
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the nature of noble gas – metal bond in silver aggregates†
Celian
Courtney
 a and
Bruno
Siberchicot
*ab
a and
Bruno
Siberchicot
*ab
aCEA, DAM, DIF, F-91297, Arpajon CEDEX, France. E-mail: bruno.siberchicot@cea.fr
bCEA, Laboratoire Matière en Conditions Extrêmes, Université Paris-Saclay, F-91680, Bruyères-le-Châtel, France
First published on 23rd August 2023
Abstract
The aim of this paper is to extend the study of the nature of the bond between noble gas to nano- and sub nanoscale silver aggregates. In the framework of DFT-PAW calculations implemented in the abinit package, we carried out a thorough investigation on the nature of the bond between the six noble gases NG (He, Ne, Ar, Kr, Xe and Rn) and numerous neutral silver aggregates Agn from the single atom Ag1 to the nanoparticle Ag147 using atoms-in-molecules (AIM) dual functional analysis,. We evaluated the impact of the silver aggregate size, the adsorption site and of the noble gas on the Ag–NG bond. Our study concluded on the favored adsorption of heavier noble gases (Kr, Xe and Rn) over that of lighter noble gases (He, Ne and Ar) on any aggregate size due to an enhanced chemical contribution in the bond. For these heavier noble gases, in accordance with studies carried out on surfaces, we noted their preferential adsorption on on-top sites rather than on hollow sites, which further evidences the chemical contribution to the bond. Moreover, the slight positive Bader charge on these heavier noble gases implies an electron transfer from the noble gas to the silver atom. Noble gas adsorption is favored on smaller, few-atom, two-dimensional clusters rather than on larger three-dimensional nanoparticles. Finally, we identified a universal power law with a unique exponent linking bond length and electronic density at the bond critical point for all aggregate sizes, noble gases and adsorption sites.
Introduction
Since the discovery of the first xenon compound in 1962,1 many experimental and theoretical studies were carried out towards a better understanding of the bonding properties of noble gases. More specifically, the noble gas – transition metal bonding has been thoroughly studied. In 1977, Nieuwenhuys2 described the adsorption bond of xenon on metals as a “combination of a chemical (Charge-transfer-no-bond (CTNB)) interaction with a bonding by dispersion forces”. A few years later, in their study of Xe adsorption on palladium surfaces, Wandelt and Gumhalter3 proposed a model that incorporates both van der Waals and charge transfer effects in the Xe–Pd bond.In the case of noble gases adsorption on metallic surfaces, considering that van der Waals interactions play the main role, high-coordination sites were expected to be preferential adsorption sites because they maximize van der Waals interaction with the surface. Nonetheless, experimental data unanimously shows that some noble gases, such as xenon, tend to preferentially adsorb on low-coordination sites of metal surfaces.4,5 Müller and al.6 attributed the preferential interaction of Xe with the on-top sites of a Pt(111) surface to a hybridization of Xe 5p orbitals with the metal d states. A decade later, Clarke et al.7 rationalized the phenomenon studying the xenon adsorption on Ag(001). They identified the presence of a bonding interaction between the Xe 5p electrons and the Ag 4d electrons that originates from spin–orbit coupling of the Xe 5p states, inducing a drastic alteration of the electronic structure of the system. The adsorption on low-symmetry sites is hence favored because it introduces an extra splitting of the 5p3/2 orbitals to m = 3/2 and m = 1/2, due to a lowering of the local symmetry. A couple of years later, Da Silva et al.8 further demonstrated the on-top adsorption preference of Xe on metal surfaces using a full-potential linearized augmented plane wave method (FLAPAW); they concluded on the importance of polarization and on site-dependent Pauli repulsion to explain this site preference. Later on, other studies also reported an on-top site adsorption preference on several metal surfaces.9,10
In addition to the adsorption of noble gases on metallic surfaces, the noble gas–metal bond in few-atom complexes has been a great subject of study. As early as 1995, Pyykkö11 predicted and identified a chemical bond between noble gas atoms (He to Xe) and cationic gold Au+. From that point forward, many studies on the interaction of metallic cations M+ with noble gases were carried out. The interaction of neutral and anionic metals with noble gases has also been a subject of study.12–14 In 2013, Jamshidi et al.15 studied the nature of the interactions between noble gas atoms (Kr, Xe and Rn) and neutral, anionic and cationic coinage metals (Cu, Ag, Au). On the basis of several quantum chemical methods, they concluded on the existence of a dative bond due to a charge transfer from the noble gas to the coinage metal.
This bonding of intermediate strength, neither covalent/ionic nor van der Waals, is reminiscent of the aurophilic interaction described by Schimdbaur.16
Among other uses, noble metal-loaded material such as zeolites are used in noble gas separation processes.17–19 In these materials, the highly dispersed metallic phase acts as the active adsorption site. Thereby, noble gas bonding on smaller metallic systems encountered in these zeolites, such as cations, charged and neutral few-atom metallic clusters (2 to 6) and small nanoparticles have been studied20–22
In this study, we aim to further investigate the nature of the noble metal – noble gas bonding in several neutral Agn systems, with n ranging from 1 to 147. Focusing on silver, we present a systematic atoms in molecules (AIM) dual functional analysis of the bonding of silver with all six noble gases: He, Ne, Ar, Kr, Xe and Rn.
