
Evaluation of DNA–protein complex structures using the deep learning method†
Chengwei
Zeng‡
a,
Yiren
Jian‡
b,
Chen
Zhuo
a,
Anbang
Li
a,
Chen
Zeng
c and
Yunjie
Zhao
 *a
*a
aInstitute of Biophysics and Department of Physics, Central China Normal University, Wuhan, 430079, China. E-mail: yjzhaowh@ccnu.edu.cn
bDepartment of Computer Science, Dartmouth College, Hanover, NH 03755, USA
cDepartment of Physics, The George Washington University, Washington, DC 20052, USA
First published on 24th November 2023
Abstract
Biological processes such as transcription, repair, and regulation require interactions between DNA and proteins. To unravel their functions, it is imperative to determine the high-resolution structures of DNA–protein complexes. However, experimental methods for this purpose are costly and technically demanding. Consequently, there is an urgent need for computational techniques to identify the structures of DNA–protein complexes. Despite technological advancements, accurately identifying DNA–protein complexes through computational methods still poses a challenge. Our team has developed a cutting-edge deep-learning approach called DDPScore that assesses DNA–protein complex structures. DDPScore utilizes a 4D convolutional neural network to overcome limited training data. This approach effectively captures local and global features while comprehensively considering the conformational changes arising from the flexibility during the DNA–protein docking process. DDPScore consistently outperformed the available methods in comprehensive DNA–protein complex docking evaluations, even for the flexible docking challenges. DDPScore has a wide range of applications in predicting and designing structures of DNA–protein complexes.
1 Introduction
The DNA–protein complex is essential for many cellular processes, such as DNA replication, RNA transcription, protein synthesis, signal transduction, gene repair, and gene regulation.1,2 It coordinates these activities and ensures they are carried out effectively and efficiently. A comprehensive understanding of the structure and function of DNA–protein complexes significantly enhances our knowledge of life processes, disease mechanisms, and potential drug targets.3–6 Understanding this concept has immense importance in various fields, such as clinical diagnosis, nanomedicine, transition metal chemistry, and other interdisciplinary areas.7–9 For example, Pax2 (paired box protein 2) is a TF (transcription factor) that regulates the differentiation of various cell types.10–12 Its functionality relies on direct DNA–protein interactions. Typically, Pax2 is downregulated, but it shows abnormal overexpression in kidney cancers.13–16 Some research indicates that depleting Pax2 has significant antineoplastic effects in renal carcinoma, rendering Pax2 an attractive target for kidney cancer chemotherapy.17,18 The lack of a crystal structure is a major obstacle to designing drugs that target this specific DNA-binding domain. Determining the structure of DNA–protein complexes through experimental techniques such as X-ray crystallography, NMR (nuclear magnetic resonance), and cryo-electron microscopy presents formidable challenges, which are expensive and time-consuming.19,20 As of August 28, 2023, it has been reported that there are already 208![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 844 structures that have been experimentally determined and deposited in the PDB (protein data bank). However, out of this number, only 6909 are DNA–protein complexes.21 The number of experimentally determined DNA–protein complex structures is significantly lower than the expected number of DNA–protein complexes formed within cells. The need for reliable and accurate theoretical methods to predict the structures of DNA–protein complexes has become increasingly urgent.
844 structures that have been experimentally determined and deposited in the PDB (protein data bank). However, out of this number, only 6909 are DNA–protein complexes.21 The number of experimentally determined DNA–protein complex structures is significantly lower than the expected number of DNA–protein complexes formed within cells. The need for reliable and accurate theoretical methods to predict the structures of DNA–protein complexes has become increasingly urgent.
From a computational perspective, methods for predicting DNA–protein complex structures can be primarily categorized into homologous template modeling and free docking. Template-based methods aim to model complexes using the structural information from homologous complexes stored in the template library. The quality of template-based prediction methods relies heavily on the quality of homologous templates. However, predicting DNA–protein complex structures using homologous modeling is limited due to the scarcity of DNA–protein complex structures. The process of the free docking method primarily comprises two steps: conformation sampling and evaluation.22,23 The shape and electrostatic complementarity of DNA and protein allow sampling and generation of all possible complex conformations. Then, the scoring function is employed in the scoring phase to rank and identify the near-native structure. The classical free docking algorithm disassembles the complex for testing, ignoring potential conformational changes in DNA and proteins during the formation of the DNA–protein complex. As a result of limited consideration for the flexibility of DNA–protein complexes, accurately predicting their structures is challenging, especially in flexible docking scenarios.
Recently, various algorithms have been developed for the molecular docking of DNA–protein complexes, such as FTDock,24 GRAMM-X,25 HEX,26 PatchDock,27 NPDock,28 HDOCK,29,30 and HADDOCK.31 However, only NPDock and HDOCK were initially tailored for nucleic acid–protein complexes. Other methods were primarily designed for protein–protein complex docking, lacking scoring functions explicitly tailored for DNA–protein complexes. Hence, these methods may need to be more effective in capturing the structure characteristics specific to DNA–protein complexes, resulting in subpar performance in the structure prediction of DNA–protein complexes. Additionally, there is currently no method for independently evaluating the conformation of DNA–protein complexes without relying on conformational sampling, which presents a significant challenge in achieving high precision. The NPDock and HDOCK algorithms are designed based on the static structure features without addressing the flexible docking challenge. HDOCK is the most popular molecular docking method to dock protein–protein and protein–DNA/RNA using a hybrid algorithm combining template-based modeling and ab initio-free docking.29,30 HDOCK employs HHSuite32 to explore the sequence similarities in the PDB database and then utilizes the homologous sequence structure for docking. HDOCK proves highly effective in rigid docking owing to its iterative knowledge-based scoring functions.33,34 Unfortunately, these methods face accuracy challenges when accommodating conformational changes caused by the flexibility of DNA docking with proteins. Therefore, there is an urgent need for a fast and accurate method to assess DNA–protein complex structures effectively.
Recently, there have been advancements in deep neural networks, which have proved to be powerful tools for image processing, disease diagnosis, natural language processing, and bioinformatics.35–46 Each deep neural network employs different feature processing techniques and frameworks tailored to specific tasks. However, there is a bottleneck of deep learning-based models applied to predicting DNA–protein complex structures due to the limited number of available DNA–protein complex structures. The multi-body interactions within DNA–protein complexes at the interface also present a formidable challenge. Such models need to be customized to suit the unique characteristics of these complexes, including their small data sets and complex interactions. Therefore, developing deep learning models tailored to the specific features of DNA–protein complexes is crucial to achieving better predictions with the limited available data and intricate interactions.
In this study, we developed a novel assessment approach, DDPScore (deep-learning-based DNA–protein complex score), employing a 4D convolutional neural network to evaluate DNA–protein complex structures. We trained a deep learning-based scoring function using decoy structures generated through physics simulations. The 4D convolutional neural network framework can holistically acquire diverse heterogeneous information, including sequence, charge, mass, interface interactions, secondary structure, and spatial distribution. This approach effectively captures local and global features while comprehensively considering the conformational changes arising from the flexibility during the DNA–protein docking process. With the comprehensive performance evaluations in the DNA–protein complex docking benchmarks, encompassing even the challenges of flexible DNA–protein complex docking, DDPScore consistently achieved superior performance. To our knowledge, this is the first work to evaluate the DNA–protein complex structures using a deep learning method that can be used independently and easily extended to other DNA–protein complex docking algorithms. We hope this approach will guide the prediction and design of DNA–protein complex structures.
2 Methods
2.1 Convolution neural network for DNA–protein complex scoring function
Our previous deep-learning-based approach, DRPScore, has successfully evaluated the structures of RNA–protein complexes.41 Our method has successfully tackled the problem of modeling the flexibility of RNA–protein complexes during the docking process. This has resulted in better outcomes for selecting native-state conformations. Since DNA and RNA share similarities in composition and conformation, and there aren’t any efficient methods for evaluating DNA–protein complex structures, it's crucial to extend our approach to assess DNA–protein complex structures. In Fig. 1 and Fig. S1 (ESI†), we demonstrate that our approach differs from previous scoring functions that depend on statistical potential energy. We address the limited data by employing physics-based simulations to focus on the DNA–protein interfaces. We begin the process by utilizing physics-based simulations to generate 300 decoy structures for each DNA–protein complex. This approach allowed us to amass a training dataset consisting of almost 0.2 million structures, which is a significant improvement compared to the usual number of structures (typically less than 300) available in traditional methods. We extract the DNA–protein interaction interface with a 6 Å cut-off for any DNA–protein complex structure. After that, we establish a local Cartesian coordinate system using specific atoms of each nucleotide/residue (C1′, O5′, C5′, N1/N9 for DNA, and CB, CA, O, C for protein). To capture atomic-level interactions, we have developed a system that creates a 32 Å × 32 Å × 32 Å grid named moving structural window around each nucleotide/residue. This local coordinate system is the basis for generating individual frames, resulting in a training dataset with over 20 million images for feature extraction.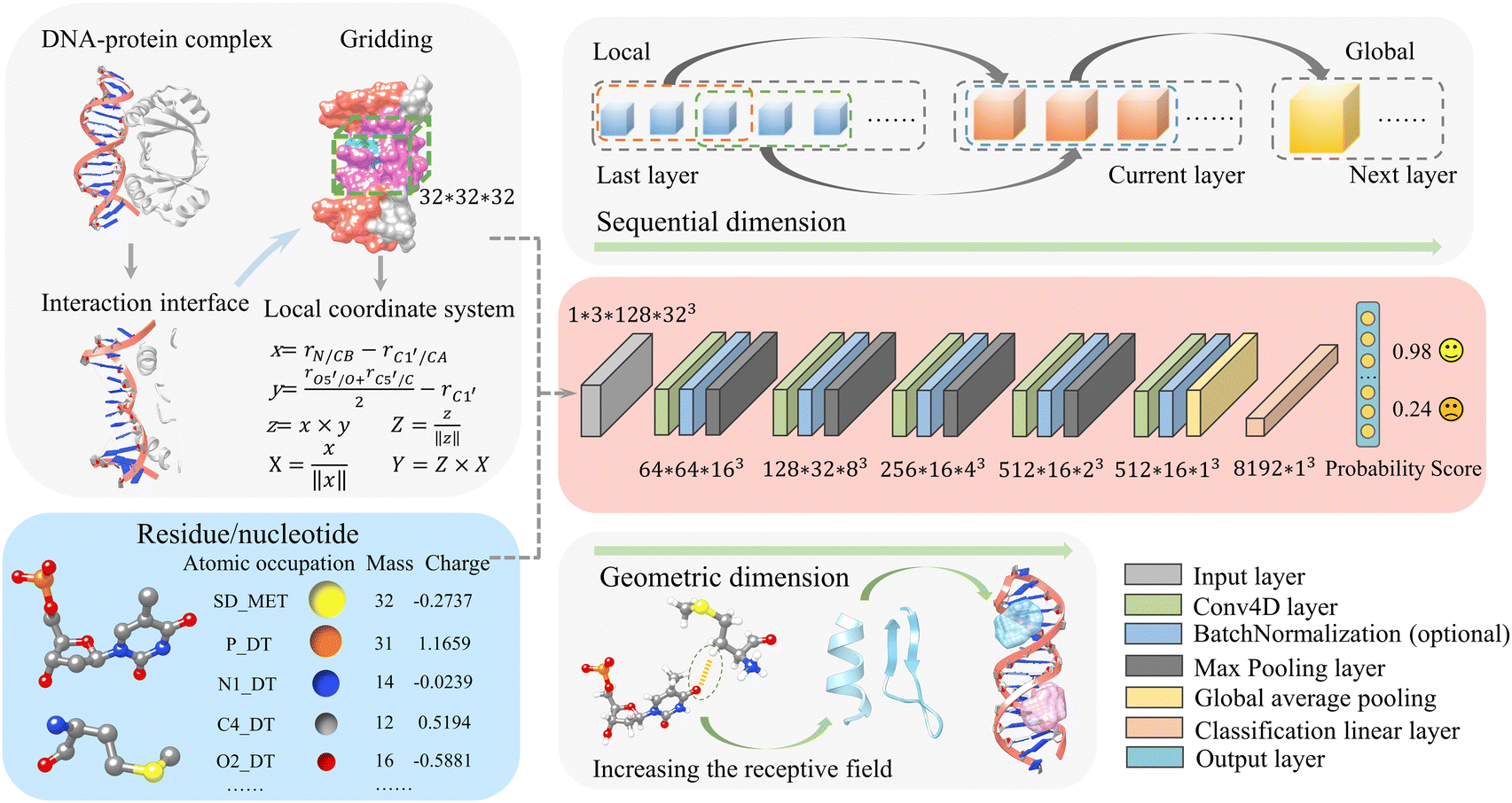 | ||
| Fig. 1 The input and architecture of the 4D convolutional neural network in this work. The network is arranged in the order of input layer, five layers comprising a Conv4d module, an optional BatchNorm module, a MaxPooling module (the last one being global average pooling), and a final layer fully connected for classification, respectively. m × s × n3 signifies the presence of m features, a maximum length of s for DNA–protein complex sequences, and n3 voxels. | ||
We then developed a method to describe molecular systems at the atomic level accurately. Our approach involves assigning precise mass and charge values to each atom through specific feature processing. To facilitate this, we have incorporated 82 atom types for DNA nucleotides and 225 atom types for protein residues, along with their respective mass and charge information (Tables S1–S3, ESI†). Then, we provide this interaction interface information to the convolutional neural network, along with accumulations of the occupation number, mass, and charge of atoms within the grid information.
