
Amplification-free CRISPR/Cas detection technology: challenges, strategies, and perspectives
Huimin
Li†
 ab,
Yi
Xie†
ab,
Fumin
Chen
ab,
Huiwen
Bai
c,
Leshan
Xiu
ab,
Xiaonong
Zhou
ab,
Xiaokui
Guo
*ab,
Qinqin
Hu
*ab and
Kun
Yin
*ab
ab,
Yi
Xie†
ab,
Fumin
Chen
ab,
Huiwen
Bai
c,
Leshan
Xiu
ab,
Xiaonong
Zhou
ab,
Xiaokui
Guo
*ab,
Qinqin
Hu
*ab and
Kun
Yin
*ab
aSchool of Global Health, Chinese Center for Tropical Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai, People's Republic of China. E-mail: xkguo@shsmu.edu.cn; qinqinhu@sjtu.edu.cn; kunyin@sjtu.edu.cn
bOne Health Center, Shanghai Jiao Tong University-The University of Edinburgh, Shanghai, People's Republic of China
cDepartment of Mechanical Engineering and Applied Mechanics, University of Pennsylvania, 220 South 33rd St., Philadelphia, Pennsylvania, USA
First published on 19th December 2022
Abstract
Rapid and accurate molecular diagnosis is a prerequisite for precision medicine, food safety, and environmental monitoring. The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)-based detection, as a cutting-edged technique, has become an immensely effective tool for molecular diagnosis because of its outstanding advantages including attomolar level sensitivity, sequence-targeted single-base specificity, and rapid turnover time. However, the CRISPR/Cas-based detection methods typically require a pre-amplification step to elevate the concentration of the analyte, which may produce non-specific amplicons, prolong the detection time, and raise the risk of carryover contamination. Hence, various strategies for target amplification-free CRISPR/Cas-based detection have been developed, aiming to minimize the sensitivity loss due to lack of pre-amplification, enable detection for non-nucleic acid targets, and facilitate integration in portable devices. In this review, the current status and challenges of target amplification-free CRISPR/Cas-based detection are first summarized, followed by highlighting the four main strategies to promote the performance of target amplification-free CRISPR/Cas-based technology. Furthermore, we discuss future perspectives that will contribute to developing more efficient amplification-free CRISPR/Cas detection systems.
 Huimin Li | Huimin Li received her Bachelor Degree of Medicine from Shanghai Jiao Tong University in 2021. In the same year, she joined the School of Global Health, Shanghai Jiao Tong University School of Medicine, as a postgraduate under the guidance of Prof. Kun Yin. Her current research interests focus on the development of CRISPR/Cas-based molecular diagnosis and rapid detection of pathogens. |
 Yi Xie | Yi Xie received her Bachelor Degree of Medicine from Xuzhou Medical University in 2017. She is currently a postgraduate under the guidance of Prof. Kun Yin in the School of Global Health, Shanghai Jiao Tong University School of Medicine. Her current research interests focus on CRISPR/Cas-based molecular diagnosis and microfluidic applications. |
 Xiaokui Guo | Xiaokui Guo received his PhD degree from Aichi Gakuin University. He is a distinguished Professor of Shanghai Jiao Tong University; Deputy Dean of the School of Global Health, Chinese Center for Tropical Diseases Research, Shanghai Jiao Tong University School of Medicine; Acting Deputy Director of Shanghai Jiao Tong University–University of Edinburgh One Health Research Center; and Vice Dean of Ottawa-Shanghai joint School of Medicine. Dr Guo focuses on the following three most important areas of One Health, including zoonotic diseases such as leptospirosis and brucellosis, safety of functional foods and drugs such as probiotics, and prevention and control of bacterial resistance. |
 Qinqin Hu | Qinqin Hu received her PhD degree in biosystem engineering from Zhejiang University, finished her postdoctoral training, and worked as an assistant professor at the Institutes of Biomedicine of Fudan University. She is now an associate professor at the School of Global Health, Shanghai Jiao Tong University School of Medicine. Dr Hu's expertise is in the areas of establishing biosensors and screening functional nucleic acids. In particular, she is interested in developing innovative optical nanomaterial-based biosensors for detection of hazards and pathogens in food and the environment, as well as selection of aptamers or DNAzymes for disease diagnosis. |
 Kun Yin | Kun Yin is a Professor in School of Global Health, Shanghai Jiao Tong University School of Medicine. He is the Department Chair of Environmental Health and Drug Resistance Control. He obtained a PhD degree from the University of Chinese Academy of Sciences and completed a postdoctoral fellowship at Ohio State University, University of Pennsylvania and University of Connecticut. Dr Yin is the Associate Editor of Science of One Health. He presided over the development and research of a series of new, fast, ultra-sensitive, high-throughput analysis and diagnosis technologies and a mobile Internet intelligent detection platform for infectious diseases. |
1. Introduction
Fast and accurate detection is vital for applications in disease diagnosis, agriculture, food safety, and environmental monitoring.1,2 Among them, nucleic acid testing (NAT) shows high sensitivity and single-base specificity because the target DNA or RNA sequence can be selectively amplified from trace amounts and be recognized via the pairing of complementary nucleotides (i.e., hybridization). In fact, the nucleic-acid-based diagnostics relying on the quantitative polymerase chain reaction (qPCR) or next-generation sequencing have been considered as gold standards for the clinical diagnosis of various infectious diseases and noninfectious chronic diseases.3–5 During the coronavirus disease 2019 (COVID-19) pandemic, the reverse transcription qPCR (RT-qPCR) technique has played an important role in the diagnosis of COVID infections for further effective pandemic response.6 These PCR-based diagnosis methods are typically implemented using bulky and sophisticated instruments with complicated procedures, such as the programmed thermal cycling, which restrict their applications toward on-site, home-based and miniaturized devices. To address this challenge, a series of isothermal amplification technologies (IATs), including recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP), and rolling circle amplification (RCA), have been established for ultra-sensitive detection of nucleic acids with no requirement of thermal cyclers.7 Nevertheless, wide deployment of IAT remains challenging due to its suboptimal specificity caused by non-specific amplification and the critical requirements for elaborate primer design. Therefore, there is an urgent need for an ideal NAT method that has ultrahigh sensitivity, single-base specificity, a short sample-to-result time, flexibility for ease of use, and the ability to be combined with point-of-care test platforms.8Recently, clustered regularly interspaced short palindromic repeat (CRISPR)-based technology has shown the potential to become an immensely effective tool for molecular diagnosis. CRISPR and CRISPR-associated (Cas) enzyme systems are fundamental parts of adaptive immune systems in bacteria and archaea, which recognize foreign nucleic acids and subsequently eliminate them using programmable endonucleases.9 Under the guidance of CRISPR RNA (crRNA) that hybridizes to a complementary sequence of a target DNA or RNA, Cas proteins enable the recognition and the cleavage of nucleic acids with high specificity. Based on high enzyme activity and specificity, CRISPR/Cas systems have been repurposed for a variety of applications, especially in molecular diagnosis.8,10,11 However, compared with ideal NAT, CRISPR/Cas-based technology shows lower sensitivity. To solve this problem, a target amplification step is performed by isothermal amplification or PCR to enhance target concentration prior to detection, such as SHERLOCK12 and DETECTR10 for ultrasensitive RNA and DNA detection, respectively. On the other hand, pre-amplification not only prolongs the detection time but also poses limitations for subsequent detection, such as non-specific amplification and interferences between primers.13 Moreover, this two-step reaction system (i.e., pre-amplification and detection) significantly increases the risk of aerosol contamination. Researchers have proposed single-tube solutions to reduce the chance of potential carryover contamination,14–16 but partly sacrificed the best performance of both the reactions.
To overcome the limitations mentioned above, various amplification-free CRISPR/Cas-based detection strategies have been explored as alternatives, which omit pre-amplification steps for elevating the target concentration and exhibit the merits of high specificity, flexibility, and short detection time (Fig. 1A). The detection targets have also been expanded from nucleic acids to other analytes such as live bacterial cells, proteins and metal ions,17–21 which have been summarized in Table 1. Moreover, because of eliminating the complicated target amplification process, the amplification-free CRISPR/Cas systems are capable of adapting to compact, portable, and easy-to-operate devices for promising field-deployable monitoring.22 However, compared with state-of-the-art techniques (SHERLOCK, DETECTR, or other gold standard methods), it is necessary to further enhance the sensitivity of target amplification-free CRISPR/Cas detection systems to the attomolar level or even single-molecule level.6 In fact, referring to the classic fluorescence-based CRISPR-detection system,10,23 various strategies have been further designed and practiced to improve the sensitivity of amplification-free CRISPR/Cas systems (Fig. 1B) and build novel molecular diagnostic platforms. Instead of relying on converting a single detection target itself to multiple targets, these approaches focus on signal amplification which lies in the formation of a multitude number of reporter molecules or the improved properties of multiple reporters and sensors. To highlight the emerging developments of the amplification-free CRISPR/Cas technique, it is of vital importance to summarize the recent advances for further innovation.
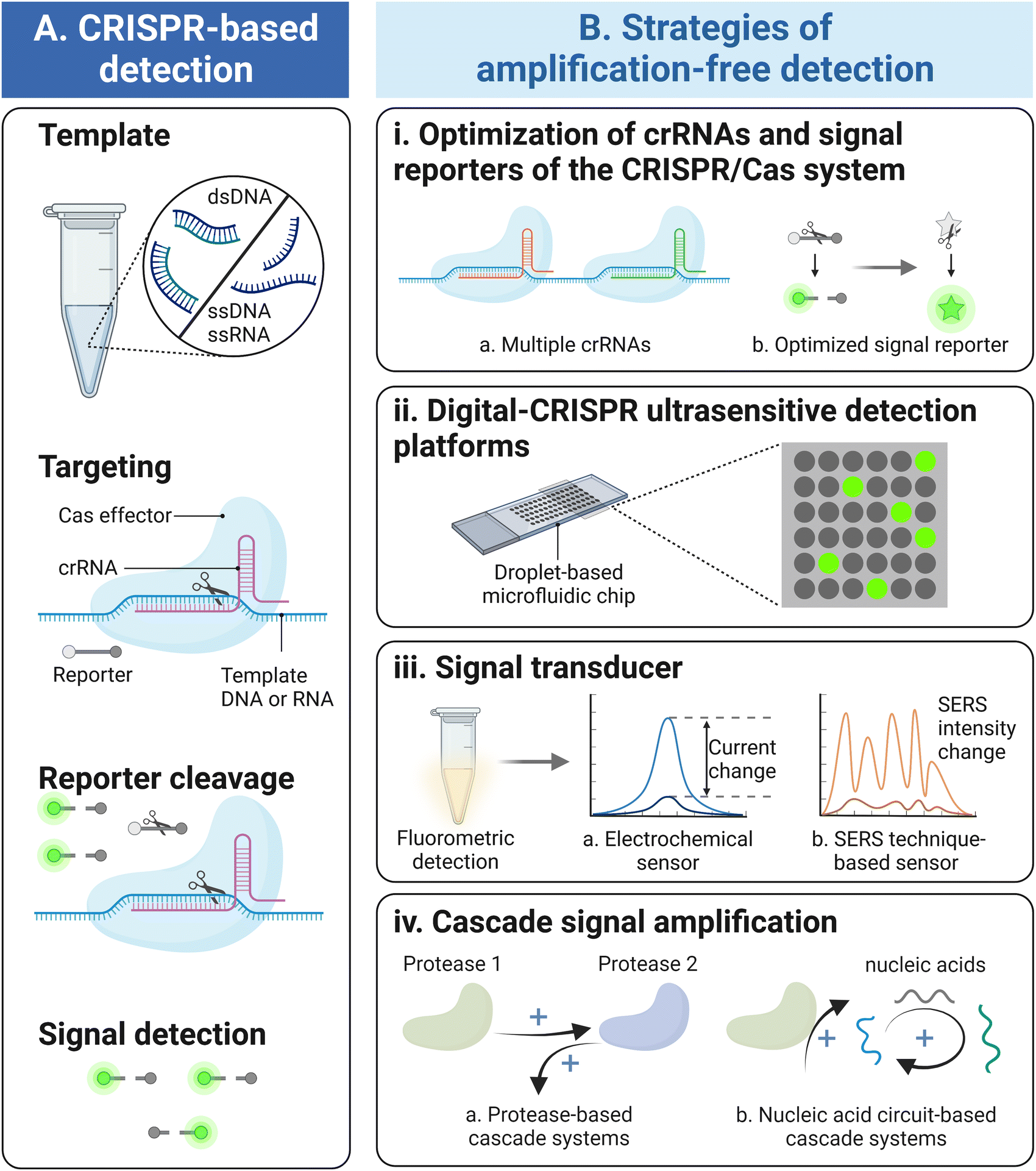 | ||
