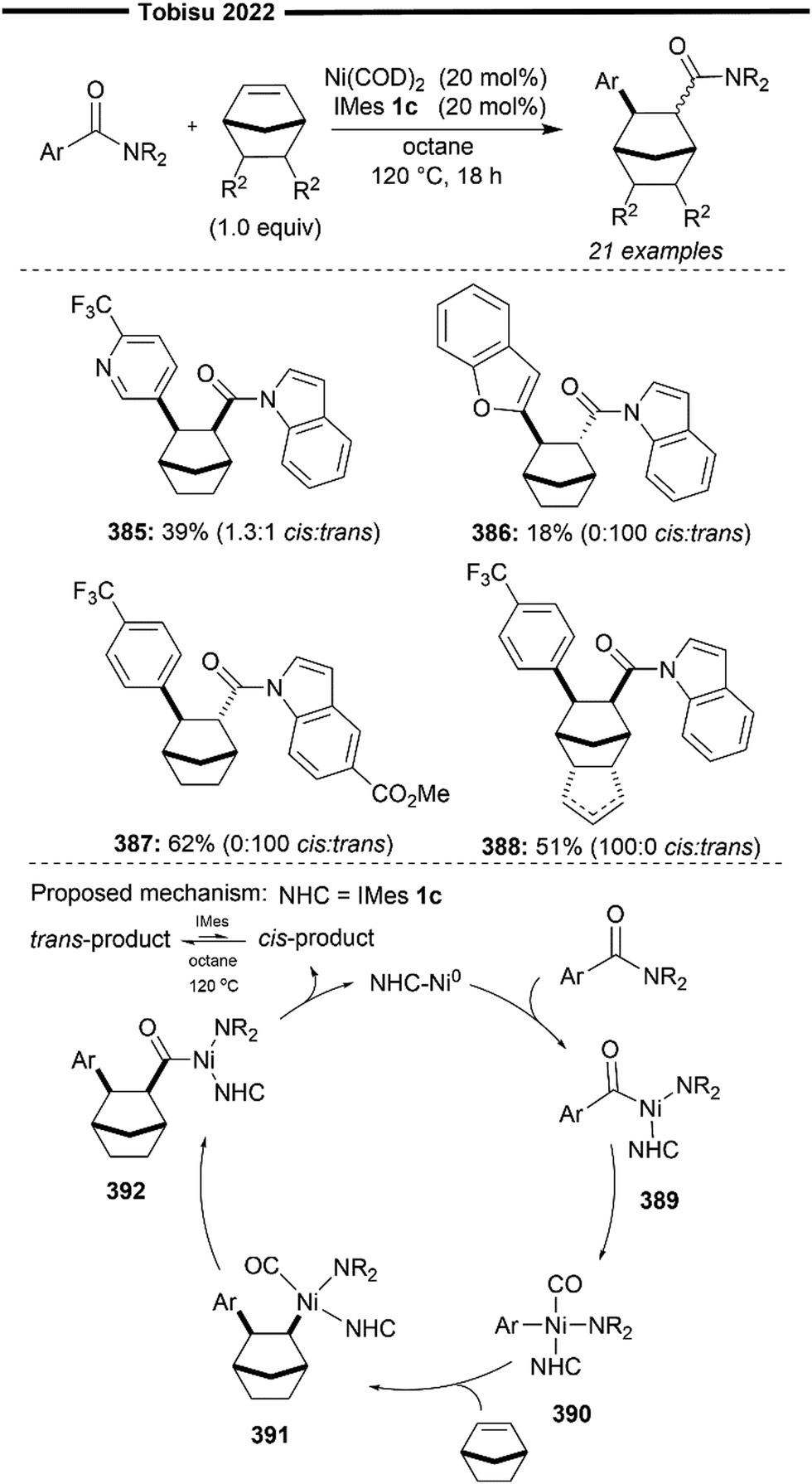 | ||
| Scheme 60 NHC/Ni-catalysed 1,2-carboaminocarbonylation of strained alkenes via C–C bond activation of amides.146 | ||
The use of NHC ligands, particularly IMes 1c, led to efficient addition reaction to yield the corresponding 1,2-carboaminocarbonylation products while phosphine ligands failed to deliver any desired product. Heteroaromatic groups of the arylamide such as pyridine (385) or benzofuran (386) were tolerated, but resulted in a lowered yield possibly due to decarbonylation side reactions. Interestingly, benzofuran-substituted arylamide (386) or the introduction of an ester group (387) led to the exclusive formation of the trans isomer. Alternative strained alkenes such as dicyclopentadiene (388) reacted readily to deliver the cis-product as the sole isomer. For the catalytic regime, certain structural requirements for the substrate are essential for effective transformation. The lack of reaction from simple amides derived from aliphatic or aromatic amines indicates the crucial requirement of an indole-type amide for effective reaction. Electronic perturbation of the aryl component of the arylamide substrate has a significant impact on the reaction's efficiency, where higher levels of electron deficiency led to improved yields.
