
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison and assessment of methods for cellulose crystallinity determination
Khandoker Samaher
Salem†
 *ab,
Nitesh Kumar
Kasera†
ac,
Md. Ashiqur
Rahman
ad,
Hasan
Jameel
b,
Youssef
Habibi
e,
Stephen J.
Eichhorn
f,
Alfred D.
French
g,
Lokendra
Pal
*b and
Lucian A.
Lucia
*bhi
*ab,
Nitesh Kumar
Kasera†
ac,
Md. Ashiqur
Rahman
ad,
Hasan
Jameel
b,
Youssef
Habibi
e,
Stephen J.
Eichhorn
f,
Alfred D.
French
g,
Lokendra
Pal
*b and
Lucian A.
Lucia
*bhi
aDepartment of Applied Chemistry and Chemical Engineering, University of Dhaka, Dhaka-1000, Bangladesh. E-mail: samaher.salem@du.ac.bd
bDepartment of Forest Biomaterials, North Carolina State University, Raleigh, NC, USA. E-mail: lpal@ncsu.edu; lalucia@ncsu.edu
cDepartment of Biological and Agricultural Engineering, North Carolina State University, Raleigh, NC, USA
dNational Institute of Textile Engineering and Research, University of Dhaka, Dhaka-1000, Bangladesh
eSustainable Materials Research Center (SUSMAT-RC), University Mohamed VI Polytechnic (UM6P), Lot 660, Hay Moulay Rachid, Benguerir 43150, Morocco
fBristol Composites Institute, School of Civil, Aerospace, and Mechanical Engineering, University of Bristol, Bristol, BS8 1TR, UK
gUnited States Department of Agriculture, Agricultural Research Service, Southern Regional Research Center USDA ARS SRRC, New Orleans, LA 70124, USA
hDepartment of Chemistry, North Carolina State University, Raleigh, CD 27695-8204, USA
iState Key Laboratory of Biobased Materials & Green Papermaking, Qilu University of Technology/Shandong Academy of Sciences, Jinan, 250353, P. R. China
First published on 17th August 2023
Abstract
The degree of crystallinity in cellulose significantly affects the physical, mechanical, and chemical properties of cellulosic materials, their processing, and their final application. Measuring the crystalline structures of cellulose is a challenging task due to inadequate consistency among the variety of analytical techniques available and the lack of absolute crystalline and amorphous standards. Our article reviews the primary methods for estimating the crystallinity of cellulose, namely, X-ray diffraction (XRD), nuclear magnetic resonance (NMR), Raman and Fourier-transform infrared (FTIR) spectroscopy, sum-frequency generation vibrational spectroscopy (SFG), as well as differential scanning calorimetry (DSC), and evolving biochemical methods using cellulose binding molecules (CBMs). The techniques are compared to better interrogate not only the requirements of each method, but also their differences, synergies, and limitations. The article highlights fundamental principles to guide the general community to initiate studies of the crystallinity of cellulosic materials.
 Khandoker Samaher Salem | Khandoker Samaher Salem is Assistant Professor of Department of Applied Chemistry and Chemical Engineering at the University of Dhaka, Bangladesh. He also maintains his appointments as a visiting scholar in the Department of Forest Biomaterials, North Carolina State University. He completed his doctoral degree from the Forest Biomaterials, North Carolina State University. His research interests focus on surface modification of lignocellulosic materials for barrier application and hygiene products, such as paper towel, tissue paper, and synthesis of biomaterials by upcycling leather waste and their valorization through sustainable applications. |
 Nitesh Kumar Kasera | Nitesh Kasera completed his PhD in Biological and Agricultural Engineering from North Carolina State University in 2022. His research interests include modifying the properties of biopolymers and biochars for environmental applications with an emphasis on structure–activity relationships. Nitesh is a graduate of Applied Chemistry and Chemical Engineering from the University of Dhaka, Bangladesh. He achieved the 2nd runner-up position in the Boyd Scott Graduate Research Competition at the 2021 Annual International Meeting of the American Society of Agricultural and Biological Engineers for a part of his doctoral research. Nitesh will be joining as a Post-Doctoral Fellow in Auburn University's Biosystems Engineering Department starting in August 2023. |
 Youssef Habibi | Professor Youssef Habibi holds a dual PhD in Polymer Chemistry from Joseph Fourier University and University of Cadi Ayyad. He joined the University Mohammed VI Polytechnic (UM6P) as Chair in Sustainable Materials, is a member of the ACS, a Fellow of the RSC, and a Fellow of the IAAM. His research focuses on new bio-derived polymers and (nano)fillers, biomaterials development, and high-performance lignocellulosic nanocomposites, biomass conversion, recycling, and novel analytical tools. He authored over 120 articles, is a Global Highly Cited Researcher, authored over 20 book chapters, edited one book, and has an H-index of 54 with over 22 |
 Stephen J. Eichhorn | Stephen Eichhorn is Professor of Materials Science and Engineering at the University of Bristol, within the Bristol Composites Institute. He has research interests in the physical properties of cellulosic materials, with particular emphasis on Raman spectroscopy. He was awarded the Swinburne Medal in 2020 from the Institute of Materials, Minerals and Mining (IOM3), the Hayashi Jisuke Prize in 2017, and the Rosenhain Medal and Award from IOM3 in 2012 for his research. Professor Eichhorn is also a Fellow of the Royal Society of Chemistry. |
 Alfred D. French | Alfred Dexter French is the son of Dexter French, who pioneered X-ray diffraction studies of starch and carbohydrate enzymology. Alfred is unofficially an Emeritus Research Scientist and serves as Editor-in-Chief of the journal Cellulose. Al is known for his work on molecular modeling of the building blocks of polysaccharides and the connection between the models and observed crystal structures. His recent work has been on powder XRD of cellulose using the Rietveld approach. He has served as the Chair of both the ACS Divisions of Cellulose and Renewable Materials and of Carbohydrate Chemistry & Chemical Glycobiology. |
 Lokendra Pal | Lokendra Pal is the E. J. Woody Rice Professor in the Department of Forest Biomaterials at NC State University. He has over 22 years of global experience in the research and technological advancement of bio-based materials. He has been granted 24 patents. Pal received a PhD in Paper and Imaging Science and Engineering from Western Michigan University. He is the NC State University Faculty Scholar and TAPPI Fellow and received the University Outstanding Teacher Award. His current research is focused on sustainable materials research and advanced manufacturing innovation, utilizing green and sustainable chemistry, engineering, and artificial intelligence/machine learning. |
 Lucian A. Lucia | Lucian Lucia is Professor and Director of The Laboratory of Soft Materials & Green Chemistry at NC State University. He maintains appointments in Biomedical Engineering, Chemistry, and Forest Biomaterials, while serving as faculty in the Fiber & Polymer Sciences program. He received his PhD in physical organic chemistry from the University of Florida with Professor Kirk Schanze, served as a postdoctoral fellow at the NSF Center for Photoinduced Charge Transfer at the University of Rochester with Professor David Whitten, and is a Fellow of the Royal Society of Chemistry. His research includes biopolymer functionalization, green chemistry reactions, and sustainable materials characterization. |
1. Introduction
Societal awareness and governmental regulations are steering the world towards a sustainable bio-economy through optimized exploitation of natural resources.1–5 Thus, biomaterials based on cellulose, the most abundant global biopolymer and one of the most important polysaccharides, are gaining significance and importance.6,7 Cellulose occurs primarily as the major component of plant cell walls in a hierarchy of structures (Fig. 1A). It is present as a high molecular weight linear-chain polymer containing three hydroxyl groups per glucose residue unit.8 An extended shape arises from the preferred torsion angles about the C1–O and O–C4 bonds of the β-(1 → 4)-glycosidic linkage.9 These linear (i.e., unbranched) macromolecules comprise 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 β-glucose residues (the correct descriptor of the repeating unit of cellulose is a glucose residue, which is often confused with anhydroglucose and sometimes with a cellobiose repeating unit).10–13 The shape of the cellulose chains allows them to closely approach each other and their hydroxyl groups form extensive intra- and intermolecular hydrogen bond networks. Further, the most common shape, that of a flat ribbon, also permits van der Waals forces and C–H⋯O hydrogen bonds to form.14 Together, these forces result in various ordered crystalline arrangements (Fig. 1B).15 The hydrogen bonds augment the stiff and straight chain nature of the cellulose molecules. Another critical factor is the presence of the C6 primary alcohol group adjacent to the linkage.16
000 β-glucose residues (the correct descriptor of the repeating unit of cellulose is a glucose residue, which is often confused with anhydroglucose and sometimes with a cellobiose repeating unit).10–13 The shape of the cellulose chains allows them to closely approach each other and their hydroxyl groups form extensive intra- and intermolecular hydrogen bond networks. Further, the most common shape, that of a flat ribbon, also permits van der Waals forces and C–H⋯O hydrogen bonds to form.14 Together, these forces result in various ordered crystalline arrangements (Fig. 1B).15 The hydrogen bonds augment the stiff and straight chain nature of the cellulose molecules. Another critical factor is the presence of the C6 primary alcohol group adjacent to the linkage.16
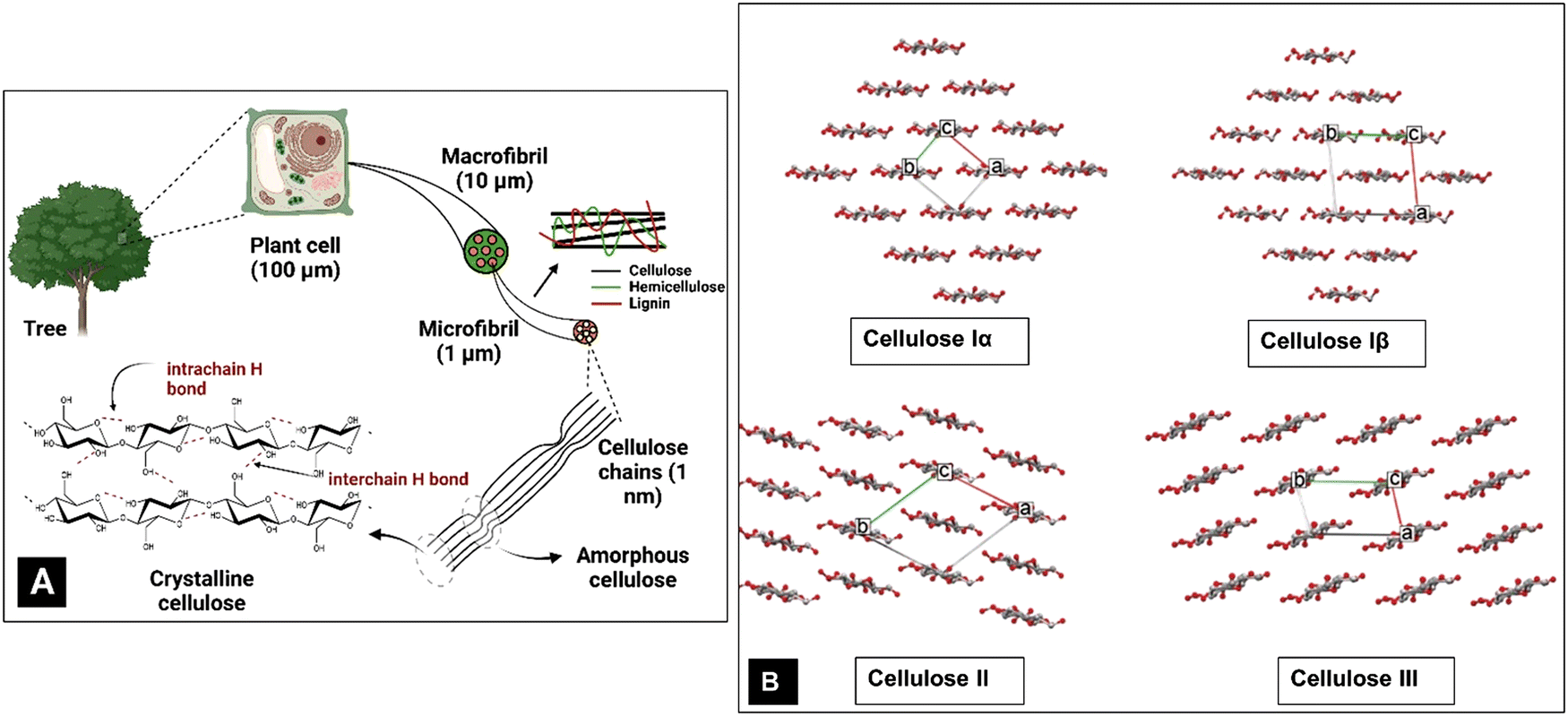 | ||
| Fig. 1 (A) Hierarchical structure of cellulose and (B) different crystalline forms of cellulose. | ||
The forces also organize the chains in parallel arrangements of crystallites, which are the elements of the secondary structure of native cellulose fibrils and cellulose fibres.11 Cellulose fibres are typically composed of nanofibrils with lengths of several micrometers and diameters ranging from 2 to 20 nm, where the cellulose crystallites are linked to each other by amorphous cellulose domains.11 Eight different crystalline allomorphs, referred to as cellulose Iα, Iβ, II, IIII, IIIII, IVI, IVII and X, have been identified using different techniques, with Iα and Iβ being the naturally occurring or native crystalline forms. Cellulose II has also been discovered in rare examples of algal17,18 and bacterial cellulose19,20 but more typically results from treatments with strong NaOH in the laboratory21,22 (mercerization) or dissolution and regeneration, as in the industrial production of rayon. The other forms are mostly of scientific interest, although there is commercial interest in cellulose IIII which results from treatment of cellulose I with amines.
Carl von Nägeli first discovered the ordered arrangement of cellulose fibrils in 1858 using polarized light microscopy,23 which was later confirmed by X-ray diffraction (XRD) patterns of ramie24 in 1926. This was more than a decade after the first diffraction pattern of cellulose was recorded in 1913, just a year after the technique itself was discovered.25–27 The 1926 paper proposed the polymeric nature of cellulose, but had 1,1 and 4,4 linkages instead of the 1,4 linkages known today. A correction to the linkages soon followed along with changes in the unit cell axes. This resulted in a structure with the correct basic nature, but left un-resolved issues of whether the two molecular chains of the unit cell had parallel or antiparallel packing. Another question left to the 2000s era was the orientation of the rotatable C6–OH group. The cellulose molecules aggregate to adopt crystalline fibrils with well-defined shapes, sizes and crystal allomorphs.26 The nature of these crystal structures, their interaction with the surrounding molecules and their further assembly into higher order structures to form long slender fibres profoundly affect the overall properties of cellulosic materials. Therefore, an understanding of cellulose crystallinity is one of the most important requisites because crystallinity affects the physical, mechanical, chemical, and morphological properties of cellulose and its surface interaction with the surroundings and hence its use in a wide range of applications including, but not limited to, biomedicine, textiles, and electronics.28–34
The crystallinity index (CI) is commonly used to measure the relative amount of the crystalline fraction of cellulosic materials and to quantify their modification following a variety of physicochemical and biological treatments.8,31,35–40 This parameter is based on a two-phase model of the material – namely a crystalline phase and an amorphous phase. This may or may not be the most correct understanding. An alternative would be to consider crystallinity based on the size, not perfection, of the crystals in the sample. Smaller crystallites would be more like amorphous material in the sense that they have a larger proportion of surface molecules than large crystallites and therefore faster reactivity. Rather than having just two distinct phases, amorphous and crystalline, the actual situation could be more of a continuous variation in the molecular order. In naturally deposited cellulose structures, this is a somewhat less attractive view, as the biosynthetic mechanisms are thought to generally create the fibrils very repetitiously.
The CI (often called CrI) is simply the percentage of the crystalline phase that is present. In native cellulose, the non-crystalline or amorphous materials are thought to be located on the surfaces of the cellulose elementary fibres as well as between the crystalline regions as indicated in Fig. 1A. The primary evidence for the interruption of the crystalline regions along the elementary fiber axes is the “leveling-off degree of polymerization”.41 Namely, hydrolysis in 2 N HCl at 80 °C rapidly reduces the molecular weight until it stops dropping so rapidly. Also, the crystallite length is much shorter than the initial molecular weight.42 Some NMR studies have indicated that the surface molecules on the cellulose crystallites have different structural details from those in the interior, and the surface molecules are expected to be less ordered based on computational molecular dynamics studies.
Several techniques have been used to measure the CI, including X-ray diffraction (XRD),21,26,36,43–51 nuclear magnetic resonance (NMR) spectroscopy,44,52–57 Fourier transform (FTIR) infrared spectroscopy,44,45,49,55,58,59 Raman spectroscopy,55,60 sum frequency generation (SFG) vibration spectroscopy,61,62 differential scanning calorimetry (DSC)63 and enzymatic cellulose binding molecules (CBMs).64 However, a common feature of all these techniques is that the crystal disorder and complex heterogeneous nature of the components in lignocellulosic biomass lead to issues in interpretation for most established methods estimating cellulose crystallinity.26,60
The amorphous and crystalline structures in cellulose have been found to govern its chemical and enzymatic reaction kinetics.65–67 Therefore, the measurement of CI has become crucial to ascertain cellulose structure, which consequently would help to design experimental conditions to achieve the desired properties of the final products for different applications. In light of this, our present review assesses the available techniques to measure cellulose crystallinity and establishes basic governing principles of XRD, NMR, IR, Raman, SFG spectroscopy, DSC thermogram, and CBM methods and their applications for cellulose structure characterization. The first part performs an individual analysis of each technique, including working principles, capabilities, and limitations. The latter section provides a comprehensive comparison among techniques and elaborates on the idea of the lack of an absolute method for measuring cellulose crystallinity. It also discusses the use of diffractograms and spectroscopic techniques complementing each other to measure cellulose crystallinity. In all cases, the review introduces fundamental principles and the application of different techniques for the measurement of cellulose crystallinity. These enabling techniques can help us better evaluate the structure of cellulosic materials, but they also have their limitations and pitfalls in applications if not undertaken with judicious care and attention.
2. X-Ray diffraction (XRD)
2.1. XRD fundamentals
The phenomenon of diffraction by crystals is based on the concept that a crystal effectively has infinite repetition of a small geometric unit in three dimensions. A small parallelepiped, the unit cell, contains all the atoms and their three-dimensional arrangement to permit the crystal to be generated along three linear axes by simple translation. It is not required to have intact atoms or molecules within the unit cell. For many crystalline substances, the atoms and molecules only become whole when the unit cell is repeated. In Fig. 1B, for example, the unit cells, indicated by the white, red, and green lines, could be cut out from the paper and moved along the unit cell boundary lines to create the rest of the crystal. A simple inorganic or metallic unit cell may only have atoms at the corners of the unit cell, but organic molecules generally will not be able to have such a simple arrangement. These unit cells have, over the years, been described under several conventions as to whether the angles among the axes at the cell origin are obtuse or acute and which axis, x, y, or z, should correspond to the molecular axis in the case of a polymer like cellulose. Our review will consider the z-axis (the c unit cell dimension) as the molecular axis, with the shorter of the other two axes being x, and all three axes being related by the “right-hand rule”. Crystal symmetry also plays a role; the triclinic P1 cell used herein is as published by Nishiyama et al. for cellulose Iα.68 The remaining space groups for neat cellulose are all monoclinic P21 (P1121 indicates the unique c-axis) with the obtuse monoclinic angle γ being between the a- and b-axes.In the earliest decades of diffraction crystal structure studies, it was not properly understood that a polymer could fit in the small unit cells indicated by the diffraction data. For example, the longest dimension of a cellulose unit cell is approximately a nanometer, while a polymer of the glucose molecule would be many times that long. The quandary was resolved by understanding that the observed unit cell dimensions along the molecular axis were the predominantly repeated unit and there were so few ends, that the additional H atom and OH group at the ends of the molecule would not be detected by the experiment.
Once a proper context of the unit cell is established, the concept of crystal planes can be discussed. These planes are useful for the identification of the various peaks on a diffraction pattern. For example, the (200) plane intersects the a-axis at one half its length; the “2” in “200” represents the reciprocal of one half. The reciprocals of the other two “Miller indices” are infinity, indicating that the (200) plane never intersects the b- or c-axis. Another important plane in cellulose crystallinity is often the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) plane, which intersects the a-axis one unit away from the origin of the unit cell. It also intersects the b-axis at one unit away in the negative direction from the origin (Fig. 2). The (
0) plane, which intersects the a-axis one unit away from the origin of the unit cell. It also intersects the b-axis at one unit away in the negative direction from the origin (Fig. 2). The (![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 10) and (10) planes are equivalent, but the convention is to list the negative Miller index value on the b term. Another important plane in cellulose, the (110) is shown in Fig. 2.
10) and (10) planes are equivalent, but the convention is to list the negative Miller index value on the b term. Another important plane in cellulose, the (110) is shown in Fig. 2.
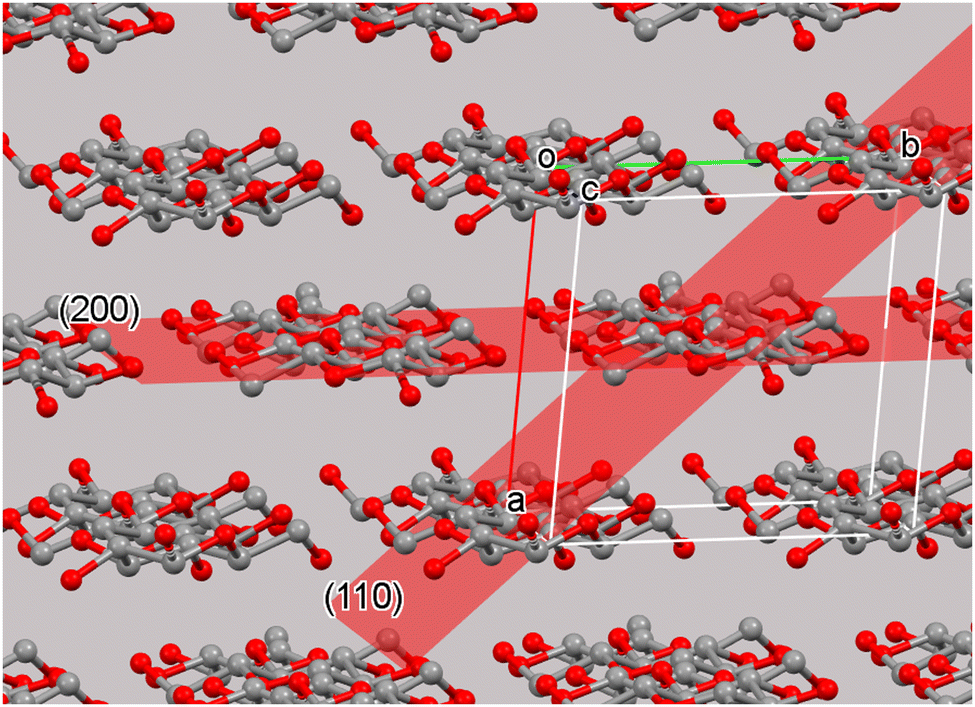 | ||
| Fig. 2 The cellulose Iβ unit cell and surrounding molecules with the crystal (200) and (110) planes. The planes extend the entire dimensions of the crystal and are found for each unit cell. The chemically repeated unit of cellulose is the glucose residue. In neat cellulose crystals the two-fold (or pseudo-two-fold) symmetry results in a crystallographic repeat distance that includes two glucose residues. Other cellulose structures have different repeat distances.12 | ||
X-Ray diffraction (XRD), being a powerful non-invasive analytical technique for characterizing crystalline materials, can provide information on structures (3-D spatial representations of the molecular structure), phases, crystal orientations, crystallinity, and other structural parameters such as average grain size, strain, and crystal defects. The very first applications of XRD techniques used polychromatic X-rays which produced ‘smeared’ diffraction patterns.27 This issue was later resolved by using monochromatic sources that resulted in distinct diffraction peaks from what are termed ‘planes of reflection’ in Fig. 2. Although the peaks result from diffraction planes, the Miller indices for planes are enclosed in parentheses, but the peaks of a diffraction pattern are not. That convention is often ignored, however. Distinct XRD peaks are produced by coherent interference of a monochromatic beam of X-rays which are scattered by atoms in periodic lattice planes in a sample.69 The atomic positions within the unit cell contribute to the peak intensities – often called the lattice and molecular transforms. It is also important to note that the diffraction pattern observed is then a combination of two transforms – the lattice and the molecular transforms. Therefore the intensities contain information about the relative atomic positions and thus about the shapes of the molecules at those lattice positions. Only crystal lattices can have planes that in turn have Miller indices; it is never appropriate to refer to the Miller indices of amorphous materials.
