
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStepwise hydration of [CH3COOMg]+ studied by cold ion trap infrared spectroscopy: insights into interactions in the magnesium channel selection filters†
Hikaru
Takayanagi‡
ab,
Jean-Xavier
Bardaud§‡
 c,
Keisuke
Hirata
ad,
Valérie
Brenner¶
c,
Eric
Gloaguen§
*c,
Shun-ichi
Ishiuchi
*ad and
Masaaki
Fujii
*abe
c,
Keisuke
Hirata
ad,
Valérie
Brenner¶
c,
Eric
Gloaguen§
*c,
Shun-ichi
Ishiuchi
*ad and
Masaaki
Fujii
*abe
aLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan. E-mail: ishiuchi.s.aa@m.titech.ac.jp; mfujii@res.titech.ac.jp
bSchool of Life Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan
cLIDYL, CEA, CNRS, Université Paris Saclay, CEA Saclay, Bât 522, Gif-sur-Yvette 91191, France. E-mail: eric.gloaguen@cnrs.fr
dDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
eIRFI/IPWR, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan
First published on 18th August 2023
Abstract
The magnesium channel controls Mg2+ concentration in the cell and plays an indispensable role in biological functions. The crystal structure of the Magnesium Transport E channel suggested that Mg2+ hydrated by 6 water molecules is transported through a selection filter consisting of COO− groups on two Asp residues. This Mg2+ motion implies successive pairing with −OOC-R and dissociation mediated by water molecules. For another divalent ion, however, it is known that RCOO−⋯Ca2+ cannot be separated even with 12 water molecules. From this discrepancy, we probe the structure of Mg2+(CH3COO−)(H2O)4–17 clusters by measuring the infrared spectra and monitoring the vibrational frequencies of COO− with the help of quantum chemistry calculations. The hydration by (H2O)6 is not enough to induce ion separation, and partially-separated or separated pairs are formed from 10 water molecules at least. These results suggest that the ion separation between Mg2+ and carboxylate ions in the selection-filter of the MgtE channel not only results from water molecules in their first hydration shell, but also from additional factors including water molecules and protein groups in the second solvation shell of Mg2+.
1. Introduction
Ion channels are membrane proteins that selectively permeate specific ions and play an important role in biological systems.1–4 The mechanism of ion selectivity depends on the type of ion channel, but the simplest understanding is a competition between the ion affinity of the channel and the solvation of the ion. For example, in the case of potassium, the selective filter motif at the channel opening has several carbonyl groups, which are responsible for ion selection.1,2 This ion selection is achieved because the interaction energy of the carbonyl group with the potassium ion is slightly more favorable than the hydration energy of the potassium ion, whereas the sodium ion cannot enter the ion channel because its hydration energy is more favorable. Further detailed understanding at the molecular level has been gained by using X-ray diffraction,1,2 2D infrared spectroscopy,5 molecular dynamics simulations,6–8 and bottom-up approaches in which the interactions between parts of the selection filter and metal ions are studied by cold ion trap laser spectroscopy.9–12The magnesium channel is one of the ion channels that controls Mg2+ concentration in the cell.13–17 The magnesium ion is the most popular divalent ion in biological cells and is essential for life because of its role in biological functions, such as RNA and protein formation, catalysis by enzymes and so on.18 Recently, the crystal structure of Magnesium Transport E (MgtE), a type of Mg2+ channel, has been revealed by Nureki's group19–21 raising questions about Mg2+ transport mechanism and selectivity still under investigation.22 According to this report, the selection filter responsible for selectivity consists of two COO− groups on two Asp residues. This anionic moiety does not bind directly to Mg2+, but binds to the water molecule-clad Mg2+ in an octahedral arrangement, thus exerting ion selectivity. In the crystal structure analysis, 13 water molecules are found in the channel, but only 6 in the vicinity of Mg2+, which is interpreted as the transportation of Mg2+ hydrated by 6 water molecules, Mg2+(H2O)6, i.e. corresponding to a complete first hydration shell for this ion. The transportation of Mg2+ hydrated by its first solvation shell seems to be preferred over the transportation of the isolated divalent ion (see Fig. 1a).21 Indeed, the hydration shell appears necessary to avoid the strong interactions that can occur between the divalent ion and the channel,10 and contributes to smooth transport.
 | ||
| Fig. 1 (a) Mg2+(H2O)6 transportation through the –COO− selection filter and (b) full contact ion pair formation with 6 H2O. The former corresponds to separated ion pair of R–COO−Mg2+ by hydration (Solvent separated Ion Pair in solution chemistry). | ||
Here, we would like to raise a new viewpoint on Mg2+ transportation in MgtE. With a small number of water molecules, Mg2+ and R–COO− can bind to form a full contact ion pair because of their strong electrostatic interaction (see Fig. 1b), but such a contact has not been observed in the crystal structure.21 Instead, the transportation of Mg2+(H2O)6 through the –COO− selection filter involves two separated ion pairs between Mg2+ and two opposite carboxylate groups where ions are separated by hydration (see Fig. 1a). One may think that the second –COO− may help to keep such a centred position of Mg2+, however the presence of a two charge system never guarantees such an equilibrium at the centre of the two charges like Fig. 1a. In general, such a two-charge centre system may also lead to a double minimum potential, which is the reason why e.g. a lithium cation prefers to bind to one of the oxygen of the carboxylate group instead of making two equal interactions with the oxygen atoms (bidentate binding) in crystals23 as well as in solution.24 Such a double minimum potential would put the Mg2+ cation in one side, like in Fig. 1b.
Such solvent induced ion pair separation has been extensively studied by gas-phase laser spectroscopy and ab initio calculations on solvated clusters, such as Li+I− and Cs+I−,25 Mg2+HO−,26 Na+AcO−,27 Na+Cl−,28,29 Mg2+RCOO−,30 Ca2+RCOO−,30–32 Ba2+AcO−32 and LiCl2−.33 These studies show that 5 or 6 solvent molecules are barely enough to separate pairs made of monovalent ions. For divalent ions, however, no separation has been observed for the cluster size investigated. Recent results on RCOO−⋯Ca2+ ion pairs31 demonstrated that even 12 water molecules are not able to separate these ions. Although the chemical properties of Ca2+ and Mg2+ are not the same,34 both are divalent ions capable of binding strongly to RCOO−. Then, it is legitimate to wonder whether six water molecules are intrinsically enough to separate –COO− and Mg2+ or not. If not, additional factors must help to keep these ions separated in the channel as observed by XRD.21
In order to solve the discrepancy between Mg2+(H2O)6 transport in MgtE and the accumulated knowledge of ion pair separation by solvent molecules, we have applied cold ion trap spectroscopy35–39 to hydrated Mg2+(CH3COO−) clusters, which serve as minimum model systems of the selection filter in MgtE. Infrared spectra can sensitively probe the coordination and H-bonds in hydrated Mg2+(CH3COO−) clusters. With the help of quantum chemistry calculations, we can discuss whether a solvent-separated ion pair state which corresponds to the Mg2+(H2O)6 transport, is formed from Mg2+(CH3COO−) by hydration of six water molecules or not.
2. Methods
The experimental procedure is described in a previous article except for an additional quadruple ion mass spectrometer (Q-MS) before the first ion trap, which generate hydrated clusters (Fig. S1, ESI†). [CH3COOMg]+ is introduced into vacuum from electrospray (ESI) source (2.5 × 10−4 M in methanol solution). The ions are collected by ion funnel and introduced into the first Q-MS. The mass-selected ions are trapped by the first clustering ion trap40,41 at 130 K, in which water vapor and He buffer gas are filled. The hydrated ions, (CH3COOMg)+·(H2O)n are further mass-selected by the second Q-MS, and then are introduced into the cryogenic quadruple ion trap (QIT) at 4 K. The QIT was cooled to 4 K by a closed cycle He refrigerator. A buffer gas (He/H2 (20%)) was injected into the QIT via a pulsed nozzle, and H2 were able to attach the hydrated complex ions. Then, a tunable IR laser (OPO/OPA: LaserVision) was introduced into the ion trap and scanned over the range of CO stretching vibrational range (1300 to 1800 cm−1). When the frequency of the IR laser is resonant to vibrational levels of H2-tagged clusters, H2 is dissociated. Then, the infrared photodissociation (IRPD) spectra, which correspond to the IR absorption spectra, can be measured by monitoring the H2-lossed ion intensity as a function of the IR laser wavenumber (H2-tagging method). Here, the fragment ions were measured by time-of-flight mass spectrometer (TOF-MS), and ion signal is digitized and integrated in PC via a digital oscilloscope. Following the investigation of [CH3COOMg]+ (H2O)n (n = 1–5) by DePalma et al.,30 we measured the infrared spectra of [CH3COOMg]+ (H2O)n from n = 4 to 17, and discussed the change of ion pair solvation state by stepwise hydration with the help of theoretical calculations (see ESI†).We performed two steps of theoretical calculations. The first calculations are simpler one to estimate the vibrational signatures that indicate the solvation condition of Mg2+ from CH3COO−, such as full-contact ion pair state (bidentate ion pair), dissociated state (solvent-shared ion pair) and semi-dissociated state (monodentate ion pair) structures. For this purpose, IR spectra of (CH3COOMg)+·(H2O)6 are calculated by density functional theory (DFT) calculations at CAM-B3LYP-D3/6-311+G(d,p)42–45 level by using Gaussian 16. Optimized structures of three different bonding states are reported for (CH3COOMg)+·(H2O)6 by the DFT calculations without dispersion correction and the theoretical spectra are not shown.46 We build the three initial structures according to the previous work, and the geometries are optimized by the dispersion corrected DFT calculations. The calculations give several conformations for a single ionic condition (such as the dissociated state). The conformational difference is relatively the minor part in relation to the ionic condition, so that we obtained the theoretical IR spectra for three typical ionic conditions. The obtained harmonic vibrational frequencies were scaled by 0.985. The performance and limitation of this well-known approach used to compare experimental and theoretical frequencies can be found e.g. in Section 5 of ref. 47 and ref. therein.
The RI-B97-D3/dhf-TZVPP42,48,49 level of calculation (TURBOMOLE package50) was used for quantitative comparison with experimental results obtained on (CH3COOMg)+·(H2O)n clusters for n = 10 and 14. The potential energy surface of these systems being too complex for an exhaustive structural exploration, only a sample of selected structures was considered for geometry optimization (see ESI† for detail). These initial structures were either built from educated guesses, or randomly chosen among previously optimized structures of analogue [CH3COONa](H2O)n (n > 14) clusters24,51 by replacing Na+ by Mg2+ and removing water molecules in excess. These initial structures were optimized by the DFT calculations and vibrational frequencies were calculated at the same level. Optimized structures were sorted according to the type of ion pair formed, i.e. either bidentate or monodentate contact ion pairs, or solvent-shared ion pairs, thus defining three families of structures for each cluster size. Their Gibbs energy are reported on Fig. S2 (ESI†) and frequencies and intensities are reported on Tables S1–S6 (ESI†). Then, an averaged spectrum for each family and each size was obtained by averaging energy-weighted frequencies and intensities of each individual structures according to a procedure detailed in Section S4 (ESI†). Finally, an arbitrary spectral width of 6 cm−1 was chosen in order to model each vibrational transition by a Gaussian function, and present their sums as full theoretical spectra for comparison with the experimental ones.
3. Results and discussion
Prior to spectroscopic measurements, DFT calculations (CAM-B3LYP-D3/6-311+G(d,p)) were performed on [CH3COOMg]+·(H2O)6 to estimate the vibrational signatures of each type of ion pair (see Section S3 in ESI†). We optimized three typical structures of [CH3COOMg]+·(H2O)6 in which COO– binds to Mg2+ either as a full-contact ion pair state (bidentate), a partially-separated state (monodentate), or a separated state (no coordination). The obtained theoretical spectra and structures are shown in Fig. 2. In the full-contact ion pair state, from the low wavenumber side, each band is assigned to δCH, vcoo−s, vcoo−as and δHOH. However, in separated and partial-dissociated states, vcoo−s and vcoo−as are no more observed and replaced with two decoupled vco modes, one of which being observed at 1430–1450 cm−1 instead of ∼1510 cm−1 for vcoo−as in the full contact ion pair. Thus, a transition at 1510 cm−1 is a signature specific to bidentate ion pairs. The intensities of the vco modes at 1430–1450 cm−1 are enhanced significantly in the separated state and mildly in the partially-separated, and can thus be used to monitor the formation of these states. Similarly, the enhancement of vco near 1550 cm−1 can be an indicator of the separated state, although it may overlap with vcoo−as in the case of a mixture of different types of structures. From these expectations, we measured the infrared spectra of [CH3COOMg]+·(H2O)n. | ||
| Fig. 2 Calculated stick spectra of [CH3COOMg]+·(H2O)6 for full-contact, partially-separated and separated ion pairs with the IRPD spectrum of [CH3COOMg]+·(H2O)6. | ||
Fig. 3 shows the infrared spectra of [CH3COOMg]+·(H2O)n (n = 4–17). The spectra of n = 4 and 5 reproduce those of a previous report,30 and the spectral changes from n = 4 to n = 9 are relatively minor. These spectra can be assigned to the full-contact ion pair state according to our calculations. They are indeed consistent with the corresponding theoretical spectrum for n = 6 in Fig. 2, where the observed vibrational bands are assigned to δCH, vcoo−s, vcoo−as and δHOH as indicated in the figure. Among the minor changes in this series, the general blue shift of vcoo−as results from the motion away from the cation in full contact, induced by hydration by an increasing number of water molecules as already observed in the case of Ca2+.31,32 The blue shift of δHOH between n = 6 and n = 9 suggests the formation of the second hydrated shell from the analogy to the infrared spectra of [CD3CD2COOCa]+(H2O)n (n ≤ 12),31 in which the ion pair COO−⋯Ca2+ keeps the full contact structure. From these infrared spectra, we conclude that Mg2+ is in full contact with COO− in [CH3COOMg]+(H2O)6, and thus that the solvent-separated ion pair (see Fig. 1a), which is involved in the transportation of Mg2+(H2O)6 in MgtE in solution, is intrinsically not stable enough to be formed with only six, or even nine water molecules. In contrast, the XRD study21 clearly shows the presence of the Mg2+(H2O)6 of octahedral hydration structure separated from COO−. Then, the formation of the separated Mg2+(H2O)6 must be attributed to additional factors, such as the presence of the second COO− group and/or the second hydration shell. As the XRD study reported the presence of the 13 water molecules in the channel, the effect of the second hydration shell should be considered. Indeed, for Mg2+ in water, a priori 6 water molecules form the first shell and the next 12 molecules the second one.21 According to the Nureki's crystal structure, the second shell in the Mg2+ channel is filled by 7 water molecules and 4 oxygen atoms in the peptide framework, instead of 12 water molecules.
 | ||
| Fig. 3 Infrared photodissociation spectra of [CH3COOMg]+(H2O)n (n = 4–17). | ||
The effect of higher hydration can be explored by the infrared spectra in Fig. 3. The spectrum changes significantly when the 10th water molecule is added to the cluster (see Fig. 3 for the change from n = 9 to 10). In the δCH range at around 1430 cm−1, the intensity of one band (green) is significantly enhanced. In contrast, vcoo−s becomes weaker. Such a clear spectral change reveals a notable modification of the hydration condition of the (Mg2+⋯COO−) ion pair. This spectral feature keeps going up to n = 13, before another clear spectral change takes place at n = 14 where vcoo−s is no more observed, leaving onlyvco-like bands. If we take the simple analogy of the calculated structures for n = 6, the disappearance of vcoo−s from n = 13 to n = 14 strongly suggests the complete transition from full-contact ion pairs to partially-separated or separated ones. Similarly, the spectral change from n = 9 to n = 10 suggests that [CH3COOMg]+·(H2O)10–13 clusters are a mixture of full-contact ion pairs with partially-separated or separated ones.
These interpretations are further examined by more sophisticated simulations for two critical cluster sizes n = 10 and 14. By investigating a set of structures representative of the diversity encountered in these clusters, the simulated spectra enable us to study the influence of the partially-filled second hydration shell. These energy-weighted averaged spectra are presented in Fig. 4 (see ESI† for details). It should be noted that the labels of the vibrational bands in Fig. 2 and 3 are slightly different from Fig. 4 because the description of the vibrational motions in terms of internal coordinates changes with the size of the clusters. The former figures present calculated systems with only 6 water molecules, while the latter show an average of various conformations for n = 10 and 14 clusters. The correspondence between the assignments in Fig. 2 and 4 are explained in ESI.†
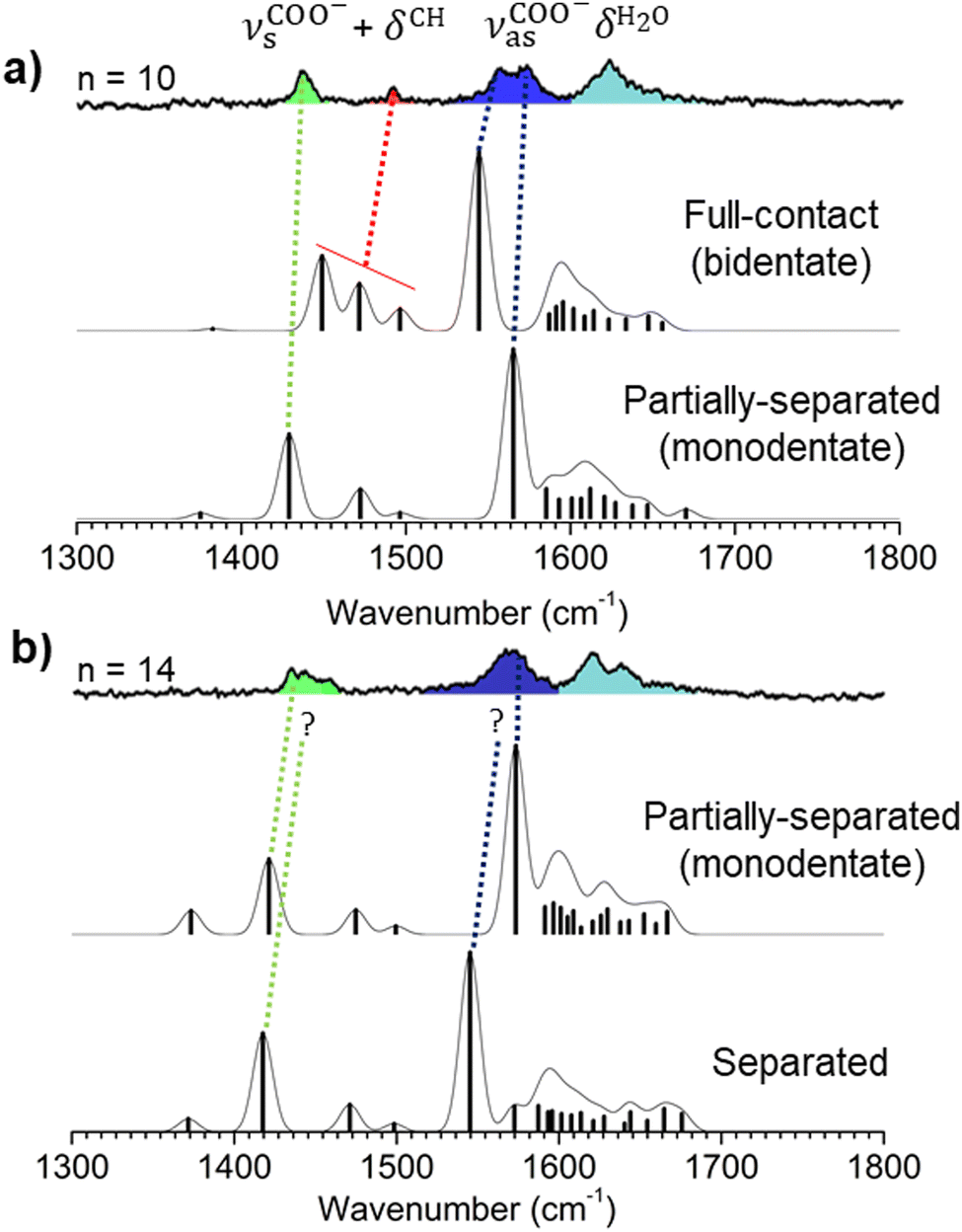 | ||
| Fig. 4 RI-B97-D3/dhf-TZVPP mode-dependent scaled harmonic vibrational spectra compared to IRPD spectra for n = 10 (a) and n = 14 (b) | ||
First, according to the theoretical spectra for n = 10 (Fig. 4a), the whole experimental spectrum is consistent with the coexistence of only two types of structures: (i) a full contact ion pair whose vibrational pattern can be continuously tracked from n = 4 (Fig. 3), the poor agreement near 1500 cm−1 only resulting from the insufficient description of the vcoo−s and δCH3 coupling30 by harmonic frequency calculations. (ii) a partially separated ion pair, which is assigned to the new spectral features popping up at n = 10, e.g. the blue-most component of the vcoo−as band (blue band) and the main red-most feature (green band) in the vcoo−s spectral region. In addition, both these ion pairs are isoenergetic (ESI†), supporting their simultaneous observation at this cluster size. In turn, separated ion pairs present the worst vcoo−a frequency and energy agreement (ESI†). For n = 14, the specific signature of the full contact ion pair disappears (red band in Fig. 3 and 4), in agreement with the significant increase in their relative energy compared to n = 10 (ESI†). In parallel, the triplet on the green band is most likely explained by the coexistence of several structures. According to calculated vcoo−as bands (Fig. 4b), partially separated ion pairs best agree with the experiment, suggesting that the IR spectrum results mainly from a mixture of several partially separated ion pairs. Alternatively, the appearance of separated ion pairs should be accompanied by a new red-shifted vcoo−as band, which is not observed. By comparing n = 10 and n = 14 spectra, however, a modest red-shift of vcoo−as (∼3 cm−1) is observed and could be interpreted as the manifestation of the presence of separated ion pairs as minor species.
4. Conclusions
Mg2+(H2O)6 transportation through RCOO− ion selection filters in MgtE corresponds to the separation of (RCOO−)2·Mg2+ ion pairs by 6 water molecules if we focus on the local structure. From accumulated knowledge on ion pair solvation, we pointed out that contact ion pairs in the channel could compete with separated ion pairs. This hydration-induced separation of RCOO−⋯Mg2+ and its dependence on the number of water molecules has been explored by cold ion trap IR laser spectroscopy in which the separation of the ion pair can be detected by the shift of IR frequencies for specific sizes of [CH3COOMg]+(H2O)n (n = 4–17) clusters. With the help of quantum chemistry calculations, the shifts of the vibrational bands in the 1300–1800 cm−1 range were interpreted along this series. The nature of the ion pair, i.e. full contact, partially-separated and separated, is monitored by following the vibrational frequencies and intensities of the CO2− stretch marker bands. No clear change of these vibrational bands has been observed from n = 4 to 9, which demonstrates that the hydration by six water molecules cannot disturb the strong interaction RCOO−⋯Mg2+. Consequently, additional factors other than these six water molecules have to be invoked to promote the transportation of Mg2+. Note that the presence of another RCOO− does not explain the central transportation path between the pair of RCOO− automatically because two charge center produces the double minimum potential in which Mg2+ ion have to be located at one of RCOO−, not at the center. The results suggest that the transportation of hexa-hydrated Mg2+ have to be rationalized not only by the interaction to the pair of RCOO− but also by the additional factors, such as the circulstance generated by other part of the channel and/or higher hydration. Considering additional water molecules in the second solvation shell of Mg2+, the change of the marker bands revealed significant structural changes upon hydration from n = 9 to n = 10, and n = 13 to n = 14. From the more sophisticated quantum chemistry calculations, the former change in the vibrational spectrum is rationalized by the formation of partially-separated ion pairs, while the latter is consistent with the disappearance of the full contact ion pair and the dominant formation of several partially-separated ion pairs together with minor separated ion pairs. It may suggest that the transportation of Mg2+ in MgtE, where separated ion pairs are found in XRD of the crystal, may require hydration by at least 14 water molecules. In other sense, Mg2+ would be transported without help of the circulstance generated by the whole protein if Mg2+ is hydrated enough. Interestingly, the XRD analysis21 of the channel shows 13 water molecules, which echo our conclusion. Overall, these results highlight the strength of our bottom-up approach using solvated ion pair clusters. They reveal the importance of various contributions of the molecular environment in Mg2+ transportation, which have not been discussed yet. The transportation of other divalent ions such as Ca2+ should also be studied with a similar approach in the future.Author contributions
E. G., S. I. and M. F. designed the project. H. T., K. H. and S. I. conducted experiment with simple calculations, whereas J. X. B., V. B. and E. G. performed sophisticated level calculations. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Michel Mons for his interest in this study and his advice. This study has been made possible thanks to the following grants: KAKENHI (JP20H00372, JP20K20446, JP21H04674, JP21K14585) and the Core-to-core program (JPJSCCA20210004) of JSPS, World Research Hub Initiatives in Tokyo Institute of Technology, the Cooperative Research Program of the ‘‘Network Joint Research Center for Materials and Devices’’ from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and the RIKEN Pioneering Project, ‘‘Fundamental Principles Underlying the Hierarchy of Matter: A Comprehensive Experimental Study’’; ANR grants IONPAIRS ANR-16-CE29-0017 and VAPOBIO ANR-20-CE29-0016. The computations were in part performed at the Research Center for Computational Science, Okazaki, Japan (21-IMS-C109).References
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS PubMed.
- Y. Zhou, J. H. Morais-Cabral, A. Kaufman and R. MacKinnon, Nature, 2001, 414, 43–48 CrossRef CAS PubMed.
- J. Payandeh, T. Scheuer, N. Zheng and W. A. Catterall, Nature, 2011, 475, 353–358 CrossRef CAS PubMed.
- L. Tang, T. M. Gamal El-Din, J. Payandeh, G. Q. Martinez, T. M. Heard, T. Scheuer, N. Zheng and W. A. Catterall, Nature, 2014, 505, 56–61 CrossRef PubMed.
- H. T. Kratochvil, J. K. Carr, K. Matulef, A. W. Annen, H. Li, M. Maj, J. Ostmeyer, A. L. Serrano, H. Raghuraman and S. D. Moran, et al. , Science, 2016, 353, 1040–1044 CrossRef CAS PubMed.
- S. Bernèche and B. Roux, Biophys. J., 2000, 78, 2900–2917 CrossRef.
- D. A. Köpfer, C. Song, T. Gruene, G. M. Sheldrick, U. Zachariae and B. L. de Groot, Science, 2014, 346, 352–355 CrossRef PubMed.
- W. Kopec, D. A. Köpfer, O. N. Vickery, A. S. Bondarenko, T. L. C. Jansen, B. L. de Groot and U. Zachariae, Nat. Chem., 2018, 10, 813–820 CrossRef CAS PubMed.
- S. Ishiuchi, Y. Sasaki, J. M. Lisy and M. Fujii, Phys. Chem. Chem. Phys., 2019, 21, 561–571 RSC.
- R. Otsuka, K. Hirata, Y. Sasaki, J. M. Lisy, S. Ishiuchi and M. Fujii, Chem. Phys. Chem., 2020, 21, 712–724 CrossRef CAS PubMed.
- T. Negoro, K. Hirata, J. M. Lisy, S. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2021, 23, 12045–12050 RSC.
- Y. Suzuki, K. Hirata, J. M. Lisy, S. Ishiuchi and M. Fujii, J. Phys. Chem. A, 2021, 125, 9609–9618 CrossRef CAS.
- J. Payandeh and E. F. Pai, EMBO J., 2006, 25, 3762–3773 CrossRef CAS.
- S. Eshaghi, D. Niegowski, A. Kohl, D. M. Molina, S. A. Lesley and P. Nordlund, Science, 2006, 313, 354–357 CrossRef CAS PubMed.
- V. V. Lunin, E. Dobrovetsky, G. Khutoreskaya, R. Zhang, A. Joachimiak, D. A. Doyle, A. Bochkarev, M. E. Maguire, A. M. Edwards and C. M. Koth, Nature, 2006, 440, 833–837 CrossRef CAS PubMed.
- A. Guskov, N. Nordin, A. Reynaud, H. Engman, A.-K. Lundbäck, O. Jong Agnes Jin, T. Cornvik, T. Phua and S. Eshaghi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18459–18464 CrossRef CAS PubMed.
- Y. Huang, F. Jin, Y. Funato, Z. Xu, W. Zhu, J. Wang, M. Sun, Y. Zhao, Y. Yu and H. Miki, et al. , Sci. Adv., 2021, 7, eabe6140 CrossRef CAS.
- M. Konrad, K. P. Schlingmann and T. Gudermann, Am. J. Phys. Renal. Phys., 2004, 286, F599–F605 CAS.
- M. Hattori, Y. Tanaka, S. Fukai, R. Ishitani and O. Nureki, Nature, 2007, 448, 1072–1075 CrossRef CAS PubMed.
- M. Hattori, N. Iwase, N. Furuya, Y. Tanaka, T. Tsukazaki, R. Ishitani, M. E. Maguire, K. Ito, A. Maturana and O. Nureki, EMBO J., 2009, 28, 3602–3612 CrossRef CAS PubMed.
- H. Takeda, M. Hattori, T. Nishizawa, K. Yamashita, S. T. A. Shah, M. Caffrey, A. D. Maturana, R. Ishitani and O. Nureki, Nat. Commun., 2014, 5, 5374 CrossRef CAS PubMed.
- X. Teng, D. Sheng, J. Wang, Y. Yu and M. Hattori, iScience, 2022, 25, 105565 CrossRef CAS PubMed.
- F. J. Martínez Casado, M. Ramos Riesco, M. I. Redondo, D. Choquesillo-Lazarte, S. López-Andrés and J. A. R. Cheda, Cryst. Growth Des., 2011, 11, 1021–1032 CrossRef.
- J. Donon, S. Habka, T. Very, F. Charnay-Pouget, M. Mons, D. J. Aitken, V. Brenner and E. Gloaguen, Chem. Phys. Chem., 2021, 22, 2442–2455 CrossRef CAS PubMed.
- R.-Z. Li, C.-W. Liu, Y. Q. Gao, H. Jiang, H.-G. Xu and W.-J. Zheng, J. Am. Chem. Soc., 2013, 135, 5190–5199 CrossRef CAS PubMed.
- C. J. Johnson, L. C. Dzugan, A. B. Wolk, C. M. Leavitt, J. A. Fournier, A. B. McCoy and M. A. Johnson, J. Phys. Chem. A, 2014, 118, 7590–7597 CrossRef CAS PubMed.
- W.-J. Zhang, G.-L. Hou, P. Wang, H.-G. Xu, G. Feng, X.-L. Xu and W.-J. Zheng, J. Chem. Phys., 2015, 143, 054302 CrossRef.
- G.-L. Hou, C.-W. Liu, R.-Z. Li, H.-G. Xu, Y. Q. Gao and W.-J. Zheng, J. Phys. Chem. Lett., 2017, 8, 13–20 CrossRef CAS.
- A. M. Sadoon, G. Sarma, E. M. Cunningham, J. Tandy, M. W. D. Hanson-Heine, N. A. Besley, S. Yang and A. M. Ellis, J. Phys. Chem. A, 2016, 120, 8085–8092 CrossRef CAS PubMed.
- J. W. DePalma, P. J. Kelleher, L. C. Tavares and M. A. Johnson, J. Phys. Chem. Lett., 2017, 8, 484–488 CrossRef CAS PubMed.
- J. K. Denton, P. J. Kelleher, M. A. Johnson, M. D. Baer, S. M. Kathmann, C. J. Mundy, R. B. A. Wellen, H. C. Allen, T. H. Choi and K. D. Jordan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14874–14880 CrossRef CAS PubMed.
- J. Donon, J.-X. Bardaud, V. Brenner, S. Ishiuchi, M. Fujii and E. Gloaguen, Phys. Chem. Chem. Phys., 2022, 24, 12121–12125 RSC.
- A. Chakraborty, S. Schmahl and K. R. Asmis, Chem. Phys. Chem., 2021, 22, 1036–1041 CrossRef CAS PubMed.
- K. D. Collins, Biophys. Chem., 2006, 119, 271–281 CrossRef CAS.
- M. Z. Kamrath, E. Garand, P. A. Jordan, C. M. Leavitt, A. B. Wolk, M. J. Van Stipdonk, S. J. Miller and M. A. Johnson, J. Am. Chem. Soc., 2011, 133, 6440–6448 CrossRef CAS PubMed.
- N. S. Nagornova, T. R. Rizzo and O. V. Boyarkin, Science, 2012, 336, 320–323 CrossRef CAS PubMed.
- J. Jašík, J. Žabka, J. Roithová and D. Gerlich, Int. J. Mass Spectrom., 2013, 354–355, 204–210 CrossRef.
- Y. Inokuchi, K. Soga, K. Hirai, M. Kida, F. Morishima and T. Ebata, J. Phys. Chem. A, 2015, 119, 8512–8518 CrossRef CAS PubMed.
- S. Ishiuchi, H. Wako, D. Kato and M. Fujii, J. Mol. Spectrosc., 2017, 332, 45–51 CrossRef CAS.
- B. M. Marsh, J. M. Voss and E. Garand, J. Chem. Phys., 2015, 143, 204201 CrossRef PubMed.
- E. Sato, K. Hirata, J. M. Lisy, S. Ishiuchi and M. Fujii, J. Phys. Chem. Lett., 2021, 12, 1754–1758 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 2008, 72, 650–654 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 2008, 72, 5639–5648 CrossRef.
- J. Paterová, J. Heyda, P. Jungwirth, C. J. Shaffer, Á. Révész, E. L. Zins and D. Schröder, J. Phys. Chem. A, 2011, 115, 6813–6819 CrossRef PubMed.
- E. Gloaguen, M. Mons, K. Schwing and M. Gerhards, Chem. Rev., 2020, 120, 12490–12562 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. Habka, T. Very, J. Donon, V. Vaquero-Vara, B. Tardivel, F. Charnay-Pouget, M. Mons, D. J. Aitken, V. Brenner and E. Gloaguen, Phys. Chem. Chem. Phys., 2019, 21, 12798–12805 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental Methods; Theoretical methods; Structures. See DOI: https://doi.org/10.1039/d3cp00992k |
| ‡ Contributed equally. |
| § Present address: Université Paris-Saclay, CNRS, ISMO, 91400 Orsay, France. |
| ¶ Present address: Direction de la recherche fondamentale, Commissariat à l'énergie atomique et aux énergies alternatives, CEA Saclay – 91191 Gif-sur-Yvette, Bât 530 – p 319B, France. |
| This journal is © the Owner Societies 2023 |