
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-covalent interactions (NCIs) in π-conjugated functional materials: advances and perspectives†
Ashanul
Haque
 *a,
Khalaf M.
Alenezi
a,
Muhammad S.
Khan
*b,
Wai-Yeung
Wong
*c and
Paul R.
Raithby
*d
*a,
Khalaf M.
Alenezi
a,
Muhammad S.
Khan
*b,
Wai-Yeung
Wong
*c and
Paul R.
Raithby
*d
aDepartment of Chemistry, College of Science, University of Hail, Kingdom of Saudi Arabia. E-mail: a.haque@uoh.edu.sa; haque_chem@yahoo.com
bDepartment of Chemistry, Sultan Qaboos University, Al-Khod, Muscat, Oman. E-mail: msk@squ.edu.om
cDepartment of Applied Biology and Chemical Technology and Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, P. R. China. E-mail: wai-yeung.wong@polyu.edu.hk
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, Avon BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk
First published on 3rd January 2023
Abstract
The design and development of functional materials with real-life applications are highly demanding. Understanding and controlling inter- and intra-molecular interactions provide opportunities to design new materials. A judicious manipulation of the molecular structure significantly alters such interactions and can boost selected properties and functions of the material. There is burgeoning evidence of the beneficial effects of non-covalent interactions (NCIs), showing that manipulating NCIs may generate functional materials with a wide variety of physical properties leading to applications in catalysis, drug delivery, crystal engineering, etc. This prompted us to review the implications of NCIs on the molecular packing, optical properties, and applications of functional π-conjugated materials. To this end, this tutorial review will cover different types of interactions (electrostatic, π-interactions, metallophilic, etc.) and their impact on π-conjugated materials. Attempts have also been made to delineate the effects of weak interactions on opto-electronic (O-E) applications.
 Ashanul Haque | Dr Haque has obtained his early education in Bihar, a bachelor's degree from Jamia Millia Islamia, New Delhi and a master's degree from Jamia Hamdard University, New Delhi. He completed his PhD in organic chemistry at Jamia Millia Islamia, New Delhi, in 2014 and conducted post-doctoral research with Prof. Khan at Sultan Qaboos University, Oman. In 2018, Dr Haque joined the University of Hail, Saudi Arabia, where he is currently an Associate Professor in the Dept. of Chemistry. He is an Associate member of the Royal Society of Chemistry (AMRSC) and was listed among the Top 2% for citation impact in a single year (2021). His research focuses on the development of functional compounds that are useful in daily life. |
 Khalaf M. Alenezi | Professor Khalaf M. Alenezi received his BSc (Chemistry, 1999) and MSc (Analytical Chemistry, 2007) degree from King Saud University, Saudi Arabia. After that he joined Prof. Chris Pickett's group at the University of East Anglia, UK and obtained his PhD degree in 2014. He is currently a Professor in the Department of Chemistry at the University of Hail, Saudi Arabia. Since 2022, he has been the Dean of the College of Sciences at University of Hail. His current research focuses on the design and development of electrocatalysts for the hydrogen evolution reaction (HER) and carbon dioxide (CO2) conversion and co-authored 41 publications. |
 Muhammad S. Khan | Professor Muhammad S. Khan was an 1851 Exhibition Scholar and received his PhD (1983) from the University of Cambridge, U.K. Currently he is a Professor of Inorganic Chemistry at the Sultan Qaboos University, Oman. He has received many medals and awards, the recent ones include the Best Researcher Award, College of Science, SQU (2005, 2017), the National Research Award, The Research Council, Oman (2018) and the Best Research Group Award, Deanship of Research, SQU. His research interests include the design of novel sustainable energy materials incorporating conjugated organic, organometallic polymers, and lanthanide coordination complexes for opto-electronic applications. |
 Wai-Yeung Wong | Professor Wai-Yeung Wong obtained his BSc(Hons) and PhD degrees from the University of Hong Kong. After postdoctoral work at Texas A&M University (with Prof. F. A. Cotton) and the University of Cambridge (with Prof. The Lord Lewis and P. R. Raithby), he joined Hong Kong Baptist University from 1998 to 2016 and now works at the Hong Kong Polytechnic University as the Dean of Faculty of Science and Chair Professor of Chemical Technology. Among his awards are the RSC Chemistry of the Transition Metals Award, FACS Distinguished Young Chemist Award, State Natural Science Award from China and RGC Senior Research Fellow Award. His research focusses on synthetic inorganic/organometallic chemistry, aiming at developing photofunctional metal-organic molecules and polymers for optoelectronic and energy-related applications. |
 Paul R. Raithby | Professor Paul Raithby is an Emeritus Professor of Inorganic Chemistry at the University of Bath. He is a Fellow of the European Academy of Sciences (EURASC) and of the Royal Society of Chemistry. He was awarded the RSC Corday Morgan Medal in 1988 and the RSC Prize for Structural Chemistry in 2008. He has published over 920 refereed research papers. His research focusses on coordination chemistry, the development of the chemistry of platinum poly-ynes as sensor materials, and he has pioneered the development of time-resolved crystallographic techniques for determining the structures of crystalline, excited state complexes with millisecond lifetimes. |
Key learning points(1) Key features and properties of NCIs in π-conjugated materials.(2) Effect of NCIs on the structural and photo-physical properties of π-conjugated materials. (3) Recent examples from the literature to highlight the design strategies. (4) Opto-electronic (O-E) applications of NCI modulated materials: from organic solar cells (OSCs) through organic light emitting diodes (OLEDs) to organic field-effect transistors (OFETs) and rechargeable batteries. (5) Prospects and challenges of exploiting NCIs. |
1. Introduction
Despite the advances made in modern science and technology, several challenges need to be addressed to realize materials suitable for real-life applications. For instance, replacing inorganic components with organic counterparts in opto-electronic (O-E) devices while maintaining flexibility and performance is one of the contemporary challenges. To tackle this, a plethora of research activities have been devoted to developing π-conjugated organic and organometallic compounds. This class of materials is attractive due to their exceptional and tuneable structural features, their variety of optical properties, and the diverse applications in which they can be used. During the design and development of these materials, several exciting discoveries related to inter- and intra-molecular processes have also been made.1 For instance, the charge transport (CT) phenomena are dependent upon conjugation length, molecular stacking, chain entanglement and multiple weak interactions in solid-state materials. Based on such knowledge, strategies have been developed to tune the properties and applications. Classically, researchers modify the chain length and the molecular spacers and introduce heavy metal ions in the main or side chains to modulate the properties of the oligomeric and polymeric materials. These strategies work well in modulating the structural, photo-physical, thermal, and mechanical properties in several cases, but not always. One modern approach is to modify the properties of π-conjugated molecular systems is by introducing non-covalent interactions (NCIs).2 Basically, NCIs are a group of weaker and kinetically labile forces that control the overall properties of a given system. Knowledge of the inherent presence of NCIs in molecular systems is centuries old, but their manipulation to realize new materials is a relatively new area of research. Today, the substantial importance of inter- and intra-molecular NCIs in both the natural (cellular signalling, transportation of small molecules, enzymatic reactions, etc.) and synthetic worlds (design and synthesis of self-assembled nanostructures and supramolecular architectures) is well established.3 It has been shown that the overall performance of a π-conjugated material can be easily fine-tuned via variation/introduction of NCIs, which alter their backbone co-planarity, molecular packing, CT properties, and excited state dynamics. The usefulness of NCIs in materials science and other research areas, such as organic and inorganic polymers, catalysis, and photo-voltaic applications, has been well reviewed by peers in the past.4–6 It is worth mentioning that the NCIs also serve as primary driving forces in supramolecular assemblies such as metal-organic frameworks (MOFs) or porous coordination polymers (PCPs),7 covalent organic frameworks (COFs) or porous organic polymers (POPs),8 hydrogen-bonded organic frameworks (HOFs),9 and nanomaterials.10 As the structural and electronic properties of these materials are a function of one or more NCIs, extensive research has been carried out recently on the understanding and applications of NCIs. A search on the Web of Science database using the keyword “non-covalent interactions” indicated 9253 publications in various fields during the period 2000–2022 (Table S1 in ESI†). Besides, citations on this topic also keep increasing.Despite the vast number of papers published, we present this tutorial review for two main reasons: (a) the rapid progress made in recent years and (b) our long-standing interest in the design and development of π-conjugated functional materials. This Tutorial Review focuses on state-of-the-art π-conjugated functional materials that can be manipulated through NCIs. This review starts with an introductory discussion on NCIs prevalent in π-conjugated organic and organometallic frameworks. This is followed by a discussion on the effect of NCIs on the structural and photo-physical properties. In this section, we describe structural features that promote NCIs. Following this, we describe NCI-modulated organic semiconductors and their applications in different research areas.
2. Non-covalent interactions (NCIs) in π-conjugated systems
Studies on NCIs have been an intriguing topic of interest recently due to their impact on wide-ranging applications. Since the concept of NCIs was introduced, an understanding of their role has been increasingly explored.11 NCIs are relatively weak and do not have the directionality of covalent bonds but can be smartly tailored to endow new properties. Various inter- and intra-molecular NCIs with different features can be found in the literature. While some are prevalent in organic systems (Fig. 1 and Table 1),2,3,12 others (e.g., interaction with metal centres) are found in organometallic and inorganic complexes. It is worth noting that, in a complex structure (e.g., oligomer or polymer), combinations of NCIs work together and define the overall properties. Herein, we briefly highlight the features and properties of NCIs in π-conjugated systems.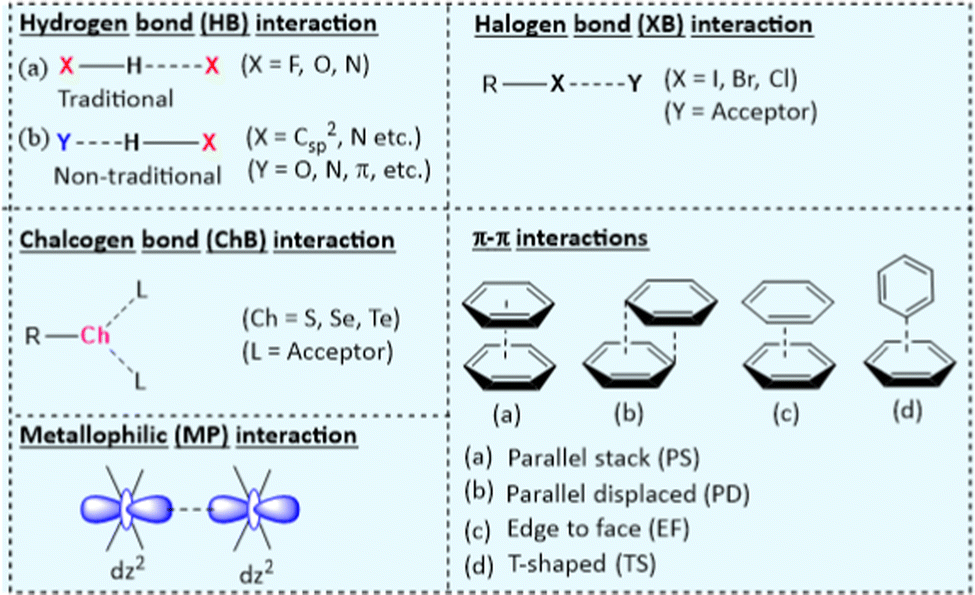 | ||
| Fig. 1 Key NCIs found in π-conjugated functional materials that are responsible for the structural features and the properties that the materials exhibit. | ||
| NCIsa | Key features | Utilities in organic semiconductor research |
|---|---|---|
| HB: H-bonding; CB: halogen bonding; ChB: chalcogen bonding; MP: metallophilic. R = alkyl, Ch = chalcogen, L = acceptor. | ||
| HB | Directional, interaction strength ranges from weak to strong, H⋯X distance (d = 1.6–2.0 Å), preferred X–H⋯X angle = 145–180°. | Crystal engineering, backbone co-planarization, conformation restriction, conjugation length increment, control of diffusional motion of the matrix, and assist supra-molecular assembly formation. |
| XB | More directional, interaction strength ranges from weak to strong, halogen-dependent interaction, hydrophobic in nature, and R–X⋯Y angle close to 180°. | Crystal engineering, enhance phosphorescence via suppressing molecular motion, promoting spin–orbit coupling (SOC) and, restricting vibrational motion of the phosphors, assist in supra-molecular assembly formation. |
| ChB | Directional, interaction strength ranges from weak to fairly strong, chalcogen-dependent interaction, hydrophobic in nature, R–Ch⋯L bond angle close to 180°. | Crystal engineering, improves electronic delocalization, facilitate CT, enhance backbone co-planarity and intermolecular coupling. |
| π–π | Directional, interaction strength ranges from weak to fairly strong, hydrophobic | Crystal engineering, prevent intermolecular aggregation, modulate molecular packing, and electron mobility. |
| MP | Directional and relatively weak to moderate strength, d = 3.0–2.5 Å in Pt(II)⋯Pt(II), 2.7–3.3 Å in Au(I)⋯Au(I). | Bestow metal–metal-to-ligand charge transfer (MMLCT) emissive state and assist the aggregate formation. |
Among a large pool of functional materials, the hydrogen bond (HB) is by far the most exploited NCI because of its directionality and simplicity. It has an electrostatic (dipole–dipole) origin, possesses a partly covalent character, and can be found between various molecular fragments.13 Hydrogen bonding can be divided into traditional (X–H⋯X, X = F, O, N) and non-traditional (O⋯H–Csp2, N⋯H–Csp2, C–H⋯π, C–F⋯H–O and C–F⋯H–N) hydrogen bonding, depending on the interacting partners. They have been exploited to engineer crystals, planarize conjugated backbones, restrict conformational changes, enhance conjugation length, lower the bandgap (Eopg) and assemble large supramolecular architectures. The role of inter- and intra-molecular hydrogen bonding in crystal engineering and materials science is well established.14 For instance, intermolecular hydrogen bonding between an organic phosphor and a solid matrix (e.g. poly(vinyl alcohol)) aids in achieving room temperature phosphorescence by restricting the vibrational/diffusional motion of the phosphors.15
When a halogen atom (X = Cl, Br, or I) is covalently linked to a group (R), it creates an electrophilic site over the X, known as σ-hole.16 This region interacts with the electron-rich site in the same or different molecule (Y, Fig. 1). The strength of the σ-hole depends on the nature of the substituent (i.e., R) and the type of halogen atom. In principle, a deeper σ-hole and a stronger XB is observed when the substituent (R) is electron-withdrawing and the halogen is larger. Well-known for their versatile interaction directionality, XBs have proven useful in altering the molecular packing, electron transport and self-assembly of the materials.17 XB is rare in fluoro-substituted aromatic compound16 due to the strong electronegativity, low polarizability and the absence of σ-hole. XBs are of similar strength and often exist in combination with hydrogen bonding. However, unlike hydrogen bonding, XB interactions are hydrophobic and do not involve polar groups or trap charges and disturb charge transfer, which is beneficial for OE applications.18 XBs can suppress molecular motions, enhance spin orbit coupling (SOC), and thereby bestow phosphorescence features.19
In contrast to XB, when the σ holes are present over a chalcogen atom (Ch), an electrostatic interaction takes place between the chalcogen (Ch = S, Se, Te) acting as an acceptor and a Lewis base (L) as the donor.20,21 These interactions have recently been used as an alternative to hydrogen bonding.3 The integration of ChB to a conjugated organic material increases backbone co-planarity and intermolecular coupling.