The diffraction of X-rays by crystal planes shows how lattice spacings are formed in the plane using Bragg's law (nλ = 2dhklsinθ) as illustrated in Fig. 3A. Here, n is an integer called the order of the reflection, λ is the wavelength of the X-rays used, d is the characteristic spacing between the crystal planes of a given specimen and θ is the incident angle of light (X-ray beam). By measuring the angles, θ, under which the constructively interfering X-rays leave the crystal, the interplanar spacings, dhkl, of these planes can be determined. In the case of cellulose powder diffraction, however, the peaks from the various planes are mostly overlapped and such determinations are more difficult. Fig. 3B shows the schematic of a typical XRD Bragg-Brentano (reflection) set up used for a powder diffractometer, where X-ray diffraction results from planes that satisfy the Bragg condition.
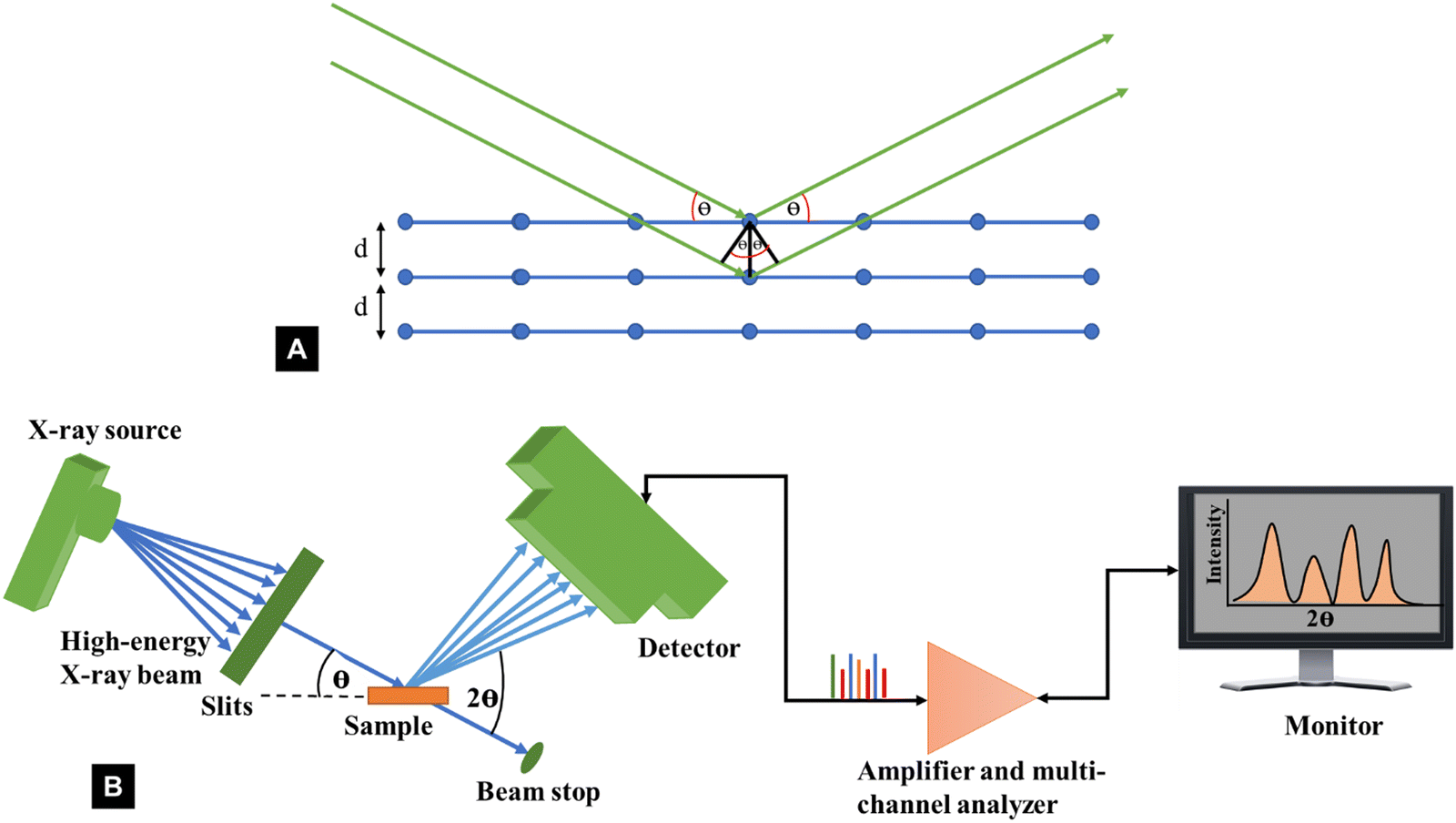 | ||
| Fig. 3 Schematic representation of (A) Bragg's law and (B) XRD instrumentation set up typically used for powder diffractometer determinations of the crystallinity index of cellulose. | ||
Again, in the first forty years of X-ray diffraction studies, the X-ray peaks were detected on photographic film. If the sample was a bundle of aligned fibres, the recorded pattern was a two-dimensional array (a “fibre pattern”) that furnishes more information about the structure. See, for example, the diffraction patterns for tunicate cellulose or highly crystallized cellulose II published by Nishiyama, Langan, and Chanzy and by Langan, Nishiyama, and Chanzy.56,70 For many samples, however, especially nanocellulose and wood pulp, it is more convenient to use powder diffraction. Unfortunately, that results in a substantial loss of information, particularly as it is important to capture as many intensities as possible to get an accurate structural analysis. The XRD part of our review is focused on powder diffraction with a powder diffractometer, which is distinct from flat plate or 2D diffraction studies. (2D data of various types can be converted to 1D data typical of powder diffractometers with software.)
Since the discovery of XRD, its non-invasive nature and relative accessibility have made it a ubiquitous technique for studying the crystallinity of cellulosic materials.2,15,64,71–77 The diffraction pattern for cellulose displays several intensities typically superimposed on a broad background. The “broad background” is composed of environmental scatter (air scatter, sample holder, shot noise,78etc.). There is also inelastic or Compton scattering that goes into the background as well as other wavelengths that get through the filter that removes K beta radiation. However, XRD requires a certain minimum crystallite size to consider a fraction in a solid as being crystalline, in which the width of the peaks in the diffractograms is known to be inversely proportional to the lateral width of the crystallites.79,80 Thus, in cellulosic materials, the crystalline peaks get broader for smaller sized crystallites.69 The material's surface also affects the crystallinity in XRD analysis. Though the surface atoms have greater mobility, and a lower degree of order compared to the bulk of the crystallites, they are captured as crystalline material during the XRD measurements. This is also the case for minor defects within crystalline regions that are too small to be detected as regions of molecular disorder.71 So, it is worth noting at this point that not all disorder can be accurately determined for cellulosic samples, if the scale of this disordered material is sufficiently small.
As previously mentioned, the crystallinity index (CI) has been used to describe the relative amount of crystalline material in cellulose. It is calculated as the contribution of the crystalline portion of the material relative to the combined areas of the crystalline and amorphous regions.55 Several XRD techniques have been proposed to determine the CI. In this sense, comparing literature data for cellulose crystallinity is challenging due to the lack of standard methods. However, the most widely applied XRD methods for studying cellulose crystallinity are Segal peak height, peak fitting (also referred to as peak deconvolution), and amorphous subtraction.15,71 However, there is a substantial amount of Rietveld crystallinity work being currently undertaken which is very attractive because it can consider all of the inputs to a diffraction pattern in a physically relevant manner. Although intellectually satisfying, it is however noted that the Rietveld method has been widely applied to inorganic materials (e.g. ceramics and metals) that make better crystals and provide more diffraction data. Even in their application to these materials that form much larger crystals, issues regarding “accuracy” of fit, and thereby calculations of peak intensity do occur. The theoretical implications behind each method, as well as limitations, are discussed in the following sections. Comparing the different techniques will elucidate how CI values change with different methods and enable an understanding of the simplest and the most widely used XRD method to measure the CI of cellulosic materials.
| Crystallinity index (CrI) = 100 × (I200 − IAM)/I200 | (1) |
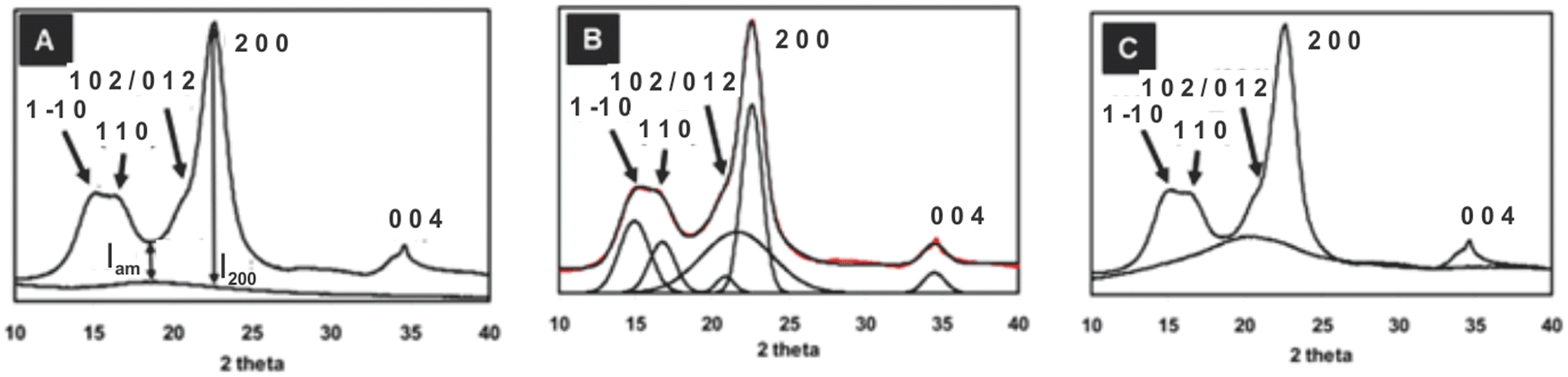 | ||
| Fig. 4 The XRD pattern for Avicel PH-101 showing three methods of crystallinity determination: (A) background-corrected Segal peak height method, (B) peak fitting (peak deconvolution) method using Voigt functions for fitting five crystalline peaks and the relatively broad amorphous contribution,14,66 and (C) the amorphous subtraction method where a ball milled amorphous sample is used as a reference15,71 (adapted from and used with permission from ref. 15. Copyright Springer-Nature, 2010). Note: the authors of the original drawing in ref. 15 opted to use the convention of b as the unique axis, rather than the typical c-axis convention for polymer diffraction that is used here. Thus, the original peak labels were 101, 10, 021 and 120, 002, and 040. | ||
A major advantage of the Segal method is its simplicity. It could be easily used on the paper chart output from the early diffractometers; no computers were needed (or otherwise available). However, there are numerous criticisms of the Segal method in the literature. Despite its simplicity, some researchers misapply the Segal method by taking the 10/110 peak (Fig. 4A) height to represent the amorphous fraction, whereas it is the trough height that must be used. Of course, that is not Segal's fault. A major intrinsic fault is that it assumes that all the intensity at the 18° point (modern work finds the minimum closer to 18.6°) is from amorphous scattering and none of it arises from the overlap of the broad peaks that result from the small crystallite sizes of cellulose. Because of peak overlap, calculated patterns for perfect but small crystals (e.g., a size similar to cotton crystallites) would only be 90% crystalline, and an experimental sample will never give a value of 0% crystallinity because I200 and IAM would have to be identical. Another little-mentioned defect in the assumptions is that samples can suffer from non-random orientation of the crystallites, based on the sample preparation and placement in the X-ray beam. Situations with preferred orientation of the crystallites place a particular plane with respect to the incident X-ray beam and therefore exaggerate those intensities, changing the ratio of I200 to IAM despite identical crystallinity. Preferred orientation is especially prevalent in samples of bacterial cellulose as well as nanocellulose that is dried in various ways.
High-end diffractometers often have spinning sample holders to assist with randomization of the crystallite orientation, but they will not generally solve all such problems. For example, samples of fibers cut to very short lengths still tend to lie down in the plane of the sample holder. Spinning the sample will make sure that the pattern does not suffer from a secondary orientation of the fibres. The fibres will be directed uniformly in 360°. However, a third type of orientation can still arise. Namely, the crystallites might be rotated about their long axes in a particular orientation. Spinning sample holders do not deal with the first or the third type of preferred orientation. It is good practice to do this anyway for all samples since the beam size may sample regions of a supposedly random sample, but local orientation effects may occur. This approach is not only valid for the Segal method, but also for all powder XRD approaches. Of course, complete characterization of the sample would include information on the extent of preferred orientation. In any case, such effects can result in invalid crystallinity measurements if not taken into consideration, and the Segal method cannot easily deal with them.
Segal's method is empirical and was not developed to compare samples of different nature, but rather to quantify differences among a single set of samples.15,71 Therefore, Segal's method is better suited to study relative changes in crystallinity resulting from exposure of a sample to different treatments and not for estimating absolute values of crystallinity and amorphous fractions in cellulosic materials.2
As many as five crystalline peaks indexed 10, 110, 012/102, 200, and 004 are often considered while using Gaussian and Voigt fit functions. The peaks indexed 10, 110 and 200 are clearly visible in most cellulose samples, whereas the 012/102 overlapped reflection might appear as a shoulder; however, these peaks might overlap with each other and could not be resolved so distinctly for some samples, e.g., bleached eucalyptus.84 Another diffraction peak is often visible at approximately 34.5° which gives a somewhat broad peak indexed as 004 although it is a composite of several neighbouring peaks.47 The Voigt (often the pseudo-Voigt) function has been used to fit the five peaks generated in the diffractogram of cellulose samples. Fig. 4B is a typical example of such a fit. However, four crystalline peaks indexed 10, 110, 200, and 004 might be used when a Lorentzian fit function is applied since the 012/102 is difficult to resolve as it appears as a shoulder in the diffractogram. The crystallinity index is calculated from the ratio of the area of all crystalline peaks to the total area using the equation
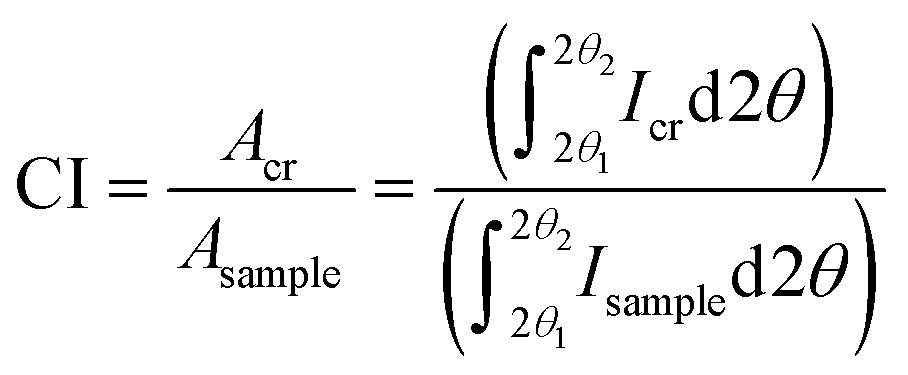 | (2) |
Because more peaks are considered than in the Segal method, a possible advantage of the peak fitting method is that it could more safely be used to compare the crystallinity index of different samples. Concern has been expressed that the peak area method does not account for all the factors that contribute to peak broadening, including crystalline disorder, crystallite size and non-uniform strain within the crystal.84 Other work had indicated that crystallite size is the dominating factor.87 Another problem is that the pattern cannot be resolved into many narrow diffraction contributions as the cellulose intensities are very broad with overlapping peaks.84,88,89
While the Segal method has an intrinsic fault, i.e., assuming that all the intensity at the 18° intensity minimum in Fig. 4A is from amorphous scattering, problems with peak deconvolution are more practical. The first is that there are many smaller peaks that are not considered. This is shown very clearly by patterns calculated assuming crystallites large enough to give very sharp peaks (see Fig. 5).47 When the size is 100 nm or more the peaks that contribute to the crystalline intensity become visible but when the modeled crystallites are realistically small, these peaks blend into what is often mistaken for background or amorphous scattering. Thus, these smaller peaks in the higher 2θ range give a reasonable basis for that broad but low intensity. No observed or calculated intensity is shown in that area of Fig. 4B despite its presence in 4A. This indicates that the intensity in that region has been subtracted as background, as done in ref. 82. In that work, the authors mention that “a detailed and rigorous treatment would include a full description or the amorphous background.” As mentioned above, it is important to not equate “amorphous scattering” with “environmental and intrinsic background.” An intrinsic limitation of peak deconvolution is that there is no crystallographic science used to link the intensities of the various peaks.
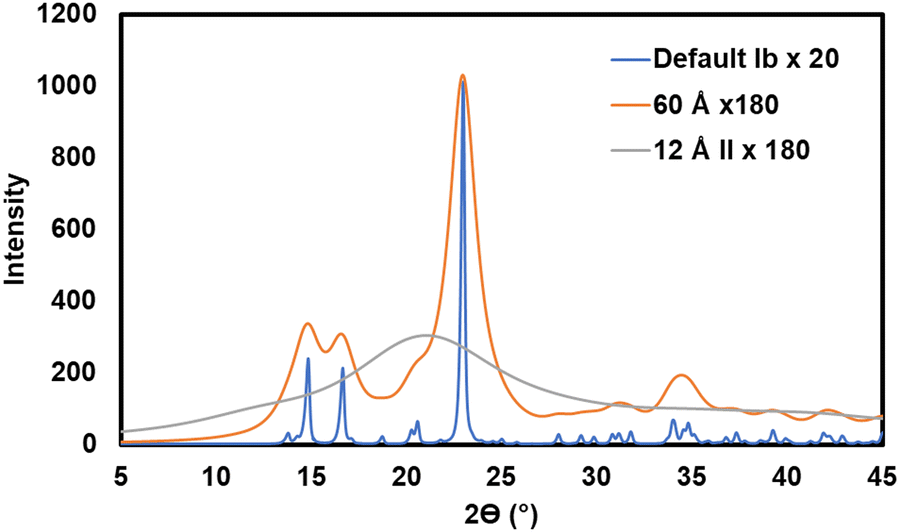 | ||
| Fig. 5 Calculated diffraction patterns. The blue pattern for cellulose Ib represents the default parameters in the MAUD90 Rietveld software including a nearly infinite crystal size of 1000 Å (the incident X-ray beam has been scaled by a factor of 20 to reach 1000 counts). To prevent excess noise in experimental data, at least 2000 counts should be accumulated at the maximum intensity for any report of the XRD pattern, with more being helpful for Rietveld work. (10000 to 100000 is not excessive with a modern XRD detector.) The orange pattern, which represents a moderately large cellulose crystal of 60 Å diameter, was scaled by a factor of 180. The grey line is a calculated pattern for cellulose II, except that it has a very small crystallite size of 12 Å, representing amorphous cellulose. It was scaled for the same incident beam intensity as the orange pattern. The area under the amorphous pattern is 88% that of the 60 Å pattern. | ||
However, if the intensity of the selected amorphous standard does not match the experimental amorphous contribution, then the fitting is allowed by some workers to exceed the experimental intensities marginally at some scattering angles to improve the fit. The area under the amorphous contribution and the total area of the samples are measured, and the crystallinity is calculated using the equation
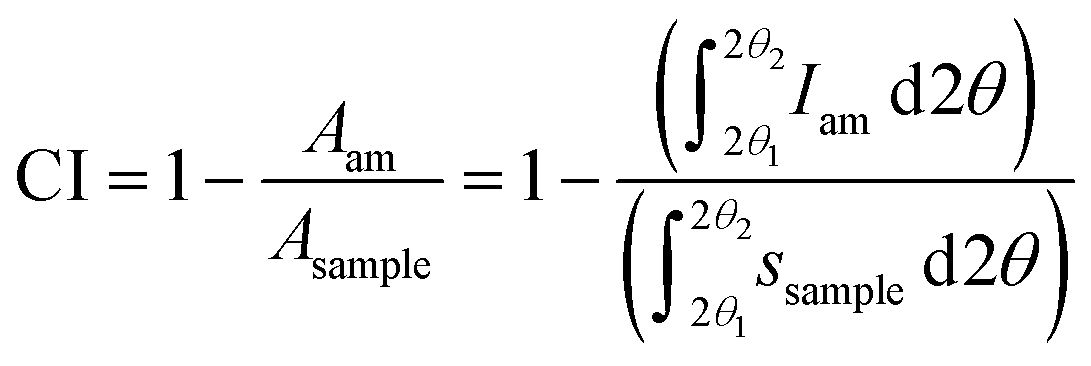 | (3) |
A wide scattering angle from 2θ1 ∼ 13° to 2θ2 ∼ 50° is used to calculate the area. The selection of an ideally amorphous standard is very challenging which limits the use of this method.15 Moreover, amorphous subtraction has the same limitation as Segal's method, which is due to assigning the intensities to the amorphous fraction without considering the presence of overlapping peaks. Nonetheless, ball-milled cellulose, regenerated cellulose, and xylan or lignin powder are widely used among several standards to fit in the measured amorphous region.40,51 As seen in Fig. 4C, the experimental data above 25° 2θ has been considered to be amorphous, except for the point near the 004 peak. A test that should be applied to results from both the deconvolution and the amorphous subtraction results is to plot just the pattern that results from what is considered as the crystalline material. Here, the subtraction would just be the amorphous from the experimental, and for deconvolution, it would be the sum of all the crystalline peaks at each 2θ step. If the data had been corrected for environmental background, then the removal of amorphous data from the pattern should result in an ideal pattern that can be compared with ideal patterns for crystallites of a given size. Alternatively, calculated ideal patterns can be analyzed by the selected method to check for 100% crystallinity.
