
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal control of organic semiconductors for trace detection of explosives†
Edward B.
Ogugu
 a,
Ross N.
Gillanders
a,
Salam
Mohammed
ab and
Graham A.
Turnbull
*a
a,
Ross N.
Gillanders
a,
Salam
Mohammed
ab and
Graham A.
Turnbull
*a
aOrganic Semiconductor Centre, SUPA, School of Physics & Astronomy, University of St Andrews, Fife KY16 9SS, UK. E-mail: gat@st-andrews.ac.uk
bSwedish EOD and Demining Centre-SWEDEC, Swedish Armed Forces, SE-575 28 Eksjö, Sweden
First published on 31st October 2023
Abstract
Organic semiconductors can be applied as ultra-sensitive fluorescent sensors for detecting trace vapours of explosives. The detection of explosives is manifest by the fluorescence quenching of the sensors. However, for many organic fluorescent sensors, the fluorescence quenching is irreversible and imposes a limitation in terms of reusability. Here we present a study of the thermal control of thin-film fluorescent sensors made from the commercial fluorescent polymer Super Yellow (SY). Thermal control of the sensor's temperature results in the desorption of the absorbed analytes, nitroaromatic explosives (2,4-DNT and DNB), and a taggant molecule (DMDNB). The amount of photoluminescence (PL) quenching and the desorption temperature of analytes provides a route to discriminate between the analytes, and additonally make the SY sensors reusable.
Introduction
Explosive detection remains critical for homeland security and humanitarian demining because of the use of improvised explosives devices (IEDs) by terrorists to wreak havoc.1 The use of explosives weapons in military conflicts has resulted in unexploded ordnances (UXOs) and landmines being left behind after conflicts, causing the deaths of security personnel and civilians.2 Moreover, besides UXOs and landmines acting as victim-activated traps for civilians, the soil where these explosives are planted is polluted, posing a severe health threat/impacting agriculture. Hence, detecting explosives remains critical in clearing lands of IEDs, UXOs, and landmines. Several commercially available devices can be used to detect bulk explosives, such as X-ray,3 radar imaging,4etc. Alternately, trace particles of explosives can be detected with high specificity using instrumentation like ion mobility spectrometry, gas chromatography,5etc. However, current laboratory-based trace detection approaches can be bulky and complex to operate, and require repeated calibration and highly-trained operators, and so are not well suited for many in-field deployment scenarios.Fluorescent organic semiconducting polymers are a promising complementary technology with potential for mobile, in-field deployment of explosives detection. They can show very high sensitivity to trace explosives molecules, with detection limits comparable to sniffer dogs.6 The polymers can be configured as thin film chemical sensors using simple solution processing, to be integrated onto a wide range of substrates. When immersed in the trace vapour of nitrated explosives molecules such as trinitrotoluene (TNT), the film shows a change in fluorescence. TNT is a major explosive found in the main charge of most landmines, and detection limits down to part per billion (ppb) to part per trillion (ppt) have been reported using organic fluorescent sensors.7,8 The optical and electronic properties of the polymers may be tuned by changing the molecular structure. Other advantages of using organic fluorescent polymers include making portable and relatively cheaper sensors compared with other sensing technologies,9 and the transduction is a direct conversion of incident chemical into optical readout. The optical readout typically manifests as a reduction in fluorescence intensity, resulting from the transfer of an electron from a photoexcited fluorescent material to the analyte (TNT, for example), forming a charge transfer state and then returning the sensor and analyte to the ground state non-radiatively,8,10 thereby reducing the fluorescence intensity of the sensor. This fluorescence quenching gives information about the presence of explosives in the environment.
However, like other sensing technologies, selectivity is challenging when using most fluorescent sensors. Common interferents, such as benzophenone used in perfume and other cosmetics, can also quench the fluorescence of an organic fluorescent sensor.11 One way to make fluorescent sensors specific to analyte detection is by using an array of fluorescent sensors. The responses from the various array elements give a fingerprint unique to a particular analyte.12,13 Recently, Campbell and Turnbull14 presented some computational and experimental results studying the interactions of explosives molecules with fluorescent films of various thicknesses. They suggested that analysis should make use of fluorescence recovery after a chemical incident instead of fluorescent quenching because this could give better information about the sensor(s)/analyte(s) interactions, thereby discriminating between analytes.