DDPScore uses a 4DCNN (4D convolutional neural network) to evaluate the conformation of DNA–protein complexes. To do this, DDPScore requires the nucleotides/residues of the DNA–protein complex within a 6Å distance at the interaction interface as input. Once the information is provided, DDPScore generates probability scores to evaluate and select the near-native structures of the DNA–protein complex.
While 3DCNNs have proven successful in modeling RNA for specific tasks,40 they ignore the critical sequential properties inherent in RNA structures. The simple averaging of independent representations of each nucleotide to create a global representation may result in the loss of essential information about nucleotide interactions. Our proposed 4D approach directly addresses this limitation by introducing an additional convolution operation along the sequential dimension. This method captures spatial and sequential information, focusing on interactions between nucleotides and residues. Our Conv4D architecture utilizes a non-overlapping moving structural window with a size of 3 nucleotides/residues, equivalent to a kernel size of 3. This configuration effectively captures interactions between the nucleotides/residues at each convolutional layer, with each layer's input derived from the preceding one's output. For example, in a two-layer CNN architecture with a kernel size of 3 and stride of 2, the first layer captures interactions among three consecutive moving structural windows. In contrast, the second layer focuses on higher-level interactions between every 3-moving structural windows. As a result, in the second layer, we model the interactions among seven consecutive moving structural windows of the DNA–protein complex. This design allows for the learning of short-range interactions initially. As the convolutional layers go deeper, they can capture long-range interactions, extending the field of view from local interactions to encompass global features.
During the pre-processing phase, each DNA–protein complex is transformed as a tensor with dimensions of 1 × 3 × L × (H × W × D). The “3” refers to the number of features: the accumulations of the occupation number, mass, and charge of the atoms in the grid box. L is fixed at 128, representing the maximum length of DNA–protein complex sequences. H, W, and D correspond to the height, width, and depth of a 3D cube for each nucleotide/residue in the DNA–protein complex sequence. Consistent with our previous work,41 we also set H = W = D = 32 in this study.
Our network consists of six layers. The final layer is fully connected for classification. Each of the first five layers includes a Conv4d module, an optional BatchNorm module, and a Max-pooling module. The last layer uses global average pooling. In these Conv4d modules, the number of channels varies: 64, 128, 256, 512, and 512. The strides used in each module vary as follows: 2, 2, 2, 1, and 1. This means that, when considering the sequential order of dimensions, the length of DNA–protein complex features is effectively halved in each of the first three blocks and remains the same in the last two blocks. Additionally, all pooling layers have a kernel size and stride of 2, halving each pooling module's height, width, and depth dimensions. A global average pooling is applied as the last step in representing a DNA–protein complex. This results in a final vector of size 8192. For example, if a DNA–protein complex is initially described by a vector O0 with the shape of 1 × 3 × 128 × 32 × 32 × 32, as illustrated in eqn (1), a 4D convolution with a stride of 2 is first applied in the sequential dimension:
| O1 = Conv4D(O0) | (1) |
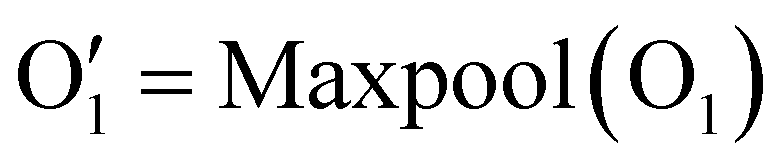 | (2) |
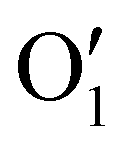 has the shape of 1 × 64 × 64 × 16 × 16 × 16. As illustrated in eqn (3), at the next layer, we also apply a 4D convolution first:
has the shape of 1 × 64 × 64 × 16 × 16 × 16. As illustrated in eqn (3), at the next layer, we also apply a 4D convolution first: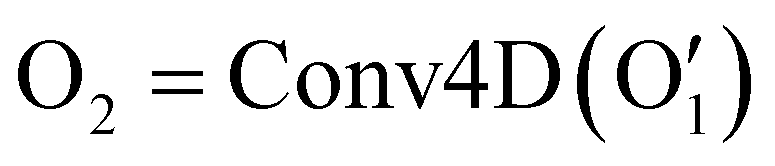 | (3) |
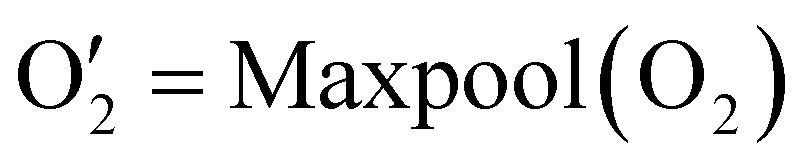 | (4) |
The output 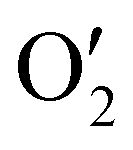 has a shape of 1 × 128 × 32 × 8 × 8 × 8. Similarly, the output
has a shape of 1 × 128 × 32 × 8 × 8 × 8. Similarly, the output 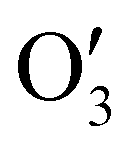 and
and 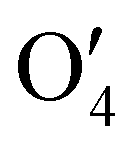 have the shape of 1 × 256 × 16 × 4 × 4 × 4 and 1 × 512 × 16 × 2 × 2 × 2, respectively.
have the shape of 1 × 256 × 16 × 4 × 4 × 4 and 1 × 512 × 16 × 2 × 2 × 2, respectively.
Finally, after applying a 4D convolution at the last layer, we have a tensor OLN(O5) with the shape of 512 × 16 × 2 × 2 × 2. Eqn (5) illustrates an adaptive spatial pooling for the final DNA–protein complex representation:
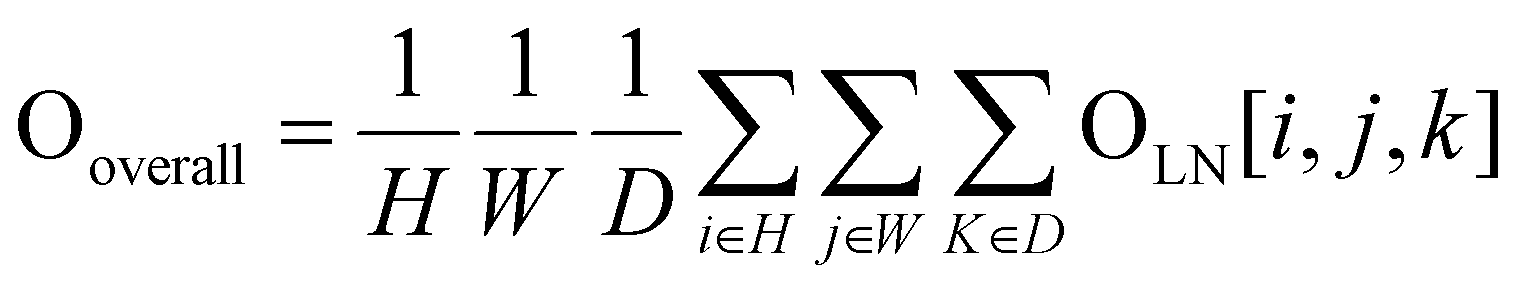 | (5) |
After flattening 512 × 16, Ooverall has the shape of 8192 × 1 × 1 × 1. After adding a linear classification layer to the model, probability scores can be generated to evaluate and select near-native DNA–protein complex structures. The advantage of using 4D convolutional neural networks is that it allows adding an extra convolution operation to the three-dimensional spatial dimension, thereby effectively capturing both the sequential and spatial information of the DNA–protein complex. This helps to account for the interactions between nucleotides/residues, giving a more complete complex analysis.
In 4DCNN, the local features are the sequence, charge, and mass of heavy atoms. On the other hand, the global features consist of secondary structure features (alpha helix, beta sheet of protein, base pairing of DNA, etc.), the distance between nucleotides and residues, and the interactions at the DNA–protein complex interaction interface (such as electrostatic interactions, hydrogen bond interactions, van der Waals interactions, etc.). DDPScore effectively learns local and global features using multi-layer convolutional neural networks.