A catalytic cycle proposed by the authors is depicted in Scheme 60. Oxidative addition of the C(acyl)–N bond of the arylamide generates the acyl-NiII(1-indolyl) complex 389, which undergoes decarbonylation to form the aryl-nickel intermediate 390 with a bound CO ligand. Subsequent carbonickelation of the norbornene substrate forms the secondary alkylnickel species 391. Re-insertion of CO into the C(sp3)–Ni bond results in the formation of an acyl-Ni complex 392, which releases the final cis-product after reductive elimination to form the desired C(acyl)–N bond. The desired 1,2-carboaminocarbonylation product can be efficiently formed despite the reversible nature of CO insertion/de-insertion, which is attributed to the energetically feasible C(acyl)–N bond forming reductive elimination process, in contrast to the C(sp3)–N bond forming process from 391, which explains the absence of the 1,2-carboamination product. In-depth experimental studies also found that the cis-isomer is the kinetically favoured product, which spontaneously isomerises to the more thermodynamically stable trans-isomer (from theoretical calculations) in the presence of a Lewis base, such as the IMes 1c free carbene.
In 2022, Koh and co-workers successfully achieved a directing group free approach for direct addition of carbogenic aryl groups across unactivated C–C π-bonds in an NHC/Ni-catalysed three-component reaction147 (Scheme 61). This novel approach solely relies on catalyst and ligand control for the efficient and highly regioselective 1,2-diarylation of unactivated aliphatic alkenes, eliminating the need for pre-installation of pendant directing auxiliaries or substrate control which hinges on the favourable intramolecular formation of a cyclic intermediate. An extensive survey of catalysts and ligands led to the selection of a bulky dimeric IPr-Ni(I) precatalyst, which was found to be more efficient than the conventional Ni(0) or Ni(II) precatalysts.
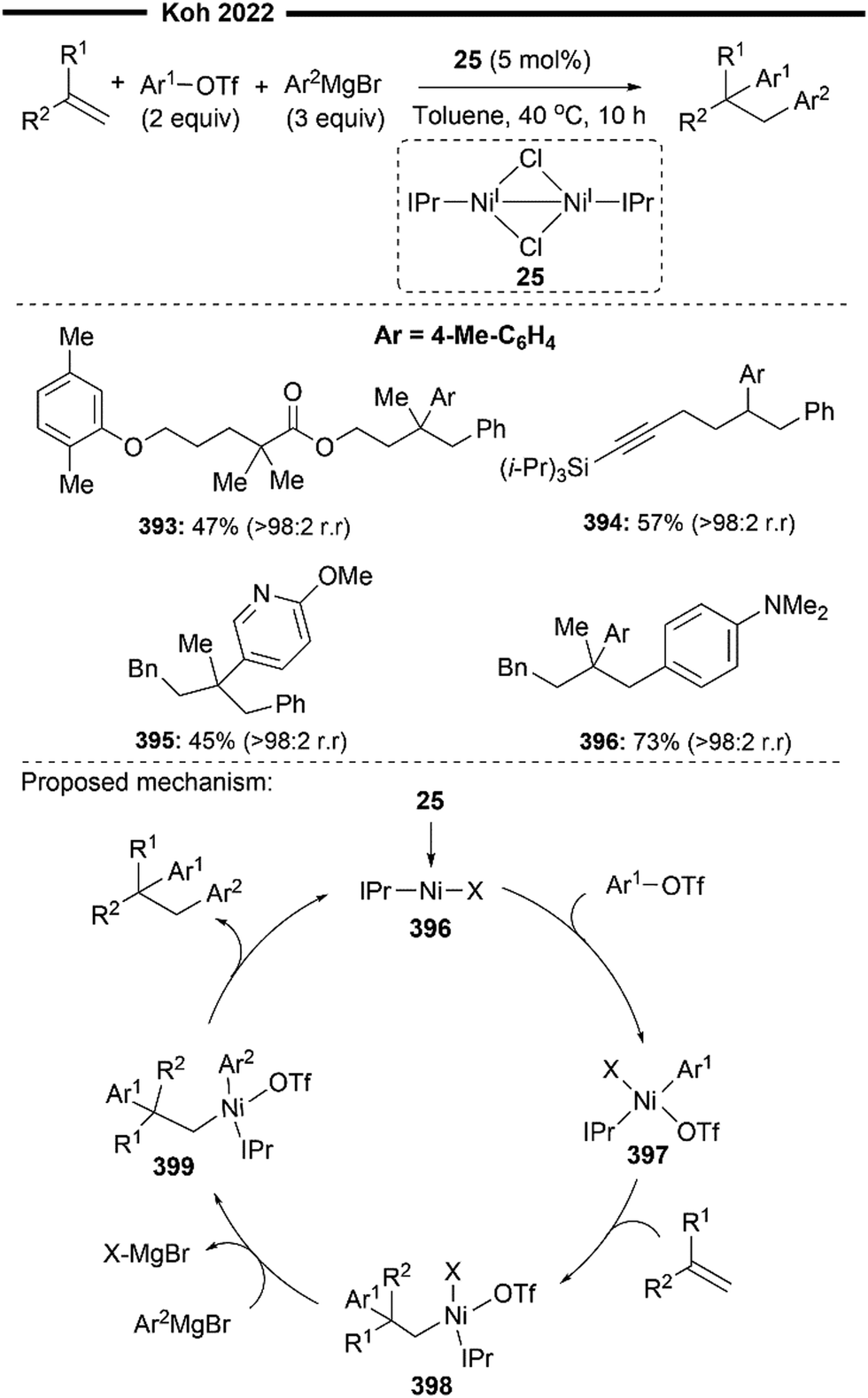 | ||
| Scheme 61 Directing group free NHC/Ni-catalysed dicarbofunctionalisation of unactivated alkenes.147 | ||