In addition to using fluorescence recovery to address the specificity challenge, recovery of a sensor's fluorescence would make the organic fluorescent sensors reusable. That is, we can reset the sensor after exposure to analytes and use them multiple times before replacement. A fluorescent sensor capable of multiple uses is essential when the sensors are used in field applications, limiting the number of fresh sensors required.
For many organic fluorescent sensors, the quenching of fluorescence is irreversible or exhibits extremely slow reversibility, which imposes a limitation in reusability. The irreversible interaction has been attributed to a strong binding interaction between the thin film of the sensors and the analytes.14,15
A promising approach to make fluorescent sensors reusable is the application of heat to thermally desorb the absorbed analytes.16,17 Tang et al.17 showed fluorescence recovery of some polydendrimers after exposure to a range of analytes. The fluorescent recovery was achieved by placing the quenched films in a stream of nitrogen and applying heat. Temperature ramps up to 120 °C desorbed the analytes and resulted in the recovery of the sensor's fluorescence. However, a detailed analysis of the dynamics of the thermal release of analytes from fluorescent sensors has not yet been reported.
Here we present a study of the thermal release of a range of explosives molecules from thin-film fluorescent sensors made from the commercial fluorescent polymer Super Yellow (SY). Thermal control of the sensor's temperature can overcome the binding interactions of the absorbed analytes and makes the sensor reusable, and additionally provides a route to confirm that the fluorescence quenching was due to the absorbed analytes. Our sensing setup was custom-built and made of stainless steel. Analytes adsorption on the internal surfaces of the chamber was minimised by using a vacuum pump to evacuate the chamber after thermal desorption, thereby ensuring no recontamination of the sensor from a previous exposure. The efficacy of PL quenching and the desorption temperature of analytes from the SY sensor may be used to discriminate between analytes.
2. Experimental methods
Fluorescent polymer Super Yellow (SY) (PDY-132) and solvents were purchased from Sigma Aldrich (Merck) and were used without further purification.To prepare sensor films, SY was dissolved in chlorobenzene at 6.50 mg ml−1. The solutions were then spin-coated at 2000 rpm for 60 seconds on glass substrates (20 × 20 mm), giving a thickness of 90 nm. The films were then annealed at 100 °C for 30 minutes, allowed to cool down to room temperature on the hot plate, and stored until usage. All processing was undertaken in a nitrogen-filled glove box. For optical absorbance measurements, fused quartz plates of 12 mm diameter (UQG optics) were used as substrates. Before spin coating, the substrates were cleaned by ultrasonicating for 10 minutes in acetone then isopropanol. The substrates were dried in a nitrogen stream and plasma ashed in 100% oxygen (Plasma Technology MiniFlecto) for 3 minutes. The effect of the processing environment on the films was investigated by preparing solutions and spin-coating films in a nitrogen-filled glove box (oxygen < 6 ppm, water < 0.1 ppm) and in ambient air, results shown in ESI† S1 and S2.
The films were characterised by measuring the optical absorbance using a Cary 300 UV-vis spectrophotometer and photoluminescence using an Edinburgh Instruments FLS980 fluorimeter. Photoluminescence quantum yield (PLQY) measurements were performed in an integrating sphere using a Hamamatsu Photonics C9920-02 measurement system with 444 nm excitation wavelength. Films thicknesses were determined using an Ellipsometer (J. AWoollam M2000U).
Explosives sensing was performed in a custom-made vacuum-tight chamber made of stainless steel with a metal ceramic heater (Thorlabs, Germany HT24S) attached to the sample holder, as shown in Fig. 1.
 | ||
| Fig. 1 Experimental setup for sensing and thermal release of explosives. | ||
A k-type thermocouple (RS components) was held in contact with the SY film and connected to a PID temperature controller with a recorder data logger (ThermoMart, PID-SSR-USB) to read and record the sensor's temperature.
For each sensing experiment, a fluorescent film was placed in the chamber, and clean nitrogen gas flowed for 60 seconds to displace oxygen from the chamber and create a nitrogen atmosphere. Then photoexcitation of the film was performed using 405 nm continuous wave laser light from a diode laser (Photonic Solutions) after attenuation of the power to 80 μW. A lens was used to expand the laser beam to illuminate an area of 1.73 ± 0.01 cm2 of the film.