2.2 Training set
A non-redundant and diverse DNA–protein complex training set was constructed from DNA–protein complex structures deposited in the RCSB PDB21 (before February 20, 2023). First, we extracted 5431 available DNA–protein complex structures with the search restrictions “X-ray crystallographic structures with a resolution better than 3.5 Å”, “DNA and protein, excluding RNA” and “no more than 6 DNA or protein chains”. Second, we removed the complex containing DNA structures with a sequence length smaller than 8 bp (base pairs). Third, we removed the absence of a double helix of DNA structures in the complex. Fourth, we removed the DNA redundancy by 80% sequence similarity cutoff using CD-HIT.47–49 Then, we also removed the redundancy between the training and testing sets by an 80% sequence similarity cutoff using CD-HIT. Finally, 585 non-redundant DNA–protein complex structures were selected from these DNA–protein structures to construct the training set (Table S4, ESI†).To compare with the current state-of-the-art method, we utilize the HDOCK developed by Huang et al. to generate docking decoys for each DNA–protein complex. HDOCK is a hybrid docking strategy for protein–protein, protein–DNA, and protein–RNA complexes and is considered one of the best molecular docking methods available today.29,30 First, the HDOCKlite global docking program employs FFT to sample binding modes, and the resulting complex structures undergo evaluation and ranking using an iterative knowledge-based scoring function. We used the command “hdock protein.pdb DNA.pdb –out Hdock.out” and “createpl Hdock.out top4392.pdb –nmax 4392 –complex –models –rmsd 0” to generate 4392 decoys for each DNA–protein complex structure, and then selected the top 300 decoys in order of RMSD from smallest to largest. Thus, the training set consisted of a total number of 176085 DNA–protein complexes.
2.3 Testing sets
To evaluate its robustness and efficacy, we tested DDPScore with two independent DNA–protein docking benchmarks developed previously. The unbound structure is defined as a structure in free form or being a binding partner in a different complex.50 Thus, an “unbound–bound” structure is defined as one of the two binding partners (DNA or protein) being either in apo form or taken from another complex. The “unbound–unbound” refers to structures in which both binding partners (DNA and protein) are either in apo form or taken from a different complex.Testing set I is the published non-redundant DNA–protein docking benchmark (version 1.2) developed by Bonvin.51 The testing set focuses on conformational changes in DNA, proteins, or both. Scoring functions face a formidable challenge due to the omnipresence of these changes. This testing set contains unbound–bound and unbound–unbound cases for each of the 47 DNA–protein complexes in which DNA is in B-DNA conformation (Table S5, ESI†). The structures of unbound DNA were generated by the DNA analysis and rebuilding program 3DNA.52 The unbound protein structures include both X-ray and solution NMR structures. These 47 DNA–protein complexes are grouped into three categories – “easy target”, “intermediate target”, and “difficult target” – based on the interface RMSD values between the bound and unbound components of the complex (Fig. S2, ESAI). An interface RMSD value between 0.0 Å and 2.0 Å is classified as an “easy target”, one between 2.0 Å and 5.0 Å is an “intermediate target”, and an interface RMSD above 5.0 Å is a “difficult target”. In this testing set, HDOCK generated 1000 decoys for each DNA–protein complex.
Testing set II is another non-redundant DNA–protein docking benchmark developed by Fernández-Recio.53 The testing set was created based on the criteria selection used in the testing set developed by Bonvin. It includes unbound–bound and unbound–unbound cases for each of the 10 DNA–protein complexes. The sequences in this set differ from those in the DNA–protein testing set I (Table S6, ESI†). For each DNA–protein complex in this testing set, 1000 decoys were also generated by HDOCK.
2.4 Implementation details
DDPScore utilized the 4DCNN to minimize the mean squared errors between the actual and predicted results. The back-propagation-based mini-batch gradient descent optimization algorithm is used for training. The initial learning rate for the 4DCNN is set to 0.000045. The training process stopped when the loss approached 0 and stabilized. In total, there were 50000 training steps, and each iteration took approximately 6 seconds, utilizing 6.6 GB of memory when using a ‘batch_size’ of one on a single NVIDIA 2080Ti GPU. Eventually, a model with 40400 training steps was selected. Evaluating 1000 DNA–protein complex structures with pre-computed features using DDPScore takes around 5 minutes.
2.5 The binding free energy calculations
We also calculate the binding free energy of the DNA–protein complex upon associating by using the Python program MM-PBSA.py54 in AMBER18.55,56 The MM-PBSA (molecular mechanics Poisson Boltzmann surface area) method calculates the difference in free energy between the bound and unbound states of the DNA–protein complexes. The binding free energy ΔG0bind,solv can be calculated as eqn (6):57| ΔG0bind,solv = ΔG0bind,vacuum + ΔG0solv,complex − (ΔG0solv,ligand + ΔG0solv,receptor) | (6) |
| ΔG0bind,vacuum = ΔG0complex,vacuum − (ΔG0receptor,vacuum + ΔG0ligand,vacuum) | (7) |
| ΔG0vacuum = ΔE0molecularmechanics − T*ΔS0normalmodeanalysis = (ΔE0int + ΔE0vdw + ΔE0ele) − T*ΔS0normalmodeanalysis | (8) |
modeanalysis is the entropy contribution, which can be obtained using normal mode analysis.
As eqn (9) shows, solvation energy can be divided into two parts: polar solvation energy and non-polar solvation energy:
| ΔG0solv = ΔG0solv,polar + ΔG0solv,nonpolar | (9) |
AMBER18 software was used to calculate the binding free energy.55,56 First, the complex structure was processed using the FF14SB force field and TIP3B model to remove the initial solvent and ions. The complex was then solvated in a cubic box with periodic boundary conditions. The negative charge of the system was neutralized by adding Na+ or Cl− ions. The side of the box size was 12.0 nm. The energy minimization was performed with the steepest descent method in two successive steps. The first minimization involved 5000 iterations, followed by a subsequent step of 10000 iterations. The system was then heated to 300 K to determine the rational orientation of the solute. The NVT ensemble underwent a 50 ps equilibration phase with position restraints using a modified Berendsen thermostat for temperature control. Following this, the NPT ensemble underwent a 200 ps equilibration phase with position restraints, utilizing the Parrinello–Rahman barostat for pressure control. Finally, a 500 ps simulation was conducted to generate a trajectory, which was then used to calculate the binding free energy.
2.6 Criteria for the assessment of the prediction quality