π-Interactions involve interactions between entities containing π orbitals. Various types of π-interactions such as H–π, π–π, π+–π, π+–π+, ion–π, π−–π, and π−–π− interactions with energy ranging from 1.5–40 kcal have been reported22 with the ability to enhance the conformational stability.23 Among these, π–π interactions (parallel stacked, parallel displaced, edge to face, T-shaped configuration) (Fig. 1) are one of the most exploited NCIs in supramolecular chemistry and are often used in combination with other NCIs. Furthermore, they are instrumental in materials science due to their facile, non-destructive, and reversible nature.24 In addition to these main NCIs, other interactions that provide driving forces in many supramolecular systems and are used to tailor the electronic structure and optical properties of conjugated systems include metallophilic (MP), hydrophobic, dipole–dipole anion–π interactions (vide infra).
3. Visualizing NCIs: a formidable task
Monitoring molecular structures and characterizing the nature of NCIs have always been challenging. Among the classical experimental techniques, NCIs in solids and solutions are characterized primarily through infrared (IR), nuclear magnetic resonance (NMR), Raman spectroscopic and crystallographic techniques.25–28 For instance, IR spectroscopy is beneficial in providing information on the presence or absence of the electrostatic interactions such as hydrogen bonding. Further information (e.g., intra-, or intermolecular nature) can be obtained by 1D and 2D NMR techniques such as variable temperature (VT) NMR and deuterium exchange experiments. X-ray diffraction (powder and single XRD) analysis is a direct method for visualising the extent of NCIs.24,29 Moreover, the valuable contribution of computational calculations cannot be ignored.30,31While studying NCIs in supramolecular assemblies, one main challenge is to map and prove the presence of a particular NCI as they are dynamic, flexible, reversible, and sensitive to minor stimuli. To overcome this, the use of state-of-the-art surface science techniques such as atomic force microscopy (AFM)32,33 scanning tunnelling microscopy (STM)34 and their variants32,35,36 as well as electron microscopy37 have emerged as very useful methods because they allow a good understanding of NCIs at the molecular level with high resolution. Not only are they able to resolve the chemical structure of a single molecule, but they also assist in identifying different on-surface synthesized products,38 and discriminate between spin states.35 In order to explore how small molecules interact with a porous material, Wei and co-workers37 employed the integrated differential phase contrast scanning transmission electron microscopy (iDPC-STEM) technique to understand the behaviour of heterocyclic compounds such as pyridine within zeolite ZSM-5. They successfully imaged both the empty ZSM-5 and pyridine-saturated channels. Besides, the adsorption/desorption behaviour of the pyridine was also studied.
To gain an in-depth understanding of the role of XB, researchers investigated the feature of XB in various organic assemblies. For instance, molecular assemblies with C–F⋯F–C interaction are relatively rare due to the lack of σ-hole over fluorine atom.16 However, theoretical calculations on a highly substituted fluorinated oligo(arylene ethynylene) revealed that the attractive interaction might prevail over the repulsive forces.16 To test this, a STM image of (bis(2,3,5,6-tetrafluoro-4-(2,3,4,5,6-pentafluorophenylethynyl) phenyl) ethyne (1, Fig. 2) on Ag(111) surface was collected (Fig. 2a). It was noted that the oligomers self-assemble to form highly directional C–F⋯F bonds (Fig. 2b and c) with “windmill” structure (Fig. 2d). However, in the case of small molecules such as tetrakis(4-fluorophenyl) methane, Kelvin probe force microscopy (KPFM) revealed that there is an isotropic negative charge on the F atom,39 in contrast to its brominated analogue, which bears σ-hole.
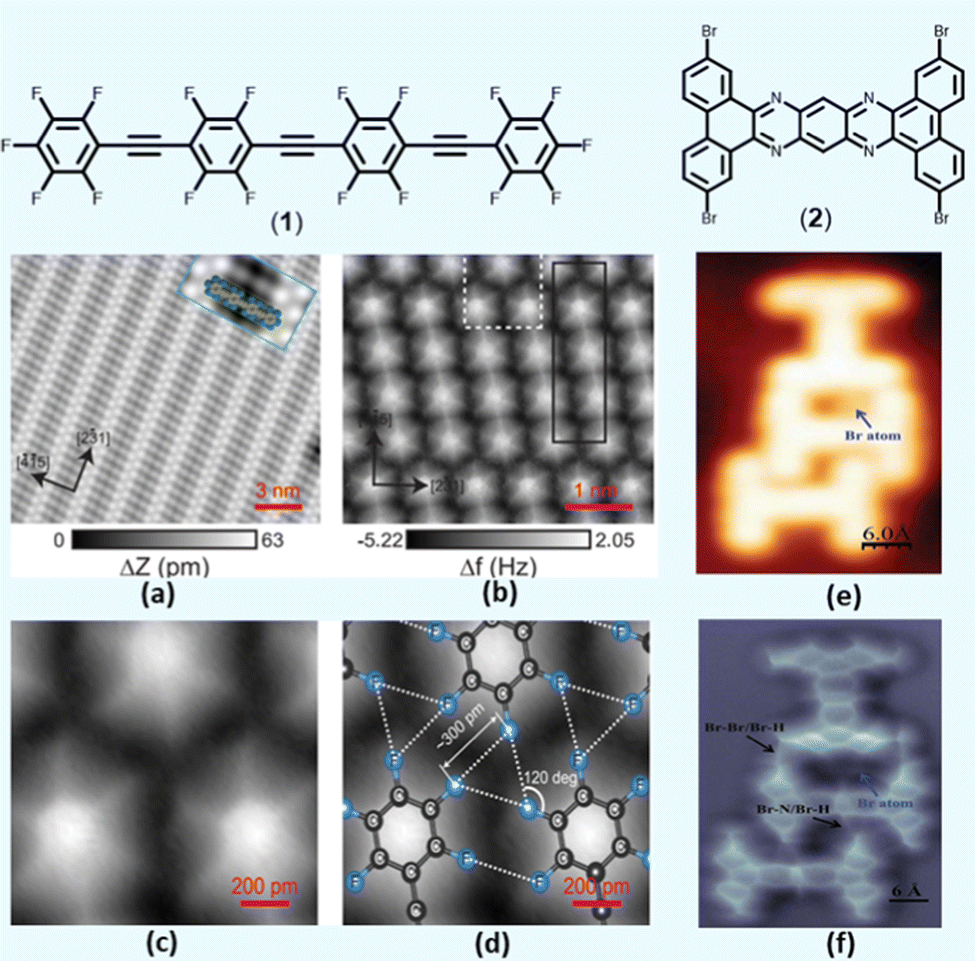 | ||
| Fig. 2 (a) STM topography of the self-assembled fluorinated phenyleneethynylene 1 on Ag(111) surface, (b and c) Frequency shift (Δf) map of the self-assembly with a unit cell in black and magnified Δf map in white rectangle, (d) magnified area with superimposed stick-and-ball drawing of the molecules. (e and f) STM and AFM images of 2 trimer stabilized by multiple halogen bonds. For additional details, please see original references. Reproduced with permission from ref. 16 (Copyright (2015) American Chemical Society) and ref. 40 (Copyright (2022) Wiley-VCH GmbH). | ||
The presence of diverse XB in 2,7,13,18-tetrabromo dibenzo[a,c]dibenzo[5,6:7,8]quinoxalino[2,3-i]phenazine (2, Fig. 2) molecular clusters has also been identified using combined AFM/STM techniques.40 It was demonstrated that the compound formed three types (Br⋯H, Br⋯Br and Br⋯N) of XBs depending on its molecular environment (Fig. 2e and f). Furthermore, a similar technique was employed to study the self-assembly process of 3D molecular clusters driven by XB, HB, and lone pair⋯π interactions.41 Overall, interaction study using surface science techniques has made a tremendous advance and offers real-space monitoring of intricate interactions and molecular assemblies at the atomic scale.
4. Impact of NCIs on the structural and photo-physical properties of π-conjugated organic materials
In π-conjugated semiconductors, the strength and orientation of the charge transfer (CT) is a function of the molecular packing conformation and can be regulated by various NCIs. Therefore, the overall O-E properties and application of a π-conjugated material can be fine-tuned by controlling NCI. In the upcoming sub-sections, we discuss the recent literature on the effect of HB, ChB, XB, dipole–dipole, metallophilic and π-interactions on the structure, photo-physical and photo-electrical properties of π-conjugated materials.4.1. H-bonding
Compounds 3 and 4 (Chart 1) are among recent examples that have been reported with HB-regulated molecular and electronic properties in the solid state.26 The presence of a 6-membered intramolecular H-bonded ring (as confirmed by NMR and absorption spectroscopic data) in 3 and 4 induced co-planarity in the backbone. Red-shifted absorption/emission spectra, lower Eopg and increased thermal stability further confirm the beneficial effect of hydrogen bonding. In a similar study, it was found that intramolecular Ar–H⋯O bonding between fluorescent dyad 5 (Chart 1) containing perylene bisimide (PBI) as the donor and naphthyl as the acceptor moieties show interesting temperature-dependent emission behaviour. The methoxy group on the naphthalene unit assists the formation of hydrogen bond with hydrogen atom of the PBI unit, leading to co-planarization, delocalization of π-electrons, and intramolecular charge transfer (ICT) induced fluorescence quenching (Fig. 3a).42 Owing to the weak nature of hydrogen bond, a rare temperature-dependent increase in emission intensity and lifetime was noted. Such a positive temperature effect is rare and attributed to the distortion in co-planarity and a decreased degree of ICT upon increasing temperature. Although various types of non-traditional hydrogen bonding (e.g., C–F⋯H–O and C–F⋯H–N) are known in the literature, the formation of C–F⋯H–C hydrogen bond remains elusive. Theoretical calculations on tetrafluorophenylene embedded ethynylene-conjugated porphyrin rings 6 (Chart 1) revealed possible C–F⋯H–C interactions (dF⋯H = ∼3 Å) between the β-protons of the porphyrin ring and the adjacent fluorophenyl ethynyl group (Fig. 3b).27 Interestingly, 1H NMR spectroscopic results unambiguously confirmed the presence of C–F⋯H–C interaction in the solution. A marginal effect on the absorption and emission was also seen. Besides, complex 6 exhibits a co-planar structure owing to the torsional restriction through C–F⋯H–C interactions.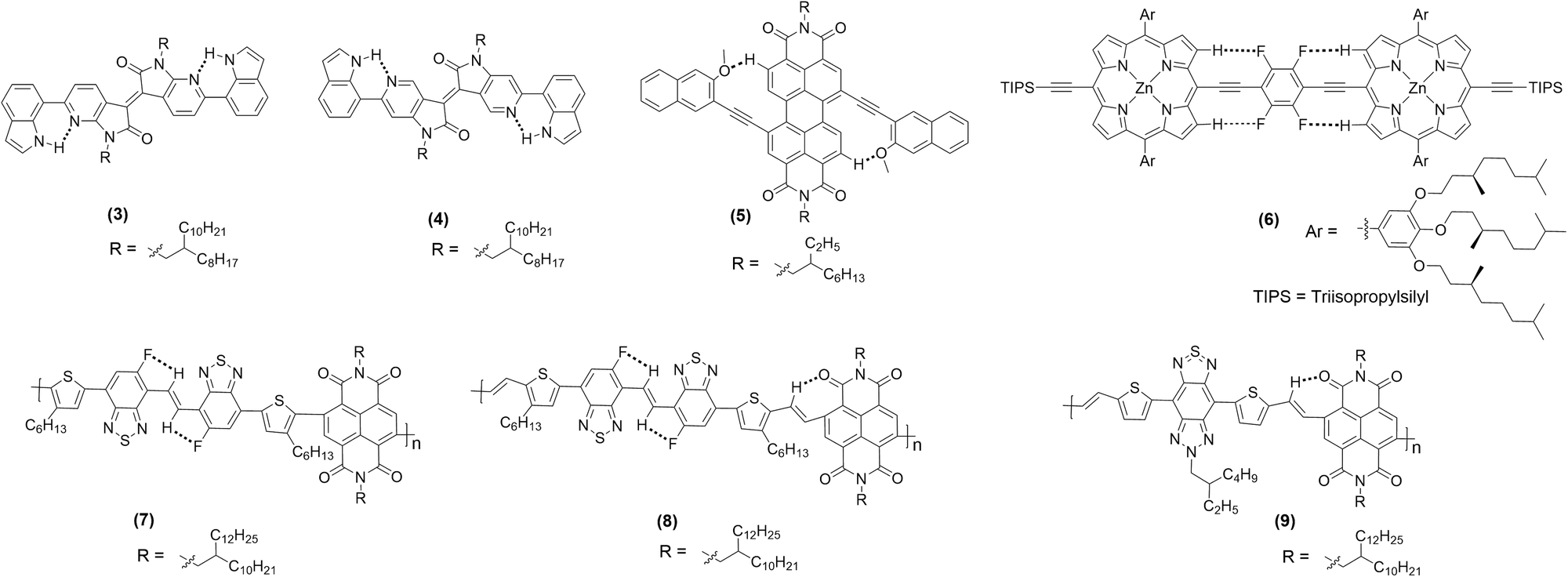 | ||
| Chart 1 Chemical structure of hydrogen bonded π-conjugated materials discussed in this review. Dotted lines depict interacting fragments. | ||
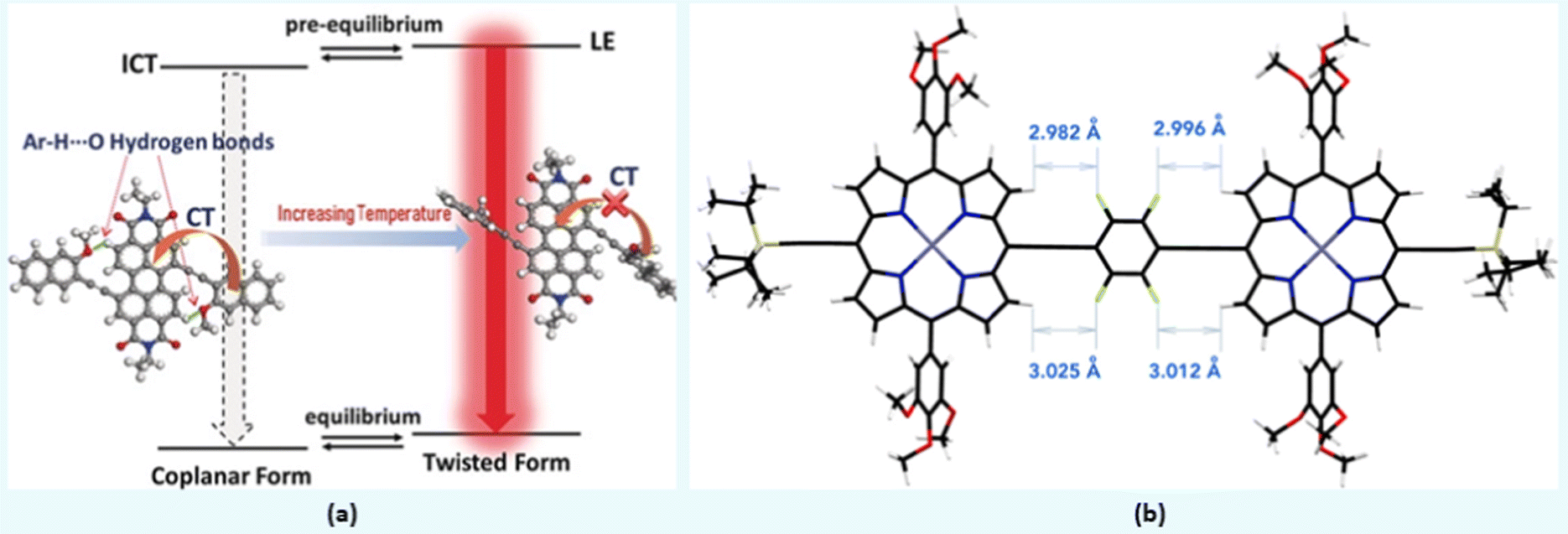 | ||
| Fig. 3 (a) The presence of coplanar and twisted structures driven by weak intramolecular hydrogen bonding in 5, (b) DFT optimized structure of 6. Reproduced with permission from ref. 27 (Copyright (2021) American Chemical Society) and ref. 42 (Copyright (2020) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Organic semiconductors such as vinylene-bridged bis(benzothiadiazole) (BBTV) are well-known electron acceptors. When linked to a suitable donor, it shows excellent electron transport properties. However, DFT calculations suggest the presence of detrimental trans–cis isomerization with inter-ring torsion angles of ∼1.2° to 2.8°. Michinobu and co-workers43 found that introducing intramolecular HB in fluorinated BBTV-based polymers can effectively restrict the rotation, induce co-planarity, enhance electron mobility, aid thin-film crystallinity, and influence molecular packing orientation. An ultraviolet (UV)-vis-near-infrared (NIR) spectroscopic study revealed that the polymer 7 (Chart 1) share absorption profile similar to the one without NCI in solution (λfilmmax = 510 nm and Eopg = 1.70 eV), while slightly red shifted peak in thin film (λfilmmax = 510 nm with a shoulder at 624 nm, Eopg = 1.62 eV), indicating better hydrogen bonding mediated solid-state packing. On the other hand, 8 and 9 (Chart 1) displayed a significantly red-shifted absorption profile (λfilmmax = 676 nm, 917 nm and Eopg = 1.50 eV, 1.08 eV, respectively), attributed to a more rigid backbone, extended π-conjugation, and O⋯H bonding. Nevertheless, the presence of additional O⋯H hydrogen bond in 9 was also responsible for the reduction in dihedral angle (40° → 20°), thereby shortening the π–π stacking distance (dπ–π = 3.40 Å). In fact, this value is one of the shortest among high-performance semiconducting polymers.