There are several lessons in Fig. 5, which shows, in blue, the ideal pattern that might greet the user at the start of the Rietveld calculation. The first is that there are many small peaks in that blue pattern that blur out when the crystallite size is diminished to match an experimental pattern. Another is that a pattern with lower peak intensities is not necessarily less crystalline in terms of crystal perfection, but in this case, it took nine times as many incident photons for the 200 peak on the 60 Å pattern to reach the same height as the 1000 Å pattern.
Different Rietveld software programs, widely available in free and commercial products, have different philosophies for handling an amorphous phase. One strategy uses a very small crystal of the same material as a model. For cellulose, it seems at present that a small crystal of cellulose I is not the best approach. Instead, both cellulose II and cellulose IV structures are used. Each can be justified. The cellulose II model is supported by the resemblance of patterns calculated from 12 Å crystallites (Fig. 5) to the ball-milled or quickly regenerated samples that are regarded as amorphous models. This is not arbitrary. Amorphous samples share the dominant orientation of O6 with cellulose II and soaking of amorphous material in water can result in cellulose II.94 Additionally, nanocellulose samples can have cellulose II that is not visible on the diffraction pattern. However, if the size of the model crystal is allowed to change during refinement, it can rise to 30 Å, indicating the presence of cellulose II. The cellulose IV model is supported by the finding that the primary cell wall is often composed of that allomorph. Another approach is to use different functions that result in a pattern resembling ball-milled or regenerated cellulose, or to include an additional arbitrary broad peak as needed.92 This is currently the frontier area of research.
The correct analysis of texture (crystallite size and orientation) also affects the CI (percentages of the crystalline and amorphous phases) of cellulose samples.95 The Rietveld method optimizes the CI measurement by using a curve fitting method, where it considers all the variables including the lower intensity peaks as well. Thus, Rietveld is a complete peak area fitting method that does not have the limitations of the simple methods discussed above. It is worth noting that this method is well-established for more crystalline inorganic and metallic materials. More pertinently, the issues with obtaining an accurate fit to diffraction data for these more crystalline materials are also well-known, even when these materials’ patterns appear to have a relative absence of pronounced amorphous backgrounds. Peak fitting itself is fraught with difficulty when it comes to diffraction patterns, especially when signal to noise and backgrounds are considered. This is a subject which has been noted in the literature but is often ignored in analysis. Another issue is on the consensus of what parameter to use to quantify the degree of fit, and the fact that an overinterpretation of small values of commonly used parameters such as the R-factor can lead to erroneous conclusions. Having reliable structural determinations on a database is key to this approach, and without due care in using these database structures, mistakes can easily be made, e.g., the use of the wrong allomorph for a native cellulose, and the fact that lattice distortions (seen as shifts in peak positions) can occur for woody plant materials versus more crystalline forms of cellulose. An over-reliance on a good fit between the refined model and the experimental data can lead to the wrong conclusions.
The precision of the Rietveld analysis has however been improved by the Cellulose Rietveld Analysis for Fine Structure (CRAFS) software which uses a crystallographic model along with a computational algorithm having only the variables which are deemed to be most practical for cellulose studies with the limited amount of information. The other advantage is that CRAFS as well as some other Rietveld software, can also handle 2-dimensional data where the 2-D fibre patterns, as mentioned above, can yield much more information than the 1-D patterns discussed herein.95,96 The advantage of 2-D patterns is shown in ref. 90 that compares both 1-D and 2-D calculated patterns. The spots on a 2-D pattern fall on different layer lines so that they are better resolved, providing a greater number of independent data points. That work also compared the 1-D patterns for both ideal crystallites of different sizes and their counterparts that had been subjected to energy minimization and molecular dynamics studies and therefore they lost their exact periodic order. As a result of this approach, it has been observed that many of the diffraction peaks formerly credited to background scattering or amorphous cellulose are actually coming from adjacent crystalline peaks.47,92 However, it is still early to say that Rietveld can produce the most accurate and reliable crystallinity index value for a broad range of cellulose allomorphs due to its capacity to consider all the crystalline peaks. However, in principle, it has much to offer if better standards are adopted and if the pitfalls encountered for more regular materials are considered. It is fair to say that the approach is in its infancy for cellulose compared to more crystalline materials, and much work is needed to ensure that robust approaches are adopted without an over-reliance on erroneous underlying assumptions.
The biggest problem with Rietveld determination is the limited amount of experimental data. Based on the structure in Fig. 6b, the variation of atomic coordinates for cellulose is futile, and simplified models of the crystallite size anisotropy must be used. Unlike single crystal diffraction studies, the ratio of data to variable parameters is not clearly defined for Rietveld work on cellulose diffraction patterns.
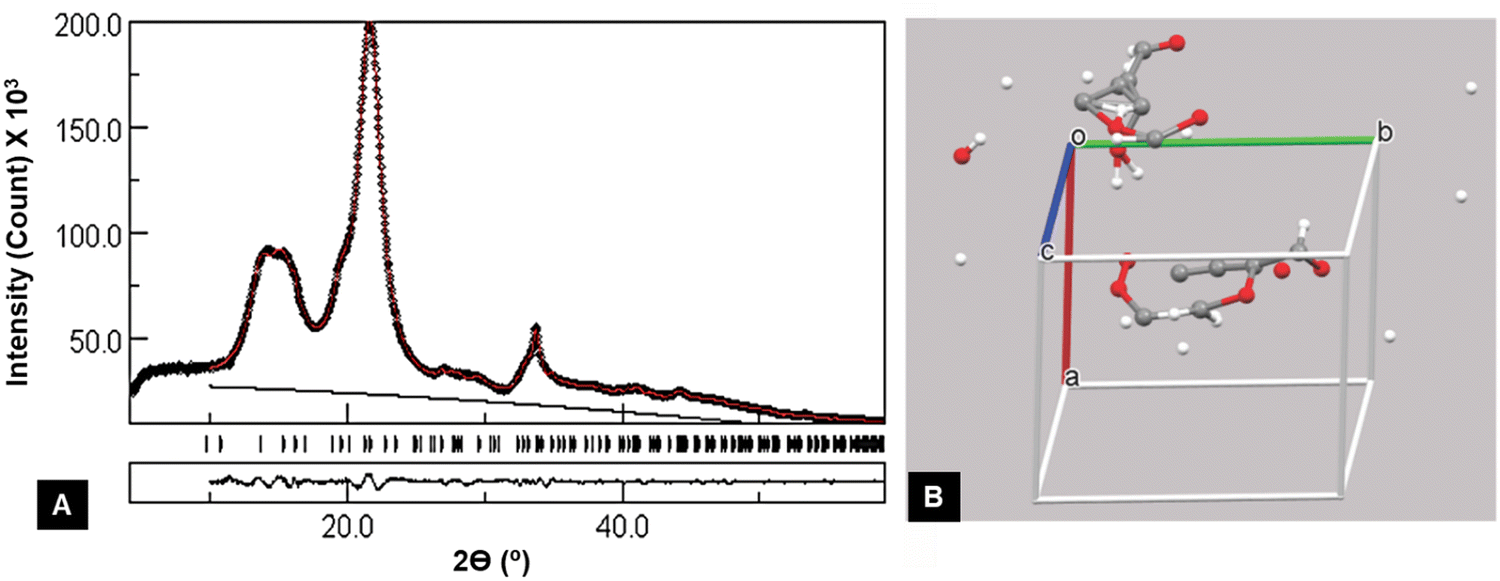 | ||
| Fig. 6 Example of a badly underdetermined structure, i.e., too many variables for the available data, that gave an excellent agreement between the observed and calculated intensities. (A) Output from the MAUD Rietveld software. The black circles represent the experimental data, and the red line shows the fit line for the adjusted cellulose Iβ pattern. The black line is the added background needed despite the previous subtraction of environmental background obtained with a blank run (see Fig. 7a). The black and red lines show that data before 2θ = 10° were not included in the analysis. The short vertical lines to the right of “Tunicate” indicate all of the peaks that contributed to the red calculated pattern. Output from the MAUD Rietveld software. (B) However, the “refined” structure with variable parameters for the unit cell dimensions and atomic coordinates resulted in a physically meaningless underlying structure. Unit cell and contents from the MAUD refinement that included atomic positions, unit cell dimensions, background, preferred orientation, and anisotropic crystallite size, displayed with the Mercury program. The 147 variables were far too many for the limited data and the results were non-physical. | ||
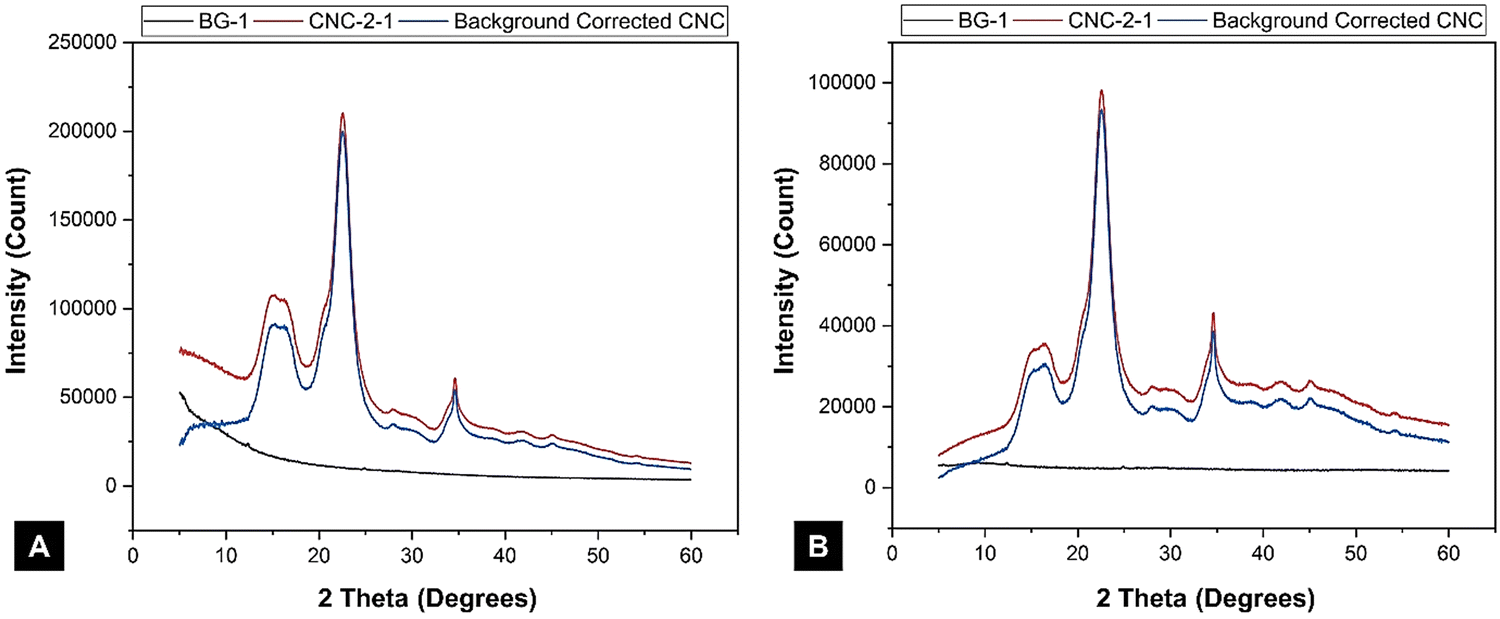 | ||
| Fig. 7 Effect of the (A) fixed slit and (B) variable slit X-ray diffractometer on the background curve and the resulting corrected intensity of a cellulose nanocrystal (CNC) sample. Rietveld analysis software typically assumes a fixed-slit diffractometer configuration. These fixed-slit data were converted from variable-slit data with the Malvern-Panalytical HighScore software. | ||
2.2. Comparison of different XRD methods
XRD is a commonly used method to characterize the crystallinity of cellulosic materials. However, each of the above-discussed methods has some limitations. One of the limitations associated with the methods is the dependence of the crystallinity index on the crystallite size.71 For fully crystalline models, all the methods have been found to show a positive linear correlation with the crystallite size.71 Segal CI calculations on model patterns showed a very strong, but non-linear correlation.48 However, all the methods showed variation in the crystallinity index as shown in Table 1. The peak fitting method exhibits the highest variation (from the ‘Maximum Difference’ parameter), while the Rietveld method the least variation.71| XRD method | CI mean (%) | St. dev. (%) | Max. difference (%) |
|---|---|---|---|
| Maximum difference is the difference between the highest and lowest crystallinity values. | |||
| Segal peak height | ∼93 | 3.7 | 10.3 |
| Peak fitting | 77–84 | 3.5 | 13.3 |
| Amorphous subtraction | 80–82 | 4.0 | 10.8 |
| Rietveld method | 0–100 | N/A | N/A |
| Ideal values | 100 | 0 | 0 |
From Table 1, it is also observed that the crystallinity index measured using all the methods is less than the ideal value. The crystallinity index measured using the Segal method is higher than that of the other methods, and this trend has been reported for several different cellulosic samples, as shown in Table 2.15,51,71,97
| Sample | Crystallinity index (%) | Rietveld method | ||
|---|---|---|---|---|
| Segal peak height method | Peak fitting method | Amorphous subtraction method | ||
| Juniper | 34–38 | 19–22 | 22–23 | 36 |
| Moso bamboo | 45–46 | 22–24 | 20–21 | 28 |
| Hemp | 75 | 40 | 49 | 60 |
| Nata de coco | 77 | 44–47 | 49–52 | 61 |
| Spruce-pine | 75–77 | 49–50 | 42–48 | 58 |
| Avicel PH-102 | 76–82 | 49–53 | 60–65 | 67 |
| Cotton linter | 85–87 | 55–56 | 62–67 | 72.1 |
The Segal method yields the highest values for crystallinity indices and has been reported to show a mean error of over 20% for crystallinity values for known samples.43 In contrast, the crystallinity indices obtained by Rietveld and peak fitting methods for different cellulosic species have been shown to be consistent.99 The high values of the crystallinity index of the cellulosic samples obtained by the Segal method are thought to be due to several reasons. The main reason is that the peaks associated with the amorphous regions in the XRD are very broad, and a simple height comparison ignoring the variation in peak width cannot provide a correct estimate of the crystallinity index.84 Besides, the trough height also varies which is not considered during the CI measurement. Therefore, when the crystallinity index is measured using the Segal method without considering the above mentioned discrepancies, it leads to overestimation of the intensity of the crystalline region and hence the total crystallinity index.100–104
The calculated degree of cellulose crystallinity can also be affected by the orientation of samples, which can be assessed by measuring the same sample using various diffractometer geometries, as well as the rotation of the samples already mentioned. For example, it was found that when a medium-density balsa sample was measured with a different geometry, the symmetrical transmission geometry produced a crystallinity index of 48%, whereas when the symmetrical reflection geometry of the same sample was used, the measured crystallinity index was almost 100%.2 This was due to optimal scattering of the sample's 200 reflection, which resulted in overestimating the contribution from the scattering pattern.105 Thus, a symmetrical transmission geometry yielded a higher Segal crystallinity index for balsa. Another issue is the presence of “environmental background” which is independent of the sample and tends to overlap with the diffraction pattern of the amorphous scattering contributed by a low intensity region. Therefore, the background and amorphous scattering must be separated to get a more reliable and meaningful crystallinity index value.39
Despite different techniques resulting in different crystallinity indices of the cellulosic samples, a good linear correlation (r2 ≥ 0.9) has been reported between samples when their relative difference in the crystallinity index was large.71 The Segal method showed a linear correlation with other methods when a single sample set was measured; nonetheless, the linearity disappeared when various samples were measured.71 This is thought to occur because the Segal method is an empirical method used to study relative changes within a single sample set. The peak fitting method yields lower crystallinity values compared to the Segal method since the peak fitting method can give unrealistic amorphous contributions if the fitting limits are too strict. Amorphous subtraction would also yield a crystallinity index founded on an erroneous assumption if an appropriate standard amorphous model cannot be used since the standard model cannot go beyond the sample intensity even if their intensities do not match in some regions of the selected scattering angle.71 To overcome this limitation, occasionally amorphous fitting is used, allowing mismatch between the intensities of the standard model and the sample. But due to different factors like the amorphous model, scattering angle, background corrections, and subtraction, the amorphous fitting model cannot be used to determine absolute values, i.e., whether a sample is 100% crystalline or not.
The Rietveld method is emerging as a useful tool to measure the crystallinity index of cellulose samples by XRD. The CRAFS software analyzes the 2D diffraction pattern and generates a few data such as unit cell parameters, crystallite size, crystalline index/intensity, and crystallite orientation. This method could overcome the two major limitations, such as overlapping of diffraction peaks and preferred crystallite orientation, which were issues with the other three techniques.95 The increased accuracy was possible because the Rietveld software usually can adjust the model pattern for preferred orientation of the cellulose, and some software also can compensate for crystallites that are anisotropic (they have different dimensions along different axes). This allows the Rietveld method to result in a plausibly more reliable determination of crystallinity than the Segal height, peak deconvolution, and amorphous subtraction techniques. However, issues exist in its application, and the choice of amorphous standard can change the results significantly. Scattering from various amorphous samples varies, and so a single, invariant standard might result in an incorrect estimation of the crystallinity index since the amorphous nature in the sample to be tested is unknown. Proper selection also of the standard crystallographic allomorph of cellulose is obviously a prerequisite to obtain accurate fits to diffraction data. While standard for most well-studied inorganic materials, Rietveld analysis is still in its infancy in the field of cellulose crystallinity determination and should be pursued with caution. One way to be cautious is to introduce the variables in several individual refinements rather than to try to refine all variables in just one refinement. Individual variables will tend to take unrealistic values and should be monitored when judging the success of the refinement.
Another factor that affects all the methods discussed above is the intensity of the radiation on the sample at different 2θ angles. It has been observed that the calculated lines are smooth, but the experimental lines contain noise which arises from the photon counting statistics. The noise in the data can be reduced when the sample is irradiated with a high intensity beam, e.g., a rotating anode tube or synchrotron radiation, or by counting for a longer time at each step with a conventional diffractometer. The standard deviation (noise) for the photon count is proportional to the square root of the number of photons counted (the intensity). The high counts that gave very smooth experimental data in Fig. 7 were obtained with a conventional laboratory diffractometer with a modern detector in a half-hour. Moreover, using fixed-slit diffractometer, a standard method to collect the data, can produce a background with quite a radical curvature, as shown in Fig. 7A. In contrast, variable slits cause the radiation to hit the sample with different intensity which produces an almost horizontal background (Fig. 7B). Researchers should also make sure that the chosen Rietveld software is programmed for the slit type and reduce the impact of the environmental background by subtracting a blank run.
XRD techniques have been widely used to determine cellulose crystallinity, although it is very challenging to use these approaches to compare the crystallinity of different samples if identical measurement and analysis protocols are not followed. All the diffraction peaks of amorphous and crystalline regions must be considered for complete quantification and optimum assessment of cellulose crystallinity. The presence of preferred orientation can be confirmed by using different sample orientations although in principle, it is not necessary as the Rietveld software can include corrections for preferred orientation. Though any XRD technique can provide relative crystallinity indices of a single set of samples, it might be beneficial to use the Rietveld method to determine and compare values for different sets of samples because it has the capability, if applied correctly, to produce reliable results if enough data are provided.
3. Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) ranks among the widely used tools to measure the crystallinity of cellulose and nanocellulose.106–112 The technique makes use of many different instrumentation types that are available for the characterization of the cellulose crystallinity index.110 The crystallinity index measured by NMR can result in more meaningful data than those obtained by DSC or FTIR since NMR is able to detect multiple conformations, whereas DSC and FTIR techniques cannot determine secondary structures or domain movements. In this sense, crystallinity measurement by NMR relates more meaningfully to a discrete structure of cellulose.3.1. NMR fundamentals
Nuclear magnetic resonance is based on the magnetic properties of the atomic nucleus. The resonance phenomenon occurs at a characteristic frequency (Larmor frequency) in the electromagnetic spectrum when the nuclei of certain atoms are placed in a strong magnetic field.28 The nuclei of certain atoms are very sensitive to slight variations in the magnetic field shielding applied by the surrounding atoms.28 Characteristic, yet slight variations occur in the electrons when the external magnetic field near the nucleus is modified.113 These slight variations that occur are called a “chemical shift” and provide critical information about the molecules. The information obtained indicates the characteristics of the type of nucleus and the atom's position within a molecule.28,113 When a constant magnetic field of strength B0 is applied, the polarization of the magnetic nuclear spin splits into two energy states (2I + 1) (Fig. 8A).28I is the magnetic spin number and I = 1/2 is from the atomic nucleus, where the sum of the proton and neutron number is odd such as 1H and 13C. One of the states has the same magnetic moment as the B0 field, and the other state has a momentum opposite to the B0 field.114 The magnetic moment, parallel to the external field, has a lower energy (m = +1/2). In contrast, the magnetic moment with the antiparallel alignment has higher energy (m = −1/2).114 In the presence of a magnetic field B0, the nucleus moves in precession around the applied field at an angular velocity defined by the Larmor frequency ω0 = ϒB0 (Fig. 8B).115,116 The constant ϒ is called the magnetogyric ratio and has a unique value for different nuclei.116 When the electromagnetic waves with frequency (ν) irradiate the system, the precessing nuclei can undergo a transition between the two energy states by the absorption of energy.28 The nuclei in the lower energy state (m = +1/2) are promoted to the higher energy state (m = −1/2). Because of the precession resonance (ν = ω0) and the frequencies of the applied radiation, the transition process is defined as nuclear magnetic resonance (NMR).116 Based on the fundamentals of NMR, when a magnetogyric ratio with a specific frequency is used to pulse the sample to a high energy state, the atomic nucleus in the sample can give a unique signal for the analytical spectra. The schematic of typical NMR instrumentation is shown in Fig. 8C.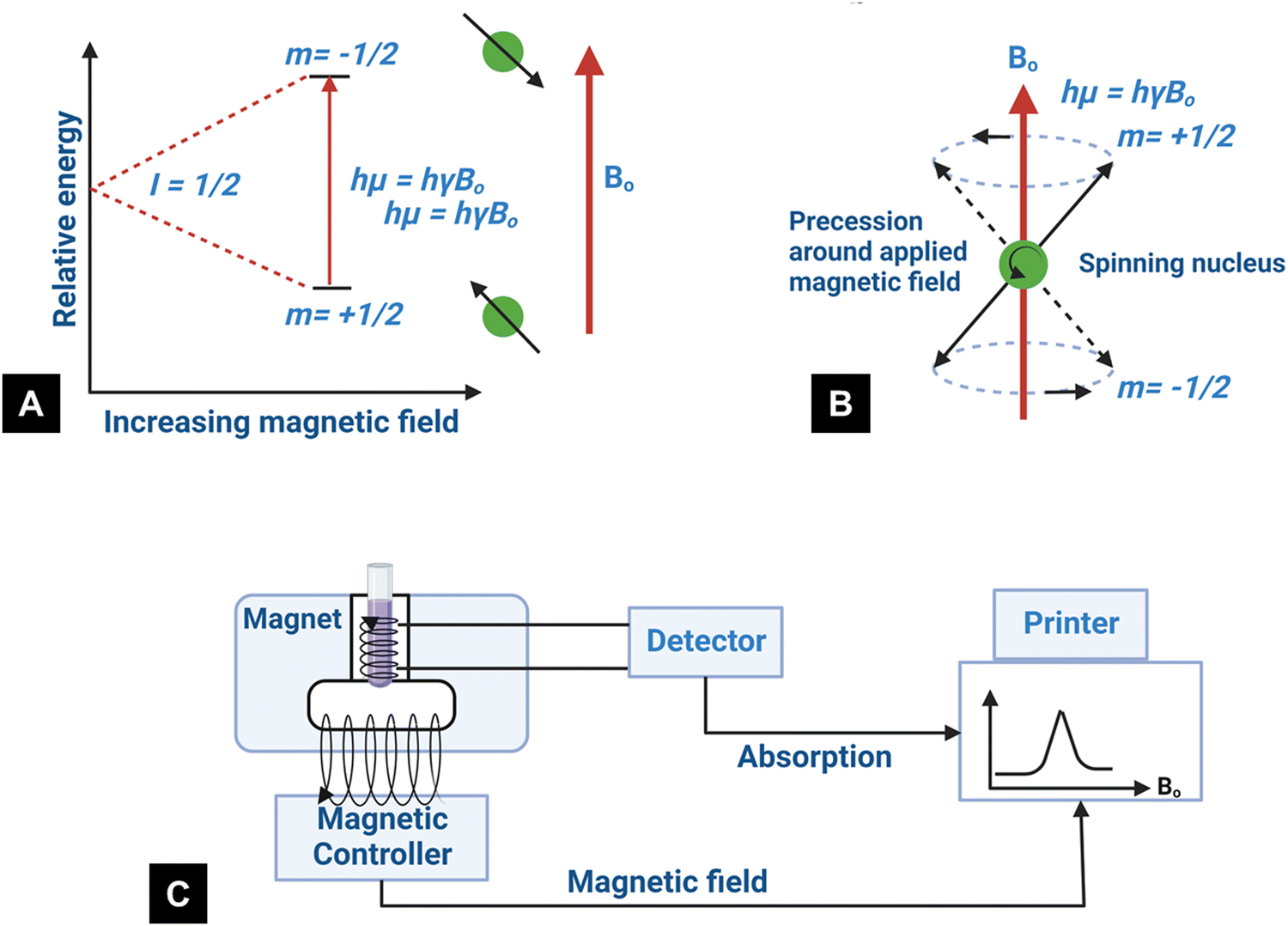 | ||
| Fig. 8 (A) State of nuclear spin under the influence of the external magnetic field, (B) graphical representation of the precession for spin-up and spin-down states at the Larmor frequency (redrawn from ref. 117), and (C) schematic of the NMR technique.118 | ||
3.2. NMR to measure crystallinity of cellulose
Cellulose exhibits limited solubility and complex chemical bonds due to its crystallinity. Thus, NMR can be used for studying cellulose crystallinity in addition to providing information about bonding, functionality, and the chemical environment. 13C CP/MAS NMR is the summation of three techniques, namely, cross-polarization, magic angle spinning, and high-power coupling.119 Cross polarization increases the 13C intensity (as it is low because of the low natural cluster and magnetogyric ratio) by transferring magnetization from the proton to the carbon. The adaptation of the proton–carbon polarization (CP) method has been found to solve the weak signal problem by increasing the 13C sensitivity and annihilating the dipolar interactions. Moreover, magic angle spinning (MAS), which spins the sample about the alleged magic angle, has solved the issue of anisotropy of solid samples in the magnetic field.28 Using 13C CP/MAS solid-state NMR, cellulose derived from variable processes such as kraft pulp, low-DP acid-hydrolyzed cellulose, and tunicate-extracted specimens exhibiting highly crystalline structures can be analyzed. These materials have been widely studied by NMR and their crystallinity is measured by calculating the variation in the chemical shift.52,120–122Several different NMR techniques are used to study cellulose crystallinity. However, one of the most widely used NMR methods is the C4 peak separation of the cellulose molecules using 13C CP/MAS NMR spectroscopy.54,120,121,123,124 In this case, doublets of C4 peaks represent two different signals in the 13C spectrum, and each peak is assigned to each cellulose structure. One of the peaks, located at ∼89 ppm, indicates the C4 carbon in ordered cellulose structures in the interior of the crystallites, and another, located at ∼84 ppm, reveals the nature of amorphous cellulose.120,121 Amorphous cellulose has been assigned by the region of amorphous C4 of cellulose.52 The reason is that the surface material of cellulose crystallites appears as a sharper doublet in the amorphous region, and xylan deposited on cellulose crystallites appears as a peak at 82.1 ppm.125,126 In addition, crystalline mannan has been reported to resonate in that area.127 The crystallinity index (CI) of cellulose can be obtained by dividing the area of the crystalline peak (∼89 ppm) by the total area of the C4 peaks,75 where the ∼89 ppm and ∼84 ppm signals represent the crystalline and amorphous (disordered) regions respectively. The crystallinity index (CrI) as a percentage can be calculated by the sum of the integral area below these signals according to the equation
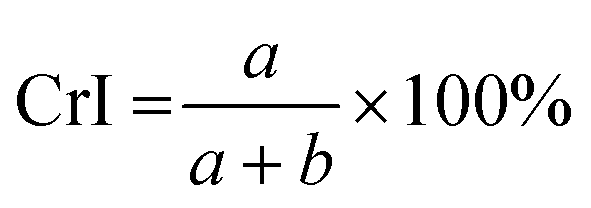 | (4) |
The 13C-CP/MAS NMR spectra of different cellulose samples have been used to understand the cellulose crystallinity by the spectral analysis of chemical shifts of different polymorphs as shown in Fig. 9A.131
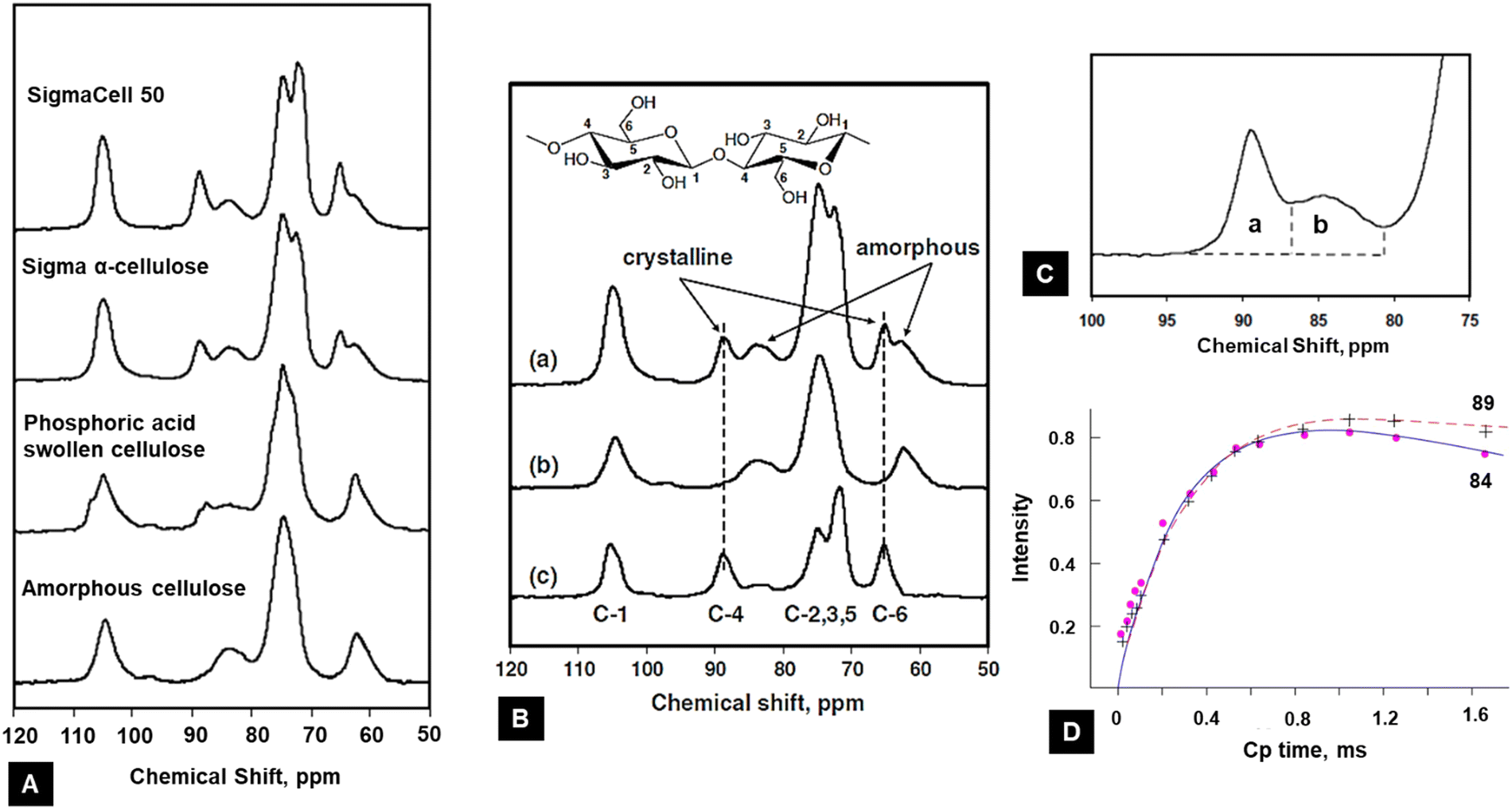 | ||
| Fig. 9 (A) NMR method to calculate the crystallinity of cellulose from different sources.131 (B) Example of the procedure of the subtraction method using 13C NMR spectra: (a) sigma α-cellulose, (b) 100% amorphous cellulose, and (c) crystalline fraction131 (reprinted with permission from ref. 131. Copyright Springer-Nature 2009). (C) Magnified version to show the peaks assigned to C4 in cellulose15 (adapted from and used with permission from ref. 15. Copyright Springer-Nature, 2010). (D) 13C CP NMR spectral signal intensities of 84 and 89 ppm lines upon CP time for microcrystalline cellulose. Curves are normalized to equal spin temperature and the number of protons at t = 0132 (reprinted with permission from ref. 132. Copyright Springer-Nature, 1987). | ||
In the spectrum of the sample, two outer and inner signals at 105 ppm represents the cellulose Iβ and Iα are apparent, respectively.125,133 The presence of the Iα allomorph is confirmed by up-field wings in the C4 signals of the fibres (see Table 3).
| Allomorphs | Chemical shift (ppm) | ||
|---|---|---|---|
| C1 | C4 | C6 | |
| Amorphous | — | 86.3–80.1 | 63.5–59.5 |
| Cellulose Iβ | 105.6, 104.0 | 89.4, 88.2 | 65.4, 64.9 |
| Cellulose Iα | 105.2 | 90.0, 89.4 | — |
| Paracrystalline | 104.8 | 88.8 | — |
A sharper resonance of C1 and C4 signals represents the crystalline region that overlaps with the up-field wings of the less ordered regions, having two different environments – one is the crystallite surface and the other is the amorphous regions.133 However, NMR spectral intensities play a valuable role in determining crystallinity, as the signals from different nuclei reduce the magnetic moment induction due to the application of a radio frequency pulse during the measurements. As the proton–carbon polarization (CP) dynamics and proton relaxation are different for different phases, the total intensities of the spectrum do not necessarily represent the actual number of nuclei under the spectrum. To overcome this situation, and get reliable results from CP/MAS NMR, the time constant is maintained in the order TCH ≪ TCP ≪ T1PH, where TCH, TCP, and T1PH are respectively proton carbon (CP) time constants, CP time intervals in the process, and proton spin–lattice relaxation times in the rotation frame. Thus, to avoid errors due to intensity distortion, the time constant is kept in the order 25TCH ≪ 5TCP ≪ T1PH.132,134 The effect of the nature of the intensity of NMR spectra of microcrystalline cellulose (with various CP times up to 20 ms) on its crystallinity is shown in Table 4.
| Spectral lines | T 1pH | T CH |
|---|---|---|
| Crystalline | 11.8 | 0.31 |
| Amorphous | 6.4 | 0.29 |
The calculation has been carried out following the logarithmic scale described for the high-resolution NMR of solids, using the equation135
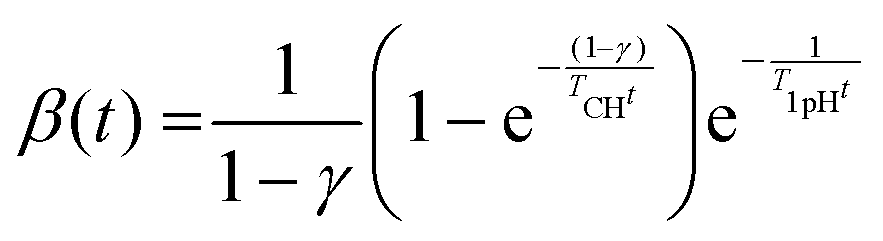 | (5) |
Another limiting factor is the proton–carbon (CP) polarization time. It has been found that a longer CP time is responsible for the increase in signal intensity difference.132 The 13C NMR intensities have been determined using eqn (5), and the 13C NMR signal intensities vary with different CP times as shown in Fig. 9D. The data signify no intensity distortion below 0.5 ms, but there is an increasing signal intensity difference between ∼89 ppm and ∼84 ppm above 0.5 ms CP time (Fig. 9D). Therefore, the CP time must be below 0.5 ms for reliable intensity characterization. The determination of spectral lines is often carried out using a curve fitting process, much the same as for XRD analysis. Curve fitting has been carried out using Gaussian and Lorentzian functions by the variation of the shapes of spectral lines. The crystallinity has then been determined by considering the line located at ∼89 ppm to the simulated C4 total intensity line.131
3.3. Other methods to measure cellulose crystallinity using NMR
The crystallinity index can also be determined from the average lateral microfibril size (L) or crystallite size as it depends on the peak intensities of C4 signals of NMR spectra by using the following equations: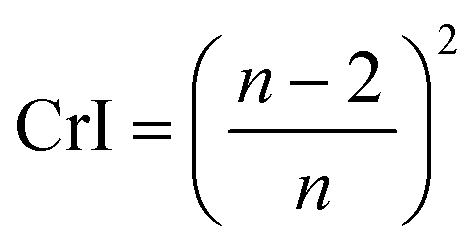 | (6) |
| L = 0.57n | (7) |
Another methodology based on 13C NMR spectroscopy is the subtraction of a standard for amorphous cellulose.137 For this method, a fully amorphous cellulose sample is prepared to estimate the signal that might be expected of the amorphous regions of the cellulose. The amount of crystalline cellulose in this sample can be obtained by subtracting the area of the amorphous cellulose from the original signal. After the subtraction, a scale factor is applied to the amorphous cellulose spectrum to eliminate a negative signal in the residual spectrum, as shown in Fig. 9(B and C). The CI is finally acquired by calculating the area ratio between the crystalline contribution and the total area.131Fig. 9(B and C) depicts the subtraction method used to calculate cellulose crystallinity. The spectrum of the α-cellulose (Fig. 9B(a)) and 100% amorphous cellulose (Fig. 9B(b)) was measured, and crystallinity fractions were calculated by subtracting the amorphous value from the α-cellulose (Fig. 9C(c)). After subtraction, a small intensity peak located at ∼84 ppm showed that the ordered crystallites present in the amorphous region had been normalized.138 The peak located at ∼89 ppm represents the crystallites of the internal crystalline region. The area under these two peaks is calculated, and the crystallinity index is calculated using eqn (4).
It was determined that amorphous C4-carbons are more than likely located at distances less than about 1 nm from the crystalline C4-carbons and that those amorphous C4-carbons are not solely localized to the microfibril surface.139 Contradictory NMR spin-diffusion results were published by Fernandes et al. that seem to support a core–shell model for the cellulose microfibril (isolated from spruce wood), where the core is more ordered than the shell.140,141
3.4. Comparison of different NMR methods
One of the major concerns with the peak separation technique is that it only deals with the C4 carbon signals and the separation in energy between the two spin states. This signal strongly depends on the content of lignin and hemicellulose and the external magnetic field strength, the latter of which is relatively weak.131 The presence of residual lignin and hemicellulose causes overlapping peaks in 13C NMR spectra, which makes it harder to separate the signals into ordered and disordered peaks when explicitly using the C4 peak separation technique. Specifically, the NMR technique relies on the size of the crystallites because only the cellulose within the interior of the crystallites is regarded as crystalline.44,52 The larger the crystallites, the more intense the NMR peak appears.Moreover, ambient Boltzmann thermal energy (E ∼ kT) is sufficient to promote large portions of nuclei into a less favorable higher energy state.115 For example, the Boltzmann distribution predicts that for one million hydrogen nuclei in a magnetic field of B0 = 1.4 T, only a few could contribute to the NMR signal. This explains the limitation of the sensitivity of NMR. Therefore, NMR needs a significant sample size (usually more than 100 mg for most MWs).28,122 NMR measurements also yield relatively weak signals that are averaged over a long time with enough signal-to-noise ratio to produce spectra. The subtraction method has some advantages over the peak separation method in measuring the crystallinity index of cellulose as this method can be used for determining this parameter for a wide range of materials.
