
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Benchmark and performance of long-range corrected time-dependent density functional tight binding (LC-TD-DFTB) on rhodopsins and light-harvesting complexes†
Beatrix M.
Bold
a,
Monja
Sokolov
a,
Sayan
Maity
b,
Marius
Wanko
c,
Philipp M.
Dohmen
a,
Julian J.
Kranz
ad,
Ulrich
Kleinekathöfer
b,
Sebastian
Höfener
a and
Marcus
Elstner
*ad
aInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany. E-mail: marcus.elstner@kit.edu
bDepartment of Physics and Earth Science, Jacobs University Bremen, 28759 Bremen, Germany
cNano-Bio Spectroscopy Group and ETSF, Dpto. Material Physics, Universidad del País Vasco, 20018 San Sebastiàn, Spain
dInstitute of Biological Interfaces (IBG2), Karlsruhe Institute of Technology (KIT), Kaiserstrasse 12, 76131 Karlsruhe, Germany
First published on 6th June 2023
Abstract
Correction for ‘Benchmark and performance of long-range corrected time-dependent density functional tight binding (LC-TD-DFTB) on rhodopsins and light-harvesting complexes’ by Beatrix M. Bold et al., Phys. Chem. Chem. Phys., 2020, 22, 10500–10518, https://doi.org/10.1039/C9CP05753F.
The authors have recognised two errors in the data for the published version of this article. The first one concerns the reported QM/MM calculations on the Fenna–Matthews–Olson (FMO) complex, and the second error affects all Coulomb couplings presented.
In brief, the most important changes are: (i) the ZINDO site energies for the individual pigments are closer to each other and the ZINDO site energy fluctuations are smaller; (ii) the couplings are smaller and now in better agreement with reference data; and (iii) as a consequence, the exciton splitting is decreased. These errors do not change the conclusions of the study but only lead to small corrections of the reported results, as detailed below.
Error description
In the originally published version of the article, the excitation energies and couplings of the pigments in an FMO monomer were computed using the time-dependent long-range corrected density functional tight binding (TD-LC-DFTB) and ZINDO/S approaches in a QM/MM fashion representing their respective environment by point charges. The environment of an individual pigment consisted of the FMO monomer, the remaining six BChl a chromophores, TIP3P water molecules, as well as 15 sodium and 16 chloride ions.Prior to the calculations of site energies and couplings along the trajectory, the coordinates of the system were processed as follows: the protein complex was centered in the simulation box with Gromacs tools in two steps. First, the protein and the chromophores were clustered using the pbc cluster option of trjconv; second, they were centered in the box using the pbc mol option of trjconv. However, it was not recognized that some counterions were placed outside the simulation box erroneously. Consequently, the MM point charges corresponding to these counterions had wrong coordinates in the QM/MM calculations of the site energies and couplings, leading to an incorrect description of the QM–MM electrostatics.
These calculations of site energies and couplings were repeated using a corrected procedure for the processing of the MM point charges. Specifically, the trajectory was centered using the Gromacs tools in a more appropriate way: in the first step, any appearing error due to the periodic boundary conditions were corrected, and the protein was centered in the box using the pbc atom option of trjconv in the second step.
Additionally, the authors have recognized an error in a script that was used to compute the Coulomb couplings. The coordinates were not converted to atomic units as needed, which resulted in a significant overestimation of the coupling values reported in the original publication. The corrected couplings are now in much better agreement with the reference data.
Corrections
All corrected results presented in this erratum were computed using the newest parameter set for excitation energies.1 In general, DFTB parameters can be divided into electronic parameters and parameters describing the repulsive potential. Excitation energy calculations as well as calculations of transition charges require only electronic parameters. These parameters can be optimized for certain properties with relatively low effort by variation of the compression radii. The parameters in the original publication were already optimized for excitation energies but the new parameter set1 derived with a small modification at the carbon compression radius provides improved absolute excitation energies. The predicted relative excitation energies of BChl a in FMO are basically the same, as shown in Fig. 12.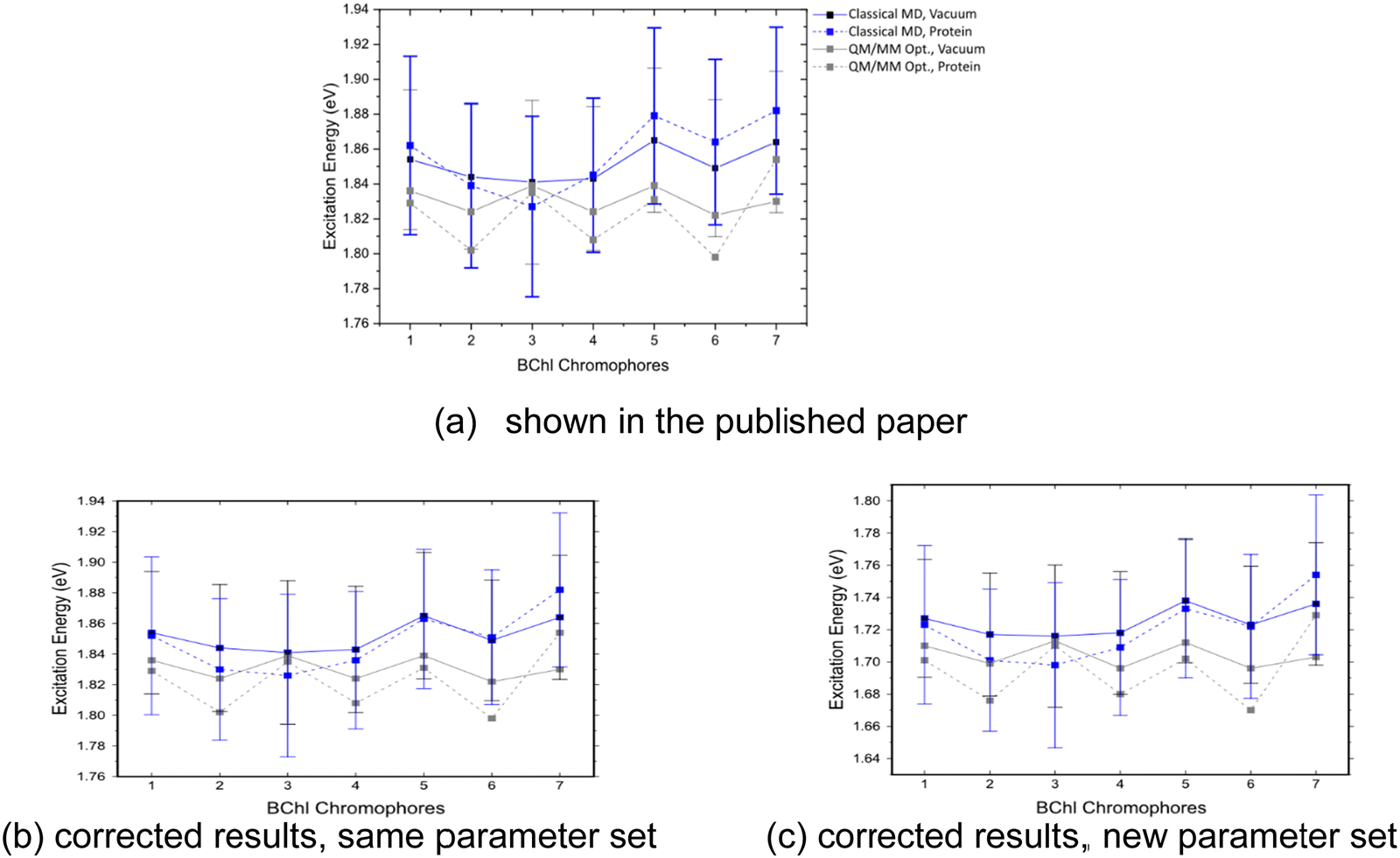 | ||
| Fig. 12 Excitation energies of the seven BChl a chromophores, computed for DFTB QM/MM optimized structures and sampled along the classical trajectories. These values were calculated in vacuum and with fixed MM point charges using LC-DFTB. | ||
The authors recalculated only a selection and comment on the expected changes for the other figures and tables. In the following, they start with the benchmark of the couplings on the BChl a dimer models, followed by exemplary couplings in the LH2 complex. Thereafter, they continue with the new site energies, couplings and the resulting exciton energies of the FMO complex. Previous results are shown in parentheses, while the new results are shown in bold. The figures and tables carry the same numbers as in the published version of the article. The corrections to the ESI have been discussed here. Please refer to revised version of the ESI (https://www.rsc.org/suppdata/c9/cp/c9cp05753f/c9cp05753f1.pdf) for the corrected tables and figures.
1 Benchmark: bacteriochlorophyll a (BChl a)-excitonic coupling I: model dimers
Table 3 shows that the correctly computed TD-LC-DFTB couplings not only have the same trend, but also the absolute values are very similar to the DFT references. Using the correction, the TD-LC-DFTB couplings are no more overestimated, but are slightly smaller than the reference values.| Distance [Å] | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|
| TrESP | B3LYP | 0.050 | 0.038 | 0.029 | 0.023 |
| CAM-B3LYP | 0.056 | 0.043 | 0.033 | 0.026 | |
| Tr-Mulliken | B3LYP | 0.047 | 0.036 | 0.028 | 0.022 |
| CAM-B3LYP | 0.054 | 0.041 | 0.032 | 0.025 | |
| — | LC-DFTB | 0.041 | 0.031 | 0.024 | 0.019 |
| (0.082) | (0.063) | (0.049) | (0.039) | ||
2 Performance of LC-DFTB in biological model systems: light-harvesting complexes: light-harvesting complex II (LH2)
The TD-LC-DFTB Coulomb couplings in LH2 are compared to the TrESP couplings in Fig. S5. The corrected TD-LC-DFTB couplings are now in much better agreement with the TrESP values. Within the B800 ring, they are slightly higher, but in the B850 ring the TD-LC-DFTB coupling values are of the same size as the TrESP couplings. The authors expect similarly smaller coupling values for all BChl a pairs in the LH2 complexes studied in the published version of the article.Performance of LC-DFTB in biological model systems: light-harvesting complexes: Fenna–Matthews–Olson (FMO) complex
The erroneous electrostatics in the QM/MM calculations in the FMO complex had only a small effect on the TD-LC-DFTB site energies and couplings, while the ZINDO/S excitation energies show slightly larger deviations.Fig. 12 depicts a slightly different trend for the corrected TD-LC-DFTB site energies compared to the ones shown in the original publication, while the range between the lowest and largest values remains basically the same (Table 9). The results in Fig. 12b were computed with the same parameter set as in the original publication and reveal the changes due to the corrected electrostatics. Fig. 12c shows the almost constant shift to lower excitation energies when the new parameter set is applied for the computations with a correct representation of the electrostatics.
| Max | Min | Shift | |
|---|---|---|---|
| Vacuum | 1.738 (1.865) | 1.716 (1.841) | −0.022 (−0.024) |
| Protein | 1.754 (1.882) | 1.698 (1.827) | −0.056 (−0.055) |
| Coupled chromophores | |||
| LC-DFTB | 1.811 (1.979) | 1.659 (1.775) | −0.152 (−0.204) |
| TrESP | 1.876 | 1.777 | −0.099 |
| Experimental | 1.563 | 1.503 | −0.060 |
The ZINDO/S approach now predicts a smaller range of the excitation energies along the MD trajectory (Fig. S7 and Table S20). It is remarkable that the excitation energy of BChl 5 is no longer overestimated as previously reported. Nonetheless, the large excitation energy of BChl 3 in the QM/MM optimized structure as well as the large energetic change in BChl 5 indicate a strong sensitivity to the electrostatics.
The corrected couplings of the BChl a pairs in the FMO complex are presented in Table S19. Their values are roughly half of those previously presented in the published version of the article.
Finally, the excitonic energies were recalculated using the corrected excitation energy and coupling values (Table S21). The range of the excitonic energies is smaller due to the smaller couplings.
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
References
- N. Schieschke, B. M. Bold, P. M. Dohmen, D. Wehl, M. Hoffmann, A. Dreuw, M. Elstner and S. Höfener, Geometry dependence of excitonic couplings and the consequences for configuration-space sampling, J. Comput. Chem., 2021, 42(20), 1402–1418 CrossRef CAS PubMed.
Footnote |
| † Corrections have also been made to the ESI, which can be reached using the following link, https://www.rsc.org/suppdata/c9/cp/c9cp05753f/c9cp05753f1.pdf |
| This journal is © the Owner Societies 2023 |