4.2. Halogen bond (XB)
Materials that phosphoresce at room temperature are in high demand due to their wide-ranging applications in the area of sensing, imaging, anti-counterfeiting, etc. Although numerous organometallic room temperature phosphorescent (RTP) materials with ultralong lifetimes are available, realizing metal-free RTPs with a high quantum efficiency (Φp) remains elusive. Fortunately, heavy halogen atoms in the organic core have been found to facilitate the intersystem crossing (ISC) process and boost the triplet exciton population via XB and the heavy atom effect. XB has also been associated with suppressed non-radiative pathways and prolonged lifetimes in 10–13 (Chart 2 and Fig. 4a). Furthermore, by simply changing the position of the halogen atom, the strength of intramolecular XB can be varied, leading to ultralong emissions that are detectable to the naked eye.1 For example, the photophysical processes were mainly governed by the heavy atom effect in 10 (Φp = 6.0%) and 11 (Φp = 3.4%), it is mainly XB mediated in 12 (Φp = 52.1%) and 13 (Φp = 27.3%) (Fig. 4b).1 Zhou et al.44 reported XB enabled RTP feature in compounds 14a–d (Chart 2). In contrast to the other naphthalene-based cores, these compounds were found non-emissive in solution. However, compounds having bromine substituents were found to phosphoresce (500–700 nm region) in the solid state (Φp = 1.4%, τp = 1.53 ms for 14b, Φp = 19.6%, τp = 1.94 ms for 14c and Φp = 9.3%, τp = 1.11 ms for 14d). The low performance of 14b and 14d clearly indicates the requirement for the optimum number of halogen atoms for structural and electronic balance. | ||
| Chart 2 Chemical structures of π-conjugated materials with XB and π–π dependent properties. | ||
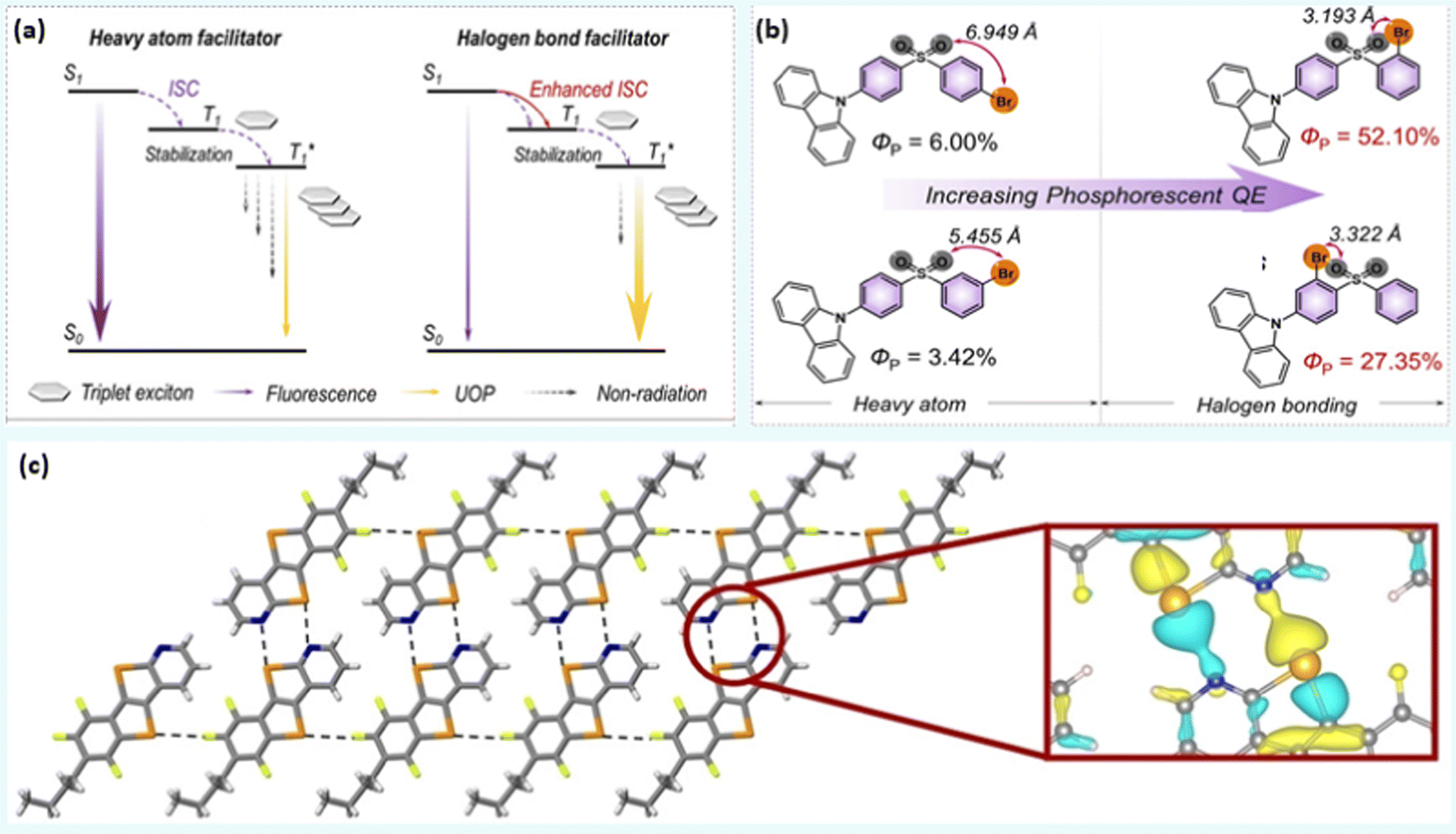 | ||
| Fig. 4 (a) Proposed mechanisms for heavy atom and halogen bonding facilitated ISC in ultralong organic phosphors, and (b) phosphorescence enhancement via XB in 10–13 (UOP = ultralong organic phosphorescence). (c) X-ray stick representation of the supramolecular nanoribbon developed by 20a. Local charge density is zoomed. Reproduced with permission from ref. 1 (Copyright (2020) Wiley-VCH GmbH) and ref. 53 (Copyright (2022) The Authors. Angewandte Chemie published by Wiley-VCH GmbH). | ||
4.3. Chalcogen-bonding (ChB)
Generally, a conjugated material bearing one or more heteroatomic spacers exhibits appealing photo-physical properties. Especially, materials containing chalcogenides (S, Se, Te)-based spacer are fascinating. This is indeed due to their ability to endow ChB interactions to the molecule and control the conformation and architecture. Materials incorporating heterocyclic spacers such as thiophenes, thienothiophenes, thiazole, 3,4-ethylenedioxythiophene (EDOT), diketopyrrolo pyrrole (DPP), etc. are often utilized in organic semiconductor research because of their ability to make five or six-membered ring involving ChB. Using a suitable side chain (e.g., OR), the substituent (e.g., F) or neighbouring aromatic ring (e.g., pyridine, pyrazine), planarization of a conjugated system can be realized. A theoretical study found that out of the six interactions (S⋯N, S⋯O, S⋯F, O⋯N, O⋯F, and N⋯F), the S⋯N interaction not only reduces Eg but also improves the transport properties.45 Besides, the EDOT core shows a lower Eg than DTT due to the presence of greater co-planarity and attractive S⋯O ChB interactions in the former. A competitive hydrogen bonding and ChB interaction may also exist, especially when a fluorene atom is present over the ring.46 In a recent study, using theoretical and experimental tools, it has been demonstrated that among several ChBs, the S⋯O interaction is far more common than others.47 Conjugated materials containing bithiazole N-oxide and N,N′-dioxide moieties in 15(a–c) (Chart 2) exhibit S⋯O ChB interactions with a profound effect on the electronic and physical properties of both the oligomers and polymers. For example, increasing the content of the N-oxide led to a bathochromic shift in absorption (λmax. = 387–416 nm and Eopg = 2.7–2.8 eV), a decrease in the Stokes shift, a decrease in solubility and an increase in thermal stability which can be further attributed to the strong S⋯O ChB interaction.47With the advancement in synthetic chemistry and the understanding of molecular features, semiconductor research has seen tremendous growth recently. Whether we consider metal-free RTPs or non-fullerene acceptors (NFAs), significant progress has been made. One example is the discovery of Y-series acceptors used in photo-voltaic (PV) research. It has been shown that the efficiency of these NFAs can be boosted via ChB too. Y6-type NFAs 16a–c (Chart 2), bearing a vinylene π-bridge-modified backbone exhibit heteroatom-dependent molecular flatness, novel photo-physical properties (λfilmmax = 853–876 nm and Eopg = 1.24–1.26 eV), and modulated energy level in the solid state.48 Remarkable differences in the photo-physical properties in solution and in the film was a clear proof of the intermolecular interaction and electronic coupling in the solid state. Among the studied NFAs, 16c exhibits the highest co-planarity leading to an absorption edge near 1000 nm. In case of NFAs 17a–c (Chart 2), it was found that as the number of Se-atoms in a conjugated system increases, the intermolecular interaction in the solid state increases with a positive effect (red-shifted and more intense absorption) on the photo-physical properties.49 Results of theoretical calculation implied that the positive potential resides on the central part of the molecule (thus the electron acceptor part) and decreases with an increase in the number of Se units, indicating that selenation reduces the acceptor strength. An absorption study reveals a bathochromic shift and an enhanced molar absorption coefficient upon increasing the selenium content, i.e.17a (λfilmmax = 835 nm, 1.56 × 105 cm−1), 17b (λfilmmax = 840 nm, 1.63 × 105 cm−1) and 17c (λfilmmax = 853 nm, 1.94 × 105 cm−1). This behaviour could be attributed to the NCI generated through Se-core, which was also supported by X-ray analysis. It revealed that the dihedral angle between donor–acceptor–donor units decreases along the series (8.4° → 7.1° → 2.3°), and the extent of NCI increases upon increasing the Se-content. Besides, a reduction in π–π distance and enhanced aggregation were also noted. Another study finds that S⋯N interaction between benzo[2,1,3]thiadiazole moieties in Y6 and its selenium analogue 18 (Chart 2, λfilmmax = 844 nm and Eopg = 1.30 eV) is important to facilitate face-to-face π-core interactions, molecular self-assembly and hence the enhanced CT features.50 Dominant π–π stacking of polymeric backbones upon the insertion of Se and halogenation leading to different crystallinity, molecular packing, electron mobility, morphology and physical properties, has also been reported by others.51 Not only the selenation and halogenation but changing the alkyl side chains by an alkoxy chain is another strategy to improve the performance of NFAs. It was demonstrated that the introduction of an alkoxy group into polymer 19 (Chart 2, λfilmmax = 676 nm and Eopg = 1.64 eV) alters the frontier orbital energy level and introduces S⋯O type NCI, leading to an increase in the co-planarity, and rigidity of the backbone with ultimate improvement in the PV performance.52 Very recently, the first example of ChB interaction-enabled nanoribbon formation by 20 (Chart 2) has been reported.53 In thin films, the ribbon would π-stack in a multi-layered architecture. X-ray analysis revealed the formation of hexagonal dimers via ChB interactions (Fig. 4c). While 20a undergoes ribbon-like organization through the formation of two double ChBs, 20b forms only herringbone-type architectures. Both exhibit similar absorption and emission profiles with phosphorescence emission (τp ≈ 700 μs) indicative of the beneficial role of Te atoms. Theoretical investigations showed the presence of local charge density over Te and N atoms in 20a while it was absent in 20b (Fig. 4c, inset). When used as a transistor, they lacked electron transport characteristics, while 20a exhibited the hole transport behaviour. Such a semiconducting feature has never been reported for any architecture supported by ChB interactions. Moreover, when 20a was assessed as hole-transport layers to build light-emitting electrochemical cells (LECs), it enhanced the stability and performance of Cu(I)-based LEC.