Photoluminescence from the sensors was measured using a fibre-coupled CCD spectrometer, taking a measurement every 3 s for 300 s (variation on this measurement protocol are specified against the experiments in the Results and discussion section). Explosive vapours were generated using the setup as shown in ref. 14 by flowing nitrogen at a rate of 6 L min−1 across 1 g of powder of each analyte to generate a continuous flow of vapour significantly lower than the saturated vapour pressure18,19 of the explosives.
Four different protocols were tested to check for fluorescence recovery. (1) A flow of clean nitrogen gas was used for flushing the analyte-exposed sensors at room temperature, (2) evacuation of the chamber using a vacuum pump (to create a low pressure of 10−2 mbar) followed by a flow of clean nitrogen at room temperature (3) the sensors were heated to 90 °C to desorb analytes followed by a flow of clean nitrogen, and (4) the sensors were heated to 90 °C to release the analytes followed by evacuation of the chamber using a vacuum pump to create a vacuum of 10−2 mbar, before a flow of nitrogen gas flushed out any remnants of the desorbed analytes from the chamber, and created a clean nitrogen atmosphere for the next experiment.
3. Results and discussion
3.1. Explosives sensing and recovery at room temperature
The fluorescence response of a 90 nm SY film exposed to 2,4-DNT vapour is shown in Fig. 2. The DNT exposure was from 30 seconds to 90 seconds, followed by a continuous flow of nitrogen at the rate of 6 L min−1 to check for recovery. During DNT exposure the PL showed a decrease in intensity but no change in spectrum (Fig. 2b). The PL continued to decrease more slowly after the vapour was replaced with a clean nitrogen flow. The sensing mechanism7,8,10,20 is shown in Fig. 2a, photoexcitation of an organic fluorescent sensor results in the promotion of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (SY polymer, for example with HOMO and LUMO,21 −5.3 eV and −2.9 eV, respectively). The promoted electron remains bound to the positive charge left behind, forming a singlet exciton; recombination of the exciton results in the sensor's photoluminescence (PL) emission. | ||
| Fig. 2 (a) The principle of explosives detection (DNT in this case) using a conjugated polymer, Super Yellow (SY). The HOMO and LUMO level of SY and DNT were obtained from ref. 21 and 22, respectively (b) experimental result showing the response of a 90 nm Super Yellow (SY) sensor to 2,4-DNT at room temperature. DNT exposure was from 30 s to 90 s. | ||
However, when a trace explosive molecule such as 2,4-DNT (with HOMO and LUMO,22 −8.5 eV and −3.8 eV, respectively) is in contact with the sensor, the excited electron is transferred from the LUMO of the sensor to the LUMO of the explosive molecule. The charge transfer/separated state decays non-radiatively, resulting in a PL reduction of the sensor and giving information about an analyte's presence. The driving force for the electron transfer is the positive offset between the LUMO of the sensor and the LUMO of the explosive molecule, and the offset should be higher than the exciton binding energy.
The continuing PL quenching during the second flow of clean nitrogen suggests that the DNT molecules remain in the SY film due to a strong binding interaction, and molecules may be diffusing further into the film.14,23
Fig. 3 presents the change in PL from the SY sensor, when exposed to vapours of DNT, DNB, and DMDNB. The red rectangular region (from 31–90 seconds) represents the period of analyte exposure to the sensor, which is followed by a period of flowing clean nitrogen, to enable a release of the analytes from the sensor. We find there is a further small decrease in the PL for all analytes during the second flow of clean nitrogen instead of a PL recovery, which may be attributed to analytes' diffusion further into the film14,23 and/or recontamination of the sensors by the gradual desorption of analytes from the walls of the tubing and sensing chamber to the sensor. SY sensors have strong binding interactions with several explosives vapours, which poses a limitation in terms of reusability.