| Fig. 1 Schematic overview of target amplification-free CRISPR/Cas-based detection. (A) The schematic for CRISPR/Cas-based detection system. On the one side, the Cas effectors recognize and specifically cleave the template nucleic acids guided by the crRNAs. On the other side, the activated Cas effectors exhibit extremely high trans-cleavage activity to cut reporters nonspecifically to generate fluorescence signal readouts. (B) Strategies of amplification-free CRISPR/Cas-based detection, including the optimization of crRNAs and signal reporters (i), droplet-based digital CRISPR/Cas detection platforms (ii), signal transducers which alter fluorescence detection to either electrochemical or SERS signal output (iii), and the cascade reaction focusing on protease-based and nucleic acid circuit-based signal amplification systems (iv). | ||
| Method | Cas type | Targets | Signal read-out | LOD | Sample to answer time (min) | Quantitative | Refs. |
|---|---|---|---|---|---|---|---|
| Notes: ASFV, African swine fever virus; BRCA-1, breast cancer susceptibility gene-1; CXCL9, chemokine ligand 9; DENV, dengue virus; DMD, Duchenne muscular dystrophy; EBV, Epstein Barr virus; EC, electrochemistry; ECL, electrochemiluminescence; FL, fluorescence; FSV, fast scan voltammetry; HBV, hepatitis B virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; MRSA, methicillin-resistant Staphylococcus aureus; N.S., not specified; PB-19, parvovirus B-19. | |||||||
| Optimization of crRNAs and signal reporters of the CRISPR/Cas system | Cas12 | Cyt b gene | FL | 2.7 ng μL−1 | >150 | Yes | 29 |
| Cas12a | BRCA-1 | FL/colorimetry | 0.34 fM per nM level | <30 | Yes | 13 | |
| Cas12a | HPV-16 | FL | 10 fM | N.S. | Yes | 36 | |
| Cas12a | CXCL9 | FL | 14 pg mL−1 | 60 | No | 47 | |
| Cas12a | Salmonella enterica | FL | 619 CFU | >75 | Yes | 18 | |
| Cas12a | HIV, HPV-16 | ECL | 30 fM, 320 fM | 120 | Yes | 51 | |
| Cas12a | SARS-CoV-2 antigen | FL | 0.17 fM (∼2 copies per μL−1) | 20 | Yes | 52 | |
| Cas12a | ASFV DNA, EBV, HBV | FL | 17.5 copies per μL−1 | 60 | Yes | 53 | |
| Cas13a | SARS-CoV-2 | FL | 100 copies per μL−1 | 30 | Yes | 15 | |
| Cas13a | SARS-CoV-2 | FL | 10 fM | N.S. | No | 58 | |
| Cas13a | Bacillus cereus | FL | 10 CFU | 20 | Yes | 22 | |
| Digital-CRISPR ultrasensitive detection platforms | Cas12a | HBV, HPV | FL | 5 fM | 90 | Yes | 34 |
| Cas12a | ASFV DNA, EBV, HBV | FL | 17.5 copies per μL | 60 | Yes | 53 | |
| Cas13a | SARS-CoV-2 | FL | 5.7 fM (3.4 × 103 copies per μL) | <5 | Yes | 63 | |
| Cas13a | miRNAs, 16S rRNAs, SARS-CoV-2 RNA | FL | 101 – 105 aM, 6 copies per μL, 6 copies per μL | N.S. | Yes | 14 | |
| Cas13a | SARS-CoV-2 | FL | 6.5 aM | 9 | Yes | 64 | |
| Signal transducer formation | Cas9 | HIV | FL | N.S. | <60 | No | 65 |
| Cas9 | exon 3 and exon 51 of DMD-associated gene | Electrical signal | 1.7 fM | 15 | Yes | 3 and 66 | |
| Signal transducer formation | Cas9 | exons 2–10 and 45–55 of DMD-associated gene | Electrical signal | 1.3 fM | 5 | Yes | 103 |
| Cas12a | PB-19 | EC | 10 fM | >60 | Yes | 79 | |
| Cas12a | HBV, HPV-16, HPV-18 | Raman | 1 aM | 20 | Yes | 97 | |
| Cas12a | genetically modified soybean SHZD32-1 gene | ECL/FSV | 0.3 fM/3 fM | 60 | Yes | 93 | |
| Cas12a | DENV-4 | EC | 100 fM | 50 | Yes | 78 | |
| Cas12a | SARS-CoV-2 | Raman | 1 fM | 30∼40 | Yes | 100 | |
| Cas12a | HPV-16 | ECL | 480 fM | 70 | Yes | 92 | |
| Cas12a | MRSA bacteria mecA gene | EC | 3.5 fM | 90 | Yes | 85 | |
| Cas12a | HIV-1 | Raman | 0.3 fM | 60 | Yes | 101 | |
| Cas12a | HPV-16 | EC | 100 fM | 180 | Yes | 83 | |
| Cas13a | miR-17 | ECL | 1 fM | >90 | Yes | 48 | |
| Cas13a | miR-17 | EC | 50 aM | 36 | Yes | 123 | |
| Cas13a | DENV-1 | EC | 0.78 fM | 80 | Yes | 122 | |
| Cas13a | SARS-CoV-2 | EC | 4.4 × 10−2 fg mL−1 | 90 | Yes | 81 | |
| Cascade signal amplification | Cas10, Csm6 | SARS-CoV-2 | FL | 200 copies μL−1 | <30 | Yes | 110 |
| Cas12a | SARS-CoV-2 | Optical signal: gas bubbles | 50 copies μL−1 | 71 | Yes | 114 | |
| Cas12a | HBV, Human bladder cancer–associated single-nucleotide mutation | FL | 5 aM | 240 | Yes | 121 | |
| Cas13a | miR-17 | EC | 50 aM | 36 | Yes | 123 | |
| Cas13a | DENV-1 | EC | 0.78 fM | 80 | Yes | 122 | |
| Cas13a | Respiratory syncytial virus | FL | 75 aM | 30 | Yes | 124 | |
| Cas13a | SARS-CoV-2 | Colorimetry | ∼400 fM | <60 | Yes | 116 | |
| Cas13a | A synthetic 71-nucleotide RNA | Colorimetry | ∼260 fM | 210–270 | Yes | 117 | |
| Cas13a, Cas14a | miR-1 | FL | 1.33 fM | N.S. | Yes | 113 | |
| Cas13a, Csm6 | SARS-CoV-2 | FL | 30 copies μL−1 | 20 | Yes | 112 | |
In this review, we have summarized the latest advancement of the amplification-free CRISPR/Cas-based technique and its applications toward diagnosis, where we survey the state-of-the-art techniques, highlight strategies for improving the detection sensitivity, and outline the future perspectives. First, we overview the current status of amplification-free CRISPR/Cas detection techniques and describe the remaining challenge for high sensitivity detection. Additionally, referring to the initial amplification-free CRISPR/Cas-based detection method which utilized a fluorescence signal output,23 four representative strategies to overcome the challenges are summarized, including optimization of key parameters of CRISPR/Cas systems, digitalization of detection assays, change of signal transducer modes, and amplification of the output signal generated by the cascade assay (Fig. 1B). This review has also discussed challenges and future perspectives of amplification-free CRISPR/Cas techniques, which will contribute to developing more efficient strategies in biosensing and hold tremendous promise to become the next-generation of detection techniques.
2. Current status and challenges
2.1. Current status of amplification-free CRISPR/Cas detection systems
CRISPR/Cas systems are attractive molecular diagnostic tools due to their programmability and capability to directly target nucleic acid sequences in genomes. As the research develops, CRISPR/Cas-based detection technology unfolds great potential for forming next-generation NAT platforms and has been chosen as one of seven technologies that look set to shake up science in 2022.24,25The novel capability of the CRISPR/Cas systems to precisely cleave certain nucleic acid sequences stems from their acquired immunity-alike biological effects against exogenous viral invasion. However, Cas enzymes are created unequally depending on different purposes of application. The currently established CRISPR/Cas-based detection strategies (class 2 CRISPR/Cas systems) are primarily classified into three categories (type II, V, and VI) that implement different types of Cas effectors based on different underlying mechanisms (summarized in Table 2). The dCas9, a Cas9 variant without endonuclease activity while keeping programmable DNA binding activity, is typically used to bind target DNA with various modules like gFET sensors and nanopores, resulting in electrical conductivity signal output.26,27 Owing to superior collateral activity on nonspecific oligonucleotides, many examples of CRISPR/Cas-based detection utilized type V or VI systems and the CRISPR-associated nucleases Cas12 or Cas13 accordingly. After binding a single crRNA with specific spacer sequences to form the nuclease-inactive ribonucleoprotein complex (RNP), Cas12 or Cas13 effectors will be guided to their target nucleic acids and triggered the collateral cleavage activity to non-specifically degrade nearby single-stranded DNA or RNA (ssDNA or ssRNA), respectively. Typically, an ssDNA- or ssRNA-fluorophore quencher (FQ)-labeled reporter is introduced into the CRISPR/Cas detection system, which will generate a fluorescence signal after collateral cleavage, and builds a relationship between the target molecule concentrations and fluorescence signal outputs.28 Notably, for dCas9 and Cas12a, the template strand with an effector protein specific protospacer adjacent motif (PAM) sequence will be better specifically bound to RNP complex. As to Cas13a, the PAM sequence is not the key factor to the system, but there exists a protospacer flanking site (PFS) where a guanine base directly following the protospacer negatively influences the complementation between crRNA and the target.28
| Type | Cas | PAM sequence | Target | Trans-cleavage capability | Strategy | Refs. |
|---|---|---|---|---|---|---|
| Notes: crRNA, CRISPR RNA; dsDNA, double-stranded DNA; PAM, protospacer adjacent motif; PFS, protospacer flanking site; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA; tracrRNA, transactivating crRNA. | ||||||
| II |
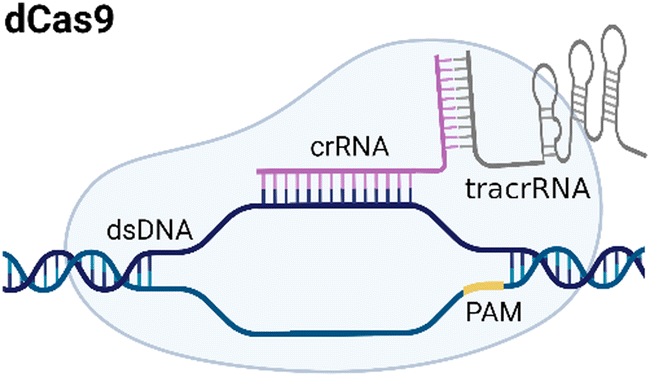
|
NGG | dsDNA | No | Based on specific binding: the selective recognition between dCas9 and target DNA can induce current signal change. | 27 |
| V |
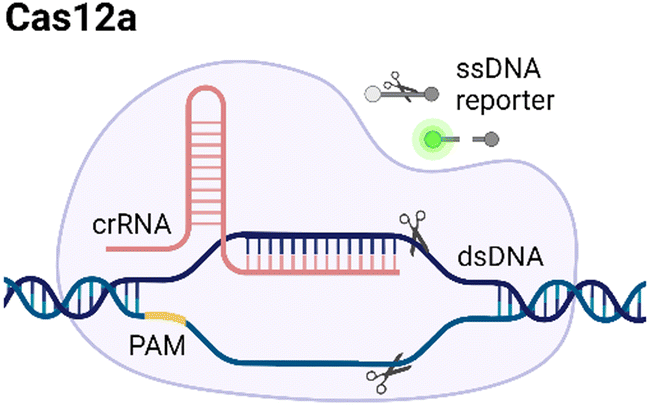
|
TTTN | dsDNA/ssDNA | Yes, ssDNA | Based on trans-cleavage: the target can bind to crRNA, activating the Cas12a and triggering trans-cleavage of quenched fluorescent reporters. | 39 |
| VI |
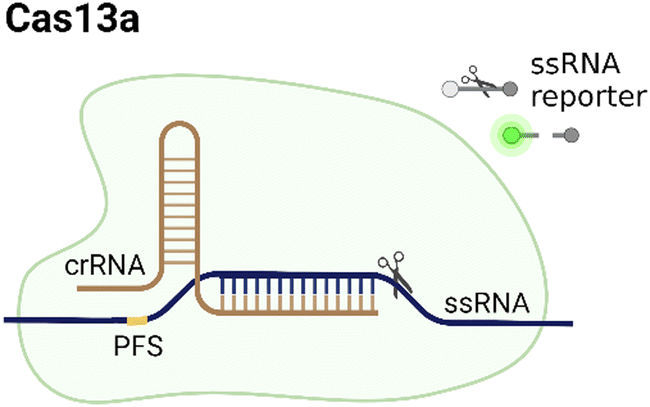
|
No, PFS-dependent | ssRNA | Yes, ssRNA | Based on trans-cleavage: the target sample binds to the complementary crRNA spacer and activates Cas13a, inducing the trans-cleavage effect on quenched fluorescent reporters. | 23 |
As a transformative technique in NAT, the CRISPR/Cas detection systems are highly specific and accurate, which benefit from the specific recognition of Cas effectors and their complementary target sequence under crRNA's guidance. Additionally, without the pre-amplification step, the amplification-free CRISPR/Cas detection can be rapidly completed within 15 min because of the highly catalyzed turnovers of Cas13a or Cas12a effectors armed with sensitivity-enhancing strategies.10,23,29 With simple steps and mild experimental conditions compared to those of conventional molecular diagnosis methods, CRISPR/Cas system-based detection can be coupled with portable devices as long as their accommodate settings (such as physiological temperature) are compatible with Cas effectors.30–32 The innovative uses of amplification-free CRISPR/Cas detection have allowed for broad sample testing categories from various areas such as clinical diagnosis, food identification, and environment monitoring, since the detection strategies for almost all types of targets can be converted to NAT. For instance, an amplification-free CRISPR/Cas12-based method can specifically discriminate pork meat from beef, mutton, and chicken, suggesting a promising test strategy for rapid halal food authentication.33 Fluoride in drinking water could be detected and quantified using a CRISPR/Cas13-based biosensor without pre-amplification, forming the advanced approach for on-site anion environmental monitoring.17 As a simple and fast terminal biosensing platform that has ‘cleavage upon activation’ capability, CRISPR/Cas detection holds tremendous promise to be combined with other novel biotechnologies and flesh out the future biosensing toolbox.
2.2. Challenges of amplification-free CRISPR/Cas detection systems
Without pre-amplification, the detection limit of CRISPR/Cas systems typically locates at the picomolar to femtomolar levels, which is far below the requirement of the ideal NAT method. Here, the main challenges that need to be addressed to improve the detection sensitivity are listed below (Fig. 2).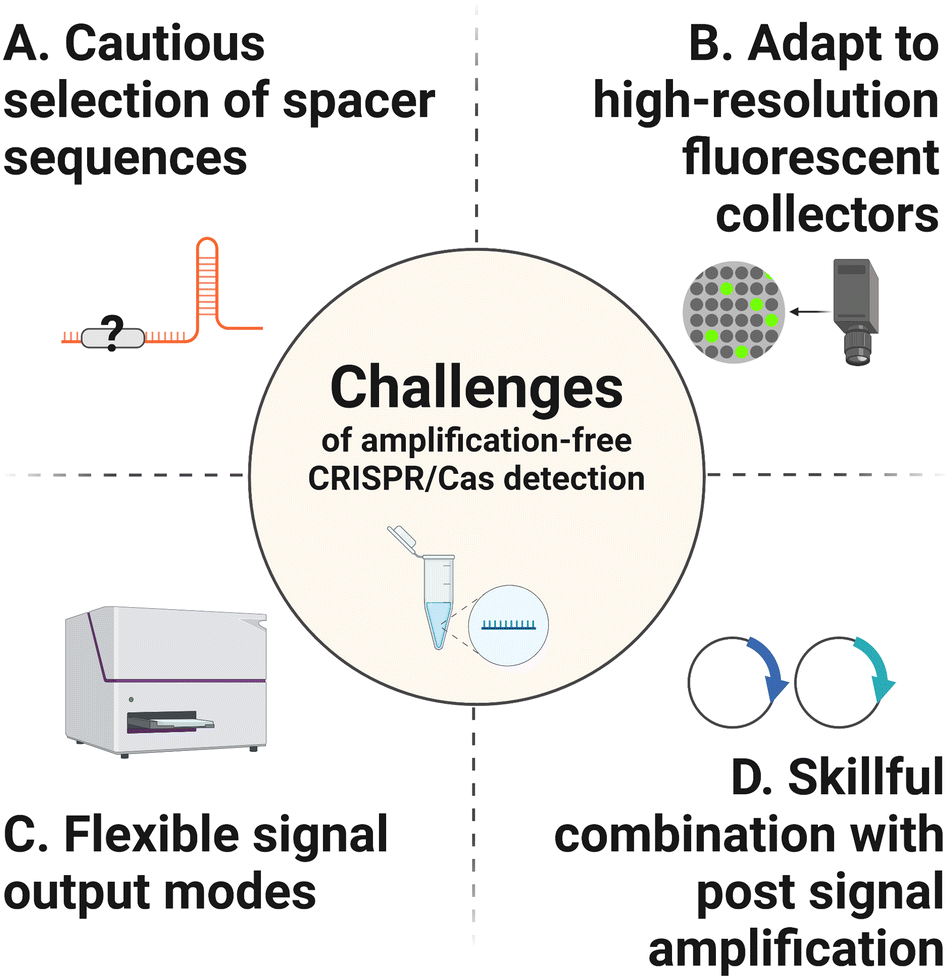 | ||
| Fig. 2 Challenges of amplification-free CRISPR/Cas detection. There are four main challenges remaining to be addressed to improve the detection sensitivity. (A) Optimize the spacer sequences of crRNAs to obtain satisfactory fluorescent signals. (B) Adapt to high-resolution fluorescent collectors to improve the detection signals of reaction system. (C) Attempt to combine CRISPR/Cas detection system with other flexible signal output modes. (D) Design post-amplification strategies to break the limitation of trans-cleavage catalytic efficiency of the Cas effectors. | ||
3. Strategies of amplification-free CRISPR/Cas-based detection
3.1. Optimization of crRNAs and signal reporters of the CRISPR/Cas system
In the CRISPR/Cas detection system, crRNA and signal reporters are two critical components. The former crRNA is vital for detection because only with its specific targeting behavior could the follow-up enzymatic cleavage be induced. Also, it determines CRISPR diagnostic a major advantage of high specificity, based on its sequence-specific recognition of the target nucleic acid. The latter signal reporter is a basic element that directly decides the performance of the signal readout. Therefore, the initial strategy to improve the sensitivity of amplification-free CRISPR/Cas detection is to optimize the crRNAs, such as the length and the base arrangement, and reporter types (ssDNA or ssRNA) (Fig. 3A). Moreover, recent research on the Cas effector kinetics has provided a fundamental theoretical basis for determining achievable LODs of CRISPR/Cas-based detection platforms.40,41 In the classic amplification-free CRISPR reaction system, Ramachandran et al. have found that the Michaelis–Menten model fits well for trans-cleavage activity of CRISPR-associated enzymes. By developing back-of-envelope analysis on reported research studies and performing experimental study, they summarized that the catalytic efficiency related parameters (kcat and KM) depend on the variations of gRNA, targets and reporters, thus further influencing the achievable assay sensitivity.40 Optimization strategies that noticeably improve the kinetic rates of CRISPR systems will be discussed in the following section.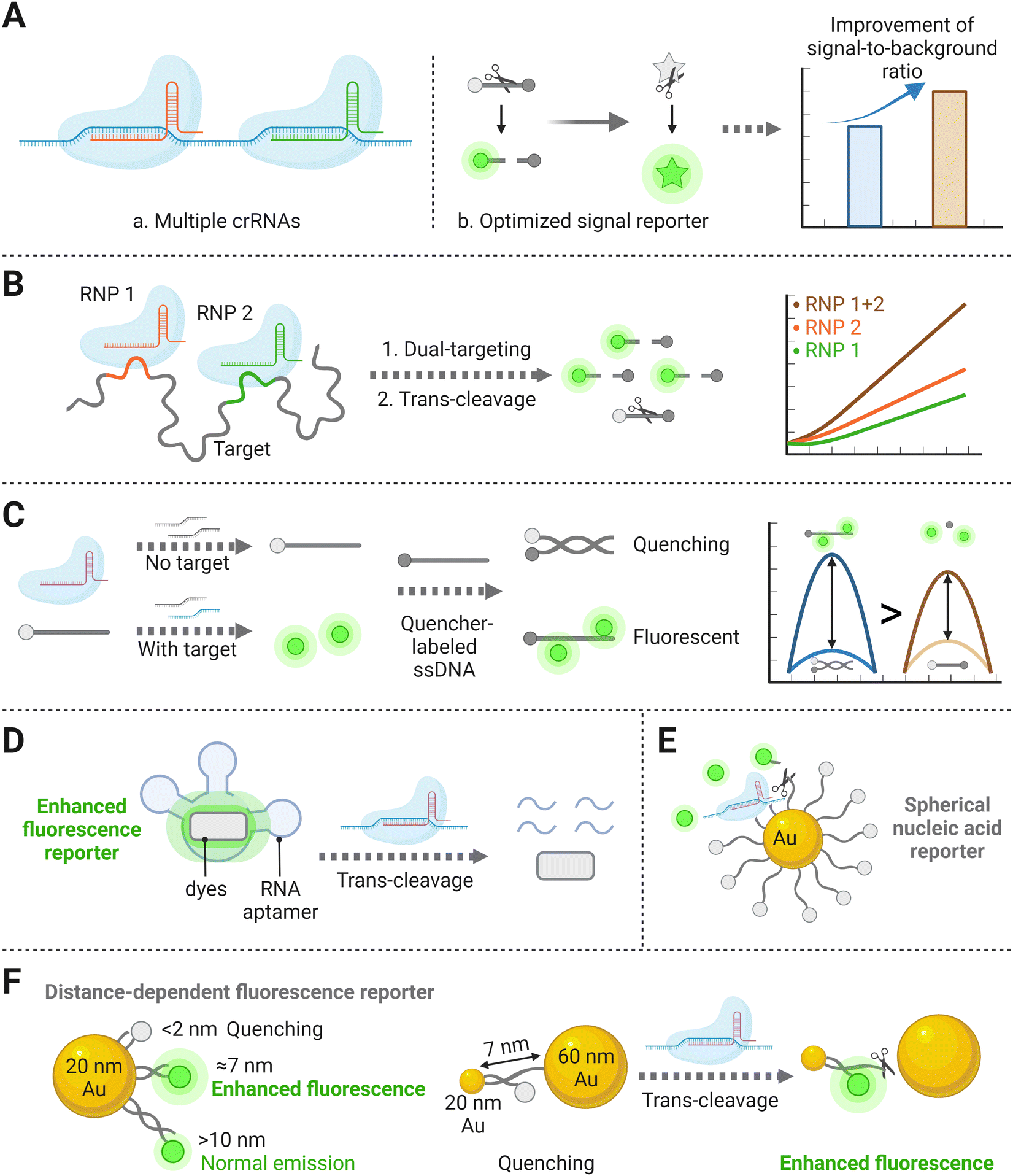 | ||
| Fig. 3 (A) Schematic representation of optimizations operated through biological processes for identification and cleavage of the CRISPR system, including multiple crRNA combinations (a), and formation of alternatively designed nucleic acid reporters (b), which can amplify the signal-to-background ratio and thereby improve the sensitivity. (B) Schematic of two RNP combination binding to different positions of the same target template, resulting in the cleavage of the signal reporter and increased fluorescence compared to single RNP.32 (C) The proximal DNA probe could yield enhanced fluorescence after being cleaved by the Cas effector.52 (D) A special RNA aptamer that could bind to specific dyes and induce fluorescence enhancement.37 (E) Illustration of a stably quenched spherical nucleic acid reporter, with a circle of fluorescence-labeled ssDNAs binding to the AuNP core.36 (F) Schematic diagram of an AuNP nanosensor inducing metal enhanced fluorescence after collateral effect of the Cas effector.13 | ||
In theory, integrating multiple crRNAs targeting different nucleotide regions of a single nucleic acid can synergistically activate Cas nucleases, resulting in more released fluorophores and thereby improve the sensitivity. For example, harnessing four targeted crRNA sequences in the CRISPR/Cas12a system displayed ∼64 times stronger signals than the conventional single-crRNA system.46 A previous study has also verified that combining two or three crRNAs can markedly increase the sensitivity of CRISPR/Cas detection systems (Fig. 3B).32 Specifically, two crRNAs inducing the greatest Cas13a activation were selected, which exhibited high sensitivity with an LOD of 270 copies per μL and low background fluorescence. Although trinal-crRNAs further enhanced the detection limit (31 copies per μL−1), more off-target events could possibly occur upon crRNA combination, resulting in false positive quantification. Consistent with the signal-amplified strategy described above, a dual-crRNA-based CRISPR/Cas13a assay performed in microchambers enabled an LOD down to 5 fM.47 These studies inspire us that an ideal crRNA combination should be formed via systematically searching across the entire target sequences, which can significantly increase the detection sensitivity of the amplification-free CRISPR/Cas-based platform.32 However, the LOD and the corresponding reaction time reported by Fozouni et al.32 reveal to be physically inconsistent with the theoretical enzyme kinetic criteria developed by Ramachandran et al.40 This conflict may be explained by the disparity of detector sensitivities, but there is still a large space to explore kinetic changes in the multi-crRNAs strategy.41
Recently, one study has demonstrated a novel alternative method by designing a proximal DNA probe. The probe consisted of a fluorophore-labeled ssDNA, which was first incubated with CRISPR reagents in the solution. After the reaction, a quencher group-labeled ssDNA was then added for the following assay. Without the target, the above two ssDNA materials hybridized with each other and showed high quenching efficiency. In contrast, in the presence of the target, the fluorophore-labeled ssDNA could be trans-cleaved in advance and inhibit the hybridization with the quencher group-labeled ssDNA, leading to the increase of the fluorescence intensity (Fig. 3C). This study has illustrated a 4-fold sensitivity improvement than other assays utilizing single-stranded dual-labeled probe.52 However, the principle of why the fluorophores near the quencher group-labeled DNA strand can increase fluorescence intensity more than the free fluorophores remains to be further discussed. Here is another research that has established a light-up RNA aptamer as the alternative signal reporter to dual-labeled nucleic acid strands.37 Without the target, the special RNA aptamer sequence remained intact and could bind to specific dyes, inducing fluorescence enhancement and significantly raising the background-to-signal ratio. In the presence of the target, the single-stranded aptamers were trans-cleaved into fragments with the activation of the Cas effector (Fig. 3D). The signaling-CRISPR/Cas13a assay allowed as low as 10 CFU of Bacillus cereus to be detected.
Besides the optimization methods mentioned above, nanomaterial-based or nanoparticle-assisted reporters can also enhance the signal intensity. To date, several studies have investigated the roles that nanoparticles play on enzyme activity, either positively or negatively, when they are anchored by nucleic acids.53,54 Owing to their high quenching efficiency when anchored with fluorophores, Au nanoparticles (AuNPs) have been creatively utilized for nucleic acid reporter constructions.55 Researchers believe that a stable and sensitive CRISPR/Cas detection system can be established by developing an extraordinary structure of metal nanoparticle-based nucleic acid reporters. Among several DNA-based nanostructures, spherical nucleic acids (SNAs) have worked as attractive tools due to their unique three-dimensional construction with a nanoparticle core densely with a nucleic acid shell.56,57 By uniformly immobilizing plenty of fluorescent dye-labeled ssDNAs (15 nt) on the AuNP core to form an SNA reporter (Fig. 3E), the target DNA as low as the femtomolar range could be sensed with a high signal-to-noise ratio, which benefited from the high quenching efficiency of AuNPs on fluorescent chromophores.58 Based on the above stable SNA structure, replacing the loaded-fluorophore with electrochemiluminescent (ECL) luminophores of carbon dots also showed an improvement in signal responses, again demonstrating that this nanostructure based on the high quenching ability of AuNPs can improve the sensitivity of the overall detection.59 Another attempt to detect cell-free DNA (cfDNA) based on an amplification-free CRISPR/Cas12a fluorescent biosensor was developed, in which metal-enhanced fluorescence (MEF) using DNA-functionalized Au nanoparticles (AuNPs) was designed to improve the sensitivity.39 In this case, two different-sized AuNP (20 and 60 nm) pairs connected by a 7 nm long dsDNA and a 2 nm long ssDNA were introduced to induce MEF with target DNA and fluorescence quenching without target DNA (Fig. 3F). In the presence of the target, the ssDNA between the 20- and 60-AuNPs was cleaved by the activated CRISPR/Cas12a complex, which resulted in the dissociation of the 60-AuNPs from fluorescein isothiocyanate (FITC) with 7 nm long DNA functionalized 20-AuNPs, thereby inducing an enhanced fluorescence signal that was proportional to the target cfDNA concentration down to femtomolar concentration ranges. Notably, because of AuNPs’ surface plasmon resonance properties, the proposed method not only showed excellent fluorescence enhancement by the CRISPR/Cas12a system but also provided the ability to intuitively assess target DNA concentrations via visual inspection of color change. The above nanoparticle-assisted reporters have shown improvement in LODs because of their high fluorescence quenching efficiency, which can be explained by the kinetic research conclusion that lower background signal facilitates the production of more cleaved reporters in fluorescence-based detection schemes.41 There is no doubt that nanomaterial-based CRISPR assays targeting certain biomolecules have exhibited excellent diagnostic performance by improving LODs for 2–4 orders of magnitude and have been successfully adapted in real clinical sample detection.39,58 Moving forward, further studies on the achievable sensitivity for different targets are expected, to expand the applicability of an amplification-free CRISPR/Cas detection system with nanomaterial-based reporters.
Researchers have also found that the detection signals of amplification-free CRISPR/Cas system can be amplified by simply increasing the number of recognition sites. For instance, an engineered DNA barcode complex containing multiple recognition sites for the Cas12a/crRNA complex was designed for the ELISA-like “immune-CRISPR” assay.18 Specifically, a DNA barcode was an oligonucleotide strand with the crRNA targeting sequence in the middle and biotin modification at both ends. Then, biotinylated DNA barcodes self-assembled onto streptavidin (SA) to generate SA-DNA barcode complexes, which increased the number and density of CRISPR/Cas12a recognition sites attached to the target protein antibody. As a result, the detection rate improved approximately 8-fold compared to using a monomeric DNA barcode. The LOD of the immune-CRISPR assay with multiple recognition sites represented a ∼7-fold improvement compared with traditional horseradish peroxidase (HRP)-based ELISA. Similarly, a CRISPR/Cas-based protein biosensor was developed by introducing aptamers to recognize the SARS-CoV-2 antigen. The platform transduced the aptamer defector signal into the reflection of trans-cleavage activity through hybrid DNA containing multiple sites for the specific touch-off of Cas12a.19 These lead to us anticipating that the extensive molecular detection methods taking advantage of programmable cleavage events from the Cas nuclease may be extended to proteins or other biomacromolecules.