4.4. π-Interactions
π-Interactions, a type of weak NCI compared to those discussed previously, play a significant role in various disciplines of science.23 A variety of π-interactions is possible in a π-conjugated material, including π–π, XH–π, cation–π, anion–π and lone-pair–π etc. They can modulate the charge transfer, photoconductivity, chiro-optical, electrochemical, and other properties of a system. In organic systems, π–π interactions are mostly related to its property or application. For example, enhanced ISC rate and phosphorescence (even RTP) in organic systems have been attributed mainly to the π–π stacking with other NCIs.54,55 Upon decoration with substituents having different electronic properties, 10-phenyl-10H-phenothiazine 5,5-dioxide derivatives 21 (Chart 2) produces persistent RTP as well as photo-induced phosphorescent compounds.56 A subtle change in the functional group brings a drastic shift in the lifetime (88–410 ms) of the phosphors (Fig. 5a). This was ascribed to the altered π–π stacking with the change in the substituents. For example, when the halogen atom (–I effect) was attached, it minimized the repulsion between two aromatic cores leading to enhanced π–π stacking. The examples above demonstrate the excellent ability of π–π interactions in tuning the luminescence properties in small organic semiconductors. However, sometimes intermolecular π–π interactions are detrimental in the solid state as excimers are formed leading to fluorescence quenching. To overcome this issue, both physical (the host-guest approach) and chemical (use of bulky substituents) modifications have been suggested. As an exception to this observation, Yang and co-workers57 found that 22 (Chart 2) forms a discrete dimer with PLQY ∼ 44%, which was nearly a 26-fold increase relative to that of the monomer. Crystallographic analysis revealed discrete π–π stacking with short interplanar distances and large overlaps. The interaction was strong enough to resist external mechanical or thermal stimuli, as there was no major change in the emission/absorption profile.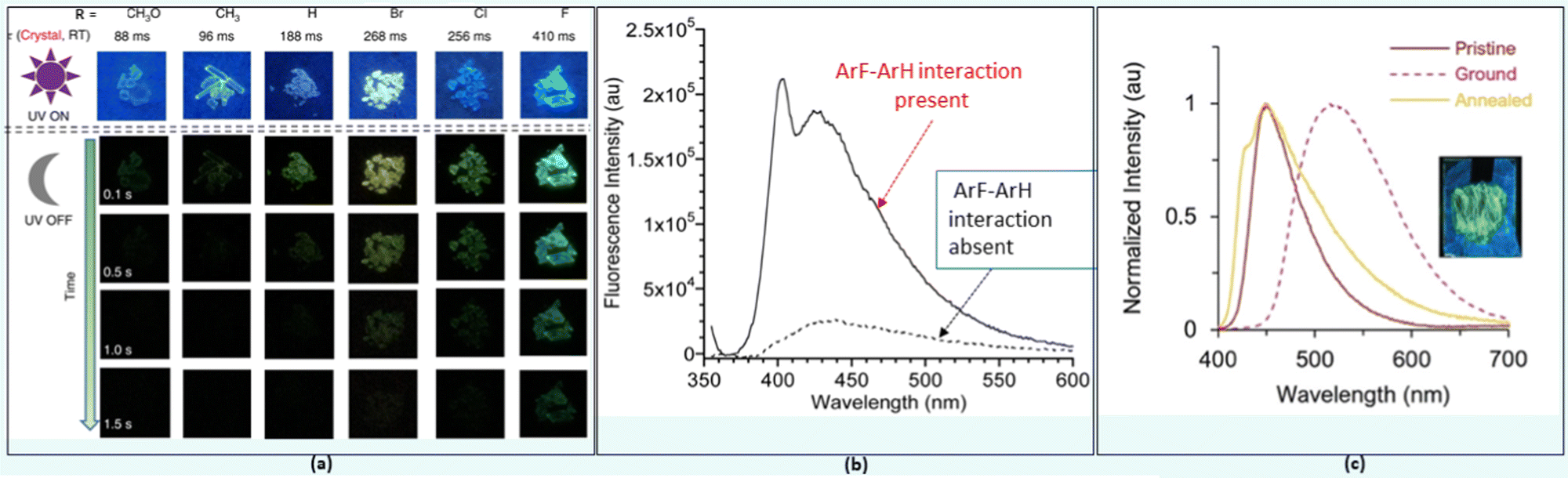 | ||
| Fig. 5 (a) The RTP behaviour and corresponding phosphorescence lifetimes at room temperature in 21, (b) fluorescence emission spectra demonstrating the effect of ArF–ArH interactions in 23, and (c) multi-fluorochromism behaviour of 24. Reproduced with permission from ref. 56 (Under a Creative Commons Attribution 4.0 International License), ref. 62 (Copyright (2019) The Royal Society of Chemistry) and ref. 63 (Copyright (2022) The Royal Society of Chemistry). | ||
In π-conjugated organic and organometallic materials, especially polymers, long alkyl (R) or alkoxy (OR) groups are often used to enhance solubility and decrease crystallinity. Other than this traditional usage, a suitable side chain can also reduce the π–π interchain length, induce a bathochromic shift in the optical absorption, and enhance hole mobility and photovoltaic performance.58 Recent studies demonstrated that these flexible groups could influence electron density and photo-physical properties.59 Moreover, they can also be used to confine the torsional rotation (co-planarity of aryl rings along the main chain) in oligomers or polymers, thereby altering CT. For example, p-arylene ethynylene (PAEs) adopts several conformations at room temperature because of the low rotational barrier. In such systems, one can modulate the electronic properties by capturing the co-planarity in a suitable conformation.60 Recently, the conformation control of PAEs utilizing co-facial electron deficient fluoroarenes (ArF) and electron-rich arenes (ArH) stacking interactions between the main chain and pendant groups in conjugated materials emerged as a powerful tool.61 It was noted that the incorporation of perfluorinated benzyl ester side chains into conjugated PAEs backbones 23 (Chart 3) inhibits the aggregation caused quenching (ACQs), leading to 5–7 times more intense emission (Fig. 5b) in solution.62 This was essentially due to the presence of intramolecular ArF–ArH stacking in the fluorinated analogues. Besides, such interactions led to the twisting of PEs backbone. It is noted that the presence or absence of ArF–ArH stacking as well as position of the absorption/emission band depends on the type of central ring selected. For example, oligomer containing bithiophene exhibit no ArF–ArH interaction but most bathochromically shifted optical features in the solid as well as in solutions. By extending this study, the same group investigated the structural and photophysical properties of a series of OPEs bearing electron rich or electron deficient heterocyclic spacers in the main chain 24 (Chart 3).63 While electron rich spacers were able to form ArF–ArH stacking, and exhibit positive solvatochromism effect, electron deficient did not. However, both series of compounds bearing fluorinated pendants exhibited multi-fluorochromism (Fig. 5c) which was absent in hydrogenated analogues, dictating the importance of NCIs. Overall, these studies strongly support the fact that the solid-state properties can also be modulated via smartly tuning NCIs.
 | ||
| Chart 3 Some PAEs with ArF–ArH interaction feature. | ||
4.5. Intermolecular dipole–dipole interactions
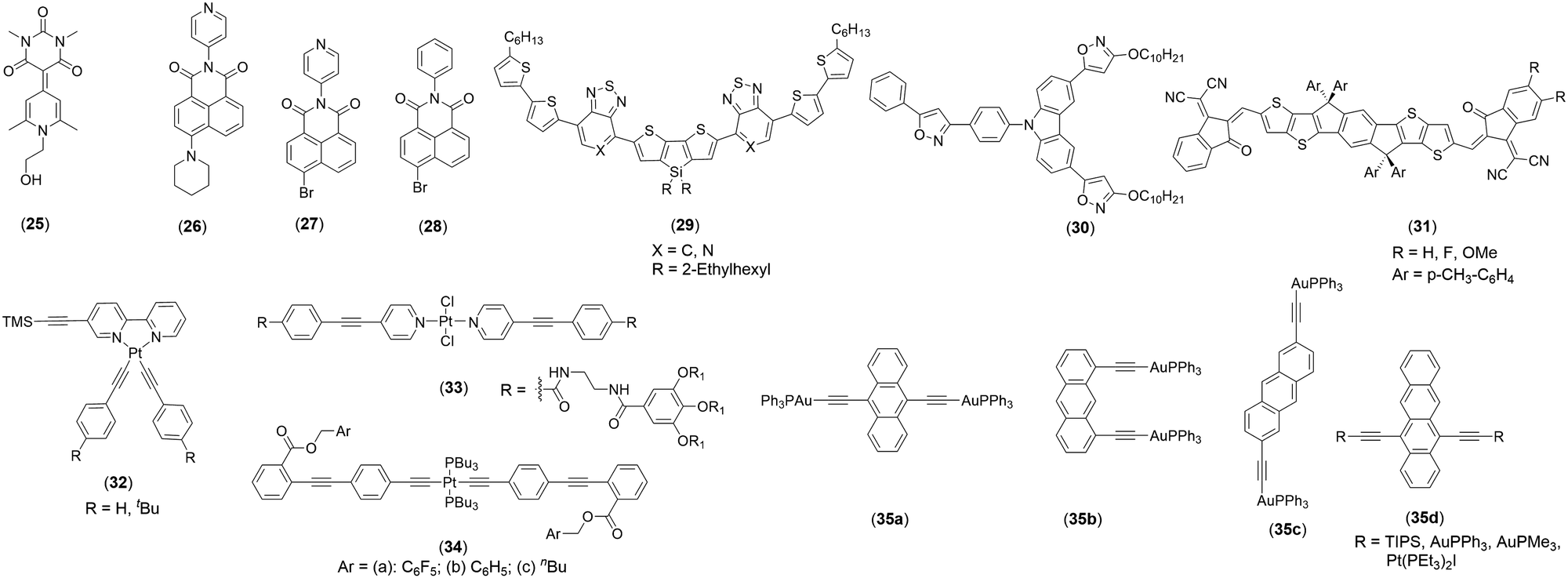
Dipole moment (μ) is another weak yet decisive electrostatic NCI that has the potential to modulate the overall photo-physical and opto-electrical properties of a π-conjugated system.64 Despite the fact that they are directional and possess relatively high binding strength (in push–pull type merocyanine dyes), the effect of intermolecular dipolar interactions has not been investigated extensively for its role in dictating self-assembly behaviour. Merocyanine dyes are well known for their high dipole moments, and in the presence of a combination of dipolar interactions with other NCIs, they form long-range ordered assemblies. The assembly of a merocyanine dye was found to be dependent on the molecular packing condition. For example, 25 (Chart 4) was found to self-assemble in a polar solvent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 dioxane:water) governed mainly by dipole–dipole interactions and solvophobic forces (Fig. 6a).28 However, in the absence of such conditions (such as in the solid state), the same dye showed hydrogen bond mediated stacking formation.28 Contrary to this finding, it was noted that naphthalene monoimide (NMI) luminogens 26–28 (Chart 4) form stimuli-responsive luminescent assemblies in solid state, which is mainly mediated by dipole–dipole interactions (Fig. 6b).25
:90 dioxane:water) governed mainly by dipole–dipole interactions and solvophobic forces (Fig. 6a).28 However, in the absence of such conditions (such as in the solid state), the same dye showed hydrogen bond mediated stacking formation.28 Contrary to this finding, it was noted that naphthalene monoimide (NMI) luminogens 26–28 (Chart 4) form stimuli-responsive luminescent assemblies in solid state, which is mainly mediated by dipole–dipole interactions (Fig. 6b).25
 | ||
| Chart 4 Some examples of organic and organometallic π-conjugated materials sensitive to dipole–dipole and other NCIs. | ||
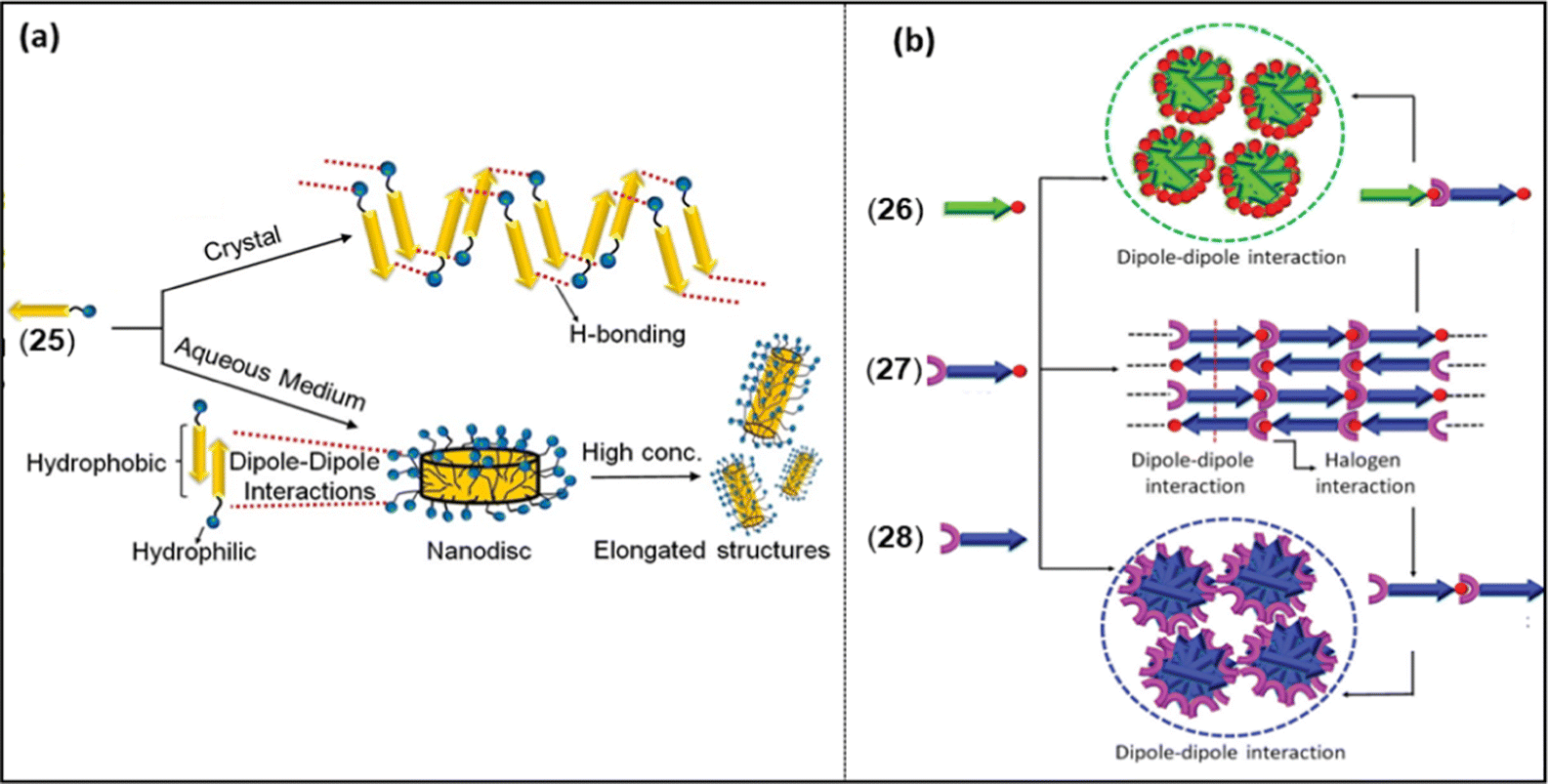 | ||
| Fig. 6 (a) Crystal packing of 25 in solid and solution, (b) proposed model for the molecular arrangement in 26–28 and their co-assemblies through dipole–dipole interactions and XB. Reproduced with permission from ref. 25 (Copyright (2021) The Royal Society of Chemistry) and ref. 28 (Copyright (2022) Wiley-VCH GmbH). | ||
For electronic and other reasons, a π-conjugated system with D–π–A configuration is highly useful for OE applications. A non-centrosymmetric molecule with such a configuration and a large μ value is known to form centrosymmetric aggregates and complex supramolecular architectures via intermolecular dipole–dipole interactions. It is quite unusual that the dipolar property, thus functionalised, could be modulated via varying donor and acceptor groups. For instance, Heeger and co-workers65 noted that compounds 29 (Chart 4) with different acceptor units exhibit a large difference in photo-physical profile, charge transport ability and PV performance. A better performance of 29 (X = N) as compared to 29 (X = C) was attributed to the poor thin-film self-assembly of the latter. This was ascribed to a large variation in the magnitude and direction of the molecular dipole in 2,1,3-benzothiadiazole-containing compound as compared to [1,2,5]thiadiazolo[3,4-c]pyridine acceptor unit. Similarly, dipole-induced formation of charge carrier transport channels was reported in small molecules 30 and 31 (Chart 4).66,67 The intermolecular dipole–dipole interactions can be derived from either the conjugated moieties or the side chains. Such interaction directs the cooperativity for the self-assembly of planar π-conjugated systems. In the past, such cooperating interactions have been reported for fluorenone68 and carbazole69 derivatives.