 | ||
| Fig. 3 SY response to nitroaromatic analytes (DNT and DNB) and a taggant (DMDMB); the normalised PL is obtained by integrating the PL intensity across the wavelengths and normalising. The rectangle strip bar indicates the duration of analytes exposure. Each quenching experiment was repeated three times, and the solid lines show the average. | ||
3.2. Fluorescence recovery through thermal desorption of analytes
The binding of explosives molecules to organic semiconductor films may be overcome, and the sorbed analytes desorbed, when the film is raised to higher temperatures. Fig. 4(a) shows the normalised PL emitted from a SY film during exposure to DNT vapour at room temperature, before the temperature was raised from room temperature (∼20 °C) to 90 °C. As we increase the temperature, we observe a slight drop in the PL of SY film which can be related to the film's intrinsic response to temperature, followed by a rapid increase of the PL starting at 138 seconds, corresponding to a temperature of 53 °C. We attribute this increase to when the DNT vapour was released from the SY film. The film's temperature was then lowered from 90 °C to 22 °C by turning off the heater and flowing clean nitrogen across the SY film. This results in an initial increase in PL from 180 seconds to 206 seconds, a response of the SY to temperature, that is, on cooling the SY film towards room temperature. However, during further cooling of the film a gradual decrease in the PL, is then observed, which we attribute to a recontamination of the SY sensor by DNT vapour from the walls of the chamber. A detailed analysis of analytes recontamination of the SY sensor and how it was resolved is presented later in Section 3.5. Fig. 4(b) shows the PL response of pristine SY films to a similar heating cycle. The temperature ramp to 90 °C results in 28% drop in PL which fully recovers on cooling the film to room temperature.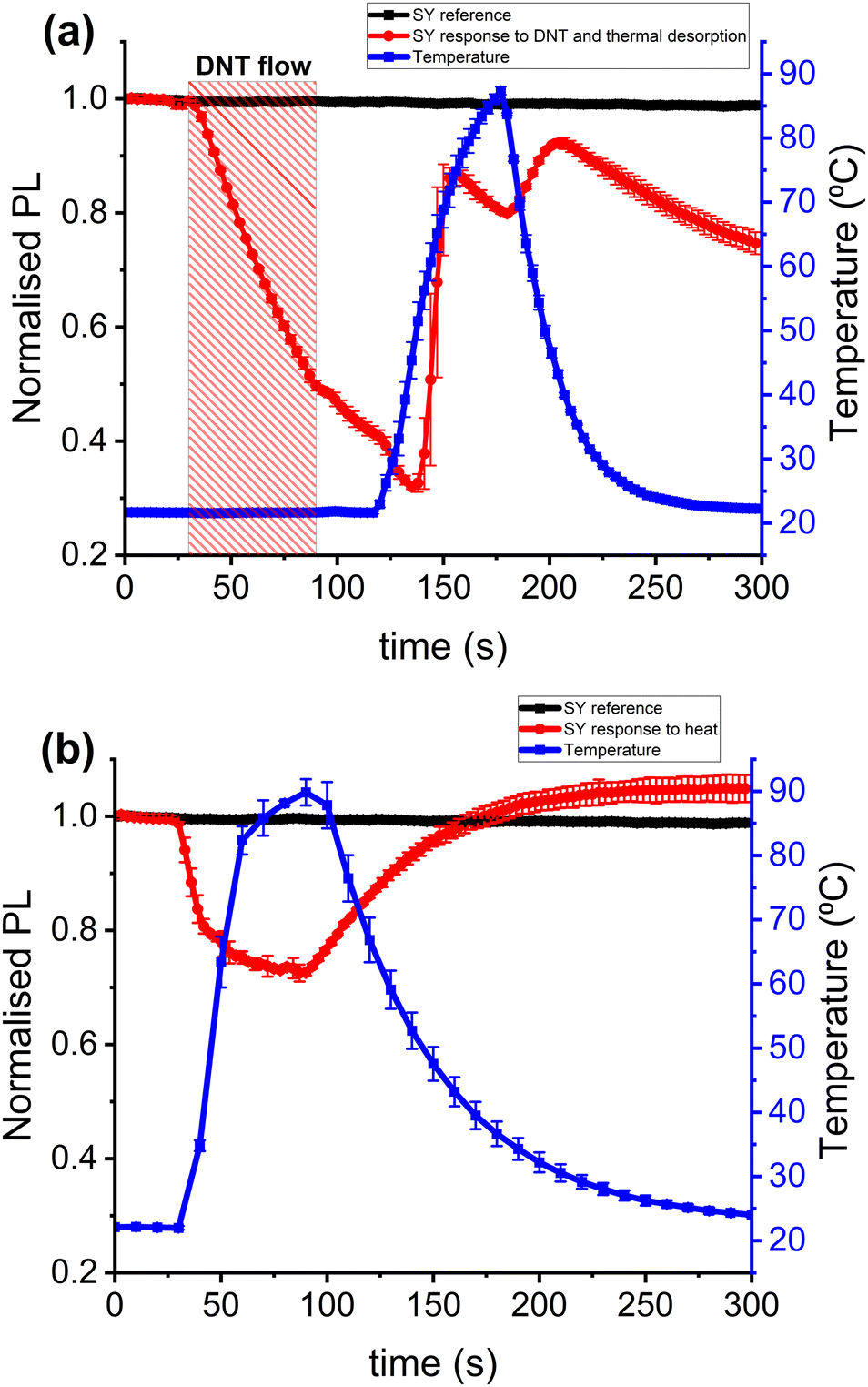 | ||
| Fig. 4 90 nm SY sensor: (a) PL quenching due to 2,4-DNT at room temperature and thermal desorption of DNT vapour from the sensor. We attribute the rise in PL starting at 138 seconds, as the time the explosive vapour is desorbed from the SY, corresponding to a temperature of 53 °C to 70 °C. (b) Response of pristine SY film to temperature ramp. | ||
3.3. Analytes desorption temperature – a route to selective detection
