
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in copper chalcogenides for CO2 electroreduction
Wenjian
Hu
 ab,
Didier
Grandjean
b,
Jan
Vaes
ac,
Deepak
Pant
*ad and
Ewald
Janssens
*b
ab,
Didier
Grandjean
b,
Jan
Vaes
ac,
Deepak
Pant
*ad and
Ewald
Janssens
*b
aSeparation and Conversion Technology, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400 Mol, Belgium. E-mail: deepak.pant@vito.be
bQuantum Solid-State Physics, Department of Physics and Astronomy, KU Leuven, Celestijnenlaan 200 D, 3001 Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be
cDepartment of Solid-state Sciences, Ghent University, Krijgslaan 281/S1, 9000 Gent, Belgium
dCenter for Advanced Process Technology for Urban Resource Recovery (CAPTURE), Frieda Saeysstraat 1, 9052 Zwijnaarde, Belgium
First published on 23rd October 2023
Abstract
Transforming CO2 through electrochemical methods into useful chemicals and energy sources may contribute to solutions for global energy and ecological challenges. Copper chalcogenides exhibit unique properties that make them potential catalysts for CO2 electroreduction. In this review, we provide an overview and comment on the latest advances made in the synthesis, characterization, and performance of copper chalcogenide materials for CO2 electroreduction, focusing on the work of the last five years. Strategies to boost their performance can be classified in three groups: (1) structural and compositional tuning, (2) leveraging on heterostructures and hybrid materials, and (3) optimizing size and morphology. Despite overall progress, concerns about selectivity and stability persist and require further investigation. This review outlines future directions for developing the next-generation of copper chalcogenide materials, emphasizing on rational design and advanced characterization techniques for efficient and selective CO2 electroreduction.
1. Introduction
The worldwide need for energy continues to grow while the alarming rise in greenhouse gas (GHG) emissions demand for sustainable and eco-friendly solutions.1–8 Carbon dioxide (CO2) is a major GHG, contributing significantly to global warming and shifts in climate patterns. Creating practical and affordable solutions to capture and transform CO2 into valuable chemicals and fuels is thus of urgent importance.9–11 Among various CO2 conversion methods that are being developed, electrochemical reduction has emerged as one of the most appealing approaches, given its notable energy efficiency, its potential for renewable energy integration, and its capacity to generate a variety of valuable products.12–27 Specifically, the process of CO2 electroreduction using Cu-based catalysts22,27–44 has garnered tremendous attention because Cu demonstrates notable activity for C–C coupling, essential to the formation of multicarbon products.Copper chalcogenides are a family of compounds that combine copper with elements from the chalcogen group, comprising sulfur, selenium, and tellurium. These materials have attracted considerable interest for multiple uses in semiconductors, photoconductors, thermoelectric materials, and solar cells.45–50 Copper chalcogenide bonds are highly covalent, although the chalcogenide is treated as a divalent anion (S2−, Se2−, or Te2−) in some cases. In a basic lattice cell, chalcogen atoms constitute the frame of a regular lattice, whereas the copper atoms are disordered, moving like a liquid and taking random sites in the chalcogen lattice.51,52 This phenomenon leads to variations in stoichiometry, which is represented by the characteristic formula Cu2−xE (E = S, Se, Te) with possible x values ranging from 0 to 1.53,54 The copper chalcogenide family encompasses various binary, ternary, and quaternary compounds, whose stoichiometries determine the crystal structure. The binary copper sulfide system has monoclinic (Cu31S16 and Cu1.94–1.97S), cubic (Cu9S5 and Cu1.8S), orthorhombic (Cu7S4 and Cu1.75S), triclinic (Cu58S32 and Cu1.81S), and hexagonal (CuS) crystal phases. The binary copper selenide system has cubic (Cu2Se and Cu1.8Se), tetragonal (Cu3Se2), hexagonal (CuSe and Cu0.87Se), and orthorhombic (Cu5Se4 and CuSe2) phases. Known crystal phases of copper tellurides are hexagonal (Cu2Te), tetragonal (Cu7Te4, Cu4Te3 and Cu3Te2), orthorhombic (CuTe), and pyrite-type (CuTe2).55–57 In the context of the electrochemical CO2 reduction, copper chalcogenides have shown an exceptional selectivity as a result of their distinct electronic and structural properties.
The synergistic effect between copper and the chalcogen elements is responsible for the remarkable electrocatalytic CO2 reduction performance of these compounds. The presence of chalcogen atoms modulates their electronic structure, in particular the location of the Cu d-band center altering the adsorption behavior of the reaction intermediates, and subsequently influencing the catalyst's specificity and effectiveness.54,58 The tunability of the copper chalcogenide properties enables the optimization of their electrocatalytic performance through various strategies, such as controlling their composition, adjusting their crystal structure and morphology, and creating heterostructures or hybrid materials. These approaches can be employed to customize the electronic configuration and catalytic performance of copper chalcogenides, enhancing their performance in CO2 electroreduction by achieving higher selectivity, improved stability, and increased energy efficiency.59
The objective of this review is to examine the latest developments in copper chalcogenide catalysts for CO2 electroreduction, focusing on the most significant contributions from the past five years. We discuss the latest approaches to enhance their efficacy as electrocatalysts, including structural and compositional tuning, exploiting heterostructures and hybrid materials, and optimizing their shape and particle size by synthesis methods.
Although significant advancements have been made in recent times, obstacles persist in increasing the preference for particular products and boosting the enduring stability of copper chalcogenide catalysts. Therefore, we also outline future perspectives on the development of next-generation copper chalcogenide materials, emphasizing the importance of rational design strategies and the integration of advanced characterization techniques to achieve efficient and selective CO2 electroreduction.
2. Synthesis strategies for copper chalcogenides
Copper chalcogenides have been studied for several decades, initially for their applications in superconductors and thermoelectric materials. Their synthesis has evolved over time with enhanced control over their composition, structure, and morphology. Solvothermal methods, vital for nanoparticle synthesis, allow the production of highly crystalline materials under controlled temperatures and pressures, with the ability to tailor compositions and structures. Electrosynthesis, offers fine control over the material properties and allows for in situ formation of compounds like copper chalcogenides on conductive substrates, but it does present challenges in scaling up. Meanwhile, cation exchange methods permit careful post-synthesis modifications, control over composition, and the synthesis of complex heterostructures and hybrid materials. By combining these techniques, researchers could overcome their individual limitations; solvothermal synthesis forms the initial nanostructures, electrosynthesis deposits additional material, and cation exchange refines their final properties, broadening the horizons for nanomaterial design and innovation (Fig. 1). Other methods, such as wet chemical method60–62 pyrolysis63 and heat treatment64,65 have also been explored to produce copper chalcogenide materials with various catalytic properties for the electrochemical CO2 reduction reaction (eCO2RR). Inspired by the water evaporation technique used in ref. 66, we could synthesize a variety of Cu2−xSe nanostructures, including nanosheets, nanoflowers, and nanowires. This was achieved by variations of the Cu/Se ratio of 3.75/19, 5/19 and 7.5/19, respectively (Fig. 2).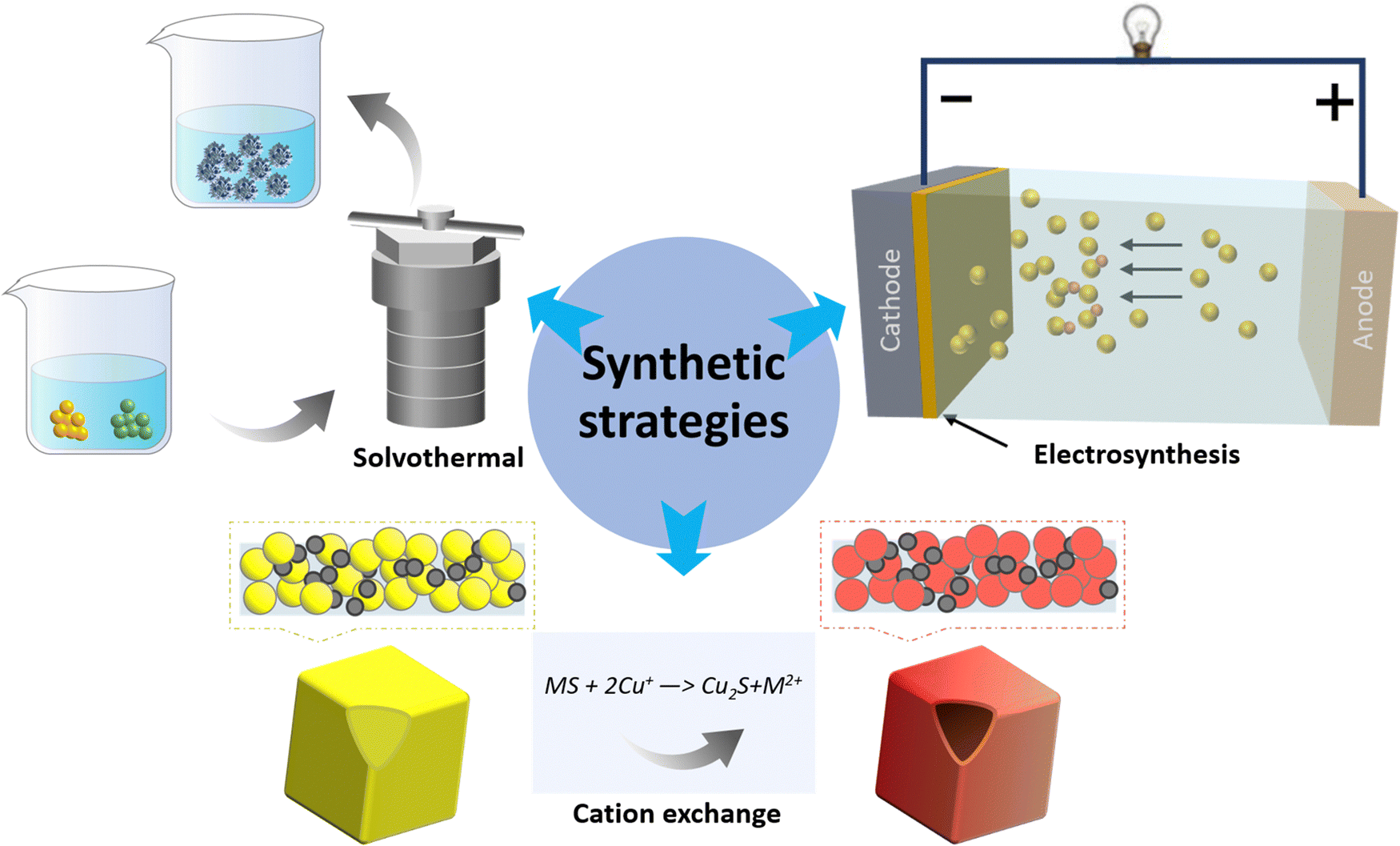 | ||
| Fig. 1 Schematic diagram of synthesis strategies, including solvothermal, electrosynthesis and cation exchange. M stands for metal. | ||
 | ||
| Fig. 2 Scanning electron microscopy images illustrating the variety of Cu2−xSe nanostructure that can be grown by changing the Cu/Se ratio. (a) Nanosheets, (b) nanoflowers, and (c) nanowires synthesized from a Cu/Se precursor ratio of 3.75/19, 5/19 and 7.5/19, respectively. | ||
2.1 Solvothermal synthesis
Solvothermal synthesis is a prominent method for preparing copper chalcogenides for eCO2RR. It uses elevated temperatures and pressures in a sealed reactor to produce highly crystalline materials with tailored properties. By adjusting the reaction parameters, various nanostructures, such as nanoparticles, nanowires, and nanosheets could be obtained.Solvothermal methods have extensively been used to synthesize copper sulfide nanostructures.63,67–82 For example, Shinagawa et al.67 reported that CuS nanocatalysts can be produced by in situ reductive reformation of CuS precursor materials. They developed a new method to create carbon-supported copper sulfide nanoparticles by mixing copper nitrate, thiourea, urea, and carbon black in water, and heating the mixture to precipitate the nanoparticles. Wang et al.73 utilized a combination of solvothermal and electrochemical techniques to create Cu2SnS3 nanosheets. This involved dissolving SnCl4·5H2O and Cu(CH3COO)2·H2O within a combined concoction of ethylene glycol and water, agitating the solution, adding thioacetamide, and heating the mixture in a Teflon-lined autoclave. The synthesis of CuS nanosheets followed a similar procedure, while SnS2 nanosheets were prepared using SnCl4·5H2O, sodium dodecyl benzenesulfonate, and L-cysteine. Following eCO2RR, the samples experienced self-adapted phase separation, resulting in the formation of SnO2@CuS and SnO2@Cu2O heterojunctions. Zhang et al.69 created CuS nanosheets by mixing 6 mL of 0.5 M thioacetamide with 25 mL of 0.2 M Cu(NO3)2 ethanol solutions. The mixture was heated to 90 °C for 3 hours, whereafter the resulting black precipitate was collected. CuS hollow particles were synthesized using a modified method in which thioacetamide dissolved in ethanol is added to a dispersion of MOF-199 in ethanol that is subsequently heated at 90 °C for 1 hour. TEM and high-angle annular dark-field scanning TEM (HAADF-STEM) images showed that CuS hollow particles have an average size of 1.3 μm and are composed of CuS nanosheets with a thickness of about 3 nm.