5. Impact of NCIs on the structural and photo-physical properties of organometallic materials
The beneficial role of NCIs on transition metal-based organometallics, as well as coordination complexes containing conjugated coordinated ligands, is well recognized. Through careful ligand design and selection of the metal centre, it is possible to fine-tune their structural, photo-physical and photo-chemical properties.70 A literature survey suggests that the most commonly adopted strategy for designing new organometallic complexes includes variation in the donor (main as well as auxiliary) ligands. To this end, structurally rigid mono-, bi- or polydentate main ligands are used, which facilitate achieving high QY and lifetime values via multiple NCIs (metallophilic, π–π, etc.). Those containing late transition metals with d8–10 configurations are prone to metallophilic interactions (viz. platinophilic, aurophilic, mercurophilic, etc.) along with the other ligand-based NCIs. Among them, organoplatinum complexes are particularly interesting. This is indeed because a square planar Pt(II) complex bears open, axial coordination sites which assist in stacking via Pt(II)⋯Pt(II) interactions leading to interesting supramolecular architectures. In fact, Pt⋯Pt interactions are the driving force for realizing supramolecular assemblies with unique spectroscopic behaviour and sensitivity towards multiple stimuli (e.g., pH, grinding, temperature, etc.). Recently, Yam and co-workers71 showed that the self-assembly of Pt(II) complex is the result of metallophilic interaction between two Pt(II) centres. Such assemblies often exhibit red-shifted absorption (along with new metal–metal-to-ligand charge transfer MMLCT transition) and emission, as well as enhanced quantum yield and longer lifetime. Metallophilic interactions in mono-, di- or tridentate organic ligand bound σ-acetylide complexes are very common and known for controlling the morphology of molecular assembly and PL properties. It has been demonstrated that the ligand attached to the metal centre dictates the extent and nature of the metallophilic interaction.72 For example, complex 32 (R = H, Chart 4) has been found to show stronger metallophilic interaction than 32 (R = tBu) due to the bulkiness of tBu group. This difference led to a large variation in the emission spectra of the complexes. Moreno and coworkers73 reported that cyclometalated Pt(II) complex supported by isocyanide ligands forms a long-range aggregate mediated by Pt⋯Pt and/or π⋯π stacking interactions. An increased MMLCT character in the solid-state aggregates was also attributed to the presence of NCIs. The resulting 1D columnar stackings exhibit external multi-stimulus responsiveness (MSR) and phosphorescent colour switching behaviour. The modulation of metallophilic and π–π interactions in cyclometalated Pt(II) complexes via XB as well as the stabilization of excited states via metal⋯halogen NCI have been studied recently.74,75 The above-mentioned results unambiguously prove that the luminescence (MMLCT) behaviour of a complex is highly sensitive to the type of ligand selected. However, Fernandez and coworkers76 reported that the luminescence and size of a supramolecular system can also be controlled through metal–metal interactions. Complex 33 (Chart 4) was found to self-assemble into a non-emissive aggregate via HB interaction. On the other hand, ligand coplanarization in the same complex led to a thermodynamically stable product with long-range metallophilic interaction and luminescence.It is to be noted that metallophiphilic interaction in the supramolecular structure is not always advantageous. Sometimes the solid-state PL property is largely hindered by aggregation and metallophilic interaction-induced quenching. To circumvent this, strategies such as the use of rigid, sterically hindered groups and enforcing co-planarity through NCI have been suggested. Motivated by the results of NCI-controlled luminescence properties in oligo-ynes, Thomas and co-workers77 demonstrated that programmed aromatic stacking interactions of pendant groups in 34 (Chart 4) could prevent intermolecular aggregation and therefore enhance phosphorescence in neat solid film, which is quite rare for platina-ynes. Complexes 34a–c, which contain a series of different ester alkoxy side chains, showed similar absorption (λabsmax = 375 nm) and emission (λflmax = 390 nm, 540 nm, Φem < 0.01) profiles in solution and in solid matrices. However, a large variation in emission profile was noted in thin films. Complexes 34b and 34c showed fluorescence (λflmax = 420 nm) only, complex 34a displayed a dual behaviour (fluorescence and phosphorescence) in the neat films.
Because of the stronger metallophilic interactions in Au(I) complexes, complexes 35a–c (Chart 4) bearing two Au(I) units attached to different positions of anthracene have been reported with unique properties. For example, the one with 9,10-positions as the point of attachment exhibit rare intermolecular Au⋯H–C NCI, high fluorescence and high quantum yield compared to 2,6- and 1,8- isomers. In fact, 35a is the first example which forms 2D-network involving intermolecular Au⋯H–C interaction.78 On the other hand, its counterparts 35b and 35c form 1D-polymeric chains via C–H⋯π and π⋯π interactions and intramolecular Au⋯H–C interactions, respectively.79 A remarkable effect of metal insertion on the structural and PL properties has been reported for anthracene-based compounds 35d. In these complexes, the presence of two metals showed a more pronounced effect compared to those with one, Pt(II) being more effective than Au(I). Besides, the Au(I) complex formed an interesting honeycomb-like structure.
6. Applications
6.1. Organic solar cells (OSCs)
One of the most promising applications of NCIs is around PV research. They are exploited to realize new NFAs and solid additives for organic solar cells (OSCs), HTMs for perovskite SCs, dye mediators in BHJ, etc. In this section, we highlight some recent applications of functional organic and organometallic NCI-mediated π-conjugated functional materials. The PV performance of conjugated systems with O⋯S, N⋯S, X⋯S (where X = Cl, Br, F), and H⋯S through-space interactions that form non-covalent conformational locks are often improved.86 Therefore, by exploiting these NCIs, high-performance donors and acceptors have been developed recently. To achieve this goal, numerous chemical modifications have been explored for establishing structure–property-performance relationships for NFAs such as Y-series, 2,2′-((2Z, 2′Z)-((4,4,9,9-tetrahexyl 4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene-2,7-diyl)bis (methanylylidene)) bis(3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalono nitrile (IDIC) etc. For instance, varying molecular packing of the active materials in the solid state could be one of the reasons for variation in device performance. It was found that fluorinated analogue 36c (Chart 5) displayed lower PCE (9.14%) and Voc (0.78 V) but higher Jsc (15.01 mA cm2) and FF (76.1) than 36a (Voc = 0.98 V, Jsc = 13.93 mA cm2, FF = 73.5%, and PCE = 10.31%).80 On the other hand, 36b, because of its asymmetric structure, exhibited the best performance owing to balanced Voc and Jsc (Voc = 0.98 V, Jsc = 13.93 mA cm2, FF = 73.5%, and PCE = 10.31%). The hole (μh) and electron mobilities (μe) of the fluorinated analogue were higher than for the non-fluorinated one (Table 2), explaining the higher FF value in the former. OSCs based on Y-type acceptors exhibited high PCEs when blended with suitable donors. Since the introduction of a chalcogenide (Se) significantly reduces the Urbach energy of organic PV materials,87 the efficiency of the resulting material is also enhanced via ChB interactions. It has been demonstrated that a delicate balance between a dissymmetric acceptor and the number of selenophenes could yield PCE as high as 17.51%.49 Increasing the number of Se atoms led to enhanced NCI, a bathochromic shift in the absorption, increased electron mobility and crystallinity. It was noted that, owing to the red-shifted and more intense absorption and favoured blend morphology, the dissymmetric 17b (Chart 2) based device fabricated using polymeric donor PM6 exhibited better performance (Voc = 0.85 V, Jsc = 26.58 mA cm2, FF = 77.5%, and PCE = 17.51%) than 17c (Voc = 0.82 V, Jsc = 26.35 mA cm2, FF = 73.41%, and PCE = 16.01%) and 17a (Voc = 0.86 V, Jsc = 25.85 mA cm2, FF = 75.28%, and PCE = 16.73%) based devices.49 It is to be noted that the increase in Se-atom downshifted the LUMO energy levels causing a decrease in Voc. The dissymmetric structure of 17b was one of the reasons for higher charge carrier mobilities and balanced μe/μh ratio. Binary all-PSCs fabricated using the 17c-type spacer (with one chlorine and additional thiophene) based polymer and a PM6 donor has also been reported with impressively high performance (Voc = 0.88 V, Jsc = 24.5 mA cm2, FF = 74.3%, and PCE = 16.1%).51 Recently, Song et al.48 reported electron-deficient fused-ring cores 16 (Chart 2) and found that devices based on the Se analogue exhibit better performance (PCE = 14.2%) than the N (PCE = 11.97%) or S (PCE = 12.56%) counterparts. An increase in Jsc and EQE, while a decrease in Voc was noted upon replacement of N by S and Se. Notably, authors used polymer electron donor (poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene)-co-(1,3-bis(5-thiophene-2-yl)-5,7-bis(2-ethylhexyl)benzo[1,2-c:4,5-c]dithiophene-4,8 dione) (PBDB-T), which is economically cheaper and easy to realize, signifying the importance of this work. Wang and co-workers81 carried out the very first study on NFAs 37a–c (Chart 5) bearing different hetero-dihalogenated terminal groups. It was found that NFAs with fluorine substituents display slightly downshifted HOMO and LUMO energies, altered intermolecular packing and helical molecular geometry.81 PCE of the device fabricated using PM6 as a donor and 37a as an acceptor yielded a PCE of 17.52%, which is one of the highest among binary devices. The other two varied in the order: 37b (16.47%) > 37c (13.61%). This significant difference was attributed to the differences in charge recombination and mobilities. Overall, halogen substitution (especially F and Cl) leads to different π–π intermolecular packing and is a useful strategy to achieve large electron mobility and thus broader photocurrent response, Jsc and FF values. | ||
| Chart 5 Some recent examples of PV materials. | ||
| Comp. | Performance parameters | Ref. | ||||
|---|---|---|---|---|---|---|
| V oc (V) | J sc (mA cm−2) | FF (%) | PCEmax (%) | Carrier mobilities (μh/μe) × 10−4 (cm2 V−1 s−1) | ||
| a ITO/PEDOT:PSS/active layer/PDIN/Ag. b ITO/PEDOT:PSS/active layer/ZnO/Al. c ITO/PEDOT:PSS/active layer/PDINO/Al. d ITO/ZnO/active layer/MoO3/Ag. | ||||||
| 16a | 0.75 | 24.87 | 64.35 | 11.97 | 4.28/1.36 | 48 |
| 16b | 0.71 | 26.50 | 66.65 | 12.56 | 2.35/1.77 | 48 |
| 16c | 0.71 | 28.66 | 69.70 | 14.20 | 4.33/3.52 | 48 |
| 17a | 0.86 | 25.85 | 75.28 | 16.73 | 6.63/5.01 | 49 |
| 17b | 0.85 | 26.58 | 77.5 | 17.51 | 8.77/7.33 | 49 |
| 17c | 0.82 | 26.35 | 73.41 | 16.01 | 4.46/2.78 | 49 |
| 36a | 0.98 | 13.93 | 73.5 | 10.31 | 4.36/5.56 | 80 |
| 36b | 0.88 | 17.02 | 75.8 | 11.67 | 10.8/7.76 | 80 |
| 36c | 0.78 | 15.01 | 76.1 | 9.14 | 6.53/10.4 | 80 |
| 37a | 0.85 | 26.45 | 77.92 | 17.52 | 8.51/4.46 | 81 |
| 37b | 0.85 | 25.83 | 75.02 | 16.47 | 6.00/2.83 | 81 |
| 37c | 0.86 | 22.09 | 71.62 | 13.61 | 3.39/1.05 | 81 |
| 38 | 0.92 | 25.79 | 73.96 | 17.57 | 1.52/4.39 | 82 |
| 39a | 0.83 | 21.91 | 60.10 | 11.00 | 2.32/1.40 | 83 |
| 39b | 0.81 | 25.25 | 70.78 | 14.53 | 2.38/3.09 | 83 |
| 39c | 0.79 | 25.76 | 64.91 | 13.24 | 2.26/3.47 | 83 |
| 40a | 0.87 | 16.54 | 66.08 | 9.49 | 9.41/3.81 | 84 |
| 40b | 0.73 | 21.95 | 69.36 | 11.16 | 12.2/6.41 | 84 |
| 40b | 0.87 | 24.02 | 59.79 | 12.55 | 93.1/24.0 | 84 |
| 41a | 0.89 | 18.95 | 63.56 | 10.72 | 1.52/1.68 | 85 |
| 41b | 0.87 | 18.43 | 61.90 | 9.92 | 1.29/1.59 | 85 |
| 41c | 0.93 | 9.56 | 48.58 | 4.32 | 0.77/1.10 | 85 |
One of the foremost factors that limits the efficiencies of many OSCs is energy loss, such as non-radiative recombination energy loss (ΔEnr). Compound 18 (Chart 2) is one of the most red-shifted NFAs based on π-extended thiadiazole reported recently.50 In contrast to previous studies on binary OSCs, a ternary device (ITO/PEDOT:PSS/(PM6:Y6/18:PC71BM)/PNDIT-F3N/Ag) yielded PCE = 17.08% with higher Jsc (27.48 mA cm2) but lower Voc (0.822 V) than the device without 18 (Voc = 0.841 V, Jsc = 25.80 mA cm2, FF = 75.70%, and PCE = 16.41%).50 This study also reinforced the fact that incorporation of Se leads to higher and more balanced mobilities (μe = 1.7 × 10−4 cm2 V−1 s−1, μh = 1.8 × 10−4 cm2 V−1 s−1). It was found that the Eloss was less (0.48 V vs. 0.55 V) when 18 was used. An acceptor 38 (Chart 5) with an asymmetric and extended π-conjugated central core exhibits upshifted HOMO (−5.62 eV), and LUMO (−3.87 eV) energy levels and a higher PL quantum yield (PLQY) compared to Y6 (−5.64/−3.91 eV). The OSC based on PM6:38 demonstrates a low ΔEnr value (0.177 eV), leading to higher Voc (0.921) and PCE (17.57%) values. In contrast, the control device PM6:Y6 gives a PCE of 16.45%, Voc = 0.921, with a ΔEnr value of 0.231 eV.82 In fact, 38-based devices exhibit the lowest ΔEnr for binary OSCs with PCE > 17%. Binary OSCs fabricated using noncovalently fused-ring electron acceptors (39a–c, Chart 5) with S⋯O intramolecular interaction display Voc = 0.79–0.83 V, Jsc = 21.91–25.76 mA cm2, FF = 60–10–70.78%, and PCE = 11.0–14.53%. In these acceptors, there was a notable effect of π-extended end-groups on the electronic properties, CT, film morphology, and energy loss.83 It was noted that the multi-fluorination into a π-extended core causes an absorption shift to the red, increases crystallinity, and enhances the electron mobility, Jsc and PCE values. The ΔEnr of 39a–c were found to be 0.28, 0.28, and 0.31 eV, respectively.