The affinity of a sensor for a specific analyte is influenced by the chemical composition of the explosive molecule, which then determines the sensor–analyte interaction. For example, the benzene ring of the DNT molecule could form a π-stacking interaction15,24 with the binding sites of SY polymer. These binding sites can be the phenyl rings of the SY backbone or those of the side chains. Because of the variability of the chemical compositions of explosives compounds, it is expected that analyte–sensor intermolecular interactions for different explosives should have different binding energy24 and, consequently, different desorption energy. So, the sensor temperature at which sorbed explosives are released reflects the desorption energy. The desorption temperature of volatile organic compounds (VOCs) has previously been used for the selective detection of VOCs,25 where they attributed the selectivity to the boiling points of the VOCs and their affinity for the material in which they are in contact. Fig. 5 shows the temperatures at which nitroaromatic explosives (2,4-DNT and DNB) and a taggant molecule (DMDNB) are released from SY sensors. In Fig. 5(a–c), SY sensors were exposed to the various analytes indicated at room temperature, which resulted in PL quenching. The degree of PL quenching depends on the vapour pressure of the explosives and how far the explosives molecules can diffuse into the SY film and interact with the fluorescence-emitting site of the sensor. An increase in the sensor's temperature results in the desorption of the sorbed explosives, and the desorption temperature could be used to support the selective detection of various explosives. Fig. 5 shows DNT is desorbed at the highest temperature (∼57 °C) and the taggant at lowest temperature (∼45 °C). A comparison of sensor response and recovery of nitroaromatic explosives with several potential interferents is presented in ESI† (Table S1 and Fig. S1). The temperature and peak rate of PL recovery are found to offer a small degree of discrimination, even between the analytes of very similar molecular structures. This indicates that the thermal release of sorbed analyte molecules has potential to be used in future to help classify/identify analyte vapours. | ||
| Fig. 5 PL quenching of 90 nm SY sensors due to reaction with various analytes and their subsequent thermal desorption: (a) shows DNT release at 60–70 °C, (b) shows the release of DNB at 45–60 °C, (c) shows the shows the release of DMDNB (a taggant) at 40–60 °C, and (d) the shows the response of pristine SY film to thermal cycling. | ||
We note that changing the speed of heating can have some effect on the desorption onset temperature measured, for example an average temperature ramp rate of 2 °C s−1 gives an onset of desorption at a temperature around 5 °C higher than when using a ramp of 1 °C s−1, while slower ramp rates give a similar onset value to 1 °C s−1. This difference illustrates the importance of slowly varying the temperature to find the threshold for analyte release, and a need to compare the desorption of different analytes at the same temperature ramp rate.