The hydrothermal synthesis was also used to produce copper selenide nanomaterials.83–89 Yang et al.83 combined CuCl2·2H2O, Na2SeO3, and hydroxylamine in a diethylenetriamine and deionized water mixture with varying volume ratios to synthesize Cu2−xSe(y) nanocatalysts. After stirring for 30 minutes, the mixture was warmed in a Teflon-coated pressure vessel at 180 °C during 15 hours and the resulting products were washed, and dried. The obtained Cu1.63 Se nanoparticles had a typical size of 50 nm (see Fig. 3). Wang et al.85 prepared CuInSe2 using a modified solvothermal method. Oleylamine was degassed and purged with N2 before combining CuCl, InCl3 and Se in a flask. The mixture was heated and stirred before the products were washed, and dried. Duan et al.88 synthesized Cu2Se nanosheets using a hydrothermal treatment. BiCuSeO nanosheets were produced by mixing Bi(NO3)2·5H2O with Cu(NO3)2·3H2O, KOH and NaOH, heating the mixture in a Teflon-lined vessel, and washing and drying the obtained precipitate. The average thickness of the BiCuSeO sheets was 9.2 nm. High-resolution TEM images display two perpendicular sets of lattice fringes with a spacing of 0.28 nm, verifying the successful synthesis of ultrathin BiCuSeO single-crystal nanosheets. Ding et al.87 synthesized K-doped Cu2Se nanosheet arrays on a Cu foam. The foam was cleaned, soaked in a mixture of Se powder, NaBH4, NaOH, deionized water, and KBr, and heated in an autoclave. Various KBr amounts were used to produce different potassium dopant contents, while undoped pure Cu2Se was obtained without KBr. 11.2% K-doped Cu2Se had a honeycomb-like structure and a thickness of 1.4 μm, while pure Cu2Se had a thickness of 1.8 μm. Those nanosheets display a plane spacing of 0.205 nm, which correlates with the (220) facet of cubic Cu2Se.
 | ||
| Fig. 3 Structural characterization of Cu1.63Se nanocatalysts. (a) SEM image with a 200 nm scale bar. The inset presents the size distribution as obtained by dynamic light scattering (DLS) measurements. (b) TEM image with a 100 nm scale bar, including elemental mappings as insets. (c) HRTEM image with a 10 nm scale bar. Copyright 2019 Nature Publishing Group.83 | ||
The latter method is widely used for the synthesis of various types of materials including nanoparticles, single crystals, and complex structures. It provides the advantage of synthesizing high-quality materials at relatively lower temperatures than conventional methods. However, the process is often time-consuming and requires careful handling due to high pressures involved. In addition, the solvents used can be harmful, requiring careful management.
2.2 Electrosynthesis
In situ electrochemical synthesis, including electrochemical deposition, recently gained traction for the synthesis of copper chalcogenide nanomaterial.75,81,90–94 Redox reactions cause selective ion deposition on either the cathode or the anode surface. Key benefits of electrochemical deposition include simplicity of the operating procedure, precise control over particle size, and environment friendly outcomes without corresponding oxidation or reduction byproducts. As the understanding of catalyst synthesis methods evolves, many catalysts are not produced using a single method but by combining solvothermal and in situ electrochemical synthesis techniques.Studies emphasize the importance of the catalysts' elemental composition and their intricate microstructures. Cheng et al.81 demonstrated that introducing surface nitrogen can notably improve the electrocatalyst's performance by raising the concentration of active sites and optimizing their electronic structure. The obtained N-enriched Sn/SnS nanosheets demonstrated a five-fold rise in current density and a 2.45-fold increase in faradaic efficiency (FE) compared to their unmodified SnS2-derived counterparts. The formate production rate was 14 times higher in the N-enriched sheets than in the unmodified ones, reaching 1358 μmol h−1 cm−2. The surface nitrogen-injection technique was also effectively employed with other precursors such as CuS and In2S3, demonstrating its potential as a broader approach. Peng et al.75 synthesized double sulfur vacancy-rich CuSx nanosheets for the electro-conversion of CO2 into C3 species. In a first stage CuS nanosheets were prepared by dissolving CuCl2·2H2O and thioacetamide in separate solutions that were then combined and subsequently heated and washed. Sulfur vacancies were then introduced in the CuS powder by mixing it with polyvinylidene fluoride and coating it on a copper foil that was used as a cathode in a lithium-ion battery. A 0.044 mA cm−2 current density was applied within a 0.01–3 V vs. Li+/Li voltage range. CuSx catalysts with various sulfur vacancy densities were obtained after 1, 10, and 100 charge–discharge cycles. HAADF-STEM images revealed that the CuSx-10-cycle nanosheets display a hexagonal phase where the 0.283 nm spacing corresponds to CuS (103) facets. The (103) planes were also seen on CuSx-1-cycle and CuSx-100-cycle edges. Deng et al.90 developed a method for the electrochemical deposition of CuSx films on polished Cu disks by utilizing square wave voltammetry. The synthesis procedure involved 2000 cycles with a plating solution containing a mixture of CuSO4·5H2O, thiourea, and HCl. Active CuSx and desulfurized CuSx were prepared for electrochemical CO2 reduction to investigate the role of sulfur. Phillips et al.91 prepared sulfide-derived-Cu working electrodes via pulsed electrodeposition. The active sulfide-derived-Cu layer was formed in situ during the first ∼15 minutes of electrolysis. Guo et al.93 synthesized Cu2S hollow nanocubes with a significant presence of sulfur vacancies by electrochemical reduction (see Fig. 4). Cu2S1−x hollow nanocubes were obtained by electroreducing Cu2S hollow nanocubes loaded on a gas diffusion electrode (GDE) at −0.3 V vs. RHE for 30 minutes. Li et al.94 prepared Ag and S co-doped Cu2O/Cu (Ag, S-Cu2O/Cu) using an electrochemical reduction method at room temperature in a conventional H-cell. Ag–Cu2S was first prepared through a cation exchange method and used as the working electrode. Electrochemical reduction was then carried out by applying a potential of −1.6 V versus a saturated calomel electrode for 30 minutes, while bubbling CO2 and stirring continuously. Cu2O/Cu, S-Cu2O/Cu, and Ag–Cu2O/Cu were fabricated using the same procedure, with Cu2O, Cu2S, and Ag–Cu2O as working electrodes, respectively.
 | ||
| Fig. 4 (a) Schematic representation of the electrochemical reduction process of CuS hollow nanocubes into their Cu2S1−x and Cu counterparts. (b) Subsequent LSV curves for the CuS hollow nanocubes (numbered LSV-1 to LSV-5). TEM images along with corresponding EDX line-scanning profiles for (c) CuS, (d) Cu2S1−x, and (e) Cu hollow nanocubes, all featuring a 30 nm scale bar. Figure adapted from ref. 93. Copyright 2022 Wiley VCH. | ||
Electrosynthesis can be more environment friendly than conventional methods since in many cases the use of electricity does not require potentially harmful chemical reagents. On the downside, it can require extensive optimization of individual processes and, for certain reactions, achieving a high selectivity can be challenging.
2.3 Cation exchange
Cation exchange, a wet chemical method,61,62,95–97 is a relatively new approach for synthesizing copper sulfide nanocatalysts for eCO2RR. By selectively replacing the cations of a host material with copper ions, CuS nanosheets and nanoparticles with controlled properties can be produced.He et al.96 introduced electrochemically-driven cation exchange (ED-CE), which enables converting a prearranged CoS2 template into Cu2S through a three-step process using a standard three-electrode system. A metal–organic framework (MOF) consisting of a Co-containing zeolitic imidazolate framework (Co-ZIF-L) grown on a carbon cloth and Cu foil is electrochemically converted into cobalt sulfides and finally cations are exchanged. In this configuration, the cobalt sulfide film functioned as working electrode and cation exchange template, while a platinum foil and a silver/silver chloride electrode saturated with potassium chloride served as counter and reference electrodes, respectively. To replace the cobalt cations of CoS2 with copper ions, a 40 mL dimethylformamide solution containing 3 mM copper nitrate trihydrate and 0.1 M lithium perchlorate was used as electrolyte. Chronoamperometry was employed to drive the cation exchange processes, which was performed first at −0.2 V (vs. Ag/AgCl) and thereafter at −0.6 V (vs. Ag/AgCl) until a targeted value of electrical charge per electrode area was reached. The synthesis procedure is schematically depicted in Fig. 5. This method ensures that the synthetized Cu2S catalyst keeps the same structure as the CoS2 template, while maintaining a high density of grain boundaries. Li et al.97 developed a simple cation exchange technique to create Ag/Cu electrocatalysts using copper sulfide nanosheets as growth template. The crystal structure of Cu2−xS nanosheets with lateral dimensions of 100 nm and a 14 nm thickness gradually transformed from Cu7S4 to Ag2S, with an Ag/Cu mass ratio varying between 0.3 and 25 upon increasing the Ag+ concentration in the exchange solution. Following the cation exchange process, the nanosheets retain their original morphology, but exhibit shape distortion with increasing Ag content.