The significance of a sufficiently bulky NHC ligand stems from several reasons. The steric influence exerted by the NHC ligand must be substantial enough to provide good regiocontrol in the carbonickelation (migratory insertion) step. Without the presence of a directing group, the vacant coordination site of the nickel complex could trigger adventitious β-H elimination, leading to the formation of Heck-type by-products.148 Having a sizeable NHC ligand could also preclude agostic Cβ–H interactions which productively leads to β-H elimination. Lastly, reductive elimination would be facilitated with a sterically encumbered metal complex, outcompeting, and suppressing any undesired side reactions.
With support from DFT calculations, a catalyst-controlled reaction pathway was suggested by the authors. Substrate or solvent assisted dissociation of the dimeric Ni(I) precatalyst 25 forms the monomeric IPr-Ni(I) active species 396. Oxidative insertion of the aryltriflate electrophile affords the arylnickel(III) intermediate 397, which undergoes a branched-selective 1,2-carbonickelation reaction across the alkene substrate, resulting in the formation of the primary alkylnickel intermediate 398. Theoretical calculations showed that the transition state leading to the linear-selective insertion intermediate was comparatively higher in energy and unfavourable due to the severe steric repulsions between the bulky IPr ligand and the olefinic substituent. Irreversible transmetalation with the organometallic nucleophile followed, resulting in an arylalkylnickel species 399. Reductive elimination of 399 furnishes the desired 1,2-diarylation product with high regioselectivity and regenerates the active Ni(I) species 396.
The generality of the reaction has been demonstrated through its application to various functionalised terminal and 1,1-disubstituted alkenes. Notably, reactions with the latter class of alkenes allowed for the formation of diarylation adducts which incorporate fully-substituted quaternary carbon centres (395 and 396). In addition, alkynylsilanes (394) and Lewis basic N-heterocycles like pyridine (395) were amenable substrates under the optimised reaction conditions. Coupling with alkene-derived bioactive molecules such as gemfibrozil 393 (a lipid-lowering agent) proceeded smoothly, thereby highlighting its utility for the synthesis of complex, pharmaceutically relevant molecules.