The quality of the DNA–protein complex prediction is evaluated by the CAPRI criterion.58,59 The Irmsd is the interface RMSD between the native and predicted structures after the superposition of corresponding proteins. The Lrmsd is the ligand (refers to DNA) RMSD between the native and predicted structures after the superposition of corresponding proteins. The definition of the RMSD is as eqn (10):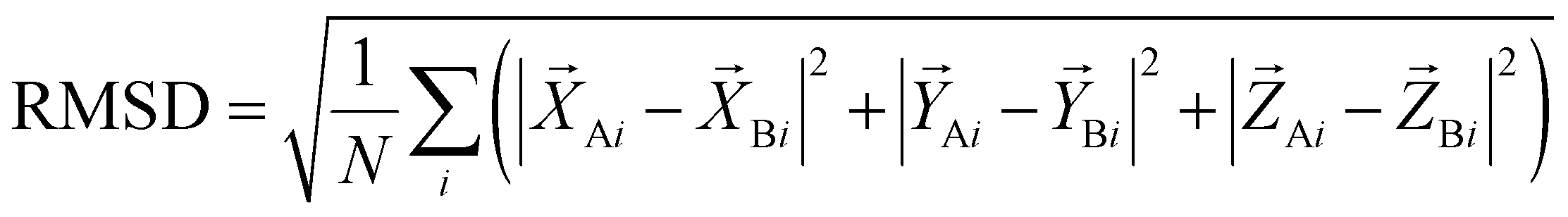 | (10) |
3 Results
3.1 Testing on the semi-flexible DNA–protein testing sets
DNA is considered much more stable than RNA due to the formation of base pairs between most nucleotides.60 When a protein binds to DNA, it often induces a conformational change to fit into the DNA structure.61 In this process, DNA typically conforms to the protein's shape through slight alterations, such as stretching, bending, and twisting. These induced conformational changes can even encompass secondary structural adjustments, such as transitioning from an irregular coil to a helix. Hence, in testing sets I and II, we immobilize DNA and allow the protein to change conformation to analyze DDPScore performance on semi-flexible DNA–protein complexes. Fig. 2(A) shows the performance of DDPScore and HDOCK on semi-flexible DNA–protein complexes on testing set I. The success rate of DDPScore is 48.94% in the top 5 predictions, compared with 42.55% for HDOCK. When the top 20 predictions are considered, the success rate of DDPScore is 63.83% compared with 57.45% for HDOCK. Fig. 2(B) shows the performance of DDPScore and HDOCK on semi-flexible DNA–protein complexes on testing set II. The success rate of DDPScore is 50.00% in the top 5 predictions, compared with 40.00% for HDOCK. When the top 20 predictions are considered, the success rate of DDPScore is 60.00% compared with 40.00% for HDOCK. The ranking and Lrmsd are shown in Tables S7 and S8 (ESI†). The results consistently show that DDPScore outperforms HDOCK in testing sets I and II, across a range of top 5 to 1000.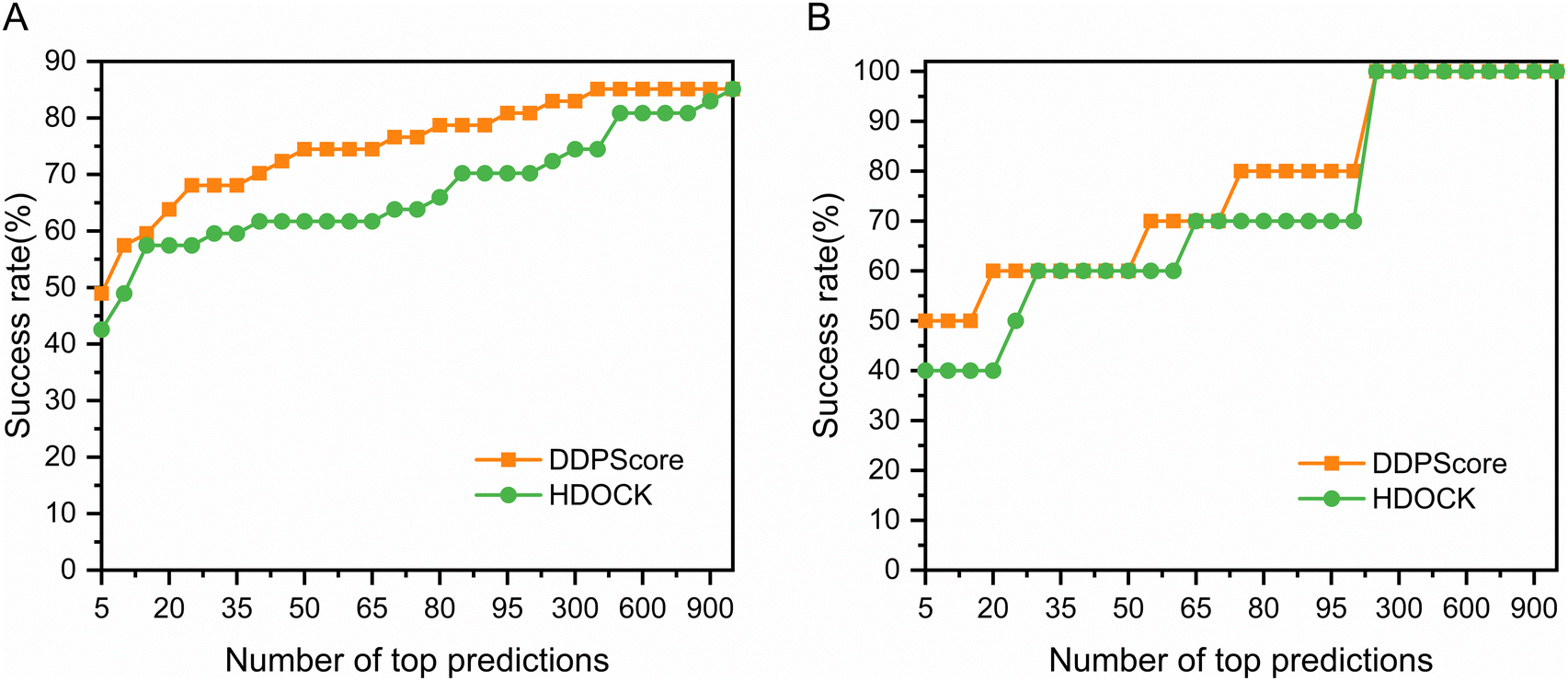 | ||
| Fig. 2 The performance of DDPScore and HDOCK on the semi-flexible DNA–protein testing sets. The performance of DDPScore (orange square) and HDOCK (green circle) on the (A) semi-flexible DNA–protein testing set I and (B) semi-flexible DNA–protein testing set II. | ||
3.2 Testing on the flexible DNA–protein testing sets
We rigorously assessed DDPScore's performance in evaluating real flexible DNA–protein complexes with significant conformational changes in DNA and proteins. The DDPScore was evaluated on the two unbound–unbound testing sets. Fig. 3(A) shows the performance of DDPScore and HDOCK on testing set I. The results show that DDPScore consistently outperforms HDOCK, regardless of the number of top hits from 5 to 1000. The success rate of DDPScore is 17.02% in the top 5 predictions, compared with 8.51% for HDOCK. When the top 20 predictions are considered, the success rate of DDPScore is 38.30% compared with 23.40% for HDOCK. The performance of DDPScore and HDOCK on testing set II is demonstrated in Fig. 3(B). The success rate of DDPScore is 30.00% in the top 5 predictions, compared with 20.00% for HDOCK. When the top 20 predictions are considered, the success rate of DDPScore is 40.00% compared with 30.00% for HDOCK. The ranking and Lrmsd are shown in Tables S9 and S10 (ESI†). Overall, DDPScore consistently outperforms HDOCK in all testing sets, regardless of their ranking from the top 5 to the top 1000.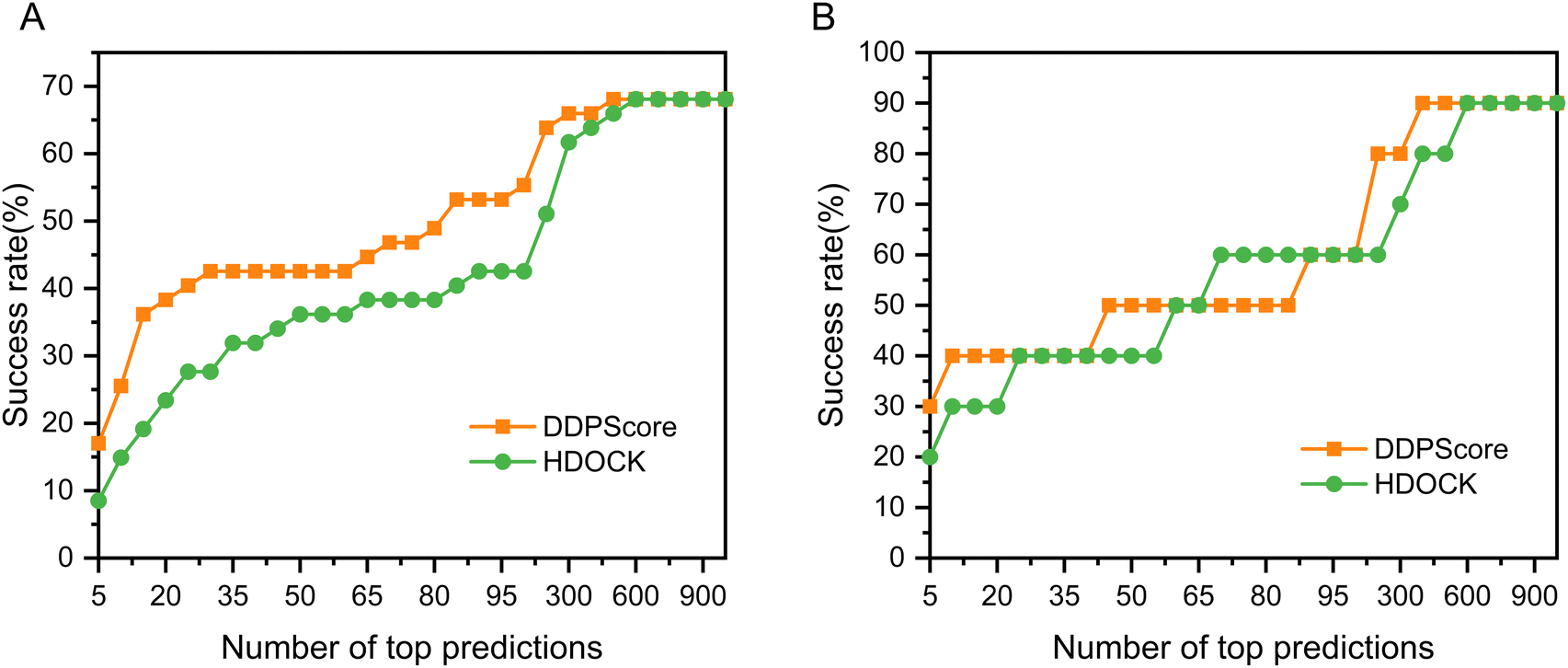 | ||
| Fig. 3 The performance of DDPScore and HDOCK on the flexible DNA–protein testing sets. The success rate of DDPScore (orange square) and HDOCK (green circle) on the (A) flexible DNA–protein testing set I and (B) flexible DNA–protein testing set II. | ||
3.3 Performance analysis on the flexible DNA–protein complexes
In the case of flexible unbound–unbound docking, the interaction interface will induce complex conformational changes. The traditional work mainly focuses on the DNA–protein rigid docking without consideration of the complex structural flexibility. Thus, we focused on analyzing the evaluation performance of DDPScore on DNA–protein complexes across three categories. The first category includes easy targets with an interface RMSD between 0.0 Å and 2.0 Å. The second category covers intermediate targets with an interface RMSD between 2.0 Å and 5.0 Å. The third category includes challenging targets with an interface RMSD above 5.0 Å. When it comes to easy targets, the monomers of the DNA–protein complex don’t undergo major changes in their shape when they bind together. The conformational changes at the DNA interface usually cause the DNA to bend or twist in the area where it's interacting with the protein. The conformational changes at the protein interface usually only involve minor adjustments to flexible loops. In intermediate targets, DNA–protein binding causes significant structural rearrangements in monomers at their interfaces. These changes involve global conformational changes in DNA and global or local domain rearrangements in proteins. When binding with challenging targets, the DNA–protein complex monomers undergo even more significant structural rearrangement. Along with the conformational changes seen in intermediate targets, these complexes also go through major domain reorientations and structural transitions in the protein. As the targets become more challenging, the Irmsd gradually increases. The interaction process between DNA and proteins becomes harder to simulate accurately due to the pronounced conformational changes that occur when they form complexes. The success rate of DDPScore is 23.08% in the top 5 predictions, compared with 7.69% for HDOCK on the easy targets (Fig. 4(A)). When the top 20 predictions are considered, the success rate of DDPScore is 53.85% compared with 38.46% for HDOCK. The success rate of DDPScore is 13.64% in the top 5 predictions, compared with 9.09% for HDOCK on the intermediate targets (Fig. 4(B)). When the top 20 predictions are considered, the success rate of DDPScore is 31.82% compared with 18.18% for HDOCK. The success rate of DDPScore is 16.67% in the top 5 predictions, compared with 8.33% for HDOCK on the difficult targets (Fig. 4(C)). When the top 20 predictions are considered, the success rate of DDPScore is 33.33% compared with 16.67% for HDOCK. DDPScore consistently outperforms HDOCK despite the decreasing performance with increased interaction interface flexibility. This shows that DDPScore is a reliable and accurate method for DNA–protein docking, even in the case of highly flexible interaction interfaces.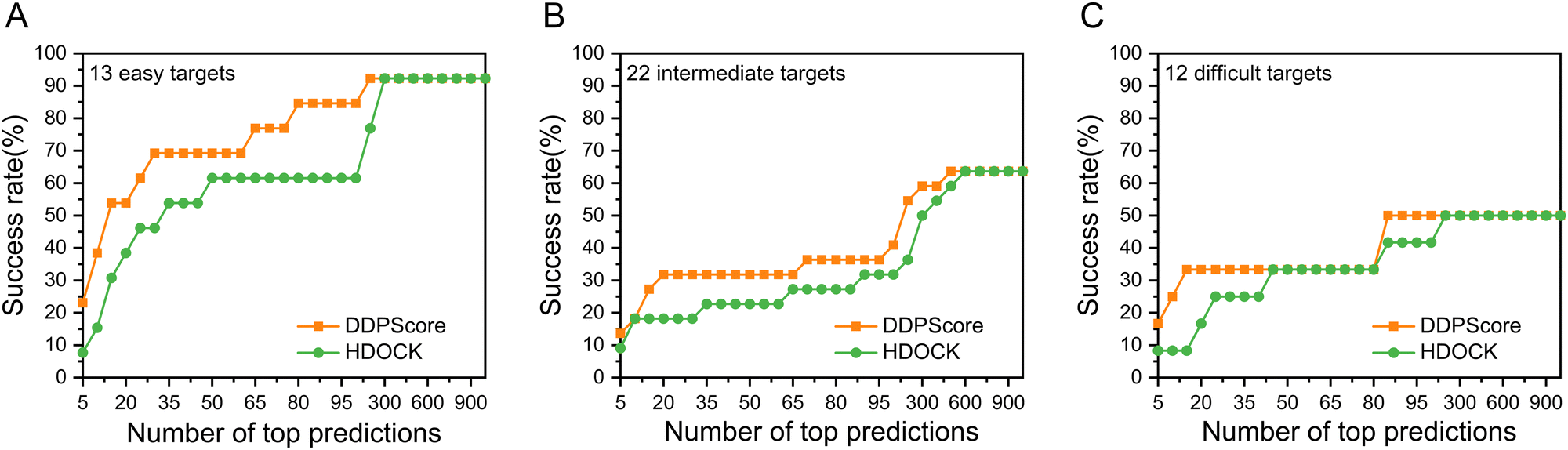 | ||
| Fig. 4 The performance of DDPScore and HDOCK on the three categories of the flexible DNA–protein testing set I. The success rate of DDPScore (orange square) and HDOCK (green circle) on the (A) easy targets, (B) intermediate targets, and (C) difficult targets of the flexible DNA–protein testing set I. | ||
3.4 Performance analysis of the structural categories