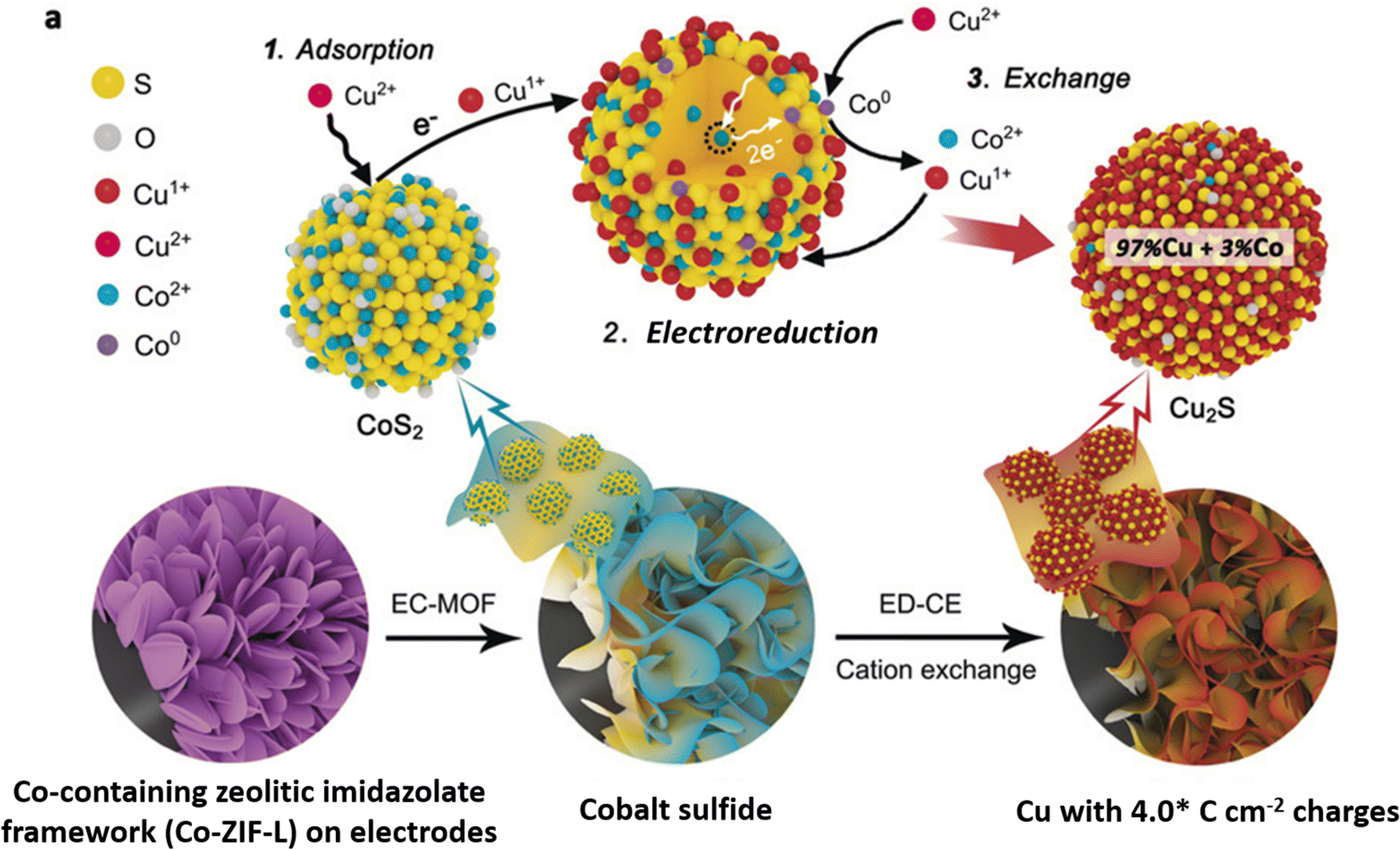 | ||
| Fig. 5 Experimental routes and the mechanisms involved in cation exchange driven by electrochemical means. Figure adapted from ref. 96. Copyright 2020 Wiley-VCH. | ||
We conclude that cation exchange is an attractive method to modify the chemical composition and hence the properties of the material without disrupting its overall structure. It has potential to fine-tune materials for specific applications. However, the method's success highly depends on the cation exchange kinetics and thermodynamics, which can be complex and hard to control.
3. Recent strategies to enhance the performance of copper sulfide in eCO2RR
Copper sulfide has emerged as an effective catalyst for the eCO2RR, but further improvement is needed. Notably, the hydrogen evolution reaction (HER) is a competitive reaction to eCO2RR that hinders its efficiency. Research has shown a linear correlation between the binding energies of COOH*/HCOO* and H* on most eCO2RR catalysts, negatively impacting the selectivity.98–102 Breaking this correlation is imperative for improving copper sulfide's effectiveness as an eCO2RR catalyst for industrial applications. Recent strategies to enhance the selectivity and activity of CuS include (i) structural and compositional tuning through sulfur vacancy engineering and foreign metal ions doping, (ii) combining CuS with other materials in heterostructures or hybrid materials to introduce new active sites and improve the catalytic performance through synergistic effects, and (iii) size and morphology control, for example to create nanosheets or hollow structures with optimized surface area and enhanced accessibility of the active sites. These approaches offer a guide for developing effective CuS catalysts for eCO2RR, advancing eco-friendly conversion methods. A summary of cutting-edge Cu sulfide catalysts, including information on their preparation method, operating conditions and product preferences, is provided in Table 1.| Preparation method | Catalysts | Electrolyte | E (V vs. RHE) | Partial current density (mA cm−2) | Product (FE) | Ref. |
|---|---|---|---|---|---|---|
| Solvothermal | Cu2O/CuS Nanocomposites | 0.1 M KHCO3 | −0.9 | 15.3 | HCOO− (67.6%) | 74 |
| N-Enriched CuS | 0.1 M KHCO3 | −0.8 | NA | HCOO− (∼88%) | 81 | |
| Copper sulfide nanocrystals | 0.1 M KHCO3 | −1 | 2.1 | HCOO− (52.2%) | 82 | |
| CuS@SnO2 nanosheets | 0.5 M KHCO3 | −1.0 | ∼150 | HCOO− (83.4%) | 73 | |
| CuS@MoS2 heterostructures | CO2 saturated electrolyte | −0.69 | NA | NA | 68 | |
| CuS/N, S-rGO | 0.5 M KHCO3 | −0.63 | ∼5 | HCOO− (82%) | 71 | |
| CuS-thiourea | 0.1 M KHCO3 | −0.51 | ∼0.8 | CO (72.67%) | 80 | |
| Cu2S nanocrystal-decorated Cu nanosheets | 0.1 M KHCO3 | −1.2 | ∼20.7 | C2H5OH (46%) | 78 | |
| Smx-CuSy | 1 M KOH | −0.52 | 276 | HCOO− (92.1%) | 77 | |
| Sulfur tailored Cu2O | 0.1 M KHCO3 | −1 | 16.3 | HCOO− (67.2%) | 70 | |
| Electrosynthesis | Active CuSx | 0.1 M KHCO3 | −0.9 | 9 | HCOO− (75%) | 90 |
| Sulfide-derived Cu | 0.1 M KHCO3 | −0.8 | ∼2.5 | HCOOH (∼60%) | 91 | |
| Cu2S1−x with abundant Cuδ+ | 0.5 M KHCO3 | −0.3 | ∼2.5 | C2H5OH (73.3%) | 93 | |
| Dual-doped Ag, S-Cu2O/Cu | BMIMBF4/H2O | −1.18 | 82.7 | CH3OH (67.4%) | 94 | |
| Cation exchange | CuSx | 0.1 M KHCO3 | −0.6 | 2.2 | HCOO− (71.8%) | 62 |
| 3D Cu2S | 0.1 M NaHCO3 | −0.9 | 19.1 | HCOO− (87.3%) | 96 | |
| Ag/Cu sulfide | 0.05 M K2CO3 | −1.2 | 5.1 | C2H4 (34%) | 97 | |
| Wet chemistry | Cu and CuS nano-particles@graphene nanoflakes | 0.1 M KHCO3 | −0.6 | 2.3 | HCOO− (42.5%) | 79 |
| SnO2 confined on CuS nanosheets | 0.1 M KHCO3 | −0.8 | ∼17 | syngas (>85%) | 72 | |
| Sulfide-derived CuCd | 0.1 M KHCO3 | −0.8 | 0.6 | C2H5OH (∼32%) | 76 | |
| Sulfur vacancy-rich CuS | 0.1 M KHCO3 | −1.05 | ∼3.3 | n-Propanol (15.4%) | 75 | |
| Hierarchical CuS hollow polyhedrons | 0.5 M K2SO4 | −0.6 | ∼16 | HCOO− (∼50%) | 69 | |
| Sulfur-modified copper catalysts | 0.1 M KHCO3 | −0.8 | ∼21 | HCOO− (80%) | 67 | |
| Other methods | ||||||
| Thermal oxidation | CuS decorated CuO heterostructure | 0.1 M KHCO3 | −0.7 | 20 | HCOO− (84%) | 95 |
| In situ crystallization | Cu1.81S particles on carbon nanotubes | 0.5 M KHCO3 | −0.67 | 3.1 | HCOO− (82%) | 60 |
| Chemical bath deposition | CuS nanosheet arrays | 0.5 M KHCO3 | −0.7 | ∼50 | HCOO− (70.2%) | 61 |
| Impregnation and annealing | Cu2−xS derived nanoparticles@C | 0.5 M KHCO3 and/or 0.5 M KCl | −0.78 | 0.18 | HCOO− (12%) | 65 |
3.1 Structural and compositional tuning
To enhance copper sulfide's eCO2RR catalytic performance, various structural and compositional tuning strategies,61,65,67,69,75,79,81,82,90,91,93,94,96,97,103 can be employed to adjust its electronic structure and morphology.We outline two key strategies. The first one is by creating sulfur vacancies in the material during or after its synthesis by controlling the preparation conditions or through post-synthesis treatments, respectively. Their presence increases the likelihood of CO* formation on the catalyst surface. The second is by doping CuS with metal cations. Ag doping, for instance, induces synergistic interactions between Ag2S and CuS that suppress HER and improve the selectivity towards CO2 reduction. Metal doping can be achieved by co-precipitation, ion exchange, or electrochemical deposition.