4. Conclusion
In this review, we have underscored the advancements made in nickel-catalysed alkene functionalisation reactions, through the utilisation of N-heterocyclic carbene (NHC) ligands and their unique associated properties. Three main reaction classes of alkene functionalisation have been presented: Heck-type reactions, hydrofunctionalisation and dicarbofunctionalisation. The reaction methodologies presented have illustrated the potential of the NHC–Ni catalytic system as a valuable tool for the efficient and selective construction of bioactive and pharmaceutically relevant molecular architectures from inexpensive and readily available olefinic precursors. The key features of NHC ligands include their strong σ-donating capacity and their large buried volume (%Vbur) because of their steric bulkiness. Modular synthesis of structurally diverse NHC precursors149 allows for convenient tuning of electronic and steric properties, which can considerably impact the efficiency, as well as the chemo-, regio- and stereo-selectivity of a reaction. The multifaceted design of NHC scaffolds permits the incorporation of stereogenicity at different positions of the NHCs, leading to the emergence of chiral NHC ligands employed in numerous enantioselective transformations facilitated with transition-metal catalysts.150 Many mono- or multi-dentate chiral NHC ligands demonstrated exceptional capacity to induce high levels of enantioselectivity, attributed to their steric influence and attractive secondary non-covalent interactions in the stereodetermining transition state.Despite the substantial progress made in the field of nickel-catalysed alkene functionalisation over the years, major challenges still remain. For instance, the regio- and enantio-selective carbofunctionalisation of unactivated alkenes and light olefins, which have weak electronic and steric bias, is yet to be achieved in the absence of directing auxiliaries. Selective hydro/carbofunctionalisation of fluorinated alkenes, which are prone to defluorinative side reactions, is also a difficult task. These obstacles could potentially be resolved via the use of nickel catalysis in combination with judiciously designed chiral NHC ligands, such as the more elusive chiral CAAC-type or mesoionic NHC ligands that have found limited applications to date.150 Another exciting prospect in this field could be the integration of NHCs as ligands for nickel/photoredox dual catalysis, which have remained largely unexplored since the seminal work for indoline synthesis by Jamison and Tasker.151 This represents a new disconnection approach to enable access to new chemical space and a border assortment of molecular scaffolds, including prized heterocyclic and hetero-substituted molecules, from simple olefin feedstocks.
Abbreviations
| 1,2-DCE | 1,2-Dichloroethane |
| Ad | Adamantyl |
| BArF | Tetrakis[3,5-bis(trifluoromethyl)phenyl]borate |
| Bn | Benzyl |
| Bpy | Bipyridine |
| Bu | Butyl |
| COD | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl |
| Me | Methyl |
| CAAC | Cyclic(alkyl/aryl)(amino)carbene |
| Cy | Cyclohexyl |
| DFT | Density functional theory |
| ee | Enantiomeric excess |
| er | Enantiomeric ratio |
| HBCat | Catecholborane |
| HBpin | Pinacolborane |
| hIEtPyO | (1-(2,6-Diethylphenyl)-3-(6-oxidopyridin-2-yl)-imidazol-2-ylidene) |
| iPr | Iso-propyl |
| IAd | 1,3-Bis(1-adamantyl)imidazol-2-ylidene |
| IPr | 1,3-Bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene |
| IPr* | 1,3-Bis(2,6-bis(diphenylmethyl)-4methylphenyl)imidazol-2-ylidene |
| ItBu | 1,3-Di-tert-butylimidazol-2-ylidene |
| LLHT | Ligand-to-ligand hydrogen transfer |
| MAD | Methylaluminium bis(2,6-di-tert-butyl-4-methylphenoxide) |
| Mes | Mesityl |
| MTBE | Methyl tert-butyl ether |
| NHC | N-Heterocyclic carbene |
| Ph | Phenyl |
| r.r. | Regioisomeric ratio |
| t-Am | tert-Amyl |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TIPS | Triisopropylsilyl |
| tBu | tert-Butyl |
| TBAB | Tetrabutylammonium bromide |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Ministry of Education of Singapore Academic Research Fund Tier 2 (A-8000941-00-00).References
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS.
- K. Dzieszkowski, I. Baranska, K. Mroczynska, M. Słotwinski and Z. Rafinski, Materials, 2020, 13, 3574 CrossRef CAS PubMed.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953–2956 CrossRef CAS.
- D. P. Allen and C. M. Crudden, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef.
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
- M. T. Haynes, II, E. P. Jackson and J. Montgomery, in N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, Germany, 2014, pp. 371–396 Search PubMed.