4. Vibrational spectroscopy methods
Along with other widely used analytical techniques such as XRD and NMR for the characterization of cellulose structure, vibrational spectroscopy methods (infrared and Raman) are also useful for investigating cellulose-like polymer structures and properties.142–146 A vibrational spectrum is obtained due to the absorption or emission of electromagnetic radiation at specific frequencies that correlate to the energy states of the molecules, which are the quantum mechanical states of electron waves confined in specific chemical bond coordinates.28 However, there are some fundamental differences in the mechanisms that give rise to IR and Raman intensities. The former relies on an existing dipole between the atoms, and the latter is due to a dipole that is induced by radiation.4.1. Vibrational spectroscopy fundamentals
The basic principle of molecular vibrations responsible for the characteristic bands observed in infrared and Raman spectroscopy can be derived by comparing a di-atomic chemical bond with a simple harmonic oscillator model (Fig. 10A).117 In this model, a chemical bond can be considered as a spring with its ends attached to two atomic species with masses m1 and m2 separated by a distance d, having a spring constant k. Hooke's law can be used to express the chemical potential near the equilibrium bond distance.106 After solving Schrödinger's wave equation, the wave functions of electrons involved in this chemical bond yield discrete vibration energy levels, which are expressed as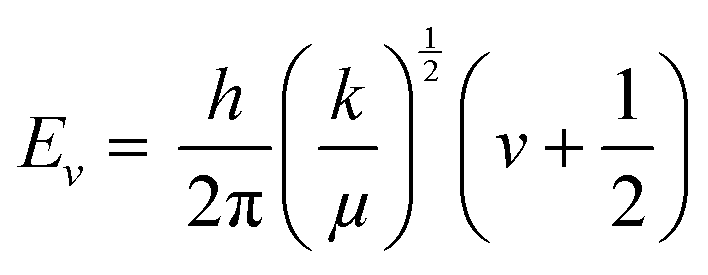 | (8) |
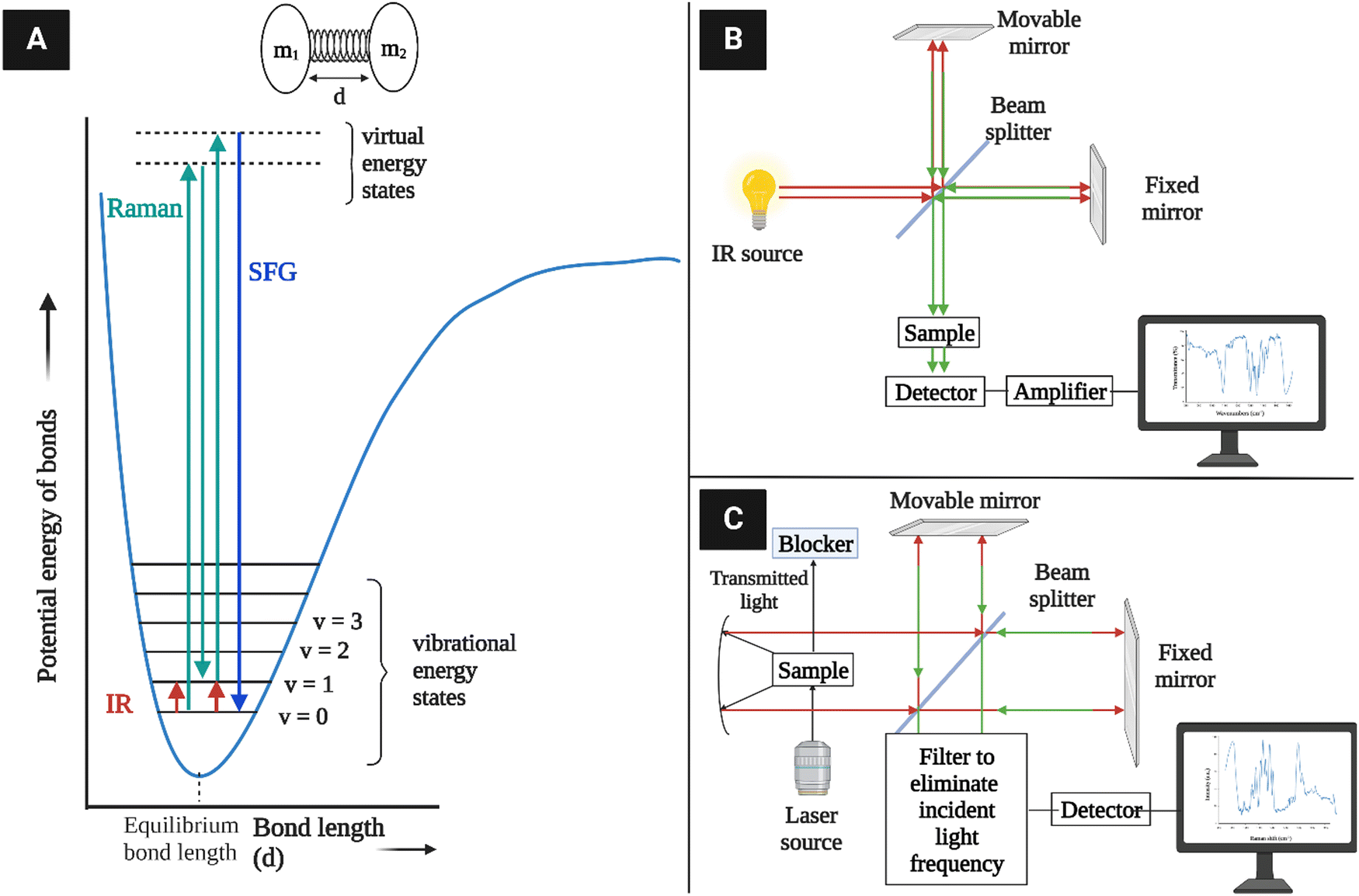 | ||
| Fig. 10 (A) Vibration energy states of diatomic molecules and electronic transitions involved in IR absorption and Raman scattering. The diatomic molecules can be modeled as two masses (m1 and m2) connected with a spring constant k117 (redrawn from ref. 117). (B) A simplified schematic of typical IR instrumentation. (C) A Raman spectrometer setup. | ||
IR spectroscopy is the measure of the intensity of a beam of electromagnetic radiation, usually a laser, observed through the absorption of radiation whose specific oscillating radiation frequency matches the natural frequency of a particular normal vibrational mode, i.e., vibrating dipole of a molecule. The spectrum is obtained by plotting the intensity (absorbance or transmittance) versus the wavenumber or frequency, which is proportional to the energy (E) difference between the ground and the excited vibrational states, mainly from the v = 0 state to the v = 1 state, which can be expressed as
| ωIR = Ev=1 − Ev=0 | (9) |
As the excited electrons tend to revert to the ground electronic state after being excited to a virtual electronic state, most of them come back to the vibrational ground state (v = 0) in elastic scattering, but a small fraction returns to the v = 1 state in inelastic scattering. The latter case is known as Raman scattering, where the energy difference between the originally absorbed photon (excitation) and the emitted photon (scattering) corresponds to the vibration energy level difference according to the equation
| ωRaman = ωexcite − ωscatter | (10) |
4.2. Measurement of crystallinity of cellulose substrates using IR spectroscopy
According to the traditional two-phase cellulose model, the polymeric chains contain both crystalline (ordered) and amorphous (less ordered) regions. A clear relationship between the interaction of hydroxyl groups and crystallinity in cellulose has been established by using FTIR.156 An empirical “crystallinity index” has been developed using the absorption ratio at 1429 cm−1 and 893 cm−1.157 As already discussed, the FTIR absorption band located at ∼1430 cm−1 is assigned to asymmetric CH2 bending vibration, and is known as the “crystallinity band.” On the other hand, the FTIR absorption band located at ∼898 cm−1 is assigned to C–O–C stretching of β-(1 → 4)-glycosidic linkages and has been designated as the “amorphous” absorption band. It has been found that crystallinity decreases as the intensity of the absorption at ∼1429 cm−1 decreases, whereas the intensity of the absorption at 893 cm−1 increases.158–160 Nelson and O’Connor introduced another ratio index, “total crystalline index,” which is the ratio of the intensities at ∼1372 cm−1 and ∼2900 cm−1. Bands centered near ∼2900 cm−1 are C–H stretching vibration bands, which conform to the characteristics of amorphous cellulose, and the absorbance band at ∼1372 cm−1 is attributed to C–H bending.161 It has been reported that the band located at ∼1336 cm−1 showed the most significant differences between crystalline cellulose II and amorphous cellulose. Later it was demonstrated that these bands were not characteristic of the amorphous regions.161,162 Both of these ratios are now used to measure the crystallinity of cellulose where these indices are assigned as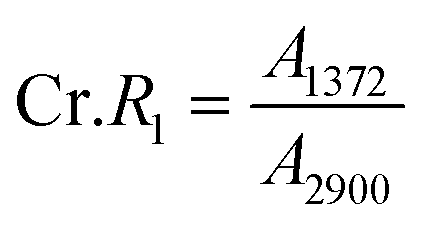 | (11) |
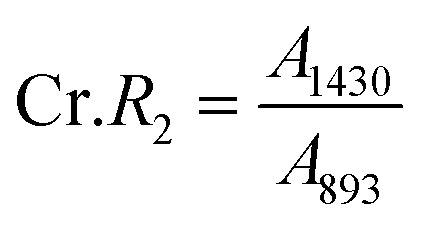 | (12) |
| Yield corrected crystalline cellulose (%) = A1370/A670 × amount of cellulose × pulp yield × 0.1 | (13) |
| Peak (cm−1) | Type of vibration |
|---|---|
| 1370 | CH bending |
| 1335 | OH in-plane bending |
| 1320 | CH2 wagging |
| 670 | OH out-of-plate bending |
| 893 | Non-symmetric out-phase ring |
| 1429 | CH2 symmetric bending |
| 2900 | CH stretching |
Hydrogen bond intensity (HBI) can also be used to interpret qualitative changes in cellulose crystallinity; for example, crystallinity decreases with the increase of HBI. An increase in HBI represents an increase in hydrogen bonding between certain hydroxyl functions in the cellulose, which is typical of the conversion of cellulose I to cellulose II; even though this represents a decrease in the overall crystallinity index.45
FTIR spectra in attenuated total reflectance (ATR) mode offer a complementary method for providing information on chemical compounds and hydrogen bonding characteristics to confirm XRD findings.164 FTIR spectra of freeze-dried bacterial cellulose samples in attenuated total reflectance (ATR) mode have been used to obtain crystallinity indices by using the two conventional methods: (1) ratios of the absorption intensities at wavenumber positions of ∼1428 and ∼897 cm−1 (Cr1 = A1428/A897), and (2) ratios of the absorption intensities at wavenumber positions of ∼1372 and ∼2898 cm−1 (Cr2 = A1372/A2898)165 (Fig. 11).
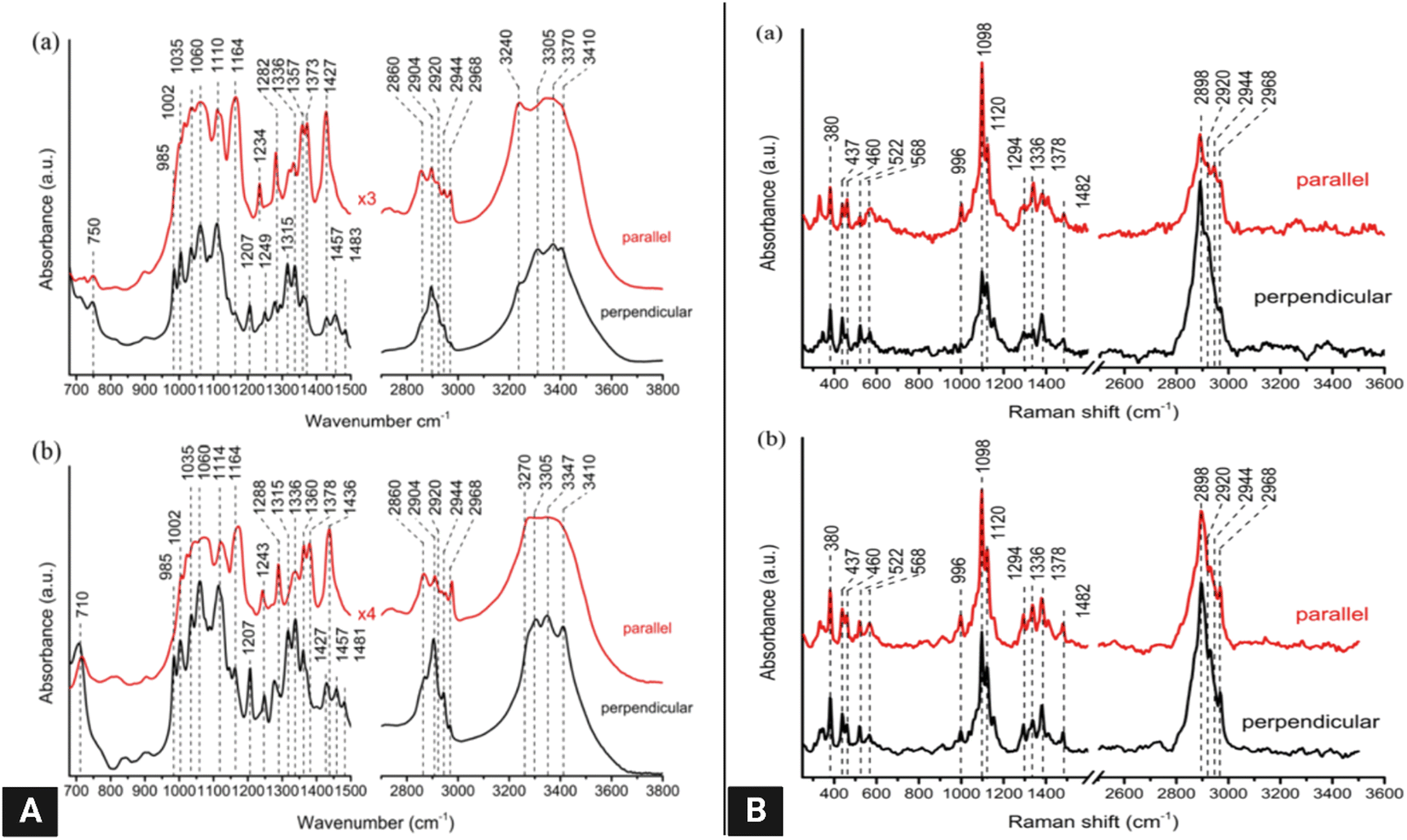 | ||
| Fig. 11 (A) Polarized FT-IR spectra of laterally packed and aligned films of (a) cellulose Iα and (b) Iβ nanocrystals. Spectra were taken with the IR polarization perpendicular to the nanocrystals’ alignment direction.117 (B) Polarized FT-Raman spectra of laterally packed and aligned films of (a) cellulose Iα and (b) Iβ nanocrystals. Spectra were taken with the excitation laser polarization perpendicular to the nanocrystals’ alignment direction. The peaks discussed in the text are highlighted with dotted lines117 (reprinted with permission from ref. 117. Copyright 2013 Korean Journal of Chemical Engineering). | ||
4.3. Measurement of crystallinity of cellulose substrates using Raman spectroscopy
Raman or its more advanced form, FT-Raman spectroscopy, has emerged as a rapid and convenient analytical method to determine cellulose crystallinity. The principle of the Raman method is to use spectral features whose intensity, bandwidth, and position are affected by cellulose crystallinity.58The FT-Raman-based quantitation method for cellulose I crystallinity is developed using the weak bands located at ∼1462 and ∼1481 cm−1 (CH2 bending modes) in conjunction with spectral deconvolution. However, the intensities of the selected bands are relatively low, and the deconvolution process is not free of the band fitting problem; hence a better approach has been sought.55 In this better approach, the crystallinity of cellulose I samples has been determined based on the intensity ratio of the bands located at ∼380 and ∼1096 cm−1.58 The peak height was calculated by choosing a minimum intensity wavenumber position near the peak (e.g., ∼358 and ∼944 cm−1 for the ∼380 and ∼1,096 cm−1 bands, respectively) and by drawing a horizontal line (from that wavenumber position) under the peak. This is called the baseline method and using this method, in the remaining spectra for bands located at ∼380 and ∼1096 cm−1, the same values of band minima (at ∼358 and ∼944 cm−1) were used for drawing the baselines. After taking the peak heights, the intensity data were used to calculate Raman band intensity ratios, and regression models were developed. To generate a calibration curve, cellulosic samples were ball-milled for different timespans to observe a change in their crystallinity. Significant effects on the peak shape and intensity at those peak regions were observed because of the mechanical treatment.
4.4. Sum-frequency generation vibrational spectroscopy (SFG)
Another vibrational method, SFG vibrational spectroscopy, is a non-linear optical phenomenon demonstrated to be an effective technique for probing interfaces.166–169 It is highly sensitive to non-centrosymmetric crystalline materials, e.g., piezocrystals and non-centrosymmetrically arranged functional groups in a medium.170,171 Due to its selective vibrational modes and sensitivity for chiral molecules in biomaterials, it is also used to probe biological molecules such as DNA and other helical proteins.172 Recently, it has been shown to selectively detect crystalline cellulose from lignocellulosic biomass.61The principle of SFG vibrational spectroscopy depends on the non-linear optical response of sum frequency generation. In this process, temporal and spatial overlap between two pulsed laser beams occurs at an interface. One of these beams has a fixed visible frequency (ωVIS) and the other has an adjustable infrared frequency (ωIR). Thus, the light is emitted as the summed frequency (ωSFG) of the two frequencies via the equation173
| ωSFG = ωVIS + ωIR | (14) |
| P(ω) = εo(X(1)E(ω) + X(2)E(ω)2 + X(3)E(ω)3 + …) | (15) |
| E(ω) = E1cosω1t + E2cosω2t | (16) |
 | (17) |
| I(ωSFG) ∝ |X(2)eff|2I(ωIR)I(ωVIS) | (18) |
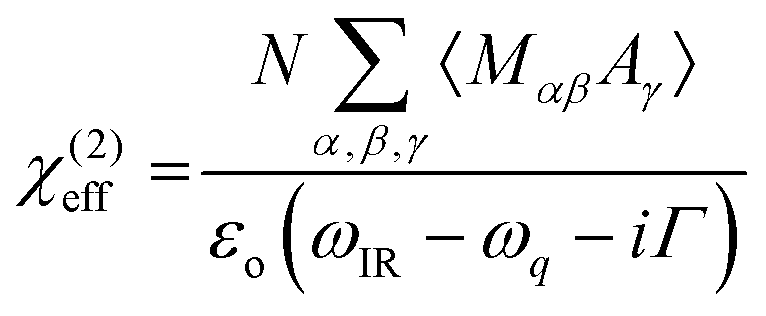 | (19) |
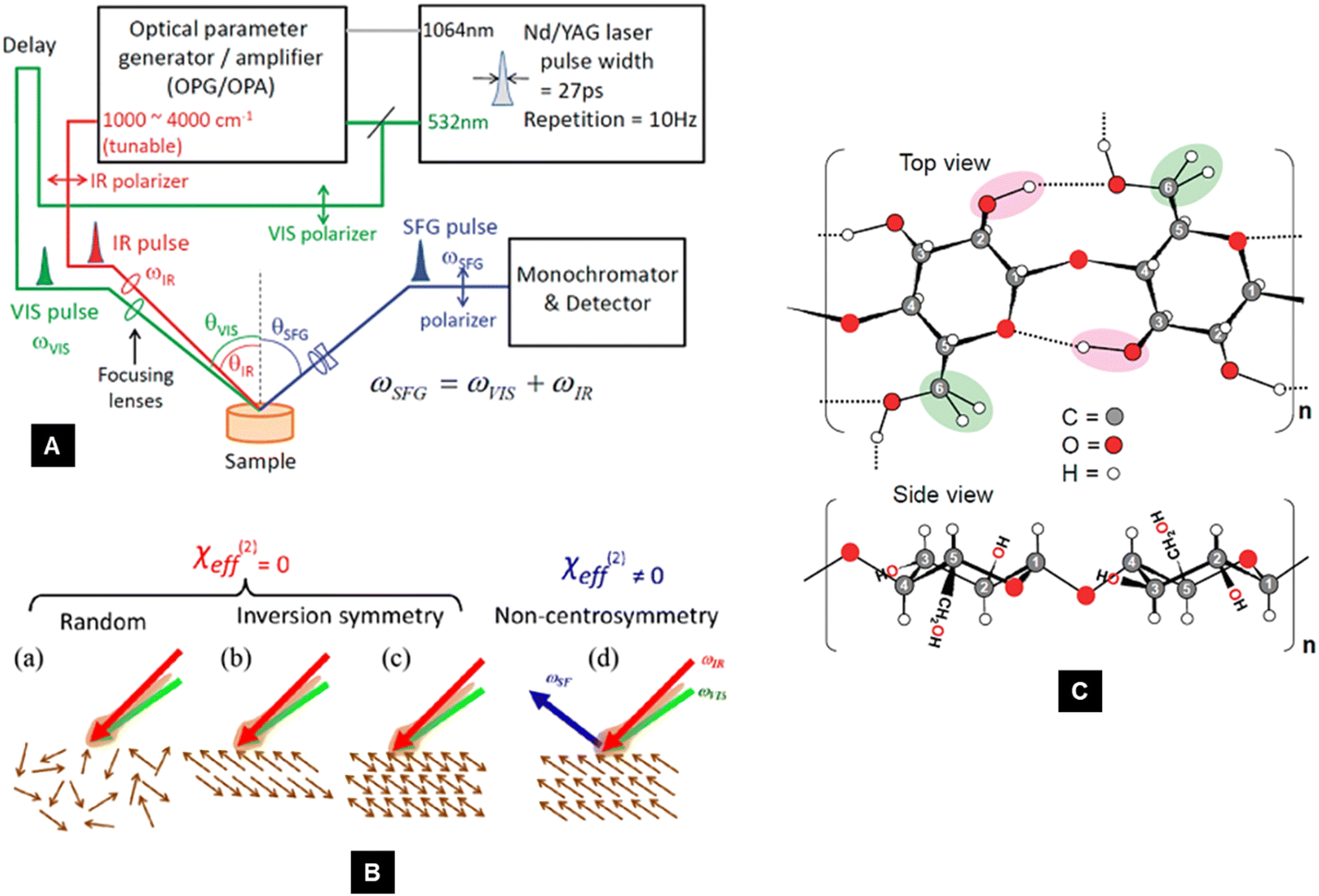 | ||
| Fig. 12 (A) Schematic diagram of an SFG system61 (reprinted with permission from ref. 61. Copyright 2011 American Chemical Society). (B) Schematic of the noncentrosymmetry requirement of X(2)eff in the SFG process.117 The directionality of vibration modes is depicted by brown arrows (reprinted with permission from ref. 117. Copyright 2013 Korean Journal of Chemical Engineering). (C) Schematic view of two glucopyranose units in a cellulose chain. The SFG-active CH2 and OH stretch peaks are highlighted in green and pink, respectively (reprinted with permission from ref. 61. Copyright 2011 American Chemical Society). | ||
As shown in Fig. 12B, when the vibration modes (shown by brown arrows) are arranged either randomly (a), or with inversion symmetry (b), or the molecule has inversion symmetry itself (c), the SFG signal cannot be generated. The only way that X(2)eff becomes non-zero is when the molecules with no inversion symmetry are arranged non-centrosymmetrically in space.28,173 Non-centrosymmetry exists over several orders of length scales in crystalline cellulose.177 At the molecular level, its carbon centers (C1, C2, C3, C4, and C5) in the glucose ring are chiral. The crystal unit cells manifest non-centrosymmetry (space groups P1 and P21).56,68 Also, cellulose Iα and Iβ have parallel glucan chains, unlike cellulose II, in which the chains have an antiparallel arrangement.178 As these glucan chains in cellulose I are unidirectional, the CH2 group of exocyclic C6H2OH side chains and OH groups that have interchain H-bonding form net dipoles across the entire crystal or multiple closely packed crystals.61,179 As a result, the characteristic SFG peaks located at 2945 cm−1 and 3320–3250 cm−1 are observed for CH2 and OH vibration modes, respectively.62,178 It has been observed that, the SFG peak intensity at 2945 cm−1 varies monotonically, but non-linearly with crystalline cellulose samples,97 based on which an Avicel-based calibration curve using XRD, IR, and Raman techniques to determine the amount of Avicel-equivalent crystalline cellulose in wood samples has been developed.
Other C–H and O–H than the aforementioned peaks are not observed because of the non-centrosymmetric selection rule. The same reason is also attributed to the SFG inactivity of hemicellulose and lignin because of their random arrangement in biomass.61 Moreover, these noncentrosymmetric crystals could be dispersed in a random, centrosymmetric, or noncentrosymmetric fashion within an amorphous matrix, which can also affect the SFG spectra over the characteristic length of SFG.180 The SFG technique is becoming increasingly popular in probing the crystal structure of cellulose. Huang et al. (2020) employed SFG vibrational spectroscopy to correlate cellulose enzyme complexes' shape and size to cellulose's crystal size using freshwater algae Micrasterias.181 The process was also able to distinguish between the surface and bulk hydroxyl groups in cellulose nanocrystals owing to its non-linear scattering.182 The SFG technique is also sensitive to changes in the inter-fibrillar distance between cellulose microfibrils and the distance between crystals.183,184 The process can distinguish between chain polarity and determine the orientation in the direction of growth of the cellulose microfibrils (unidirectional or bidirectional).28,185–187 This technique is also able to distinguish different cellulose polymorphs.178 Moreover, the technique has enabled researchers to probe into the mesoscale assembly of cellulose microfibrils.179,180
The SFG technique can be used to quantify cellulose crystallinity, given that the crystalline packing pattern of cellulose remains reasonably constant.97 Current quantification of cellulose crystallinity by the SFG method involves a calibration curve between a known cellulose concentration and the SFG peak intensity.188,189 However, the quantification becomes challenging when the packing of crystalline cellulose changes drastically with different samples.177,180
5. Differential scanning calorimetry (DSC)
Calorimetry measures thermal energy absorbed or released when a change in state variables is encountered due to phase transitions, chemical reactions, or physical changes. The simple equation for measuring heat change as a function of temperature can be expressed as| dQ = m × c × ΔT | (20) |
5.1. Heat-flux DSC
In a heat-flux DSC system, the reference and the sample are heated in a single furnace, where two separate sensors are used to measure their temperatures. A purge gas allows a uniform heat distribution and expels unwanted substances like oxygen. Thus, both the sample and reference are given equal heat transfer, and their temperature difference is measured. The temperature difference is translated using the heat flow equation to heat flux.190 However, the heat-flux DSC systems suffer from the disadvantage of the significant time constant. Hence, they are typically not used to study semi-crystalline polymers.191 A simplified schematic is shown in Fig. 13A.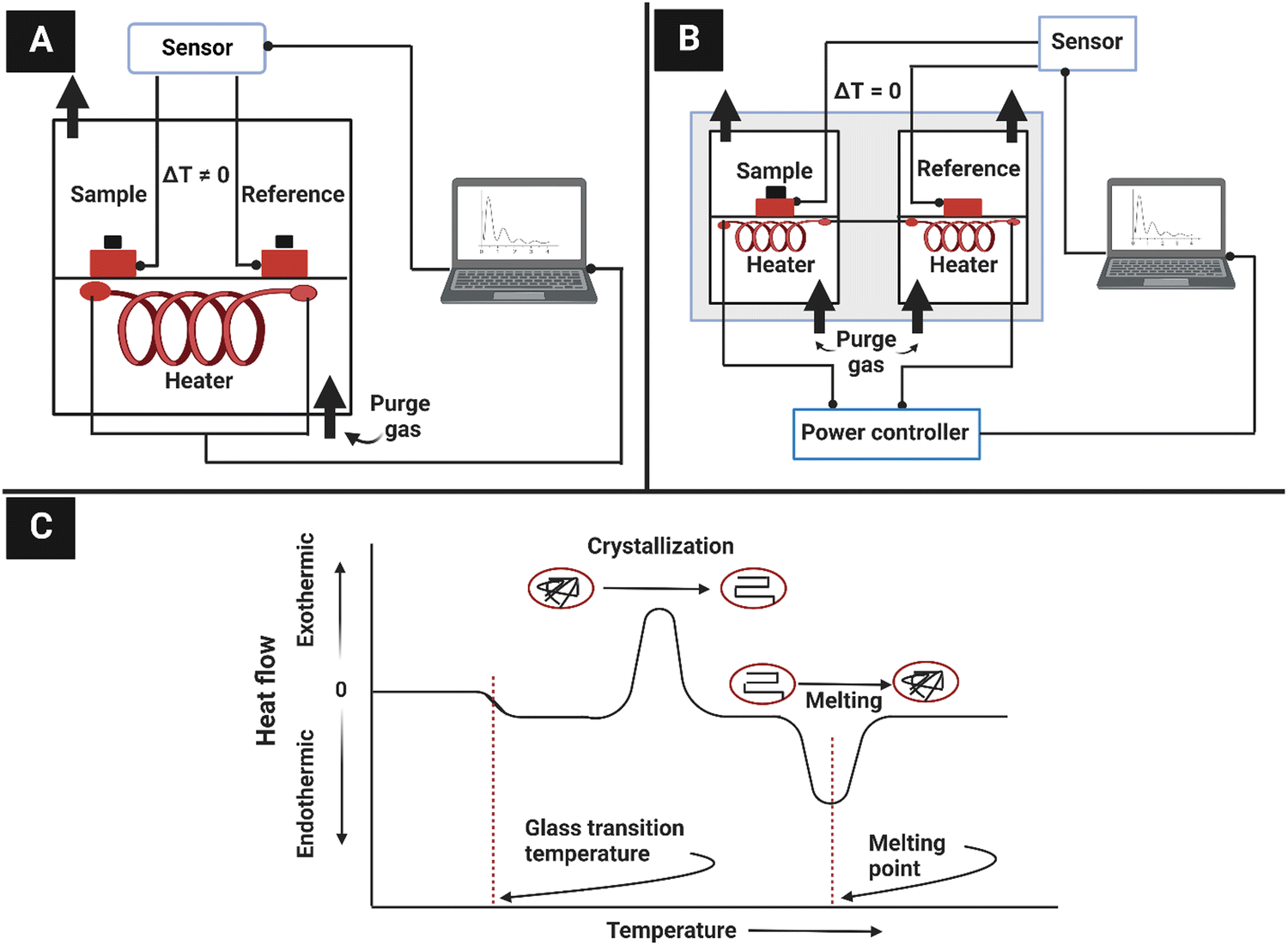 | ||
| Fig. 13 (A) Schematic of heat-flux DSC, (B) schematic diagram of power-compensated DSC, and (C) general DSC output for semi-crystalline polymers. | ||
5.2. Power-compensated DSC
In power-compensated DSC systems, two separate chambers are used for the sample and the reference. Two individual heaters heat the sample and the reference through a differential power controller to maintain a zero-temperature difference between them. The temperature is increased linearly with time. Thus, in power-compensated DSC systems, the energy flow rate varies to keep the same temperature in both chambers. The purge gas is used to allow for uniform heat distribution and the removal of unwanted substances. The difference in power can be determined using the equation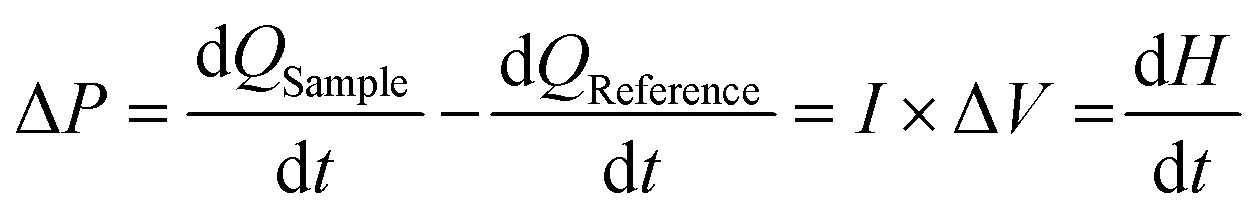 | (21) |
Both methods produce an output of the heat-flow curve with respect to temperature. From the curve, an inference about material properties can be drawn. Fig. 13C shows the general DSC output of a semi-crystalline polymer along with possible interpretations. The enthalpy change is proportional to the area under the peaks. The enthalpy change (ΔH) can be expressed as
| ΔH = K × A | (22) |
5.3. Measurement of crystallinity of cellulose using DSC
Conventionally, relative crystallinity in polymers is determined by taking the ratio of the melting enthalpy of the sample obtained from a DSC run to the melting enthalpy of a crystalline sample.194,195 It can be expressed as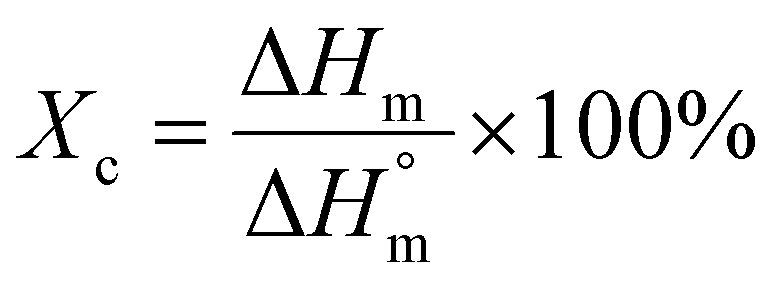 | (23) |
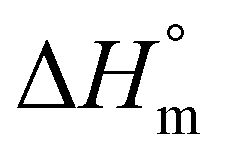 is the melting enthalpy of a 100% crystalline sample of the same polymer.
is the melting enthalpy of a 100% crystalline sample of the same polymer.
Cellulose has a mixture of well-defined crystalline and amorphous structures and the amorphous regions allow aqueous reagents to penetrate inside the structure.196 The crystalline domains are very closely packed and resist water absorption. Hence, it is thought that the free hydroxyl groups present in the amorphous regions are responsible for almost complete water absorption. Based on these considerations, Dale proposed a method to determine the cellulose crystallinity that considers the dehydration endothermic peak area.196 It is reported that cellulose fibres typically show an endothermic peak between 273 and 400 K, which is attributed to dehydration.197 Thus, the authors hypothesized that the endothermic peak of cellulose dehydration is related to the amorphous fraction of cellulose.196 The cellulose crystallinity (CC%) could be determined from the equation
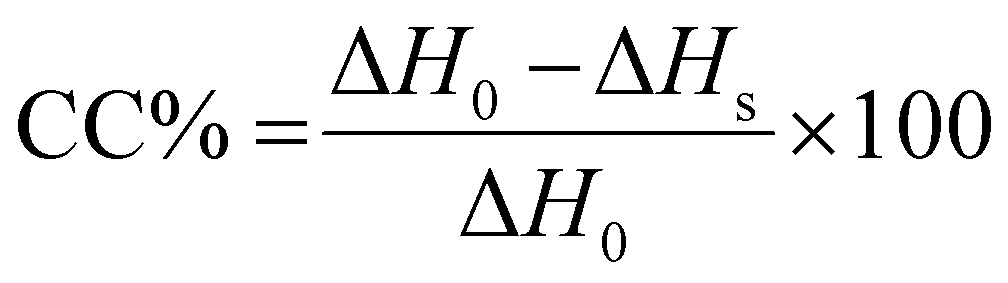 | (24) |
There are some other limitations associated with DSC since this method assumes that the amorphous regions of cellulose are entirely responsible for water absorption. However, a small portion of water adheres to the crystalline part of cellulose, which cannot be avoided for samples with high crystallinity. Thus, the method is most suitable for low crystallinity cellulose samples.196 It is also difficult to estimate the baseline for the peak area measurement.191,194,198 Although the DSC technique has gained attention among polymer scientists regarding its use in determining crystallinity, it has been used in only a limited way in cellulose research. The main reason for this is that in conventional semicrystalline polymers one can measure the heat of crystallization through melting. Since cellulose does not melt, it is not possible to measure this property. Moreover, the transitions of cellulose due to moisture are also influenced by other factors and other polymers present in the samples. It has been used to probe only the thermal behavior, which can be understood from the literature survey on determining cellulose crystallinity by the DSC method.199,200 Further research is necessary to address the limitations associated with the DSC technique of assessing cellulose crystallinity.
6. Cellulose crystallinity determination by carbohydrate-binding modules (CBM)
Biochemical routes have also been developed to determine the cellulose crystallinity in addition to getting information on the morphology of cellulosic structures. Initially, these methods were developed to quantitatively determine the accessibility of cellulose fibres subjected to enzymatic hydrolysis. However, the degree of cellulose crystallinity may not influence the yield during enzymatic hydrolysis but can affect the kinetics of hydrolysis.201 For example, it has been shown that paracrystalline cellulosic regions hydrolyze faster than crystalline regions.202 However, as cellulose crystallinity is one of the critical factors influencing cellulose accessibility, qualitative information on the crystalline cellulosic part can be obtained through these methods.These methods take advantage of carbohydrate-binding modules (CBMs), which are polypeptide sequences found in carbohydrate-active enzymes with specific binding abilities to different carbohydrate structures. Depending on the molecular structures and amino acid sequences, CBMs can be classified into 68 families and based on their three-dimensional structures and activity, they are classified as type A, type B, and type C.201,203 Type A CBMs are surface binding type proteins with a greater affinity towards crystalline cellulose. Unlike type A, type B proteins show affinity towards the amorphous cellulosic regions by binding to glycan chains. Yet, type B CBMs cannot recognize glycan chains consisting of less than three sugar units while type C CBMs exhibit binding specificity for 1–3 sugar units.201,203
Moreover, fusion proteins have also been developed by combining CBMs with green fluorescent proteins (GFPs) to visualize cellulose morphology easily. For example, Hong et al. (2007) prepared one such fusion protein to quantitatively determine cellulose accessibility to cellulase with a hydrolysis temperature of 50 °C.204 Similarly, Li et al. (2018) designed a recombinant protein by combining CBMs with a di-green fluorescent protein to quantitatively determine cellulose accessibility to cellulase.205 Kljun et al. (2011) used CBMs to assess changes in cellulose crystallinity during the mercerization process of cotton fibres (Fig. 14A).206 The authors used two type A and two type B CBMs coupled with fluorescence imaging to visualize the changes in the crystallinity at various stages of the mercerization process, which agreed with the results obtained from traditional XRD and FTIR techniques as represented in Fig. 14B.206 The binding intensity of the CBMs was calculated by measuring the total dark areas for the amorphous region and total green/bright areas for the crystalline region. The total fibre area was also measured, and the bright or dark area ratio to the total fibre area was taken to calculate the percentage of bound and unbound CBMs to the fibre. They also estimated the lateral order index (LOI) and hydrogen bond intensity (HBI) from an FTIR trace. LOI and HBI were determined from the intensity of the absorbance bands located at 1429 to 893 cm−1 and the ratio of the intensities of the 3336 to 1336 cm−1 bands, respectively.207,208 They found that the LOI followed a similar trend as the relative intensity of the binding capacity of the CBMs onto the crystalline cellulose for both the Coker and FM966 fibres. On the other hand, HBI increased with an increase in the amorphous region of the fibre samples. It also displayed similar trends to the relative intensity of the CBM which shows affinity towards the amorphous cellulosic region, as shown in Fig. 14C.
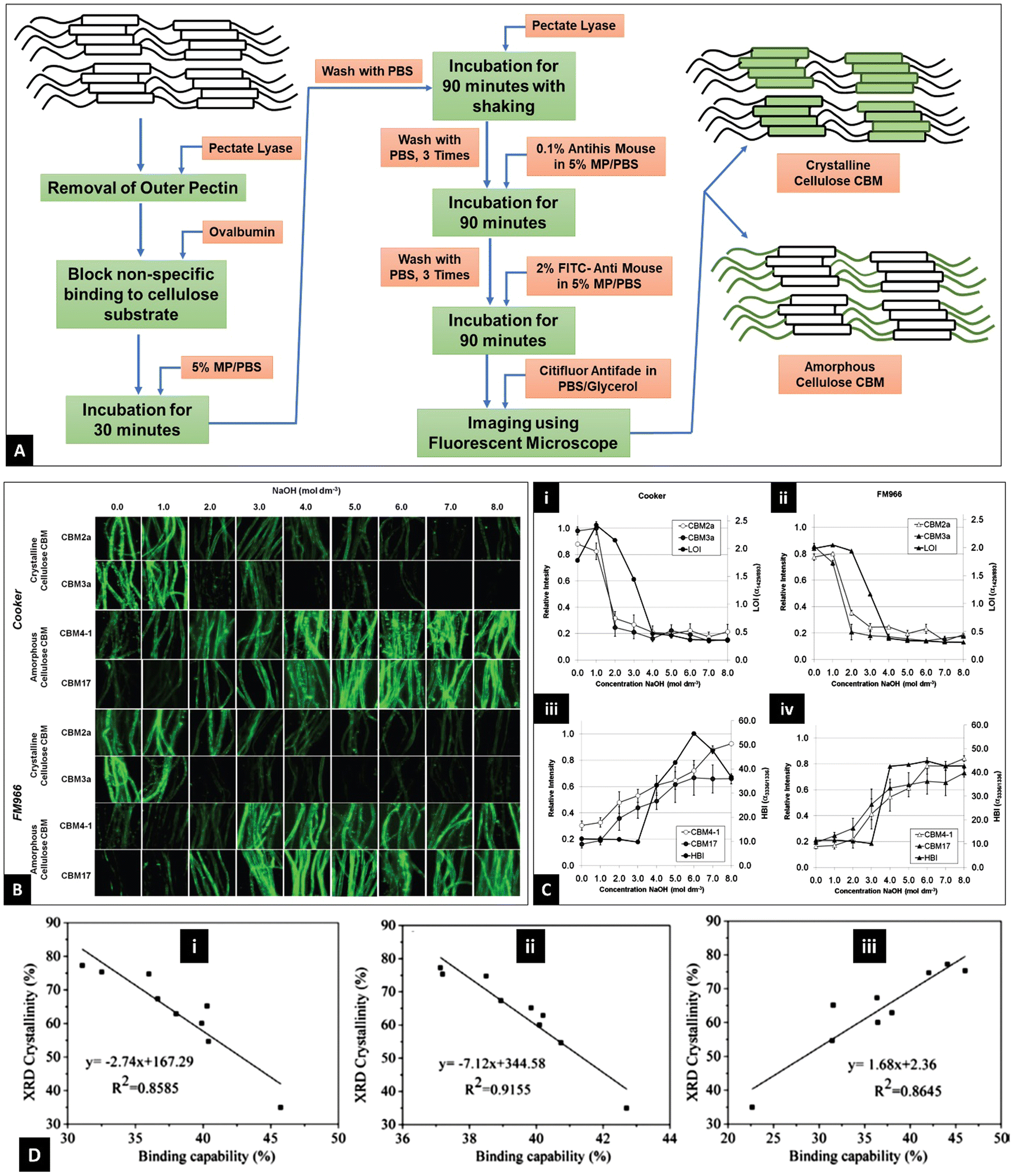 | ||
| Fig. 14 (A) Schematic of cellulose crystallinity determination using CBMs.206,208 (B) Fluorescence imaging of the binding of four CBMs to different cotton fibres after treatments with a range of NaOH concentrations (0–8 M). (C) Quantification of CBM binding to cotton fibres and FTIR analyses in response to NaOH treatments. (i) and (ii) Comparison of LOI with the binding intensity of crystalline directed CBMs, (iii) and (iv) comparison of HBI with the binding intensity of amorphous directed CBMs. (Parts B and C are reprinted with permission from ref. 206. Copyright 2011 American Chemical Society.) (D) Correlation between CBM binding capacity and crystallinity index of different cellulose samples where (i) and (ii) CBMs show affinity towards amorphous cellulose and (iii) CBM has an affinity for crystalline cellulose (reprinted with permission from ref. 209, 2019. All rights reserved by Springer International Publishing). | ||
Guo et al. (2019) constructed a CBM-GFP-directed protein probe to quantitatively determine cellulose crystallinity. The binding concentrations of the different probes were determined using a fluorescence spectrophotometer, and the binding capability of the probes to substrates was calculated from the relative intensity using the equation209,210
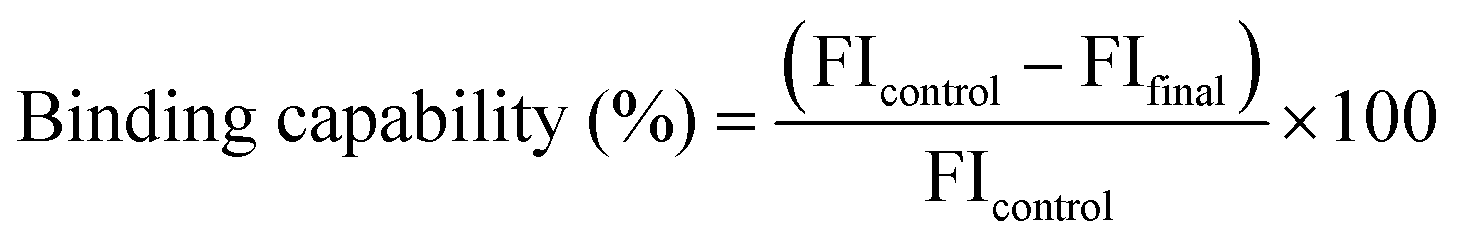 | (25) |
7. Other methods
Several physicochemical properties of cellulose such as specific gravity,211,212 enthalpy of wetting,213 water vapor sorption,214 iodine sorption,215 chemical reactivity,216 and others could also be used to estimate the crystalline fraction in the cellulose polymer. Some of these are discussed in the following.Specific gravity
Specific gravity is defined as the density of a substance compared to the density of water. Direct measurement of specific gravity of amorphous cellulose can be challenging due to the dependence of cellulose structure on factors such as the presence of impurities and residual moisture, different oxidation degrees, and drying methods. Thus, cellulose samples with known degrees of crystallinity can be used to estimate the crystallinity of the unknown samples. It is known that the relationship between the specific gravity of semi-crystalline polymers and their degree of crystallinity (X) can be expressed as follows: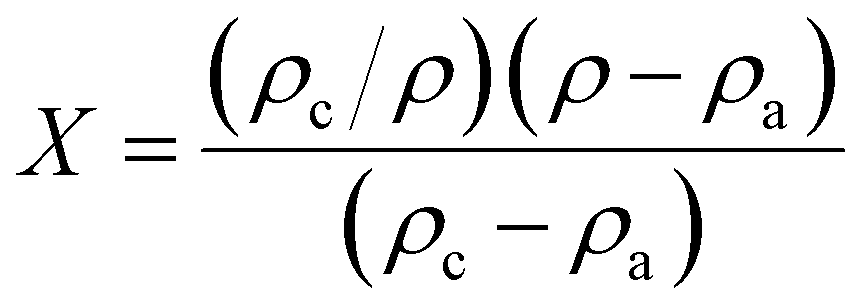 | (26) |
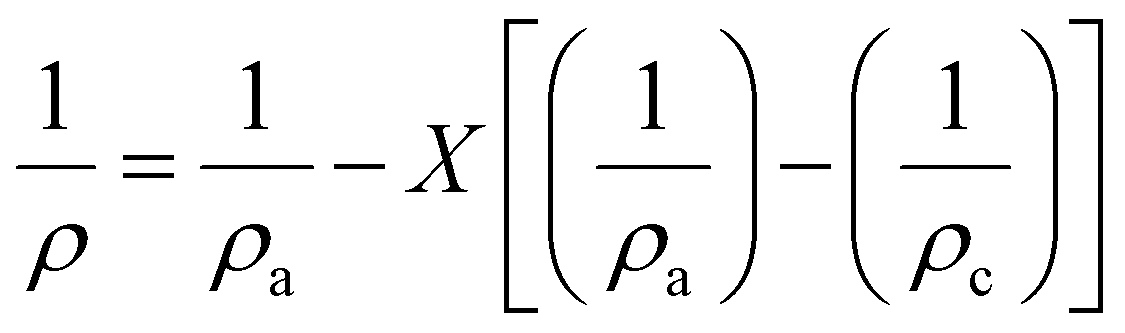 | (27) |
| V = Va − KX | (28) |
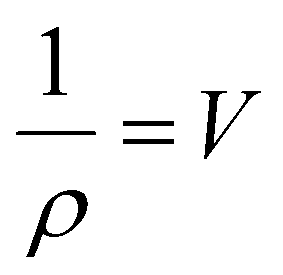 is the sample's specific volume and
is the sample's specific volume and 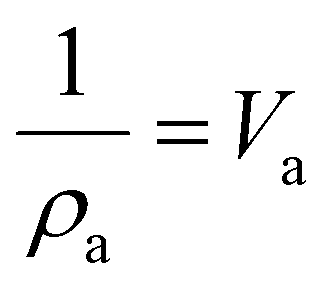 is the specific volume of the amorphous region, and
is the specific volume of the amorphous region, and 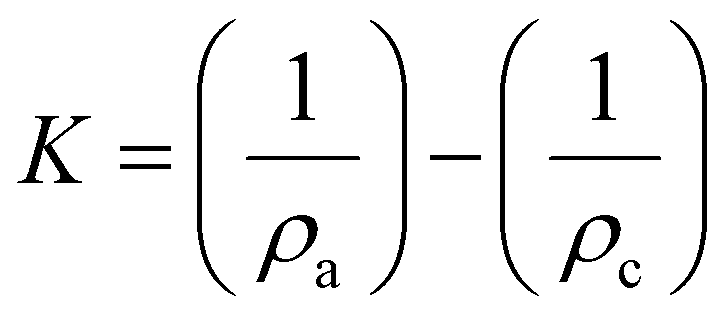 .
.
Thus, by measuring the specific gravity of some cellulose samples of known crystallinity, it is possible to obtain a straight line and calculate the specific gravity of the crystalline and amorphous regions. Ioelovich et al. (2010) used ρc = 1.62 g cm−3 and ρa = 1.44 g cm−3 to measure the cellulose crystallinity with this method. The results obtained agreed with the XRD results.211
Wetting enthalpy
It has been mentioned before that, the cellulose–water interaction is attributed to the amorphous regions of cellulose. Thus, the associated exothermic heat generation with this phenomenon or wetting enthalpy (ΔH) is directly proportional to the amorphous content (Y) of cellulose.| ΔH = ΔHAmY | (29) |
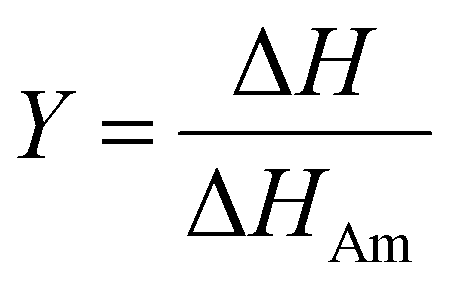 | (30) |
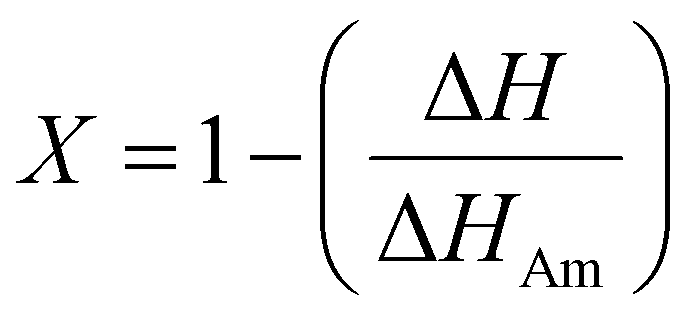 | (31) |
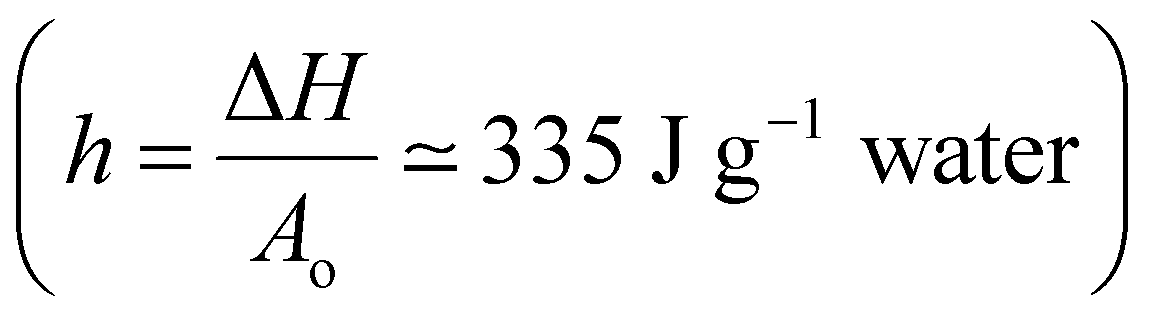 .
.
Thus, considering maximal absorption for amorphous cellulose, ΔHAm was determined as follows:
| ΔHAm = hAo | (32) |
8. Comparison among different techniques for measuring cellulose crystallinity
Cellulose crystallinity has long been playing an important role in defining a material's physical and chemical properties. Thus, the study of the crystalline structure of cellulose has received attention for its diversified application. Different techniques have also been used to measure the cellulose crystallinity index, all of which yield different values. These are now discussed in this section and compared, as shown in Fig. 15.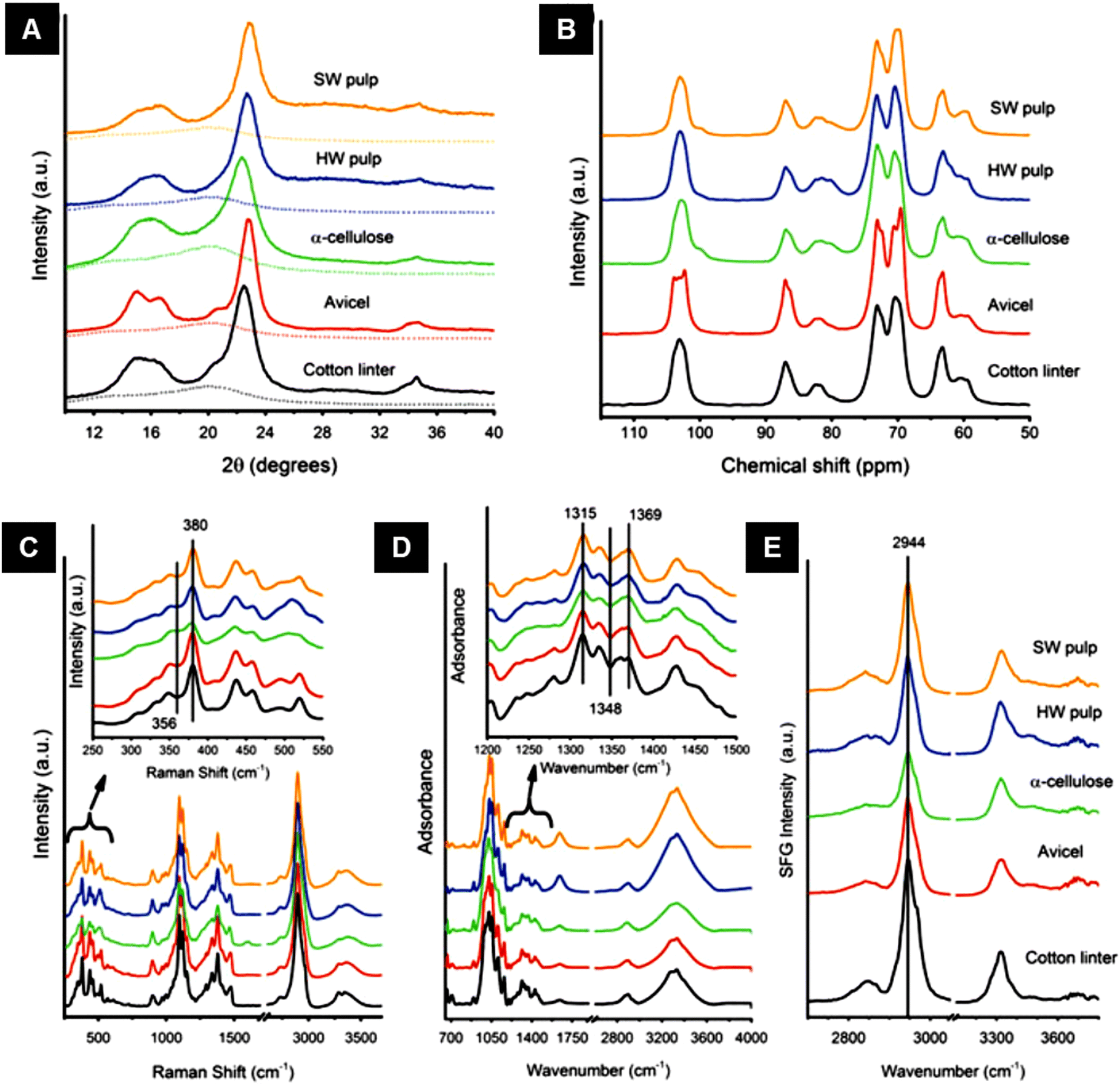 | ||
| Fig. 15 Crystallinity measurement of cellulose samples using (A) XRD, (B) 13C NMR, (C) Raman, (D) IR, and (E) SFG. Samples are cotton linter (black), Avicel (red), α-cellulose (green), hardwood (HW) pulp (blue), and softwood (SW) pulp (orange). In (A), the amorphous standard is plotted as a dotted line. The insets in figures (C) and (D) show the spectral regions used for CI calculations. All spectra are offset for clarity (Reprinted with permission from ref. 217, 2015. All rights reserved by Springer International Publishing). | ||
Cellulose pulps from different sources having different purity were taken and analyzed using XRD, NMR, IR, Raman, SFG, and DSC to measure their crystallinity index.177 Cotton linter (one of the purest sources of cellulose), which contain more than 95% of cellulose by dry mass, Avicel with a low degree of polymerization, α-cellulose (mostly cellulose Iβ), bleached hardwood (HW), and softwood (SW) pulps were taken for comparison. Fig. 15 shows the graphical representation of data produced while measuring the CI of cellulose using a range of different techniques, namely XRD, NMR, IR, Raman, and SFG. The CI values measured from XRD were calculated using the peak height (PH) and amorphous subtraction (AS) method, as shown in Table 6. For 13C NMR, the CI was measured by the relative intensity of the C4 peak chemical shifts of amorphous (80–85 ppm) and crystalline (85–89 ppm) cellulose. It can be observed from Table 6 that the methods used to measure the cellulose CI values show the same qualitative trends for samples, i.e., CI is the highest for cotton linter and lowest for α-cellulose. However, the absolute CI values differ for different methods, limiting direct quantitative comparison of the CI values estimated using different approaches. So, the cellulose crystallinity index measured for different samples using any technique can only be compared qualitatively.
| Sample | Calculated crystallinitya (%) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| XRD(PH) | XRD(AS) | NMR | Raman | IR | SGF | DSC | ||
| a Values obtained from Raman, IR, and SFG were correlated relative to the XRD values obtained using the peak height method (PH) and the amorphous subtraction method (AS). | ||||||||
| Cotton linter | 85 | 72 | 65 | 78 | 80 | 77 | 83.8 | 196,217 |
| Avicel | 81 | 70 | 58 | 77 | 75 | 71 | 80.37 | 196,217 |
| α-Cellulose | 65 | 48 | 44 | 65 | 67 | 65 | 63.73 | 196,217 |
| Hardwood fibre | 84 | 81 | 45 | 73 | 72 | 71 | 79.75 | 159,217 |
| Softwood fibre | 84 | 84 | 52 | 76 | 73 | 73 | 82.23 | 159,217 |
The CI values of HW and SW measured using the XRD AS method are very close to those measured using the PH method. However, there is a large difference in the CI values for other cellulose sources, as shown in Table 6. This was due to the high intensity of the amorphous background of the HW and SW fibres at a 2θ angle greater than 25°, whereas the other cellulosic sources exhibited low intensity in this region. As a result of this, the AS method was not able to subtract the amorphous background in the high 2θ region, and instead it effectively added the difference in the crystalline portion, which led to much higher CI values than the true mass fraction of crystalline cellulose over the total mass for HW and SW than the other samples.
Different CI values are obtained from different techniques for several reasons. NMR is likely to yield a much lower CI value compared to that calculated from XRD, even if the CI is measured by the AS method, as shown in Table 6. It has been found from different literature sources that the XRD method gives more precise data for the crystalline phase and shows a lesser sensitivity to the amorphous phase, consequently producing data having an inevitable inclination towards crystalline phases.15,217 The cellulose structure also contains paracrystalline cellulose, a more complicated structure than the crystalline and amorphous cellulose, which also interferes with the CI value measurement.125 In contrast, NMR shows equal sensitivity to cellulose structure's crystalline and amorphous phases. The difference in detection sensitivity between these two methods affects the CI values and is responsible for the discrepancy, as shown in Table 6.
The CI values measured using DSC were very close to those measured using XRD. Still, DSC has some limitations as it only takes account of the moisture present in the amorphous region of the cellulose and completely neglects the presence of moisture at the surface of the crystalline regions. Another limiting factor of DSC is that it measures only the sum of all heat flow events occurring at a particular time and temperature, which might result in hiding the exothermic crystallization of others. Thus, the method is most suitable for cellulose having low crystallinity.196 Therefore, it is clear from the discussion that none of the crystallinity measuring techniques can be used to obtain an absolute value. However, all the methods display similar trends in measuring the crystallinity values of cellulose samples. Therefore, the techniques overviewed can be used to provide qualitative data and a relative measurement of cellulose crystallinity.
9. Conclusions
The importance of defining the crystallinity of cellulose has been discussed in many reports and over many decades since the very early beginning of our understanding of this complex and ubiquitous polymer. Understanding the structure of cellulose facilitates physical modification to render high-performance products and increase product yields. This review paper has covered the different techniques used to determine cellulose crystallinity, namely X-ray diffraction (XRD), nuclear magnetic resonance (NMR), Fourier-transform infrared spectroscopy (FTIR, Raman), differential scanning calorimetry (DSC), sum-frequency generation vibrational spectroscopy (SFG), cellulose binding molecule (CBM) methods, specific gravity, and wetting enthalpy. Each crystallinity index can take different values depending on the technique selected for the analysis. Typically, CI values obtained from NMR are lower than values obtained from XRD measurements. However, relative crystallinity values and crystallinity trends are common for all techniques. For example, cotton linter, known as almost a fully crystalline structure, yields the highest CI according to the different methods presented. Moreover, other factors such as size of the crystallite, intensity of the incident beam (in the case of XRD), source and pretreatment of the sample before the tests also affect the crystallinity value. In this sense, despite many efforts to determine cellulose crystallinity, there is no agreement in the literature on the absolute technique for determining the crystallinity value and studying the cellulose structure. This fact reflects that cellulose crystallinity cannot be estimated using a single technique; therefore, combining different techniques to calculate the CI is likely the best approach. The use of software or computational analysis might be a vital tool in addition to the experimental data since the software can be designed to calculate and predict the intricate details and refine the data which is not possible experimentally. Consequently, they can complement the experimental findings by assisting the interpretation of the experimental data.Conflicts of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.Acknowledgements
This work is in part supported by McIntire-Stennis grant number NI21MSCFRXXXG054 from the USDA National Institute of Food and Agriculture. The work of Dr. Lokendra Pal is in part supported by E. J. “Woody” Rice Professorship Endowment. The authors also acknowledge the funding support from Ministry of Science and Technology (Project ID: SRG-222388), Bangladesh and University Grants Commision's research grant (2022-2023) for the faculty of University of Dhaka while carrying out the work of Dr. Salem partially in the Department of Applied Chemistry and Chemical Engineering, University of Dhaka. We also wish to acknowledge Dr. Peter W. Hart of WestRock for his generous financial support for the current review. The authors gratefully thank Mrittika Debnath for her support during the writing of this manuscript.References
- M. Brodin, M. Vallejos, M. T. Opedal, M. C. Area and G. Chinga-Carrasco, J. Cleaner Prod., 2017, 162, 646–664 CrossRef CAS.
- K. S. Salem, H. R. Starkey, L. Pal, L. Lucia and H. Jameel, ACS Sustainable Chem. Eng., 2020, 8, 1471–1478 CrossRef CAS.
- K. S. Salem, V. Naithani, H. Jameel, L. Lucia and L. Pal, Global chall., 2021, 5, 2000065 CrossRef PubMed.
- K. S. Salem, V. Naithani, H. Jameel, L. Lucia and L. Pal, Carbohydr. Polym., 2022, 295, 119856 CrossRef CAS PubMed.
- C. Chen, Y. Kuang, S. Zhu, I. Burgert, T. Keplinger, A. Gong, T. Li, L. Berglund, S. J. Eichhorn and L. Hu, Nat. Rev. Mater., 2020, 5, 642–666 CrossRef CAS.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- A. Etale, A. J. Onyianta, S. R. Turner and S. J. Eichhorn, Chem. Rev., 2023, 123, 2016–2048 CrossRef CAS PubMed.
- X. Ju, M. Bowden, E. E. Brown and X. Zhang, Carbohydr. Polym., 2015, 123, 476–481 CrossRef CAS PubMed.
- S. Rongpipi, D. Ye, E. D. Gomez and E. W. Gomez, Front. Plant Sci., 2019, 9, 1894 CrossRef PubMed.
- N. S. Çetin, P. Tingaut, N. Özmen, N. Henry, D. Harper, M. Dadmun and G. Sèbe, Macromol. Biosci., 2009, 9, 997–1003 CrossRef PubMed.
- W. Stelte and A. R. Sanadi, Ind. Eng. Chem. Res., 2009, 48, 11211–11219 CrossRef CAS.
- A. D. French, Cellulose, 2017, 24, 4605–4609 CrossRef CAS.
- E. Toumpanaki, D. U. Shah and S. J. Eichhorn, Adv. Mater., 2021, 33, 2001613 CrossRef CAS PubMed.
- A. D. French, M. Concha, M. K. Dowd and E. D. Stevens, Cellulose, 2014, 21, 1051–1063 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1–10 CrossRef PubMed.
- A. D. French, D. W. Montgomery, N. T. Prevost, J. V. Edwards and R. J. Woods, Carbohydr. Polym., 2021, 264, 118004 CrossRef CAS PubMed.
- Y. B. Park, K. Kafle, C. M. Lee, D. J. Cosgrove and S. H. Kim, Cellulose, 2015, 22, 3531–3540 CrossRef CAS.
- W. A. Sisson, Science, 1938, 87, 350 CrossRef CAS PubMed.
- A. Hirai, M. Tsuji and F. Horii, Cellulose, 2002, 9, 105–113 CrossRef CAS.
- A. Hirai, M. Tsuji and F. Horii, Cellulose, 1997, 4, 239–245 CrossRef CAS.
- J. Hayashi, A. Sufoka, J. Ohkita and S. Watanabe, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 23–27 CrossRef CAS.
- E. S. Gardiner and A. Sarko, Can. J. Chem., 1985, 63, 173–180 CrossRef CAS.
- J. S. Wilkie, Nature, 1961, 190, 1145–1150 CrossRef.
- O. L. Sponsler, J. Gen. Physiol., 1926, 9, 677–695 CrossRef CAS PubMed.
- K. H. Meyer and L. Misch, Helv. Chim. Acta, 1937, 20, 232–244 CrossRef CAS.
- A. D. French and P. Langan, Cellulose, 2014, 21, 1087–1089 CrossRef.
- S. Nishikawa and S. Ono, Proc. Tokyo Math.-Phys. Soc., 1913, 7, 131 CAS.
- S. H. Kim, C. M. Lee and K. Kafle, Korean J. Chem. Eng., 2013, 30, 2127–2141 CrossRef CAS.
- P. J. Weimer, J. M. Hackney and A. D. French, Biotechnol. Bioeng., 1995, 48, 169–178 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- S. Andersson, R. Serimaa, T. Paakkari, P. Saranpää and E. Pesonen, J. Wood Sci., 2003, 49, 531–537 CrossRef.
- M. Debnath, K. S. Salem, V. Naithani, E. Musten, M. A. Hubbe and L. Pal, Cellulose, 2021, 12, 7981–7994 CrossRef.
- X. Sun, S. Agate, K. S. Salem, L. Lucia and L. Pal, ACS Appl. Bio Mater., 2020, 4, 140–162 CrossRef PubMed.
- P. Tyagi, K. S. Salem, M. A. Hubbe and L. Pal, Trends Food Sci. Technol., 2021, 115, 461–485 CrossRef CAS.
- S. Al-Zuhair, Bioresour. Technol., 2008, 99, 4078–4085 CrossRef CAS PubMed.
- Y. Cao and H. Tan, Enzyme Microb. Technol., 2005, 36, 314–317 CrossRef CAS.
- N. Lavoine, I. Desloges, A. Dufresne and J. Bras, Carbohydr. Polym., 2012, 90, 735–764 CrossRef CAS PubMed.
- M. Hall, P. Bansal, J. H. Lee, M. J. Realff and A. S. Bommarius, FEBS J., 2010, 277, 1571–1582 CrossRef CAS PubMed.
- A. French, BioResources, 2022, 17, 5557–5561 CAS.
- Z. Ling, T. Wang, M. Makarem, M. Santiago Cintrón, H. N. Cheng, X. Kang, M. Bacher, A. Potthast, T. Rosenau, H. King, C. D. Delhom, S. Nam, J. Vincent Edwards, S. H. Kim, F. Xu and A. D. French, Cellulose, 2019, 26, 305–328 CrossRef CAS.
- M. Borrega, P. Ahvenainen and E. Kontturi, Cellulose, 2018, 25, 6811–6818 CrossRef CAS.
- Y. Nishiyama, U.-J. Kim, D.-Y. Kim, K. S. Katsumata, R. P. May and P. Langan, Biomacromolecules, 2003, 4, 1013–1017 CrossRef CAS PubMed.
- P. Bansal, M. Hall, M. J. Realff, J. H. Lee and A. S. Bommarius, Bioresour. Technol., 2010, 101, 4461–4471 CrossRef CAS PubMed.
- R. Evans, A. F. A. Wallis, R. H. Newman, U. C. Roick and I. D. Suckling, Holzforschung, 1995, 49, 498–504 CrossRef CAS.
- A. Kljun, T. A. S. Benians, F. Goubet, F. Meulewaeter, J. P. Knox and R. S. Blackburn, Biomacromolecules, 2011, 12, 4121–4126 CrossRef CAS PubMed.
- C. Driemeier and G. A. Calligaris, J. Appl. Crystallogr., 2011, 44, 184–192 CrossRef CAS.
- A. D. French, Cellulose, 2014, 21, 885–896 CrossRef CAS.
- A. D. French and M. Santiago Cintrón, Cellulose, 2013, 20, 583–588 CrossRef CAS.
- R. C. Rowe, A. G. McKillop and D. Bray, Int. J. Pharm., 1994, 101, 169–172 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- A. Thygesen, J. Oddershede, H. Lilholt, A. B. Thomsen and K. Ståhl, Cellulose, 2005, 12, 563–576 CrossRef CAS.
- T. Liitiä, S. L. Maunu and B. Hortling, Holzforschung, 2000, 54, 618–624 Search PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- F. Horii, A. Hirai and R. Kitamaru, Macromolecules, 1987, 20, 2117–2120 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1–10 CrossRef PubMed.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- A. Kirui, Z. Ling, X. Kang, M. C. Dickwella Widanage, F. Mentink-Vigier, A. D. French and T. Wang, Cellulose, 2019, 26, 329–339 CrossRef CAS PubMed.
- U. P. Agarwal, R. S. Reiner and S. A. Ralph, Cellulose, 2010, 17, 721–733 CrossRef CAS.
- S. H. D. Hulleman, J. M. Van Hazendonk and J. E. G. Van Dam, Carbohydr. Res., 1994, 261, 163–172 CrossRef CAS.
- U. P. Agarwal, R. R. Reiner and S. A. Ralph, J. Agric. Food Chem., 2013, 61, 103–113 CrossRef CAS PubMed.
- A. L. Barnette, L. C. Bradley, B. D. Veres, E. P. Schreiner, Y. B. Park, J. Park, S. Park and S. H. Kim, Biomacromolecules, 2011, 12, 2434–2439 CrossRef CAS PubMed.
- C. M. Lee, N. M. A. Mohamed, H. D. Watts, J. D. Kubicki and S. H. Kim, J. Phys. Chem. B, 2013, 117, 6681–6692 CrossRef CAS PubMed.
- M. S. Bertran and B. E. Dale, J. Appl. Polym. Sci., 1986, 32, 4241–4253 CrossRef CAS.
- A. Kljun, T. A. S. Benians, F. Goubet, F. Meulewaeter, J. P. Knox and R. S. Blackburn, Biomacromolecules, 2011, 12, 4121–4126 CrossRef CAS PubMed.
- M. M. Rehman, M. Zeeshan, K. Shaker and Y. Nawab, Cellulose, 2019, 26, 9057–9069 CrossRef CAS.
- H.-J. Kim, S. Roy and J.-W. Rhim, J. Environ. Chem. Eng., 2021, 9, 106043 CrossRef CAS.
- L. T. Fan, Y.-H. Lee and D. H. Beardmore, Biotechnol. Bioeng., 1980, 22, 177–199 CrossRef CAS.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed.
- A. D. French, Cellulose, 2020, 27, 5445–5448 CrossRef.
- P. Langan, Y. Nishiyama and H. Chanzy, Biomacromolecules, 2001, 2, 410–416 CrossRef CAS PubMed.
- P. Ahvenainen, I. Kontro and K. Svedström, Cellulose, 2016, 23, 1073–1086 CrossRef CAS.
- K. S. Salem, H. Jameel, L. Lucia and L. Pal, Mater. Today Sust., 2023, 22, 100342 Search PubMed.
- W. Yao, Y. Weng and J. M. Catchmark, Cellulose, 2020, 27, 5563–5579 CrossRef CAS.
- W. R. Kunusa, I. Isa, L. A. Laliyo and H. Iyabu, J. Phys.: Conf. Ser., 2018, 1028, 012199 CrossRef.
- S. Y. Oh, I. Y. Dong, Y. Shin, C. K. Hwan, Y. K. Hak, S. C. Yong, H. P. Won and H. Y. Ji, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef CAS PubMed.
- R. P. Oliveira and C. Driemeier, J. Appl. Crystallogr., 2013, 46, 1196–1210 CrossRef CAS.
- K. Das, D. Ray, N. R. Bandyopadhyay and S. Sengupta, J. Polym. Environ., 2010, 18, 355–363 CrossRef CAS.
- M. Holtz, C. Hauf, J. Weisshaupt, A.-A. H. Salvador, M. Woerner and T. Elsaesser, Struct. Dyn., 2017, 4, 054304 CrossRef PubMed.
- V. W. Tripp, Cellulose and cellulose derivatives, 1971, vol. 5, pp. 305–323 Search PubMed.
- P. Scherrer, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 1918, 98–100 Search PubMed.
- Y. Wang, Y. Zhao and Y. Deng, Carbohydr. Polym., 2008, 72, 178–184 CrossRef CAS.
- G. V. Gusev, Polym. Sci. U.S.S.R., 1978, 20, 1295–1297 CrossRef.
- L. E. Hult, T. Iversen and J. Sugiyama, Cellulose, 2003, 10, 103–110 CrossRef.
- C. J. Garvey, I. H. Parker and G. P. Simon, Macromol. Chem. Phys., 2005, 206, 1568–1575 CrossRef CAS.
- J. He, S. Cui and S-Y. Wang, J. Appl. Polym. Sci., 2008, 107, 1029–1038 CrossRef CAS.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed.
- H.-P. Fink, D. Hofmann and B. Philipp, Cellulose, 1995, 2, 51–70 CrossRef CAS.
- J. He, S. Cui and S. Wang, J. Appl. Polym. Sci., 2008, 107, 1029–1038 CrossRef CAS.
- E. Gümüskaya, M. Usta and H. Kirci, Polym. Degrad. Stab., 2003, 81, 559–564 CrossRef.
- L. Lutterotti, M. Bortolotti, G. Ischia, I. Lonardelli and H.-R. Wenk, Zeitschrift für Kristallographie Supplements, 2007, 26, 125–130 CrossRef.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. D. French, Cellulose, 2020, 27, 5445–5448 CrossRef.
- M. Wada, T. Okano and J. Sugiyama, J. Wood Sci., 2001, 47, 124–128 CrossRef CAS.
- A. Isogai and R. H. Atalla, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 113–119 CrossRef CAS.
- C. Driemeier, Cellulose, 2014, 21, 1065–1073 CrossRef CAS.
- Y. Nishiyama, G. P. Johnson and A. D. French, Cellulose, 2012, 19, 319–336 CrossRef CAS.
- A. L. Barnette, C. Lee, L. C. Bradley, E. P. Schreiner, Y. B. Park, H. Shin, D. J. Cosgrove, S. Park and S. H. Kim, Carbohydr. Polym., 2012, 89, 802–809 CrossRef CAS PubMed.
- S. Wu, X. Jiang, H. Jiang, S. Wu, S. Ding and Y. Jin, Cellulose, 2021, 28, 1947–1959 CrossRef CAS.
- N. Terinte, R. Ibbett and K. C. Schuster, Lenzinger Ber., 2011, 89, 118–131 CAS.
- U. P. Agarwal, S. A. Ralph, C. Baez, R. S. Reiner and S. P. Verrill, Cellulose, 2017, 24, 1971–1984 CrossRef CAS.
- S. Nam, A. D. French, B. D. Condon and M. Concha, Carbohydr. Polym., 2016, 135, 1–9 CrossRef CAS PubMed.
- Z. Ling, S. Chen, X. Zhang, K. Takabe and F. Xu, Sci. Rep., 2017, 7, 1–11 CrossRef CAS PubMed.
- Z. Ling, S. Chen, X. Zhang and F. Xu, Bioresour. Technol., 2017, 224, 611–617 CrossRef CAS PubMed.
- X. Guo, F. Yang, H. Liu, Y. Hou, Y. Wang, J. Sun, X. Chen, Y. Liu and X. Li, Macromol. Res., 2019, 27, 377–385 CrossRef CAS.
- T. Paakkari, M. Blomberg, R. Serimaa and M. Järvinen, J. Appl. Crystallogr., 1988, 21, 393–397 CrossRef CAS.
- W. Ren, F. Guo, J. Zhu, M. Cao, H. Wang and Y. Yu, Cellulose, 2021, 28, 5993–6005 CrossRef CAS.
- S. Nomura, Y. Kugo and T. Erata, Cellulose, 2020, 27, 3553–3563 CrossRef CAS.
- S. Haslinger, S. Hietala, M. Hummel, S. L. Maunu and H. Sixta, Carbohydr. Polym., 2019, 207, 11–16 CrossRef CAS PubMed.
- M. Tyufekchiev, A. Kolodziejczak, P. Duan, M. Foston, K. Schmidt-Rohr and M. T. Timko, Green Chem., 2019, 21, 5541–5555 RSC.
- T. Sparrman, L. Svenningsson, K. Sahlin-Sjövold, L. Nordstierna, G. Westman and D. Bernin, Cellulose, 2019, 26, 8993–9003 CrossRef CAS.
- H. Zhao, J. H. Kwak, Z. Conrad Zhang, H. M. Brown, B. W. Arey and J. E. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS.
- M. Foston, Curr. Opin. Biotechnol, 2014, 27, 176–184 CrossRef CAS PubMed.
- N. E. Jacobsen, NMR spectroscopy explained: simplified theory, applications and examples for organic chemistry and structural biology, John Wiley & Sons, 2007 Search PubMed.
- J. W. Akitt and B. E. Mann, NMR and Chemistry: An introduction to modern NMR spectroscopy, CRC Press, 2017 Search PubMed.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid-state NMR: Basic principles and practice, Momentum Press, 2012 Search PubMed.
- L. Schröder and C. Faber, In vivo NMR Imaging, Humana Press, 2011 Search PubMed.
- S. H. Kim, C. M. Lee and K. Kafle, Korean J. Chem. Eng., 2013, 30, 2127–2141 CrossRef CAS.
- K. Zia, T. Siddiqui, S. Ali, I. Farooq, M. S. Zafar and Z. Khurshid, Eur. J. Dent., 2019, 13, 124–128 CrossRef PubMed.
- Introduction to Solid-State NMR Spectroscopy|Wiley, https://www.wiley.com/en-us/Introduction+to+Solid+State+NMR+Spectroscopy-p-9781405109147, accessed April 4, 2023.
- W. L. Earl and D. L. VanderHart, Macromolecules, 1981, 14, 570–574 CrossRef CAS.
- D. L. VanderHart and R. H. Atalla, Macromolecules, 1984, 17, 1465–1472 CrossRef CAS.
- M. Foston, Curr. Opin. Biotechnol, 2014, 27, 176–184 CrossRef CAS PubMed.
- R. H. Newman and J. A. Hemmingson, Holzforschung, 1990, 44, 351–356 CrossRef CAS.
- H. Yang, T. Wang, D. Oehme, L. Petridis, M. Hong and J. D. Kubicki, Cellulose, 2018, 25, 23–36 CrossRef CAS.
- P. T. Larsson, K. Wickholm and T. Iversen, Carbohydr. Res., 1997, 302, 19–25 CrossRef CAS.
- K. Wickholm, P. T. Larsson and T. Iversen, Carbohydr. Res., 1998, 312, 123–129 CrossRef CAS.
- C. Y. Liang and R. H. Marchessault, J. Polym. Sci., 1959, 39, 269–278 CrossRef CAS.
- M. El Hariri El Nokab, M. H. Habib, Y. A. Alassmy, M. M. Abduljawad, K. M. Alshamrani and K. O. Sebakhy, Polymers, 2022, 14, 1049 CrossRef CAS PubMed.
- M. Ghosh, B. P. Prajapati, R. K. Suryawanshi, K. Kishor Dey and N. Kango, Chem. Phys. Lett., 2019, 727, 105–115 CrossRef CAS.
- A. Palme, A. Idström, L. Nordstierna and H. Brelid, Cellulose, 2014, 21, 4681–4691 CrossRef CAS.
- S. Park, D. K. Johnson, C. I. Ishizawa, P. A. Parilla and M. F. Davis, Cellulose, 2009, 16, 641–647 CrossRef CAS.
- R. Teeäär, R. Serimaa and T. Paakkarl, Polym. Bull., 1987, 17, 231–237 CrossRef.
- R. H. Atalla and D. L. VanderHart, Solid State Nucl. Magn. Reson., 1999, 15, 1–19 CrossRef CAS PubMed.
- F. Horii, A. Hirai and R. Kitamaru, J. Carbohydr. Chem., 1984, 3, 641–662 CrossRef CAS.
- S. Sternhell and M. Mehring, Org. Magn. Reson., 1983, 21, 770 CrossRef.
- C. Rondeau-Mouro, B. Bouchet, B. Pontoire, P. Robert, J. Mazoyer and A. Buléon, Carbohydr. Polym., 2003, 53, 241–252 CrossRef CAS.
- L. R. Schroeder, V. M. Gentile and R. H. Atalla, J. Wood Chem. Technol., 1986, 6, 1–14 CrossRef CAS.
- R. H. Newman and J. A. Hemmingson, Cellulose, 1995, 2, 95–110 CrossRef CAS.
- K. Masuda, M. Adachi, A. Hirai, H. Yamamoto, H. Kaji and F. Horii, Solid State Nucl. Magn. Reson., 2003, 23, 198–212 CrossRef CAS PubMed.
- A. N. Fernandes, L. H. Thomas, C. M. Altaner, P. Callow, V. T. Forsyth, D. C. Apperley, C. J. Kennedy and M. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1195–E1203 CrossRef PubMed.
- M. Foston, R. Katahira, E. Gjersing, M. F. Davis and A. J. Ragauskas, J. Agric. Food Chem., 2012, 60, 1419–1427 CrossRef CAS PubMed.
- I. Jankowska, P. Ławniczak, K. Pogorzelec-Glaser, A. Łapiński, R. Pankiewicz and J. Tritt-Goc, Mater. Chem. Phys., 2020, 239, 122056 CrossRef CAS.
- G. Cavallaro, A. Agliolo Gallitto, L. Lisuzzo and G. Lazzara, Cellulose, 2019, 26, 8853–8865 CrossRef.
- A. L. P. Queiroz, B. M. Kerins, J. Yadav, F. Farag, W. Faisal, M. E. Crowley, S. E. Lawrence, H. A. Moynihan, A.-M. Healy, S. Vucen and A. M. Crean, Cellulose, 2021, 28, 8971–8985 CrossRef CAS PubMed.
- U. P. Agarwal, S. A. Ralph, C. Baez and R. S. Reiner, Cellulose, 2021, 28, 9069–9079 CrossRef CAS.
- U. P. Agarwal, S. A. Ralph, C. Baez and R. S. Reiner, Biomacromolecules, 2021, 22, 1357–1373 CrossRef CAS PubMed.
- B. Stuart, Analytical techniques in the sciences. Infrared Spectroscopy: Fundamentals and Applications, 2004 Search PubMed.
- C. N. Banwell and M. Elaine, Fundamentals of molecular spectroscopy, McGraw-Hill, New York, 1994 Search PubMed.
- P. Larkin, Infrared and Raman spectroscopy: principles and spectral interpretation, Elsevier, 2017 Search PubMed.
- C. L. Haynes, A. D. McFarland and R. P. Van Duyne, Anal. Chem., 2005, 338–346 CrossRef.
- P. Rostron, S. Gaber and D. Gaber, Laser, 2016, 21, 24.
- D. Srivastava, M. S. Kuklin, J. Ahopelto and A. J. Karttunen, Carbohydr. Polym., 2020, 243, 116440 CrossRef CAS PubMed.
- A. Jorio, R. Saito, J. H. Hafner, C. M. Lieber, M. Hunter, T. McClure, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. Lett., 2001, 86, 1118–1121 CrossRef CAS PubMed.
- S. J. Eichhorn and R. J. Young, Compos. Sci. Technol., 2004, 64, 767–772 CrossRef CAS.
- R. J. Young, D. Lu and R. J. Day, Polym. Int., 1991, 24, 71–76 CrossRef CAS.
- J. Široký, R. S. Blackburn, T. Bechtold, J. Taylor and P. White, Cellulose, 2010, 17, 103–115 CrossRef.
- A. A. Saapan, S. H. Kandil and A. M. Habib, Text. Res. J., 1984, 54, 863–867 CrossRef.
- R. T. O’Connor, E. F. DuPré and D. Mitcham, Text. Res. J., 1958, 28, 382–392 CrossRef.
- D. Ciolacu, F. Ciolacu and V. I. Popa, Cellul. Chem. Technol., 2011, 45, 13 CAS.
- M. Fan, D. Dai and B. Huang, Fourier transform-materials analysis, 2012 Search PubMed.
- M. L. Nelson and R. T. O’Connor, J. Appl. Polym. Sci., 1964, 8, 1325–1341 CrossRef CAS.
- T. Kondo and C. Sawatari, Polymer, 1996, 37, 393–399 CrossRef CAS.
- U. Richter, T. Krause and W. Schempp, Die Angewandte Makromolekulare Chemie, Appl. Macromol. Chem. Phys., 1991, 185, 155–167 Search PubMed.
- J. Li, X. Wei, Q. Wang, J. Chen, G. Chang, L. Kong, J. Su and Y. Liu, Carbohydr. Polym., 2012, 90, 1609–1613 CrossRef CAS PubMed.
- I. Reiniati, A. N. Hrymak and A. Margaritis, Biochem. Eng. J., 2017, 127, 21–31 CrossRef CAS.
- M. J. Shultz, Advances In Multi-Photon Processes And Spectroscopy, 2008, pp. 133–199 Search PubMed.
- M. Makarem, C. M. Lee, K. Kafle, S. Huang, I. Chae, H. Yang, J. D. Kubicki and S. H. Kim, Cellulose, 2019, 26, 35–79 CrossRef CAS.
- J. Choi, J. Lee, M. Makarem, S. Huang and S. H. Kim, J. Phys. Chem. B, 2022, 126, 6629–6641 CrossRef CAS PubMed.
- I. Chae, S. M. Q. Bokhari, X. Chen, R. Zu, K. Liu, A. Borhan, V. Gopalan, J. M. Catchmark and S. H. Kim, Carbohydr. Polym., 2021, 255, 117328 CrossRef CAS PubMed.
- Y. R. Shen, Nature, 1989, 337, 519 CrossRef CAS.
- S. A. Denev, T. T. A. Lummen, E. Barnes, A. Kumar and V. Gopalan, J. Am. Ceram. Soc., 2011, 94, 2699–2727 CrossRef CAS.
- H. C. Hieu, N. A. Tuan, H. Li, Y. Miyauchi and G. Mizutani, Appl. Spectrosc., 2011, 65, 1254–1259 CrossRef CAS PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS.
- V. Cedric, C. Yves and P. Andre, Biosensors – Emerging Materials and Applications, 2011 Search PubMed.
- G. L. Richmond, Chem. Rev., 2002, 102, 2693–2724 CrossRef CAS PubMed.
- C. T. Williams and D. A. Beattie, Surf. Sci., 2002, 500, 545–576 CrossRef CAS.
- O. J. Rojas, Cellulose Chemistry and Properties: Fibers, Nanocelluloses and Advanced Materials, 2016 Search PubMed.
- C. M. Lee, A. Mittal, A. L. Barnette, K. Kafle, Y. B. Park, H. Shin, D. K. Johnson, S. Park and S. H. Kim, Cellulose, 2013, 20, 991–1000 CrossRef CAS.
- Y. B. Park, C. M. Lee, B. W. Koo, S. Park, D. J. Cosgrove and S. H. Kim, Plant Physiol., 2013, 163, 907–913 CrossRef CAS PubMed.
- C. M. Lee, K. Kafle, Y. B. Park and S. H. Kim, Phys. Chem. Chem. Phys., 2014, 16, 10844–10853 RSC.
- S. Huang, S. N. Kiemle, M. Makarem and S. H. Kim, Cellulose, 2020, 27, 57–69 CrossRef CAS.
- M. Makarem, C. M. Lee, D. Sawada, H. M. O’Neill and S. H. Kim, J. Phys. Chem. Lett., 2018, 9, 70–75 CrossRef CAS PubMed.
- T. De Assis, S. Huang, C. E. Driemeier, B. S. Donohoe, C. Kim, S. H. Kim, R. Gonzalez, H. Jameel and S. Park, Biotechnol. Biofuels, 2018, 11, 1–11 CrossRef PubMed.
- M. Makarem, D. Sawada, H. M. O’Neill, C. M. Lee, K. Kafle, Y. B. Park, A. Mittal and S. H. Kim, J. Phys. Chem. C, 2017, 121, 10249–10257 CrossRef CAS.
- X. Chen, C. M. Lee, H. F. Wang, L. Jensen and S. H. Kim, J. Phys. Chem. C, 2017, 121, 18876–18886 CrossRef CAS.
- K. Kafle, R. Shi, C. M. Lee, A. Mittal, Y. B. Park, Y. H. Sun, S. Park, V. Chiang and S. H. Kim, Cellulose, 2014, 21, 2219–2231 CrossRef CAS.
- L. Kong, C. Lee, S. H. Kim and G. R. Ziegler, J. Phys. Chem. B, 2014, 118, 1775–1783 CrossRef CAS PubMed.
- P. P. Handakumbura, D. A. Matos, K. S. Osmont, M. J. Harrington, K. Heo, K. Kafle, S. H. Kim, T. I. Baskin and S. P. Hazen, BMC Plant Biol., 2013, 13, 1–6 CrossRef PubMed.
- K. Kafle, C. M. Lee, H. Shin, J. Zoppe, D. K. Johnson, S. H. Kim and S. Park, BioEnergy Res., 2015, 8, 1750–1758 CrossRef CAS.
- J. Xie, Y. Li, W. Wang, S. Pan, N. Cui and J. Liu, Adv. Mech. Eng., 2020, 44, 125001 Search PubMed.
- C. Schick, Anal. Bioanal. Chem., 2009, 395, 1589–1611 CrossRef CAS PubMed.
- G. W. H. Hljhne, Thermochim. Acta, 1991, 187, 283–292 CrossRef.
- H. J. Flammersheim, W. F. Hemminger and G. W. H. Hohne, Differential Scanning Calorimetry, 2003 Search PubMed.
- Y. Kong and J. N. Hay, Polymer, 2002, 43, 3873–3878 CrossRef CAS.
- S. Chapi, S. Raghu and H. Devendrappa, Ionics, 2016, 22, 803–814 CrossRef CAS.
- M. S. Bertran and B. E. Dale, J. Appl. Polym. Sci., 1986, 32, 4241–4253 CrossRef CAS.
- T. Hatakeyama and H. Hatakeyama, Thermal Properties of Green Polymers, Hot Topics in Thermal Analysis and Calorimetry, 2005 Search PubMed.
- D. J. Blundell, D. R. Beckett and P. H. Willcocks, Polymer, 1981, 22, 704–707 CrossRef CAS.
- V. B. F. Mathot and M. F. J. Pijpers, Thermochim. Acta, 1989, 151, 241–259 CrossRef CAS.
- L. Szcześniak, A. Rachocki and J. Tritt-Goc, Cellulose, 2008, 15, 445–451 CrossRef.
- K. Aïssa, Doctoral dissertation, University of British Columbia, 2020.
- G. T. Beckham, J. F. Matthews, B. Peters, Y. J. Bomble, M. E. Himmel and M. F. Crowley, J. Phys. Chem. B, 2011, 115, 4118–4127 CrossRef CAS PubMed.
- S. Gao, C. You, S. Renneckar, J. Bao and Y.-H. P. Zhang, Biotechnol. Biofuels, 2014, 7, 24 CrossRef PubMed.
- J. Hong, X. Ye and Y.-H. P. Zhang, Langmuir, 2007, 23, 12535–12540 CrossRef CAS PubMed.
- T. Li, N. Liu, X. Ou, X. Zhao, F. Qi, J. Huang and D. Liu, Biotechnol. Biofuels, 2018, 11, 105 CrossRef PubMed.
- A. Kljun, T. A. S. Benians, F. Goubet, F. Meulewaeter, J. P. Knox and R. S. Blackburn, Biomacromolecules, 2011, 12, 4121–4126 CrossRef CAS PubMed.
- J. Široký, R. S. Blackburn, T. Bechtold, J. Taylor and P. White, Cellulose, 2010, 17, 103–115 CrossRef.
- J. Široký, T. A. S. Benians, S. J. Russell, T. Bechtold, J. Paul Knox and R. S. Blackburn, Carbohydr. Polym., 2012, 89, 213–221 CrossRef PubMed.
- X. Guo, F. Yang, H. Liu, Y. Hou, Y. Wang, J. Sun, X. Chen, Y. Liu and X. Li, Macromol. Res., 2019, 27, 377–385 CrossRef CAS.
- T. Kawakubo, S. Karita, Y. Araki, S. Watanabe, M. Oyadomari, R. Takada, F. Tanaka, K. Abe, T. Watanabe, Y. Honda and T. Watanabe, Biotechnol. Bioeng., 2010, 105, 499–508 CrossRef CAS PubMed.
- M. Ioelovich, A. Leykin and O. Figovsky, BioResources, 2010, 5, 3 Search PubMed.
- J. W. Hearle and W. E. Morton, Physical properties of textile fibres, Elsevier, 2008 Search PubMed.
- M. Ioelovich, Am. J. BioSci., 2014, 2, 6 CAS.
- M. Ioelovich, ChemXpress, 2016, 9, 245–251 CAS.
- L. E. Hessler and R. E. Power, Text. Res. J., 1954, 24, 822–827 CrossRef CAS.
- M. Ioelovich, BioResources, 2009, 4, 3 Search PubMed.
- C. Lee, K. Dazen, K. Kafle, A. Moore, D. K. Johnson, S. Park and S. H. Kim, in Cellulose Chemistry and Properties: Fibers, Nanocelluloses and Advanced Materials, ed. O. J. Rojas, Springer International Publishing, Cham, 2016, pp. 115–131 Search PubMed.
Footnote |
| † Author KSS and NKK have equally contributed to the conceptualizations, writing, drawing figures and editing. |
| This journal is © The Royal Society of Chemistry 2023 |