Cheng et al.81 found that surface nitrogen-injection can increase the number of active sites and optimize the material's electronic structure. N-Enriched Sn/SnS nanosheets demonstrated significant improvements in current density and FE compared to their pristine versions. The improved catalytic activity was confirmed through density functional theory (DFT) calculations to be a result of the alteration in the valence of the Sn sites. The approach is applicable to other metal sulfide precursors as well. Peng et al.75 showed that double sulfur vacancies (DSVs) in the CuS(100) planes could serve as active electrocatalytic centers for CO2RR by favoring the creation of n-propanol's key *C3 intermediate. They applied a lithium electrochemical tuning method, using the CuS as the cathode and a lithium foil as the anode, yielding promising results for the controlled creation of ion vacancies (see Fig. 6). DFT calculations suggest that the creation processes of *HCOOH and CO* are in competition with the presence of sulfur vacancies, increasing the formation probability of the latter one. During continuous electrochemical CO2RR testing, the CuSx-DSV catalyst maintained a FEn-PrOH of 12.1% for 10 hours at −1.05 V vs. RHE, corresponding to a retention of approximately 78%. Meanwhile, an increase in the hydrogen production activity was observed, indicated by a continuous rise in the current. Deng et al.90 found that sulfur plays a crucial role in this process, with active CuSx exhibiting high selectivity and activity towards formate (FE HCOO− = 75% and j HCOO = −9.0 mA cm−2 at −0.9 V). Operando Raman spectroscopy revealed that sulfur dopants suppress the formation of CO intermediate during eCO2RR, resulting in a decreased FE for CO, hydrocarbons, and alcohols. Supporting DFT calculations indicated that the presence of sulfur on the copper surface modified the adsorption strengths of HCOO*, promoting formate production while suppressing that of CO. Phillips et al.91 suggested that the selective formation of formate is due to the stronger binding of the CO intermediate originating from the sulfur atoms. Guo et al.93 reported a Cu2S1−x catalyst, featuring numerous Cuδ+ species that can produce ethanol at ultralow overpotential with an outstanding selectivity and FE with a 6-hour stability. The integrated findings from in situ attenuated total reflection Fourier transformed infrared spectroscopy and DFT calculations demonstrate that the robust electron-donating ability of Cuδ+ sites contributes to reducing the Gibbs free energy for the C–C coupling step involved in generating ethanol, while concurrently enhancing that associated with formate formation.
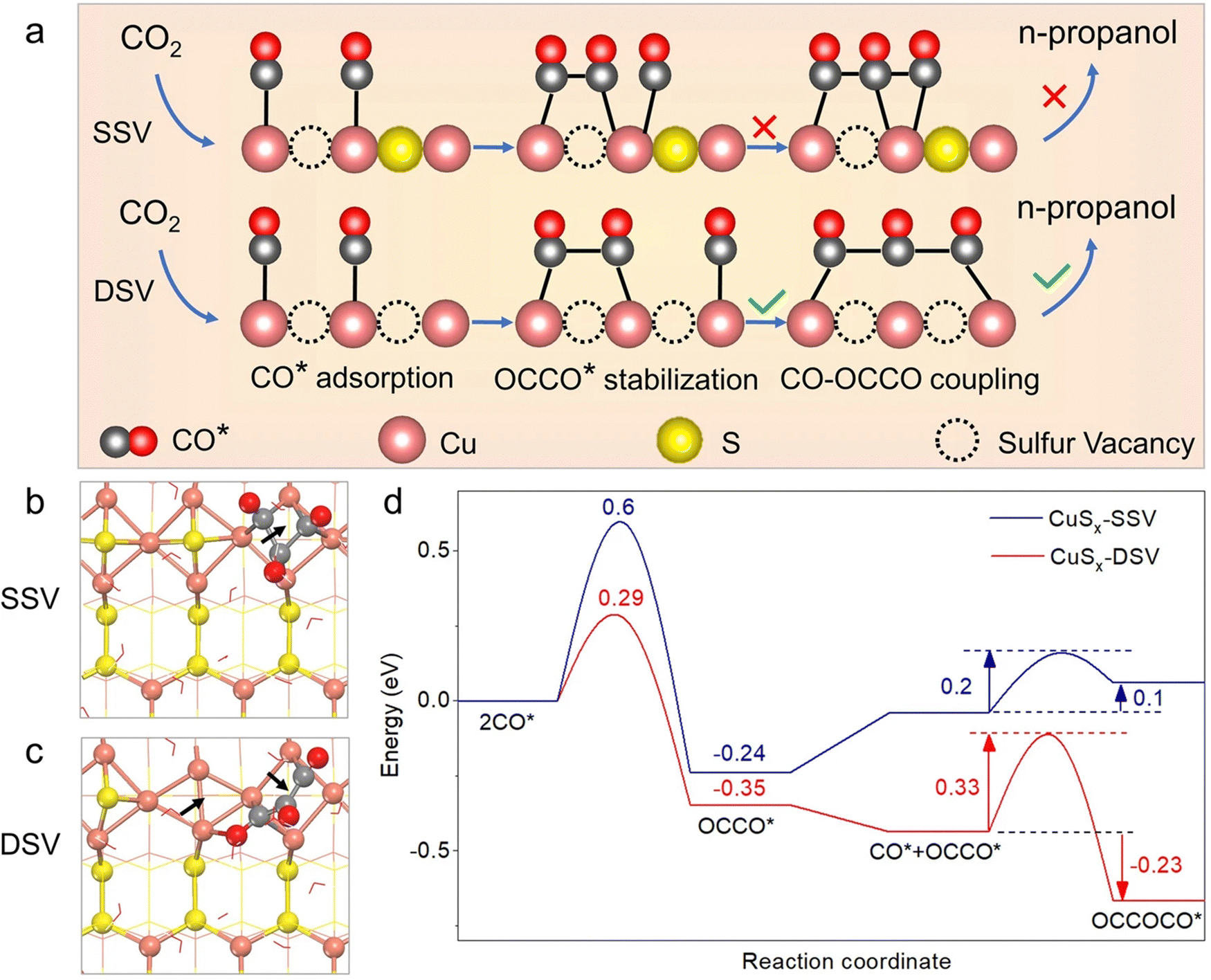 | ||
| Fig. 6 (a) Schematic illustration of the conversion of CO2 to n-propanol on CuSx with double sulfur vacancies (DSV) and related calculations. The process involves n-propanol formation on adjacent CuSx-DSV through CO–CO dimerization and CO–OCCO coupling. Structural diagrams displaying the optimized OCCOCO* intermediate configurations on the (100) surface of CuSx with (b) a single sulfur vacancy (SSV) and (c) CuSx-DSV. Arrows indicate sulfur vacancy positions. (d) Energy diagrams for CuSx-SSV (blue curve) and CuSx-DSV (red curve) at 0 V vs. RHE. Copyright 2021 Nature Publishing Group.75 | ||
Li et al.94 found that the synergistic interactions between S2− anions and Ag+ cations dopants and the Cu2O/Cu host material, not only enhanced the Ag, S-Cu2O/Cu electrocatalyst selectivity and current density for methanol formation but also ensured its stability over 24 hours. More specifically, S anions were found to adjust the electronic structure and morphology of the material, while according to their DFT calculations Ag+ cations suppressed HER. Zhang et al.69 studied the eCO2RR catalytic properties of CuS hollow polyhedra that experience structural transformation, as they evolve into a sulfur-doped metallic Cu catalyst. The in situ evolved catalyst exhibited a high durability and formate selectivity, maintaining stable operation for 36 hours at a formate partial current density of approximately 16 mA cm−2 (at −0.6 V vs. RHE). This stability and selectivity were underscored by DFT calculations, which highlighted the significant role of sulfur-doped Cu (111) facets in its performance.
3.2 Synergistic effects of heterostructures and hybrid materials
Advancements in material science have demonstrated that heterostructures, comprising multilayer materials, and hybrid materials allow to optimize the catalytic CO2RR performance, selectivity, and stability through synergistic effects.68,71–74,76–78,95 For example, CuS-based heterostructures with other semiconductors or metal catalysts can have optimized charge transfer, energy band alignment, and beneficial interfacial interactions to enhance their eCO2RR performance. The same applies to hybrid materials that combine CuS with conductive polymers, metal–organic frameworks, or graphene-based materials.Wang et al.73 discovered that Cu2SnS3 nanosheets undergo a self-adaptive phase separation during the electrochemical process, resulting in the formation of stable SnO2@CuS and SnS2@Cu2O heterojunctions. Because Sn4+ cations can be easily drawn by the negative potential, they migrate from the core to the surface of the nanosheet, replacing coordinated S atoms with O to form SnO2 nanoparticles. Meanwhile, Cu+ ions, carrying S atoms, concentrate in the center, forming CuS (Fig. 7). They identified that activated Sn is responsible for CO2 reduction in CuS@SnO2 ternary sulfides, which facilitates a self-adaptive transformation. By harnessing the synergistic effect of a stable heterojunction structure and electron self-flow, the optimized catalysts efficiently convert CO2 to formate with 84.1% FE for 10 hours. This is supported by DFT calculations demonstrating that at the SnO2 and CuS/Cu2O interface, delocalized Sn4+ electrons transfer to Cu+, thereby enhancing the HCOO* affinity, promoting H2O dissociation, and stabilizing Sn4+ sites, which collectively improve the CO2RR activity and selectivity. This work revealed the active center of the ternary sulfide, offering insights for designing better electrocatalysts. Wang et al.74 presented an innovative Cu2O/CuS composite catalyst that exhibits a 67.6% FE and a notable partial current density of 15.3 mA cm−2 at −0.9 V versus RHE for formate. This catalyst maintains an average FE of 62.9% for at least 30 hours at the same potential, outperforming other Cu, CuS, and Cu2O catalysts in terms of selectivity and stability. The incorporation of CuS is believed to stabilize Cu2O during CO2 reduction. DFT calculations imply that the CuS (110) surface favors the formation of formate over CO. In another study, Wang et al.72 designed a novel hierarchical structure combining SnO2 nanoparticles with CuS nanosheets, improving the electroreduction of CO2 into a versatile syngas mixture (CO/H2 ratio varying in the range of 0.11–3.86). The material achieved a remarkable FE >85%, a significant turnover frequency of 96.1 h−1, and a durability of more than 24 hours. Experimental characterization and theoretical calculations demonstrated that various factors like size, morphology and the density of the SnO2–CuS interface contribute to the enhanced catalytic performance. In a study by Mosali et al.,76 a near 30% FE for ethanol production for 6 hours was reported (at −0.8 V versus RHE), using the sulfide-derived-CuCd2 catalyst in 0.1 M KHCO3. This notable ethanol selectivity was attributed to the abundant distribution of CuS/Cu2S–CdS boundaries and Cu2S sites within the sulfide-derived-CuCd2 catalyst identified through electrochemical, spectroscopic, and microscopic analyses. The electrochemically stable CdS phase plays a crucial role in creating the favorable catalyst structure, which stimulated C–C coupling and further reduction to ethanol. Li et al.78 investigated an electroreductive CO2-to-ethanol conversion process, catalyzed by Cu nanosheets that are uniformly decorated with small Cu2S nanocrystals. The system, housed in an H-cell containing 0.1 M KHCO3, achieved a 20-hour stable high ethanol current density of approximately 20.7 mA cm−2 with a large FE of 46% (at 1.2 V vs. RHE). Nanocrystal-decorated Cu nanosheets offer several advantages over heteroatom doping, Cu sulfide/oxide/nitride, and the parent catalyst, including: (i) a relatively high positive local charge of Cuδ+; (ii) effective interfaces between Cuδ+ and Cu0; and (iii) a non-flat, stepped surface. These features improve CO2 adsorption, enhance eCO2RR kinetics, facilitate the dissociation of the *COOH intermediate into *CO, strengthen the affinity of *CO on the catalyst, and bring the energy barrier for C–C coupling below the desorption energy of *CO. Liu et al.77 found that incorporating Sm2O3 into an S-doped Cu matrix creates a mixed phase of heterogeneous Sm and homogeneous S co-doped Cu, which stabilizes Cuδ+ species and encourages the HCOOH formation pathway. With the Sm0.06-CuS0.03 catalyst, they achieved a 92.1% FE for HCOOH production for 24 hours and an impressive current density of 300 mA cm−2 at a low reduction potential of −0.52 V vs. RHE. This good electrocatalytic performance could, based on material characterizations and in situ experiments, be attributed to synergistic interaction between heterogeneously doped Sm species and homogeneously doped S elements. To explore the synergy between Sm and S, homogeneous S doping in the Cu active phase has been studied. This modification alters the inherent electronic structure of Cu, generating a surplus of Cuδ+ (δ = +0.61). The presence of heterogeneous Sm species not only stabilizes Cuδ+ sites but also raises the concentration of H ions, which enhances the adsorption of oxygen-containing intermediates (*OCHO) during HCOOH formation.
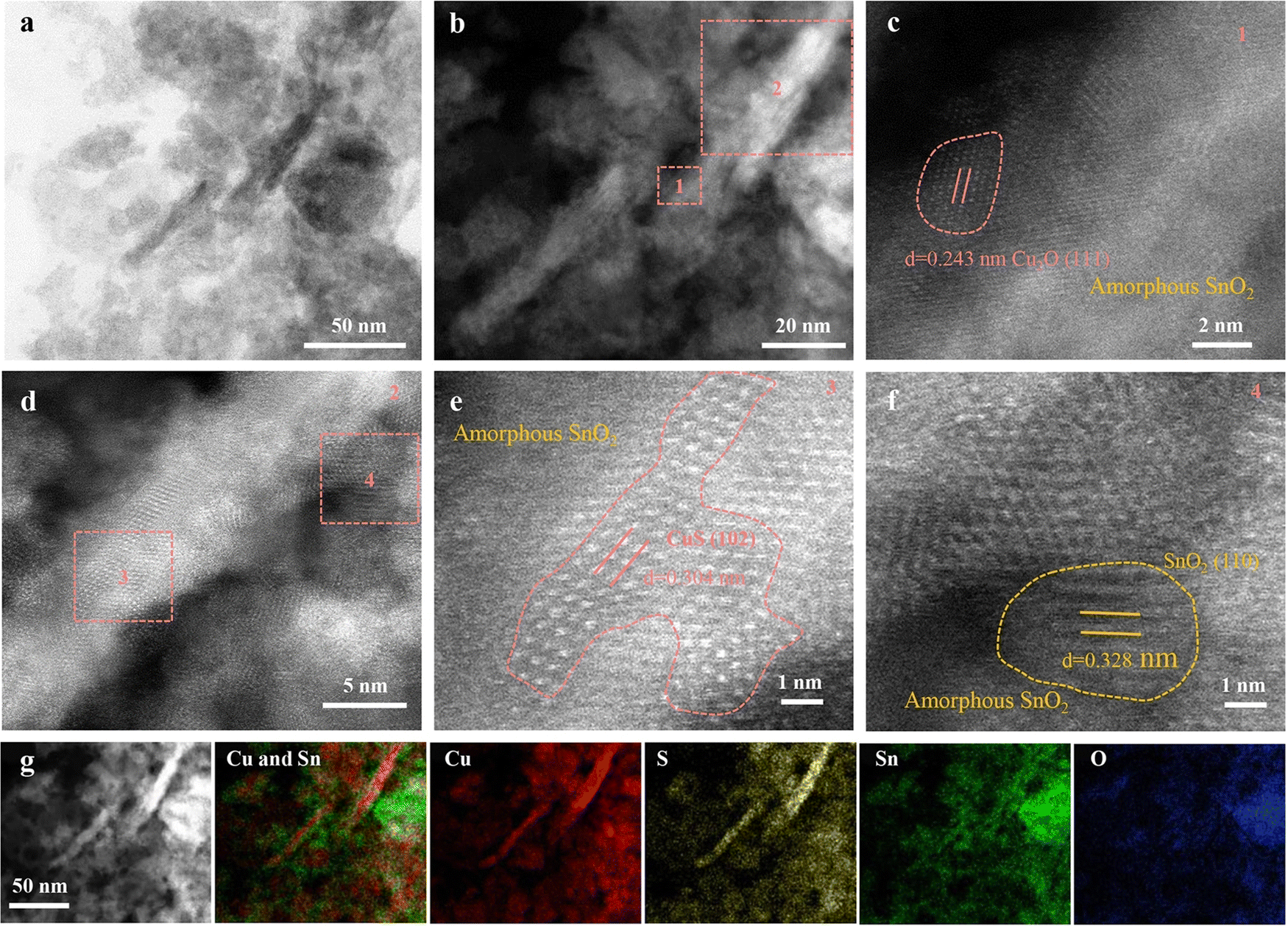 | ||
| Fig. 7 Characterization of Cu2SnS3 after CO2RR (R-Cu2SnS3). (a) TEM image. (b)–(f) HAAD-STEM images. (g) HAAD-STEM image and EDX element mapping. Copyright 2021 Wiley-VCH.73 | ||
3.3 Size and morphology engineering
This section comments on recent approaches to customize CuS60,62,67,70,80,82,96 catalyst properties by particle size and morphology engineering. Through the optimization of these aspects, researchers continue to unveil new opportunities of copper sulfides in electrochemical CO2 reduction.Shinagawa et al.67 identified a relationship between the particle size and the formate selectivity in sulfur-modified copper catalysts. The particle size varies between fresh and used samples. After the electrochemical test, the initial average particle size of about 15 ± 7 nm for a particular CuS samples reduced to approximately 3 nm, while that of another sample decreased from roughly 42 nm to around 5 nm. They found that CuS-4 with larger particle sizes and higher sulfur contents resulted in increased FE for formate. Submicron-sized CuS catalysts, prepared solvothermally, outperformed their nanosized counterparts. In regard to stability, we note that the Cu–S catalyst sustained its exclusive formate production over 12 hours at −0.8 V vs. RHE. He et al.96 uncovered a relationship between electrochemical parameters like the applied potential and charge passed and the extent of electrochemically-driven cation exchange reactions through structural and compositional analyses. This allowed them to systematically adjust binary cation ratios in nanocrystalline films, while preserving their initial 3D structural characteristics and engineer highly reactive catalytic sites by generating ultra-small structures and controlling surface oxidation states. Using this approach, they synthesized a Cu2S electrocatalyst with high selectivity and activity for eCO2RR, achieving a CO2 partial current density over 19 mA cm−2 for formate production at about −0.9 V vs. RHE with a FE of 87.3% (stable for 9 hours at −0.82 V vs. NHE). This performance exceeds that of all Cu-based catalysts previously reported, providing insights into targeting specific reactive sites within nanocrystalline metal sulfides for eCO2RR and suggesting a method for designing advanced electrocatalysts for various energy conversion processes. In their research, Lim et al.62 developed highly selective and stable copper sulfide (CuSx) catalysts capable of converting industrial CO2 into formate. The copper foil spontaneously reacted with sulfur species, increasing the average size and surface density of the CuSx nanoparticles as the H2S concentration rose, resulting in an increase of the formate FE from 22.7% to 71.8%. CuSx catalysts demonstrated efficacy in both industrial and pure CO2 saturated electrolytes and maintained stability for 12 hours of continuous operation. The high FE for formate achieved by the CuSx catalysts was due to the significant sulfur content, enhanced size and augmented surface density of CuSx nanoparticles. Gao et al.80 investigated the synthesis of CuS catalysts using various sulfur precursors for electrochemical CO2 reduction, and found that these precursors influence the catalyst performance due to different morphological features and CO selectivity. The CuS thiourea catalyst, with its distinctive morphology resembling nanoflowers, outperformed others by achieving the highest CO FE of 72.67% and favorable CO2 reduction kinetics.
Generally, the strategic engineering of catalysts’ size and morphology selectively enhances CO2 reduction, while minimizing HER. This approach entails increasing exposure of boundary density and active sites, controlling exposed catalyst facets, and adjusting particle sizes. Furthermore, a refined morphology facilitates better mass transfer and encourages favorable kinetics for CO2 reduction reactions.
4. Recent strategies to enhance the performance of copper selenide in eCO2RR
Similar as for CuS, recent strategies to improve the performance of copper selenide in the electrochemical CO2 reduction include structural and compositional tuning,64,83–85,87,88,103,104 size and morphology engineering,83,89,92 and heterostructure design.86Table 2 provides a compact summary of the latest results obtained on CuSe-based eCO2RR catalysts, detailing their composition, preparation method, performance and product selectivity according to the electrolyte used. A comparison of the CO2 electroreduction efficiency of copper chalcogenides prepared by alternative methods to those employed for the current leading catalysts is depicted in Fig. 9.| Preparation method | Catalysts | Electrolyte | E (V vs. RHE) | Partial current density (mA cm−2) | Product and FE | Ref. |
|---|---|---|---|---|---|---|
| Solvothermal | BiCuSeO superlattices | 0.5 M KHCO3 | −0.9 | 18.7 | HCOO−(∼93.4%) | 88 |
| Cu1.63Se | [BMIM]PF6 (30 wt%)/CH3CN/H2O (5 wt%) | −2.1 V vs. Ag/Ag+ | 32.2 | CH3OH (77.6%) | 83 | |
| Se-defective CuInSe2 | 0.5 M KHCO3 | −0.7 | 112 | CO (91%) | 85 | |
| Nanostructured Cu2Se | 0.3 M NaHCO3 | −1.3 | NA | HCOO−(94.2%) | 84 | |
| 2D CuSe/g-C3N4 | 0.1 M KHCO3 | −1.2 | ∼8.4 | CO (85.28%) | 86 | |
| K11.2%-Cu2Se nanosheets | 0.1 M KHCO3 | −0.8 | 35.8 | C2H5OH (70.3%) | 87 | |
| Cu1.22V0.19Se nanotubes | 0.1 M KHCO3 | −0.8 | 207.9 | C2H5OH (68.3%) | 105 | |
| Cu3Se2 nanosheet on Cu foam | 0.1 M KHCO3 | −1.2 | 11.9 | CO (>40%) | 89 | |
| 2D Cu2−xSe with numerous Se vacancies | 0.5 M KHCO3 | −0.8 | 7.44 | C2H5OH (68.1%) | 106 | |
| Wet chemistry and annealing | Cu1.82Se nanowires | 0.1 M KHCO3 | −1.1 | 10.2 | C2H4 (55%) | 64 |
| N-CuSe | 0.5 M KHCO3 | −1.1 | 25.8 | HCOO− (55.2%) | 104 | |
| Electro-synthesis | 3D CuSe nanocubes | 1.0 M KHCO3 or 0.1 M [BMIM]PF6 | −1.3 | 10 | NA | 92 |
The most common strategy for designing better catalysts is to optimize the catalyst's structure and composition. Wang et al.85 designed bimetallic CuInSe2 with Se vacancies to mimic the eCO2RR catalytic properties of Au, resulting in an efficient conversion to CO accompanied by a suppressed HER activity. The orbital interaction of both metals with Se vacancies played a critical role in the eCO2RR pathway. Se-defective CuInSe2 (V-CuInSe2) nanosheets exhibited an effective performance, illustrating the potential of cost-effective, noble-metal-like catalysts with unique electronic, structural, and catalytic properties. Duan et al.88 suggested using natural superlattices with alternating active/conductive layers to stabilize the metal oxidation states, improving the eCO2RR performance. They utilized BiCuSeO that features alternating conductive [Cu2Se2]2− and insulating [Bi2O2]2+ sublayers as a representative example. It was demonstrated that the bismuth oxide layers played a significant role in activating CO2, with created electrons being transferred quickly through the [Cu2Se2]2− sublayers. DFT computations suggested that the Bi–O coordination in [Bi2O2]2+ effectively activates and stabilizes OCHO* intermediates, which facilitates the selective CO2 electroreduction to formate. As a result, the naturally occurring BiCuSeO superlattices generate formate with a FE over 90% across a broad potential range between −0.4 and −1.1 V in a neutral electrolyte. This work demonstrates the use of natural superlattices for enhanced eCO2RR selectivity and highlights the importance of specific coordination structures (see design strategies in Fig. 8). Ding et al.87 found that exposed (220) crystal planes facilitate the adsorption of a key bridge-bonded *COB intermediate. Potassium doping enhances the adsorption of linear *COL and bridge *COB intermediates on the catalyst surface, promoting C–C coupling to produce ethanol by protecting Cu(I) sites during the electrocatalysis. The optimal K11.2%-Cu2Se nanosheet array achieves a 70.3% FE and a partial current density of 35.8 mA cm−2 for ethanol at −0.8 V in 0.1 M KHCO3, while remaining stable for 130 hours. With a FE of ethanol exceeding 50% in a wide potential range from −0.6 to −1.2 V, this approach demonstrates the good potential of these superlattices for real-world implementations. This study presents a novel method to enhance CO2RR to ethanol through alkali metal doping and offers a rational strategy for designing catalysts with high product selectivity for multiple proton-coupled electron transfer reactions. Sun et al.105 developed V-doped Cu2Se nanotubes for flow-cell CO2RR, efficiently producing ethanol. The introduction of V4+ ions into Cu2Se provides diverse active sites, prevents Cu+ reduction, and facilitates different CO intermediates’ adsorption, leading to ethanol formation, as confirmed by in situ DRIFTS and DFT calculations. The optimized Cu1.22V0.19Se nanotubes achieve a 68.3% FEC2H5OH and a −207.9 mA cm−2 partial current density for ethanol at −0.8 V in 1 m KOH. Hao et al.89 presented self-supported Cu3Se2 featuring a hierarchical structure composed of nanosheet-assembled fibers whose properties are modulated by anions. This structure delivers a flexible and broad range of H2-to-CO ratios (from 0.8 to 6.0) within an extensive potential window and boasts an impressive turnover frequency (TOF) of 1303 h−1. Wang et al.106 prepared 2D Cu2−xSe with numerous Se vacancies, enhancing CO2 to ethanol conversion activity and selectivity during eCO2RR. The optimal Cu-Cu spacing in this ultra-thin material, obtained by the presence of Se vacancies, promotes C–C coupling, resulting in efficient ethanol production. Specifically, VSe-Cu2−xSe, with a 2.51 Å Cu–Cu distance, achieves a 68.1% faradaic efficiency and 7.44 mA cm−2 partial current density for ethanol in 0.5 M KHCO3, within a −0.4 to −1.6 V potential range. In situ Fourier transform infrared spectroscopy and DFT calculations confirm that this spacing optimizes the Gibbs free energy for both ethanol generation and formate formation steps.
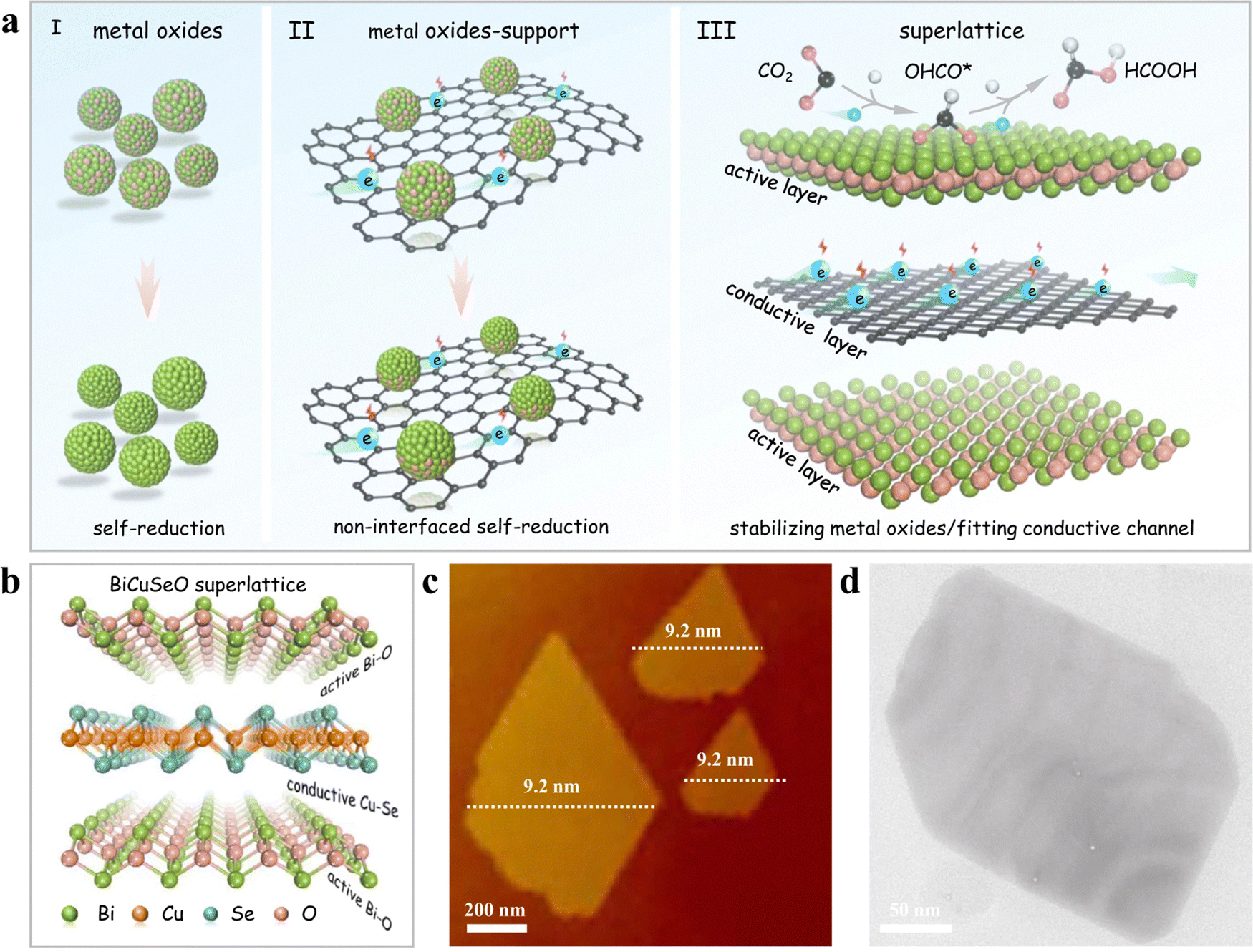 | ||
| Fig. 8 Development of BiCuSeO superlattice nanosheets: design strategy and structural analysis. (a) Visual representation of single-phase materials using alternating active and conductive layer arrangements, including (I) pure metal oxides, (II) metal oxides interacting with a support, and (III) a superlattice made up of vertically stacked active (metal oxides) and conductive CuSe layers in a one-to-one ratio. (b) Structural representation of the BiCuSeO superlattice, (c) AFM image, and (d) TEM image. Copyright 2022 Nature Publishing Group.88 | ||
Yang et al.83 emphasized that the copper selenide catalysts' performance was strongly influenced by their size and morphology, which could be fine-tuned using the volume ratio of diethylenetriamine and water (VDETA/VH2O). They found that the catalysts synthesized at a VDETA/VH2O ratio of 1/3 had the smallest size, resulting in the highest current density and FE. The Cu1.63Se(1/3) nanocatalysts achieved an exceptional current density of 41.5 mA cm−2 and an FE of 77.6% at a potential of −2.1 V versus Ag/Ag+.
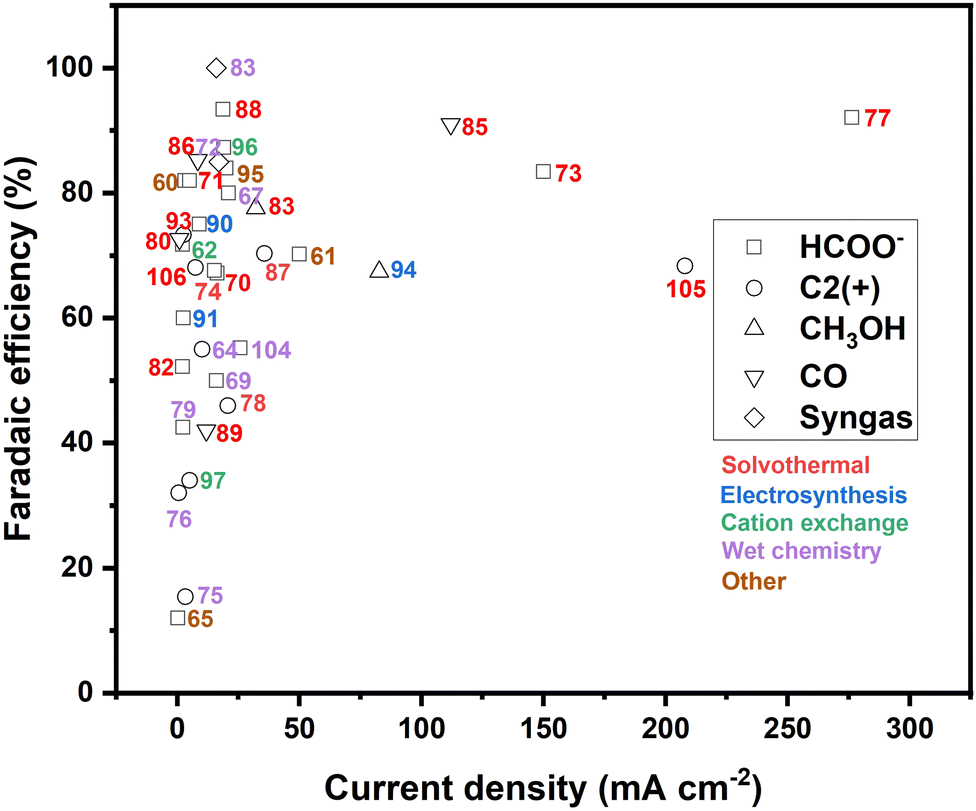 | ||
| Fig. 9 Comparison of the CO2 electroreduction performance of copper chalcogenides in different reported state-of-art catalysts. The numbers in the figure are the corresponding reference numbers. The symbols reflect the main eCO2RR product of each catalyst, and the color its fabrication method. | ||
Since the first application of copper selenide in the electrocatalytic carbon dioxide reduction in 2019,83 the field has seen substantial progress. This advancement is highlighted by the diversification of the reaction products that now include methanol, formic acid, carbon monoxide (CO), and notably, ethanol.87,105,106 Additionally, there has been a remarkable increase in electrical current efficiency, rising from 41 mA cm−2 to 207.9 mA cm−2. These improvements that are largely due to the implementation of new defect engineering and doping strategies will drive further research interest in this area.
5. Challenges and future directions
Among the family of copper chalcogenides, copper sulfide and copper selenide have been extensively utilized for electrochemical CO2 reduction while copper telluride has only been employed for water splitting applications.107,108 Copper telluride, due to its distinct electronic structure and stability compared to its predecessors, may bring more possibilities for future eCO2RR research.Despite significant progress in the application of copper chalcogenide materials as efficient electro-catalysts for eCO2RR, moving from products like CO, formate to methanol and C2+ products, and the transition from low to high current densities up to 300 mA cm−2, numerous challenges remain to be tackled. These include the use of more rational design strategies for increasing selectivity towards specific products and enhancing the catalysts’ long-term stability under operational conditions. Understanding the relationship between catalyst structure, product selectivity, and long-term stability is crucial. In situ and operando characterization techniques can contribute to the understanding of structure-performance relationships and the identification of active sites in these materials.
As the synthesis of catalysts typically does not involve a unique synthetic method but multiple ones, it is vital to explore and optimize synergistic effects that can be obtained by integration of various synthetic strategies. This can result in the advancement of novel copper chalcogenide materials with unique properties and improved eCO2RR performance.
A major challenge in eCO2RR using copper chalcogenides is achieving a high selectivity towards specific products such as formate, CO or hydrocarbons. Subsequent investigations should prioritize the development of strategies for tailoring the electronic properties of copper chalcogenide materials using elemental doping and adjusting their geometric properties through specific chemical synthesis methods, as these can directly influence their catalytic activity and selectivity.
Another important point is the stability of copper chalcogenide catalysts during long-term eCO2RR operation, which is crucial for practical applications. Degradation, agglomeration, and morphological changes can lead to performance deterioration over time. Addressing these stability issues requires the development of novel catalyst architectures and protective strategies, such as encapsulating the catalysts in robust matrices or engineering support materials to prevent degradation.
Optimizing the performance of copper chalcogenide materials requires developing rational design strategies that can be achieved by integrating advanced characterization techniques, including in situ and operando methods, with computational modeling to acquire understanding of active sites and reaction mechanisms. Such an approach will enable the strategic development of advanced copper chalcogenide materials with enhanced eCO2RR performance.
For practical applications of copper chalcogenide catalysts in large-scale eCO2RR, it is essential to address the challenges associated with their scalability and environmental impact. Future research should focus on developing cost-effective and environmentally friendly synthetic methods, as well as evaluating the life cycle and techno-economic aspects of these materials.
In conclusion, overcoming the challenges in enhancing the selectivity and stability of copper chalcogenide materials will require a combination of rational design strategies, advanced characterization techniques, and the integration of various synthetic methods. Addressing these challenges is crucial for realizing the full potential of copper chalcogenides in CO2 electroreduction and contributing towards a sustainable future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 MSCA-ITN programme under grant agreement No 955650 (CATCHY).References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Adv. Mater., 2020, 32, e1908398 CrossRef PubMed.
- L. Zhang, I. Merino-Garcia, J. Albo and C. M. Sánchez-Sánchez, Curr. Opin. Electrochem., 2020, 23, 65–73 CrossRef CAS.
- J. Zhao, S. Xue, J. Barber, Y. Zhou, J. Meng and X. Ke, J. Mater. Chem. A, 2020, 8, 4700–4734 RSC.
- S. Bhardwaj, A. Biswas, M. Das and R. S. Dey, Sustain. Energy Fuels., 2021, 5, 2393–2414 RSC.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Mater., 2022, 1, 100012 CrossRef.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- K. Van Daele, B. De Mot, M. Pupo, N. Daems, D. Pant, R. Kortlever and T. Breugelmans, ACS Energy Lett., 2021, 6, 4317–4327 CrossRef CAS.
- O. Gutiérrez-Sánchez, B. de Mot, N. Daems, M. Bulut, J. Vaes, D. Pant and T. Breugelmans, Energy Fuels, 2022, 36, 13115–13123 CrossRef.
- J. Mertens, C. Breyer, K. Arning, A. Bardow, R. Belmans, A. Dibenedetto, S. Erkman, J. Gripekoven, G. Léonard, S. Nizou, D. Pant, A. S. Reis-Machado, P. Styring, J. Vente, M. Webber and C. J. Sapart, Joule, 2023, 7, 442–449 CrossRef.
- C. Liu, J. Gong, Z. Gao, L. Xiao, G. Wang, J. Lu and L. Zhuang, Sci. China: Chem., 2021, 64, 1660–1678 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- W. Quan, Y. Lin, Y. Luo and Y. Huang, Adv. Sci., 2021, 8, 2101597 CrossRef CAS PubMed.
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- B. Talukdar, S. Mendiratta, M. H. Huang and C. H. Kuo, Chem. – Asian J., 2021, 16, 2168–2184 CrossRef CAS PubMed.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- Y. Xin, K. Yu, L. Zhang, Y. Yang, H. Yuan, H. Li, L. Wang and J. Zeng, Adv. Mater., 2021, 33, e2008145 CrossRef PubMed.
- B. Deng, X. Zhao, Y. Li, M. Huang, S. Zhang and F. Dong, Sci. China: Chem., 2022, 66, 78–95 CrossRef.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustain. Chem., 2022, 34, 100589 CrossRef CAS.
- S. Mohan, B. Honnappa, A. Augustin, M. Shanmugam, C. Chuaicham, K. Sasaki, B. Ramasamy and K. Sekar, Catalysts, 2022, 12, 445 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- L. Zaza, K. Rossi and R. Buonsanti, ACS Energy Lett., 2022, 7, 1284–1291 CrossRef CAS.
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z. L. Wang, Small, 2022, 18, 2107450 CrossRef CAS PubMed.
- C. Zhu, S. Zhao, G. Shi and L. Zhang, ChemSusChem, 2022, 15, e202200068 CrossRef CAS PubMed.
- Y. Lei, Z. Wang, A. Bao, X. Tang, X. Huang, H. Yi, S. Zhao, T. Sun, J. Wang and F. Gao, J. Chem. Eng., 2023, 453, 139663 CrossRef CAS.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijoo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruna and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- J. Bu, Z. Liu, W. Ma, L. Zhang, T. Wang, H. Zhang, Q. Zhang, X. Feng and J. Zhang, Nat. Catal., 2021, 4, 557–564 CrossRef CAS.
- C. Durante, Nat. Catal., 2021, 4, 537–538 CrossRef CAS.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- F. Li, X. V. Medvedeva, J. J. Medvedev, E. Khairullina, H. Engelhardt, S. Chandrasekar, Y. Guo, J. Jin, A. Lee, H. Thérien-Aubin, A. Ahmed, Y. Pang and A. Klinkova, Nat. Catal., 2021, 4, 479–487 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- R. Shi, Z. Wang, Y. Zhao, G. I. N. Waterhouse, Z. Li, B. Zhang, Z. Sun, C. Xia, H. Wang and T. Zhang, Nat. Catal., 2021, 4, 565–574 CrossRef CAS.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C. W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng, L. Yin, Z. Weng and Q. H. Yang, Nat. Commun., 2022, 13, 3158 CrossRef CAS PubMed.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O’Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- B. Seger, M. Robert and F. Jiao, Nat. Sustain., 2023, 6, 236–238 CrossRef.
- D. R. Brown, T. Day, K. A. Borup, S. Christensen, B. B. Iversen and G. J. Snyder, APL Mater., 2013, 1, 052107 CrossRef.
- C. Han, Q. Sun, Z. Li and S. X. Dou, Adv. Energy Mater., 2016, 6, 1600498 CrossRef.
- Y. Shi, C. Sturm and H. Kleinke, J. Solid State Chem., 2019, 270, 273–279 CrossRef CAS.
- T. Deng, T. Xing, M. K. Brod, Y. Sheng, P. Qiu, I. Veremchuk, Q. Song, T.-R. Wei, J. Yang, G. J. Snyder, Y. Grin, L. Chen and X. Shi, Energy Environ. Sci., 2020, 13, 3041–3053 RSC.
- J. Jiang, H. Zhu, Y. Niu, Q. Zhu, S. Song, T. Zhou, C. Wang and Z. Ren, J. Mater. Chem. A, 2020, 8, 4790–4799 RSC.
- M. M. Kubenova, K. A. Kuterbekov, M. K. Balapanov, R. K. Ishembetov, A. M. Kabyshev and K. Z. Bekmyrza, Nanomater., 2021, 11, 2238 CrossRef CAS PubMed.
- J. R.-F. Ilka Kriegel, A. Wisnet, H. Zhang, C. Waurisch, A. Eychmüller, A. Dubavik, A. O. Govorov and J. Feldmann, ACS Nano, 2013, 7, 4367–4377 CrossRef PubMed.
- B. Hamawandi, S. Ballikaya, M. Rasander, J. Halim, L. Vinciguerra, J. Rosen, M. Johnsson and M. Toprak, Nanomater, 2020, 10, 854 CrossRef CAS PubMed.
- T. Willhammar, K. Sentosun, S. Mourdikoudis, B. Goris, M. Kurttepeli, M. Bercx, D. Lamoen, B. Partoens, I. Pastoriza-Santos, J. Perez-Juste, L. M. Liz-Marzan, S. Bals and G. Van Tendeloo, Nat. Commun., 2017, 8, 14925 CrossRef CAS PubMed.
- X. Chen, J. Yang, T. Wu, L. Li, W. Luo, W. Jiang and L. Wang, Nanoscale, 2018, 10, 15130–15163 RSC.
- W. Li, A. Shavel, R. Guzman, J. Rubio-Garcia, C. Flox, J. Fan, D. Cadavid, M. Ibanez, J. Arbiol, J. R. Morante and A. Cabot, Chem. Commun., 2011, 47, 10332–10334 RSC.
- Y. Zhao and C. Burda, Energy Environ. Sci., 2012, 5, 5564–5576 RSC.
- C. Coughlan, M. Ibanez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- B. Yun, H. Zhu, J. Yuan, Q. Sun and Z. Li, J. Mater. Chem. B, 2020, 8, 4778–4812 RSC.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- B. Zhang, M. Wang, J. Ding, Y. Li, G. Cao, M. T. Bernards, Y. He and Y. Shi, J. CO2 Util., 2020, 39, 101169 CrossRef CAS.
- T. Dou, Y. Qin, F. Zhang and X. Lei, ACS Appl. Energy Mater., 2021, 4, 4376–4384 CrossRef CAS.
- J. W. Lim, W. J. Dong, J. Y. Park, D. M. Hong and J. L. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 22891–22900 CrossRef CAS PubMed.
- K. Sun, S. Song, A. Kumar, Z. Shang, X. Duan, S. Wang, S. Tian, J. Tang, D. Zhou and X. Sun, ACS Appl. Nano Mater., 2021, 4, 2357–2364 CrossRef CAS.
- Y. Mi, X. Peng, X. Liu and J. Luo, ACS Appl. Energy Mater., 2018, 1, 5119–5123 CAS.
- C. H. M. van Oversteeg, M. Tapia Rosales, K. H. Helfferich, M. Ghiasi, J. D. Meeldijk, N. J. Firet, P. Ngene, C. de Mello Donegá and P. E. de Jongh, Catal. Today, 2021, 377, 157–165 CrossRef CAS.
- J. Xu, W. Zhang, Z. Yang, S. Ding, C. Zeng, L. Chen, Q. Wang and S. Yang, Adv. Funct. Mater., 2009, 19, 1759–1766 CrossRef CAS.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS.
- F. Liu, R. Xu, L. Hong, Z. Yang, Y. Li, C. Wang, H. Jia and C. Yang, ChemistrySelect, 2020, 5, 360–368 CrossRef CAS.
- X. Zhang, R. Sa, F. Zhou, Y. Rui, R. Liu, Z. Wen and R. Wang, CCS Chem., 2021, 3, 199–207 CrossRef CAS.
- H. Yuan, Z. Liu, S. Sang and X. Wang, Appl. Surf. Sci., 2023, 613, 156130 CrossRef CAS.
- Z. Wu, J. Yu, K. Wu, J. Song, H. Gao, H. Shen, X. Xia, W. Lei and Q. Hao, Appl. Surf. Sci., 2022, 575, 151796 CrossRef CAS.
- X. Wang, J. Lv, J. Zhang, X. L. Wang, C. Xue, G. Bian, D. Li, Y. Wang and T. Wu, Nanoscale, 2020, 12, 772–784 RSC.
- W. Wang, Z. Wang, R. Yang, J. Duan, Y. Liu, A. Nie, H. Li, B. Y. Xia and T. Zhai, Angew. Chem., Int. Ed., 2021, 60, 22940–22947 CrossRef CAS PubMed.
- S. Wang, T. Kou, J. B. Varley, S. A. Akhade, S. E. Weitzner, S. E. Baker, E. B. Duoss and Y. Li, ACS Mater. Lett., 2020, 3, 100–109 CrossRef.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- V. S. S. Mosali, X. Zhang, Y. Liang, L. Li, G. Puxty, M. D. Horne, A. Brajter-Toth, A. M. Bond and J. Zhang, ChemSusChem, 2021, 14, 2924–2934 CrossRef CAS PubMed.
- J. Liu, P. Li, J. Bi, Y. Wang, Q. Zhu, X. Sun, J. Zhang, Z. Liu and B. Han, Chin. J. Chem., 2023, 41, 1443–1449 CrossRef CAS.
- Y. Li, Y. Chen, T. Chen, G. Shi, L. Zhu, Y. Sun and M. Yu, ACS Appl. Mater. Interfaces, 2023, 15, 18857–18866 CrossRef CAS PubMed.
- U. Legrand, R. Boudreault and J. L. Meunier, Electrochim. Acta, 2019, 318, 142–150 CrossRef CAS.
- Y. Gao, Y. Guo, Y. Zou, W. Liu, Y. Luo, B. Liu and C. Zhao, ACS Appl. Energy Mater., 2023, 6, 1340–1354 CrossRef CAS.
- H. Cheng, S. Liu, J. Zhang, T. Zhou, N. Zhang, X. S. Zheng, W. Chu, Z. Hu, C. Wu and Y. Xie, Nano Lett., 2020, 20, 6097–6103 CrossRef CAS PubMed.
- Y. T. J. Chen, Y. Zou, X. Li and J. Jiang, Mater. Lett., 2021, 284, 128919 CrossRef.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- A. Saxena, W. Liyanage, J. Masud, S. Kapila and M. Nath, J. Mater. Chem. A, 2021, 9, 7150–7161 RSC.
- J. Wang, X. Zheng, G. Wang, Y. Cao, W. Ding, J. Zhang, H. Wu, J. Ding, H. Hu, X. Han, T. Ma, Y. Deng and W. Hu, Adv. Mater., 2021, 34, e2106354 CrossRef PubMed.
- H. Zhang, T. Ouyang, J. Li, M. Mu and X. Yin, Electrochim. Acta, 2021, 390, 138766 CrossRef CAS.
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed.
- J. Duan, T. Liu, Y. Zhao, R. Yang, Y. Zhao, W. Wang, Y. Liu, H. Li, Y. Li and T. Zhai, Nat. Commun., 2022, 13, 2039 CrossRef CAS PubMed.
- Y. Hao, Y. Sun, H. Wang, J. Xue, J. Ren, A. A. S. Devi, M. Y. Maximov, F. Hu and S. Peng, Electrochim. Acta, 2023, 449, 142213 CrossRef CAS.
- Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 CrossRef CAS PubMed.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS PubMed.
- R. Elakkiya and G. Maduraiveeran, Sustain. Energy Fuels., 2021, 5, 6430–6440 RSC.
- C. Guo, Y. Guo, Y. Shi, X. Lan, Y. Wang, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202205909 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang and B. Han, Nat. Commun., 2022, 13, 1965 CrossRef CAS PubMed.
- A. W. Kahsay, K. B. Ibrahim, M.-C. Tsai, M. K. Birhanu, S. A. Chala, W.-N. Su and B.-J. Hwang, Catal. Lett., 2019, 149, 860–869 CrossRef CAS.
- W. He, I. Liberman, I. Rozenberg, R. Ifraemov and I. Hod, Angew. Chem., Int. Ed., 2020, 59, 8262–8269 CrossRef CAS PubMed.
- J. Li, J. Li, C. Dun, W. Chen, D. Zhang, J. Gu, J. J. Urban and J. W. Ager, RSC Adv., 2021, 11, 23948–23959 RSC.
- Z. Zhao and G. Lu, Chem. Sci., 2022, 13, 3880–3887 RSC.
- Z. Zhao and G. Lu, Adv. Energy Mater., 2023, 13, 2203138 CrossRef CAS.
- M.-J. Cheng, E. L. Clark, H. H. Pham, A. T. Bell and M. Head-Gordon, ACS Catal., 2016, 6, 7769–7777 CrossRef CAS.
- M. Karamad, V. Tripkovic and J. Rossmeisl, ACS Catal., 2014, 4, 2268–2273 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Norskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- C. Brea and G. Hu, J. Mater. Chem. A, 2022, 10, 10162–10170 RSC.
- Y. Li, G. Shi, T. Chen, L. Zhu, D. Yu, Y. Sun, F. Besenbacher and M. Yu, Appl. Catal., B, 2022, 305, 121080 CrossRef CAS.
- W. Sun, P. Wang, Y. Jiang, Z. Jiang, R. Long, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Adv. Mater., 2022, 34, e2207691 CrossRef PubMed.
- H. Wang, X. Bi, Y. Yan, Y. Zhao, Z. Yang, H. Ning and M. Wu, Adv. Funct. Mater., 2023, 33, 2214846 Search PubMed.
- X. Shao, X. Zhang, Y. Liu, J. Qiao, X.-D. Zhou, N. Xu, J. L. Malcombe, J. Yi and J. Zhang, J. Mater. Chem. A, 2021, 9, 2526–2559 RSC.
- S. Kumaravel, K. Karthick, P. Thiruvengetam, J. M. Johny, S. S. Sankar and S. Kundu, Inorg. Chem., 2020, 59, 11129–11141 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2023 |