- F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293–294, 48–79 CrossRef CAS.
- K. Matsubara, Chem. Rec., 2021, 21, 3925–3942 CrossRef CAS PubMed.
- T. S. Andreas, A. Danopoulos and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef PubMed.
- K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS PubMed.
- C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed.
- M. H. Vincent Ritleng and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef.
- A. P. Prakasham and P. Ghosh, Inorg. Chim. Acta, 2015, 431, 61–100 CrossRef CAS.
- R. S. Manan, P. Kilaru and P. Zhao, J. Am. Chem. Soc., 2015, 137, 6136–6139 CrossRef CAS PubMed.
- Y. Cai, J.-W. Zhang, F. Li, J.-M. Liu and S.-L. Shi, ACS Catal., 2019, 9, 1–6 CrossRef.
- Y.-Q. Li, F. Li and S.-L. Shi, Chin. J. Chem., 2020, 38, 1035–1039 CrossRef CAS.
- J. S. E. Ahlin, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 13229–13233 CrossRef CAS PubMed.
- W.-B. Zhang, G. Chen and S.-L. Shi, J. Am. Chem. Soc., 2022, 144, 130–136 CrossRef CAS PubMed.
- S. Cañellas, J. Montgomery and M. À. Pericàs, J. Am. Chem. Soc., 2018, 140, 17349–17355 CrossRef PubMed.
- R. Kumar, E. Tamai, A. Ohnishi, A. Nishimura, Y. Hoshimoto, M. Ohashi and S. Ogoshi, Synthesis, 2016, 2789–2794 CAS.
- R. Kumar, H. Tokura, A. Nishimura, T. Mori, Y. Hoshimoto, M. Ohashi and S. Ogoshi, Org. Lett., 2015, 17, 6018–6021 CrossRef CAS PubMed.
- R. Kumar, Y. Hoshimoto, E. Tamai, M. Ohashi and S. Ogoshi, Nat. Commun., 2017, 8, 32 CrossRef.
- X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS.
- J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- C. Frech, Angew. Chem., Int. Ed., 2009, 48, 6947 CrossRef CAS.
- The Nobel Prize in Chemistry 2010, https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2010/ (accessed January 6, 2023).
- S. Bhaktaa and T. Ghosh, Adv. Synth. Catal., 2020, 362, 5257–5274 CrossRef.
- B.-L. Lin, L. Liu, Y. Fu, S.-W. Luo, Q. Chen and Q.-X. Guo, Organometallics, 2004, 23, 2114–2123 CrossRef CAS.
- T. M. Gøgsig, J. Kleimark, S. O. N. Lill, S. Korsager, A. T. Lindhardt, P.-O. Norrby and T. Skrydstrup, J. Am. Chem. Soc., 2012, 134, 443–452 CrossRef PubMed.
- K. Inamoto, J.-i Kuroda, T. Danjo and T. Sakamoto, Synlett, 2005, 1624–1626 CrossRef CAS.
- K. D. Schleicher and T. F. Jamison, Org. Lett., 2007, 9, 875–878 CrossRef CAS PubMed.
- H. Hoberg and D. Guhl, J. Organomet. Chem., 1990, 384, C43–C46 CrossRef CAS.
- C.-Y. Ho and T. F. Jamison, Angew. Chem., 2007, 119, 796–799 CrossRef.
- F. Strieth-Kalthoff, A. R. Longstreet, J. M. Weber and T. F. Jamison, ChemCatChem, 2018, 10, 2873–2877 CrossRef CAS PubMed.
- C.-F. Liu, H. Wang, R. T. Martin, H. Zhao, O. Gutierrez and M. J. Koh, Nat. Catal., 2021, 4, 674–683 CrossRef CAS.
- K. Yoshida and T. Hayashi, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. Carreras, A. Caballero and P. J. Perez, Chem. – Asian J., 2019, 14, 329–343 CrossRef CAS PubMed.
- Y. Zhao, C.-F. Liu, L. Q. H. Lin, A. S. C. Chan and M. J. Koh, Angew. Chem., Int. Ed., 2022, 61, e202202674 CAS.
- X.-Y. Sun, B.-Y. Yao, B. Xuan, L.-J. Xiao and Q.-L. Zhou, Chem. Catal., 2022, 2, 3140–3162 CrossRef CAS.