As mentioned in Section 3.2, there is evidence of recontamination of the sensors when the sensors are cooled to room temperature, with DNT showing a higher poisoning of the sensor. Fig. 5(d) shows the response of a clean SY sensor to temperature showing a PL quenching due to heat and PL recovery on cooling. A detailed analysis of the thermal response of SY films prepared in various environments is presented in ESI† Fig. S2 and S3. A blue shift in the PL spectrum with increasing temperature is observed, with about a 9 nm shift in the electronic 0–0 excitation peak (from 549 nm to 540 nm) for a temperature change of 20 °C to 90 °C respectively. This blue shift in the spectrum may be attributed to increasing thermal disorder in the polymer chain, which reduces the effective conjugation length.26,27
3.4. Temperature effect on adsorption and desorption of explosives from SY sensor
As there is an intrinsic change in PL efficiency of the polymer film with temperature, it may be convenient to operate at a fixed elevated temperature. In addition, in real-world detection of explosives, the sensors would be deployed in varied environmental conditions which may affect how they respond to explosives. Therefore, it is essential to understand the range of temperatures that can result in a binding event of explosives vapour to a sensor. We show that the SY sensor can detect DNT vapour even at temperatures above 21 °C, as shown in Fig. 6. | ||
| Fig. 6 Sensing of 2,4-DNT using SY films at various temperatures. The grey rectangular stripes from 120 to 600 seconds represents when the heater was turned on at a fixed temperature as indicated against each curve, and the red bars indicate the two consecutive periods of DNT exposure. Sensing at 40 to 50 °C shows a reversible quenching, in contrast to temperatures at 35 °C and below, which only show PL quenching at each exposure. The sensing at 60 °C and above shows little or no response to the analyte. | ||
For each sensing experiment, a new SY film was placed in the chamber, followed by a flow of clean nitrogen at 6 L min−1 throughout the experiment, from 0 to 900 seconds, except for the period of DNT exposure. The grey rectangular hatched bar from 120 seconds to 600 seconds represents when the heater was turned on, while the two red rectangle stripes represent the DNT exposure periods. The responses of SY films at various temperatures can be broadly categorised into three regions: for temperatures from 21 °C to 35 °C, there is accumulative PL quenching in the two periods of DNT exposures. In the second regime (from 40 °C to 50 °C), there is a reversible binding of DNT vapour to the SY films. This indicates that SY sensors can be utilised even in countries with high temperatures. The third regime (from 65 °C to 70 °C), there is no evidence of a binding event of DNT molecules to the SY sensors. This indicates the importance of molecular binding to an observable change in PL.
3.5. Thermal control of fluorescent sensors for multiple usages
As mentioned in Section 3.2, we attribute the gradual PL quenching after the thermal desorption of the analyte in Fig. 4(a) to a recontamination of the SY sensor to DNT vapour. Explosives molecules are known to adhere strongly to various surfaces. Trace explosives particles and vapour can adhere to various materials such as glass, aluminium, textile, and stainless steel.28,29 The explosives vapour generator and the sensing chamber used in these experiments are made of stainless steel, and the chamber windows (see Fig. 1) are made of fused quartz. We hypothesise that DNT molecules adhered to the internal surface of the stainless-steel tube used to deliver explosive vapour to the chamber and on the fused quartz windows and chamber wall surfaces. Therefore, the adhered DNT molecules are gradually desorbed during the flow of clean nitrogen. Some molecules could interact with the sensor while propagating out of the chamber via the nitrogen stream.Evidence of DNT molecules being stuck in the stainless-steel tubing and chamber after DNT exposure is shown in ESI† S4(a) and (b). A roughing pump was connected to the chamber outlet, and a pristine SY film was placed in the chamber to check for DNT adsorption before and after sensing experiments. We observed a rapid PL quenching of the SY films when the chamber was pumped down to a vacuum of ∼10−2 mbar in post sensing experiment checks, suggesting the adsorbed DNT molecule are being released.
To minimise the adsorption of explosives vapour, Grate et al.30 used thermal control to maintain the flow stream system of an explosives vapour generator at elevated temperatures, thereby reducing or preventing explosives adsorption on the tubing. However, operating the vapour generator or the flow stream tubing at elevated temperatures will also affect the explosives' vapor pressure.19 It can also affect the temperature of the fluorescent sensor because the stream of analytes reaching the sensor would be at an elevated temperature. Collins et al.29 mentioned that they significantly reduced the adsorption of RDX by coating a chamber made of stainless steel with “SilcoNert 2000” without heating the flow stream system.