In the quest for high-performance PV materials, non-fused ring NFAs have also been investigated. Like the previous example, a large dependency of the performance on the structure was noted. For example, 40a and 40b (Chart 5), which differ in the π-bridge and end-groups, showed different PV performance and CT features. For example, the device fabricated using 40b (PCE = 11.16%) as an acceptor and PBDB-T as a donor showed better performance than 40a (PCE = 9.49%), owing to the enhanced crystallinity and higher CT mobility of the blend with the former. An improvement in performance was noted when donor PBDB-T was replaced by poly[(2,6-(4,8 bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′] dithiophene))-alt-5,5′-(5,8-bis(4-(2-butyloctyl)thiophen-2-yl) di thieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5]thiadiazole)] (D18) (PCE = 12.55%).84 The ΔEnr value was 0.27 and 0.29 eV for 40a and 40b, respectively. The CT property can also be modulated via an isomerization strategy. In a study involving adjustment of the topology of the central naphthalene core has been found to alter the molecular conformation as well as π–π stacking features. It was noted that the topology also influences the morphology and charge mobility of the active layer. Acceptors 41a–c (Chart 5) composed of naphthalene isomer-based central core flanked by thiophene and dithieno[3,2-b:2′,3′-d]pyrrole are narrow Eopg (ca. 1.62–2.04 eV) materials with λonset beyond the visible range. As-cast PBDB-T donor based OSCs yielded a PCE in the range of 4.32–10.72%, with 41a being the most efficient.85
In addition to the design and development of these state-of-the-art NFAs for OSC, attempts have also been made to improve the performance by modulating the morphology of the fullerene based OSCs, which is usually a function of the additive used. 1,8-Diiodooctane (DIO) and 1-chloronaphthalene (1-CN) are the mostly used additives but they impart a negative effect on the device lifetime. To circumvent this challenge, the use of solid additives has been suggested. However, maintaining trade-off between the volatility and intermolecular interactions of the additive remains a real challenge. Hou and co-workers88 found that a solid additive 2,2′-(perfluoro-1,4-phenylene)dithiophene (DTBF) exhibits very high volatility with a strong quadrupole moment. Interestingly, BHJ based on a conjugated polymer donor yielded a PCE = 17.1% (Voc = 0.0.846 V, Jsc = 26.2 mA cm2, FF = 0.770) in the presence of 40% DTBF while additive-free device yielded a PCE = 14.8% (Voc = 0.855 V, Jsc = 24.5 mA cm2, FF = 0.706). Besides, the addition of this fluorinated additive also enhanced the stability of the device. Indeed, this is due to the regular and condensed molecular packing of the active layer as well as strong charge quadrupole interaction between the acceptor and additive. Another additive fluorinated bi (perfluorophenyl)pimelate (BF7), has also been found to effectively improve the performance of the fullerene-free OSCs.89 It was noted that the use of 0.5 wt% BF7 in solution improves the performance of PM6:Y6 (PCE = 15.16% vs. 17.01%) as well as PM6:IT-4F (PCE = 12.34% vs. 13.32%) OSCs (IT-4F: 3,9 bis(2-methylene-((3-(1,1-dicyanomethylene)-6,7-difluoro)-ind anone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene). This improvement was ascribed to the improved fractal-like network structures and F–π interaction. Prior to this work, a “σ-hole”-containing volatile solid additive 1,4-diiodotetrafluoro benzene was reported, which assists in achieving a PCE value of 16.5% in the YM6/PM6 based OSC.90 Enhanced performance was attributed to the synergistic XB interactions, more condensed and ordered molecular arrangement, augmented CT process with suppressed charge recombination. The similar encouraging results have been reported for the additives 1,3-diiodobenzene (1,3-DIB)91 and 1,4-diiodobenzene (1,4-DIB)92 based OSCs and PSCs. One additional benefit of the use of 1,4-DIB is its solubility in alcohol (an important factor when considering post-processing). Benefitting from S⋯O NCI ability, additive 42 (Chart 5) was found to enhance the PV performance via inserting between the donor molecules leading to improved packing, morphology and charge mobility.93
NCIs have also been exploited to improve the performance of metal halide perovskite solar cells, which are at the forefront of PV research. In perovskite solar cells, among others, HTM plays a crucial role in achieving high efficiency. An HTM with suitable hole transport ability and self-assembly features are highly promising to realize high-performance solar cells. Yang et al.94 reported a small molecular HTM featuring intramolecular NCIs (S⋯O). The introduction of S⋯O ChB interaction led to self-planarized backbones, tuned energy levels, enhanced thermal properties, and afforded high film crystallinity and hole mobilities. Compared to 43a (without NCI, Chart 5), compounds 43b (Chart 5) and 43c (Chart 5) exhibited higher PCE values (18.4% vs. 20.27–21.1%). By looking closely at the molecular structure, it is quite clear that the incorporation of the alkoxy group improved the backbone co-planarity of 43b and 43cvia S⋯O interactions. Similarly, phenoxazine-based HTM 44 (Chart 5) was found to undergo NCI-mediated self-assembly process and possessed enhanced hole mobility, surface properties, and perovskite growth.95 A stable inverted perovskite using this HTM yielded PCE = 21.85%. Hydrogen-bonded (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) self-assembly in HTM 45a (Chart 5) has also been reported (Fig. 7a).96 It was noted that 45a exhibited three folds more hole mobility compared to the control 45b (without HB ability). This led to an enhanced PCE (15.1%). Moreover, the HB network has also been linked with the higher stability of the device.
C) self-assembly in HTM 45a (Chart 5) has also been reported (Fig. 7a).96 It was noted that 45a exhibited three folds more hole mobility compared to the control 45b (without HB ability). This led to an enhanced PCE (15.1%). Moreover, the HB network has also been linked with the higher stability of the device.
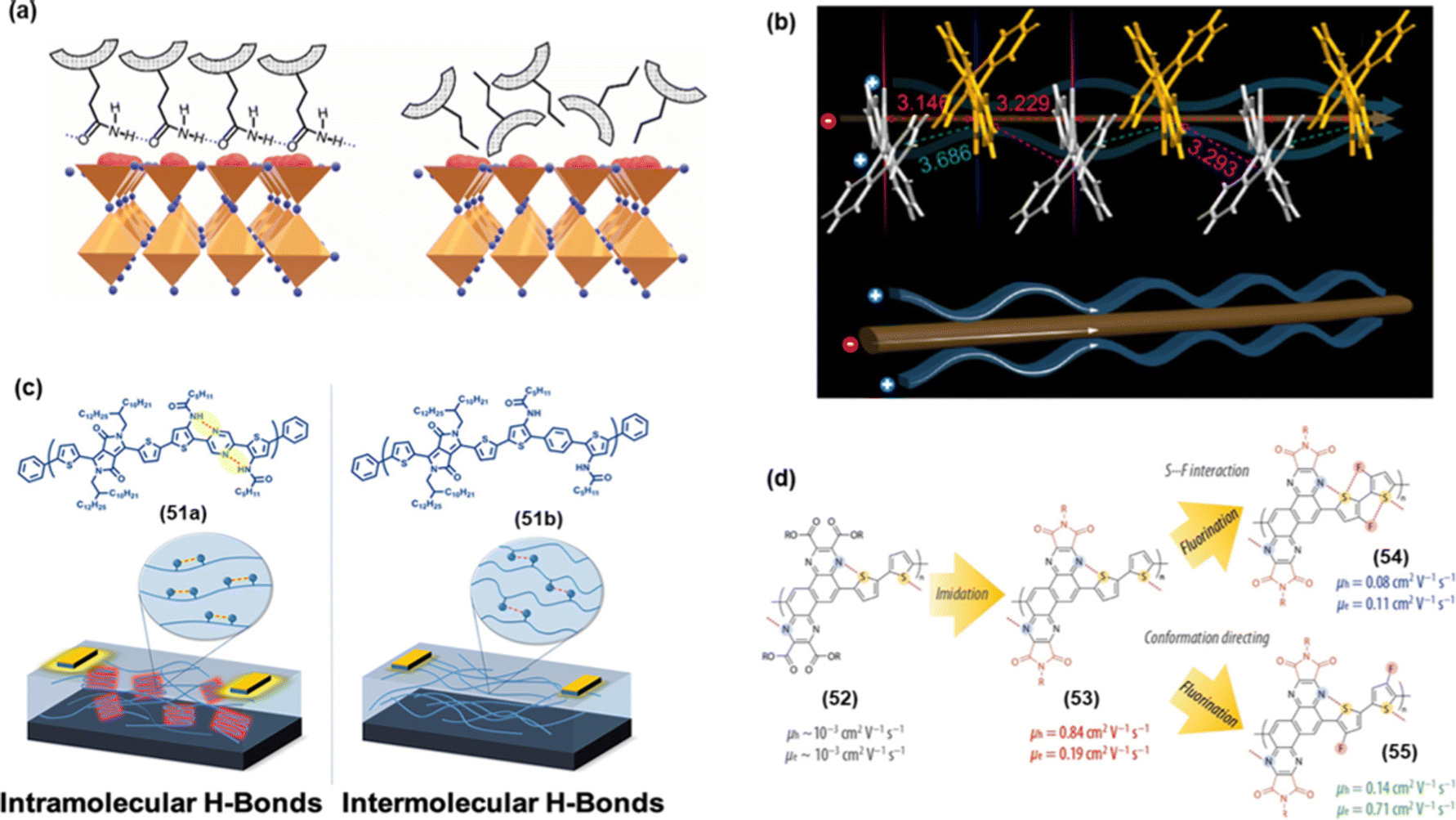 | ||
| Fig. 7 (a) The effect of HB on the possible arrangement of 45a and 45b on the perovskite surface, (b) drawing of possible charge flux styles speculated from single-crystal packing modes of 46, (c) effect of inter- and intramolecular HB in 51a and 51b on OFET devices, and (d) effect on hole–electron mobility in 52–55. Reproduced with permission from ref. 96, 97, 102 and 103. (Under a Creative Commons Attribution 4.0 International License, Copyright (2021) Elsevier B.V., Copyright (2021) American Chemical Society and Copyright (2022) American Chemical Society) | ||
6.2. Organic light emitting devices (OLEDs) and organic field-effect transistors (OFETs)
Thermally activated delayed fluorescent (TADF) materials are considered an excellent host for the next generation of organic light-emitting devices (OLEDs). To further improve the performance and rigidify the backbone of these materials, researchers are now exploiting the power of NCI. For example, 1,3-bis(N-carbazolyl)benzene is a phosphorescent host material which is often used in OLED research. It was noted that when the inner phenyl core of this host is replaced by a triazine derivative, it yields a new host material 46 with blue and white electroluminescence.97 It was suggested that such replacement bestowed intramolecular NCI feature (C–H⋯N of carbazole and triazine units) to the host leading to improved molecular and electrical attributes. Besides, it also generated multi-dimensional channels for efficient intermolecular carrier flux (Fig. 7b). Device fabricated using 46 (Chart 6) yielded white TADF OLEDs with maximum luminance (L) = 12770 cd m−2, external quantum efficiency (EQE) = 23.2%, current efficiency (CE) = 66.9 cd A−1 and power efficiencies (PE) = 70.0 lm W−1. Besides, sky-blue (EQE = 21.0%) and blue PhOLED (EQE = 14.2%) devices were also realized. It is noted that the efficiencies of white TADF OLEDs are one of the best values among white TADF OLEDs with low turn-on voltage. Zuo-Quan and co-workers98 reported a π-stacked TADF material 47 (Chart 6) with MSR properties. The reported compound exhibited L = 5997 cd m−2, EQE = 28.4%, CE = 60.0 cd A−1 and PE = 35.5 lm W−1, making it one of the most efficient TADF OLEDs.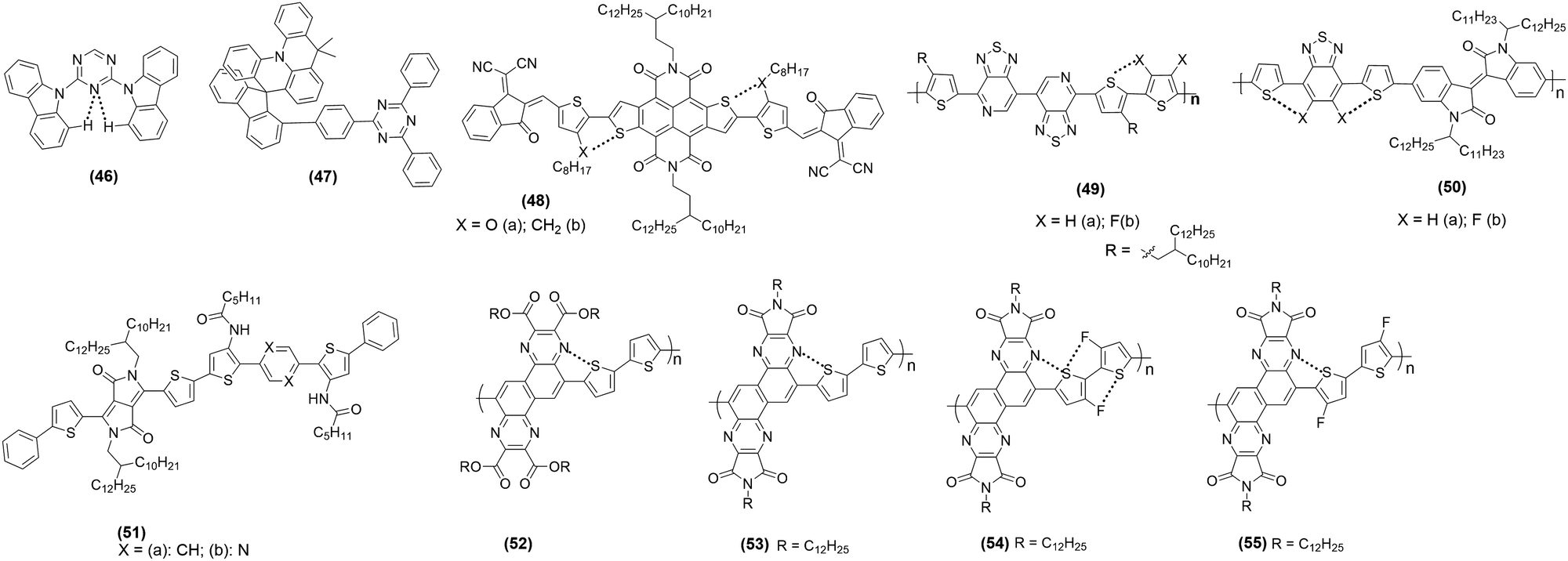 | ||
| Chart 6 Some recent examples of OLED and OFET active materials. | ||
In the field of organic field-effect transistor (OFET) research, the development of organic semiconductors has seen an excellent progress. Both unipolar and ambipolar OFETs utilizing NCIs have been reported recently. Especially n-type semiconductors are in high demand due to their diverse utility. However, the synthesis of high-performing air-stable n-type OFETs could be challenging. n-Type OFET with lowered LUMO level to reduce electron injection barrier and effectively improve electron injection is indeed in high demand. Hu and co-workers99 developed π-conjugated backbones 48a and 48b (Chart 6) with low LUMO energy levels (HOMO/LUMO = −4.37/−5.93 vs. −4.45/−6.15 eV). OFET devices with a bottom-gate-top-contact (BGTC) configuration under atmospheric conditions show that both molecules exhibited unipolar n-channel OFET performance (Table 3). As expected, 48a exhibited higher electron mobility than 48b owing to the difference in structure (S⋯O interlock and π–π stacking in 48a). Liu and co-workers100 noted that the backbone co-planarity, electronic properties, film organization and the CT properties of semiconducting polymers could be largely modulated by fluorination strategy, which led to weak F⋯S non-covalent conformation lock. An ambipolar device based on 49a (Chart 6) (μh/μe = 0.332/1.602 cm2 V−1 s−1) showed better performance than the non-fluorinated counterpart 49b (μh/μe = 0.0135/0.0191 cm2 V−1 s−1) attributed to enhanced backbone co-planarity and fluorine substitutes. It was noted that the device exhibited the best performance upon thermal annealing at 160 and 200 °C, respectively. Fluorination-mediated enhanced backbone co-planarity and ambipolar charge carrier transport behaviour have also been reported for isoindigo (IID)-based copolymers 50a (Chart 6) (μh/μe = 0.053/0.013 cm2 V−1 s−1) and 50b (μh/μe = 0.29/0.1 cm2 V−1 s−1) in the BGTC device structures, putting the latter among the best-performing materials.101
| S. no. | Hole transport | Electron transport | Ref. | ||||
|---|---|---|---|---|---|---|---|
| μ h (cm2 V−1 s−1) | I on/Ioff | V th (V) | μ e (cm2 V−1 s−1) | I on/Ioff | V th (V) | ||
| a Annealing temperature = 150 °C. b Annealing temperature = 160 °C. c Annealing temperature = 220 °C. d Annealing temperature = 200 °C. e Annealing temperature = 170 °C. | |||||||
| 48a | — | — | — | 0.17 | 1.0 × 104 | 11.6 | 99 |
| 48b | — | — | — | 0.085 | 5.6 × 106 | 10.1 | 99 |
| 49a | 1.35 × 10−2 | 2.0 × 103 | −34 | 1.91 × 10−2 | 5.0 × 102 | 72 | 100 |
| 49b | 0.332 | 8.0 × 103 | −36 | 1.602 | 2.0 × 105 | 65 | 100 |
| 50a | 0.053 | 102–103 | −25 to −30 | 0.013 | 101–102 | 41–55 | 101 |
| 50b | 0.29 | 102–104 | −35 to −53 | 0.1 | 102–104 | 36–46 | 101 |
| 51a | 0.163 | 105 | −19.50 | — | — | — | 102 |
| 51b | 0.00023 | 106 | −15.56 | — | — | — | 102 |
| 52 | 0.0010 | — | −35 | 0.0016 | — | 45 | 103 |
| 53 | 0.84 | — | −35 | 0.19 | — | 43 | 103 |
| 54 | 0.075 | — | −49 | 0.11 | — | 29 | 103 |
| 55 | 0.14 | — | −52 | 0.71 | — | 28 | 103 |
Liu et al.102 evaluated the effect of the NCIs configurations (intra vs. intermolecular hydrogen bonding) on OFET applications. To this end, they realized polymers 51a and 51b (Fig. 7c). It was noted that devices fabricated using 51a exhibited higher performance as intramolecular HB interaction led to backbone coplanarization, and crystallinity enhanced effective conjugation. Whereas 51a showed a field-effect mobility of 2.2 × 10−4 cm2 V−1 s−1, its counterpart with intermolecular hydrogen bonding showed a much lower (one-third) value. These results are very important as they highlight the pros and cons of a particular NCI in device fabrication. OFET device fabricated using pyrazine-based polymer 52 showed almost similar hole and electron mobilities. However, it was also noted that via suitable functionalization (imidation and fluorination, 53–55), its ambipolar characteristics could be significantly improved (Fig. 7d). Indeed, the enhanced performance could be attributed to the greater extent of inter and intramolecular NCIs and regular backbone.103
6.3. Energy storage materials and others
For the fabrication of “green batteries” suitable for future usage, organic polymers as cathode materials have gained tremendous interest. The fact that π-conjugated materials exhibit multiple weak interactions effectively promotes the electron transfer in the material and lowers the energy barrier of the ions.104 Despite this, one current challenge with the use of organic cathode materials is their inferior reversible capacity and rate performance. To improve the cycling stability and charge transport ability, Esser and co-workers105 suggested using a redox-active polymer as the cathode material. A Li-ion battery with cathode-active material based on poly(3-vinyl-N-methylphenothiazine) exhibited 93% and 52% of the initial capacity retained even after 10000 cycles at a C rate of 10 C and 100 C, respectively. The higher performance of the battery was attributed to the reduction in stacking distance upon oxidation leading to enhanced π–π stacking interactions. Besides, redox- active polymers based on dianhydride-derived polyimides (PIs) have been found suitable for storing Mg2+ ions.106 Jin and co-workers107 found that π–π stacking interactions between PI cores can help improve the Mg2+ storage performances.
In addition to the aforementioned potential applications of NCIs in OE research, they have also been utilized in areas such as nanophotonic, bioimaging, sensing, catalysis, and fuel storage research108–112 The potential of NCI in single-molecule electronics has also been tested.113 In molecular electronics, dimer formed by π-stacking is detrimental and lowers the electrical conductance. However, a recent paper showed that π-stacking significantly increases conductance.114 Hein et al.115 showed that the ChB potency of the conjugated system could be significantly reversible when modulated electrochemically.
7. Prospects and challenges
The quest for nanoarchitectonics has led to the discovery of several new classes of functional materials. Considering the diverse applications in organic semiconductors, exhaustive research has been carried out using π-conjugated materials. Since NCIs have a direct effect on the physical and chemical properties, numerous potential π-conjugated systems (small through medium to large) with modulated features have been reported. As we highlighted, these secondary interactions are most effective in combination and realized through the conjugated organic core, non-conjugated side arm/pendant group, metal centre, etc. Several recent studies have demonstrated that, with the aid of HB, XB and vdW (van der Waals) interactions, π-conjugated organics could show attractive luminescence properties, including RTPs. Interactions such as hydrogen bonding and metallophilicity control the patterning of aromatic cores in the solid state.Although outstanding progress has already been made, there are still some issues that need to be addressed. For instance, designing a new material controlled by a specific NCI would be challenging as the materials property is a subject of whole range of manipulations, involving covalent and non-covalent bonding. When more than one potential NCI is present in the molecule, a competitive interaction may also exist thereby reducing the effect of one or the other. Similarly, many of the materials have only been examined at small laboratory scale. Demonstration of applicability at large scale, stability, reliability, and reproducibility under normal condition and in an economical way is essential. It is also important to note that NCI-based organometallic materials design may lead to materials that could surpass/compete with their current organic counterparts.
8. Conclusions
In this tutorial review, we underlined the importance and merits of introducing NCIs in π-conjugated semiconducting materials. Using pertinent examples, we highlighted the properties, features and applications of various electrostatic and hydrophobic NCIs operating at the atomic and molecular levels. Advances made in the NCI characterization has been discussed. A critical evaluation of structure–property relationship with respect to NCIs in π-conjugated semiconducting materials has been made. Using several recent references, we have shown that NCI modulation is an intriguing strategy to realize high-performance materials in an economical and easy way. Although weak and often work in combination, NCIs have the ability to tune photo-physical and opto-electrical properties, which is remarkable. It has been demonstrated how swiftly interplay of the NCIs can solve problems inherent with the π-conjugated materials, especially polymers such as aggregation-induced emission quenching.Overall, research in the area of NCIs is a vibrant and active field, with new developments emerging at an impressive pace. It can be anticipated that a more sophisticated understanding of the unique features of NCIs could help us further in developing new materials. We expect that this review will stimulate further interest in the development of NCI-modulated materials and their applications in the near future. It will be useful to synthetic as well as theoretical chemists interested in designing and study the role of non-bonding interactions on the properties and conformation of a material.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deputy for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number RDO-2001.Notes and references
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich and A. J. Pombeiro, Non-covalent interactions in the synthesis and design of new compounds, John Wiley & Sons, 2016 Search PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. G. da Silva and A. J. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- P. J. Costa, Phys. Sci. Rev., 2017, 2, 20170136 Search PubMed.
- S. Yu, A. Peng, S. Zhang and H. Huang, Sci. China: Chem., 2018, 61, 1359–1367 CrossRef CAS.
- A. Del Prado, D. González-Rodríguez and Y. L. Wu, ChemistryOpen, 2020, 9, 409–430 CrossRef CAS PubMed.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 24, 10663–10687 CrossRef PubMed.
- V. R. Cooper, C. N. Lam, Y. Wang and B. G. Sumpter, Non-Covalent Interactions in Quantum Chemistry and Physics, Elsevier, 2017, pp. 417–451 Search PubMed.
- J. D. van der Waals, Over de Continuiteit van den Gas-en Vloeistoftoestand, Sijthoff, 1873 Search PubMed.
- R. Bishop, CrystEngComm, 2015, 17, 7448–7460 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef CAS PubMed.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., 2014, 126, 11359–11363 CrossRef.
- S. Kawai, A. Sadeghi, F. Xu, L. Peng, A. Orita, J. Otera, S. Goedecker and E. Meyer, ACS Nano, 2015, 9, 2574–2583 CrossRef CAS PubMed.
- Y. Kumar, S. Kumar, S. Kumar Keshri, J. Shukla, S. S. Singh, T. S. Thakur, M. Denti, A. Facchetti and P. Mukhopadhyay, Org. Lett., 2016, 18, 472–475 CrossRef CAS PubMed.
- R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- L. Bai, P. Bose, Q. Gao, Y. Li, R. Ganguly and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 436–441 CrossRef CAS PubMed.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo and P. Metrangolo, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- J. Noh, S. Jung, D. G. Koo, G. Kim, K. S. Choi, J. Park, T. J. Shin, C. Yang and J. Park, Sci. Rep., 2018, 8, 1–11 Search PubMed.
- N. J. Singh, S. K. Min, D. Y. Kim and K. S. Kim, J. Chem. Theory Comput., 2009, 5, 515–529 CrossRef CAS PubMed.
- M. Y. Jin, Q. Zhen, D. Xiao, G. Tao, X. Xing, P. Yu and C. Xu, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- A. Jamadar, A. K. Singh, L. Roy and A. Das, J. Mater. Chem. C, 2021, 9, 11893–11904 RSC.
- A. U. Mu, Y.-J. Kim, O. Miranda, M. Vazquez, J. Strzalka, J. Xu and L. Fang, ACS Mater. Lett., 2022, 4, 1270–1278 CrossRef CAS.
- M. Morisue, M. Kawanishi, I. Ueno, T. Nakamura, T. Nabeshima, K. Imamura and K. Nozaki, J. Phys. Chem. B, 2021, 125, 9286–9295 CrossRef CAS PubMed.
- A. Rajak, A. Kumar Singh, L. Roy and A. Das, ChemNanoMat, 2022, e202200082 CAS.
- S. A. Harry, M. Kazim, P. M. Nguyen, A. Zhu, M. R. Xiang, J. Catazaro, M. Siegler and T. Lectka, Angew. Chem., Int. Ed., 2022, 61, e202207966 CrossRef CAS PubMed.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- Y. S. Al-Hamdani and A. Tkatchenko, J. Chem. Phys., 2019, 150, 010901 CrossRef PubMed.
- J. Zhang, P. Chen, B. Yuan, W. Ji, Z. Cheng and X. Qiu, Science, 2013, 342, 611–614 CrossRef CAS PubMed.
- J. Nie, Y. Deng, F. Tian, S. Shi and P. Zheng, Nano Res., 2022, 15, 4251–4257 CrossRef CAS PubMed.
- S. Zhang, Y. Geng, Y. Fan, W. Duan, K. Deng, D. Zhao and Q. Zeng, Phys. Chem. Chem. Phys., 2017, 19, 31284–31289 RSC.
- S. Kawai, A. S. Foster, T. Björkman, S. Nowakowska, J. Björk, F. F. Canova, L. H. Gade, T. A. Jung and E. Meyer, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- S. Kawai, T. Nishiuchi, T. Kodama, P. Spijker, R. Pawlak, T. Meier, J. Tracey, T. Kubo, E. Meyer and A. S. Foster, Sci. Adv., 2017, 3, e1603258 CrossRef PubMed.
- B. Shen, H. Wang, H. Xiong, X. Chen, E. G. Bosch, I. Lazić, W. Qian and F. Wei, Nature, 2022, 607, 703–707 CrossRef CAS PubMed.
- S. Kawai, O. Krejci, A. S. Foster, R. Pawlak, F. Xu, L. Peng, A. Orita and E. Meyer, ACS Nano, 2018, 12, 8791–8797 CrossRef CAS PubMed.
- B. Mallada, A. Gallardo, M. Lamanec, B. De La Torre, V. Špirko, P. Hobza and P. Jelinek, Science, 2021, 374, 863–867 CrossRef CAS PubMed.
- D. Wang, Z. Wang, W. Liu, S. Zhong, Y. P. Feng, K. P. Loh and A. T. S. Wee, Small, 2022, 18, 2202368 CrossRef CAS PubMed.
- S. Song, L. Wang, J. Su, Z. Xu, C.-H. Hsu, C. Hua, P. Lyu, J. Li, X. Peng and T. Kojima, Chem. Sci., 2021, 12, 11659–11667 RSC.
- C. Shang, G. Wang, K. Liu, Q. Jiang, F. Liu, P. T. Chou and Y. Fang, Angew. Chem., Int. Ed., 2020, 59, 8579–8585 CrossRef CAS PubMed.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS PubMed.
- J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y.-L. Wu, Chem. Sci., 2021, 12, 767–773 RSC.
- B. Liu, D. Rocca, H. Yan and D. Pan, JACS Au, 2021, 1, 2182–2187 CrossRef CAS PubMed.
- T. Kharandiuk, E. J. Hussien, J. Cameron, R. Petrina, N. J. Findlay, R. Naumov, W. T. Klooster, S. J. Coles, Q. Ai and S. Goodlett, Chem. Mater., 2019, 31, 7070–7079 CrossRef CAS.
- G. S. Sinclair, R. C. Claridge, A. J. Kukor, W. S. Hopkins and D. J. Schipper, Chem. Sci., 2021, 12, 2304–2312 RSC.
- Y. Song, Z. Zhong, L. Li, X. Liu, J. Huang, H. Wu, M. Li, Z. Lu, J. Yu and J. Hai, Chem. Eng. J., 2022, 430, 132830 CrossRef CAS.
- C. Yang, Q. An, H. R. Bai, H. F. Zhi, H. S. Ryu, A. Mahmood, X. Zhao, S. Zhang, H. Y. Woo and J. L. Wang, Angew. Chem., Int. Ed., 2021, 60, 19241–19252 CrossRef CAS PubMed.
- F. Lin, K. Jiang, W. Kaminsky, Z. Zhu and A. K.-Y. Jen, J. Am. Chem. Soc., 2020, 142, 15246–15251 CrossRef CAS PubMed.
- Q. Fan, H. Fu, Z. Luo, J. Oh, B. Fan, F. Lin, C. Yang and A. K.-Y. Jen, Nano Energy, 2022, 92, 106718 CrossRef CAS.
- J. Wu, Y. Xu, Z. Yang, Y. Chen, X. Sui, L. Yang, P. Ye, T. Zhu, X. Wu and X. Liu, Adv. Energy Mater., 2019, 9, 1803012 CrossRef.
- D. Romito, E. Fresta, L. M. Cavinato, H. Kählig, H. Amenitsch, L. Caputo, Y. Chen, P. Samorì, J.-C. Charlier and R. Costa, Angew. Chem., Int. Ed., 2022, 61, e202202137 CrossRef CAS PubMed.
- J. Yu, H. Ma, W. Huang, Z. Liang, K. Zhou, A. Lv, X.-G. Li and Z. He, JACS Au, 2021, 1, 1694–1699 CrossRef CAS PubMed.
- Y. Wen, H. Liu, S. Zhang, Y. Gao, Y. Yan and B. Yang, J. Mater. Chem. C, 2019, 7, 12502–12508 RSC.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- X. Zhang, Z. Fu, X. Jiang, Z. Yang, S. Wang, K. Wang, Z. Wu, S.-T. Zhang, H. Liu and B. Yang, Chem. Commun., 2022, 58, 8250–8253 RSC.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357–4363 CrossRef CAS.
- J. Liu, L.-K. Ma, F. K. Sheong, L. Zhang, H. Hu, J.-X. Zhang, J. Zhang, Z. Li, C. Ma and X. Han, J. Mater. Chem. A, 2018, 6, 16874–16881 RSC.
- Z. R. Kehoe, G. R. Woller, E. D. Speetzen, J. B. Lawrence, E. Bosch and N. P. Bowling, J. Org. Chem., 2018, 83, 6142–6150 CrossRef CAS PubMed.
- S. A. Sharber, W. J. Mullin and S. W. Thomas III, Chem. Mater., 2021, 33, 6640–6661 CrossRef CAS.
- W. J. Mullin, R. H. Pawle, S. A. Sharber, P. Müller and S. W. Thomas, J. Mater. Chem. C, 2019, 7, 1198–1207 RSC.
- W. J. Mullin, P. Müller, A. J. Schaefer, E. Guzman, S. E. Wheeler and S. W. Thomas III, J. Mater. Chem. C, 2022, 10, 11199–11210 RSC.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- C. J. Takacs, Y. Sun, G. C. Welch, L. A. Perez, X. Liu, W. Wen, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 16597–16606 CrossRef CAS PubMed.
- M. Li, Y. Zhou, J. Zhang, J. Song and Z. Bo, J. Mater. Chem. A, 2019, 7, 8889–8896 RSC.
- D. Yang, Y. Jiao, L. Yang, Y. Chen, S. Mizoi, Y. Huang, X. Pu, Z. Lu, H. Sasabe and J. Kido, J. Mater. Chem. A, 2015, 3, 17704–17712 RSC.
- M. Dong, K. Miao, Y. Hu, J. Wu, J. Li, P. Pang, X. Miao and W. Deng, Phys. Chem. Chem. Phys., 2017, 19, 31113–31120 RSC.
- T. Ikeda, T. Iijima, R. Sekiya, O. Takahashi and T. Haino, J. Org. Chem., 2016, 81, 6832–6837 CrossRef CAS PubMed.
- A. Haque, R. A. Al-Balushi, I. J. Al-Busaidi, M. S. Khan and P. R. Raithby, Chem. Rev., 2018, 118, 8474–8597 CrossRef CAS PubMed.
- X. Zheng, M. H.-Y. Chan, A. K.-W. Chan, S. Cao, M. Ng, F. K. Sheong, C. Li, E. C. Goonetilleke, W. W. Y. Lam and T.-C. Lau, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116543119 CrossRef CAS PubMed.
- J. Ni, X. Zhang, N. Qiu, Y. H. Wu, L. Y. Zhang, J. Zhang and Z. N. Chen, Inorg. Chem., 2011, 50, 9090–9096 CrossRef CAS PubMed.
- M. Martínez-Junquera, E. Lalinde and M. T. Moreno, Inorg. Chem., 2022, 61, 10898–10914 CrossRef PubMed.
- E. Bulatov, T. Eskelinen, A. Y. Ivanov, P. M. Tolstoy, E. Kalenius, P. Hirva and M. Haukka, ChemPhysChem, 2021, 22, 2044–2049 CrossRef CAS PubMed.
- V. Sivchik, A. Kochetov, T. Eskelinen, K. S. Kisel, A. I. Solomatina, E. V. Grachova, S. P. Tunik, P. Hirva and I. O. Koshevoy, Chem. – Eur. J., 2021, 27, 1787–1794 CrossRef CAS PubMed.
- J. Matern, I. Maisuls, C. A. Strassert and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202208436 CAS.
- W. J. Mullin, H. Qin, T. Mani, P. Muller, M. J. Panzer and S. W. Thomas, Chem. Commun., 2020, 56, 6854–6857 RSC.
- V. Mishra, A. Raghuvanshi, A. K. Saini and S. M. Mobin, J. Organomet. Chem., 2016, 813, 103–109 CrossRef CAS.
- M.-H. Nguyen and J. H. Yip, Organometallics, 2010, 29, 2422–2429 CrossRef CAS.
- Y. Zhang, Y. Wang, T. Shan and H. Zhong, ACS Appl. Energy Mater., 2022, 5, 11283–11291 CrossRef CAS.
- L. Wang, Q. An, L. Yan, H.-R. Bai, M. Jiang, A. Mahmood, C. Yang, H. Zhi and J.-L. Wang, Energy Environ. Sci., 2022, 15, 320–333 RSC.
- J. Wang, H. Chen, X. Xu, Z. Ma, Z. Zhang, C. Li, Y. Yang, J. Wang, Y. Zhao and M. Zhang, J. Mater. Chem. A, 2022, 10, 16714–16721 RSC.
- X. Zhang, L. Qin, J. Yu, Y. Li, Y. Wei, X. Liu, X. Lu, F. Gao and H. Huang, Angew. Chem., 2021, 133, 12583–12589 CrossRef.
- Y. Zhang, C. e Zhang, A. Zhang, H. Wu, G. Ran, Y. Zhou, X. Wang, C. Li, Y. Liu and C. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 21287–21294 CrossRef CAS PubMed.
- S. He, Z. Lin, F. Du, X. Wang, Y. Liu and W. Tang, Chem. Eng. J., 2022, 441, 135973 CrossRef CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, G. Cai, Y. Zhang, X. Lu and Y. Lin, J. Am. Chem. Soc., 2020, 142, 18741–18745 CrossRef CAS PubMed.
- R. Yu, H. Yao, Y. Xu, J. Li, L. Hong, T. Zhang, Y. Cui, Z. Peng, M. Gao and L. Ye, Adv. Funct. Mater., 2021, 31, 2010535 CrossRef CAS.
- K. E. Hung, Y. S. Lin, Y. J. Xue, H. R. Yang, Y. Y. Lai, J. W. Chang, C. J. Su, A. C. Su, C. S. Hsu and U. S. Jeng, Adv. Energy Mater., 2022, 12, 2103702 CrossRef CAS.
- J. Fu, S. Chen, K. Yang, S. Jung, J. Lv, L. Lan, H. Chen, D. Hu, Q. Yang and T. Duan, iScience, 2020, 23, 100965 CrossRef CAS PubMed.
- Q. Yu, J. Fu, H. Chen, S. Chen, D. Hu, K. Yang, Z. Kan, K. Sun, S. Lu and Z. Xiao, Org. Electron., 2021, 93, 106161 CrossRef CAS.
- Y. Xie, H. S. Ryu, L. Han, Y. Cai, X. Duan, D. Wei, H. Y. Woo and Y. Sun, Sci. China: Chem., 2021, 64, 2161–2168 CrossRef CAS.
- C. Li, X. Gu, Z. Chen, X. Han, N. Yu, Y. Wei, J. Gao, H. Chen, M. Zhang and A. Wang, J. Am. Chem. Soc., 2022, 144, 14731–14739 CrossRef CAS PubMed.
- K. Yang, Q. Liao, J. Huang, Z. Zhang, M. Su, Z. Chen, Z. Wu, D. Wang, Z. Lai, H. Y. Woo, Y. Cao, P. Gao and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202113749 CAS.
- N. Cai, F. Li, Y. Chen, R. Luo, T. Hu, F. Lin, S. M. Yiu, D. Liu, D. Lei and Z. Zhu, Angew. Chem., Int. Ed., 2021, 60, 20437–20442 CrossRef CAS PubMed.
- C. Wang, M. Liu, S. Rahman, H. P. Pasanen, J. Tian, J. Li, Z. Deng, H. Zhang and P. Vivo, Nano Energy, 2022, 101, 107604 CrossRef CAS.
- H. Li, Y. Wang, L. Yu, C. Liu, C. Zhou, S. Sun, M. Li, Y. Tao, G. Xie and H. Xu, Chem. Eng. J., 2021, 425, 131487 CrossRef CAS.
- S. Y. Yang, Z. Q. Feng, Z. Fu, K. Zhang, S. Chen, Y. J. Yu, B. Zou, K. Wang, L. S. Liao and Z. Q. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202206861 CAS.
- H. Ran, F. Li, R. Zheng, W. Ni, Z. Lei, F. Xie, X. Duan, R. Han, N. Pan and J.-Y. Hu, ACS Appl. Electron. Mater., 2021, 3, 5573–5583 CrossRef CAS.
- X. Tao, Y. Liu, L. Du, Y. Yan, Z. Wu, Y. Zhao, Y. Guo, H. Chen and Y. Liu, J. Mater. Chem. C, 2021, 9, 15083–15094 RSC.
- X. Qiao, Q. Wei, H. Wu and H. Li, Macromol. Chem. Phys., 2020, 221, 2000189 CrossRef CAS.
- M. U. Ocheje, R. B. Goodman, K.-T. Lu, Y. Wang, L. A. Galuska, L. Soullard, Z. Cao, S. Zhang, M. Yadiki and X. Gu, Chem. Mater., 2021, 33, 8267–8277 CrossRef CAS.
- T. Mikie, K. Okamoto, Y. Iwasaki, T. Koganezawa, M. Sumiya, T. Okamoto and I. Osaka, Chem. Mater., 2022, 34, 2717–2729 CrossRef CAS.
- H. Zhang, Y. Fang, F. Yang, X. Liu and X. Lu, Energy Environ. Sci., 2020, 13, 2515–2523 RSC.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- H. Zhang, Y. Fang, F. Yang, X. Liu and X. Lu, Energy Environ. Sci., 2020, 13, 2515–2523 RSC.
- Y. Wang, Z. Liu, C. Wang, Y. Hu, H. Lin, W. Kong, J. Ma and Z. Jin, Energy Storage Mater., 2020, 26, 494–502 CrossRef.
- Y. Lv, D. Li, A. Ren, Z. Xiong, Y. Yao, K. Cai, S. Xiang, Z. Zhang and Y. S. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 28662–28667 CrossRef CAS PubMed.
- R. D. Mule, R. Roy, K. Mandal, D. Chopra, T. Dutta, S. P. Sancheti, P. S. Shinde, S. Banerjee, A. Lal Koner and R. Bhowal, Chem. – Eur. J., 2022, 28, e202200632 CrossRef CAS PubMed.
- D. Chen, W. Yang, L. Jiao, L. Li, S. H. Yu and H. L. Jiang, Adv. Mater., 2020, 32, 2000041 CrossRef CAS PubMed.
- L. Li, Z. Li, W. Yang, Y. Huang, G. Huang, Q. Guan, Y. Dong, J. Lu, S.-H. Yu and H.-L. Jiang, Chem, 2021, 7, 686–698 CAS.
- L. Jiao, J. Wang and H.-L. Jiang, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS.
- B. Limburg, J. O. Thomas, G. Holloway, H. Sadeghi, S. Sangtarash, I. C. Y. Hou, J. Cremers, A. Narita, K. Müllen and C. J. Lambert, Adv. Funct. Mater., 2018, 28, 1803629 CrossRef.
- P. Li, S. Hou, B. Alharbi, Q. Wu, Y. Chen, L. Zhou, T. Gao, R. Li, L. Yang, X. Chang, G. Dong, X. Liu, S. Decurtins, S. X. Liu, W. Hong, C. J. Lambert, C. Jia and X. Guo, J. Am. Chem. Soc., 2022, 144, 15689–15697 CrossRef CAS PubMed.
- R. Hein, A. Docker, J. J. Davis and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 8827–8836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cs00262k |
| This journal is © The Royal Society of Chemistry 2023 |