- Z. Zhang, S. Bera, C. Fan and X. Hu, J. Am. Chem. Soc., 2022, 144, 7015–7029 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- H. Gao, L. Hu, Y. Hu, X. Lv, Y.-B. Wu and G. Lu, Chem. Commun., 2022, 58, 8650–8653 RSC.
- J. Guihaumé, S. Halbert, O. Eisenstein and R. N. Perutz, Organometallics, 2012, 31, 1300–1314 CrossRef.
- Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 16170–16171 CrossRef CAS PubMed.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373–8426 CrossRef CAS PubMed.
- S. M. Khake and N. Chatani, Chem., 2020, 6, 1056–1081 CAS.
- C.-F. Liu, X. Luo, H. Wang and M. J. Koh, J. Am. Chem. Soc., 2021, 143, 9498–9506 CrossRef CAS PubMed.
- Y. L. Zheng and S. G. Newman, Angew. Chem., Int. Ed., 2019, 58, 18159–18164 CrossRef CAS PubMed.
- S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 RSC.
- C.-Y. Ho and L. He, Angew. Chem., Int. Ed., 2010, 49, 9182–9186 CrossRef CAS PubMed.
- C.-Y. Ho, C.-W. Chan and L. He, Angew. Chem., Int. Ed., 2015, 54, 4512–4516 CrossRef CAS PubMed.
- C.-Y. Ho and L. He, Chem. Commun., 2012, 48, 1481–1483 RSC.
- C.-Y. Ho and L. He, J. Org. Chem., 2014, 79, 11873–11884 CrossRef CAS PubMed.
- X. Lian, W. Chen, L. Dang, Y. Li and C.-Y. Ho, Angew. Chem., Int. Ed., 2017, 56, 9048–9052 CrossRef CAS PubMed.
- S. Biswas, A. Zhang, B. Raya and T. V. RajanBabua, Adv. Synth. Catal., 2014, 356, 2281–2292 CrossRef CAS PubMed.
- Y. Chen, L. Dang and C.-Y. Ho, Nat. Commun., 2020, 11, 2269 CrossRef CAS PubMed.
- W. Chen, Y. Li, Y. Chen and C.-Y. Ho, Angew. Chem., Int. Ed., 2018, 57, 2677–2681 CrossRef CAS PubMed.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 11, 5245–5287 CrossRef PubMed.
- X. Yong, W. Gao, X. Lin and C.-Y. Ho, Commun. Chem., 2020, 3, 50 CrossRef CAS PubMed.
- J. Kischel, I. Jovel, K. Mertins, A. Zapf and M. Beller, Org. Lett., 2006, 8, 19–22 CrossRef CAS PubMed.
- L. J. Oxtoby, J. A. Gurak, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Trends Chem., 2019, 1, 572–587 CrossRef CAS PubMed.
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 15996–15997 CrossRef CAS PubMed.
- Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4451–4454 CrossRef CAS PubMed.
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed.
- J.-B. Ma, X. Zhao, D. Zhang and S.-L. Shi, J. Am. Chem. Soc., 2022, 144, 13643–13651 CrossRef CAS PubMed.
- Y. Cai, X.-T. Yang, D. S.-Q. Zhang, F. Li, Y.-Q. Li, L.-X. Ruan, X. Hong and S.-L. Shi, Angew. Chem., Int. Ed., 2018, 57, 1376–1380 CrossRef CAS PubMed.
- P. Naweephattana, B. Sawatlon and P. Surawatanawong, J. Org. Chem., 2020, 85, 11340–11349 CrossRef CAS PubMed.
- R. Tamura, Y. Yamada, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2012, 51, 5679–5682 CrossRef CAS PubMed.
- W.-C. Shih, W.-C. Chen, Y.-C. Lai, M.-S. Yu, J.-J. Ho, G. P. A. Yap and T.-G. Ong, Org. Lett., 2012, 14, 2046–2049 CrossRef CAS PubMed.
- W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, Org. Lett., 2013, 15, 5358–5361 CrossRef CAS PubMed.
- S. Krompiec, M. Krompiec, R. Penczek and H. Ignasiak, Coord. Chem. Rev., 2008, 252, 1819–1841 CrossRef CAS.
- W.-C. Lee, C.-H. Chen, C.-Y. Liu, M.-S. Yu, Y.-H. Lina and T.-G. Ong, Chem. Commun., 2015, 51, 17104–17107 RSC.
- G. Vijaykumar, A. Jose, P. K. Vardhanapu, P. Sreejyothi and S. K. Mandal, Organometallics, 2017, 36, 4753–4758 CrossRef CAS.
- H. Xie, L. Zhao, L. Yang, Q. Lei, W. Fang and C. Xiong, J. Org. Chem., 2014, 79, 4517–4527 CrossRef CAS PubMed.
- O. V. Khazipov, K. E. Shepelenko, D. V. Pasyukov, V. V. Chesnokov, S. B. Soliev, V. M. Chernyshev and V. P. Ananikov, Org. Chem. Front., 2021, 8, 2515–2524 RSC.
- X. Xi, T. Chen, J.-S. Zhang and L.-B. Han, Chem. Commun., 2018, 54, 1521–1524 RSC.
- J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef CAS PubMed.
- Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215–12218 CrossRef CAS PubMed.
- N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- M. Chen and J. Montgomery, ACS Catal., 2022, 12, 11015–11023 CrossRef CAS.
- P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 633–637 CAS.
- J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489–4493 CrossRef CAS PubMed.
- A. Albright and R. E. Gawley, J. Am. Chem. Soc., 2011, 133, 19680–19683 CrossRef CAS PubMed.
- K. V. Vasudevan, R. R. Butorac, C. D. Abernethy and A. H. Cowley, Dalton Trans., 2010, 39, 7401–7408 RSC.
- Y. Cai, X. Ye, S. Liu and S.-L. Shi, Angew. Chem., Int. Ed., 2019, 58, 13433–13437 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- W.-B. Zhang, X.-T. Yang, J.-B. Ma, Z.-M. Su and S.-L. Shi, J. Am. Chem. Soc., 2019, 141, 5628–5634 CrossRef CAS PubMed.
- D. Shen, W.-B. Zhang, Z. Li, S.-L. Shi and Y. Xu, Adv. Synth. Catal., 2020, 362, 1125–1130 CrossRef CAS.
- J. Diesel, D. Grosheva, S. Kodama and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 11044–11048 CrossRef CAS PubMed.
- J.-F. Li, W.-W. Xu, R.-H. Wang, Y. Li, G. Yin and M. Ye, Nat. Commun., 2021, 12, 3070 CrossRef CAS PubMed.
- J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- C.-F. Liu, Z.-C. Wang, X. Luo, J. Lu, C. H. M. Ko, S.-L. Shi and M. J. Koh, Nat. Catal., 2022, 5, 934–942 CrossRef CAS.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- S. Okumura, T. Komine, E. Shigeki, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2018, 57, 929–932 CrossRef CAS PubMed.
- G. Zhang, C.-Y. Zhao, X.-T. Min, Y. Li, X.-X. Zhang, H. Liu, D.-W. Ji, Y.-C. Hu and Q.-A. Chen, Nat. Catal., 2022, 5, 708–715 CrossRef CAS.
- S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386–390 CrossRef CAS PubMed.
- X. Yu, H. Zhao, S. Xi, Z. Chen, X. Wang, L. Wang, L. Q. H. Lin, K. P. Loh and M. J. Koh, Nat. Catal., 2020, 3, 585–592 CrossRef CAS.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- S. Manna, K. K. Das, S. Nandy, D. Aich, S. Paul and S. Panda, Coord. Chem. Rev., 2021, 448, 214165 CrossRef CAS.
- E. E. Touney, R. V. Hoveln, C. T. Buttke, M. D. Freidberg, I. A. Guzei and J. M. Schomaker, Organometallics, 2016, 35, 3436–3439 CrossRef CAS.
- F. Ulm, Y. Cornaton, J.-P. Djukic, M. J. Chetcuti and V. Ritleng, Chem. – Eur. J., 2020, 26, 8916–8925 CrossRef CAS PubMed.
- M. Afandiyeva, A. A. Kadam, X. Wu, W. W. Brennessel and C. R. Kennedy, Organometallics, 2022, 41, 3014–3023 CrossRef CAS.
- Y. Nakajima and S. Shimada, RSC. Adv., 2015, 5, 20603–20616 RSC.
- X. Wu, G. Ding, W. Lu, L. Yang, J. Wang, Y. Zhang, X. Xie and Z. Zhang, Org. Lett., 2021, 23, 1434–1439 CrossRef CAS PubMed.
- L. B. Junquera, M. Carmen Puerta and P. Valerga, Organometallics, 2012, 31, 2175–2183 CrossRef CAS.
- Z. D. Miller, W. Li, T. S. R. Belderrain and J. Montgomery, J. Am. Chem. Soc., 2013, 135, 15282–15285 CrossRef CAS PubMed.
- Z. D. Miller, R. Dorel and J. Montgomery, Angew. Chem., Int. Ed., 2015, 54, 9088–9091 CrossRef CAS PubMed.
- S.-S. Ng and T. F. Jamison, J. Am. Chem. Soc., 2005, 127, 7320–7321 CrossRef CAS PubMed.
- A. S.-M. Chang, K. E. Kawamura, H. S. Henness, V. M. Salpino, Jack C. Greene, L. N. Zakharov and A. K. Cook, ACS Catal., 2022, 12, 11002–11014 CrossRef CAS.
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16–21 CrossRef CAS.
- T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- K. C. Hultzsch, Org. Biomol. Chem., 2005, 3, 1819–1824 RSC.
- C. Dash, M. M. Shaikh, R. J. Butcherc and P. Ghosh, Dalton Trans., 2010, 39, 2515–2524 RSC.
- R. Kumar, M. Katari, A. Choudhary, G. Rajaraman and P. Ghosh, Inorg. Chem., 2017, 56, 14859–14869 CrossRef CAS PubMed.
- A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker and L. M. Stanley, Org. Chem. Front., 2016, 3, 639–644 RSC.
- Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2012, 51, 10812–10815 CrossRef CAS PubMed.
- Q. Meng and F. Wang, J. Mol. Model., 2017, 23, 11 CrossRef PubMed.
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed.
- Y. Hayashi, Y. Hoshimoto, R. Kumar, M. Ohashia and S. Ogoshi, Chem. Commun., 2016, 52, 6237–6240 RSC.
- N. Saito, T. Katayama and Y. Sato, Org. Lett., 2008, 10, 3829–3832 CrossRef CAS PubMed.
- A. Ghosh, J. James, A. Walker, A. Ellern and L. M. Stanley, ACS Catal., 2016, 6, 2673–2680 CrossRef CAS.
- J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem., 2019, 5, 526–552 CAS.
- Y. Miyazaki, Y. Y. Y. Nakao and T. Hiyama, Chem. Lett., 2012, 41, 298–300 CrossRef CAS.
- J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890–3908 CrossRef CAS PubMed.
- Y.-C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371–1394 CrossRef CAS.
- B. C. Lee, L. Lin, C. Ko and M. J. Koh, in Science of Synthesis: Base-Metal Catalysis, ed. N. Yoshikai, Thieme, Stuttgart, 2022, vol. 1, p. 469 Search PubMed.
- M. Ohashi, H. Shirataki, K. Kikushima and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 6496–6499 CrossRef CAS PubMed.
- J. S. E. Ahlin and N. Cramer, Org. Lett., 2016, 18, 3242–3245 CrossRef CAS PubMed.
- J. James, A. Walker, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228–10231 CrossRef PubMed.
- L. Hie, N. F. F. Nathel, X. Hong, Y.-F. Yang, P. K. N. Houk and P. N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810–2814 CrossRef CAS PubMed.
- J. M. Medina, J. Moreno, S. Racine, S. Du and N. K. Garg, Angew. Chem., Int. Ed., 2017, 56, 6567–6571 CrossRef CAS PubMed.
- H. Wang, C.-F. Liu, T.-D. Tan, K. R. B. Khoo and M. J. Koh, ACS Catal., 2022, 12, 724–732 CrossRef CAS.
- Y. Xiong, Y. Sun and G. Zhang, Tetrahedron Lett., 2018, 59, 347–355 CrossRef CAS.
- Y. Ito, S. Nakatani, R. Shiraki, T. Kodama and M. Tobisu, J. Am. Chem. Soc., 2022, 144, 662–666 CrossRef CAS PubMed.
- H. Wang, C.-F. Liu, R. T. Martin, O. Gutierrez and M. J. Koh, Nat. Chem., 2022, 14, 188–195 CrossRef CAS PubMed.
- B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS PubMed.
- Z. Szabo, M. Timari, R. Kassai, B. Szokol, A. C. Benyei, T. Gáti, A. Paczal and A. Kotschy, Organometallics, 2020, 39, 3572–3589 CrossRef CAS.
- D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531–9534 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |