
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A trick of the tail: computing the entropic contribution to the energetics of quinone-protein unbindung†
Jetmir
Haxhija
,
Felix
Guischard
and
Thorsten
Koslowski
 *
*
Institut für Physikalische Chemie, Universität Freiburg, Albertstrasse 21, 79104 Freiburg, Germany. E-mail: Thorsten.Koslowski@physchem.uni-freiburg.de
First published on 30th September 2023
Abstract
We estimate the entropic contributions to the free energy of quinone unbinding in bacterial and mitochondrial respiratory chains using molecular dynamics (MD) and Monte Carlo (MC) computer simulations. For a varying length of the isoprenoid side chain, MD simulations in lipid bilayers and in unpolar solvents are used to assess the dihedral angle distributions along the chain. These form the basis of a MC estimate of the number of molecular structures that do not exhibit steric self-overlap and that are confined to the bilayer. We obtain an entropy drive of TΔS = 1.4 kcal mol−1 for each isoprene unit, which in sum is comparable to the redox potential differences involved in respiratory chain electron transfer. We postulate an entropy-driven zipper for quinone unbinding and discuss it in the context of the bioenergetics and the structure of complex I, and we indicate possible consequences of our findings for MD-based free energy computations.
1. Introduction
The appropriately named ubiquinones play a central role in the respiratory chain of many organisms1–4 They can be reduced by accepting two electrons and two protons, with semiquinone as an intermediate. In the respiratory protein complex I, the redox potential difference between the NADH/NAD+ redox pair and a quinone/hydroquinone acceptor drives conformational changes that enable the complex to transfer protons against a proton gradient as the first step in the biosynthesis of ATP.5 Ubiquinone also acts as an electron shuttle between the respiratory complexes I, II and III and is the namesake of the Q cycle in the latter protein complex6Ubiquinones have 1,4-benzoquinone as a structural basis, which is substituted at the positions 5 and 6 by methoxy groups. These molecules carry an isoprenoid side chain at position 3 and a methyl group at position 2. Ubiquinones are classified according to the degree of polymerization of the side chain, n, as UQ n. In human mitochondria, UQ10 is operative as an electron and proton carrier, rodents have UQ9 and yeast UQ6.
Menaquinones are electron carriers used primarily by bacteria. Here, the methoxy groups attached to the positions 5 and 6 are replaced by a benzyl ring, cf.Fig. 1 (note that the number of atom 6 changes to 10). The introduction of methoxy groups makes the head group of the quinone slightly more demanding from a steric point of view, and also slightly more hydrophobic. Menaquinones are found in the inner membrane of bacteria and are involved in the transfer of electrons in cellular respiration and other metabolic processes. Examples include Bacillus subtilis, Streptococcus pneumoniae and Mycobacterium tuberculosis.
 | ||
| Fig. 1 Structure formulae of menaquinone and menaquinol, with the enumeration of atoms as used in the text. | ||
In photosynthesis, plastoquinones play a similar role. They are located in the thylakoid membrane of chloroplasts and are involved in the transfer of electrons from photosystem II to photosystem I. This electron transfer is part of the reactions that generate ATP and NADPH, which are used in the light-independent reactions of photosynthesis. Plastoquinones do not carry a methyl group in position 2, but a simple hydrogen. The two methoxy moieties become methyl groups.
Ubiquinone is involved in several human diseases and disorders. Q10 levels are often low in individuals with cardiovascular disease7,8 and diabetes,9 although cause and effect are not always clear. As ubiquinone is vital to the respiratory chain, deficiencies can lead to mitochondrial disorders such as mitochondrial encephalomyopathy.10
We identify the quinones by their chemical structure (menaquinone MK, ubiquinone UQ or plastoquinone PQ), state of reduction (the respective hydroquinones or quinoles are referenced as MHK, UHQ and PHQ) and the chain length, which will be added to the identifier as an integer. We focus on MKn with n = 4, 6 and 8 in the MD simulations described below, and on n = 2 to 10 in the MC approach.
In the last years considerable effort has been devoted to investigate the exact mechanism of the respiratory complex I. Mechanistically and structurally, the complex can be divided into two domains. In the hydrophilic part, electrons are transferred from NADH to FMN, which transfers them to ubiquinone via FeS clusters.11,12 In the membrane part, four protons are then translocalized to the periplasmatic side. The interplay of both mechanisms is referred to as proton coupled electron transfer, although proton pumping is still not fully understood. Due to the availability of high resolution structures and the advances in computing power, molecular dynamics simulations have been widely used to investigate the exact mechanism of respiratory complex I.13,14 Varying proposals for the proton–pumping mechanism have been reported.15–17 In combination with biochemical data the reduction of quinone actives a reaction cascade combined of electrostatic and conformational changes that lead to the proton transfer.16
Complementary to these large-scale MD simulations, we focus on a single aspect of the bioenergetics of quinones, the contribution of the conformational entropy of the isoprenoid side chain to the free energy of protein–hydroquinone unbinding. We consider the confining environment of a lipid bilayer an assume that the number of conformations in the quinone binding pocket of the protein is small compared to those within the layer. In this way, we are able to estimate a lower boundary to the entropy.
The remaining part of the article is organized as follows. In the next section we describe molecular dynamics simulations of menaquinones embedded in a lipid bilayer and in n-hexane. The resulting dihedral angle distributions of the menaquinones’ oligoisoprene chain and the concentration profiles are used as the input to a Monte Carlo microcanonical computation of the entropic contributions to ubiquinone unbinding. We discuss our findings and draw conclusions in the final section of this paper.
2. Molecular dynamics
2.1. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of POPC and POPE. The membrane was created with packmol-memgen. Each 146 × 146 × 150 Å simulation box contains, besides the menaquinol, 65361 water and 1932 lipid molecules. Additional simulations were performed in a uniform hydrophobic environment, where menaquinol-6 was centered in a 100 × 100 × 100 Å simulation box with 4100 n-hexane molecules as solvent using the packmol program. The partial charges for the different menaquinones were calculated using the RESP method on the R.E.D. server.21,22 The remaining missing menaquinone parameters were completed using the general AMBER force field version 2 (gaff2). For lipid and water molecules the lipid14 force field23 and the TIP3P model24 were used, respectively.
:1 ratio of POPC and POPE. The membrane was created with packmol-memgen. Each 146 × 146 × 150 Å simulation box contains, besides the menaquinol, 65361 water and 1932 lipid molecules. Additional simulations were performed in a uniform hydrophobic environment, where menaquinol-6 was centered in a 100 × 100 × 100 Å simulation box with 4100 n-hexane molecules as solvent using the packmol program. The partial charges for the different menaquinones were calculated using the RESP method on the R.E.D. server.21,22 The remaining missing menaquinone parameters were completed using the general AMBER force field version 2 (gaff2). For lipid and water molecules the lipid14 force field23 and the TIP3P model24 were used, respectively.
2.2. Results
From the molecular dynamics simulations, we obtain two main results. First, snapshots of the simulation permit the calculation of density profiles, which indicate the position of the benzoquinone head group in the membrane, which is also required for the microcanonical entropy estimates. In addition, the profiles already hint at the structure of the isoprenoid side chain and possible spatial constraints. Second, we get the distribution of the dihedral angles, which constitute the input of the microcanonical calculation of the entropy, as decribed below. A simulation snapshot is shown in Fig. 2.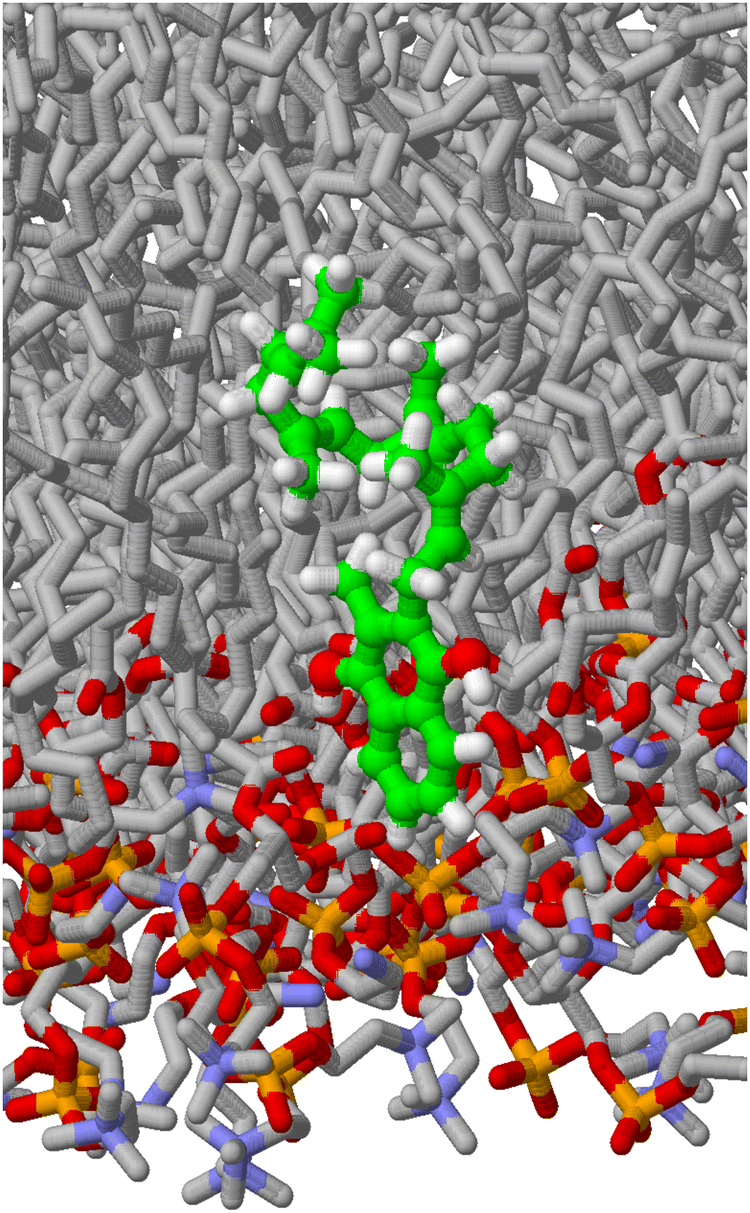 | ||
| Fig. 2 A snapshot of the molecular dynamics simulation of lipid molecules and a MHK4 menaquinol. MHK4 is shown as a ball and stick model with CPK coloring and green carbons, lipid molecules as stick models and CPK coloring, lipid hydrogens are not shown. Water molecules have been omitted for convenience. Horizontally, the figure extends to one half of the lipid bilayer. | ||
As described above, molecular dynamics simulations have been performed for MHK4, MHK6 and MHK8 in a lipid bilayer, which is embedded into an aqueous environment. To address the question to which extent geometrical constraints play a role, MHK6 was also simulated in n-hexane as a hydrophobic solvent. Test simulations have also been performed for the oxidized quinones, we find no significant differences to the hydroquinones.
Concentration profiles for the MHK8 menaquinol simulations within a lipid bilayer are shown in Fig. 3. The arrangement of lipid and water molecules shows the familiar picture of a lipid double layer in contact with the aqueous phase via the hydrophobic head groups and a hydrophobic core formed by a double layer of the hydrophobic lipid tails. For the quinone, we find a binodal profile. The first maximum – representing the quinone head group – is embedded in a neighbourhood of lipid molecule head groups and water molecules. The second, corresponding to the oligoisoprene chain, is slightly broader and is located ∼7 Å deeper within the bilayer, i.e. in a purely hydrophobic environment. It does not show any overlap with the water or the hydrophilic head group profile. As a consequence, we consider the isoprenoid side chain of MHK8 to be confined to the hydophobic core of the lipid bilayer. The side chain is attached to a naphtoquinone that is localized in an amphipilic environement between the head and the tail groups of the lipid. We will use this information as input to the Monte Carlo model described below. Throughout the simulations, no flips of the quinone molecules have been observed. The side chain profile is typical of large-chain polymers confined to two dimensions, where the probability distribution strongly peaks at the center.30
 | ||
| Fig. 3 Concentration profile of a molecular dynamics simulation of the MHK8 menaquinol embedded into a lipid bilayer and water environment. We display nonhydrogen atom counts as a function of the z coordinate, which is orthogonal to the bilayer. Water (blue), lipid head groups (green), lipid tail groups (black) and menaquinol (red). Symbols correspond to counts with histogram bins of 1 Å, the lines serve as a guide to the eye. Note the altered scale for the menaquinol profile. | ||
In the following, we focus on the dihedral angles of the isoprenoid chain beyond the first double bond. Within the accuracy of the simulations and the analysis, they reflect the periodicity of the chemical structure. Sample distributions are displayed in Fig. 4. The complete side chain distributions are supplied as ESI.† As expected, the conformational space of the molecules is not fully explored for all but the smallest side chains, an effect that has also been described for proteins.31 On the time scale of the simulation, phase space is not sampled properly, a phenomenon sometimes referred as ergodicity breaking. This behaviour is evident from the number of unique dihedral angle conformations as a function of the simulation time, as presented in the ESI.† This finding is part of our motivation for switching from molecular dynamics to Monte Carlo simulations.
 | ||
| Fig. 4 Distributions of the dihedral angles ϕadcb (a), ϕbadc (b) and ϕdcba (c) from the simulation of MHK6. The distributions are drawn as histograms with a binning of one degree, and the analysis has been performed for the second isoprene unit of the chain. | ||
The first dihedral angle, ϕdcba, always exhibits two maxima at ∼80 and ∼272 degrees. Regardless of the environment – lipid or n-hexane – an additional maximum shows up at zero degrees for MHK6 and MHK8, whichs is missing for MHK4. As the latter dihedral angle corresponds to a U turn of the chain, we assume that its introduction to a small chain is sterically prohibited, while it can evade this situation for longer chains. We are mainly interested in the energetics of the larger chains, and will consequently use all three dihedral angles ϕdcba ∈ {0,86,272} in the Monte Carlo setup. As the maxima are all of a similar height, we can safely assume that they belong to isoenergetic minima of the dihedral angle potential energy surface.
For ϕadcb, we find three well-separated maxima of the angle probability distribution at ∼62, ∼178 and ∼295 degrees with a dominant peak at 178 degrees. As the maxima exhibit different heights, we have to check whether this non-uniformity stems from a steric overlap that is also taken into account in the Monte Carlo model, or whether it has a different origin. As we will demonstrate below, the first assumption is valid, and we consistently recover the weights of the maxima in the Monte Carlo simulations. Hence, we can use all three dihedral angles, idealized as ϕadcb ∈ {60,180,300} in the microcanonical Monte Carlo setup with an equal weight.
For the dihedral angle ϕbadc, we observe two maxima at ∼80 and ∼272 degrees for MHK6 (lipid bilayer and n-hexane environment) and MHK8. Only for MHK4, an additional maximum around 180 degrees can be observed. For larger chains, vestiges can be found as a small nonzero contribution to the dihedral angle distribution, which renders the dominant parts asymmetric. Focussing on the larger chains, we can conservatively use idealized angles ϕbadc ∈ {90,270} in the Monte Carlo setup.
The dihedral angle ϕcbad is always fixed due to the presence of a double bond. For the same reason, any dihedral angle involving e is also fixed, such as ϕebad.
3. Monte Carlo simulations
3.1. Methods
Inspecting the degrees of freedom of an isoprenoid side chain, we find two fixed, well-defined dihedral angles per isoprene unit, which reflect the fact that no thermally acitivated rotations around double bonds exist. For each of the three remaining dihedral angles, either three (ϕdcba, ϕadcb) or two ϕbadc maxima in the probability distribution can be observed. To prevent immediate overlap with the benzoquinone moiety, we fix the first two dihedral angles at 60 and 180 degrees, and note that a variation of the final dihedral angle in the chain from one minimum in the potential energy to another does not lead to new entropically relevant conformations. With reference to the maxima of the distributions – or the minima of the free energy surface of dihedral angle rotation – we have 3 × 3 × 2 conformations per isoprene unit, leading to a total of m = 18n−1 conformations. For the largest biologically relevant n = 10, we have m ≃ 1011 individual conformations to be studied, a task clearly out the range of today's computational capabilities. As a consequence, we have to resort to an approximate sampling method, which also requires an approximate geometrical model of the molecule and its environment. We note that simplified dihedral angle rotational models, as described below, are frequently used in molecular modeling, e.g. in the widespread AutoDock Vina program32,33As a model for the molecule, we use a rigid benzoquinone as a basis to randomly grow a chain. Motivated by the localization patterns obtained from the MD simulations, we place the two oxygens in the x–y-plane, which we define as separating the hydrophilic head and the hydrophobic tail groups of the lipids. The minor axis of rotation is oriented along the z axis, with the methyl group and the chain pointing towards negative z values. This orientation is motivated by our analysis of the MD simulations for MHK4, where we find a direction cosine of the minor rotational axis of z/r = 0.92 ± 0.02. To grow the chain, we make use of standard single (1.54 Å) and double (1.34 Å) bond lengths and idealized sp2 and sp3 bond angles of 120 and 109.45 degrees, respectively. For each carbon atom added to the chain, the dihedral angle is either fixed (if involving a double bond) or randomly drawn from a set of two or three values. They stem from our analysis of the MD simulations and are – again idealized – given by ϕdcba ∈ {0,90,270} ϕadcb ∈ {60,180,300} and ϕbadc ∈ {90,270} degrees. The geometries are build making use of the xyzatm subroutine of the TINKER molecular modeling program package.34 Hydrogen positions are not generated explicitely, as we consider these atoms in the frame of a united atom approach. They are characterized by fused parameters with the carbon atoms, and they share their positions. For each fully grown chain we check for a steric overlap within the chain or of the chain and the benzoquinone. Optionally, we record whether a chain atom is located within a layer of −20 Å < z < 0, corresponding to a very conservative estimate of the size of the hydrophobic core of the lipid bilayer, cf.Fig. 3. As an overlap criterion, we take a united atom carbon–hydrogen van der Waals diameter of 3.5 Å, as for example used in the OPLS/UA force field.35 Overlaps up to and including 1–4 interactions are not taken into account.
We assume that all non-overlapping chains have the same energy. This implies that – within statistical inaccuracies – all dihedral angle distributions originating from the MD simulations have pronounced maxima of the same height (or that non-uniform distributions are self-consistently recovered), that the dihedral angles are uncorrelated and that details of the interactions do not play a significant role. In this regime, we have two energies, one that is identical for all non-overlapping conformations and can be set to the zero of the energy scale, and one that is infinite for all overlapping conformations. As a consequence, the system can be described thermodynamically by a microcanonical ensemble of Ω non-overlapping microstates, and the entropy is given by the Boltzmann expression S = kBlnΩ. Due to the large number of states, this expression can not be evaluated directly, but we have to resort to sampling a randomly generated subset. We make use of a simple hit-and-miss approach36 with mshot attempts to create a chain, which leads to mhit chains that are accepted w.r.t. to the criteria listed above. We can then estimate Ω ≃ m × mhit/mshot or
| lnΩ ≃ (n − 1) ln18 + lnmhit − lnmshot | (1) |
Self-overlapping configurations and those protruding out of the hydrophobic core of the lipid bilayer do not contribute to mhit, and we are confident that to reject them is correct, as none of these are encountered in the MD simulations, either.
A sample realization of a hit-and-miss algorithm is provided by Allen and Tildesley as a Fortran program (code 4.1.).36 Applications of hit-and-miss methods date back to Lazzarini's experimental approach to Buffon's needle problem, a geometrical problem related to the determination of the number π.37 Apocryphically, Ulam's idea to use a hit and miss method to estimate the number of decks that lead to a successful solution of the solitaire card game has initialized Monte Carlo methods on electronic computers.38
3.2. Results
For each chain length n, we have made mshot = 5 × 106 attempts to grow a chain. For chains that fulfill the requirement of being embedded in the hydrophilic core of the lipid bilayer, the numerical results are listed in Table 1. Entropies are converted to free energies at room temperature (298 K) and are given in kcal mol−1. We note that for small chains, all conformations can be generated deterministically, and we have mshot = m without the noise inherent to the Monte Carlo procedure.| n | m hit | −TS (kcal mol−1) |
|---|---|---|
| 2 | 2221080 | −1.49 |
| 3 | 955493 | −2.96 |
| 4 | 399675 | −4.42 |
| 5 | 149060 | −5.80 |
| 6 | 56240 | −7.19 |
| 7 | 20620 | −8.57 |
| 8 | 7757 | −9.96 |
| 9 | 2913 | −11.35 |
| 10 | 1129 | −12.76 |
A least squares linear regression analysis assuming −kBTS = a − bn leads to a = 1.24 ± 0.03 kcal mol−1 and b = 1.402 ± 0.005 kcal mol−1. As expected, the entropy vanishes for a chain consisting of a single isoprene unit, as we fix the first two and the final dihedral angle of each chain. As an entropic contribution to the free energy of quinone unbinding from a situation in which all dihedral angles are fixed, we observe a gain of 1.4 kcal mol−1 per isoprene unit. The analysis is shown in the ESI,† including the theoretical limit of a model that permits overlap and is not confined.
The confinement of the chain to the lipid bilayer has a small effect on the entropy. For n = 10 – the maximum chain length studied here – we observe mhit = 1129 out of 5 × 106 configurations that do neither exhibit collisional overlap nor protrude out of the hydrophilic core of the membrane. Dispensing with confinement to the membrane, we find mhit = 3340, which amounts to a free energy difference of 0.65 kcal mol−1, which is close to kBT at room temperature.
We recover the non-uniform distribution of the dihedral angle ϕadcb in the number of ideal dihedral angles that contribute to successful realizations of the isoprenoid random walk model. For n = 10, we have 1168 hits for ϕadcb = 90 degrees, 1142 hits for ϕadcb = 270 degrees and 7851 hits for ϕadcb = 180 degrees. We note that ϕbadc is symmetric within the limit of fluctuations with 5151 hits for 90 degrees and 5010 hits for 270 degrees. Notably, for ϕdcba a zero dihedral angle can be less frequently observed (2616 hits) than those of 90 and 270 degrees (3855 and 3690 hits, respectively). On the other hand, the MD simulations show ϕdcba dihedral angle maxima of roughly equal height. Thus, the MC model exhibits less U turns than the MD simulations, which suggests that overlap is slightly penalized too strong in our MC model.
4. Discussion and conclusions
We have presented a simple model to estimate the entropic contributions to the free energy of menaquinol unbinding. We assume that bound quinone tighly fits into a protein binding pocket with a single, unique distribution of dihedral angles and an accordingly vanishing entropy. Upon reduction and protonation, menaquinol is released into the lipid bilayer embedding the protein. We have investigated the latter situation using all-atom molecular dynamics simulations, which provide the input data for a Monte Carlo procedure via the distribution of dihedral angles of the quinol isoprenoid side chain and the position and orientation of the benzohydroquinone moiety. While there are many approaches to compute entropies from MD simulations,31 the procedure used here has been motivated by the sheer size of the conformational space (∼1011 states for n = 10), of which only a very small subset is scanned during a MD simulation. Furthermore, spatial constraints can be easily varied and implemented, and the computations are efficient and transparent in each step.Using a simple hit-and-miss algorithm, we have constructed and counted non-overlapping isoprenoid random walks confined to a lipid bilayer, thus counting the number of accessible dihedral angle microstates from a subset of all possible states. By the Boltzmann relation, we find a corresponding free energy release of 1.4 kcal mol−1 per isoprenoid unit.
In the following, this number is compared to other energy scales relevant to charge transfer in complex I, e.g. the redox potential, which is proportional to the thermodynamic driving force, −eΔE0 = ΔG0 = ΔH0 − TΔS, or to the strength of hydrogen bonds. From our calculations, we cannot provide a serious estimate of ΔH. This is due to the lack of an experimentally determined structure with a quinone localized within the protein binding pocket and its chemically specific interactions, and to the possible dissipation of energy along the electron transport chain.
In terms of the redox potential, the entropic contribution to quinone unbinding amounts to a substantial value of 61 mV per isoprene unit. Hence, quinone redox potentials in protein binding–unbinding reactions are not only affected by quinone–amino acid interactions, but also by a release of conformational entropy. Other factors may also play a role in reducing the number of conformations during the release of hydroquinones from a protein: the isoprenoid chain may overlap with atoms from the host protein, or other protein complexes from a hypothetical supercomplex. Consequently, our computations serve as an estimate of the entropy in a more complex environment.
The entropy contribution to the free energy generated by releasing menaquinoles is considerably larger than the redox potential differences of the redox pair of NAD+/NADH and MK/MHK. Although the exact numerical value of this difference in a complex biological environment is not yet known, it is save to assume that it is close to the corresponding difference in the standard potentials:39–41 we have an electronic driving force of ∼400 meV or ∼8 kcal mol−1, a value slightly smaller than the dihedral angle entropy of the MHK10 isoprenoid chain. This implies that the release of conformational entropy has to be coupled to an endergonic process in the vicinity of the MK binding pocket, which has to occur almost simultaneously. Breaking the two hydrogen bonds between the menaquinol hydroxyl groups and their coordinating Nqo4 amino acids amounts to an estimated 12 kcal mol∼, as we have a O⋯H–O bond to Tyr87 and a O⋯H–N bond to His38 with ∼7 and 5 kcal mol−1, respectively.42
There is, however, an additional angle, as the driving force is an entropic one. In each cycle of MK reduction, 2–3 protons are pumped against a pH gradient. These protons are in turn required to catalytically convert one ADP into an ATP, a reaction that stores ∼6 kcal mol−1 and that takes place in a different enzyme. It is difficult to imagine that ATP synthesis is accompanied by a similar entropic waste generated by the MHK release from its binding pocket. Consequently, the MHK release has not only to fulfill a free energy balance, as outlined above, but also an entropic one. This is subject to the same criteria, viz. those of a spatial proximity and a immediate coupling in the chain of chemical reactions or structural rearrangements.
In their determination of the structure of the entire complex I of Th. thermophilus, Baradaran et al. have noted an unusual feature of the MK binding funnel:13 it is lined with amino acids that have a charged or strongly dipolar character: they find His34, Arg216, Glu223, Glu225, Glu248, Tyr249, Arg294 and Arg299, in the Nqo8 subunit and His1, Arg36 and Asp62 in the Nqo6 subunit of the complex (cf. Fig. 4(d) of ref. 13). The latter amino acid is not resolved in the protein database (PDB) entry 4HEA. These amino acids may be of some assistance in transiently binding to the MHK head group in an energetically downhill cascade while leaving the binding site. Their interaction with the hydrophobic tail, however, can only be assumed as weak, unspecific and non-directional. Only upon the release of MHK, they are likely to form pairs or other aggregates with a reduced number of degrees of freedom, simultaneously compensating the monomer-by-monomer entropy production of an oligomer chain leaving its binding pocket. We refer to this model as an entropic zipper, and its essential elements are illustrated in Fig. 5 as a cartoon model. Verification or falsification of this hypothesis has to await structural models that compare vacant binding pockets to those populated by long-chain quinones or comparable inhibitors.
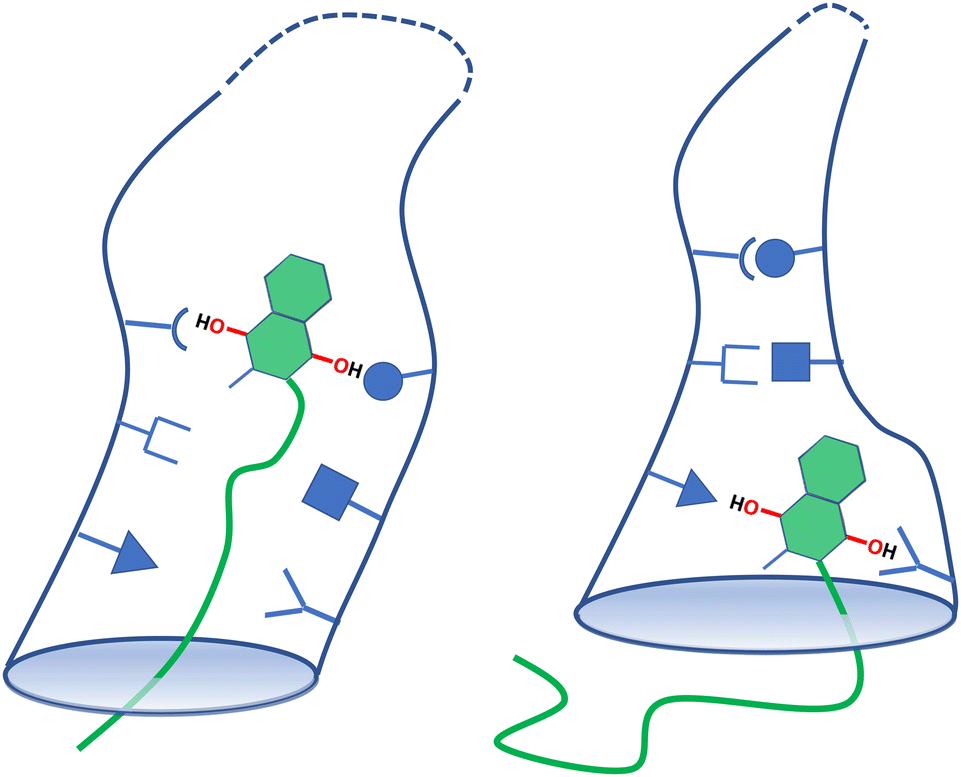 | ||
| Fig. 5 Cartoon model of the postulated entropy-driven zipper mechanism of menaquinol release from its binding site. Left: Early stage of unbinding with a constrained low entropy oligoisoprene chain and a high entropy binding pocket lining. Right: Late stage of unbinding with a free high entropy oligoisoprene chain and a low entropy binding pocket coating. | ||
Finally, we note that entropy – or heat – production is essential for endothermic organisms like mammals or birds, which may explain an evolutionary drive towards elongated isoprenoid chains for these classes of animals.
In molecular dynamics simulations, free energies can be computed by thermodynamic integration or umbrella sampling techniques. The latter requires the introduction of a potential – usually harmonic – to a subset of the atoms. In this way, an oligomer chain can be gradually pulled out of its binding pocket. It is crucial to select the atoms affected by the external potential in a way that retains the full flexibility of the chain upon unbinding to recover the full entropic contribution to the free energy. As an alternative, our computations may serve as a correction, e.g. for the unbinding of a stretched chain.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft via the grants DFG 278002225/RTG 2202 (JH, TK) and FOR 5099 (FG, TK). It is a pleasure to thank Thorsten Friedrich, Thorsten Hugel, Nora Kremer, Mike Castellano, Christine Koslowski and Bizan Balzer for many fruitful discussions and helpful comments.Notes and references
- H. Nohl, W. Jordan and R. J. Youngman, Quinones in Biology: Functions in electron transfer and oxygen activation, Adv. Free Radical Biol. Med., 1986, 2(1), 211–279 CrossRef CAS.
- U. Brandt, Energy Converting NADH:Quinone Oxidoreductase (Complex I), Annu. Rev. Biochem., 2006, 75, 69–92 CrossRef CAS PubMed.
- N. El-Najjar, H. Gali-Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, The chemical and biological activities of quinones: overview and implications in analytical detection, Phytochem. Rev., 2011, 10, 353–370 CrossRef CAS.
- C. S. Coates, J. Ziegler, K. Manz, J. Good, B. Kang and S. Milikisiyants, et al., The Structure and Function of Quinones in Biological Solar Energy Transduction: A Cyclic Voltammetry, EPR, and Hyperfine Sub-Level Correlation (HYSCORE) Spectroscopy Study of Model Naphthoquinones, J. Phys. Chem. B, 2013, 117(24), 7210–7220 CrossRef CAS PubMed.
- P. Mitchell, Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism, Nature, 1961, 191, 144–148 CrossRef CAS PubMed.
- P. Mitchell, The protonmotive Q cycle: a general formulation, FEBS Lett., 1975, 59, 137–139 CrossRef CAS PubMed.
- B. Sarter, Coenzyme Q10 and Cardiovascular Disease: A Review, J Cardiovasc Nurs., 2002, 16, 9–20 CrossRef PubMed.
- T. Rundek, A. Naini, R. Sacco, K. Coates and S. DiMauro, Atorvastatin Decreases the Coenzyme Q10 Level in the Blood of Patients at Risk for Cardiovascular Disease and Stroke, Arch. Neurol., 2004, 61(6), 889–892 CrossRef PubMed.
- D. Mantle, Coenzyme Q10 supplementation for diabetes and its complications: an overview, Br. J. Diabetes, 2017, 17, 145–148 CrossRef.
- S. DiMauro and M. Hirano, Mitochondrial encephalomyopathies: an update, Neuromuscular Disord., 2005, 15, 276–286 CrossRef PubMed.
- D. R. Martin and D. V. Matyushov, Electron-transfer chain in respiratory complex I, Sci. Rep., 2017, 7(1), 5495 CrossRef PubMed.
- S. Na, S. Jurkovic, T. Friedrich and T. Koslowski, Charge transfer through a fragment of the respiratory complex I and its regulation: an atomistic simulation approach, Phys. Chem. Chem. Phys., 2018, 20(30), 20023–20032 RSC.
- R. Baradaran, J. M. Berrisford, G. S. Minhas and L. A. Sazanov, Crystal structure of the entire respiratory complex I, Nature, 2013, 494(7438), 443–448 CrossRef CAS PubMed.
- R. G. Efremov, R. Baradaran and L. A. Sazanov, The architecture of respiratory complex I, Nature, 2010, 465(7297), 441–445 CrossRef CAS PubMed.
- V. R. Kaila, Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I, J. R. Soc., Interface, 2018, 15(141), 20170916 CrossRef PubMed.
- V. Sharma, G. Belevich, A. P. Gamiz-Hernandez, T. Róg, I. Vattulainen and M. L. Verkhovskaya, et al., Redox-induced activation of the proton pump in the respiratory complex I, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(37), 11571–11576 CrossRef CAS PubMed.
- K. Parey, O. Haapanen, V. Sharma, H. Köfeler, T. Züllig and S. Prinz, et al., High-resolution cryo-EM structures of respiratory complex I: Mechanism, assembly, and disease, Sci. Adv., 2019, 5(12), eaax9484 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. A. Ben-Shalom, J. T. Berryman and S. R. Brozell, et al., AMBER 2022, San Francisco, 2023 Search PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, An overview of the Amber biomolecular simulation package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3(2), 198–210 CAS.
- C. Tian, K. Kasavajhala, K. A. Belfon, L. Raguette, H. Huang and A. N. Migues, et al., ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution, J. Chem. Theory Comput., 2019, 16(1), 528–552 CrossRef PubMed.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak and J. C. Delepine, et al., RED Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments, Nucleic Acids Res., 2011, 39(suppl_2), W511–W517 CrossRef CAS PubMed.
- F. Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong and N. Grivel, et al., The REd. Tools: Advances in RESP and ESP charge derivation and force field library building, Phys. Chem. Chem. Phys., 2010, 12(28), 7821–7839 RSC.
- C. J. Dickson, B. D. Madej, Å. A. Skjevik, R. M. Betz, K. Teigen and I. R. Gould, et al., Lipid14: the amber lipid force field, J. Chem. Theory Comput., 2014, 10(2), 865–879 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. W. Götz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born, J. Chem. Theory Comput., 2012, 8(5), 1542–1555 CrossRef PubMed.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald, J. Chem. Theory Comput., 2013, 9(9), 3878–3888 CrossRef CAS PubMed.
- S. Le Grand, A. W. Götz and R. C. Walker, SPFP: Speed without compromise: A mixed precision model for GPU accelerated molecular dynamics simulations, Comput. Phys. Commun., 2013, 184(2), 374–380 CrossRef CAS.
- D. R. Roe and T. E. Cheatham, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data, Wiley Online Library, 2018 Search PubMed.
- A. Sikorski and P. Romiszowski, Computer simulations of polymers in a confined environment, J. Phys.: Condens. Matter, 2007, 19, 205136 CrossRef.
- S. Genheden and U. Ryde, Will molecular dynamics simulations of proteins ever reach equilibrium?, Phys. Chem. Chem. Phys., 2012, 14, 8662–8677 RSC.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- J. W. Ponder and D. A. Case, Force Fields for Protein Simulations, Protein Simulations, Academic Press, 2003, vol. 66 of Advances in Protein Chemistry, pp.27–85 Search PubMed.
- W. L. Jorgensen and J. Tirado-Rives, The OPLS Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- M. P. Allen and D. J. Tildesley, Computer simulation of liquids, Oxford University Press, Oxford, 2nd edn, 2017, p.147 Search PubMed.
- M. Aigner and G. M. Ziegler, Proofs from THE BOOK, Springer, Berlin, Heidelberg, 2018, pp.189–192 Search PubMed.
- M. Metropolis, The beginning of the Monte Carlo method, Los Alamos Science, 1987, Special Issue, pp.125–130 Search PubMed.
- M. Wikström and G. Hummer, Stoichiometry of proton translocation by respiratory complex I and its mechanistic implications, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4431–4436 CrossRef PubMed.
- F. S. Saleh, M. R. Rahman, T. Okajima, L. Mao and T. Ohsaka, Determination of formal potential of NADH/NAD+ redox couple and catalytic oxidation of NADH using poly(phenosafranin)-modified carbon electrodes, Bioelectrochemistry, 2011, 80, 121–127 CrossRef CAS PubMed.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, Quinone 1 e− and 2 e−/H+ Reduction Potentials: Identification and Analysis of Deviations from Systematic Scaling Relationships, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed.
- D. T. Haynie, Biological Thermodynamics, Cambridge University Press, 2001, ch. 2, p.37 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03466f |
| This journal is © the Owner Societies 2023 |