We explored using a vacuum pump to remove the adhered analytes from the tubing and sensing chamber after thermally desorbing the sorbed analytes from the SY films. This procedure makes the SY film reusable without any recontamination during nitrogen flow. Fig. 7(a) shows an experiment where a SY film was exposed to DNT at room temperature from 30 s to 90 s which resulted in a 40% drop in the PL, followed by a further drop of ∼5% during clean nitrogen flow. On heating, there is a further drop in PL of 10% due to the increase in the sensor's temperature, followed by a rapid release of the sorbed DNT molecules from the film. Turning the vacuum pump on results in an initial drop in the sensor's temperature from ∼90 °C to 64 °C, which can be attributed to the sensor losing some of its thermal energy to the surrounding air leaving the chamber. As mentioned before, turning on the vacuum from ∼10−2 to 10−3 mbar releases the adsorbed analytes from the tubing and chamber, and the released analytes could recontaminate the sensor as they are being drawn out of the chamber; there is no evidence of recontamination of the SY by the desorbed DNT vapour (red curve). This is because the DNT vapour cannot bind to the sensor at high temperatures (as shown in Fig. 6), between 90 °C and 64 °C in this case. This protocol has been used to demonstrate multiple uses of a SY sensor for detecting DNT, as shown in the Fig. 7(b). Multiple uses of the SY sensor for DNT sensing using thermal control without a vacuum pump result in DNT poisoning of the SY film after the exposure cycle, as shown in ESI† S5.
 | ||
| Fig. 7 (a) PL quenching due to 2,4-DNT at room temperature and thermal desorption of DNT vapour from the sensor (red curve), using a vacuum pump to prevent or minimise recontamination. The red and blue rectangular stripes are the periods of DNT exposure and vacuum pump, respectively. (b) Result of multiple uses of SY for sensing DNT using thermal control and a pump. The blue curve shows the temperature cycling, the red curve shows the multiple quenching and recovery of the SY sensor PL, and the black curve is the reference showing the stability of SY film under nitrogen flow of 6 L min−1. The red and blue rectangle stripes are the periods of DNT exposure and vacuum pump, respectively. | ||
Conclusions
We have demonstrated that the commercially available conjugated polymer Super Yellow (SY) can be used as a highly sensitive and reusable sensor for detecting nitroaromatic explosives and a taggant. An increase in the sensor temperature overcomes the analyte binding interaction and allows the sorbed analytes to diffuse out of the thin film, which results in the recovery of the PL of the sensors. We show the degree of PL quenching and the temperatures at which the analytes are desorbed may be used to address the challenge of selective detection. Analyte recontamination of the SY sensors was minimised by vacuum pumping in conjunction with controlling the sensor temperature, which provides a route for multiple uses of the SY sensors. This protocol can be applied to other organic semiconductors, thereby addressing the challenge of selectivity and reusability of fluorescent sensors for trace detection of explosives.Author contributions
Edward B. Ogugu: conceptualisation, methodology, software, formal analysis, investigation, data curation, writing – original draft preparation, visualisation, writing – review and editing. Ross N. Gillanders: conceptualisation, methodology, formal analysis, validation, writing – review and editing, funding acquisition. Salam Mohammed: formal analysis, validation, writing – review and editing. Graham A. Turnbull: conceptualisation, methodology, formal analysis, validation, writing – review and editing, funding acquisition, supervision, project administration. All authors have read and agreed to the published version of the manuscript.Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/603e5964-f6e3-4ab4-b481-f23bb086c2eb.31Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge scientific discussions with Prof I. D. W. Samuel. The authors acknowledge funding from NATO Science for Peace & Security under grant agreement MYP G5355. Edward B. Ogugu acknowledges funding from the Commonwealth Scholarship Commission and the Foreign, Commonwealth and Development Office in the UK. He is grateful for their support. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.Notes and references
- J.-P. Pham, Boko Haram's evolving threat, Africa Center for Strategic Studies Washington, WA, 2012 Search PubMed.
- E. Lin, Am. J. Political Sci., 2022, 66, 222–237 CrossRef.
- K. Wells and D. A. Bradley, Appl. Radiat. Isot., 2012, 70, 1729–1746 CrossRef CAS PubMed.
- D. J. Daniels, Sens. Imaging., 2006, 7, 90–123 CrossRef.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS.
- C. J. Cumming, C. Aker, M. Fisher, M. Fok, M. J. L. Grone, D. Reust, M. G. Rockley, T. M. Swager, E. Towers and V. Williams, IEEE Trans. Geosci. Electron., 2001, 39, 1119–1128 Search PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- R. N. Gillanders, I. D. W. Samuel and G. A. Turnbull, Sens. Actuators, B, 2017, 245, 334–340 CrossRef CAS.
- A. Räupke, A. Palma-Cando, E. Shkura, P. Teckhausen, A. Polywka, P. Görrn, U. Scherf and T. Riedl, Sci. Rep., 2016, 6, 29118 CrossRef PubMed.
- S. W. Thomas Iii, J. P. Amara, R. E. Bjork and T. M. Swager, Chem. Commun., 2005, 4572–4574, 10.1039/B508408C.
- N. Bolse, R. Eckstein, M. Schend, A. Habermehl, C. Eschenbaum, G. Hernandez-Sosa and U. Lemmer, Flexible Printed Electron., 2017, 2, 024001 CrossRef.
- J. R. Askim, M. Mahmoudi and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 8649–8682 RSC.
- I. A. Campbell and G. A. Turnbull, Phys. Chem. Chem. Phys., 2021, 23, 10791–10798 RSC.
- D. Zhao and T. M. Swager, Macromolecules, 2005, 38, 9377–9384 CrossRef CAS.
- P. E. Shaw, H. Cavaye, S. S. Y. Chen, M. James, I. R. Gentle and P. L. Burn, Phys. Chem. Chem. Phys., 2013, 15, 9845–9853 RSC.
- G. Tang, S. S. Y. Chen, P. E. Shaw, K. Hegedus, X. Wang, P. L. Burn and P. Meredith, Polym. Chem., 2011, 2, 2360–2368 RSC.
- R. G. Ewing, M. J. Waltman, D. A. Atkinson, J. W. Grate and P. J. Hotchkiss, TrAC, Trends Anal. Chem., 2013, 42, 35–48 CrossRef CAS.
- P. A. Pella, Anal. Chem., 1976, 48, 1632–1637 CrossRef CAS.
- P. E. Shaw and P. L. Burn, Phys. Chem. Chem. Phys., 2017, 19, 29714–29730 RSC.
- B. Walker, M. Ullah, G. J. Chae, P. L. Burn, S. Cho, J. Y. Kim, E. B. Namdas and J. H. Seo, Appl. Phys. Lett., 2014, 105, 183302 CrossRef.
- D. Dinda, A. Gupta, B. K. Shaw, S. Sadhu and S. K. Saha, ACS Appl. Mater. Interfaces, 2014, 6, 10722–10728 CrossRef CAS PubMed.
- M. A. Ali, S. S. Y. Chen, H. Cavaye, A. R. G. Smith, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, Sens. Actuators, B, 2015, 210, 550–557 Search PubMed.
- L. Liu, J. Hao, Y. Shi, J. Qiu and C. Hao, RSC Adv., 2015, 5, 3045–3053 RSC.
- W. Winter, C. Day, J. Prestage and T. Hutter, Analyst, 2021, 146, 109–117 RSC.
- F. A. C. Oliveira, L. A. Cury, A. Righi, R. L. Moreira, P. S. S. Guimarães, F. M. Matinaga, M. A. Pimenta and R. A. Nogueira, J. Chem. Phys., 2003, 119, 9777–9782 CrossRef CAS.
- E. W. Snedden, L. A. Cury, K. N. Bourdakos and A. P. Monkman, Chem. Phys. Lett., 2010, 490, 76–79 CrossRef CAS.
- H. A. Yu, T. Becker, N. Nic Daeid and S. W. Lewis, Forensic Sci. Int., 2017, 273, 88–95 CrossRef CAS PubMed.
- G. E. Collins, B. C. Giordano, V. Sivaprakasam, R. Ananth, M. Hammond, C. D. Merritt, J. E. Tucker, M. Malito, J. D. Eversole and S. Rose-Pehrsson, Rev. Sci. Instrum., 2014, 85, 054101 CrossRef PubMed.
- J. W. Grate, R. G. Ewing and D. A. Atkinson, TrAC, Trends Anal. Chem., 2012, 41, 1–14 CrossRef CAS.
- E. B. Ogugu, R. N. Gillanders and G. A. Turnbull, Dataset, University of St Andrews Research Portal, 2023 DOI:10.17630/603e5964-f6e3-4ab4-b481-f23bb086c2ebI.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02868b |
| This journal is © the Owner Societies 2023 |