3.2. Digital-CRISPR ultrasensitive detection platforms
The digital assay is the technology based on reaction system segmentation and has shown great application potential in biomedical analysis. The whole system is divided into massive reaction units, followed by counting the result of each micro-reaction unit in the final statistic processing. Compared with the regular detection in tubes, reaction units formed in the digital assay are very small with the droplets at the micro-nano level inside.60 The main types of methods for droplet generation can be divided into two categories, one of which is to submerge water in oil (Fig. 4A(a)).61 With the continuous oil flowing in from both sides of the microchannels, the dispersed water in the middle is squeezed, forced to shrink and deform, and finally formed droplets under the basis of entrainment focusing.60 The alternative method that produces microdroplets relies on the passive distribution of the fluid by microchannel geometry (Fig. 4A(b)).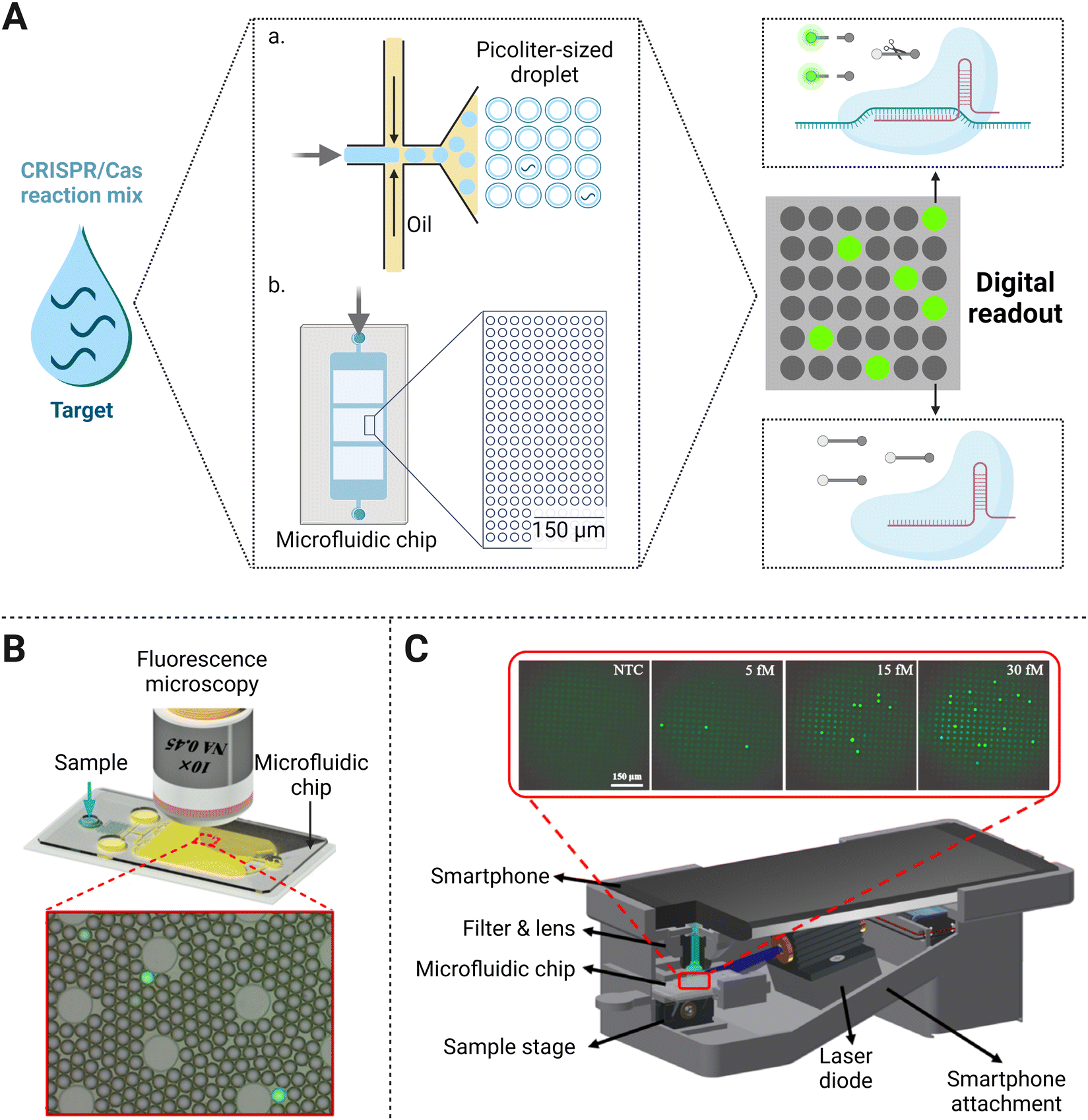 | ||
| Fig. 4 (A) Principal schematic representation of the digital assay. The bulk sample and CRISPR/Cas reaction mixture are emulsified with oil (a) or added to a microfluidic chip (b) to form a large number of microvolume droplets. In the presence of a single target molecule in each volume, fluorescent reporters could be trans-cleaved and yield a signal positive droplet. (B) Use of fluorescence microscopy for fluorescence imaging-based droplet counting on the microfluidic droplet chip. Adapted with permission from ref.65, Copyright 2021 American Chemical Society. (C) Illustration of the smartphone platform as a signal reader for the biosensing of nucleic acids. Reproduced with permission from ref.34, Copyright 2021 American Institute of Chemical Engineers. | ||
NAT using digital assay has highlighted its advantages of small sample requirement and rapid reaction speed. Droplet generation is fast and significantly improves the mixing efficiency while containing mixed small volumes of fluids.62 The individual reaction in each micro-unit instead of typical detection in tubes can significantly improve the sensitivity of biomedical detection and provide a novel capability of quantification. For example, droplet digital PCR (ddPCR) allows single template molecule amplification, providing a new strategy of NAT for pathogen detection, personalized cancer treatment, drug resistance, prenatal diagnosis, and other fields.63
As a next-generation NAT method, the CRISPR/Cas system combined with droplet digital technology allows the establishment of an ultra-sensitive biosensing platform for direct molecular detection (Fig. 4A). One platform named CRISPR/Cas-based amplification-free digital RNA detection (SATORI) was devised upon the combination of CRISPR/Cas13 and microchamber-array technologies.47 The induced cleavage provided a signal read-out with the detection of targets at as low as 5 fM concentration within 5 min, which was much more sensitive than other amplification-free CRISPR/Cas13a biosensing platforms (∼50 pM or 3 × 1010 copies per mL).24 They further established an automated detection platform on SATORI (opn-SATORI) integrated with magnetic-bead enrichment module, which not only eliminated the human operation errors but also lowered the LOD down to 6.5 aM (about 1000-fold higher than the former SATORI assay).64 By confining the target and other reaction reagents in picoliter-sized droplets, an ultra-localized amplification-free CRISPR/Cas13a assay format was performed with the LOD of SARS-CoV-2 down to ∼6 copies per μL. Once Cas13a was activated by the single-target RNA in one droplet, the cleavage of reporters produced a fluorescence signal. Then the “lighted” microdroplet was assigned as “one” for digital counting, otherwise “zero” would be recorded (Fig. 4B).65 The disparity of LODs between the above two strategies may be caused by their different experimental parameters. One difference lies in that the SATORI exhibited the whole assay in ∼3 fL droplets, while the latter performed in pL-sized reactors. Besides, SATORI two crRNAs were additionally used to simultaneously target one template for sensitivity improvement. With the same CRISPR/Cas system in a microdroplet-based assay, the reason why the latter strategy yielded higher sensitivity than the former one remains to be further studied.
In terms of molecular diagnostic applications, Cas12a-based collateral cleavage can detect as low as the pM range, which is about 1–2 orders of magnitude weaker than the CRISPR/Cas13a system. Therefore, it is a critical task to build a droplet-mediated single-molecule nucleic acid detection platform by amplification-free CRISPR/Cas12a system with satisfactory sensitivity. Under the optimal stoichiometric ratio of the Cas nuclease and crRNA, the CRISPR/Cas12a detection system without pre-amplification achieved an LOD of 8 pM in bulk solution. Further enhancement of sensitivity down to 5 fM was acquired after the reaction was switched into droplet-based digital assays (Fig. 4C).66 Optimization of multiple crRNAs, which has proven to be valid for sensitivity enhancement before, can also be coupled with a digital assay to achieve single-molecule detection. For example, a dual-crRNA targeted droplet-powered CRISPR/Cas12a detection platform was capable of ultrasensitive DNA detection at the single-molecule level, which exhibited a hundred-fold improvement compared with digital assays alone.67 Considering the former CRISPR/Cas12a reaction performed in the same pL-sized volume, this strategy proves that leveraging powerful optimization strategies of the CRISPR assay tailored for droplet-based analysis endows better detection efficiency.
3.3. Signal transducer formation
Apart from detecting fluorescence intensity changes caused by reporter cleavage, researchers have designed amplification-free CRISPR/Cas-mediated biosensing strategies with a variety of signal output modes, such as current changes and transformation of Raman scattering intensities (Fig. 5A). Electronic sensors are typically created under two main principles based on CRISPR/Cas systems. One is on account of target recognition. Specifically, the combination of the target strand and CRISPR/Cas complex hinders the transfection of electrons on the electrode and thus induces current response change. Other electronic sensors use the trans-cleavage effect on ssDNA/ssRNA reporters immobilized on the electrode surface, and their follow-up configuration changes can be transferred into the current change, because of the removal of the signal transduction tag modified on the reporters. Similarly, surface-enhanced Raman scattering (SERS)-integrated CRISPR/Cas biosensors rely on the change of Raman intensity caused by fragmentation of single-strand probe binding to SERS-active array. And this deactivates the so-called hot-spots effect, revealing a significant signal decrease compared to distinct SERS enhancement without trans-cleavage. The above biosensing devices utilize low-cost materials for the fabrication of compact platforms, and the sample-to-result time is short, usually within 60 min. Therefore, they are suitable for point-of-care testing (POCT), endowing amplification-free detection methods to fit resource-limited areas where well-equipped laboratories are not accessible. Additionally, the trials on biosensing strategies have also expanded the applied range of CRISPR/Cas-based detection methods for non-nucleic acid targets, via the specific alteration of the recognition element. In terms of sensitivity, the above biosensing strategies are mostly at the fM–pM level, around 3 orders of magnitude lower than the digital-based assay. But the detection sensitivity of biosensors can reach an LOD of the aM level by taking advantage of nanomaterials and post-amplification. In this section, we will discuss signal transducer-based strategies for amplification-free CRISPR/Cas detection systems.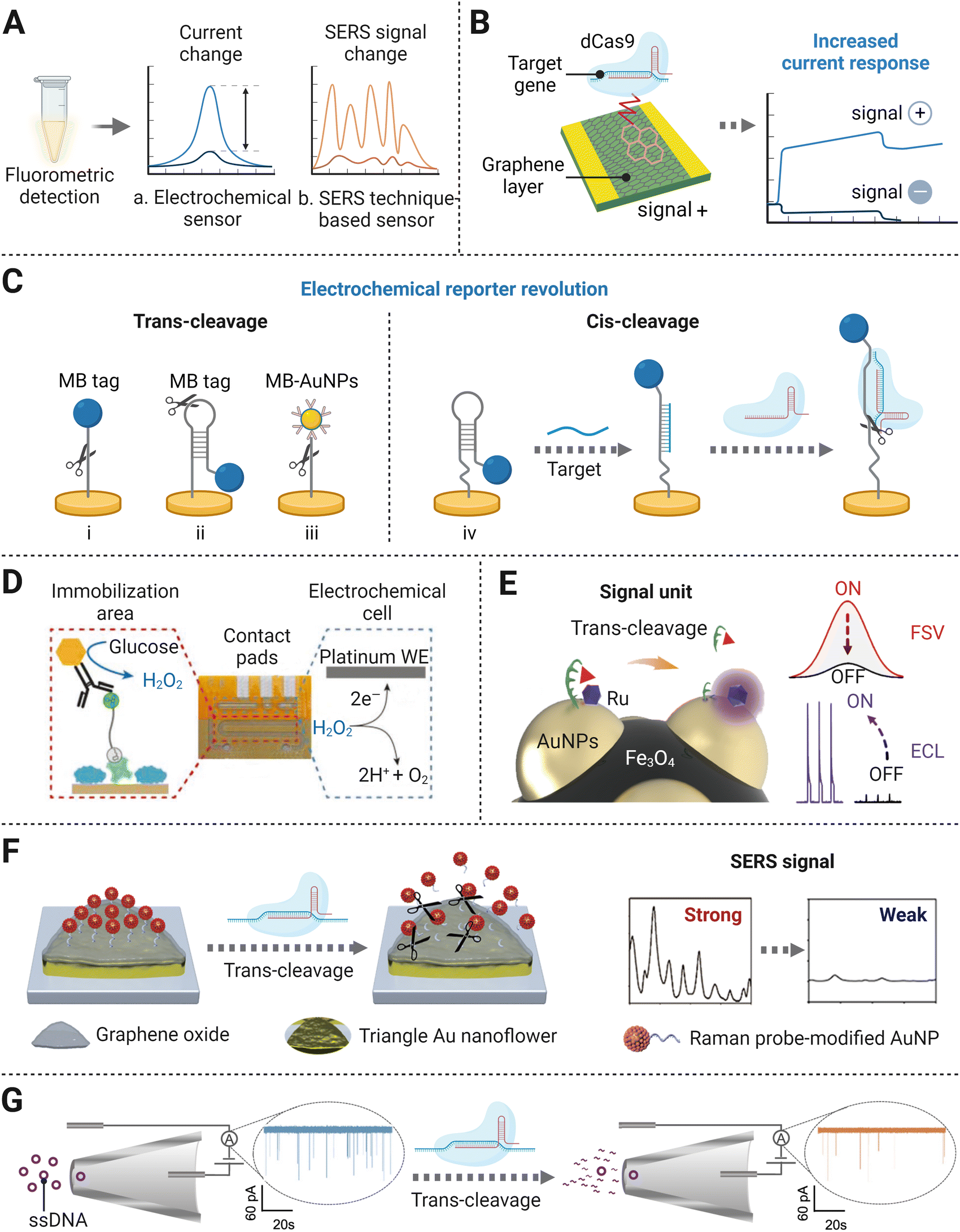 | ||
| Fig. 5 (A) Principal schematic representation of the different signal readout modes. (B) Illustration of a CRISPR/Cas-gFET biosensor based on target recognition by the catalytic dCas9 complex.30 (C) An overview of electrochemical/electrochemiluminescence signal transducers: electrochemical sensors coupled with the MB tag via ssDNA (i),76 hairpin DNA probe (ii),77 AuNP modified MB tag via ssDNA (iii),78 and hairpin probe using both DNA sensor induced conformation change and cis-cleavage of the Cas effector (iv);95 (D) An electrochemical sensor with a glucose oxidase tag. Reproduced from ref. 86 according to Creative Commons Attribution License (CC BY 4.0). (E) The dual-mode electrochemical sensor using Fe3O4@AuNPs/DNA-Fc&Ru as a signal unit. Adapted with permission from ref. 93, Copyright 2021 American Chemical Society. (F) Schematic diagram of the formation of SERS-based signal transducer. Adapted with permission from ref. 97, Copyright 2021 American Chemical Society. (G) CRISPR trans-cleavage-based glass nanopore sensors. Adapted with permission from ref. 106, Copyright 2020 American Chemical Society. | ||
Following the above method, a series of detection systems have been set up with a few alterations on electrochemical tags to improve sensitivity. Instead of using linear ssDNA as the linker, an optimized hairpin DNA electrochemical probe enabled lower concentration target detection (Fig. 5C(ii)).77 The development of multiple MB electrochemical tags conjugated to streptavidin-AuNPs as a whole, which was linked to Au electrode via linear SH-ssDNA-Biotin, was found to reduce the LOD to 0.1 pM without the pre-amplification process (Fig. 5C(iii)).78 The use of AuNPs expands the number of MB labels per reporter, indicating a stronger capability in increasing the conductivity of the system than a single MB molecule. Furthermore, a CRISPR/Cas system enhanced electrochemical DNA (E-DNA) sensor showed unprecedented sensitivity and accuracy.79 In this study, the target first induced a conformational change of the surface signalling probe from hairpin to linear structure, leading to the variation of the electron transfer rate of the electrochemical tag. The followed introduction of Cas cleavage activity into the E-DNA sensor dramatically induced a more apparent signal change between with and without the presence of the target (Fig. 5C(iv)). Such dual reduction of current outputs enhanced the sensitivity of the E-DNA sensor to the femtomolar level, which was capable of detecting a single mutation under unknown concentration conditions.
Nano noble-metal materials not only have been used for the modification of electrochemical tags but also acted as working electrodes, due to their exclusive electrical properties.78 The employment of noble-metal nanomaterials (such as gold nanostructures) and DNA nanostructures modified electrodes can highly improve the sensitivity by increasing the conductivity and the sensing areas, as well as providing an upper electrochemical platform with high affinity to the electrode surface.80 For instance, with the modification of Au nanoflower (AuNF)-like nanoparticles on the electrode surface, the developed CRISPR/Cas13a electrochemical biosensing platform enabled the detection of the ORF gene of SARS-CoV-2 at a concentration down to 4.4 × 10−2 fg mL−1.81 In another case, framework nucleic acids (FNAs, especially tetrahedral DNA nanostructures) were used as scaffolds with one face anchoring on the electrode interface. Due to their special 3D structure, FNAs can effectively weaken the surface crowing effect and well control the density of extending DNA probes to capture the target ssDNAs, achieving optimal detection sensitivity.82 Based on the recognition of the complementary target through PAM confirmation and crRNA matching, the Cas12a/crRNA complex got access to the interfaces, and the cis and trans-cleavages of Cas12a were activated at the same time. In combination with the enzyme-catalyzed reaction, the electrochemical platform with AuNPs-FNA modification achieved an ultralow LOD of 100 fM in HPV-16 detection without pre-amplification.83 Robust development of the electroactive silver nanowire metallization has also enhanced the current disparity. Based on the electrostatic interaction between negatively charged DNA probes and positively charged Ag+, a mass of Ag+ was deposited on the electrode surface. Compared with the bare electrode only modified with DNA probes, Ag+ metallization offered a large improvement in the electrochemical signal response.84 In the presence of the target, DNA probes would be trans-cleaved by the Cas effector, which led to a low current response. Notably, the degree of silver metallization was proportional to the quantity of ssDNA target, thereby enabling the quantitative measurement of the target.85
Besides for the evaluation of the current response directly caused by the degradation of ssDNA on the metal electrode, a novel tool has been manufactured by tuning the detection of Cas protease cleavage activity into glucose oxidase activity monitoring. In the absence of the target molecule, glucose oxidase bound to the intact reporter RNA was therefore immobilized, resulting in a catalytic reaction producing H2O2 (Fig. 5D). Otherwise, cleaved reporter RNA would fail to seize the glucose oxidase as the result of the trans-cleavage activity of the Cas effector. The enriched product concentration could be detected by the amperometric method.86 The above assay was performed in small sample volumes, and the biosensor chip could be stored for several months under refrigerated conditions. Without the requirement for pre-amplification of the target, the above properties endowed the capability of POCT application. Further optimization stretched its capability on multiplexed biosensing for miRNA, by subdividing microfluidic channels and optimizing related microfluidic dynamics.87 The compatible detection of the amplification-free CRISPR/Cas platform greatly improves its POCT detection potential because of its cost-effectiveness, flexibility, and simplicity.
Taking advantage of both electrochemistry and chemiluminescence, ECL techniques have drawn much attention in molecular diagnostic and bioanalysis, featuring low background noise, wide dynamic range, and requiring simple devices.88–90 The initial version of the CRISPR/Cas detection method implementing an ECL signal readout was established on a bipolar electrode electrical conductor in dielectric fluid and was sensitive to the femtomolar level range for miRNA sensing.48 Once trans-cleaved by Cas13a, a pre-trigger probe degraded into two fragments, one of which served as the initiator for follow-up isothermal exponential amplification (EXPAR). Upon the addition of ruthenium (Ru)-complexes and their subsequent oxidation reaction with the amplified dsDNA product, an intense luminescence signal was induced, proportional to the target concentration. This portable platform allows for simple experimental procedures, averting electrode modification and washing process. However, Ru-complexes used in this work serving as the ECL emitter are costly and have limitations in the immobilization capacity.91 As a cost-effective alternative to Ru-complexes, L-methionine stabilized gold nanoclusters (Met-AuNCs) were further used as substrate emitters to formulate an ECL transducer. At the beginning, Met-AuNCs were quenched by the ferrocene (Fc)-tagged thiolated ssDNA tethered on their surface. Upon non-specific ssDNA degradation by the CRISPR/Cas12a system, the Met-AuNCs modified on the electrode were released from the quenching states. The recording recovered ECL signals demonstrated an LOD of 0.48 pM for target HPV-16 DNA biosensing.92 Further research on a dual signal mode detection platform was achieved by synthesizing a functionalized composite bionanomaterial Fe3O4@AuNPs/DNA-Fc&Ru as a signal unit (Fig. 5E).93 A signal unit consisted of one Fe3O4 particle with several AuNPs deposited on its surface, and each AuNP was modified by a Ru-complex emitter and a short ssDNA labeled with Fc. The ssDNA–Fc in the signal unit was cleaved due to the nonspecific cleavage ability of the CRISPR/Cas12a system, which resulted in the decrease in the fast scan voltammetric (FSV) signal from Fc and the increase in the ECL signal from the Ru-complex which was formerly inhibited by Fc. The strategy not only showed high sensitivity with LODs of 0.3 fM and 3 fM for ECL signal and FSV signal, respectively, but also had satisfactory cross-validating ability and reliability in the rapid detection of genetically modified crops.
Another similar advancement in the utilization of SERS-based biosensing strategy obtained 1 fM detection of SARS-CoV-2 by a SERS probe consisting of Ag nanoparticles and the MB tag linked through ssDNA.100 Further development focused on the portability improvement by developing an ultra-sensitive SERS-based CRISPR/Cas12a lateral flow assay with an LOD of 0.3 fM, making it possible to detect biomolecules even in resource-limited settings.101 Compared with the first SERS-based CRISPR biosensor capable of detecting single molecules, these two methods own sensitivity merely at the fM level, but they are well-designed POCT approaches due to their portable signal read-out system. Above all, the schematic to simultaneously improve the sensitivity and stability of the portable SERS detection platform still needs further exploration and validation.
Surface plasmon resonance (SPR) sensing platforms are promising technologies for monitoring molecular interactions and enabling a wide range of applications in the field of clinical diagnostics and biological analysis. The SPR phenomenon is where the light beam hits at a specific incident angle that can excite the electrons at the interface between a dielectric and metal and transfer the resonance energy parallel to the metal surface layer.102 When the light source stays constant, the certain angle that triggers SPR is sensitively influenced by the surface refractive index change, which can therefore be utilized for analyte detection. This approach was integrated with dCas9 to capture specific DNA sequences and realized sensitive NAT without pre-amplification.103 In this case, the SPR sensing surface was modified with graphdiyne film coupling with the Au metal layer, which enabled a powerful dCas9 loading capacity through linkers and enhancement in changes of incident angles when the surface refractive index was altered by the presence of the dCas9-target DNA complex. By monitoring the change in the angle, the target molecule could be successfully detected with real-time quantification, allowing for an LOD of 1.3 fM. Future improvements of SPR platforms should focus on exploring other materials for metal layer modification and Cas effector optimization to improve the sensitivity and ensure the reusability of the sensing system to overcome their high-cost construction.
The specific targeting activity of dCas9 can also be integrated with an ion concentration polarization (ICP) phenomenon-based sensor for direct nucleic acid detection.107 This was achieved under the interaction between the electrophoretic force, which was formed by the charge-selective nano-junction, and the convective drag force from the flow in the channel. Since the electrophoretic mobility of biomolecules varied, the complexes of dCas9 and target DNA were separated from the sample and kept to accumulate in the vicinity of an ion boundary nanolayer, and other free DNAs in the analytes propagated towards the opposite direction. Using a fluorescence-labeled DNA sample, the two distinguishable behaviors revealed different optical profiles, thereby realizing the direct detection method of the target biomolecule.
3.4. Cascade signal amplification
Currently, the LOD of CRISPR/Cas mediated amplification-free nucleic acid detection is usually at the pM level, which cannot meet the requirement of most diagnostic cases (attomolar sensitivity). Except as mentioned above, a series of studies with cascade signal amplification strategies have shown great application prospects of improving the sensitivity of CRISPR/Cas-based detection methods. Typically, the major development is focused on protease-based cascade signal amplification systems and nucleic acid circuit-based cascade signal amplification systems (Fig. 6A). The former system containing a tandem protease reaction amplified the signal by leveraging products of the first nuclease activity as the substrate for the next enzymatic reaction. In virtue of the high catalytic activity of both enzymes, the final detection signal can be improved by 2–3 orders of magnitude. In the nucleic acid circuit-based cascade system, probes with a certain structure are usually designed as the intermediate. After being activated by the target biomolecule, the Cas effectors nonspecifically cut intermediates, leading to their transformation and thus bringing the subsequent nucleic acid cycle circuit, such as catalytic hairpin assembly and CRISPR/Cas-only amplification network (CONAN), which realizes exponential signal amplification. Here, we will discuss in detail the cascade signal amplification-based amplification-free CRISPR/Cas methods for ultrasensitive detection.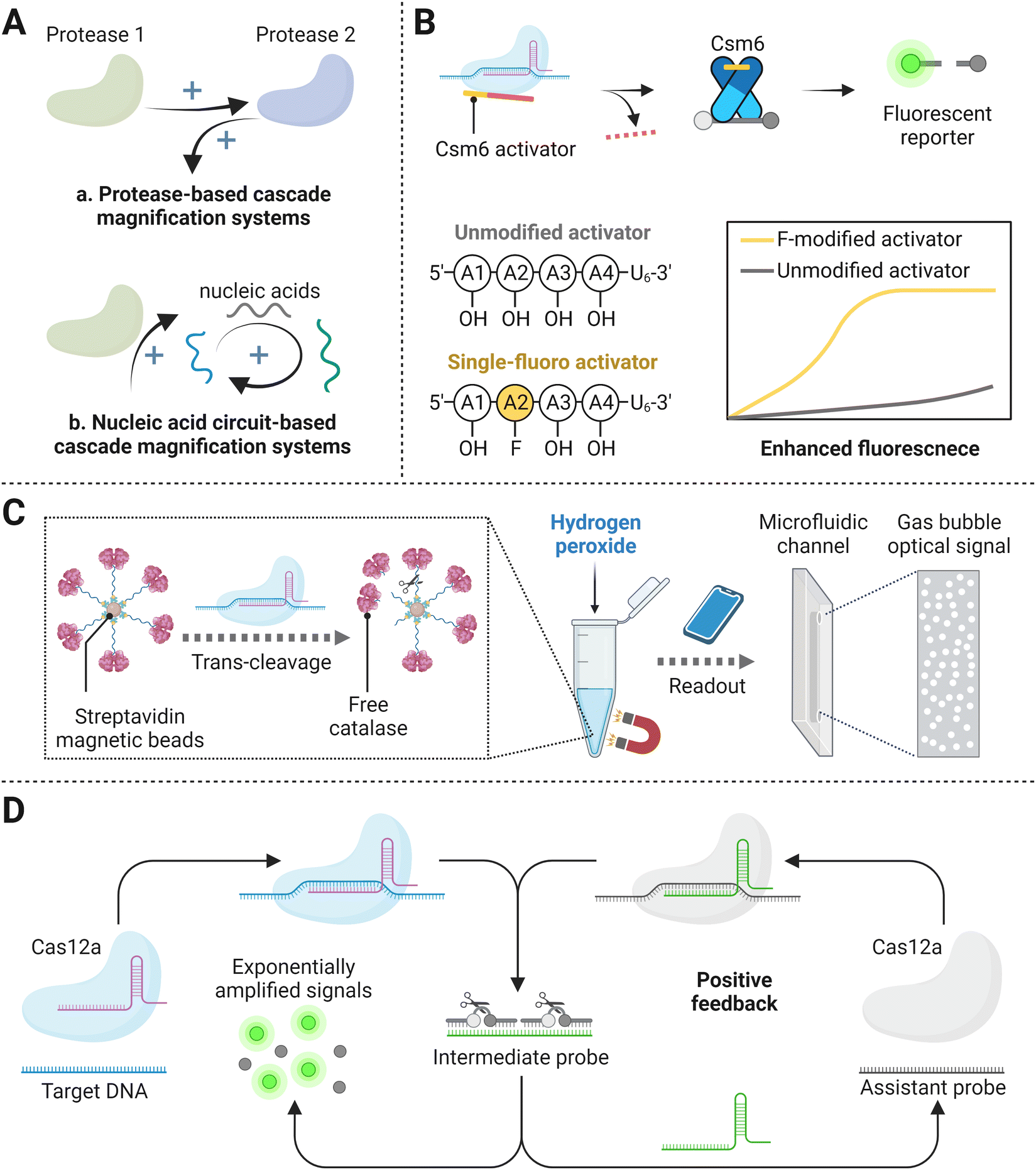 | ||
| Fig. 6 (A) Schematic representation of cascade signal amplification mechanisms, including protease-based cascade signal amplification systems (a) and nucleic acid circuit-based cascade signal amplification systems (b). (B) Tandem CRISPR nucleases-based detection.112 (C) The catalase-assisted CRISPR/Cas13a detection.114 (D) CRISPR/Cas-powered catalytic nucleic acid circuit.121 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 104 copies per μL, constituting around 3 orders of improvement in sensitivity over that of the former Csm complex based detection system. Furthermore, the inclusion of a set of eight optimized crRNAs in the Cas13a-Csm6 tandem reaction incredibly enhanced the LOD of 31 copies per μL within 20 min. Instead of employing Csm6 to achieve final signal generation, another strategy combined Cas13a and Cas14a for sensitive target RNA biosensing without pre-amplification.113 After target-activated Cas13a collateral cleaving of the phosphodiester bond of RNA molecule featuring a stable hairpin structure, the resultant products interacted with Cas14a through strand displacement, and thereby triggered the trans-cleavage activity of Cas14a and released fluorescence signals. It is worth noting that apart from designing and selecting the most proper structure of hairpin DNA oligonucleotides (i.e., length ratios of the stem to loop), researchers also compared Cas13a–Cas14a to Cas13a–Cas12a tandem series and found that the former version possessed better cleavage efficiency. The final amplified signal provides a lower LOD of 1.33 fM for microRNA detection, compared with the relatively weaker detection capability of Cas13a alone.113
× 104 copies per μL, constituting around 3 orders of improvement in sensitivity over that of the former Csm complex based detection system. Furthermore, the inclusion of a set of eight optimized crRNAs in the Cas13a-Csm6 tandem reaction incredibly enhanced the LOD of 31 copies per μL within 20 min. Instead of employing Csm6 to achieve final signal generation, another strategy combined Cas13a and Cas14a for sensitive target RNA biosensing without pre-amplification.113 After target-activated Cas13a collateral cleaving of the phosphodiester bond of RNA molecule featuring a stable hairpin structure, the resultant products interacted with Cas14a through strand displacement, and thereby triggered the trans-cleavage activity of Cas14a and released fluorescence signals. It is worth noting that apart from designing and selecting the most proper structure of hairpin DNA oligonucleotides (i.e., length ratios of the stem to loop), researchers also compared Cas13a–Cas14a to Cas13a–Cas12a tandem series and found that the former version possessed better cleavage efficiency. The final amplified signal provides a lower LOD of 1.33 fM for microRNA detection, compared with the relatively weaker detection capability of Cas13a alone.113
Proofs have also shown that combining Cas proteins with other non-nucleic acid catalytic effectors can render superior detection sensitivity.86 A study termed cellphone-based amplification-free system with CRISPR/Cas-dependent enzymatic (CASCADE) has been created, where a catalase–ssDNA probe was devised to bound to streptavidin beads with a high loading capacity (Fig. 6C).114 Upon the trans-cleavage effect of Cas12a protein triggered by target SARS-CoV-2 reverse-transcribed DNA, catalase was released from conjugated magnetic beads and was able to freely perform catalytic reaction after magnetic separation by the interaction between beads and magnetic rack. Free catalase then decomposed H2O2 and produced final optical gas bubble signals, featuring the platform with an LOD of 50 copies per μL in 71 min. The enzymatic reaction based on catalase could reveal direct optical signal readouts detected by portable smartphones, whereas the stability of Cas12a and catalase reagents and the thermal influence on H2O2 must be taken into consideration. HRP is one of the most commonly chosen enzymatic reporter because of its ubiquitous use in a variety of commercial assay formats and ability to be detected with high sensitivity via several different signaling substrates.115,116 Recently, Samanta et al. designed an HRP-based amplified signal strategy in the CRISPR/Cas detection system. In this study, target recognition activated the CRISPR/Cas complex, leading to catalytic cleavage and release of HRP-labeled oligonucleotides from the surface of microbeads. The HRP in solution could be monitored through colorimetric, fluorometric, or luminescent approaches, yielding up to a ∼75-fold turn-on signal and limits of detection (LODs) as low as ∼10 fM, which was ∼30-fold higher than the conventional fluorophore/quencher reporter in CRISPR/Cas detection systems. In addition, this study expanded its scope to non-nucleic acid targets with the aid of aptamers, showing commercially comparable detection sensitivity in complex biological media (i.e., human serum sample). Similarly, Marta Broto et al. have studied the combination of a CRISPR/Cas-based reaction with a nanozyme-linked immunosorbent assay (CrisprZyme), allowing for the quantitative and colorimetric readout of Cas13-mediated RNA detection.117 In principle, nanozymes have a higher substrate conversion rate than HRP.118 However, CrisprZyme has failed to show much improvement on the sensitivity compared with HRP-coupled CRISPR/Cas detection platform, probably because short target sequence restricted crRNA and non-specific immunosorbent.117,118
Briefly, the protease-based cascade signal amplification systems have proved their capability of improving sensitivity, but they still require for further optimization to achieve compatible multi-reaction and real clinical applications.
Taking advantage of Cas13a to directly recognize the target RNA and non-specifically cleave the probe for follow-up EXPAR reaction, multiple dsDNA products were synthesized and later interacted with the ECL emitter, which ultimately induced high intensity of fluorescence, allowing for target detection as low as 1 fM.48 One of the most common signal amplification circuits, catalytic hairpin assembly, was also incorporated with a CRISPR/Cas13a-based detection assay. A swing arm-DNA walker (DW) was devised to initiate subsequent signal amplification, enabling the cascade platform to obtain a detection limit of 0.78 fM.122 The DW machine was an ssDNA anchored to the electrode with a blocker-silenced walking strand. In the presence of the target, the activated Cas13a trans-cleaved blocker and released the walking arm, realizing catalytic signal amplification.122 In another Cas-catalytic hairpin DNA circuit (CHDC)-powered cascade system, Cas13a was utilized to trans-cleave hairpin probes, which were then degraded to intermediate for the following toehold-mediated strand displacement reactions.123 After multiple cycles of strand displacement reactions, numerous product molecules with MB tag were added to an electrochemical cell to interact with ferrocene-modified nucleic acid strand and thus perform a high level of the current response. After the measurement, an enzyme mixture of uracil-DNA glycosylase and endonuclease IV joined the assay and broke down the interaction between the two molecules, leaving only ferrocene-modified nucleic acid strands laid on the gold electrode for the next measurement. This CRISPR/Cas13a-mediated CHDC amplification system exhibited high sensitivity, allowing the LOD down to 50 aM, with its cost-effectiveness in reusable materials. Apart from the above CRISPR-triggered cascaded systems which were fabricated based on nucleic acid circuits exclusive of Cas effectors, an excellent study termed CONAN that contained an ordered integration of nucleic acid circuits for rapid DNA detection was established, exponentially amplifying signals by a Cas12a protein autocatalysis-driven feedback loop (Fig. 6D).121 Specifically, the target DNA was first combined with one CRISPR/Cas12a, followed by the activated nuclease cleaving the inhibitor DNA hybridizing a well-designed gRNA as the intermediate. The fragments of prior caged gRNA had two important functions: one served as the crRNA for another Cas12a effector, while the other two fragments turned into a fluorescence signal. With one of the intermediate fragments assembled with a Cas12a effector-assistant dsDNA probe system, this Cas12a nuclease could be activated and trans-cleave another gRNA, sequentially generating new intermediate fragments and providing positive feedback. Hence, multiple cleavage turnovers yielded abundant fluorescence signals. Exploiting CONAN for genomic DNA detection, concentrations as low as aM level could be sensed in unamplified samples. Another Cas13a cascade cyclic reaction based on a similar nucleic acid circuit principle also showed a low LOD of 75 aM.124 After the target RNA recognized by the first Cas13a, it can non-specifically digest fluorescent reporters as well as one designed mediator molecule. The mediator came with double-stranded RNA with a toehold, and it could activate another Cas13a effector after being first digested. This induced more and more active Cas13a and cleaved mediator molecule, thereby generating amplified signal. The above studies exhibit the promising future of nucleic acid circuits for rapid biomolecule detection.
While the ability of the CRISPR/Cas system to specifically recognize the target makes up for the artificial nucleic acid circuits, a CRISPR biosensing platform coupled with nucleic acid control circuits significantly improves the detection sensitivity. The programmability and precision of DNA hybridization could be employed to endow self-assembled DNA nanostructures, allowing for spatiotemporal regulation of biomolecular reactions.120 Also, the sophisticated process of designing diverse intermediate nucleic acid modules is needed to build CRISPR-nucleic acid circuits with higher performance, and the standardization and automation of the biosensing platform will contribute to enhancing the stability of clinical implementation.
4. Conclusions and future perspectives
Over the past few decades, CRISPR/Cas system-based programmed NAT has become a broadly accessible and popularized technology. The repurposing binding or cleavage event from bacterial CRISPR/Cas systems endows the design of ideal detection tools for molecule biosensing. While masses of CRISPR/Cas-based detection modalities are facilitated with target pre-amplification procedure in solution for sensitivity enhancement,125 they also raise the risk of non-specific amplification and cross-carryover contamination. Additionally, the two-step operation is not friendly for users, which is also a problem for field-deployable detection. To address these challenges, a series of amplification-free CRISPR/Cas-based detection platforms have been developed using strategies mentioned below. First, the optimally engineered crRNAs and signal reporters have been utilized to improve the signal-to-background ratio. Additionally, various biosensors with practicable modifications, ultrasensitive digital detection platforms, and cascade signal amplification systems also have allowed finer detection resolution and greater signal responses.Partial improvements have contributed amplification-free CRISPR/Cas-based detection approaches to have comparable sensitivity to gold standard techniques. For example, confining the CRISPR/Cas NAT reaction within one pL-sized microdroplet enabled detection sensitivity as low as a single molecule level.65 Generally, the sensitivity requirements of detection methods are different depending on specific targets and detection purposes. For example, viral kinetic modeling has shown that a detection approach with an LOD of 100 SAR-CoV-2 RNA copies per μL was needed for screening during the COVID-19 pandemic.51 Therefore, an LOD of the aM∼fM level is typically required to detect infectious disease. However, for non-communicable diseases like cancers, the concentration of cancer biomarker cfDNA higher than 127 ng mL−1 (∼108 copies per μL) may be a predictive indicator of death for patients in the intensive care units (ICUs).126 Abnormal concentration of ATP molecule in human venous plasma, usually higher than 28 nM probably indicates the occurrence of various physiological and pathophysiological events such as hypoxia and ischemia.127–129 When monitoring these targets, the detection limit that reaches their physiological concentration is enough to diagnose real clinical samples. For example, an electrochemical amplification-free CRISPR/Cas-based detection approach has been used to detect miR-19b from serum samples of children, with an LOD of 10 pM.86 Therefore, strategies that only improve the sensitivity by a few times can also be deployed for amplification-free systems when they apply for specific detection targets.
Because of the mild reaction conditions and short detection time given by Cas nuclease, amplification-free CRISPR/Cas-based detection techniques have shown excellent compatibility with portable devices and promising field-deployable application and home-based diagnostics. Connecting with microfluidic-chip reactors and portable signal collectors such as smartphones as a signal analyzer, or colorimetric lateral flow assay techniques, amplification-free CRISPR/Cas-based detection approaches can be easily used to develop universal POCT platforms.19,32,66,101,112 To date, CRISPR/Cas-based detection methods without pre-amplification have achieved a sample-to-answer process time within 5 min,47 which is much shorter than gold standards (usually 4–6 h). During the global pandemic of COVID-19, such a fast turnaround time of amplification-free CRISPR/Cas-based detection techniques was able to cope with the rapid epidemic expansion and helped to break the pandemic.51 With increasing efforts on innovative uses of amplification-free CRISPR/Cas-based detection technology in diverse areas including but not limited to clinical diagnosis, precision medicine, and environmental monitoring, it is reasonable to assume that this developing technology will provide more breakthroughs shortly.
Despite the tremendous promise of amplification-free CRISPR/Cas-based biosensing platforms, certain obstacles remain to be addressed to make these technologies universal tools for real-life applications in POCT and at-home settings. The first challenge is the limitation of target sequence selection. A specific PAM sequence on the target array is crucial for most of the Cas nucleases’ activity. Pre-amplification of target molecules can introduce PAM sequences and thus allows CRISPR/Cas detection platforms to work without considering the particular sequence fragment in targets. Moreover, target types can be converted with the target amplification process, enabling CRISPR/Cas biosensors to detect various analytes including nucleic acids and biological molecules. However, the amplification-free CRISPR/Cas techniques are currently unsatisfactory to the features mentioned above, resulting in a certain limited range of target options and compromised universality and flexibility of detection.
To become an ideal NAT platform, amplification-free CRISPR/Cas detection techniques must overcome the drawbacks associated with conventional methods and be suitable for POCT without requiring ancillary complicated equipment. The main issues requiring continuous improvement, which may help to shape a simple and practical workflow of CRISPR/Cas-based biosensors are listed below:
4.1. Standardization
Standardization of reaction conditions, such as buffer, temperature, and pH values, is vital for CRISPR/Cas-based detection methods applied in real sample, which might interfere with the efficiency of assembly between crRNA and effectors and further influence the detection performance.76 This issue may be solved by adding internal references or establishing calibration curves for samples with known concentrations before each independent test.130 Furthermore, turning results of CRISPR/Cas-based detection to pH paper or color chart may be an ideal improvement.1304.2. Cost-effective detection
It is necessary to consider the production cost of CRISPR/Cas components and portable devices, from the view of long-term cost-effective employment. The ingenious design of nucleic acid circuits in CRISPR/Cas systems might reduce the cost, by reusing reaction nucleic acid materials or regenerating the electrochemical sensing interfaces.123 For digital-CRISPR detection platforms that usually perform on microsystems, small size reaction chamber and volume feature the advantage of low cost.47,65–67 Further efforts may focus on reducing the cost of ultrasensitive and portable signal collectors in digital-CRISPR detection assays, to meet the market demand.4.3. Integrated platform
Always, sample pretreatment step is the rate-limiting part of POCT, which is also one of the key issues in amplification-free CRISPR/Cas-based detection techniques.8,16,30 Also, the transfer process from sampling to detection steps may result in biological contamination.131 The majority of amplification-free detection methods use pre-treated samples to perform well in real applications, but the prerequisite involves much time and complexity as well. Rapid and portable DNA purification kits have been explored to minimize or even eliminate the extraction step.86 We look forward to the emergence of more sample pretreatment methods or products in the future, to expand on-site applications of amplification-free CRISPR/Cas-based detection techniques.184.4. Multiplexed biosensing
The demonstration of multiplexing capability has grown to be a future focus influenced by recent trends. In some circumstances, the sole biomarker is not adequate for an appropriate diagnosis of a disease or confirmed abnormal test results. For example, most laboratory diagnosis methods of COVID-19 are based on positive results of RT-qPCR of two distinctive genes. Also, the simultaneous screening of various analytes will enable low-cost, rapid, and accurate detection. For amplification-free CRISPR/Cas-based detection methods, it is challenging to distinguish multiple targets in a single reaction volume because of the responded signal leveraged by non-specific collateral cleavage. Introduction of microfluidic technology based on physical separation may be an effective solution. For example, deploying spatially multiplexed chips to obtain different targets in each reaction space can contribute to multiple detection.65,97 Besides, employing orthogonal effectors of LwaCas13a, PsmCas13b, CcaCas13b, and AsCas12a, which show different dinucleotide cleavage motif preferences, may also achieve the detection of different targets at the same time.11With improvements to gradually become an ideal NAT tool, the pioneering amplification-free CRISPR/Cas-based detection technology will not only serve as a fundamental component of the biomedical toolkit but also spread to the the brink of every aspect of human lives.
Author contributions
Kun Yin, Qinqin Hu and Xiaokui Guo conceived the scope of the review. Huimin Li, Qinqin Hu and Kun Yin drafted the initial idea and proposal for the review. Huimin Li and Yi Xie wrote the initial draft, supervised by Kun Yin, Qinqin Hu and Xiaokui Guo. Fumin Chen, Huiwen Bai, Leshan Xiu, Xiaonong Zhou and Xiaokui Guo revised and provided suggestions to the first draft. All the authors read, revised, and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22104090), the Natural Science Foundation of Shanghai (22ZR1436200), and the Open Project of National Facility for Translational Medicine (Shanghai) (TMSK-2021-114). Unless otherwise stated in the figure captions, all images in the figures were adapted with permission from Biorender.com with a paid academic subscription.References
- J. R. Choi, J. Hu, R. Tang, Y. Gong, S. Feng, H. Ren, T. Wen, X. Li, W. A. Wan Abas, B. Pingguan-Murphy and F. Xu, Lab Chip, 2016, 16, 611–621 RSC.
- R. A. Mumford, R. Macarthur and N. Boonham, Food Secur., 2016, 8, 103–109 CrossRef.
- G. Siravegna, S. Marsoni, S. Siena and A. Bardelli, Nat. Rev. Clin. Oncol., 2017, 14, 531–548 CrossRef CAS PubMed.
- N. D. Grubaugh, J. T. Ladner, P. Lemey, O. G. Pybus, A. Rambaut, E. C. Holmes and K. G. Andersen, Nat. Microbiol., 2019, 4, 10–19 CrossRef CAS PubMed.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS PubMed.
- C. B. F. Vogels, A. F. Brito, A. L. Wyllie, J. R. Fauver, I. M. Ott, C. C. Kalinich, M. E. Petrone, A. Casanovas-Massana, M. Catherine Muenker, A. J. Moore, J. Klein, P. Lu, A. Lu-Culligan, X. Jiang, D. J. Kim, E. Kudo, T. Mao, M. Moriyama, J. E. Oh, A. Park, J. Silva, E. Song, T. Takahashi, M. Taura, M. Tokuyama, A. Venkataraman, O. E. Weizman, P. Wong, Y. Yang, N. R. Cheemarla, E. B. White, S. Lapidus, R. Earnest, B. Geng, P. Vijayakumar, C. Odio, J. Fournier, S. Bermejo, S. Farhadian, C. S. Dela Cruz, A. Iwasaki, A. I. Ko, M. L. Landry, E. F. Foxman and N. D. Grubaugh, Nat. Microbiol., 2020, 5, 1299–1305 CrossRef CAS PubMed.
- R. Martzy, C. Kolm, R. Krska, R. L. Mach, A. H. Farnleitner and G. H. Reischer, Anal. Bioanal. Chem., 2019, 411, 1695–1702 CrossRef CAS PubMed.
- C. Myhrvold, C. A. Freije, J. S. Gootenberg, O. O. Abudayyeh, H. C. Metsky, A. F. Durbin, M. J. Kellner, A. L. Tan, L. M. Paul, L. A. Parham, K. F. Garcia, K. G. Barnes, B. Chak, A. Mondini, M. L. Nogueira, S. Isern, S. F. Michael, I. Lorenzana, N. L. Yozwiak, B. L. MacInnis, I. Bosch, L. Gehrke, F. Zhang and P. C. Sabeti, Science, 2018, 360, 444–448 CrossRef CAS PubMed.
- R. Sorek, V. Kunin and P. Hugenholtz, Nat. Rev. Microbiol., 2008, 6, 181–186 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439–444 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- H. Zaghloul and M. El-Shahat, World J. Hepatol., 2014, 6, 916–922 CrossRef PubMed.
- L. Li, S. Li, N. Wu, J. Wu, G. Wang, G. Zhao and J. Wang, ACS Synth. Biol., 2019, 8, 2228–2237 CrossRef CAS PubMed.
- B. Wang, R. Wang, D. Wang, J. Wu, J. Li, J. Wang, H. Liu and Y. Wang, Anal. Chem., 2019, 91, 12156–12161 CrossRef CAS PubMed.
- J. Joung, A. Ladha, M. Saito, N. G. Kim, A. E. Woolley, M. Segel, R. P. J. Barretto, A. Ranu, R. K. Macrae, G. Faure, E. I. Ioannidi, R. N. Krajeski, R. Bruneau, M. W. Huang, X. G. Yu, J. Z. Li, B. D. Walker, D. T. Hung, A. L. Greninger, K. R. Jerome, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, N. Engl. J. Med., 2020, 383, 1492–1494 CrossRef CAS PubMed.
- Y. Ma, Q. Mou, P. Yan, Z. Yang, Y. Xiong, D. Yan, C. Zhang, X. Zhu and Y. Lu, Chem. Sci., 2021, 12, 11740–11747 RSC.
- I. Lee, S. J. Kwon, M. Sorci, P. S. Heeger and J. S. Dordick, Anal. Chem., 2021, 93, 16528–16534 CrossRef CAS PubMed.
- X. Zhao, Z. Wang, B. Yang, Z. Li, Y. Tong, Y. Bi, Z. Li, X. Xia, X. Chen, L. Zhang, W. Wang and G. Y. Tan, Synth. Syst. Biotechnol., 2021, 6, 283–291 CrossRef CAS PubMed.
- Y. Wei, Z. Tao, L. Wan, C. Zong, J. Wu, X. Tan, B. Wang, Z. Guo, L. Zhang, H. Yuan, P. Wang, Z. Yang and Y. Wan, Biosens. Bioelectron., 2022, 211, 114282 CrossRef CAS PubMed.
- W. Kang, L. Liu, P. Yu, T. Zhang, C. Lei and Z. Nie, Biosens. Bioelectron., 2022, 213, 114468 CrossRef CAS PubMed.
- P. Qin, M. Park, K. J. Alfson, M. Tamhankar, R. Carrion, J. L. Patterson, A. Griffiths, Q. He, A. Yildiz, R. Mathies and K. Du, ACS Sens., 2019, 4, 1048–1054 CrossRef CAS PubMed.
- A. East-Seletsky, M. R. O’Connell, S. C. Knight, D. Burstein, J. H. Cate, R. Tjian and J. A. Doudna, Nature, 2016, 538, 270–273 CrossRef CAS PubMed.
- Y. Li, S. Li, J. Wang and G. Liu, Trends Biotechnol., 2019, 37, 730–743 CrossRef CAS PubMed.
- M. Eisenstein, Nature, 2022, 601, 658–661 CrossRef CAS PubMed.
- Y. Zhang, L. Qian, W. Wei, Y. Wang, B. Wang, P. Lin, W. Liu, L. Xu, X. Li, D. Liu, S. Cheng, J. Li, Y. Ye, H. Li, X. Zhang, Y. Dong, X. Zhao, C. Liu, H. M. Zhang, Q. Ouyang and C. Lou, ACS Synth. Biol., 2017, 6, 211–216 CrossRef CAS.
- R. Hajian, S. Balderston, T. Tran, T. deBoer, J. Etienne, M. Sandhu, N. A. Wauford, J. Y. Chung, J. Nokes, M. Athaiya, J. Paredes, R. Peytavi, B. Goldsmith, N. Murthy, I. M. Conboy and K. Aran, Nat. Biomed. Eng., 2019, 3, 427–437 CrossRef CAS PubMed.
- O. O. Abudayyeh, J. S. Gootenberg, S. Konermann, J. Joung, I. M. Slaymaker, D. B. Cox, S. Shmakov, K. S. Makarova, E. Semenova, L. Minakhin, K. Severinov, A. Regev, E. S. Lander, E. V. Koonin and F. Zhang, Science, 2016, 353, aaf5573 CrossRef PubMed.
- L. T. Nguyen, B. M. Smith and P. K. Jain, Nat. Commun., 2020, 11, 4906 CrossRef CAS PubMed.
- J. Arizti-Sanz, C. A. Freije, A. C. Stanton, B. A. Petros, C. K. Boehm, S. Siddiqui, B. M. Shaw, G. Adams, T. F. Kosoko-Thoroddsen, M. E. Kemball, J. N. Uwanibe, F. V. Ajogbasile, P. E. Eromon, R. Gross, L. Wronka, K. Caviness, L. E. Hensley, N. H. Bergman, B. L. MacInnis, C. T. Happi, J. E. Lemieux, P. C. Sabeti and C. Myhrvold, Nat. Commun., 2020, 11, 5921 CrossRef CAS PubMed.
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C. Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS PubMed.
- P. Fozouni, S. Son, M. Diaz de Leon Derby, G. J. Knott, C. N. Gray, M. V. D’Ambrosio, C. Zhao, N. A. Switz, G. R. Kumar, S. I. Stephens, D. Boehm, C. L. Tsou, J. Shu, A. Bhuiya, M. Armstrong, A. R. Harris, P. Y. Chen, J. M. Osterloh, A. Meyer-Franke, B. Joehnk, K. Walcott, A. Sil, C. Langelier, K. S. Pollard, E. D. Crawford, A. S. Puschnik, M. Phelps, A. Kistler, J. L. DeRisi, J. A. Doudna, D. A. Fletcher and M. Ott, Cell, 2021, 184(323–333), e329 Search PubMed.
- Y. Wu, Y. Dong, Y. Shi, H. Yang, J. Zhang, M. R. Khan, S. Deng, G. He, Q. He, Y. Lv and R. Deng, J. Agric. Food Chem., 2021, 69, 10321–10328 CrossRef CAS PubMed.
- L. X. Li, S. Y. Li, N. Wu, J. C. Wu, G. Wang, G. P. Zhao and J. Wang, ACS Synth. Biol., 2019, 8, 2228–2237 CrossRef CAS PubMed.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- P. Fozouni, S. Son, M. D. de León Derby, G. J. Knott, C. N. Gray, M. V. D’Ambrosio, C. Zhao, N. A. Switz, G. R. Kumar and S. I. Stephens, Cell, 2021, 184(323–333), e329 Search PubMed.
- T. Zhang, W. Zhou, X. Lin, M. R. Khan, S. Deng, M. Zhou, G. He, C. Wu, R. Deng and Q. He, Biosens. Bioelectron., 2021, 176, 112906 CrossRef CAS PubMed.
- L. Liu, Z. Wang, Y. Wang, J. Luan, J. J. Morrissey, R. R. Naik and S. Singamaneni, Adv. Healthcare Mater., 2021, 10, e2100956 CrossRef PubMed.
- J. H. Choi, J. Lim, M. Shin, S. H. Paek and J. W. Choi, Nano Lett., 2021, 21, 693–699 CrossRef CAS PubMed.
- A. Ramachandran and J. G. Santiago, Anal. Chem., 2021, 93, 7456–7464 CrossRef CAS.
- D. A. Huyke, A. Ramachandran, V. I. Bashkirov, E. K. Kotseroglou, T. Kotseroglou and J. G. Santiago, Anal. Chem., 2022, 94, 9826–9834 CrossRef CAS PubMed.
- L. Liu, X. Li, J. Wang, M. Wang, P. Chen, M. Yin, J. Li, G. Sheng and Y. Wang, Cell, 2017, 168(121–134), e112 Search PubMed.
- L. Liu, X. Li, J. Ma, Z. Li, L. You, J. Wang, M. Wang, X. Zhang and Y. Wang, Cell, 2017, 170(714–726), e710 Search PubMed.
- Y. Shan, X. Zhou, R. Huang and D. Xing, Anal. Chem., 2019, 91, 5278–5285 CrossRef CAS PubMed.
- E. A. Nalefski, N. Patel, P. J. Y. Leung, Z. Islam, R. M. Kooistra, I. Parikh, E. Marion, G. J. Knott, J. A. Doudna, A. M. Le Ny and D. Madan, iScience, 2021, 24, 102996 CrossRef CAS PubMed.
- M. Zeng, Y. Ke, Z. Zhuang, C. Qin, L. Y. Li, G. Sheng, Z. Li, H. Meng and X. Ding, Anal. Chem., 2022, 94, 10805–10812 CrossRef CAS PubMed.
- H. Shinoda, Y. Taguchi, R. Nakagawa, A. Makino, S. Okazaki, M. Nakano, Y. Muramoto, C. Takahashi, I. Takahashi, J. Ando, T. Noda, O. Nureki, H. Nishimasu and R. Watanabe, Commun. Biol., 2021, 4, 476 CrossRef CAS PubMed.
- T. Zhou, R. Huang, M. Huang, J. Shen, Y. Shan and D. Xing, Adv. Sci., 2020, 7, 1903661 CrossRef CAS PubMed.
- Q. He, Q. Chen, F. Li, X. Yuan, C. Wang, C. Liu, L. Xu, X. Zhong, J. Wei, V. Pandey, D. Yu, Y. Dong, Y. Zhang, L. Deng, K. Du and P. Qin, bioRxiv, 2021, 2021.2007.2017.452803 DOI:10.1101/2021.07.17.452803.
- M. Rossetti, R. Merlo, N. Bagheri, D. Moscone, A. Valenti, A. Saha, P. R. Arantes, R. Ippodrino, F. Ricci, I. Treglia, E. Delibato, J. van der Oost, G. Palermo, G. Perugino and A. Porchetta, Nucleic Acids Res., 2022, 50, 8377–8391 CrossRef PubMed.
- D. B. Larremore, B. Wilder, E. Lester, S. Shehata, J. M. Burke, J. A. Hay, M. Tambe, M. J. Mina and R. Parker, Sci. Adv., 2021, 7, eabd5393 CrossRef CAS PubMed.
- T. Zhang, H. T. Li, X. Xia, J. Liu, Y. Lu, M. R. Khan, S. Deng, R. Busquets, G. He, Q. He, J. Zhang and R. Deng, J. Agric. Food Chem., 2021, 69, 12828–12836 CrossRef CAS PubMed.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308–311 CrossRef CAS PubMed.
- X. Qu, D. Zhu, G. Yao, S. Su, J. Chao, H. Liu, X. Zuo, L. Wang, J. Shi, L. Wang, W. Huang, H. Pei and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- S. B. Ebrahimi, D. Samanta and C. A. Mirkin, J. Am. Chem. Soc., 2020, 142, 11343–11356 CrossRef CAS PubMed.
- D. Samanta, S. B. Ebrahimi and C. A. Mirkin, Adv. Mater., 2020, 32, e1901743 CrossRef PubMed.
- X. Fu, Y. Shi, F. Peng, M. Zhou, Y. Yin, Y. Tan, M. Chen, X. Yin, G. Ke and X. B. Zhang, Anal. Chem., 2021, 93, 4967–4974 CrossRef CAS PubMed.
- K.-R. Zhao, L. Wang, P.-F. Liu, X.-M. Hang, H.-Y. Wang, S.-Y. Ye, Z.-J. Liu and G.-X. Liang, Sens. Actuators, B, 2021, 346, 130485 CrossRef CAS.