To thoroughly evaluate the effectiveness of DDPScore in flexible docking for various DNA–protein complexes, the 47 DNA–protein complexes in the first testing set were divided into six categories based on the protein structures present in each complex, using the pre-existing classification categories.62 Members of the same group share a structural attribute for DNA recognition but vary in relatedness to each other. There are 16 cases of helix-turn-helix, 3 cases of zinc-coordinating, 5 cases of α-helix, 2 cases of β-sheet, 4 cases of β-hairpin/ribbon, and 17 cases of enzyme. The performance of DDPScore and HDOCK in various categories is presented in Fig. 5(A). The first successful HDOCK and DDPScore prediction rankings are shown on the horizontal and vertical axes. The results show that DDPScore outperforms HDOCK in predicting helix-turn-helix and β-hairpin/ribbon DNA–protein complexes while achieving comparable results in zinc-coordinating, α-helix, β-sheet, and enzyme complexes. The detailed data are shown in Table S11 (ESI†). DDPScore's success in predicting complex types significantly improves its overall performance.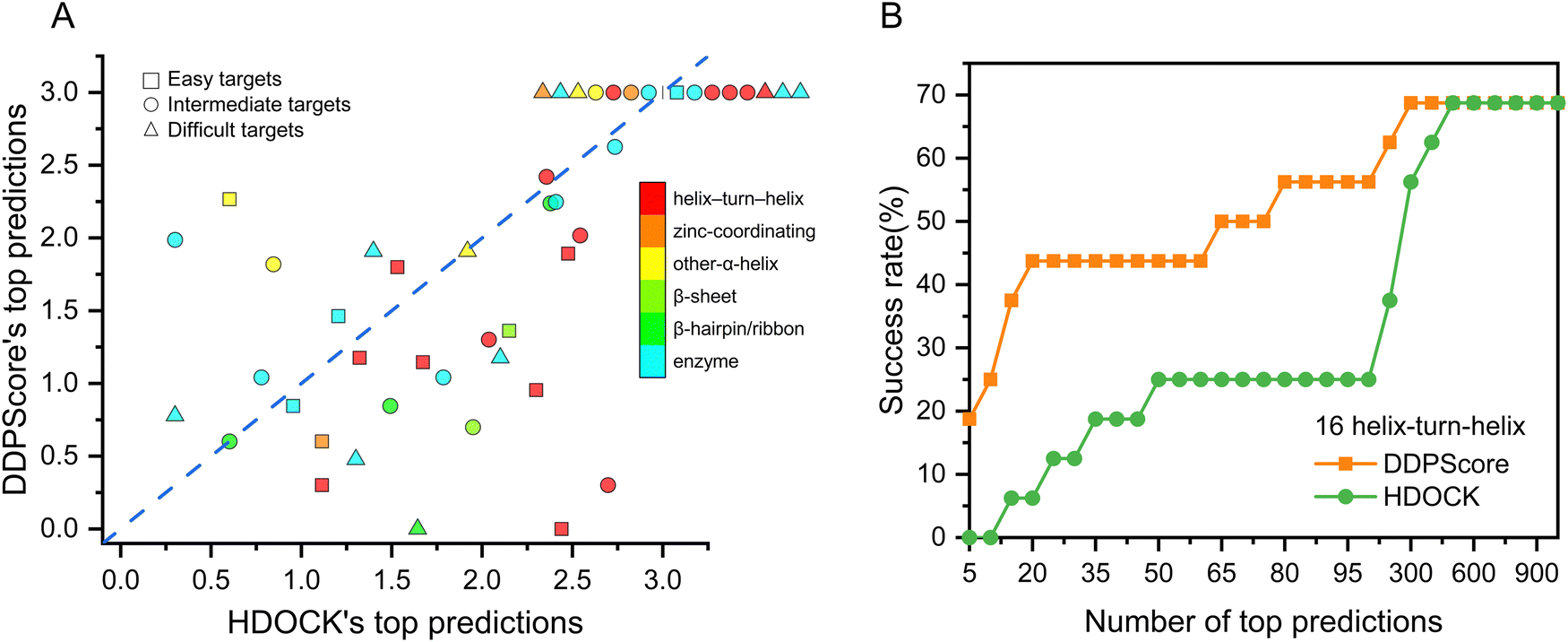 | ||
| Fig. 5 Detailed analysis of the structural categories of flexible DNA–protein testing set I. (A) Distributions of the first successful DDPScore prediction rankings relative to the first successful DDPScore prediction rankings on the easy targets (square), intermediate targets (circle), and difficult targets (triangle) are shown in double logarithmic axes (log–log plot). DDPScore outperforms HDOCK in predicting helix-turn-helix (red) and β-hairpin/ribbon (green) DNA–protein complexes while achieving comparable results in zinc-coordinating (orange), α-helix (yellow), β-sheet (light green), and enzyme (cyan) complexes. (B) The performance of DDPScore (orange square) and HDOCK (green circle) on the helix-turn-helix targets of the flexible DNA–protein testing set I. | ||
To assess the performance of specific helix-turn-helix DNA–protein complexes, we compared the success rate of DDPScore and HDOCK in 17 cases (Fig. 5(B)). The success rate of DDPScore is 18.75% in the top 5 predictions, compared with 0% for HDOCK. When the top 20 predictions are considered, the success rate of DDPScore is 43.75% compared with 6.25% for HDOCK. Overall, DDPScore performs significantly better than HDOCK. The helix-turn-helix motif consists of two α-helices and a four-residue β-turn. It is used by transcription regulators and enzymes in prokaryotes and eukaryotes for recognition. In most complexes, helix-turn-helix motifs are bound in large grooves of DNA by inserting the second helix, called the probe, into the grooves. This results in direct contact between the side chains of amino acids and the bases of nucleotides. The geometries of DNA–protein complexes are complex and diverse. Alpha helices and major grooves play a crucial role in their interactions. Although a close fit is achieved between the two, there is enough flexibility to allow for distinct conformations of both DNA and protein. This results in multispecific complementarity, which enables complex conformational changes at the interaction interface. By utilizing convolutional layers to simulate longer-range interactions, DDPScore accurately predicts interactions between nucleotides and residues. This allows for the effective capture of both intra- (local) and inter-nucleotide/residue (global) information, including the α-helix and β-sheet of the protein. As a result, the binding mechanism of the helix-turn-helix motif, which exposes the helix on the surface of the protein for specific recognition with DNA, can be effectively identified. Despite exhibiting considerable variations in residue sequence and geometry, these structures have evolved independently to suit their specific functions. DDPScore can effectively recognize DNA–protein complex binding characteristics and consistently achieves success in analyzing such special motifs.
3.5 DNA–protein flexible docking evaluation examples
To showcase the effectiveness, we selected three typical DNA–protein complexes from easy, intermediate, and difficult categories in testing set I. Fig. 6 shows the RMSD of the structure with the lowest RMSD among the top 5 structures selected using DDPScore and HDOCK in flexible docking. It displays the changes in RMSD for the complex interface, DNA, and protein throughout the docking process. Fig. 6(A) shows the complex of the fadR transcription factor from E. coli with the FadB operator, which is an easy target (PDB ID: 1H9T). The interface RMSDs between the bound and unbound components of the complex is 1.63 Å, the DNA RMSD is 3.88 Å, and the protein RMSD is 0.77 Å, respectively. The RMSD value for the DDPScore model is 9.05 Å, while HDOCK's is 33.79 Å. Fig. 6(B) shows the crystal structure of the Adenovirus major late promoter TATA box bound to wild-type TBP (PDB ID: 1QNE), which is the intermediate target. The interface RMSDs between the bound and unbound components of the complex is 4.57 Å, the DNA RMSD is 8.54 Å, and the protein RMSD is 0.89 Å, respectively. The DDPScore model has a lower RMSD value of 4.85 Å than HDOCK's value of 25.05 Å. Fig. 6(C) shows the cytosine-specific methyltransferase HhaI/DNA complex (PDB ID: 7MHT), which is a difficult target. The interface RMSDs between the bound and unbound components of the complex is 6.71 Å, the DNA RMSD is 2.55 Å, and the protein RMSD is 3.84 Å, respectively. The lowest RMSD model of DDPScore is 9.62 Å, compared with 25.59 Å for HDOCK. Generally, DDPScore can achieve a favorable predictive performance across a range of targets with varying difficulty. When comparing the extent of conformational changes in these three DNA–protein complexes, it is noteworthy that the DNA in easy and intermediate targets had a larger RMSD than the protein. This indicates that the DNA experienced greater conformational changes. However, the interaction regions between the DNA and protein only experienced minor conformational changes. In cases of difficult targets, the protein undergoes a big conformational change in the interaction region, resulting in a large RMSD value. The results suggest that the conformational alterations of proteins at the interaction interface are more influenced by local or global domain rearrangements than the relatively stable double helix structure of DNA.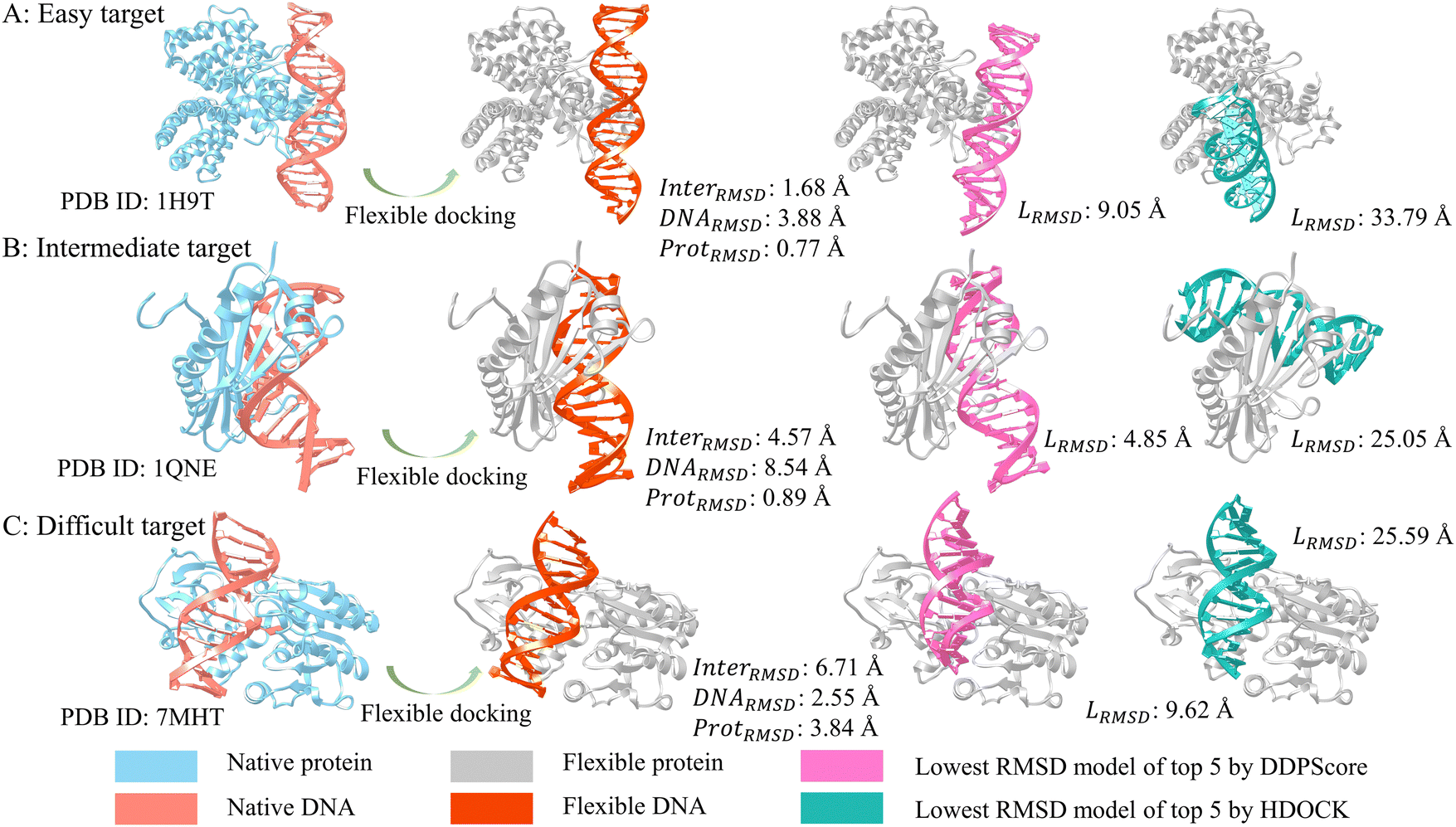 | ||
| Fig. 6 Three examples of DNA–protein flexible docking evaluation analysis. In flexible docking, from (A) easy target (PDB ID: 1H9T), (B) intermediate target (PDB ID: 1QNE) to (C) difficult target (PDB ID: 7MHT), the flexible DNA (colored in orange) and flexible protein (colored in gray) undergo an increasing degree of conformational change compared to the native DNA (colored in salmon) and native protein (colored in blue). The lowest RMSD models in the top 5 predictions by DDPScore (DNA in orchid) are more similar to native complexes (DNA in salmon) than HDOCK (DNA in green). | ||
3.6 Physics-based interaction contributions
The most popular methods for evaluating DNA–protein complex structures rely on statistical potential functions derived from the inverse Boltzmann distribution. In contrast to conventional 3DCNN models, DDPScore uses a deep learning approach to evaluate the structure of the DNA–protein complex and benefits from an additional sequential order dimension. This advantage allows DDPScore to capture complex multi-body interactions at the interface, facilitating the transition from local to global features and resulting in accurate identification of near-native structures. We calculated the binding free energy of DNA–protein complex structures selected by both HDOCK and DDPScore using three examples as representatives. Table 1 shows the top 5 predictions chosen by HDOCK based on free energy and DDPScore utilizing a deep learning-based function. The three structures chosen by HDOCK have an average binding free energy of −35.46 kcal mol−1, −36.07 kcal mol−1, and −51.93 kcal mol−1, respectively. On the other hand, DDPScore's chosen structures have an average binding free energy of −72.10 kcal mol−1, −47.49 kcal mol−1, and −59.19 kcal mol−1, respectively. Although DDPScore-selected structures have a slightly lower binding free energy than HDOCK-selected structures, the RMSD measurements between them differ significantly. These results are indeed quite intriguing. HDOCK and DDPScore have different strategies for selecting structures. HDOCK uses an inverse Boltzmann statistical distribution method to choose structures with evenly distributed probabilities. Meanwhile, DDPScore considers both spatial and sequential features of structures, learning complex multi-body interactions that capture the binding dynamics between DNA and proteins in native structures. This approach enables DDPScore to identify decoys that closely resemble the native structure, even in cases where binding strength is not particularly high.| Complex | HDOCK |
|
DDPScore | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔGa | RMSDb | ΔG | RMSD | ΔG | RMSD | ΔG | RMSD | ΔG | RMSD | ΔG | RMSD | ||
| a The binding free energy in kcal mol−1. b Root mean square deviation in Å. | |||||||||||||
| 1H9T | −41.51 | 34.50 | −46.27 | 33.79 | −19.76 | 43.47 | −31.48 | 43.52 | −38.29 | 43.69 | −35.46 | −72.10 | 9.05 |
| 1QNE | −38.99 | 25.17 | −32.82 | 25.23 | −33.76 | 25.05 | −33.00 | 26.04 | −41.78 | 29.93 | −36.07 | −47.49 | 4.85 |
| 7MHT | −48.63 | 25.61 | −55.04 | 25.60 | −58.35 | 25.61 | −41.79 | 25.63 | −55.86 | 25.59 | −51.93 | −59.19 | 9.62 |
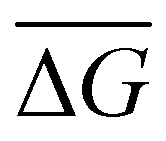
To evaluate the accuracy of DDPScore in capturing the complex interactions occurring within the interface of DNA and protein interactions, we utilized an example of the fadR transcription factor, which is responsive to fatty acids and found in E. coli, in conjunction with the FadB operator (identified by PDB ID: 1H9T). We utilized HBPLUS (version 3.2)63 to identify the hydrogen bond interactions between DNA and protein at the interaction interface. Additionally, we utilized PyMOL (version 1.8.0.3) to analyze the electrostatic interactions between DNA and protein. Fig. 7(A) and (B) present the lowest RMSD model among the top 5 models selected by DDPScore and HDOCK. It is noted that in the model selected by DDPScore, DNA is bound to a positively charged protein region. However, the region where DNA binds has a significant negative charge in the model selected by HDOCK. It is recognized that DNA has a strong negative charge, and it should ideally bind to the positively charged region of the protein. Fig. 7(C) and (D) show the hydrogen bonds in two models. The detailed data are shown in the ESI,† Table S12. The structure chosen by DDPScore has 5 hydrogen bonds that interact with both DNA and protein, while the HDOCK-selected structure has 9 hydrogen bonds in these interactions. It is noted that even though flexible docking leads to significant conformational changes in both DNA and protein, DDPScore identifies 60% of the hydrogen bonds that align with the native structure. However, HDOCK fails to identify any of the native hydrogen bonds. The structures selected by HDOCK may exhibit more hydrogen bond interactions using geometric matching with higher probability statistical scores, leading to missing real interactions. On the other hand, DDPScore successfully learns the complex multi-interaction pattern between DNA and protein, including native hydrogen bonds and electrostatic interactions.
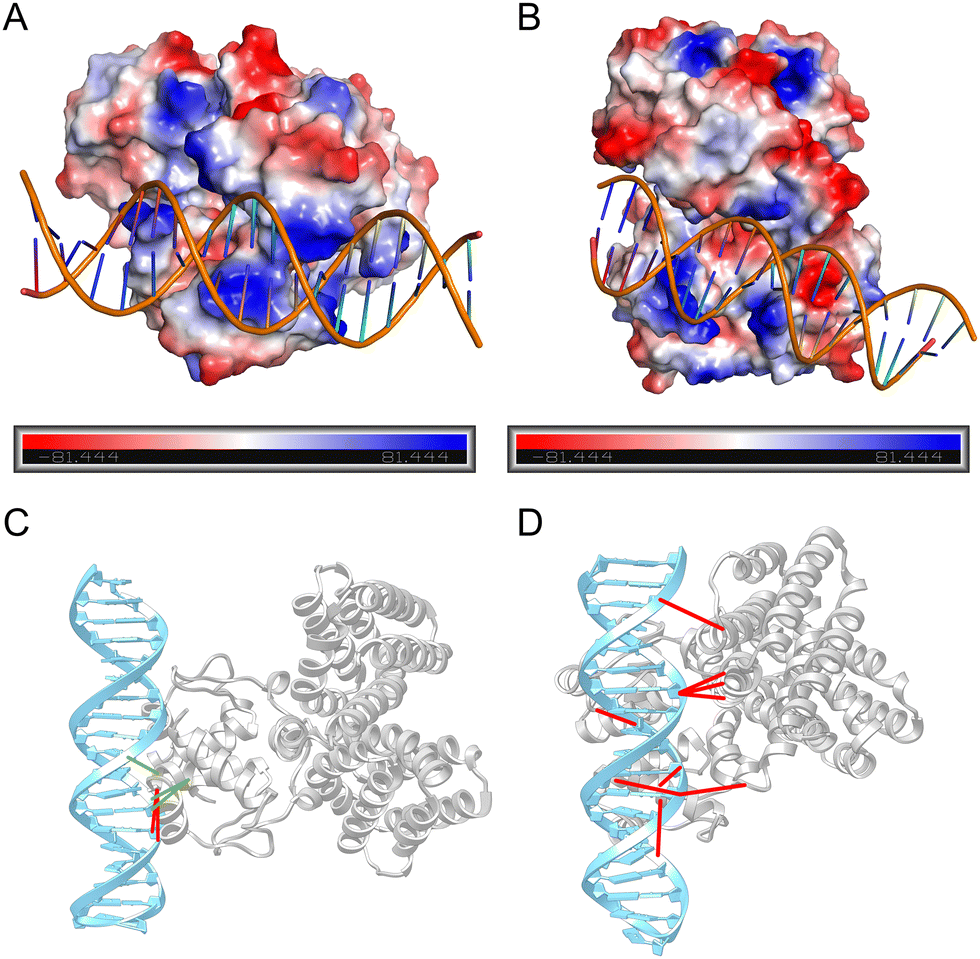 | ||
| Fig. 7 Physics-based interaction distribution. The electrostatic distribution on the lowest RMSD model among the top 5 models selected by DDPScore (A) and HDOCK (B), respectively (PDB ID: 1H9T). Positively charged regions are colored in blue, while negatively charged regions are colored in red. The hydrogen bond distribution on the lowest RMSD model among the top 5 models selected by DDPScore (C) and HDOCK (D), respectively (PDB ID: 1H9T). Hydrogen bond interactions consistent with native patterns are connected by green links, while those that deviate from native hydrogen bond interactions are connected by red links. | ||
3.7 Advances compared to the traditional deep learning model
We have compared DDPScore, our deep learning model, with the traditional 3D CNN model to demonstrate its superiority. The 3D CNN model captures information from the X, Y, and Z coordinates and consolidates it into a single image but fails to establish connections between multiple images. It is good at representing local structural features but cannot capture global structural information. In simpler terms, it captures intra-nucleotide/residue information but neglects inter-nucleotide/residue interactions. This method can only calculate the energy like traditional methods rely on physics-based energy functions or the knowledge-based energy functions from inverse Boltzmann distribution. Due to the limited available data, this method may not be as accurate as traditional methods.Our model can capture local (intra) and global (inter) nucleotide/residue information, making it stand out from other models. It does this by incorporating convolutional layers along the sequential dimension. Each layer progressively models interactions spanning longer ranges between nucleotides/residues, making it an expert in extracting and learning local and global structural features. This includes interactions related to secondary structures. Fig. S3 (ESI†) shows the success rate for DDPScore, HDOCK, and 3DCNN (trained on 300 and 100 decoys per DNA–protein complex, respectively) on the semi-flexible DNA–protein testing set I. Interestingly, 3DCNN has a limitation in accurately identifying near-native DNA–protein complexes. Despite employing physical-based molecular docking to generate additional structures, achieving satisfactory accuracy remains challenging for the traditional 3DCNN network.
4 Discussions and conclusion
Predicting the structure of DNA–protein complexes is pivotal for comprehending biological mechanisms and designing new drugs. The existing scoring functions for DNA–protein prediction rely on statistical potential energy from inverse Boltzmann distribution. Only a few of these methods were specifically designed for DNA–protein complexes. Thus, their ability to predict the structure of DNA–protein complexes has reached a bottleneck in terms of performance. Local interactions involve various factors, including hydrogen bonding and electrostatic interactions, which are a subset of these factors. Fig. 8 illustrates the differences between the structures selected by DDPScore and HDOCK in three examples to compare their ability to capture local interaction features. Specifically, it focuses on the residue–nucleotide pairs within the 6 Å range of the native bound structures. The red and black dots indicate the comparison of residue–nucleotide pairs added or reduced in the native DNA–protein structure. DDPScore captures 84, 77, and 87 residue–nucleotide pairs within a 6 Å range, while HDOCK captures 92, 88, and 101. Although HDOCK appears to capture more interactions, it's important to note that DDPScore captures interactions primarily located near the native interactions. In contrast, HDOCK captures interactions that are farther from the native interactions. This difference is due to the scoring function of HDOCK, which relies on the inverse Boltzmann distribution, resulting in a statistical bias in terms of geometric matching and the count of residue–nucleotide pairs. The 4D convolutional neural network DDPScore is highly efficient in recognizing the local features of DNA–protein complex structures. This enables it to accurately capture binding patterns, even when the number of interactions is limited. As a result, the DDPScore can effectively distinguish and select structures that closely resemble the native state.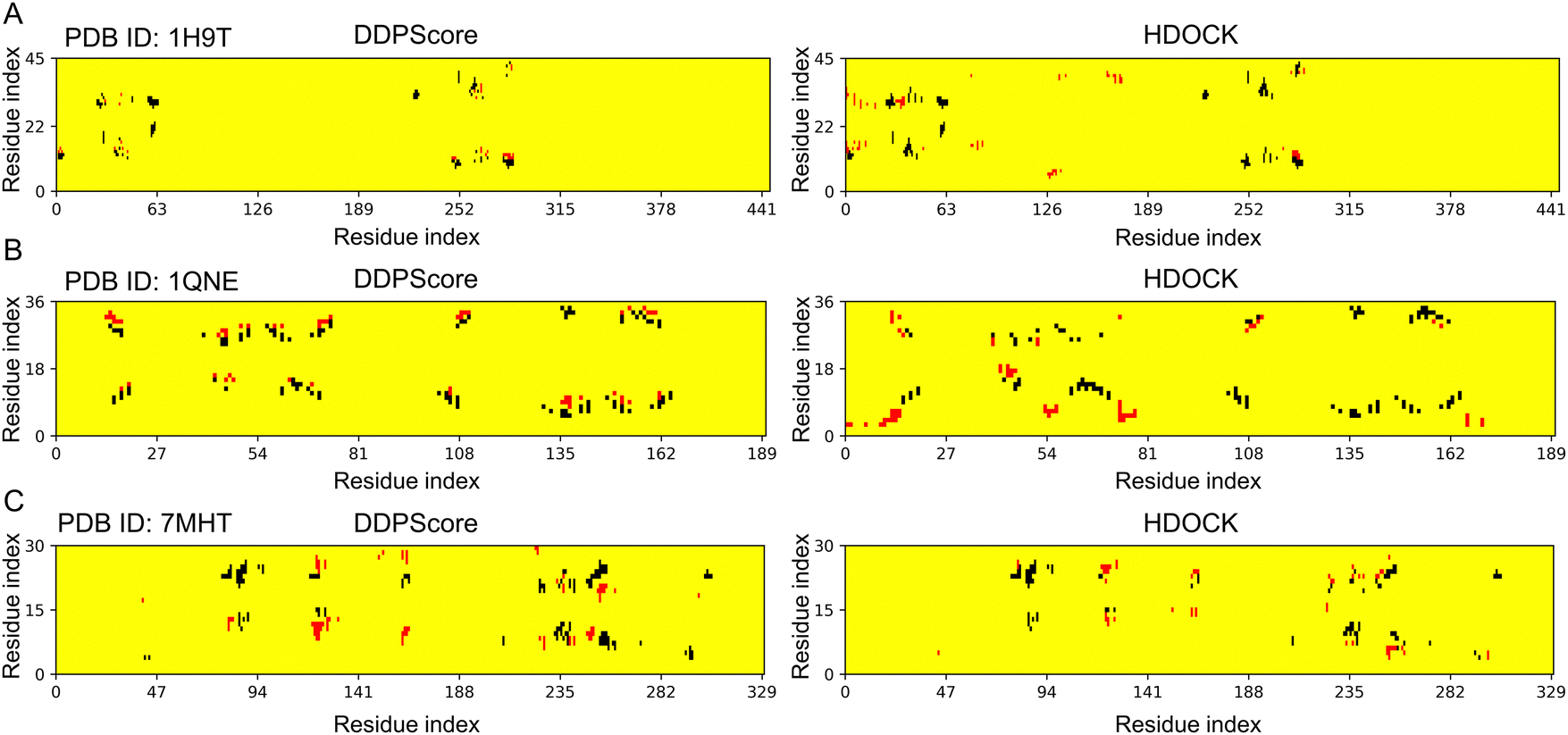 | ||
| Fig. 8 Contact distributions on three flexible docking examples. The contact maps (left to right) are the interactions between residues and nucleotides within the 6 Å range in the lowest RMSD model among the top 5 models selected by DDPScore and HDOCK respectively in (A) PDB ID: 1H9T, (B) PDB ID: 1QNE, and (C) PDB ID: 7MHT. The red and black represent the residue–nucleotide pairs that have been added and reduced by the models selected via DDPScore and HDOCK, compared to the native DNA–protein complex structure. | ||
DDPScore uses a 4D convolutional neural network that simulates a wider range of interactions between nucleotides and residues with each layer. This allows it to learn global features effectively. One example is its ability to capture the binding features of the helix-turn-helix motif. This motif exposes the helix on the protein's surface, which enables it to be specifically recognized by DNA. Thus, DDPScore is able to accurately identify the binding pattern between DNA and protein complexes at the secondary structure level. The current statistical potential-based functions utilize native structures to discern the features of DNA–protein interactions. In contrast, DDPScore is trained with a substantial dataset of decoys obtained through physics-based simulations. As a result, it implicitly covers the dynamic features of DNA–protein interactions. This breakthrough sheds new light on the challenges faced by DNA–protein prediction. DDPScore is particularly effective due to two key factors: (i) the 4DCNN method, which was specifically developed, is able to learn both local and global features of these complexes, and (ii) including a significant number of decoys has provided valuable insights into the dynamic nature of DNA–protein complexes.
DDPScore is a highly efficient tool for evaluating structures of DNA–protein complexes (Table 2 and Fig. S4, ESI†). On a single NVIDIA 2080Ti GPU, it only takes an average of 4.65 seconds to assess 5 DNA–protein complex structures. When scaled up, evaluating 20 DNA–protein complex structures takes an average of 9.03 seconds, while assessing 100 structures takes approximately 32.08 seconds. Even more impressive is that 1000 DNA–protein complex structures are evaluated in less than 5 minutes. We compared the operating efficiency of DDPScore with the traditional 3DCNN model on testing set I (Table S13, ESI†). It is worth noting that DDPScore exhibits a comparable speed compared to 3DCNN. This highlights DDPScore's impressive operational speed and ability to handle large amounts of structural data efficiently. It can even manage the many decoys produced during molecular docking and molecular dynamics trajectories, enabling the exploration of dynamic DNA–protein binding patterns.
Moving forward, we plan to focus on three aspects in future research: (1) Obtaining a more comprehensive understanding of the possible conformations of DNA–protein complexes during the docking process remains an open challenge. To achieve this, we have generated a large dataset of almost 0.2 million decoys through physics-based simulations, covering the full range of potential interactions between DNA and protein surface potentials across a vast spatial landscape. In addition, we plan to expand this dataset further to improve the accuracy of our models using scaling techniques. (2) Improving the accuracy of our DNA–protein docking analysis by using higher-resolution data remains an unsolved problem. To achieve this, we plan to use molecular dynamics to simulate the subtle conformational changes resulting from the docking process. This will allow us to create a more diverse and comprehensive dataset for model training. To do this, we will use a combination of physical simulation-based sampling and molecular dynamics simulations. (3) Developing a hybrid model to extract local and global features effectively. It is possible to integrate a transformer-based model into an optimized 4D convolutional neural network framework. This integration may enable the consideration of sequential and spatial information simultaneously, including local features (hydrogen bonds and electrostatic interactions) and global features (secondary structure and pocket information). This will help us account for the conformational changes induced by flexibility during DNA–protein docking. Overall, this integration promises to enhance the accuracy of conformational evaluation.
In summary, this study introduces DDPScore, a deep learning-based scoring function to evaluate DNA–protein structures. We utilized 4D convolutional neural networks to learn the local and global features from a substantial dataset of decoys derived from physics-based simulations. After a thorough evaluation, it was found that DDPScore performed better than methods that relied on statistical potential energy, especially when dealing with flexible docking. These significant improvements show that DDPScore effectively addresses the challenge of flexibility in assessing DNA–protein complex structures. Our solution, DDPScore, is a fast, flexible, and accurate tool that can provide valuable insights for biologists investigating the mechanisms behind mutational diseases and chemists working on targeted drug design.
Data availability
A full list of the PDB codes used in this study is available in Tables S4–S6 (ESI†). All PDB data sets used in this paper can be downloaded from the Protein Data Bank (https://www.rcsb.org/), Bonvin lab (https://www.bonvinlab.org/), and https://model3dbio.csic.es/pydockdna. The training set that supports the findings of this study is available to download at https://www.zhaoserver.com.cn/DDPScore/DDPScore.html.Code availability
The DDPScore code is freely available for academic or non-commercial users viahttps://www.zhaoserver.com.cn/DDPScore/DDPScore.html.Abbreviations
| Pax2 | Paired box protein 2 |
| TF | Transcription factor |
| NMR | Nuclear magnetic resonance |
| PDB | Protein data bank |
| DDPScore | Deep-learning-based DNA–protein complex score |
| CNN | Convolution neural network |
| 4DCNN | 4-Dimensional convolutional neural network |
| 3DCNN | 3-Dimensional convolutional neural network |
| FFT | Fast Fourier transform |
| RMSD | Root mean square deviation |
| GPU | Graphic processing unit |
| MMPBSA | Molecular mechanics Poisson Boltzmann surface area |
Author contributions
C. Z. (Chengwei Zeng) performed the majority of the computational analysis; Y. J. built the deep learning model; C. Z. (Chen Zhuo) and A. L. helped with the computational analyzing; C. Z. (Chen Zeng) helped with the deep learning modeling; Y. Z. designed the project and supervised the overall study. All authors have read, edited, and approved the final manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the National Natural Science Foundation of China 12175081 (YZ), the Fundamental Research Funds for the Central Universities CCNU22QN004 (YZ), and the Central China Normal University's excellent postgraduate education innovation funding project (no. 30106230507).References
- V. Charoensawan, D. Wilson and S. A. Teichmann, Nucleic Acids Res., 2010, 38, 7364–7377 CrossRef CAS PubMed.
- S. A. Lambert, A. Jolma, L. F. Campitelli, P. K. Das, Y. Yin, M. Albu, X. Chen, J. Taipale, T. R. Hughes and M. T. Weirauch, Cell, 2018, 172, 650–665 CrossRef CAS PubMed.
- R. Kumar, M. A. Corbett, B. W. van Bon, J. A. Woenig, L. Weir, E. Douglas, K. L. Friend, A. Gardner, M. Shaw, L. A. Jolly, C. Tan, M. F. Hunter, A. Hackett, M. Field, E. E. Palmer, M. Leffler, C. Rogers, J. Boyle, M. Bienek, C. Jensen, G. Van Buggenhout, H. Van Esch, K. Hoffmann, M. Raynaud, H. Zhao, R. Reed, H. Hu, S. A. Haas, E. Haan, V. M. Kalscheuer and J. Gecz, Am. J. Hum. Genet., 2015, 97, 302–310 CrossRef CAS PubMed.
- S. Wang, K. Liang, Q. Hu, P. Li, J. Song, Y. Yang, J. Yao, L. S. Mangala, C. Li, W. Yang, P. K. Park, D. H. Hawke, J. Zhou, Y. Zhou, W. Xia, M. C. Hung, J. R. Marks, G. E. Gallick, G. Lopez-Berestein, E. R. Flores, A. K. Sood, S. Huang, D. Yu, L. Yang and C. Lin, J. Clin. Invest., 2017, 127, 4498–4515 CrossRef PubMed.
- P. Schmidtke and X. Barril, J. Med. Chem., 2010, 53, 5858–5867 CrossRef CAS PubMed.
- M. Xu, T. Ran and H. Chen, J. Chem. Inf. Model., 2021, 61, 3240–3254 CrossRef CAS PubMed.
- X. Ma, P. L. Truong, N. H. Anh and S. J. Sim, Biosens. Bioelectron., 2015, 67, 59–65 CrossRef CAS PubMed.
- M. J. Campolongo, S. J. Tan, J. Xu and D. Luo, Adv. Drug Delivery Rev., 2010, 62, 606–616 CrossRef CAS PubMed.
- Z. Zhou and S. Dong, Nanoscale, 2015, 7, 1296–1300 RSC.
- M. Radaeva, A. T. Ton, M. Hsing, F. Q. Ban and A. Cherkasov, Drug Discovery Today, 2021, 26, 2660–2679 CrossRef CAS PubMed.
- J. Favor, R. Sandulache, A. Neuhauser-Klaus, W. Pretsch, B. Chatterjee, E. Senft, W. Wurst, V. Blanquet, P. Grimes, R. Sporle and K. Schughart, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13870–13875 CrossRef CAS PubMed.
- M. Torres, E. Gomez-Pardo, G. R. Dressler and P. Gruss, Development, 1995, 121, 4057–4065 CrossRef CAS PubMed.
- G. Ryan, V. Steele-Perkins, J. F. Morris, F. J. Rauscher, 3rd and G. R. Dressler, Development, 1995, 121, 867–875 CrossRef CAS PubMed.
- J. R. Gnarra and G. R. Dressler, Cancer Res., 1995, 55, 4092–4098 CAS.
- G. R. Dressler and E. C. Douglass, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1179–1183 CrossRef CAS PubMed.
- M. R. Eccles, L. J. Wallis, A. E. Fidler, N. K. Spurr, P. J. Goodfellow and A. E. Reeve, Cell Growth Differ., 1992, 3, 279–289 CAS.
- P. A. Hueber, D. Iglesias, L. L. Chu, M. Eccles and P. Goodyer, Cancer Lett., 2008, 265, 148–155 CrossRef CAS PubMed.
- P. A. Hueber, P. Waters, P. Clarke, M. Eccles and P. Goodyer, Kidney Int., 2006, 69, 1139–1145 CrossRef CAS PubMed.
- C. A. Orengo, A. D. Michie, S. Jones, D. T. Jones, M. B. Swindells and J. M. Thornton, Structure, 1997, 5, 1093–1108 CrossRef CAS PubMed.
- K. Szpotkowski, K. Wojcik and A. Kurzynska-Kokorniak, Comput. Struct. Biotechnol. J., 2023, 21, 2858–2872 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- S. Y. Huang, Drug Discovery Today, 2014, 19, 1081–1096 CrossRef CAS PubMed.
- S. Vajda, D. R. Hall and D. Kozakov, Proteins, 2013, 81, 1874–1884 CrossRef CAS PubMed.
- H. A. Gabb, R. M. Jackson and M. J. Sternberg, J. Mol. Biol., 1997, 272, 106–120 CrossRef CAS PubMed.
- A. Tovchigrechko and I. A. Vakser, Nucleic Acids Res., 2006, 34, W310–W314 CrossRef CAS PubMed.
- G. Macindoe, L. Mavridis, V. Venkatraman, M. D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, W445–W449 CrossRef CAS PubMed.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef CAS PubMed.
- I. Tuszynska, M. Magnus, K. Jonak, W. Dawson and J. M. Bujnicki, Nucleic Acids Res., 2015, 43, W425–430 CrossRef CAS PubMed.
- Y. M. Yan, D. Zhang, P. Zhou, B. T. Li and S. Y. Huang, Nucleic Acids Res., 2017, 45, W365–W373 CrossRef CAS PubMed.
- Y. M. Yan, H. Y. Tao, J. H. He and S. Y. Huang, Nat. Protoc., 2020, 15, 1829–1852 CrossRef CAS PubMed.
- G. C. P. van Zundert, J. Rodrigues, M. Trellet, C. Schmitz, P. L. Kastritis, E. Karaca, A. S. J. Melquiond, M. van Dijk, S. J. de Vries and A. Bonvin, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS PubMed.
- M. Remmert, A. Biegert, A. Hauser and J. Soding, Nat. Methods, 2011, 9, 173–175 CrossRef PubMed.
- S. Y. Huang and X. Zou, Nucleic Acids Res., 2014, 42, e55 CrossRef CAS PubMed.
- S. Y. Huang and X. Q. Zou, Proteins: Struct., Funct., Bioinf., 2008, 72, 557–579 CrossRef CAS PubMed.
- R. J. L. Townshend, S. Eismann, A. M. Watkins, R. Rangan, M. Karelina, R. Das and R. O. Dror, Science, 2021, 373, 1047–1051 CrossRef CAS PubMed.
- K. Sato, M. Akiyama and Y. Sakakibara, Nat. Commun., 2021, 12, 941 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Zidek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS PubMed.
- J. Li, W. Zhu, J. Wang, W. Li, S. Gong, J. Zhang and W. Wang, PLoS Comput. Biol., 2018, 14, e1006514 CrossRef PubMed.
- C. W. Zeng, Y. R. Jian, S. Vosoughi, C. Zeng and Y. J. Zhao, Nat. Commun., 2023, 14, 1060 CrossRef CAS PubMed.
- L. Yuan, J. Wang, L. C. Yu and X. J. Zhang, Inf. Process. Manage., 2022, 59, 103048 CrossRef.
- S. Yang, D. Zhou, J. Cao and Y. Guo, IEEE Signal Process. Lett., 2022, 29, 1082–1086 Search PubMed.
- W. Li, W. Liu, Y. Guo, B. Wang and H. Qing, Chin. J. Electron., 2023, 32, 868–881 Search PubMed.
- Y. Guo, D. Zhou, P. Li, C. Li and J. Cao, IEEE Trans. Neural Netw. Learn. Syst., 2022, 1–13 Search PubMed.
- S. Yang, D. Zhou, J. Cao and Y. Guo, IEEE Trans. Comput. Imaging, 2023, 9, 29–42 Search PubMed.
- W. Li and A. Godzik, Bioinformatics, 2006, 22, 1658–1659 CrossRef CAS PubMed.
- W. Li, L. Jaroszewski and A. Godzik, Bioinformatics, 2001, 17, 282–283 CrossRef CAS PubMed.
- W. Li, L. Jaroszewski and A. Godzik, Bioinformatics, 2002, 18, 77–82 CrossRef CAS PubMed.
- S. Y. Huang and X. Zou, J. Comput. Chem., 2013, 34, 311–318 CrossRef CAS PubMed.
- M. van Dijk and A. M. J. J. Bonvin, Nucleic Acids Res., 2008, 36, e88 CrossRef PubMed.
- X. J. Lu and W. K. Olson, Nucleic Acids Res., 2003, 31, 5108–5121 CrossRef CAS PubMed.
- L. A. Rodriguez-Lumbreras, B. Jimenez-Garcia, S. Gimenez-Santamarina and J. Fernandez-Recio, Front. Mol. Biosci., 2022, 9, 988996 CrossRef CAS PubMed.
- B. R. Miller, 3rd, T. D. McGee, Jr., J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed.
- D. A. Case, T. E. Cheatham, 3rd, T. Darden, H. Gohlke, R. Luo, K. M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CAS.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- J. Janin, K. Henrick, J. Moult, L. T. Eyck, M. J. Sternberg, S. Vajda, I. Vakser and S. J. Wodak, Proteins, 2003, 52, 2–9 CrossRef CAS PubMed.
- R. Mendez, R. Leplae, M. F. Lensink and S. J. Wodak, Proteins, 2005, 60, 150–169 CrossRef CAS PubMed.
- C. Zhuo, C. W. Zeng, R. Yang, H. Q. Liu and Y. J. Zhao, Int. J. Mol. Sci., 2023, 24, 5497 CrossRef CAS PubMed.
- T. Sunami and H. Kono, PLoS One, 2013, 8, e56080 CrossRef CAS PubMed.
- N. M. Luscombe, S. E. Austin, H. M. Berman and J. M. Thornton, Genome Biol., 2000, 1, REVIEWS001 CrossRef CAS PubMed.
- I. K. McDonald and J. M. Thornton, J. Mol. Biol., 1994, 238, 777–793 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04980a |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2024 |