Theoretical approach
Clusters and nanoparticles
We selected nine different Agn aggregates (n = 1, 2, 3, 4, 5, 6, 13, 55 and 147). In the case of Ag147, we only considered Xe adsorption due to computer costs. For flat, two-dimensional clusters Ag2 to Ag4, we started from the geometries detailed in Yumura et al.20 on which we performed structural relaxation.Around five to six atoms, metallic clusters can start to organize in three dimensions; in that regard, we selected several two-dimensional and three-dimensional structures for Ag5 and Ag6 aggregates, as a few metastable structures coexist for these sizes.23 Structures of clusters are found in Fig. S1 (ESI†). Regarding nanoparticles, we decided to use the Mackay icosahedron geometry as a starting point for structural relaxation of Ag13, Ag55 and Ag147. One has to bear in mind that, for Ag13, the Mackay icosahedron geometry is one of the many shapes that a 13-atom agglomerate can take, and is not necessarily the most stable. However, we made the choice to impose this geometry since it has the added benefit of retaining the particle geometry through the particle size: each next cluster is built by adding an atomic layer, or shell, on top of the previous one.24 In addition, Nelli et al.25 reported the presence of inequivalent on-top sites in these magic-number icosahedral clusters. The number of inequivalent sites progressively increases with the shell size from shell 3 (55 atoms) upwards. The Ag13 nanoparticle contains a single type of five-fold coordinated on-top site and a single type of hollow site. The Ag55 nanoparticle contains two types of inequivalent on-top sites: the five-fold symmetry S5 Vertex site and the six-fold symmetry S6 Edge site; the Ag147 cluster contains an additional site, the six-fold symmetry Facet. Ag55 and Ag147 clusters also contain two inequivalent hollow sites, which, in adequacy with plane formalism, are referred to as FCC and HCP sites. In summary, Ag13 contains a single type of on-top site, akin to Vertex, and one type of hollow site, akin to FCC. Ag55 contains two types of on-top sites and two types of hollow-sites. Finally, Ag147 contains three types of on-top sites and two types of hollow sites. A schematic representation of the nanoparticles and their inequivalent adsorption sites is displayed on Fig. 1.
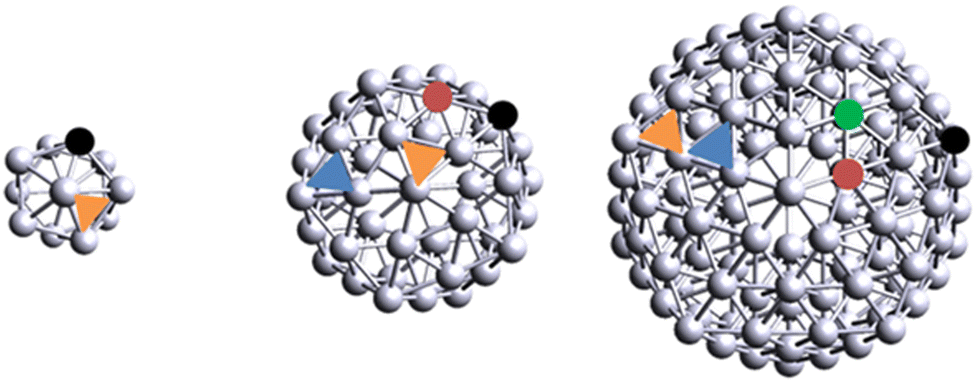 | ||
| Fig. 1 Schematic representation of Ag13, Ag55 and Ag147. Inequivalent adsorption sites are emphasized with colors. Black = Vertex, orange = FCC, red = Edge, blue = HCP and green = Facet. | ||
In both clusters and nanoparticles cases, we performed structural optimization and used periodic boundary conditions as implemented in the ABINIT package in the projector augmented wave (PAW) formalism.26,27 The pseudopotentials of silver and the six noble gases have been generated using the ATOMPAW code. The first-principles total energy calculations were performed with the Generalized Gradient Approximation Density Functional of Perdew, Burke and Ernzerhof (GGA-PBE).28 Spin orbit coupling (SOC) and the dispersion correction term according to the formalism of Grimme corrected by Becke and Johnson (DFT-D3 BJ)29,30 are both taken into account, as they are paramount to accurately describe the Ag–Noble gas bond. Several published studies have reported that the choice of the correct van der Waals correction functional for metallic systems is rather tricky. Most notably, it was reported that vdW-DFT-D2 approach was found to overestimate the dispersion interactions, sometimes leading to an erroneous adsorption site preference on metal surfaces.31 In our case, DFT-D3 BJ seems to be the best compromise. We neglected the effects of magnetism in our systems, in accordance with previous studies.32
First, the bare silver cluster is placed in a cubic box and relaxed. The periodic nature of ABINIT requires an essentially empty DFT mesh to effectively isolate the system from its periodic images. We adapted the size of the boxes with that of the system so that the vacuum space between adjacent replicas be at least 15 Å in each direction. The Brillouin zone was sampled with a k-point mesh of 27 points. The plane-wave expansion of the Bloch functions was truncated at an energy cutoff of 820 eV and the self-consistent convergence parameter on energy was set to 10−10 Ha. Forces were converged to 0.01 Ha bohr−1 for the atomic relaxations, so that we ensure that all forces are fully converged for all systems. Second, for each system in the presence of a noble gas atom, the potential energy surfaces were calculated by freezing the relaxed metal aggregates and varying the noble gas-silver distance rAg–NG between 2 and 5 Å. The electron density file at the local minimum was then used in quantum theory of atoms in molecules (QTAIM)33 calculation using the AIM software implemented in the ABINIT code.
Bader's quantum theory of atoms in molecules (QTAIM)
QTAIM is based on the topological analysis of the electron density (ED) ρ(r), wherein the space between atoms of a molecule or a crystal is divided in sub-systems called basins, separated from “zero-flux” surfaces for which the first derivative of the electron density is equal to zero:| ∇ρ(r)·n(r) = 0 | (1) |
The curvature of the electron density reveals critical points, which correspond to extrema defined by the sign of the second derivative of the ED at that critical point. The calculation of the Hessian matrix and its eigenvalues allows for the comprehensive definition of the curvature at these points. The three eigenvalues of the Hessian matrix λ1, λ2 and λ3 correspond to the electron density curvature and the Laplacian ∇2ρ(rc) is the the sum of the eigenvalues. The couple (ω, σ) defines each critical point, where ω is the rank and σ is the signature of the critical point. The rank is the number of non-zero eigenvalues and the signature is the algebraic sum of the sign of the eigenvalues. A couple of values (3,−1) is characteristic of a bond critical point (BCP), where ρ(r) is maximal in a plane defined by two negative eigenvalues λ1 and λ2, and minimal in the third axis, perpendicular to the plane, defined by the positive eigenvalue λ3. The critical point is located between the two nuclei and characterizes the bond.
The Laplacian ∇2ρ(rc) holds a very important role in the characterization of the chemical bond. Its sign indicates a charge concentration (∇2ρ(rc) < 0) or depletion (∇2ρ(rc) > 0), which allows a discrimination of chemical bonds in two groups: shared–shell (SS) interactions and closed–shell (CS) interactions. SS interactions are typical of covalent systems and are characterized by a high ρ(r) and ∇2ρ(rc) < 0, whereas CS interactions are typical in ionic or van der Waals bonds and hold a low ρ(r) and ∇2ρ(rc) > 0. These two cases act as limits; a whole spectrum of other types of bonds lies in between.34
Other parameters can be obtained from the electronic density and its Laplacian. The local form of the virial theorem allows the calculation of the electron energy densities at the bond critical point in reduced units (ħ/m = 1):
| 0.25 × ∇2ρ(rc) = 2 × Gb(rc) + Vb(rc) | (2) |
| Hb(rc) = Gb(rc) + Vb(rc) | (3) |
| Gb(rc) = 0.3 × (3π2)2/3·ρ(rc)5/3 + 1/6∇2ρ(rc) | (4) |
These topological parameters are also used to describe the nature of bonding: the balance of Gb(rc) and Vb(rc) reveals the nature of the interaction. For instance, |Vb(rc)| > Gb(rc), i.e. Hb(rc) < 0 characterizes a shared–shell interaction. Numerous publications have reported several criteria for describing the nature of bonding using these topological parameters. Among them, the dual functional analysis by plotting Hb(rc) as a function of ∇2ρ(rc) reported by36 is a very efficient tool in understanding the nature of bonding in systems such as ours. Likewise, it is convenient to use polar coordinates to characterize the nature of bonds. In this case, Hb(rc) must be plotted vs. Hb(rc) − Vb(rc)/2 instead of ∇2ρ(rc), with Hb(rc) − Vb(rc)/2 = 1/8 ∇2ρ(rc) so that both y- and x-axis units are energy units. Because of this transformation, the second plot is superimposed to the first one with the x-axis values divided by 8. In this plot, the couple of coordinates (R,θ) is used to discriminate bonds of different nature, where R and θ are defined as:
Hb(rc) − Vb(rc)/2 = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ sinθ | (5) |
| Hb(rc) = Rcosθ | (6) |
| R = [2 × (Hb(rc) − (Vb(rc)/4)2 + (Vb(rc)2)/8]0.5 | (7) |
For any type of bond, the value of θ is exclusively limited to the interval [45°:206.6°], which can be subdivided to categorize the bond. As appears on Fig. 2, a value of θ = [45°:90°] is typical of pure close–shell interactions, a value of θ = [90°:180°] is typical of regular close–shell interaction and a value of θ = [180°:206.6°] is typical of shared–shell interaction. The added benefit of using polar coordinates is the fact that θ increases monotonously from the weakest to the strongest types of bonds, whereas comparing absolute values of Hb(rc) can be confusing as it merges increase in positive values of Hb(rc) and decrease in negative values of Hb(rc), which do not have the same meaning.
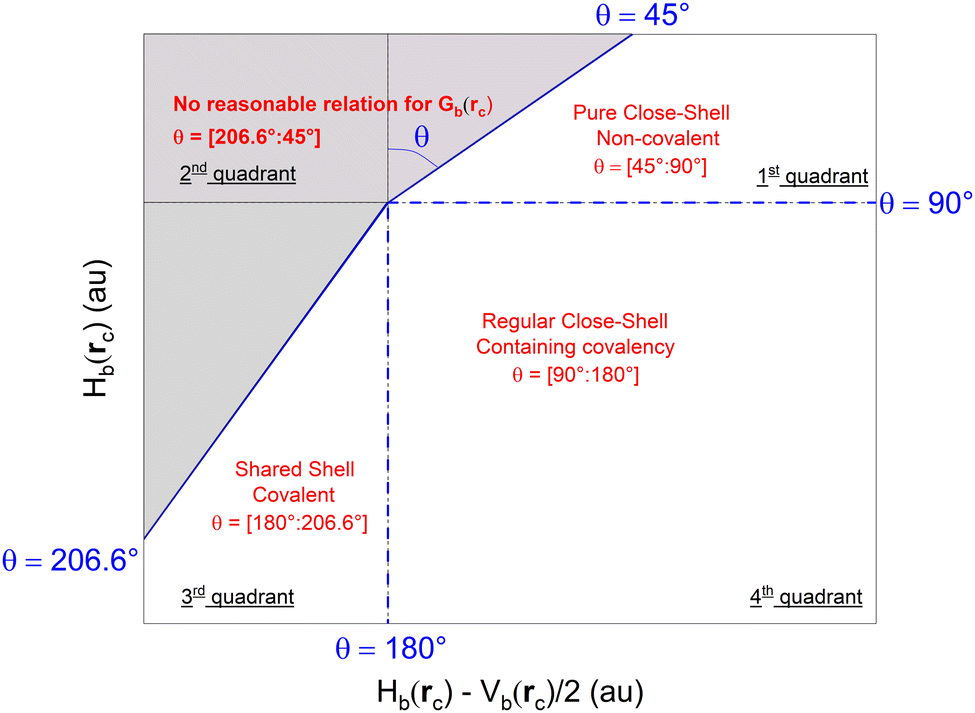 | ||
| Fig. 2 Classification of chemical bonds and interactions in the plot of Hb(rc) vs. Hb(rc) − V(rc)/2 and nature of the bond associated with each sub-range. No points can be found the gray zone. Adapted from ref. 36. | ||
Results and discussion
Fig. 3 presents the evolution of the total electron energy density Hb(rc) at the bond critical point between the rare gas atom and the silver atom involved in the bonding with the second derivative of the electronic density (Laplacian) at the bond critical point ∇2ρ(rc). All points organize along six streams, one for each noble gas. Values can be found in Tables S1 to S9 (ESI†). We can arbitrarily sort the studied Ag systems in two categories: atom & clusters (Ag, Ag2, Ag3, Ag4, Ag5 and Ag6) and nanoparticles (Ag13, Ag55 and Ag147).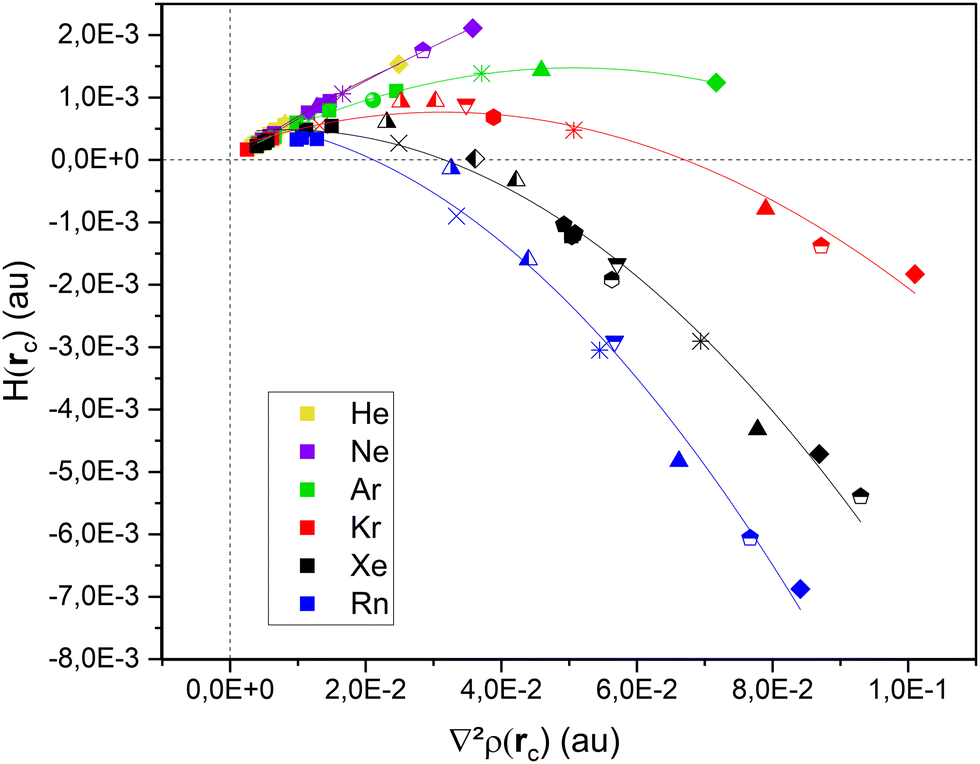 | ||
| Fig. 3 H b(rc) vs. ∇2ρ(rc) for Ag–NG bonding (NG = He → Rn) in Agn systems. Legend: cross (Ag1), double-cross (Ag2), solid triangle (Ag3), solid diamond (Ag4), solid pentagon (2D Ag5), half-colored pentagon (3D Ag5), solid hexagon (2D Ag6), half-colored hexagon (3D Ag6), down point half-colored triangle (Ag13 on-top), up point half-colored triangles (Ag55 Vertex and Edge), half-colored diamond (Ag147 Vertex). | ||
Trends in gases
For all six noble gases and regardless of aggregate size, Ag–NG bonding on hollow sites are pure close–shell, non-covalent interactions, as evidenced by their weak and positive value of Hb(rc) and ∇2ρ(rc) and θ ≈ 60°. On the contrary, the nature of Ag–NG bonding on the on-top sites is very dependent on the noble gas. For the three lightest noble gases (He, Ne and Ar), the Ag–NG bonding is purely close–shell. For the three heaviest noble gases (Kr, Xe and Rn), the Ag–NG bonding can be either pure close–shell or regular close–shell. These values are in accordance with the results of Jamshidi et al.37 who also observed positive values of ∇2ρ(rc) and negative values of Hb(rc) for Me2-NG (Me = Au, Ag, Cu; Rg = Kr, Xe, Rn)The fact that only Ag–NG bonding on on-top sites of nanoparticles displays a covalent contribution for the three heaviest noble gases corroborates the observations of Clarke et al.7 of on-top sites being favored over hollow sites for xenon adsorption. Indeed, spin–orbit coupling (SOC) on the noble gases p-states is a relativistic effect that appears in rather heavy elements. When plotting both the polarizability and the SOC constant (Fig. 4) for all six noble gases, a clear trend appears:
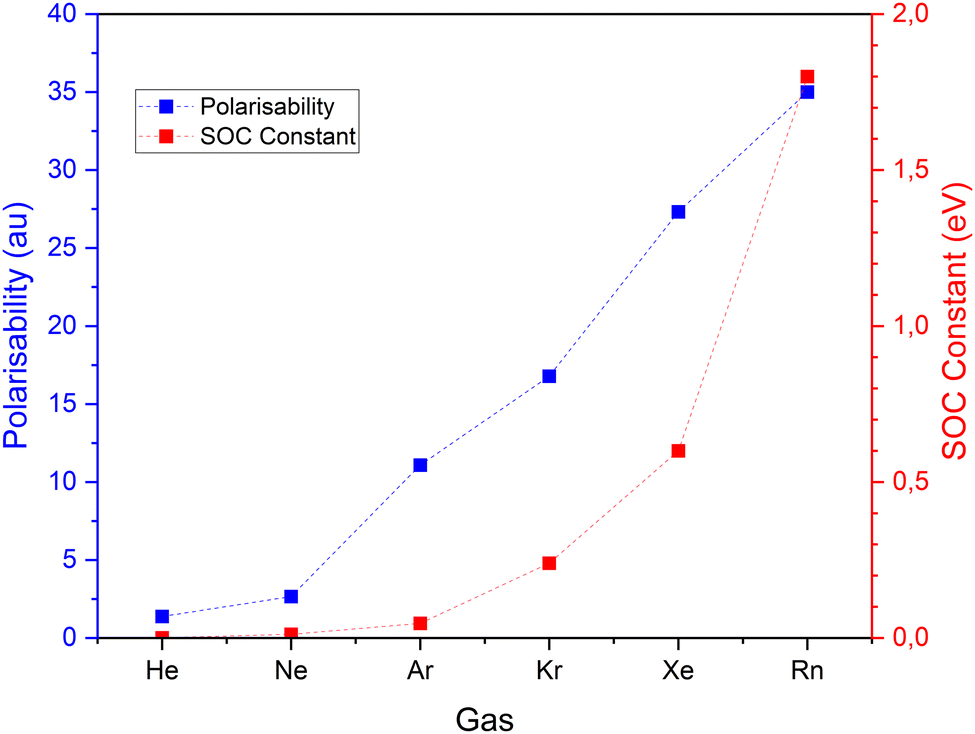 | ||
| Fig. 4 Polarizability and spin–orbit coupling (SOC) constant values for the six noble gases. Values of SOC constant from,41 value of polarizability from ref. 42 and 43. | ||
• In lighter noble gases, and more particularly helium, both polarizability and SOC constant are very low. There is no adsorption site preference.
• Going down the column, for neon and argon the polarizability increases while the SOC constant is close to zero. The supremacy of polarizability in the bond corroborates the preferential adsorption of these gases on hollow sites.
• For krypton and heavier noble gases, the polarizability increases but so does the SOC constant. For these gases, the growing contribution of the chemical effect of SOC induces an adsorption preference on on-top sites.
To sum up, as long as the SOC constant is close to zero, the main parameter governing the bond (adsorption site and bond length) is the polarizability. From Kr and beyond, the chemical contribution enhanced by SOC surpasses the polarizability effects. Then, the bond properties are characterized by the chemical contribution.
In addition, the theoretical Hb(rc) vs. ∇2ρ(rc) curve always presents an helical behavior.36 This implies a maximum value for Hb(rc) in the first quadrant. In the case of noble gas bonding with silver, this maximal Hb(rc) value is reached for neon, in accordance with its highest Mulliken electronegativity compared to other noble gases.38–40 Some peculiarities in neon bonding have also been suggested.49,50
To summarize, independently from site geometry and cluster size, the Ag–He, Ag–Ne and Ag–Ar bondings are always pure close–shell. For the three heaviest noble gases, the behavior is as follows:
• The Ag–Kr bonding is regular close–shell in Ag3 and Ag4.
• The Ag–Xe bonding is regular close–shell in the Ag2, Ag3, Ag4, 2D and 3D Ag5, 2D and 3D Ag6, the on-top site of Ag13 and the vertex site of Ag55.
• The Ag–Rn bond is regular close–shell in the single atom, Ag2, Ag3, Ag4, 2D and 3D Ag5, 2D and 3D Ag6 and all on-top sites of nanoparticles.
These conclusions are to be linked with the evolution of the Bader charge of the noble gas atom involved in the Ag–NG bond. In lighter noble gases (He, Ne and Ar), the Bader charge of the noble gas is close to zero and the Ag–NG bond in both on-top and hollow adsorption sites is pure close–shell. This describes a bond with little to no electron transfer, stabilized by dispersion forces. On the other hand, the Bader charge becomes slightly but increasingly positive for heavier noble gases as we go down the column. The slight positive value of the Bader charge indicates an electron transfer from the noble gas atom to the silver atom, in accordance with previous studies.11,37,44 In addition, for a given noble gas, the Bader charge is systematically larger by an order of magnitude when the noble gas is adsorbed on on-top sites compared to hollow sites.
Trends in adsorption sites on nanoparticles
As stated in the previous paragraphs, the adsorption on on-top sites is systematically favored for krypton, xenon and radon. However, the inequivalent nature of on-top sites induces an additional site preference between on-top sites. For a given gas (Kr, Xe or Rn) and a given nanoparticle size, the adsorption on the five-fold coordinated site is always favored over its six-fold coordinated counterparts (cf. Table S11, ESI†). This is evidenced by the increase in both θ and ∇2ρ(rc) values, which characterizes a stronger chemical contribution in the Ag–NG bonding, as shown in Fig. 2.This corroborates the conclusions of Clarke et al.,7 who associate the extent of the Ng p orbital splitting to a decrease in local symmetry of the adsorption site. In Ag147, a difference in adsorption preference is also observed for xenon between the two six-fold coordinated on-top sites Edge and Facet. Indeed, the xenon adsorption is slightly favored on the Edge site over the Facet site. As seen on Fig. 1, the Edge site of Ag147 is surrounded by atoms that are not in the same plane, whereas all six atoms that surround the Facet atom are coplanar. It appears that the decrease in symmetry due to the non-planarity of the neighboring atoms of the Edge site slightly enhances the bonding with the heavier noble gases.
When considering bigger, three-dimensional systems Ag13, Ag55 and Ag147, the nanoparticle surface curvature decreases with particle size. Thus, for an equivalent on-top adsorption site across sizes such as vertex, the breaking of symmetry is more significant in smaller particles. Moreover, the van der Waals interaction between an atom and a convex surface is known to decrease with its curvature.45 Both effects reduce the strength of the Ng–Ag bond as the particle size increases. This corroborates the decrease in θ and ∇2ρ(rc) values with the increase in nanoparticle size.
Trends in Ag system size
Fig. 5 shows the evolution of the θ value of the Ag–NG bonding with the size of the silver aggregate (note: for nanoparticles, only the adsorption on the five-fold coordinated on-top site is represented). All Ag–NG bonds are enclosed in a narrow range of 60° < θ < 125° and 0 au < ∇2ρ(rc) < 0.02 au. In small Ag systems (Ag to Ag4), θ and ∇2ρ(rc) increase with gas atomic number. For a given gas, the θ and ∇2ρ(rc) values increase with aggregate size from Ag1 to Ag4 but then decrease with aggregate size from Ag4 to Ag147. We hypothesize that the shift occurs when the silver aggregates start organizing in three dimensions, which can happen when the cluster contains five atoms or more. The use of polar coordinate greatly helps visualizing this shift, as evidenced by the cusp of the curves. To verify our hypothesis, we simulated the adsorption of noble gases on several 2D and 3D Ag5 and Ag6 clusters.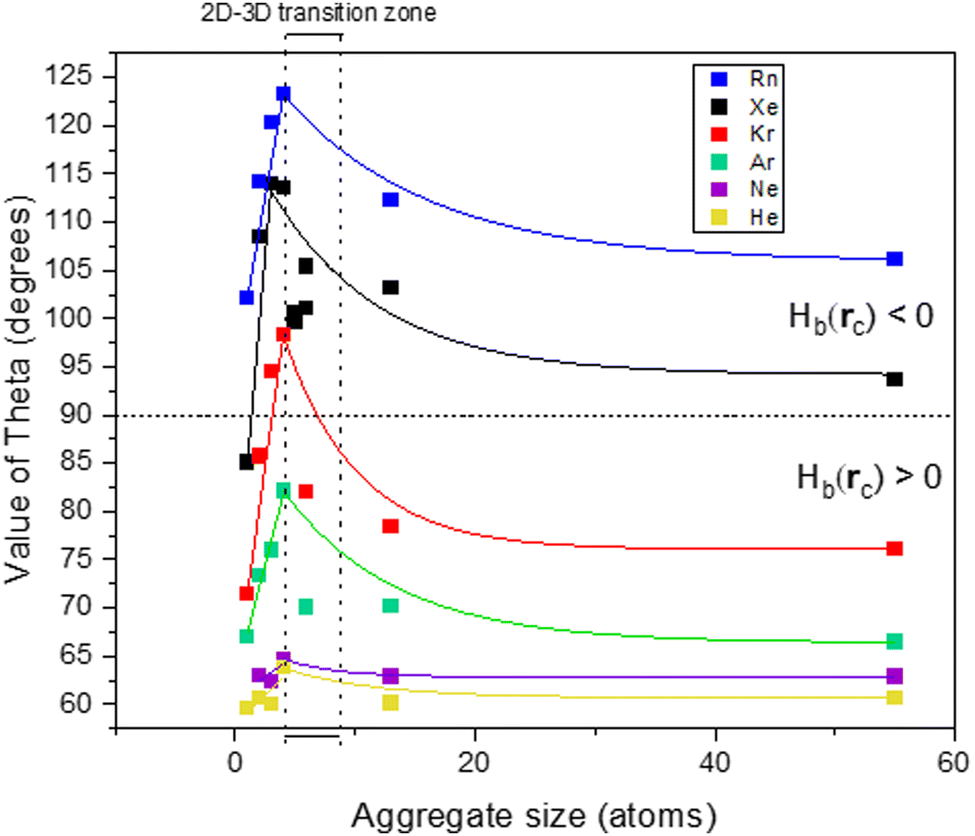 | ||
| Fig. 5 Evolution of the value of θ with aggregate size. For Ag13 and Ag55, adsorption on Vertex sites only. The horizontal dashed line marks the boundary between positive and negative values of Hb(rc). The dotted zone displays the number of atoms at which an aggregate can start to organize in three dimensions. Solid lines act as a visual aid. | ||
Independently from the gas, the value of θ increases up to its maximum for a flat Ag4 aggregate, then decreases with particle size. However, the decrease in θ is not monotonous for clusters of 5 atoms and more. For a given particle size, the θ value of Ag5–Xe and Ag6–Xe is different depending on the chosen geometry. One must bear in mind that clusters between 5 and 13 atoms can take on many geometries with close energies,23 and the choice of the geometry seems to affect the nature of the bond; what happens in the 2D–3D transition zone is rather ill defined.
Nevertheless, the maximal value of θ increases with the atomic number of the noble gas. Moreover, the difference in minimal and maximal value of θ for a single gas also tends to increase with noble gas atomic number.
To summarize, only noble gas adsorption on on-top sites can contain an amount of covalence, and it is dependent on both noble gas atomic number and Agn system size. The heaviest noble gas – radon – also has the highest spin–orbit coupling constant and polarizability values and its bonding with on-top sites displays a chemical contribution throughout all Agn systems. Going up the Ng column, xenon being lighter and thus having a lower SOC constant value has only some bonding with on-top sites that are regular close–shell. The binding of krypton on the on-top sites of nanoparticles is pure close–shell, but is regular close–shell on a few clusters, namely Ag3 and Ag4. A shift happens when clusters start to organize in three dimensions (i.e. five atoms and above), but the relationship between dimensionality and bond nature in that transition zone remains unclear.
Power-law relationship between bond length and electronic density
Power law relationships linking bond length and bond strength have been used as early as 1973.46 In 1987, Gibbs et al.47 studied the relationship between bond length and bond strength for the M–O bond in various oxide molecules and crystals. Ten years later,48 they expanded their study to consider the electronic density determined by Bader's AIM theory. Most interestingly, they demonstrated the existence of a power law liking bond length and electron density whose exponent was the same as the one of the power law linking bond strength and bond length. Their conclusions lead to think that, for a given bond, the value of that exponent is universal and characteristic of the nature of the bond.Following the same formalism, we plotted the bond length R against the electronic density at the bond critical point ρ(rc) for every noble gas, aggregate size and adsorption site. The scatter plot is reported in Fig. 6.
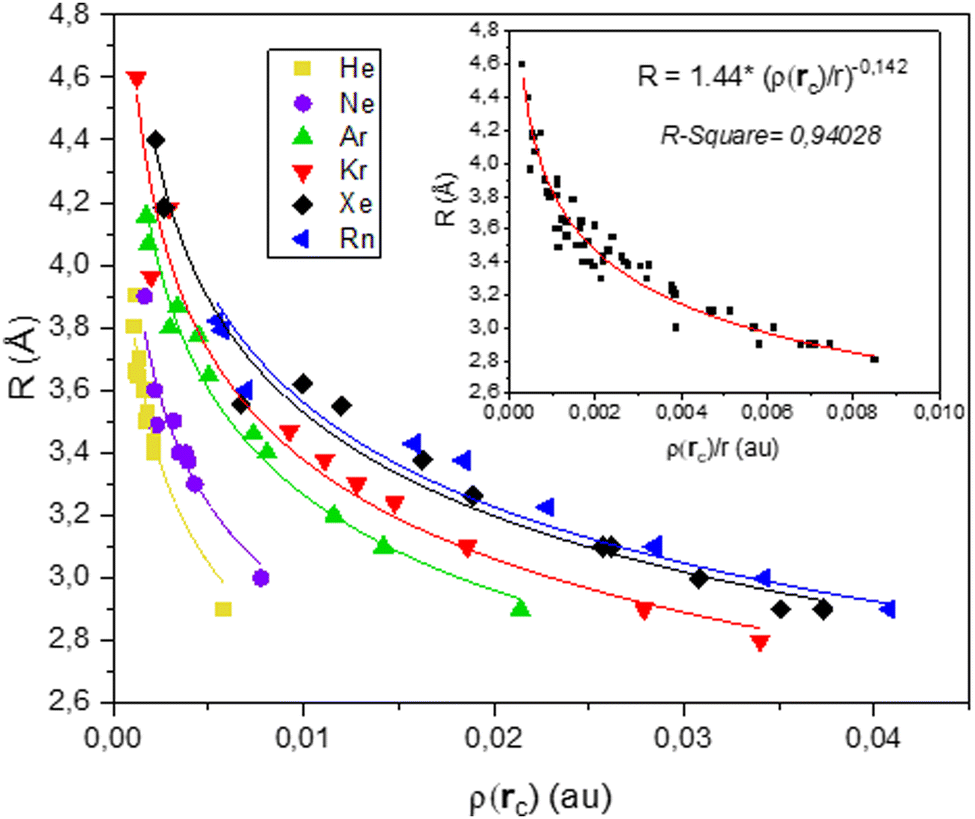 | ||
| Fig. 6 Plot of the Ag–NG bond length as a function of the electronic density at the bond critical point. Each noble gas is plotted separately and can be modelled with a power law R = R0·ρ(rc)−N. Inset: Plot of Ag–NG bond length as a function of ρ(rc)/r, with r the row number of the noble gas. | ||
Each Ag–NG bond is fitted with a power law of the form R = R0·ρ(rc)−N. The difference in the Ag–NG bond is expressed by a unique value of R0 for each noble gas, whose values are reported in Table S9 (ESI†). This emphasizes the differences in the Ag–NG bond from the lightest to the heaviest noble gas. On the other hand, a value of N = 0.142 offers a great fit for each single Ag–NG curve.
All six noble gases are brought together in a single curve when plotting the bond length R against the ratio of the electronic density at the bond critical point and the row number r of the noble gas ρ(rc)/r (see Fig. 6 inset). This emphasizes on the homogeneity of the silver-noble gas bond.
In accordance with what Gibbs et al. intuited a couple decades prior, there seems to exist a universal power law R = R0·ρ(rc)−N whose exponent solely depends on the nature of the bond. For the Ag–NG bond, this value of N is 0.142.
In summary, these results show that the Ag–NG bond is homogenous and the relation between bond length and electronic density can be expressed as a power law with an exponent term N = 0.142, common between all noble gases. The difference in silver bonding with each noble gas is evidenced by the unique value of R0, which increases with the gas atomic number. The value of the R0 parameter can be compared with that of θ, which also increases with the atomic gas number, all other things kept equal. A physical interpretation of that increase in R0 could be the growing covalent contribution found in the bonds between silver and the heaviest noble gases.
Conclusions
Based on DFT in the frame of the abinit package and using the Atoms in Molecules dual functional analysis, we were able to thoroughly study the nature of the bonding of the series of noble gases (He to Rn) with silver nanoaggregates of size ranging from a single atom to a 147-atom nanoparticle. We studied the influence of several parameters, namely that of the aggregate size, of the site of adsorption and of the noble gas.We come to the conclusion that the adsorption of heavier noble gases (Kr, Xe and Rn) is favored over the adsorption of lighter noble gases (He, Ne and Ar) on any aggregate size due to a chemical contribution enhanced by spin–orbit coupling (SOC). For these heavier noble gases, we also noted their preferential adsorption on on-top sites rather than on hollow sites, which further evidences the chemical contribution to the bond. On the contrary, lighter noble gases are adsorbed quasi-indifferently on on-top and hollow sites through a pure close–shell bond. Moreover, the positive Bader charge on these heavier noble gases implies an electron transfer from the noble gas to the silver atom. Finally, noble gas adsorption is favored on smaller, few-atom, two-dimensional clusters rather than on larger three-dimensional nanoparticles. It seems that the transition to three-dimensional structures induces a shift is some of the Ag–NG bond descriptors, such as the theta angle.
We also investigated the relation between the Ag–NG bond length and electronic density using the universal R = R0·ρ(rc)−N power law described by Gibbs et al. We found a unique value of the exponent N that offers a great fit for every noble gas regardless of aggregate size and adsorption site.
Author contributions
Celian Courtney: writing – original draft, methodology, investigation, formal analysis, data curation, conceptualization. Bruno Siberchicot: writing – review & editing, investigation, data curation, supervision, formal analysis, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jean Aupiais, Bruno Chaudret, Gabriel Couchaux, Sylvain Topin and David Farrusseng for the fruitful discussions on the subject of noble gas adsorption.Notes and references
- N. Bartlett, Proc. R. Chem. Soc., 1962, 218 CAS.
- B. E. Nieuwenhuys and W. M. H. Sachtler, J. Colloid Interface Sci., 1977, 58, 65–75 CrossRef CAS.
- K. Wandelt and B. Gumhalter, Surf. Sci., 1984, 140, 355–376 CrossRef CAS.
- T. Seyller, M. Caragiu, R. D. Diehl, P. Kaukasoina and M. Lindroos, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 11084–11088 CrossRef CAS.
- R. Pérez, F. J. Garcia-Vidal, P. L. de Andrés and F. Flores, Surf. Sci., 1994, 307, 704–709 CrossRef.
- J. E. Müller, Phys. Rev. Lett., 1990, 65, 3021–3024 CrossRef PubMed.
- S. Clarke, G. Bihlmayer and S. Blügel, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 085416 CrossRef.
- J. L. F. D. Silva, C. Stampfl and M. Scheffler, Phys. Rev. Lett., 2003, 90, 066104 CrossRef PubMed.
- P. L. Silvestrelli, A. Ambrosetti, S. Grubisiĉ and F. Ancilotto, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165405 CrossRef.
- P. L. Silvestrelli and A. Ambrosetti, J. Low Temp. Phys., 2016, 185, 183–197 CrossRef CAS.
- P. Pyykkoe, J. Am. Chem. Soc., 1995, 117, 2067–2070 CrossRef CAS.
- L. A. Mancera and D. M. Benoit, J. Phys. Chem. A, 2015, 119, 3075–3088 CrossRef CAS PubMed.
- D. Luo, Q. Yuan and L. Cheng, Chem. Phys., 2023, 565, 111743 CrossRef CAS.
- H. G. Nguyen, G. Konya, E. M. Eyring, D. B. Hunter and T. N. Truong, J. Phys. Chem. C, 2009, 113, 12818–12825 CrossRef CAS.
- Z. Jamshidi, K. Eskandari and S. M. Azami, Int. J. Quantum Chem., 2013, 113, 1981–1991 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- M. Aldener, A. Axelsson, T. Fritioff, J. Kastlander and A. Ringbom, J. Environ. Radioact., 2023, 262, 107159 CrossRef CAS PubMed.
- S. Topin, C. Greau, L. Delière, A. Hovesepian, T. Taffary, G. Le Petit, G. Douysset and C. Moulin, J. Environ. Radioact., 2015, 149, 43–50 CrossRef CAS.
- S. Topin, P. Gross, P. Achim, S. Generoso, A. Cagniant, O. Delaune, M. Morin, T. Philippe, J.-P. Fontaine, C. Moulin, G. Douysset and G. L. Petit, J. Environ. Radioact., 2020, 225, 106442 CrossRef CAS.
- T. Yumura, M. Kumondai, Y. Kuroda, T. Wakasugi and H. Kobayashi, RSC Adv., 2017, 7, 4950–4959 RSC.
- T. Yumura, T. Nanba, H. Torigoe, Y. Kuroda and H. Kobayashi, Inorg. Chem., 2011, 50, 6533–6542 CrossRef CAS PubMed.
- T. Yumura, H. Yamashita, H. Torigoe, H. Kobayashi and Y. Kuroda, Phys. Chem. Chem. Phys., 2010, 12, 2392–2400 RSC.
- R. Fournier, J. Chem. Phys., 2001, 115, 2165–2177 CrossRef CAS.
- M. J. Piotrowski, C. G. Ungureanu, P. Tereshchuk, K. E. A. Batista, A. S. Chaves, D. Guedes-Sobrinho and D. A. J. L. F. Silva, J. Phys. Chem. C, 2016, 120, 28844–28856 CrossRef CAS.
- D. Nelli, F. Petrucci and R. Ferrando, J. Chem. Phys., 2021, 155, 144304 CrossRef CAS PubMed.
- X. Gonze, B. Amadon, G. Antonius, F. Arnardi, L. Baguet, J.-M. Beuken, J. Bieder, F. Bottin, J. Bouchet, E. Bousquet, N. Brouwer, F. Bruneval, G. Brunin, T. Cavignac, J.-B. Charraud, W. Chen, M. Côté, S. Cottenier, J. Denier, G. Geneste, P. Ghosez, M. Giantomassi, Y. Gillet, O. Gingras, D. R. Hamann, G. Hautier, X. He, N. Helbig, N. Holzwarth, Y. Jia, F. Jollet, W. Lafargue-Dit-Hauret, K. Lejaeghere, M. A. L. Marques, A. Martin, C. Martins, H. P. C. Miranda, F. Naccarato, K. Persson, G. Petretto, V. Planes, Y. Pouillon, S. Prokhorenko, F. Ricci, G.-M. Rignanese, A. H. Romero, M. M. Schmitt, M. Torrent, J. van Setten, M. B. V. Troeye, M. J. Verstraete, G. Zérah and J. W. Zwanziger, Comput. Phys. Commun., 2020, 248, 107042 CrossRef CAS.
- M. Torrent, F. Jollet, F. Bottin, G. Zérah and X. Gonze, Comput. Mater. Sci., 2008, 42, 337–351 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed.
- B. Van Troeye, M. Torrent and X. Gonze, Phys. Rev. B, 2016, 93, 144304 CrossRef.
- D.-L. Chen, W. A. Al-Saidi and J. K. Johnson, J. Phys. Condens. Matter., 2012, 24, 424211 CrossRef PubMed.
- A. Monpezat, J. Aupiais and B. Siberchicot, ACS Omega, 2021, 6, 31513–31519 CrossRef CAS PubMed.
- R. Bader, Atoms in Molecules: A Quantum Theory, 1990.
- W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2009, 113, 10050–10057 CrossRef CAS PubMed.
- Y. A. Abramov, Acta Crystallogr., Sect. A: Found. Crystallogr., 1997, 53, 264–272 CrossRef.
- W. Nakanishi and S. Hayashi, Curr. Org. Chem., 2010, 14, 181–197 CrossRef CAS.
- Z. Jamshidi, M. F. Far and A. Maghari, J. Phys. Chem. A, 2012, 116, 12510–12517 CrossRef CAS PubMed.
- L. C. Allen, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
- S. G. Bratsch, J. Chem. Educ., 1988, 65, 34–41 CrossRef CAS.
- J. Furtado, F. De Proft and P. Geerlings, J. Phys. Chem. A, 2015, 119, 1339–1346 CrossRef CAS PubMed.
- W. C. Martin, J. Res. Natl. Bur. Stand., 1971, 75A(2), 109–111 CrossRef CAS PubMed.
- C. Hättig and B. A. Hess, J. Phys. Chem., 1996, 100, 6243–6248 CrossRef.
- J. E. Rice, P. R. Taylor, T. J. Lee and J. Almlöf, J. Chem. Phys., 1991, 94, 4972–4979 CrossRef CAS.
- A. M. Gardner, R. J. Plowright, M. J. Watkins, T. G. Wright and W. H. Breckenridge, J. Chem. Phys., 2010, 132, 184301 CrossRef.
- K. J. Schmidt and K. Christmann, Surf. Sci., 2001, 492, 167–184 CrossRef CAS.
- I. D. T. Brown and R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266–282 CrossRef CAS.
- G. V. Gibbs, L. W. Finger and M. B. Boisen, Phys. Chem. Miner., 1987, 14, 327–331 CrossRef CAS.
- G. V. Gibbs, F. C. Hill, M. B. Boisen and R. T. Downs, Phys. Chem. Miner., 1998, 25, 585–590 CrossRef CAS.
- F. Grandinetti, Nat. Chem., 2013, 5, 438 CrossRef CAS PubMed.
- W. Grochala, Found. Chem., 2018, 20, 191–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03416j |
| This journal is © the Owner Societies 2023 |