- W. B. Han and X. Y. Chen, J. Braz. Soc. Mech. Sci. Eng., 2021, 43, 247 CrossRef.
- O. J. Miller, A. El Harrak, T. Mangeat, J. C. Baret, L. Frenz, B. El Debs, E. Mayot, M. L. Samuels, E. K. Rooney, P. Dieu, M. Galvan, D. R. Link and A. D. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS PubMed.
- V. Cristini and Y. C. Tan, Lab Chip, 2004, 4, 257–264 RSC.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- H. Shinoda, T. Iida, A. Makino, M. Yoshimura, J. Ishikawa, J. Ando, K. Murai, K. Sugiyama, Y. Muramoto, M. Nakano, K. Kiga, L. Cui, O. Nureki, H. Takeuchi, T. Noda, H. Nishimasu and R. Watanabe, Commun. Biol., 2022, 5, 473 CrossRef CAS PubMed.
- T. Tian, B. Shu, Y. Jiang, M. Ye, L. Liu, Z. Guo, Z. Han, Z. Wang and X. Zhou, ACS Nano, 2021, 15, 1167–1178 CrossRef CAS PubMed.
- T. Yu, S. W. Zhang, R. Matei, W. Marx, C. L. Beisel and Q. S. Wei, AIChE J., 2021, 67, e17365 CAS.
- H. Yue, B. Shu, T. Tian, E. Xiong, M. Huang, D. Zhu, J. Sun, Q. Liu, S. Wang, Y. Li and X. Zhou, Nano Lett., 2021, 21, 4643–4653 CrossRef CAS PubMed.
- D. Reddy, L. F. Register, G. D. Carpenter and S. K. Banerjee, J. Phys. D: Appl. Phys., 2011, 44, 313001 CrossRef.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- M. I. Katsnelson, K. S. Novoselov and A. K. Geim, Nat. Phys., 2006, 2, 620–625 Search PubMed.
- S. Balderston, J. J. Taulbee, E. Celaya, K. Fung, A. Jiao, K. Smith, R. Hajian, G. Gasiunas, S. Kutanovas, D. Kim, J. Parkinson, K. Dickerson, J. J. Ripoll, R. Peytavi, H. W. Lu, F. Barron, B. R. Goldsmith, P. G. Collins, I. M. Conboy, V. Siksnys and K. Aran, Nat. Biomed. Eng., 2021, 5, 713–725 Search PubMed.
- V. Pattanayak, S. Lin, J. P. Guilinger, E. Ma, J. A. Doudna and D. R. Liu, Nat. Biotechnol., 2013, 31, 839–843 CrossRef CAS PubMed.
- D. Singh, S. H. Sternberg, J. Fei, J. A. Doudna and T. Ha, Nat. Commun., 2016, 7, 12778 CrossRef CAS PubMed.
- M. Yang, S. Peng, R. Sun, J. Lin, N. Wang and C. Chen, Cell Rep., 2018, 22, 372–382 CrossRef CAS PubMed.
- H. Li, J. Yang, G. Wu, Z. Weng, Y. Song, Y. Zhang, J. A. Vanegas, L. Avery, Z. Gao, H. Sun, Y. Chen, K. D. Dieckhaus, X. Gao and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202203826 CAS.
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 17399–17405 CrossRef CAS PubMed.
- D. Zhang, Y. Yan, H. Que, T. Yang, X. Cheng, S. Ding, X. Zhang and W. Cheng, ACS Sens., 2020, 5, 557–562 CrossRef CAS PubMed.
- Y. Lee, J. Choi, H. K. Han, S. Park, S. Y. Park, C. Park, C. Baek, T. Lee and J. Min, Sens. Actuators, B, 2021, 326, 128677 CrossRef CAS.
- W. Xu, T. Jin, Y. Dai and C. C. Liu, Biosens. Bioelectron., 2020, 155, 112100 CrossRef CAS PubMed.
- L. Wang, X. Chen, X. Wang, X. Han, S. Liu and C. Zhao, Biosens. Bioelectron., 2011, 30, 151–157 CrossRef CAS PubMed.
- W. Heo, K. Lee, S. Park, K. A. Hyun and H. I. Jung, Biosens. Bioelectron., 2022, 201, 113960 CrossRef CAS PubMed.
- M. Lin, J. Wang, G. Zhou, J. Wang, N. Wu, J. Lu, J. Gao, X. Chen, J. Shi, X. Zuo and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 2151–2155 CrossRef CAS PubMed.
- J. Su, Y. Ke, N. Maboyi, X. Zhi, S. Yan, F. Li, B. Zhao, X. Jia, S. Song and X. Ding, Small Methods, 2021, 5, e2100935 CrossRef PubMed.
- A. Suea-Ngam, P. D. Howes, C. E. Stanley and A. J. deMello, ACS Sens., 2019, 4, 1560–1568 CrossRef CAS PubMed.
- A. Suea-Ngam, P. D. Howes and A. J. deMello, Chem. Sci., 2021, 12, 12733–12743 RSC.
- R. Bruch, J. Baaske, C. Chatelle, M. Meirich, S. Madlener, W. Weber, C. Dincer and G. A. Urban, Adv. Mater., 2019, 31, e1905311 CrossRef PubMed.
- R. Bruch, M. Johnston, A. Kling, T. Mattmuller, J. Baaske, S. Partel, S. Madlener, W. Weber, G. A. Urban and C. Dincer, Biosens. Bioelectron., 2021, 177, 112887 CrossRef CAS PubMed.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- H. Peng, Z. Huang, Y. Sheng, X. Zhang, H. Deng, W. Chen and J. Liu, Angew. Chem., Int. Ed., 2019, 58, 11691–11694 CrossRef CAS PubMed.
- W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef CAS PubMed.
- L. Y. Yao, F. Yang, G. B. Hu, Y. Yang, W. Huang, W. B. Liang, R. Yuan and D. R. Xiao, Biosens. Bioelectron., 2020, 155, 112099 CrossRef CAS PubMed.
- P. F. Liu, K. R. Zhao, Z. J. Liu, L. Wang, S. Y. Ye and G. X. Liang, Biosens. Bioelectron., 2021, 176, 112954 CrossRef CAS PubMed.
- H. Ge, X. Wang, J. Xu, H. Lin, H. Zhou, T. Hao, Y. Wu and Z. Guo, Anal. Chem., 2021, 93, 14885–14891 CrossRef CAS PubMed.
- X. Dai, W. Fu, H. Chi, V. S. D. Mesias, H. Zhu, C. W. Leung, W. Liu and J. Huang, Nat. Commun., 2021, 12, 1292 CrossRef CAS PubMed.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguie, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. Garcia de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Kall, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhofer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlucker, L. L. Tay, K. G. Thomas, Z. Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzan, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- C. Zong, M. Xu, L. J. Xu, T. Wei, X. Ma, X. S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- J. H. Choi, M. Shin, L. Yang, B. Conley, J. Yoon, S. N. Lee, K. B. Lee and J. W. Choi, ACS Nano, 2021, 15, 13475–13485 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, Z. C. Dong, S. Jiang, C. Zhang, L. G. Chen, L. Zhang, Y. Liao, J. Aizpurua, Y. Luo, J. L. Yang and J. G. Hou, Nature, 2013, 498, 82–86 CrossRef CAS PubMed.
- X. Y. Zhang, D. Han, N. Ma, R. Gao, A. Zhu, S. Guo, Y. Zhang, Y. Wang, J. Yang and L. Chen, J. Phys. Chem. Lett., 2018, 9, 6047–6051 CrossRef CAS PubMed.
- J. Liang, P. Teng, W. Xiao, G. He, Q. Song, Y. Zhang, B. Peng, G. Li, L. Hu, D. Cao and Y. Tang, J. Nanobiotechnol., 2021, 19, 273 CrossRef CAS PubMed.
- Y. Pang, Q. Li, C. Wang, S. zhen, Z. Sun and R. Xiao, Chem. Eng. J., 2022, 429, 132109 CrossRef CAS.
- X. Zhu and T. Gao, in Nano-Inspired Biosensors for Protein Assay with Clinical Applications, ed. G. Li, Elsevier, 2019, pp. 237–264 DOI:10.1016/B978-0-12-815053-5.00010-6.
- F. Zheng, Z. Chen, J. Li, R. Wu, B. Zhang, G. Nie, Z. Xie and H. Zhang, Adv. Sci., 2022, 9, e2105231 CrossRef PubMed.
- R. W. DeBlois and C. P. Bean, Rev. Sci. Instrum., 1970, 41, 909–916 CrossRef.
- W. Yang, L. Restrepo-Perez, M. Bengtson, S. J. Heerema, A. Birnie, J. van der Torre and C. Dekker, Nano Lett., 2018, 18, 6469–6474 CrossRef CAS PubMed.
- R. Nouri, Y. Jiang, X. L. Lian and W. Guan, ACS Sens., 2020, 5, 1273–1280 CrossRef CAS PubMed.
- H. Lee, J. Choi, E. Jeong, S. Baek, H. C. Kim, J. H. Chae, Y. Koh, S. W. Seo, J. S. Kim and S. J. Kim, Nano Lett., 2018, 18, 7642–7650 CrossRef CAS PubMed.
- P. Samai, N. Pyenson, W. Jiang, G. W. Goldberg, A. Hatoum-Aslan and L. A. Marraffini, Cell, 2015, 161, 1164–1174 CrossRef CAS PubMed.
- M. Kazlauskiene, G. Kostiuk, C. Venclovas, G. Tamulaitis and V. Siksnys, Science, 2017, 357, 605–609 CrossRef CAS PubMed.
- A. Santiago-Frangos, L. N. Hall, A. Nemudraia, A. Nemudryi, P. Krishna, T. Wiegand, R. A. Wilkinson, D. T. Snyder, J. F. Hedges, C. Cicha, H. H. Lee, A. Graham, M. A. Jutila, M. P. Taylor and B. Wiedenheft, Cell Rep. Med., 2021, 2, 100319 CrossRef CAS PubMed.
- C. Garcia-Doval, F. Schwede, C. Berk, J. T. Rostol, O. Niewoehner, O. Tejero, J. Hall, L. A. Marraffini and M. Jinek, Nat. Commun., 2020, 11, 1596 CrossRef CAS PubMed.
- T. Y. Liu, G. J. Knott, D. C. J. Smock, J. J. Desmarais, S. Son, A. Bhuiya, S. Jakhanwal, N. Prywes, S. Agrawal, M. D. D. Derby, N. A. Switz, M. Armstrong, A. R. Harris, E. J. Charles, B. W. Thornton, P. Fozouni, J. Shu, S. I. Stephens, G. R. Kumar, C. Y. Zhao, A. Mok, A. T. Iavarone, A. M. Escajeda, R. McIntosh, S. E. Kim, E. J. Dugan, K. S. Pollard, M. X. Tan, M. Ott, D. A. Fletcher, L. F. Lareau, P. D. Hsu, D. F. Savage, J. A. Doudna and I. T. Consortium, Nat. Chem. Biol., 2021, 17, 982–988 CrossRef CAS PubMed.
- Y. Sha, R. Huang, M. Huang, H. Yue, Y. Shan, J. Hu and D. Xing, Chem. Commun., 2021, 57, 247–250 RSC.
- F. S. R. Silva, E. Erdogmus, A. Shokr, H. Kandula, P. Thirumalaraju, M. K. Kanakasabapathy, J. M. Hardie, L. G. C. Pacheco, J. Z. Li, D. R. Kuritzkes and H. Shafiee, Adv. Mater. Technol., 2021, 6, 2100602 CrossRef CAS PubMed.
- D. Calabria, M. M. Calabretta, M. Zangheri, E. Marchegiani, I. Trozzi, M. Guardigli, E. Michelini, F. Di Nardo, L. Anfossi, C. Baggiani and M. Mirasoli, Sensors, 2021, 21, 3358 CrossRef CAS PubMed.
- D. Samanta, S. B. Ebrahimi, N. Ramani and C. A. Mirkin, J. Am. Chem. Soc., 2022, 144, 16310–16315 CrossRef CAS PubMed.
- M. Broto, M. M. Kaminski, C. Adrianus, N. Kim, R. Greensmith, S. Dissanayake-Perera, A. J. Schubert, X. Tan, H. Kim, A. S. Dighe, J. J. Collins and M. M. Stevens, Nat. Nanotechnol., 2022, 17, 1120–1126 CrossRef CAS PubMed.
- C. N. Loynachan, M. R. Thomas, E. R. Gray, D. A. Richards, J. Kim, B. S. Miller, J. C. Brookes, S. Agarwal, V. Chudasama, R. A. McKendry and M. M. Stevens, ACS Nano, 2018, 12, 279–288 CrossRef CAS PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- J. Li, A. A. Green, H. Yan and C. Fan, Nat. Chem., 2017, 9, 1056–1067 CrossRef CAS PubMed.
- K. Shi, S. Xie, R. Tian, S. Wang, Q. Lu, D. Gao, C. Lei, H. Zhu and Z. Nie, Sci. Adv., 2021, 7, eabc7802 CrossRef CAS PubMed.
- J. Wang, Q. Xia, J. Wu, Y. Lin and H. Ju, Anal. Chim. Acta, 2021, 1187, 339131 CrossRef CAS PubMed.
- Y. Sheng, T. Zhang, S. Zhang, M. Johnston, X. Zheng, Y. Shan, T. Liu, Z. Huang, F. Qian, Z. Xie, Y. Ai, H. Zhong, T. Kuang, C. Dincer, G. A. Urban and J. Hu, Biosens. Bioelectron., 2021, 178, 113027 CrossRef CAS PubMed.
- H. Zeng, P. Zhang, X. Jiang, C. Duan, Y. Yu, Q. Wu and X. Yang, Anal. Chim. Acta, 2022, 1217, 340009 CrossRef CAS PubMed.
- O. O. Abudayyeh and J. S. Gootenberg, Science, 2021, 372, 914–915 CrossRef CAS PubMed.
- S. Volik, M. Alcaide, R. D. Morin and C. Collins, Mol. Cancer Res., 2016, 14, 898–908 CrossRef CAS PubMed.
- M. W. Gorman, E. O. Feigl and C. W. Buffington, Clin. Chem., 2007, 53, 318–325 CrossRef CAS PubMed.
- B. M. Paddle and G. Burnstock, Blood Vessels, 1974, 11, 110–119 CAS.
- M. M. Borst and J. Schrader, Circ. Res., 1991, 68, 797–806 CrossRef CAS PubMed.
- J. E. van Dongen, J. T. W. Berendsen, R. D. M. Steenbergen, R. M. F. Wolthuis, J. C. T. Eijkel and L. I. Segerink, Biosens. Bioelectron., 2020, 166, 112445 CrossRef CAS PubMed.
- K. Karthik, R. P. Aravindh Babu, K. Dhama, M. A. Chitra, G. Kalaiselvi, T. M. Alagesan Senthilkumar and G. D. Raj, Arch. Med. Res., 2020, 51, 623–630 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |